User login
COMMENT & CONTROVERSY
Misoprostol: Clinical pharmacology in obstetrics and gynecology
ROBERT L. BARBIERI, MD (JULY 2022)
Outcomes from my practice’s pilot study
In his recent editorial, Dr. Barbieri addressed the important topic of office-based cervical ripening prior to inpatient induction of labor. In order to decrease the length of labor and increase the success of vaginal delivery, the cervical factor is of prime importance. Patients with an unfavorable cervix (Bishop score of ≥6) are more likely to experience longer labor, risk of infection, fetal distress, etc, and may end up with an unwanted cesarean delivery. To prevent the above, numerous approaches (mechanical methods, double-balloon catheter, laminaria, misoprostol among others) have been discussed.
The inclusion criteria for office-based cervical ripening are low-risk patients, singleton pregnancies between 39 and 40 weeks of gestation, and cephalic presentation. The details of inclusion and exclusion criteria have to be determined by each practice individually. Our practice went a step further. We performed a small pilot study to assess the safety and efficacy of office cervical ripening in low-risk primigravid patients with low Bishop scores who were not scheduled for induction in anticipation of labor. Ten primigravid patients with poor Bishop scores (6 or less) were administered 50 µg misoprostol at 39+ weeks of pregnancy in the office setting. Bishop scores were taken twice per week until delivery. In 7 out of 10 patients, the Bishop score became favorable within a week of treatment, and in 3 patients the Bishop score remained the same. Three out of 10 patients experienced self-limited episodes of uterine contractility, and 2 of the patients went into labor within 3 days of using misoprostol. All patients were delivered within 2 weeks of treatment without an induction: 8 delivered vaginally, and 2 by cesarean delivery.2
Cesarean delivery was done for fetal distress (1 case) and prolonged second stage of labor (1 case). All neonates were born in satisfactory condition with Apgar scores between 7 and 10. Our preliminary results demonstrated marked improvement in cervical ripening judged by the Bishop score in 70% of patients.2
A prospective randomized study should be performed with the following agenda:
- Does late pregnancy medical cervical ripening in low-risk patients affect labor course and cesarean delivery rate?
- What is the optimal dose and route of administration of misoprostol?3,4
References
- Barbieri R. Office-based ambulatory cervical ripening prior to in patient induction of labor. OBG Manag. 2021;33:9-13.
- Petrikovsky B. Should cervical ripening become routine in primigravid low risk patients [In press]. Neonat Int Care. 2022:1, 4-6.
- Sharami SH, Milani F, Faraji R. Comparison of 25 µg sublingual and 50 µg intravaginal misoprostol for cervical ripening and labor: a randomized controlled equivalence trial. Arch Med. 2014:10:653-656.
- Barbieri R. Misoprostol: clinical pharmacology in obstetrics and gynecology. OBG Manag. 2022:34:7, 8-12.
B. Petrikovsky, MD, PhD
New Hyde Park, New York
Dr. Barbieri responds
I appreciate that Dr. Petrikovsky took time from a busy practice to provide our readers with his very innovative idea. I agree with him that a clinical trial is warranted to test the effects of late pregnancy medical cervical ripening in low-risk patients on labor course and birth outcome. Maybe one of our readers will take on the challenge to complete such a trial! ●
Misoprostol: Clinical pharmacology in obstetrics and gynecology
ROBERT L. BARBIERI, MD (JULY 2022)
Outcomes from my practice’s pilot study
In his recent editorial, Dr. Barbieri addressed the important topic of office-based cervical ripening prior to inpatient induction of labor. In order to decrease the length of labor and increase the success of vaginal delivery, the cervical factor is of prime importance. Patients with an unfavorable cervix (Bishop score of ≥6) are more likely to experience longer labor, risk of infection, fetal distress, etc, and may end up with an unwanted cesarean delivery. To prevent the above, numerous approaches (mechanical methods, double-balloon catheter, laminaria, misoprostol among others) have been discussed.
The inclusion criteria for office-based cervical ripening are low-risk patients, singleton pregnancies between 39 and 40 weeks of gestation, and cephalic presentation. The details of inclusion and exclusion criteria have to be determined by each practice individually. Our practice went a step further. We performed a small pilot study to assess the safety and efficacy of office cervical ripening in low-risk primigravid patients with low Bishop scores who were not scheduled for induction in anticipation of labor. Ten primigravid patients with poor Bishop scores (6 or less) were administered 50 µg misoprostol at 39+ weeks of pregnancy in the office setting. Bishop scores were taken twice per week until delivery. In 7 out of 10 patients, the Bishop score became favorable within a week of treatment, and in 3 patients the Bishop score remained the same. Three out of 10 patients experienced self-limited episodes of uterine contractility, and 2 of the patients went into labor within 3 days of using misoprostol. All patients were delivered within 2 weeks of treatment without an induction: 8 delivered vaginally, and 2 by cesarean delivery.2
Cesarean delivery was done for fetal distress (1 case) and prolonged second stage of labor (1 case). All neonates were born in satisfactory condition with Apgar scores between 7 and 10. Our preliminary results demonstrated marked improvement in cervical ripening judged by the Bishop score in 70% of patients.2
A prospective randomized study should be performed with the following agenda:
- Does late pregnancy medical cervical ripening in low-risk patients affect labor course and cesarean delivery rate?
- What is the optimal dose and route of administration of misoprostol?3,4
References
- Barbieri R. Office-based ambulatory cervical ripening prior to in patient induction of labor. OBG Manag. 2021;33:9-13.
- Petrikovsky B. Should cervical ripening become routine in primigravid low risk patients [In press]. Neonat Int Care. 2022:1, 4-6.
- Sharami SH, Milani F, Faraji R. Comparison of 25 µg sublingual and 50 µg intravaginal misoprostol for cervical ripening and labor: a randomized controlled equivalence trial. Arch Med. 2014:10:653-656.
- Barbieri R. Misoprostol: clinical pharmacology in obstetrics and gynecology. OBG Manag. 2022:34:7, 8-12.
B. Petrikovsky, MD, PhD
New Hyde Park, New York
Dr. Barbieri responds
I appreciate that Dr. Petrikovsky took time from a busy practice to provide our readers with his very innovative idea. I agree with him that a clinical trial is warranted to test the effects of late pregnancy medical cervical ripening in low-risk patients on labor course and birth outcome. Maybe one of our readers will take on the challenge to complete such a trial! ●
Misoprostol: Clinical pharmacology in obstetrics and gynecology
ROBERT L. BARBIERI, MD (JULY 2022)
Outcomes from my practice’s pilot study
In his recent editorial, Dr. Barbieri addressed the important topic of office-based cervical ripening prior to inpatient induction of labor. In order to decrease the length of labor and increase the success of vaginal delivery, the cervical factor is of prime importance. Patients with an unfavorable cervix (Bishop score of ≥6) are more likely to experience longer labor, risk of infection, fetal distress, etc, and may end up with an unwanted cesarean delivery. To prevent the above, numerous approaches (mechanical methods, double-balloon catheter, laminaria, misoprostol among others) have been discussed.
The inclusion criteria for office-based cervical ripening are low-risk patients, singleton pregnancies between 39 and 40 weeks of gestation, and cephalic presentation. The details of inclusion and exclusion criteria have to be determined by each practice individually. Our practice went a step further. We performed a small pilot study to assess the safety and efficacy of office cervical ripening in low-risk primigravid patients with low Bishop scores who were not scheduled for induction in anticipation of labor. Ten primigravid patients with poor Bishop scores (6 or less) were administered 50 µg misoprostol at 39+ weeks of pregnancy in the office setting. Bishop scores were taken twice per week until delivery. In 7 out of 10 patients, the Bishop score became favorable within a week of treatment, and in 3 patients the Bishop score remained the same. Three out of 10 patients experienced self-limited episodes of uterine contractility, and 2 of the patients went into labor within 3 days of using misoprostol. All patients were delivered within 2 weeks of treatment without an induction: 8 delivered vaginally, and 2 by cesarean delivery.2
Cesarean delivery was done for fetal distress (1 case) and prolonged second stage of labor (1 case). All neonates were born in satisfactory condition with Apgar scores between 7 and 10. Our preliminary results demonstrated marked improvement in cervical ripening judged by the Bishop score in 70% of patients.2
A prospective randomized study should be performed with the following agenda:
- Does late pregnancy medical cervical ripening in low-risk patients affect labor course and cesarean delivery rate?
- What is the optimal dose and route of administration of misoprostol?3,4
References
- Barbieri R. Office-based ambulatory cervical ripening prior to in patient induction of labor. OBG Manag. 2021;33:9-13.
- Petrikovsky B. Should cervical ripening become routine in primigravid low risk patients [In press]. Neonat Int Care. 2022:1, 4-6.
- Sharami SH, Milani F, Faraji R. Comparison of 25 µg sublingual and 50 µg intravaginal misoprostol for cervical ripening and labor: a randomized controlled equivalence trial. Arch Med. 2014:10:653-656.
- Barbieri R. Misoprostol: clinical pharmacology in obstetrics and gynecology. OBG Manag. 2022:34:7, 8-12.
B. Petrikovsky, MD, PhD
New Hyde Park, New York
Dr. Barbieri responds
I appreciate that Dr. Petrikovsky took time from a busy practice to provide our readers with his very innovative idea. I agree with him that a clinical trial is warranted to test the effects of late pregnancy medical cervical ripening in low-risk patients on labor course and birth outcome. Maybe one of our readers will take on the challenge to complete such a trial! ●
Options and outcomes for uterine preservation at the time of prolapse surgery
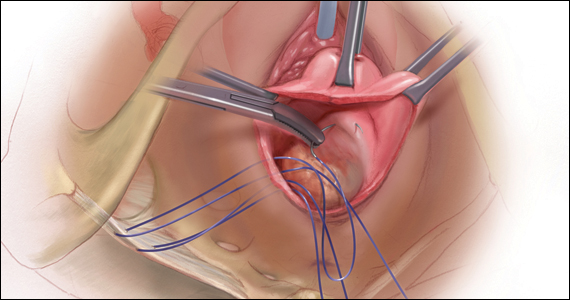
CASE Patient desires prolapse repair
A 65-year-old postmenopausal patient (G3P3) presents to your office with symptoms of a vaginal bulge for more than 1 year. She has no urinary incontinence symptoms and no bowel dysfunction symptoms. On examination, you diagnose stage 2 uterovaginal prolapse with both anterior and apical defects. The patient declines expectant and pessary management and desires surgery, but she states that she feels her uterus “is important for me to keep, as my babies grew in there and it is part of me.” She denies any family or personal history of breast, endometrial, or ovarian cancer and has no history of abnormal cervical cancer screening or postmenopausal bleeding. What are the options for this patient?
Who is the appropriate hysteropexy patient, and how do we counsel her?
Uterine prolapse is the third leading cause of benign hysterectomy, with approximately 70,000 procedures performed each year in the United States. It has long been acknowledged that the uterus is a passive bystander to the prolapse process,1 but modern practice often involves a hysterectomy as part of addressing apical prolapse. However, more and more uterine-preserving surgeries are being performed, with one study showing an increase from 1.8% to 5% from 2002 and 2012.2
When presented with the option to keep or remove their uterus during the time of prolapse surgery, 36% of patients indicated that they would prefer to keep their uterus with similar outcomes while 21% would still prefer uterine preservation even if outcomes were inferior compared with hysterectomy.3 Another study showed that 60% of patients would decline concurrent hysterectomy if there were equal surgical outcomes,4 and popular platforms, such as Health magazine (www.health.com) and AARP magazine (www.aarp.org), have listed benign hysterectomy as a “top surgery to avoid.”
Patients desire uterine preservation for many reasons, including concerns about sexual function and pleasure, the uterus being important to their sense of identity or womanhood, and concerns around menopausal symptoms. Early patient counseling and discussion of surgical goals can help clinicians fully understand a patient’s thoughts toward uterine preservation. Women who identified their uterus as important to their sense of self had a 28.2-times chance of preferring uterine preservation.3 Frequently, concerns about menopausal symptoms are more directly related to hormones and ovary removal, not uterus removal, but clinicians should be careful to also counsel patients on the increased risk of menopause in the 5 years after hysterectomy, even with ovarian preservation.5
There are some patients for whom experts do not recommend uterine preservation.6 Patients with an increased risk of cervical or endometrial pathology should be counseled on the benefits of hysterectomy. Additionally, patients who have abnormal uterine bleeding from benign pathology should consider hysterectomy to treat these issues and avoid future workups (TABLE). For postmenopausal patients with recent postmenopausal bleeding, we encourage hysterectomy. A study of patients undergoing hysterectomy at the time of prolapse repair found a rate of 13% unanticipated endometrial pathology with postmenopausal bleeding and negative preoperative workup.7
At this time, a majority of clinicians consider the desire for future fertility to be a relative contraindication to surgical prolapse repair and advise conservative management with pessary until childbearing is complete. This is reasonable, given the paucity of safety data in subsequent pregnancies as well as the lack of prolapse outcomes after those pregnancies.8,9 Lastly, cervical elongation is considered a relative contraindication, as it represents a risk for surgical failure.10,11 This may be counteracted with trachelectomy at the time of hysteropexy or surgeries such as the Manchester repair, which involve a trachelectomy routinely,12 but currently there is no strong evidence for this as routine practice.
Continue to: Uterine preservation surgical techniques and outcomes...
Uterine preservation surgical techniques and outcomes
Le Fort colpocleisis
First described in 1840 by Neugebauer of Poland and later by Le Fort in Paris in 1877, the Le Fort colpocleisis repair technique remains the most reliable prolapse surgery to date.14 The uterus is left in place while the vagina is narrowed and shortened. It typically also is performed with a levator plication to reduce the genital hiatus.
This procedure is quick and effective, with a 90% to 95% success rate. If necessary, it can be performed under local or regional anesthesia, making it a good option for medically frail patients. It is not an option for everyone, however, as penetrative intercourse is no longer an option after surgery. Studies suggest an approximately 13% dissatisfaction rate after the procedure, with most of that coming from postoperative urinary symptoms, such as urgency or stress incontinence,15 and some studies show a dissatisfaction rate as low as 0% in a well-counseled patient population.16,17
Vaginal native tissue hysteropexy
Many patients who elect for uterine preservation at the time of prolapse surgery are “minimalists,” meaning that a vaginal native tissue procedure appeals to them due to the lack of abdominal incisions, decreased operating room time, and lack of permanent graft materials.
Of all the hysteropexy procedures, sacrospinous hysteropexy (SSHP) has the most robust data available. The approach to SSHP can be tailored to the patient’s anatomy and it is performed in a manner similar to posthysterectomy sacrospinous ligament fixation. The traditional posterior approach can be used with predominantly posterior prolapse, while an apical approach through a semilunar paracervical incision can be used for predominantly apical prolapse. Expert surgeons agree that one key to success is anchoring the suspension sutures through the cervical stroma, not just the vaginal epithelium.
Researchers in the Netherlands published the 5-year outcomes of a randomized trial that compared SSHP with vaginal hysterectomy with uterosacral ligament suspension.18 Their data showed no difference between groups in composite failure, reoperation rates, quality of life measures, and postoperative sexual function. Adverse events were very similar to those reported for posthysterectomy sacrospinous ligament fixation, including 15% transient buttock pain. Of note, the same authors explored risk factors for recurrence after SSHP and found that higher body mass index, smoking, and a large point Ba measurement were risk factors for prolapse recurrence.19
A randomized, controlled trial in the United Kingdom (the VUE trial) compared vaginal hysterectomy with apical suspension to uterine preservation with a variety of apical suspension techniques, mostly SSHP, and demonstrated no significant differences in outcomes.20 Overall, SSHP is an excellent option for many patients interested in uterine preservation.
Uterosacral ligament hysteropexy (USHP), when performed vaginally, is very similar to uterosacral ligament suspension at the time of vaginal hysterectomy, with entry into the peritoneal cavity through a posterior colpotomy. The uterosacral ligaments are grasped and delayed absorbable suture placed through the ligaments and anchored into the posterior cervical stroma. Given the maintenance of the normal axis of the vagina, USHP is a good technique for patients with isolated apical defects. Unfortunately, the least amount of quality data is available for USHP at this time. Currently, evidence suggests that complications are rare and that the procedure may offer acceptable anatomic and symptomatic outcomes.21 Some surgeons approach the uterosacral suspension laparoscopically, which also has mixed results in the literature, with failure rates between 8% and 27% and few robust studies.22–24
The Manchester-Fothergill operation, currently not common in the United States but popular in Europe, primarily is considered a treatment for cervical elongation when the uterosacral ligaments are intact. In this procedure, trachelectomy is performed and the uterosacral ligaments are plicated to the uterine body. Sturmdorf sutures are frequently placed to close off the endometrial canal, which can lead to hematometra and other complications of cervical stenosis. Previous unmatched studies have shown similar outcomes with the Manchester procedure compared with vaginal hysterectomy.25,26
The largest study currently available is a registry study from Denmark, with matched cohort populations, that compared the Manchester procedure, SSHP, and total vaginal hysterectomy with uterosacral ligament suspension.27 This study indicated less morbidity related to the Manchester procedure, decreased anterior recurrence compared with SSHP, and a 7% reoperation rate.27 The same authors also established better cost-effectiveness with the Manchester procedure as opposed to vaginal hysterectomy with uterosacral ligament suspension.28
Continue to: Vaginal mesh hysteropexy...
Vaginal mesh hysteropexy
Hysteropexy using vaginal mesh is limited in the United States given the removal of vaginal mesh kits from the market by the US Food and Drug Administration in 2019. However, a Pelvic Floor Disorders Network randomized trial compared vaginal mesh hysteropexy using the Uphold LITE transvaginal mesh support system (Boston Scientific) and vaginal hysterectomy with uterosacral ligament suspension.29 At 5 years, mesh hysteropexy had fewer failures than hysterectomy (37% vs 54%) and there was no difference in retreatment (9% vs 13%). The authors noted an 8% mesh exposure rate in the mesh hysteropexy group but 12% granulation tissue and 21% suture exposure rate in the hysterectomy group.29
While vaginal mesh hysteropexy was effective in the treatment of apical prolapse, the elevated mesh exposure rate and postoperative complications ultimately led to its removal from the market.
Sacrohysteropexy
Lastly, prolapse surgery with uterine preservation may be accomplished abdominally, most commonly laparoscopically with or without robotic assistance.
Sacrohysteropexy (SHP) involves the attachment of permanent synthetic mesh posteriorly to the posterior vagina and cervix with or without the additional placement of mesh to the anterior vagina and cervix. When the anterior mesh is placed, the arms are typically routed through the broad ligament bilaterally and joined with the posterior mesh for attachment to the anterior longitudinal ligament, overlying the sacrum.
Proponents of this technique endorse the use of mesh to augment already failing native tissues and propose similarities to the durability of sacrocolpopexy. While no randomized controlled trials have compared hysterectomy with sacrocolpopexy or supracervical hysterectomy with sacrocolpopexy to sacrohysteropexy, a meta-analysis suggests that sacrohysteropexy may have a decreased risk of mesh exposure but a higher reoperation rate with lower anatomic success.9 Randomized trials that compared abdominal sacrohysteropexy with vaginal hysterectomy and suspension indicate that apical support may be improved with sacrohysteropexy,30 but reoperations, postoperative pain and disability, and urinary dysfunction was higher with SHP.31,32
What further research is needed?
With the increasing patient and clinician interest in uterine preservation, more research is needed to improve patient counseling and surgical planning. Much of the current research compares hysteropexy outcomes with those of traditional prolapse repairs with hysterectomy, with only a few randomized trials. We are lacking robust, prospective comparison studies between hysteropexy methods, especially vaginal native tissue techniques, long-term follow-up on the prevalence of uterine or cervical pathology after hysteropexy, and pregnancy or postpartum outcomes following uterine preservation surgery.
Currently, work is underway to validate and test the effectiveness of a questionnaire to evaluate the uterus’s importance to the patient seeking prolapse surgery in order to optimize counseling. The VUE trial, which randomizes women to vaginal hysterectomy with suspension versus various prolapse surgeries with uterine preservation, is continuing its 6-year follow-up.20 In the Netherlands, an ongoing randomized, controlled trial (the SAM trial) is comparing the Manchester procedure with sacrospinous hysteropexy and will follow patients up to 24 months.33 Fortunately, both of these trials are rigorously assessing both objective and patient-centered outcomes.
CASE Counseling helps the patient weigh surgical options
After thorough review of her surgical options, the patient elects for a uterine-preserving prolapse repair. She would like to have the most minimally invasive procedure and does not want any permanent mesh used. You suggest, and she agrees to, a sacrospinous ligament hysteropexy, as it is the current technique with the most robust data. ●
- DeLancey JO. Anatomic aspects of vaginal eversion after hysterectomy. Am J Obstet Gynecol. 1992;166(6 pt 1):1717-1724; discussion 1724-1728. doi:10.1016/0002-9378(92)91562-o.
- Madsen AM, Raker C, Sung VW. Trends in hysteropexy and apical support for uterovaginal prolapse in the United States from 2002 to 2012. Female Pelvic Med Reconstr Surg. 2017;23:365-371. doi:10.1097/SPV.0000000000000426.
- Korbly NB, Kassis NC, Good MM, et al. Patient preferences for uterine preservation and hysterectomy in women with pelvic organ prolapse. Am J Obstet Gynecol. 2013;209:470.e16. doi:10.1016/j.ajog.2013.08.003.
- Frick AC, Barber MD, Paraiso MF, et al. Attitudes toward hysterectomy in women undergoing evaluation for uterovaginal prolapse. Female Pelvic Med Reconstr Surg. 2013;19:103-109. doi:10.1097/SPV.0b013e31827d8667.
- Farquhar CM, Sadler L, Harvey SA, et al. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112:956-962. doi:10.1111/j.1471-0528.2005.00696.x
- Gutman R, Maher C. Uterine-preserving POP surgery. Int Urogynecol J. 2013;24:1803-1813. doi:10.1007/s00192-0132171-2.
- Frick AC, Walters MD, Larkin KS, et al. Risk of unanticipated abnormal gynecologic pathology at the time of hysterectomy for uterovaginal prolapse. Am J Obstet Gynecol. 2010;202:507. e1-4. doi:10.1016/j.ajog.2010.01.077.
- Meriwether KV, Balk EM, Antosh DD, et al. Uterine-preserving surgeries for the repair of pelvic organ prolapse: a systematic review with meta-analysis and clinical practice guidelines. Int Urogynecol J. 2019;30:505-522. doi:10.1007/s00192-01903876-2.
- Meriwether KV, Antosh DD, Olivera CK, et al. Uterine preservation vs hysterectomy in pelvic organ prolapse surgery: a systematic review with meta-analysis and clinical practice guidelines. Am J Obstet Gynecol. 2018;219:129-146. e2. doi:10.1016/j.ajog.2018.01.018.
- Lin TY, Su TH, Wang YL, et al. Risk factors for failure of transvaginal sacrospinous uterine suspension in the treatment of uterovaginal prolapse. J Formos Med Assoc. 2005;104:249-253.
- Hyakutake MT, Cundiff GW, Geoffrion R. Cervical elongation following sacrospinous hysteropexy: a case series. Int Urogynecol J. 2014;25:851-854. doi:10.1007/s00192-013-2258-9.
- Thys SD, Coolen AL, Martens IR, et al. A comparison of long-term outcome between Manchester Fothergill and vaginal hysterectomy as treatment for uterine descent. Int Urogynecol J. 2011;22:1171-1178. doi:10.1007/s00192-011-1422-3.
- Ridgeway BM, Meriwether KV. Uterine preservation in pelvic organ prolapse surgery. In: Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier, Inc; 2022:358-373.
- FitzGerald MP, Richter HE, Siddique S, et al; for the Pelvic Floor Disorders Network. Colpocleisis: a review. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:261-271. doi:10.1007/s00192005-1339-9.
- Winkelman WD, Haviland MJ, Elkadry EA. Long-term pelvic f loor symptoms, recurrence, satisfaction, and regret following colpocleisis. Female Pelvic Med Reconstr Surg. 2020;26:558562. doi:10.1097/SPV.000000000000602.
- Lu M, Zeng W, Ju R, et al. Long-term clinical outcomes, recurrence, satisfaction, and regret after total colpocleisis with concomitant vaginal hysterectomy: a retrospective single-center study. Female Pelvic Med Reconstr Surg. 2021;27(4):e510-e515. doi:10.1097/SPV.0000000000000900.
- Wang X, Chen Y, Hua K. Pelvic symptoms, body image, and regret after LeFort colpocleisis: a long-term follow-up. J Minim Invasive Gynecol. 2017;24:415-419. doi:10.1016/j. jmig.2016.12.015.
- Schulten SFM, Detollenaere RJ, Stekelenburg J, et al. Sacrospinous hysteropexy versus vaginal hysterectomy with uterosacral ligament suspension in women with uterine prolapse stage 2 or higher: observational followup of a multicentre randomised trial. BMJ. 2019;366:I5149. doi:10.1136/bmj.l5149.
- Schulten SF, Detollenaere RJ, IntHout J, et al. Risk factors for pelvic organ prolapse recurrence after sacrospinous hysteropexy or vaginal hysterectomy with uterosacral ligament suspension. Am J Obstet Gynecol. 2022;227:252.e1252.e9. doi:10.1016/j.ajog.2022.04.017.
- Hemming C, Constable L, Goulao B, et al. Surgical interventions for uterine prolapse and for vault prolapse: the two VUE RCTs. Health Technol Assess. 2020;24:1-220. doi:10.3310/hta24130.
- Romanzi LJ, Tyagi R. Hysteropexy compared to hysterectomy for uterine prolapse surgery: does durability differ? Int Urogynecol J. 2012;23:625-631. doi:10.1007/s00192-011-1635-5.
- Rosen DM, Shukla A, Cario GM, et al. Is hysterectomy necessary for laparoscopic pelvic floor repair? A prospective study. J Minim Invasive Gynecol. 2008;15:729-734. doi:10.1016/j.jmig.2008.08.010.
- Bedford ND, Seman EI, O’Shea RT, et al. Effect of uterine preservation on outcome of laparoscopic uterosacral suspension. J Minim Invasive Gynecol. 2013;20(2):172-177. doi:10.1016/j.jmig.2012.10.014.
- Diwan A, Rardin CR, Strohsnitter WC, et al. Laparoscopic uterosacral ligament uterine suspension compared with vaginal hysterectomy with vaginal vault suspension for uterovaginal prolapse. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:79-83. doi:10.1007/s00192-005-1346-x.
- de Boer TA, Milani AL, Kluivers KB, et al. The effectiveness of surgical correction of uterine prolapse: cervical amputation with uterosacral ligament plication (modified Manchester) versus vaginal hysterectomy with high uterosacral ligament plication. Int Urogynecol J Pelvic Floor Dysfunct. 2009;20:13131319. doi:10.1007/s00192-009-0945-3.
- Thomas AG, Brodman ML, Dottino PR, et al. Manchester procedure vs. vaginal hysterectomy for uterine prolapse. A comparison. J Reprod Med. 1995;40:299-304.
- Husby KR, Larsen MD, Lose G, et al. Surgical treatment of primary uterine prolapse: a comparison of vaginal native tissue surgical techniques. Int Urogynecol J. 2019;30:18871893. doi:10.1007/s00192-019-03950-9.
- Husby KR, Tolstrup CK, Lose G, et al. Manchester-Fothergill procedure versus vaginal hysterectomy with uterosacral ligament suspension: an activity-based costing analysis. Int Urogynecol J. 2018;29:1161-1171. doi:10.1007/s00192-0183575-9.
- Nager CW, Visco AG, Richter HE, et al; National Institute of Child Health and Human Development Pelvic Floor Disorders Network. Effect of sacrospinous hysteropexy with graft vs vaginal hysterectomy with uterosacral ligament suspension on treatment failure in women with uterovaginal prolapse: 5-year results of a randomized clinical trial. Am J Obstet Gynecol. 2021;225:153.e1-153.e31. doi:10.1016/j. ajog.2021.03.012.
- Rahmanou P, Price N, Jackson SR. Laparoscopic hysteropexy versus vaginal hysterectomy for the treatment of uterovaginal prolapse: a prospective randomized pilot study. Int Urogynecol J. 2015;26:1687-1694. doi:10.1007/s00192-0152761-2.
- Roovers JP, van der Vaart CH, van der Bom JG, et al. A randomised controlled trial comparing abdominal and vaginal prolapse surgery: effects on urogenital function. BJOG. 2004;111:50-56. doi:10.1111/j.1471-0528.2004.00001.x.
- Roovers JP, van der Bom JG, van der Vaart CH, et al. A randomized comparison of post-operative pain, quality of life, and physical performance during the first 6 weeks after abdominal or vaginal surgical correction of descensus uteri. Neurourol Urodyn. 2005;24:334-340. doi:10.1002/nau.20104.
- Schulten SFM, Enklaar RA, Kluivers KB, et al. Evaluation of two vaginal, uterus sparing operations for pelvic organ prolapse: modified Manchester operation (MM) and sacrospinous hysteropexy (SSH), a study protocol for a multicentre randomized non-inferiority trial (the SAM study). BMC Womens Health. 20192;19:49. doi:10.1186/ s12905-019-0749-7.

Dr. Woodburn is Assistant Professor, Department of Urology/Female Pelvic Health, Atrium Health Wake Forest Baptist, Winston-Salem, North Carolina.
Dr. Meriwether is Associate Professor, Division Chief, Division of Urogynecology, Department of Obstetrics and Gynecology, University of New Mexico, Albuquerque.
Dr. Meriwether reports receiving grant or research support from Cook Medical and Caldera Medical and serving as a consultant to RBI Medical. Dr. Woodburn reports no financial relationships relevant to this article.
Dr. Woodburn is Assistant Professor, Department of Urology/Female Pelvic Health, Atrium Health Wake Forest Baptist, Winston-Salem, North Carolina.
Dr. Meriwether is Associate Professor, Division Chief, Division of Urogynecology, Department of Obstetrics and Gynecology, University of New Mexico, Albuquerque.
Dr. Meriwether reports receiving grant or research support from Cook Medical and Caldera Medical and serving as a consultant to RBI Medical. Dr. Woodburn reports no financial relationships relevant to this article.
Dr. Woodburn is Assistant Professor, Department of Urology/Female Pelvic Health, Atrium Health Wake Forest Baptist, Winston-Salem, North Carolina.
Dr. Meriwether is Associate Professor, Division Chief, Division of Urogynecology, Department of Obstetrics and Gynecology, University of New Mexico, Albuquerque.
Dr. Meriwether reports receiving grant or research support from Cook Medical and Caldera Medical and serving as a consultant to RBI Medical. Dr. Woodburn reports no financial relationships relevant to this article.
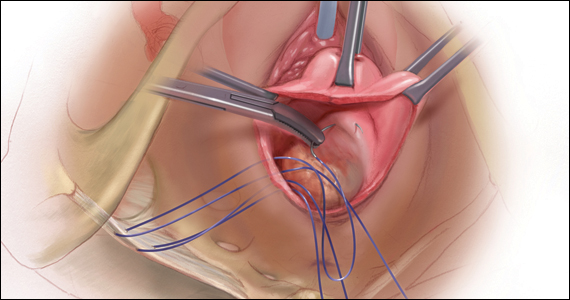
CASE Patient desires prolapse repair
A 65-year-old postmenopausal patient (G3P3) presents to your office with symptoms of a vaginal bulge for more than 1 year. She has no urinary incontinence symptoms and no bowel dysfunction symptoms. On examination, you diagnose stage 2 uterovaginal prolapse with both anterior and apical defects. The patient declines expectant and pessary management and desires surgery, but she states that she feels her uterus “is important for me to keep, as my babies grew in there and it is part of me.” She denies any family or personal history of breast, endometrial, or ovarian cancer and has no history of abnormal cervical cancer screening or postmenopausal bleeding. What are the options for this patient?
Who is the appropriate hysteropexy patient, and how do we counsel her?
Uterine prolapse is the third leading cause of benign hysterectomy, with approximately 70,000 procedures performed each year in the United States. It has long been acknowledged that the uterus is a passive bystander to the prolapse process,1 but modern practice often involves a hysterectomy as part of addressing apical prolapse. However, more and more uterine-preserving surgeries are being performed, with one study showing an increase from 1.8% to 5% from 2002 and 2012.2
When presented with the option to keep or remove their uterus during the time of prolapse surgery, 36% of patients indicated that they would prefer to keep their uterus with similar outcomes while 21% would still prefer uterine preservation even if outcomes were inferior compared with hysterectomy.3 Another study showed that 60% of patients would decline concurrent hysterectomy if there were equal surgical outcomes,4 and popular platforms, such as Health magazine (www.health.com) and AARP magazine (www.aarp.org), have listed benign hysterectomy as a “top surgery to avoid.”
Patients desire uterine preservation for many reasons, including concerns about sexual function and pleasure, the uterus being important to their sense of identity or womanhood, and concerns around menopausal symptoms. Early patient counseling and discussion of surgical goals can help clinicians fully understand a patient’s thoughts toward uterine preservation. Women who identified their uterus as important to their sense of self had a 28.2-times chance of preferring uterine preservation.3 Frequently, concerns about menopausal symptoms are more directly related to hormones and ovary removal, not uterus removal, but clinicians should be careful to also counsel patients on the increased risk of menopause in the 5 years after hysterectomy, even with ovarian preservation.5
There are some patients for whom experts do not recommend uterine preservation.6 Patients with an increased risk of cervical or endometrial pathology should be counseled on the benefits of hysterectomy. Additionally, patients who have abnormal uterine bleeding from benign pathology should consider hysterectomy to treat these issues and avoid future workups (TABLE). For postmenopausal patients with recent postmenopausal bleeding, we encourage hysterectomy. A study of patients undergoing hysterectomy at the time of prolapse repair found a rate of 13% unanticipated endometrial pathology with postmenopausal bleeding and negative preoperative workup.7
At this time, a majority of clinicians consider the desire for future fertility to be a relative contraindication to surgical prolapse repair and advise conservative management with pessary until childbearing is complete. This is reasonable, given the paucity of safety data in subsequent pregnancies as well as the lack of prolapse outcomes after those pregnancies.8,9 Lastly, cervical elongation is considered a relative contraindication, as it represents a risk for surgical failure.10,11 This may be counteracted with trachelectomy at the time of hysteropexy or surgeries such as the Manchester repair, which involve a trachelectomy routinely,12 but currently there is no strong evidence for this as routine practice.
Continue to: Uterine preservation surgical techniques and outcomes...
Uterine preservation surgical techniques and outcomes
Le Fort colpocleisis
First described in 1840 by Neugebauer of Poland and later by Le Fort in Paris in 1877, the Le Fort colpocleisis repair technique remains the most reliable prolapse surgery to date.14 The uterus is left in place while the vagina is narrowed and shortened. It typically also is performed with a levator plication to reduce the genital hiatus.
This procedure is quick and effective, with a 90% to 95% success rate. If necessary, it can be performed under local or regional anesthesia, making it a good option for medically frail patients. It is not an option for everyone, however, as penetrative intercourse is no longer an option after surgery. Studies suggest an approximately 13% dissatisfaction rate after the procedure, with most of that coming from postoperative urinary symptoms, such as urgency or stress incontinence,15 and some studies show a dissatisfaction rate as low as 0% in a well-counseled patient population.16,17
Vaginal native tissue hysteropexy
Many patients who elect for uterine preservation at the time of prolapse surgery are “minimalists,” meaning that a vaginal native tissue procedure appeals to them due to the lack of abdominal incisions, decreased operating room time, and lack of permanent graft materials.
