User login
Preventing early-onset group B streptococcal disease in newborns
In 1992, the American College of Obstetricians and Gynecologists (ACOG) and the American Academy of Pediatrics (AAP) published their first joint guidelines on the prevention of early-onset neonatal group B streptococcal (GBS) infection.1 In this initial statement, the organizations recommended universal culturing of obstetric patients at 28 weeks’ gestation and treatment of colonized women during labor if they had a recognized risk factor for neonatal GBS infection.
In 1996, the Centers for Disease Control and Prevention (CDC) published its first set of official guidelines on the topic and suggested that both universal screening and a risk-factor–based approach were reasonable options.2 The 2002 update of the CDC guidelines strongly recommended universal screening of all pregnant women at 35 to 37 weeks’ gestation and intrapartum prophylaxis for all colonized women regardless of risk factors.3
The third set of CDC guidelines was published in 2010.4 The key features of this version were the elimination of erythromycin as an alternative to penicillin in patients who are allergic to beta-lactam antibiotics and the establishment of 4 hours as the critical interval for administration of prophylaxis prior to delivery. The 2010 publication was the last such report from the CDC. Since then ACOG and AAP have been tasked with providing updated practice guidelines. To that end, ACOG recently issued a new Committee Opinion on “Prevention of Group B Streptococcal Early-Onset Disease in Newborns.”5 Here we will highlight the key features of our current strategy for preventing neonatal GBS infection.
CASE Pregnant patient presents with many questions about GBS
A 26-year-old primigravid woman presents for her first prenatal appointment at 9 weeks’ gestation. Her older sister recently delivered a term infant that died in the first week of life from GBS sepsis. Understandably, she has many questions.
1. Your patient first wants to know, “What is this streptococcal organism and how likely am I to have this infection?”
Streptococcus agalactiae, also known as GBS, is a gram-positive encapsulated bacterium that produces beta hemolysis when grown on blood agar. Approximately 25% of pregnant women harbor this organism in the lower genital tract and/or rectum.6
GBS is one of the most important causes of neonatal infection, particularly in preterm infants. The frequency of infection is now 0.23 per 1,000 live births in the US.5
Neonatal infection can be divided into early-onset infection (occurring within the first 7 days of life) and late-onset infection (occurring from after the first week until the third month of life). Approximately 80% to 85% of cases of neonatal GBS infections are early in onset. Virtually all of the early-onset infections result from vertical transmission during delivery from a colonized mother to her infant.5-7
2. “How dangerous is this infection to my baby and me? Are there certain factors that increase the risk of my baby becoming infected?”
GBS is responsible for approximately 2% to 3% of cases of either asymptomatic bacteriuria or acute cystitis. Women with urinary tract infections caused by GBS are at increased risk for preterm premature rupture of membranes and preterm delivery. Genital tract colonization also increases a woman’s risk for chorioamnionitis and endometritis, particularly after cesarean delivery (CD). In addition, GBS can be part of the polymicrobial flora in women who have a wound (incisional site) infection following CD.6,7
Continue to: In colonized women, several risk factors...
In colonized women, several risk factors have been identified that increase the probability of early-onset neonatal GBS infection. These factors include: preterm labor, especially when complicated by premature rupture of membranes; intrapartum maternal fever (usually due to chorioamnionitis); rupture of membranes greater than 18 hours before delivery; previous delivery of an infected infant; young age; and black or Hispanic ethnicity. Approximately 25% of colonized women will have one of these risk factors.5-7
These risk factors have a profound impact on neonatal attack rates and mortality. Without the interventions outlined below, the neonatal infection rate is 40% to 50% in the presence of a risk factor and less than 5% in the absence of a risk factor. In infected infants, neonatal mortality approaches 30% to 35% when a maternal risk factor is present, but is less than 5% when risk factors are absent.5-7
3. “What will you do to determine if I am colonized with this organism?”
The current guidelines set forth in the ACOG Committee Opinion recommend that selected high-risk patients (patients with preterm labor or preterm premature rupture of membranes) be tested for GBS at the time of initial presentation. All other women should be tested for GBS during the interval 36 0/7 to 37 6/7 weeks’ gestation.5 Testing at this point in pregnancy is almost 90% sensitive for identifying patients who will be colonized at the time of admission for labor if no more than 5 weeks elapse between the time the culture is obtained and labor begins. The positive predictive value of this test is 87%, and the negative predictive value is 96%.8
ACOG’s previous guidelines provided for testing at 35 rather than 36 weeks. The change in the recommendations was based on 2 factors. First, all women with unknown GBS status who may deliver before 37 weeks already should be targeted for prophylaxis. Second, the new 5-week window now will include women who deliver up to 41 weeks’ gestation. Given current obstetric practice in the US, delivery beyond 41 weeks is unlikely.5
At the present time, the best test for identification of GBS colonization is bacteriologic culture. A cotton swab is placed into the lower third of the vagina, streaked along the perineum, and then placed into the rectum. The swab is withdrawn, placed in a culturette tube, and transported to the laboratory. In the laboratory, the swab is cultured for approximately 24 hours in a nutrient broth and then subcultured on a selective blood agar plate. Failure to sample both the vagina and rectum or failure to use selective broth and selective blood agar will reduce the yield of positive cultures by approximately 50%.5-7
In recent years, researchers have become interested in the use of rapid nucleic acid amplification tests for the identification of GBS. These tests perform well if the test protocol provides for an 18- to 24-hour incubation in nutrient broth prior to application of the nucleic acid probe. When the tests are performed without this enrichment phase, sensitivities are inferior to those associated with bacteriologic culture. In addition, because the rapid tests do not isolate the organisms, they do not allow for antibiotic sensitivity testing.5-7
Continue to: “If I test positive for GBS, how and when will you treat me?”...
4. “If I test positive for GBS, how and when will you treat me?”
The current ACOG guidelines recommend that all colonized women be treated intrapartum with prophylactic antibiotics regardless of whether risk factors are present. Treatment should be started at the time of admission and continued until the infant is delivered.5
The drugs of choice for intrapartum prophylaxis are intravenous penicillin or ampicillin. If the patient has a mild allergy to penicillin, cefazolin is the appropriate alternative. If the patient has a severe allergy to penicillin, the 2 options are vancomycin or clindamycin. If the latter drug is used, the laboratory must perform sensitivity testing because 13% to 20% of strains of GBS may be resistant to clindamycin. The frequency of resistance to erythromycin now ranges from 25% to 32%. Thus, erythromycin is no longer used for intrapartum prophylaxis.5-7,9
The appropriate intravenous dosages of these antibiotics are listed in the TABLE.5 The new ACOG guidelines have revised the previous recommendations for dosing of penicillin, eliminating the 2.5 million-unit dose. They also have revised the dosing recommendations for vancomyin, eliminating the previous recommendation of 1 g every 12 hours.5 The new recommendations regarding vancomycin are particularly important and are based, at least in part, on an interesting report from Onwuchuruba and colleagues.10 These authors studied maternal and cord blood concentrations of vancomycin in mother-infant dyads receiving either the original recommended dosage of vancomycin (1 g every 12 hours) or a dosage of 15 to 20 mg/kg every 8 hours. With standard dosing, only 9% of neonates had therapeutic vancomycin serum concentrations at delivery. With the 20 mg/kg dose of vancomycin, the percent of neonates with therapeutic serum concentrations of vancomycin increased to 80%.

5. “For how long must I be treated in labor before my baby will be protected by the antibiotics?”
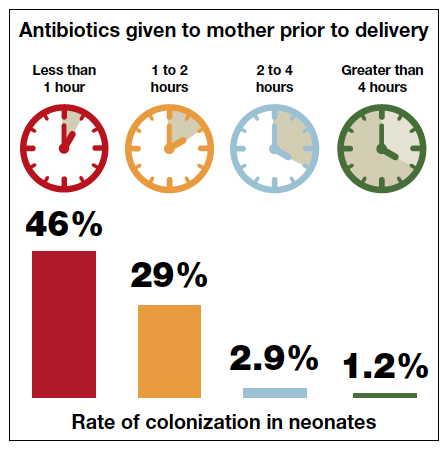
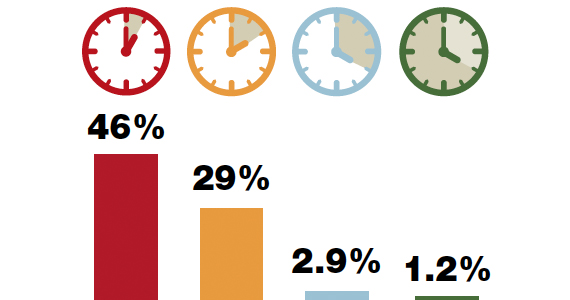
The current ACOG Committee Opinion stresses the importance of treating the colonized mother for at least 4 hours prior to delivery.5 This recommendation is based primarily on the landmark report by De Cueto and colleagues.11 These authors evaluated colonized women who received intrapartum prophylaxis at varying times prior to delivery. Their primary endpoint was the percentage of newborns who were colonized with GBS. If the mothers had received antibiotics for less than 1 hour prior to delivery, 46% of neonates were colonized. This figure was equal to the rate of colonization in neonates whose mothers received no antibiotics. When the interval was 1 to 2 hours, the percentage was 29%. When mothers had received antibiotics for 2 to 4 hours, the neonatal colonization rate fell to 2.9%. When antibiotics had been administered for greater than 4 hours, the rate of neonatal colonization was only 1.2%.
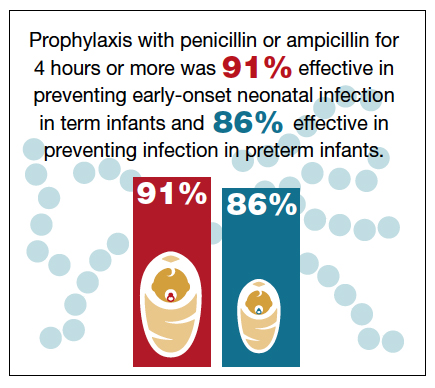
Fairlie and colleagues recently reported the results of another interesting investigation comparing the effectiveness of prophylaxis based on duration of treatment and choice of individual antibiotics.12 Prophylaxis with penicillin or ampicillin for 4 hours or more was 91% effective in preventing early-onset neonatal infection in term infants and 86% effective in preventing infection in preterm infants. These outcomes were superior to the outcomes in both term and preterm infants who received penicillin or ampicillin for less than 4 hours.
These observations agree with the findings of McNanley and colleagues who evaluated vaginal colony counts of GBS following different periods of antibiotic administration.13 These authors noted that mean colony counts decreased 5-fold within 2 hours of penicillin administration, 50-fold within 4 hours, and 1,000-fold within 6 hours.
Despite these compelling findings, the ACOG Committee Opinion stresses that obstetric interventions such as amniotomy and oxytocin augmentation should not be delayed simply to permit a certain time period of antibiotic administration.5
Continue to: “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”...
6. “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”
If a mother is scheduled to have a CD, for example because of a prior cesarean or because of a persistent fetal malpresentation, she should still have a GBS culture at 36 0/7 to 37 6/7 weeks’ gestation. The information obtained from this culture may be of value to both the obstetrician and pediatrician if the patient experiences labor or rupture of membranes prior to her scheduled surgery. If she does not experience spontaneous labor prior to her scheduled date of surgery, she does not require specific GBS prophylaxis at the time of her operation.5 Rather, she should receive prophylactic antibiotics to prevent post–cesarean infection, ideally, the combination of cefazolin (2 g IV) plus azithromycin (500 mg IV).14 Cefazolin, of course, provides excellent coverage of GBS.
7. “If I am colonized with GBS and I receive treatment during labor, will my baby be safe after delivery?”
The interventions outlined above will prevent almost 90% of early-onset GBS infections, but they are not foolproof.5-7,15,16 Successful management of the neonate is dependent upon several factors, including:5-7
- gestational age
- presence of maternal chorioamnionitis
- presence or absence of risk factors for early-onset infection
- duration (adequacy) of maternal treatment during labor
- presence of immediate clinical signs of infection in the neonate (such as fever, lethargy, hemodynamic instability, respiratory distress, or elevated or decreased white blood cell count).
If the mother is at term and receives intrapartum prophylaxis for at least 4 hours prior to delivery, the neonate usually will not require any special tests and simply will be observed for 24 to 48 hours for signs of infection.
If the mother delivers preterm and receives appropriate intrapartum prophylaxis, the pediatricians typically will obtain a complete blood count (CBC) and treat with prophylactic antibiotics (ampicillin plus gentamicin) for 48 hours if abnormalities are noted on the CBC or the baby exhibits signs of infection. If the CBC is normal and the baby shows no signs of infection, no treatment is indicated.
Regardless of gestational age, if the mother does not receive prophylaxis for at least 4 hours before delivery, the pediatricians usually will obtain a CBC and closely observe the baby in the hospital for signs of infection. If such signs develop or the CBC is abnormal, blood and cerebrospinal fluid cultures will be obtained. Antibiotic therapy (usually ampicillin plus gentamicin) is then initiated, and the drugs are continued until cultures return with no growth. If either culture is positive, antibiotics will then be continued for 7 to 10 days.
If the mother has documented chorioamnionitis and receives treatment intrapartum with appropriate antibiotics (usually ampicillin plus gentamicin), the pediatricians usually will obtain a CBC, C-reactive protein (CRP) level, and blood cultures and then start the infant on antibiotics, pending the result of the laboratory tests. If the CBC and CRP are reassuring, the cultures are negative after 48 hours, and the infant demonstrates no signs of clinical infection, many pediatricians will then discontinue antibiotics. Others may still continue the antibiotics for 7 to 10 days.
- Committee on Infectious Diseases and Committee on Fetus and Newborn. Guidelines for prevention of group B streptococcal (GBS) infection by chemoprophylaxis. Pediatrics. 1992;90:775-778.
- CDC. Prevention of perinatal group B streptococcal disease: a public health perspective. MMWR Recomm Rep. 1996;45(RR-7):1-24.
- Schrag S, Gorwitz R, Fultz-Butts K, et al. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. MMWR Recomm Rep. 2002;51(RR-11):1-22.
- Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC). MMWR Recomm Rep. 2010;59:1-36.
- Prevention of group B streptococcal early-onset disease in newborns. ACOG Committee Opinion Summary, Number 782. Obstet Gynecol. 2019;134:206-210.
- Duff P, Birsner M. Maternal and perinatal infection in pregnancy: bacteria. In: Gabbe SG, Niebyl JR, Simpson JL, et al, eds. Obstetrics. Normal and Problem Pregnancies. 7th ed. Philadelphia, PA: Elsevier; 2017.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik's Maternal-Fetal Medicine: Principles and Practice. 8th ed. Philadelphia, PA: Elsevier; 2019.
- Yancey MK, Schuchat A, Brown LK, et al. The accuracy of late antenatal screening cultures in predicting genital group B streptococcal colonization at delivery. Obstet Gynecol. 1996;88:811-815.
- Edwards RK, Clark P, Duff P. Intrapartum antibiotic prophylaxis 2: positive predictive value of antenatal group B streptococci cultures and antibiotic susceptibility of clinical isolates. Obstet Gynecol. 2002;100:540-544.
- Onwuchuruba CN, Towers CV, Howard BC, et al. Transplacental passage of vancomycin from mother to neonate. Am J Obstet Gynecol. 2014;210:352.e1-352.e4.
- de Cueto M, Sanchez MJ, Sampedro A, et al. Timing of intrapartum ampicillin and prevention of vertical transmission of group B streptococcus. Obstet Gynecol. 1998;91:112-114.
- Fairlie T, Zell ER, Schrag S. Effectiveness of intrapartum antibiotic prophylaxis for prevention of early-onset group B streptococcal disease. Obstet Gynecol. 2013;121:570-577.
- McNanley AR, Glantz JC, Hardy DJ, et al. The effect of intrapartum penicillin on vaginal group B streptococcus colony counts. Am J Obstet Gynecol. 2007;197:583.e1-583.e4.
- Tita AT, Szychowski JM, Boggess K, et al. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375:1231-1241.
- Brozanski BS, Jones JG, Krohn MA, et al. Effect of a screening-based prevention policy on prevalence of early-onset group B streptococcal sepsis. Obstet Gynecol. 2000;95:496-501.
- Rosenstein NE, Schuchat A. Opportunities for prevention of perinatal group B streptococcal disease: a multistate surveillance analysis. The National Group B Streptococcal Disease Study Group. Obstet Gynecol. 1997;90:901-906.

Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
In 1992, the American College of Obstetricians and Gynecologists (ACOG) and the American Academy of Pediatrics (AAP) published their first joint guidelines on the prevention of early-onset neonatal group B streptococcal (GBS) infection.1 In this initial statement, the organizations recommended universal culturing of obstetric patients at 28 weeks’ gestation and treatment of colonized women during labor if they had a recognized risk factor for neonatal GBS infection.
In 1996, the Centers for Disease Control and Prevention (CDC) published its first set of official guidelines on the topic and suggested that both universal screening and a risk-factor–based approach were reasonable options.2 The 2002 update of the CDC guidelines strongly recommended universal screening of all pregnant women at 35 to 37 weeks’ gestation and intrapartum prophylaxis for all colonized women regardless of risk factors.3
The third set of CDC guidelines was published in 2010.4 The key features of this version were the elimination of erythromycin as an alternative to penicillin in patients who are allergic to beta-lactam antibiotics and the establishment of 4 hours as the critical interval for administration of prophylaxis prior to delivery. The 2010 publication was the last such report from the CDC. Since then ACOG and AAP have been tasked with providing updated practice guidelines. To that end, ACOG recently issued a new Committee Opinion on “Prevention of Group B Streptococcal Early-Onset Disease in Newborns.”5 Here we will highlight the key features of our current strategy for preventing neonatal GBS infection.
CASE Pregnant patient presents with many questions about GBS
A 26-year-old primigravid woman presents for her first prenatal appointment at 9 weeks’ gestation. Her older sister recently delivered a term infant that died in the first week of life from GBS sepsis. Understandably, she has many questions.
1. Your patient first wants to know, “What is this streptococcal organism and how likely am I to have this infection?”
Streptococcus agalactiae, also known as GBS, is a gram-positive encapsulated bacterium that produces beta hemolysis when grown on blood agar. Approximately 25% of pregnant women harbor this organism in the lower genital tract and/or rectum.6
GBS is one of the most important causes of neonatal infection, particularly in preterm infants. The frequency of infection is now 0.23 per 1,000 live births in the US.5
Neonatal infection can be divided into early-onset infection (occurring within the first 7 days of life) and late-onset infection (occurring from after the first week until the third month of life). Approximately 80% to 85% of cases of neonatal GBS infections are early in onset. Virtually all of the early-onset infections result from vertical transmission during delivery from a colonized mother to her infant.5-7
2. “How dangerous is this infection to my baby and me? Are there certain factors that increase the risk of my baby becoming infected?”
