User login
Bipolar disorder or borderline personality disorder?
Although evidence suggests that bipolar disorder (BD) and borderline personality disorder (BPD) are distinct entities, their differential diagnosis is often challenging as a result of considerable overlap of phenotypical features. Moreover, BD and BPD frequently co-occur, which makes it even more difficult to differentiate these 2 conditions. Strategies for improving diagnostic accuracy are critical to optimizing patients’ clinical outcomes and long-term prognosis. Misdiagnosing these 2 conditions can be particularly deleterious, and failure to recognize their co-occurrence can result in additional burden to typically complex and severe clinical presentations.
This article describes key aspects of the differential diagnosis between BD and BPD, emphasizing core features and major dissimilarities between these 2 conditions, and discusses the implications of misdiagnosis. The goal is to highlight the clinical and psychopathological aspects of BD and BPD to help clinicians properly distinguish these 2 disorders.
Psychopathological and sociodemographic correlates
Bipolar disorder is a chronic and severe mental illness that is classified based on clusters of symptoms—manic, hypomanic, and depressive.1 It is among the 10 leading causes of disability worldwide, with significant morbidity arising from acute affective episodes and subacute states.2 Data suggest the lifetime prevalence of BPD is 2.1%, and subthreshold forms may affect an additional 2.4% of the US population.3 The onset of symptoms typically occurs during late adolescence or early adulthood, and mood lability and cyclothymic temperament are the most common prodromal features.4
In contrast, personality disorders, such as BPD, are characteristically pervasive and maladaptive patterns of emotional responses that usually deviate from an individual’s stage of development and cultural background.1 These disorders tend to cause significant impairment, particularly in personal, occupational, and social domains. Environmental factors, such as early childhood trauma, seem to play an important role in the genesis of personality disorders, which may be particularly relevant in BPD, a disorder characterized by marked impulsivity and a pattern of instability in personal relationships, self-image, and affect.1,5,6 Similarly to BD, BPD is also chronic and highly disabling.
According to the National Survey on Alcohol and Related Conditions (NESARC), approximately 15% of US adults were found to have at least one type of personality disorder, and 6% met criteria for a cluster B personality disorder (antisocial, borderline, narcissistic, and histrionic).7 The lifetime prevalence of BPD is nearly 2%, with higher estimates observed in psychiatric settings.7,8
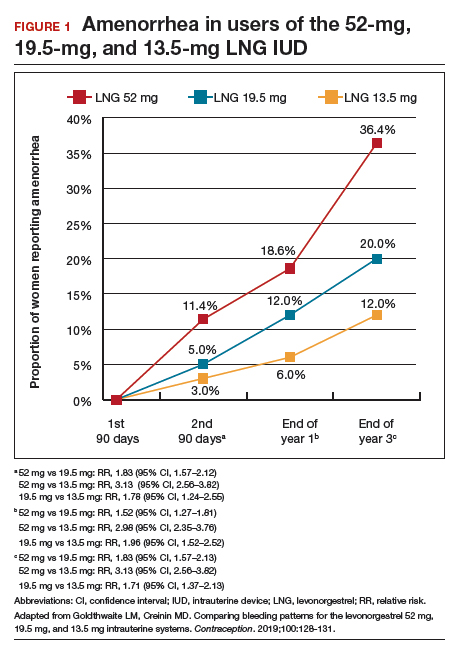
As a result of the phenotypical resemblance between BD and BPD (Figure), the differential diagnosis is often difficult. Recent studies suggest that co-occurrence of BD and BPD is common, with rates of comorbid BPD as high as 29% in BD I and 24% in BD II.8,9 On the other hand, nearly 20% of individuals with BPD seem to have comorbid BD.8,9 Several studies suggest that comorbid personality disorders represent a negative prognostic factor in the course of mood disorders, and the presence of BPD in patients with BD seems to be associated with more severe clinical presentations, greater treatment complexity, a higher number of depressive episodes, poor inter-episode functioning, and higher rates of other comorbidities, such as substance use disorders (SUDs).8-11 The effect of BD on the course of BPD is unclear and fairly unexplored, although it has been suggested that better control of mood symptoms may lead to more stable psychosocial functioning in BPD.9

Whether BD and BPD are part of the same spectrum is a matter for debate.12-14 Multidimensional approaches have been proposed to better characterize these disorders in at-risk populations, based on structured interviews, self-administered and clinician-rated clinical scales (Table 1), neuroimaging studies, biological markers, and machine-learning models.15,16 Compelling evidence suggests that BD and BPD have distinct underlying neurobiological and psychopathological mechanisms12,13; however, the differential diagnosis still relies on phenotypical features, since the search for biological markers has not yet identified specific biomarkers that can be used in clinical practice.

Continue to: Core features of BPD...
Core features of BPD, such as mood lability, impulsivity, and risk-taking behaviors, are also part of the diagnostic criteria for BD (Table 2).1 Similarly, depressive symptoms prevail in the course of BD.17,18 This adds complexity to the differential because “depressivity” is also part of the diagnostic criteria for BPD.1 Therefore, comprehensive psychiatric assessments and longitudinal observations are critical to diagnostic accuracy and treatment planning. Further characterization of symptoms, such as onset patterns, clinical course, phenomenology of symptoms (eg, timing, frequency, duration, triggers), and personality traits, will provide information to properly distinguish these 2 syndromes when, for example, it is unclear if the “mood swings” and impulsivity are part of a mood or a personality disorder (Table 3).

Clinical features: A closer look
Borderline personality disorder. Affect dysregulation, emotional instability, impoverished and unstable self-image, and chronic feelings of emptiness are core features of BPD.1,5,19 These characteristics, when combined with a fear of abandonment or rejection, a compromised ability to recognize the feelings and needs of others, and extremes of idealization-devaluation, tend to culminate in problematic and chaotic relationships.6,19 Individuals with BPD may become suspicious or paranoid under stressful situations. Under these circumstances, individuals with BPD may also experience depersonalization and other dissociative symptoms.6,20 The mood lability and emotional instability observed in patients with BPD usually are in response to environmental factors, and although generally intense and out of proportion, they tend to be ephemeral and short-lived, typically lasting a few hours.1,5 The anxiety and depressive symptoms reported by patients with BPD frequently are associated with feelings of “falling apart” or “losing control,” pessimism, shame, and low self-esteem. Coping strategies tend to be poorly developed and/or maladaptive, and individuals with BPD usually display a hostile and antagonistic demeanor and engage in suicidal or nonsuicidal self-injury (NSSI) behaviors as means to alleviate overwhelming emotional distress. Impulsivity, disinhibition, poor tolerance to frustration, and risk-taking behaviors are also characteristic of BPD.1,5 As a result, BPD is usually associated with significant impairment in functioning, multiple hospitalizations, and high rates of comorbid mood disorders, posttraumatic stress disorder (PTSD), SUDs, and death by suicide.

Bipolar disorder. Conversely, the fluctuations in mood and affect observed in patients with BD are usually episodic rather than pervasive, and tend to last longer (typically days to weeks) compared with the transient mood shifts observed in patients with BPD.4,17,18 The impulsivity, psychomotor agitation, and increased goal-directed activity reported by patients with BD are usually seen in the context of an acute affective episode, and are far less common during periods of stability or euthymic affect.4,17,18 Grandiosity and inflated self-esteem—hallmarks of a manic or hypomanic state—seem to oppose the unstable self-image observed in BPD, although indecisiveness and low self-worth may be observed in individuals with BD during depressive episodes. Antidepressant-induced mania or hypomania, atypical depressive episodes, and disruptions in sleep and circadian rhythms may be predictors of BD.4,21 Furthermore, although psychosocial stressors may be associated with acute affective episodes in early stages of bipolar illness, over time minimal stressors are necessary to ignite new affective episodes.22,23 Although BD is associated with high rates of suicide, suicide attempts are usually seen in the context of an acute depressive episode, and NSSI behaviors are less common among patients with BD.24
Lastly, other biographical data, such as a history of early life trauma, comorbidity, and a family history of psychiatric illnesses, can be particularly helpful in establishing the differential diagnosis between BD and BPD.25 For instance, evidence suggests that the heritability of BD may be as high as 70%, which usually translates into an extensive family history of bipolar and related disorders.26 In addition, studies suggest a high co-occurrence of anxiety disorders, attention-deficit/hyperactivity disorder, and SUDs in patients with BD, whereas PTSD, SUDs, and eating disorders tend to be highly comorbid with BPD.27 Childhood adversity (ie, a history of physical, sexual, or emotional abuse, or neglect) seems to be pivotal in the genesis of BPD and may predispose these individuals to psychotic and dissociative symptoms, particularly those with a history of sexual abuse, while playing a more secondary role in BD.28-31
Implications of misdiagnosis
In the view of the limitations of the existing models, multidimensional approaches are necessary to improve diagnostic accuracy. Presently, the differential diagnosis of BD and BPD continues to rely on clinical findings and syndromic classifications. Misdiagnosing BD and BPD has adverse therapeutic and prognostic implications.32 For instance, while psychotropic medications and neuromodulatory therapies (eg, electroconvulsive therapy, repetitive transcranial magnetic stimulation) are considered first-line treatments for patients with BD, psychosocial interventions tend to be adjunctive treatments in BD.33 Conversely, although pharmacotherapy might be helpful for patients with BPD, psychosocial and behavioral interventions are the mainstay treatment for this disorder, with the strongest evidence supporting cognitive-behavioral therapy, dialectical behavioral therapy, mentalization-based therapy, and transference-focused therapy.34-36 Thus, misdiagnosing BD as BPD with comorbid depression may result in the use of antidepressants, which can be detrimental in BD. Antidepressant treatment of BD, particularly as monotherapy, has been associated with manic or hypomanic switch, mixed states, and frequent cycling.21 Moreover, delays in diagnosis and proper treatment of BD may result in protracted mood symptoms, prolonged affective episodes, higher rates of disability, functional impairment, and overall worse clinical outcomes.24 In addition, because behavioral and psychosocial interventions are usually adjunctive therapies rather than first-line interventions for patients with BD, misdiagnosing BPD as BD may ultimately prevent these individuals from receiving proper treatment, likely resulting in more severe functional impairment, multiple hospitalizations, self-inflicted injuries, and suicide attempts, since psychotropic medications are not particularly effective for improving self-efficacy and coping strategies, nor for correcting cognitive distortions, particularly in self-image, and pathological personality traits, all of which are critical aspects of BPD treatment.
Continue to: Several factors might...
Several factors might make clinicians reluctant to diagnose BPD, or bias them to diagnose BD more frequently. These include a lack of familiarity with the diagnostic criteria for BPD, the phenotypical resemblance between BP and BPD, or even concerns about the stigma and negative implications that are associated with a BPD diagnosis.32,37,38
Whereas BD is currently perceived as a condition with a strong biological basis, there are considerable misconceptions regarding BPD and its nature.4-6,22,26 As a consequence, individuals with BPD tend to be perceived as “difficult-to-treat,” “uncooperative,” or “attention-seeking.” These misconceptions may result in poor clinician-patient relationships, unmet clinical and psychiatric needs, and frustration for both clinicians and patients.37
Through advances in biological psychiatry, precision medicine may someday be a part of psychiatric practice. Biological “signatures” may eventually help clinicians in diagnosing and treating psychiatric disorders. Presently, however, rigorous history-taking and comprehensive clinical assessments are still the most powerful tools a clinician can use to accomplish these goals. Finally, destigmatizing psychiatric disorders and educating patients and clinicians are also critical to improving clinical outcomes and promoting mental health in a compassionate and empathetic fashion.
Bottom Line
Despite the phenotypical resemblance between bipolar disorder (BP) and borderline personality disorder (BPD), the 2 are independent conditions with distinct neurobiological and psychopathological underpinnings. Clinicians can use a rigorous assessment of pathological personality traits and characterization of symptoms, such as onset patterns, clinical course, and phenomenology, to properly distinguish between BP and BPD.
Related Resources
- Fiedorowicz JG, Black DW. Borderline, bipolar, or both? Frame your diagnosis on the patient history. Current Psychiatry. 2010; 9(1):21-24,29-32.
- Zimmerman M. Improving the recognition of borderline personality disorder. Current Psychiatry. 2017;16(10):13-19.
- National Institute of Mental Health. Overview on bipolar disorder. www.nimh.nih.gov/health/publications/bipolar-disorder/index.shtml. Revised October 2018.
- National Institute of Mental Health. Overview on borderline personality disorder. www.nimh.nih.gov/health/publications/borderline-personality-disorder-fact-sheet/index.shtml.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575-1586.
3. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry. 2007;64(5):543-552.
4. Malhi GS, Bargh DM, Coulston CM, et al. Predicting bipolar disorder on the basis of phenomenology: implications for prevention and early intervention. Bipolar Disord. 2014;16(5):455-470.
5. Skodol AE, Gunderson JG, Pfohl B, et al. The borderline diagnosis I: psychopathology. Biol Psychiatry. 2002;51(12):936-950.
6. Skodol AE, Siever LJ, Livesley WJ, et al. The borderline diagnosis II: biology, genetics, and clinical course. Biol Psychiatry. 2002;51(12):951-963.
7. Hasin DS, Grant BF. The National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Waves 1 and 2: review and summary of findings. Soc Psychiatry Psychiatr Epidemiol. 2015;50(11):1609-1640.
8. McDermid J, Sareen J, El-Gabalawy R, et al. Co-morbidity of bipolar disorder and borderline personality disorder: findings from the National Epidemiologic Survey on Alcohol and Related Conditions. Compr Psychiatry. 2015;58:18-28.
9. Gunderson JG, Weinberg I, Daversa MT, et al. Descriptive and longitudinal observations on the relationship of borderline personality disorder and bipolar disorder. Am J Psychiatry. 2006;163(7):1173-1178.
10. Swartz HA, Pilkonis PA, Frank E, et al. Acute treatment outcomes in patients with bipolar I disorder and co-morbid borderline personality disorder receiving medication and psychotherapy. Bipolar Disord. 2005;7(2):192-197.
11. Riemann G, Weisscher N, Post RM, et al. The relationship between self-reported borderline personality features and prospective illness course in bipolar disorder. Int J Bipolar Disord. 2017;5(1):31.
12. de la Rosa I, Oquendo MA, García G, et al. Determining if borderline personality disorder and bipolar disorder are alternative expressions of the same disorder. J Clin Psychiatry. 2017;778(8):e994-e999. doi: 10.4088/JCP.16m11190.
13. di Giacomo E, Aspesi F, Fotiadou M, et al. Unblending borderline personality and bipolar disorders. J Psychiatr Res. 2017;91:90-97.
14. Parker G, Bayes A, McClure G, et al. Clinical status of comorbid bipolar disorder and borderline personality disorder. Br J Psychiatry. 2016;209(3):209-215.
15. Perez Arribas I, Goodwin GM, Geddes JR, et al. A signature-based machine learning model for distinguishing bipolar disorder and borderline personality disorder. Transl Psychiatry. 2018;8(1):274.
16. Insel T, Cuthbert B, Garvey M, et al. Research Domain Criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748-751.
17. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60(3):261-269.
19. Oldham JM, Skodol AE, Bender DS. A current integrative perspective on personality disorders. American Psychiatric Publishing, Inc. 2005.
20. Herzog JI, Schmahl C. Adverse childhood experiences and the consequences on neurobiological, psychosocial, and somatic conditions across the lifespan. Front Psychiatry. 2018;9:420.
21. Barbuti M, Pacchiarotti I, Vieta E, et al. Antidepressant-induced hypomania/mania in patients with major depression: evidence from the BRIDGE-II-MIX study. J Affect Disord. 2017;219:187-192.
22. Post RM. Mechanisms of illness progression in the recurrent affective disorders. Neurotox Res. 2010;18(3-4):256-271.
23. da Costa SC, Passos IC, Lowri C, et al. Refractory bipolar disorder and neuroprogression. Prog Neuro-Psychopharmacology Biol Psychiatry. 2016;70:103-110.
24. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
25. Zimmerman M, Martinez JH, Morgan TA, et al. Distinguishing bipolar II depression from major depressive disorder with comorbid borderline personality disorder: demographic, clinical, and family history differences. J Clin Psychiatry. 2013;74(9):880-886.
26. Hasler G, Drevets WC, Gould TD, et al. Toward constructing an endophenotype strategy for bipolar disorders. Biol Psychiatry. 2006;60(2):93-105.
27. Brieger P, Ehrt U, Marneros A. Frequency of comorbid personality disorders in bipolar and unipolar affective disorders. Compr Psychiatry. 2003;44(1):28-34.
28. Leverich GS, McElroy SL, Suppes T, et al. Early physical and sexual abuse associated with an adverse course of bipolar illness. Biol Psychiatry. 2002;51(4):288-297.
29. Leverich GS, Post RM. Course of bipolar illness after history of childhood trauma. Lancet. 2006;367(9516):1040-1042.
30. Golier JA, Yehuda R, Bierer LM, et al. The relationship of borderline personality disorder to posttraumatic stress disorder and traumatic events. Am J Psychiatry. 2003;160(11):2018-2024.
31. Nicol K, Pope M, Romaniuk L, et al. Childhood trauma, midbrain activation and psychotic symptoms in borderline personality disorder. Transl Psychiatry. 2015;5:e559. doi:10.1038/tp.2015.53.
32. Ruggero CJ, Zimmerman M, Chelminski I, et al. Borderline personality disorder and the misdiagnosis of bipolar disorder. J Psychiatr Res. 2010;44(6):405-408.
33. Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet. 2013;381(9878):1672-1682.
34. McMain S, Korman LM, Dimeff L. Dialectical behavior therapy and the treatment of emotion dysregulation. J Clin Psychol. 2001;57(2):183-196.
35. Cristea IA, Gentili C, Cotet CD, et al. Efficacy of psychotherapies for borderline personality disorder: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(4):319-328.
36. Linehan MM, Korslund KE, Harned MS, et al. Dialectical behavior therapy for high suicide risk in individuals with borderline personality disorder. JAMA Psychiatry. 2015;72(75);475-482.
37. LeQuesne ER, Hersh RG. Disclosure of a diagnosis of borderline personality disorder. J Psychiatr Pract. 2004:10(3):170-176.
38. Young AH. Bipolar disorder: diagnostic conundrums and associated comorbidities. J Clin Psychiatry. 2009;70(8):e26. doi:10.4088/jcp.7067br6c.
Although evidence suggests that bipolar disorder (BD) and borderline personality disorder (BPD) are distinct entities, their differential diagnosis is often challenging as a result of considerable overlap of phenotypical features. Moreover, BD and BPD frequently co-occur, which makes it even more difficult to differentiate these 2 conditions. Strategies for improving diagnostic accuracy are critical to optimizing patients’ clinical outcomes and long-term prognosis. Misdiagnosing these 2 conditions can be particularly deleterious, and failure to recognize their co-occurrence can result in additional burden to typically complex and severe clinical presentations.
This article describes key aspects of the differential diagnosis between BD and BPD, emphasizing core features and major dissimilarities between these 2 conditions, and discusses the implications of misdiagnosis. The goal is to highlight the clinical and psychopathological aspects of BD and BPD to help clinicians properly distinguish these 2 disorders.
Psychopathological and sociodemographic correlates
Bipolar disorder is a chronic and severe mental illness that is classified based on clusters of symptoms—manic, hypomanic, and depressive.1 It is among the 10 leading causes of disability worldwide, with significant morbidity arising from acute affective episodes and subacute states.2 Data suggest the lifetime prevalence of BPD is 2.1%, and subthreshold forms may affect an additional 2.4% of the US population.3 The onset of symptoms typically occurs during late adolescence or early adulthood, and mood lability and cyclothymic temperament are the most common prodromal features.4
In contrast, personality disorders, such as BPD, are characteristically pervasive and maladaptive patterns of emotional responses that usually deviate from an individual’s stage of development and cultural background.1 These disorders tend to cause significant impairment, particularly in personal, occupational, and social domains. Environmental factors, such as early childhood trauma, seem to play an important role in the genesis of personality disorders, which may be particularly relevant in BPD, a disorder characterized by marked impulsivity and a pattern of instability in personal relationships, self-image, and affect.1,5,6 Similarly to BD, BPD is also chronic and highly disabling.
According to the National Survey on Alcohol and Related Conditions (NESARC), approximately 15% of US adults were found to have at least one type of personality disorder, and 6% met criteria for a cluster B personality disorder (antisocial, borderline, narcissistic, and histrionic).7 The lifetime prevalence of BPD is nearly 2%, with higher estimates observed in psychiatric settings.7,8
As a result of the phenotypical resemblance between BD and BPD (Figure), the differential diagnosis is often difficult. Recent studies suggest that co-occurrence of BD and BPD is common, with rates of comorbid BPD as high as 29% in BD I and 24% in BD II.8,9 On the other hand, nearly 20% of individuals with BPD seem to have comorbid BD.8,9 Several studies suggest that comorbid personality disorders represent a negative prognostic factor in the course of mood disorders, and the presence of BPD in patients with BD seems to be associated with more severe clinical presentations, greater treatment complexity, a higher number of depressive episodes, poor inter-episode functioning, and higher rates of other comorbidities, such as substance use disorders (SUDs).8-11 The effect of BD on the course of BPD is unclear and fairly unexplored, although it has been suggested that better control of mood symptoms may lead to more stable psychosocial functioning in BPD.9
Whether BD and BPD are part of the same spectrum is a matter for debate.12-14 Multidimensional approaches have been proposed to better characterize these disorders in at-risk populations, based on structured interviews, self-administered and clinician-rated clinical scales (Table 1), neuroimaging studies, biological markers, and machine-learning models.15,16 Compelling evidence suggests that BD and BPD have distinct underlying neurobiological and psychopathological mechanisms12,13; however, the differential diagnosis still relies on phenotypical features, since the search for biological markers has not yet identified specific biomarkers that can be used in clinical practice.
Continue to: Core features of BPD...
Core features of BPD, such as mood lability, impulsivity, and risk-taking behaviors, are also part of the diagnostic criteria for BD (Table 2).1 Similarly, depressive symptoms prevail in the course of BD.17,18 This adds complexity to the differential because “depressivity” is also part of the diagnostic criteria for BPD.1 Therefore, comprehensive psychiatric assessments and longitudinal observations are critical to diagnostic accuracy and treatment planning. Further characterization of symptoms, such as onset patterns, clinical course, phenomenology of symptoms (eg, timing, frequency, duration, triggers), and personality traits, will provide information to properly distinguish these 2 syndromes when, for example, it is unclear if the “mood swings” and impulsivity are part of a mood or a personality disorder (Table 3).
Clinical features: A closer look
Borderline personality disorder. Affect dysregulation, emotional instability, impoverished and unstable self-image, and chronic feelings of emptiness are core features of BPD.1,5,19 These characteristics, when combined with a fear of abandonment or rejection, a compromised ability to recognize the feelings and needs of others, and extremes of idealization-devaluation, tend to culminate in problematic and chaotic relationships.6,19 Individuals with BPD may become suspicious or paranoid under stressful situations. Under these circumstances, individuals with BPD may also experience depersonalization and other dissociative symptoms.6,20 The mood lability and emotional instability observed in patients with BPD usually are in response to environmental factors, and although generally intense and out of proportion, they tend to be ephemeral and short-lived, typically lasting a few hours.1,5 The anxiety and depressive symptoms reported by patients with BPD frequently are associated with feelings of “falling apart” or “losing control,” pessimism, shame, and low self-esteem. Coping strategies tend to be poorly developed and/or maladaptive, and individuals with BPD usually display a hostile and antagonistic demeanor and engage in suicidal or nonsuicidal self-injury (NSSI) behaviors as means to alleviate overwhelming emotional distress. Impulsivity, disinhibition, poor tolerance to frustration, and risk-taking behaviors are also characteristic of BPD.1,5 As a result, BPD is usually associated with significant impairment in functioning, multiple hospitalizations, and high rates of comorbid mood disorders, posttraumatic stress disorder (PTSD), SUDs, and death by suicide.
Bipolar disorder. Conversely, the fluctuations in mood and affect observed in patients with BD are usually episodic rather than pervasive, and tend to last longer (typically days to weeks) compared with the transient mood shifts observed in patients with BPD.4,17,18 The impulsivity, psychomotor agitation, and increased goal-directed activity reported by patients with BD are usually seen in the context of an acute affective episode, and are far less common during periods of stability or euthymic affect.4,17,18 Grandiosity and inflated self-esteem—hallmarks of a manic or hypomanic state—seem to oppose the unstable self-image observed in BPD, although indecisiveness and low self-worth may be observed in individuals with BD during depressive episodes. Antidepressant-induced mania or hypomania, atypical depressive episodes, and disruptions in sleep and circadian rhythms may be predictors of BD.4,21 Furthermore, although psychosocial stressors may be associated with acute affective episodes in early stages of bipolar illness, over time minimal stressors are necessary to ignite new affective episodes.22,23 Although BD is associated with high rates of suicide, suicide attempts are usually seen in the context of an acute depressive episode, and NSSI behaviors are less common among patients with BD.24
Lastly, other biographical data, such as a history of early life trauma, comorbidity, and a family history of psychiatric illnesses, can be particularly helpful in establishing the differential diagnosis between BD and BPD.25 For instance, evidence suggests that the heritability of BD may be as high as 70%, which usually translates into an extensive family history of bipolar and related disorders.26 In addition, studies suggest a high co-occurrence of anxiety disorders, attention-deficit/hyperactivity disorder, and SUDs in patients with BD, whereas PTSD, SUDs, and eating disorders tend to be highly comorbid with BPD.27 Childhood adversity (ie, a history of physical, sexual, or emotional abuse, or neglect) seems to be pivotal in the genesis of BPD and may predispose these individuals to psychotic and dissociative symptoms, particularly those with a history of sexual abuse, while playing a more secondary role in BD.28-31
Implications of misdiagnosis
In the view of the limitations of the existing models, multidimensional approaches are necessary to improve diagnostic accuracy. Presently, the differential diagnosis of BD and BPD continues to rely on clinical findings and syndromic classifications. Misdiagnosing BD and BPD has adverse therapeutic and prognostic implications.32 For instance, while psychotropic medications and neuromodulatory therapies (eg, electroconvulsive therapy, repetitive transcranial magnetic stimulation) are considered first-line treatments for patients with BD, psychosocial interventions tend to be adjunctive treatments in BD.33 Conversely, although pharmacotherapy might be helpful for patients with BPD, psychosocial and behavioral interventions are the mainstay treatment for this disorder, with the strongest evidence supporting cognitive-behavioral therapy, dialectical behavioral therapy, mentalization-based therapy, and transference-focused therapy.34-36 Thus, misdiagnosing BD as BPD with comorbid depression may result in the use of antidepressants, which can be detrimental in BD. Antidepressant treatment of BD, particularly as monotherapy, has been associated with manic or hypomanic switch, mixed states, and frequent cycling.21 Moreover, delays in diagnosis and proper treatment of BD may result in protracted mood symptoms, prolonged affective episodes, higher rates of disability, functional impairment, and overall worse clinical outcomes.24 In addition, because behavioral and psychosocial interventions are usually adjunctive therapies rather than first-line interventions for patients with BD, misdiagnosing BPD as BD may ultimately prevent these individuals from receiving proper treatment, likely resulting in more severe functional impairment, multiple hospitalizations, self-inflicted injuries, and suicide attempts, since psychotropic medications are not particularly effective for improving self-efficacy and coping strategies, nor for correcting cognitive distortions, particularly in self-image, and pathological personality traits, all of which are critical aspects of BPD treatment.
Continue to: Several factors might...
Several factors might make clinicians reluctant to diagnose BPD, or bias them to diagnose BD more frequently. These include a lack of familiarity with the diagnostic criteria for BPD, the phenotypical resemblance between BP and BPD, or even concerns about the stigma and negative implications that are associated with a BPD diagnosis.32,37,38
Whereas BD is currently perceived as a condition with a strong biological basis, there are considerable misconceptions regarding BPD and its nature.4-6,22,26 As a consequence, individuals with BPD tend to be perceived as “difficult-to-treat,” “uncooperative,” or “attention-seeking.” These misconceptions may result in poor clinician-patient relationships, unmet clinical and psychiatric needs, and frustration for both clinicians and patients.37
Through advances in biological psychiatry, precision medicine may someday be a part of psychiatric practice. Biological “signatures” may eventually help clinicians in diagnosing and treating psychiatric disorders. Presently, however, rigorous history-taking and comprehensive clinical assessments are still the most powerful tools a clinician can use to accomplish these goals. Finally, destigmatizing psychiatric disorders and educating patients and clinicians are also critical to improving clinical outcomes and promoting mental health in a compassionate and empathetic fashion.
Bottom Line
Despite the phenotypical resemblance between bipolar disorder (BP) and borderline personality disorder (BPD), the 2 are independent conditions with distinct neurobiological and psychopathological underpinnings. Clinicians can use a rigorous assessment of pathological personality traits and characterization of symptoms, such as onset patterns, clinical course, and phenomenology, to properly distinguish between BP and BPD.
Related Resources
- Fiedorowicz JG, Black DW. Borderline, bipolar, or both? Frame your diagnosis on the patient history. Current Psychiatry. 2010; 9(1):21-24,29-32.
- Zimmerman M. Improving the recognition of borderline personality disorder. Current Psychiatry. 2017;16(10):13-19.
- National Institute of Mental Health. Overview on bipolar disorder. www.nimh.nih.gov/health/publications/bipolar-disorder/index.shtml. Revised October 2018.
- National Institute of Mental Health. Overview on borderline personality disorder. www.nimh.nih.gov/health/publications/borderline-personality-disorder-fact-sheet/index.shtml.
Although evidence suggests that bipolar disorder (BD) and borderline personality disorder (BPD) are distinct entities, their differential diagnosis is often challenging as a result of considerable overlap of phenotypical features. Moreover, BD and BPD frequently co-occur, which makes it even more difficult to differentiate these 2 conditions. Strategies for improving diagnostic accuracy are critical to optimizing patients’ clinical outcomes and long-term prognosis. Misdiagnosing these 2 conditions can be particularly deleterious, and failure to recognize their co-occurrence can result in additional burden to typically complex and severe clinical presentations.
This article describes key aspects of the differential diagnosis between BD and BPD, emphasizing core features and major dissimilarities between these 2 conditions, and discusses the implications of misdiagnosis. The goal is to highlight the clinical and psychopathological aspects of BD and BPD to help clinicians properly distinguish these 2 disorders.
Psychopathological and sociodemographic correlates
Bipolar disorder is a chronic and severe mental illness that is classified based on clusters of symptoms—manic, hypomanic, and depressive.1 It is among the 10 leading causes of disability worldwide, with significant morbidity arising from acute affective episodes and subacute states.2 Data suggest the lifetime prevalence of BPD is 2.1%, and subthreshold forms may affect an additional 2.4% of the US population.3 The onset of symptoms typically occurs during late adolescence or early adulthood, and mood lability and cyclothymic temperament are the most common prodromal features.4
In contrast, personality disorders, such as BPD, are characteristically pervasive and maladaptive patterns of emotional responses that usually deviate from an individual’s stage of development and cultural background.1 These disorders tend to cause significant impairment, particularly in personal, occupational, and social domains. Environmental factors, such as early childhood trauma, seem to play an important role in the genesis of personality disorders, which may be particularly relevant in BPD, a disorder characterized by marked impulsivity and a pattern of instability in personal relationships, self-image, and affect.1,5,6 Similarly to BD, BPD is also chronic and highly disabling.
According to the National Survey on Alcohol and Related Conditions (NESARC), approximately 15% of US adults were found to have at least one type of personality disorder, and 6% met criteria for a cluster B personality disorder (antisocial, borderline, narcissistic, and histrionic).7 The lifetime prevalence of BPD is nearly 2%, with higher estimates observed in psychiatric settings.7,8
As a result of the phenotypical resemblance between BD and BPD (Figure), the differential diagnosis is often difficult. Recent studies suggest that co-occurrence of BD and BPD is common, with rates of comorbid BPD as high as 29% in BD I and 24% in BD II.8,9 On the other hand, nearly 20% of individuals with BPD seem to have comorbid BD.8,9 Several studies suggest that comorbid personality disorders represent a negative prognostic factor in the course of mood disorders, and the presence of BPD in patients with BD seems to be associated with more severe clinical presentations, greater treatment complexity, a higher number of depressive episodes, poor inter-episode functioning, and higher rates of other comorbidities, such as substance use disorders (SUDs).8-11 The effect of BD on the course of BPD is unclear and fairly unexplored, although it has been suggested that better control of mood symptoms may lead to more stable psychosocial functioning in BPD.9
Whether BD and BPD are part of the same spectrum is a matter for debate.12-14 Multidimensional approaches have been proposed to better characterize these disorders in at-risk populations, based on structured interviews, self-administered and clinician-rated clinical scales (Table 1), neuroimaging studies, biological markers, and machine-learning models.15,16 Compelling evidence suggests that BD and BPD have distinct underlying neurobiological and psychopathological mechanisms12,13; however, the differential diagnosis still relies on phenotypical features, since the search for biological markers has not yet identified specific biomarkers that can be used in clinical practice.
Continue to: Core features of BPD...
Core features of BPD, such as mood lability, impulsivity, and risk-taking behaviors, are also part of the diagnostic criteria for BD (Table 2).1 Similarly, depressive symptoms prevail in the course of BD.17,18 This adds complexity to the differential because “depressivity” is also part of the diagnostic criteria for BPD.1 Therefore, comprehensive psychiatric assessments and longitudinal observations are critical to diagnostic accuracy and treatment planning. Further characterization of symptoms, such as onset patterns, clinical course, phenomenology of symptoms (eg, timing, frequency, duration, triggers), and personality traits, will provide information to properly distinguish these 2 syndromes when, for example, it is unclear if the “mood swings” and impulsivity are part of a mood or a personality disorder (Table 3).
Clinical features: A closer look
Borderline personality disorder. Affect dysregulation, emotional instability, impoverished and unstable self-image, and chronic feelings of emptiness are core features of BPD.1,5,19 These characteristics, when combined with a fear of abandonment or rejection, a compromised ability to recognize the feelings and needs of others, and extremes of idealization-devaluation, tend to culminate in problematic and chaotic relationships.6,19 Individuals with BPD may become suspicious or paranoid under stressful situations. Under these circumstances, individuals with BPD may also experience depersonalization and other dissociative symptoms.6,20 The mood lability and emotional instability observed in patients with BPD usually are in response to environmental factors, and although generally intense and out of proportion, they tend to be ephemeral and short-lived, typically lasting a few hours.1,5 The anxiety and depressive symptoms reported by patients with BPD frequently are associated with feelings of “falling apart” or “losing control,” pessimism, shame, and low self-esteem. Coping strategies tend to be poorly developed and/or maladaptive, and individuals with BPD usually display a hostile and antagonistic demeanor and engage in suicidal or nonsuicidal self-injury (NSSI) behaviors as means to alleviate overwhelming emotional distress. Impulsivity, disinhibition, poor tolerance to frustration, and risk-taking behaviors are also characteristic of BPD.1,5 As a result, BPD is usually associated with significant impairment in functioning, multiple hospitalizations, and high rates of comorbid mood disorders, posttraumatic stress disorder (PTSD), SUDs, and death by suicide.
Bipolar disorder. Conversely, the fluctuations in mood and affect observed in patients with BD are usually episodic rather than pervasive, and tend to last longer (typically days to weeks) compared with the transient mood shifts observed in patients with BPD.4,17,18 The impulsivity, psychomotor agitation, and increased goal-directed activity reported by patients with BD are usually seen in the context of an acute affective episode, and are far less common during periods of stability or euthymic affect.4,17,18 Grandiosity and inflated self-esteem—hallmarks of a manic or hypomanic state—seem to oppose the unstable self-image observed in BPD, although indecisiveness and low self-worth may be observed in individuals with BD during depressive episodes. Antidepressant-induced mania or hypomania, atypical depressive episodes, and disruptions in sleep and circadian rhythms may be predictors of BD.4,21 Furthermore, although psychosocial stressors may be associated with acute affective episodes in early stages of bipolar illness, over time minimal stressors are necessary to ignite new affective episodes.22,23 Although BD is associated with high rates of suicide, suicide attempts are usually seen in the context of an acute depressive episode, and NSSI behaviors are less common among patients with BD.24
Lastly, other biographical data, such as a history of early life trauma, comorbidity, and a family history of psychiatric illnesses, can be particularly helpful in establishing the differential diagnosis between BD and BPD.25 For instance, evidence suggests that the heritability of BD may be as high as 70%, which usually translates into an extensive family history of bipolar and related disorders.26 In addition, studies suggest a high co-occurrence of anxiety disorders, attention-deficit/hyperactivity disorder, and SUDs in patients with BD, whereas PTSD, SUDs, and eating disorders tend to be highly comorbid with BPD.27 Childhood adversity (ie, a history of physical, sexual, or emotional abuse, or neglect) seems to be pivotal in the genesis of BPD and may predispose these individuals to psychotic and dissociative symptoms, particularly those with a history of sexual abuse, while playing a more secondary role in BD.28-31
Implications of misdiagnosis
In the view of the limitations of the existing models, multidimensional approaches are necessary to improve diagnostic accuracy. Presently, the differential diagnosis of BD and BPD continues to rely on clinical findings and syndromic classifications. Misdiagnosing BD and BPD has adverse therapeutic and prognostic implications.32 For instance, while psychotropic medications and neuromodulatory therapies (eg, electroconvulsive therapy, repetitive transcranial magnetic stimulation) are considered first-line treatments for patients with BD, psychosocial interventions tend to be adjunctive treatments in BD.33 Conversely, although pharmacotherapy might be helpful for patients with BPD, psychosocial and behavioral interventions are the mainstay treatment for this disorder, with the strongest evidence supporting cognitive-behavioral therapy, dialectical behavioral therapy, mentalization-based therapy, and transference-focused therapy.34-36 Thus, misdiagnosing BD as BPD with comorbid depression may result in the use of antidepressants, which can be detrimental in BD. Antidepressant treatment of BD, particularly as monotherapy, has been associated with manic or hypomanic switch, mixed states, and frequent cycling.21 Moreover, delays in diagnosis and proper treatment of BD may result in protracted mood symptoms, prolonged affective episodes, higher rates of disability, functional impairment, and overall worse clinical outcomes.24 In addition, because behavioral and psychosocial interventions are usually adjunctive therapies rather than first-line interventions for patients with BD, misdiagnosing BPD as BD may ultimately prevent these individuals from receiving proper treatment, likely resulting in more severe functional impairment, multiple hospitalizations, self-inflicted injuries, and suicide attempts, since psychotropic medications are not particularly effective for improving self-efficacy and coping strategies, nor for correcting cognitive distortions, particularly in self-image, and pathological personality traits, all of which are critical aspects of BPD treatment.
Continue to: Several factors might...
Several factors might make clinicians reluctant to diagnose BPD, or bias them to diagnose BD more frequently. These include a lack of familiarity with the diagnostic criteria for BPD, the phenotypical resemblance between BP and BPD, or even concerns about the stigma and negative implications that are associated with a BPD diagnosis.32,37,38
Whereas BD is currently perceived as a condition with a strong biological basis, there are considerable misconceptions regarding BPD and its nature.4-6,22,26 As a consequence, individuals with BPD tend to be perceived as “difficult-to-treat,” “uncooperative,” or “attention-seeking.” These misconceptions may result in poor clinician-patient relationships, unmet clinical and psychiatric needs, and frustration for both clinicians and patients.37
Through advances in biological psychiatry, precision medicine may someday be a part of psychiatric practice. Biological “signatures” may eventually help clinicians in diagnosing and treating psychiatric disorders. Presently, however, rigorous history-taking and comprehensive clinical assessments are still the most powerful tools a clinician can use to accomplish these goals. Finally, destigmatizing psychiatric disorders and educating patients and clinicians are also critical to improving clinical outcomes and promoting mental health in a compassionate and empathetic fashion.
Bottom Line
Despite the phenotypical resemblance between bipolar disorder (BP) and borderline personality disorder (BPD), the 2 are independent conditions with distinct neurobiological and psychopathological underpinnings. Clinicians can use a rigorous assessment of pathological personality traits and characterization of symptoms, such as onset patterns, clinical course, and phenomenology, to properly distinguish between BP and BPD.
Related Resources
- Fiedorowicz JG, Black DW. Borderline, bipolar, or both? Frame your diagnosis on the patient history. Current Psychiatry. 2010; 9(1):21-24,29-32.
- Zimmerman M. Improving the recognition of borderline personality disorder. Current Psychiatry. 2017;16(10):13-19.
- National Institute of Mental Health. Overview on bipolar disorder. www.nimh.nih.gov/health/publications/bipolar-disorder/index.shtml. Revised October 2018.
- National Institute of Mental Health. Overview on borderline personality disorder. www.nimh.nih.gov/health/publications/borderline-personality-disorder-fact-sheet/index.shtml.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575-1586.
3. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry. 2007;64(5):543-552.
4. Malhi GS, Bargh DM, Coulston CM, et al. Predicting bipolar disorder on the basis of phenomenology: implications for prevention and early intervention. Bipolar Disord. 2014;16(5):455-470.
5. Skodol AE, Gunderson JG, Pfohl B, et al. The borderline diagnosis I: psychopathology. Biol Psychiatry. 2002;51(12):936-950.
6. Skodol AE, Siever LJ, Livesley WJ, et al. The borderline diagnosis II: biology, genetics, and clinical course. Biol Psychiatry. 2002;51(12):951-963.
7. Hasin DS, Grant BF. The National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Waves 1 and 2: review and summary of findings. Soc Psychiatry Psychiatr Epidemiol. 2015;50(11):1609-1640.
8. McDermid J, Sareen J, El-Gabalawy R, et al. Co-morbidity of bipolar disorder and borderline personality disorder: findings from the National Epidemiologic Survey on Alcohol and Related Conditions. Compr Psychiatry. 2015;58:18-28.
9. Gunderson JG, Weinberg I, Daversa MT, et al. Descriptive and longitudinal observations on the relationship of borderline personality disorder and bipolar disorder. Am J Psychiatry. 2006;163(7):1173-1178.
10. Swartz HA, Pilkonis PA, Frank E, et al. Acute treatment outcomes in patients with bipolar I disorder and co-morbid borderline personality disorder receiving medication and psychotherapy. Bipolar Disord. 2005;7(2):192-197.
11. Riemann G, Weisscher N, Post RM, et al. The relationship between self-reported borderline personality features and prospective illness course in bipolar disorder. Int J Bipolar Disord. 2017;5(1):31.
12. de la Rosa I, Oquendo MA, García G, et al. Determining if borderline personality disorder and bipolar disorder are alternative expressions of the same disorder. J Clin Psychiatry. 2017;778(8):e994-e999. doi: 10.4088/JCP.16m11190.
13. di Giacomo E, Aspesi F, Fotiadou M, et al. Unblending borderline personality and bipolar disorders. J Psychiatr Res. 2017;91:90-97.
14. Parker G, Bayes A, McClure G, et al. Clinical status of comorbid bipolar disorder and borderline personality disorder. Br J Psychiatry. 2016;209(3):209-215.
15. Perez Arribas I, Goodwin GM, Geddes JR, et al. A signature-based machine learning model for distinguishing bipolar disorder and borderline personality disorder. Transl Psychiatry. 2018;8(1):274.
16. Insel T, Cuthbert B, Garvey M, et al. Research Domain Criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748-751.
17. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60(3):261-269.
19. Oldham JM, Skodol AE, Bender DS. A current integrative perspective on personality disorders. American Psychiatric Publishing, Inc. 2005.
20. Herzog JI, Schmahl C. Adverse childhood experiences and the consequences on neurobiological, psychosocial, and somatic conditions across the lifespan. Front Psychiatry. 2018;9:420.
21. Barbuti M, Pacchiarotti I, Vieta E, et al. Antidepressant-induced hypomania/mania in patients with major depression: evidence from the BRIDGE-II-MIX study. J Affect Disord. 2017;219:187-192.
22. Post RM. Mechanisms of illness progression in the recurrent affective disorders. Neurotox Res. 2010;18(3-4):256-271.
23. da Costa SC, Passos IC, Lowri C, et al. Refractory bipolar disorder and neuroprogression. Prog Neuro-Psychopharmacology Biol Psychiatry. 2016;70:103-110.
24. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
25. Zimmerman M, Martinez JH, Morgan TA, et al. Distinguishing bipolar II depression from major depressive disorder with comorbid borderline personality disorder: demographic, clinical, and family history differences. J Clin Psychiatry. 2013;74(9):880-886.
26. Hasler G, Drevets WC, Gould TD, et al. Toward constructing an endophenotype strategy for bipolar disorders. Biol Psychiatry. 2006;60(2):93-105.
27. Brieger P, Ehrt U, Marneros A. Frequency of comorbid personality disorders in bipolar and unipolar affective disorders. Compr Psychiatry. 2003;44(1):28-34.
28. Leverich GS, McElroy SL, Suppes T, et al. Early physical and sexual abuse associated with an adverse course of bipolar illness. Biol Psychiatry. 2002;51(4):288-297.
29. Leverich GS, Post RM. Course of bipolar illness after history of childhood trauma. Lancet. 2006;367(9516):1040-1042.
30. Golier JA, Yehuda R, Bierer LM, et al. The relationship of borderline personality disorder to posttraumatic stress disorder and traumatic events. Am J Psychiatry. 2003;160(11):2018-2024.
31. Nicol K, Pope M, Romaniuk L, et al. Childhood trauma, midbrain activation and psychotic symptoms in borderline personality disorder. Transl Psychiatry. 2015;5:e559. doi:10.1038/tp.2015.53.
32. Ruggero CJ, Zimmerman M, Chelminski I, et al. Borderline personality disorder and the misdiagnosis of bipolar disorder. J Psychiatr Res. 2010;44(6):405-408.
33. Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet. 2013;381(9878):1672-1682.
34. McMain S, Korman LM, Dimeff L. Dialectical behavior therapy and the treatment of emotion dysregulation. J Clin Psychol. 2001;57(2):183-196.
35. Cristea IA, Gentili C, Cotet CD, et al. Efficacy of psychotherapies for borderline personality disorder: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(4):319-328.
36. Linehan MM, Korslund KE, Harned MS, et al. Dialectical behavior therapy for high suicide risk in individuals with borderline personality disorder. JAMA Psychiatry. 2015;72(75);475-482.
37. LeQuesne ER, Hersh RG. Disclosure of a diagnosis of borderline personality disorder. J Psychiatr Pract. 2004:10(3):170-176.
38. Young AH. Bipolar disorder: diagnostic conundrums and associated comorbidities. J Clin Psychiatry. 2009;70(8):e26. doi:10.4088/jcp.7067br6c.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575-1586.
3. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry. 2007;64(5):543-552.
4. Malhi GS, Bargh DM, Coulston CM, et al. Predicting bipolar disorder on the basis of phenomenology: implications for prevention and early intervention. Bipolar Disord. 2014;16(5):455-470.
5. Skodol AE, Gunderson JG, Pfohl B, et al. The borderline diagnosis I: psychopathology. Biol Psychiatry. 2002;51(12):936-950.
6. Skodol AE, Siever LJ, Livesley WJ, et al. The borderline diagnosis II: biology, genetics, and clinical course. Biol Psychiatry. 2002;51(12):951-963.
7. Hasin DS, Grant BF. The National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Waves 1 and 2: review and summary of findings. Soc Psychiatry Psychiatr Epidemiol. 2015;50(11):1609-1640.
8. McDermid J, Sareen J, El-Gabalawy R, et al. Co-morbidity of bipolar disorder and borderline personality disorder: findings from the National Epidemiologic Survey on Alcohol and Related Conditions. Compr Psychiatry. 2015;58:18-28.
9. Gunderson JG, Weinberg I, Daversa MT, et al. Descriptive and longitudinal observations on the relationship of borderline personality disorder and bipolar disorder. Am J Psychiatry. 2006;163(7):1173-1178.
10. Swartz HA, Pilkonis PA, Frank E, et al. Acute treatment outcomes in patients with bipolar I disorder and co-morbid borderline personality disorder receiving medication and psychotherapy. Bipolar Disord. 2005;7(2):192-197.
11. Riemann G, Weisscher N, Post RM, et al. The relationship between self-reported borderline personality features and prospective illness course in bipolar disorder. Int J Bipolar Disord. 2017;5(1):31.
12. de la Rosa I, Oquendo MA, García G, et al. Determining if borderline personality disorder and bipolar disorder are alternative expressions of the same disorder. J Clin Psychiatry. 2017;778(8):e994-e999. doi: 10.4088/JCP.16m11190.
13. di Giacomo E, Aspesi F, Fotiadou M, et al. Unblending borderline personality and bipolar disorders. J Psychiatr Res. 2017;91:90-97.
14. Parker G, Bayes A, McClure G, et al. Clinical status of comorbid bipolar disorder and borderline personality disorder. Br J Psychiatry. 2016;209(3):209-215.
15. Perez Arribas I, Goodwin GM, Geddes JR, et al. A signature-based machine learning model for distinguishing bipolar disorder and borderline personality disorder. Transl Psychiatry. 2018;8(1):274.
16. Insel T, Cuthbert B, Garvey M, et al. Research Domain Criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748-751.
17. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530-537.
18. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60(3):261-269.
19. Oldham JM, Skodol AE, Bender DS. A current integrative perspective on personality disorders. American Psychiatric Publishing, Inc. 2005.
20. Herzog JI, Schmahl C. Adverse childhood experiences and the consequences on neurobiological, psychosocial, and somatic conditions across the lifespan. Front Psychiatry. 2018;9:420.
21. Barbuti M, Pacchiarotti I, Vieta E, et al. Antidepressant-induced hypomania/mania in patients with major depression: evidence from the BRIDGE-II-MIX study. J Affect Disord. 2017;219:187-192.
22. Post RM. Mechanisms of illness progression in the recurrent affective disorders. Neurotox Res. 2010;18(3-4):256-271.
23. da Costa SC, Passos IC, Lowri C, et al. Refractory bipolar disorder and neuroprogression. Prog Neuro-Psychopharmacology Biol Psychiatry. 2016;70:103-110.
24. Crump C, Sundquist K, Winkleby MA, et al. Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry. 2013;70(9):931-939.
25. Zimmerman M, Martinez JH, Morgan TA, et al. Distinguishing bipolar II depression from major depressive disorder with comorbid borderline personality disorder: demographic, clinical, and family history differences. J Clin Psychiatry. 2013;74(9):880-886.
26. Hasler G, Drevets WC, Gould TD, et al. Toward constructing an endophenotype strategy for bipolar disorders. Biol Psychiatry. 2006;60(2):93-105.
27. Brieger P, Ehrt U, Marneros A. Frequency of comorbid personality disorders in bipolar and unipolar affective disorders. Compr Psychiatry. 2003;44(1):28-34.
28. Leverich GS, McElroy SL, Suppes T, et al. Early physical and sexual abuse associated with an adverse course of bipolar illness. Biol Psychiatry. 2002;51(4):288-297.
29. Leverich GS, Post RM. Course of bipolar illness after history of childhood trauma. Lancet. 2006;367(9516):1040-1042.
30. Golier JA, Yehuda R, Bierer LM, et al. The relationship of borderline personality disorder to posttraumatic stress disorder and traumatic events. Am J Psychiatry. 2003;160(11):2018-2024.
31. Nicol K, Pope M, Romaniuk L, et al. Childhood trauma, midbrain activation and psychotic symptoms in borderline personality disorder. Transl Psychiatry. 2015;5:e559. doi:10.1038/tp.2015.53.
32. Ruggero CJ, Zimmerman M, Chelminski I, et al. Borderline personality disorder and the misdiagnosis of bipolar disorder. J Psychiatr Res. 2010;44(6):405-408.
33. Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet. 2013;381(9878):1672-1682.
34. McMain S, Korman LM, Dimeff L. Dialectical behavior therapy and the treatment of emotion dysregulation. J Clin Psychol. 2001;57(2):183-196.
35. Cristea IA, Gentili C, Cotet CD, et al. Efficacy of psychotherapies for borderline personality disorder: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(4):319-328.
36. Linehan MM, Korslund KE, Harned MS, et al. Dialectical behavior therapy for high suicide risk in individuals with borderline personality disorder. JAMA Psychiatry. 2015;72(75);475-482.
37. LeQuesne ER, Hersh RG. Disclosure of a diagnosis of borderline personality disorder. J Psychiatr Pract. 2004:10(3):170-176.
38. Young AH. Bipolar disorder: diagnostic conundrums and associated comorbidities. J Clin Psychiatry. 2009;70(8):e26. doi:10.4088/jcp.7067br6c.