Of all the hysteropexy procedures, sacrospinous hysteropexy (SSHP) has the most robust data available. The approach to SSHP can be tailored to the patient’s anatomy and it is performed in a manner similar to posthysterectomy sacrospinous ligament fixation. The traditional posterior approach can be used with predominantly posterior prolapse, while an apical approach through a semilunar paracervical incision can be used for predominantly apical prolapse. Expert surgeons agree that one key to success is anchoring the suspension sutures through the cervical stroma, not just the vaginal epithelium.
Researchers in the Netherlands published the 5-year outcomes of a randomized trial that compared SSHP with vaginal hysterectomy with uterosacral ligament suspension.18 Their data showed no difference between groups in composite failure, reoperation rates, quality of life measures, and postoperative sexual function. Adverse events were very similar to those reported for posthysterectomy sacrospinous ligament fixation, including 15% transient buttock pain. Of note, the same authors explored risk factors for recurrence after SSHP and found that higher body mass index, smoking, and a large point Ba measurement were risk factors for prolapse recurrence.19
A randomized, controlled trial in the United Kingdom (the VUE trial) compared vaginal hysterectomy with apical suspension to uterine preservation with a variety of apical suspension techniques, mostly SSHP, and demonstrated no significant differences in outcomes.20 Overall, SSHP is an excellent option for many patients interested in uterine preservation.
Uterosacral ligament hysteropexy (USHP), when performed vaginally, is very similar to uterosacral ligament suspension at the time of vaginal hysterectomy, with entry into the peritoneal cavity through a posterior colpotomy. The uterosacral ligaments are grasped and delayed absorbable suture placed through the ligaments and anchored into the posterior cervical stroma. Given the maintenance of the normal axis of the vagina, USHP is a good technique for patients with isolated apical defects. Unfortunately, the least amount of quality data is available for USHP at this time. Currently, evidence suggests that complications are rare and that the procedure may offer acceptable anatomic and symptomatic outcomes.21 Some surgeons approach the uterosacral suspension laparoscopically, which also has mixed results in the literature, with failure rates between 8% and 27% and few robust studies.22–24
The Manchester-Fothergill operation, currently not common in the United States but popular in Europe, primarily is considered a treatment for cervical elongation when the uterosacral ligaments are intact. In this procedure, trachelectomy is performed and the uterosacral ligaments are plicated to the uterine body. Sturmdorf sutures are frequently placed to close off the endometrial canal, which can lead to hematometra and other complications of cervical stenosis. Previous unmatched studies have shown similar outcomes with the Manchester procedure compared with vaginal hysterectomy.25,26
The largest study currently available is a registry study from Denmark, with matched cohort populations, that compared the Manchester procedure, SSHP, and total vaginal hysterectomy with uterosacral ligament suspension.27 This study indicated less morbidity related to the Manchester procedure, decreased anterior recurrence compared with SSHP, and a 7% reoperation rate.27 The same authors also established better cost-effectiveness with the Manchester procedure as opposed to vaginal hysterectomy with uterosacral ligament suspension.28
Continue to: Vaginal mesh hysteropexy...
Vaginal mesh hysteropexy
Hysteropexy using vaginal mesh is limited in the United States given the removal of vaginal mesh kits from the market by the US Food and Drug Administration in 2019. However, a Pelvic Floor Disorders Network randomized trial compared vaginal mesh hysteropexy using the Uphold LITE transvaginal mesh support system (Boston Scientific) and vaginal hysterectomy with uterosacral ligament suspension.29 At 5 years, mesh hysteropexy had fewer failures than hysterectomy (37% vs 54%) and there was no difference in retreatment (9% vs 13%). The authors noted an 8% mesh exposure rate in the mesh hysteropexy group but 12% granulation tissue and 21% suture exposure rate in the hysterectomy group.29
While vaginal mesh hysteropexy was effective in the treatment of apical prolapse, the elevated mesh exposure rate and postoperative complications ultimately led to its removal from the market.
Sacrohysteropexy
Lastly, prolapse surgery with uterine preservation may be accomplished abdominally, most commonly laparoscopically with or without robotic assistance.
Sacrohysteropexy (SHP) involves the attachment of permanent synthetic mesh posteriorly to the posterior vagina and cervix with or without the additional placement of mesh to the anterior vagina and cervix. When the anterior mesh is placed, the arms are typically routed through the broad ligament bilaterally and joined with the posterior mesh for attachment to the anterior longitudinal ligament, overlying the sacrum.
Proponents of this technique endorse the use of mesh to augment already failing native tissues and propose similarities to the durability of sacrocolpopexy. While no randomized controlled trials have compared hysterectomy with sacrocolpopexy or supracervical hysterectomy with sacrocolpopexy to sacrohysteropexy, a meta-analysis suggests that sacrohysteropexy may have a decreased risk of mesh exposure but a higher reoperation rate with lower anatomic success.9 Randomized trials that compared abdominal sacrohysteropexy with vaginal hysterectomy and suspension indicate that apical support may be improved with sacrohysteropexy,30 but reoperations, postoperative pain and disability, and urinary dysfunction was higher with SHP.31,32
What further research is needed?
With the increasing patient and clinician interest in uterine preservation, more research is needed to improve patient counseling and surgical planning. Much of the current research compares hysteropexy outcomes with those of traditional prolapse repairs with hysterectomy, with only a few randomized trials. We are lacking robust, prospective comparison studies between hysteropexy methods, especially vaginal native tissue techniques, long-term follow-up on the prevalence of uterine or cervical pathology after hysteropexy, and pregnancy or postpartum outcomes following uterine preservation surgery.
Currently, work is underway to validate and test the effectiveness of a questionnaire to evaluate the uterus’s importance to the patient seeking prolapse surgery in order to optimize counseling. The VUE trial, which randomizes women to vaginal hysterectomy with suspension versus various prolapse surgeries with uterine preservation, is continuing its 6-year follow-up.20 In the Netherlands, an ongoing randomized, controlled trial (the SAM trial) is comparing the Manchester procedure with sacrospinous hysteropexy and will follow patients up to 24 months.33 Fortunately, both of these trials are rigorously assessing both objective and patient-centered outcomes.
CASE Counseling helps the patient weigh surgical options
After thorough review of her surgical options, the patient elects for a uterine-preserving prolapse repair. She would like to have the most minimally invasive procedure and does not want any permanent mesh used. You suggest, and she agrees to, a sacrospinous ligament hysteropexy, as it is the current technique with the most robust data. ●
CASE Patient desires prolapse repair
A 65-year-old postmenopausal patient (G3P3) presents to your office with symptoms of a vaginal bulge for more than 1 year. She has no urinary incontinence symptoms and no bowel dysfunction symptoms. On examination, you diagnose stage 2 uterovaginal prolapse with both anterior and apical defects. The patient declines expectant and pessary management and desires surgery, but she states that she feels her uterus “is important for me to keep, as my babies grew in there and it is part of me.” She denies any family or personal history of breast, endometrial, or ovarian cancer and has no history of abnormal cervical cancer screening or postmenopausal bleeding. What are the options for this patient?
Who is the appropriate hysteropexy patient, and how do we counsel her?
Uterine prolapse is the third leading cause of benign hysterectomy, with approximately 70,000 procedures performed each year in the United States. It has long been acknowledged that the uterus is a passive bystander to the prolapse process,1 but modern practice often involves a hysterectomy as part of addressing apical prolapse. However, more and more uterine-preserving surgeries are being performed, with one study showing an increase from 1.8% to 5% from 2002 and 2012.2
When presented with the option to keep or remove their uterus during the time of prolapse surgery, 36% of patients indicated that they would prefer to keep their uterus with similar outcomes while 21% would still prefer uterine preservation even if outcomes were inferior compared with hysterectomy.3 Another study showed that 60% of patients would decline concurrent hysterectomy if there were equal surgical outcomes,4 and popular platforms, such as Health magazine (www.health.com) and AARP magazine (www.aarp.org), have listed benign hysterectomy as a “top surgery to avoid.”
Patients desire uterine preservation for many reasons, including concerns about sexual function and pleasure, the uterus being important to their sense of identity or womanhood, and concerns around menopausal symptoms. Early patient counseling and discussion of surgical goals can help clinicians fully understand a patient’s thoughts toward uterine preservation. Women who identified their uterus as important to their sense of self had a 28.2-times chance of preferring uterine preservation.3 Frequently, concerns about menopausal symptoms are more directly related to hormones and ovary removal, not uterus removal, but clinicians should be careful to also counsel patients on the increased risk of menopause in the 5 years after hysterectomy, even with ovarian preservation.5
There are some patients for whom experts do not recommend uterine preservation.6 Patients with an increased risk of cervical or endometrial pathology should be counseled on the benefits of hysterectomy. Additionally, patients who have abnormal uterine bleeding from benign pathology should consider hysterectomy to treat these issues and avoid future workups (TABLE). For postmenopausal patients with recent postmenopausal bleeding, we encourage hysterectomy. A study of patients undergoing hysterectomy at the time of prolapse repair found a rate of 13% unanticipated endometrial pathology with postmenopausal bleeding and negative preoperative workup.7
At this time, a majority of clinicians consider the desire for future fertility to be a relative contraindication to surgical prolapse repair and advise conservative management with pessary until childbearing is complete. This is reasonable, given the paucity of safety data in subsequent pregnancies as well as the lack of prolapse outcomes after those pregnancies.8,9 Lastly, cervical elongation is considered a relative contraindication, as it represents a risk for surgical failure.10,11 This may be counteracted with trachelectomy at the time of hysteropexy or surgeries such as the Manchester repair, which involve a trachelectomy routinely,12 but currently there is no strong evidence for this as routine practice.
Continue to: Uterine preservation surgical techniques and outcomes...
Uterine preservation surgical techniques and outcomes
Le Fort colpocleisis
First described in 1840 by Neugebauer of Poland and later by Le Fort in Paris in 1877, the Le Fort colpocleisis repair technique remains the most reliable prolapse surgery to date.14 The uterus is left in place while the vagina is narrowed and shortened. It typically also is performed with a levator plication to reduce the genital hiatus.
This procedure is quick and effective, with a 90% to 95% success rate. If necessary, it can be performed under local or regional anesthesia, making it a good option for medically frail patients. It is not an option for everyone, however, as penetrative intercourse is no longer an option after surgery. Studies suggest an approximately 13% dissatisfaction rate after the procedure, with most of that coming from postoperative urinary symptoms, such as urgency or stress incontinence,15 and some studies show a dissatisfaction rate as low as 0% in a well-counseled patient population.16,17
Vaginal native tissue hysteropexy
Many patients who elect for uterine preservation at the time of prolapse surgery are “minimalists,” meaning that a vaginal native tissue procedure appeals to them due to the lack of abdominal incisions, decreased operating room time, and lack of permanent graft materials.
Of all the hysteropexy procedures, sacrospinous hysteropexy (SSHP) has the most robust data available. The approach to SSHP can be tailored to the patient’s anatomy and it is performed in a manner similar to posthysterectomy sacrospinous ligament fixation. The traditional posterior approach can be used with predominantly posterior prolapse, while an apical approach through a semilunar paracervical incision can be used for predominantly apical prolapse. Expert surgeons agree that one key to success is anchoring the suspension sutures through the cervical stroma, not just the vaginal epithelium.
Researchers in the Netherlands published the 5-year outcomes of a randomized trial that compared SSHP with vaginal hysterectomy with uterosacral ligament suspension.18 Their data showed no difference between groups in composite failure, reoperation rates, quality of life measures, and postoperative sexual function. Adverse events were very similar to those reported for posthysterectomy sacrospinous ligament fixation, including 15% transient buttock pain. Of note, the same authors explored risk factors for recurrence after SSHP and found that higher body mass index, smoking, and a large point Ba measurement were risk factors for prolapse recurrence.19
A randomized, controlled trial in the United Kingdom (the VUE trial) compared vaginal hysterectomy with apical suspension to uterine preservation with a variety of apical suspension techniques, mostly SSHP, and demonstrated no significant differences in outcomes.20 Overall, SSHP is an excellent option for many patients interested in uterine preservation.
Uterosacral ligament hysteropexy (USHP), when performed vaginally, is very similar to uterosacral ligament suspension at the time of vaginal hysterectomy, with entry into the peritoneal cavity through a posterior colpotomy. The uterosacral ligaments are grasped and delayed absorbable suture placed through the ligaments and anchored into the posterior cervical stroma. Given the maintenance of the normal axis of the vagina, USHP is a good technique for patients with isolated apical defects. Unfortunately, the least amount of quality data is available for USHP at this time. Currently, evidence suggests that complications are rare and that the procedure may offer acceptable anatomic and symptomatic outcomes.21 Some surgeons approach the uterosacral suspension laparoscopically, which also has mixed results in the literature, with failure rates between 8% and 27% and few robust studies.22–24
The Manchester-Fothergill operation, currently not common in the United States but popular in Europe, primarily is considered a treatment for cervical elongation when the uterosacral ligaments are intact. In this procedure, trachelectomy is performed and the uterosacral ligaments are plicated to the uterine body. Sturmdorf sutures are frequently placed to close off the endometrial canal, which can lead to hematometra and other complications of cervical stenosis. Previous unmatched studies have shown similar outcomes with the Manchester procedure compared with vaginal hysterectomy.25,26
The largest study currently available is a registry study from Denmark, with matched cohort populations, that compared the Manchester procedure, SSHP, and total vaginal hysterectomy with uterosacral ligament suspension.27 This study indicated less morbidity related to the Manchester procedure, decreased anterior recurrence compared with SSHP, and a 7% reoperation rate.27 The same authors also established better cost-effectiveness with the Manchester procedure as opposed to vaginal hysterectomy with uterosacral ligament suspension.28
Continue to: Vaginal mesh hysteropexy...
Vaginal mesh hysteropexy
Hysteropexy using vaginal mesh is limited in the United States given the removal of vaginal mesh kits from the market by the US Food and Drug Administration in 2019. However, a Pelvic Floor Disorders Network randomized trial compared vaginal mesh hysteropexy using the Uphold LITE transvaginal mesh support system (Boston Scientific) and vaginal hysterectomy with uterosacral ligament suspension.29 At 5 years, mesh hysteropexy had fewer failures than hysterectomy (37% vs 54%) and there was no difference in retreatment (9% vs 13%). The authors noted an 8% mesh exposure rate in the mesh hysteropexy group but 12% granulation tissue and 21% suture exposure rate in the hysterectomy group.29
While vaginal mesh hysteropexy was effective in the treatment of apical prolapse, the elevated mesh exposure rate and postoperative complications ultimately led to its removal from the market.
Sacrohysteropexy
Lastly, prolapse surgery with uterine preservation may be accomplished abdominally, most commonly laparoscopically with or without robotic assistance.
Sacrohysteropexy (SHP) involves the attachment of permanent synthetic mesh posteriorly to the posterior vagina and cervix with or without the additional placement of mesh to the anterior vagina and cervix. When the anterior mesh is placed, the arms are typically routed through the broad ligament bilaterally and joined with the posterior mesh for attachment to the anterior longitudinal ligament, overlying the sacrum.
Proponents of this technique endorse the use of mesh to augment already failing native tissues and propose similarities to the durability of sacrocolpopexy. While no randomized controlled trials have compared hysterectomy with sacrocolpopexy or supracervical hysterectomy with sacrocolpopexy to sacrohysteropexy, a meta-analysis suggests that sacrohysteropexy may have a decreased risk of mesh exposure but a higher reoperation rate with lower anatomic success.9 Randomized trials that compared abdominal sacrohysteropexy with vaginal hysterectomy and suspension indicate that apical support may be improved with sacrohysteropexy,30 but reoperations, postoperative pain and disability, and urinary dysfunction was higher with SHP.31,32
What further research is needed?
With the increasing patient and clinician interest in uterine preservation, more research is needed to improve patient counseling and surgical planning. Much of the current research compares hysteropexy outcomes with those of traditional prolapse repairs with hysterectomy, with only a few randomized trials. We are lacking robust, prospective comparison studies between hysteropexy methods, especially vaginal native tissue techniques, long-term follow-up on the prevalence of uterine or cervical pathology after hysteropexy, and pregnancy or postpartum outcomes following uterine preservation surgery.
Currently, work is underway to validate and test the effectiveness of a questionnaire to evaluate the uterus’s importance to the patient seeking prolapse surgery in order to optimize counseling. The VUE trial, which randomizes women to vaginal hysterectomy with suspension versus various prolapse surgeries with uterine preservation, is continuing its 6-year follow-up.20 In the Netherlands, an ongoing randomized, controlled trial (the SAM trial) is comparing the Manchester procedure with sacrospinous hysteropexy and will follow patients up to 24 months.33 Fortunately, both of these trials are rigorously assessing both objective and patient-centered outcomes.
CASE Counseling helps the patient weigh surgical options
After thorough review of her surgical options, the patient elects for a uterine-preserving prolapse repair. She would like to have the most minimally invasive procedure and does not want any permanent mesh used. You suggest, and she agrees to, a sacrospinous ligament hysteropexy, as it is the current technique with the most robust data. ●
- DeLancey JO. Anatomic aspects of vaginal eversion after hysterectomy. Am J Obstet Gynecol. 1992;166(6 pt 1):1717-1724; discussion 1724-1728. doi:10.1016/0002-9378(92)91562-o.
- Madsen AM, Raker C, Sung VW. Trends in hysteropexy and apical support for uterovaginal prolapse in the United States from 2002 to 2012. Female Pelvic Med Reconstr Surg. 2017;23:365-371. doi:10.1097/SPV.0000000000000426.
- Korbly NB, Kassis NC, Good MM, et al. Patient preferences for uterine preservation and hysterectomy in women with pelvic organ prolapse. Am J Obstet Gynecol. 2013;209:470.e16. doi:10.1016/j.ajog.2013.08.003.
- Frick AC, Barber MD, Paraiso MF, et al. Attitudes toward hysterectomy in women undergoing evaluation for uterovaginal prolapse. Female Pelvic Med Reconstr Surg. 2013;19:103-109. doi:10.1097/SPV.0b013e31827d8667.
- Farquhar CM, Sadler L, Harvey SA, et al. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112:956-962. doi:10.1111/j.1471-0528.2005.00696.x
- Gutman R, Maher C. Uterine-preserving POP surgery. Int Urogynecol J. 2013;24:1803-1813. doi:10.1007/s00192-0132171-2.
- Frick AC, Walters MD, Larkin KS, et al. Risk of unanticipated abnormal gynecologic pathology at the time of hysterectomy for uterovaginal prolapse. Am J Obstet Gynecol. 2010;202:507. e1-4. doi:10.1016/j.ajog.2010.01.077.
- Meriwether KV, Balk EM, Antosh DD, et al. Uterine-preserving surgeries for the repair of pelvic organ prolapse: a systematic review with meta-analysis and clinical practice guidelines. Int Urogynecol J. 2019;30:505-522. doi:10.1007/s00192-01903876-2.
- Meriwether KV, Antosh DD, Olivera CK, et al. Uterine preservation vs hysterectomy in pelvic organ prolapse surgery: a systematic review with meta-analysis and clinical practice guidelines. Am J Obstet Gynecol. 2018;219:129-146. e2. doi:10.1016/j.ajog.2018.01.018.
- Lin TY, Su TH, Wang YL, et al. Risk factors for failure of transvaginal sacrospinous uterine suspension in the treatment of uterovaginal prolapse. J Formos Med Assoc. 2005;104:249-253.
- Hyakutake MT, Cundiff GW, Geoffrion R. Cervical elongation following sacrospinous hysteropexy: a case series. Int Urogynecol J. 2014;25:851-854. doi:10.1007/s00192-013-2258-9.
- Thys SD, Coolen AL, Martens IR, et al. A comparison of long-term outcome between Manchester Fothergill and vaginal hysterectomy as treatment for uterine descent. Int Urogynecol J. 2011;22:1171-1178. doi:10.1007/s00192-011-1422-3.
- Ridgeway BM, Meriwether KV. Uterine preservation in pelvic organ prolapse surgery. In: Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier, Inc; 2022:358-373.
- FitzGerald MP, Richter HE, Siddique S, et al; for the Pelvic Floor Disorders Network. Colpocleisis: a review. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:261-271. doi:10.1007/s00192005-1339-9.
- Winkelman WD, Haviland MJ, Elkadry EA. Long-term pelvic f loor symptoms, recurrence, satisfaction, and regret following colpocleisis. Female Pelvic Med Reconstr Surg. 2020;26:558562. doi:10.1097/SPV.000000000000602.
- Lu M, Zeng W, Ju R, et al. Long-term clinical outcomes, recurrence, satisfaction, and regret after total colpocleisis with concomitant vaginal hysterectomy: a retrospective single-center study. Female Pelvic Med Reconstr Surg. 2021;27(4):e510-e515. doi:10.1097/SPV.0000000000000900.
- Wang X, Chen Y, Hua K. Pelvic symptoms, body image, and regret after LeFort colpocleisis: a long-term follow-up. J Minim Invasive Gynecol. 2017;24:415-419. doi:10.1016/j. jmig.2016.12.015.
- Schulten SFM, Detollenaere RJ, Stekelenburg J, et al. Sacrospinous hysteropexy versus vaginal hysterectomy with uterosacral ligament suspension in women with uterine prolapse stage 2 or higher: observational followup of a multicentre randomised trial. BMJ. 2019;366:I5149. doi:10.1136/bmj.l5149.
- Schulten SF, Detollenaere RJ, IntHout J, et al. Risk factors for pelvic organ prolapse recurrence after sacrospinous hysteropexy or vaginal hysterectomy with uterosacral ligament suspension. Am J Obstet Gynecol. 2022;227:252.e1252.e9. doi:10.1016/j.ajog.2022.04.017.
- Hemming C, Constable L, Goulao B, et al. Surgical interventions for uterine prolapse and for vault prolapse: the two VUE RCTs. Health Technol Assess. 2020;24:1-220. doi:10.3310/hta24130.
- Romanzi LJ, Tyagi R. Hysteropexy compared to hysterectomy for uterine prolapse surgery: does durability differ? Int Urogynecol J. 2012;23:625-631. doi:10.1007/s00192-011-1635-5.
- Rosen DM, Shukla A, Cario GM, et al. Is hysterectomy necessary for laparoscopic pelvic floor repair? A prospective study. J Minim Invasive Gynecol. 2008;15:729-734. doi:10.1016/j.jmig.2008.08.010.
- Bedford ND, Seman EI, O’Shea RT, et al. Effect of uterine preservation on outcome of laparoscopic uterosacral suspension. J Minim Invasive Gynecol. 2013;20(2):172-177. doi:10.1016/j.jmig.2012.10.014.
- Diwan A, Rardin CR, Strohsnitter WC, et al. Laparoscopic uterosacral ligament uterine suspension compared with vaginal hysterectomy with vaginal vault suspension for uterovaginal prolapse. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:79-83. doi:10.1007/s00192-005-1346-x.
- de Boer TA, Milani AL, Kluivers KB, et al. The effectiveness of surgical correction of uterine prolapse: cervical amputation with uterosacral ligament plication (modified Manchester) versus vaginal hysterectomy with high uterosacral ligament plication. Int Urogynecol J Pelvic Floor Dysfunct. 2009;20:13131319. doi:10.1007/s00192-009-0945-3.
- Thomas AG, Brodman ML, Dottino PR, et al. Manchester procedure vs. vaginal hysterectomy for uterine prolapse. A comparison. J Reprod Med. 1995;40:299-304.
- Husby KR, Larsen MD, Lose G, et al. Surgical treatment of primary uterine prolapse: a comparison of vaginal native tissue surgical techniques. Int Urogynecol J. 2019;30:18871893. doi:10.1007/s00192-019-03950-9.
- Husby KR, Tolstrup CK, Lose G, et al. Manchester-Fothergill procedure versus vaginal hysterectomy with uterosacral ligament suspension: an activity-based costing analysis. Int Urogynecol J. 2018;29:1161-1171. doi:10.1007/s00192-0183575-9.
- Nager CW, Visco AG, Richter HE, et al; National Institute of Child Health and Human Development Pelvic Floor Disorders Network. Effect of sacrospinous hysteropexy with graft vs vaginal hysterectomy with uterosacral ligament suspension on treatment failure in women with uterovaginal prolapse: 5-year results of a randomized clinical trial. Am J Obstet Gynecol. 2021;225:153.e1-153.e31. doi:10.1016/j. ajog.2021.03.012.
- Rahmanou P, Price N, Jackson SR. Laparoscopic hysteropexy versus vaginal hysterectomy for the treatment of uterovaginal prolapse: a prospective randomized pilot study. Int Urogynecol J. 2015;26:1687-1694. doi:10.1007/s00192-0152761-2.
- Roovers JP, van der Vaart CH, van der Bom JG, et al. A randomised controlled trial comparing abdominal and vaginal prolapse surgery: effects on urogenital function. BJOG. 2004;111:50-56. doi:10.1111/j.1471-0528.2004.00001.x.
- Roovers JP, van der Bom JG, van der Vaart CH, et al. A randomized comparison of post-operative pain, quality of life, and physical performance during the first 6 weeks after abdominal or vaginal surgical correction of descensus uteri. Neurourol Urodyn. 2005;24:334-340. doi:10.1002/nau.20104.
- Schulten SFM, Enklaar RA, Kluivers KB, et al. Evaluation of two vaginal, uterus sparing operations for pelvic organ prolapse: modified Manchester operation (MM) and sacrospinous hysteropexy (SSH), a study protocol for a multicentre randomized non-inferiority trial (the SAM study). BMC Womens Health. 20192;19:49. doi:10.1186/ s12905-019-0749-7.
- DeLancey JO. Anatomic aspects of vaginal eversion after hysterectomy. Am J Obstet Gynecol. 1992;166(6 pt 1):1717-1724; discussion 1724-1728. doi:10.1016/0002-9378(92)91562-o.
- Madsen AM, Raker C, Sung VW. Trends in hysteropexy and apical support for uterovaginal prolapse in the United States from 2002 to 2012. Female Pelvic Med Reconstr Surg. 2017;23:365-371. doi:10.1097/SPV.0000000000000426.
- Korbly NB, Kassis NC, Good MM, et al. Patient preferences for uterine preservation and hysterectomy in women with pelvic organ prolapse. Am J Obstet Gynecol. 2013;209:470.e16. doi:10.1016/j.ajog.2013.08.003.
- Frick AC, Barber MD, Paraiso MF, et al. Attitudes toward hysterectomy in women undergoing evaluation for uterovaginal prolapse. Female Pelvic Med Reconstr Surg. 2013;19:103-109. doi:10.1097/SPV.0b013e31827d8667.
- Farquhar CM, Sadler L, Harvey SA, et al. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112:956-962. doi:10.1111/j.1471-0528.2005.00696.x
- Gutman R, Maher C. Uterine-preserving POP surgery. Int Urogynecol J. 2013;24:1803-1813. doi:10.1007/s00192-0132171-2.
- Frick AC, Walters MD, Larkin KS, et al. Risk of unanticipated abnormal gynecologic pathology at the time of hysterectomy for uterovaginal prolapse. Am J Obstet Gynecol. 2010;202:507. e1-4. doi:10.1016/j.ajog.2010.01.077.
- Meriwether KV, Balk EM, Antosh DD, et al. Uterine-preserving surgeries for the repair of pelvic organ prolapse: a systematic review with meta-analysis and clinical practice guidelines. Int Urogynecol J. 2019;30:505-522. doi:10.1007/s00192-01903876-2.
- Meriwether KV, Antosh DD, Olivera CK, et al. Uterine preservation vs hysterectomy in pelvic organ prolapse surgery: a systematic review with meta-analysis and clinical practice guidelines. Am J Obstet Gynecol. 2018;219:129-146. e2. doi:10.1016/j.ajog.2018.01.018.
- Lin TY, Su TH, Wang YL, et al. Risk factors for failure of transvaginal sacrospinous uterine suspension in the treatment of uterovaginal prolapse. J Formos Med Assoc. 2005;104:249-253.
- Hyakutake MT, Cundiff GW, Geoffrion R. Cervical elongation following sacrospinous hysteropexy: a case series. Int Urogynecol J. 2014;25:851-854. doi:10.1007/s00192-013-2258-9.
- Thys SD, Coolen AL, Martens IR, et al. A comparison of long-term outcome between Manchester Fothergill and vaginal hysterectomy as treatment for uterine descent. Int Urogynecol J. 2011;22:1171-1178. doi:10.1007/s00192-011-1422-3.
- Ridgeway BM, Meriwether KV. Uterine preservation in pelvic organ prolapse surgery. In: Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier, Inc; 2022:358-373.
- FitzGerald MP, Richter HE, Siddique S, et al; for the Pelvic Floor Disorders Network. Colpocleisis: a review. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:261-271. doi:10.1007/s00192005-1339-9.
- Winkelman WD, Haviland MJ, Elkadry EA. Long-term pelvic f loor symptoms, recurrence, satisfaction, and regret following colpocleisis. Female Pelvic Med Reconstr Surg. 2020;26:558562. doi:10.1097/SPV.000000000000602.
- Lu M, Zeng W, Ju R, et al. Long-term clinical outcomes, recurrence, satisfaction, and regret after total colpocleisis with concomitant vaginal hysterectomy: a retrospective single-center study. Female Pelvic Med Reconstr Surg. 2021;27(4):e510-e515. doi:10.1097/SPV.0000000000000900.
- Wang X, Chen Y, Hua K. Pelvic symptoms, body image, and regret after LeFort colpocleisis: a long-term follow-up. J Minim Invasive Gynecol. 2017;24:415-419. doi:10.1016/j. jmig.2016.12.015.
- Schulten SFM, Detollenaere RJ, Stekelenburg J, et al. Sacrospinous hysteropexy versus vaginal hysterectomy with uterosacral ligament suspension in women with uterine prolapse stage 2 or higher: observational followup of a multicentre randomised trial. BMJ. 2019;366:I5149. doi:10.1136/bmj.l5149.
- Schulten SF, Detollenaere RJ, IntHout J, et al. Risk factors for pelvic organ prolapse recurrence after sacrospinous hysteropexy or vaginal hysterectomy with uterosacral ligament suspension. Am J Obstet Gynecol. 2022;227:252.e1252.e9. doi:10.1016/j.ajog.2022.04.017.
- Hemming C, Constable L, Goulao B, et al. Surgical interventions for uterine prolapse and for vault prolapse: the two VUE RCTs. Health Technol Assess. 2020;24:1-220. doi:10.3310/hta24130.
- Romanzi LJ, Tyagi R. Hysteropexy compared to hysterectomy for uterine prolapse surgery: does durability differ? Int Urogynecol J. 2012;23:625-631. doi:10.1007/s00192-011-1635-5.
- Rosen DM, Shukla A, Cario GM, et al. Is hysterectomy necessary for laparoscopic pelvic floor repair? A prospective study. J Minim Invasive Gynecol. 2008;15:729-734. doi:10.1016/j.jmig.2008.08.010.
- Bedford ND, Seman EI, O’Shea RT, et al. Effect of uterine preservation on outcome of laparoscopic uterosacral suspension. J Minim Invasive Gynecol. 2013;20(2):172-177. doi:10.1016/j.jmig.2012.10.014.
- Diwan A, Rardin CR, Strohsnitter WC, et al. Laparoscopic uterosacral ligament uterine suspension compared with vaginal hysterectomy with vaginal vault suspension for uterovaginal prolapse. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:79-83. doi:10.1007/s00192-005-1346-x.
- de Boer TA, Milani AL, Kluivers KB, et al. The effectiveness of surgical correction of uterine prolapse: cervical amputation with uterosacral ligament plication (modified Manchester) versus vaginal hysterectomy with high uterosacral ligament plication. Int Urogynecol J Pelvic Floor Dysfunct. 2009;20:13131319. doi:10.1007/s00192-009-0945-3.
- Thomas AG, Brodman ML, Dottino PR, et al. Manchester procedure vs. vaginal hysterectomy for uterine prolapse. A comparison. J Reprod Med. 1995;40:299-304.
- Husby KR, Larsen MD, Lose G, et al. Surgical treatment of primary uterine prolapse: a comparison of vaginal native tissue surgical techniques. Int Urogynecol J. 2019;30:18871893. doi:10.1007/s00192-019-03950-9.
- Husby KR, Tolstrup CK, Lose G, et al. Manchester-Fothergill procedure versus vaginal hysterectomy with uterosacral ligament suspension: an activity-based costing analysis. Int Urogynecol J. 2018;29:1161-1171. doi:10.1007/s00192-0183575-9.
- Nager CW, Visco AG, Richter HE, et al; National Institute of Child Health and Human Development Pelvic Floor Disorders Network. Effect of sacrospinous hysteropexy with graft vs vaginal hysterectomy with uterosacral ligament suspension on treatment failure in women with uterovaginal prolapse: 5-year results of a randomized clinical trial. Am J Obstet Gynecol. 2021;225:153.e1-153.e31. doi:10.1016/j. ajog.2021.03.012.
- Rahmanou P, Price N, Jackson SR. Laparoscopic hysteropexy versus vaginal hysterectomy for the treatment of uterovaginal prolapse: a prospective randomized pilot study. Int Urogynecol J. 2015;26:1687-1694. doi:10.1007/s00192-0152761-2.
- Roovers JP, van der Vaart CH, van der Bom JG, et al. A randomised controlled trial comparing abdominal and vaginal prolapse surgery: effects on urogenital function. BJOG. 2004;111:50-56. doi:10.1111/j.1471-0528.2004.00001.x.
- Roovers JP, van der Bom JG, van der Vaart CH, et al. A randomized comparison of post-operative pain, quality of life, and physical performance during the first 6 weeks after abdominal or vaginal surgical correction of descensus uteri. Neurourol Urodyn. 2005;24:334-340. doi:10.1002/nau.20104.
- Schulten SFM, Enklaar RA, Kluivers KB, et al. Evaluation of two vaginal, uterus sparing operations for pelvic organ prolapse: modified Manchester operation (MM) and sacrospinous hysteropexy (SSH), a study protocol for a multicentre randomized non-inferiority trial (the SAM study). BMC Womens Health. 20192;19:49. doi:10.1186/ s12905-019-0749-7.
2022 Update on contraception
On June 24, 2022, the US Supreme Court ruled in Dobbs v Jackson to overturn the landmark Roe v Wade decision, deeming that abortion is not protected by statutes that provide the right to privacy, liberty, or autonomy. With this historic ruling, other rights founded on the same principles, including the freedom to use contraception, may be called into question in the future. Clinics that provide abortion care typically play a vital role in providing contraception services. Due to abortion restriction across the country, many of these clinics are predicted to close and many have already closed. Within one month of the Dobbs decision, 43 clinics in 11 states had shut their doors to patients, reducing access to basic contraception services.1 It is more important now than ever that clinicians address barriers and lead the effort to improve and ensure that patients have access to contraceptive services.
In this Update, we review recent evidence that may help aid patients in obtaining contraception more easily and for longer periods of time. We review strategies demonstrated to improve contraceptive access, including how to increase prescribing rates of 1-year contraceptive supplies and pharmacist-prescribed contraception. We also review new data on extended use of the levonorgestrel 52 mg intrauterine device (LNG 52 mg IUD).
One-year prescribing of hormonal contraception decreases an access barrier
Uhm S, Chen MJ, Cutler ED, et al. Twelve-month prescribing of contraceptive pill, patch, and ring before and after a standardized electronic medical record order change. Contraception. 2021;103:60-63.