GBS is responsible for approximately 2% to 3% of cases of either asymptomatic bacteriuria or acute cystitis. Women with urinary tract infections caused by GBS are at increased risk for preterm premature rupture of membranes and preterm delivery. Genital tract colonization also increases a woman’s risk for chorioamnionitis and endometritis, particularly after cesarean delivery (CD). In addition, GBS can be part of the polymicrobial flora in women who have a wound (incisional site) infection following CD.6,7
Continue to: In colonized women, several risk factors...
In colonized women, several risk factors have been identified that increase the probability of early-onset neonatal GBS infection. These factors include: preterm labor, especially when complicated by premature rupture of membranes; intrapartum maternal fever (usually due to chorioamnionitis); rupture of membranes greater than 18 hours before delivery; previous delivery of an infected infant; young age; and black or Hispanic ethnicity. Approximately 25% of colonized women will have one of these risk factors.5-7
These risk factors have a profound impact on neonatal attack rates and mortality. Without the interventions outlined below, the neonatal infection rate is 40% to 50% in the presence of a risk factor and less than 5% in the absence of a risk factor. In infected infants, neonatal mortality approaches 30% to 35% when a maternal risk factor is present, but is less than 5% when risk factors are absent.5-7
3. “What will you do to determine if I am colonized with this organism?”
The current guidelines set forth in the ACOG Committee Opinion recommend that selected high-risk patients (patients with preterm labor or preterm premature rupture of membranes) be tested for GBS at the time of initial presentation. All other women should be tested for GBS during the interval 36 0/7 to 37 6/7 weeks’ gestation.5 Testing at this point in pregnancy is almost 90% sensitive for identifying patients who will be colonized at the time of admission for labor if no more than 5 weeks elapse between the time the culture is obtained and labor begins. The positive predictive value of this test is 87%, and the negative predictive value is 96%.8
ACOG’s previous guidelines provided for testing at 35 rather than 36 weeks. The change in the recommendations was based on 2 factors. First, all women with unknown GBS status who may deliver before 37 weeks already should be targeted for prophylaxis. Second, the new 5-week window now will include women who deliver up to 41 weeks’ gestation. Given current obstetric practice in the US, delivery beyond 41 weeks is unlikely.5
At the present time, the best test for identification of GBS colonization is bacteriologic culture. A cotton swab is placed into the lower third of the vagina, streaked along the perineum, and then placed into the rectum. The swab is withdrawn, placed in a culturette tube, and transported to the laboratory. In the laboratory, the swab is cultured for approximately 24 hours in a nutrient broth and then subcultured on a selective blood agar plate. Failure to sample both the vagina and rectum or failure to use selective broth and selective blood agar will reduce the yield of positive cultures by approximately 50%.5-7
In recent years, researchers have become interested in the use of rapid nucleic acid amplification tests for the identification of GBS. These tests perform well if the test protocol provides for an 18- to 24-hour incubation in nutrient broth prior to application of the nucleic acid probe. When the tests are performed without this enrichment phase, sensitivities are inferior to those associated with bacteriologic culture. In addition, because the rapid tests do not isolate the organisms, they do not allow for antibiotic sensitivity testing.5-7
Continue to: “If I test positive for GBS, how and when will you treat me?”...
4. “If I test positive for GBS, how and when will you treat me?”
The current ACOG guidelines recommend that all colonized women be treated intrapartum with prophylactic antibiotics regardless of whether risk factors are present. Treatment should be started at the time of admission and continued until the infant is delivered.5
The drugs of choice for intrapartum prophylaxis are intravenous penicillin or ampicillin. If the patient has a mild allergy to penicillin, cefazolin is the appropriate alternative. If the patient has a severe allergy to penicillin, the 2 options are vancomycin or clindamycin. If the latter drug is used, the laboratory must perform sensitivity testing because 13% to 20% of strains of GBS may be resistant to clindamycin. The frequency of resistance to erythromycin now ranges from 25% to 32%. Thus, erythromycin is no longer used for intrapartum prophylaxis.5-7,9
The appropriate intravenous dosages of these antibiotics are listed in the TABLE.5 The new ACOG guidelines have revised the previous recommendations for dosing of penicillin, eliminating the 2.5 million-unit dose. They also have revised the dosing recommendations for vancomyin, eliminating the previous recommendation of 1 g every 12 hours.5 The new recommendations regarding vancomycin are particularly important and are based, at least in part, on an interesting report from Onwuchuruba and colleagues.10 These authors studied maternal and cord blood concentrations of vancomycin in mother-infant dyads receiving either the original recommended dosage of vancomycin (1 g every 12 hours) or a dosage of 15 to 20 mg/kg every 8 hours. With standard dosing, only 9% of neonates had therapeutic vancomycin serum concentrations at delivery. With the 20 mg/kg dose of vancomycin, the percent of neonates with therapeutic serum concentrations of vancomycin increased to 80%.
5. “For how long must I be treated in labor before my baby will be protected by the antibiotics?”
The current ACOG Committee Opinion stresses the importance of treating the colonized mother for at least 4 hours prior to delivery.5 This recommendation is based primarily on the landmark report by De Cueto and colleagues.11 These authors evaluated colonized women who received intrapartum prophylaxis at varying times prior to delivery. Their primary endpoint was the percentage of newborns who were colonized with GBS. If the mothers had received antibiotics for less than 1 hour prior to delivery, 46% of neonates were colonized. This figure was equal to the rate of colonization in neonates whose mothers received no antibiotics. When the interval was 1 to 2 hours, the percentage was 29%. When mothers had received antibiotics for 2 to 4 hours, the neonatal colonization rate fell to 2.9%. When antibiotics had been administered for greater than 4 hours, the rate of neonatal colonization was only 1.2%.
Fairlie and colleagues recently reported the results of another interesting investigation comparing the effectiveness of prophylaxis based on duration of treatment and choice of individual antibiotics.12 Prophylaxis with penicillin or ampicillin for 4 hours or more was 91% effective in preventing early-onset neonatal infection in term infants and 86% effective in preventing infection in preterm infants. These outcomes were superior to the outcomes in both term and preterm infants who received penicillin or ampicillin for less than 4 hours.
These observations agree with the findings of McNanley and colleagues who evaluated vaginal colony counts of GBS following different periods of antibiotic administration.13 These authors noted that mean colony counts decreased 5-fold within 2 hours of penicillin administration, 50-fold within 4 hours, and 1,000-fold within 6 hours.
Despite these compelling findings, the ACOG Committee Opinion stresses that obstetric interventions such as amniotomy and oxytocin augmentation should not be delayed simply to permit a certain time period of antibiotic administration.5
Continue to: “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”...
6. “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”
If a mother is scheduled to have a CD, for example because of a prior cesarean or because of a persistent fetal malpresentation, she should still have a GBS culture at 36 0/7 to 37 6/7 weeks’ gestation. The information obtained from this culture may be of value to both the obstetrician and pediatrician if the patient experiences labor or rupture of membranes prior to her scheduled surgery. If she does not experience spontaneous labor prior to her scheduled date of surgery, she does not require specific GBS prophylaxis at the time of her operation.5 Rather, she should receive prophylactic antibiotics to prevent post–cesarean infection, ideally, the combination of cefazolin (2 g IV) plus azithromycin (500 mg IV).14 Cefazolin, of course, provides excellent coverage of GBS.
7. “If I am colonized with GBS and I receive treatment during labor, will my baby be safe after delivery?”
The interventions outlined above will prevent almost 90% of early-onset GBS infections, but they are not foolproof.5-7,15,16 Successful management of the neonate is dependent upon several factors, including:5-7
- gestational age
- presence of maternal chorioamnionitis
- presence or absence of risk factors for early-onset infection
- duration (adequacy) of maternal treatment during labor
- presence of immediate clinical signs of infection in the neonate (such as fever, lethargy, hemodynamic instability, respiratory distress, or elevated or decreased white blood cell count).
If the mother is at term and receives intrapartum prophylaxis for at least 4 hours prior to delivery, the neonate usually will not require any special tests and simply will be observed for 24 to 48 hours for signs of infection.
If the mother delivers preterm and receives appropriate intrapartum prophylaxis, the pediatricians typically will obtain a complete blood count (CBC) and treat with prophylactic antibiotics (ampicillin plus gentamicin) for 48 hours if abnormalities are noted on the CBC or the baby exhibits signs of infection. If the CBC is normal and the baby shows no signs of infection, no treatment is indicated.
Regardless of gestational age, if the mother does not receive prophylaxis for at least 4 hours before delivery, the pediatricians usually will obtain a CBC and closely observe the baby in the hospital for signs of infection. If such signs develop or the CBC is abnormal, blood and cerebrospinal fluid cultures will be obtained. Antibiotic therapy (usually ampicillin plus gentamicin) is then initiated, and the drugs are continued until cultures return with no growth. If either culture is positive, antibiotics will then be continued for 7 to 10 days.
If the mother has documented chorioamnionitis and receives treatment intrapartum with appropriate antibiotics (usually ampicillin plus gentamicin), the pediatricians usually will obtain a CBC, C-reactive protein (CRP) level, and blood cultures and then start the infant on antibiotics, pending the result of the laboratory tests. If the CBC and CRP are reassuring, the cultures are negative after 48 hours, and the infant demonstrates no signs of clinical infection, many pediatricians will then discontinue antibiotics. Others may still continue the antibiotics for 7 to 10 days.
In 1992, the American College of Obstetricians and Gynecologists (ACOG) and the American Academy of Pediatrics (AAP) published their first joint guidelines on the prevention of early-onset neonatal group B streptococcal (GBS) infection.1 In this initial statement, the organizations recommended universal culturing of obstetric patients at 28 weeks’ gestation and treatment of colonized women during labor if they had a recognized risk factor for neonatal GBS infection.
In 1996, the Centers for Disease Control and Prevention (CDC) published its first set of official guidelines on the topic and suggested that both universal screening and a risk-factor–based approach were reasonable options.2 The 2002 update of the CDC guidelines strongly recommended universal screening of all pregnant women at 35 to 37 weeks’ gestation and intrapartum prophylaxis for all colonized women regardless of risk factors.3
The third set of CDC guidelines was published in 2010.4 The key features of this version were the elimination of erythromycin as an alternative to penicillin in patients who are allergic to beta-lactam antibiotics and the establishment of 4 hours as the critical interval for administration of prophylaxis prior to delivery. The 2010 publication was the last such report from the CDC. Since then ACOG and AAP have been tasked with providing updated practice guidelines. To that end, ACOG recently issued a new Committee Opinion on “Prevention of Group B Streptococcal Early-Onset Disease in Newborns.”5 Here we will highlight the key features of our current strategy for preventing neonatal GBS infection.
CASE Pregnant patient presents with many questions about GBS
A 26-year-old primigravid woman presents for her first prenatal appointment at 9 weeks’ gestation. Her older sister recently delivered a term infant that died in the first week of life from GBS sepsis. Understandably, she has many questions.
1. Your patient first wants to know, “What is this streptococcal organism and how likely am I to have this infection?”
Streptococcus agalactiae, also known as GBS, is a gram-positive encapsulated bacterium that produces beta hemolysis when grown on blood agar. Approximately 25% of pregnant women harbor this organism in the lower genital tract and/or rectum.6
GBS is one of the most important causes of neonatal infection, particularly in preterm infants. The frequency of infection is now 0.23 per 1,000 live births in the US.5
Neonatal infection can be divided into early-onset infection (occurring within the first 7 days of life) and late-onset infection (occurring from after the first week until the third month of life). Approximately 80% to 85% of cases of neonatal GBS infections are early in onset. Virtually all of the early-onset infections result from vertical transmission during delivery from a colonized mother to her infant.5-7
2. “How dangerous is this infection to my baby and me? Are there certain factors that increase the risk of my baby becoming infected?”
GBS is responsible for approximately 2% to 3% of cases of either asymptomatic bacteriuria or acute cystitis. Women with urinary tract infections caused by GBS are at increased risk for preterm premature rupture of membranes and preterm delivery. Genital tract colonization also increases a woman’s risk for chorioamnionitis and endometritis, particularly after cesarean delivery (CD). In addition, GBS can be part of the polymicrobial flora in women who have a wound (incisional site) infection following CD.6,7
Continue to: In colonized women, several risk factors...
In colonized women, several risk factors have been identified that increase the probability of early-onset neonatal GBS infection. These factors include: preterm labor, especially when complicated by premature rupture of membranes; intrapartum maternal fever (usually due to chorioamnionitis); rupture of membranes greater than 18 hours before delivery; previous delivery of an infected infant; young age; and black or Hispanic ethnicity. Approximately 25% of colonized women will have one of these risk factors.5-7
These risk factors have a profound impact on neonatal attack rates and mortality. Without the interventions outlined below, the neonatal infection rate is 40% to 50% in the presence of a risk factor and less than 5% in the absence of a risk factor. In infected infants, neonatal mortality approaches 30% to 35% when a maternal risk factor is present, but is less than 5% when risk factors are absent.5-7
3. “What will you do to determine if I am colonized with this organism?”
The current guidelines set forth in the ACOG Committee Opinion recommend that selected high-risk patients (patients with preterm labor or preterm premature rupture of membranes) be tested for GBS at the time of initial presentation. All other women should be tested for GBS during the interval 36 0/7 to 37 6/7 weeks’ gestation.5 Testing at this point in pregnancy is almost 90% sensitive for identifying patients who will be colonized at the time of admission for labor if no more than 5 weeks elapse between the time the culture is obtained and labor begins. The positive predictive value of this test is 87%, and the negative predictive value is 96%.8
ACOG’s previous guidelines provided for testing at 35 rather than 36 weeks. The change in the recommendations was based on 2 factors. First, all women with unknown GBS status who may deliver before 37 weeks already should be targeted for prophylaxis. Second, the new 5-week window now will include women who deliver up to 41 weeks’ gestation. Given current obstetric practice in the US, delivery beyond 41 weeks is unlikely.5
At the present time, the best test for identification of GBS colonization is bacteriologic culture. A cotton swab is placed into the lower third of the vagina, streaked along the perineum, and then placed into the rectum. The swab is withdrawn, placed in a culturette tube, and transported to the laboratory. In the laboratory, the swab is cultured for approximately 24 hours in a nutrient broth and then subcultured on a selective blood agar plate. Failure to sample both the vagina and rectum or failure to use selective broth and selective blood agar will reduce the yield of positive cultures by approximately 50%.5-7
In recent years, researchers have become interested in the use of rapid nucleic acid amplification tests for the identification of GBS. These tests perform well if the test protocol provides for an 18- to 24-hour incubation in nutrient broth prior to application of the nucleic acid probe. When the tests are performed without this enrichment phase, sensitivities are inferior to those associated with bacteriologic culture. In addition, because the rapid tests do not isolate the organisms, they do not allow for antibiotic sensitivity testing.5-7
Continue to: “If I test positive for GBS, how and when will you treat me?”...
4. “If I test positive for GBS, how and when will you treat me?”
The current ACOG guidelines recommend that all colonized women be treated intrapartum with prophylactic antibiotics regardless of whether risk factors are present. Treatment should be started at the time of admission and continued until the infant is delivered.5
The drugs of choice for intrapartum prophylaxis are intravenous penicillin or ampicillin. If the patient has a mild allergy to penicillin, cefazolin is the appropriate alternative. If the patient has a severe allergy to penicillin, the 2 options are vancomycin or clindamycin. If the latter drug is used, the laboratory must perform sensitivity testing because 13% to 20% of strains of GBS may be resistant to clindamycin. The frequency of resistance to erythromycin now ranges from 25% to 32%. Thus, erythromycin is no longer used for intrapartum prophylaxis.5-7,9
The appropriate intravenous dosages of these antibiotics are listed in the TABLE.5 The new ACOG guidelines have revised the previous recommendations for dosing of penicillin, eliminating the 2.5 million-unit dose. They also have revised the dosing recommendations for vancomyin, eliminating the previous recommendation of 1 g every 12 hours.5 The new recommendations regarding vancomycin are particularly important and are based, at least in part, on an interesting report from Onwuchuruba and colleagues.10 These authors studied maternal and cord blood concentrations of vancomycin in mother-infant dyads receiving either the original recommended dosage of vancomycin (1 g every 12 hours) or a dosage of 15 to 20 mg/kg every 8 hours. With standard dosing, only 9% of neonates had therapeutic vancomycin serum concentrations at delivery. With the 20 mg/kg dose of vancomycin, the percent of neonates with therapeutic serum concentrations of vancomycin increased to 80%.
5. “For how long must I be treated in labor before my baby will be protected by the antibiotics?”
The current ACOG Committee Opinion stresses the importance of treating the colonized mother for at least 4 hours prior to delivery.5 This recommendation is based primarily on the landmark report by De Cueto and colleagues.11 These authors evaluated colonized women who received intrapartum prophylaxis at varying times prior to delivery. Their primary endpoint was the percentage of newborns who were colonized with GBS. If the mothers had received antibiotics for less than 1 hour prior to delivery, 46% of neonates were colonized. This figure was equal to the rate of colonization in neonates whose mothers received no antibiotics. When the interval was 1 to 2 hours, the percentage was 29%. When mothers had received antibiotics for 2 to 4 hours, the neonatal colonization rate fell to 2.9%. When antibiotics had been administered for greater than 4 hours, the rate of neonatal colonization was only 1.2%.
Fairlie and colleagues recently reported the results of another interesting investigation comparing the effectiveness of prophylaxis based on duration of treatment and choice of individual antibiotics.12 Prophylaxis with penicillin or ampicillin for 4 hours or more was 91% effective in preventing early-onset neonatal infection in term infants and 86% effective in preventing infection in preterm infants. These outcomes were superior to the outcomes in both term and preterm infants who received penicillin or ampicillin for less than 4 hours.
These observations agree with the findings of McNanley and colleagues who evaluated vaginal colony counts of GBS following different periods of antibiotic administration.13 These authors noted that mean colony counts decreased 5-fold within 2 hours of penicillin administration, 50-fold within 4 hours, and 1,000-fold within 6 hours.
Despite these compelling findings, the ACOG Committee Opinion stresses that obstetric interventions such as amniotomy and oxytocin augmentation should not be delayed simply to permit a certain time period of antibiotic administration.5
Continue to: “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”...
6. “If I were to have a scheduled CD before the onset of labor and/or ruptured membranes, would I still need to receive antibiotics?”