2019 Update on minimally invasive gynecologic surgery
Through the years, the surgical approach to hysterectomy has expanded from its early beginnings of being performed only through an abdominal or transvaginal route with traditional surgical clamps and suture. The late 1980s saw the advent of the laparoscopic-assisted vaginal hysterectomy (LAVH), and from that point forward several additional hysterectomy methods evolved, including today’s robotic approaches.
Although clinical evidence and societal endorsements support vaginal hysterectomy as a superior high-value modality, it remains one of the least performed among all available routes.1-3 In an analysis of inpatient hysterectomies published by Wright and colleagues in 2013, 16.7% of hysterectomies were performed vaginally, a number that essentially has remained steady throughout the ensuing years.4
Attempts to improve the application of vaginal hysterectomy have been made.5 These include the development of various curriculum and simulation-based medical education programs on vaginal surgical skills training and acquisition in the hopes of improving utilization.6 An interesting recent development is the rethinking of vaginal hysterectomy by several surgeons globally who are applying facets of the various hysterectomy methods to a transvaginal approach known as vaginal natural orifice transluminal endoscopic surgery (vNOTES).7,8 Unique to this thinking is the incorporation of conventional laparoscopic instrumentation.
Although I have not yet incorporated this approach in my surgical armamentarium at Columbia University Medical Center/New York–Presbyterian Hospital, I am intrigued by the possibility that this technique may serve as a rescue for vaginal hysterectomies that are at risk of conversion or of not being performed at all.9
At this time, vNOTES is not a standard of care and should be performed only by highly specialized surgeons. However, in the spirit of this Update on minimally invasive surgery and to keep our readers abreast of burgeoning techniques, I am delighted to bring you this overview by Dr. Xiaoming Guan, one of the pioneers of this surgical approach, and Dr. Tamisa Koythong and Dr. Juan Liu. I hope you find this recent development in hysterectomy of interest.
—Arnold P. Advincula, MD
Continue to: Development and evolution of NOTES...
Development and evolution of NOTES
Over the past few decades, emphasis has shifted from laparotomy to minimally invasive surgery because of its proven significant advantages in patient care, such as improved cosmesis, shorter hospital stay, shorter postoperative recovery, and decreased postoperative pain and blood loss.10 Advances in laparoendoscopic surgery and instrumentation, including robot-assisted laparoscopy (RAL), single-incision laparoscopic surgery (SILS), and most recently natural orifice transluminal endoscopic surgery (NOTES), reflect ongoing innovative developments in the field of minimally invasive surgery.
Here, we provide a brief literature review of the NOTES technique, focus on its application in gynecologic surgery, and describe how we perform NOTES at our institution.
NOTES application in gynecology
With NOTES, peritoneal access is gained through a natural orifice (such as the mouth, vagina, urethra, or anus) to perform endoscopic surgery, occasionally without requiring an abdominal incision. First described in 2004, transgastric peritoneoscopy was performed in a porcine model, and shortly thereafter the first transgastric appendectomy was performed in humans.11,12 The technique has further been adopted in cholecystectomy, appendectomy, gastrectomy, and nephrectomy procedures.13
Given rapid interest in a possible paradigm shift in the field of minimally invasive surgery, the Natural Orifice Surgery Consortiumfor Assessment and Research (NOSCAR) was formed, and the group published an article on potential barriers to accepted practice and adoption of NOTES as a realistic alternative to traditional laparoscopic surgery.14
While transgastric and transanal access to the peritoneum were initially more popular, the risk of anastomotic leaks associated with incomplete closure and subsequent infection were thought to be prohibitively high.15 Transvaginal access was considered a safer and simpler alternative, allowing for complete closure without increased risk of infection, and this is now the route through which the majority of NOTES procedures are completed.16,17
The eventual application of NOTES in the field of gynecology seemed inevitable. The American College of Obstetricians and Gynecologists stated that transvaginal surgery is the most minimally invasive and preferred surgical route in the management of patients with benign gynecologic diseases.18 However, performing it can be challenging at times due to limited visualization and lack of the required skills for single-site surgery. NOTES allows a gynecologic surgeon to improve visualization through the use of laparoendoscopic instruments and to complete surgery through a transvaginal route.
In 2012, Ahn and colleagues demonstrated the feasibility of the NOTES technique in gynecologic surgery after using it to successfully complete benign adnexal surgery in 10 patients.19 Vaginal NOTES (vNOTES) has since been further developed to include successful hysterectomy, myomectomy, sacrocolpopexy, tubal anastomosis, and even lymphadenectomy in the treatment of early- stage endometrial carcinoma.20-26 vNOTES also can be considered a rescue approach for traditional vaginal hysterectomy in instances in which it is necessary to evaluate adnexal pathology.9 Most recently, vNOTES hysterectomy has been reported with da Vinci Si or Xi robotic platforms.27,28
Continue to: Operative time, post-op stay shorter in NAOC-treated patients...
Operative time, post-op stay shorter in NAOC-treated patients
Few studies have compared outcomes with vNOTES to those with traditional laparoscopy. In 2016, Wang and colleagues compared surgical outcomes between NOTES-assisted ovarian cystectomy (NAOC) and laparoscopic ovarian cystectomy (LOC) in a case-matched study that included 277 patients.29 Although mean (SD) blood loss in patients who underwent LOC was significantly less compared with those who underwent NAOC (21.4 [14.7] mL vs 31.6 [24.1] mL; P = .028), absolute blood loss in both groups was deemed minimal. Additionally, mean (SD) operative time and postoperative stay were significantly less in patients undergoing NAOC compared with those having LOC (38.23 [10.19] minutes vs 53.82 [18.61] minutes; P≤.001; and 1.38 [0.55] days vs 1.82 [0.52] days; P≤.001; respectively).29
How vNOTES hysterectomy stacked up against TLH
In 2018, Baekelandt and colleagues compared outcomes between vNOTES hysterectomy and total laparoscopic hysterectomy (TLH) in a noninferiority single-blinded trial of 70 women.8 Compared with TLH, vNOTES hysterectomy was associated with shorter operative time (41 vs 75 minutes; P<.001), shorter hospital stay (0.8 vs 1.3 days; P = .004), and lower postoperative analgesic requirement (8 vs 14 U; P = .006). Additionally, there were no differences between the 2 groups in postoperative infection rate, intraoperative complications, or hospital readmissions within 6 weeks.8
Clearly, vNOTES is the next exciting development in minimally invasive surgery, improving patient outcomes and satisfaction with truly scarless surgery. Compared with traditional transvaginal surgery, vNOTES has the advantage of improved visualization with laparoendoscopic guidance, and it may be beneficial even for patients previously thought to have relative contraindications to successful completion of transvaginal surgery, such as nulliparity or a narrow introitus.
Approach for performing vNOTES procedures
At our institution, Baylor College of Medicine, the majority of gynecologic surgeries are performed via either transumbilical robot-assisted single-incision laparoscopy or vNOTES. Preoperative selection of appropriate candidates for vNOTES includes:
- low suspicion for or prior diagnosis of endometriosis with obliteration of the posterior cul-de-sac
- no surgical history suggestive of severe adhesive disease, and
- adequate vaginal sidewall access and sufficient descent for instrumentation for entry into the peritoneal cavity.
In general, a key concept in vNOTES is "vaginal pull, laparoscopic push," which means that the surgeon must pull the cervix while performing vaginal entry and then push the uterus back in the peritoneal cavity to increase surgical space during laparoscopic surgery.
Continue to: Overview of vNOTES steps...
Overview of vNOTES steps
Below we break down a description of vNOTES in 6 sections. Our patients are always placed in dorsal lithotomy position with TrenGuard (D.A. Surgical) Trendelenburg restraint. We prep the abdomen in case we need to convert to transabdominal surgery via transumbilical single-incision laparoscopic surgery or traditional laparoscopic surgery.
1. Vaginal entry
Accessing the peritoneal cavity through the vagina initially proceeds like a vaginal hysterectomy. We inject dilute vasopressin (20 U in 20 mL of normal saline) circumferentially in the cervix (for hysterectomy) or in the posterior cervix in the cervicovaginal junction (for adnexal surgery without hysterectomy) for vasoconstriction and hydrodissection.
We then incise the vaginal mucosa circumferentially with electrosurgical cautery and follow with posterior colpotomy. We find that reapproximating the posterior peritoneum to the posterior vagina with either figure-of-8 stitches or a running stitch of polyglactin 910 suture (2-0 Vicryl) assists in port placement, bleeding at the peritoneal edge, and closure of the cuff or colpotomy at the end of the case. We tag this suture with a curved hemostat.
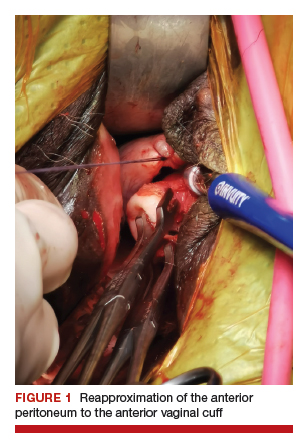
Depending on whether a hysterectomy is being performed, anterior colpotomy is made. Again, the anterior peritoneum is then tagged to the anterior vaginal cuff in similar fashion, and this suture is tagged with a different instrument; we typically use a straight hemostat or Sarot clamp (FIGURE 1).
2. Traditional vaginal hysterectomy
After colpotomy, we prefer to perform progressive clamping of the broad ligament from the uterosacral and cardinal ligaments to the level of uterine artery as in traditional vaginal hysterectomy, if feasible.
3. Single-site port placement
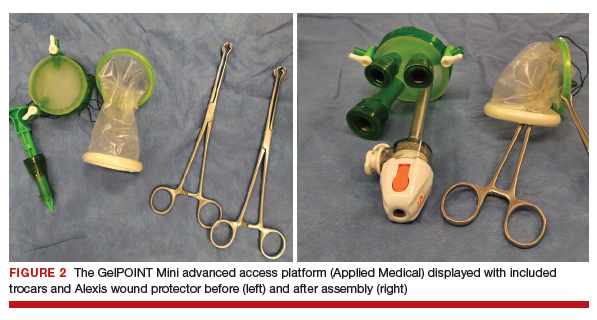
The assembled GelPOINT Mini advanced access platform (Applied Medical) (FIGURE 2) is introduced through the vagina after the Alexis wound protector (included with the kit) is first placed through the colpotomy with assistance of Babcock clamps (FIGURE 3).