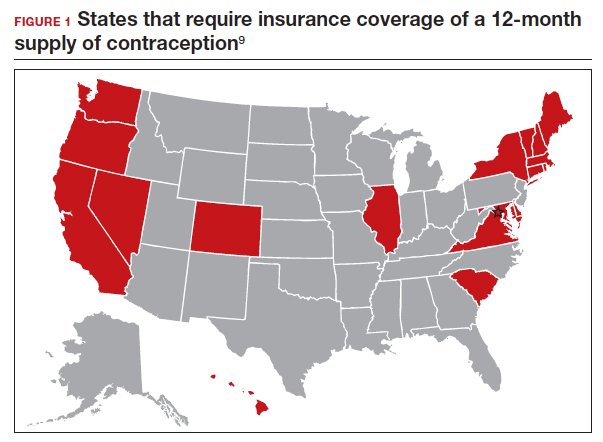
Providing a 1-year supply of self-administered contraception can lead to higher likelihood of continued use and is associated with reduced cost, unintended pregnancy, and abortion rates.2-4 Although some patients may not use a full year’s supply of pills, rings, or patches under such programs, the lower rates of unintended pregnancy result in significant cost savings as compared with the unused contraceptives.2,3 Accordingly, the Centers for Disease Control and Prevention (CDC) advises dispensing a 1-year supply of self-administered hormonal contraception.5 Insurance coverage and providers’ prescribing practices can be barriers to patients obtaining a year’s supply of hormonal contraception. Currently, 18 states and the District of Columbia legally require insurers to cover a 12-month supply of prescription contraceptives (FIGURE 1). Despite these laws and the CDC recommendation, studies show that most people continue to receive only a 1- to 3-month supply.6-8 One strategy to increase the number of 1-year supplies of self-administered contraception is institutional changes to default prescription orders.
Study design
In California, legislation enacted in January 2017 required commercial and medical assistance health plans to cover up to 12 months of US Food and Drug Administration (FDA)-approved self-administered hormonal contraceptives dispensed at 1 time as prescribed or requested. To better serve patients, a multidisciplinary team from the University of California Davis Health worked with the institution’s pharmacy to institute an electronic medical record (EMR) default order change from dispensing 1-month with refills to dispensing 12-month quantities for all combined and progestin-only pills, patches, and rings on formulary.
After this EMR order change in December 2019, Uhm and colleagues conducted a retrospective pre-post study using outpatient prescription data that included nearly 5,000 contraceptive pill, patch, and ring prescriptions over an 8-month period. They compared the frequency of 12-month prescriptions for each of these methods 4 months before and 4 months after the default order change. They compared the proportion of 12-month prescriptions by prescriber department affiliation and by clinic location. Department affiliation was categorized as obstetrics-gynecology or non–obstetrics-gynecology. Clinic location was categorized as medical center campus or community clinics.
Increase in 12-month prescriptions
The authors found an overall increase in 12-month prescriptions, from 11% to 27%, after the EMR order change. Prescribers at the medical center campus clinics more frequently ordered a 12-month supply compared with prescribers at community clinics both before (33% vs 4%, respectively) and after (53% vs 19%, respectively) the EMR change. The only group of providers without a significant increase in 12-month prescriptions was among obstetrics-gynecology providers at community clinics (4% before vs 6% after).
The system EMR change modified only the standard facility order settings and did not affect individual favorite orders, which may help explain the differences in prescribing practices. While this study found an increase in 12-month prescriptions, there were no data on the actual number of supplies a patient received or on reimbursement.
The study by Uhm and colleagues showed that making a relatively simple change to default EMR orders can increase 12-month contraception prescribing and lead to greater patient-centered care. Evidence shows that providers and pharmacists are not necessarily aware of laws that require 12-month supply coverage and routinely prescribe smaller supplies.6,7,9 For clinicians in states that have these laws (FIGURE 1), we urge you to provide as full a supply of contraceptives as possible as this approach is both evidence based and patient centered. Although this study shows the benefit of universal system change to the EMR, individual clinicians also must be sure to modify personal order preferences. In addition, pharmacists can play an important role by updating policies that comply with these laws and by increasing pharmacy stocks of contraception supplies.7 For those living in states that do not currently have these laws, we encourage you to reach out to your legislators to advocate for similar laws as the data show clear medical and cost benefits for patients and society.
Continue to: Pharmacist prescription of hormonal contraception is safe and promotes continuation...
Pharmacist prescription of hormonal contraception is safe and promotes continuation
Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
Patients often face difficulty obtaining both new and timely refills of self-administered contraception.10,11 To expand contraception access, Oregon became the first state (in 2016) to enact legislation to authorize direct pharmacist prescribing of hormonal contraceptives.12 Currently, 17 states and the District of Columbia have protocols for pharmacist prescribing privileges (FIGURE 2), and proposed legislation is pending in another 14 states.10,12 These protocols vary, but basic processes include screening, documentation, monitoring, and referrals when necessary. Typically, protocols require a pharmacist to review a patient’s medical history, pregnancy status, medication use, and blood pressure, followed by contraceptive counseling.10 Pharmacies are generally located in the community they serve, have extended hours, and usually do not require an appointment.8,13,14
Pharmacist prescribing increases the number of new contraceptive users, and pharmacists are more likely to prescribe a 6-month or longer supply of contraceptives compared with clinicians.8,13,15 Also, pharmacist prescribing is safe, with adherence rates to the CDC’s US Medical Eligibility Criteria for Contraceptive Use similar to those of prescriptions provided by a clinician.13
Authors of a recent multi-state study further assessed the impact of pharmacist prescribing by evaluating 12-month continuation and perfect use rates.
Study design
Rodriguez and colleagues evaluated the results of a 1-year prospective cohort study conducted in 2019 that included 388 participants who sought contraception in California, Colorado, Hawaii, and Oregon. All these states had laws permitting pharmacist prescribing and 12-month supply of hormonal contraception. Participants received prescriptions directly from a pharmacist at 1 of 139 pharmacies (n = 149) or filled a prescription provided by a clinician (n = 239). The primary outcomes were continuation of an effective method and perfect use of contraception across 12 months.
Participant demographics were similar between the 2 groups except for education and insurance status. Participants who received a prescription from a clinician reported higher levels of education. A greater proportion of uninsured participants received a prescription from a pharmacist (11%) compared with from a clinician (3%).
Contraceptive continuation rates
Participants were surveyed 3 times during the 12-month study about their current contraceptive method, if they had switched methods, or if they had any missed days of contraception.
Overall, 340 participants (88%) completed a full 12 months of follow-up. Continuation rates were similar between the 2 groups: 89% in the clinician-prescribed and 90% in the pharmacist-prescribed group (P=.86). Participants in the 2 groups also reported similar rates of perfect use (no missed days: 54% and 47%, respectively [P=.69]). Additionally, the authors reported that 29 participants changed from a tier 2 (pill, patch, ring, injection) to a tier 1 (intrauterine device or implant) method during follow-up, with no difference in switch rates for participants who received care from a clinician (10%) or a pharmacist (7%).
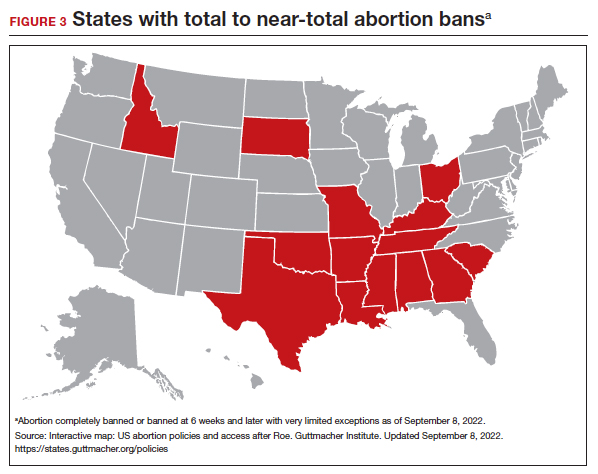
Patients have difficulties in obtaining both an initial contraceptive prescription and refills in time to avoid breaks in coverage.16 Pharmacist prescription of contraception is a proven strategy to increase access to contraception for new users or to promote continuation among current users. This practice is evidence based, decreases unintended pregnancy rates, and is safe.8,13,15,17
Promoting universal pharmacist prescribing is even more important given the overruling of Roe v Wade. With abortion restrictions, many family planning clinics that also play a vital role in providing contraception will close. Most states that are limiting abortion care (FIGURE 3) are the same states without pharmacist-prescribing provisions (FIGURE 2). As patient advocates, we need to continue to support this evidence-based practice in states where it is available and push legislators in states where it is not. Pharmacists should receive support to complete the training and certification needed to not only provide this service but also to receive appropriate reimbursements. Restrictions, such as requiring patients to be 18 years or older or to have prior consultation with a physician, should be limited as these are not necessary to provide self-administered contraception safely. Clinicians and pharmacists should inform patients, in states where this is available, that they can access initial or refill prescriptions at their local pharmacy if that is more convenient or their preference. Clinicians who live in states without these laws can advocate for their community by encouraging their legislators to pass laws that allow this evidence-based practice.
Continue to: LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use...
LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use
Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;S00029378(22)00366-0.
Given the potential difficulty accessing contraceptive and abortion services due to state restrictions, patients may be more motivated to maintain long-acting reversible contraceptives for maximum periods of time. The LNG 52 mg IUD was first marketed as a 5-year product, but multiple studies suggested that it had potential longer duration of efficacy and safety.18,19 The most recent clinical trial report shows that the LNG 52 mg IUD has at least 8 years of efficacy and safety.
Evidence supports 8 years’ use
The ACCESS IUS (A Comprehensive Contraceptive Efficacy and Safety Study of an IUS) phase 3 trial was designed to assess the safety and efficacy of a LNG 52 mg IUD (Liletta) for up to 10 years of use. The recent publication by Creinin and colleagues extends the available data from this study from 6 to 8 years.
Five-hundred and sixty-nine participants started year 7; 478 completed year 7 and 343 completed year 8 by the time the study was discontinued. Two pregnancies occurred in year 7 and no pregnancies occurred in year 8. One of the pregnancies in year 7 was determined by ultrasound examination to have implantation on day 4 after LNG IUD removal. According to the FDA, any pregnancy that occurs within 7 days of discontinuation is included as on-treatment, whereas the European Medicines Agency (EMA) has a 2-day cutoff. Over 8 years, 11 pregnancies occurred. The cumulative life-table pregnancy rate in the primary efficacy population through year 8 was 1.32% (95% confidence interval [CI], 0.69–2.51) under FDA rules and 1.09% (95% CI, 0.56–2.13) according to EMA guidance.
Absence of bleeding/spotting rates and adverse events
Rates of absence of bleeding/spotting remained relatively stable in years 7 and 8 at around 40%, similar to the rates during years 3 to 8 (FIGURE 4). Overall, only 2.6% of participants discontinued LNG IUD use because of bleeding problems, with a total of 4 participants discontinuing for this reason in years 7 and 8. Expulsion rates remained low at a rate of approximately 0.5% in years 7 and 8. Vulvovaginal infections were the most common adverse effect during year 7–8 of use. These findings are consistent with those found at 6 years.20 ●
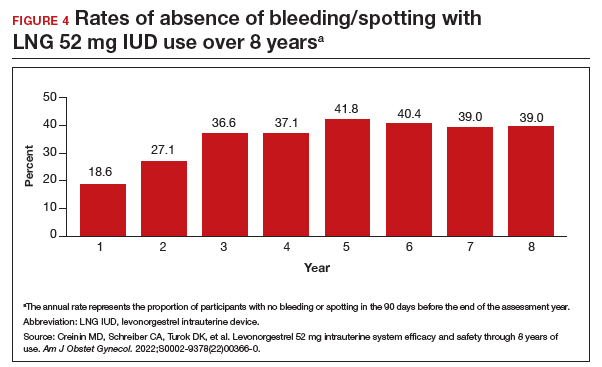
As abortion and contraception services become more difficult to access, patients may be more motivated to initiate or maintain an intrauterine device for longer. The ACCESS IUS trial provides contemporary data that are generalizable across the US population. Clinicians should educate patients about the efficacy, low incidence of new adverse events, and the steady rate at which patients experience absence of bleeding/spotting. The most recent data analysis supports continued use of LNG 52 mg IUD products for up to 8 years with an excellent extended safety profile. While some providers may express concern that patients may experience more bleeding with prolonged use, this study demonstrated low discontinuation rates due to bleeding in years 7 and 8. Perforations were diagnosed only during the first year, meaning that they most likely are related to the insertion process. Additionally, in this long-term study, expulsions occurred most frequently in the first year after placement. This study, which shows that the LNG IUD can continue to be used for longer than before, is important because it means that many patients will need fewer removals and reinsertions over their lifetime, reducing a patient’s risks and discomfort associated with these procedures. Sharing these data is important, as longer LNG IUD retention may reduce burdens faced by patients who desire long-acting reversible contraception.
- Kirstein M, Jones RK, Philbin J. One month post-Roe: at least 43 abortion clinics across 11 states have stopped offering abortion care. Guttmacher Institute. July 28, 2022. Accessed September 14, 2022. https://www.guttmacher.org /article/2022/07/one-month-post-roe-least-43-abortion-clinics-across -11-states-have-stopped-offering
- Foster DG, Hulett D, Bradsberry M, et al. Number of oral contraceptive pill packages dispensed and subsequent unintended pregnancies. Obstet Gynecol. 2011;117:566-572.
- Foster DG, Parvataneni R, de Bocanegra HT, et al. Number of oral contraceptive pill packages dispensed, method continuation, and costs. Obstet Gynecol. 2006;108:1107-114.
- Niu F, Cornelius J, Aboubechara N, et al. Real world outcomes related to providing an annual supply of short-acting hormonal contraceptives. Contraception. 2022;107:58-61.
- Curtis KM, Jatlaoui TC, Tepper NK, et al. US selected practice recommendations for contraceptive use, 2016. MMWR Recomm Rep. 2016;65:1-66.
- Women’s sexual and reproductive health services: key findings from the 2017 Kaiser Women’s Health Survey. KFF: Kaiser Family Foundation. March 13, 2018. Accessed September 14, 2022. https://www.kff.org/womens-health-policy /issue-brief/womens-sexual-and-reproductive-health-services-key-findings -from-the-2017-kaiser-womens-health-survey/
- Nikpour G, Allen A, Rafie S, et al. Pharmacy implementation of a new law allowing year-long hormonal contraception supplies. Pharmacy (Basel). 2020;8:E165.
- Rodriguez MI, Edelman AB, Skye M, et al. Association of pharmacist prescription with dispensed duration of hormonal contraception. JAMA Netw Open. 2020;3:e205252.
- Insurance coverage of contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state-policy /explore/insurance-coverage-contraceptives
- Chim C, Sharma P. Pharmacists prescribing hormonal contraceptives: a status update. US Pharm. 2021;46:45-49.
- Rodriguez MI, Hersh A, Anderson LB, et al. Association of pharmacist prescription of hormonal contraception with unintended pregnancies and Medicaid costs. Obstet Gynecol. 2019;133:1238-1246.
- Pharmacist-prescribed contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state -policy/explore/pharmacist-prescribed-contraceptives
- Anderson L, Hartung DM, Middleton L, et al. Pharmacist provision of hormonal contraception in the Oregon Medicaid population. Obstet Gynecol. 2019;133:1231-1237.
- Rodriguez MI, Edelman AB, Skye M, et al. Reasons for and experience in obtaining pharmacist prescribed contraception. Contraception. 2020;102:259-261.
- Rodriguez MI, Manibusan B, Kaufman M, et al. Association of pharmacist prescription of contraception with breaks in coverage. Obstet Gynecol. 2022;139:781-787.
- Pittman ME, Secura GM, Allsworth JE, et al. Understanding prescription adherence: pharmacy claims data from the Contraceptive CHOICE Project. Contraception. 2011;83:340-345.
- Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
- Secura GM, Allsworth JE, Madden T, et al. The Contraceptive CHOICE Project: reducing barriers to long-acting reversible contraception. Am J Obstet Gynecol. 2010;203:115.e1-7.
- Rowe P, Farley T, Peregoudov A, et al. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93:498-506.
- Westhoff CL, Keder LM, Gangestad A, et al. Six-year contraceptive efficacy and continued safety of a levonorgestrel 52 mg intrauterine system. Contraception. 2020;101:159-161.
On June 24, 2022, the US Supreme Court ruled in Dobbs v Jackson to overturn the landmark Roe v Wade decision, deeming that abortion is not protected by statutes that provide the right to privacy, liberty, or autonomy. With this historic ruling, other rights founded on the same principles, including the freedom to use contraception, may be called into question in the future. Clinics that provide abortion care typically play a vital role in providing contraception services. Due to abortion restriction across the country, many of these clinics are predicted to close and many have already closed. Within one month of the Dobbs decision, 43 clinics in 11 states had shut their doors to patients, reducing access to basic contraception services.1 It is more important now than ever that clinicians address barriers and lead the effort to improve and ensure that patients have access to contraceptive services.
In this Update, we review recent evidence that may help aid patients in obtaining contraception more easily and for longer periods of time. We review strategies demonstrated to improve contraceptive access, including how to increase prescribing rates of 1-year contraceptive supplies and pharmacist-prescribed contraception. We also review new data on extended use of the levonorgestrel 52 mg intrauterine device (LNG 52 mg IUD).
One-year prescribing of hormonal contraception decreases an access barrier
Uhm S, Chen MJ, Cutler ED, et al. Twelve-month prescribing of contraceptive pill, patch, and ring before and after a standardized electronic medical record order change. Contraception. 2021;103:60-63.
Providing a 1-year supply of self-administered contraception can lead to higher likelihood of continued use and is associated with reduced cost, unintended pregnancy, and abortion rates.2-4 Although some patients may not use a full year’s supply of pills, rings, or patches under such programs, the lower rates of unintended pregnancy result in significant cost savings as compared with the unused contraceptives.2,3 Accordingly, the Centers for Disease Control and Prevention (CDC) advises dispensing a 1-year supply of self-administered hormonal contraception.5 Insurance coverage and providers’ prescribing practices can be barriers to patients obtaining a year’s supply of hormonal contraception. Currently, 18 states and the District of Columbia legally require insurers to cover a 12-month supply of prescription contraceptives (FIGURE 1). Despite these laws and the CDC recommendation, studies show that most people continue to receive only a 1- to 3-month supply.6-8 One strategy to increase the number of 1-year supplies of self-administered contraception is institutional changes to default prescription orders.
Study design
In California, legislation enacted in January 2017 required commercial and medical assistance health plans to cover up to 12 months of US Food and Drug Administration (FDA)-approved self-administered hormonal contraceptives dispensed at 1 time as prescribed or requested. To better serve patients, a multidisciplinary team from the University of California Davis Health worked with the institution’s pharmacy to institute an electronic medical record (EMR) default order change from dispensing 1-month with refills to dispensing 12-month quantities for all combined and progestin-only pills, patches, and rings on formulary.
After this EMR order change in December 2019, Uhm and colleagues conducted a retrospective pre-post study using outpatient prescription data that included nearly 5,000 contraceptive pill, patch, and ring prescriptions over an 8-month period. They compared the frequency of 12-month prescriptions for each of these methods 4 months before and 4 months after the default order change. They compared the proportion of 12-month prescriptions by prescriber department affiliation and by clinic location. Department affiliation was categorized as obstetrics-gynecology or non–obstetrics-gynecology. Clinic location was categorized as medical center campus or community clinics.
Increase in 12-month prescriptions
The authors found an overall increase in 12-month prescriptions, from 11% to 27%, after the EMR order change. Prescribers at the medical center campus clinics more frequently ordered a 12-month supply compared with prescribers at community clinics both before (33% vs 4%, respectively) and after (53% vs 19%, respectively) the EMR change. The only group of providers without a significant increase in 12-month prescriptions was among obstetrics-gynecology providers at community clinics (4% before vs 6% after).
The system EMR change modified only the standard facility order settings and did not affect individual favorite orders, which may help explain the differences in prescribing practices. While this study found an increase in 12-month prescriptions, there were no data on the actual number of supplies a patient received or on reimbursement.
The study by Uhm and colleagues showed that making a relatively simple change to default EMR orders can increase 12-month contraception prescribing and lead to greater patient-centered care. Evidence shows that providers and pharmacists are not necessarily aware of laws that require 12-month supply coverage and routinely prescribe smaller supplies.6,7,9 For clinicians in states that have these laws (FIGURE 1), we urge you to provide as full a supply of contraceptives as possible as this approach is both evidence based and patient centered. Although this study shows the benefit of universal system change to the EMR, individual clinicians also must be sure to modify personal order preferences. In addition, pharmacists can play an important role by updating policies that comply with these laws and by increasing pharmacy stocks of contraception supplies.7 For those living in states that do not currently have these laws, we encourage you to reach out to your legislators to advocate for similar laws as the data show clear medical and cost benefits for patients and society.
Continue to: Pharmacist prescription of hormonal contraception is safe and promotes continuation...
Pharmacist prescription of hormonal contraception is safe and promotes continuation
Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
Patients often face difficulty obtaining both new and timely refills of self-administered contraception.10,11 To expand contraception access, Oregon became the first state (in 2016) to enact legislation to authorize direct pharmacist prescribing of hormonal contraceptives.12 Currently, 17 states and the District of Columbia have protocols for pharmacist prescribing privileges (FIGURE 2), and proposed legislation is pending in another 14 states.10,12 These protocols vary, but basic processes include screening, documentation, monitoring, and referrals when necessary. Typically, protocols require a pharmacist to review a patient’s medical history, pregnancy status, medication use, and blood pressure, followed by contraceptive counseling.10 Pharmacies are generally located in the community they serve, have extended hours, and usually do not require an appointment.8,13,14
Pharmacist prescribing increases the number of new contraceptive users, and pharmacists are more likely to prescribe a 6-month or longer supply of contraceptives compared with clinicians.8,13,15 Also, pharmacist prescribing is safe, with adherence rates to the CDC’s US Medical Eligibility Criteria for Contraceptive Use similar to those of prescriptions provided by a clinician.13
Authors of a recent multi-state study further assessed the impact of pharmacist prescribing by evaluating 12-month continuation and perfect use rates.
Study design
Rodriguez and colleagues evaluated the results of a 1-year prospective cohort study conducted in 2019 that included 388 participants who sought contraception in California, Colorado, Hawaii, and Oregon. All these states had laws permitting pharmacist prescribing and 12-month supply of hormonal contraception. Participants received prescriptions directly from a pharmacist at 1 of 139 pharmacies (n = 149) or filled a prescription provided by a clinician (n = 239). The primary outcomes were continuation of an effective method and perfect use of contraception across 12 months.
Participant demographics were similar between the 2 groups except for education and insurance status. Participants who received a prescription from a clinician reported higher levels of education. A greater proportion of uninsured participants received a prescription from a pharmacist (11%) compared with from a clinician (3%).
Contraceptive continuation rates
Participants were surveyed 3 times during the 12-month study about their current contraceptive method, if they had switched methods, or if they had any missed days of contraception.
Overall, 340 participants (88%) completed a full 12 months of follow-up. Continuation rates were similar between the 2 groups: 89% in the clinician-prescribed and 90% in the pharmacist-prescribed group (P=.86). Participants in the 2 groups also reported similar rates of perfect use (no missed days: 54% and 47%, respectively [P=.69]). Additionally, the authors reported that 29 participants changed from a tier 2 (pill, patch, ring, injection) to a tier 1 (intrauterine device or implant) method during follow-up, with no difference in switch rates for participants who received care from a clinician (10%) or a pharmacist (7%).
Patients have difficulties in obtaining both an initial contraceptive prescription and refills in time to avoid breaks in coverage.16 Pharmacist prescription of contraception is a proven strategy to increase access to contraception for new users or to promote continuation among current users. This practice is evidence based, decreases unintended pregnancy rates, and is safe.8,13,15,17
Promoting universal pharmacist prescribing is even more important given the overruling of Roe v Wade. With abortion restrictions, many family planning clinics that also play a vital role in providing contraception will close. Most states that are limiting abortion care (FIGURE 3) are the same states without pharmacist-prescribing provisions (FIGURE 2). As patient advocates, we need to continue to support this evidence-based practice in states where it is available and push legislators in states where it is not. Pharmacists should receive support to complete the training and certification needed to not only provide this service but also to receive appropriate reimbursements. Restrictions, such as requiring patients to be 18 years or older or to have prior consultation with a physician, should be limited as these are not necessary to provide self-administered contraception safely. Clinicians and pharmacists should inform patients, in states where this is available, that they can access initial or refill prescriptions at their local pharmacy if that is more convenient or their preference. Clinicians who live in states without these laws can advocate for their community by encouraging their legislators to pass laws that allow this evidence-based practice.
Continue to: LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use...
LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use
Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;S00029378(22)00366-0.
Given the potential difficulty accessing contraceptive and abortion services due to state restrictions, patients may be more motivated to maintain long-acting reversible contraceptives for maximum periods of time. The LNG 52 mg IUD was first marketed as a 5-year product, but multiple studies suggested that it had potential longer duration of efficacy and safety.18,19 The most recent clinical trial report shows that the LNG 52 mg IUD has at least 8 years of efficacy and safety.
Evidence supports 8 years’ use
The ACCESS IUS (A Comprehensive Contraceptive Efficacy and Safety Study of an IUS) phase 3 trial was designed to assess the safety and efficacy of a LNG 52 mg IUD (Liletta) for up to 10 years of use. The recent publication by Creinin and colleagues extends the available data from this study from 6 to 8 years.
Five-hundred and sixty-nine participants started year 7; 478 completed year 7 and 343 completed year 8 by the time the study was discontinued. Two pregnancies occurred in year 7 and no pregnancies occurred in year 8. One of the pregnancies in year 7 was determined by ultrasound examination to have implantation on day 4 after LNG IUD removal. According to the FDA, any pregnancy that occurs within 7 days of discontinuation is included as on-treatment, whereas the European Medicines Agency (EMA) has a 2-day cutoff. Over 8 years, 11 pregnancies occurred. The cumulative life-table pregnancy rate in the primary efficacy population through year 8 was 1.32% (95% confidence interval [CI], 0.69–2.51) under FDA rules and 1.09% (95% CI, 0.56–2.13) according to EMA guidance.
Absence of bleeding/spotting rates and adverse events
Rates of absence of bleeding/spotting remained relatively stable in years 7 and 8 at around 40%, similar to the rates during years 3 to 8 (FIGURE 4). Overall, only 2.6% of participants discontinued LNG IUD use because of bleeding problems, with a total of 4 participants discontinuing for this reason in years 7 and 8. Expulsion rates remained low at a rate of approximately 0.5% in years 7 and 8. Vulvovaginal infections were the most common adverse effect during year 7–8 of use. These findings are consistent with those found at 6 years.20 ●
As abortion and contraception services become more difficult to access, patients may be more motivated to initiate or maintain an intrauterine device for longer. The ACCESS IUS trial provides contemporary data that are generalizable across the US population. Clinicians should educate patients about the efficacy, low incidence of new adverse events, and the steady rate at which patients experience absence of bleeding/spotting. The most recent data analysis supports continued use of LNG 52 mg IUD products for up to 8 years with an excellent extended safety profile. While some providers may express concern that patients may experience more bleeding with prolonged use, this study demonstrated low discontinuation rates due to bleeding in years 7 and 8. Perforations were diagnosed only during the first year, meaning that they most likely are related to the insertion process. Additionally, in this long-term study, expulsions occurred most frequently in the first year after placement. This study, which shows that the LNG IUD can continue to be used for longer than before, is important because it means that many patients will need fewer removals and reinsertions over their lifetime, reducing a patient’s risks and discomfort associated with these procedures. Sharing these data is important, as longer LNG IUD retention may reduce burdens faced by patients who desire long-acting reversible contraception.
On June 24, 2022, the US Supreme Court ruled in Dobbs v Jackson to overturn the landmark Roe v Wade decision, deeming that abortion is not protected by statutes that provide the right to privacy, liberty, or autonomy. With this historic ruling, other rights founded on the same principles, including the freedom to use contraception, may be called into question in the future. Clinics that provide abortion care typically play a vital role in providing contraception services. Due to abortion restriction across the country, many of these clinics are predicted to close and many have already closed. Within one month of the Dobbs decision, 43 clinics in 11 states had shut their doors to patients, reducing access to basic contraception services.1 It is more important now than ever that clinicians address barriers and lead the effort to improve and ensure that patients have access to contraceptive services.
In this Update, we review recent evidence that may help aid patients in obtaining contraception more easily and for longer periods of time. We review strategies demonstrated to improve contraceptive access, including how to increase prescribing rates of 1-year contraceptive supplies and pharmacist-prescribed contraception. We also review new data on extended use of the levonorgestrel 52 mg intrauterine device (LNG 52 mg IUD).
One-year prescribing of hormonal contraception decreases an access barrier
Uhm S, Chen MJ, Cutler ED, et al. Twelve-month prescribing of contraceptive pill, patch, and ring before and after a standardized electronic medical record order change. Contraception. 2021;103:60-63.
Providing a 1-year supply of self-administered contraception can lead to higher likelihood of continued use and is associated with reduced cost, unintended pregnancy, and abortion rates.2-4 Although some patients may not use a full year’s supply of pills, rings, or patches under such programs, the lower rates of unintended pregnancy result in significant cost savings as compared with the unused contraceptives.2,3 Accordingly, the Centers for Disease Control and Prevention (CDC) advises dispensing a 1-year supply of self-administered hormonal contraception.5 Insurance coverage and providers’ prescribing practices can be barriers to patients obtaining a year’s supply of hormonal contraception. Currently, 18 states and the District of Columbia legally require insurers to cover a 12-month supply of prescription contraceptives (FIGURE 1). Despite these laws and the CDC recommendation, studies show that most people continue to receive only a 1- to 3-month supply.6-8 One strategy to increase the number of 1-year supplies of self-administered contraception is institutional changes to default prescription orders.
Study design
In California, legislation enacted in January 2017 required commercial and medical assistance health plans to cover up to 12 months of US Food and Drug Administration (FDA)-approved self-administered hormonal contraceptives dispensed at 1 time as prescribed or requested. To better serve patients, a multidisciplinary team from the University of California Davis Health worked with the institution’s pharmacy to institute an electronic medical record (EMR) default order change from dispensing 1-month with refills to dispensing 12-month quantities for all combined and progestin-only pills, patches, and rings on formulary.
After this EMR order change in December 2019, Uhm and colleagues conducted a retrospective pre-post study using outpatient prescription data that included nearly 5,000 contraceptive pill, patch, and ring prescriptions over an 8-month period. They compared the frequency of 12-month prescriptions for each of these methods 4 months before and 4 months after the default order change. They compared the proportion of 12-month prescriptions by prescriber department affiliation and by clinic location. Department affiliation was categorized as obstetrics-gynecology or non–obstetrics-gynecology. Clinic location was categorized as medical center campus or community clinics.
Increase in 12-month prescriptions
The authors found an overall increase in 12-month prescriptions, from 11% to 27%, after the EMR order change. Prescribers at the medical center campus clinics more frequently ordered a 12-month supply compared with prescribers at community clinics both before (33% vs 4%, respectively) and after (53% vs 19%, respectively) the EMR change. The only group of providers without a significant increase in 12-month prescriptions was among obstetrics-gynecology providers at community clinics (4% before vs 6% after).
The system EMR change modified only the standard facility order settings and did not affect individual favorite orders, which may help explain the differences in prescribing practices. While this study found an increase in 12-month prescriptions, there were no data on the actual number of supplies a patient received or on reimbursement.
The study by Uhm and colleagues showed that making a relatively simple change to default EMR orders can increase 12-month contraception prescribing and lead to greater patient-centered care. Evidence shows that providers and pharmacists are not necessarily aware of laws that require 12-month supply coverage and routinely prescribe smaller supplies.6,7,9 For clinicians in states that have these laws (FIGURE 1), we urge you to provide as full a supply of contraceptives as possible as this approach is both evidence based and patient centered. Although this study shows the benefit of universal system change to the EMR, individual clinicians also must be sure to modify personal order preferences. In addition, pharmacists can play an important role by updating policies that comply with these laws and by increasing pharmacy stocks of contraception supplies.7 For those living in states that do not currently have these laws, we encourage you to reach out to your legislators to advocate for similar laws as the data show clear medical and cost benefits for patients and society.
Continue to: Pharmacist prescription of hormonal contraception is safe and promotes continuation...
Pharmacist prescription of hormonal contraception is safe and promotes continuation
Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
Patients often face difficulty obtaining both new and timely refills of self-administered contraception.10,11 To expand contraception access, Oregon became the first state (in 2016) to enact legislation to authorize direct pharmacist prescribing of hormonal contraceptives.12 Currently, 17 states and the District of Columbia have protocols for pharmacist prescribing privileges (FIGURE 2), and proposed legislation is pending in another 14 states.10,12 These protocols vary, but basic processes include screening, documentation, monitoring, and referrals when necessary. Typically, protocols require a pharmacist to review a patient’s medical history, pregnancy status, medication use, and blood pressure, followed by contraceptive counseling.10 Pharmacies are generally located in the community they serve, have extended hours, and usually do not require an appointment.8,13,14
Pharmacist prescribing increases the number of new contraceptive users, and pharmacists are more likely to prescribe a 6-month or longer supply of contraceptives compared with clinicians.8,13,15 Also, pharmacist prescribing is safe, with adherence rates to the CDC’s US Medical Eligibility Criteria for Contraceptive Use similar to those of prescriptions provided by a clinician.13
Authors of a recent multi-state study further assessed the impact of pharmacist prescribing by evaluating 12-month continuation and perfect use rates.
Study design
Rodriguez and colleagues evaluated the results of a 1-year prospective cohort study conducted in 2019 that included 388 participants who sought contraception in California, Colorado, Hawaii, and Oregon. All these states had laws permitting pharmacist prescribing and 12-month supply of hormonal contraception. Participants received prescriptions directly from a pharmacist at 1 of 139 pharmacies (n = 149) or filled a prescription provided by a clinician (n = 239). The primary outcomes were continuation of an effective method and perfect use of contraception across 12 months.
Participant demographics were similar between the 2 groups except for education and insurance status. Participants who received a prescription from a clinician reported higher levels of education. A greater proportion of uninsured participants received a prescription from a pharmacist (11%) compared with from a clinician (3%).
Contraceptive continuation rates
Participants were surveyed 3 times during the 12-month study about their current contraceptive method, if they had switched methods, or if they had any missed days of contraception.
Overall, 340 participants (88%) completed a full 12 months of follow-up. Continuation rates were similar between the 2 groups: 89% in the clinician-prescribed and 90% in the pharmacist-prescribed group (P=.86). Participants in the 2 groups also reported similar rates of perfect use (no missed days: 54% and 47%, respectively [P=.69]). Additionally, the authors reported that 29 participants changed from a tier 2 (pill, patch, ring, injection) to a tier 1 (intrauterine device or implant) method during follow-up, with no difference in switch rates for participants who received care from a clinician (10%) or a pharmacist (7%).