If a mother is scheduled to have a CD, for example because of a prior cesarean or because of a persistent fetal malpresentation, she should still have a GBS culture at 36 0/7 to 37 6/7 weeks’ gestation. The information obtained from this culture may be of value to both the obstetrician and pediatrician if the patient experiences labor or rupture of membranes prior to her scheduled surgery. If she does not experience spontaneous labor prior to her scheduled date of surgery, she does not require specific GBS prophylaxis at the time of her operation.5 Rather, she should receive prophylactic antibiotics to prevent post–cesarean infection, ideally, the combination of cefazolin (2 g IV) plus azithromycin (500 mg IV).14 Cefazolin, of course, provides excellent coverage of GBS.
7. “If I am colonized with GBS and I receive treatment during labor, will my baby be safe after delivery?”
The interventions outlined above will prevent almost 90% of early-onset GBS infections, but they are not foolproof.5-7,15,16 Successful management of the neonate is dependent upon several factors, including:5-7
- gestational age
- presence of maternal chorioamnionitis
- presence or absence of risk factors for early-onset infection
- duration (adequacy) of maternal treatment during labor
- presence of immediate clinical signs of infection in the neonate (such as fever, lethargy, hemodynamic instability, respiratory distress, or elevated or decreased white blood cell count).
If the mother is at term and receives intrapartum prophylaxis for at least 4 hours prior to delivery, the neonate usually will not require any special tests and simply will be observed for 24 to 48 hours for signs of infection.
If the mother delivers preterm and receives appropriate intrapartum prophylaxis, the pediatricians typically will obtain a complete blood count (CBC) and treat with prophylactic antibiotics (ampicillin plus gentamicin) for 48 hours if abnormalities are noted on the CBC or the baby exhibits signs of infection. If the CBC is normal and the baby shows no signs of infection, no treatment is indicated.
Regardless of gestational age, if the mother does not receive prophylaxis for at least 4 hours before delivery, the pediatricians usually will obtain a CBC and closely observe the baby in the hospital for signs of infection. If such signs develop or the CBC is abnormal, blood and cerebrospinal fluid cultures will be obtained. Antibiotic therapy (usually ampicillin plus gentamicin) is then initiated, and the drugs are continued until cultures return with no growth. If either culture is positive, antibiotics will then be continued for 7 to 10 days.
If the mother has documented chorioamnionitis and receives treatment intrapartum with appropriate antibiotics (usually ampicillin plus gentamicin), the pediatricians usually will obtain a CBC, C-reactive protein (CRP) level, and blood cultures and then start the infant on antibiotics, pending the result of the laboratory tests. If the CBC and CRP are reassuring, the cultures are negative after 48 hours, and the infant demonstrates no signs of clinical infection, many pediatricians will then discontinue antibiotics. Others may still continue the antibiotics for 7 to 10 days.
- Committee on Infectious Diseases and Committee on Fetus and Newborn. Guidelines for prevention of group B streptococcal (GBS) infection by chemoprophylaxis. Pediatrics. 1992;90:775-778.
- CDC. Prevention of perinatal group B streptococcal disease: a public health perspective. MMWR Recomm Rep. 1996;45(RR-7):1-24.
- Schrag S, Gorwitz R, Fultz-Butts K, et al. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. MMWR Recomm Rep. 2002;51(RR-11):1-22.
- Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC). MMWR Recomm Rep. 2010;59:1-36.
- Prevention of group B streptococcal early-onset disease in newborns. ACOG Committee Opinion Summary, Number 782. Obstet Gynecol. 2019;134:206-210.
- Duff P, Birsner M. Maternal and perinatal infection in pregnancy: bacteria. In: Gabbe SG, Niebyl JR, Simpson JL, et al, eds. Obstetrics. Normal and Problem Pregnancies. 7th ed. Philadelphia, PA: Elsevier; 2017.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik's Maternal-Fetal Medicine: Principles and Practice. 8th ed. Philadelphia, PA: Elsevier; 2019.
- Yancey MK, Schuchat A, Brown LK, et al. The accuracy of late antenatal screening cultures in predicting genital group B streptococcal colonization at delivery. Obstet Gynecol. 1996;88:811-815.
- Edwards RK, Clark P, Duff P. Intrapartum antibiotic prophylaxis 2: positive predictive value of antenatal group B streptococci cultures and antibiotic susceptibility of clinical isolates. Obstet Gynecol. 2002;100:540-544.
- Onwuchuruba CN, Towers CV, Howard BC, et al. Transplacental passage of vancomycin from mother to neonate. Am J Obstet Gynecol. 2014;210:352.e1-352.e4.
- de Cueto M, Sanchez MJ, Sampedro A, et al. Timing of intrapartum ampicillin and prevention of vertical transmission of group B streptococcus. Obstet Gynecol. 1998;91:112-114.
- Fairlie T, Zell ER, Schrag S. Effectiveness of intrapartum antibiotic prophylaxis for prevention of early-onset group B streptococcal disease. Obstet Gynecol. 2013;121:570-577.
- McNanley AR, Glantz JC, Hardy DJ, et al. The effect of intrapartum penicillin on vaginal group B streptococcus colony counts. Am J Obstet Gynecol. 2007;197:583.e1-583.e4.
- Tita AT, Szychowski JM, Boggess K, et al. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375:1231-1241.
- Brozanski BS, Jones JG, Krohn MA, et al. Effect of a screening-based prevention policy on prevalence of early-onset group B streptococcal sepsis. Obstet Gynecol. 2000;95:496-501.
- Rosenstein NE, Schuchat A. Opportunities for prevention of perinatal group B streptococcal disease: a multistate surveillance analysis. The National Group B Streptococcal Disease Study Group. Obstet Gynecol. 1997;90:901-906.
- Committee on Infectious Diseases and Committee on Fetus and Newborn. Guidelines for prevention of group B streptococcal (GBS) infection by chemoprophylaxis. Pediatrics. 1992;90:775-778.
- CDC. Prevention of perinatal group B streptococcal disease: a public health perspective. MMWR Recomm Rep. 1996;45(RR-7):1-24.
- Schrag S, Gorwitz R, Fultz-Butts K, et al. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. MMWR Recomm Rep. 2002;51(RR-11):1-22.
- Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC). MMWR Recomm Rep. 2010;59:1-36.
- Prevention of group B streptococcal early-onset disease in newborns. ACOG Committee Opinion Summary, Number 782. Obstet Gynecol. 2019;134:206-210.
- Duff P, Birsner M. Maternal and perinatal infection in pregnancy: bacteria. In: Gabbe SG, Niebyl JR, Simpson JL, et al, eds. Obstetrics. Normal and Problem Pregnancies. 7th ed. Philadelphia, PA: Elsevier; 2017.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik's Maternal-Fetal Medicine: Principles and Practice. 8th ed. Philadelphia, PA: Elsevier; 2019.
- Yancey MK, Schuchat A, Brown LK, et al. The accuracy of late antenatal screening cultures in predicting genital group B streptococcal colonization at delivery. Obstet Gynecol. 1996;88:811-815.
- Edwards RK, Clark P, Duff P. Intrapartum antibiotic prophylaxis 2: positive predictive value of antenatal group B streptococci cultures and antibiotic susceptibility of clinical isolates. Obstet Gynecol. 2002;100:540-544.
- Onwuchuruba CN, Towers CV, Howard BC, et al. Transplacental passage of vancomycin from mother to neonate. Am J Obstet Gynecol. 2014;210:352.e1-352.e4.
- de Cueto M, Sanchez MJ, Sampedro A, et al. Timing of intrapartum ampicillin and prevention of vertical transmission of group B streptococcus. Obstet Gynecol. 1998;91:112-114.
- Fairlie T, Zell ER, Schrag S. Effectiveness of intrapartum antibiotic prophylaxis for prevention of early-onset group B streptococcal disease. Obstet Gynecol. 2013;121:570-577.
- McNanley AR, Glantz JC, Hardy DJ, et al. The effect of intrapartum penicillin on vaginal group B streptococcus colony counts. Am J Obstet Gynecol. 2007;197:583.e1-583.e4.
- Tita AT, Szychowski JM, Boggess K, et al. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375:1231-1241.
- Brozanski BS, Jones JG, Krohn MA, et al. Effect of a screening-based prevention policy on prevalence of early-onset group B streptococcal sepsis. Obstet Gynecol. 2000;95:496-501.
- Rosenstein NE, Schuchat A. Opportunities for prevention of perinatal group B streptococcal disease: a multistate surveillance analysis. The National Group B Streptococcal Disease Study Group. Obstet Gynecol. 1997;90:901-906.
Black-box warnings: How they can improve your clinical practice
Recently, the FDA issued “black-box” warnings, its most prominent drug safety statements, for esketamine,1 which is indicated for treatment-resistant depression, and the Z-drugs, which are indicated for insomnia2 (Table 1). A black-box warning also comes with brexanolone, which was recently approved for postpartum depression.3 While these newly issued warnings serve as a timely reminder of the importance of black-box warnings, older black-box warnings also cover large areas of psychiatric prescribing, including all medications indicated for treating psychosis or schizophrenia (increased mortality in patients with dementia), and all psychotropic medications with a depression indication (suicidality in younger people).

In this article, we help busy prescribers navigate the landscape of black-box warnings by providing a concise review of how to use them in clinical practice, and where to find information to keep up-to-date.
What are black-box warnings?
A black-box warning is a summary of the potential serious or life-threatening risks of a specific prescription medication. The black-box warning is formatted within a black border found at the top of the manufacturer’s prescribing information document (also known as the package insert or product label). Below the black-box warning, potential risks appear in descending order in sections titled “Contraindications,” “Warnings and Precautions,” and “Adverse Reactions.”4 The FDA issues black-box warnings either during drug development, to take effect upon approval of a new agent, or (more commonly) based on post-marketing safety information,5 which the FDA continuously gathers from reports by patients, clinicians, and industry.6 Federal law mandates the existence of black-box warnings, stating in part that, “special problems, particularly those that may lead to death or serious injury, may be required by the [FDA] to be placed in a prominently displayed box” (21 CFR 201.57(e)).
When is a black-box warning necessary?
The FDA issues a black-box warning based upon its judgment of the seriousness of the adverse effect. However, by definition, these risks do not inherently outweigh the benefits a medication may offer to certain patients. According to the FDA,7 black-box warnings are placed when:
- an adverse reaction so significant exists that this potential negative effect must be considered in risks and benefits when prescribing the medication
- a serious adverse reaction exists that can be prevented, or the risk reduced, by appropriate use of the medication
- the FDA has approved the medication with restrictions to ensure safe use.
Table 2 shows examples of scenarios where black-box warnings have been issued.8 Black-box warnings may be placed on an individual agent or on an entire class of medications. For example, both antipsychotics and antidepressants have class-wide warnings. Finally, black-box warnings are not static, and their content may change; in a study of black-box warnings issued from 2007 to 2015, 29% were entirely new, 32% were considered major updates to existing black-box warnings, and 40% were minor updates.5

Critiques of black-box warnings focus on the absence of published, formal criteria for instituting such warnings, the lack of a consistent approach in their content, and the infrequent inclusion of any information on the relative size of the risk.9 Suggestions for improvement include offering guidance on how to implement the black-box warnings in a patient-centered, shared decision-making model by adding evidence profiles and implementation guides.10 Less frequently considered, black-box warnings may be discontinued if new evidence demonstrates that the risk is lower than previously appreciated; however, similarly to their placement, no explicit criteria for the removal of black-box warnings have been made public.11
When a medication poses an especially high safety risk, the FDA may require the manufacturer to implement a Risk Evaluation and Mitigation Strategy (REMS) program. These programs can describe specific steps to improve medication safety, known as elements to assure safe use (ETASU).4 A familiar example is the clozapine REMS. In order to reduce the risk of severe neutropenia, the clozapine REMS requires prescribers (and pharmacists) to complete specialized training (making up the ETASU). Surprisingly, not every medication with a REMS has a corresponding black-box warning12; more understandably, many medications with black-box warnings do not have an associated REMS, because their risks are evaluated to be manageable by an individual prescriber’s clinical judgment. Most recently, esketamine carries both a black-box warning and a REMS. The black-box warning focuses on adverse effects (Table 1), while the REMS focuses on specific steps used to lessen these risks, including requiring use of a patient enrollment and monitoring form, a fact sheet for patients, and health care setting and pharmacy enrollment forms.13
Continue to: Psychotropic medications and black-box warnings
Psychotropic medications and black-box warnings
Psychotropic medications have a large number of black-box warnings.14 Because it is difficult to find black-box warnings for multiple medications in one place, we have provided 2 convenient resources to address this gap: a concise summary guide (Table 3) and a more detailed database (Table 4, Table 5, Table 6, Table 7, and Table 8). In these Tables, the possible risk mitigations, off-label uses, and monitoring are not meant to be formal recommendations or endorsements but are for independent clinician consideration only.

The information in these Tables was drawn from publicly available data, primarily the Micromedex and FDA web sites (see Related Resources). Because this information changes over time, at the end of this article we suggest ways for clinicians to stay updated with black-box warnings and build on the information provided in this article. These tools can be useful for day-to-day clinical practice in addition to studying for professional examinations. The following are selected high-profile black-box warnings.
Antidepressants and suicide risk. As a class, antidepressants carry a black-box warning on suicide risk in patients age ≤24. Initially issued in 2005, this warning was extended in 2007 to indicate that depression itself is associated with an increased risk of suicide. This black-box warning is used for an entire class of medications as well as for a specific patient population (age ≤24). Moreover, it indicates that suicide rates in patients age >65 were lower among patients using antidepressants.
Among psychotropic medication black-box warnings, this warning has perhaps been the most controversial. For example, it has been suggested that this black-box warning may have inadvertently increased suicide rates by discouraging clinicians from prescribing antidepressants,15 although this also has been called into question.16 This black-box warning illustrates that the consequences of issuing black-box warnings can be very difficult to assess, which makes their clinical effects highly complex and challenging to evaluate.14
Antipsychotics and dementia-related psychosis. This warning was initially issued in 2005 for second-generation antipsychotics and extended to first-generation antipsychotics in 2008. Antipsychotics as a class carry a black-box warning for increased risk of death in patients with dementia (major neurocognitive disorder). This warning extends to the recently approved antipsychotic pimavanserin, even though this agent’s proposed mechanism of action differs from that of other antipsychotics.17 However, it specifically allows for use in Parkinson’s disease psychosis, which is pimavanserin’s indication.18 In light of recent research suggesting pimavanserin is effective in dementia-related psychosis,19 it bears watching whether this agent becomes the first antipsychotic to have this warning removed.
Continue to: This class warning has...
This class warning has had widespread effects. For example, it has prompted less use of antipsychotics in nursing home facilities, as a result of stricter Centers for Medicare and Medicaid Services regulations20; overall, there is some evidence that there has been reduced prescribing of antipsychotics in general.21 Additionally, this black-box warning is unusual in that it warns about a specific off-label indication, which is itself poorly supported by evidence.21 Concomitantly, few other treatment options are available for this clinical situation. These medications are often seen as the only option for patients with dementia complicated by severe behavioral disturbance, and thus this black-box warning reflects real-world practices.14
Varenicline and neuropsychiatric complications. The withdrawal of the black-box warning on potential neuropsychiatric complications of using varenicline for smoking cessation shows that black-box warnings are not static and can, though infrequently, be removed as more safety data accumulates.11 As additional post-marketing information emerged on this risk, this black-box warning was reconsidered and withdrawn in 2016.22 Its withdrawal could potentially make clinicians more comfortable prescribing varenicline and in turn, help to reduce smoking rates.
How to use black-box warnings
To enhance their clinical practice, prescribers can use black-box warnings to inform safe prescribing practices, to guide shared decision-making, and to improve documentation of their treatment decisions.
Informing safe prescribing practices. A prescriber should be aware of the main safety concerns contained in a medication’s black-box warning; at the same time, these warnings are not meant to unduly limit use when crucial treatment is needed.14 In issuing a black-box warning, the FDA has clearly stated the priority and seriousness of its concern. These safety issues must be balanced against the medication’s utility for a given patient, at the prescriber’s clinical judgment.
Guiding shared decision-making. Clinicians are not required to disclose black-box warnings to patients, and there are no criteria that clearly define the role of these warnings in patient care. As is often noted, the FDA does not regulate the practice of medicine.6 However, given the seriousness of the potential adverse effects delineated by black-box warnings, it is reasonable for clinicians to have a solid grasp of black-box warnings for all medications they prescribe, and to be able to relate these warnings to patients, in appropriate language. This patient-centered discussion should include weighing the risks and benefits with the patient and educating the patient about the risks and strategies to mitigate those risks. This discussion can be augmented by patient handouts, which are often offered by pharmaceutical manufacturers, and by shared decision-making tools. A proactive discussion with patients and families about black-box warnings and other risks discussed in product labels can help reduce fears associated with taking medications and may improve adherence.
Continue to: Improving documentation of treatment decisions
Improving documentation of treatment decisions. Fluent knowledge of black-box warnings may help clinicians improve documentation of their treatment decisions, particularly the risks and benefits of their medication choices. Fluency with black-box warnings will help clinicians accurately document both their awareness of these risks, and how these risks informed their risk-benefit analysis in specific clinical situations.
Despite the clear importance the FDA places on black-box warnings, they are not often a topic of study in training or in postgraduate continuing education, and as a result, not all clinicians may be equally conversant with black-box warnings. While black-box warnings do change over time, many psychotropic medication black-box warnings are long-standing and well-established, and they evolve slowly enough to make mastering these warnings worthwhile in order to make the most informed clinical decisions for patient care.
Keeping up-to-date
There are practical and useful ways for busy clinicians to stay up-to-date with black-box warnings. Although these resources exist in multiple locations, together they provide convenient ways to keep current.
The FDA provides access to black-box warnings via its comprehensive database, DRUGS@FDA (https://www.accessdata.fda.gov/scripts/cder/daf/). Detailed information about REMS (and corresponding ETASU and other information related to REMS programs) is available at REMS@FDA (https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm). Clinicians can make safety reports that may contribute to FDA decision-making on black-box warnings by contacting MedWatch (https://www.fda.gov/safety/medwatch-fda-safety-information-and-adverse-event-reporting-program), the FDA’s adverse events reporting system. MedWatch releases safety information reports, which can be followed on Twitter @FDAMedWatch. Note that FDA information generally is organized by specific drug, and not into categories, such as psychotropic medications.
BlackBoxRx (www.blackboxrx.com) is a subscription-based web service that some clinicians may have access to via facility or academic resources as part of a larger FormWeb software package. Individuals also can subscribe (currently, $89/year).