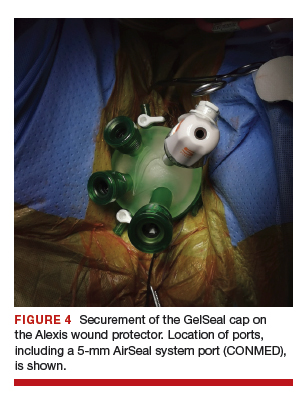
After ensuring that the green rigid ring of the Alexis wound protector is contained and completely expanded within the peritoneal cavity, we cross our previously tagged sutures as we find this helps with preventing the GelPOINT Mini access platform from inadvertently shifting out of the peritoneal cavity during surgery. The GelSeal cap is then secured and pneumoperitoneum is established (FIGURE 4).
Continue to: 4. Laparoendoscopic surgery...
4. Laparoendoscopic surgery
Instruments used in our surgeries include a 10-mm rigid 30° 43-cm working length laparoscope; a 44-cm LigaSure device (Medtronic); a 5-mm, 37-cm laparoscopic cobra grasping forceps and fenestrated grasper (Karl Storz); and a 5-mm, 45-cm laparoscopic suction with hydrodissection tip (Stryker) (FIGURE 5).
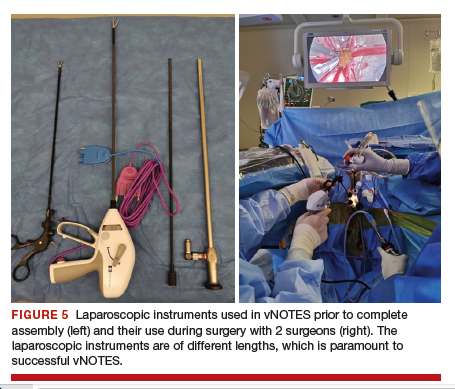
vNOTES allows a gynecologic surgeon the unique ability to survey the upper abdomen. The remainder of the surgery proceeds using basic laparoscopic single-site skills.
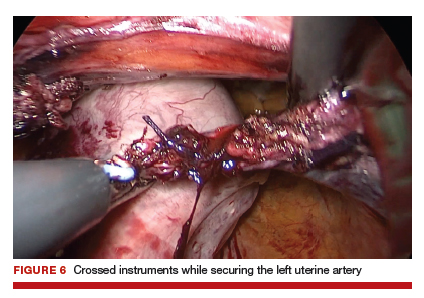
During vNOTES, as with all single-site surgical procedures, understanding the optimal placement of crossed instruments is important for successful completion. For example, when securing the right uterine artery, the surgeon needs to push the cervix toward the patient's left and slightly into the peritoneal cavity using a laparoscopic cobra grasper with his or her left hand while then securing the uterine pedicle using the LigaSure device with his or her right hand. This is then reversed when securing the left uterine artery, where the assistant surgeon pushes the cervix toward the patient's right while the surgeon secures the pedicle ("vaginal pull, laparoscopic push") (FIGURE 6).
This again is reiterated in securing the ovarian pedicles, which are pushed into the peritoneal cavity while being secured with the LigaSure device.
5. Specimen removal
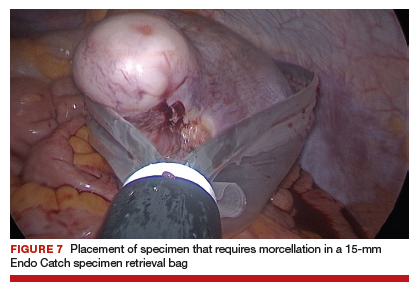
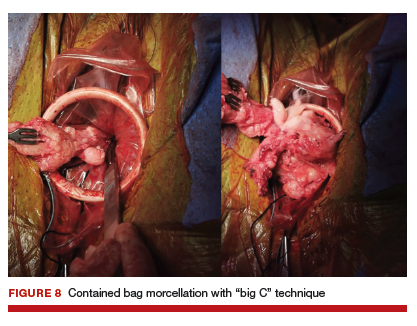
For large uteri or specimens that need morcellation, a 15-mm Endo Catch specimen retrieval bag (Medtronic) is introduced through the GelPOINT Mini system. The specimen is then placed in the bag and delivered to the vagina, where contained bag morcellation is performed in standard fashion (FIGURES 7 AND 8). We utilized the "big C" technique by first grasping the specimen with a penetrating clamp. The clamp is then held in our nondominant hand and a No. 10 blade scalpel is used to create a reverse c-incision, keeping one surface of the specimen intact. This is continued until the specimen can be completely delivered through the vagina.
Specimens that do not require morcellation can be grasped laparoscopically, brought to the GelPOINT Mini port, which is quickly disassembled, and delivered. The GelSeal cap is then reassembled.
6. Vaginal cuff closure
The colpotomy or vaginal cuff is closed with barbed suture continuously, as in traditional vaginal hysterectomy cuff closure. Uterosacral ligament suspension should be performed for vaginal cuff support.
vNOTES is the most recent innovative development in the field of minimally invasive surgery, and it has demonstrated feasibility and safety in the fields of general surgery, urology, and gynecology. Adopting vNOTES in clinical practice can improve patient satisfaction and cosmesis as well as surgical outcomes. Gynecologic surgeons can think of vNOTES hysterectomy as "placing an eye" in the vagina while performing transvaginal hysterectomy. The surgical principle of "vaginal pull, laparoscopic push" facilitates the learning process.
1. ACOG Committee on Gynecologic Practice. Committee opinion no. 444. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
2. AAGL Advancing Minimally Invasive Gynecology Worldwide. AAGL position statement: route of hysterectomy to treat benign uterine disease. J Minim Invasive Gynecol. 2011;18:1-3.
3. Whiteside JL, Kaeser CT, Ridgeway B. Achieving high value in the surgical approach to hysterectomy. Am J Obstet Gynecol. 2019;220:242-245.
4. Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
5. Moen M, Walter A, Harmanli O, et al. Considerations to improve the evidence-based use of vaginal hysterectomy in benign gynecology. Obstet Gynecol. 2014;124:585-588.
6. Balgobin S, Owens DM, Florian-Rodriguez ME, et al. Vaginal hysterectomy suturing skills training model and curriculum. Obstet Gynecol. 2019;134:553-558.
7. Baekelandt J. Total vaginal NOTES hysterectomy: a new approach to hysterectomy. J Minim Invasive Gynecol. 2015;22:1088-1094.
8. Baekelandt JF, De Mulder PA, Le Roy I, et al. Hysterectomy by transvaginal natural orifice transluminal endoscopic surgery versus laparoscopy as a day-care procedure: a randomised controlled trial. BJOG. 2019;126:105-113.
9. Guan X, Bardawil E, Liu J, et al. Transvaginal natural orifice transluminal endoscopic surgery as a rescue for total vaginal hysterectomy. J Minim Invasive Gynecol. 2018;25:1135-1136.
10. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;3:CD003677.
11. Kalloo AN, Singh VK, Jagannath SB, et al. Flexible transgastric peritoneoscopy: a novel approach to diagnostic and therapeutic interventions in the peritoneal cavity. Gastrointest Endosc. 2004;60:114-117.
12. Reddy N, Rao P. Per oral transgastric endoscopic appendectomy in human. Paper Presented at: 45th Annual Conference of the Society of Gastrointestinal Endoscopy of India; February 28-29, 2004; Jaipur, India.
13. Clark MP, Qayed ES, Kooby DA, et al. Natural orifice translumenal endoscopic surgery in humans: a review. Minim Invasive Surg. 2012;189296.
14. Rattner D, Kalloo A; ASGE/SAGES Working Group. ASGE/ SAGES Working Group on natural orifice translumenal endoscopic surgery, October 2005. Surg Endosc. 2006;20:329-333.
15. Autorino R, Yakoubi R, White WM, et al. Natural orifice transluminal endoscopic surgery (NOTES): where are we going? A bibliometric assessment. BJU Int. 2013;111:11-16.
16. Santos BF, Hungness ES. Natural orifice transluminal endoscopic surgery: progress in humans since the white paper. World J Gastroenterol. 2011;17:1655-1665.
17. Tolcher MC, Kalogera E, Hopkins MR, et al. Safety of culdotomy as a surgical approach: implications for natural orifice transluminal endoscopic surgery. JSLS. 2012;16:413-420.
18. ACOG Committee on Gynecologic Practice. Committee opinion no. 701. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017:129:e155-e159.
19. Ahn KH, Song JY, Kim SH, et al. Transvaginal single-port natural orifice transluminal endoscopic surgery for benign uterine adnexal pathologies. J Minim Invasive Gynecol. 2012;19:631-635.
20. Liu J, Kohn J, Sun B, et al. Transvaginal natural orifice transluminal endoscopic surgery sacrocolpopexy: tips and tricks. Minim Invasive Gynecol. 2019;26:38-39.
21. Liu J, Kohn J, Fu H, et al. Transvaginal natural orifice transluminal endoscopic surgery for sacrocolpopexy: a pilot study of 26 cases. J Minim Invasive Gynecol. 2019;26:748-753.
22. Su H, Yen CF, Wu KY, et al. Hysterectomy via transvaginal natural orifice transluminal endoscopic surgery (NOTES): feasibility of an innovative approach. Taiwan J Obstet Gynecol. 2012;51:217-221.
23. Lee CL, Huang CY, Wu KY, et al. Natural orifice transvaginal endoscopic surgery myomectomy: an innovative approach to myomectomy. Gynecol Minim Invasive Ther. 2014;3:127-130.
24. Chen Y, Li J, Zhang Y, et al. Transvaginal single-port laparoscopy sacrocolpopexy. J Minim Invasive Gynecol. 2018;25:585- 588.
25. Lee CL, Wu KY, Tsao FY, et al. Natural orifice transvaginal endoscopic surgery for endometrial cancer. Gynecol Minim Invasive Ther. 2014;3:89-92.
26. Leblanc E, Narducci F, Bresson L, et al. Fluorescence-assisted sentinel (SND) and pelvic node dissections by single-port transvaginal laparoscopic surgery, for the management of an endometrial carcinoma (EC) in an elderly obese patient. Gynecol Oncol. 2016;143:686-687.
27. Lee CL, Wu KY, Su H, et al. Robot-assisted natural orifice transluminal endoscopic surgery for hysterectomy. Taiwan J Obstet Gynecol. 2015;54:761-765.
28. Rezai S, Giovane RA, Johnson SN, et al. Robotic natural orifice transluminal endoscopic surgery (R-NOTES) in gynecologic surgeries, a case report and review of literature. Obstet Gynecol Int J. 2019;10:287-289.
29. Wang CJ, Wu PY, Kuo HH, et al. Natural orifice transluminal endoscopic surgery-assisted versus laparoscopic ovarian cystectomy (NAOC vs. LOC): a case-matched study. Surg Endosc. 2016;30:1227-1234.
Through the years, the surgical approach to hysterectomy has expanded from its early beginnings of being performed only through an abdominal or transvaginal route with traditional surgical clamps and suture. The late 1980s saw the advent of the laparoscopic-assisted vaginal hysterectomy (LAVH), and from that point forward several additional hysterectomy methods evolved, including today’s robotic approaches.
Although clinical evidence and societal endorsements support vaginal hysterectomy as a superior high-value modality, it remains one of the least performed among all available routes.1-3 In an analysis of inpatient hysterectomies published by Wright and colleagues in 2013, 16.7% of hysterectomies were performed vaginally, a number that essentially has remained steady throughout the ensuing years.4
Attempts to improve the application of vaginal hysterectomy have been made.5 These include the development of various curriculum and simulation-based medical education programs on vaginal surgical skills training and acquisition in the hopes of improving utilization.6 An interesting recent development is the rethinking of vaginal hysterectomy by several surgeons globally who are applying facets of the various hysterectomy methods to a transvaginal approach known as vaginal natural orifice transluminal endoscopic surgery (vNOTES).7,8 Unique to this thinking is the incorporation of conventional laparoscopic instrumentation.
Although I have not yet incorporated this approach in my surgical armamentarium at Columbia University Medical Center/New York–Presbyterian Hospital, I am intrigued by the possibility that this technique may serve as a rescue for vaginal hysterectomies that are at risk of conversion or of not being performed at all.9
At this time, vNOTES is not a standard of care and should be performed only by highly specialized surgeons. However, in the spirit of this Update on minimally invasive surgery and to keep our readers abreast of burgeoning techniques, I am delighted to bring you this overview by Dr. Xiaoming Guan, one of the pioneers of this surgical approach, and Dr. Tamisa Koythong and Dr. Juan Liu. I hope you find this recent development in hysterectomy of interest.
—Arnold P. Advincula, MD
Continue to: Development and evolution of NOTES...
Development and evolution of NOTES
Over the past few decades, emphasis has shifted from laparotomy to minimally invasive surgery because of its proven significant advantages in patient care, such as improved cosmesis, shorter hospital stay, shorter postoperative recovery, and decreased postoperative pain and blood loss.10 Advances in laparoendoscopic surgery and instrumentation, including robot-assisted laparoscopy (RAL), single-incision laparoscopic surgery (SILS), and most recently natural orifice transluminal endoscopic surgery (NOTES), reflect ongoing innovative developments in the field of minimally invasive surgery.
Here, we provide a brief literature review of the NOTES technique, focus on its application in gynecologic surgery, and describe how we perform NOTES at our institution.
NOTES application in gynecology
With NOTES, peritoneal access is gained through a natural orifice (such as the mouth, vagina, urethra, or anus) to perform endoscopic surgery, occasionally without requiring an abdominal incision. First described in 2004, transgastric peritoneoscopy was performed in a porcine model, and shortly thereafter the first transgastric appendectomy was performed in humans.11,12 The technique has further been adopted in cholecystectomy, appendectomy, gastrectomy, and nephrectomy procedures.13
Given rapid interest in a possible paradigm shift in the field of minimally invasive surgery, the Natural Orifice Surgery Consortiumfor Assessment and Research (NOSCAR) was formed, and the group published an article on potential barriers to accepted practice and adoption of NOTES as a realistic alternative to traditional laparoscopic surgery.14
While transgastric and transanal access to the peritoneum were initially more popular, the risk of anastomotic leaks associated with incomplete closure and subsequent infection were thought to be prohibitively high.15 Transvaginal access was considered a safer and simpler alternative, allowing for complete closure without increased risk of infection, and this is now the route through which the majority of NOTES procedures are completed.16,17
The eventual application of NOTES in the field of gynecology seemed inevitable. The American College of Obstetricians and Gynecologists stated that transvaginal surgery is the most minimally invasive and preferred surgical route in the management of patients with benign gynecologic diseases.18 However, performing it can be challenging at times due to limited visualization and lack of the required skills for single-site surgery. NOTES allows a gynecologic surgeon to improve visualization through the use of laparoendoscopic instruments and to complete surgery through a transvaginal route.
In 2012, Ahn and colleagues demonstrated the feasibility of the NOTES technique in gynecologic surgery after using it to successfully complete benign adnexal surgery in 10 patients.19 Vaginal NOTES (vNOTES) has since been further developed to include successful hysterectomy, myomectomy, sacrocolpopexy, tubal anastomosis, and even lymphadenectomy in the treatment of early- stage endometrial carcinoma.20-26 vNOTES also can be considered a rescue approach for traditional vaginal hysterectomy in instances in which it is necessary to evaluate adnexal pathology.9 Most recently, vNOTES hysterectomy has been reported with da Vinci Si or Xi robotic platforms.27,28
Continue to: Operative time, post-op stay shorter in NAOC-treated patients...
Operative time, post-op stay shorter in NAOC-treated patients
Few studies have compared outcomes with vNOTES to those with traditional laparoscopy. In 2016, Wang and colleagues compared surgical outcomes between NOTES-assisted ovarian cystectomy (NAOC) and laparoscopic ovarian cystectomy (LOC) in a case-matched study that included 277 patients.29 Although mean (SD) blood loss in patients who underwent LOC was significantly less compared with those who underwent NAOC (21.4 [14.7] mL vs 31.6 [24.1] mL; P = .028), absolute blood loss in both groups was deemed minimal. Additionally, mean (SD) operative time and postoperative stay were significantly less in patients undergoing NAOC compared with those having LOC (38.23 [10.19] minutes vs 53.82 [18.61] minutes; P≤.001; and 1.38 [0.55] days vs 1.82 [0.52] days; P≤.001; respectively).29
How vNOTES hysterectomy stacked up against TLH
In 2018, Baekelandt and colleagues compared outcomes between vNOTES hysterectomy and total laparoscopic hysterectomy (TLH) in a noninferiority single-blinded trial of 70 women.8 Compared with TLH, vNOTES hysterectomy was associated with shorter operative time (41 vs 75 minutes; P<.001), shorter hospital stay (0.8 vs 1.3 days; P = .004), and lower postoperative analgesic requirement (8 vs 14 U; P = .006). Additionally, there were no differences between the 2 groups in postoperative infection rate, intraoperative complications, or hospital readmissions within 6 weeks.8
Clearly, vNOTES is the next exciting development in minimally invasive surgery, improving patient outcomes and satisfaction with truly scarless surgery. Compared with traditional transvaginal surgery, vNOTES has the advantage of improved visualization with laparoendoscopic guidance, and it may be beneficial even for patients previously thought to have relative contraindications to successful completion of transvaginal surgery, such as nulliparity or a narrow introitus.
Approach for performing vNOTES procedures
At our institution, Baylor College of Medicine, the majority of gynecologic surgeries are performed via either transumbilical robot-assisted single-incision laparoscopy or vNOTES. Preoperative selection of appropriate candidates for vNOTES includes:
- low suspicion for or prior diagnosis of endometriosis with obliteration of the posterior cul-de-sac
- no surgical history suggestive of severe adhesive disease, and
- adequate vaginal sidewall access and sufficient descent for instrumentation for entry into the peritoneal cavity.
In general, a key concept in vNOTES is "vaginal pull, laparoscopic push," which means that the surgeon must pull the cervix while performing vaginal entry and then push the uterus back in the peritoneal cavity to increase surgical space during laparoscopic surgery.
Continue to: Overview of vNOTES steps...
Overview of vNOTES steps
Below we break down a description of vNOTES in 6 sections. Our patients are always placed in dorsal lithotomy position with TrenGuard (D.A. Surgical) Trendelenburg restraint. We prep the abdomen in case we need to convert to transabdominal surgery via transumbilical single-incision laparoscopic surgery or traditional laparoscopic surgery.
1. Vaginal entry
Accessing the peritoneal cavity through the vagina initially proceeds like a vaginal hysterectomy. We inject dilute vasopressin (20 U in 20 mL of normal saline) circumferentially in the cervix (for hysterectomy) or in the posterior cervix in the cervicovaginal junction (for adnexal surgery without hysterectomy) for vasoconstriction and hydrodissection.
We then incise the vaginal mucosa circumferentially with electrosurgical cautery and follow with posterior colpotomy. We find that reapproximating the posterior peritoneum to the posterior vagina with either figure-of-8 stitches or a running stitch of polyglactin 910 suture (2-0 Vicryl) assists in port placement, bleeding at the peritoneal edge, and closure of the cuff or colpotomy at the end of the case. We tag this suture with a curved hemostat.
Depending on whether a hysterectomy is being performed, anterior colpotomy is made. Again, the anterior peritoneum is then tagged to the anterior vaginal cuff in similar fashion, and this suture is tagged with a different instrument; we typically use a straight hemostat or Sarot clamp (FIGURE 1).
2. Traditional vaginal hysterectomy
After colpotomy, we prefer to perform progressive clamping of the broad ligament from the uterosacral and cardinal ligaments to the level of uterine artery as in traditional vaginal hysterectomy, if feasible.
3. Single-site port placement
The assembled GelPOINT Mini advanced access platform (Applied Medical) (FIGURE 2) is introduced through the vagina after the Alexis wound protector (included with the kit) is first placed through the colpotomy with assistance of Babcock clamps (FIGURE 3).
After ensuring that the green rigid ring of the Alexis wound protector is contained and completely expanded within the peritoneal cavity, we cross our previously tagged sutures as we find this helps with preventing the GelPOINT Mini access platform from inadvertently shifting out of the peritoneal cavity during surgery. The GelSeal cap is then secured and pneumoperitoneum is established (FIGURE 4).
Continue to: 4. Laparoendoscopic surgery...
4. Laparoendoscopic surgery
Instruments used in our surgeries include a 10-mm rigid 30° 43-cm working length laparoscope; a 44-cm LigaSure device (Medtronic); a 5-mm, 37-cm laparoscopic cobra grasping forceps and fenestrated grasper (Karl Storz); and a 5-mm, 45-cm laparoscopic suction with hydrodissection tip (Stryker) (FIGURE 5).
vNOTES allows a gynecologic surgeon the unique ability to survey the upper abdomen. The remainder of the surgery proceeds using basic laparoscopic single-site skills.
During vNOTES, as with all single-site surgical procedures, understanding the optimal placement of crossed instruments is important for successful completion. For example, when securing the right uterine artery, the surgeon needs to push the cervix toward the patient's left and slightly into the peritoneal cavity using a laparoscopic cobra grasper with his or her left hand while then securing the uterine pedicle using the LigaSure device with his or her right hand. This is then reversed when securing the left uterine artery, where the assistant surgeon pushes the cervix toward the patient's right while the surgeon secures the pedicle ("vaginal pull, laparoscopic push") (FIGURE 6).
This again is reiterated in securing the ovarian pedicles, which are pushed into the peritoneal cavity while being secured with the LigaSure device.
5. Specimen removal
For large uteri or specimens that need morcellation, a 15-mm Endo Catch specimen retrieval bag (Medtronic) is introduced through the GelPOINT Mini system. The specimen is then placed in the bag and delivered to the vagina, where contained bag morcellation is performed in standard fashion (FIGURES 7 AND 8). We utilized the "big C" technique by first grasping the specimen with a penetrating clamp. The clamp is then held in our nondominant hand and a No. 10 blade scalpel is used to create a reverse c-incision, keeping one surface of the specimen intact. This is continued until the specimen can be completely delivered through the vagina.
Specimens that do not require morcellation can be grasped laparoscopically, brought to the GelPOINT Mini port, which is quickly disassembled, and delivered. The GelSeal cap is then reassembled.
6. Vaginal cuff closure
The colpotomy or vaginal cuff is closed with barbed suture continuously, as in traditional vaginal hysterectomy cuff closure. Uterosacral ligament suspension should be performed for vaginal cuff support.
vNOTES is the most recent innovative development in the field of minimally invasive surgery, and it has demonstrated feasibility and safety in the fields of general surgery, urology, and gynecology. Adopting vNOTES in clinical practice can improve patient satisfaction and cosmesis as well as surgical outcomes. Gynecologic surgeons can think of vNOTES hysterectomy as "placing an eye" in the vagina while performing transvaginal hysterectomy. The surgical principle of "vaginal pull, laparoscopic push" facilitates the learning process.
Through the years, the surgical approach to hysterectomy has expanded from its early beginnings of being performed only through an abdominal or transvaginal route with traditional surgical clamps and suture. The late 1980s saw the advent of the laparoscopic-assisted vaginal hysterectomy (LAVH), and from that point forward several additional hysterectomy methods evolved, including today’s robotic approaches.
Although clinical evidence and societal endorsements support vaginal hysterectomy as a superior high-value modality, it remains one of the least performed among all available routes.1-3 In an analysis of inpatient hysterectomies published by Wright and colleagues in 2013, 16.7% of hysterectomies were performed vaginally, a number that essentially has remained steady throughout the ensuing years.4
Attempts to improve the application of vaginal hysterectomy have been made.5 These include the development of various curriculum and simulation-based medical education programs on vaginal surgical skills training and acquisition in the hopes of improving utilization.6 An interesting recent development is the rethinking of vaginal hysterectomy by several surgeons globally who are applying facets of the various hysterectomy methods to a transvaginal approach known as vaginal natural orifice transluminal endoscopic surgery (vNOTES).7,8 Unique to this thinking is the incorporation of conventional laparoscopic instrumentation.
Although I have not yet incorporated this approach in my surgical armamentarium at Columbia University Medical Center/New York–Presbyterian Hospital, I am intrigued by the possibility that this technique may serve as a rescue for vaginal hysterectomies that are at risk of conversion or of not being performed at all.9
At this time, vNOTES is not a standard of care and should be performed only by highly specialized surgeons. However, in the spirit of this Update on minimally invasive surgery and to keep our readers abreast of burgeoning techniques, I am delighted to bring you this overview by Dr. Xiaoming Guan, one of the pioneers of this surgical approach, and Dr. Tamisa Koythong and Dr. Juan Liu. I hope you find this recent development in hysterectomy of interest.
—Arnold P. Advincula, MD
Continue to: Development and evolution of NOTES...
Development and evolution of NOTES
Over the past few decades, emphasis has shifted from laparotomy to minimally invasive surgery because of its proven significant advantages in patient care, such as improved cosmesis, shorter hospital stay, shorter postoperative recovery, and decreased postoperative pain and blood loss.10 Advances in laparoendoscopic surgery and instrumentation, including robot-assisted laparoscopy (RAL), single-incision laparoscopic surgery (SILS), and most recently natural orifice transluminal endoscopic surgery (NOTES), reflect ongoing innovative developments in the field of minimally invasive surgery.
Here, we provide a brief literature review of the NOTES technique, focus on its application in gynecologic surgery, and describe how we perform NOTES at our institution.
NOTES application in gynecology
With NOTES, peritoneal access is gained through a natural orifice (such as the mouth, vagina, urethra, or anus) to perform endoscopic surgery, occasionally without requiring an abdominal incision. First described in 2004, transgastric peritoneoscopy was performed in a porcine model, and shortly thereafter the first transgastric appendectomy was performed in humans.11,12 The technique has further been adopted in cholecystectomy, appendectomy, gastrectomy, and nephrectomy procedures.13
Given rapid interest in a possible paradigm shift in the field of minimally invasive surgery, the Natural Orifice Surgery Consortiumfor Assessment and Research (NOSCAR) was formed, and the group published an article on potential barriers to accepted practice and adoption of NOTES as a realistic alternative to traditional laparoscopic surgery.14
While transgastric and transanal access to the peritoneum were initially more popular, the risk of anastomotic leaks associated with incomplete closure and subsequent infection were thought to be prohibitively high.15 Transvaginal access was considered a safer and simpler alternative, allowing for complete closure without increased risk of infection, and this is now the route through which the majority of NOTES procedures are completed.16,17
The eventual application of NOTES in the field of gynecology seemed inevitable. The American College of Obstetricians and Gynecologists stated that transvaginal surgery is the most minimally invasive and preferred surgical route in the management of patients with benign gynecologic diseases.18 However, performing it can be challenging at times due to limited visualization and lack of the required skills for single-site surgery. NOTES allows a gynecologic surgeon to improve visualization through the use of laparoendoscopic instruments and to complete surgery through a transvaginal route.
In 2012, Ahn and colleagues demonstrated the feasibility of the NOTES technique in gynecologic surgery after using it to successfully complete benign adnexal surgery in 10 patients.19 Vaginal NOTES (vNOTES) has since been further developed to include successful hysterectomy, myomectomy, sacrocolpopexy, tubal anastomosis, and even lymphadenectomy in the treatment of early- stage endometrial carcinoma.20-26 vNOTES also can be considered a rescue approach for traditional vaginal hysterectomy in instances in which it is necessary to evaluate adnexal pathology.9 Most recently, vNOTES hysterectomy has been reported with da Vinci Si or Xi robotic platforms.27,28
Continue to: Operative time, post-op stay shorter in NAOC-treated patients...
Operative time, post-op stay shorter in NAOC-treated patients
Few studies have compared outcomes with vNOTES to those with traditional laparoscopy. In 2016, Wang and colleagues compared surgical outcomes between NOTES-assisted ovarian cystectomy (NAOC) and laparoscopic ovarian cystectomy (LOC) in a case-matched study that included 277 patients.29 Although mean (SD) blood loss in patients who underwent LOC was significantly less compared with those who underwent NAOC (21.4 [14.7] mL vs 31.6 [24.1] mL; P = .028), absolute blood loss in both groups was deemed minimal. Additionally, mean (SD) operative time and postoperative stay were significantly less in patients undergoing NAOC compared with those having LOC (38.23 [10.19] minutes vs 53.82 [18.61] minutes; P≤.001; and 1.38 [0.55] days vs 1.82 [0.52] days; P≤.001; respectively).29
How vNOTES hysterectomy stacked up against TLH
In 2018, Baekelandt and colleagues compared outcomes between vNOTES hysterectomy and total laparoscopic hysterectomy (TLH) in a noninferiority single-blinded trial of 70 women.8 Compared with TLH, vNOTES hysterectomy was associated with shorter operative time (41 vs 75 minutes; P<.001), shorter hospital stay (0.8 vs 1.3 days; P = .004), and lower postoperative analgesic requirement (8 vs 14 U; P = .006). Additionally, there were no differences between the 2 groups in postoperative infection rate, intraoperative complications, or hospital readmissions within 6 weeks.8
Clearly, vNOTES is the next exciting development in minimally invasive surgery, improving patient outcomes and satisfaction with truly scarless surgery. Compared with traditional transvaginal surgery, vNOTES has the advantage of improved visualization with laparoendoscopic guidance, and it may be beneficial even for patients previously thought to have relative contraindications to successful completion of transvaginal surgery, such as nulliparity or a narrow introitus.
Approach for performing vNOTES procedures
At our institution, Baylor College of Medicine, the majority of gynecologic surgeries are performed via either transumbilical robot-assisted single-incision laparoscopy or vNOTES. Preoperative selection of appropriate candidates for vNOTES includes:
- low suspicion for or prior diagnosis of endometriosis with obliteration of the posterior cul-de-sac
- no surgical history suggestive of severe adhesive disease, and
- adequate vaginal sidewall access and sufficient descent for instrumentation for entry into the peritoneal cavity.
In general, a key concept in vNOTES is "vaginal pull, laparoscopic push," which means that the surgeon must pull the cervix while performing vaginal entry and then push the uterus back in the peritoneal cavity to increase surgical space during laparoscopic surgery.
Continue to: Overview of vNOTES steps...
Overview of vNOTES steps
Below we break down a description of vNOTES in 6 sections. Our patients are always placed in dorsal lithotomy position with TrenGuard (D.A. Surgical) Trendelenburg restraint. We prep the abdomen in case we need to convert to transabdominal surgery via transumbilical single-incision laparoscopic surgery or traditional laparoscopic surgery.
1. Vaginal entry
Accessing the peritoneal cavity through the vagina initially proceeds like a vaginal hysterectomy. We inject dilute vasopressin (20 U in 20 mL of normal saline) circumferentially in the cervix (for hysterectomy) or in the posterior cervix in the cervicovaginal junction (for adnexal surgery without hysterectomy) for vasoconstriction and hydrodissection.
We then incise the vaginal mucosa circumferentially with electrosurgical cautery and follow with posterior colpotomy. We find that reapproximating the posterior peritoneum to the posterior vagina with either figure-of-8 stitches or a running stitch of polyglactin 910 suture (2-0 Vicryl) assists in port placement, bleeding at the peritoneal edge, and closure of the cuff or colpotomy at the end of the case. We tag this suture with a curved hemostat.
Depending on whether a hysterectomy is being performed, anterior colpotomy is made. Again, the anterior peritoneum is then tagged to the anterior vaginal cuff in similar fashion, and this suture is tagged with a different instrument; we typically use a straight hemostat or Sarot clamp (FIGURE 1).
2. Traditional vaginal hysterectomy
After colpotomy, we prefer to perform progressive clamping of the broad ligament from the uterosacral and cardinal ligaments to the level of uterine artery as in traditional vaginal hysterectomy, if feasible.
3. Single-site port placement
The assembled GelPOINT Mini advanced access platform (Applied Medical) (FIGURE 2) is introduced through the vagina after the Alexis wound protector (included with the kit) is first placed through the colpotomy with assistance of Babcock clamps (FIGURE 3).
After ensuring that the green rigid ring of the Alexis wound protector is contained and completely expanded within the peritoneal cavity, we cross our previously tagged sutures as we find this helps with preventing the GelPOINT Mini access platform from inadvertently shifting out of the peritoneal cavity during surgery. The GelSeal cap is then secured and pneumoperitoneum is established (FIGURE 4).
Continue to: 4. Laparoendoscopic surgery...
4. Laparoendoscopic surgery
Instruments used in our surgeries include a 10-mm rigid 30° 43-cm working length laparoscope; a 44-cm LigaSure device (Medtronic); a 5-mm, 37-cm laparoscopic cobra grasping forceps and fenestrated grasper (Karl Storz); and a 5-mm, 45-cm laparoscopic suction with hydrodissection tip (Stryker) (FIGURE 5).
vNOTES allows a gynecologic surgeon the unique ability to survey the upper abdomen. The remainder of the surgery proceeds using basic laparoscopic single-site skills.
During vNOTES, as with all single-site surgical procedures, understanding the optimal placement of crossed instruments is important for successful completion. For example, when securing the right uterine artery, the surgeon needs to push the cervix toward the patient's left and slightly into the peritoneal cavity using a laparoscopic cobra grasper with his or her left hand while then securing the uterine pedicle using the LigaSure device with his or her right hand. This is then reversed when securing the left uterine artery, where the assistant surgeon pushes the cervix toward the patient's right while the surgeon secures the pedicle ("vaginal pull, laparoscopic push") (FIGURE 6).
This again is reiterated in securing the ovarian pedicles, which are pushed into the peritoneal cavity while being secured with the LigaSure device.
5. Specimen removal
For large uteri or specimens that need morcellation, a 15-mm Endo Catch specimen retrieval bag (Medtronic) is introduced through the GelPOINT Mini system. The specimen is then placed in the bag and delivered to the vagina, where contained bag morcellation is performed in standard fashion (FIGURES 7 AND 8). We utilized the "big C" technique by first grasping the specimen with a penetrating clamp. The clamp is then held in our nondominant hand and a No. 10 blade scalpel is used to create a reverse c-incision, keeping one surface of the specimen intact. This is continued until the specimen can be completely delivered through the vagina.
Specimens that do not require morcellation can be grasped laparoscopically, brought to the GelPOINT Mini port, which is quickly disassembled, and delivered. The GelSeal cap is then reassembled.
6. Vaginal cuff closure
The colpotomy or vaginal cuff is closed with barbed suture continuously, as in traditional vaginal hysterectomy cuff closure. Uterosacral ligament suspension should be performed for vaginal cuff support.
vNOTES is the most recent innovative development in the field of minimally invasive surgery, and it has demonstrated feasibility and safety in the fields of general surgery, urology, and gynecology. Adopting vNOTES in clinical practice can improve patient satisfaction and cosmesis as well as surgical outcomes. Gynecologic surgeons can think of vNOTES hysterectomy as "placing an eye" in the vagina while performing transvaginal hysterectomy. The surgical principle of "vaginal pull, laparoscopic push" facilitates the learning process.
1. ACOG Committee on Gynecologic Practice. Committee opinion no. 444. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
2. AAGL Advancing Minimally Invasive Gynecology Worldwide. AAGL position statement: route of hysterectomy to treat benign uterine disease. J Minim Invasive Gynecol. 2011;18:1-3.
3. Whiteside JL, Kaeser CT, Ridgeway B. Achieving high value in the surgical approach to hysterectomy. Am J Obstet Gynecol. 2019;220:242-245.
4. Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
5. Moen M, Walter A, Harmanli O, et al. Considerations to improve the evidence-based use of vaginal hysterectomy in benign gynecology. Obstet Gynecol. 2014;124:585-588.
6. Balgobin S, Owens DM, Florian-Rodriguez ME, et al. Vaginal hysterectomy suturing skills training model and curriculum. Obstet Gynecol. 2019;134:553-558.
7. Baekelandt J. Total vaginal NOTES hysterectomy: a new approach to hysterectomy. J Minim Invasive Gynecol. 2015;22:1088-1094.
8. Baekelandt JF, De Mulder PA, Le Roy I, et al. Hysterectomy by transvaginal natural orifice transluminal endoscopic surgery versus laparoscopy as a day-care procedure: a randomised controlled trial. BJOG. 2019;126:105-113.
9. Guan X, Bardawil E, Liu J, et al. Transvaginal natural orifice transluminal endoscopic surgery as a rescue for total vaginal hysterectomy. J Minim Invasive Gynecol. 2018;25:1135-1136.
10. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;3:CD003677.
11. Kalloo AN, Singh VK, Jagannath SB, et al. Flexible transgastric peritoneoscopy: a novel approach to diagnostic and therapeutic interventions in the peritoneal cavity. Gastrointest Endosc. 2004;60:114-117.
12. Reddy N, Rao P. Per oral transgastric endoscopic appendectomy in human. Paper Presented at: 45th Annual Conference of the Society of Gastrointestinal Endoscopy of India; February 28-29, 2004; Jaipur, India.
13. Clark MP, Qayed ES, Kooby DA, et al. Natural orifice translumenal endoscopic surgery in humans: a review. Minim Invasive Surg. 2012;189296.
14. Rattner D, Kalloo A; ASGE/SAGES Working Group. ASGE/ SAGES Working Group on natural orifice translumenal endoscopic surgery, October 2005. Surg Endosc. 2006;20:329-333.
15. Autorino R, Yakoubi R, White WM, et al. Natural orifice transluminal endoscopic surgery (NOTES): where are we going? A bibliometric assessment. BJU Int. 2013;111:11-16.
16. Santos BF, Hungness ES. Natural orifice transluminal endoscopic surgery: progress in humans since the white paper. World J Gastroenterol. 2011;17:1655-1665.
17. Tolcher MC, Kalogera E, Hopkins MR, et al. Safety of culdotomy as a surgical approach: implications for natural orifice transluminal endoscopic surgery. JSLS. 2012;16:413-420.
18. ACOG Committee on Gynecologic Practice. Committee opinion no. 701. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017:129:e155-e159.
19. Ahn KH, Song JY, Kim SH, et al. Transvaginal single-port natural orifice transluminal endoscopic surgery for benign uterine adnexal pathologies. J Minim Invasive Gynecol. 2012;19:631-635.
20. Liu J, Kohn J, Sun B, et al. Transvaginal natural orifice transluminal endoscopic surgery sacrocolpopexy: tips and tricks. Minim Invasive Gynecol. 2019;26:38-39.
21. Liu J, Kohn J, Fu H, et al. Transvaginal natural orifice transluminal endoscopic surgery for sacrocolpopexy: a pilot study of 26 cases. J Minim Invasive Gynecol. 2019;26:748-753.
22. Su H, Yen CF, Wu KY, et al. Hysterectomy via transvaginal natural orifice transluminal endoscopic surgery (NOTES): feasibility of an innovative approach. Taiwan J Obstet Gynecol. 2012;51:217-221.
23. Lee CL, Huang CY, Wu KY, et al. Natural orifice transvaginal endoscopic surgery myomectomy: an innovative approach to myomectomy. Gynecol Minim Invasive Ther. 2014;3:127-130.
24. Chen Y, Li J, Zhang Y, et al. Transvaginal single-port laparoscopy sacrocolpopexy. J Minim Invasive Gynecol. 2018;25:585- 588.
25. Lee CL, Wu KY, Tsao FY, et al. Natural orifice transvaginal endoscopic surgery for endometrial cancer. Gynecol Minim Invasive Ther. 2014;3:89-92.
26. Leblanc E, Narducci F, Bresson L, et al. Fluorescence-assisted sentinel (SND) and pelvic node dissections by single-port transvaginal laparoscopic surgery, for the management of an endometrial carcinoma (EC) in an elderly obese patient. Gynecol Oncol. 2016;143:686-687.
27. Lee CL, Wu KY, Su H, et al. Robot-assisted natural orifice transluminal endoscopic surgery for hysterectomy. Taiwan J Obstet Gynecol. 2015;54:761-765.
28. Rezai S, Giovane RA, Johnson SN, et al. Robotic natural orifice transluminal endoscopic surgery (R-NOTES) in gynecologic surgeries, a case report and review of literature. Obstet Gynecol Int J. 2019;10:287-289.
29. Wang CJ, Wu PY, Kuo HH, et al. Natural orifice transluminal endoscopic surgery-assisted versus laparoscopic ovarian cystectomy (NAOC vs. LOC): a case-matched study. Surg Endosc. 2016;30:1227-1234.
1. ACOG Committee on Gynecologic Practice. Committee opinion no. 444. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
2. AAGL Advancing Minimally Invasive Gynecology Worldwide. AAGL position statement: route of hysterectomy to treat benign uterine disease. J Minim Invasive Gynecol. 2011;18:1-3.
3. Whiteside JL, Kaeser CT, Ridgeway B. Achieving high value in the surgical approach to hysterectomy. Am J Obstet Gynecol. 2019;220:242-245.
4. Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
5. Moen M, Walter A, Harmanli O, et al. Considerations to improve the evidence-based use of vaginal hysterectomy in benign gynecology. Obstet Gynecol. 2014;124:585-588.
6. Balgobin S, Owens DM, Florian-Rodriguez ME, et al. Vaginal hysterectomy suturing skills training model and curriculum. Obstet Gynecol. 2019;134:553-558.
7. Baekelandt J. Total vaginal NOTES hysterectomy: a new approach to hysterectomy. J Minim Invasive Gynecol. 2015;22:1088-1094.
8. Baekelandt JF, De Mulder PA, Le Roy I, et al. Hysterectomy by transvaginal natural orifice transluminal endoscopic surgery versus laparoscopy as a day-care procedure: a randomised controlled trial. BJOG. 2019;126:105-113.
9. Guan X, Bardawil E, Liu J, et al. Transvaginal natural orifice transluminal endoscopic surgery as a rescue for total vaginal hysterectomy. J Minim Invasive Gynecol. 2018;25:1135-1136.
10. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;3:CD003677.
11. Kalloo AN, Singh VK, Jagannath SB, et al. Flexible transgastric peritoneoscopy: a novel approach to diagnostic and therapeutic interventions in the peritoneal cavity. Gastrointest Endosc. 2004;60:114-117.
12. Reddy N, Rao P. Per oral transgastric endoscopic appendectomy in human. Paper Presented at: 45th Annual Conference of the Society of Gastrointestinal Endoscopy of India; February 28-29, 2004; Jaipur, India.
13. Clark MP, Qayed ES, Kooby DA, et al. Natural orifice translumenal endoscopic surgery in humans: a review. Minim Invasive Surg. 2012;189296.
14. Rattner D, Kalloo A; ASGE/SAGES Working Group. ASGE/ SAGES Working Group on natural orifice translumenal endoscopic surgery, October 2005. Surg Endosc. 2006;20:329-333.
15. Autorino R, Yakoubi R, White WM, et al. Natural orifice transluminal endoscopic surgery (NOTES): where are we going? A bibliometric assessment. BJU Int. 2013;111:11-16.
16. Santos BF, Hungness ES. Natural orifice transluminal endoscopic surgery: progress in humans since the white paper. World J Gastroenterol. 2011;17:1655-1665.
17. Tolcher MC, Kalogera E, Hopkins MR, et al. Safety of culdotomy as a surgical approach: implications for natural orifice transluminal endoscopic surgery. JSLS. 2012;16:413-420.
18. ACOG Committee on Gynecologic Practice. Committee opinion no. 701. Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017:129:e155-e159.
19. Ahn KH, Song JY, Kim SH, et al. Transvaginal single-port natural orifice transluminal endoscopic surgery for benign uterine adnexal pathologies. J Minim Invasive Gynecol. 2012;19:631-635.
20. Liu J, Kohn J, Sun B, et al. Transvaginal natural orifice transluminal endoscopic surgery sacrocolpopexy: tips and tricks. Minim Invasive Gynecol. 2019;26:38-39.
21. Liu J, Kohn J, Fu H, et al. Transvaginal natural orifice transluminal endoscopic surgery for sacrocolpopexy: a pilot study of 26 cases. J Minim Invasive Gynecol. 2019;26:748-753.
22. Su H, Yen CF, Wu KY, et al. Hysterectomy via transvaginal natural orifice transluminal endoscopic surgery (NOTES): feasibility of an innovative approach. Taiwan J Obstet Gynecol. 2012;51:217-221.
23. Lee CL, Huang CY, Wu KY, et al. Natural orifice transvaginal endoscopic surgery myomectomy: an innovative approach to myomectomy. Gynecol Minim Invasive Ther. 2014;3:127-130.
24. Chen Y, Li J, Zhang Y, et al. Transvaginal single-port laparoscopy sacrocolpopexy. J Minim Invasive Gynecol. 2018;25:585- 588.
25. Lee CL, Wu KY, Tsao FY, et al. Natural orifice transvaginal endoscopic surgery for endometrial cancer. Gynecol Minim Invasive Ther. 2014;3:89-92.
26. Leblanc E, Narducci F, Bresson L, et al. Fluorescence-assisted sentinel (SND) and pelvic node dissections by single-port transvaginal laparoscopic surgery, for the management of an endometrial carcinoma (EC) in an elderly obese patient. Gynecol Oncol. 2016;143:686-687.
27. Lee CL, Wu KY, Su H, et al. Robot-assisted natural orifice transluminal endoscopic surgery for hysterectomy. Taiwan J Obstet Gynecol. 2015;54:761-765.
28. Rezai S, Giovane RA, Johnson SN, et al. Robotic natural orifice transluminal endoscopic surgery (R-NOTES) in gynecologic surgeries, a case report and review of literature. Obstet Gynecol Int J. 2019;10:287-289.
29. Wang CJ, Wu PY, Kuo HH, et al. Natural orifice transluminal endoscopic surgery-assisted versus laparoscopic ovarian cystectomy (NAOC vs. LOC): a case-matched study. Surg Endosc. 2016;30:1227-1234.

Medical management of abnormal uterine bleeding in reproductive-age women
Case 1 Multiparous woman presents with heavy regular menses
Over the past several years, a 34-year-old woman has noted increasing intensity and duration of menstrual flow, which now persists for 8 days and includes clots “the size of quarters” and soaks a pad within 1 hour. Sometimes she misses or leaves work on her heaviest days of flow. She reports that menstrual cramps prior to and during flow are increasingly bothersome and do not respond adequately to ibuprofen. She intermittently uses condoms for contraception. She does not wish to be pregnant currently; however, she recently entered into a new relationship and may wish to conceive in the future.
On bimanual examination, the uterus appears bulky. Her hemoglobin is 10.9 g/dL with low mean corpuscular volume and a serum ferritin level indicating iron depletion. Pelvic ultrasonography suggests uterine adenomyosis; no fibroids are imaged (FIGURE 1).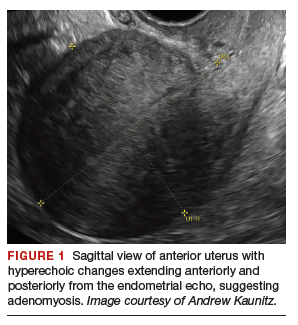
You advise the patient to take ferrous sulfate 325 mg every other day. After discussion with the patient regarding different treatment options, she chooses to proceed with placement of a 52-mg levonorgestrel (LNG) intrauterine device (IUD; Mirena or Liletta).
Case 2 Older adolescent presents with irregular bleeding
A 19-year-old patient reports approximately 6 bleeding episodes each year. She reports the duration of her bleeding as variable, and sometimes the bleeding is heavy with small clots passed. She has been previously diagnosed with polycystic ovary syndrome (PCOS). Combination estrogen-progestin oral contraceptives have been prescribed several times in the past, but she always has discontinued them due to nausea. The patient is in a same-sex relationship and does not anticipate being sexually active with a male. She reports having to shave her mustache and chin twice weekly for the past 1 to 2 years.
On physical examination, the patient is obese (body mass index [BMI], 32 kg/m2), facial acne and hirsutism are present, and hair extends from the mons toward the umbilicus. Bimanual examination reveals a normal size, mobile, nontender uterus without obvious adnexal pathology. Pelvic ultrasonography demonstrates a normal-appearing uterus with multiplanar endometrium (consistent with proliferative changes) (FIGURE 2). Ovarian imaging demonstrates ≥12 follicles per image (FIGURE 3).
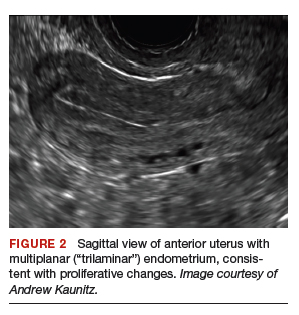

After reviewing various treatment options, you prescribe oral medroxyprogesterone acetate 20 mg (two 10-mg tablets) daily in a continuous fashion. You counsel her that she should not be surprised or concerned if frequent or even continuous bleeding occurs initially, and that she should continue this medication despite the occurrence of such.
About one-third of all women experience abnormal uterine bleeding (AUB) sometime during their lifetime and AUB can impair quality of life.1 Surgical management, including hysterectomy and endometrial ablation, plays an important role in the management of AUB in patients who do not desire future pregnancies. However, many cases of AUB occur in women who may not have completed childbearing or in women who prefer to avoid surgery.2 AUB can be managed effectively medically in most cases.1 Accordingly, in this review, we focus on nonsurgical management of AUB.
Continue to: Because previously used terms, including...
Because previously used terms, including menorrhagia and meno-metrorrhagia, were inconsistently defined and confusing, the International Federation of Gynecology and Obstetrics introduced updated terminology in 2011 to better describe and characterize AUB in nonpregnant women. Heavy menstrual bleeding (HMB) refers to ovulatory (cyclic) bleeding that is more than 8 days’ duration, or sufficiently heavy to impair a woman’s quality of life. HMB is a pattern of AUB distinct from the irregular bleeding pattern typically caused by ovulatory dysfunction (AUB-O).1
Clinical evaluation
Obtain menstrual history. In addition to a medical, surgical, and gynecologic history, a thorough menstrual history should be obtained to further characterize the patient’s bleeding pattern. In contrast to the cyclical or ovulatory bleeding seen with HMB, bleeding associated with inconsistent ovulation (AUB-O) is unpredictable or irregular, and is commonly associated with PCOS. AUB-O is also encountered in recently menarchal girls (secondary to immaturity of the hypothalamic-pituitary-gonadal axis) and in those who are perimenopausal. In addition, medications that can induce hyperprolactinemia (such as certain antipsychotics) can cause AUB-O.
Evaluate for all sources of bleeding. Be sure to evaluate for extrauterine causes of bleeding, including the cervix, vagina, vulva, or the urinary or gastrointestinal tracts for bleeding. Intermenstrual bleeding occurring between normal regular menses may be caused by an endometrial polyp, submucosal fibroid, endometritis, or an IUD. The patient report of postcoital bleeding suggests that cervical disease (cervicitis, polyp, or malignancy) may be present. Uterine leiomyoma or adenomyosis represent common causes of HMB. However, HMB also may be caused by a copper IUD, coagulation disorders (including von Willebrand disease), or use of anticoagulant medications. Hormonal contraceptives also can cause irregular bleeding.
Perform a pelvic examination and measure vital signs. The presence of fever suggests the possible presence of pelvic inflammatory disease (PID), while orthostatic hypotension raises the possibility of hypovolemia. When vaginal speculum examination is performed, a cervical cause of abnormal bleeding may be noted. The presence of fresh or old blood or finding clots in the vaginal vault or at the cervical os are all consistent with AUB. A bimanual examination that reveals an enlarged or lobular uterus suggests leiomyoma or adenomyosis. Cervical or adnexal tenderness is often noted in women with PID, which itself may be associated with endometritis. The presence of hyperandrogenic signs on physical examination (eg, acne, hirsutism, or clitoromegaly) suggests PCOS. The finding of galactorrhea suggests that hyperprolactinemia may be present.
Laboratory assessment
Test for pregnancy, cervical disease, and sexually transmitted infection when appropriate. Pregnancy testing is appropriate for women with AUB aged 55 years or younger. If patients with AUB are not up to date with normal cervical cancer screening results, cervical cytology and/or human papillomavirus testing should be performed. Testing for Chlamydia trachomatis, Neisseria gonorrhoeae, and Trichomonas vaginalis should be performed in patients:
- younger than 25 years
- when the history indicates new or multiple sexual partners, or
- when vaginal discharge, cervicitis, cervical motion, or adnexal tenderness is present.
Continue to: Obtain a complete blood count and serum ferritin levels...
Obtain a complete blood count and serum ferritin levels. In women presenting with HMB, iron depletion and iron deficiency anemia are common. The finding of leukocytosis raises the possibility of PID or postpartum endometritis. In women with presumptive AUB-O, checking the levels of thyroid-stimulating hormone, free T4, and prolactin should be performed.
Screen for a hemostasis disorder. Women with excessive menstrual bleeding should be clinically screened for an underlying disorder of hemostasis (TABLE 1).3 When a hemostasis disorder is suspected, initial laboratory evaluation includes a partial thromboplastin time, prothrombin time, activated partial thromboplastin time, and fibrinogen. Women who have a positive clinical screen for a possible bleeding disorder or abnormal initial laboratory test results for disorders of hemostasis should undergo further laboratory evaluation, including von Willebrand factor antigen, ristocetin cofactor assay, and factor VIII. Consultation with a hematologist should be considered in these cases.

Perform endometrial biopsy when indicated
After excluding pregnancy, endometrial biopsy (through pipelle biospy or brush sampling; FIGURE 4) should be performed in women with AUB who are at increased risk for endometrial neoplasia. The prevalence of endometrial neoplasia is substantially higher among women ≥45 years of age4 and among patients with AUB who are also obese (BMI, ≥30 kg/m2).5 In addition, AUB patients with unopposed estrogen exposure (presumed anovulation/PCOS), as well as those with persistent AUB or failed medical management, should undergo endometrial biopsy.6
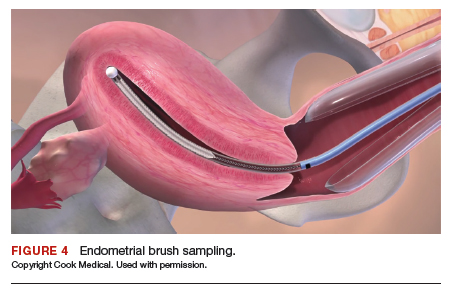
Utilize transvaginal ultrasonography
Transvaginal ultrasonography is often useful in the evaluation of patients with AUB, as it may identify uterine fibroids or adenomyosis, suggest intracavitary pathology (such as an endometrial polyp or submucosal fibroid), or raise the possibility of PCOS. In virginal patients or those in whom vaginal ultrasound is not appropriate, abdominal pelvic ultrasonography represents appropriate imaging. If unenhanced ultrasound suggests endometrial polyps or fibroids within the endometrial cavity, an office-based saline infusion sonogram (sonohysterogram) (FIGURE 5) or hysteroscopy should be performed. Targeted endometrial sampling and biopsy of intracavitary pathology can be performed at the time of hysteroscopy.
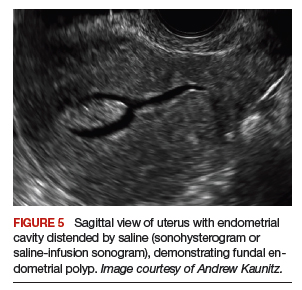
Treatment
When HMB impairs quality of life, is bothersome to the patient, or results in anemia, treatment is appropriate. Although bleeding episodes in women with AUB-O may be infrequent (as with Case 2), treatment prevents heavy or prolonged bleeding episodes as well as endometrial neoplasia that may otherwise occur in anovulatory women.
Many women with AUB can be managed medically. However, treatment choices will vary with respect to the patient’s desire for future fertility, medical comorbidities, personal preferences, and financial barriers. While many women may prefer outpatient medical management (TABLE 2),7-14 others might desire surgical therapy, including endometrial ablation or hysterectomy.