Patients have difficulties in obtaining both an initial contraceptive prescription and refills in time to avoid breaks in coverage.16 Pharmacist prescription of contraception is a proven strategy to increase access to contraception for new users or to promote continuation among current users. This practice is evidence based, decreases unintended pregnancy rates, and is safe.8,13,15,17
Promoting universal pharmacist prescribing is even more important given the overruling of Roe v Wade. With abortion restrictions, many family planning clinics that also play a vital role in providing contraception will close. Most states that are limiting abortion care (FIGURE 3) are the same states without pharmacist-prescribing provisions (FIGURE 2). As patient advocates, we need to continue to support this evidence-based practice in states where it is available and push legislators in states where it is not. Pharmacists should receive support to complete the training and certification needed to not only provide this service but also to receive appropriate reimbursements. Restrictions, such as requiring patients to be 18 years or older or to have prior consultation with a physician, should be limited as these are not necessary to provide self-administered contraception safely. Clinicians and pharmacists should inform patients, in states where this is available, that they can access initial or refill prescriptions at their local pharmacy if that is more convenient or their preference. Clinicians who live in states without these laws can advocate for their community by encouraging their legislators to pass laws that allow this evidence-based practice.
Continue to: LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use...
LNG 52 mg IUD demonstrates efficacy and safety through 8 years of use
Creinin MD, Schreiber CA, Turok DK, et al. Levonorgestrel 52 mg intrauterine system efficacy and safety through 8 years of use. Am J Obstet Gynecol. 2022;S00029378(22)00366-0.
Given the potential difficulty accessing contraceptive and abortion services due to state restrictions, patients may be more motivated to maintain long-acting reversible contraceptives for maximum periods of time. The LNG 52 mg IUD was first marketed as a 5-year product, but multiple studies suggested that it had potential longer duration of efficacy and safety.18,19 The most recent clinical trial report shows that the LNG 52 mg IUD has at least 8 years of efficacy and safety.
Evidence supports 8 years’ use
The ACCESS IUS (A Comprehensive Contraceptive Efficacy and Safety Study of an IUS) phase 3 trial was designed to assess the safety and efficacy of a LNG 52 mg IUD (Liletta) for up to 10 years of use. The recent publication by Creinin and colleagues extends the available data from this study from 6 to 8 years.
Five-hundred and sixty-nine participants started year 7; 478 completed year 7 and 343 completed year 8 by the time the study was discontinued. Two pregnancies occurred in year 7 and no pregnancies occurred in year 8. One of the pregnancies in year 7 was determined by ultrasound examination to have implantation on day 4 after LNG IUD removal. According to the FDA, any pregnancy that occurs within 7 days of discontinuation is included as on-treatment, whereas the European Medicines Agency (EMA) has a 2-day cutoff. Over 8 years, 11 pregnancies occurred. The cumulative life-table pregnancy rate in the primary efficacy population through year 8 was 1.32% (95% confidence interval [CI], 0.69–2.51) under FDA rules and 1.09% (95% CI, 0.56–2.13) according to EMA guidance.
Absence of bleeding/spotting rates and adverse events
Rates of absence of bleeding/spotting remained relatively stable in years 7 and 8 at around 40%, similar to the rates during years 3 to 8 (FIGURE 4). Overall, only 2.6% of participants discontinued LNG IUD use because of bleeding problems, with a total of 4 participants discontinuing for this reason in years 7 and 8. Expulsion rates remained low at a rate of approximately 0.5% in years 7 and 8. Vulvovaginal infections were the most common adverse effect during year 7–8 of use. These findings are consistent with those found at 6 years.20 ●
As abortion and contraception services become more difficult to access, patients may be more motivated to initiate or maintain an intrauterine device for longer. The ACCESS IUS trial provides contemporary data that are generalizable across the US population. Clinicians should educate patients about the efficacy, low incidence of new adverse events, and the steady rate at which patients experience absence of bleeding/spotting. The most recent data analysis supports continued use of LNG 52 mg IUD products for up to 8 years with an excellent extended safety profile. While some providers may express concern that patients may experience more bleeding with prolonged use, this study demonstrated low discontinuation rates due to bleeding in years 7 and 8. Perforations were diagnosed only during the first year, meaning that they most likely are related to the insertion process. Additionally, in this long-term study, expulsions occurred most frequently in the first year after placement. This study, which shows that the LNG IUD can continue to be used for longer than before, is important because it means that many patients will need fewer removals and reinsertions over their lifetime, reducing a patient’s risks and discomfort associated with these procedures. Sharing these data is important, as longer LNG IUD retention may reduce burdens faced by patients who desire long-acting reversible contraception.
- Kirstein M, Jones RK, Philbin J. One month post-Roe: at least 43 abortion clinics across 11 states have stopped offering abortion care. Guttmacher Institute. July 28, 2022. Accessed September 14, 2022. https://www.guttmacher.org /article/2022/07/one-month-post-roe-least-43-abortion-clinics-across -11-states-have-stopped-offering
- Foster DG, Hulett D, Bradsberry M, et al. Number of oral contraceptive pill packages dispensed and subsequent unintended pregnancies. Obstet Gynecol. 2011;117:566-572.
- Foster DG, Parvataneni R, de Bocanegra HT, et al. Number of oral contraceptive pill packages dispensed, method continuation, and costs. Obstet Gynecol. 2006;108:1107-114.
- Niu F, Cornelius J, Aboubechara N, et al. Real world outcomes related to providing an annual supply of short-acting hormonal contraceptives. Contraception. 2022;107:58-61.
- Curtis KM, Jatlaoui TC, Tepper NK, et al. US selected practice recommendations for contraceptive use, 2016. MMWR Recomm Rep. 2016;65:1-66.
- Women’s sexual and reproductive health services: key findings from the 2017 Kaiser Women’s Health Survey. KFF: Kaiser Family Foundation. March 13, 2018. Accessed September 14, 2022. https://www.kff.org/womens-health-policy /issue-brief/womens-sexual-and-reproductive-health-services-key-findings -from-the-2017-kaiser-womens-health-survey/
- Nikpour G, Allen A, Rafie S, et al. Pharmacy implementation of a new law allowing year-long hormonal contraception supplies. Pharmacy (Basel). 2020;8:E165.
- Rodriguez MI, Edelman AB, Skye M, et al. Association of pharmacist prescription with dispensed duration of hormonal contraception. JAMA Netw Open. 2020;3:e205252.
- Insurance coverage of contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state-policy /explore/insurance-coverage-contraceptives
- Chim C, Sharma P. Pharmacists prescribing hormonal contraceptives: a status update. US Pharm. 2021;46:45-49.
- Rodriguez MI, Hersh A, Anderson LB, et al. Association of pharmacist prescription of hormonal contraception with unintended pregnancies and Medicaid costs. Obstet Gynecol. 2019;133:1238-1246.
- Pharmacist-prescribed contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state -policy/explore/pharmacist-prescribed-contraceptives
- Anderson L, Hartung DM, Middleton L, et al. Pharmacist provision of hormonal contraception in the Oregon Medicaid population. Obstet Gynecol. 2019;133:1231-1237.
- Rodriguez MI, Edelman AB, Skye M, et al. Reasons for and experience in obtaining pharmacist prescribed contraception. Contraception. 2020;102:259-261.
- Rodriguez MI, Manibusan B, Kaufman M, et al. Association of pharmacist prescription of contraception with breaks in coverage. Obstet Gynecol. 2022;139:781-787.
- Pittman ME, Secura GM, Allsworth JE, et al. Understanding prescription adherence: pharmacy claims data from the Contraceptive CHOICE Project. Contraception. 2011;83:340-345.
- Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
- Secura GM, Allsworth JE, Madden T, et al. The Contraceptive CHOICE Project: reducing barriers to long-acting reversible contraception. Am J Obstet Gynecol. 2010;203:115.e1-7.
- Rowe P, Farley T, Peregoudov A, et al. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93:498-506.
- Westhoff CL, Keder LM, Gangestad A, et al. Six-year contraceptive efficacy and continued safety of a levonorgestrel 52 mg intrauterine system. Contraception. 2020;101:159-161.
- Kirstein M, Jones RK, Philbin J. One month post-Roe: at least 43 abortion clinics across 11 states have stopped offering abortion care. Guttmacher Institute. July 28, 2022. Accessed September 14, 2022. https://www.guttmacher.org /article/2022/07/one-month-post-roe-least-43-abortion-clinics-across -11-states-have-stopped-offering
- Foster DG, Hulett D, Bradsberry M, et al. Number of oral contraceptive pill packages dispensed and subsequent unintended pregnancies. Obstet Gynecol. 2011;117:566-572.
- Foster DG, Parvataneni R, de Bocanegra HT, et al. Number of oral contraceptive pill packages dispensed, method continuation, and costs. Obstet Gynecol. 2006;108:1107-114.
- Niu F, Cornelius J, Aboubechara N, et al. Real world outcomes related to providing an annual supply of short-acting hormonal contraceptives. Contraception. 2022;107:58-61.
- Curtis KM, Jatlaoui TC, Tepper NK, et al. US selected practice recommendations for contraceptive use, 2016. MMWR Recomm Rep. 2016;65:1-66.
- Women’s sexual and reproductive health services: key findings from the 2017 Kaiser Women’s Health Survey. KFF: Kaiser Family Foundation. March 13, 2018. Accessed September 14, 2022. https://www.kff.org/womens-health-policy /issue-brief/womens-sexual-and-reproductive-health-services-key-findings -from-the-2017-kaiser-womens-health-survey/
- Nikpour G, Allen A, Rafie S, et al. Pharmacy implementation of a new law allowing year-long hormonal contraception supplies. Pharmacy (Basel). 2020;8:E165.
- Rodriguez MI, Edelman AB, Skye M, et al. Association of pharmacist prescription with dispensed duration of hormonal contraception. JAMA Netw Open. 2020;3:e205252.
- Insurance coverage of contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state-policy /explore/insurance-coverage-contraceptives
- Chim C, Sharma P. Pharmacists prescribing hormonal contraceptives: a status update. US Pharm. 2021;46:45-49.
- Rodriguez MI, Hersh A, Anderson LB, et al. Association of pharmacist prescription of hormonal contraception with unintended pregnancies and Medicaid costs. Obstet Gynecol. 2019;133:1238-1246.
- Pharmacist-prescribed contraceptives. Guttmacher Institute. Updated August 1, 2022. Accessed September 14, 2022. https://www.guttmacher.org/state -policy/explore/pharmacist-prescribed-contraceptives
- Anderson L, Hartung DM, Middleton L, et al. Pharmacist provision of hormonal contraception in the Oregon Medicaid population. Obstet Gynecol. 2019;133:1231-1237.
- Rodriguez MI, Edelman AB, Skye M, et al. Reasons for and experience in obtaining pharmacist prescribed contraception. Contraception. 2020;102:259-261.
- Rodriguez MI, Manibusan B, Kaufman M, et al. Association of pharmacist prescription of contraception with breaks in coverage. Obstet Gynecol. 2022;139:781-787.
- Pittman ME, Secura GM, Allsworth JE, et al. Understanding prescription adherence: pharmacy claims data from the Contraceptive CHOICE Project. Contraception. 2011;83:340-345.
- Rodriguez MI, Skye M, Edelman AB, et al. Association of pharmacist prescription and 12-month contraceptive continuation rates. Am J Obstet Gynecol. 2021;225:647.e1-647.e9.
- Secura GM, Allsworth JE, Madden T, et al. The Contraceptive CHOICE Project: reducing barriers to long-acting reversible contraception. Am J Obstet Gynecol. 2010;203:115.e1-7.
- Rowe P, Farley T, Peregoudov A, et al. Safety and efficacy in parous women of a 52-mg levonorgestrel-medicated intrauterine device: a 7-year randomized comparative study with the TCu380A. Contraception. 2016;93:498-506.
- Westhoff CL, Keder LM, Gangestad A, et al. Six-year contraceptive efficacy and continued safety of a levonorgestrel 52 mg intrauterine system. Contraception. 2020;101:159-161.
Nonsurgical treatments for patients with urinary incontinence
CASE Patient has urine leakage that worsens with exercise
At her annual preventative health visit, a 39-year-old woman reports that she has leakage of urine. She states that she drinks “a gallon of water daily” to help her lose the 20 lb she gained during the COVID-19 pandemic. She wants to resume Zumba fitness classes, but exercise makes her urine leakage worse. She started wearing protective pads because she finds herself often leaking urine on the way to the bathroom.
What nonsurgical treatment options are available for this patient?
Nearly half of all women experience urinary incontinence (UI), the involuntary loss of urine, and the condition increases with age.1 This common condition negatively impacts physical and psychological health and has been associated with social isolation, sexual dysfunction, and reduced independence.2,3 Symptoms of UI are underreported, and therefore universal screening is recommended for women of all ages.4 The diversity of available treatments (TABLE 1) provides patients and clinicians an opportunity to develop a plan that aligns with their symptom severity, goals, preferences, and resources.
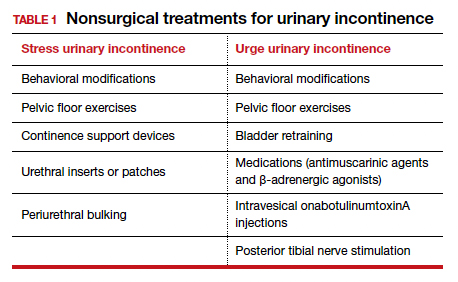
Types of UI
The most common types of UI are stress urinary incontinence (SUI) and urgency urinary incontinence (UUI). Mixed urinary incontinence (MUI) occurs when symptoms of both SUI and UUI are present. Although the mechanisms that lead to urine leakage vary by the type of incontinence, many primary interventions improve both types of leakage, so a clinical diagnosis is sufficient to initiate treatment.
Stress urinary incontinence results from an impaired or weakened sphincter, which leads to involuntary, yet predictable, urine loss during increased abdominal pressure, such as coughing, laughing, sneezing, lifting, or physical activity.5 In UUI, involuntary loss of urine often accompanies the sudden urge to void. UUI is associated with overactive bladder (OAB), defined as urinary urgency, with or without urinary incontinence, usually accompanied by urinary frequency and/or nocturia (urination that interrupts sleep).6
In OAB, the detrusor muscle contracts randomly, leading to a sudden urge to void. When bladder pressure exceeds urethral sphincter closure pressure, urine leakage occurs. Women describe the urgency episodes as unpredictable, the urine leakage as prolonged with large volumes, and often occurring as they seek the toilet. Risk factors include age, obesity, parity, history of vaginal delivery, family history, ethnicity/race, medical comorbidities, menopausal status, and tobacco use.5
Making a diagnosis
A basic office evaluation is the most key step for diagnostic accuracy that leads to treatment success. This includes a detailed history, assessment of symptom severity, physical exam, pelvic exam, urinalysis, postvoid residual (to rule out urinary retention), and a cough stress test (to demonstrate SUI). The goal is to assess symptom severity, determine the type of UI, and identify contributing and potentially reversible factors, such as a urinary tract infection, medications, pelvic organ prolapse, incomplete bladder emptying, or impaired neurologic status. In the absence of the latter, advanced diagnostic tests, such as urodynamics, contribute little toward discerning the type of incontinence or changing first-line treatment plans.7
During the COVID-19 pandemic, abbreviated, virtual assessments for urinary symptoms were associated with high degrees of satisfaction (91% for fulfillment of personal needs, 94% overall satisfaction).8 This highlights the value of validated symptom questionnaires that help establish a working diagnosis and treatment plan in the absence of a physical exam. Questionnaire-based diagnoses have acceptable accuracy for classifying UUI and SUI among women with uncomplicated medical and surgical histories and for initiating low-risk therapies for defined intervals.
The 3 incontinence questions (3IQ) screen is an example of a useful, quick diagnostic tool designed for the primary care setting (FIGURE 1).9 It has been used in pharmaceutical treatment trials for UUI, with low frequency of misdiagnosis (1%–4%), resulting in no harm by the drug treatment prescribed or by the delay in appropriate care.10 Due to the limitations of an abbreviated remote evaluation, however, clinicians should assess patient response to primary interventions in a timely window. Patients who fail to experience satisfactory symptom reduction within 6 to 12 weeks should complete their evaluation in person or through a referral to a urogynecology program.

Continue to: Primary therapies for UI...
Primary therapies for UI
Primary therapies for UUI and SUI target strength training of the pelvic floor muscles, moderation of fluid intake, and adjustment in voiding behaviors and medications. Any functional barriers to continence also should be identified and addressed. Simple interventions, including a daily bowel regimen to address constipation, a bedside commode, and scheduled voiding, may reduce incontinence episodes without incurring significant cost or risk. For women suspected of having MUI, the treatment plan should prioritize their most bothersome symptoms.
Lifestyle and behavioral modifications
Everyday habits, medical comorbidities, and medications may exacerbate the severity of both SUI and UUI. Behavioral therapy alone or in combination with other interventions effectively reduces both SUI and UUI symptoms and has been shown to improve the efficacy of continence surgery.11 Information gained from a 3-day bladder diary (FIGURE 2)12 can guide clinicians on personalized patient recommendations, such as reducing excessive consumption of fluids and bladder irritants, limiting late evening drinking in the setting of bothersome nocturia, and scheduling voids (every 2–3 hours) to preempt incontinence episodes.

Weight loss
Obesity is a strong, independent, modifiable risk factor for both SUI and UUI. Each 5 kg/m2 increase in body mass index (BMI) has been associated with a 20% to 70% increased risk of UI, while weight loss of 5% or greater in overweight or obese women can lead to at least a 50% decrease in UI frequency.13
Reducing fluid intake and bladder irritants
Overactive bladder symptoms often respond to moderation of excessive fluid intake and reduction of bladder irritants (caffeine, carbonated beverages, diet beverages, and alcohol). While there is no established definition of excess caffeine intake, one study categorized high caffeine intake as greater than 400 mg/day (approximately four 8-oz cups of coffee).14
Information provided in a bladder diary can guide individualized recommendations for reducing fluid intake, particularly when 24-hour urine production exceeds the normative range (> 50–60 oz or 1.5-1.8 L/day).15 Hydration needs vary by activity, environment, and food; some general guidelines suggest 48 to 64 oz/day.5,16
Continue to: Pelvic floor muscle training...
Pelvic floor muscle training
An effective treatment for both UUI and SUI symptoms, pelvic floor muscle training (PFMT) leads to high degrees of patient satisfaction and improvement in quality of life.17 The presumed mechanisms of action of PFMT include improved urethral closure pressure and inhibition of detrusor muscle contractions.
Common exercise protocols recommend 3 sets of 10 contractions, held for 6 to 10 seconds per day, in varying positions of sitting, standing, and lying. While many women may be familiar with Kegel exercises, poor technique with straining and recruitment of gluteal and abdominal muscles can undermine the effect of PFMT. Clinicians can confirm successful pelvic muscle contractions by placing a finger in the vagina to appreciate contraction around and elevation of the finger toward the pubic symphysis in the absence of pushing.
Referral to supervised physical therapy and use of such teaching aid tools as booklets, mobile applications, and biofeedback can improve exercise adherence and outcomes.18,19 Systematic reviews report initial cure or improvement of incontinence symptoms as high as 74%, although little information is available about the long-term duration of effect.17
Vaginal pessaries
Vaginal continence support pessaries and devices work by stabilizing urethral mobility and compression of the bladder neck. Continence devices are particularly effective for situational SUI (such as during exercise).
The reusable medical grade silicone pessaries are available in numerous shapes and sizes and are fitted by a health care clinician (FIGURE 3). Uresta is a self-fitted intravaginal device that women can purchase online with a prescription. The Poise Impressa bladder support is a disposable intravaginal device marketed for incontinence and available over-the-counter, without a prescription (FIGURE 4). Anecdotally, many women find that menstrual tampons provide a similar effect, but outcome data are lacking.
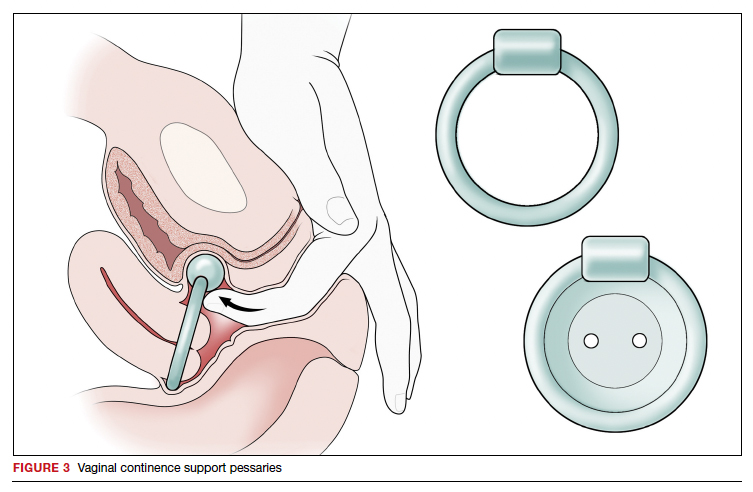
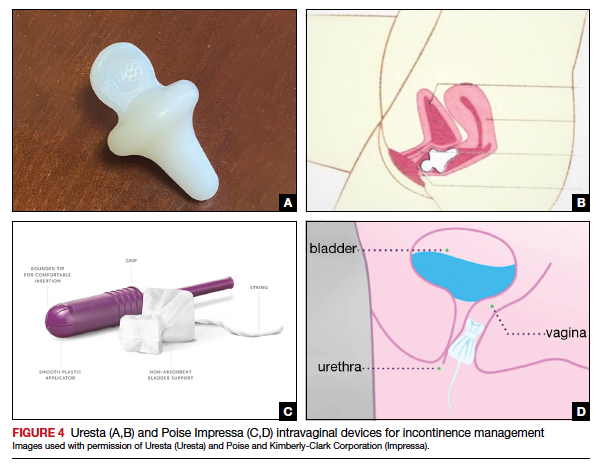
In a comparative effectiveness trial of a continence pessary and behavior therapy, behavioral therapy was more likely to result in no bothersome incontinence symptoms (49% vs 33%, P = .006) and greater treatment satisfaction at 3 months.20 However, these short-term group differences did not persist at 12 months, presumably due to waning adherence.
UUI-specific nonsurgical treatments
Drug therapy
All medications approved by the US Food and Drug Administration (FDA) for UI are for the indications of OAB or UUI. These second-line treatments are most effective as adjuncts to behavioral modifications and PFMT.
A multicenter randomized trial that evaluated the efficacy of drug therapy alone compared with drug therapy in combination with behavioral modification, PFMT, urge suppression strategies, timed voiding, and fluid management for UUI found that combined therapy was more successful in achieving greater than 70% reduction in incontinence episodes (58% for drug therapy vs 69% for combined therapy).21
Of the 8 medications currently marketed in the United States for OAB or UUI, 6 are anticholinergic agents that block muscarinic receptors in the smooth muscle of the bladder, leading to inhibition of detrusor contractions, and 2 are β-adrenergic receptor agonists that promote bladder storage capacity by relaxing the detrusor muscle (TABLE 2). Similar efficacies lead most clinicians to initiate drug therapy based on formulary coverage and tolerance for adverse effects. Patients can expect a 53% to 80% reduction in UUI episodes and a 12% to 32% reduction in urinary frequency.22
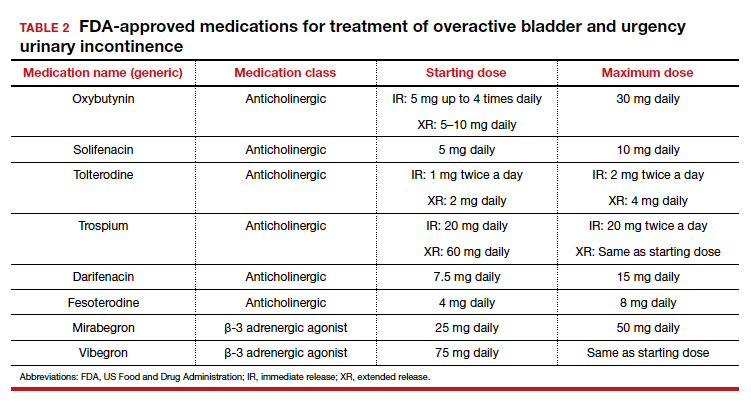
Extended-release formulations are associated with reduced anticholinergic side effects (dry mouth, constipation, somnolence, dry eyes), leading to improved adherence. Notably, the anticholinergic medications are contraindicated in patients with untreated narrow-angle glaucoma, gastric retention, and supraventricular tachycardia. Mirabegron should be used with caution in patients with poorly controlled hypertension. 5 Due to concerns regarding the association between cumulative anticholinergic burden and the development of dementia, clinicians may consider avoiding the anticholinergic medications in older and at-risk patients.23
Continue to: UUI office-based procedure treatments...
UUI office-based procedure treatments
If behavioral therapies and medications are ineffective, contraindicated, or not the patient’s preference, additional FDA-approved therapies for UUI are available, typically through referral to a urogynecologist, urologist, or continence center.
Posterior tibial nerve stimulation (PTNS) is a nondrug treatment that delivers electrical stimulation using an acupuncture needle for 12 weekly 30-minute sessions followed by monthly maintenance for responders. The time commitment for this treatment plan can be a barrier for some patients. However, patients who adhere to the recommended protocol can expect a 60% improvement in symptoms, with minimal adverse events. Treatment efficacy is comparable to that of anticholinergic medication.24
OnabotulinumtoxinA injections into the bladder muscle are performed cystoscopically under local anesthetic. The toxin blocks the presynaptic release of acetylcholine at the neuromuscular junction, resulting in temporary muscle paralysis. This treatment is associated with high satisfaction. Efficacy varies by study population and outcome measure.
In one US comparative effectiveness trial, 67% of study participants with UUI symptoms refractory to oral medication reported a greater than 50% reduction in OAB symptoms at 6 months, 20% reported complete resolution of UUI, and 72% requested a second injection within 24 months.25 The interval between the first and second injection was nearly 1 year (350 days).Risks include urinary tract infection (12% within 1 month of the procedure and 35% through 6 months); urinary retention requiring catheterization has decreased to 6% with recognition that most moderate retention is tolerated by patients.
Some insurers limit onabotulinumtoxinA treatment coverage to patients who have failed to achieve symptom control with first- and second-line treatments.
SUI-specific nonsurgical treatments
Cystoscopic injection of urethral bulking agents into the urethral submucosa is designed to improve urethral coaptation. It is a minor procedure that can be performed in an ambulatory setting under local anesthetic with or without sedation.
Various bulking agents have been approved for use in the United States, some of which have been withdrawn due to complications of migration, erosion, and pseudoabscess formation. Cure or improvement after bulking agent injection was found to be superior to a home pelvic floor exercise program but inferior to a midurethral sling procedure for cure (9% vs 89%).26
The durability of currently available urethral bulking agents beyond 1 year is unknown. Complications are typically minor and transient and include pain at the injection site, urinary retention, de novo urgency, and implant leakage. The advantages include no postprocedure activity restrictions.
CASE Symptom presentation guides treatment plan
Our patient described symptoms of stress-predominant MUI. She was counseled to moderate her fluid intake to 2 L per day and to strategically time voids (before exercise, and at least every 4 hours). The patient was fitted with an incontinence pessary, and she elected to pursue a course of supervised physical therapy for pelvic floor muscle strengthening. Her follow-up visit is scheduled in 3 months to determine if other interventions are warranted. ●
1. Lee UJ, Feinstein L, Ward JB, et al. Prevalence of urinary incontinence among a nationally representative sample of women, 2005–2016: findings from the Urologic Diseases in America Project. J Urol. 2021;205:1718-1724. doi:10.1097 /JU.0000000000001634
2. Sims J, Browning C, Lundgren-Lindquist B, et al. Urinary incontinence in a community sample of older adults: prevalence and impact on quality of life. Disabil Rehabil. 2011;33:1389-1398. doi:10.3109/09638288.2010.532284
3. Sarikaya S, Yildiz FG, Senocak C, et al. Urinary incontinence as a cause of depression and sexual dysfunction: questionnaire-based study. Rev Int Androl. 2020:18:50-54. doi:10.1016 /j.androl.2018.08.003
4. O’Reilly N, Nelson HD, Conry JM, et al; Women’s Preventive Services Initiative. Screening for urinary incontinence in women: a recommendation from the Women’s Preventive Services Initiative. Ann Intern Med. 2018;169(5):320-328. doi:10.7326/M18-0595
5. Barber MD, Walters MD, Karram MM, et al. Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier Saunders; 2021.
6. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Int Urogynecol J. 2010;21: 5-26. doi:10.1007/s00192-009-0976-9
7. ACOG practice bulletin no. 155. Urinary incontinence in women. Obstet Gynecol. 2015;126:e66-e81. doi:10.1097 /AOG.0000000000001148
8. Sansone S, Lu J, Drangsholt S, et al. No pelvic exam, no problem: patient satisfaction following the integration of comprehensive urogynecology telemedicine. Int Urogynecol J. 2022;1:3. doi:10.1007/s00192-022-05104-w
9. Brown JS, Bradley CS, Subak LL, et al; Diagnostic Aspects of Incontinence Study (DAISy) Research Group. The sensitivity and specificity of a simple test to distinguish between urge and stress urinary incontinence. Ann Intern Med. 2006;144:715723. doi:10.7326/0003-4819-144-10-200605160-00005
10. Hess R, Huang AJ, Richter HE, et al. Long-term efficacy and safety of questionnaire-based initiation of urgency urinary incontinence treatment. Am J Obstet Gynecol. 2013;209:244. e1-9. doi:10.1016/j.ajog.2013.05.008
11. Sung VW, Borello-France D, Newman DK, et al; NICHD Pelvic Floor Disorders Network. Effect of behavioral and pelvic floor muscle therapy combined with surgery vs surgery alone on incontinence symptoms among women with mixed urinary incontinence. JAMA. 2019;322:1066-1076. doi:10.1001 /jama.2019.12467
12. American Urogynecologic Society. Voices for PFD: intake and voiding diary. Accessed August 11, 2022. https://www .voicesforpfd.org/assets/2/6/Voiding_Diary.pdf
13. Subak LL, Richter HE, Hunskaar S. Obesity and urinary incontinence: epidemiology and clinical research update. J Urol. 2009;182(6 suppl):S2-7. doi:10.1016/j.juro.2009.08.071
14. Arya LA, Myers DL, Jackson ND. Dietary caffeine intake and the risk for detrusor instability: a case-control study. Obstet Gynecol. 2000;96:85-89. doi:10.1016/s0029-7844(00)00808-5
15. Wyman JF, Zhou J, LaCoursiere DY, et al. Normative noninvasive bladder function measurements in healthy women: a systematic review and meta-analysis. Neurourol Urodyn. 2020;39:507-522. doi:10.1002/nau.24265
16. Hashim H, Al Mousa R. Management of fluid intake in patients with overactive bladder. Curr Urol Rep. 2009;10: 428-433. doi:10.1007/s11934-009-0068-x
17. Dumoulin C, Cacciari LP, Hay-Smith EJC. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women. Cochrane Database Syst Rev. 2018;10:CD005654. doi:10.1002/14651858.CD005654.pub4
18. Araujo CC, de A Marques A, Juliato CRT. The adherence of home pelvic floor muscles training using a mobile device application for women with urinary incontinence: a randomized controlled trial. Female Pelvic Med Reconstr Surg. 2020;26:697-703. doi:10.1097/SPV.0000000000000670
19. Sjöström M, Umefjord G, Stenlund H, et al. Internet-based treatment of stress urinary incontinence: a randomized controlled study with focus on pelvic floor muscle training. BJU Int. 2013;112:362-372. doi:10.1111/j.1464 -410X.2012.11713.x
20. Richter HE, Burgio KL, Brubaker L, et al; Pelvic Floor Disorders Network. Continence pessary compared with behavioral therapy or combined therapy for stress incontinence: a randomized controlled trial. Obstet Gynecol. 2010;115:609617. doi:10.1097/AOG.0b013e3181d055d4
21. Burgio KL, Kraus SR, Menefee S, et al. Behavioral therapy to enable women with urge incontinence to discontinue drug treatment: a randomized trial. Ann Intern Med. 2008;149(3): 161-169. doi:10.7326/0003-4819-149-3-200808050 -00005
22. Lukacz ES, Santiago-Lastra Y, Albo ME, et al. Urinary incontinence in women: a review. JAMA. 2017;318:1592-1604. doi:10.1001/jama.2017.12137
23. Welk B, Richardson K, Panicker JN. The cognitive effect of anticholinergics for patients with overactive bladder. Nat Rev Urol. 2021;18:686-700. doi:10.1038/s41585-021-00504-x
24. Burton C, Sajja A, Latthe PM. Effectiveness of percutaneous posterior tibial nerve stimulation for overactive bladder: a systematic review and meta-analysis. Neurourol Urodyn. 2012;31:1206-1216. doi:10.1002/nau.22251
25. Amundsen CL, Richter HE, Menefee SA, et al. OnabotulinumtoxinA vs sacral neuromodulation on refractory urgency urinary incontinence in women: A randomized clinical trial. JAMA. 2016;316:1366-1374. doi:10.1001/jama.2016.14617
26. Kirchin V, Page T, Keegan PE, et al. Urethral injection therapy for urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD003881. doi:10.1002/14651858.CD003881.pub4

Dr. Murillo is Fellow, Female Pelvic Medicine and Reconstructive Surgery at UPMC, Pittsburgh, Pennsylvania.

Dr. Zyczynski is Professor, Obstetrics, Gynecology and Reproductive Sciences in the Division of Urogynecology and Pelvic Reconstructive Surgery, University of Pittsburgh School of Medicine.
Dr. Zyczynski reports serving as a consultant to Amara Therapeutics Scientific Advisory Board. Dr. Murillo reports no financial relationships relevant to this article.
Dr. Murillo is Fellow, Female Pelvic Medicine and Reconstructive Surgery at UPMC, Pittsburgh, Pennsylvania.
Dr. Zyczynski is Professor, Obstetrics, Gynecology and Reproductive Sciences in the Division of Urogynecology and Pelvic Reconstructive Surgery, University of Pittsburgh School of Medicine.
Dr. Zyczynski reports serving as a consultant to Amara Therapeutics Scientific Advisory Board. Dr. Murillo reports no financial relationships relevant to this article.
Dr. Murillo is Fellow, Female Pelvic Medicine and Reconstructive Surgery at UPMC, Pittsburgh, Pennsylvania.
Dr. Zyczynski is Professor, Obstetrics, Gynecology and Reproductive Sciences in the Division of Urogynecology and Pelvic Reconstructive Surgery, University of Pittsburgh School of Medicine.
Dr. Zyczynski reports serving as a consultant to Amara Therapeutics Scientific Advisory Board. Dr. Murillo reports no financial relationships relevant to this article.
CASE Patient has urine leakage that worsens with exercise
At her annual preventative health visit, a 39-year-old woman reports that she has leakage of urine. She states that she drinks “a gallon of water daily” to help her lose the 20 lb she gained during the COVID-19 pandemic. She wants to resume Zumba fitness classes, but exercise makes her urine leakage worse. She started wearing protective pads because she finds herself often leaking urine on the way to the bathroom.
What nonsurgical treatment options are available for this patient?
Nearly half of all women experience urinary incontinence (UI), the involuntary loss of urine, and the condition increases with age.1 This common condition negatively impacts physical and psychological health and has been associated with social isolation, sexual dysfunction, and reduced independence.2,3 Symptoms of UI are underreported, and therefore universal screening is recommended for women of all ages.4 The diversity of available treatments (TABLE 1) provides patients and clinicians an opportunity to develop a plan that aligns with their symptom severity, goals, preferences, and resources.
Types of UI
The most common types of UI are stress urinary incontinence (SUI) and urgency urinary incontinence (UUI). Mixed urinary incontinence (MUI) occurs when symptoms of both SUI and UUI are present. Although the mechanisms that lead to urine leakage vary by the type of incontinence, many primary interventions improve both types of leakage, so a clinical diagnosis is sufficient to initiate treatment.