Continue to: Micromedex
Micromedex (www.micromedex.com), which is widely available through medical libraries, is a subscription-based web service that provides black-box warning information from a separate tab that is easily accessed in each drug’s information front page. There is also an alphabetical list of black-box warnings under a separate tab on the Micromedex landing page.
ePocrates (www.epocrates.com) is a subscription-based service that provides extensive drug information, including black-box warnings, in a convenient mobile app.
Bottom Line
Black-box warnings are the most prominent drug safety warnings issued by the FDA. Many psychotropic medications carry black-box warnings that are crucial to everyday psychiatric prescribing. A better understanding of blackbox warnings can enhance your clinical practice by informing safe prescribing practices, guiding shared decision-making, and improving documentation of your treatment decisions.
Related Resources
- US Food and Drug Administration. DRUGS@FDA: FDAapproved drug products. www.accessdata.fda.gov/scripts/cder/daf/.
- US Food and Drug Administration. Drug safety and availability. www.fda.gov/drugs/drug-safety-and-availability. Updated October 10, 2019.
- BlackBoxRx. www.blackboxrx.com. (Subscription required.)
- Mircromedex. www.micromedex.com. (Subscription required.)
- ePocrates. www.epocrates.com. (Subscription required.)
Drug Brand Names
Amitriptyline • Elavil, Vanatrip
Amoxatine • Strattera
Amoxapine • Asendin
Aripiprazole • Abilify
Asenapine • Saphris
Brexanolone • Zulresso
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dexmethylphenidate • Focalin
Dextroamphetamine/amphetamine • Adderall
Disulfiram • Antabuse
Doxepin • Prudoxin, Silenor
Droperidol • Inapsine
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Eszopiclone • Lunesta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imipramine • Tofranil
Isocarboxazid • Marplan
Lamotrigine • Lamictal
Levomilnacipran • Fetzima
Levothyroxine • Synthroid
Linezolid • Zyvox
Lisdexamfetamine • Vyvanse
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Lurasidone • Latuda
Maprotiline • Ludiomil
Methadone • Dolophine, Methadose
Methylphenidate • Ritalin, Concerta
Midazolam • Versed
Milnacipran • Savella
Mirtazapine • Remeron
Naltrexone • Revia, Vivitrol
Nefazodone • Serzone
Nortriptyline • Aventyl, Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenelzine • Nardil
Pimavanserin • Nuplazid
Prochlorperazine • Compro
Protriptyline • Vivactil
Quetiapine • Seroquel
Risperidone • Risperdal
Selegiline • Emsam
Sertraline • Zoloft
Thioridazine • Mellaril
Thiothixene • Navane
Tranylcypromine • Parnate
Trazodone • Desyrel, Oleptro
Trifluoperazine • Stelazine
Trimipramine • Surmontil
Valproate • Depakote
Varenicline • Chantix, Wellbutrin
Vilazodone • Viibryd
Venlafaxine • Effexor
Vortioxetine • Trintellix
Zaleplon • Sonata
Ziprasidone • Geodon
Zolpidem • Ambien
1. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceutical Companies; 2019.
2. U.S. Food and Drug Administration. FDA drug safety announcement: FDA adds boxed warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia. Published April 30, 2019. Accessed October 28, 2019.
3. Zulresso [package insert]. Cambridge, Mass.: Sage Therapeutics Inc.; 2019.
4. Gassman AL, Nguyen CP, Joffe HV. FDA regulation of prescription drugs. N Engl J Med. 2017;376(7):674-682.
5. Solotke MT, Dhruva SS, Downing NS, et al. New and incremental FDA black box warnings from 2008 to 2015. Expert Opin Drug Saf. 2018;17(2):117-123.
6. Murphy S, Roberts R. “Black box” 101: how the Food and Drug Administration evaluates, communicates, and manages drug benefit/risk. J Allergy Clin Immunol. 2006;117(1):34-39.
7. U.S. Food and Drug Administration. Guidance document: Warnings and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products – content and format. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/warnings-and-precautions-contraindications-and-boxed-warning-sections-labeling-human-prescription. Published October 2011. Accessed October 28, 2019.
8. Beach JE, Faich GA, Bormel FG, et al. Black box warnings in prescription drug labeling: results of a survey of 206 drugs. Food Drug Law J. 1998;53(3):403-411.
9. Matlock A, Allan N, Wills B, et al. A continuing black hole? The FDA boxed warning: an appeal to improve its clinical utility. Clinical Toxicol (Phila). 2011;49(6):443-447.
10. Elraiyah T, Gionfriddo MR, Montori VM, et al. Content, consistency, and quality of black box warnings: time for a change. Ann Intern Med. 2015;163(11):875-876.
11. Yeh JS, Sarpatwari A, Kesselheim AS. Ethical and practical considerations in removing black box warnings from drug labels. Drug Saf. 2016;39(8):709-714.
12. Boudes PF. Risk Evaluation and Mitigation Strategies (REMSs): are they improving drug safety? A critical review of REMSs requiring Elements to Assure Safe Use (ETASU). Drugs R D. 2017;17(2):245-254.
13. U.S. Food and Drug Administration. Approved risk evaluation mitigation strategies (REMS): Spravato (esketamine) REMS program. https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=IndvRemsDetails.page&REMS=386. Updated June 25, 2019. Accessed October 28, 2018.
14. Stevens JR, Jarrahzadeh T, Brendel RW, et al. Strategies for the prescription of psychotropic drugs with black box warnings. Psychosomatics. 2014;55(2):123-133.
15. Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668.
16. Stone MB. The FDA warning on antidepressants and suicidality--why the controversy? N Engl J Med. 2014;371(18):1668-1671.
17. Mathis MV, Muoio BM, Andreason P, et al. The US Food and Drug Administration’s perspective on the new antipsychotic pimavanserin. J Clin Psychiatry. 2017;78(6):e668-e673. doi: 10.4088/JCP.16r11119.
18. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; May 2019.
19. Ballard C, Banister C, Khan Z, et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 2018;17(3):213-222.
20. Maust DT, Kim HM, Chiang C, et al. Association of the Centers for Medicare & Medicaid Services’ National Partnership to Improve Dementia Care with the use of antipsychotics and other psychotropics in long-term care in the United States from 2009 to 2014. JAMA Intern Med. 2018;178(5):640-647.
21. Dorsey ER, Rabbani A, Gallagher SA, et al. Impact of FDA black box advisory on antipsychotic medication use. Arch Intern Med. 2010;170(1):96-103.
22. U.S. Food and Drug Administration. FDA drug safety communication: FDA revises description of mental health side effects of the stop-smoking medicines Chantix (varenicline) and Zyban (bupropion) to reflect clinical trial findings. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-revises-description-mental-health-side-effects-stop-smoking. Published December 16, 2016. Accessed October 28, 2019.
Recently, the FDA issued “black-box” warnings, its most prominent drug safety statements, for esketamine,1 which is indicated for treatment-resistant depression, and the Z-drugs, which are indicated for insomnia2 (Table 1). A black-box warning also comes with brexanolone, which was recently approved for postpartum depression.3 While these newly issued warnings serve as a timely reminder of the importance of black-box warnings, older black-box warnings also cover large areas of psychiatric prescribing, including all medications indicated for treating psychosis or schizophrenia (increased mortality in patients with dementia), and all psychotropic medications with a depression indication (suicidality in younger people).
In this article, we help busy prescribers navigate the landscape of black-box warnings by providing a concise review of how to use them in clinical practice, and where to find information to keep up-to-date.
What are black-box warnings?
A black-box warning is a summary of the potential serious or life-threatening risks of a specific prescription medication. The black-box warning is formatted within a black border found at the top of the manufacturer’s prescribing information document (also known as the package insert or product label). Below the black-box warning, potential risks appear in descending order in sections titled “Contraindications,” “Warnings and Precautions,” and “Adverse Reactions.”4 The FDA issues black-box warnings either during drug development, to take effect upon approval of a new agent, or (more commonly) based on post-marketing safety information,5 which the FDA continuously gathers from reports by patients, clinicians, and industry.6 Federal law mandates the existence of black-box warnings, stating in part that, “special problems, particularly those that may lead to death or serious injury, may be required by the [FDA] to be placed in a prominently displayed box” (21 CFR 201.57(e)).
When is a black-box warning necessary?
The FDA issues a black-box warning based upon its judgment of the seriousness of the adverse effect. However, by definition, these risks do not inherently outweigh the benefits a medication may offer to certain patients. According to the FDA,7 black-box warnings are placed when:
- an adverse reaction so significant exists that this potential negative effect must be considered in risks and benefits when prescribing the medication
- a serious adverse reaction exists that can be prevented, or the risk reduced, by appropriate use of the medication
- the FDA has approved the medication with restrictions to ensure safe use.
Table 2 shows examples of scenarios where black-box warnings have been issued.8 Black-box warnings may be placed on an individual agent or on an entire class of medications. For example, both antipsychotics and antidepressants have class-wide warnings. Finally, black-box warnings are not static, and their content may change; in a study of black-box warnings issued from 2007 to 2015, 29% were entirely new, 32% were considered major updates to existing black-box warnings, and 40% were minor updates.5
Critiques of black-box warnings focus on the absence of published, formal criteria for instituting such warnings, the lack of a consistent approach in their content, and the infrequent inclusion of any information on the relative size of the risk.9 Suggestions for improvement include offering guidance on how to implement the black-box warnings in a patient-centered, shared decision-making model by adding evidence profiles and implementation guides.10 Less frequently considered, black-box warnings may be discontinued if new evidence demonstrates that the risk is lower than previously appreciated; however, similarly to their placement, no explicit criteria for the removal of black-box warnings have been made public.11
When a medication poses an especially high safety risk, the FDA may require the manufacturer to implement a Risk Evaluation and Mitigation Strategy (REMS) program. These programs can describe specific steps to improve medication safety, known as elements to assure safe use (ETASU).4 A familiar example is the clozapine REMS. In order to reduce the risk of severe neutropenia, the clozapine REMS requires prescribers (and pharmacists) to complete specialized training (making up the ETASU). Surprisingly, not every medication with a REMS has a corresponding black-box warning12; more understandably, many medications with black-box warnings do not have an associated REMS, because their risks are evaluated to be manageable by an individual prescriber’s clinical judgment. Most recently, esketamine carries both a black-box warning and a REMS. The black-box warning focuses on adverse effects (Table 1), while the REMS focuses on specific steps used to lessen these risks, including requiring use of a patient enrollment and monitoring form, a fact sheet for patients, and health care setting and pharmacy enrollment forms.13
Continue to: Psychotropic medications and black-box warnings
Psychotropic medications and black-box warnings
Psychotropic medications have a large number of black-box warnings.14 Because it is difficult to find black-box warnings for multiple medications in one place, we have provided 2 convenient resources to address this gap: a concise summary guide (Table 3) and a more detailed database (Table 4, Table 5, Table 6, Table 7, and Table 8). In these Tables, the possible risk mitigations, off-label uses, and monitoring are not meant to be formal recommendations or endorsements but are for independent clinician consideration only.
The information in these Tables was drawn from publicly available data, primarily the Micromedex and FDA web sites (see Related Resources). Because this information changes over time, at the end of this article we suggest ways for clinicians to stay updated with black-box warnings and build on the information provided in this article. These tools can be useful for day-to-day clinical practice in addition to studying for professional examinations. The following are selected high-profile black-box warnings.
Antidepressants and suicide risk. As a class, antidepressants carry a black-box warning on suicide risk in patients age ≤24. Initially issued in 2005, this warning was extended in 2007 to indicate that depression itself is associated with an increased risk of suicide. This black-box warning is used for an entire class of medications as well as for a specific patient population (age ≤24). Moreover, it indicates that suicide rates in patients age >65 were lower among patients using antidepressants.
Among psychotropic medication black-box warnings, this warning has perhaps been the most controversial. For example, it has been suggested that this black-box warning may have inadvertently increased suicide rates by discouraging clinicians from prescribing antidepressants,15 although this also has been called into question.16 This black-box warning illustrates that the consequences of issuing black-box warnings can be very difficult to assess, which makes their clinical effects highly complex and challenging to evaluate.14
Antipsychotics and dementia-related psychosis. This warning was initially issued in 2005 for second-generation antipsychotics and extended to first-generation antipsychotics in 2008. Antipsychotics as a class carry a black-box warning for increased risk of death in patients with dementia (major neurocognitive disorder). This warning extends to the recently approved antipsychotic pimavanserin, even though this agent’s proposed mechanism of action differs from that of other antipsychotics.17 However, it specifically allows for use in Parkinson’s disease psychosis, which is pimavanserin’s indication.18 In light of recent research suggesting pimavanserin is effective in dementia-related psychosis,19 it bears watching whether this agent becomes the first antipsychotic to have this warning removed.
Continue to: This class warning has...
This class warning has had widespread effects. For example, it has prompted less use of antipsychotics in nursing home facilities, as a result of stricter Centers for Medicare and Medicaid Services regulations20; overall, there is some evidence that there has been reduced prescribing of antipsychotics in general.21 Additionally, this black-box warning is unusual in that it warns about a specific off-label indication, which is itself poorly supported by evidence.21 Concomitantly, few other treatment options are available for this clinical situation. These medications are often seen as the only option for patients with dementia complicated by severe behavioral disturbance, and thus this black-box warning reflects real-world practices.14
Varenicline and neuropsychiatric complications. The withdrawal of the black-box warning on potential neuropsychiatric complications of using varenicline for smoking cessation shows that black-box warnings are not static and can, though infrequently, be removed as more safety data accumulates.11 As additional post-marketing information emerged on this risk, this black-box warning was reconsidered and withdrawn in 2016.22 Its withdrawal could potentially make clinicians more comfortable prescribing varenicline and in turn, help to reduce smoking rates.
How to use black-box warnings
To enhance their clinical practice, prescribers can use black-box warnings to inform safe prescribing practices, to guide shared decision-making, and to improve documentation of their treatment decisions.
Informing safe prescribing practices. A prescriber should be aware of the main safety concerns contained in a medication’s black-box warning; at the same time, these warnings are not meant to unduly limit use when crucial treatment is needed.14 In issuing a black-box warning, the FDA has clearly stated the priority and seriousness of its concern. These safety issues must be balanced against the medication’s utility for a given patient, at the prescriber’s clinical judgment.
Guiding shared decision-making. Clinicians are not required to disclose black-box warnings to patients, and there are no criteria that clearly define the role of these warnings in patient care. As is often noted, the FDA does not regulate the practice of medicine.6 However, given the seriousness of the potential adverse effects delineated by black-box warnings, it is reasonable for clinicians to have a solid grasp of black-box warnings for all medications they prescribe, and to be able to relate these warnings to patients, in appropriate language. This patient-centered discussion should include weighing the risks and benefits with the patient and educating the patient about the risks and strategies to mitigate those risks. This discussion can be augmented by patient handouts, which are often offered by pharmaceutical manufacturers, and by shared decision-making tools. A proactive discussion with patients and families about black-box warnings and other risks discussed in product labels can help reduce fears associated with taking medications and may improve adherence.
Continue to: Improving documentation of treatment decisions
Improving documentation of treatment decisions. Fluent knowledge of black-box warnings may help clinicians improve documentation of their treatment decisions, particularly the risks and benefits of their medication choices. Fluency with black-box warnings will help clinicians accurately document both their awareness of these risks, and how these risks informed their risk-benefit analysis in specific clinical situations.
Despite the clear importance the FDA places on black-box warnings, they are not often a topic of study in training or in postgraduate continuing education, and as a result, not all clinicians may be equally conversant with black-box warnings. While black-box warnings do change over time, many psychotropic medication black-box warnings are long-standing and well-established, and they evolve slowly enough to make mastering these warnings worthwhile in order to make the most informed clinical decisions for patient care.
Keeping up-to-date
There are practical and useful ways for busy clinicians to stay up-to-date with black-box warnings. Although these resources exist in multiple locations, together they provide convenient ways to keep current.
The FDA provides access to black-box warnings via its comprehensive database, DRUGS@FDA (https://www.accessdata.fda.gov/scripts/cder/daf/). Detailed information about REMS (and corresponding ETASU and other information related to REMS programs) is available at REMS@FDA (https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm). Clinicians can make safety reports that may contribute to FDA decision-making on black-box warnings by contacting MedWatch (https://www.fda.gov/safety/medwatch-fda-safety-information-and-adverse-event-reporting-program), the FDA’s adverse events reporting system. MedWatch releases safety information reports, which can be followed on Twitter @FDAMedWatch. Note that FDA information generally is organized by specific drug, and not into categories, such as psychotropic medications.
BlackBoxRx (www.blackboxrx.com) is a subscription-based web service that some clinicians may have access to via facility or academic resources as part of a larger FormWeb software package. Individuals also can subscribe (currently, $89/year).
Continue to: Micromedex
Micromedex (www.micromedex.com), which is widely available through medical libraries, is a subscription-based web service that provides black-box warning information from a separate tab that is easily accessed in each drug’s information front page. There is also an alphabetical list of black-box warnings under a separate tab on the Micromedex landing page.
ePocrates (www.epocrates.com) is a subscription-based service that provides extensive drug information, including black-box warnings, in a convenient mobile app.
Bottom Line
Black-box warnings are the most prominent drug safety warnings issued by the FDA. Many psychotropic medications carry black-box warnings that are crucial to everyday psychiatric prescribing. A better understanding of blackbox warnings can enhance your clinical practice by informing safe prescribing practices, guiding shared decision-making, and improving documentation of your treatment decisions.
Related Resources
- US Food and Drug Administration. DRUGS@FDA: FDAapproved drug products. www.accessdata.fda.gov/scripts/cder/daf/.
- US Food and Drug Administration. Drug safety and availability. www.fda.gov/drugs/drug-safety-and-availability. Updated October 10, 2019.
- BlackBoxRx. www.blackboxrx.com. (Subscription required.)
- Mircromedex. www.micromedex.com. (Subscription required.)
- ePocrates. www.epocrates.com. (Subscription required.)