Oral contraceptives
Combination estrogen-progestin oral contraceptives represent appropriate initial therapy for many women in the reproductive-age group with AUB, whether women have HMB or AUB-O. However, contraceptive doses of estrogen are not appropriate for some women with risk factors for cardiovascular disease, including those who smoke cigarettes and are age ≥35 years or those who have hypertension (TABLE 3).15,16
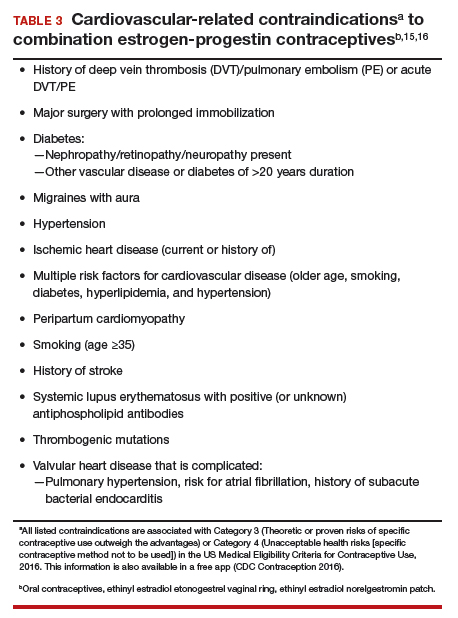
Continue to: Menopausal dosages of HT...
Menopausal dosages of HT
If use of contraceptive doses of estrogen is not appropriate, continuous off-label use of menopausal combination formulations (physiologic dosage) of hormonal therapy (HT; ie, lower doses of estrogen than contraceptives) may be effective in reducing or eliminating AUB. Options for menopausal combination formulations include generic ethinyl estradiol 5 µg/norethindrone acetate 1 mg or estradiol 1 mg/norethindrone acetate 0.5 mg.7 High-dose oral progestin therapy (norethindrone acetate 5 mg tablet once daily or medroxyprogesterone acetate 10 mg tablets 1–3 times daily) also can be used when combination contraceptives are contraindicated and may be more effective than lower-dose combination formulations.
Package labeling, as well as some guidelines, indicate that oral progestins used to treat AUB should be taken cyclically.8 However, continuous daily use is easier for many patients and may be more effective in reducing bleeding. Accordingly, we counsel patients with AUB who are using progestins and who do not wish to conceive to take these medications continuously. High-dose oral progestin therapy may cause bloating, dysphoria, and increased appetite/weight gain. Women initiating hormonal management (including the progestin IUDs detailed below) for AUB should be counseled that irregular or even continuous light bleeding/spotting is common initially, but this bleeding pattern typically decreases with continued use.
IUDs
The LNG 52 mg IUD (Mirena or Liletta) effectively treats HMB, reducing bleeding in a manner comparable to that of endometrial ablation.9,10 The Mirena IUD is approved for treatment of HMB in women desiring intrauterine contraception. In contrast to oral medications, use of progestin IUDs does not involve daily administration and may represent an attractive option for women with HMB who would like to avoid surgery or preserve fertility. With ongoing use, continuous oral or intrauterine hormonal management may result in amenorrhea in some women with AUB.
When the LNG 52 mg IUD is used to treat HMB, the menstrual suppression impact may begin to attenuate after approximately 4 years of use; in this setting, replacing the IUD often restores effective menstrual suppression.11 The LNG 52 mg IUD effectively suppresses menses in women with coagulation disorders; if menstrual suppression with the progestin IUD is not adequate in this setting, it may be appropriate to add an oral combination estrogen-progestin contraceptive or high-dose oral progestin.11,12
NSAIDs and tranexamic acid
Off-label use of nonsteroidal anti-inflammatory drugs (naproxen 500–1,000 mg daily for 5 days beginning at the onset of menstrual flow or tranexamic acid two 650-mg tablets 3 times daily for up to 5 days during episodes of heavy flow) can suppress HMB and is useful for women who prefer to avoid or have contraindications to hormonal treatments.13,14 Unfortunately, these agents are not as effective as hormonal management in treating AUB.
Iron supplementation is often needed
Iron depletion commonly results from HMB, often resulting in iron deficiency anemia. When iron depletion (readily identified by checking a serum ferritin level) or iron deficiency anemia is identified, iron supplementation should be recommended. Every-other-day administration of iron supplements maximizes iron absorption while minimizing the adverse effects of unabsorbed iron, such as nausea. Sixty mg of elemental iron (ferrous sulfate 325 mg) administered every other day represents an inexpensive and effective treatment for iron deficiency/anemia.17 In patients who cannot tolerate oral iron supplementation or for those in whom oral therapy is not appropriate or effective, newer intravenous iron formulations are safe and effective.18
Continue to: Case 1 Follow-up...
Case 1 Follow-up
The patient noted marked improvement in her menstrual cramps following LNG-containing IUD placement. Although she also reported that she no longer experienced heavy menstrual flow or cramps, she was bothered by frequent, unpredictable light bleeding/spotting. You prescribed norethindrone acetate (NETA) 5-mg tablet orally once daily, to be used in addition to her IUD. After using the IUD with concomitant NETA for 2 months’ duration, she noted that her bleeding/spotting almost completely resolved; however, she did report feeling irritable with use of the progestin tablets. She subsequently stopped the NETA tablets and, after 6 months of additional follow-up, reported only minimal spotting and no cramps.
At this later follow-up visit, you noted that her hemoglobin level increased to 12.6 g/dL, and the ferritin level no longer indicated iron depletion. After the IUD had been in place for 4 years, she reported that she was beginning to experience frequent light bleeding again. A follow-up vaginal sonogram noted a well-positioned IUD, there was no suggestion of intracavitary pathology, and adenomyosis continued to be imaged. She underwent IUD removal and placement of a new LNG 52 mg IUD. This resulted in marked reduction in her bleeding.
Case 2 Follow-up
Two weeks after beginning continuous oral progestin therapy, the patient called reporting frequent irregular bleeding. She was reassured that this was not unexpected and encouraged to continue oral progestin therapy. During a 3-month follow-up visit, the patient noted little, if any, bleeding over the previous 2 months and was pleased with this result. She continued to note acne and hirsutism and asked about the possibility of adding spironolactone to her oral progestin regimen.
- Munro MG, Critchley HOD, Fraser IS; FIGO Menstrual Disorders Committee. The two FIGO systems for normal and abnormal uterine bleeding symptoms and classification of causes of abnormal uterine bleeding in the reproductive years: 2018 revisions. Int J Gynecol Obstet. 2018;143:393-408.
- Kaunitz AM. Abnormal uterine bleeding in reproductive-age women. JAMA. 2019;321:2126-2127.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 557: management of acute abnormal uterine bleeding in nonpregnant reproductive-aged women. Obstet Gynecol. 2013;121:891-896.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Uterine Cancer. http://seer.cancer.gov/statfacts/html/corp.html. Accessed October 10, 2019.
- Wise MR, Gill P, Lensen S, et al. Body mass index trumps age in decision for endometrial biopsy: cohort study of symptomatic premenopausal women. Am J Obstet Gynecol. 2016;215:598.e1-598.e8.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
- The North American Menopause Society. Menopause Practice–A Clinician’s Guide. 5th ed. NAMS: Mayfield Heights, OH; 2014.
- National Institute for Health and Care Excellence. Heavy menstrual bleeding: assessment and management. https://www.nice.org.uk/guidance/ng88. Accessed October 10, 2019.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrel-releasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:625-632.
- Kaunitz AM, Meredith S, Inki P, et al. Levonorgestrel-releasing intrauterine system and endometrial ablation in heavy menstrual bleeding: a systematic review and meta-analysis. Obstet Gynecol. 2009;113:1104-1116.
- Kaunitz AM, Inki P. The levonorgestrel-releasing intrauterine system in heavy menstrual bleeding: a benefit-risk review. Drugs. 2012;72:193-215.
- James AH, Kouides PA, Abdul-Kadir R, et al. Von Willebrand disease and other bleeding disorders in women: consensus on diagnosis and management from an international expert panel. Am J Obstet Gynecol. 2009;201:12.e1-8.
- Ylikorkala O, Pekonen F. Naproxen reduces idiopathic but not fibromyoma-induced menorrhagia. Obstet Gynecol. 1986;68:10-12.
- Lukes AS, Moore KA, Muse KN, et al. Tranexamic acid treatment for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:865-875.
- Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1–103.
- ACOG Practice Bulletin no. 206: use of hormonal contraception in women with coexisting medical conditions. Obstet Gynecol. 2019;133:e128-e150.
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4:e524–e533.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91:31-38.

Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women’s Health Specialists at Emerson, Jacksonville. He serves on the OBG

Dr. McCullough is Assistant Professor, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville.

Dr. Burnett is Assistant Professor, Department of Obstetrics and Gynecology; Chief, Maternal Fetal Medicine; Program Director, Ultrasound and Prenatal Diagnosis; and Clerkship Director, University of Florida College of Medicine-Jacksonville.
Dr. Kaunitz reports receiving grant or research support from Allergan, Bayer, and Medicines360 and being a consultant to Pfizer. Drs. McCullough and Burnett report no financial relationships relevant to this article.
Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women’s Health Specialists at Emerson, Jacksonville. He serves on the OBG
Dr. McCullough is Assistant Professor, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville.
Dr. Burnett is Assistant Professor, Department of Obstetrics and Gynecology; Chief, Maternal Fetal Medicine; Program Director, Ultrasound and Prenatal Diagnosis; and Clerkship Director, University of Florida College of Medicine-Jacksonville.
Dr. Kaunitz reports receiving grant or research support from Allergan, Bayer, and Medicines360 and being a consultant to Pfizer. Drs. McCullough and Burnett report no financial relationships relevant to this article.
Dr. Kaunitz is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women’s Health Specialists at Emerson, Jacksonville. He serves on the OBG
Dr. McCullough is Assistant Professor, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville.
Dr. Burnett is Assistant Professor, Department of Obstetrics and Gynecology; Chief, Maternal Fetal Medicine; Program Director, Ultrasound and Prenatal Diagnosis; and Clerkship Director, University of Florida College of Medicine-Jacksonville.
Dr. Kaunitz reports receiving grant or research support from Allergan, Bayer, and Medicines360 and being a consultant to Pfizer. Drs. McCullough and Burnett report no financial relationships relevant to this article.
Case 1 Multiparous woman presents with heavy regular menses
Over the past several years, a 34-year-old woman has noted increasing intensity and duration of menstrual flow, which now persists for 8 days and includes clots “the size of quarters” and soaks a pad within 1 hour. Sometimes she misses or leaves work on her heaviest days of flow. She reports that menstrual cramps prior to and during flow are increasingly bothersome and do not respond adequately to ibuprofen. She intermittently uses condoms for contraception. She does not wish to be pregnant currently; however, she recently entered into a new relationship and may wish to conceive in the future.
On bimanual examination, the uterus appears bulky. Her hemoglobin is 10.9 g/dL with low mean corpuscular volume and a serum ferritin level indicating iron depletion. Pelvic ultrasonography suggests uterine adenomyosis; no fibroids are imaged (FIGURE 1).
You advise the patient to take ferrous sulfate 325 mg every other day. After discussion with the patient regarding different treatment options, she chooses to proceed with placement of a 52-mg levonorgestrel (LNG) intrauterine device (IUD; Mirena or Liletta).
Case 2 Older adolescent presents with irregular bleeding
A 19-year-old patient reports approximately 6 bleeding episodes each year. She reports the duration of her bleeding as variable, and sometimes the bleeding is heavy with small clots passed. She has been previously diagnosed with polycystic ovary syndrome (PCOS). Combination estrogen-progestin oral contraceptives have been prescribed several times in the past, but she always has discontinued them due to nausea. The patient is in a same-sex relationship and does not anticipate being sexually active with a male. She reports having to shave her mustache and chin twice weekly for the past 1 to 2 years.
On physical examination, the patient is obese (body mass index [BMI], 32 kg/m2), facial acne and hirsutism are present, and hair extends from the mons toward the umbilicus. Bimanual examination reveals a normal size, mobile, nontender uterus without obvious adnexal pathology. Pelvic ultrasonography demonstrates a normal-appearing uterus with multiplanar endometrium (consistent with proliferative changes) (FIGURE 2). Ovarian imaging demonstrates ≥12 follicles per image (FIGURE 3).
After reviewing various treatment options, you prescribe oral medroxyprogesterone acetate 20 mg (two 10-mg tablets) daily in a continuous fashion. You counsel her that she should not be surprised or concerned if frequent or even continuous bleeding occurs initially, and that she should continue this medication despite the occurrence of such.
About one-third of all women experience abnormal uterine bleeding (AUB) sometime during their lifetime and AUB can impair quality of life.1 Surgical management, including hysterectomy and endometrial ablation, plays an important role in the management of AUB in patients who do not desire future pregnancies. However, many cases of AUB occur in women who may not have completed childbearing or in women who prefer to avoid surgery.2 AUB can be managed effectively medically in most cases.1 Accordingly, in this review, we focus on nonsurgical management of AUB.
Continue to: Because previously used terms, including...
Because previously used terms, including menorrhagia and meno-metrorrhagia, were inconsistently defined and confusing, the International Federation of Gynecology and Obstetrics introduced updated terminology in 2011 to better describe and characterize AUB in nonpregnant women. Heavy menstrual bleeding (HMB) refers to ovulatory (cyclic) bleeding that is more than 8 days’ duration, or sufficiently heavy to impair a woman’s quality of life. HMB is a pattern of AUB distinct from the irregular bleeding pattern typically caused by ovulatory dysfunction (AUB-O).1
Clinical evaluation
Obtain menstrual history. In addition to a medical, surgical, and gynecologic history, a thorough menstrual history should be obtained to further characterize the patient’s bleeding pattern. In contrast to the cyclical or ovulatory bleeding seen with HMB, bleeding associated with inconsistent ovulation (AUB-O) is unpredictable or irregular, and is commonly associated with PCOS. AUB-O is also encountered in recently menarchal girls (secondary to immaturity of the hypothalamic-pituitary-gonadal axis) and in those who are perimenopausal. In addition, medications that can induce hyperprolactinemia (such as certain antipsychotics) can cause AUB-O.
Evaluate for all sources of bleeding. Be sure to evaluate for extrauterine causes of bleeding, including the cervix, vagina, vulva, or the urinary or gastrointestinal tracts for bleeding. Intermenstrual bleeding occurring between normal regular menses may be caused by an endometrial polyp, submucosal fibroid, endometritis, or an IUD. The patient report of postcoital bleeding suggests that cervical disease (cervicitis, polyp, or malignancy) may be present. Uterine leiomyoma or adenomyosis represent common causes of HMB. However, HMB also may be caused by a copper IUD, coagulation disorders (including von Willebrand disease), or use of anticoagulant medications. Hormonal contraceptives also can cause irregular bleeding.
Perform a pelvic examination and measure vital signs. The presence of fever suggests the possible presence of pelvic inflammatory disease (PID), while orthostatic hypotension raises the possibility of hypovolemia. When vaginal speculum examination is performed, a cervical cause of abnormal bleeding may be noted. The presence of fresh or old blood or finding clots in the vaginal vault or at the cervical os are all consistent with AUB. A bimanual examination that reveals an enlarged or lobular uterus suggests leiomyoma or adenomyosis. Cervical or adnexal tenderness is often noted in women with PID, which itself may be associated with endometritis. The presence of hyperandrogenic signs on physical examination (eg, acne, hirsutism, or clitoromegaly) suggests PCOS. The finding of galactorrhea suggests that hyperprolactinemia may be present.
Laboratory assessment
Test for pregnancy, cervical disease, and sexually transmitted infection when appropriate. Pregnancy testing is appropriate for women with AUB aged 55 years or younger. If patients with AUB are not up to date with normal cervical cancer screening results, cervical cytology and/or human papillomavirus testing should be performed. Testing for Chlamydia trachomatis, Neisseria gonorrhoeae, and Trichomonas vaginalis should be performed in patients:
- younger than 25 years
- when the history indicates new or multiple sexual partners, or
- when vaginal discharge, cervicitis, cervical motion, or adnexal tenderness is present.
Continue to: Obtain a complete blood count and serum ferritin levels...
Obtain a complete blood count and serum ferritin levels. In women presenting with HMB, iron depletion and iron deficiency anemia are common. The finding of leukocytosis raises the possibility of PID or postpartum endometritis. In women with presumptive AUB-O, checking the levels of thyroid-stimulating hormone, free T4, and prolactin should be performed.
Screen for a hemostasis disorder. Women with excessive menstrual bleeding should be clinically screened for an underlying disorder of hemostasis (TABLE 1).3 When a hemostasis disorder is suspected, initial laboratory evaluation includes a partial thromboplastin time, prothrombin time, activated partial thromboplastin time, and fibrinogen. Women who have a positive clinical screen for a possible bleeding disorder or abnormal initial laboratory test results for disorders of hemostasis should undergo further laboratory evaluation, including von Willebrand factor antigen, ristocetin cofactor assay, and factor VIII. Consultation with a hematologist should be considered in these cases.
Perform endometrial biopsy when indicated
After excluding pregnancy, endometrial biopsy (through pipelle biospy or brush sampling; FIGURE 4) should be performed in women with AUB who are at increased risk for endometrial neoplasia. The prevalence of endometrial neoplasia is substantially higher among women ≥45 years of age4 and among patients with AUB who are also obese (BMI, ≥30 kg/m2).5 In addition, AUB patients with unopposed estrogen exposure (presumed anovulation/PCOS), as well as those with persistent AUB or failed medical management, should undergo endometrial biopsy.6
Utilize transvaginal ultrasonography
Transvaginal ultrasonography is often useful in the evaluation of patients with AUB, as it may identify uterine fibroids or adenomyosis, suggest intracavitary pathology (such as an endometrial polyp or submucosal fibroid), or raise the possibility of PCOS. In virginal patients or those in whom vaginal ultrasound is not appropriate, abdominal pelvic ultrasonography represents appropriate imaging. If unenhanced ultrasound suggests endometrial polyps or fibroids within the endometrial cavity, an office-based saline infusion sonogram (sonohysterogram) (FIGURE 5) or hysteroscopy should be performed. Targeted endometrial sampling and biopsy of intracavitary pathology can be performed at the time of hysteroscopy.
Treatment
When HMB impairs quality of life, is bothersome to the patient, or results in anemia, treatment is appropriate. Although bleeding episodes in women with AUB-O may be infrequent (as with Case 2), treatment prevents heavy or prolonged bleeding episodes as well as endometrial neoplasia that may otherwise occur in anovulatory women.
Many women with AUB can be managed medically. However, treatment choices will vary with respect to the patient’s desire for future fertility, medical comorbidities, personal preferences, and financial barriers. While many women may prefer outpatient medical management (TABLE 2),7-14 others might desire surgical therapy, including endometrial ablation or hysterectomy.
Oral contraceptives
Combination estrogen-progestin oral contraceptives represent appropriate initial therapy for many women in the reproductive-age group with AUB, whether women have HMB or AUB-O. However, contraceptive doses of estrogen are not appropriate for some women with risk factors for cardiovascular disease, including those who smoke cigarettes and are age ≥35 years or those who have hypertension (TABLE 3).15,16
Continue to: Menopausal dosages of HT...
Menopausal dosages of HT
If use of contraceptive doses of estrogen is not appropriate, continuous off-label use of menopausal combination formulations (physiologic dosage) of hormonal therapy (HT; ie, lower doses of estrogen than contraceptives) may be effective in reducing or eliminating AUB. Options for menopausal combination formulations include generic ethinyl estradiol 5 µg/norethindrone acetate 1 mg or estradiol 1 mg/norethindrone acetate 0.5 mg.7 High-dose oral progestin therapy (norethindrone acetate 5 mg tablet once daily or medroxyprogesterone acetate 10 mg tablets 1–3 times daily) also can be used when combination contraceptives are contraindicated and may be more effective than lower-dose combination formulations.
Package labeling, as well as some guidelines, indicate that oral progestins used to treat AUB should be taken cyclically.8 However, continuous daily use is easier for many patients and may be more effective in reducing bleeding. Accordingly, we counsel patients with AUB who are using progestins and who do not wish to conceive to take these medications continuously. High-dose oral progestin therapy may cause bloating, dysphoria, and increased appetite/weight gain. Women initiating hormonal management (including the progestin IUDs detailed below) for AUB should be counseled that irregular or even continuous light bleeding/spotting is common initially, but this bleeding pattern typically decreases with continued use.
IUDs
The LNG 52 mg IUD (Mirena or Liletta) effectively treats HMB, reducing bleeding in a manner comparable to that of endometrial ablation.9,10 The Mirena IUD is approved for treatment of HMB in women desiring intrauterine contraception. In contrast to oral medications, use of progestin IUDs does not involve daily administration and may represent an attractive option for women with HMB who would like to avoid surgery or preserve fertility. With ongoing use, continuous oral or intrauterine hormonal management may result in amenorrhea in some women with AUB.
When the LNG 52 mg IUD is used to treat HMB, the menstrual suppression impact may begin to attenuate after approximately 4 years of use; in this setting, replacing the IUD often restores effective menstrual suppression.11 The LNG 52 mg IUD effectively suppresses menses in women with coagulation disorders; if menstrual suppression with the progestin IUD is not adequate in this setting, it may be appropriate to add an oral combination estrogen-progestin contraceptive or high-dose oral progestin.11,12
NSAIDs and tranexamic acid
Off-label use of nonsteroidal anti-inflammatory drugs (naproxen 500–1,000 mg daily for 5 days beginning at the onset of menstrual flow or tranexamic acid two 650-mg tablets 3 times daily for up to 5 days during episodes of heavy flow) can suppress HMB and is useful for women who prefer to avoid or have contraindications to hormonal treatments.13,14 Unfortunately, these agents are not as effective as hormonal management in treating AUB.
Iron supplementation is often needed
Iron depletion commonly results from HMB, often resulting in iron deficiency anemia. When iron depletion (readily identified by checking a serum ferritin level) or iron deficiency anemia is identified, iron supplementation should be recommended. Every-other-day administration of iron supplements maximizes iron absorption while minimizing the adverse effects of unabsorbed iron, such as nausea. Sixty mg of elemental iron (ferrous sulfate 325 mg) administered every other day represents an inexpensive and effective treatment for iron deficiency/anemia.17 In patients who cannot tolerate oral iron supplementation or for those in whom oral therapy is not appropriate or effective, newer intravenous iron formulations are safe and effective.18
Continue to: Case 1 Follow-up...
Case 1 Follow-up
The patient noted marked improvement in her menstrual cramps following LNG-containing IUD placement. Although she also reported that she no longer experienced heavy menstrual flow or cramps, she was bothered by frequent, unpredictable light bleeding/spotting. You prescribed norethindrone acetate (NETA) 5-mg tablet orally once daily, to be used in addition to her IUD. After using the IUD with concomitant NETA for 2 months’ duration, she noted that her bleeding/spotting almost completely resolved; however, she did report feeling irritable with use of the progestin tablets. She subsequently stopped the NETA tablets and, after 6 months of additional follow-up, reported only minimal spotting and no cramps.
At this later follow-up visit, you noted that her hemoglobin level increased to 12.6 g/dL, and the ferritin level no longer indicated iron depletion. After the IUD had been in place for 4 years, she reported that she was beginning to experience frequent light bleeding again. A follow-up vaginal sonogram noted a well-positioned IUD, there was no suggestion of intracavitary pathology, and adenomyosis continued to be imaged. She underwent IUD removal and placement of a new LNG 52 mg IUD. This resulted in marked reduction in her bleeding.
Case 2 Follow-up
Two weeks after beginning continuous oral progestin therapy, the patient called reporting frequent irregular bleeding. She was reassured that this was not unexpected and encouraged to continue oral progestin therapy. During a 3-month follow-up visit, the patient noted little, if any, bleeding over the previous 2 months and was pleased with this result. She continued to note acne and hirsutism and asked about the possibility of adding spironolactone to her oral progestin regimen.
Case 1 Multiparous woman presents with heavy regular menses
Over the past several years, a 34-year-old woman has noted increasing intensity and duration of menstrual flow, which now persists for 8 days and includes clots “the size of quarters” and soaks a pad within 1 hour. Sometimes she misses or leaves work on her heaviest days of flow. She reports that menstrual cramps prior to and during flow are increasingly bothersome and do not respond adequately to ibuprofen. She intermittently uses condoms for contraception. She does not wish to be pregnant currently; however, she recently entered into a new relationship and may wish to conceive in the future.
On bimanual examination, the uterus appears bulky. Her hemoglobin is 10.9 g/dL with low mean corpuscular volume and a serum ferritin level indicating iron depletion. Pelvic ultrasonography suggests uterine adenomyosis; no fibroids are imaged (FIGURE 1).
You advise the patient to take ferrous sulfate 325 mg every other day. After discussion with the patient regarding different treatment options, she chooses to proceed with placement of a 52-mg levonorgestrel (LNG) intrauterine device (IUD; Mirena or Liletta).
Case 2 Older adolescent presents with irregular bleeding
A 19-year-old patient reports approximately 6 bleeding episodes each year. She reports the duration of her bleeding as variable, and sometimes the bleeding is heavy with small clots passed. She has been previously diagnosed with polycystic ovary syndrome (PCOS). Combination estrogen-progestin oral contraceptives have been prescribed several times in the past, but she always has discontinued them due to nausea. The patient is in a same-sex relationship and does not anticipate being sexually active with a male. She reports having to shave her mustache and chin twice weekly for the past 1 to 2 years.
On physical examination, the patient is obese (body mass index [BMI], 32 kg/m2), facial acne and hirsutism are present, and hair extends from the mons toward the umbilicus. Bimanual examination reveals a normal size, mobile, nontender uterus without obvious adnexal pathology. Pelvic ultrasonography demonstrates a normal-appearing uterus with multiplanar endometrium (consistent with proliferative changes) (FIGURE 2). Ovarian imaging demonstrates ≥12 follicles per image (FIGURE 3).
After reviewing various treatment options, you prescribe oral medroxyprogesterone acetate 20 mg (two 10-mg tablets) daily in a continuous fashion. You counsel her that she should not be surprised or concerned if frequent or even continuous bleeding occurs initially, and that she should continue this medication despite the occurrence of such.
About one-third of all women experience abnormal uterine bleeding (AUB) sometime during their lifetime and AUB can impair quality of life.1 Surgical management, including hysterectomy and endometrial ablation, plays an important role in the management of AUB in patients who do not desire future pregnancies. However, many cases of AUB occur in women who may not have completed childbearing or in women who prefer to avoid surgery.2 AUB can be managed effectively medically in most cases.1 Accordingly, in this review, we focus on nonsurgical management of AUB.
Continue to: Because previously used terms, including...
Because previously used terms, including menorrhagia and meno-metrorrhagia, were inconsistently defined and confusing, the International Federation of Gynecology and Obstetrics introduced updated terminology in 2011 to better describe and characterize AUB in nonpregnant women. Heavy menstrual bleeding (HMB) refers to ovulatory (cyclic) bleeding that is more than 8 days’ duration, or sufficiently heavy to impair a woman’s quality of life. HMB is a pattern of AUB distinct from the irregular bleeding pattern typically caused by ovulatory dysfunction (AUB-O).1
Clinical evaluation
Obtain menstrual history. In addition to a medical, surgical, and gynecologic history, a thorough menstrual history should be obtained to further characterize the patient’s bleeding pattern. In contrast to the cyclical or ovulatory bleeding seen with HMB, bleeding associated with inconsistent ovulation (AUB-O) is unpredictable or irregular, and is commonly associated with PCOS. AUB-O is also encountered in recently menarchal girls (secondary to immaturity of the hypothalamic-pituitary-gonadal axis) and in those who are perimenopausal. In addition, medications that can induce hyperprolactinemia (such as certain antipsychotics) can cause AUB-O.
Evaluate for all sources of bleeding. Be sure to evaluate for extrauterine causes of bleeding, including the cervix, vagina, vulva, or the urinary or gastrointestinal tracts for bleeding. Intermenstrual bleeding occurring between normal regular menses may be caused by an endometrial polyp, submucosal fibroid, endometritis, or an IUD. The patient report of postcoital bleeding suggests that cervical disease (cervicitis, polyp, or malignancy) may be present. Uterine leiomyoma or adenomyosis represent common causes of HMB. However, HMB also may be caused by a copper IUD, coagulation disorders (including von Willebrand disease), or use of anticoagulant medications. Hormonal contraceptives also can cause irregular bleeding.
Perform a pelvic examination and measure vital signs. The presence of fever suggests the possible presence of pelvic inflammatory disease (PID), while orthostatic hypotension raises the possibility of hypovolemia. When vaginal speculum examination is performed, a cervical cause of abnormal bleeding may be noted. The presence of fresh or old blood or finding clots in the vaginal vault or at the cervical os are all consistent with AUB. A bimanual examination that reveals an enlarged or lobular uterus suggests leiomyoma or adenomyosis. Cervical or adnexal tenderness is often noted in women with PID, which itself may be associated with endometritis. The presence of hyperandrogenic signs on physical examination (eg, acne, hirsutism, or clitoromegaly) suggests PCOS. The finding of galactorrhea suggests that hyperprolactinemia may be present.
Laboratory assessment
Test for pregnancy, cervical disease, and sexually transmitted infection when appropriate. Pregnancy testing is appropriate for women with AUB aged 55 years or younger. If patients with AUB are not up to date with normal cervical cancer screening results, cervical cytology and/or human papillomavirus testing should be performed. Testing for Chlamydia trachomatis, Neisseria gonorrhoeae, and Trichomonas vaginalis should be performed in patients:
- younger than 25 years
- when the history indicates new or multiple sexual partners, or
- when vaginal discharge, cervicitis, cervical motion, or adnexal tenderness is present.
Continue to: Obtain a complete blood count and serum ferritin levels...
Obtain a complete blood count and serum ferritin levels. In women presenting with HMB, iron depletion and iron deficiency anemia are common. The finding of leukocytosis raises the possibility of PID or postpartum endometritis. In women with presumptive AUB-O, checking the levels of thyroid-stimulating hormone, free T4, and prolactin should be performed.
Screen for a hemostasis disorder. Women with excessive menstrual bleeding should be clinically screened for an underlying disorder of hemostasis (TABLE 1).3 When a hemostasis disorder is suspected, initial laboratory evaluation includes a partial thromboplastin time, prothrombin time, activated partial thromboplastin time, and fibrinogen. Women who have a positive clinical screen for a possible bleeding disorder or abnormal initial laboratory test results for disorders of hemostasis should undergo further laboratory evaluation, including von Willebrand factor antigen, ristocetin cofactor assay, and factor VIII. Consultation with a hematologist should be considered in these cases.
Perform endometrial biopsy when indicated
After excluding pregnancy, endometrial biopsy (through pipelle biospy or brush sampling; FIGURE 4) should be performed in women with AUB who are at increased risk for endometrial neoplasia. The prevalence of endometrial neoplasia is substantially higher among women ≥45 years of age4 and among patients with AUB who are also obese (BMI, ≥30 kg/m2).5 In addition, AUB patients with unopposed estrogen exposure (presumed anovulation/PCOS), as well as those with persistent AUB or failed medical management, should undergo endometrial biopsy.6
Utilize transvaginal ultrasonography
Transvaginal ultrasonography is often useful in the evaluation of patients with AUB, as it may identify uterine fibroids or adenomyosis, suggest intracavitary pathology (such as an endometrial polyp or submucosal fibroid), or raise the possibility of PCOS. In virginal patients or those in whom vaginal ultrasound is not appropriate, abdominal pelvic ultrasonography represents appropriate imaging. If unenhanced ultrasound suggests endometrial polyps or fibroids within the endometrial cavity, an office-based saline infusion sonogram (sonohysterogram) (FIGURE 5) or hysteroscopy should be performed. Targeted endometrial sampling and biopsy of intracavitary pathology can be performed at the time of hysteroscopy.
Treatment
When HMB impairs quality of life, is bothersome to the patient, or results in anemia, treatment is appropriate. Although bleeding episodes in women with AUB-O may be infrequent (as with Case 2), treatment prevents heavy or prolonged bleeding episodes as well as endometrial neoplasia that may otherwise occur in anovulatory women.
Many women with AUB can be managed medically. However, treatment choices will vary with respect to the patient’s desire for future fertility, medical comorbidities, personal preferences, and financial barriers. While many women may prefer outpatient medical management (TABLE 2),7-14 others might desire surgical therapy, including endometrial ablation or hysterectomy.
Oral contraceptives
Combination estrogen-progestin oral contraceptives represent appropriate initial therapy for many women in the reproductive-age group with AUB, whether women have HMB or AUB-O. However, contraceptive doses of estrogen are not appropriate for some women with risk factors for cardiovascular disease, including those who smoke cigarettes and are age ≥35 years or those who have hypertension (TABLE 3).15,16
Continue to: Menopausal dosages of HT...
Menopausal dosages of HT
If use of contraceptive doses of estrogen is not appropriate, continuous off-label use of menopausal combination formulations (physiologic dosage) of hormonal therapy (HT; ie, lower doses of estrogen than contraceptives) may be effective in reducing or eliminating AUB. Options for menopausal combination formulations include generic ethinyl estradiol 5 µg/norethindrone acetate 1 mg or estradiol 1 mg/norethindrone acetate 0.5 mg.7 High-dose oral progestin therapy (norethindrone acetate 5 mg tablet once daily or medroxyprogesterone acetate 10 mg tablets 1–3 times daily) also can be used when combination contraceptives are contraindicated and may be more effective than lower-dose combination formulations.
Package labeling, as well as some guidelines, indicate that oral progestins used to treat AUB should be taken cyclically.8 However, continuous daily use is easier for many patients and may be more effective in reducing bleeding. Accordingly, we counsel patients with AUB who are using progestins and who do not wish to conceive to take these medications continuously. High-dose oral progestin therapy may cause bloating, dysphoria, and increased appetite/weight gain. Women initiating hormonal management (including the progestin IUDs detailed below) for AUB should be counseled that irregular or even continuous light bleeding/spotting is common initially, but this bleeding pattern typically decreases with continued use.
IUDs
The LNG 52 mg IUD (Mirena or Liletta) effectively treats HMB, reducing bleeding in a manner comparable to that of endometrial ablation.9,10 The Mirena IUD is approved for treatment of HMB in women desiring intrauterine contraception. In contrast to oral medications, use of progestin IUDs does not involve daily administration and may represent an attractive option for women with HMB who would like to avoid surgery or preserve fertility. With ongoing use, continuous oral or intrauterine hormonal management may result in amenorrhea in some women with AUB.
When the LNG 52 mg IUD is used to treat HMB, the menstrual suppression impact may begin to attenuate after approximately 4 years of use; in this setting, replacing the IUD often restores effective menstrual suppression.11 The LNG 52 mg IUD effectively suppresses menses in women with coagulation disorders; if menstrual suppression with the progestin IUD is not adequate in this setting, it may be appropriate to add an oral combination estrogen-progestin contraceptive or high-dose oral progestin.11,12
NSAIDs and tranexamic acid
Off-label use of nonsteroidal anti-inflammatory drugs (naproxen 500–1,000 mg daily for 5 days beginning at the onset of menstrual flow or tranexamic acid two 650-mg tablets 3 times daily for up to 5 days during episodes of heavy flow) can suppress HMB and is useful for women who prefer to avoid or have contraindications to hormonal treatments.13,14 Unfortunately, these agents are not as effective as hormonal management in treating AUB.
Iron supplementation is often needed
Iron depletion commonly results from HMB, often resulting in iron deficiency anemia. When iron depletion (readily identified by checking a serum ferritin level) or iron deficiency anemia is identified, iron supplementation should be recommended. Every-other-day administration of iron supplements maximizes iron absorption while minimizing the adverse effects of unabsorbed iron, such as nausea. Sixty mg of elemental iron (ferrous sulfate 325 mg) administered every other day represents an inexpensive and effective treatment for iron deficiency/anemia.17 In patients who cannot tolerate oral iron supplementation or for those in whom oral therapy is not appropriate or effective, newer intravenous iron formulations are safe and effective.18
Continue to: Case 1 Follow-up...
Case 1 Follow-up
The patient noted marked improvement in her menstrual cramps following LNG-containing IUD placement. Although she also reported that she no longer experienced heavy menstrual flow or cramps, she was bothered by frequent, unpredictable light bleeding/spotting. You prescribed norethindrone acetate (NETA) 5-mg tablet orally once daily, to be used in addition to her IUD. After using the IUD with concomitant NETA for 2 months’ duration, she noted that her bleeding/spotting almost completely resolved; however, she did report feeling irritable with use of the progestin tablets. She subsequently stopped the NETA tablets and, after 6 months of additional follow-up, reported only minimal spotting and no cramps.
At this later follow-up visit, you noted that her hemoglobin level increased to 12.6 g/dL, and the ferritin level no longer indicated iron depletion. After the IUD had been in place for 4 years, she reported that she was beginning to experience frequent light bleeding again. A follow-up vaginal sonogram noted a well-positioned IUD, there was no suggestion of intracavitary pathology, and adenomyosis continued to be imaged. She underwent IUD removal and placement of a new LNG 52 mg IUD. This resulted in marked reduction in her bleeding.
Case 2 Follow-up
Two weeks after beginning continuous oral progestin therapy, the patient called reporting frequent irregular bleeding. She was reassured that this was not unexpected and encouraged to continue oral progestin therapy. During a 3-month follow-up visit, the patient noted little, if any, bleeding over the previous 2 months and was pleased with this result. She continued to note acne and hirsutism and asked about the possibility of adding spironolactone to her oral progestin regimen.
- Munro MG, Critchley HOD, Fraser IS; FIGO Menstrual Disorders Committee. The two FIGO systems for normal and abnormal uterine bleeding symptoms and classification of causes of abnormal uterine bleeding in the reproductive years: 2018 revisions. Int J Gynecol Obstet. 2018;143:393-408.
- Kaunitz AM. Abnormal uterine bleeding in reproductive-age women. JAMA. 2019;321:2126-2127.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 557: management of acute abnormal uterine bleeding in nonpregnant reproductive-aged women. Obstet Gynecol. 2013;121:891-896.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Uterine Cancer. http://seer.cancer.gov/statfacts/html/corp.html. Accessed October 10, 2019.
- Wise MR, Gill P, Lensen S, et al. Body mass index trumps age in decision for endometrial biopsy: cohort study of symptomatic premenopausal women. Am J Obstet Gynecol. 2016;215:598.e1-598.e8.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
- The North American Menopause Society. Menopause Practice–A Clinician’s Guide. 5th ed. NAMS: Mayfield Heights, OH; 2014.
- National Institute for Health and Care Excellence. Heavy menstrual bleeding: assessment and management. https://www.nice.org.uk/guidance/ng88. Accessed October 10, 2019.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrel-releasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:625-632.
- Kaunitz AM, Meredith S, Inki P, et al. Levonorgestrel-releasing intrauterine system and endometrial ablation in heavy menstrual bleeding: a systematic review and meta-analysis. Obstet Gynecol. 2009;113:1104-1116.
- Kaunitz AM, Inki P. The levonorgestrel-releasing intrauterine system in heavy menstrual bleeding: a benefit-risk review. Drugs. 2012;72:193-215.
- James AH, Kouides PA, Abdul-Kadir R, et al. Von Willebrand disease and other bleeding disorders in women: consensus on diagnosis and management from an international expert panel. Am J Obstet Gynecol. 2009;201:12.e1-8.
- Ylikorkala O, Pekonen F. Naproxen reduces idiopathic but not fibromyoma-induced menorrhagia. Obstet Gynecol. 1986;68:10-12.
- Lukes AS, Moore KA, Muse KN, et al. Tranexamic acid treatment for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:865-875.
- Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1–103.
- ACOG Practice Bulletin no. 206: use of hormonal contraception in women with coexisting medical conditions. Obstet Gynecol. 2019;133:e128-e150.
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4:e524–e533.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91:31-38.
- Munro MG, Critchley HOD, Fraser IS; FIGO Menstrual Disorders Committee. The two FIGO systems for normal and abnormal uterine bleeding symptoms and classification of causes of abnormal uterine bleeding in the reproductive years: 2018 revisions. Int J Gynecol Obstet. 2018;143:393-408.
- Kaunitz AM. Abnormal uterine bleeding in reproductive-age women. JAMA. 2019;321:2126-2127.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 557: management of acute abnormal uterine bleeding in nonpregnant reproductive-aged women. Obstet Gynecol. 2013;121:891-896.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Uterine Cancer. http://seer.cancer.gov/statfacts/html/corp.html. Accessed October 10, 2019.
- Wise MR, Gill P, Lensen S, et al. Body mass index trumps age in decision for endometrial biopsy: cohort study of symptomatic premenopausal women. Am J Obstet Gynecol. 2016;215:598.e1-598.e8.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins—Gynecology. Practice bulletin no. 128: diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
- The North American Menopause Society. Menopause Practice–A Clinician’s Guide. 5th ed. NAMS: Mayfield Heights, OH; 2014.
- National Institute for Health and Care Excellence. Heavy menstrual bleeding: assessment and management. https://www.nice.org.uk/guidance/ng88. Accessed October 10, 2019.
- Kaunitz AM, Bissonnette F, Monteiro I, et al. Levonorgestrel-releasing intrauterine system or medroxyprogesterone for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:625-632.
- Kaunitz AM, Meredith S, Inki P, et al. Levonorgestrel-releasing intrauterine system and endometrial ablation in heavy menstrual bleeding: a systematic review and meta-analysis. Obstet Gynecol. 2009;113:1104-1116.
- Kaunitz AM, Inki P. The levonorgestrel-releasing intrauterine system in heavy menstrual bleeding: a benefit-risk review. Drugs. 2012;72:193-215.
- James AH, Kouides PA, Abdul-Kadir R, et al. Von Willebrand disease and other bleeding disorders in women: consensus on diagnosis and management from an international expert panel. Am J Obstet Gynecol. 2009;201:12.e1-8.
- Ylikorkala O, Pekonen F. Naproxen reduces idiopathic but not fibromyoma-induced menorrhagia. Obstet Gynecol. 1986;68:10-12.
- Lukes AS, Moore KA, Muse KN, et al. Tranexamic acid treatment for heavy menstrual bleeding: a randomized controlled trial. Obstet Gynecol. 2010;116:865-875.
- Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. 2016;65:1–103.
- ACOG Practice Bulletin no. 206: use of hormonal contraception in women with coexisting medical conditions. Obstet Gynecol. 2019;133:e128-e150.
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4:e524–e533.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91:31-38.
Autoimmune Hemolytic Anemia: Treatment of Common Types
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.

Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.

Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44

Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.

Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune Hemolytic Anemia: Evaluation and Diagnosis
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.

Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.

Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
Aplastic Anemia: Diagnosis and Treatment
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7

Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.

Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68

Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7
Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.
Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68
Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
From the Oregon Health and Science University, Portland, OR.
Abstract
- Objective: To describe the current approach to diagnosis and treatment of aplastic anemia.
- Methods: Review of the literature.
- Results: Aplastic anemia can be acquired or associated with an inherited marrow failure syndrome (IMFS), and the treatment and prognosis vary dramatically between these 2 etiologies. Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to life-threatening neutropenic infections or bleeding. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia.
- Conclusion: Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marro
w transplant program to evaluate for early transplantation is the new standard of care for aplastic anemia.
Keywords: inherited marrow failure syndrome; Fanconi anemia; immunosuppression; transplant; stem cell.
Aplastic anemia is a clinical and pathological entity of bone marrow failure that causes progressive loss of hematopoietic progenitor stem cells (HPSC), resulting in pancytopenia.1 Patients may present along a spectrum, ranging from being asymptomatic with incidental findings on peripheral blood testing to having life-threatening neutropenic infections or bleeding. Aplastic anemia results from either inherited or acquired causes, and the pathophysiology and treatment approach vary significantly between these 2 causes. Therefore, recognition of inherited marrow failure diseases, such as Fanconi anemia and telomere biology disorders, is critical to establishing the management plan.
Epidemiology
Aplastic anemia is a rare disorder, with an incidence of approximately 1.5 to 7 cases per million individuals per year.2,3 A recent Scandinavian study reported that the incidence of aplastic anemia among the Swedish population is 2.3 cases per million individuals per year, with a median age at diagnosis of 60 years and a slight female predominance (52% versus 48%, respectively).2 This data is congruent with prior observations made in Barcelona, where the incidence was 2.34 cases per million individuals per year, albeit with a slightly higher incidence in males compared to females (2.54 versus 2.16, respectively).4 The incidence of aplastic anemia varies globally, with a disproportionate increase in incidence seen among Asian populations, with rates as high as 8.8 per million individuals per year.3-5 This variation in incidence in Asia versus other countries has not been well explained. There appears to be a bimodal distribution, with incidence peaks seen in young adults and in older adults.2,3,6
Pathophysiology
Acquired Aplastic Anemia
The leading hypothesis as to the cause of most cases of acquired aplastic anemia is that a dysregulated immune system destroys HPSCs. Inciting etiologies implicated in the development of acquired aplastic anemia include pregnancy, infection, medications, and exposure to certain chemicals, such as benzene.1,7 The historical understanding of acquired aplastic anemia implicates cytotoxic T-lymphocyte–mediated destruction of CD34+ hematopoietic stem cells.1,8,9 This hypothesis served as the basis for treatment of acquired aplastic anemia with immunosuppressive therapy, predominantly anti-thymocyte globulin (ATG) combined with cyclosporine A.1,8 More recent work has focused on cytokine interactions, particularly the suppressive role of interferon (IFN)-γ on hematopoietic stem cells independent of T-lymphocyte–mediated destruction, which has been demonstrated in a murine model.8 The interaction of IFN-γ with the hematopoietic stem cell pool is dynamic. IFN-γ levels are elevated during an acute inflammatory response, such as a viral infection, providing further basis for the immune-mediated nature of the acquired disease.10 Specifically, in vitro studies suggest the effects of IFN-γ on HPSC may be secondary to interruption of thrombopoietin and its respective signaling pathways, which play a key role in hematopoietic stem cell renewal.11 Eltrombopag, a thrombopoietin receptor antagonist, has shown promise in the treatment of refractory aplastic anemia, with studies indicating that its effectiveness is independent of IFN-γ levels.11,12
Inherited Aplastic Anemia
The inherited marrow failure syndromes (IMFSs) are a group of disorders characterized by cellular maintenance and repair defects, leading to cytopenias, increased cancer risk, structural defects, and risk of end organ damage, such as liver cirrhosis and pulmonary fibrosis.13-15 The most common diseases include Fanconi anemia, dyskeratosis congenita/telomere biology disorders, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome, but with the advent of whole exome sequencing, new syndromes continue to be discovered. While classically these disorders present in children, adult presentations are now commonplace. Broadly, the pathophysiology of inherited aplastic anemia relates to the defective HPSCs and an accelerated decline of the hematopoietic stem cell compartment.
The most common IMFSs, Fanconi anemia and telomere biology disorders, are associated with numerous mutations in DNA damage repair pathways and telomere maintenance pathways. TERT, DKC, and TERC mutations are most commonly associated with dyskeratosis congenita, but may also be found infrequently in patients with aplastic anemia presenting at an older age in the absence of the classic phenotypical features.1,16,17 The recognition of an underlying genetic disorder or telomere biology disorder leading to constitutional aplastic anemia is significant, as these conditions are associated not only with marrow failure, but also with endocrinopathies, organ fibrosis, and and hematopoietic and solid organ malignancies.13-15 In particular, TERT and TERC gene mutations have been associated with dyskeratosis congenita as well as pulmonary fibrosis and cirrhosis.18,19 The implications of early diagnosis of an IMFS lie in the approach to treatment and prognosis.
Clonal Disorders and Secondary Malignancies
Myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) are 2 clonal disorders that may arise from a background of aplastic anemia.9,20,21 Hypoplastic MDS can be difficult to differentiate from aplastic anemia at diagnosis based on morphology alone, although recent work has demonstrated that molecular testing for somatic mutations in ASXL1, DNMT3A, and BCOR can aid in differentiating a subset of aplastic anemia patients who are more likely to progress to MDS.21 Clonal populations of cells harboring 6p uniparental disomy are seen in more than 10% of patients with aplastic anemia on cytogenetic analysis, which can help differentiate the diseases.9 Yoshizato and colleagues found lower rates of ASXL1 and DNMT3A mutations in patients with aplastic anemia as compared with patients with MDS or AML. In this study, patients with aplastic anemia had higher rates of mutations in PIGA (reflecting the increased paroxysmal nocturnal hemoglobinuria [PNH] clonality seen in aplastic anemia) and BCOR.9 Mutations were also found in genes commonly mutated in MDS and AML, including TET2, RUNX1, TP53, and JAK2, albeit at lower frequencies.9 These mutations as a whole have not predicted response to therapy or prognosis. However, when performing survival analysis in patients with specific mutations, those commonly encountered in MDS/AML (ASXL1, DNMT3A, TP53, RUNX1, CSMD1) are associated with faster progression to overt MDS/AML and decreased overall survival (OS),20,21 suggesting these mutations may represent early clonality that can lead to clonal evolution and the development of secondary malignancies. Conversely, mutations in BCOR and BCORL appear to identify patients who may have a favorable outcome in response to immunosuppressive therapy and, similar to patients with PIGA mutations, improved OS.9
Paroxysmal Nocturnal Hemoglobinuria
In addition to having an increased risk of myelodysplasia and malignancy due to the development of a dominant pre-malignant clone, patients with aplastic anemia often harbor progenitor cell clones associated with PNH.1,17 PNH clones have been identified in more than 50% of patients with aplastic anemia.22,23 PNH represents a clonal disorder of hematopoiesis in which cells harbor X-linked somatic mutations in the PIGA gene; this gene encodes a protein responsible for the synthesis of glycosylphosphatidylinositol anchors on the cell surface.22,24 The lack of these cell surface proteins, specifically CD55 (also known as decay accelerating factor) and CD59 (also known as membrane inhibitor of reactive lysis), predisposes red cells to increased complement-mediated lysis.25 The exact mechanism for the development of these clones in patients with aplastic anemia is not fully understood. Current theories hypothesize that the clones are protected from the immune-mediated destruction of normal hematopoietic stem cells due to the absence of the cell surface proteins.1,20 The role of these clones over time in patients with aplastic anemia is less clear, though recent work demonstrated that despite differences in clonality over the disease course, aplastic anemia patients with small PNH clones are less likely to develop overt hemolysis and larger PNH clones compared to patients harboring larger (≥ 50%) PNH clones at diagnosis.23,26,27 Additionally, PNH clones in patients with aplastic anemia infrequently become clinically significant.27 It should be noted that these conditions exist along a continuum; that is, patients with aplastic anemia may develop PNH clones, while conversely patients with PNH may develop aplastic anemia.20 Patients with PNH clones should be followed via peripheral blood flow cytometry and complete blood count to track clonal stability and identify clinically significant PNH among aplastic anemia patients.28
Clinical Presentation
Patients with aplastic anemia typically are diagnosed either due to asymptomatic cytopenias found on peripheral blood sampling, symptomatic anemia, bleeding secondary to thrombocytopenia, or wound healing and infectious complications related to neutropenia.29 A thorough history to understand the timing of symptoms, recent infectious symptoms/exposure, habits, and chemical or toxin exposures (including medications, travel, and supplements) helps guide diagnostic testing. Family history is also critical, with attention given to premature graying; pulmonary, renal, and liver disease; and blood disorders.
Patients with an IMFS (eg, Fanconi anemia or dyskeratosis congenita) may have associated phenotypical findings such as urogenital abnormalities or short stature; in addition, those with dyskeratosis congenita may present with the classic triad of oral leukoplakia, lacy skin pigmentation, and dystrophic nails.7 However, classic phenotypical findings may be lacking in up to 30% to 40% of patients with an IMFS.7 As described previously, while congenital malformations are common in Fanconi anemia and dyskeratosis congenita, a third of patients may have no or only subtle phenotypical abnormalities, including alterations in skin or hair pigmentation, skeletal and growth abnormalities, and endocrine disorders.30 The International Fanconi Anemia Registry identified central nervous system, genitourinary, skin and musculoskeletal, ophthalmic, and gastrointestinal system malformations among children with Fanconi anemia.31,32 Patients with dyskeratosis congenita may present with pulmonary fibrosis, hepatic cirrhosis, or premature graying, as highlighted in a recent study by DiNardo and colleagues.33 Therefore, physicians must have a heightened index of suspicion in patients with subtle phenotypical findings and associated cytopenias.
Diagnosis
The diagnosis of aplastic anemia should be suspected in any patient presenting with pancytopenia. Aplastic anemia is a diagnosis of exclusion.34 Other conditions associated with peripheral blood pancytopenia should be considered, including infections (HIV, hepatitis, parvovirus B19, cytomegalovirus, Epstein-Barr virus, varicella-zoster virus), nutritional deficiencies (vitamin B12, folate, copper, zinc), autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, hemophagocytic lymphohistiocytosis), hypersplenism, marrow-occupying diseases (eg, leukemia, lymphoma, MDS), solid malignancies, and fibrosis (Table).7
Diagnostic Evaluation
The workup for aplastic anemia should include a thorough history and physical exam to search simultaneously for alternative diagnoses and clues pointing to potential etiologic agents.7 Diagnostic tests to be performed include a complete blood count with differential, reticulocyte count, immature platelet fraction, flow cytometry (to rule out lymphoproliferative disorders and atypical myeloid cells and to evaluate for PNH), and bone marrow biopsy with subsequent cytogenetic, immunohistochemical, and molecular testing.35 Typical findings in aplastic anemia include peripheral blood pancytopenia without dysplastic features and bone marrow biopsy demonstrating a hypocellular marrow.7 A relative lymphocytosis in the peripheral blood is common.7 In patients with a significant PNH clone, a macrocytosis along with elevated lactate dehydrogenase and elevated reticulocyte and granulocyte counts may be present.36
The diagnosis (based on the Camitta criteria37 and modified Camitta criteria38 for severe aplastic anemia) requires 2 of the following findings on peripheral blood samples:
- Absolute neutrophil count (ANC) < 500 cells/µL
- Platelet count < 20,000 cells/µL
- Reticulocyte count < 1% corrected or < 20,000 cells/µL.35
In addition to peripheral blood findings, bone marrow biopsy is essential for the diagnosis, and should demonstrate a markedly hypocellular marrow (cellularity < 25%), occasionally with an increase in T lymphocytes.7,39 Because marrow cellularity varies with age and can be challenging to assess, additional biopsies may be needed to confirm the diagnosis.29 A 1- to 2-cm core biopsy is necessary to confirm hypocellularity, as small areas of residual hematopoiesis may be present and obscure the diagnosis.35
Excluding Hypocellular MDS and IMFS
Excluding hypocellular MDS is challenging, especially in the older adult presenting with aplastic anemia, as patients with aplastic anemia may have some degree of erythroid dysplasia on bone marrow morphology.36 The presence of a PNH clone on flow cytometry can aid in diagnosing aplastic anemia and excluding MDS,34 although PNH clones can be present in refractory anemia MDS. Patients with aplastic anemia have a lower ratio of CD34+ cells compared to those with hypoplastic MDS, with 1 study demonstrating a mean CD34+ percentage of < 0.5% in aplastic anemia versus 3.7% in hypoplastic MDS.40 Cytogenetic and molecular testing can also aid in making this distinction by identifying mutations commonly implicated in MDS.7 The presence of monosomy 7 (-7) in aplastic anemia patients is associated with a poor overall prognosis.34,41
Peripheral blood screening using chromosome breakage analysis (done using either mitomycin C or diepoxybutane as in vitro DNA-crosslinking agents)42 and telomere length testing (of peripheral blood leukocytes) is necessary to exclude the main IMFSs, Fanconi anemia and telomere biology disorders, respectively. Ruling out these conditions is imperative, as the approach to treatment varies significantly between IMFS and aplastic anemia. Patients with shortened telomeres should undergo genetic screening for mutations in the telomere maintenance genes to evaluate the underlying defect leading to shortened telomeres. Patients with increased peripheral blood breakage should have genetic testing to detect mutations associated with Fanconi anemia.
Classification
Once the diagnosis of aplastic anemia has been made, the patient should be classified according to the severity of their disease. Disease severity is determined based on peripheral blood ANC: non-severe aplastic anemia (NSAA), ANC > 500 polymorphonuclear neutrophils (PMNs)/µL; severe aplastic anemia (SAA), 200–500 PMNs/µL; and very severe aplastic anemia (VSAA), 0–200 PMNs/µL.4,34 Disease classification is important, as VSAA is associated with a decreased OS compared to SAA.2 Disease classification may affect treatment decisions, as patients with NSAA may be observed for a short period of time, while, conversely, patients with SAA have a worse prognosis with delays in therapy.43-45
Treatment of Inherited Aplastic Anemia
First-line treatment options for patients with IMFS are androgen therapy and hematopoietic stem cell transplant (HSCT). When evaluating patients for HSCT, it is critical to identify the presence of an IMFS, as the risk and mortality associated with the conditioning regimen, stem cell source, graft-versus-host disease (GVHD), and secondary malignancies differ between patients with IMFS and those with acquired marrow failure syndromes or hematologic malignancies.
Potential sibling donors need to be screened for donor candidacy as well as for the inherited defect. Among patients with Fanconi anemia or a telomere biology disorder, the stem cell source must be considered, with multiple studies in IMFSs and SAA showing superior outcomes with a bone marrow product compared to peripheral blood stem cells.46-48 In IMFS patients, the donor cell type may affect the choice of conditioning regimen.5,6 Reduced-intensity conditioning in lieu of myeloablative conditioning without total body irradiation has proved feasible in patients with Fanconi anemia, and is associated with a reduced risk of secondary malignancies.49,50 Incorporation of fludarabine in the conditioning regimen of patients without a matched sibling donor is associated with superior engraftment and survival46,49,51 compared to cyclophosphamide conditioning, which was historically used in matched related donors.50,52 Adding fludarabine appears to be especially beneficial in older patients, in whom its use is associated with lower rates of graft failure, likely due to increased immunosuppression at the time of engraftment.51,53 Fludarabine has also been incorporated into conditioning regimens for patients with a telomere biology disorder, but outcomes data are limited.5
For patients presenting with AML or a high-risk MDS who are subsequently diagnosed with an IMFS, treatment can be more complex, as these patients are at high risk for toxicity from standard chemotherapy. Limited data suggest that induction therapy and transplantation are feasible in this group of patients, and this approach is associated with increased OS, despite lower OS rates than those of IMFS patients who present prior to the development of MDS or AML.54,55 Further work is needed to determine the optimal induction regimen that balances the risks of treatment-related mortality and complications associated with conditioning regimens, risk of relapse, and risk of secondary malignancies, especially in the cohort of patients diagnosed at an older age.
Treatment of Acquired Aplastic Anemia
Supportive Care
While the workup and treatment plan are being established, attention should be directed at supportive care for prevention of complications. The most common complications leading to death in patients with significant pancytopenia and neutropenia are opportunistic infections and hemorrhagic complications.2
Transfusion support is critical to avoid symptomatic anemia and hemorrhagic complications related to thrombocytopenia, which typically occur with platelet counts lower than 10,000 cells/µL. However, transfusion carries the risk of alloimmunization (which may persist for years following transfusion) and transfusion-related graft versus host disease (trGVHD), and thus use of transfusion should be minimized when possible.56,57 All blood products given to patients with aplastic anemia should be irradiated and leukoreduced to reduce the risk of both alloimmunization and trGVHD. Guidelines from the British Society for Haematology recommend routine screening for Rh and Kell antibodies to reduce the risk of alloimmunization.58 Infectious complications remain a common cause of morbidity and mortality in patients with aplastic anemia who have prolonged neutropenia (defined as an ANC < 500 cells/µL).59-62 Therefore, patients should receive broad-spectrum antibiotics with antipseudomonal coverage. In a study evaluating the role of granulocyte-colony stimulating factor (G-CSF) in patients with SAA receiving immunosuppressive therapy, 55% of all patient deaths were secondary to infection.63 There was no OS benefit seen in patients who received G-CSF, though a significantly lower rate of infection was observed in the G-CSF arm compared to those not receiving G-CSF (56% versus 81%, P = 0.006). This difference was largely driven by a decrease in infectious episodes in patients with VSAA treated with G-CSF as compared to those who did not receive this therapy (22% versus 48%, P = 0.014).63
Angio-invasive pulmonary aspergillosis and Zygomycetes (eg, Rhizopus, Mucor species) remain major causes of mortality related to opportunistic mycotic infections in patients with aplastic anemia.18 The infectious risk is directly related to the duration and severity of neutropenia, with one study demonstrating a significant increase in risk in AML patients with neutropenia lasting longer than 3 weeks.64 Invasive fungal infections carry a high mortality in patients with severe neutropenia, though due to earlier recognition and empiric antifungal therapy with extended-spectrum azoles, overall mortality secondary to invasive fungal infections is declining.62,65
While neutropenia related to cytotoxic chemotherapy is commonly associated with gram-negative bacteria due to disruption of mucosal barriers, patients with aplastic anemia have an increased incidence of gram-positive bacteremia with staphylococcal species compared to other neutropenic populations.61,62 This appears to be changing with time. Valdez et al demonstrated a decrease in prevalence of coagulase-negative staphylococcal infections, increased prevalence of gram-positive bacilli bacteremia, and no change in prevalence of gram-negative bacteremia in patients with aplastic anemia treated between 1989 and 2008.65 Gram-negative bacteremia caused by Stenotrophomonas maltophila, Escherichia coli, Klebsiella pneumoniae, Citrobacter, and Proteus has also been reported.62 Despite a lack of clinical trials investigating the role of antifungal and antibacterial prophylaxis for patients with aplastic anemia, most centers initiate antifungal prophylaxis in patients with SAA or VSAA with an anti-mold agent such as voriconazole or posaconazole (which has the additional benefit compared to voriconazole of covering Mucor species).60,66 This is especially true for patients who have received ATG or undergone HSCT. For antimicrobial prophylaxis, a fluoroquinolone antibiotic with a spectrum of activity against Pseudomonas should be considered for patients with an ANC < 500 cells/µL.60 Acyclovir or valacyclovir prophylaxis is recommended for varicella-zoster virus and herpes simplex virus. Cytomegalovirus reactivation is minimal in patients with aplastic anemia, unless multiple courses of ATG are used.
Iron overload is another complication the provider must be aware of in the setting of increased transfusions in aplastic anemia patients. Lee and colleagues showed that iron chelation therapy using deferasirox is effective at reducing serum ferritin levels in patients with aplastic anemia (median ferritin level of 3254 ng/mL prior to therapy, 1854 ng/mL following), and is associated with no serious adverse events (most common adverse events included nausea, diarrhea, vomiting, and rash).67 Approximately 25% of patients in this trial had an increase in creatinine, with patients taking concomitant cyclosporine affected to a greater degree than those on chelation therapy alone. For patients following HSCT or with improved hematopoiesis following immunosuppressive therapy, phlebotomy can be used to treat iron overload in lieu of chelation therapy.58
Approach to Therapy
The main treatment options for SAA and VSAA include allogeneic bone marrow transplant and immunosuppression. The deciding factors as to which treatment is best initially depends on the availability of HLA-matched related donors and age (Figure 1 and Figure 2). Survival is decreased in patients with SAA or VSAA who delay initiation of therapy, and therefore prompt referral for HLA typing and evaluation for bone marrow transplant is a very important first step in managing aplastic anemia.
Matched Sibling Donor Transplant. Current standards of care recommend HLA-matched sibling donor transplant for patients with SAA or VSAA who are younger than 50 years, with the caveat that integration of fludarabine and reduced cyclophosphamide dosing along with ATG shows the best overall outcomes. Locasciulli and colleagues examined outcomes in patients given either immunosuppressive therapy or sibling HSCT between 1991-1996 and 1997-2002, respectively, and found that sibling HSCT was associated with a superior 10-year OS compared to immunosuppressive therapy (73% versus 68%).43 Interestingly in this study, there was no OS improvement seen with immunosuppressive therapy alone (69% versus 73%) between the 2 time periods, despite increased OS in both sibling HSCT (74% and 80%) and MUD HSCT (38% and 65%).43 Though total body irradiation has been used in the past, it is typically not included in current conditioning regimens for matched related donor transplants.68
Current conditioning regimens typically use a combination of cyclophosphamide and ATG,69,70 with or without fludarabine. Fludarabine-based conditioning regimens have shown promise in patients undergoing sibling HSCT. Maury and colleagues evaluated the role of fludarabine in addition to low-dose cyclophosphamide and ATG compared to cyclophosphamide alone or in combination with ATG in patients over age 30 undergoing sibling HSCT.53 There was a nonsignificant improvement in 5-year OS in the fludarabine arm compared to controls (77% ± 8% versus 60% ± 3%, P = 0.14) in the pooled analysis, but when adjusted for age the fludarabine arm had a significantly lower relative risk (RR) of death (0.44; P = 0.04) compared to the control arm. Shin et al reported outcomes with fludarabine/cyclophosphamide/ATG, with excellent overall outcomes and no difference in patients older or younger than 40 years.71
Kim et al evaluated their experience with patients older than 40 years receiving matched related donors, finding comparable outcomes in those ages 41 to 50 years compared to younger patients. Outcomes declined in those over the age of 50 years.72 Long-term data for matched related donor transplant for aplastic anemia show excellent long-term outcomes, with minimal chronic GVHD and good performance status.73 Hence, these factors support the role of matched related donor transplant as the initial treatment in SAA and VSAA.
Regarding the role of transplant for patients who lack a matched related donor, a growing body of literature demonstrating identical outcomes between matched related and MUD transplants for pediatric patients74,75 supports recent recommendations for upfront unrelated donor transplantation for aplastic anemia.76,77
Immunosuppressive Therapy. For patients without an HLA-matched sibling donor or those who are older than 50 years of age, immunosuppressive therapy is the first-line therapy. ATG and cyclosporine A are the treatments of choice.78 The potential effectiveness of immunosuppressive therapy in treating aplastic anemia was initially observed in patients in whom autologous transplant failed but who still experienced hematopoietic reconstitution despite the failed graft; this observation led to the hypothesis that the conditioning regimen may have an effect on hematopoiesis.59,78,79
Immunosuppressive therapy with ATG has been used for the treatment of aplastic anemia since the 1980s.80 Historically, rabbit ATG had been used, but a 2011 study of horse ATG demonstrated superior hematological response at 6 months compared to rabbit ATG (68% versus 37%).59 Superior survival was also seen with horse ATG compared to rabbit ATG (3-year OS, 96% versus 76%). Due to these results, horse ATG is preferred over rabbit ATG. ATG should be used in combination with cyclosporine A to optimize outcomes.
Early studies also demonstrated the efficacy of cyclosporine A in the treatment of aplastic anemia, with response rates equivalent to that of ATG monotherapy.81 Recent publications still note the efficacy of cyclosporine A in the treatment of aplastic anemia. Its role as an affordable option for single-agent therapy in developing countries is intriguing.81 The combination of ATG and cyclosporine A was proven superior to either agent alone in a study by Frickhofen et al.79 In this study, patients were randomly assigned to a control arm that received ATG plus methylprednisolone or to an arm that received ATG plus cyclosporine A and methylprednisolone. At 6 months, 70% of patients in the cyclosporine A arm had a complete remission (CR) or partial remission compared to 46% in the control arm.82 Further work confirmed the long-term efficacy of this regimen, reporting a 7-year OS of 55%.83 Among a pediatric population, immunosuppressive therapy was associated with an 83% 10-year OS.84
It is recommended that patients remain on cyclosporine therapy for a minimum of 6 months, after which a gradual taper may be considered, although there is variation among practitioners, with some continuing immunosuppressive therapy for a minimum of 12 months due to a proportion of patients being cyclosporine dependent.34,84 A study found that within a population of patients who responded to immunosuppressive therapy, 18% became cyclosporine dependent.84 The median duration of cyclosporine A treatment at full dose was 12 months, with tapering completed over a median of 19 months after patients had been in a stable CR for a minimum of 3 months. Relapse occurred more often when patients were tapered quickly (decrease ≥ 0.8 mg/kg/month) compared to slowly (0.4-0.7 mg/kg/month) or very slowly (< 0.3 mg/kg/month).
Townsley and colleagues recently investigated incorporating the use of the thrombopoietin receptor agonist eltrombopag with immunosuppressive therapy as first-line therapy in aplastic anemia.85 When given at a dose of 150 mg daily in patients ages 12 years and older or 75 mg daily in patients younger than 12 years, in conjunction with cyclosporine A and ATG, patients demonstrated markedly improved hematological response compared to historical treatment with standard immunosuppressive therapy alone.45 In the patient cohort administered eltrombopag starting on day 1 and continuing for 6 months, the complete response rate was 58%. Eltrombopag led to improvement in all cell lines among all treatment subgroups, and OS (censored for patients who proceeded to transplant) was 99% at 2 years.12 Overall, toxicities associated with this therapy were low, with liver enzyme elevations most commonly observed.85 Recently, a phase 2 trial of immunosuppressive therapy with or without eltrombopag was reported. Of the 38 patients enrolled, overall response, complete response, and time to response were not statistically different.86 With this recent finding, the role of eltrombopag in addition to immunosuppressive therapy is not clearly defined, and further studies are warranted.
OS for patients who do not respond to immunosuppressive therapy is approximately 57% at 5 years, largely due to improved supportive measures among this patient population.48,65 Therefore, it is important to recognize those patients who have a low chance of response so that second-line therapy can be pursued to improve outcomes.
Matched Unrelated Donor Transplant. For patients with refractory disease following immunosuppressive therapy who lack a matched sibling donor, MUD HSCT is considered standard therapy given the marked improvement in overall outcomes with modulating conditioning regimens and high-resolution HLA typing. A European Society for Blood and Marrow Transplantation (EBMT) analysis comparing matched sibling HSCT to MUD HSCT noted significantly higher rates of acute grade II-IV and grade III-IV GVHD (grade II-IV 13% versus 25%, grade III-IV 5% versus 10%) among patients undergoing MUD transplant.47 Chronic GVHD rates were 14% in the sibling group, as compared to 26% in the MUD group. Factors associated with improved survival in this analysis include transplant under age 20 years (84% versus 72%), transplant within 6 months of diagnosis (85% versus 72%), the use of ATG in the conditioning regimen (81% versus 73%), and cytomegalovirus-negative donor and recipient as compared to other combinations (82% versus 76%).87 Interestingly, this study demonstrated that OS was not significantly increased when using a sibling HSCT compared to a MUD HSCT, likely as a result of improved understanding of conditioning regimens, GVHD prophylaxis, and supportive care.
Additional studies of MUD HSCT have shown outcomes similar to those seen in sibling HSCT.34,48 A French study found a significant increase in survival in patients undergoing MUD HSCT compared to historical cohorts (2000-2005: OS 52%; 2006-2012: OS 74%).75 The majority of patients underwent conditioning with cyclophosphamide or a combination of busulfan and cyclophosphamide, with or without fludarabine; 81% of patients underwent in vivo T-cell depletion, and a bone marrow donor source was utilized. OS was significantly lower in patients over age 30 years undergoing MUD HSCT (57%) compared to those under age 30 years (70%). Improved OS was also seen when patients underwent transplant within 1 year of diagnosis and when a 10/10 matched donor (compared to a 9/10 mismatched donor) was utilized.48
A 2015 study investigated the role of MUD HSCT as frontline therapy instead of immunosuppressive therapy in patients without a matched sibling donor.75 The 2-year OS was 96% in the MUD HSCT cohort compared to 91%, 94%, and 74% in historical cohorts of sibling HSCT, frontline immunosuppressive therapy, and second-line MUD HSCT following failed immunosuppressive therapy, respectively. Additionally, event-free survival in the MUD HSCT cohort (defined by the authors as death, lack of response, relapse, occurrence of clonal evolution/clinical PNH, malignancies developing over follow‐up, and transplant for patients receiving immunosuppressive therapy frontline) was similar compared to sibling HSCT and superior to frontline immunosuppressive therapy and second-line MUD HSCT. Furthermore, Samarasinghe et al highlighted the importance of in vivo T-cell depletion with either ATG or alemtuzumab (anti-CD52 monoclonal antibody) in the prevention of acute and chronic GVHD in both sibling HSCT and MUD HSCT.88
With continued improvement of less toxic and more immunomodulating conditioning regimens,utilization of bone marrow as a donor cell source, in vivo T-cell depletion, and use of GVHD and antimicrobial prophylaxis, more clinical evidence supports elevating MUD HSCT in the treatment plan for patients without a matched sibling donor.89 However, there is still a large population of patients without matched sibling or unrelated donor options. Given the need to expand the transplant pool and thus avoid clonal hematopoiesis, clinically significant PNH, and relapsed aplastic anemia, more work continues to recognize the expanding role of alternative donor transplants (cord blood and haploidentical) as another viable treatment strategy for aplastic anemia after immunosuppressive therapy failure.90
Summary
Aplastic anemia is a rare but potentially life-threatening disorder with pancytopenia and a marked reduction in the HSC compartment. It can be acquired or associated with an IMFS, and the treatment and prognosis vary dramatically between these 2 etiologies. Workup and diagnosis involves investigating IMFSs and ruling out malignant or infectious etiologies for pancytopenia. Treatment outcomes are excellent with modern supportive care and the current approach to allogeneic transplantation, and therefore referral to a bone marrow transplant program to evaluate for early transplantation is the new standard of care.
Corresponding author: Gabrielle Meyers, MD, 3181 SW Sam Jackson Park Road, Mail Code UHN73C, Portland, OR 97239.
Financial disclosures: None.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.
1. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
2. Vaht K, Göransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica. 2017;102:1683-1690.
3. Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718-1721.
4. Montané E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93:518-523.
5. Ohta A, Nagai M, Nishina M, et al. Incidence of aplastic anemia in Japan: analysis of data from a nationwide registration system. Int J Epidemiol. 2015; 44(suppl_1):i178.
6. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
7. Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol. 2013;139:9-29.
8. Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoiesis stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699-3708.
9. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35-47.
10. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479-2486.
11. Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870.
12. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11-19.
13. Townsley DM, Dumitriu B, Young NS, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922-1931.
14. Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43:598-608.
15. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775-2783.
16. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.
17. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446-4455.
18. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
19. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48:1721-1731.
20. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337-347.
21. Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124:2698-2704.
22. Mukhina GL, Buckley JT, Barber JP, et al. Multilineage glycosylphosphatidylinositol anchor‐deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476-482.
23. Pu JJ, Mukhina G, Wang H, et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87:37-45.
24. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-5340.
25. Devalet B, Mullier F, Chatelain B, et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190-198.
26. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314.
27. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95:1075-1080.
28. Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-3709.
29. Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76-81.
30. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genetics. 1997;68:58-61.
31. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4-10.
32. Giampietro PF, Davis JG, Adler-Brecher B, et al. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116-1120.
33. DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk. 2016;16:417-428.
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129:1428-1436.
35. DeZern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol. 2011;4:221-230.
36. Tichelli A, Gratwohl A, Nissen C, et al. Morphology in patients with severe aplastic anemia treated with antilymphocyte globulin. Blood. 1992;80:337-345.
37. Camitta BM, Storb R, Thomas ED. Aplastic anemia: pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306:645-652.
38. Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70:177-182.
39. Brodsky RA, Chen AR, Dorr D, et al. High-dose cyclophosphamide for severe aplastic anemia: long-term follow-up. Blood. 2010;115:2136-2141.
40. Matsui WH, Brodsky RA, Smith BD, et al. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia. 2006;20:458-462.
41. Maciejewski JP, Risitano AM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
42. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1-17.
43. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92:11-18.
44. Passweg JR, Socié G, Hinterberger W, et al. Bone marrow transplantation for severe aplastic anemia: has outcome improved? Blood. 1997;90:858-864.
45. Gupta V, Eapen M, Brazauskas R, et al. Impact of age on outcomes after transplantation for acquired aplastic anemia using HLA-identical sibling donors. Haematologica. 2010;95:2119-2125.
46. Peffault de Latour R, Le Rademacher J, Antin JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279-4286.
47. Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118:2618-2621.
48. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the Severe Aplastic Anemia Working Party of EBMT. Haematologica. 2016;101:884-890.
49. Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50:1168-1172.
50. De Medeiros CR, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849-852.
51. Mohanan E, Panetta JC, Lakshmi KM, et al. Population pharmacokinetics of fludarabine in patients with aplastic anemia and Fanconi anemia undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:977-983.
52. Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856-2862.
53. Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94:1312-1315.
54. Talbot A, Peffault de Latour R, Raffoux E, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199-e200.
55. Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for Fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669-1676.
56. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010;2010:36-42.
57. Laundy GJ, Bradley BA, Rees BM, et al. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44:814-825.
58. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172:187-207.
59. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430-438.
60. Höchsmann B, Moicean A, Risitano A, et al. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48:168-173.
61. Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269-276.
62. Torres HA, Bodey GP, Rolston KV, et al. Infections in patients with aplastic anemia: experience at a tertiary care cancer center. Cancer. 2003;98:86-93.
63. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117:4434-4441.
64. Gerson SL, Talbot GH, Hurwitz S, et al. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med. 1984;100:345-351.
65. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726-735.
66. Robenshtok E, Gafter-Gvili A, Goldberg E, et al. Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol. 2007;25:5471-5489.
67. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: a subgroup analysis of 116 patients from the EPIC trial. Blood. 2010;116:2448-2454.
68. Deeg HJ, Amylon MD, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208-215.
69. Kahl C, Leisenring W, Joachim Deeg H, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long‐term follow‐up. Br J Haematol. 2005;130:747-751.
70. Socié G. Allogeneic BM transplantation for the treatment of aplastic anemia: current results and expanding donor possibilities. Hematology Am Soc Hematol Educ Program. 2013;2013:82-86.
71. Shin SH, Jeon YW, Yoon JH, et al. Comparable outcomes between younger (<40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016;51:1456-1463.
72. Kim H, Lee KH, Yoon SS, et al; Korean Society of Blood and Marrow Transplantation. Allogeneic hematopoietic stem cell transplant for adults over 40 years old with acquired aplastic anemia. Biol Blood Marrow Transplant. 2012;18:1500-1508.
73. Mortensen BK, Jacobsen N, Heilmann C, Sengelov H. Allogeneic hematopoietic cell transplantation for severe aplastic anemia: similar long-term overall survival after transplantation with related donors compared to unrelated donors. Bone Marrow Transplant. 2016;51:288-290.
74. Dufour C, Svahn J, Bacigalupo A. Front-line immunosuppressive treatment of acquired aplastic anemia. Bone Marrow Transplant. 2013;48:174-177.
75. Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on the behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of the EBMT. Br J Haematol. 2015;171:585-594.
76. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2:2020-2028.
77. Yoshida N, Kojima S. Updated guidelines for the treatment of acquired aplastic anemia in children. Curr Oncol Rep. 2018;20:67.
78. Mathe G, Amiel JL, Schwarzenberg L, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J. 1970;2:131-136.
79. Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al; German Aplastic Anemia Study Group. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. N Engl J Med. 1991;324:1297-1304.
80. Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J. 1981;282:860-863.
81. Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK, et al. Cyclosporine monotherapy for severe aplastic anemia: a developing country experience. Ann Saudi Med. 2005;25:375-379.
82. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185-1196.
83. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130-1135.
84. Saracco P, Quarello P, Iori AP, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long‐term observation follow‐up. Br J Haematol. 2008;140:197-205.
85. Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540-1550.
86. Assi R, Garcia-Manero G, Ravandi F, et al. Addition of eltrombopag to immunosuppressive therapy in patients with newly diagnosed aplastic anemia. Cancer. 2018;124:4192-4201.
87. Bacigalupo A, Socié G, Hamladji RM, et al. Current outcome of HLA identical sibling vs. unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100:696-702.
88. Samarasinghe S, Iacobelli S, Knol C, et al. Impact of different in vivo T cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anaemia: a study on behalf of the EBMT SAA Working Party. Am J Hematol. 2019; 94:80-86.
89. Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619-628.
90. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am. 2018;32:629-642.
Role of Yoga Across the Cancer Care Continuum: From Diagnosis Through Survivorship
From the University of Texas MD Anderson Cancer Center, Houston, TX (Drs. Narayanan, Lopez, Chaoul, Liu, Milbury, and Cohen, and Ms. Mallaiah); the University of Texas Health Science Center at Tyler (Dr. Meegada); and Texas Tech University Health Sciences Center, Lubbock, TX (Ms. Francisco).
Abstract
- Objective: To review the effects of yoga as an adjunct supportive care modality alongside conventional cancer treatment on quality of life (QOL), physical and mental health outcomes, and physiological and biological measures of cancer survivors.
- Methods: Nonsystematic review of the literature.
- Results: Yoga therapy, one of the most frequently used mind-body modalities, has been studied extensively in cancer survivors (from the time of diagnosis through long-term recovery). Yoga affects human physiology on multiple levels, including psychological outcomes, immune and endocrine function, and cardiovascular parameters, as well as multiple areas of QOL. It has been found to reduce psychological stress and fatigue and improve QOL in cancer patients and survivors. Yoga has also been used to manage symptoms such as arthralgia, fatigue, and insomnia. In addition, yoga offers benefits not only for cancer survivors but also for their caregivers.
- Conclusion: As part of an integrative, evidence-informed approach to cancer care, yoga may provide benefits that support the health of cancer survivors and caregivers.
Keywords: fatigue; cancer; proinflammatory cytokines; integrative; mind-body practices; meditation; DNA damage; stress; psychoneuro-immunoendocrine axis; lymphedema; insomnia.
A diagnosis of cancer and adverse effects related to its treatment may have negative effects on quality of life (QOL), contributing to emotional and physical distress in patients and caregivers. Many patients express an interest in pursuing nonpharmacological options, alone or as an adjunct to conventional therapy, to help manage symptoms. The use of complementary medicine approaches to health, including nonpharmacological approaches to symptom management, is highest among individuals with cancer.1 According to a published expert consensus, integrative oncology is defined as a “patient-centered, evidence-informed field of cancer care that utilizes mind and body practices, natural products, and/or lifestyle modifications from different traditions alongside conventional cancer treatments. Integrative oncology aims to optimize health, QOL, and clinical outcomes across the cancer care continuum and to empower people to prevent cancer and become active participants before, during, and beyond cancer treatment.”2 A key component of this definition, often misunderstood in the field of oncology, is that these modalities and treatments are used alongside conventional cancer treatments and not as an alternative. In an attempt to meet patients’ needs and appropriately use these approaches, integrative oncology programs are now part of most cancer centers in the United States.3-6
Because of their overall safety, mind-body therapies are commonly used by patients and recommended by clinicians. Mind-body therapies include yoga, tai chi, qigong, meditation, and relaxation. Expressive arts such as journaling and music, art, and dance therapies also fall in the mind-body category.7 Yoga is a movement-based mind-body practice that focuses on synchronizing body, breath, and mind. Yoga has been increasingly used by patients for health benefits,8 and numerous studies have evaluated yoga as a complementary intervention for individuals with cancer.9-14 Here, we review the physiological basis of yoga in oncology and the effects of yoga on biological processes, QOL, and symptoms during and after cancer treatment.
Physiological Basis
Many patients may use mind-body programs such as yoga to help manage the psychological and physiological consequences of unmanaged chronic stress and improve their overall QOL. The central nervous system, endocrine system, and immune system influence and interact with each other in a complex manner in response to chronic stress.15,16 In a stressful situation, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) are activated. HPA axis stimulation leads to adrenocorticotrophic hormone production by the pituitary gland, which releases glucocorticoid hormones. SNS axis stimulation leads to epinephrine and norepinephrine production by the adrenal gland.17,18 Recently, studies have explored modulation of signal transduction between the nervous and immune systems and how that may impact tumor growth and metastasis.19 Multiple studies, controlled for prognosis, disease stage, and other factors, have shown that patients experiencing more distress or higher levels of depressive symptoms do not live as long as their counterparts with low distress or depression levels.20 Both the meditative and physical components of yoga can lead to enhanced relaxation, reduced SNS activation, and greater parasympathetic tone, countering the negative physiological effects of chronic stress. The effects of yoga on the HPA axis and SNS, proinflammatory cytokines, immune function, and DNA damage are discussed below.
Biological Processes
Nervous System
The effects of yoga and other forms of meditation on brain functions have been established through several studies. Yoga seems to influence basal ganglia function by improving circuits that are involved in complex cognitive functions, motor coordination, and somatosensory and emotional processes.21,22 Additionally, changes in neurotransmitter levels have been observed after yoga practice. For instance, in a 12-week yoga intervention in healthy subjects, increased levels of thalamic gamma-aminobutyric acid (GABA) in the yoga group were reported to have a positive correlation with improved mood and decreased anxiety compared with a group who did metabolically matched walking exercise.23 Levels of GABA, an inhibitory neurotransmitter, are decreased in conditions such as anxiety, depression, and epilepsy.24 Yoga therapy has been shown to improve symptoms of mood disorders and epilepsy, which leads to the hypothesis that the mechanism driving the benefits of yoga may work through stimulation of vagal efferents and an increase in GABA-mediated cortical-inhibitory tone.24,25
HPA Axis
Stress activates the HPA/SNS axis, which releases hormones such as cortisol and norepinephrine. These hormones may play a role in angiogenesis, inflammation, immune suppression, and other physiological functions, and may even reduce the effect of chemotherapeutic agents.26,27 Regular yoga practice has been shown to reduce SNS and HPA axis activity, most likely by increasing parasympathetic dominance through vagal stimulation, as demonstrated through increases in heart rate variability.28 One indicator of HPA axis dysregulation, diurnal salivary cortisol rhythm, was shown to predict survival in patients with advanced breast and renal cancer.29-33 Yoga has been shown to lead to less cortisol dysregulation due to radiotherapy and to reductions in mean cortisol levels and early morning cortisol levels in breast cancer patients undergoing radiotherapy.34 This lends support to the hypothesis that yoga helps restore HPA axis balance.
Proinflammatory Cytokines
Cancer patients tend to have increased levels of inflammatory markers such as interleukin (IL)-4, IL-10, tumor necrosis factor (TNF), interferon-γ, and C-reactive protein. This increase in inflammation is associated with worse outcomes in cancer.35 This association becomes highly relevant because the effect of inflammation on host cells in the tumor microenvironment is connected to disease progression.26 Inflammatory cytokines are also implicated in cancer-related symptoms such as fatigue, cognitive dysfunction, peripheral neuropathy, and sleep disturbances.36
Yoga is known to reduce stress and may directly or indirectly decrease inflammatory cytokines. A randomized clinical trial of a 12-week hatha yoga intervention among breast cancer survivors demonstrated decreases in IL-6, IL-1β, TNF, corticotropin-releasing factor, and cognitive complaints in the yoga group compared with those in the standard care group after 3 months.37,38 Furthermore, Carlson et al showed that, after mindfulness-based stress reduction involving a combination of gentle yoga, meditation techniques, and relaxation exercises, breast and prostate cancer patients had reduced levels of proinflammatory cytokines and cortisol.39 These reductions translated into patients reporting decreased stress levels and enhanced QOL.
Immune Function
The effects of yoga practice on the immune system have been studied in both healthy individuals and individuals with cancer. The effects on T and B lymphocytes, natural killer (NK) cells, and other immune effector cells demonstrate that meditation and yoga have beneficial effects on immune activity.40 Hormones such as catecholamines and glucocorticoids are thought to influence the availability and function of NK cells, and, as noted above, yoga has been shown to modulate stress hormones and lead to reduced immune suppression in patients with early-stage breast cancer undergoing chemotherapy.41 Additional evidence supports the ability of yoga to reduce immune suppression in the postsurgical setting, with no observed decrease in NK cell percentage after surgery for those in a yoga group compared with a control group.42 This finding is relevant to patients undergoing surgical management of their cancer and highlights the impact of yoga on the immune system.
DNA Damage
Radiation damages DNA in the peripheral blood lymphocytes of patients undergoing treatment.43,44 This damage is significant in breast cancer patients undergoing radiotherapy.45 Stress additionally causes DNA damage46 and is correlated to impaired DNA repair capacity.47,48 In a study conducted by Banerjee et al, breast cancer patients were randomly assigned to a yoga group or a supportive therapy group for 6 weeks during radiotherapy.49 Prior to the intervention, patients in the study had significant genomic instability. After treatment, patients in the yoga group experienced not only a significant reduction in anxiety and depression levels, but also a reduction in DNA damage due to radiotherapy.
Yoga in Quality of Life and Symptom Management
There is evidence showing that yoga therapy improves multiple aspects of QOL, including physical functioning, emotional health outcomes, and the symptoms cancer patients may experience, such as sleep disturbances, fatigue, and pain. Danhauer et al systematically reviewed both nonrandomized trials and randomized controlled trials involving yoga during cancer treatment.50 They found that yoga improved depression and anxiety as well as sleep and fatigue. Benefits of yoga in cancer based on randomized controlled trials are summarized in the Table. The role of yoga in improving QOL and managing symptoms patients experience during and after treatment is discussed in the following sections.