Stress urinary incontinence results from an impaired or weakened sphincter, which leads to involuntary, yet predictable, urine loss during increased abdominal pressure, such as coughing, laughing, sneezing, lifting, or physical activity.5 In UUI, involuntary loss of urine often accompanies the sudden urge to void. UUI is associated with overactive bladder (OAB), defined as urinary urgency, with or without urinary incontinence, usually accompanied by urinary frequency and/or nocturia (urination that interrupts sleep).6
In OAB, the detrusor muscle contracts randomly, leading to a sudden urge to void. When bladder pressure exceeds urethral sphincter closure pressure, urine leakage occurs. Women describe the urgency episodes as unpredictable, the urine leakage as prolonged with large volumes, and often occurring as they seek the toilet. Risk factors include age, obesity, parity, history of vaginal delivery, family history, ethnicity/race, medical comorbidities, menopausal status, and tobacco use.5
Making a diagnosis
A basic office evaluation is the most key step for diagnostic accuracy that leads to treatment success. This includes a detailed history, assessment of symptom severity, physical exam, pelvic exam, urinalysis, postvoid residual (to rule out urinary retention), and a cough stress test (to demonstrate SUI). The goal is to assess symptom severity, determine the type of UI, and identify contributing and potentially reversible factors, such as a urinary tract infection, medications, pelvic organ prolapse, incomplete bladder emptying, or impaired neurologic status. In the absence of the latter, advanced diagnostic tests, such as urodynamics, contribute little toward discerning the type of incontinence or changing first-line treatment plans.7
During the COVID-19 pandemic, abbreviated, virtual assessments for urinary symptoms were associated with high degrees of satisfaction (91% for fulfillment of personal needs, 94% overall satisfaction).8 This highlights the value of validated symptom questionnaires that help establish a working diagnosis and treatment plan in the absence of a physical exam. Questionnaire-based diagnoses have acceptable accuracy for classifying UUI and SUI among women with uncomplicated medical and surgical histories and for initiating low-risk therapies for defined intervals.
The 3 incontinence questions (3IQ) screen is an example of a useful, quick diagnostic tool designed for the primary care setting (FIGURE 1).9 It has been used in pharmaceutical treatment trials for UUI, with low frequency of misdiagnosis (1%–4%), resulting in no harm by the drug treatment prescribed or by the delay in appropriate care.10 Due to the limitations of an abbreviated remote evaluation, however, clinicians should assess patient response to primary interventions in a timely window. Patients who fail to experience satisfactory symptom reduction within 6 to 12 weeks should complete their evaluation in person or through a referral to a urogynecology program.
Continue to: Primary therapies for UI...
Primary therapies for UI
Primary therapies for UUI and SUI target strength training of the pelvic floor muscles, moderation of fluid intake, and adjustment in voiding behaviors and medications. Any functional barriers to continence also should be identified and addressed. Simple interventions, including a daily bowel regimen to address constipation, a bedside commode, and scheduled voiding, may reduce incontinence episodes without incurring significant cost or risk. For women suspected of having MUI, the treatment plan should prioritize their most bothersome symptoms.
Lifestyle and behavioral modifications
Everyday habits, medical comorbidities, and medications may exacerbate the severity of both SUI and UUI. Behavioral therapy alone or in combination with other interventions effectively reduces both SUI and UUI symptoms and has been shown to improve the efficacy of continence surgery.11 Information gained from a 3-day bladder diary (FIGURE 2)12 can guide clinicians on personalized patient recommendations, such as reducing excessive consumption of fluids and bladder irritants, limiting late evening drinking in the setting of bothersome nocturia, and scheduling voids (every 2–3 hours) to preempt incontinence episodes.
Weight loss
Obesity is a strong, independent, modifiable risk factor for both SUI and UUI. Each 5 kg/m2 increase in body mass index (BMI) has been associated with a 20% to 70% increased risk of UI, while weight loss of 5% or greater in overweight or obese women can lead to at least a 50% decrease in UI frequency.13
Reducing fluid intake and bladder irritants
Overactive bladder symptoms often respond to moderation of excessive fluid intake and reduction of bladder irritants (caffeine, carbonated beverages, diet beverages, and alcohol). While there is no established definition of excess caffeine intake, one study categorized high caffeine intake as greater than 400 mg/day (approximately four 8-oz cups of coffee).14
Information provided in a bladder diary can guide individualized recommendations for reducing fluid intake, particularly when 24-hour urine production exceeds the normative range (> 50–60 oz or 1.5-1.8 L/day).15 Hydration needs vary by activity, environment, and food; some general guidelines suggest 48 to 64 oz/day.5,16
Continue to: Pelvic floor muscle training...
Pelvic floor muscle training
An effective treatment for both UUI and SUI symptoms, pelvic floor muscle training (PFMT) leads to high degrees of patient satisfaction and improvement in quality of life.17 The presumed mechanisms of action of PFMT include improved urethral closure pressure and inhibition of detrusor muscle contractions.
Common exercise protocols recommend 3 sets of 10 contractions, held for 6 to 10 seconds per day, in varying positions of sitting, standing, and lying. While many women may be familiar with Kegel exercises, poor technique with straining and recruitment of gluteal and abdominal muscles can undermine the effect of PFMT. Clinicians can confirm successful pelvic muscle contractions by placing a finger in the vagina to appreciate contraction around and elevation of the finger toward the pubic symphysis in the absence of pushing.
Referral to supervised physical therapy and use of such teaching aid tools as booklets, mobile applications, and biofeedback can improve exercise adherence and outcomes.18,19 Systematic reviews report initial cure or improvement of incontinence symptoms as high as 74%, although little information is available about the long-term duration of effect.17
Vaginal pessaries
Vaginal continence support pessaries and devices work by stabilizing urethral mobility and compression of the bladder neck. Continence devices are particularly effective for situational SUI (such as during exercise).
The reusable medical grade silicone pessaries are available in numerous shapes and sizes and are fitted by a health care clinician (FIGURE 3). Uresta is a self-fitted intravaginal device that women can purchase online with a prescription. The Poise Impressa bladder support is a disposable intravaginal device marketed for incontinence and available over-the-counter, without a prescription (FIGURE 4). Anecdotally, many women find that menstrual tampons provide a similar effect, but outcome data are lacking.
In a comparative effectiveness trial of a continence pessary and behavior therapy, behavioral therapy was more likely to result in no bothersome incontinence symptoms (49% vs 33%, P = .006) and greater treatment satisfaction at 3 months.20 However, these short-term group differences did not persist at 12 months, presumably due to waning adherence.
UUI-specific nonsurgical treatments
Drug therapy
All medications approved by the US Food and Drug Administration (FDA) for UI are for the indications of OAB or UUI. These second-line treatments are most effective as adjuncts to behavioral modifications and PFMT.
A multicenter randomized trial that evaluated the efficacy of drug therapy alone compared with drug therapy in combination with behavioral modification, PFMT, urge suppression strategies, timed voiding, and fluid management for UUI found that combined therapy was more successful in achieving greater than 70% reduction in incontinence episodes (58% for drug therapy vs 69% for combined therapy).21
Of the 8 medications currently marketed in the United States for OAB or UUI, 6 are anticholinergic agents that block muscarinic receptors in the smooth muscle of the bladder, leading to inhibition of detrusor contractions, and 2 are β-adrenergic receptor agonists that promote bladder storage capacity by relaxing the detrusor muscle (TABLE 2). Similar efficacies lead most clinicians to initiate drug therapy based on formulary coverage and tolerance for adverse effects. Patients can expect a 53% to 80% reduction in UUI episodes and a 12% to 32% reduction in urinary frequency.22
Extended-release formulations are associated with reduced anticholinergic side effects (dry mouth, constipation, somnolence, dry eyes), leading to improved adherence. Notably, the anticholinergic medications are contraindicated in patients with untreated narrow-angle glaucoma, gastric retention, and supraventricular tachycardia. Mirabegron should be used with caution in patients with poorly controlled hypertension. 5 Due to concerns regarding the association between cumulative anticholinergic burden and the development of dementia, clinicians may consider avoiding the anticholinergic medications in older and at-risk patients.23
Continue to: UUI office-based procedure treatments...
UUI office-based procedure treatments
If behavioral therapies and medications are ineffective, contraindicated, or not the patient’s preference, additional FDA-approved therapies for UUI are available, typically through referral to a urogynecologist, urologist, or continence center.
Posterior tibial nerve stimulation (PTNS) is a nondrug treatment that delivers electrical stimulation using an acupuncture needle for 12 weekly 30-minute sessions followed by monthly maintenance for responders. The time commitment for this treatment plan can be a barrier for some patients. However, patients who adhere to the recommended protocol can expect a 60% improvement in symptoms, with minimal adverse events. Treatment efficacy is comparable to that of anticholinergic medication.24
OnabotulinumtoxinA injections into the bladder muscle are performed cystoscopically under local anesthetic. The toxin blocks the presynaptic release of acetylcholine at the neuromuscular junction, resulting in temporary muscle paralysis. This treatment is associated with high satisfaction. Efficacy varies by study population and outcome measure.
In one US comparative effectiveness trial, 67% of study participants with UUI symptoms refractory to oral medication reported a greater than 50% reduction in OAB symptoms at 6 months, 20% reported complete resolution of UUI, and 72% requested a second injection within 24 months.25 The interval between the first and second injection was nearly 1 year (350 days).Risks include urinary tract infection (12% within 1 month of the procedure and 35% through 6 months); urinary retention requiring catheterization has decreased to 6% with recognition that most moderate retention is tolerated by patients.
Some insurers limit onabotulinumtoxinA treatment coverage to patients who have failed to achieve symptom control with first- and second-line treatments.
SUI-specific nonsurgical treatments
Cystoscopic injection of urethral bulking agents into the urethral submucosa is designed to improve urethral coaptation. It is a minor procedure that can be performed in an ambulatory setting under local anesthetic with or without sedation.
Various bulking agents have been approved for use in the United States, some of which have been withdrawn due to complications of migration, erosion, and pseudoabscess formation. Cure or improvement after bulking agent injection was found to be superior to a home pelvic floor exercise program but inferior to a midurethral sling procedure for cure (9% vs 89%).26
The durability of currently available urethral bulking agents beyond 1 year is unknown. Complications are typically minor and transient and include pain at the injection site, urinary retention, de novo urgency, and implant leakage. The advantages include no postprocedure activity restrictions.
CASE Symptom presentation guides treatment plan
Our patient described symptoms of stress-predominant MUI. She was counseled to moderate her fluid intake to 2 L per day and to strategically time voids (before exercise, and at least every 4 hours). The patient was fitted with an incontinence pessary, and she elected to pursue a course of supervised physical therapy for pelvic floor muscle strengthening. Her follow-up visit is scheduled in 3 months to determine if other interventions are warranted. ●
CASE Patient has urine leakage that worsens with exercise
At her annual preventative health visit, a 39-year-old woman reports that she has leakage of urine. She states that she drinks “a gallon of water daily” to help her lose the 20 lb she gained during the COVID-19 pandemic. She wants to resume Zumba fitness classes, but exercise makes her urine leakage worse. She started wearing protective pads because she finds herself often leaking urine on the way to the bathroom.
What nonsurgical treatment options are available for this patient?
Nearly half of all women experience urinary incontinence (UI), the involuntary loss of urine, and the condition increases with age.1 This common condition negatively impacts physical and psychological health and has been associated with social isolation, sexual dysfunction, and reduced independence.2,3 Symptoms of UI are underreported, and therefore universal screening is recommended for women of all ages.4 The diversity of available treatments (TABLE 1) provides patients and clinicians an opportunity to develop a plan that aligns with their symptom severity, goals, preferences, and resources.
Types of UI
The most common types of UI are stress urinary incontinence (SUI) and urgency urinary incontinence (UUI). Mixed urinary incontinence (MUI) occurs when symptoms of both SUI and UUI are present. Although the mechanisms that lead to urine leakage vary by the type of incontinence, many primary interventions improve both types of leakage, so a clinical diagnosis is sufficient to initiate treatment.
Stress urinary incontinence results from an impaired or weakened sphincter, which leads to involuntary, yet predictable, urine loss during increased abdominal pressure, such as coughing, laughing, sneezing, lifting, or physical activity.5 In UUI, involuntary loss of urine often accompanies the sudden urge to void. UUI is associated with overactive bladder (OAB), defined as urinary urgency, with or without urinary incontinence, usually accompanied by urinary frequency and/or nocturia (urination that interrupts sleep).6
In OAB, the detrusor muscle contracts randomly, leading to a sudden urge to void. When bladder pressure exceeds urethral sphincter closure pressure, urine leakage occurs. Women describe the urgency episodes as unpredictable, the urine leakage as prolonged with large volumes, and often occurring as they seek the toilet. Risk factors include age, obesity, parity, history of vaginal delivery, family history, ethnicity/race, medical comorbidities, menopausal status, and tobacco use.5
Making a diagnosis
A basic office evaluation is the most key step for diagnostic accuracy that leads to treatment success. This includes a detailed history, assessment of symptom severity, physical exam, pelvic exam, urinalysis, postvoid residual (to rule out urinary retention), and a cough stress test (to demonstrate SUI). The goal is to assess symptom severity, determine the type of UI, and identify contributing and potentially reversible factors, such as a urinary tract infection, medications, pelvic organ prolapse, incomplete bladder emptying, or impaired neurologic status. In the absence of the latter, advanced diagnostic tests, such as urodynamics, contribute little toward discerning the type of incontinence or changing first-line treatment plans.7
During the COVID-19 pandemic, abbreviated, virtual assessments for urinary symptoms were associated with high degrees of satisfaction (91% for fulfillment of personal needs, 94% overall satisfaction).8 This highlights the value of validated symptom questionnaires that help establish a working diagnosis and treatment plan in the absence of a physical exam. Questionnaire-based diagnoses have acceptable accuracy for classifying UUI and SUI among women with uncomplicated medical and surgical histories and for initiating low-risk therapies for defined intervals.
The 3 incontinence questions (3IQ) screen is an example of a useful, quick diagnostic tool designed for the primary care setting (FIGURE 1).9 It has been used in pharmaceutical treatment trials for UUI, with low frequency of misdiagnosis (1%–4%), resulting in no harm by the drug treatment prescribed or by the delay in appropriate care.10 Due to the limitations of an abbreviated remote evaluation, however, clinicians should assess patient response to primary interventions in a timely window. Patients who fail to experience satisfactory symptom reduction within 6 to 12 weeks should complete their evaluation in person or through a referral to a urogynecology program.
Continue to: Primary therapies for UI...
Primary therapies for UI
Primary therapies for UUI and SUI target strength training of the pelvic floor muscles, moderation of fluid intake, and adjustment in voiding behaviors and medications. Any functional barriers to continence also should be identified and addressed. Simple interventions, including a daily bowel regimen to address constipation, a bedside commode, and scheduled voiding, may reduce incontinence episodes without incurring significant cost or risk. For women suspected of having MUI, the treatment plan should prioritize their most bothersome symptoms.
Lifestyle and behavioral modifications
Everyday habits, medical comorbidities, and medications may exacerbate the severity of both SUI and UUI. Behavioral therapy alone or in combination with other interventions effectively reduces both SUI and UUI symptoms and has been shown to improve the efficacy of continence surgery.11 Information gained from a 3-day bladder diary (FIGURE 2)12 can guide clinicians on personalized patient recommendations, such as reducing excessive consumption of fluids and bladder irritants, limiting late evening drinking in the setting of bothersome nocturia, and scheduling voids (every 2–3 hours) to preempt incontinence episodes.
Weight loss
Obesity is a strong, independent, modifiable risk factor for both SUI and UUI. Each 5 kg/m2 increase in body mass index (BMI) has been associated with a 20% to 70% increased risk of UI, while weight loss of 5% or greater in overweight or obese women can lead to at least a 50% decrease in UI frequency.13
Reducing fluid intake and bladder irritants
Overactive bladder symptoms often respond to moderation of excessive fluid intake and reduction of bladder irritants (caffeine, carbonated beverages, diet beverages, and alcohol). While there is no established definition of excess caffeine intake, one study categorized high caffeine intake as greater than 400 mg/day (approximately four 8-oz cups of coffee).14
Information provided in a bladder diary can guide individualized recommendations for reducing fluid intake, particularly when 24-hour urine production exceeds the normative range (> 50–60 oz or 1.5-1.8 L/day).15 Hydration needs vary by activity, environment, and food; some general guidelines suggest 48 to 64 oz/day.5,16
Continue to: Pelvic floor muscle training...
Pelvic floor muscle training
An effective treatment for both UUI and SUI symptoms, pelvic floor muscle training (PFMT) leads to high degrees of patient satisfaction and improvement in quality of life.17 The presumed mechanisms of action of PFMT include improved urethral closure pressure and inhibition of detrusor muscle contractions.
Common exercise protocols recommend 3 sets of 10 contractions, held for 6 to 10 seconds per day, in varying positions of sitting, standing, and lying. While many women may be familiar with Kegel exercises, poor technique with straining and recruitment of gluteal and abdominal muscles can undermine the effect of PFMT. Clinicians can confirm successful pelvic muscle contractions by placing a finger in the vagina to appreciate contraction around and elevation of the finger toward the pubic symphysis in the absence of pushing.
Referral to supervised physical therapy and use of such teaching aid tools as booklets, mobile applications, and biofeedback can improve exercise adherence and outcomes.18,19 Systematic reviews report initial cure or improvement of incontinence symptoms as high as 74%, although little information is available about the long-term duration of effect.17
Vaginal pessaries
Vaginal continence support pessaries and devices work by stabilizing urethral mobility and compression of the bladder neck. Continence devices are particularly effective for situational SUI (such as during exercise).
The reusable medical grade silicone pessaries are available in numerous shapes and sizes and are fitted by a health care clinician (FIGURE 3). Uresta is a self-fitted intravaginal device that women can purchase online with a prescription. The Poise Impressa bladder support is a disposable intravaginal device marketed for incontinence and available over-the-counter, without a prescription (FIGURE 4). Anecdotally, many women find that menstrual tampons provide a similar effect, but outcome data are lacking.
In a comparative effectiveness trial of a continence pessary and behavior therapy, behavioral therapy was more likely to result in no bothersome incontinence symptoms (49% vs 33%, P = .006) and greater treatment satisfaction at 3 months.20 However, these short-term group differences did not persist at 12 months, presumably due to waning adherence.
UUI-specific nonsurgical treatments
Drug therapy
All medications approved by the US Food and Drug Administration (FDA) for UI are for the indications of OAB or UUI. These second-line treatments are most effective as adjuncts to behavioral modifications and PFMT.
A multicenter randomized trial that evaluated the efficacy of drug therapy alone compared with drug therapy in combination with behavioral modification, PFMT, urge suppression strategies, timed voiding, and fluid management for UUI found that combined therapy was more successful in achieving greater than 70% reduction in incontinence episodes (58% for drug therapy vs 69% for combined therapy).21
Of the 8 medications currently marketed in the United States for OAB or UUI, 6 are anticholinergic agents that block muscarinic receptors in the smooth muscle of the bladder, leading to inhibition of detrusor contractions, and 2 are β-adrenergic receptor agonists that promote bladder storage capacity by relaxing the detrusor muscle (TABLE 2). Similar efficacies lead most clinicians to initiate drug therapy based on formulary coverage and tolerance for adverse effects. Patients can expect a 53% to 80% reduction in UUI episodes and a 12% to 32% reduction in urinary frequency.22
Extended-release formulations are associated with reduced anticholinergic side effects (dry mouth, constipation, somnolence, dry eyes), leading to improved adherence. Notably, the anticholinergic medications are contraindicated in patients with untreated narrow-angle glaucoma, gastric retention, and supraventricular tachycardia. Mirabegron should be used with caution in patients with poorly controlled hypertension. 5 Due to concerns regarding the association between cumulative anticholinergic burden and the development of dementia, clinicians may consider avoiding the anticholinergic medications in older and at-risk patients.23
Continue to: UUI office-based procedure treatments...
UUI office-based procedure treatments
If behavioral therapies and medications are ineffective, contraindicated, or not the patient’s preference, additional FDA-approved therapies for UUI are available, typically through referral to a urogynecologist, urologist, or continence center.
Posterior tibial nerve stimulation (PTNS) is a nondrug treatment that delivers electrical stimulation using an acupuncture needle for 12 weekly 30-minute sessions followed by monthly maintenance for responders. The time commitment for this treatment plan can be a barrier for some patients. However, patients who adhere to the recommended protocol can expect a 60% improvement in symptoms, with minimal adverse events. Treatment efficacy is comparable to that of anticholinergic medication.24
OnabotulinumtoxinA injections into the bladder muscle are performed cystoscopically under local anesthetic. The toxin blocks the presynaptic release of acetylcholine at the neuromuscular junction, resulting in temporary muscle paralysis. This treatment is associated with high satisfaction. Efficacy varies by study population and outcome measure.
In one US comparative effectiveness trial, 67% of study participants with UUI symptoms refractory to oral medication reported a greater than 50% reduction in OAB symptoms at 6 months, 20% reported complete resolution of UUI, and 72% requested a second injection within 24 months.25 The interval between the first and second injection was nearly 1 year (350 days).Risks include urinary tract infection (12% within 1 month of the procedure and 35% through 6 months); urinary retention requiring catheterization has decreased to 6% with recognition that most moderate retention is tolerated by patients.
Some insurers limit onabotulinumtoxinA treatment coverage to patients who have failed to achieve symptom control with first- and second-line treatments.
SUI-specific nonsurgical treatments
Cystoscopic injection of urethral bulking agents into the urethral submucosa is designed to improve urethral coaptation. It is a minor procedure that can be performed in an ambulatory setting under local anesthetic with or without sedation.
Various bulking agents have been approved for use in the United States, some of which have been withdrawn due to complications of migration, erosion, and pseudoabscess formation. Cure or improvement after bulking agent injection was found to be superior to a home pelvic floor exercise program but inferior to a midurethral sling procedure for cure (9% vs 89%).26
The durability of currently available urethral bulking agents beyond 1 year is unknown. Complications are typically minor and transient and include pain at the injection site, urinary retention, de novo urgency, and implant leakage. The advantages include no postprocedure activity restrictions.
CASE Symptom presentation guides treatment plan
Our patient described symptoms of stress-predominant MUI. She was counseled to moderate her fluid intake to 2 L per day and to strategically time voids (before exercise, and at least every 4 hours). The patient was fitted with an incontinence pessary, and she elected to pursue a course of supervised physical therapy for pelvic floor muscle strengthening. Her follow-up visit is scheduled in 3 months to determine if other interventions are warranted. ●
1. Lee UJ, Feinstein L, Ward JB, et al. Prevalence of urinary incontinence among a nationally representative sample of women, 2005–2016: findings from the Urologic Diseases in America Project. J Urol. 2021;205:1718-1724. doi:10.1097 /JU.0000000000001634
2. Sims J, Browning C, Lundgren-Lindquist B, et al. Urinary incontinence in a community sample of older adults: prevalence and impact on quality of life. Disabil Rehabil. 2011;33:1389-1398. doi:10.3109/09638288.2010.532284
3. Sarikaya S, Yildiz FG, Senocak C, et al. Urinary incontinence as a cause of depression and sexual dysfunction: questionnaire-based study. Rev Int Androl. 2020:18:50-54. doi:10.1016 /j.androl.2018.08.003
4. O’Reilly N, Nelson HD, Conry JM, et al; Women’s Preventive Services Initiative. Screening for urinary incontinence in women: a recommendation from the Women’s Preventive Services Initiative. Ann Intern Med. 2018;169(5):320-328. doi:10.7326/M18-0595
5. Barber MD, Walters MD, Karram MM, et al. Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier Saunders; 2021.
6. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Int Urogynecol J. 2010;21: 5-26. doi:10.1007/s00192-009-0976-9
7. ACOG practice bulletin no. 155. Urinary incontinence in women. Obstet Gynecol. 2015;126:e66-e81. doi:10.1097 /AOG.0000000000001148
8. Sansone S, Lu J, Drangsholt S, et al. No pelvic exam, no problem: patient satisfaction following the integration of comprehensive urogynecology telemedicine. Int Urogynecol J. 2022;1:3. doi:10.1007/s00192-022-05104-w
9. Brown JS, Bradley CS, Subak LL, et al; Diagnostic Aspects of Incontinence Study (DAISy) Research Group. The sensitivity and specificity of a simple test to distinguish between urge and stress urinary incontinence. Ann Intern Med. 2006;144:715723. doi:10.7326/0003-4819-144-10-200605160-00005
10. Hess R, Huang AJ, Richter HE, et al. Long-term efficacy and safety of questionnaire-based initiation of urgency urinary incontinence treatment. Am J Obstet Gynecol. 2013;209:244. e1-9. doi:10.1016/j.ajog.2013.05.008
11. Sung VW, Borello-France D, Newman DK, et al; NICHD Pelvic Floor Disorders Network. Effect of behavioral and pelvic floor muscle therapy combined with surgery vs surgery alone on incontinence symptoms among women with mixed urinary incontinence. JAMA. 2019;322:1066-1076. doi:10.1001 /jama.2019.12467
12. American Urogynecologic Society. Voices for PFD: intake and voiding diary. Accessed August 11, 2022. https://www .voicesforpfd.org/assets/2/6/Voiding_Diary.pdf
13. Subak LL, Richter HE, Hunskaar S. Obesity and urinary incontinence: epidemiology and clinical research update. J Urol. 2009;182(6 suppl):S2-7. doi:10.1016/j.juro.2009.08.071
14. Arya LA, Myers DL, Jackson ND. Dietary caffeine intake and the risk for detrusor instability: a case-control study. Obstet Gynecol. 2000;96:85-89. doi:10.1016/s0029-7844(00)00808-5
15. Wyman JF, Zhou J, LaCoursiere DY, et al. Normative noninvasive bladder function measurements in healthy women: a systematic review and meta-analysis. Neurourol Urodyn. 2020;39:507-522. doi:10.1002/nau.24265
16. Hashim H, Al Mousa R. Management of fluid intake in patients with overactive bladder. Curr Urol Rep. 2009;10: 428-433. doi:10.1007/s11934-009-0068-x
17. Dumoulin C, Cacciari LP, Hay-Smith EJC. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women. Cochrane Database Syst Rev. 2018;10:CD005654. doi:10.1002/14651858.CD005654.pub4
18. Araujo CC, de A Marques A, Juliato CRT. The adherence of home pelvic floor muscles training using a mobile device application for women with urinary incontinence: a randomized controlled trial. Female Pelvic Med Reconstr Surg. 2020;26:697-703. doi:10.1097/SPV.0000000000000670
19. Sjöström M, Umefjord G, Stenlund H, et al. Internet-based treatment of stress urinary incontinence: a randomized controlled study with focus on pelvic floor muscle training. BJU Int. 2013;112:362-372. doi:10.1111/j.1464 -410X.2012.11713.x
20. Richter HE, Burgio KL, Brubaker L, et al; Pelvic Floor Disorders Network. Continence pessary compared with behavioral therapy or combined therapy for stress incontinence: a randomized controlled trial. Obstet Gynecol. 2010;115:609617. doi:10.1097/AOG.0b013e3181d055d4
21. Burgio KL, Kraus SR, Menefee S, et al. Behavioral therapy to enable women with urge incontinence to discontinue drug treatment: a randomized trial. Ann Intern Med. 2008;149(3): 161-169. doi:10.7326/0003-4819-149-3-200808050 -00005
22. Lukacz ES, Santiago-Lastra Y, Albo ME, et al. Urinary incontinence in women: a review. JAMA. 2017;318:1592-1604. doi:10.1001/jama.2017.12137
23. Welk B, Richardson K, Panicker JN. The cognitive effect of anticholinergics for patients with overactive bladder. Nat Rev Urol. 2021;18:686-700. doi:10.1038/s41585-021-00504-x
24. Burton C, Sajja A, Latthe PM. Effectiveness of percutaneous posterior tibial nerve stimulation for overactive bladder: a systematic review and meta-analysis. Neurourol Urodyn. 2012;31:1206-1216. doi:10.1002/nau.22251
25. Amundsen CL, Richter HE, Menefee SA, et al. OnabotulinumtoxinA vs sacral neuromodulation on refractory urgency urinary incontinence in women: A randomized clinical trial. JAMA. 2016;316:1366-1374. doi:10.1001/jama.2016.14617
26. Kirchin V, Page T, Keegan PE, et al. Urethral injection therapy for urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD003881. doi:10.1002/14651858.CD003881.pub4
1. Lee UJ, Feinstein L, Ward JB, et al. Prevalence of urinary incontinence among a nationally representative sample of women, 2005–2016: findings from the Urologic Diseases in America Project. J Urol. 2021;205:1718-1724. doi:10.1097 /JU.0000000000001634
2. Sims J, Browning C, Lundgren-Lindquist B, et al. Urinary incontinence in a community sample of older adults: prevalence and impact on quality of life. Disabil Rehabil. 2011;33:1389-1398. doi:10.3109/09638288.2010.532284
3. Sarikaya S, Yildiz FG, Senocak C, et al. Urinary incontinence as a cause of depression and sexual dysfunction: questionnaire-based study. Rev Int Androl. 2020:18:50-54. doi:10.1016 /j.androl.2018.08.003
4. O’Reilly N, Nelson HD, Conry JM, et al; Women’s Preventive Services Initiative. Screening for urinary incontinence in women: a recommendation from the Women’s Preventive Services Initiative. Ann Intern Med. 2018;169(5):320-328. doi:10.7326/M18-0595
5. Barber MD, Walters MD, Karram MM, et al. Walters & Karram Urogynecology and Reconstructive Pelvic Surgery. 5th ed. Elsevier Saunders; 2021.
6. Haylen BT, de Ridder D, Freeman RM, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction. Int Urogynecol J. 2010;21: 5-26. doi:10.1007/s00192-009-0976-9
7. ACOG practice bulletin no. 155. Urinary incontinence in women. Obstet Gynecol. 2015;126:e66-e81. doi:10.1097 /AOG.0000000000001148
8. Sansone S, Lu J, Drangsholt S, et al. No pelvic exam, no problem: patient satisfaction following the integration of comprehensive urogynecology telemedicine. Int Urogynecol J. 2022;1:3. doi:10.1007/s00192-022-05104-w
9. Brown JS, Bradley CS, Subak LL, et al; Diagnostic Aspects of Incontinence Study (DAISy) Research Group. The sensitivity and specificity of a simple test to distinguish between urge and stress urinary incontinence. Ann Intern Med. 2006;144:715723. doi:10.7326/0003-4819-144-10-200605160-00005
10. Hess R, Huang AJ, Richter HE, et al. Long-term efficacy and safety of questionnaire-based initiation of urgency urinary incontinence treatment. Am J Obstet Gynecol. 2013;209:244. e1-9. doi:10.1016/j.ajog.2013.05.008
11. Sung VW, Borello-France D, Newman DK, et al; NICHD Pelvic Floor Disorders Network. Effect of behavioral and pelvic floor muscle therapy combined with surgery vs surgery alone on incontinence symptoms among women with mixed urinary incontinence. JAMA. 2019;322:1066-1076. doi:10.1001 /jama.2019.12467
12. American Urogynecologic Society. Voices for PFD: intake and voiding diary. Accessed August 11, 2022. https://www .voicesforpfd.org/assets/2/6/Voiding_Diary.pdf
13. Subak LL, Richter HE, Hunskaar S. Obesity and urinary incontinence: epidemiology and clinical research update. J Urol. 2009;182(6 suppl):S2-7. doi:10.1016/j.juro.2009.08.071
14. Arya LA, Myers DL, Jackson ND. Dietary caffeine intake and the risk for detrusor instability: a case-control study. Obstet Gynecol. 2000;96:85-89. doi:10.1016/s0029-7844(00)00808-5
15. Wyman JF, Zhou J, LaCoursiere DY, et al. Normative noninvasive bladder function measurements in healthy women: a systematic review and meta-analysis. Neurourol Urodyn. 2020;39:507-522. doi:10.1002/nau.24265
16. Hashim H, Al Mousa R. Management of fluid intake in patients with overactive bladder. Curr Urol Rep. 2009;10: 428-433. doi:10.1007/s11934-009-0068-x
17. Dumoulin C, Cacciari LP, Hay-Smith EJC. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women. Cochrane Database Syst Rev. 2018;10:CD005654. doi:10.1002/14651858.CD005654.pub4
18. Araujo CC, de A Marques A, Juliato CRT. The adherence of home pelvic floor muscles training using a mobile device application for women with urinary incontinence: a randomized controlled trial. Female Pelvic Med Reconstr Surg. 2020;26:697-703. doi:10.1097/SPV.0000000000000670
19. Sjöström M, Umefjord G, Stenlund H, et al. Internet-based treatment of stress urinary incontinence: a randomized controlled study with focus on pelvic floor muscle training. BJU Int. 2013;112:362-372. doi:10.1111/j.1464 -410X.2012.11713.x
20. Richter HE, Burgio KL, Brubaker L, et al; Pelvic Floor Disorders Network. Continence pessary compared with behavioral therapy or combined therapy for stress incontinence: a randomized controlled trial. Obstet Gynecol. 2010;115:609617. doi:10.1097/AOG.0b013e3181d055d4
21. Burgio KL, Kraus SR, Menefee S, et al. Behavioral therapy to enable women with urge incontinence to discontinue drug treatment: a randomized trial. Ann Intern Med. 2008;149(3): 161-169. doi:10.7326/0003-4819-149-3-200808050 -00005
22. Lukacz ES, Santiago-Lastra Y, Albo ME, et al. Urinary incontinence in women: a review. JAMA. 2017;318:1592-1604. doi:10.1001/jama.2017.12137
23. Welk B, Richardson K, Panicker JN. The cognitive effect of anticholinergics for patients with overactive bladder. Nat Rev Urol. 2021;18:686-700. doi:10.1038/s41585-021-00504-x
24. Burton C, Sajja A, Latthe PM. Effectiveness of percutaneous posterior tibial nerve stimulation for overactive bladder: a systematic review and meta-analysis. Neurourol Urodyn. 2012;31:1206-1216. doi:10.1002/nau.22251
25. Amundsen CL, Richter HE, Menefee SA, et al. OnabotulinumtoxinA vs sacral neuromodulation on refractory urgency urinary incontinence in women: A randomized clinical trial. JAMA. 2016;316:1366-1374. doi:10.1001/jama.2016.14617
26. Kirchin V, Page T, Keegan PE, et al. Urethral injection therapy for urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD003881. doi:10.1002/14651858.CD003881.pub4
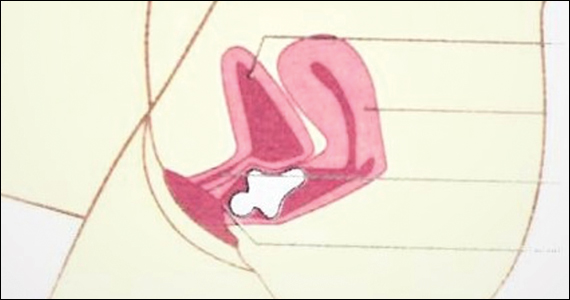
2022 Update on abnormal uterine bleeding
In this Update, we focus on therapies for abnormal uterine bleeding (AUB) that include a new formulation of a progesterone-only pill (POP), drospirenone 4 mg in a 24/4 regimen (24 days of drospirenone/4 days of inert tablets), which recently showed benefit over the use of desogestrel in a European randomized clinical trial (RCT). Two other commonly used treatments for AUB— the levonorgestrel-releasing intrauterine system (LNG IUS) and endometrial ablation—were studied in terms of cost-effectiveness as well as whether they should be used in combination for added efficacy. In addition, although at times either COVID-19 disease or the COVID-19 vaccine has been blamed for societal and medical problems, one study showed that it is unlikely that significant changes in the menstrual cycle are a result of the COVID-19 vaccine.