Drug Brand Names
Amitriptyline • Elavil, Vanatrip
Amoxatine • Strattera
Amoxapine • Asendin
Aripiprazole • Abilify
Asenapine • Saphris
Brexanolone • Zulresso
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dexmethylphenidate • Focalin
Dextroamphetamine/amphetamine • Adderall
Disulfiram • Antabuse
Doxepin • Prudoxin, Silenor
Droperidol • Inapsine
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Eszopiclone • Lunesta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imipramine • Tofranil
Isocarboxazid • Marplan
Lamotrigine • Lamictal
Levomilnacipran • Fetzima
Levothyroxine • Synthroid
Linezolid • Zyvox
Lisdexamfetamine • Vyvanse
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Lurasidone • Latuda
Maprotiline • Ludiomil
Methadone • Dolophine, Methadose
Methylphenidate • Ritalin, Concerta
Midazolam • Versed
Milnacipran • Savella
Mirtazapine • Remeron
Naltrexone • Revia, Vivitrol
Nefazodone • Serzone
Nortriptyline • Aventyl, Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenelzine • Nardil
Pimavanserin • Nuplazid
Prochlorperazine • Compro
Protriptyline • Vivactil
Quetiapine • Seroquel
Risperidone • Risperdal
Selegiline • Emsam
Sertraline • Zoloft
Thioridazine • Mellaril
Thiothixene • Navane
Tranylcypromine • Parnate
Trazodone • Desyrel, Oleptro
Trifluoperazine • Stelazine
Trimipramine • Surmontil
Valproate • Depakote
Varenicline • Chantix, Wellbutrin
Vilazodone • Viibryd
Venlafaxine • Effexor
Vortioxetine • Trintellix
Zaleplon • Sonata
Ziprasidone • Geodon
Zolpidem • Ambien
Recently, the FDA issued “black-box” warnings, its most prominent drug safety statements, for esketamine,1 which is indicated for treatment-resistant depression, and the Z-drugs, which are indicated for insomnia2 (Table 1). A black-box warning also comes with brexanolone, which was recently approved for postpartum depression.3 While these newly issued warnings serve as a timely reminder of the importance of black-box warnings, older black-box warnings also cover large areas of psychiatric prescribing, including all medications indicated for treating psychosis or schizophrenia (increased mortality in patients with dementia), and all psychotropic medications with a depression indication (suicidality in younger people).
In this article, we help busy prescribers navigate the landscape of black-box warnings by providing a concise review of how to use them in clinical practice, and where to find information to keep up-to-date.
What are black-box warnings?
A black-box warning is a summary of the potential serious or life-threatening risks of a specific prescription medication. The black-box warning is formatted within a black border found at the top of the manufacturer’s prescribing information document (also known as the package insert or product label). Below the black-box warning, potential risks appear in descending order in sections titled “Contraindications,” “Warnings and Precautions,” and “Adverse Reactions.”4 The FDA issues black-box warnings either during drug development, to take effect upon approval of a new agent, or (more commonly) based on post-marketing safety information,5 which the FDA continuously gathers from reports by patients, clinicians, and industry.6 Federal law mandates the existence of black-box warnings, stating in part that, “special problems, particularly those that may lead to death or serious injury, may be required by the [FDA] to be placed in a prominently displayed box” (21 CFR 201.57(e)).
When is a black-box warning necessary?
The FDA issues a black-box warning based upon its judgment of the seriousness of the adverse effect. However, by definition, these risks do not inherently outweigh the benefits a medication may offer to certain patients. According to the FDA,7 black-box warnings are placed when:
- an adverse reaction so significant exists that this potential negative effect must be considered in risks and benefits when prescribing the medication
- a serious adverse reaction exists that can be prevented, or the risk reduced, by appropriate use of the medication
- the FDA has approved the medication with restrictions to ensure safe use.
Table 2 shows examples of scenarios where black-box warnings have been issued.8 Black-box warnings may be placed on an individual agent or on an entire class of medications. For example, both antipsychotics and antidepressants have class-wide warnings. Finally, black-box warnings are not static, and their content may change; in a study of black-box warnings issued from 2007 to 2015, 29% were entirely new, 32% were considered major updates to existing black-box warnings, and 40% were minor updates.5
Critiques of black-box warnings focus on the absence of published, formal criteria for instituting such warnings, the lack of a consistent approach in their content, and the infrequent inclusion of any information on the relative size of the risk.9 Suggestions for improvement include offering guidance on how to implement the black-box warnings in a patient-centered, shared decision-making model by adding evidence profiles and implementation guides.10 Less frequently considered, black-box warnings may be discontinued if new evidence demonstrates that the risk is lower than previously appreciated; however, similarly to their placement, no explicit criteria for the removal of black-box warnings have been made public.11
When a medication poses an especially high safety risk, the FDA may require the manufacturer to implement a Risk Evaluation and Mitigation Strategy (REMS) program. These programs can describe specific steps to improve medication safety, known as elements to assure safe use (ETASU).4 A familiar example is the clozapine REMS. In order to reduce the risk of severe neutropenia, the clozapine REMS requires prescribers (and pharmacists) to complete specialized training (making up the ETASU). Surprisingly, not every medication with a REMS has a corresponding black-box warning12; more understandably, many medications with black-box warnings do not have an associated REMS, because their risks are evaluated to be manageable by an individual prescriber’s clinical judgment. Most recently, esketamine carries both a black-box warning and a REMS. The black-box warning focuses on adverse effects (Table 1), while the REMS focuses on specific steps used to lessen these risks, including requiring use of a patient enrollment and monitoring form, a fact sheet for patients, and health care setting and pharmacy enrollment forms.13
Continue to: Psychotropic medications and black-box warnings
Psychotropic medications and black-box warnings
Psychotropic medications have a large number of black-box warnings.14 Because it is difficult to find black-box warnings for multiple medications in one place, we have provided 2 convenient resources to address this gap: a concise summary guide (Table 3) and a more detailed database (Table 4, Table 5, Table 6, Table 7, and Table 8). In these Tables, the possible risk mitigations, off-label uses, and monitoring are not meant to be formal recommendations or endorsements but are for independent clinician consideration only.
The information in these Tables was drawn from publicly available data, primarily the Micromedex and FDA web sites (see Related Resources). Because this information changes over time, at the end of this article we suggest ways for clinicians to stay updated with black-box warnings and build on the information provided in this article. These tools can be useful for day-to-day clinical practice in addition to studying for professional examinations. The following are selected high-profile black-box warnings.
Antidepressants and suicide risk. As a class, antidepressants carry a black-box warning on suicide risk in patients age ≤24. Initially issued in 2005, this warning was extended in 2007 to indicate that depression itself is associated with an increased risk of suicide. This black-box warning is used for an entire class of medications as well as for a specific patient population (age ≤24). Moreover, it indicates that suicide rates in patients age >65 were lower among patients using antidepressants.
Among psychotropic medication black-box warnings, this warning has perhaps been the most controversial. For example, it has been suggested that this black-box warning may have inadvertently increased suicide rates by discouraging clinicians from prescribing antidepressants,15 although this also has been called into question.16 This black-box warning illustrates that the consequences of issuing black-box warnings can be very difficult to assess, which makes their clinical effects highly complex and challenging to evaluate.14
Antipsychotics and dementia-related psychosis. This warning was initially issued in 2005 for second-generation antipsychotics and extended to first-generation antipsychotics in 2008. Antipsychotics as a class carry a black-box warning for increased risk of death in patients with dementia (major neurocognitive disorder). This warning extends to the recently approved antipsychotic pimavanserin, even though this agent’s proposed mechanism of action differs from that of other antipsychotics.17 However, it specifically allows for use in Parkinson’s disease psychosis, which is pimavanserin’s indication.18 In light of recent research suggesting pimavanserin is effective in dementia-related psychosis,19 it bears watching whether this agent becomes the first antipsychotic to have this warning removed.
Continue to: This class warning has...
This class warning has had widespread effects. For example, it has prompted less use of antipsychotics in nursing home facilities, as a result of stricter Centers for Medicare and Medicaid Services regulations20; overall, there is some evidence that there has been reduced prescribing of antipsychotics in general.21 Additionally, this black-box warning is unusual in that it warns about a specific off-label indication, which is itself poorly supported by evidence.21 Concomitantly, few other treatment options are available for this clinical situation. These medications are often seen as the only option for patients with dementia complicated by severe behavioral disturbance, and thus this black-box warning reflects real-world practices.14
Varenicline and neuropsychiatric complications. The withdrawal of the black-box warning on potential neuropsychiatric complications of using varenicline for smoking cessation shows that black-box warnings are not static and can, though infrequently, be removed as more safety data accumulates.11 As additional post-marketing information emerged on this risk, this black-box warning was reconsidered and withdrawn in 2016.22 Its withdrawal could potentially make clinicians more comfortable prescribing varenicline and in turn, help to reduce smoking rates.
How to use black-box warnings
To enhance their clinical practice, prescribers can use black-box warnings to inform safe prescribing practices, to guide shared decision-making, and to improve documentation of their treatment decisions.
Informing safe prescribing practices. A prescriber should be aware of the main safety concerns contained in a medication’s black-box warning; at the same time, these warnings are not meant to unduly limit use when crucial treatment is needed.14 In issuing a black-box warning, the FDA has clearly stated the priority and seriousness of its concern. These safety issues must be balanced against the medication’s utility for a given patient, at the prescriber’s clinical judgment.
Guiding shared decision-making. Clinicians are not required to disclose black-box warnings to patients, and there are no criteria that clearly define the role of these warnings in patient care. As is often noted, the FDA does not regulate the practice of medicine.6 However, given the seriousness of the potential adverse effects delineated by black-box warnings, it is reasonable for clinicians to have a solid grasp of black-box warnings for all medications they prescribe, and to be able to relate these warnings to patients, in appropriate language. This patient-centered discussion should include weighing the risks and benefits with the patient and educating the patient about the risks and strategies to mitigate those risks. This discussion can be augmented by patient handouts, which are often offered by pharmaceutical manufacturers, and by shared decision-making tools. A proactive discussion with patients and families about black-box warnings and other risks discussed in product labels can help reduce fears associated with taking medications and may improve adherence.
Continue to: Improving documentation of treatment decisions
Improving documentation of treatment decisions. Fluent knowledge of black-box warnings may help clinicians improve documentation of their treatment decisions, particularly the risks and benefits of their medication choices. Fluency with black-box warnings will help clinicians accurately document both their awareness of these risks, and how these risks informed their risk-benefit analysis in specific clinical situations.
Despite the clear importance the FDA places on black-box warnings, they are not often a topic of study in training or in postgraduate continuing education, and as a result, not all clinicians may be equally conversant with black-box warnings. While black-box warnings do change over time, many psychotropic medication black-box warnings are long-standing and well-established, and they evolve slowly enough to make mastering these warnings worthwhile in order to make the most informed clinical decisions for patient care.
Keeping up-to-date
There are practical and useful ways for busy clinicians to stay up-to-date with black-box warnings. Although these resources exist in multiple locations, together they provide convenient ways to keep current.
The FDA provides access to black-box warnings via its comprehensive database, DRUGS@FDA (https://www.accessdata.fda.gov/scripts/cder/daf/). Detailed information about REMS (and corresponding ETASU and other information related to REMS programs) is available at REMS@FDA (https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm). Clinicians can make safety reports that may contribute to FDA decision-making on black-box warnings by contacting MedWatch (https://www.fda.gov/safety/medwatch-fda-safety-information-and-adverse-event-reporting-program), the FDA’s adverse events reporting system. MedWatch releases safety information reports, which can be followed on Twitter @FDAMedWatch. Note that FDA information generally is organized by specific drug, and not into categories, such as psychotropic medications.
BlackBoxRx (www.blackboxrx.com) is a subscription-based web service that some clinicians may have access to via facility or academic resources as part of a larger FormWeb software package. Individuals also can subscribe (currently, $89/year).
Continue to: Micromedex
Micromedex (www.micromedex.com), which is widely available through medical libraries, is a subscription-based web service that provides black-box warning information from a separate tab that is easily accessed in each drug’s information front page. There is also an alphabetical list of black-box warnings under a separate tab on the Micromedex landing page.
ePocrates (www.epocrates.com) is a subscription-based service that provides extensive drug information, including black-box warnings, in a convenient mobile app.
Bottom Line
Black-box warnings are the most prominent drug safety warnings issued by the FDA. Many psychotropic medications carry black-box warnings that are crucial to everyday psychiatric prescribing. A better understanding of blackbox warnings can enhance your clinical practice by informing safe prescribing practices, guiding shared decision-making, and improving documentation of your treatment decisions.
Related Resources
- US Food and Drug Administration. DRUGS@FDA: FDAapproved drug products. www.accessdata.fda.gov/scripts/cder/daf/.
- US Food and Drug Administration. Drug safety and availability. www.fda.gov/drugs/drug-safety-and-availability. Updated October 10, 2019.
- BlackBoxRx. www.blackboxrx.com. (Subscription required.)
- Mircromedex. www.micromedex.com. (Subscription required.)
- ePocrates. www.epocrates.com. (Subscription required.)
Drug Brand Names
Amitriptyline • Elavil, Vanatrip
Amoxatine • Strattera
Amoxapine • Asendin
Aripiprazole • Abilify
Asenapine • Saphris
Brexanolone • Zulresso
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dexmethylphenidate • Focalin
Dextroamphetamine/amphetamine • Adderall
Disulfiram • Antabuse
Doxepin • Prudoxin, Silenor
Droperidol • Inapsine
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Eszopiclone • Lunesta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imipramine • Tofranil
Isocarboxazid • Marplan
Lamotrigine • Lamictal
Levomilnacipran • Fetzima
Levothyroxine • Synthroid
Linezolid • Zyvox
Lisdexamfetamine • Vyvanse
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Lurasidone • Latuda
Maprotiline • Ludiomil
Methadone • Dolophine, Methadose
Methylphenidate • Ritalin, Concerta
Midazolam • Versed
Milnacipran • Savella
Mirtazapine • Remeron
Naltrexone • Revia, Vivitrol
Nefazodone • Serzone
Nortriptyline • Aventyl, Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenelzine • Nardil
Pimavanserin • Nuplazid
Prochlorperazine • Compro
Protriptyline • Vivactil
Quetiapine • Seroquel
Risperidone • Risperdal
Selegiline • Emsam
Sertraline • Zoloft
Thioridazine • Mellaril
Thiothixene • Navane
Tranylcypromine • Parnate
Trazodone • Desyrel, Oleptro
Trifluoperazine • Stelazine
Trimipramine • Surmontil
Valproate • Depakote
Varenicline • Chantix, Wellbutrin
Vilazodone • Viibryd
Venlafaxine • Effexor
Vortioxetine • Trintellix
Zaleplon • Sonata
Ziprasidone • Geodon
Zolpidem • Ambien
1. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceutical Companies; 2019.
2. U.S. Food and Drug Administration. FDA drug safety announcement: FDA adds boxed warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia. Published April 30, 2019. Accessed October 28, 2019.
3. Zulresso [package insert]. Cambridge, Mass.: Sage Therapeutics Inc.; 2019.
4. Gassman AL, Nguyen CP, Joffe HV. FDA regulation of prescription drugs. N Engl J Med. 2017;376(7):674-682.
5. Solotke MT, Dhruva SS, Downing NS, et al. New and incremental FDA black box warnings from 2008 to 2015. Expert Opin Drug Saf. 2018;17(2):117-123.
6. Murphy S, Roberts R. “Black box” 101: how the Food and Drug Administration evaluates, communicates, and manages drug benefit/risk. J Allergy Clin Immunol. 2006;117(1):34-39.
7. U.S. Food and Drug Administration. Guidance document: Warnings and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products – content and format. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/warnings-and-precautions-contraindications-and-boxed-warning-sections-labeling-human-prescription. Published October 2011. Accessed October 28, 2019.
8. Beach JE, Faich GA, Bormel FG, et al. Black box warnings in prescription drug labeling: results of a survey of 206 drugs. Food Drug Law J. 1998;53(3):403-411.
9. Matlock A, Allan N, Wills B, et al. A continuing black hole? The FDA boxed warning: an appeal to improve its clinical utility. Clinical Toxicol (Phila). 2011;49(6):443-447.
10. Elraiyah T, Gionfriddo MR, Montori VM, et al. Content, consistency, and quality of black box warnings: time for a change. Ann Intern Med. 2015;163(11):875-876.
11. Yeh JS, Sarpatwari A, Kesselheim AS. Ethical and practical considerations in removing black box warnings from drug labels. Drug Saf. 2016;39(8):709-714.
12. Boudes PF. Risk Evaluation and Mitigation Strategies (REMSs): are they improving drug safety? A critical review of REMSs requiring Elements to Assure Safe Use (ETASU). Drugs R D. 2017;17(2):245-254.
13. U.S. Food and Drug Administration. Approved risk evaluation mitigation strategies (REMS): Spravato (esketamine) REMS program. https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=IndvRemsDetails.page&REMS=386. Updated June 25, 2019. Accessed October 28, 2018.
14. Stevens JR, Jarrahzadeh T, Brendel RW, et al. Strategies for the prescription of psychotropic drugs with black box warnings. Psychosomatics. 2014;55(2):123-133.
15. Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668.
16. Stone MB. The FDA warning on antidepressants and suicidality--why the controversy? N Engl J Med. 2014;371(18):1668-1671.
17. Mathis MV, Muoio BM, Andreason P, et al. The US Food and Drug Administration’s perspective on the new antipsychotic pimavanserin. J Clin Psychiatry. 2017;78(6):e668-e673. doi: 10.4088/JCP.16r11119.
18. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; May 2019.
19. Ballard C, Banister C, Khan Z, et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 2018;17(3):213-222.
20. Maust DT, Kim HM, Chiang C, et al. Association of the Centers for Medicare & Medicaid Services’ National Partnership to Improve Dementia Care with the use of antipsychotics and other psychotropics in long-term care in the United States from 2009 to 2014. JAMA Intern Med. 2018;178(5):640-647.
21. Dorsey ER, Rabbani A, Gallagher SA, et al. Impact of FDA black box advisory on antipsychotic medication use. Arch Intern Med. 2010;170(1):96-103.
22. U.S. Food and Drug Administration. FDA drug safety communication: FDA revises description of mental health side effects of the stop-smoking medicines Chantix (varenicline) and Zyban (bupropion) to reflect clinical trial findings. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-revises-description-mental-health-side-effects-stop-smoking. Published December 16, 2016. Accessed October 28, 2019.
1. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceutical Companies; 2019.
2. U.S. Food and Drug Administration. FDA drug safety announcement: FDA adds boxed warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia. Published April 30, 2019. Accessed October 28, 2019.
3. Zulresso [package insert]. Cambridge, Mass.: Sage Therapeutics Inc.; 2019.
4. Gassman AL, Nguyen CP, Joffe HV. FDA regulation of prescription drugs. N Engl J Med. 2017;376(7):674-682.
5. Solotke MT, Dhruva SS, Downing NS, et al. New and incremental FDA black box warnings from 2008 to 2015. Expert Opin Drug Saf. 2018;17(2):117-123.
6. Murphy S, Roberts R. “Black box” 101: how the Food and Drug Administration evaluates, communicates, and manages drug benefit/risk. J Allergy Clin Immunol. 2006;117(1):34-39.
7. U.S. Food and Drug Administration. Guidance document: Warnings and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products – content and format. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/warnings-and-precautions-contraindications-and-boxed-warning-sections-labeling-human-prescription. Published October 2011. Accessed October 28, 2019.