Quality of Life
Danhauer et al’s systematic review of trials involving yoga during cancer treatment found that yoga improved multiple aspects of QOL.50 For example, yoga has been shown to improve QOL in breast cancer patients undergoing radiotherapy. In a study by Chandwani et al, yoga (60-minute sessions twice a week for 6 weeks) was associated with better general health perception and physical functioning scores as well as greater benefit finding, or finding meaning in their experience, after radiotherapy compared with a wait-list group.51 The yoga group had an increase in intrusive thoughts, believed to be due to a more thorough processing of the cancer experience, which helps to improve patients’ outlook on life.52 The benefits of yoga extend beyond psychological measures during radiation treatment. Yoga was found to increase physical functioning compared with stretching in breast cancer patients undergoing radiotherapy.53
Cognitive Function
Cancer-related cognitive impairment commonly occurs during cancer treatments (eg, chemotherapy, radiotherapy, surgery, hormone therapy) and persists for months or years in survivors.54 Impairment of memory, executive function, attention, and concentration are commonly reported. In a trial of a combined hatha and restorative yoga program called Yoga for Cancer Survivors (YOCAS), which was designed by researchers at the University of Rochester, patients in the yoga arm had less memory difficulty than did patients in the standard care arm.55 However, the primary aim of the trial was to treat insomnia, so this secondary outcome needs to be interpreted with caution. Deficits in attention, memory, and executive function are often seen in cancer-related cognitive impairment, and the meditative aspect of yoga may have behavioral and neurophysical benefits that could improve cognitive functions.56 More evidence is needed to understand the role of yoga in improving cognitive functioning.
Emotional Health
Psychosocial stress is high among breast cancer patients and survivors.57,58 This causes circadian rhythm and cortisol regulation abnormalities, which are reported in women with breast cancer.59-64 Yoga is known to help stress and psychosocial and physical functioning in patients with cancer.65 Yoga was also shown to be equivalent to cognitive behavioral therapy in stress management in a population of patients without cancer.66 Daily yoga sessions lasting 60 minutes were shown to reduce reactive anxiety and trait anxiety in early-stage breast cancer patients undergoing conventional radiotherapy and chemotherapy compared with patients receiving supportive therapy, highlighting the role of yoga in managing anxiety related to treatment.67 In a study done by Culos-Reed et al, 20 cancer survivors who did 75 minutes of yoga per week for 7 weeks were compared with 18 cancer survivors who served as a control group.68 The intervention group reported significant improvement in emotional well-being, depression, concentration, and mood disturbances. In a longitudinal study by Mackenzie et al, 66 cancer survivors completed a 7-week yoga program and were assessed at baseline, immediately after the final yoga session, and at 3 and 6 months after the final session.69 Participants had significantly improved energy levels and affect. They also had moderate improvement in mindfulness and a moderate decrease in stress. Breast cancer patients who underwent restorative yoga sessions found improvements in mental health, depression, positive affect, and spirituality (peace/meaning).70 This was more pronounced in women with higher negative affect and lower emotional well-being at baseline. In a study of patients with ovarian cancer receiving chemotherapy, patients were instructed to perform up to 15-minute sessions including awareness, body movement, and breathing.71 Even with just 1 session of yoga intervention, patients experienced decreased anxiety.
Fatigue
Studies on yoga show improvement in fatigue both during and after treatment. In breast cancer patients undergoing chemotherapy, yoga was shown to benefit cognitive fatigue.72 Older cancer survivors also seem to benefit from yoga interventions.73 In a trial of a DVD-based yoga program, the benefits of yoga were similar to those of strengthening exercises, and both interventions helped decrease fatigue and improve QOL during the first year after diagnosis in early-stage breast cancer patients with cancer-related fatigue.74 Bower et al also showed that, for breast cancer survivors experiencing persistent chronic fatigue, a targeted yoga intervention led to significant improvements in fatigue and vigor over a 3-month follow-up compared with controls.75 Fatigue is commonly seen in breast cancer patients who are receiving adjuvant chemotherapy. In a study by Taso et al, women with breast cancer receiving chemotherapy were assigned to 60-minute yoga sessions incorporating Anusara yoga, gentle stretching, and relaxation twice a week for 8 weeks.76 By week 4, patients with low pretest fatigue in the yoga group experienced a reduction in fatigue. By week 8, all patients in the yoga group experienced a reduction in fatigue. Four weeks after the yoga intervention, patients in the group maintained the reduction in fatigue. This study shows the feasibility of an 8-week yoga program for women undergoing breast cancer therapy by improving fatigue. Yoga recently was added to National Comprehensive Cancer Network (NCCN) guidelines for management of cancer-related fatigue (level 1 evidence).77 However, the evidence was based on studies in women with breast cancer and survivors; therefore, more studies are needed in men and women with other cancers.
Surgical Setting/Postoperative Distress
Distress surrounding surgery in patients with breast cancer can impact postoperative outcomes. Yoga interventions, including breathing exercises, regulated breathing, and yogic relaxation techniques, improved several postsurgical measures such as length of hospital stay, drain retention, and suture removal.78 In this study, patients who practiced yoga also experienced a decrease in plasma TNF and better wound healing. Symptoms of anxiety and distress that occur preoperatively can lead to impaired immune function in addition to decreased QOL. In a study of yoga in early-stage breast cancer patients undergoing surgery, the benefit of yoga was seen not only with stress reduction but also with immune enhancement.42
Yoga has been shown to help alleviate acute pain and distress in women undergoing major surgery for gynecological cancer. A regimen of 3 15-minute sessions of yoga, including awareness meditation, coordination of breath with movement, and relaxation breathing, was shown to reduce acute pain and distress in such patients in an inpatient setting.79
Menopausal Symptoms
Breast cancer survivors have more severe menopausal symptoms compared with women without cancer.80,81 Hot flashes cause sleep disturbances and worsen fatigue and QOL.82 Tamoxifen and aromatase inhibitors significantly worsen menopausal symptoms such as hot flashes.81 Carson et al conducted a study of yoga that included postures, breathing techniques, didactic presentations, and group discussions.83 The yoga awareness regimen consisted of 8 weekly 120-minute group classes. Patients in the yoga arm had statistically significant improvements in the frequency, severity, and number of hot flashes. There were also improvements in arthralgia (joint pain), fatigue, sleep disturbance, vigor, and acceptance.
Arthralgia
Joint pain can be a major side effect that interferes with daily functions and activities in postmenopausal breast cancer survivors who receive aromatase inhibitor therapy.84 Arthralgia is reported in up to 50% of patients treated with aromatase inhibitors.84,85 It can affect functional status and lead to discontinuation of aromatase inhibitor therapy, jeopardizing clinical outcomes.86 Yoga as a complementary therapy has been shown to improve conditions such as low back pain87 and knee osteoarthritis88 in patients who do not have cancer. In a single-arm pilot trial by Galantino et al, breast cancer patients with aromatase inhibitor–related joint pain were provided with twice-weekly yoga sessions for 8 weeks. There were statistically significant improvements in balance, flexibility, pain severity, and health-related QOL.89 As noted above, improvement in arthralgia was also found in the study conducted by Carson et al.83
Insomnia
Insomnia is common among cancer patients and survivors90,91 and leads to increased fatigue and depression, decreased adherence to cancer treatments, and poor physical function and QOL.90-92 Management of insomnia consists of pharmacologic therapies such as benzodiazepines93,94 and nonpharmacologic options such as cognitive behavioral therapy.95
The first study of yoga found to improve sleep quality was conducted at MD Anderson Cancer Center in lymphoma patients.96 The effects of Tibetan yoga practices incorporating controlled breathing and visualization, mindfulness techniques, and low-impact postures were studied. Patients in the Tibetan yoga group had better subjective sleep quality, faster sleep latency, longer sleep duration, and less use of sleep medications. Mustian et al conducted a large yoga study in cancer survivors in which patients reporting chronic sleep disturbances were randomly assigned to the YOCAS program, which consisted of pranayama (breath control), 16 gentle hatha and restorative yoga postures, and meditation, or to usual care.92 The study reported improvements in global sleep quality, subjective sleep quality, actigraphy measures (wake after sleep onset, sleep efficiency), daytime dysfunction, and use of sleep medication after the yoga intervention compared with participants who received standard care.
Yoga to Address Other Symptoms
There is preliminary evidence supporting yoga as an integrative therapy for other symptoms unique to cancer survivors. For example, in head and neck cancer survivors, soft tissue damage involving the jaw, neck, shoulders, and chest results in swallowing issues, trismus, and aspiration, which are more pronounced in patients treated with conventional radiotherapy than in those treated with intensity-modulated radiotherapy.97 Some late effects of radiotherapy for head and neck cancer—such as pain, anxiety, and impaired shoulder function—were shown to be improved through the practice of hatha yoga in 1 study.98 Similarly, in a randomized controlled pilot study of patients with stage I to III breast cancer 6 months after treatment, participants in an 8-week yoga program experienced a reduction in arm induration and improvement in a QOL subscale of lymphedema symptoms. However, more evidence is needed to support the use of yoga as a therapeutic measure for breast cancer lymphedema.99,100
Yoga for Caregivers
Along with cancer patients, caregivers face psychological and physical burdens as well as deterioration in their QOL. Caregivers tend to report clinical levels of anxiety, depression, sleep disturbance, and fatigue and have similar or in fact higher levels than those of the patients for whom they are caring.101,102 Yoga has been found to help caregivers of patients with cancer. Recently, MD Anderson researchers conducted a trial in patients with high-grade glioma and their caregivers as dyads.103,104 Each dyad attended 2 or 3 60-minute weekly Vivekananda yoga sessions involving breathing exercises, physical exercises, relaxation, and meditation. The researchers found that the yoga program was safe, feasible, acceptable, and subjectively useful for patients with high-grade glioma and their caregivers. Preliminary evidence of QOL improvement for both patients and caregivers was noted. An improvement in QOL was also demonstrated in another preliminary study of yoga in patients undergoing thoracic radiotherapy and their caregivers.105
Another study by the group at MD Anderson evaluated a couple-based Tibetan yoga program that emphasized breathing exercises, gentle movements, guided visualizations, and emotional connectedness during radiotherapy for lung cancer.106 This study included 10 patient‐caregiver dyads and found the program to be feasible, safe, and acceptable. The researchers also found preliminary evidence of improved QOL by the end of radiotherapy relative to baseline—specifically in the areas of spiritual well‐being for patients, fatigue for caregivers, and sleep disturbances and mental health issues such as anxiety and depressive symptoms for both patients and caregivers. This is noteworthy, as QOL typically deteriorates during the course of radiotherapy, and the yoga program was able to buffer these changes.
Conclusion
Yoga therapy has been used successfully as an adjunct modality to improve QOL and cancer-related symptoms. As a part of an integrative medicine approach, yoga is commonly recommended for patients undergoing cancer treatment. Danhauer et al reviewed randomized controlled trials during and after treatment and concluded that the evidence is clearly positive for QOL, fatigue, and perceived stress.107 Results are less consistent but supportive for psychosocial outcomes such as benefit finding and spirituality. Evidence is mixed for sleep, anxiety, and depression. Post-treatment studies demonstrate improvements in fatigue, sleep, and multiple QOL domains. Yoga has been included in NCCN guidelines for fatigue management. Yoga, if approved by a physician, is also included among the behavioral therapies for anticipatory emesis and prevention and treatment of nausea in the recent update of the NCCN guidelines.108 The Society for Integrative Oncology guidelines include yoga for anxiety/stress reduction as a part of integrative treatment in breast cancer patients during and after therapy, which was endorsed by the American Society of Clinical Oncology.109
Because of the strong evidence for its benefits and a low side-effect profile, yoga is offered in group-class settings for patients during and after treatment and/or for caregivers in our institution. We often prescribe yoga as a therapeutic modality for selected groups of patients in our clinical practice. However, some patients may have restrictions after surgery that must be considered. In general, yoga has an excellent safety profile, the evidence base is strong, and we recommend that yoga therapy should be part of the standard of care as an integrative approach for patients with cancer undergoing active treatment as well as for cancer survivors and caregivers.
Acknowledgement: The authors thank Bryan Tutt for providing editorial assistance.
Corresponding author: Santhosshi Narayanan, MD, Department of Palliative, Rehabilitation, and Integrative Medicine, Unit 1414, The University of Texas MD Anderson Cancer Center, 1400 Pressler St., Houston, TX 77030; [email protected].
Financial disclosures: None.
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Reports. 2008;(12):1-23.
2. Witt CM, Balneaves LG, Cardoso MJ, et al. A comprehensive definition for integrative oncology. NCI Monographs. 2017;2017(52):3-8.
3. Brauer JA, El Sehamy A, Metz JM, Mao JJ. Complementary and alternative medicine and supportive care at leading cancer centers: a systematic analysis of websites. J Altern Complement Med. 2010;16:183-186.
4. Lopez G, McQuade J, Cohen L, et al. Integrative oncology physician consultations at a comprehensive cancer center: analysis of demographic, clinical and patient reported outcomes. J Cancer. 2017;8:395-402.
5. Latte-Naor S, Mao JJ. Putting integrative oncology into practice: concepts and approaches. J Oncol Pract. 2019;15:7-14.
6. Richardson MA, Sanders T, Palmer JL, et al. Complementary/alternative medicine use in a comprehensive cancer center and the implications for oncology. J Clin Oncol. 2000;18:2505-2514.
7. Chaoul A, Milbury K, Sood AK, et al. Mind-body practices in cancer care. Curr Oncol Rep. 2014;16:417.
8. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10:44-49.
9. Smith KB, Pukall CF. An evidence-based review of yoga as a complementary intervention for patients with cancer. Psychooncology. 2009;18:465-475.
10. Buffart LM, van Uffelen JG, Riphagen II, et al. Physical and psychosocial benefits of yoga in cancer patients and survivors, a systematic review and meta-analysis of randomized controlled trials. BMC Cancer. 2012;12:559.
11. Cramer H, Lange S, Klose P, et al. Yoga for breast cancer patients and survivors: a systematic review and meta-analysis. BMC Cancer. 2012;12:412.
12. Felbel S, Meerpohl JJ, Monsef I, et al. Yoga in addition to standard care for patients with haematological malignancies. Cochrane Database Syst Rev. 2014;(6):CD010146.
13. Greenlee H, Balneaves LG, Carlson LE, et al. Clinical practice guidelines on the use of integrative therapies as supportive care in patients treated for breast cancer. NCI Monographs. 2014;2014:346-358.
14. Sharma M, Haider T, Knowlden AP. Yoga as an alternative and complementary treatment for cancer: a systematic review. J Altern Complement Med. 2013;19:870-875.
15. Kordon C, Bihoreau C. Integrated communication between the nervous, endocrine and immune systems. Horm Res. 1989;31:100-104.
16. Gonzalez-Diaz SN, Arias-Cruz A, Elizondo-Villarreal B, Monge-Ortega OP. Psychoneuroimmunoendocrinology: clinical implications. World Allergy Organ J. 2017;10:19.
17. Godoy LD, Rossignoli MT, Delfino-Pereira P, et al. A comprehensive overview on stress neurobiology: basic concepts and clinical implications. Front Behav Neurosci. 2018;12:127.
18. Goldstein DS. Adrenal responses to stress. Cell Mol Neurobiol. 2010;30:1433-1440.
19. Cole SW, Nagaraja AS, Lutgendorf SK, et al. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563-567.
20. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
21. Arsalidou M, Duerden EG, Taylor MJ. The centre of the brain: topographical model of motor, cognitive, affective, and somatosensory functions of the basal ganglia. Hum Brain Mapp. 2013;34:3031-3054.
22. Gard T, Taquet M, Dixit R, et al. Greater widespread functional connectivity of the caudate in older adults who practice kripalu yoga and vipassana meditation than in controls. Front Hum Neurosci. 2015;9:137.
23. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
24. Brambilla P, Perez J, Barale F, et al. GABAergic dysfunction in mood disorders. Mol Psychiatry. 2003;8:721-737.
25. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
26. Lutgendorf SK, Sood AK. Biobehavioral factors and cancer progression: physiological pathways and mechanisms. Psychosom Med. 2011;73:724-730.
27. Armaiz-Pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain Behav Immun. 2013;30(suppl):S19-S25.
28. Tyagi A, Cohen M. Yoga and heart rate variability: A comprehensive review of the literature. Int J Yoga. 2016;9:97-113.
29. Sephton SE, Lush E, Dedert EA, et al. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain Behav Immun. 2013;30 (suppl):S163-S170.
30. Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst. 2000;92:994-1000.
31. Arafah BM, Nishiyama FJ, Tlaygeh H, Hejal R. Measurement of salivary cortisol concentration in the assessment of adrenal function in critically ill subjects: a surrogate marker of the circulating free cortisol. J Clin Endocrinol Metab. 2007;92:2965-2971.
32. Cohen L, de Moor C, Devine D, et al. Endocrine levels at the start of treatment are associated with subsequent psychological adjustment in cancer patients with metastatic disease. Psychosom Med. 2001;63:951-958.
33. Cohen L, Cole SW, Sood AK, et al. Depressive symptoms and cortisol rhythmicity predict survival in patients with renal cell carcinoma: role of inflammatory signaling. PloS One. 2012;7:e42324.
34. Vadiraja HS, Raghavendra RM, Nagarathna R, et al. Effects of a yoga program on cortisol rhythm and mood states in early breast cancer patients undergoing adjuvant radiotherapy: a randomized controlled trial. Integr Cancer Ther. 2009;8:37-46.
35. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345:164-173.
36. Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun. 2013;30(suppl):S48-S57.
37. Derry HM, Jaremka LM, Bennett JM, et al. Yoga and self-reported cognitive problems in breast cancer survivors: a randomized controlled trial. Psychooncology. 2015;24:958-966.
38. Kiecolt-Glaser JK, Christian L, Preston H, et al. Stress, inflammation, and yoga practice. Psychosom Med. 2010;72:113-121.
39. Carlson LE, Speca M, Faris P, Patel KD. One year pre-post intervention follow-up of psychological, immune, endocrine and blood pressure outcomes of mindfulness-based stress reduction (MBSR) in breast and prostate cancer outpatients. Brain Behav Immun. 2007;21:1038-1049.
40. Infante JR, Peran F, Rayo JI, et al. Levels of immune cells in transcendental meditation practitioners. Int J Yoga. 2014;7:147-151.
41. Rao RM, Telles S, Nagendra HR, et al. Effects of yoga on natural killer cell counts in early breast cancer patients undergoing conventional treatment. Comment to: recreational music-making modulates natural killer cell activity, cytokines, and mood states in corporate employees Masatada Wachi, Masahiro Koyama, Masanori Utsuyama, Barry B. Bittman, Masanobu Kitagawa, Katsuiku Hirokawa Med Sci Monit, 2007; 13(2): CR57-70. Med Sci Monit. 2008;14:LE3-4.
42. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on mood states, distress, quality of life and immune outcomes in early stage breast cancer patients undergoing surgery. Int J Yoga. 2008;1:11-20.
43. Mozdarani H, Mansouri Z, Haeri SA. Cytogenetic radiosensitivity of g0-lymphocytes of breast and esophageal cancer patients as determined by micronucleus assay. J Radiat Res. 2005;46:111-116.
44. Scott D, Barber JB, Levine EL, et al. Radiation-induced micronucleus induction in lymphocytes identifies a high frequency of radiosensitive cases among breast cancer patients: a test for predisposition? Br J Cancer. 1998;77:614-620.
45. Banerjee B, Sharma S, Hegde S, Hande MP. Analysis of telomere damage by fluorescence in situ hybridisation on micronuclei in lymphocytes of breast carcinoma patients after radiotherapy. Breast Cancer Res Treat. 2008;107:25-31.
46. Epel ES, Blackburn EH, Lin J, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312-17315.
47. Glaser R, Thorn BE, Tarr KL, et al. Effects of stress on methyltransferase synthesis: an important DNA repair enzyme. Health Psychol. 1985;4:403-412.
48. Kiecolt-Glaser JK, Stephens RE, Lipetz PD, et al. Distress and DNA repair in human lymphocytes. J Behav Med. 1985;8:311-320.
49. Banerjee B, Vadiraj HS, Ram A, et al. Effects of an integrated yoga program in modulating psychological stress and radiation-induced genotoxic stress in breast cancer patients undergoing radiotherapy. Integr Cancer Ther. 2007;6:242-250.
50. Danhauer SC, Addington EL, Sohl SJ, et al. Review of yoga therapy during cancer treatment. Support Care Cancer. 2017;25:1357-1372.
51. Chandwani KD, Thornton B, Perkins GH, et al. Yoga improves quality of life and benefit finding in women undergoing radiotherapy for breast cancer. J Soc Integr Oncol. 2010;8:43-55.
52. Ratcliff CG, Milbury K, Chandwani KD, et al. Examining mediators and moderators of yoga for women with breast cancer undergoing radiotherapy. Integr Cancer Ther. 2016;15:250-262.
53. Chandwani KD, Perkins G, Nagendra HR, et al. Randomized, controlled trial of yoga in women with breast cancer undergoing radiotherapy. J Clin Oncol. 2014;32:1058-1065.
54. Janelsins MC, Kesler SR, Ahles TA, Morrow GR. Prevalence, mechanisms, and management of cancer-related cognitive impairment. Int Rev Psychiatry. 2014;26:102-113.
55. Janelsins MC, Peppone LJ, Heckler CE, et al. YOCAS(c)(R) yoga reduces self-reported memory difficulty in cancer survivors in a nationwide randomized clinical trial: investigating relationships between memory and sleep. Integr Cancer Ther. 2016;15:263-271.
56. Biegler KA, Chaoul MA, Cohen L. Cancer, cognitive impairment, and meditation. Acta Oncol. 2009;48:18-26.
57. Carlson LE, Angen M, Cullum J, et al. High levels of untreated distress and fatigue in cancer patients. Br J Cancer. 2004;90:2297-2304.
58. Herschbach P, Keller M, Knight L, et al. Psychological problems of cancer patients: a cancer distress screening with a cancer-specific questionnaire. Br J Cancer. 2004;91:504-511.
59. Abercrombie HC, Giese-Davis J, Sephton S, et al. Flattened cortisol rhythms in metastatic breast cancer patients. Psychoneuroendocrinology. 2004;29:1082-1092.
60. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med. 2005;67:277-280.
61. Bower JE, Ganz PA, Dickerson SS, et al. Diurnal cortisol rhythm and fatigue in breast cancer survivors. Psychoneuroendocrinology. 2005;30:92-100.
62. Giese-Davis J, Sephton SE, Abercrombie HC, et al. Repression and high anxiety are associated with aberrant diurnal cortisol rhythms in women with metastatic breast cancer. Health Psychol. 2004;23:645-650.
63. Giese-Davis J, DiMiceli S, Sephton S, Spiegel D. Emotional expression and diurnal cortisol slope in women with metastatic breast cancer in supportive-expressive group therapy: a preliminary study. Biol Psychol. 2006;73:190-198.
64. Stone AA, Schwartz JE, Smyth J, et al. Individual differences in the diurnal cycle of salivary free cortisol: a replication of flattened cycles for some individuals. Psychoneuroendocrinology. 2001;26:295-306.
65. Bower JE, Woolery A, Sternlieb B, Garet D. Yoga for cancer patients and survivors. Cancer Control. 2005;12:165-171.
66. Granath J, Ingvarsson S, von Thiele U, Lundberg U. Stress management: a randomized study of cognitive behavioural therapy and yoga. Cogn Behav Therap. 2006;35:3-10.
67. Rao MR, Raghuram N, Nagendra HR, et al. Anxiolytic effects of a yoga program in early breast cancer patients undergoing conventional treatment: a randomized controlled trial. Complement Ther Med. 2009;17:1-8.
68. Culos-Reed SN, Carlson LE, Daroux LM, Hately-Aldous S. A pilot study of yoga for breast cancer survivors: physical and psychological benefits. Psychooncology. 2006;15:891-897.
69. Mackenzie MJ, Carlson LE, Ekkekakis P, et al. Affect and mindfulness as predictors of change in mood disturbance, stress symptoms, and quality of life in a community-based yoga program for cancer survivors. Evid Based Complement Alternat Med. 2013;2013:419496.
70. Danhauer SC, Mihalko SL, Russell GB, et al. Restorative yoga for women with breast cancer: findings from a randomized pilot study. Psycho-oncology. 2009;18:360-368.
71. Sohl SJ, Danhauer SC, Schnur JB, et al. Feasibility of a brief yoga intervention during chemotherapy for persistent or recurrent ovarian cancer. Explore (NY). 2012;8:197-198.
72. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer. 2016;24:4005-4015.
73. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol. 2015;6:8-14.
74. Wang G, Wang S, Jiang P, Zeng C. Effect of yoga on cancer related fatigue in breast cancer patients with chemotherapy [in Chinese]. Zhong Nan Da Xue Bao Yi Xue Ban. 2014;39:1077-1082.
75. Bower JE, Garet D, Sternlieb B, et al. Yoga for persistent fatigue in breast cancer survivors: a randomized controlled trial. Cancer. 2012;118:3766-3775.
76. Taso CJ, Lin HS, Lin WL, et al. The effect of yoga exercise on improving depression, anxiety, and fatigue in women with breast cancer: a randomized controlled trial. J Nurs Res. 2014;22:155-164.
77. Berger AM, Mooney K, Alvarez-Perez A, et al. Cancer-related fatigue, Version 2.2015. J Natl Compr Canc Netw. 2015;13:1012-1039.
78. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on postoperative outcomes and wound healing in early operable breast cancer patients undergoing surgery. Int J Yoga. 2008;1:33-41.
79. Sohl SJ, Avis NE, Stanbery K, et al. Feasibility of a brief yoga intervention for improving acute pain and distress post gynecologic surgery. Int J Yoga Therap. 2016;26:43-47.
80. Gupta P, Sturdee DW, Palin SL, et al. Menopausal symptoms in women treated for breast cancer: the prevalence and severity of symptoms and their perceived effects on quality of life. Climacteric. 2006;9:49-58.
81. Canney PA, Hatton MQ. The prevalence of menopausal symptoms in patients treated for breast cancer. Clin Oncol (R Coll Radiol). 1994;6:297-299.
82. Carpenter JS, Johnson D, Wagner L, Andrykowski M. Hot flashes and related outcomes in breast cancer survivors and matched comparison women. Oncol Nurs Forum. 2002;29:E16-25.
83. Carson JW, Carson KM, Porter LS, et al. Yoga of Awareness program for menopausal symptoms in breast cancer survivors: results from a randomized trial. Support Care Cancer. 2009;17:1301-1309.
84. Burstein HJ. Aromatase inhibitor-associated arthralgia syndrome. Breast. 2007;16:223-234.
85. Mao JJ, Stricker C, Bruner D, et al. Patterns and risk factors associated with aromatase inhibitor-related arthralgia among breast cancer survivors. Cancer. 2009;115:3631-3639.
86. Presant CA, Bosserman L, Young T, et al. Aromatase inhibitor-associated arthralgia and/or bone pain: frequency and characterization in non-clinical trial patients. Clin Breast Cancer. 2007;7:775-778.
87. Saper RB, Sherman KJ, Cullum-Dugan D, et al. Yoga for chronic low back pain in a predominantly minority population: a pilot randomized controlled trial. Altern Ther Health Med. 2009;15:18-27.
88. Kolasinski SL, Garfinkel M, Tsai AG, et al. Iyengar yoga for treating symptoms of osteoarthritis of the knees: a pilot study. J Altern Complement Med. 2005;11:689-693.
89. Galantino ML, Desai K, Greene L, et al. Impact of yoga on functional outcomes in breast cancer survivors with aromatase inhibitor-associated arthralgias. Integr Cancer Ther. 2012;11:313-320.
90. Ancoli-Israel S. Recognition and treatment of sleep disturbances in cancer. J Clin Oncol. 2009;27:5864-5866.
91. Savard J, Ivers H, Villa J, et al. Natural course of insomnia comorbid with cancer: an 18-month longitudinal study. J Clin Oncol. 2011;29:3580-3586.
92. Mustian KM, Sprod LK, Janelsins M, et al. Multicenter, randomized controlled trial of yoga for sleep quality among cancer survivors. J Clin Oncol. 2013;31:3233-3241.
93. Moore TA, Berger AM, Dizona P. Sleep aid use during and following breast cancer adjuvant chemotherapy. Psychooncology. 2011;20:321-325.
94. Omvik S, Pallesen S, Bjorvatn B, et al. Patient characteristics and predictors of sleep medication use. Int Clin Psychopharmacol. 2010;25:91-100.
95. Qaseem A, Kansagara D, Forciea MA, et al. Management of chronic insomnia disorder in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;165:125-133.
96. Cohen L, Warneke C, Fouladi RT, et al. Psychological adjustment and sleep quality in a randomized trial of the effects of a Tibetan yoga intervention in patients with lymphoma. Cancer. 2004;100:2253-2260.
97. Kraaijenga SA, Oskam IM, van der Molen L, et al. Evaluation of long term (10-years+) dysphagia and trismus in patients treated with concurrent chemo-radiotherapy for advanced head and neck cancer. Oral Oncol. 2015;51:787-794.
98. Adair M, Murphy B, Yarlagadda S, et al. Feasibility and preliminary efficacy of tailored yoga in survivors of head and neck cancer: a pilot study. Integr Cancer Ther. 2018;17:774-784.
99. Loudon A, Barnett T, Williams A. Yoga, breast cancer-related lymphoedema and well-being: A descriptive report of women’s participation in a clinical trial. J Clin Nurs. 2017;26:4685-4695.
100. Loudon A, Barnett T, Piller N, et al. The effects of yoga on shoulder and spinal actions for women with breast cancer-related lymphoedema of the arm: A randomised controlled pilot study. BMC Complement Altern Med. 2016;16:343.
101. Petruzzi A, Finocchiaro CY, Lamperti E, Salmaggi A. Living with a brain tumor: reaction profiles in patients and their caregivers. Support Care Cancer. 2013;21:1105-1111.
102. Pawl JD, Lee SY, Clark PC, Sherwood PR. Sleep characteristics of family caregivers of individuals with a primary malignant brain tumor. Oncol Nurs Forum. 2013;40:171-179.
103. Milbury K, Mallaiah S, Mahajan A, et al. Yoga program for high-grade glioma patients undergoing radiotherapy and their family caregivers. Integr Cancer Ther. 2018;17:332-336.
104. Milbury K, Li J, Weathers S-P, et al. Pilot randomized controlled trial of a dyadic yoga program for glioma patients undergoing radiotherapy and their family caregivers. Neurooncol Pract. 2019;6:311-320.
105. Milbury K, Liao Z, Shannon V, et al. Dyadic yoga program for patients undergoing thoracic radiotherapy and their family caregivers: Results of a pilot randomized controlled trial. Psychooncology. 2019;28:615-621.
106. Milbury K, Chaoul A, Engle R, et al. Couple-based Tibetan yoga program for lung cancer patients and their caregivers. Psychooncology. 2015;24:117-120.
107. Danhauer SC, Addington EL, Cohen L, et al. Yoga for symptom management in oncology: A review of the evidence base and future directions for research. Cancer. 2019;125:1979-1989.
108. National Comprehensive Cancer Center. Flash Update: NCCN Guidelines® and NCCN Compendium® for Antiemesis. NCCN website. Accessed August 29, 2019.
109. Lyman GH, Greenlee H, Bohlke K, et al. Integrative therapies during and after breast cancer treatment: ASCO endorsement of the SIO clinical practice guideline. J Clin Oncol. 2018;36:2647-2655.
From the University of Texas MD Anderson Cancer Center, Houston, TX (Drs. Narayanan, Lopez, Chaoul, Liu, Milbury, and Cohen, and Ms. Mallaiah); the University of Texas Health Science Center at Tyler (Dr. Meegada); and Texas Tech University Health Sciences Center, Lubbock, TX (Ms. Francisco).
Abstract
- Objective: To review the effects of yoga as an adjunct supportive care modality alongside conventional cancer treatment on quality of life (QOL), physical and mental health outcomes, and physiological and biological measures of cancer survivors.
- Methods: Nonsystematic review of the literature.
- Results: Yoga therapy, one of the most frequently used mind-body modalities, has been studied extensively in cancer survivors (from the time of diagnosis through long-term recovery). Yoga affects human physiology on multiple levels, including psychological outcomes, immune and endocrine function, and cardiovascular parameters, as well as multiple areas of QOL. It has been found to reduce psychological stress and fatigue and improve QOL in cancer patients and survivors. Yoga has also been used to manage symptoms such as arthralgia, fatigue, and insomnia. In addition, yoga offers benefits not only for cancer survivors but also for their caregivers.
- Conclusion: As part of an integrative, evidence-informed approach to cancer care, yoga may provide benefits that support the health of cancer survivors and caregivers.
Keywords: fatigue; cancer; proinflammatory cytokines; integrative; mind-body practices; meditation; DNA damage; stress; psychoneuro-immunoendocrine axis; lymphedema; insomnia.
A diagnosis of cancer and adverse effects related to its treatment may have negative effects on quality of life (QOL), contributing to emotional and physical distress in patients and caregivers. Many patients express an interest in pursuing nonpharmacological options, alone or as an adjunct to conventional therapy, to help manage symptoms. The use of complementary medicine approaches to health, including nonpharmacological approaches to symptom management, is highest among individuals with cancer.1 According to a published expert consensus, integrative oncology is defined as a “patient-centered, evidence-informed field of cancer care that utilizes mind and body practices, natural products, and/or lifestyle modifications from different traditions alongside conventional cancer treatments. Integrative oncology aims to optimize health, QOL, and clinical outcomes across the cancer care continuum and to empower people to prevent cancer and become active participants before, during, and beyond cancer treatment.”2 A key component of this definition, often misunderstood in the field of oncology, is that these modalities and treatments are used alongside conventional cancer treatments and not as an alternative. In an attempt to meet patients’ needs and appropriately use these approaches, integrative oncology programs are now part of most cancer centers in the United States.3-6
Because of their overall safety, mind-body therapies are commonly used by patients and recommended by clinicians. Mind-body therapies include yoga, tai chi, qigong, meditation, and relaxation. Expressive arts such as journaling and music, art, and dance therapies also fall in the mind-body category.7 Yoga is a movement-based mind-body practice that focuses on synchronizing body, breath, and mind. Yoga has been increasingly used by patients for health benefits,8 and numerous studies have evaluated yoga as a complementary intervention for individuals with cancer.9-14 Here, we review the physiological basis of yoga in oncology and the effects of yoga on biological processes, QOL, and symptoms during and after cancer treatment.
Physiological Basis
Many patients may use mind-body programs such as yoga to help manage the psychological and physiological consequences of unmanaged chronic stress and improve their overall QOL. The central nervous system, endocrine system, and immune system influence and interact with each other in a complex manner in response to chronic stress.15,16 In a stressful situation, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) are activated. HPA axis stimulation leads to adrenocorticotrophic hormone production by the pituitary gland, which releases glucocorticoid hormones. SNS axis stimulation leads to epinephrine and norepinephrine production by the adrenal gland.17,18 Recently, studies have explored modulation of signal transduction between the nervous and immune systems and how that may impact tumor growth and metastasis.19 Multiple studies, controlled for prognosis, disease stage, and other factors, have shown that patients experiencing more distress or higher levels of depressive symptoms do not live as long as their counterparts with low distress or depression levels.20 Both the meditative and physical components of yoga can lead to enhanced relaxation, reduced SNS activation, and greater parasympathetic tone, countering the negative physiological effects of chronic stress. The effects of yoga on the HPA axis and SNS, proinflammatory cytokines, immune function, and DNA damage are discussed below.
Biological Processes
Nervous System
The effects of yoga and other forms of meditation on brain functions have been established through several studies. Yoga seems to influence basal ganglia function by improving circuits that are involved in complex cognitive functions, motor coordination, and somatosensory and emotional processes.21,22 Additionally, changes in neurotransmitter levels have been observed after yoga practice. For instance, in a 12-week yoga intervention in healthy subjects, increased levels of thalamic gamma-aminobutyric acid (GABA) in the yoga group were reported to have a positive correlation with improved mood and decreased anxiety compared with a group who did metabolically matched walking exercise.23 Levels of GABA, an inhibitory neurotransmitter, are decreased in conditions such as anxiety, depression, and epilepsy.24 Yoga therapy has been shown to improve symptoms of mood disorders and epilepsy, which leads to the hypothesis that the mechanism driving the benefits of yoga may work through stimulation of vagal efferents and an increase in GABA-mediated cortical-inhibitory tone.24,25
HPA Axis
Stress activates the HPA/SNS axis, which releases hormones such as cortisol and norepinephrine. These hormones may play a role in angiogenesis, inflammation, immune suppression, and other physiological functions, and may even reduce the effect of chemotherapeutic agents.26,27 Regular yoga practice has been shown to reduce SNS and HPA axis activity, most likely by increasing parasympathetic dominance through vagal stimulation, as demonstrated through increases in heart rate variability.28 One indicator of HPA axis dysregulation, diurnal salivary cortisol rhythm, was shown to predict survival in patients with advanced breast and renal cancer.29-33 Yoga has been shown to lead to less cortisol dysregulation due to radiotherapy and to reductions in mean cortisol levels and early morning cortisol levels in breast cancer patients undergoing radiotherapy.34 This lends support to the hypothesis that yoga helps restore HPA axis balance.
Proinflammatory Cytokines
Cancer patients tend to have increased levels of inflammatory markers such as interleukin (IL)-4, IL-10, tumor necrosis factor (TNF), interferon-γ, and C-reactive protein. This increase in inflammation is associated with worse outcomes in cancer.35 This association becomes highly relevant because the effect of inflammation on host cells in the tumor microenvironment is connected to disease progression.26 Inflammatory cytokines are also implicated in cancer-related symptoms such as fatigue, cognitive dysfunction, peripheral neuropathy, and sleep disturbances.36
Yoga is known to reduce stress and may directly or indirectly decrease inflammatory cytokines. A randomized clinical trial of a 12-week hatha yoga intervention among breast cancer survivors demonstrated decreases in IL-6, IL-1β, TNF, corticotropin-releasing factor, and cognitive complaints in the yoga group compared with those in the standard care group after 3 months.37,38 Furthermore, Carlson et al showed that, after mindfulness-based stress reduction involving a combination of gentle yoga, meditation techniques, and relaxation exercises, breast and prostate cancer patients had reduced levels of proinflammatory cytokines and cortisol.39 These reductions translated into patients reporting decreased stress levels and enhanced QOL.
Immune Function
The effects of yoga practice on the immune system have been studied in both healthy individuals and individuals with cancer. The effects on T and B lymphocytes, natural killer (NK) cells, and other immune effector cells demonstrate that meditation and yoga have beneficial effects on immune activity.40 Hormones such as catecholamines and glucocorticoids are thought to influence the availability and function of NK cells, and, as noted above, yoga has been shown to modulate stress hormones and lead to reduced immune suppression in patients with early-stage breast cancer undergoing chemotherapy.41 Additional evidence supports the ability of yoga to reduce immune suppression in the postsurgical setting, with no observed decrease in NK cell percentage after surgery for those in a yoga group compared with a control group.42 This finding is relevant to patients undergoing surgical management of their cancer and highlights the impact of yoga on the immune system.
DNA Damage
Radiation damages DNA in the peripheral blood lymphocytes of patients undergoing treatment.43,44 This damage is significant in breast cancer patients undergoing radiotherapy.45 Stress additionally causes DNA damage46 and is correlated to impaired DNA repair capacity.47,48 In a study conducted by Banerjee et al, breast cancer patients were randomly assigned to a yoga group or a supportive therapy group for 6 weeks during radiotherapy.49 Prior to the intervention, patients in the study had significant genomic instability. After treatment, patients in the yoga group experienced not only a significant reduction in anxiety and depression levels, but also a reduction in DNA damage due to radiotherapy.
Yoga in Quality of Life and Symptom Management
There is evidence showing that yoga therapy improves multiple aspects of QOL, including physical functioning, emotional health outcomes, and the symptoms cancer patients may experience, such as sleep disturbances, fatigue, and pain. Danhauer et al systematically reviewed both nonrandomized trials and randomized controlled trials involving yoga during cancer treatment.50 They found that yoga improved depression and anxiety as well as sleep and fatigue. Benefits of yoga in cancer based on randomized controlled trials are summarized in the Table. The role of yoga in improving QOL and managing symptoms patients experience during and after treatment is discussed in the following sections.
Quality of Life
Danhauer et al’s systematic review of trials involving yoga during cancer treatment found that yoga improved multiple aspects of QOL.50 For example, yoga has been shown to improve QOL in breast cancer patients undergoing radiotherapy. In a study by Chandwani et al, yoga (60-minute sessions twice a week for 6 weeks) was associated with better general health perception and physical functioning scores as well as greater benefit finding, or finding meaning in their experience, after radiotherapy compared with a wait-list group.51 The yoga group had an increase in intrusive thoughts, believed to be due to a more thorough processing of the cancer experience, which helps to improve patients’ outlook on life.52 The benefits of yoga extend beyond psychological measures during radiation treatment. Yoga was found to increase physical functioning compared with stretching in breast cancer patients undergoing radiotherapy.53
Cognitive Function
Cancer-related cognitive impairment commonly occurs during cancer treatments (eg, chemotherapy, radiotherapy, surgery, hormone therapy) and persists for months or years in survivors.54 Impairment of memory, executive function, attention, and concentration are commonly reported. In a trial of a combined hatha and restorative yoga program called Yoga for Cancer Survivors (YOCAS), which was designed by researchers at the University of Rochester, patients in the yoga arm had less memory difficulty than did patients in the standard care arm.55 However, the primary aim of the trial was to treat insomnia, so this secondary outcome needs to be interpreted with caution. Deficits in attention, memory, and executive function are often seen in cancer-related cognitive impairment, and the meditative aspect of yoga may have behavioral and neurophysical benefits that could improve cognitive functions.56 More evidence is needed to understand the role of yoga in improving cognitive functioning.
Emotional Health
Psychosocial stress is high among breast cancer patients and survivors.57,58 This causes circadian rhythm and cortisol regulation abnormalities, which are reported in women with breast cancer.59-64 Yoga is known to help stress and psychosocial and physical functioning in patients with cancer.65 Yoga was also shown to be equivalent to cognitive behavioral therapy in stress management in a population of patients without cancer.66 Daily yoga sessions lasting 60 minutes were shown to reduce reactive anxiety and trait anxiety in early-stage breast cancer patients undergoing conventional radiotherapy and chemotherapy compared with patients receiving supportive therapy, highlighting the role of yoga in managing anxiety related to treatment.67 In a study done by Culos-Reed et al, 20 cancer survivors who did 75 minutes of yoga per week for 7 weeks were compared with 18 cancer survivors who served as a control group.68 The intervention group reported significant improvement in emotional well-being, depression, concentration, and mood disturbances. In a longitudinal study by Mackenzie et al, 66 cancer survivors completed a 7-week yoga program and were assessed at baseline, immediately after the final yoga session, and at 3 and 6 months after the final session.69 Participants had significantly improved energy levels and affect. They also had moderate improvement in mindfulness and a moderate decrease in stress. Breast cancer patients who underwent restorative yoga sessions found improvements in mental health, depression, positive affect, and spirituality (peace/meaning).70 This was more pronounced in women with higher negative affect and lower emotional well-being at baseline. In a study of patients with ovarian cancer receiving chemotherapy, patients were instructed to perform up to 15-minute sessions including awareness, body movement, and breathing.71 Even with just 1 session of yoga intervention, patients experienced decreased anxiety.
Fatigue
Studies on yoga show improvement in fatigue both during and after treatment. In breast cancer patients undergoing chemotherapy, yoga was shown to benefit cognitive fatigue.72 Older cancer survivors also seem to benefit from yoga interventions.73 In a trial of a DVD-based yoga program, the benefits of yoga were similar to those of strengthening exercises, and both interventions helped decrease fatigue and improve QOL during the first year after diagnosis in early-stage breast cancer patients with cancer-related fatigue.74 Bower et al also showed that, for breast cancer survivors experiencing persistent chronic fatigue, a targeted yoga intervention led to significant improvements in fatigue and vigor over a 3-month follow-up compared with controls.75 Fatigue is commonly seen in breast cancer patients who are receiving adjuvant chemotherapy. In a study by Taso et al, women with breast cancer receiving chemotherapy were assigned to 60-minute yoga sessions incorporating Anusara yoga, gentle stretching, and relaxation twice a week for 8 weeks.76 By week 4, patients with low pretest fatigue in the yoga group experienced a reduction in fatigue. By week 8, all patients in the yoga group experienced a reduction in fatigue. Four weeks after the yoga intervention, patients in the group maintained the reduction in fatigue. This study shows the feasibility of an 8-week yoga program for women undergoing breast cancer therapy by improving fatigue. Yoga recently was added to National Comprehensive Cancer Network (NCCN) guidelines for management of cancer-related fatigue (level 1 evidence).77 However, the evidence was based on studies in women with breast cancer and survivors; therefore, more studies are needed in men and women with other cancers.
Surgical Setting/Postoperative Distress
Distress surrounding surgery in patients with breast cancer can impact postoperative outcomes. Yoga interventions, including breathing exercises, regulated breathing, and yogic relaxation techniques, improved several postsurgical measures such as length of hospital stay, drain retention, and suture removal.78 In this study, patients who practiced yoga also experienced a decrease in plasma TNF and better wound healing. Symptoms of anxiety and distress that occur preoperatively can lead to impaired immune function in addition to decreased QOL. In a study of yoga in early-stage breast cancer patients undergoing surgery, the benefit of yoga was seen not only with stress reduction but also with immune enhancement.42
Yoga has been shown to help alleviate acute pain and distress in women undergoing major surgery for gynecological cancer. A regimen of 3 15-minute sessions of yoga, including awareness meditation, coordination of breath with movement, and relaxation breathing, was shown to reduce acute pain and distress in such patients in an inpatient setting.79
Menopausal Symptoms
Breast cancer survivors have more severe menopausal symptoms compared with women without cancer.80,81 Hot flashes cause sleep disturbances and worsen fatigue and QOL.82 Tamoxifen and aromatase inhibitors significantly worsen menopausal symptoms such as hot flashes.81 Carson et al conducted a study of yoga that included postures, breathing techniques, didactic presentations, and group discussions.83 The yoga awareness regimen consisted of 8 weekly 120-minute group classes. Patients in the yoga arm had statistically significant improvements in the frequency, severity, and number of hot flashes. There were also improvements in arthralgia (joint pain), fatigue, sleep disturbance, vigor, and acceptance.
Arthralgia
Joint pain can be a major side effect that interferes with daily functions and activities in postmenopausal breast cancer survivors who receive aromatase inhibitor therapy.84 Arthralgia is reported in up to 50% of patients treated with aromatase inhibitors.84,85 It can affect functional status and lead to discontinuation of aromatase inhibitor therapy, jeopardizing clinical outcomes.86 Yoga as a complementary therapy has been shown to improve conditions such as low back pain87 and knee osteoarthritis88 in patients who do not have cancer. In a single-arm pilot trial by Galantino et al, breast cancer patients with aromatase inhibitor–related joint pain were provided with twice-weekly yoga sessions for 8 weeks. There were statistically significant improvements in balance, flexibility, pain severity, and health-related QOL.89 As noted above, improvement in arthralgia was also found in the study conducted by Carson et al.83
Insomnia
Insomnia is common among cancer patients and survivors90,91 and leads to increased fatigue and depression, decreased adherence to cancer treatments, and poor physical function and QOL.90-92 Management of insomnia consists of pharmacologic therapies such as benzodiazepines93,94 and nonpharmacologic options such as cognitive behavioral therapy.95
The first study of yoga found to improve sleep quality was conducted at MD Anderson Cancer Center in lymphoma patients.96 The effects of Tibetan yoga practices incorporating controlled breathing and visualization, mindfulness techniques, and low-impact postures were studied. Patients in the Tibetan yoga group had better subjective sleep quality, faster sleep latency, longer sleep duration, and less use of sleep medications. Mustian et al conducted a large yoga study in cancer survivors in which patients reporting chronic sleep disturbances were randomly assigned to the YOCAS program, which consisted of pranayama (breath control), 16 gentle hatha and restorative yoga postures, and meditation, or to usual care.92 The study reported improvements in global sleep quality, subjective sleep quality, actigraphy measures (wake after sleep onset, sleep efficiency), daytime dysfunction, and use of sleep medication after the yoga intervention compared with participants who received standard care.
Yoga to Address Other Symptoms
There is preliminary evidence supporting yoga as an integrative therapy for other symptoms unique to cancer survivors. For example, in head and neck cancer survivors, soft tissue damage involving the jaw, neck, shoulders, and chest results in swallowing issues, trismus, and aspiration, which are more pronounced in patients treated with conventional radiotherapy than in those treated with intensity-modulated radiotherapy.97 Some late effects of radiotherapy for head and neck cancer—such as pain, anxiety, and impaired shoulder function—were shown to be improved through the practice of hatha yoga in 1 study.98 Similarly, in a randomized controlled pilot study of patients with stage I to III breast cancer 6 months after treatment, participants in an 8-week yoga program experienced a reduction in arm induration and improvement in a QOL subscale of lymphedema symptoms. However, more evidence is needed to support the use of yoga as a therapeutic measure for breast cancer lymphedema.99,100
Yoga for Caregivers
Along with cancer patients, caregivers face psychological and physical burdens as well as deterioration in their QOL. Caregivers tend to report clinical levels of anxiety, depression, sleep disturbance, and fatigue and have similar or in fact higher levels than those of the patients for whom they are caring.101,102 Yoga has been found to help caregivers of patients with cancer. Recently, MD Anderson researchers conducted a trial in patients with high-grade glioma and their caregivers as dyads.103,104 Each dyad attended 2 or 3 60-minute weekly Vivekananda yoga sessions involving breathing exercises, physical exercises, relaxation, and meditation. The researchers found that the yoga program was safe, feasible, acceptable, and subjectively useful for patients with high-grade glioma and their caregivers. Preliminary evidence of QOL improvement for both patients and caregivers was noted. An improvement in QOL was also demonstrated in another preliminary study of yoga in patients undergoing thoracic radiotherapy and their caregivers.105
Another study by the group at MD Anderson evaluated a couple-based Tibetan yoga program that emphasized breathing exercises, gentle movements, guided visualizations, and emotional connectedness during radiotherapy for lung cancer.106 This study included 10 patient‐caregiver dyads and found the program to be feasible, safe, and acceptable. The researchers also found preliminary evidence of improved QOL by the end of radiotherapy relative to baseline—specifically in the areas of spiritual well‐being for patients, fatigue for caregivers, and sleep disturbances and mental health issues such as anxiety and depressive symptoms for both patients and caregivers. This is noteworthy, as QOL typically deteriorates during the course of radiotherapy, and the yoga program was able to buffer these changes.
Conclusion
Yoga therapy has been used successfully as an adjunct modality to improve QOL and cancer-related symptoms. As a part of an integrative medicine approach, yoga is commonly recommended for patients undergoing cancer treatment. Danhauer et al reviewed randomized controlled trials during and after treatment and concluded that the evidence is clearly positive for QOL, fatigue, and perceived stress.107 Results are less consistent but supportive for psychosocial outcomes such as benefit finding and spirituality. Evidence is mixed for sleep, anxiety, and depression. Post-treatment studies demonstrate improvements in fatigue, sleep, and multiple QOL domains. Yoga has been included in NCCN guidelines for fatigue management. Yoga, if approved by a physician, is also included among the behavioral therapies for anticipatory emesis and prevention and treatment of nausea in the recent update of the NCCN guidelines.108 The Society for Integrative Oncology guidelines include yoga for anxiety/stress reduction as a part of integrative treatment in breast cancer patients during and after therapy, which was endorsed by the American Society of Clinical Oncology.109
Because of the strong evidence for its benefits and a low side-effect profile, yoga is offered in group-class settings for patients during and after treatment and/or for caregivers in our institution. We often prescribe yoga as a therapeutic modality for selected groups of patients in our clinical practice. However, some patients may have restrictions after surgery that must be considered. In general, yoga has an excellent safety profile, the evidence base is strong, and we recommend that yoga therapy should be part of the standard of care as an integrative approach for patients with cancer undergoing active treatment as well as for cancer survivors and caregivers.
Acknowledgement: The authors thank Bryan Tutt for providing editorial assistance.
Corresponding author: Santhosshi Narayanan, MD, Department of Palliative, Rehabilitation, and Integrative Medicine, Unit 1414, The University of Texas MD Anderson Cancer Center, 1400 Pressler St., Houston, TX 77030; [email protected].
Financial disclosures: None.
From the University of Texas MD Anderson Cancer Center, Houston, TX (Drs. Narayanan, Lopez, Chaoul, Liu, Milbury, and Cohen, and Ms. Mallaiah); the University of Texas Health Science Center at Tyler (Dr. Meegada); and Texas Tech University Health Sciences Center, Lubbock, TX (Ms. Francisco).
Abstract
- Objective: To review the effects of yoga as an adjunct supportive care modality alongside conventional cancer treatment on quality of life (QOL), physical and mental health outcomes, and physiological and biological measures of cancer survivors.
- Methods: Nonsystematic review of the literature.
- Results: Yoga therapy, one of the most frequently used mind-body modalities, has been studied extensively in cancer survivors (from the time of diagnosis through long-term recovery). Yoga affects human physiology on multiple levels, including psychological outcomes, immune and endocrine function, and cardiovascular parameters, as well as multiple areas of QOL. It has been found to reduce psychological stress and fatigue and improve QOL in cancer patients and survivors. Yoga has also been used to manage symptoms such as arthralgia, fatigue, and insomnia. In addition, yoga offers benefits not only for cancer survivors but also for their caregivers.
- Conclusion: As part of an integrative, evidence-informed approach to cancer care, yoga may provide benefits that support the health of cancer survivors and caregivers.
Keywords: fatigue; cancer; proinflammatory cytokines; integrative; mind-body practices; meditation; DNA damage; stress; psychoneuro-immunoendocrine axis; lymphedema; insomnia.