COVID-19 vaccination had minimal effects on menstrual cycle length
Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
Does receiving the COVID-19 vaccination result in abnormal menstrual cycles? Patients often ask this question, and it has been a topic of social media discussion (including NPR) and concerns about the possibility of vaccine hesitancy,1,2 as the menstrual cycle is often considered a sign of health and fertility.
To better understand this possible association, Edelman and colleagues conducted a study that prospectively tracked menstrual cycle data using the digital app Natural Cycles in US residents aged 18 to 45 years for 3 consecutive cycles in both a vaccinated and an unvaccinated cohort.3 Almost 4,000 individuals were studied; 2,403 were vaccinated and 1,556 were unvaccinated. The study vaccine types included the BioNTech (Pfizer), Moderna, Johnson & Johnson/Janssen, and unspecified vaccines.
The primary outcome was the within-individual change in cycle length in days, comparing a 3-cycle postvaccine average to a 3-cycle prevaccination average in the 2 groups. (For the unvaccinated group, cycles 1, 2, and 3 were considered the equivalent of prevaccination cycles; cycle 4 was designated as the artificial first vaccine dose-cycle and cycle 5 as the artificial second-dose cycle.)
Increase in cycle length clinically negligible
The investigators found that the vaccinated cohort had less than a 1-day unadjusted increase in the length of their menstrual cycle, which was essentially a 0.71-day increase (98.75% confidence interval [CI], 0.47–0.94). Although this is considered statistically significant, it is likely clinically insignificant in that the overlaid histograms comparing the distribution of change showed a cycle length distribution in vaccinated individuals that is essentially equivalent to that in unvaccinated individuals. After adjusting for confounders, the difference in cycle length was reduced to a 0.64 day (98.75% CI, 0.27–1.01).
An interesting finding was that a subset of individuals who received both vaccine doses in a single cycle had, on average, an adjusted 2-day increase in their menstrual cycle compared with unvaccinated individuals. To explain this slightly longer cycle length, the authors postulated that mRNA vaccines create an immune response, or stressor, which could temporarily affect the hypothalamic-pituitary-ovarian axis if timed correctly. It is certainly possible for an individual to receive 2 doses in a single cycle, which could have both been administered in the early follicular phase. Such cycle length variability can be caused by events, including stressors, that affect the recruitment and maturation of the dominant follicle.
Counseling takeaway
This study provides reassurance to most individuals who receive a COVID-19 vaccine that it likely will not affect their menstrual cycle in a clinically significant manner.
This robust study by Edelman and colleagues on COVID-19 vaccination effects on menstrual cycle length had more than 99% power to detect an unadjusted 1-day difference in cycle length. However, given that most of the study participants were White and had access to the Natural Cycles app, the results may not be generalizable to all individuals who receive the vaccine.
Continue to: Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel...
Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel
Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
A new POP, marketed under the name Slynd, recently came to market. It contains the progestin drospirenone (DRSP) 4 mg in a 24/4 regimen. This formulation has the advantage of being an antiandrogenic progestin, with a long enough half-life to allow for managing a missed pill in the same fashion as combined oral contraceptives (COCs).
Investigators in Europe conducted a double-blind, randomized trial to assess discontinuation rates due to adverse events (mainly bleeding disorders) in participants taking DRSP 4 mg in a 24/4 regimen compared with those taking the POP desogestrel (DSG) 0.075 mg, which is commonly used in Europe.4 Regidor and colleagues compared 858 women with 6,691 DRSP treatment cycles with 332 women with 2,487 DSG treatment cycles.
Top reasons for stopping a POP
The discontinuation rate for abnormal bleeding was 3.7% in the DRSP group versus 7.3% in the DSG group (55.7% lower). The most common reasons for stopping either POP formulation were vaginal bleeding and acne. Both of these adverse events were less common in the DRSP group. Pill discontinuation due to vaginal bleeding was 2.6% in the DRSP group versus 5.4% in the DSG group, while discontinuation due to acne occurred in 1% in the DRSP group versus 2.7% in the DSG group.
New oral contraception option
This study shows improved acceptability and bleeding profiles in women using this new DRSP contraception pill regimen.
Adherence to a contraceptive method is influenced by patient satisfaction, and this is particularly important in patients who cannot take COCs. It also should be noted that the discontinuation rate for DRSP as a POP used in this 24/4 regimen was similar to discontinuation rates for COCs containing 20 µg and 30 µg of ethinyl estradiol. Cost, however, may be an issue with DRSP, depending on a patient’s insurance coverage.
Continue to: Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy...
Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy
Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. The combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
Over the years, a smattering of articles have suggested that a reduction in uterine bleeding was associated with placement of an LNG IUS at the conclusion of endometrial ablation. We now have a systematic review of this surgical modification.
Oderkerk and colleagues sifted through 747 articles to find 7 publications that could provide meaningful data on the impact of combined use of endometrial ablation and LNG IUS insertion for women with heavy menstrual bleeding.5 These included 4 retrospective cohort studies with control groups, 2 retrospective studies without control groups, and 1 case series. The primary outcome was the hysterectomy rate after therapy.
Promising results for combined therapy
Although no statistically significant intergroup differences were seen in the combined treatment group versus the endometrial ablation alone group for the first 6 months of treatment, significant differences existed at the 12- and 24-month mark. Hysterectomy rates after combined treatment varied from 0% to 11% versus 9.4% to 24% after endometrial ablation alone. Complication rates for combined treatment did not appear higher than those for endometrial ablation alone.
The authors postulated that the failure of endometrial ablation is generally caused by either remaining or regenerating endometrial tissue and that the addition of an LNG IUS allows for suppression of endometrial tissue. Also encouraging was that, in general, the removal of the LNG IUS was relatively simple. A single difficult removal was described due to uterine synechiae, but hysteroscopic resection was not necessary. The authors acknowledged that the data from these 7 retrospective studies are limited and that high-quality research from prospective studies is needed.
Bottom line
The data available from this systematic review suggest that placement of an LNG IUS at the completion of an endometrial ablation may result in lower hysterectomy rates, without apparent risk, and without significantly difficult LNG IUS removal when needed.
The data provided by Oderkerk and colleagues’ systematic review are promising and, although not studied in the reviewed publications, the potential may exist to reduce the risk of endometrial hyperplasia and endometrial cancer by adding an LNG IUS.
Continue to: LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows...
LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows
van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.
To assess the cost-effectiveness of the LNG IUS versus endometrial ablation in the treatment of heavy menstrual bleeding, van den Brink and colleagues conducted a randomized, noninferiority trial.6
Part of the rationale for this study was to better understand the cost differences between the LNG IUS and second-generation endometrial ablation. Some data have suggested that the LNG IUS is cost-effective when compared with first-generation endometrial ablation; however, definitive evidence about its cost compared with second-generation endometrial ablation is lacking, as these procedures should be less expensive than first-generation endometrial ablation since they frequently are performed in the office rather than in an operating room.
Cost-effectiveness and noninferiority assessed
A total of 270 women were randomly assigned to 1 of 2 treatment strategies. Eventually, 132 women were treated first with the 52-mg LNG IUS, and 138 were treated first with endometrial ablation by radiofrequency ablation. Menstrual blood loss after 24 months was the primary outcome.
At 24 months, the mean pictorial blood loss assessment chart (PBAC) scores were 64.8 in the LNG IUS group compared with 14.2 in the endometrial ablation group. Given that the noninferiority margin was defined as 25 points, noninferiority could not be demonstrated. However, when looking at PBAC scores less than 75 points, the LNG IUS group met this secondary end point in 87% of women versus 94% in the endometrial ablation group. When satisfaction was assessed, 74% of women in the LNG IUS group were satisfied compared with 84% in the endometrial ablation group.
Overall, the total costs per patient were €2,285 in the LNG IUS strategy and €3,465 in the endometrial ablation strategy (costs convert to $2,285 and $3,465 as of this writing).
Key takeaway
Treatment of heavy menstrual bleeding starting with the LNG IUS is cheaper, but it is slightly less effective than endometrial ablation. ●
It is interesting that there are minimal differences between satisfaction rates and PBAC scores less than 75, yet the mean PBAC scores were significantly more favorable for endometrial ablation. This study’s results support the use of a sequential therapy of a less invasive therapy, such as the LNG IUS, prior to performing endometrial ablation.
- Blumfiel G. Why reports of menstrual changes after COVID vaccine are tough to study. NPR. August 9, 2021. Accessed August 30, 2022. https://www.npr.org/sections/health-shots/2021/08/09/1024190379/covid-vaccine-period-menstrual-cycle-research
- Lee KMN, Junkins EJ, Fatima UA, et al. Characterizing menstrual bleeding changes occurring after SARSCoV-2 vaccinations. MedRxiv. February 11, 2022. doi:10.1101/2021.10.11.21264863
- Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
- Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
- Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. T he combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
- van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.

Dr. Sharp is Jon M. Huntsman Presidential Endowed Professor, Vice Chair for Clinical Activities, Department of Obstetrics and Gynecology, University of Utah Health, Salt Lake City, Utah.

Dr. Adelman is Associate Professor, Department of Obstetrics and Gynecology, University of Utah Health.
Dr. Sharp reports being an author and editor and receiving royalties from UpToDate, Inc. Dr. Adelman reports no financial relationships relevant to this article.
Dr. Sharp is Jon M. Huntsman Presidential Endowed Professor, Vice Chair for Clinical Activities, Department of Obstetrics and Gynecology, University of Utah Health, Salt Lake City, Utah.
Dr. Adelman is Associate Professor, Department of Obstetrics and Gynecology, University of Utah Health.
Dr. Sharp reports being an author and editor and receiving royalties from UpToDate, Inc. Dr. Adelman reports no financial relationships relevant to this article.
Dr. Sharp is Jon M. Huntsman Presidential Endowed Professor, Vice Chair for Clinical Activities, Department of Obstetrics and Gynecology, University of Utah Health, Salt Lake City, Utah.
Dr. Adelman is Associate Professor, Department of Obstetrics and Gynecology, University of Utah Health.
Dr. Sharp reports being an author and editor and receiving royalties from UpToDate, Inc. Dr. Adelman reports no financial relationships relevant to this article.
In this Update, we focus on therapies for abnormal uterine bleeding (AUB) that include a new formulation of a progesterone-only pill (POP), drospirenone 4 mg in a 24/4 regimen (24 days of drospirenone/4 days of inert tablets), which recently showed benefit over the use of desogestrel in a European randomized clinical trial (RCT). Two other commonly used treatments for AUB— the levonorgestrel-releasing intrauterine system (LNG IUS) and endometrial ablation—were studied in terms of cost-effectiveness as well as whether they should be used in combination for added efficacy. In addition, although at times either COVID-19 disease or the COVID-19 vaccine has been blamed for societal and medical problems, one study showed that it is unlikely that significant changes in the menstrual cycle are a result of the COVID-19 vaccine.
COVID-19 vaccination had minimal effects on menstrual cycle length
Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
Does receiving the COVID-19 vaccination result in abnormal menstrual cycles? Patients often ask this question, and it has been a topic of social media discussion (including NPR) and concerns about the possibility of vaccine hesitancy,1,2 as the menstrual cycle is often considered a sign of health and fertility.
To better understand this possible association, Edelman and colleagues conducted a study that prospectively tracked menstrual cycle data using the digital app Natural Cycles in US residents aged 18 to 45 years for 3 consecutive cycles in both a vaccinated and an unvaccinated cohort.3 Almost 4,000 individuals were studied; 2,403 were vaccinated and 1,556 were unvaccinated. The study vaccine types included the BioNTech (Pfizer), Moderna, Johnson & Johnson/Janssen, and unspecified vaccines.
The primary outcome was the within-individual change in cycle length in days, comparing a 3-cycle postvaccine average to a 3-cycle prevaccination average in the 2 groups. (For the unvaccinated group, cycles 1, 2, and 3 were considered the equivalent of prevaccination cycles; cycle 4 was designated as the artificial first vaccine dose-cycle and cycle 5 as the artificial second-dose cycle.)
Increase in cycle length clinically negligible
The investigators found that the vaccinated cohort had less than a 1-day unadjusted increase in the length of their menstrual cycle, which was essentially a 0.71-day increase (98.75% confidence interval [CI], 0.47–0.94). Although this is considered statistically significant, it is likely clinically insignificant in that the overlaid histograms comparing the distribution of change showed a cycle length distribution in vaccinated individuals that is essentially equivalent to that in unvaccinated individuals. After adjusting for confounders, the difference in cycle length was reduced to a 0.64 day (98.75% CI, 0.27–1.01).
An interesting finding was that a subset of individuals who received both vaccine doses in a single cycle had, on average, an adjusted 2-day increase in their menstrual cycle compared with unvaccinated individuals. To explain this slightly longer cycle length, the authors postulated that mRNA vaccines create an immune response, or stressor, which could temporarily affect the hypothalamic-pituitary-ovarian axis if timed correctly. It is certainly possible for an individual to receive 2 doses in a single cycle, which could have both been administered in the early follicular phase. Such cycle length variability can be caused by events, including stressors, that affect the recruitment and maturation of the dominant follicle.
Counseling takeaway
This study provides reassurance to most individuals who receive a COVID-19 vaccine that it likely will not affect their menstrual cycle in a clinically significant manner.
This robust study by Edelman and colleagues on COVID-19 vaccination effects on menstrual cycle length had more than 99% power to detect an unadjusted 1-day difference in cycle length. However, given that most of the study participants were White and had access to the Natural Cycles app, the results may not be generalizable to all individuals who receive the vaccine.
Continue to: Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel...
Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel
Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
A new POP, marketed under the name Slynd, recently came to market. It contains the progestin drospirenone (DRSP) 4 mg in a 24/4 regimen. This formulation has the advantage of being an antiandrogenic progestin, with a long enough half-life to allow for managing a missed pill in the same fashion as combined oral contraceptives (COCs).
Investigators in Europe conducted a double-blind, randomized trial to assess discontinuation rates due to adverse events (mainly bleeding disorders) in participants taking DRSP 4 mg in a 24/4 regimen compared with those taking the POP desogestrel (DSG) 0.075 mg, which is commonly used in Europe.4 Regidor and colleagues compared 858 women with 6,691 DRSP treatment cycles with 332 women with 2,487 DSG treatment cycles.
Top reasons for stopping a POP
The discontinuation rate for abnormal bleeding was 3.7% in the DRSP group versus 7.3% in the DSG group (55.7% lower). The most common reasons for stopping either POP formulation were vaginal bleeding and acne. Both of these adverse events were less common in the DRSP group. Pill discontinuation due to vaginal bleeding was 2.6% in the DRSP group versus 5.4% in the DSG group, while discontinuation due to acne occurred in 1% in the DRSP group versus 2.7% in the DSG group.
New oral contraception option
This study shows improved acceptability and bleeding profiles in women using this new DRSP contraception pill regimen.
Adherence to a contraceptive method is influenced by patient satisfaction, and this is particularly important in patients who cannot take COCs. It also should be noted that the discontinuation rate for DRSP as a POP used in this 24/4 regimen was similar to discontinuation rates for COCs containing 20 µg and 30 µg of ethinyl estradiol. Cost, however, may be an issue with DRSP, depending on a patient’s insurance coverage.
Continue to: Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy...
Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy
Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. The combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
Over the years, a smattering of articles have suggested that a reduction in uterine bleeding was associated with placement of an LNG IUS at the conclusion of endometrial ablation. We now have a systematic review of this surgical modification.
Oderkerk and colleagues sifted through 747 articles to find 7 publications that could provide meaningful data on the impact of combined use of endometrial ablation and LNG IUS insertion for women with heavy menstrual bleeding.5 These included 4 retrospective cohort studies with control groups, 2 retrospective studies without control groups, and 1 case series. The primary outcome was the hysterectomy rate after therapy.
Promising results for combined therapy
Although no statistically significant intergroup differences were seen in the combined treatment group versus the endometrial ablation alone group for the first 6 months of treatment, significant differences existed at the 12- and 24-month mark. Hysterectomy rates after combined treatment varied from 0% to 11% versus 9.4% to 24% after endometrial ablation alone. Complication rates for combined treatment did not appear higher than those for endometrial ablation alone.
The authors postulated that the failure of endometrial ablation is generally caused by either remaining or regenerating endometrial tissue and that the addition of an LNG IUS allows for suppression of endometrial tissue. Also encouraging was that, in general, the removal of the LNG IUS was relatively simple. A single difficult removal was described due to uterine synechiae, but hysteroscopic resection was not necessary. The authors acknowledged that the data from these 7 retrospective studies are limited and that high-quality research from prospective studies is needed.
Bottom line
The data available from this systematic review suggest that placement of an LNG IUS at the completion of an endometrial ablation may result in lower hysterectomy rates, without apparent risk, and without significantly difficult LNG IUS removal when needed.
The data provided by Oderkerk and colleagues’ systematic review are promising and, although not studied in the reviewed publications, the potential may exist to reduce the risk of endometrial hyperplasia and endometrial cancer by adding an LNG IUS.
Continue to: LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows...
LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows
van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.
To assess the cost-effectiveness of the LNG IUS versus endometrial ablation in the treatment of heavy menstrual bleeding, van den Brink and colleagues conducted a randomized, noninferiority trial.6
Part of the rationale for this study was to better understand the cost differences between the LNG IUS and second-generation endometrial ablation. Some data have suggested that the LNG IUS is cost-effective when compared with first-generation endometrial ablation; however, definitive evidence about its cost compared with second-generation endometrial ablation is lacking, as these procedures should be less expensive than first-generation endometrial ablation since they frequently are performed in the office rather than in an operating room.
Cost-effectiveness and noninferiority assessed
A total of 270 women were randomly assigned to 1 of 2 treatment strategies. Eventually, 132 women were treated first with the 52-mg LNG IUS, and 138 were treated first with endometrial ablation by radiofrequency ablation. Menstrual blood loss after 24 months was the primary outcome.
At 24 months, the mean pictorial blood loss assessment chart (PBAC) scores were 64.8 in the LNG IUS group compared with 14.2 in the endometrial ablation group. Given that the noninferiority margin was defined as 25 points, noninferiority could not be demonstrated. However, when looking at PBAC scores less than 75 points, the LNG IUS group met this secondary end point in 87% of women versus 94% in the endometrial ablation group. When satisfaction was assessed, 74% of women in the LNG IUS group were satisfied compared with 84% in the endometrial ablation group.
Overall, the total costs per patient were €2,285 in the LNG IUS strategy and €3,465 in the endometrial ablation strategy (costs convert to $2,285 and $3,465 as of this writing).
Key takeaway
Treatment of heavy menstrual bleeding starting with the LNG IUS is cheaper, but it is slightly less effective than endometrial ablation. ●
It is interesting that there are minimal differences between satisfaction rates and PBAC scores less than 75, yet the mean PBAC scores were significantly more favorable for endometrial ablation. This study’s results support the use of a sequential therapy of a less invasive therapy, such as the LNG IUS, prior to performing endometrial ablation.
In this Update, we focus on therapies for abnormal uterine bleeding (AUB) that include a new formulation of a progesterone-only pill (POP), drospirenone 4 mg in a 24/4 regimen (24 days of drospirenone/4 days of inert tablets), which recently showed benefit over the use of desogestrel in a European randomized clinical trial (RCT). Two other commonly used treatments for AUB— the levonorgestrel-releasing intrauterine system (LNG IUS) and endometrial ablation—were studied in terms of cost-effectiveness as well as whether they should be used in combination for added efficacy. In addition, although at times either COVID-19 disease or the COVID-19 vaccine has been blamed for societal and medical problems, one study showed that it is unlikely that significant changes in the menstrual cycle are a result of the COVID-19 vaccine.
COVID-19 vaccination had minimal effects on menstrual cycle length
Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
Does receiving the COVID-19 vaccination result in abnormal menstrual cycles? Patients often ask this question, and it has been a topic of social media discussion (including NPR) and concerns about the possibility of vaccine hesitancy,1,2 as the menstrual cycle is often considered a sign of health and fertility.
To better understand this possible association, Edelman and colleagues conducted a study that prospectively tracked menstrual cycle data using the digital app Natural Cycles in US residents aged 18 to 45 years for 3 consecutive cycles in both a vaccinated and an unvaccinated cohort.3 Almost 4,000 individuals were studied; 2,403 were vaccinated and 1,556 were unvaccinated. The study vaccine types included the BioNTech (Pfizer), Moderna, Johnson & Johnson/Janssen, and unspecified vaccines.
The primary outcome was the within-individual change in cycle length in days, comparing a 3-cycle postvaccine average to a 3-cycle prevaccination average in the 2 groups. (For the unvaccinated group, cycles 1, 2, and 3 were considered the equivalent of prevaccination cycles; cycle 4 was designated as the artificial first vaccine dose-cycle and cycle 5 as the artificial second-dose cycle.)
Increase in cycle length clinically negligible
The investigators found that the vaccinated cohort had less than a 1-day unadjusted increase in the length of their menstrual cycle, which was essentially a 0.71-day increase (98.75% confidence interval [CI], 0.47–0.94). Although this is considered statistically significant, it is likely clinically insignificant in that the overlaid histograms comparing the distribution of change showed a cycle length distribution in vaccinated individuals that is essentially equivalent to that in unvaccinated individuals. After adjusting for confounders, the difference in cycle length was reduced to a 0.64 day (98.75% CI, 0.27–1.01).
An interesting finding was that a subset of individuals who received both vaccine doses in a single cycle had, on average, an adjusted 2-day increase in their menstrual cycle compared with unvaccinated individuals. To explain this slightly longer cycle length, the authors postulated that mRNA vaccines create an immune response, or stressor, which could temporarily affect the hypothalamic-pituitary-ovarian axis if timed correctly. It is certainly possible for an individual to receive 2 doses in a single cycle, which could have both been administered in the early follicular phase. Such cycle length variability can be caused by events, including stressors, that affect the recruitment and maturation of the dominant follicle.
Counseling takeaway
This study provides reassurance to most individuals who receive a COVID-19 vaccine that it likely will not affect their menstrual cycle in a clinically significant manner.
This robust study by Edelman and colleagues on COVID-19 vaccination effects on menstrual cycle length had more than 99% power to detect an unadjusted 1-day difference in cycle length. However, given that most of the study participants were White and had access to the Natural Cycles app, the results may not be generalizable to all individuals who receive the vaccine.
Continue to: Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel...
Drospirenone improved bleeding profiles, lowered discontinuation rates compared with desogestrel
Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
A new POP, marketed under the name Slynd, recently came to market. It contains the progestin drospirenone (DRSP) 4 mg in a 24/4 regimen. This formulation has the advantage of being an antiandrogenic progestin, with a long enough half-life to allow for managing a missed pill in the same fashion as combined oral contraceptives (COCs).
Investigators in Europe conducted a double-blind, randomized trial to assess discontinuation rates due to adverse events (mainly bleeding disorders) in participants taking DRSP 4 mg in a 24/4 regimen compared with those taking the POP desogestrel (DSG) 0.075 mg, which is commonly used in Europe.4 Regidor and colleagues compared 858 women with 6,691 DRSP treatment cycles with 332 women with 2,487 DSG treatment cycles.
Top reasons for stopping a POP
The discontinuation rate for abnormal bleeding was 3.7% in the DRSP group versus 7.3% in the DSG group (55.7% lower). The most common reasons for stopping either POP formulation were vaginal bleeding and acne. Both of these adverse events were less common in the DRSP group. Pill discontinuation due to vaginal bleeding was 2.6% in the DRSP group versus 5.4% in the DSG group, while discontinuation due to acne occurred in 1% in the DRSP group versus 2.7% in the DSG group.
New oral contraception option
This study shows improved acceptability and bleeding profiles in women using this new DRSP contraception pill regimen.
Adherence to a contraceptive method is influenced by patient satisfaction, and this is particularly important in patients who cannot take COCs. It also should be noted that the discontinuation rate for DRSP as a POP used in this 24/4 regimen was similar to discontinuation rates for COCs containing 20 µg and 30 µg of ethinyl estradiol. Cost, however, may be an issue with DRSP, depending on a patient’s insurance coverage.
Continue to: Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy...
Placing an LNG IUS after endometrial ablation for heavy menstrual bleeding reduced risk of hysterectomy
Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. The combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
Over the years, a smattering of articles have suggested that a reduction in uterine bleeding was associated with placement of an LNG IUS at the conclusion of endometrial ablation. We now have a systematic review of this surgical modification.
Oderkerk and colleagues sifted through 747 articles to find 7 publications that could provide meaningful data on the impact of combined use of endometrial ablation and LNG IUS insertion for women with heavy menstrual bleeding.5 These included 4 retrospective cohort studies with control groups, 2 retrospective studies without control groups, and 1 case series. The primary outcome was the hysterectomy rate after therapy.
Promising results for combined therapy
Although no statistically significant intergroup differences were seen in the combined treatment group versus the endometrial ablation alone group for the first 6 months of treatment, significant differences existed at the 12- and 24-month mark. Hysterectomy rates after combined treatment varied from 0% to 11% versus 9.4% to 24% after endometrial ablation alone. Complication rates for combined treatment did not appear higher than those for endometrial ablation alone.
The authors postulated that the failure of endometrial ablation is generally caused by either remaining or regenerating endometrial tissue and that the addition of an LNG IUS allows for suppression of endometrial tissue. Also encouraging was that, in general, the removal of the LNG IUS was relatively simple. A single difficult removal was described due to uterine synechiae, but hysteroscopic resection was not necessary. The authors acknowledged that the data from these 7 retrospective studies are limited and that high-quality research from prospective studies is needed.
Bottom line
The data available from this systematic review suggest that placement of an LNG IUS at the completion of an endometrial ablation may result in lower hysterectomy rates, without apparent risk, and without significantly difficult LNG IUS removal when needed.
The data provided by Oderkerk and colleagues’ systematic review are promising and, although not studied in the reviewed publications, the potential may exist to reduce the risk of endometrial hyperplasia and endometrial cancer by adding an LNG IUS.
Continue to: LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows...
LNG IUS is less expensive, and less effective, than endometrial ablation for heavy menstrual bleeding, cost analysis shows
van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.
To assess the cost-effectiveness of the LNG IUS versus endometrial ablation in the treatment of heavy menstrual bleeding, van den Brink and colleagues conducted a randomized, noninferiority trial.6
Part of the rationale for this study was to better understand the cost differences between the LNG IUS and second-generation endometrial ablation. Some data have suggested that the LNG IUS is cost-effective when compared with first-generation endometrial ablation; however, definitive evidence about its cost compared with second-generation endometrial ablation is lacking, as these procedures should be less expensive than first-generation endometrial ablation since they frequently are performed in the office rather than in an operating room.
Cost-effectiveness and noninferiority assessed
A total of 270 women were randomly assigned to 1 of 2 treatment strategies. Eventually, 132 women were treated first with the 52-mg LNG IUS, and 138 were treated first with endometrial ablation by radiofrequency ablation. Menstrual blood loss after 24 months was the primary outcome.
At 24 months, the mean pictorial blood loss assessment chart (PBAC) scores were 64.8 in the LNG IUS group compared with 14.2 in the endometrial ablation group. Given that the noninferiority margin was defined as 25 points, noninferiority could not be demonstrated. However, when looking at PBAC scores less than 75 points, the LNG IUS group met this secondary end point in 87% of women versus 94% in the endometrial ablation group. When satisfaction was assessed, 74% of women in the LNG IUS group were satisfied compared with 84% in the endometrial ablation group.
Overall, the total costs per patient were €2,285 in the LNG IUS strategy and €3,465 in the endometrial ablation strategy (costs convert to $2,285 and $3,465 as of this writing).
Key takeaway
Treatment of heavy menstrual bleeding starting with the LNG IUS is cheaper, but it is slightly less effective than endometrial ablation. ●
It is interesting that there are minimal differences between satisfaction rates and PBAC scores less than 75, yet the mean PBAC scores were significantly more favorable for endometrial ablation. This study’s results support the use of a sequential therapy of a less invasive therapy, such as the LNG IUS, prior to performing endometrial ablation.
- Blumfiel G. Why reports of menstrual changes after COVID vaccine are tough to study. NPR. August 9, 2021. Accessed August 30, 2022. https://www.npr.org/sections/health-shots/2021/08/09/1024190379/covid-vaccine-period-menstrual-cycle-research
- Lee KMN, Junkins EJ, Fatima UA, et al. Characterizing menstrual bleeding changes occurring after SARSCoV-2 vaccinations. MedRxiv. February 11, 2022. doi:10.1101/2021.10.11.21264863
- Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
- Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
- Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. T he combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
- van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.
- Blumfiel G. Why reports of menstrual changes after COVID vaccine are tough to study. NPR. August 9, 2021. Accessed August 30, 2022. https://www.npr.org/sections/health-shots/2021/08/09/1024190379/covid-vaccine-period-menstrual-cycle-research
- Lee KMN, Junkins EJ, Fatima UA, et al. Characterizing menstrual bleeding changes occurring after SARSCoV-2 vaccinations. MedRxiv. February 11, 2022. doi:10.1101/2021.10.11.21264863
- Edelman A, Boniface ER, Benhar W, et al. Association between menstrual cycle length and coronavirus disease 2019 (COVID-19) vaccination: a US cohort. Obstet Gynecol. 2022;139:481-489.
- Regidor PA, Colli E, Palacios S. Overall and bleeding-related discontinuation rates of a new oral contraceptive containing 4 mg drospirenone only in a 24/4 regimen and comparison to 0.075 mg desogestrel. Gynecol Endocrinol. 2021;37:1121-1127.
- Oderkerk TJ, van de Kar MMA, van der Zanden CHM, et al. T he combined use of endometrial ablation or resection and levonorgestrel-releasing intrauterine system in women with heavy menstrual bleeding: a systematic review. Acta Obstet Gynecol Scand. 2021;100:1779-1787.
- van den Brink MJ, Beelen P, Herman MC, et al. The levonorgestrel intrauterine system versus endometrial ablation for heavy menstrual bleeding: a cost-effectiveness analysis. BJOG. 2021;128:2003-2011.
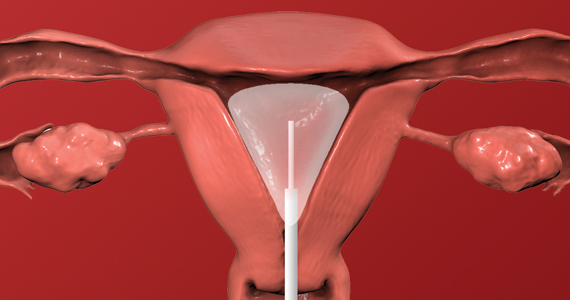
ObGyn: A leader in academic medicine, with progress still to be made in diversity
The nation’s population is quickly diversifying, making racial/ethnic disparities in health care outcomes even more apparent. Minority and non-English-speaking populations have grown and may become a majority in the next generation.1 A proposed strategy to reduce disparities in health care is to recruit more practitioners who better reflect the patient populations.2 Improved access to care with racial concordance between physicians and patients has been reported.3
Being increasingly aware of access-to-care data, more patients are advocating and asking for physicians of color to be their providers.4 Despite progress (ie, more women entering the medical profession), the proportion of physicians who are underrepresented in medicine (URiM—eg, Black, Hispanic, and Native American) still lags US population demographics.3
Why diversity in medicine matters
In addition to improving access to care, diversity in medicine offers other benefits. Working within diverse learning environments has demonstrated educational advantages.5,6 Medical students and residents from diverse backgrounds are less likely to report depression symptoms, regardless of their race. Diversity may accelerate advancements in health care as well, since it is well-established that diverse teams outperform nondiverse teams when it comes to innovation and productivity.7 Finally, as a profession committed to equity, advocacy, and justice, physicians are positioned to lead the way toward racial equity.
Overall, racial and gender diversity in all clinical specialties is improving, but not at the same pace. While the diversity of US medical students and residents by sex and race/ethnicity is greater than among faculty, change in racial diversity has been slow for all 3 groups.8 During the past 40 years the number of full-time faculty has increased 6-fold for females and more than tripled for males.8 However, this rise has not favored URiM faculty, because their proportion is still underrepresented relative to their group in the general population. Clinical departments that are making the most progress in recruiting URiM residents and faculty are often primary or preventive care specialties rather than surgical or service or hospital-based specialties.8,9 ObGyn has consistently had a proportion of URiM residents (18%) that is highest in the surgical specialties and comparable to family medicine and pediatrics.10
When examining physician workforce diversity, it is important to “drill down” to individual specialties to obtain a clearer understanding of trends. The continued need for increased resident and faculty diversity prompted us to examine ObGyn departments. The most recent nationwide data were gathered about full-time faculty from the 2021 AAMC Faculty Roster, residents from the 2021 Accreditation Counsel for Graduate Medical Education (ACGME) Data Resource Book, medical student matriculants from 2021 AAMC, and US adult women (defined arbitrarily as 15 years or older) from the 2019 American Community Survey.11-13
Increase in female faculty and residents
The expanding numbers of faculty and residents over a 40-year period (from 1973 to 2012) led to more women and underrepresented minorities in ObGyn than in other major clinical departments.14,15 Women now constitute two-thirds of all ObGyn faculty and are more likely to be junior rather than senior faculty.9 When looking at junior faculty, a higher proportion of junior faculty who are URiM are female. While more junior faculty and residents are female, male faculty are also racially and ethnically diverse.9
- ObGyn is a leader in racial/ethnic diversity in academic medicine.
- The rapid rise of faculty numbers in the past has not favored underrepresented faculty.
- The rise in ObGyn faculty and residents, who were predominantly female, has contributed to greater racial/ethnic diversity.
- Improved patient outcomes with racial concordance between physicians and patients have been reported.
- More patients are advocating and asking for physicians of color to be their clinicians.
- Racial/ethnic diversity of junior faculty and residents is similar to medical students.
- The most underrepresented group is Hispanic, due in part to its rapid growth in the US population.
Continue to: Growth of URiM physicians in ObGyn...
Growth of URiM physicians in ObGyn
The distribution of racial/ethnic groups in 2021 were compared between senior and junior ObGyn faculty and residents with the US adult female population.9 As shown in the FIGURE, the proportion of ObGyn faculty who are White approximates the White US adult female population. The most rapidly growing racial/ethnic group in the US population is Hispanic. Although Hispanic is the best represented ethnicity among junior faculty, the proportions of Hispanics among faculty and residents lag well behind the US population. The proportion of ObGyn faculty who are Black has consistently been less than in the US adult female population. ObGyns who are Asian constitute higher proportions of faculty and residents than in the US adult female population. This finding about Asians is consistent across all clinical specialties.7
Recruiting URiM students into ObGyn is important. Racial and ethnic representation in surgical and nonsurgical residency programs has not substantially improved in the past decade and continues to lag the changing demographics of the US population.10 More students than residents and faculty are Hispanic, which represents a much-needed opportunity for recruitment. By contrast, junior ObGyn faculty are more likely to be Black than residents and students. Native Americans constitute less than 1% of all faculty, residents, students, and US adult females.9 Lastly, race/ethnicity being self-reported as “other” or “unknown” is most common among students and residents, which perhaps represents greater diversity.