8. Beach JE, Faich GA, Bormel FG, et al. Black box warnings in prescription drug labeling: results of a survey of 206 drugs. Food Drug Law J. 1998;53(3):403-411.
9. Matlock A, Allan N, Wills B, et al. A continuing black hole? The FDA boxed warning: an appeal to improve its clinical utility. Clinical Toxicol (Phila). 2011;49(6):443-447.
10. Elraiyah T, Gionfriddo MR, Montori VM, et al. Content, consistency, and quality of black box warnings: time for a change. Ann Intern Med. 2015;163(11):875-876.
11. Yeh JS, Sarpatwari A, Kesselheim AS. Ethical and practical considerations in removing black box warnings from drug labels. Drug Saf. 2016;39(8):709-714.
12. Boudes PF. Risk Evaluation and Mitigation Strategies (REMSs): are they improving drug safety? A critical review of REMSs requiring Elements to Assure Safe Use (ETASU). Drugs R D. 2017;17(2):245-254.
13. U.S. Food and Drug Administration. Approved risk evaluation mitigation strategies (REMS): Spravato (esketamine) REMS program. https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=IndvRemsDetails.page&REMS=386. Updated June 25, 2019. Accessed October 28, 2018.
14. Stevens JR, Jarrahzadeh T, Brendel RW, et al. Strategies for the prescription of psychotropic drugs with black box warnings. Psychosomatics. 2014;55(2):123-133.
15. Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668.
16. Stone MB. The FDA warning on antidepressants and suicidality--why the controversy? N Engl J Med. 2014;371(18):1668-1671.
17. Mathis MV, Muoio BM, Andreason P, et al. The US Food and Drug Administration’s perspective on the new antipsychotic pimavanserin. J Clin Psychiatry. 2017;78(6):e668-e673. doi: 10.4088/JCP.16r11119.
18. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; May 2019.
19. Ballard C, Banister C, Khan Z, et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 2018;17(3):213-222.
20. Maust DT, Kim HM, Chiang C, et al. Association of the Centers for Medicare & Medicaid Services’ National Partnership to Improve Dementia Care with the use of antipsychotics and other psychotropics in long-term care in the United States from 2009 to 2014. JAMA Intern Med. 2018;178(5):640-647.
21. Dorsey ER, Rabbani A, Gallagher SA, et al. Impact of FDA black box advisory on antipsychotic medication use. Arch Intern Med. 2010;170(1):96-103.
22. U.S. Food and Drug Administration. FDA drug safety communication: FDA revises description of mental health side effects of the stop-smoking medicines Chantix (varenicline) and Zyban (bupropion) to reflect clinical trial findings. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-revises-description-mental-health-side-effects-stop-smoking. Published December 16, 2016. Accessed October 28, 2019.

Considerations on the mode of delivery for pregnant women with hepatitis C infection
CASE Pregnant woman with chronic opioid use and HIV, recently diagnosed with HCV
A 34-year-old primigravid woman at 35 weeks' gestation has a history of chronic opioid use. She previously was diagnosed with human immunodeficiency virus (HIV) infection and has been treated with a 3-drug combination antiretroviral regimen. Her most recent HIV viral load was 750 copies/mL. Three weeks ago, she tested positive for hepatitis C virus (HCV) infection. Liver function tests showed mild elevations in transaminase levels. The viral genotype is 1, and the viral load is 2.6 million copies/mL.
How should this patient be delivered? Should she be encouraged to breastfeed her neonate?
The scope of HCV infection
Hepatitis C virus is a positive-sense, enveloped, single-stranded RNA virus that belongs to the Flaviviridae family.1 There are 7 confirmed major genotypes of HCV and 67 confirmed subtypes.2 HCV possesses several important virulence factors. First, the virus's replication is prone to frequent mutations because its RNA polymerase lacks proofreading activity, resulting in significant genetic diversity. The great degree of heterogeneity among HCV leads to high antigenic variability, which is one of the main reasons there is not yet a vaccine for HCV.3 Additionally, HCV's genomic plasticity plays a role in the emergence of drug-resistant variants.4
Virus transmission. Worldwide, approximately 130 to 170 million people are infected with HCV.5 HCV infections are caused primarily by exposure to infected blood, through sharing needles for intravenous drug injection and through receiving a blood transfusion.6 Other routes of transmission include exposure through sexual contact, occupational injury, and perinatal acquisition.
The risk of acquiring HCV varies for each of these transmission mechanisms. Blood transfusion is no longer a common mechanism of transmission in places where blood donations are screened for HCV antibodies and viral RNA. Additionally, unintentional needle-stick injury is the only occupational risk factor associated with HCV infection, and health care workers do not have a greater prevalence of HCV than the general population. Moreover, sexual transmission is not a particularly efficient mechanism for spread of HCV.7 Therefore, unsafe intravenous injections are now the leading cause of HCV infection.6
Consequences of HCV infection. Once infected with HCV, about 25% of people spontaneously clear the virus and approximately 75% progress to chronic HCV infection.5 The consequences of long-term infection with HCV include end-stage liver disease, cirrhosis, and hepatocellular carcinoma.
Approximately 30% of people infected with HCV will develop cirrhosis and another 2% will develop hepatocellular carcinoma.8 Liver transplant is the only treatment option for patients with decompensated cirrhosis or hepatocellular carcinoma as a result of HCV infection. Currently, HCV infection is the leading indication for liver transplant in the United States.9
Continue to: Risk of perinatal HCV transmission...
Risk of perinatal HCV transmission
Approximately 1% to 8% of pregnant women worldwide are infected with HCV.10 In the United States, 1% to 2.5% of pregnant women are infected.11 Of these, about 6% transmit the infection to their offspring. The risk of HCV vertical transmission increases to about 11% if the mother is co-infected with HIV.12 Vertical transmission is the primary method by which children become infected with HCV.13
Several risk factors increase the likelihood of HCV transmission from mother to child, including HIV co-infection, internal fetal monitoring, and longer duration of membrane rupture.14 The effect that mode of delivery has on vertical transmission rates, however, is still debated, and a Cochrane Review found that there were no randomized controlled trials assessing the effect of mode of delivery on mother-to-infant HCV transmission.15
Serology and genotyping used in diagnosis
The serological enzyme immunoassay is the first test used in screening for HCV infection. Currently, third- and fourth-generation enzyme immunoassays are used in the United States.16 However, even these newer serological assays cannot consistently and precisely distinguish between acute and chronic HCV infections.17 After the initial diagnosis is made with serology, it usually is confirmed by assays that detect the virus's genomic RNA in the patient's serum or plasma.
The patient's HCV genotype should be identified so that the best treatment options can be determined. HCV genotyping can be accomplished using reverse transcription quantitative polymerase chain reaction (RT-qPCR) amplification. Three different RT-qPCR assessments usually are performed using different primers and probes specific to different genotypes of HCV. While direct sequencing of the HCV genome also can be performed, this method is usually not used clinically due to its technical complexity.16
Modern treatments are effective
Introduced in 2011, direct-acting antiviral therapies are now the recommended treatment for HCV infection. These drugs inhibit the virus's replication by targeting different proteins involved in the HCV replication cycle. They are remarkably successful and have achieved sustained virologic response (SVR) rates greater than 90%.11 The World Health Organization recommends several pangenotypic (that is, agents that work against all genotypes) direct-acting antiviral regimens for the treatment of chronic HCV infection in adults without cirrhosis (TABLE 1).18,19
Unfortunately, experience with these drugs in pregnant women is lacking. Many direct-acting antiviral agents have not been tested systematically in pregnant women, and, accordingly, most information about their effects in pregnant women comes from animal models.11
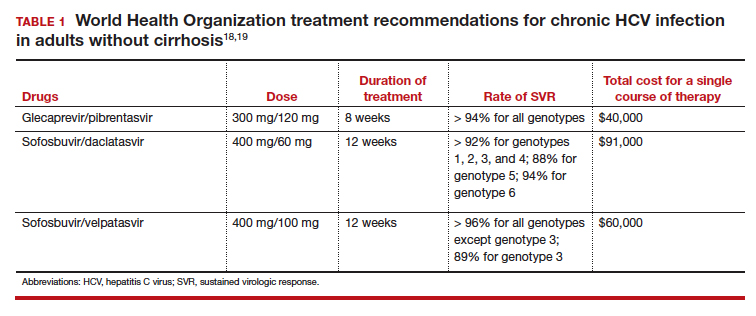
Continue to: Perinatal transmission rates and effect of mode of delivery...
Perinatal transmission rates and effect of mode of delivery
We compiled data from 11 studies that reported the perinatal transmission rate of HCV associated with various modes of delivery. These studies were selected from a MEDLINE literature review from 1999 to 2019. The studies were screened first by title and, subsequently, by abstract. Inclusion was restricted to randomized controlled trials, cohort studies, and case-control studies written in English. Study quality was assessed as good, fair, or poor based on the study design, sample size, and statistical analyses performed. The results from the total population of each study are reported in TABLE 2.14,20-29
Three studies separated data based on the mother's HIV status. The perinatal transmission rates of HCV for mothers co-infected with HIV are reported in TABLE 3.23,27 The results for HIV-negative mothers are reported in TABLE 4.14,23
Finally, 2 studies grouped mothers according to their HCV viral load. All of the mothers in these studies were anti-HCV antibody positive, and the perinatal transmission rates for the total study populations were reported previously in TABLE 2. The results for mothers who had detectable HCV RNA are reported in TABLE 5.20,21 High viral load was defined as
≥ 2.5 x 106 Eq/mL in the study by Okamoto and colleagues, which is equivalent to ≥ 6.0 x 105 IU/mL in the study by Murakami and colleagues due to the different assays that were used.20,21 The perinatal transmission rates for mothers with a high viral load are presented in TABLE 6.20,21
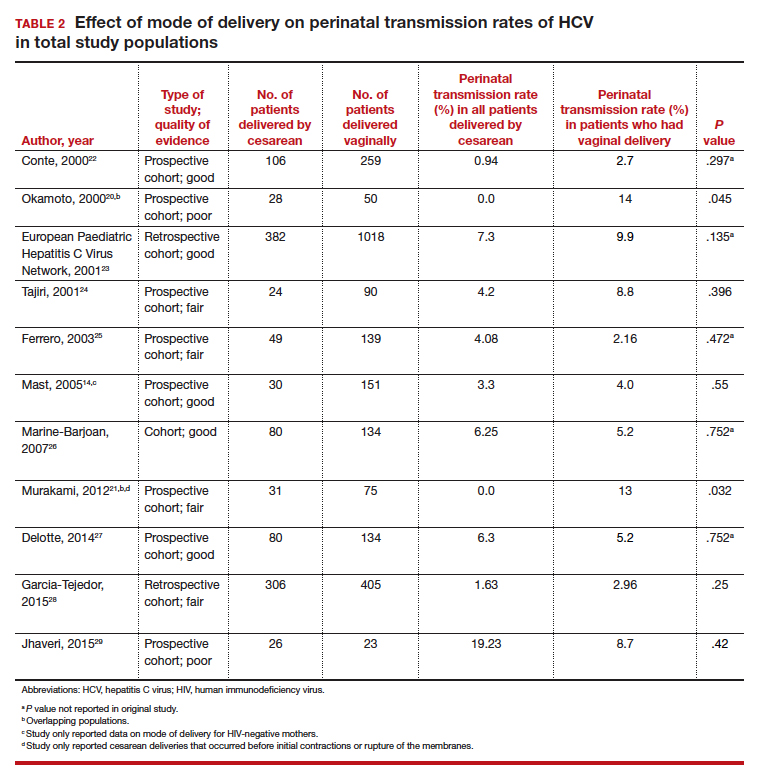
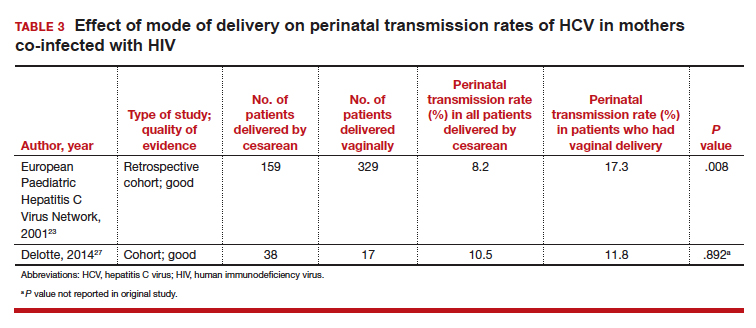
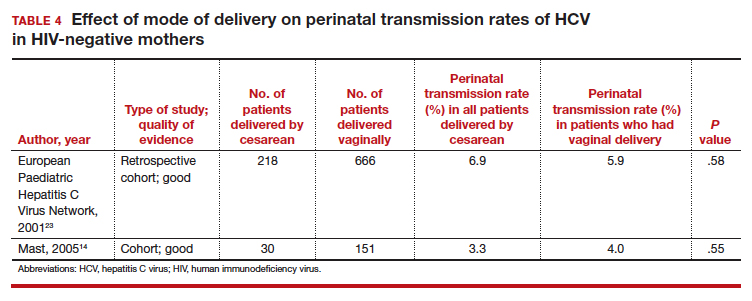
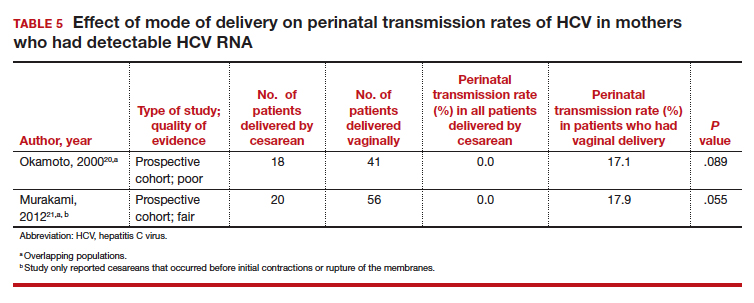

Continue to: For most, CD does not reduce HCV transmission...
For most, CD does not reduce HCV transmission
Nine of the 11 studies found that the mode of delivery did not have a statistically significant impact on the vertical transmission rate of HCV in the total study populations.14,22-29 The remaining 2 studies found that the perinatal transmission rate of HCV was lower with cesarean delivery (CD) than with vaginal delivery.20,21 When considered together, the results of these 11 studies indicate that CD does not provide a significant reduction in the HCV transmission rate in the general population.
Our review confirms the findings of others, including a systematic review by the US Preventive Services Task Force.30 That investigation also failed to demonstrate any measurable increase in risk of HCV transmission as a result of breastfeeding.
Cesarean delivery may benefit 2 groups. Careful assessment of these studies, however, suggests that 2 select groups of patients with HCV may benefit from CD:
- mothers co-infected with HIV, and
- mothers with high viral loads of HCV.
In both of these populations, the vertical transmission rate of HCV was significantly reduced with CD compared with vaginal delivery. Therefore, CD should be strongly considered in mothers with HCV who are co-infected with HIV and/or in mothers who have a high viral load of HCV.
CASE Our recommendation for mode of delivery
The patient in our case scenario has both HIV infection and a very high HCV viral load. We would therefore recommend a planned CD at 38 to 39 weeks' gestation, prior to the onset of labor or membrane rupture. Although HCV infection is not a contraindication to breastfeeding, the mother's HIV infection is a distinct contraindication.
- Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol. 2014;61(1 suppl):S3-S13.
- Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318-327.
- Rossi LM, Escobar-Gutierrez A, Rahal P. Advanced molecular surveillance of hepatitis C virus. Viruses. 2015;7:1153-1188.
- Dustin LB, Bartolini B, Capobianchi MR, et al. Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin Microbiol Infect. 2016;22:826-832.
- Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10:553-562.
- Thomas DL. Global elimination of chronic hepatitis. N Engl J Med. 2019;380:2041-2050.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep. 1998;47(RR19):1-39.
- Gonzalez-Grande R, Jimenez-Perez M, Gonzalez Arjona C, et al. New approaches in the treatment of hepatitis C. World J Gastroenterol. 2016;22:1421-1432.
- Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61(1 suppl): S58-S68.
- Spera AM, Eldin TK, Tosone G, et al. Antiviral therapy for hepatitis C: has anything changed for pregnant/lactating women? World J Hepatol. 2016;8:557-565.
- Society for Maternal-Fetal Medicine; Hughes BL, Page CM, Kuller JA. Hepatitis C in pregnancy: screening, treatment, and management. Am J Obstet Gynecol. 2017;217:B2-B12.
- Benova L, Mohamoud YA, Calvert C, et al. Vertical transmission of hepatitis C virus: systematic review and meta-analysis. Clin Infect Dis. 2014;59:765-773.
- Ghamar Chehreh ME, Tabatabaei SV, Khazanehdari S, et al. Effect of cesarean section on the risk of perinatal transmission of hepatitis C virus from HCV-RNA+/HIV- mothers: a meta-analysis. Arch Gynecol Obstet. 2011;283:255-260.
- Mast EE, Hwang LY, Seto DS, et al. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J Infect Dis. 2005;192:1880-1889.
- McIntyre PG, Tosh K, McGuire W. Caesarean section versus vaginal delivery for preventing mother to infant hepatitis C virus transmission. Cochrane Database Syst Rev. 2006;(4):CD005546.
- Mukherjee R, Burns A, Rodden D, et al. Diagnosis and management of hepatitis C virus infection. J Lab Autom. 2015;20:519-538.
- Araujo AC, Astrakhantseva IV, Fields HA, et al. Distinguishing acute from chronic hepatitis C virus (HCV) infection based on antibody reactivities to specific HCV structural and nonstructural proteins. J Clin Microbiol. 2011;49:54-57.
- World Health Organization. Guidelines for the Care and Treatment of Persons Diagnosed with Chronic Hepatitis C Virus Infection. Geneva, Switzerland: World Health Organization; 2018.
- CADTH Common Drug Review. Pharmacoeconomic Review Report: Sofosbuvir/Velpatasvir/Voxilaprevir (Vosevi) (Gilead Sciences Canada, Inc): Indication: Hepatitis C infection genotype 1 to 6. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2018.
- Okamoto M, Nagata I, Murakami J, et al. Prospective reevaluation of risk factors in mother-to-child transmission of hepatitis C virus: high virus load, vaginal delivery, and negative anti-NS4 antibody. J Infect Dis. 2000;182:1511-1514.
- Murakami J, Nagata I, Iitsuka T, et al. Risk factors for mother-to-child transmission of hepatitis C virus: maternal high viral load and fetal exposure in the birth canal. Hepatol Res. 2012;42:648-657.
- Conte D, Fraquelli M, Prati D, et al. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology. 2000;31:751-755.