A diagnosis of cancer and adverse effects related to its treatment may have negative effects on quality of life (QOL), contributing to emotional and physical distress in patients and caregivers. Many patients express an interest in pursuing nonpharmacological options, alone or as an adjunct to conventional therapy, to help manage symptoms. The use of complementary medicine approaches to health, including nonpharmacological approaches to symptom management, is highest among individuals with cancer.1 According to a published expert consensus, integrative oncology is defined as a “patient-centered, evidence-informed field of cancer care that utilizes mind and body practices, natural products, and/or lifestyle modifications from different traditions alongside conventional cancer treatments. Integrative oncology aims to optimize health, QOL, and clinical outcomes across the cancer care continuum and to empower people to prevent cancer and become active participants before, during, and beyond cancer treatment.”2 A key component of this definition, often misunderstood in the field of oncology, is that these modalities and treatments are used alongside conventional cancer treatments and not as an alternative. In an attempt to meet patients’ needs and appropriately use these approaches, integrative oncology programs are now part of most cancer centers in the United States.3-6
Because of their overall safety, mind-body therapies are commonly used by patients and recommended by clinicians. Mind-body therapies include yoga, tai chi, qigong, meditation, and relaxation. Expressive arts such as journaling and music, art, and dance therapies also fall in the mind-body category.7 Yoga is a movement-based mind-body practice that focuses on synchronizing body, breath, and mind. Yoga has been increasingly used by patients for health benefits,8 and numerous studies have evaluated yoga as a complementary intervention for individuals with cancer.9-14 Here, we review the physiological basis of yoga in oncology and the effects of yoga on biological processes, QOL, and symptoms during and after cancer treatment.
Physiological Basis
Many patients may use mind-body programs such as yoga to help manage the psychological and physiological consequences of unmanaged chronic stress and improve their overall QOL. The central nervous system, endocrine system, and immune system influence and interact with each other in a complex manner in response to chronic stress.15,16 In a stressful situation, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) are activated. HPA axis stimulation leads to adrenocorticotrophic hormone production by the pituitary gland, which releases glucocorticoid hormones. SNS axis stimulation leads to epinephrine and norepinephrine production by the adrenal gland.17,18 Recently, studies have explored modulation of signal transduction between the nervous and immune systems and how that may impact tumor growth and metastasis.19 Multiple studies, controlled for prognosis, disease stage, and other factors, have shown that patients experiencing more distress or higher levels of depressive symptoms do not live as long as their counterparts with low distress or depression levels.20 Both the meditative and physical components of yoga can lead to enhanced relaxation, reduced SNS activation, and greater parasympathetic tone, countering the negative physiological effects of chronic stress. The effects of yoga on the HPA axis and SNS, proinflammatory cytokines, immune function, and DNA damage are discussed below.
Biological Processes
Nervous System
The effects of yoga and other forms of meditation on brain functions have been established through several studies. Yoga seems to influence basal ganglia function by improving circuits that are involved in complex cognitive functions, motor coordination, and somatosensory and emotional processes.21,22 Additionally, changes in neurotransmitter levels have been observed after yoga practice. For instance, in a 12-week yoga intervention in healthy subjects, increased levels of thalamic gamma-aminobutyric acid (GABA) in the yoga group were reported to have a positive correlation with improved mood and decreased anxiety compared with a group who did metabolically matched walking exercise.23 Levels of GABA, an inhibitory neurotransmitter, are decreased in conditions such as anxiety, depression, and epilepsy.24 Yoga therapy has been shown to improve symptoms of mood disorders and epilepsy, which leads to the hypothesis that the mechanism driving the benefits of yoga may work through stimulation of vagal efferents and an increase in GABA-mediated cortical-inhibitory tone.24,25
HPA Axis
Stress activates the HPA/SNS axis, which releases hormones such as cortisol and norepinephrine. These hormones may play a role in angiogenesis, inflammation, immune suppression, and other physiological functions, and may even reduce the effect of chemotherapeutic agents.26,27 Regular yoga practice has been shown to reduce SNS and HPA axis activity, most likely by increasing parasympathetic dominance through vagal stimulation, as demonstrated through increases in heart rate variability.28 One indicator of HPA axis dysregulation, diurnal salivary cortisol rhythm, was shown to predict survival in patients with advanced breast and renal cancer.29-33 Yoga has been shown to lead to less cortisol dysregulation due to radiotherapy and to reductions in mean cortisol levels and early morning cortisol levels in breast cancer patients undergoing radiotherapy.34 This lends support to the hypothesis that yoga helps restore HPA axis balance.
Proinflammatory Cytokines
Cancer patients tend to have increased levels of inflammatory markers such as interleukin (IL)-4, IL-10, tumor necrosis factor (TNF), interferon-γ, and C-reactive protein. This increase in inflammation is associated with worse outcomes in cancer.35 This association becomes highly relevant because the effect of inflammation on host cells in the tumor microenvironment is connected to disease progression.26 Inflammatory cytokines are also implicated in cancer-related symptoms such as fatigue, cognitive dysfunction, peripheral neuropathy, and sleep disturbances.36
Yoga is known to reduce stress and may directly or indirectly decrease inflammatory cytokines. A randomized clinical trial of a 12-week hatha yoga intervention among breast cancer survivors demonstrated decreases in IL-6, IL-1β, TNF, corticotropin-releasing factor, and cognitive complaints in the yoga group compared with those in the standard care group after 3 months.37,38 Furthermore, Carlson et al showed that, after mindfulness-based stress reduction involving a combination of gentle yoga, meditation techniques, and relaxation exercises, breast and prostate cancer patients had reduced levels of proinflammatory cytokines and cortisol.39 These reductions translated into patients reporting decreased stress levels and enhanced QOL.
Immune Function
The effects of yoga practice on the immune system have been studied in both healthy individuals and individuals with cancer. The effects on T and B lymphocytes, natural killer (NK) cells, and other immune effector cells demonstrate that meditation and yoga have beneficial effects on immune activity.40 Hormones such as catecholamines and glucocorticoids are thought to influence the availability and function of NK cells, and, as noted above, yoga has been shown to modulate stress hormones and lead to reduced immune suppression in patients with early-stage breast cancer undergoing chemotherapy.41 Additional evidence supports the ability of yoga to reduce immune suppression in the postsurgical setting, with no observed decrease in NK cell percentage after surgery for those in a yoga group compared with a control group.42 This finding is relevant to patients undergoing surgical management of their cancer and highlights the impact of yoga on the immune system.
DNA Damage
Radiation damages DNA in the peripheral blood lymphocytes of patients undergoing treatment.43,44 This damage is significant in breast cancer patients undergoing radiotherapy.45 Stress additionally causes DNA damage46 and is correlated to impaired DNA repair capacity.47,48 In a study conducted by Banerjee et al, breast cancer patients were randomly assigned to a yoga group or a supportive therapy group for 6 weeks during radiotherapy.49 Prior to the intervention, patients in the study had significant genomic instability. After treatment, patients in the yoga group experienced not only a significant reduction in anxiety and depression levels, but also a reduction in DNA damage due to radiotherapy.
Yoga in Quality of Life and Symptom Management
There is evidence showing that yoga therapy improves multiple aspects of QOL, including physical functioning, emotional health outcomes, and the symptoms cancer patients may experience, such as sleep disturbances, fatigue, and pain. Danhauer et al systematically reviewed both nonrandomized trials and randomized controlled trials involving yoga during cancer treatment.50 They found that yoga improved depression and anxiety as well as sleep and fatigue. Benefits of yoga in cancer based on randomized controlled trials are summarized in the Table. The role of yoga in improving QOL and managing symptoms patients experience during and after treatment is discussed in the following sections.
Quality of Life
Danhauer et al’s systematic review of trials involving yoga during cancer treatment found that yoga improved multiple aspects of QOL.50 For example, yoga has been shown to improve QOL in breast cancer patients undergoing radiotherapy. In a study by Chandwani et al, yoga (60-minute sessions twice a week for 6 weeks) was associated with better general health perception and physical functioning scores as well as greater benefit finding, or finding meaning in their experience, after radiotherapy compared with a wait-list group.51 The yoga group had an increase in intrusive thoughts, believed to be due to a more thorough processing of the cancer experience, which helps to improve patients’ outlook on life.52 The benefits of yoga extend beyond psychological measures during radiation treatment. Yoga was found to increase physical functioning compared with stretching in breast cancer patients undergoing radiotherapy.53
Cognitive Function
Cancer-related cognitive impairment commonly occurs during cancer treatments (eg, chemotherapy, radiotherapy, surgery, hormone therapy) and persists for months or years in survivors.54 Impairment of memory, executive function, attention, and concentration are commonly reported. In a trial of a combined hatha and restorative yoga program called Yoga for Cancer Survivors (YOCAS), which was designed by researchers at the University of Rochester, patients in the yoga arm had less memory difficulty than did patients in the standard care arm.55 However, the primary aim of the trial was to treat insomnia, so this secondary outcome needs to be interpreted with caution. Deficits in attention, memory, and executive function are often seen in cancer-related cognitive impairment, and the meditative aspect of yoga may have behavioral and neurophysical benefits that could improve cognitive functions.56 More evidence is needed to understand the role of yoga in improving cognitive functioning.
Emotional Health
Psychosocial stress is high among breast cancer patients and survivors.57,58 This causes circadian rhythm and cortisol regulation abnormalities, which are reported in women with breast cancer.59-64 Yoga is known to help stress and psychosocial and physical functioning in patients with cancer.65 Yoga was also shown to be equivalent to cognitive behavioral therapy in stress management in a population of patients without cancer.66 Daily yoga sessions lasting 60 minutes were shown to reduce reactive anxiety and trait anxiety in early-stage breast cancer patients undergoing conventional radiotherapy and chemotherapy compared with patients receiving supportive therapy, highlighting the role of yoga in managing anxiety related to treatment.67 In a study done by Culos-Reed et al, 20 cancer survivors who did 75 minutes of yoga per week for 7 weeks were compared with 18 cancer survivors who served as a control group.68 The intervention group reported significant improvement in emotional well-being, depression, concentration, and mood disturbances. In a longitudinal study by Mackenzie et al, 66 cancer survivors completed a 7-week yoga program and were assessed at baseline, immediately after the final yoga session, and at 3 and 6 months after the final session.69 Participants had significantly improved energy levels and affect. They also had moderate improvement in mindfulness and a moderate decrease in stress. Breast cancer patients who underwent restorative yoga sessions found improvements in mental health, depression, positive affect, and spirituality (peace/meaning).70 This was more pronounced in women with higher negative affect and lower emotional well-being at baseline. In a study of patients with ovarian cancer receiving chemotherapy, patients were instructed to perform up to 15-minute sessions including awareness, body movement, and breathing.71 Even with just 1 session of yoga intervention, patients experienced decreased anxiety.
Fatigue
Studies on yoga show improvement in fatigue both during and after treatment. In breast cancer patients undergoing chemotherapy, yoga was shown to benefit cognitive fatigue.72 Older cancer survivors also seem to benefit from yoga interventions.73 In a trial of a DVD-based yoga program, the benefits of yoga were similar to those of strengthening exercises, and both interventions helped decrease fatigue and improve QOL during the first year after diagnosis in early-stage breast cancer patients with cancer-related fatigue.74 Bower et al also showed that, for breast cancer survivors experiencing persistent chronic fatigue, a targeted yoga intervention led to significant improvements in fatigue and vigor over a 3-month follow-up compared with controls.75 Fatigue is commonly seen in breast cancer patients who are receiving adjuvant chemotherapy. In a study by Taso et al, women with breast cancer receiving chemotherapy were assigned to 60-minute yoga sessions incorporating Anusara yoga, gentle stretching, and relaxation twice a week for 8 weeks.76 By week 4, patients with low pretest fatigue in the yoga group experienced a reduction in fatigue. By week 8, all patients in the yoga group experienced a reduction in fatigue. Four weeks after the yoga intervention, patients in the group maintained the reduction in fatigue. This study shows the feasibility of an 8-week yoga program for women undergoing breast cancer therapy by improving fatigue. Yoga recently was added to National Comprehensive Cancer Network (NCCN) guidelines for management of cancer-related fatigue (level 1 evidence).77 However, the evidence was based on studies in women with breast cancer and survivors; therefore, more studies are needed in men and women with other cancers.
Surgical Setting/Postoperative Distress
Distress surrounding surgery in patients with breast cancer can impact postoperative outcomes. Yoga interventions, including breathing exercises, regulated breathing, and yogic relaxation techniques, improved several postsurgical measures such as length of hospital stay, drain retention, and suture removal.78 In this study, patients who practiced yoga also experienced a decrease in plasma TNF and better wound healing. Symptoms of anxiety and distress that occur preoperatively can lead to impaired immune function in addition to decreased QOL. In a study of yoga in early-stage breast cancer patients undergoing surgery, the benefit of yoga was seen not only with stress reduction but also with immune enhancement.42
Yoga has been shown to help alleviate acute pain and distress in women undergoing major surgery for gynecological cancer. A regimen of 3 15-minute sessions of yoga, including awareness meditation, coordination of breath with movement, and relaxation breathing, was shown to reduce acute pain and distress in such patients in an inpatient setting.79
Menopausal Symptoms
Breast cancer survivors have more severe menopausal symptoms compared with women without cancer.80,81 Hot flashes cause sleep disturbances and worsen fatigue and QOL.82 Tamoxifen and aromatase inhibitors significantly worsen menopausal symptoms such as hot flashes.81 Carson et al conducted a study of yoga that included postures, breathing techniques, didactic presentations, and group discussions.83 The yoga awareness regimen consisted of 8 weekly 120-minute group classes. Patients in the yoga arm had statistically significant improvements in the frequency, severity, and number of hot flashes. There were also improvements in arthralgia (joint pain), fatigue, sleep disturbance, vigor, and acceptance.
Arthralgia
Joint pain can be a major side effect that interferes with daily functions and activities in postmenopausal breast cancer survivors who receive aromatase inhibitor therapy.84 Arthralgia is reported in up to 50% of patients treated with aromatase inhibitors.84,85 It can affect functional status and lead to discontinuation of aromatase inhibitor therapy, jeopardizing clinical outcomes.86 Yoga as a complementary therapy has been shown to improve conditions such as low back pain87 and knee osteoarthritis88 in patients who do not have cancer. In a single-arm pilot trial by Galantino et al, breast cancer patients with aromatase inhibitor–related joint pain were provided with twice-weekly yoga sessions for 8 weeks. There were statistically significant improvements in balance, flexibility, pain severity, and health-related QOL.89 As noted above, improvement in arthralgia was also found in the study conducted by Carson et al.83
Insomnia
Insomnia is common among cancer patients and survivors90,91 and leads to increased fatigue and depression, decreased adherence to cancer treatments, and poor physical function and QOL.90-92 Management of insomnia consists of pharmacologic therapies such as benzodiazepines93,94 and nonpharmacologic options such as cognitive behavioral therapy.95
The first study of yoga found to improve sleep quality was conducted at MD Anderson Cancer Center in lymphoma patients.96 The effects of Tibetan yoga practices incorporating controlled breathing and visualization, mindfulness techniques, and low-impact postures were studied. Patients in the Tibetan yoga group had better subjective sleep quality, faster sleep latency, longer sleep duration, and less use of sleep medications. Mustian et al conducted a large yoga study in cancer survivors in which patients reporting chronic sleep disturbances were randomly assigned to the YOCAS program, which consisted of pranayama (breath control), 16 gentle hatha and restorative yoga postures, and meditation, or to usual care.92 The study reported improvements in global sleep quality, subjective sleep quality, actigraphy measures (wake after sleep onset, sleep efficiency), daytime dysfunction, and use of sleep medication after the yoga intervention compared with participants who received standard care.
Yoga to Address Other Symptoms
There is preliminary evidence supporting yoga as an integrative therapy for other symptoms unique to cancer survivors. For example, in head and neck cancer survivors, soft tissue damage involving the jaw, neck, shoulders, and chest results in swallowing issues, trismus, and aspiration, which are more pronounced in patients treated with conventional radiotherapy than in those treated with intensity-modulated radiotherapy.97 Some late effects of radiotherapy for head and neck cancer—such as pain, anxiety, and impaired shoulder function—were shown to be improved through the practice of hatha yoga in 1 study.98 Similarly, in a randomized controlled pilot study of patients with stage I to III breast cancer 6 months after treatment, participants in an 8-week yoga program experienced a reduction in arm induration and improvement in a QOL subscale of lymphedema symptoms. However, more evidence is needed to support the use of yoga as a therapeutic measure for breast cancer lymphedema.99,100
Yoga for Caregivers
Along with cancer patients, caregivers face psychological and physical burdens as well as deterioration in their QOL. Caregivers tend to report clinical levels of anxiety, depression, sleep disturbance, and fatigue and have similar or in fact higher levels than those of the patients for whom they are caring.101,102 Yoga has been found to help caregivers of patients with cancer. Recently, MD Anderson researchers conducted a trial in patients with high-grade glioma and their caregivers as dyads.103,104 Each dyad attended 2 or 3 60-minute weekly Vivekananda yoga sessions involving breathing exercises, physical exercises, relaxation, and meditation. The researchers found that the yoga program was safe, feasible, acceptable, and subjectively useful for patients with high-grade glioma and their caregivers. Preliminary evidence of QOL improvement for both patients and caregivers was noted. An improvement in QOL was also demonstrated in another preliminary study of yoga in patients undergoing thoracic radiotherapy and their caregivers.105
Another study by the group at MD Anderson evaluated a couple-based Tibetan yoga program that emphasized breathing exercises, gentle movements, guided visualizations, and emotional connectedness during radiotherapy for lung cancer.106 This study included 10 patient‐caregiver dyads and found the program to be feasible, safe, and acceptable. The researchers also found preliminary evidence of improved QOL by the end of radiotherapy relative to baseline—specifically in the areas of spiritual well‐being for patients, fatigue for caregivers, and sleep disturbances and mental health issues such as anxiety and depressive symptoms for both patients and caregivers. This is noteworthy, as QOL typically deteriorates during the course of radiotherapy, and the yoga program was able to buffer these changes.
Conclusion
Yoga therapy has been used successfully as an adjunct modality to improve QOL and cancer-related symptoms. As a part of an integrative medicine approach, yoga is commonly recommended for patients undergoing cancer treatment. Danhauer et al reviewed randomized controlled trials during and after treatment and concluded that the evidence is clearly positive for QOL, fatigue, and perceived stress.107 Results are less consistent but supportive for psychosocial outcomes such as benefit finding and spirituality. Evidence is mixed for sleep, anxiety, and depression. Post-treatment studies demonstrate improvements in fatigue, sleep, and multiple QOL domains. Yoga has been included in NCCN guidelines for fatigue management. Yoga, if approved by a physician, is also included among the behavioral therapies for anticipatory emesis and prevention and treatment of nausea in the recent update of the NCCN guidelines.108 The Society for Integrative Oncology guidelines include yoga for anxiety/stress reduction as a part of integrative treatment in breast cancer patients during and after therapy, which was endorsed by the American Society of Clinical Oncology.109
Because of the strong evidence for its benefits and a low side-effect profile, yoga is offered in group-class settings for patients during and after treatment and/or for caregivers in our institution. We often prescribe yoga as a therapeutic modality for selected groups of patients in our clinical practice. However, some patients may have restrictions after surgery that must be considered. In general, yoga has an excellent safety profile, the evidence base is strong, and we recommend that yoga therapy should be part of the standard of care as an integrative approach for patients with cancer undergoing active treatment as well as for cancer survivors and caregivers.
Acknowledgement: The authors thank Bryan Tutt for providing editorial assistance.
Corresponding author: Santhosshi Narayanan, MD, Department of Palliative, Rehabilitation, and Integrative Medicine, Unit 1414, The University of Texas MD Anderson Cancer Center, 1400 Pressler St., Houston, TX 77030; [email protected].
Financial disclosures: None.
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Reports. 2008;(12):1-23.
2. Witt CM, Balneaves LG, Cardoso MJ, et al. A comprehensive definition for integrative oncology. NCI Monographs. 2017;2017(52):3-8.
3. Brauer JA, El Sehamy A, Metz JM, Mao JJ. Complementary and alternative medicine and supportive care at leading cancer centers: a systematic analysis of websites. J Altern Complement Med. 2010;16:183-186.
4. Lopez G, McQuade J, Cohen L, et al. Integrative oncology physician consultations at a comprehensive cancer center: analysis of demographic, clinical and patient reported outcomes. J Cancer. 2017;8:395-402.
5. Latte-Naor S, Mao JJ. Putting integrative oncology into practice: concepts and approaches. J Oncol Pract. 2019;15:7-14.
6. Richardson MA, Sanders T, Palmer JL, et al. Complementary/alternative medicine use in a comprehensive cancer center and the implications for oncology. J Clin Oncol. 2000;18:2505-2514.
7. Chaoul A, Milbury K, Sood AK, et al. Mind-body practices in cancer care. Curr Oncol Rep. 2014;16:417.
8. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10:44-49.
9. Smith KB, Pukall CF. An evidence-based review of yoga as a complementary intervention for patients with cancer. Psychooncology. 2009;18:465-475.
10. Buffart LM, van Uffelen JG, Riphagen II, et al. Physical and psychosocial benefits of yoga in cancer patients and survivors, a systematic review and meta-analysis of randomized controlled trials. BMC Cancer. 2012;12:559.
11. Cramer H, Lange S, Klose P, et al. Yoga for breast cancer patients and survivors: a systematic review and meta-analysis. BMC Cancer. 2012;12:412.
12. Felbel S, Meerpohl JJ, Monsef I, et al. Yoga in addition to standard care for patients with haematological malignancies. Cochrane Database Syst Rev. 2014;(6):CD010146.
13. Greenlee H, Balneaves LG, Carlson LE, et al. Clinical practice guidelines on the use of integrative therapies as supportive care in patients treated for breast cancer. NCI Monographs. 2014;2014:346-358.
14. Sharma M, Haider T, Knowlden AP. Yoga as an alternative and complementary treatment for cancer: a systematic review. J Altern Complement Med. 2013;19:870-875.
15. Kordon C, Bihoreau C. Integrated communication between the nervous, endocrine and immune systems. Horm Res. 1989;31:100-104.
16. Gonzalez-Diaz SN, Arias-Cruz A, Elizondo-Villarreal B, Monge-Ortega OP. Psychoneuroimmunoendocrinology: clinical implications. World Allergy Organ J. 2017;10:19.
17. Godoy LD, Rossignoli MT, Delfino-Pereira P, et al. A comprehensive overview on stress neurobiology: basic concepts and clinical implications. Front Behav Neurosci. 2018;12:127.
18. Goldstein DS. Adrenal responses to stress. Cell Mol Neurobiol. 2010;30:1433-1440.
19. Cole SW, Nagaraja AS, Lutgendorf SK, et al. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563-567.
20. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
21. Arsalidou M, Duerden EG, Taylor MJ. The centre of the brain: topographical model of motor, cognitive, affective, and somatosensory functions of the basal ganglia. Hum Brain Mapp. 2013;34:3031-3054.
22. Gard T, Taquet M, Dixit R, et al. Greater widespread functional connectivity of the caudate in older adults who practice kripalu yoga and vipassana meditation than in controls. Front Hum Neurosci. 2015;9:137.
23. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
24. Brambilla P, Perez J, Barale F, et al. GABAergic dysfunction in mood disorders. Mol Psychiatry. 2003;8:721-737.
25. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
26. Lutgendorf SK, Sood AK. Biobehavioral factors and cancer progression: physiological pathways and mechanisms. Psychosom Med. 2011;73:724-730.
27. Armaiz-Pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain Behav Immun. 2013;30(suppl):S19-S25.
28. Tyagi A, Cohen M. Yoga and heart rate variability: A comprehensive review of the literature. Int J Yoga. 2016;9:97-113.
29. Sephton SE, Lush E, Dedert EA, et al. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain Behav Immun. 2013;30 (suppl):S163-S170.
30. Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst. 2000;92:994-1000.
31. Arafah BM, Nishiyama FJ, Tlaygeh H, Hejal R. Measurement of salivary cortisol concentration in the assessment of adrenal function in critically ill subjects: a surrogate marker of the circulating free cortisol. J Clin Endocrinol Metab. 2007;92:2965-2971.
32. Cohen L, de Moor C, Devine D, et al. Endocrine levels at the start of treatment are associated with subsequent psychological adjustment in cancer patients with metastatic disease. Psychosom Med. 2001;63:951-958.
33. Cohen L, Cole SW, Sood AK, et al. Depressive symptoms and cortisol rhythmicity predict survival in patients with renal cell carcinoma: role of inflammatory signaling. PloS One. 2012;7:e42324.
34. Vadiraja HS, Raghavendra RM, Nagarathna R, et al. Effects of a yoga program on cortisol rhythm and mood states in early breast cancer patients undergoing adjuvant radiotherapy: a randomized controlled trial. Integr Cancer Ther. 2009;8:37-46.
35. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345:164-173.
36. Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun. 2013;30(suppl):S48-S57.
37. Derry HM, Jaremka LM, Bennett JM, et al. Yoga and self-reported cognitive problems in breast cancer survivors: a randomized controlled trial. Psychooncology. 2015;24:958-966.
38. Kiecolt-Glaser JK, Christian L, Preston H, et al. Stress, inflammation, and yoga practice. Psychosom Med. 2010;72:113-121.
39. Carlson LE, Speca M, Faris P, Patel KD. One year pre-post intervention follow-up of psychological, immune, endocrine and blood pressure outcomes of mindfulness-based stress reduction (MBSR) in breast and prostate cancer outpatients. Brain Behav Immun. 2007;21:1038-1049.
40. Infante JR, Peran F, Rayo JI, et al. Levels of immune cells in transcendental meditation practitioners. Int J Yoga. 2014;7:147-151.
41. Rao RM, Telles S, Nagendra HR, et al. Effects of yoga on natural killer cell counts in early breast cancer patients undergoing conventional treatment. Comment to: recreational music-making modulates natural killer cell activity, cytokines, and mood states in corporate employees Masatada Wachi, Masahiro Koyama, Masanori Utsuyama, Barry B. Bittman, Masanobu Kitagawa, Katsuiku Hirokawa Med Sci Monit, 2007; 13(2): CR57-70. Med Sci Monit. 2008;14:LE3-4.
42. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on mood states, distress, quality of life and immune outcomes in early stage breast cancer patients undergoing surgery. Int J Yoga. 2008;1:11-20.
43. Mozdarani H, Mansouri Z, Haeri SA. Cytogenetic radiosensitivity of g0-lymphocytes of breast and esophageal cancer patients as determined by micronucleus assay. J Radiat Res. 2005;46:111-116.
44. Scott D, Barber JB, Levine EL, et al. Radiation-induced micronucleus induction in lymphocytes identifies a high frequency of radiosensitive cases among breast cancer patients: a test for predisposition? Br J Cancer. 1998;77:614-620.
45. Banerjee B, Sharma S, Hegde S, Hande MP. Analysis of telomere damage by fluorescence in situ hybridisation on micronuclei in lymphocytes of breast carcinoma patients after radiotherapy. Breast Cancer Res Treat. 2008;107:25-31.
46. Epel ES, Blackburn EH, Lin J, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312-17315.
47. Glaser R, Thorn BE, Tarr KL, et al. Effects of stress on methyltransferase synthesis: an important DNA repair enzyme. Health Psychol. 1985;4:403-412.
48. Kiecolt-Glaser JK, Stephens RE, Lipetz PD, et al. Distress and DNA repair in human lymphocytes. J Behav Med. 1985;8:311-320.
49. Banerjee B, Vadiraj HS, Ram A, et al. Effects of an integrated yoga program in modulating psychological stress and radiation-induced genotoxic stress in breast cancer patients undergoing radiotherapy. Integr Cancer Ther. 2007;6:242-250.
50. Danhauer SC, Addington EL, Sohl SJ, et al. Review of yoga therapy during cancer treatment. Support Care Cancer. 2017;25:1357-1372.
51. Chandwani KD, Thornton B, Perkins GH, et al. Yoga improves quality of life and benefit finding in women undergoing radiotherapy for breast cancer. J Soc Integr Oncol. 2010;8:43-55.
52. Ratcliff CG, Milbury K, Chandwani KD, et al. Examining mediators and moderators of yoga for women with breast cancer undergoing radiotherapy. Integr Cancer Ther. 2016;15:250-262.
53. Chandwani KD, Perkins G, Nagendra HR, et al. Randomized, controlled trial of yoga in women with breast cancer undergoing radiotherapy. J Clin Oncol. 2014;32:1058-1065.
54. Janelsins MC, Kesler SR, Ahles TA, Morrow GR. Prevalence, mechanisms, and management of cancer-related cognitive impairment. Int Rev Psychiatry. 2014;26:102-113.
55. Janelsins MC, Peppone LJ, Heckler CE, et al. YOCAS(c)(R) yoga reduces self-reported memory difficulty in cancer survivors in a nationwide randomized clinical trial: investigating relationships between memory and sleep. Integr Cancer Ther. 2016;15:263-271.
56. Biegler KA, Chaoul MA, Cohen L. Cancer, cognitive impairment, and meditation. Acta Oncol. 2009;48:18-26.
57. Carlson LE, Angen M, Cullum J, et al. High levels of untreated distress and fatigue in cancer patients. Br J Cancer. 2004;90:2297-2304.
58. Herschbach P, Keller M, Knight L, et al. Psychological problems of cancer patients: a cancer distress screening with a cancer-specific questionnaire. Br J Cancer. 2004;91:504-511.
59. Abercrombie HC, Giese-Davis J, Sephton S, et al. Flattened cortisol rhythms in metastatic breast cancer patients. Psychoneuroendocrinology. 2004;29:1082-1092.
60. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med. 2005;67:277-280.
61. Bower JE, Ganz PA, Dickerson SS, et al. Diurnal cortisol rhythm and fatigue in breast cancer survivors. Psychoneuroendocrinology. 2005;30:92-100.
62. Giese-Davis J, Sephton SE, Abercrombie HC, et al. Repression and high anxiety are associated with aberrant diurnal cortisol rhythms in women with metastatic breast cancer. Health Psychol. 2004;23:645-650.
63. Giese-Davis J, DiMiceli S, Sephton S, Spiegel D. Emotional expression and diurnal cortisol slope in women with metastatic breast cancer in supportive-expressive group therapy: a preliminary study. Biol Psychol. 2006;73:190-198.
64. Stone AA, Schwartz JE, Smyth J, et al. Individual differences in the diurnal cycle of salivary free cortisol: a replication of flattened cycles for some individuals. Psychoneuroendocrinology. 2001;26:295-306.
65. Bower JE, Woolery A, Sternlieb B, Garet D. Yoga for cancer patients and survivors. Cancer Control. 2005;12:165-171.
66. Granath J, Ingvarsson S, von Thiele U, Lundberg U. Stress management: a randomized study of cognitive behavioural therapy and yoga. Cogn Behav Therap. 2006;35:3-10.
67. Rao MR, Raghuram N, Nagendra HR, et al. Anxiolytic effects of a yoga program in early breast cancer patients undergoing conventional treatment: a randomized controlled trial. Complement Ther Med. 2009;17:1-8.
68. Culos-Reed SN, Carlson LE, Daroux LM, Hately-Aldous S. A pilot study of yoga for breast cancer survivors: physical and psychological benefits. Psychooncology. 2006;15:891-897.
69. Mackenzie MJ, Carlson LE, Ekkekakis P, et al. Affect and mindfulness as predictors of change in mood disturbance, stress symptoms, and quality of life in a community-based yoga program for cancer survivors. Evid Based Complement Alternat Med. 2013;2013:419496.
70. Danhauer SC, Mihalko SL, Russell GB, et al. Restorative yoga for women with breast cancer: findings from a randomized pilot study. Psycho-oncology. 2009;18:360-368.
71. Sohl SJ, Danhauer SC, Schnur JB, et al. Feasibility of a brief yoga intervention during chemotherapy for persistent or recurrent ovarian cancer. Explore (NY). 2012;8:197-198.
72. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer. 2016;24:4005-4015.
73. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol. 2015;6:8-14.
74. Wang G, Wang S, Jiang P, Zeng C. Effect of yoga on cancer related fatigue in breast cancer patients with chemotherapy [in Chinese]. Zhong Nan Da Xue Bao Yi Xue Ban. 2014;39:1077-1082.
75. Bower JE, Garet D, Sternlieb B, et al. Yoga for persistent fatigue in breast cancer survivors: a randomized controlled trial. Cancer. 2012;118:3766-3775.
76. Taso CJ, Lin HS, Lin WL, et al. The effect of yoga exercise on improving depression, anxiety, and fatigue in women with breast cancer: a randomized controlled trial. J Nurs Res. 2014;22:155-164.
77. Berger AM, Mooney K, Alvarez-Perez A, et al. Cancer-related fatigue, Version 2.2015. J Natl Compr Canc Netw. 2015;13:1012-1039.
78. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on postoperative outcomes and wound healing in early operable breast cancer patients undergoing surgery. Int J Yoga. 2008;1:33-41.
79. Sohl SJ, Avis NE, Stanbery K, et al. Feasibility of a brief yoga intervention for improving acute pain and distress post gynecologic surgery. Int J Yoga Therap. 2016;26:43-47.
80. Gupta P, Sturdee DW, Palin SL, et al. Menopausal symptoms in women treated for breast cancer: the prevalence and severity of symptoms and their perceived effects on quality of life. Climacteric. 2006;9:49-58.
81. Canney PA, Hatton MQ. The prevalence of menopausal symptoms in patients treated for breast cancer. Clin Oncol (R Coll Radiol). 1994;6:297-299.
82. Carpenter JS, Johnson D, Wagner L, Andrykowski M. Hot flashes and related outcomes in breast cancer survivors and matched comparison women. Oncol Nurs Forum. 2002;29:E16-25.
83. Carson JW, Carson KM, Porter LS, et al. Yoga of Awareness program for menopausal symptoms in breast cancer survivors: results from a randomized trial. Support Care Cancer. 2009;17:1301-1309.
84. Burstein HJ. Aromatase inhibitor-associated arthralgia syndrome. Breast. 2007;16:223-234.
85. Mao JJ, Stricker C, Bruner D, et al. Patterns and risk factors associated with aromatase inhibitor-related arthralgia among breast cancer survivors. Cancer. 2009;115:3631-3639.
86. Presant CA, Bosserman L, Young T, et al. Aromatase inhibitor-associated arthralgia and/or bone pain: frequency and characterization in non-clinical trial patients. Clin Breast Cancer. 2007;7:775-778.
87. Saper RB, Sherman KJ, Cullum-Dugan D, et al. Yoga for chronic low back pain in a predominantly minority population: a pilot randomized controlled trial. Altern Ther Health Med. 2009;15:18-27.
88. Kolasinski SL, Garfinkel M, Tsai AG, et al. Iyengar yoga for treating symptoms of osteoarthritis of the knees: a pilot study. J Altern Complement Med. 2005;11:689-693.
89. Galantino ML, Desai K, Greene L, et al. Impact of yoga on functional outcomes in breast cancer survivors with aromatase inhibitor-associated arthralgias. Integr Cancer Ther. 2012;11:313-320.
90. Ancoli-Israel S. Recognition and treatment of sleep disturbances in cancer. J Clin Oncol. 2009;27:5864-5866.
91. Savard J, Ivers H, Villa J, et al. Natural course of insomnia comorbid with cancer: an 18-month longitudinal study. J Clin Oncol. 2011;29:3580-3586.
92. Mustian KM, Sprod LK, Janelsins M, et al. Multicenter, randomized controlled trial of yoga for sleep quality among cancer survivors. J Clin Oncol. 2013;31:3233-3241.
93. Moore TA, Berger AM, Dizona P. Sleep aid use during and following breast cancer adjuvant chemotherapy. Psychooncology. 2011;20:321-325.
94. Omvik S, Pallesen S, Bjorvatn B, et al. Patient characteristics and predictors of sleep medication use. Int Clin Psychopharmacol. 2010;25:91-100.
95. Qaseem A, Kansagara D, Forciea MA, et al. Management of chronic insomnia disorder in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;165:125-133.
96. Cohen L, Warneke C, Fouladi RT, et al. Psychological adjustment and sleep quality in a randomized trial of the effects of a Tibetan yoga intervention in patients with lymphoma. Cancer. 2004;100:2253-2260.
97. Kraaijenga SA, Oskam IM, van der Molen L, et al. Evaluation of long term (10-years+) dysphagia and trismus in patients treated with concurrent chemo-radiotherapy for advanced head and neck cancer. Oral Oncol. 2015;51:787-794.
98. Adair M, Murphy B, Yarlagadda S, et al. Feasibility and preliminary efficacy of tailored yoga in survivors of head and neck cancer: a pilot study. Integr Cancer Ther. 2018;17:774-784.
99. Loudon A, Barnett T, Williams A. Yoga, breast cancer-related lymphoedema and well-being: A descriptive report of women’s participation in a clinical trial. J Clin Nurs. 2017;26:4685-4695.
100. Loudon A, Barnett T, Piller N, et al. The effects of yoga on shoulder and spinal actions for women with breast cancer-related lymphoedema of the arm: A randomised controlled pilot study. BMC Complement Altern Med. 2016;16:343.
101. Petruzzi A, Finocchiaro CY, Lamperti E, Salmaggi A. Living with a brain tumor: reaction profiles in patients and their caregivers. Support Care Cancer. 2013;21:1105-1111.
102. Pawl JD, Lee SY, Clark PC, Sherwood PR. Sleep characteristics of family caregivers of individuals with a primary malignant brain tumor. Oncol Nurs Forum. 2013;40:171-179.
103. Milbury K, Mallaiah S, Mahajan A, et al. Yoga program for high-grade glioma patients undergoing radiotherapy and their family caregivers. Integr Cancer Ther. 2018;17:332-336.
104. Milbury K, Li J, Weathers S-P, et al. Pilot randomized controlled trial of a dyadic yoga program for glioma patients undergoing radiotherapy and their family caregivers. Neurooncol Pract. 2019;6:311-320.
105. Milbury K, Liao Z, Shannon V, et al. Dyadic yoga program for patients undergoing thoracic radiotherapy and their family caregivers: Results of a pilot randomized controlled trial. Psychooncology. 2019;28:615-621.
106. Milbury K, Chaoul A, Engle R, et al. Couple-based Tibetan yoga program for lung cancer patients and their caregivers. Psychooncology. 2015;24:117-120.
107. Danhauer SC, Addington EL, Cohen L, et al. Yoga for symptom management in oncology: A review of the evidence base and future directions for research. Cancer. 2019;125:1979-1989.
108. National Comprehensive Cancer Center. Flash Update: NCCN Guidelines® and NCCN Compendium® for Antiemesis. NCCN website. Accessed August 29, 2019.
109. Lyman GH, Greenlee H, Bohlke K, et al. Integrative therapies during and after breast cancer treatment: ASCO endorsement of the SIO clinical practice guideline. J Clin Oncol. 2018;36:2647-2655.
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Reports. 2008;(12):1-23.
2. Witt CM, Balneaves LG, Cardoso MJ, et al. A comprehensive definition for integrative oncology. NCI Monographs. 2017;2017(52):3-8.
3. Brauer JA, El Sehamy A, Metz JM, Mao JJ. Complementary and alternative medicine and supportive care at leading cancer centers: a systematic analysis of websites. J Altern Complement Med. 2010;16:183-186.
4. Lopez G, McQuade J, Cohen L, et al. Integrative oncology physician consultations at a comprehensive cancer center: analysis of demographic, clinical and patient reported outcomes. J Cancer. 2017;8:395-402.
5. Latte-Naor S, Mao JJ. Putting integrative oncology into practice: concepts and approaches. J Oncol Pract. 2019;15:7-14.
6. Richardson MA, Sanders T, Palmer JL, et al. Complementary/alternative medicine use in a comprehensive cancer center and the implications for oncology. J Clin Oncol. 2000;18:2505-2514.
7. Chaoul A, Milbury K, Sood AK, et al. Mind-body practices in cancer care. Curr Oncol Rep. 2014;16:417.
8. Saper RB, Eisenberg DM, Davis RB, et al. Prevalence and patterns of adult yoga use in the United States: results of a national survey. Altern Ther Health Med. 2004;10:44-49.
9. Smith KB, Pukall CF. An evidence-based review of yoga as a complementary intervention for patients with cancer. Psychooncology. 2009;18:465-475.
10. Buffart LM, van Uffelen JG, Riphagen II, et al. Physical and psychosocial benefits of yoga in cancer patients and survivors, a systematic review and meta-analysis of randomized controlled trials. BMC Cancer. 2012;12:559.
11. Cramer H, Lange S, Klose P, et al. Yoga for breast cancer patients and survivors: a systematic review and meta-analysis. BMC Cancer. 2012;12:412.
12. Felbel S, Meerpohl JJ, Monsef I, et al. Yoga in addition to standard care for patients with haematological malignancies. Cochrane Database Syst Rev. 2014;(6):CD010146.
13. Greenlee H, Balneaves LG, Carlson LE, et al. Clinical practice guidelines on the use of integrative therapies as supportive care in patients treated for breast cancer. NCI Monographs. 2014;2014:346-358.
14. Sharma M, Haider T, Knowlden AP. Yoga as an alternative and complementary treatment for cancer: a systematic review. J Altern Complement Med. 2013;19:870-875.
15. Kordon C, Bihoreau C. Integrated communication between the nervous, endocrine and immune systems. Horm Res. 1989;31:100-104.
16. Gonzalez-Diaz SN, Arias-Cruz A, Elizondo-Villarreal B, Monge-Ortega OP. Psychoneuroimmunoendocrinology: clinical implications. World Allergy Organ J. 2017;10:19.
17. Godoy LD, Rossignoli MT, Delfino-Pereira P, et al. A comprehensive overview on stress neurobiology: basic concepts and clinical implications. Front Behav Neurosci. 2018;12:127.
18. Goldstein DS. Adrenal responses to stress. Cell Mol Neurobiol. 2010;30:1433-1440.
19. Cole SW, Nagaraja AS, Lutgendorf SK, et al. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563-567.
20. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
21. Arsalidou M, Duerden EG, Taylor MJ. The centre of the brain: topographical model of motor, cognitive, affective, and somatosensory functions of the basal ganglia. Hum Brain Mapp. 2013;34:3031-3054.
22. Gard T, Taquet M, Dixit R, et al. Greater widespread functional connectivity of the caudate in older adults who practice kripalu yoga and vipassana meditation than in controls. Front Hum Neurosci. 2015;9:137.
23. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
24. Brambilla P, Perez J, Barale F, et al. GABAergic dysfunction in mood disorders. Mol Psychiatry. 2003;8:721-737.
25. Streeter CC, Gerbarg PL, Saper RB, et al. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses. 2012;78:571-579.
26. Lutgendorf SK, Sood AK. Biobehavioral factors and cancer progression: physiological pathways and mechanisms. Psychosom Med. 2011;73:724-730.
27. Armaiz-Pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain Behav Immun. 2013;30(suppl):S19-S25.
28. Tyagi A, Cohen M. Yoga and heart rate variability: A comprehensive review of the literature. Int J Yoga. 2016;9:97-113.
29. Sephton SE, Lush E, Dedert EA, et al. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain Behav Immun. 2013;30 (suppl):S163-S170.
30. Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst. 2000;92:994-1000.
31. Arafah BM, Nishiyama FJ, Tlaygeh H, Hejal R. Measurement of salivary cortisol concentration in the assessment of adrenal function in critically ill subjects: a surrogate marker of the circulating free cortisol. J Clin Endocrinol Metab. 2007;92:2965-2971.
32. Cohen L, de Moor C, Devine D, et al. Endocrine levels at the start of treatment are associated with subsequent psychological adjustment in cancer patients with metastatic disease. Psychosom Med. 2001;63:951-958.
33. Cohen L, Cole SW, Sood AK, et al. Depressive symptoms and cortisol rhythmicity predict survival in patients with renal cell carcinoma: role of inflammatory signaling. PloS One. 2012;7:e42324.
34. Vadiraja HS, Raghavendra RM, Nagarathna R, et al. Effects of a yoga program on cortisol rhythm and mood states in early breast cancer patients undergoing adjuvant radiotherapy: a randomized controlled trial. Integr Cancer Ther. 2009;8:37-46.
35. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345:164-173.
36. Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun. 2013;30(suppl):S48-S57.
37. Derry HM, Jaremka LM, Bennett JM, et al. Yoga and self-reported cognitive problems in breast cancer survivors: a randomized controlled trial. Psychooncology. 2015;24:958-966.
38. Kiecolt-Glaser JK, Christian L, Preston H, et al. Stress, inflammation, and yoga practice. Psychosom Med. 2010;72:113-121.
39. Carlson LE, Speca M, Faris P, Patel KD. One year pre-post intervention follow-up of psychological, immune, endocrine and blood pressure outcomes of mindfulness-based stress reduction (MBSR) in breast and prostate cancer outpatients. Brain Behav Immun. 2007;21:1038-1049.
40. Infante JR, Peran F, Rayo JI, et al. Levels of immune cells in transcendental meditation practitioners. Int J Yoga. 2014;7:147-151.
41. Rao RM, Telles S, Nagendra HR, et al. Effects of yoga on natural killer cell counts in early breast cancer patients undergoing conventional treatment. Comment to: recreational music-making modulates natural killer cell activity, cytokines, and mood states in corporate employees Masatada Wachi, Masahiro Koyama, Masanori Utsuyama, Barry B. Bittman, Masanobu Kitagawa, Katsuiku Hirokawa Med Sci Monit, 2007; 13(2): CR57-70. Med Sci Monit. 2008;14:LE3-4.
42. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on mood states, distress, quality of life and immune outcomes in early stage breast cancer patients undergoing surgery. Int J Yoga. 2008;1:11-20.
43. Mozdarani H, Mansouri Z, Haeri SA. Cytogenetic radiosensitivity of g0-lymphocytes of breast and esophageal cancer patients as determined by micronucleus assay. J Radiat Res. 2005;46:111-116.
44. Scott D, Barber JB, Levine EL, et al. Radiation-induced micronucleus induction in lymphocytes identifies a high frequency of radiosensitive cases among breast cancer patients: a test for predisposition? Br J Cancer. 1998;77:614-620.
45. Banerjee B, Sharma S, Hegde S, Hande MP. Analysis of telomere damage by fluorescence in situ hybridisation on micronuclei in lymphocytes of breast carcinoma patients after radiotherapy. Breast Cancer Res Treat. 2008;107:25-31.
46. Epel ES, Blackburn EH, Lin J, et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312-17315.
47. Glaser R, Thorn BE, Tarr KL, et al. Effects of stress on methyltransferase synthesis: an important DNA repair enzyme. Health Psychol. 1985;4:403-412.
48. Kiecolt-Glaser JK, Stephens RE, Lipetz PD, et al. Distress and DNA repair in human lymphocytes. J Behav Med. 1985;8:311-320.
49. Banerjee B, Vadiraj HS, Ram A, et al. Effects of an integrated yoga program in modulating psychological stress and radiation-induced genotoxic stress in breast cancer patients undergoing radiotherapy. Integr Cancer Ther. 2007;6:242-250.
50. Danhauer SC, Addington EL, Sohl SJ, et al. Review of yoga therapy during cancer treatment. Support Care Cancer. 2017;25:1357-1372.
51. Chandwani KD, Thornton B, Perkins GH, et al. Yoga improves quality of life and benefit finding in women undergoing radiotherapy for breast cancer. J Soc Integr Oncol. 2010;8:43-55.
52. Ratcliff CG, Milbury K, Chandwani KD, et al. Examining mediators and moderators of yoga for women with breast cancer undergoing radiotherapy. Integr Cancer Ther. 2016;15:250-262.
53. Chandwani KD, Perkins G, Nagendra HR, et al. Randomized, controlled trial of yoga in women with breast cancer undergoing radiotherapy. J Clin Oncol. 2014;32:1058-1065.
54. Janelsins MC, Kesler SR, Ahles TA, Morrow GR. Prevalence, mechanisms, and management of cancer-related cognitive impairment. Int Rev Psychiatry. 2014;26:102-113.
55. Janelsins MC, Peppone LJ, Heckler CE, et al. YOCAS(c)(R) yoga reduces self-reported memory difficulty in cancer survivors in a nationwide randomized clinical trial: investigating relationships between memory and sleep. Integr Cancer Ther. 2016;15:263-271.
56. Biegler KA, Chaoul MA, Cohen L. Cancer, cognitive impairment, and meditation. Acta Oncol. 2009;48:18-26.
57. Carlson LE, Angen M, Cullum J, et al. High levels of untreated distress and fatigue in cancer patients. Br J Cancer. 2004;90:2297-2304.
58. Herschbach P, Keller M, Knight L, et al. Psychological problems of cancer patients: a cancer distress screening with a cancer-specific questionnaire. Br J Cancer. 2004;91:504-511.
59. Abercrombie HC, Giese-Davis J, Sephton S, et al. Flattened cortisol rhythms in metastatic breast cancer patients. Psychoneuroendocrinology. 2004;29:1082-1092.
60. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med. 2005;67:277-280.
61. Bower JE, Ganz PA, Dickerson SS, et al. Diurnal cortisol rhythm and fatigue in breast cancer survivors. Psychoneuroendocrinology. 2005;30:92-100.
62. Giese-Davis J, Sephton SE, Abercrombie HC, et al. Repression and high anxiety are associated with aberrant diurnal cortisol rhythms in women with metastatic breast cancer. Health Psychol. 2004;23:645-650.
63. Giese-Davis J, DiMiceli S, Sephton S, Spiegel D. Emotional expression and diurnal cortisol slope in women with metastatic breast cancer in supportive-expressive group therapy: a preliminary study. Biol Psychol. 2006;73:190-198.
64. Stone AA, Schwartz JE, Smyth J, et al. Individual differences in the diurnal cycle of salivary free cortisol: a replication of flattened cycles for some individuals. Psychoneuroendocrinology. 2001;26:295-306.
65. Bower JE, Woolery A, Sternlieb B, Garet D. Yoga for cancer patients and survivors. Cancer Control. 2005;12:165-171.
66. Granath J, Ingvarsson S, von Thiele U, Lundberg U. Stress management: a randomized study of cognitive behavioural therapy and yoga. Cogn Behav Therap. 2006;35:3-10.
67. Rao MR, Raghuram N, Nagendra HR, et al. Anxiolytic effects of a yoga program in early breast cancer patients undergoing conventional treatment: a randomized controlled trial. Complement Ther Med. 2009;17:1-8.
68. Culos-Reed SN, Carlson LE, Daroux LM, Hately-Aldous S. A pilot study of yoga for breast cancer survivors: physical and psychological benefits. Psychooncology. 2006;15:891-897.
69. Mackenzie MJ, Carlson LE, Ekkekakis P, et al. Affect and mindfulness as predictors of change in mood disturbance, stress symptoms, and quality of life in a community-based yoga program for cancer survivors. Evid Based Complement Alternat Med. 2013;2013:419496.
70. Danhauer SC, Mihalko SL, Russell GB, et al. Restorative yoga for women with breast cancer: findings from a randomized pilot study. Psycho-oncology. 2009;18:360-368.
71. Sohl SJ, Danhauer SC, Schnur JB, et al. Feasibility of a brief yoga intervention during chemotherapy for persistent or recurrent ovarian cancer. Explore (NY). 2012;8:197-198.
72. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer. 2016;24:4005-4015.
73. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol. 2015;6:8-14.
74. Wang G, Wang S, Jiang P, Zeng C. Effect of yoga on cancer related fatigue in breast cancer patients with chemotherapy [in Chinese]. Zhong Nan Da Xue Bao Yi Xue Ban. 2014;39:1077-1082.
75. Bower JE, Garet D, Sternlieb B, et al. Yoga for persistent fatigue in breast cancer survivors: a randomized controlled trial. Cancer. 2012;118:3766-3775.
76. Taso CJ, Lin HS, Lin WL, et al. The effect of yoga exercise on improving depression, anxiety, and fatigue in women with breast cancer: a randomized controlled trial. J Nurs Res. 2014;22:155-164.
77. Berger AM, Mooney K, Alvarez-Perez A, et al. Cancer-related fatigue, Version 2.2015. J Natl Compr Canc Netw. 2015;13:1012-1039.
78. Rao RM, Nagendra HR, Raghuram N, et al. Influence of yoga on postoperative outcomes and wound healing in early operable breast cancer patients undergoing surgery. Int J Yoga. 2008;1:33-41.
79. Sohl SJ, Avis NE, Stanbery K, et al. Feasibility of a brief yoga intervention for improving acute pain and distress post gynecologic surgery. Int J Yoga Therap. 2016;26:43-47.
80. Gupta P, Sturdee DW, Palin SL, et al. Menopausal symptoms in women treated for breast cancer: the prevalence and severity of symptoms and their perceived effects on quality of life. Climacteric. 2006;9:49-58.
81. Canney PA, Hatton MQ. The prevalence of menopausal symptoms in patients treated for breast cancer. Clin Oncol (R Coll Radiol). 1994;6:297-299.
82. Carpenter JS, Johnson D, Wagner L, Andrykowski M. Hot flashes and related outcomes in breast cancer survivors and matched comparison women. Oncol Nurs Forum. 2002;29:E16-25.
83. Carson JW, Carson KM, Porter LS, et al. Yoga of Awareness program for menopausal symptoms in breast cancer survivors: results from a randomized trial. Support Care Cancer. 2009;17:1301-1309.
84. Burstein HJ. Aromatase inhibitor-associated arthralgia syndrome. Breast. 2007;16:223-234.
85. Mao JJ, Stricker C, Bruner D, et al. Patterns and risk factors associated with aromatase inhibitor-related arthralgia among breast cancer survivors. Cancer. 2009;115:3631-3639.
86. Presant CA, Bosserman L, Young T, et al. Aromatase inhibitor-associated arthralgia and/or bone pain: frequency and characterization in non-clinical trial patients. Clin Breast Cancer. 2007;7:775-778.
87. Saper RB, Sherman KJ, Cullum-Dugan D, et al. Yoga for chronic low back pain in a predominantly minority population: a pilot randomized controlled trial. Altern Ther Health Med. 2009;15:18-27.
88. Kolasinski SL, Garfinkel M, Tsai AG, et al. Iyengar yoga for treating symptoms of osteoarthritis of the knees: a pilot study. J Altern Complement Med. 2005;11:689-693.
89. Galantino ML, Desai K, Greene L, et al. Impact of yoga on functional outcomes in breast cancer survivors with aromatase inhibitor-associated arthralgias. Integr Cancer Ther. 2012;11:313-320.
90. Ancoli-Israel S. Recognition and treatment of sleep disturbances in cancer. J Clin Oncol. 2009;27:5864-5866.
91. Savard J, Ivers H, Villa J, et al. Natural course of insomnia comorbid with cancer: an 18-month longitudinal study. J Clin Oncol. 2011;29:3580-3586.
92. Mustian KM, Sprod LK, Janelsins M, et al. Multicenter, randomized controlled trial of yoga for sleep quality among cancer survivors. J Clin Oncol. 2013;31:3233-3241.
93. Moore TA, Berger AM, Dizona P. Sleep aid use during and following breast cancer adjuvant chemotherapy. Psychooncology. 2011;20:321-325.
94. Omvik S, Pallesen S, Bjorvatn B, et al. Patient characteristics and predictors of sleep medication use. Int Clin Psychopharmacol. 2010;25:91-100.
95. Qaseem A, Kansagara D, Forciea MA, et al. Management of chronic insomnia disorder in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;165:125-133.
96. Cohen L, Warneke C, Fouladi RT, et al. Psychological adjustment and sleep quality in a randomized trial of the effects of a Tibetan yoga intervention in patients with lymphoma. Cancer. 2004;100:2253-2260.
97. Kraaijenga SA, Oskam IM, van der Molen L, et al. Evaluation of long term (10-years+) dysphagia and trismus in patients treated with concurrent chemo-radiotherapy for advanced head and neck cancer. Oral Oncol. 2015;51:787-794.
98. Adair M, Murphy B, Yarlagadda S, et al. Feasibility and preliminary efficacy of tailored yoga in survivors of head and neck cancer: a pilot study. Integr Cancer Ther. 2018;17:774-784.
99. Loudon A, Barnett T, Williams A. Yoga, breast cancer-related lymphoedema and well-being: A descriptive report of women’s participation in a clinical trial. J Clin Nurs. 2017;26:4685-4695.
100. Loudon A, Barnett T, Piller N, et al. The effects of yoga on shoulder and spinal actions for women with breast cancer-related lymphoedema of the arm: A randomised controlled pilot study. BMC Complement Altern Med. 2016;16:343.
101. Petruzzi A, Finocchiaro CY, Lamperti E, Salmaggi A. Living with a brain tumor: reaction profiles in patients and their caregivers. Support Care Cancer. 2013;21:1105-1111.
102. Pawl JD, Lee SY, Clark PC, Sherwood PR. Sleep characteristics of family caregivers of individuals with a primary malignant brain tumor. Oncol Nurs Forum. 2013;40:171-179.
103. Milbury K, Mallaiah S, Mahajan A, et al. Yoga program for high-grade glioma patients undergoing radiotherapy and their family caregivers. Integr Cancer Ther. 2018;17:332-336.
104. Milbury K, Li J, Weathers S-P, et al. Pilot randomized controlled trial of a dyadic yoga program for glioma patients undergoing radiotherapy and their family caregivers. Neurooncol Pract. 2019;6:311-320.
105. Milbury K, Liao Z, Shannon V, et al. Dyadic yoga program for patients undergoing thoracic radiotherapy and their family caregivers: Results of a pilot randomized controlled trial. Psychooncology. 2019;28:615-621.
106. Milbury K, Chaoul A, Engle R, et al. Couple-based Tibetan yoga program for lung cancer patients and their caregivers. Psychooncology. 2015;24:117-120.
107. Danhauer SC, Addington EL, Cohen L, et al. Yoga for symptom management in oncology: A review of the evidence base and future directions for research. Cancer. 2019;125:1979-1989.
108. National Comprehensive Cancer Center. Flash Update: NCCN Guidelines® and NCCN Compendium® for Antiemesis. NCCN website. Accessed August 29, 2019.
109. Lyman GH, Greenlee H, Bohlke K, et al. Integrative therapies during and after breast cancer treatment: ASCO endorsement of the SIO clinical practice guideline. J Clin Oncol. 2018;36:2647-2655.
The electronic medical record’s role in ObGyn burnout and patient care

Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.

Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.

2019 Update on contraception
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
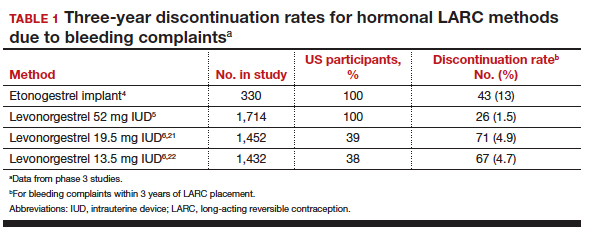
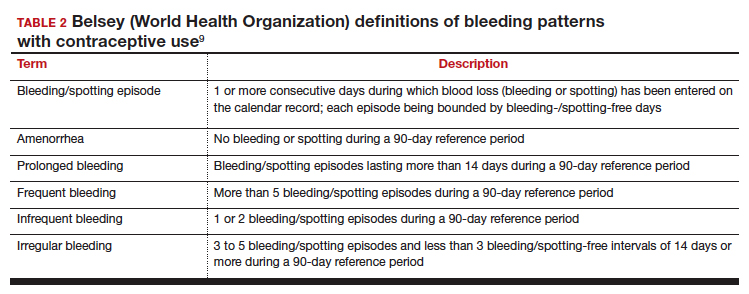
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
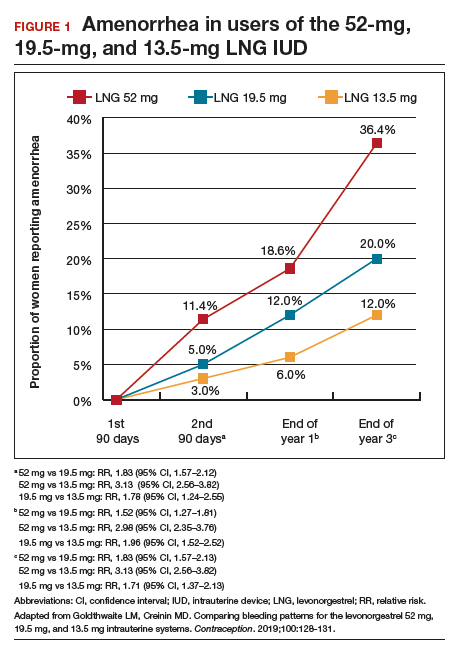
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
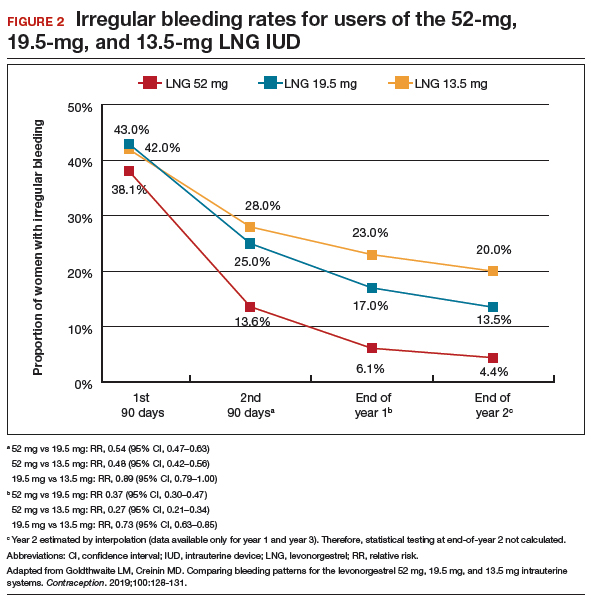
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.