Looking back
Increasing diversity in medicine and in ObGyn has not happened by accident. Transformational change requires rectifying any factors that detrimentally affect the racial/ethnic diversity of our medical students, residents, and faculty. For example, biases inherent in key residency application metrics are being recognized, and use of holistic review is increasing. Change is also accelerated by an explicit and public commitment from national organizations. In 2009, the Liaison Committee of Medical Education (LCME) mandated that medical schools engage in practices that focus on recruitment and retention of a diverse workforce. Increases in Black and Hispanic medical students were noted after implementation of this new mandate.16 The ACGME followed suit with similar guidelines in 2019.10
Diversity is one of the foundational strengths of the ObGyn specialty. Important aspects of the specialty are built upon the contributions of women of color, some voluntary and some not. One example is the knowledge of gynecology that was gained through the involuntary and nonanesthetized surgeries performed on
Moving forward
Advancing diversity in ObGyn offers advantages: better representation of patient populations, improving public health by better access to care, enhancing learning in medical education, building more comprehensive research agendas, and driving institutional excellence. While progress has been made, significant work is still to be done. We must continue to critically examine the role of biases and structural racism that are embedded in evaluating medical students, screening of residency applicants, and selecting and retaining faculty. In future work, we should explore the hypothesis that continued change in racial/ethnic diversity of faculty will only occur once more URiM students, especially the growing number of Hispanics, are admitted into medical schools and recruited for residency positions. We should also examine whether further diversity improves patient outcomes.
It is encouraging to realize that ObGyn departments are leaders in racial/ethnic diversity at US medical schools. It is also critical that the specialty commits to the progress that still needs to be made, including increasing diversity among faculty and institutional leadership. To maintain diversity that mirrors the US adult female population, the specialty of ObGyn will require active surveillance and continued recruitment of Black and, especially Hispanic, faculty and residents.19 The national strategies aimed at building medical student and residency diversity are beginning to yield results. For those gains to help faculty diversity, institutional and departmental leaders will need to implement best practices for recruiting, retaining, and advancing URiM faculty.19 Those practices would include making workforce diversity an explicit priority, building diverse applicant pools, and establishing infrastructure and mentorship to advance URiM faculty to senior leadership positions.20
In conclusion
Building a physician workforce that is more representative of the US population should aid in addressing inequalities in health and health care. Significant strides have been made in racial/ethnic diversity in ObGyn. This has resulted in a specialty that is among the most diverse in academic medicine. At the same time, there is more work to be done. For example, the specialty is far from reaching racial equity for Hispanic physicians. Also, continued efforts are necessary to advance URiM faculty to leadership positions. The legacy of racial/ethnic diversity in ObGyn did not happen by accident and will not be maintained without intention. ●
- Hummes KR, Jones NA, Ramierez RR. United States Census: overview of race and Hispanic origin: 2010. http//www. census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed May 22, 2022.
- Xierali IM, Castillo-Page L, Zhang K, et al. AM last page: the urgency of physician workforce diversity. Acad Med. 2014;89:1192.
- Association of American Medical College. Diversity in the physician workforce. Facts & figures 2014. http://www .aamcdiversityfactsandfigures.org. Accessed April 9, 2022.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: Diversifying the physician workforce may be key in addressing health disparities. JAMA Int Med. 2014;174:289-291.
- Amalba A, Abantanga FA, Scherpbier AJ, et al. Community-based education: The influence of role modeling on career choice and practice location. Med Teac. 2017;39:174-180.
- Umbach PD. The contribution of faculty of color to undergraduate education. Res High Educ. 2006;47:317-345.
- Gonzalo JD, Chuang CH, Glod SA, et al. General internists as change agents: opportunities and barriers to leadership in health systems and medical education transformation. J Gen Intern Med. 2020;35:1865-1869.
- Xierali IM, Fair MA, Nivet MA. Faculty diversity in U.S. medical schools: Progress and gaps coexist. AAMC Analysis in Brief. 2016;16. https://www.aamc.org/system/files/reports/1/decem ber2016facultydiversityinu.s.medicalschoolsprogressandga ps.pdf. Accessed May 4, 2022.
- Rayburn WF, Xierali IM, McDade WA. Racial-ethnic diversity of obstetrics and gynecology faculty at medical schools in the United States. Am J Obstet Gynecol. 2022;S00029378(22)00106-5. doi: 10.1016/j.ajog.2022.02.007.
- Hucko L, Al-khersan H, Lopez Dominguez J, et al. Racial and ethnic diversity of U.S. residency programs, 2011-2019. N Engl J Med. 2022;386:22-23.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme.org/globalassets/pfassets /publicationsbooks/2020-2021_acgme_databook _document.pdf. Accessed October 24, 2021
- United States Census Bureau. The 2019 American Community Survey 5-Year Public Use Microdata Sample (PUMS) Files.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme .org/globalassets/pfassets/publicationsbooks/2020-2021 _acgme_databook_document.pdf. Accessed October 24, 2021.
- Rayburn WF, Liu CQ, Elwell EC, et al. Diversity of physician faculty in obstetrics and gynecology. J Reprod Med. 2016;61:22-26.
- Xierali IM, Nivet MA, Rayburn WF. Full-time faculty in clinical and basic science departments by sex and underrepresented in medicine status: A 40-year review. Acad Med. 2021;96: 568-575.
- Boatright DH, Samuels EA, Cramer LJ, et al. Association between the Liaison Committee on Medical Education’s Diversity Standards and Changes in percentage of medical student sex, race, and ethnicity. JAMA. 2018;320:2267-2269.
- United States National Library of Medicine. Changing the face of medicine.
- https://cfmedicine.nlm.nih.gov/physicians/biography_82. html. Accessed May 5, 2022.
- Christmas M. #SayHerName: Should obstetrics and gynecology reckon with the legacy of JM Sims? Reprod Sci. 2021;28:3282-3284.
- Morgan HK, Winkel AF, Bands E, et al. Promoting diversity, equity, and inclusion in the selection of obstetrician-gynecologists. Obstet Gynecol. 2021;138:272-277.
- Peek ME, Kim KE, Johnson JK, et al. “URM candidates are encouraged to apply”: a national study to identify effective strategies to enhance racial and ethnic faculty diversity in academic departments of medicine. Acad Med. 2013;88:405-412.
The nation’s population is quickly diversifying, making racial/ethnic disparities in health care outcomes even more apparent. Minority and non-English-speaking populations have grown and may become a majority in the next generation.1 A proposed strategy to reduce disparities in health care is to recruit more practitioners who better reflect the patient populations.2 Improved access to care with racial concordance between physicians and patients has been reported.3
Being increasingly aware of access-to-care data, more patients are advocating and asking for physicians of color to be their providers.4 Despite progress (ie, more women entering the medical profession), the proportion of physicians who are underrepresented in medicine (URiM—eg, Black, Hispanic, and Native American) still lags US population demographics.3
Why diversity in medicine matters
In addition to improving access to care, diversity in medicine offers other benefits. Working within diverse learning environments has demonstrated educational advantages.5,6 Medical students and residents from diverse backgrounds are less likely to report depression symptoms, regardless of their race. Diversity may accelerate advancements in health care as well, since it is well-established that diverse teams outperform nondiverse teams when it comes to innovation and productivity.7 Finally, as a profession committed to equity, advocacy, and justice, physicians are positioned to lead the way toward racial equity.
Overall, racial and gender diversity in all clinical specialties is improving, but not at the same pace. While the diversity of US medical students and residents by sex and race/ethnicity is greater than among faculty, change in racial diversity has been slow for all 3 groups.8 During the past 40 years the number of full-time faculty has increased 6-fold for females and more than tripled for males.8 However, this rise has not favored URiM faculty, because their proportion is still underrepresented relative to their group in the general population. Clinical departments that are making the most progress in recruiting URiM residents and faculty are often primary or preventive care specialties rather than surgical or service or hospital-based specialties.8,9 ObGyn has consistently had a proportion of URiM residents (18%) that is highest in the surgical specialties and comparable to family medicine and pediatrics.10
When examining physician workforce diversity, it is important to “drill down” to individual specialties to obtain a clearer understanding of trends. The continued need for increased resident and faculty diversity prompted us to examine ObGyn departments. The most recent nationwide data were gathered about full-time faculty from the 2021 AAMC Faculty Roster, residents from the 2021 Accreditation Counsel for Graduate Medical Education (ACGME) Data Resource Book, medical student matriculants from 2021 AAMC, and US adult women (defined arbitrarily as 15 years or older) from the 2019 American Community Survey.11-13
Increase in female faculty and residents
The expanding numbers of faculty and residents over a 40-year period (from 1973 to 2012) led to more women and underrepresented minorities in ObGyn than in other major clinical departments.14,15 Women now constitute two-thirds of all ObGyn faculty and are more likely to be junior rather than senior faculty.9 When looking at junior faculty, a higher proportion of junior faculty who are URiM are female. While more junior faculty and residents are female, male faculty are also racially and ethnically diverse.9
- ObGyn is a leader in racial/ethnic diversity in academic medicine.
- The rapid rise of faculty numbers in the past has not favored underrepresented faculty.
- The rise in ObGyn faculty and residents, who were predominantly female, has contributed to greater racial/ethnic diversity.
- Improved patient outcomes with racial concordance between physicians and patients have been reported.
- More patients are advocating and asking for physicians of color to be their clinicians.
- Racial/ethnic diversity of junior faculty and residents is similar to medical students.
- The most underrepresented group is Hispanic, due in part to its rapid growth in the US population.
Continue to: Growth of URiM physicians in ObGyn...
Growth of URiM physicians in ObGyn
The distribution of racial/ethnic groups in 2021 were compared between senior and junior ObGyn faculty and residents with the US adult female population.9 As shown in the FIGURE, the proportion of ObGyn faculty who are White approximates the White US adult female population. The most rapidly growing racial/ethnic group in the US population is Hispanic. Although Hispanic is the best represented ethnicity among junior faculty, the proportions of Hispanics among faculty and residents lag well behind the US population. The proportion of ObGyn faculty who are Black has consistently been less than in the US adult female population. ObGyns who are Asian constitute higher proportions of faculty and residents than in the US adult female population. This finding about Asians is consistent across all clinical specialties.7
Recruiting URiM students into ObGyn is important. Racial and ethnic representation in surgical and nonsurgical residency programs has not substantially improved in the past decade and continues to lag the changing demographics of the US population.10 More students than residents and faculty are Hispanic, which represents a much-needed opportunity for recruitment. By contrast, junior ObGyn faculty are more likely to be Black than residents and students. Native Americans constitute less than 1% of all faculty, residents, students, and US adult females.9 Lastly, race/ethnicity being self-reported as “other” or “unknown” is most common among students and residents, which perhaps represents greater diversity.
Looking back
Increasing diversity in medicine and in ObGyn has not happened by accident. Transformational change requires rectifying any factors that detrimentally affect the racial/ethnic diversity of our medical students, residents, and faculty. For example, biases inherent in key residency application metrics are being recognized, and use of holistic review is increasing. Change is also accelerated by an explicit and public commitment from national organizations. In 2009, the Liaison Committee of Medical Education (LCME) mandated that medical schools engage in practices that focus on recruitment and retention of a diverse workforce. Increases in Black and Hispanic medical students were noted after implementation of this new mandate.16 The ACGME followed suit with similar guidelines in 2019.10
Diversity is one of the foundational strengths of the ObGyn specialty. Important aspects of the specialty are built upon the contributions of women of color, some voluntary and some not. One example is the knowledge of gynecology that was gained through the involuntary and nonanesthetized surgeries performed on
Moving forward
Advancing diversity in ObGyn offers advantages: better representation of patient populations, improving public health by better access to care, enhancing learning in medical education, building more comprehensive research agendas, and driving institutional excellence. While progress has been made, significant work is still to be done. We must continue to critically examine the role of biases and structural racism that are embedded in evaluating medical students, screening of residency applicants, and selecting and retaining faculty. In future work, we should explore the hypothesis that continued change in racial/ethnic diversity of faculty will only occur once more URiM students, especially the growing number of Hispanics, are admitted into medical schools and recruited for residency positions. We should also examine whether further diversity improves patient outcomes.
It is encouraging to realize that ObGyn departments are leaders in racial/ethnic diversity at US medical schools. It is also critical that the specialty commits to the progress that still needs to be made, including increasing diversity among faculty and institutional leadership. To maintain diversity that mirrors the US adult female population, the specialty of ObGyn will require active surveillance and continued recruitment of Black and, especially Hispanic, faculty and residents.19 The national strategies aimed at building medical student and residency diversity are beginning to yield results. For those gains to help faculty diversity, institutional and departmental leaders will need to implement best practices for recruiting, retaining, and advancing URiM faculty.19 Those practices would include making workforce diversity an explicit priority, building diverse applicant pools, and establishing infrastructure and mentorship to advance URiM faculty to senior leadership positions.20
In conclusion
Building a physician workforce that is more representative of the US population should aid in addressing inequalities in health and health care. Significant strides have been made in racial/ethnic diversity in ObGyn. This has resulted in a specialty that is among the most diverse in academic medicine. At the same time, there is more work to be done. For example, the specialty is far from reaching racial equity for Hispanic physicians. Also, continued efforts are necessary to advance URiM faculty to leadership positions. The legacy of racial/ethnic diversity in ObGyn did not happen by accident and will not be maintained without intention. ●
The nation’s population is quickly diversifying, making racial/ethnic disparities in health care outcomes even more apparent. Minority and non-English-speaking populations have grown and may become a majority in the next generation.1 A proposed strategy to reduce disparities in health care is to recruit more practitioners who better reflect the patient populations.2 Improved access to care with racial concordance between physicians and patients has been reported.3
Being increasingly aware of access-to-care data, more patients are advocating and asking for physicians of color to be their providers.4 Despite progress (ie, more women entering the medical profession), the proportion of physicians who are underrepresented in medicine (URiM—eg, Black, Hispanic, and Native American) still lags US population demographics.3
Why diversity in medicine matters
In addition to improving access to care, diversity in medicine offers other benefits. Working within diverse learning environments has demonstrated educational advantages.5,6 Medical students and residents from diverse backgrounds are less likely to report depression symptoms, regardless of their race. Diversity may accelerate advancements in health care as well, since it is well-established that diverse teams outperform nondiverse teams when it comes to innovation and productivity.7 Finally, as a profession committed to equity, advocacy, and justice, physicians are positioned to lead the way toward racial equity.
Overall, racial and gender diversity in all clinical specialties is improving, but not at the same pace. While the diversity of US medical students and residents by sex and race/ethnicity is greater than among faculty, change in racial diversity has been slow for all 3 groups.8 During the past 40 years the number of full-time faculty has increased 6-fold for females and more than tripled for males.8 However, this rise has not favored URiM faculty, because their proportion is still underrepresented relative to their group in the general population. Clinical departments that are making the most progress in recruiting URiM residents and faculty are often primary or preventive care specialties rather than surgical or service or hospital-based specialties.8,9 ObGyn has consistently had a proportion of URiM residents (18%) that is highest in the surgical specialties and comparable to family medicine and pediatrics.10
When examining physician workforce diversity, it is important to “drill down” to individual specialties to obtain a clearer understanding of trends. The continued need for increased resident and faculty diversity prompted us to examine ObGyn departments. The most recent nationwide data were gathered about full-time faculty from the 2021 AAMC Faculty Roster, residents from the 2021 Accreditation Counsel for Graduate Medical Education (ACGME) Data Resource Book, medical student matriculants from 2021 AAMC, and US adult women (defined arbitrarily as 15 years or older) from the 2019 American Community Survey.11-13
Increase in female faculty and residents
The expanding numbers of faculty and residents over a 40-year period (from 1973 to 2012) led to more women and underrepresented minorities in ObGyn than in other major clinical departments.14,15 Women now constitute two-thirds of all ObGyn faculty and are more likely to be junior rather than senior faculty.9 When looking at junior faculty, a higher proportion of junior faculty who are URiM are female. While more junior faculty and residents are female, male faculty are also racially and ethnically diverse.9
- ObGyn is a leader in racial/ethnic diversity in academic medicine.
- The rapid rise of faculty numbers in the past has not favored underrepresented faculty.
- The rise in ObGyn faculty and residents, who were predominantly female, has contributed to greater racial/ethnic diversity.
- Improved patient outcomes with racial concordance between physicians and patients have been reported.
- More patients are advocating and asking for physicians of color to be their clinicians.
- Racial/ethnic diversity of junior faculty and residents is similar to medical students.
- The most underrepresented group is Hispanic, due in part to its rapid growth in the US population.
Continue to: Growth of URiM physicians in ObGyn...
Growth of URiM physicians in ObGyn
The distribution of racial/ethnic groups in 2021 were compared between senior and junior ObGyn faculty and residents with the US adult female population.9 As shown in the FIGURE, the proportion of ObGyn faculty who are White approximates the White US adult female population. The most rapidly growing racial/ethnic group in the US population is Hispanic. Although Hispanic is the best represented ethnicity among junior faculty, the proportions of Hispanics among faculty and residents lag well behind the US population. The proportion of ObGyn faculty who are Black has consistently been less than in the US adult female population. ObGyns who are Asian constitute higher proportions of faculty and residents than in the US adult female population. This finding about Asians is consistent across all clinical specialties.7
Recruiting URiM students into ObGyn is important. Racial and ethnic representation in surgical and nonsurgical residency programs has not substantially improved in the past decade and continues to lag the changing demographics of the US population.10 More students than residents and faculty are Hispanic, which represents a much-needed opportunity for recruitment. By contrast, junior ObGyn faculty are more likely to be Black than residents and students. Native Americans constitute less than 1% of all faculty, residents, students, and US adult females.9 Lastly, race/ethnicity being self-reported as “other” or “unknown” is most common among students and residents, which perhaps represents greater diversity.
Looking back
Increasing diversity in medicine and in ObGyn has not happened by accident. Transformational change requires rectifying any factors that detrimentally affect the racial/ethnic diversity of our medical students, residents, and faculty. For example, biases inherent in key residency application metrics are being recognized, and use of holistic review is increasing. Change is also accelerated by an explicit and public commitment from national organizations. In 2009, the Liaison Committee of Medical Education (LCME) mandated that medical schools engage in practices that focus on recruitment and retention of a diverse workforce. Increases in Black and Hispanic medical students were noted after implementation of this new mandate.16 The ACGME followed suit with similar guidelines in 2019.10
Diversity is one of the foundational strengths of the ObGyn specialty. Important aspects of the specialty are built upon the contributions of women of color, some voluntary and some not. One example is the knowledge of gynecology that was gained through the involuntary and nonanesthetized surgeries performed on
Moving forward
Advancing diversity in ObGyn offers advantages: better representation of patient populations, improving public health by better access to care, enhancing learning in medical education, building more comprehensive research agendas, and driving institutional excellence. While progress has been made, significant work is still to be done. We must continue to critically examine the role of biases and structural racism that are embedded in evaluating medical students, screening of residency applicants, and selecting and retaining faculty. In future work, we should explore the hypothesis that continued change in racial/ethnic diversity of faculty will only occur once more URiM students, especially the growing number of Hispanics, are admitted into medical schools and recruited for residency positions. We should also examine whether further diversity improves patient outcomes.
It is encouraging to realize that ObGyn departments are leaders in racial/ethnic diversity at US medical schools. It is also critical that the specialty commits to the progress that still needs to be made, including increasing diversity among faculty and institutional leadership. To maintain diversity that mirrors the US adult female population, the specialty of ObGyn will require active surveillance and continued recruitment of Black and, especially Hispanic, faculty and residents.19 The national strategies aimed at building medical student and residency diversity are beginning to yield results. For those gains to help faculty diversity, institutional and departmental leaders will need to implement best practices for recruiting, retaining, and advancing URiM faculty.19 Those practices would include making workforce diversity an explicit priority, building diverse applicant pools, and establishing infrastructure and mentorship to advance URiM faculty to senior leadership positions.20
In conclusion
Building a physician workforce that is more representative of the US population should aid in addressing inequalities in health and health care. Significant strides have been made in racial/ethnic diversity in ObGyn. This has resulted in a specialty that is among the most diverse in academic medicine. At the same time, there is more work to be done. For example, the specialty is far from reaching racial equity for Hispanic physicians. Also, continued efforts are necessary to advance URiM faculty to leadership positions. The legacy of racial/ethnic diversity in ObGyn did not happen by accident and will not be maintained without intention. ●
- Hummes KR, Jones NA, Ramierez RR. United States Census: overview of race and Hispanic origin: 2010. http//www. census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed May 22, 2022.
- Xierali IM, Castillo-Page L, Zhang K, et al. AM last page: the urgency of physician workforce diversity. Acad Med. 2014;89:1192.
- Association of American Medical College. Diversity in the physician workforce. Facts & figures 2014. http://www .aamcdiversityfactsandfigures.org. Accessed April 9, 2022.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: Diversifying the physician workforce may be key in addressing health disparities. JAMA Int Med. 2014;174:289-291.
- Amalba A, Abantanga FA, Scherpbier AJ, et al. Community-based education: The influence of role modeling on career choice and practice location. Med Teac. 2017;39:174-180.
- Umbach PD. The contribution of faculty of color to undergraduate education. Res High Educ. 2006;47:317-345.
- Gonzalo JD, Chuang CH, Glod SA, et al. General internists as change agents: opportunities and barriers to leadership in health systems and medical education transformation. J Gen Intern Med. 2020;35:1865-1869.
- Xierali IM, Fair MA, Nivet MA. Faculty diversity in U.S. medical schools: Progress and gaps coexist. AAMC Analysis in Brief. 2016;16. https://www.aamc.org/system/files/reports/1/decem ber2016facultydiversityinu.s.medicalschoolsprogressandga ps.pdf. Accessed May 4, 2022.
- Rayburn WF, Xierali IM, McDade WA. Racial-ethnic diversity of obstetrics and gynecology faculty at medical schools in the United States. Am J Obstet Gynecol. 2022;S00029378(22)00106-5. doi: 10.1016/j.ajog.2022.02.007.
- Hucko L, Al-khersan H, Lopez Dominguez J, et al. Racial and ethnic diversity of U.S. residency programs, 2011-2019. N Engl J Med. 2022;386:22-23.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme.org/globalassets/pfassets /publicationsbooks/2020-2021_acgme_databook _document.pdf. Accessed October 24, 2021
- United States Census Bureau. The 2019 American Community Survey 5-Year Public Use Microdata Sample (PUMS) Files.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme .org/globalassets/pfassets/publicationsbooks/2020-2021 _acgme_databook_document.pdf. Accessed October 24, 2021.
- Rayburn WF, Liu CQ, Elwell EC, et al. Diversity of physician faculty in obstetrics and gynecology. J Reprod Med. 2016;61:22-26.
- Xierali IM, Nivet MA, Rayburn WF. Full-time faculty in clinical and basic science departments by sex and underrepresented in medicine status: A 40-year review. Acad Med. 2021;96: 568-575.
- Boatright DH, Samuels EA, Cramer LJ, et al. Association between the Liaison Committee on Medical Education’s Diversity Standards and Changes in percentage of medical student sex, race, and ethnicity. JAMA. 2018;320:2267-2269.
- United States National Library of Medicine. Changing the face of medicine.
- https://cfmedicine.nlm.nih.gov/physicians/biography_82. html. Accessed May 5, 2022.
- Christmas M. #SayHerName: Should obstetrics and gynecology reckon with the legacy of JM Sims? Reprod Sci. 2021;28:3282-3284.
- Morgan HK, Winkel AF, Bands E, et al. Promoting diversity, equity, and inclusion in the selection of obstetrician-gynecologists. Obstet Gynecol. 2021;138:272-277.
- Peek ME, Kim KE, Johnson JK, et al. “URM candidates are encouraged to apply”: a national study to identify effective strategies to enhance racial and ethnic faculty diversity in academic departments of medicine. Acad Med. 2013;88:405-412.
- Hummes KR, Jones NA, Ramierez RR. United States Census: overview of race and Hispanic origin: 2010. http//www. census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed May 22, 2022.
- Xierali IM, Castillo-Page L, Zhang K, et al. AM last page: the urgency of physician workforce diversity. Acad Med. 2014;89:1192.
- Association of American Medical College. Diversity in the physician workforce. Facts & figures 2014. http://www .aamcdiversityfactsandfigures.org. Accessed April 9, 2022.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: Diversifying the physician workforce may be key in addressing health disparities. JAMA Int Med. 2014;174:289-291.
- Amalba A, Abantanga FA, Scherpbier AJ, et al. Community-based education: The influence of role modeling on career choice and practice location. Med Teac. 2017;39:174-180.
- Umbach PD. The contribution of faculty of color to undergraduate education. Res High Educ. 2006;47:317-345.
- Gonzalo JD, Chuang CH, Glod SA, et al. General internists as change agents: opportunities and barriers to leadership in health systems and medical education transformation. J Gen Intern Med. 2020;35:1865-1869.
- Xierali IM, Fair MA, Nivet MA. Faculty diversity in U.S. medical schools: Progress and gaps coexist. AAMC Analysis in Brief. 2016;16. https://www.aamc.org/system/files/reports/1/decem ber2016facultydiversityinu.s.medicalschoolsprogressandga ps.pdf. Accessed May 4, 2022.
- Rayburn WF, Xierali IM, McDade WA. Racial-ethnic diversity of obstetrics and gynecology faculty at medical schools in the United States. Am J Obstet Gynecol. 2022;S00029378(22)00106-5. doi: 10.1016/j.ajog.2022.02.007.
- Hucko L, Al-khersan H, Lopez Dominguez J, et al. Racial and ethnic diversity of U.S. residency programs, 2011-2019. N Engl J Med. 2022;386:22-23.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme.org/globalassets/pfassets /publicationsbooks/2020-2021_acgme_databook _document.pdf. Accessed October 24, 2021
- United States Census Bureau. The 2019 American Community Survey 5-Year Public Use Microdata Sample (PUMS) Files.
- Accreditation Council for Graduate Medical Education. Data Resource Book Academic Year 2020-2021. https://www.acgme .org/globalassets/pfassets/publicationsbooks/2020-2021 _acgme_databook_document.pdf. Accessed October 24, 2021.
- Rayburn WF, Liu CQ, Elwell EC, et al. Diversity of physician faculty in obstetrics and gynecology. J Reprod Med. 2016;61:22-26.
- Xierali IM, Nivet MA, Rayburn WF. Full-time faculty in clinical and basic science departments by sex and underrepresented in medicine status: A 40-year review. Acad Med. 2021;96: 568-575.
- Boatright DH, Samuels EA, Cramer LJ, et al. Association between the Liaison Committee on Medical Education’s Diversity Standards and Changes in percentage of medical student sex, race, and ethnicity. JAMA. 2018;320:2267-2269.
- United States National Library of Medicine. Changing the face of medicine.
- https://cfmedicine.nlm.nih.gov/physicians/biography_82. html. Accessed May 5, 2022.
- Christmas M. #SayHerName: Should obstetrics and gynecology reckon with the legacy of JM Sims? Reprod Sci. 2021;28:3282-3284.
- Morgan HK, Winkel AF, Bands E, et al. Promoting diversity, equity, and inclusion in the selection of obstetrician-gynecologists. Obstet Gynecol. 2021;138:272-277.
- Peek ME, Kim KE, Johnson JK, et al. “URM candidates are encouraged to apply”: a national study to identify effective strategies to enhance racial and ethnic faculty diversity in academic departments of medicine. Acad Med. 2013;88:405-412.
Vismodegib for Basal Cell Carcinoma and Beyond: What Dermatologists Need to Know
Basal cell carcinomas (BCCs) are considered the most common cutaneous cancers. Approximately 80% of nonmelanoma skin cancers are BCCs.1,2 Surgical management is the gold standard for early-stage and localized BCCs; it may include simple excision vs Mohs micrographic surgery.3,4 However, if left untreated, these lesions can progress to an advanced stage (locally advanced BCC) or infrequently may spread to distant sites (metastatic BCC). In the advanced stage, the lesions are no longer manageable by surgery or radiation therapy.5,6 Recently, inhibitors targeting the hedgehog (Hh) pathway have shown great promise for these patients. The first drug approved by the US Food and Drug Administration (FDA) for locally advanced and metastatic BCC is vismodegib.7 In this article, we provide a clinical review of vismodegib for the management of BCC, including a discussion of the Hh pathway in BCC, adverse effects of vismodegib, use of vismodegib in adnexal skin tumors, recommended doses for vismodegib therapy in BCC, and management of the side effects of treatment.
Hh Pathway in BCC
In embryonic development, the Hh signaling pathway is crucial across a broad spectrum of species, including humans. Various members of the Hh family have been recognized, all working as secreted regulatory proteins.8 The name of the Hh signaling pathway is derived from a polypeptide ligand called hedgehog found in some fruit flies. Mutations in the gene led to fruit fly larvae that had a spiky hairy pattern of denticles similar to hedgehogs, leading to the name of this molecule.9 The transmembrane protein smoothened (SMO) is the main component of the Hh signaling pathway and initiates a signaling cascade that in turn leads to an increased expression of target genes, such as GLI1. Patched (PTCH), also a transmembrane protein and a cell-surface receptor for the secreted Hh ligand, suppresses the signaling capacity of SMO. Upon binding of the Hh ligand to the PTCH receptor, the suppression of SMO is relieved and a signal is propagated, evoking a cellular response.10 Molecular and genetic studies have reported that genetic alterations in the Hh signaling pathway are almost universally present in all BCCs, leading to an aberrant activation of the pathway and an uncontrolled proliferation of the basal cells. Frequently, these alterations have been shown to cause loss of function of PTCH homologue 1, which usually acts to inhibit the SMO homologue signaling activity.11,12
Because of the potential importance of Hh signaling in other solid malignancies and the failure of topical inhibition of SMO,13 subsequent studies on the development of Hh pathway inhibitors have mostly focused on the systemic approach. A multitude of Hh pathway inhibitors have been developed thus far, such as SANT1-SANT4, GDC-0449, IPI-926, BMS-833923 (XL139), HhAntag-691, and MK-4101.14 Many of these inhibitors have been clinically investigated.13,15,16
Systemic SMO Inhibitor: Vismodegib
Vismodegib was the earliest systemic SMO inhibitor to fulfill early clinical evaluation15,16 and the first drug to receive FDA approval for the management of advanced or metastatic BCC. Vismodegib is a small-molecule SMO inhibitor used for the management of selected locally advanced BCC and metastatic BCC in adults.3,17 Although there is a possibility of recurrence following drug withdrawal, vismodegib constitutes a new therapeutic strategy presenting positive benefits to patients. It may provide superior improvement over sunitinib, which has shown efficacy in a few patients; however, the efficacy and tolerance of sunitinib have been shown to be limited.18,19
Adverse Effects of Vismodegib Therapy
Adverse events with vismodegib use have been reported in 98% of patients (N=491); most of these were mild to moderate.20 However, the frequency of adverse events could prove to be a therapeutic challenge for patients requiring extended treatment. The most frequently reported reversible side effects were muscle spasms (64%), alopecia (62%), weight loss (33%), fatigue (28%), decreased appetite (25%), diarrhea (17%), nausea (16%), dysgeusia (54%), and ageusia (22%).20 In clinical trials, amenorrhea was noticed in 30% (3/10) of females with reproductive potential.2 Apart from alopecia and possibly amenorrhea, these side effects are reversible.17 Alkeraye et al17 reported 3 clinical cases of persistent alopecia following the use of vismodegib. Amenorrhea is a possible side effect of unknown reversibility.7
Vismodegib is a pregnancy category D medication.4 Severe birth defects, including craniofacial abnormalities, retardations in normal growth, open perineum, and absence of digital fusion at a corresponding 20% of the recommended daily dose, were found in rat studies. Embryo-fetal death was noted when rats were exposedto concentrations comparable to the recommended human dose.4
Hepatic events with the use of vismodegib have been reported. The use of vismodegib in randomized controlled trials resulted in elevation of both alanine aminotransferase and aspartate aminotransferase levels compared with placebo.21 Moreover, severe hepatotoxicity with vismodegib has been reported.22-24 A study conducted by Edwards et al25 concluded that the use of vismodegib in patients with severe liver disease must include thorough risk-benefit assessment, with caution in using other concomitant hepatotoxic medications.
Rare adverse events also have been reported in the literature, including vismodegib-induced pancreatitis in a 79-year-old patient treated for locally advanced, recurrent BCC that was cleared following cessation of therapy.26 Additionally, atypical fibroxanthoma was observed in an 83-year-old patient after 30 days of treatment with vismodegib for multiple BCCs.27 The development of other secondary malignancies, such as squamous cell carcinoma, melanoma, keratoacanthomas, and pilomatricomas, during or after the long-term use of vismodegib also have been described.28-35
Use of Vismodegib for Adnexal Skin Tumors
The role of the sonic Hh–PTCH pathway in the pathogenesis of adnexal tumors varies in the literature. Some studies propose the involvement of this pathway in the formation of adnexal tumors such as trichoblastoma, trichoepithelioma, and cylindroma, as in BCC. Various lines of evidence support this involvement. Firstly, in mice, the spontaneous generation of numerous BCCs, trichoblastomas, trichoepitheliomas, and cylindromas has been observed following constitutive activation of the sonic Hh–PTCH pathway.36 Secondly, in trichoepitheliomas, there have been positive results in molecular research into the tumor suppressor gene PTCH homologue 1, PTCH1, whose mutations cause constitutive activation of the sonic Hh–PTCH pathway.37 Thirdly, GLI138 and SOX939 transcription factors associated with the signaling pathway of sonic Hh–PTCH appear to have increased levels in adnexal carcinomas.19 Lepesant et al19 reported a notable clinical response to vismodegib in trichoblastic carcinoma. Baur et al40 reported successful treatment of multiple familial trichoepitheliomas with vismodegib. Nonetheless, more studies are required to assess the efficacy and reliability of vismodegib in the management of adnexal tumors.
Recommended Dose of Vismodegib Therapy
The vismodegib dosage that is approved by the FDA is 150 mg/d until disease progression or the development of intolerable side effects.4 Higher dosing regimens were evaluated with 270 mg/d and 540 mg/d. No added therapeutic benefit was noted with the increase in the dose, and no dose-limiting toxic effects were observed.41
Management of Vismodegib Side Effects
Managing patient expectations is a crucial step in improving dysgeusia. The experience of dysgeusia varies among patients; thus, patients should be instructed to adjust their diets according to their level of dysgeusia, which can be achieved by changing ingredients or dressings used with their diet. This step has been proven to be effective in overcoming vismodegib-related dysgeusia. Also, fluid taste distortion may lead to dehydration and an increase in creatine level. Thus, patients should be encouraged to monitor fluid intake. Moreover, a treatment hiatus of 2 to 8 months results in near-complete improvement of taste distortion.
For muscle spasms, quinine, treatment break for 1 month, gentle exercise of affected areas, or muscle relaxants such as baclofen and temazepam all are effective methods. For vismodegib-related alopecia, managing patient expectations is key; patients should be aware that hair may take 6 to 12 months or even longer to regrow. In addition, shaving less frequently helps improve alopecia.
For gastrointestinal disorders, loperamide with or without codeine phosphate is effective in resolving diarrhea, and metoclopramide is mostly adequate in treating nausea. Another adverse event is weight loss; weight loss of 5% or more of total body weight prompts dietetic referral. If weight loss persists, a treatment break might be needed to regain weight.
Overall, treatment breaks are sufficient to resolve adverse events caused by vismodegib and do not compromise efficacy of treatment. The duration of a treatment break should be considered before initiation. In one clinical trial, a longer treatment break was associated with fewer adverse effects without affecting the efficacy of treatment.42
Conclusion
Vismodegib provides an effective alternative to surgical intervention in the management of BCC. However, patients must be monitored vigilantly, as adverse events are common (>90%).
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Cirrone F, Harris CS. Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin Ther. 2012;34:2039-2050.