- European Paediatric Hepatitis C Virus Network. Effects of mode of delivery and infant feeding on the risk of mother-to-child transmission of hepatitis C virus. BJOG. 2001;108:371-377.
- Tajiri H, Miyoshi Y, Funada S, et al. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J. 2001;20:10-14.
- Ferrero S, Lungaro P, Bruzzone BM, et al. Prospective study of mother-to-infant transmission of hepatitis C virus: a 10-year survey (1990-2000). Acta Obstet Gynecol Scand. 2003;82:229-234.
- Marine-Barjoan E, Berrebi A, Giordanengo V, et al. HCV/HIV co-infection, HCV viral load and mode of delivery: risk factors for mother-to-child transmission of hepatitis C virus? AIDS. 2007;21:1811-1815.
- Delotte J, Barjoan EM, Berrebi A, et al. Obstetric management does not influence vertical transmission of HCV infection: results of the ALHICE group study. J Matern Fetal Neonatal Med. 2014;27:664-670.
- Garcia-Tejedor A, Maiques-Montesinos V, Diago-Almela VJ, et al. Risk factors for vertical transmission of hepatitis C virus: a single center experience with 710 HCV-infected mothers. Eur J Obstet Gynecol Reprod Biol. 2015;194:173-177.
- Jhaveri R, Hashem M, El-Kamary SS, et al. Hepatitis C virus (HCV) vertical transmission in 12-month-old infants born to HCV-infected women and assessment of maternal risk factors. Open Forum Infect Dis. 2015;2:ofv089.
- Cottrell EB, Chou R, Wasson N, et al. Reducing risk for mother-to-infant transmission of hepatitis C virus: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2013;158:109-113.
CASE Pregnant woman with chronic opioid use and HIV, recently diagnosed with HCV
A 34-year-old primigravid woman at 35 weeks' gestation has a history of chronic opioid use. She previously was diagnosed with human immunodeficiency virus (HIV) infection and has been treated with a 3-drug combination antiretroviral regimen. Her most recent HIV viral load was 750 copies/mL. Three weeks ago, she tested positive for hepatitis C virus (HCV) infection. Liver function tests showed mild elevations in transaminase levels. The viral genotype is 1, and the viral load is 2.6 million copies/mL.
How should this patient be delivered? Should she be encouraged to breastfeed her neonate?
The scope of HCV infection
Hepatitis C virus is a positive-sense, enveloped, single-stranded RNA virus that belongs to the Flaviviridae family.1 There are 7 confirmed major genotypes of HCV and 67 confirmed subtypes.2 HCV possesses several important virulence factors. First, the virus's replication is prone to frequent mutations because its RNA polymerase lacks proofreading activity, resulting in significant genetic diversity. The great degree of heterogeneity among HCV leads to high antigenic variability, which is one of the main reasons there is not yet a vaccine for HCV.3 Additionally, HCV's genomic plasticity plays a role in the emergence of drug-resistant variants.4
Virus transmission. Worldwide, approximately 130 to 170 million people are infected with HCV.5 HCV infections are caused primarily by exposure to infected blood, through sharing needles for intravenous drug injection and through receiving a blood transfusion.6 Other routes of transmission include exposure through sexual contact, occupational injury, and perinatal acquisition.
The risk of acquiring HCV varies for each of these transmission mechanisms. Blood transfusion is no longer a common mechanism of transmission in places where blood donations are screened for HCV antibodies and viral RNA. Additionally, unintentional needle-stick injury is the only occupational risk factor associated with HCV infection, and health care workers do not have a greater prevalence of HCV than the general population. Moreover, sexual transmission is not a particularly efficient mechanism for spread of HCV.7 Therefore, unsafe intravenous injections are now the leading cause of HCV infection.6
Consequences of HCV infection. Once infected with HCV, about 25% of people spontaneously clear the virus and approximately 75% progress to chronic HCV infection.5 The consequences of long-term infection with HCV include end-stage liver disease, cirrhosis, and hepatocellular carcinoma.
Approximately 30% of people infected with HCV will develop cirrhosis and another 2% will develop hepatocellular carcinoma.8 Liver transplant is the only treatment option for patients with decompensated cirrhosis or hepatocellular carcinoma as a result of HCV infection. Currently, HCV infection is the leading indication for liver transplant in the United States.9
Continue to: Risk of perinatal HCV transmission...
Risk of perinatal HCV transmission
Approximately 1% to 8% of pregnant women worldwide are infected with HCV.10 In the United States, 1% to 2.5% of pregnant women are infected.11 Of these, about 6% transmit the infection to their offspring. The risk of HCV vertical transmission increases to about 11% if the mother is co-infected with HIV.12 Vertical transmission is the primary method by which children become infected with HCV.13
Several risk factors increase the likelihood of HCV transmission from mother to child, including HIV co-infection, internal fetal monitoring, and longer duration of membrane rupture.14 The effect that mode of delivery has on vertical transmission rates, however, is still debated, and a Cochrane Review found that there were no randomized controlled trials assessing the effect of mode of delivery on mother-to-infant HCV transmission.15
Serology and genotyping used in diagnosis
The serological enzyme immunoassay is the first test used in screening for HCV infection. Currently, third- and fourth-generation enzyme immunoassays are used in the United States.16 However, even these newer serological assays cannot consistently and precisely distinguish between acute and chronic HCV infections.17 After the initial diagnosis is made with serology, it usually is confirmed by assays that detect the virus's genomic RNA in the patient's serum or plasma.
The patient's HCV genotype should be identified so that the best treatment options can be determined. HCV genotyping can be accomplished using reverse transcription quantitative polymerase chain reaction (RT-qPCR) amplification. Three different RT-qPCR assessments usually are performed using different primers and probes specific to different genotypes of HCV. While direct sequencing of the HCV genome also can be performed, this method is usually not used clinically due to its technical complexity.16
Modern treatments are effective
Introduced in 2011, direct-acting antiviral therapies are now the recommended treatment for HCV infection. These drugs inhibit the virus's replication by targeting different proteins involved in the HCV replication cycle. They are remarkably successful and have achieved sustained virologic response (SVR) rates greater than 90%.11 The World Health Organization recommends several pangenotypic (that is, agents that work against all genotypes) direct-acting antiviral regimens for the treatment of chronic HCV infection in adults without cirrhosis (TABLE 1).18,19
Unfortunately, experience with these drugs in pregnant women is lacking. Many direct-acting antiviral agents have not been tested systematically in pregnant women, and, accordingly, most information about their effects in pregnant women comes from animal models.11
Continue to: Perinatal transmission rates and effect of mode of delivery...
Perinatal transmission rates and effect of mode of delivery
We compiled data from 11 studies that reported the perinatal transmission rate of HCV associated with various modes of delivery. These studies were selected from a MEDLINE literature review from 1999 to 2019. The studies were screened first by title and, subsequently, by abstract. Inclusion was restricted to randomized controlled trials, cohort studies, and case-control studies written in English. Study quality was assessed as good, fair, or poor based on the study design, sample size, and statistical analyses performed. The results from the total population of each study are reported in TABLE 2.14,20-29
Three studies separated data based on the mother's HIV status. The perinatal transmission rates of HCV for mothers co-infected with HIV are reported in TABLE 3.23,27 The results for HIV-negative mothers are reported in TABLE 4.14,23
Finally, 2 studies grouped mothers according to their HCV viral load. All of the mothers in these studies were anti-HCV antibody positive, and the perinatal transmission rates for the total study populations were reported previously in TABLE 2. The results for mothers who had detectable HCV RNA are reported in TABLE 5.20,21 High viral load was defined as
≥ 2.5 x 106 Eq/mL in the study by Okamoto and colleagues, which is equivalent to ≥ 6.0 x 105 IU/mL in the study by Murakami and colleagues due to the different assays that were used.20,21 The perinatal transmission rates for mothers with a high viral load are presented in TABLE 6.20,21
Continue to: For most, CD does not reduce HCV transmission...
For most, CD does not reduce HCV transmission
Nine of the 11 studies found that the mode of delivery did not have a statistically significant impact on the vertical transmission rate of HCV in the total study populations.14,22-29 The remaining 2 studies found that the perinatal transmission rate of HCV was lower with cesarean delivery (CD) than with vaginal delivery.20,21 When considered together, the results of these 11 studies indicate that CD does not provide a significant reduction in the HCV transmission rate in the general population.
Our review confirms the findings of others, including a systematic review by the US Preventive Services Task Force.30 That investigation also failed to demonstrate any measurable increase in risk of HCV transmission as a result of breastfeeding.
Cesarean delivery may benefit 2 groups. Careful assessment of these studies, however, suggests that 2 select groups of patients with HCV may benefit from CD:
- mothers co-infected with HIV, and
- mothers with high viral loads of HCV.
In both of these populations, the vertical transmission rate of HCV was significantly reduced with CD compared with vaginal delivery. Therefore, CD should be strongly considered in mothers with HCV who are co-infected with HIV and/or in mothers who have a high viral load of HCV.
CASE Our recommendation for mode of delivery
The patient in our case scenario has both HIV infection and a very high HCV viral load. We would therefore recommend a planned CD at 38 to 39 weeks' gestation, prior to the onset of labor or membrane rupture. Although HCV infection is not a contraindication to breastfeeding, the mother's HIV infection is a distinct contraindication.
CASE Pregnant woman with chronic opioid use and HIV, recently diagnosed with HCV
A 34-year-old primigravid woman at 35 weeks' gestation has a history of chronic opioid use. She previously was diagnosed with human immunodeficiency virus (HIV) infection and has been treated with a 3-drug combination antiretroviral regimen. Her most recent HIV viral load was 750 copies/mL. Three weeks ago, she tested positive for hepatitis C virus (HCV) infection. Liver function tests showed mild elevations in transaminase levels. The viral genotype is 1, and the viral load is 2.6 million copies/mL.
How should this patient be delivered? Should she be encouraged to breastfeed her neonate?
The scope of HCV infection
Hepatitis C virus is a positive-sense, enveloped, single-stranded RNA virus that belongs to the Flaviviridae family.1 There are 7 confirmed major genotypes of HCV and 67 confirmed subtypes.2 HCV possesses several important virulence factors. First, the virus's replication is prone to frequent mutations because its RNA polymerase lacks proofreading activity, resulting in significant genetic diversity. The great degree of heterogeneity among HCV leads to high antigenic variability, which is one of the main reasons there is not yet a vaccine for HCV.3 Additionally, HCV's genomic plasticity plays a role in the emergence of drug-resistant variants.4
Virus transmission. Worldwide, approximately 130 to 170 million people are infected with HCV.5 HCV infections are caused primarily by exposure to infected blood, through sharing needles for intravenous drug injection and through receiving a blood transfusion.6 Other routes of transmission include exposure through sexual contact, occupational injury, and perinatal acquisition.
The risk of acquiring HCV varies for each of these transmission mechanisms. Blood transfusion is no longer a common mechanism of transmission in places where blood donations are screened for HCV antibodies and viral RNA. Additionally, unintentional needle-stick injury is the only occupational risk factor associated with HCV infection, and health care workers do not have a greater prevalence of HCV than the general population. Moreover, sexual transmission is not a particularly efficient mechanism for spread of HCV.7 Therefore, unsafe intravenous injections are now the leading cause of HCV infection.6
Consequences of HCV infection. Once infected with HCV, about 25% of people spontaneously clear the virus and approximately 75% progress to chronic HCV infection.5 The consequences of long-term infection with HCV include end-stage liver disease, cirrhosis, and hepatocellular carcinoma.
Approximately 30% of people infected with HCV will develop cirrhosis and another 2% will develop hepatocellular carcinoma.8 Liver transplant is the only treatment option for patients with decompensated cirrhosis or hepatocellular carcinoma as a result of HCV infection. Currently, HCV infection is the leading indication for liver transplant in the United States.9
Continue to: Risk of perinatal HCV transmission...
Risk of perinatal HCV transmission
Approximately 1% to 8% of pregnant women worldwide are infected with HCV.10 In the United States, 1% to 2.5% of pregnant women are infected.11 Of these, about 6% transmit the infection to their offspring. The risk of HCV vertical transmission increases to about 11% if the mother is co-infected with HIV.12 Vertical transmission is the primary method by which children become infected with HCV.13
Several risk factors increase the likelihood of HCV transmission from mother to child, including HIV co-infection, internal fetal monitoring, and longer duration of membrane rupture.14 The effect that mode of delivery has on vertical transmission rates, however, is still debated, and a Cochrane Review found that there were no randomized controlled trials assessing the effect of mode of delivery on mother-to-infant HCV transmission.15
Serology and genotyping used in diagnosis
The serological enzyme immunoassay is the first test used in screening for HCV infection. Currently, third- and fourth-generation enzyme immunoassays are used in the United States.16 However, even these newer serological assays cannot consistently and precisely distinguish between acute and chronic HCV infections.17 After the initial diagnosis is made with serology, it usually is confirmed by assays that detect the virus's genomic RNA in the patient's serum or plasma.
The patient's HCV genotype should be identified so that the best treatment options can be determined. HCV genotyping can be accomplished using reverse transcription quantitative polymerase chain reaction (RT-qPCR) amplification. Three different RT-qPCR assessments usually are performed using different primers and probes specific to different genotypes of HCV. While direct sequencing of the HCV genome also can be performed, this method is usually not used clinically due to its technical complexity.16
Modern treatments are effective
Introduced in 2011, direct-acting antiviral therapies are now the recommended treatment for HCV infection. These drugs inhibit the virus's replication by targeting different proteins involved in the HCV replication cycle. They are remarkably successful and have achieved sustained virologic response (SVR) rates greater than 90%.11 The World Health Organization recommends several pangenotypic (that is, agents that work against all genotypes) direct-acting antiviral regimens for the treatment of chronic HCV infection in adults without cirrhosis (TABLE 1).18,19
Unfortunately, experience with these drugs in pregnant women is lacking. Many direct-acting antiviral agents have not been tested systematically in pregnant women, and, accordingly, most information about their effects in pregnant women comes from animal models.11
Continue to: Perinatal transmission rates and effect of mode of delivery...
Perinatal transmission rates and effect of mode of delivery
We compiled data from 11 studies that reported the perinatal transmission rate of HCV associated with various modes of delivery. These studies were selected from a MEDLINE literature review from 1999 to 2019. The studies were screened first by title and, subsequently, by abstract. Inclusion was restricted to randomized controlled trials, cohort studies, and case-control studies written in English. Study quality was assessed as good, fair, or poor based on the study design, sample size, and statistical analyses performed. The results from the total population of each study are reported in TABLE 2.14,20-29
Three studies separated data based on the mother's HIV status. The perinatal transmission rates of HCV for mothers co-infected with HIV are reported in TABLE 3.23,27 The results for HIV-negative mothers are reported in TABLE 4.14,23
Finally, 2 studies grouped mothers according to their HCV viral load. All of the mothers in these studies were anti-HCV antibody positive, and the perinatal transmission rates for the total study populations were reported previously in TABLE 2. The results for mothers who had detectable HCV RNA are reported in TABLE 5.20,21 High viral load was defined as
≥ 2.5 x 106 Eq/mL in the study by Okamoto and colleagues, which is equivalent to ≥ 6.0 x 105 IU/mL in the study by Murakami and colleagues due to the different assays that were used.20,21 The perinatal transmission rates for mothers with a high viral load are presented in TABLE 6.20,21
Continue to: For most, CD does not reduce HCV transmission...
For most, CD does not reduce HCV transmission
Nine of the 11 studies found that the mode of delivery did not have a statistically significant impact on the vertical transmission rate of HCV in the total study populations.14,22-29 The remaining 2 studies found that the perinatal transmission rate of HCV was lower with cesarean delivery (CD) than with vaginal delivery.20,21 When considered together, the results of these 11 studies indicate that CD does not provide a significant reduction in the HCV transmission rate in the general population.
Our review confirms the findings of others, including a systematic review by the US Preventive Services Task Force.30 That investigation also failed to demonstrate any measurable increase in risk of HCV transmission as a result of breastfeeding.
Cesarean delivery may benefit 2 groups. Careful assessment of these studies, however, suggests that 2 select groups of patients with HCV may benefit from CD:
- mothers co-infected with HIV, and
- mothers with high viral loads of HCV.
In both of these populations, the vertical transmission rate of HCV was significantly reduced with CD compared with vaginal delivery. Therefore, CD should be strongly considered in mothers with HCV who are co-infected with HIV and/or in mothers who have a high viral load of HCV.
CASE Our recommendation for mode of delivery
The patient in our case scenario has both HIV infection and a very high HCV viral load. We would therefore recommend a planned CD at 38 to 39 weeks' gestation, prior to the onset of labor or membrane rupture. Although HCV infection is not a contraindication to breastfeeding, the mother's HIV infection is a distinct contraindication.
- Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol. 2014;61(1 suppl):S3-S13.
- Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318-327.
- Rossi LM, Escobar-Gutierrez A, Rahal P. Advanced molecular surveillance of hepatitis C virus. Viruses. 2015;7:1153-1188.
- Dustin LB, Bartolini B, Capobianchi MR, et al. Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin Microbiol Infect. 2016;22:826-832.
- Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10:553-562.
- Thomas DL. Global elimination of chronic hepatitis. N Engl J Med. 2019;380:2041-2050.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep. 1998;47(RR19):1-39.
- Gonzalez-Grande R, Jimenez-Perez M, Gonzalez Arjona C, et al. New approaches in the treatment of hepatitis C. World J Gastroenterol. 2016;22:1421-1432.
- Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61(1 suppl): S58-S68.
- Spera AM, Eldin TK, Tosone G, et al. Antiviral therapy for hepatitis C: has anything changed for pregnant/lactating women? World J Hepatol. 2016;8:557-565.
- Society for Maternal-Fetal Medicine; Hughes BL, Page CM, Kuller JA. Hepatitis C in pregnancy: screening, treatment, and management. Am J Obstet Gynecol. 2017;217:B2-B12.
- Benova L, Mohamoud YA, Calvert C, et al. Vertical transmission of hepatitis C virus: systematic review and meta-analysis. Clin Infect Dis. 2014;59:765-773.
- Ghamar Chehreh ME, Tabatabaei SV, Khazanehdari S, et al. Effect of cesarean section on the risk of perinatal transmission of hepatitis C virus from HCV-RNA+/HIV- mothers: a meta-analysis. Arch Gynecol Obstet. 2011;283:255-260.
- Mast EE, Hwang LY, Seto DS, et al. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J Infect Dis. 2005;192:1880-1889.
- McIntyre PG, Tosh K, McGuire W. Caesarean section versus vaginal delivery for preventing mother to infant hepatitis C virus transmission. Cochrane Database Syst Rev. 2006;(4):CD005546.