- Ruiz-Salas V, Alegre M, López-Ferrer A, et al. Vismodegib: a review [article in English, Spanish]. Actas Dermosifiliogr. 2014;105:744-751.
- Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353:2262-2269.
- Cusack CA, Nijhawan R, Miller B, et al. Vismodegib for locally advanced basal cell carcinoma in a heart transplant patient. JAMA Dermatol. 2015;151:70-72.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998;110:885-888.
- Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46:3-12.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103-115.
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423-434.
- Sekulic A, Mangold AR, Northfelt DW, et al. Advanced basal cell carcinoma of the skin: targeting the hedgehog pathway. Curr Opin Oncol. 2013;25:218-223.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nature Rev Genet. 2006;7:841-850.
- Alkeraye S, Maire C, Desmedt E, et al. Persistent alopecia induced by vismodegib. Br J Dermatol. 2015;172:1671-1672.
- Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24:199-203.
- Lepesant P, Crinquette M, Alkeraye S, et al. Vismodegib induces significant clinical response in locally advanced trichoblastic carcinoma. Br J Dermatol. 2015;173:1059-1062.
- Basset-Seguin N, Hauschild A, Grob JJ, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-plannedinterim analysis of an international, open-label trial. Lancet Oncol. 2015;16:729-736.
- Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284-4292.
- Sanchez BE, Hajjafar L. Severe hepatotoxicity in a patient treated with hedgehog inhibitor: first case report. Gastroenterology. 2011;140:S974-S975.
- Ly P, Wolf K, Wilson J. A case of hepatotoxicity associated with vismodegib. JAAD Case Rep. 2018;5:57-59.
- Eiger-Moscovich M, Reich E, Tauber G, et al. Efficacy of vismodegib for the treatment of orbital and advanced periocular basal cell carcinoma. Am J Ophthalmol. 2019;207:62-70.
- Edwards BJ, Raisch DW, Saraykar SS, et al. Hepatotoxicity with vismodegib: an MD Anderson Cancer Center and Research on Adverse Drug Events and Reports Project. Drugs R D. 2017;17:211-218.
- Velter C, Blanc J, Robert C. Acute pancreatitis after vismodegib for basal cell carcinoma: a causal relation? Eur J Cancer. 2019;118:67-69.
- Giorgini C, Barbaccia V, Croci GA, et al. Rapid development of atypical fibroxanthoma during vismodegib treatment. Clin Exp Dermatol. 2019;44:86-88.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Zhu GA, Sundram U, Chang ALS. Two different scenarios of squamous cell carcinoma within advanced basal cell carcinomas: cases illustrating the importance of serial biopsy during vismodegib usage. JAMA Dermatol. 2014;150:970-973.
- Poulalhon N, Dalle S, Balme B, et al. Fast-growing cutaneous squamous cell carcinoma in a patient treated with vismodegib. Dermatology. 2015;230:101-104.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:E33-E34.
- Giuffrida R, Kashofer K, Dika E, et al. Fast growing melanoma following treatment with vismodegib for locally advanced basal cell carcinomas: report of two cases. Eur J Cancer. 2018;91:177-179.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Magdaleno-Tapial J, Valenzuela-Oñate C, Ortiz-Salvador JM, et al. Pilomatricomas secondary to treatment with vismodegib. JAAD Case Rep. 2018;5:12-14.
- Nilsson M, Undèn AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438-3443.
- Vorechovský I, Undén AB, Sandstedt B, et al. Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: support for a gatekeeper mechanism in skin tumorigenesis. Cancer Res. 1997;57:4677-4681.
- Hatta N, Hirano T, Kimura T, et al. Molecular diagnosis of basal cell carcinoma and other basaloid cell neoplasms of the skin by the quantification of Gli1 transcript levels. J Cutan Pathol. 2005;32:131-136.
- Vidal VP, Ortonne N, Schedl A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J Cutan Pathol. 2008;35:373-379.
- Baur V, Papadopoulos T, Kazakov DV, et al. A case of multiple familial trichoepitheliomas responding to treatment with the hedgehog signaling pathway inhibitor vismodegib. Virchows Arch. 2018;473:241-246.
- LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502-2511.
- Fife K, Herd R, Lalondrelle S, et al. Managing adverse events associated with vismodegib in the treatment of basal cell carcinoma. Future Oncol. 2017;13:175-184.
Basal cell carcinomas (BCCs) are considered the most common cutaneous cancers. Approximately 80% of nonmelanoma skin cancers are BCCs.1,2 Surgical management is the gold standard for early-stage and localized BCCs; it may include simple excision vs Mohs micrographic surgery.3,4 However, if left untreated, these lesions can progress to an advanced stage (locally advanced BCC) or infrequently may spread to distant sites (metastatic BCC). In the advanced stage, the lesions are no longer manageable by surgery or radiation therapy.5,6 Recently, inhibitors targeting the hedgehog (Hh) pathway have shown great promise for these patients. The first drug approved by the US Food and Drug Administration (FDA) for locally advanced and metastatic BCC is vismodegib.7 In this article, we provide a clinical review of vismodegib for the management of BCC, including a discussion of the Hh pathway in BCC, adverse effects of vismodegib, use of vismodegib in adnexal skin tumors, recommended doses for vismodegib therapy in BCC, and management of the side effects of treatment.
Hh Pathway in BCC
In embryonic development, the Hh signaling pathway is crucial across a broad spectrum of species, including humans. Various members of the Hh family have been recognized, all working as secreted regulatory proteins.8 The name of the Hh signaling pathway is derived from a polypeptide ligand called hedgehog found in some fruit flies. Mutations in the gene led to fruit fly larvae that had a spiky hairy pattern of denticles similar to hedgehogs, leading to the name of this molecule.9 The transmembrane protein smoothened (SMO) is the main component of the Hh signaling pathway and initiates a signaling cascade that in turn leads to an increased expression of target genes, such as GLI1. Patched (PTCH), also a transmembrane protein and a cell-surface receptor for the secreted Hh ligand, suppresses the signaling capacity of SMO. Upon binding of the Hh ligand to the PTCH receptor, the suppression of SMO is relieved and a signal is propagated, evoking a cellular response.10 Molecular and genetic studies have reported that genetic alterations in the Hh signaling pathway are almost universally present in all BCCs, leading to an aberrant activation of the pathway and an uncontrolled proliferation of the basal cells. Frequently, these alterations have been shown to cause loss of function of PTCH homologue 1, which usually acts to inhibit the SMO homologue signaling activity.11,12
Because of the potential importance of Hh signaling in other solid malignancies and the failure of topical inhibition of SMO,13 subsequent studies on the development of Hh pathway inhibitors have mostly focused on the systemic approach. A multitude of Hh pathway inhibitors have been developed thus far, such as SANT1-SANT4, GDC-0449, IPI-926, BMS-833923 (XL139), HhAntag-691, and MK-4101.14 Many of these inhibitors have been clinically investigated.13,15,16
Systemic SMO Inhibitor: Vismodegib
Vismodegib was the earliest systemic SMO inhibitor to fulfill early clinical evaluation15,16 and the first drug to receive FDA approval for the management of advanced or metastatic BCC. Vismodegib is a small-molecule SMO inhibitor used for the management of selected locally advanced BCC and metastatic BCC in adults.3,17 Although there is a possibility of recurrence following drug withdrawal, vismodegib constitutes a new therapeutic strategy presenting positive benefits to patients. It may provide superior improvement over sunitinib, which has shown efficacy in a few patients; however, the efficacy and tolerance of sunitinib have been shown to be limited.18,19
Adverse Effects of Vismodegib Therapy
Adverse events with vismodegib use have been reported in 98% of patients (N=491); most of these were mild to moderate.20 However, the frequency of adverse events could prove to be a therapeutic challenge for patients requiring extended treatment. The most frequently reported reversible side effects were muscle spasms (64%), alopecia (62%), weight loss (33%), fatigue (28%), decreased appetite (25%), diarrhea (17%), nausea (16%), dysgeusia (54%), and ageusia (22%).20 In clinical trials, amenorrhea was noticed in 30% (3/10) of females with reproductive potential.2 Apart from alopecia and possibly amenorrhea, these side effects are reversible.17 Alkeraye et al17 reported 3 clinical cases of persistent alopecia following the use of vismodegib. Amenorrhea is a possible side effect of unknown reversibility.7
Vismodegib is a pregnancy category D medication.4 Severe birth defects, including craniofacial abnormalities, retardations in normal growth, open perineum, and absence of digital fusion at a corresponding 20% of the recommended daily dose, were found in rat studies. Embryo-fetal death was noted when rats were exposedto concentrations comparable to the recommended human dose.4
Hepatic events with the use of vismodegib have been reported. The use of vismodegib in randomized controlled trials resulted in elevation of both alanine aminotransferase and aspartate aminotransferase levels compared with placebo.21 Moreover, severe hepatotoxicity with vismodegib has been reported.22-24 A study conducted by Edwards et al25 concluded that the use of vismodegib in patients with severe liver disease must include thorough risk-benefit assessment, with caution in using other concomitant hepatotoxic medications.
Rare adverse events also have been reported in the literature, including vismodegib-induced pancreatitis in a 79-year-old patient treated for locally advanced, recurrent BCC that was cleared following cessation of therapy.26 Additionally, atypical fibroxanthoma was observed in an 83-year-old patient after 30 days of treatment with vismodegib for multiple BCCs.27 The development of other secondary malignancies, such as squamous cell carcinoma, melanoma, keratoacanthomas, and pilomatricomas, during or after the long-term use of vismodegib also have been described.28-35
Use of Vismodegib for Adnexal Skin Tumors
The role of the sonic Hh–PTCH pathway in the pathogenesis of adnexal tumors varies in the literature. Some studies propose the involvement of this pathway in the formation of adnexal tumors such as trichoblastoma, trichoepithelioma, and cylindroma, as in BCC. Various lines of evidence support this involvement. Firstly, in mice, the spontaneous generation of numerous BCCs, trichoblastomas, trichoepitheliomas, and cylindromas has been observed following constitutive activation of the sonic Hh–PTCH pathway.36 Secondly, in trichoepitheliomas, there have been positive results in molecular research into the tumor suppressor gene PTCH homologue 1, PTCH1, whose mutations cause constitutive activation of the sonic Hh–PTCH pathway.37 Thirdly, GLI138 and SOX939 transcription factors associated with the signaling pathway of sonic Hh–PTCH appear to have increased levels in adnexal carcinomas.19 Lepesant et al19 reported a notable clinical response to vismodegib in trichoblastic carcinoma. Baur et al40 reported successful treatment of multiple familial trichoepitheliomas with vismodegib. Nonetheless, more studies are required to assess the efficacy and reliability of vismodegib in the management of adnexal tumors.
Recommended Dose of Vismodegib Therapy
The vismodegib dosage that is approved by the FDA is 150 mg/d until disease progression or the development of intolerable side effects.4 Higher dosing regimens were evaluated with 270 mg/d and 540 mg/d. No added therapeutic benefit was noted with the increase in the dose, and no dose-limiting toxic effects were observed.41
Management of Vismodegib Side Effects
Managing patient expectations is a crucial step in improving dysgeusia. The experience of dysgeusia varies among patients; thus, patients should be instructed to adjust their diets according to their level of dysgeusia, which can be achieved by changing ingredients or dressings used with their diet. This step has been proven to be effective in overcoming vismodegib-related dysgeusia. Also, fluid taste distortion may lead to dehydration and an increase in creatine level. Thus, patients should be encouraged to monitor fluid intake. Moreover, a treatment hiatus of 2 to 8 months results in near-complete improvement of taste distortion.
For muscle spasms, quinine, treatment break for 1 month, gentle exercise of affected areas, or muscle relaxants such as baclofen and temazepam all are effective methods. For vismodegib-related alopecia, managing patient expectations is key; patients should be aware that hair may take 6 to 12 months or even longer to regrow. In addition, shaving less frequently helps improve alopecia.
For gastrointestinal disorders, loperamide with or without codeine phosphate is effective in resolving diarrhea, and metoclopramide is mostly adequate in treating nausea. Another adverse event is weight loss; weight loss of 5% or more of total body weight prompts dietetic referral. If weight loss persists, a treatment break might be needed to regain weight.
Overall, treatment breaks are sufficient to resolve adverse events caused by vismodegib and do not compromise efficacy of treatment. The duration of a treatment break should be considered before initiation. In one clinical trial, a longer treatment break was associated with fewer adverse effects without affecting the efficacy of treatment.42
Conclusion
Vismodegib provides an effective alternative to surgical intervention in the management of BCC. However, patients must be monitored vigilantly, as adverse events are common (>90%).
Basal cell carcinomas (BCCs) are considered the most common cutaneous cancers. Approximately 80% of nonmelanoma skin cancers are BCCs.1,2 Surgical management is the gold standard for early-stage and localized BCCs; it may include simple excision vs Mohs micrographic surgery.3,4 However, if left untreated, these lesions can progress to an advanced stage (locally advanced BCC) or infrequently may spread to distant sites (metastatic BCC). In the advanced stage, the lesions are no longer manageable by surgery or radiation therapy.5,6 Recently, inhibitors targeting the hedgehog (Hh) pathway have shown great promise for these patients. The first drug approved by the US Food and Drug Administration (FDA) for locally advanced and metastatic BCC is vismodegib.7 In this article, we provide a clinical review of vismodegib for the management of BCC, including a discussion of the Hh pathway in BCC, adverse effects of vismodegib, use of vismodegib in adnexal skin tumors, recommended doses for vismodegib therapy in BCC, and management of the side effects of treatment.
Hh Pathway in BCC
In embryonic development, the Hh signaling pathway is crucial across a broad spectrum of species, including humans. Various members of the Hh family have been recognized, all working as secreted regulatory proteins.8 The name of the Hh signaling pathway is derived from a polypeptide ligand called hedgehog found in some fruit flies. Mutations in the gene led to fruit fly larvae that had a spiky hairy pattern of denticles similar to hedgehogs, leading to the name of this molecule.9 The transmembrane protein smoothened (SMO) is the main component of the Hh signaling pathway and initiates a signaling cascade that in turn leads to an increased expression of target genes, such as GLI1. Patched (PTCH), also a transmembrane protein and a cell-surface receptor for the secreted Hh ligand, suppresses the signaling capacity of SMO. Upon binding of the Hh ligand to the PTCH receptor, the suppression of SMO is relieved and a signal is propagated, evoking a cellular response.10 Molecular and genetic studies have reported that genetic alterations in the Hh signaling pathway are almost universally present in all BCCs, leading to an aberrant activation of the pathway and an uncontrolled proliferation of the basal cells. Frequently, these alterations have been shown to cause loss of function of PTCH homologue 1, which usually acts to inhibit the SMO homologue signaling activity.11,12
Because of the potential importance of Hh signaling in other solid malignancies and the failure of topical inhibition of SMO,13 subsequent studies on the development of Hh pathway inhibitors have mostly focused on the systemic approach. A multitude of Hh pathway inhibitors have been developed thus far, such as SANT1-SANT4, GDC-0449, IPI-926, BMS-833923 (XL139), HhAntag-691, and MK-4101.14 Many of these inhibitors have been clinically investigated.13,15,16
Systemic SMO Inhibitor: Vismodegib
Vismodegib was the earliest systemic SMO inhibitor to fulfill early clinical evaluation15,16 and the first drug to receive FDA approval for the management of advanced or metastatic BCC. Vismodegib is a small-molecule SMO inhibitor used for the management of selected locally advanced BCC and metastatic BCC in adults.3,17 Although there is a possibility of recurrence following drug withdrawal, vismodegib constitutes a new therapeutic strategy presenting positive benefits to patients. It may provide superior improvement over sunitinib, which has shown efficacy in a few patients; however, the efficacy and tolerance of sunitinib have been shown to be limited.18,19
Adverse Effects of Vismodegib Therapy
Adverse events with vismodegib use have been reported in 98% of patients (N=491); most of these were mild to moderate.20 However, the frequency of adverse events could prove to be a therapeutic challenge for patients requiring extended treatment. The most frequently reported reversible side effects were muscle spasms (64%), alopecia (62%), weight loss (33%), fatigue (28%), decreased appetite (25%), diarrhea (17%), nausea (16%), dysgeusia (54%), and ageusia (22%).20 In clinical trials, amenorrhea was noticed in 30% (3/10) of females with reproductive potential.2 Apart from alopecia and possibly amenorrhea, these side effects are reversible.17 Alkeraye et al17 reported 3 clinical cases of persistent alopecia following the use of vismodegib. Amenorrhea is a possible side effect of unknown reversibility.7
Vismodegib is a pregnancy category D medication.4 Severe birth defects, including craniofacial abnormalities, retardations in normal growth, open perineum, and absence of digital fusion at a corresponding 20% of the recommended daily dose, were found in rat studies. Embryo-fetal death was noted when rats were exposedto concentrations comparable to the recommended human dose.4
Hepatic events with the use of vismodegib have been reported. The use of vismodegib in randomized controlled trials resulted in elevation of both alanine aminotransferase and aspartate aminotransferase levels compared with placebo.21 Moreover, severe hepatotoxicity with vismodegib has been reported.22-24 A study conducted by Edwards et al25 concluded that the use of vismodegib in patients with severe liver disease must include thorough risk-benefit assessment, with caution in using other concomitant hepatotoxic medications.
Rare adverse events also have been reported in the literature, including vismodegib-induced pancreatitis in a 79-year-old patient treated for locally advanced, recurrent BCC that was cleared following cessation of therapy.26 Additionally, atypical fibroxanthoma was observed in an 83-year-old patient after 30 days of treatment with vismodegib for multiple BCCs.27 The development of other secondary malignancies, such as squamous cell carcinoma, melanoma, keratoacanthomas, and pilomatricomas, during or after the long-term use of vismodegib also have been described.28-35
Use of Vismodegib for Adnexal Skin Tumors
The role of the sonic Hh–PTCH pathway in the pathogenesis of adnexal tumors varies in the literature. Some studies propose the involvement of this pathway in the formation of adnexal tumors such as trichoblastoma, trichoepithelioma, and cylindroma, as in BCC. Various lines of evidence support this involvement. Firstly, in mice, the spontaneous generation of numerous BCCs, trichoblastomas, trichoepitheliomas, and cylindromas has been observed following constitutive activation of the sonic Hh–PTCH pathway.36 Secondly, in trichoepitheliomas, there have been positive results in molecular research into the tumor suppressor gene PTCH homologue 1, PTCH1, whose mutations cause constitutive activation of the sonic Hh–PTCH pathway.37 Thirdly, GLI138 and SOX939 transcription factors associated with the signaling pathway of sonic Hh–PTCH appear to have increased levels in adnexal carcinomas.19 Lepesant et al19 reported a notable clinical response to vismodegib in trichoblastic carcinoma. Baur et al40 reported successful treatment of multiple familial trichoepitheliomas with vismodegib. Nonetheless, more studies are required to assess the efficacy and reliability of vismodegib in the management of adnexal tumors.
Recommended Dose of Vismodegib Therapy
The vismodegib dosage that is approved by the FDA is 150 mg/d until disease progression or the development of intolerable side effects.4 Higher dosing regimens were evaluated with 270 mg/d and 540 mg/d. No added therapeutic benefit was noted with the increase in the dose, and no dose-limiting toxic effects were observed.41
Management of Vismodegib Side Effects
Managing patient expectations is a crucial step in improving dysgeusia. The experience of dysgeusia varies among patients; thus, patients should be instructed to adjust their diets according to their level of dysgeusia, which can be achieved by changing ingredients or dressings used with their diet. This step has been proven to be effective in overcoming vismodegib-related dysgeusia. Also, fluid taste distortion may lead to dehydration and an increase in creatine level. Thus, patients should be encouraged to monitor fluid intake. Moreover, a treatment hiatus of 2 to 8 months results in near-complete improvement of taste distortion.
For muscle spasms, quinine, treatment break for 1 month, gentle exercise of affected areas, or muscle relaxants such as baclofen and temazepam all are effective methods. For vismodegib-related alopecia, managing patient expectations is key; patients should be aware that hair may take 6 to 12 months or even longer to regrow. In addition, shaving less frequently helps improve alopecia.
For gastrointestinal disorders, loperamide with or without codeine phosphate is effective in resolving diarrhea, and metoclopramide is mostly adequate in treating nausea. Another adverse event is weight loss; weight loss of 5% or more of total body weight prompts dietetic referral. If weight loss persists, a treatment break might be needed to regain weight.
Overall, treatment breaks are sufficient to resolve adverse events caused by vismodegib and do not compromise efficacy of treatment. The duration of a treatment break should be considered before initiation. In one clinical trial, a longer treatment break was associated with fewer adverse effects without affecting the efficacy of treatment.42
Conclusion
Vismodegib provides an effective alternative to surgical intervention in the management of BCC. However, patients must be monitored vigilantly, as adverse events are common (>90%).
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Cirrone F, Harris CS. Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin Ther. 2012;34:2039-2050.
- Ruiz-Salas V, Alegre M, López-Ferrer A, et al. Vismodegib: a review [article in English, Spanish]. Actas Dermosifiliogr. 2014;105:744-751.
- Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353:2262-2269.
- Cusack CA, Nijhawan R, Miller B, et al. Vismodegib for locally advanced basal cell carcinoma in a heart transplant patient. JAMA Dermatol. 2015;151:70-72.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998;110:885-888.
- Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46:3-12.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103-115.
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423-434.
- Sekulic A, Mangold AR, Northfelt DW, et al. Advanced basal cell carcinoma of the skin: targeting the hedgehog pathway. Curr Opin Oncol. 2013;25:218-223.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nature Rev Genet. 2006;7:841-850.
- Alkeraye S, Maire C, Desmedt E, et al. Persistent alopecia induced by vismodegib. Br J Dermatol. 2015;172:1671-1672.
- Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24:199-203.
- Lepesant P, Crinquette M, Alkeraye S, et al. Vismodegib induces significant clinical response in locally advanced trichoblastic carcinoma. Br J Dermatol. 2015;173:1059-1062.
- Basset-Seguin N, Hauschild A, Grob JJ, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-plannedinterim analysis of an international, open-label trial. Lancet Oncol. 2015;16:729-736.
- Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284-4292.
- Sanchez BE, Hajjafar L. Severe hepatotoxicity in a patient treated with hedgehog inhibitor: first case report. Gastroenterology. 2011;140:S974-S975.
- Ly P, Wolf K, Wilson J. A case of hepatotoxicity associated with vismodegib. JAAD Case Rep. 2018;5:57-59.
- Eiger-Moscovich M, Reich E, Tauber G, et al. Efficacy of vismodegib for the treatment of orbital and advanced periocular basal cell carcinoma. Am J Ophthalmol. 2019;207:62-70.
- Edwards BJ, Raisch DW, Saraykar SS, et al. Hepatotoxicity with vismodegib: an MD Anderson Cancer Center and Research on Adverse Drug Events and Reports Project. Drugs R D. 2017;17:211-218.
- Velter C, Blanc J, Robert C. Acute pancreatitis after vismodegib for basal cell carcinoma: a causal relation? Eur J Cancer. 2019;118:67-69.
- Giorgini C, Barbaccia V, Croci GA, et al. Rapid development of atypical fibroxanthoma during vismodegib treatment. Clin Exp Dermatol. 2019;44:86-88.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Zhu GA, Sundram U, Chang ALS. Two different scenarios of squamous cell carcinoma within advanced basal cell carcinomas: cases illustrating the importance of serial biopsy during vismodegib usage. JAMA Dermatol. 2014;150:970-973.
- Poulalhon N, Dalle S, Balme B, et al. Fast-growing cutaneous squamous cell carcinoma in a patient treated with vismodegib. Dermatology. 2015;230:101-104.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:E33-E34.
- Giuffrida R, Kashofer K, Dika E, et al. Fast growing melanoma following treatment with vismodegib for locally advanced basal cell carcinomas: report of two cases. Eur J Cancer. 2018;91:177-179.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Magdaleno-Tapial J, Valenzuela-Oñate C, Ortiz-Salvador JM, et al. Pilomatricomas secondary to treatment with vismodegib. JAAD Case Rep. 2018;5:12-14.
- Nilsson M, Undèn AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438-3443.
- Vorechovský I, Undén AB, Sandstedt B, et al. Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: support for a gatekeeper mechanism in skin tumorigenesis. Cancer Res. 1997;57:4677-4681.
- Hatta N, Hirano T, Kimura T, et al. Molecular diagnosis of basal cell carcinoma and other basaloid cell neoplasms of the skin by the quantification of Gli1 transcript levels. J Cutan Pathol. 2005;32:131-136.
- Vidal VP, Ortonne N, Schedl A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J Cutan Pathol. 2008;35:373-379.
- Baur V, Papadopoulos T, Kazakov DV, et al. A case of multiple familial trichoepitheliomas responding to treatment with the hedgehog signaling pathway inhibitor vismodegib. Virchows Arch. 2018;473:241-246.
- LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502-2511.
- Fife K, Herd R, Lalondrelle S, et al. Managing adverse events associated with vismodegib in the treatment of basal cell carcinoma. Future Oncol. 2017;13:175-184.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283-287.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Cirrone F, Harris CS. Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin Ther. 2012;34:2039-2050.
- Ruiz-Salas V, Alegre M, López-Ferrer A, et al. Vismodegib: a review [article in English, Spanish]. Actas Dermosifiliogr. 2014;105:744-751.
- Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353:2262-2269.
- Cusack CA, Nijhawan R, Miller B, et al. Vismodegib for locally advanced basal cell carcinoma in a heart transplant patient. JAMA Dermatol. 2015;151:70-72.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998;110:885-888.
- Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46:3-12.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103-115.
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423-434.
- Sekulic A, Mangold AR, Northfelt DW, et al. Advanced basal cell carcinoma of the skin: targeting the hedgehog pathway. Curr Opin Oncol. 2013;25:218-223.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nature Rev Genet. 2006;7:841-850.
- Alkeraye S, Maire C, Desmedt E, et al. Persistent alopecia induced by vismodegib. Br J Dermatol. 2015;172:1671-1672.
- Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24:199-203.
- Lepesant P, Crinquette M, Alkeraye S, et al. Vismodegib induces significant clinical response in locally advanced trichoblastic carcinoma. Br J Dermatol. 2015;173:1059-1062.
- Basset-Seguin N, Hauschild A, Grob JJ, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-plannedinterim analysis of an international, open-label trial. Lancet Oncol. 2015;16:729-736.
- Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284-4292.
- Sanchez BE, Hajjafar L. Severe hepatotoxicity in a patient treated with hedgehog inhibitor: first case report. Gastroenterology. 2011;140:S974-S975.
- Ly P, Wolf K, Wilson J. A case of hepatotoxicity associated with vismodegib. JAAD Case Rep. 2018;5:57-59.
- Eiger-Moscovich M, Reich E, Tauber G, et al. Efficacy of vismodegib for the treatment of orbital and advanced periocular basal cell carcinoma. Am J Ophthalmol. 2019;207:62-70.
- Edwards BJ, Raisch DW, Saraykar SS, et al. Hepatotoxicity with vismodegib: an MD Anderson Cancer Center and Research on Adverse Drug Events and Reports Project. Drugs R D. 2017;17:211-218.
- Velter C, Blanc J, Robert C. Acute pancreatitis after vismodegib for basal cell carcinoma: a causal relation? Eur J Cancer. 2019;118:67-69.
- Giorgini C, Barbaccia V, Croci GA, et al. Rapid development of atypical fibroxanthoma during vismodegib treatment. Clin Exp Dermatol. 2019;44:86-88.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Zhu GA, Sundram U, Chang ALS. Two different scenarios of squamous cell carcinoma within advanced basal cell carcinomas: cases illustrating the importance of serial biopsy during vismodegib usage. JAMA Dermatol. 2014;150:970-973.
- Poulalhon N, Dalle S, Balme B, et al. Fast-growing cutaneous squamous cell carcinoma in a patient treated with vismodegib. Dermatology. 2015;230:101-104.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:E33-E34.
- Giuffrida R, Kashofer K, Dika E, et al. Fast growing melanoma following treatment with vismodegib for locally advanced basal cell carcinomas: report of two cases. Eur J Cancer. 2018;91:177-179.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Magdaleno-Tapial J, Valenzuela-Oñate C, Ortiz-Salvador JM, et al. Pilomatricomas secondary to treatment with vismodegib. JAAD Case Rep. 2018;5:12-14.
- Nilsson M, Undèn AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438-3443.
- Vorechovský I, Undén AB, Sandstedt B, et al. Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: support for a gatekeeper mechanism in skin tumorigenesis. Cancer Res. 1997;57:4677-4681.
- Hatta N, Hirano T, Kimura T, et al. Molecular diagnosis of basal cell carcinoma and other basaloid cell neoplasms of the skin by the quantification of Gli1 transcript levels. J Cutan Pathol. 2005;32:131-136.
- Vidal VP, Ortonne N, Schedl A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J Cutan Pathol. 2008;35:373-379.
- Baur V, Papadopoulos T, Kazakov DV, et al. A case of multiple familial trichoepitheliomas responding to treatment with the hedgehog signaling pathway inhibitor vismodegib. Virchows Arch. 2018;473:241-246.
- LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502-2511.
- Fife K, Herd R, Lalondrelle S, et al. Managing adverse events associated with vismodegib in the treatment of basal cell carcinoma. Future Oncol. 2017;13:175-184.
Practice Points
- The recommended dosage of vismodegib is 150 mg/d until unendurable side effects develop or disease progression occurs.
- The efficacy of vismodegib in the management of locally advanced basal cell carcinoma (BCC) and metastatic BCC is promising. Thus, it is now considered an effective substitute to surgical therapy.
- Patients using vismodegib must be closely monitored, as it is commonly associated with adverse events.
Utilization and Clinical Benefit of Immune Checkpoint Inhibitor in Veterans With Microsatellite Instability-High Prostate Cancer
Background
The utilization of immune checkpoint inhibitors (ICI) in prostate cancer (PC) can be very effective for patients with mismatch repair-deficiency (as identified by MSI-H by PCR/NGS or dMMR IHC). The use of ICI in this patient population has been associated with high rates of durable response. There is limited published data on factors that may influence patient response and outcomes. The aim of this study is to describe the utilization of and tumor response to ICI in this patient population.
Methods
This is a retrospective study of men with MSI-H PC reported by somatic genomic testing from April 1, 2015 to March 31, 2022 through the VA National Precision Oncology Program (NPOP), who received at least one dose of ICI. The primary objectives are to describe the incidence of MSI-H PC and the utilization of ICI. Descriptive statistics and Kaplan- Meier estimator were used for secondary objectives to determine the prostate-specific antigen decline of at least 50% (PSA50), clinical progression free survival (cPFS), time on ICI as a function of number of prior therapies, the extent of metastasis prior to initiation of ICI, and the correlation of MMR genetic alterations with treatment response.
Results
66 patients with MSI-H PC were identified (1.5% of a total of 4267 patients with PC tested through NPOP). 23 patients (35%) received at least one dose of ICI. 12 of 23 patients (52%) had PSA response. PSA50 responses occurred in 6 patients (50%) and 5 continued to have durable PSA50 at six months. Median cPFS was 280 days (95% CI: 105 days-not reached) and the estimated PFS at six months was 72.2% (95% CI: 35.7%-90.2%). 8 of 12 (67%) responders have received multiple lines of therapy for M1 PC. 8 of 12 patients (67%) had high-volume disease at ICI initiation. Of those patients with a MMR genetic alteration, patients with MLH1 (3/3) and MSH2 (6/8) alterations responded more frequently than those with MSH6 alterations (1/4).
Conclusions
MSI-H PC is rare but response rates to ICI are high and durable. Patients with MLH1 and MSH2 alterations appeared to respond more frequently than those with MSH6. Additional follow-up is ongoing.
Background
The utilization of immune checkpoint inhibitors (ICI) in prostate cancer (PC) can be very effective for patients with mismatch repair-deficiency (as identified by MSI-H by PCR/NGS or dMMR IHC). The use of ICI in this patient population has been associated with high rates of durable response. There is limited published data on factors that may influence patient response and outcomes. The aim of this study is to describe the utilization of and tumor response to ICI in this patient population.
Methods
This is a retrospective study of men with MSI-H PC reported by somatic genomic testing from April 1, 2015 to March 31, 2022 through the VA National Precision Oncology Program (NPOP), who received at least one dose of ICI. The primary objectives are to describe the incidence of MSI-H PC and the utilization of ICI. Descriptive statistics and Kaplan- Meier estimator were used for secondary objectives to determine the prostate-specific antigen decline of at least 50% (PSA50), clinical progression free survival (cPFS), time on ICI as a function of number of prior therapies, the extent of metastasis prior to initiation of ICI, and the correlation of MMR genetic alterations with treatment response.
Results
66 patients with MSI-H PC were identified (1.5% of a total of 4267 patients with PC tested through NPOP). 23 patients (35%) received at least one dose of ICI. 12 of 23 patients (52%) had PSA response. PSA50 responses occurred in 6 patients (50%) and 5 continued to have durable PSA50 at six months. Median cPFS was 280 days (95% CI: 105 days-not reached) and the estimated PFS at six months was 72.2% (95% CI: 35.7%-90.2%). 8 of 12 (67%) responders have received multiple lines of therapy for M1 PC. 8 of 12 patients (67%) had high-volume disease at ICI initiation. Of those patients with a MMR genetic alteration, patients with MLH1 (3/3) and MSH2 (6/8) alterations responded more frequently than those with MSH6 alterations (1/4).
Conclusions
MSI-H PC is rare but response rates to ICI are high and durable. Patients with MLH1 and MSH2 alterations appeared to respond more frequently than those with MSH6. Additional follow-up is ongoing.
Background
The utilization of immune checkpoint inhibitors (ICI) in prostate cancer (PC) can be very effective for patients with mismatch repair-deficiency (as identified by MSI-H by PCR/NGS or dMMR IHC). The use of ICI in this patient population has been associated with high rates of durable response. There is limited published data on factors that may influence patient response and outcomes. The aim of this study is to describe the utilization of and tumor response to ICI in this patient population.
Methods
This is a retrospective study of men with MSI-H PC reported by somatic genomic testing from April 1, 2015 to March 31, 2022 through the VA National Precision Oncology Program (NPOP), who received at least one dose of ICI. The primary objectives are to describe the incidence of MSI-H PC and the utilization of ICI. Descriptive statistics and Kaplan- Meier estimator were used for secondary objectives to determine the prostate-specific antigen decline of at least 50% (PSA50), clinical progression free survival (cPFS), time on ICI as a function of number of prior therapies, the extent of metastasis prior to initiation of ICI, and the correlation of MMR genetic alterations with treatment response.
Results
66 patients with MSI-H PC were identified (1.5% of a total of 4267 patients with PC tested through NPOP). 23 patients (35%) received at least one dose of ICI. 12 of 23 patients (52%) had PSA response. PSA50 responses occurred in 6 patients (50%) and 5 continued to have durable PSA50 at six months. Median cPFS was 280 days (95% CI: 105 days-not reached) and the estimated PFS at six months was 72.2% (95% CI: 35.7%-90.2%). 8 of 12 (67%) responders have received multiple lines of therapy for M1 PC. 8 of 12 patients (67%) had high-volume disease at ICI initiation. Of those patients with a MMR genetic alteration, patients with MLH1 (3/3) and MSH2 (6/8) alterations responded more frequently than those with MSH6 alterations (1/4).
Conclusions
MSI-H PC is rare but response rates to ICI are high and durable. Patients with MLH1 and MSH2 alterations appeared to respond more frequently than those with MSH6. Additional follow-up is ongoing.