- Mukherjee R, Burns A, Rodden D, et al. Diagnosis and management of hepatitis C virus infection. J Lab Autom. 2015;20:519-538.
- Araujo AC, Astrakhantseva IV, Fields HA, et al. Distinguishing acute from chronic hepatitis C virus (HCV) infection based on antibody reactivities to specific HCV structural and nonstructural proteins. J Clin Microbiol. 2011;49:54-57.
- World Health Organization. Guidelines for the Care and Treatment of Persons Diagnosed with Chronic Hepatitis C Virus Infection. Geneva, Switzerland: World Health Organization; 2018.
- CADTH Common Drug Review. Pharmacoeconomic Review Report: Sofosbuvir/Velpatasvir/Voxilaprevir (Vosevi) (Gilead Sciences Canada, Inc): Indication: Hepatitis C infection genotype 1 to 6. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2018.
- Okamoto M, Nagata I, Murakami J, et al. Prospective reevaluation of risk factors in mother-to-child transmission of hepatitis C virus: high virus load, vaginal delivery, and negative anti-NS4 antibody. J Infect Dis. 2000;182:1511-1514.
- Murakami J, Nagata I, Iitsuka T, et al. Risk factors for mother-to-child transmission of hepatitis C virus: maternal high viral load and fetal exposure in the birth canal. Hepatol Res. 2012;42:648-657.
- Conte D, Fraquelli M, Prati D, et al. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology. 2000;31:751-755.
- European Paediatric Hepatitis C Virus Network. Effects of mode of delivery and infant feeding on the risk of mother-to-child transmission of hepatitis C virus. BJOG. 2001;108:371-377.
- Tajiri H, Miyoshi Y, Funada S, et al. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J. 2001;20:10-14.
- Ferrero S, Lungaro P, Bruzzone BM, et al. Prospective study of mother-to-infant transmission of hepatitis C virus: a 10-year survey (1990-2000). Acta Obstet Gynecol Scand. 2003;82:229-234.
- Marine-Barjoan E, Berrebi A, Giordanengo V, et al. HCV/HIV co-infection, HCV viral load and mode of delivery: risk factors for mother-to-child transmission of hepatitis C virus? AIDS. 2007;21:1811-1815.
- Delotte J, Barjoan EM, Berrebi A, et al. Obstetric management does not influence vertical transmission of HCV infection: results of the ALHICE group study. J Matern Fetal Neonatal Med. 2014;27:664-670.
- Garcia-Tejedor A, Maiques-Montesinos V, Diago-Almela VJ, et al. Risk factors for vertical transmission of hepatitis C virus: a single center experience with 710 HCV-infected mothers. Eur J Obstet Gynecol Reprod Biol. 2015;194:173-177.
- Jhaveri R, Hashem M, El-Kamary SS, et al. Hepatitis C virus (HCV) vertical transmission in 12-month-old infants born to HCV-infected women and assessment of maternal risk factors. Open Forum Infect Dis. 2015;2:ofv089.
- Cottrell EB, Chou R, Wasson N, et al. Reducing risk for mother-to-infant transmission of hepatitis C virus: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2013;158:109-113.
- Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol. 2014;61(1 suppl):S3-S13.
- Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318-327.
- Rossi LM, Escobar-Gutierrez A, Rahal P. Advanced molecular surveillance of hepatitis C virus. Viruses. 2015;7:1153-1188.
- Dustin LB, Bartolini B, Capobianchi MR, et al. Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin Microbiol Infect. 2016;22:826-832.
- Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10:553-562.
- Thomas DL. Global elimination of chronic hepatitis. N Engl J Med. 2019;380:2041-2050.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep. 1998;47(RR19):1-39.
- Gonzalez-Grande R, Jimenez-Perez M, Gonzalez Arjona C, et al. New approaches in the treatment of hepatitis C. World J Gastroenterol. 2016;22:1421-1432.
- Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61(1 suppl): S58-S68.
- Spera AM, Eldin TK, Tosone G, et al. Antiviral therapy for hepatitis C: has anything changed for pregnant/lactating women? World J Hepatol. 2016;8:557-565.
- Society for Maternal-Fetal Medicine; Hughes BL, Page CM, Kuller JA. Hepatitis C in pregnancy: screening, treatment, and management. Am J Obstet Gynecol. 2017;217:B2-B12.
- Benova L, Mohamoud YA, Calvert C, et al. Vertical transmission of hepatitis C virus: systematic review and meta-analysis. Clin Infect Dis. 2014;59:765-773.
- Ghamar Chehreh ME, Tabatabaei SV, Khazanehdari S, et al. Effect of cesarean section on the risk of perinatal transmission of hepatitis C virus from HCV-RNA+/HIV- mothers: a meta-analysis. Arch Gynecol Obstet. 2011;283:255-260.
- Mast EE, Hwang LY, Seto DS, et al. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J Infect Dis. 2005;192:1880-1889.
- McIntyre PG, Tosh K, McGuire W. Caesarean section versus vaginal delivery for preventing mother to infant hepatitis C virus transmission. Cochrane Database Syst Rev. 2006;(4):CD005546.
- Mukherjee R, Burns A, Rodden D, et al. Diagnosis and management of hepatitis C virus infection. J Lab Autom. 2015;20:519-538.
- Araujo AC, Astrakhantseva IV, Fields HA, et al. Distinguishing acute from chronic hepatitis C virus (HCV) infection based on antibody reactivities to specific HCV structural and nonstructural proteins. J Clin Microbiol. 2011;49:54-57.
- World Health Organization. Guidelines for the Care and Treatment of Persons Diagnosed with Chronic Hepatitis C Virus Infection. Geneva, Switzerland: World Health Organization; 2018.
- CADTH Common Drug Review. Pharmacoeconomic Review Report: Sofosbuvir/Velpatasvir/Voxilaprevir (Vosevi) (Gilead Sciences Canada, Inc): Indication: Hepatitis C infection genotype 1 to 6. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2018.
- Okamoto M, Nagata I, Murakami J, et al. Prospective reevaluation of risk factors in mother-to-child transmission of hepatitis C virus: high virus load, vaginal delivery, and negative anti-NS4 antibody. J Infect Dis. 2000;182:1511-1514.
- Murakami J, Nagata I, Iitsuka T, et al. Risk factors for mother-to-child transmission of hepatitis C virus: maternal high viral load and fetal exposure in the birth canal. Hepatol Res. 2012;42:648-657.
- Conte D, Fraquelli M, Prati D, et al. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology. 2000;31:751-755.
- European Paediatric Hepatitis C Virus Network. Effects of mode of delivery and infant feeding on the risk of mother-to-child transmission of hepatitis C virus. BJOG. 2001;108:371-377.
- Tajiri H, Miyoshi Y, Funada S, et al. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J. 2001;20:10-14.
- Ferrero S, Lungaro P, Bruzzone BM, et al. Prospective study of mother-to-infant transmission of hepatitis C virus: a 10-year survey (1990-2000). Acta Obstet Gynecol Scand. 2003;82:229-234.
- Marine-Barjoan E, Berrebi A, Giordanengo V, et al. HCV/HIV co-infection, HCV viral load and mode of delivery: risk factors for mother-to-child transmission of hepatitis C virus? AIDS. 2007;21:1811-1815.
- Delotte J, Barjoan EM, Berrebi A, et al. Obstetric management does not influence vertical transmission of HCV infection: results of the ALHICE group study. J Matern Fetal Neonatal Med. 2014;27:664-670.
- Garcia-Tejedor A, Maiques-Montesinos V, Diago-Almela VJ, et al. Risk factors for vertical transmission of hepatitis C virus: a single center experience with 710 HCV-infected mothers. Eur J Obstet Gynecol Reprod Biol. 2015;194:173-177.
- Jhaveri R, Hashem M, El-Kamary SS, et al. Hepatitis C virus (HCV) vertical transmission in 12-month-old infants born to HCV-infected women and assessment of maternal risk factors. Open Forum Infect Dis. 2015;2:ofv089.
- Cottrell EB, Chou R, Wasson N, et al. Reducing risk for mother-to-infant transmission of hepatitis C virus: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2013;158:109-113.
Head & neck cancers: What you’ll see, how to proceed
The statistics reveal a serious problem: This year, an estimated 63,030 Americans will be given a diagnosis of head and neck cancer (which includes laryngeal, oropharyngeal, sinonasal, nasopharyngeal, and salivary gland cancer1); approximately 13,360 of them will die. Furthermore, thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an estimated 56,870 cases in 2017.1,2 Major risk factors for head and neck cancer are tobacco and alcohol exposure and infection with Epstein-Barr virus and human papillomavirus (HPV).3
In this article, we review the background for each of the principal types of head and neck cancer with which you should be familiar. We also discuss how to evaluate signs and symptoms that raise suspicion of these neoplasms; outline the diagnostic strategy in the face of such suspicion; and summarize accepted therapeutic approaches. Last, we describe the important role that you, the family physician, play in providing posttreatment care for these patients, especially prevention and management of late adverse effects of radiation therapy.
General characterizationsof these cancers
Approximately one-half of patients with head and neck cancer present initially with a nonspecific, persistent neck mass that should be deemed malignant until proven otherwise, because a delay in diagnosis is associated with a worse outcome.4 In a series of 100 patients with head and neck cancer, for example, delay in diagnosis occurred in nearly 25%—most often because of time spent providing inappropriate antibiotic treatment.5 Guidelines for management of neck masses recommend against the use of antibiotics in patients who do not have evidence of infection.6
Patients with a neck mass that has been present for longer than 2 weeks or that is ulcerated, fixed to underlying tissues, of firm consistency, or > 1.5 cm should have a physical examination that includes visualization of the base of tongue, pharynx, and larynx. The mass should be evaluated with fine-needle aspiration (FNA) biopsy, which has a positive predictive value of 96% and negative predictive value of 90% for the diagnosis of a head and neck mass. (Note: Anticoagulation therapy is not an absolute contraindication to FNA, which is not associated with an increased risk of bleeding.6)
Laryngeal cancer
What you need to know. More than 90% of laryngeal cancers are squamous cell carcinoma (SCC). Smoking or heavy drinking (> 8 drinks/d), compared to neither behavior, is associated with an increased risk of laryngeal cancer (odds ratio, 9.4 and 2.5, respectively).7 The risk of cancer is directly proportional to the degree of tobacco exposure.
Laryngeal cancer occurs in the supraglottic region in one-third of patients; in the glottic region in one-half; and in the subglottic region in a very few.8 Glottic cancer presents earlier than supraglottic cancer with hoarseness, whereas supraglottic cancer presents with more advanced disease, causing stridor, dysphagia, and throat pain. (Note: Guidelines recommend against prescribing acid suppressants in patients with hoarseness who do not have symptoms of reflux.9)
Stage 1 and Stage 2 laryngeal cancers are localized; Stages 3-4B are locally advanced or involve lymph nodes, or both; Stage 4C is metastatic disease. Overall, 60% of patients have Stage 3 or Stage 4 disease at diagnosis.10
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Laryngoscopy should be performed before computed tomography (CT) or magnetic resonance imaging is considered in a patient with hoarseness that does not resolve after 3 months—or sooner, if there is suspicion of malignancy.
How is it treated? Most patients presenting with Stage 1 or Stage 2 cancer can be treated with local radiation or, less commonly, larynx-preserving surgery. Patients with Stage 3 or Stage 4 disease can be treated with a combination of radiation and chemotherapy, which, compared to radiation alone, confers a decreased risk of local recurrence and increased laryngectomy-free survival.11 Patients whose vocal cords are destroyed or who have recurrence following radiation and chemotherapy might need total laryngectomy and formation of a tracheostomy and prosthetic for voice creation.
Five-year overall survival for Stage 1 and Stage 2 supraglottic and glottic cancers is 80%—lower, however, for later-presenting subglottic cancers.12
Oropharyngeal cancer
What you need to know. The lifetime risk for cancer of the oropharynx is approximately 1%.13 SCC is responsible for approximately 90% of these cancers. Early detection is important: The 5-year survival rate is more than twice as high for localized disease (83%) than it is for metastatic disease (39%) at detection.13
At any given time, 7% of the US population has HPV infection of the oropharynx. Most of these cases clear spontaneously, but persistent high-risk HPV infection led to a 225% increase in HPV-positive oropharyngeal SCC from 1988 to 2004.14 The representative case of HPV-positive oropharyngeal SCC is a middle-aged (40- to 59-year-old) white male with a history of multiple sexual partners and with little or no tobacco exposure and low alcohol consumption.
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Oral cancers present with a lesion, often ulcerative, that should be examined by palpation with a gloved finger to describe the presence, color, and number of lesions; any tenderness; tissue consistency (soft, firm, hard); and fixation to underlying structures.15 The oropharynx should be examined without protrusion of the tongue, which obscures the oropharynx and can make it harder to depress the posterior part of the tongue.
A finding of leukoplakia (white plaques) and erythroplakia (red plaques) of the oropharynx might reflect benign hyperkeratosis or premalignant lesions; the plaques do not wipe off on examination. Referral to a dentist or otorhinolaryngologist for biopsy is indicated for all erythroplakia and leukoplakia, and for ulcers that persist longer than 2 weeks.16
(Note: Evidence is insufficient to support screening asymptomatic patients for oral and oropharyngeal cancers by physical examination. There is no US Food and Drug Administration-approved screening test for oral HPV infection.17)
How is it treated? A diagnosis of moderate dysplasia or carcinoma in situ should be treated with surgical excision to clear margins followed by routine monitoring every 3 to 6 months, for life.18 Topical medication, electrocautery, laser ablation, and cryosurgery are management options for less severe dysplasia.
Sinonasal cancer
What you need to know. Worldwide, sinonasal cancer accounts for approximately 0.7% of all new cancers but demonstrates strong genetic and regional associations, particularly among the Cantonese population of southern China.19 One-half of new sinonasal malignancies are SCC; the rest are adenocarcinoma, lymphoepithelial carcinoma, and rare subtypes.20
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Presentation tends to mimic common, nonmalignant conditions, such as sinusitis, until invasion into adjacent structures. When sinonasal passages are involved, the history might include epistaxis or nasal discharge; facial or dental pain; unilateral nasal obstruction with unexplained onset later in life; and failure to respond to treatment of presumed rhinosinusitis. Physical examination should include assessment of cranial nerves, palpation of the sinuses, and anterior rhinoscopy.
Thin-cut CT of the paranasal sinuses is the first-line imaging study. Sinonasal endoscopy, with targeted biopsy of suspicious lesions, is the evaluation of choice when malignancy is suspected.
How is it treated? Surgery is the treatment of choice, with postoperative radiation for patients at higher risk of recurrence because of more extensive disase.12 Five-year survival for advanced disease is poor (35%); only 15% of cases are diagnosed at a localized stage because presenting symptoms are nonspecific.21
Nasopharyngeal cancer
What you need to know. Nasopharyngeal cancer is rare in the United States and Europe, compared with China, where it is endemic (and where a variety of risk factors, including intake of salt-preserved fish, have been proposed22). Epstein-Barr virus infection and a history of smoking increase the risk.
Patients with nasopharyngeal cancer can present with epistaxis, nasal obstruction, and auditory symptoms, such as serous otitis media. Direct extension of the tumor can lead to cranial-nerve palsy, most commonly III, V, VI, and XII.23
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Three-quarters of patients present with a neck mass from lymph-node metastases. Patients with the risk factors for nasopharyngeal cancer noted above who present with concerning symptoms should have nasoendoscopy with biopsy.
How is it treated? Radiation is the primary treatment, which is combined with chemotherapy for more advanced disease.23 Screening high-risk populations for antibodies to Epstein-Barr virus and performing nasopharyngeal endoscopy on patients who screen positive increases the detection rate of nasopharyngeal cancer; however, this strategy has not been shown to improve survival.9
Salivary gland tumors
What you need to know. Salivary gland neoplasms are a rare and heterogeneous entity, comprising 6% to 8% of head and neck cancers.24 More than 70% of these tumors are located in the parotid gland; 8%, in the submandibular glands; 1%, in the sublingual glands; and the rest, in the minor salivary glands. Most salivary gland tumors are benign; the most prevalent malignant tumors are mucoepidermoid carcinoma (30%) and adenoid cystic carcinoma (10%).25 Additional identified risk factors for a salivary gland tumor include irradiation, prior head and neck cancer, and environmental exposures, including hairdressing, rubber manufacturing, and exposure to nickel compounds.26
What is the diagnostic strategy? The history and physical exam are essential to distinguish a salivary gland tumor from an infectious cause and sialolithiasis. Parotid tumors most commonly present as asymptomatic parotid swelling, although pain can be present in as many as 40% of malignant parotid tumors.25 Facial nerve weakness is found in 25% of parotid tumors; although the differential diagnosis of facial nerve palsy is broad, suspicion of malignancy should be raised in the presence of a parotid mass, progressive unilateral symptoms, hemifacial spasm progressing to weakness, and a history of skin cancer on the face or scalp. Additional characteristics that favor a neoplastic cause are trismus and nontender lymphadenopathy.25
In contrast, sialolithiasis is associated with intermittent pain caused by eating and is more common in the settings of dehydration and poor dental hygiene. Sialadenitis should be suspected when the presentation is fever, increased pain and swelling, erythema, and expression of pus from the salivary gland.
Continue to: If malignancy is suspected...
If malignancy is suspected, the initial diagnostic evaluation should include ultrasonography (US); concurrent FNA biopsy should be performed if a mass is detected.27 US-guided FNA has a sensitivity of 73% to 86% for salivary neoplasm.7 CT and magnetic resonance imaging are useful for further characterization of tumors and can be advantageous for surgical planning.
How is it treated? Treatment of a salivary gland tumor involves surgical resection, followed by radiotherapy for patients in whom disease is more extensive or who exhibit high-risk pathology. Primary radiotherapy can be used in patients with an unresectable tumor. Typically, chemotherapy is used only for palliative purposes in relapsing disease, when a tumor is not amenable to radiotherapy, and in metastatic disease.25
Prognosis varies by histotype but is generally favorable. The survival rates for a malignant salivary gland tumor are 83% at 1 year, 69% at 3 years, and 65% at 5 years.28 Distant metastases are the most common cause of death, occurring primarily in the lungs (80%), bone (15%), and liver.27 Factors that indicate poor prognosis include facial nerve involvement, trismus, a tumor > 4 cm, bone involvement, nodal spread, and recurrence.25
Thyroid cancer
What you need to know. Thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an annual incidence of 4.5%.1 In the United States, most thyroid cancers are differentiated thyroid cancer (DTC), which includes papillary and follicular cancers. Less-differentiated medullary thyroid cancer (MTC), typically associated with multiple endocrine neoplasia (MEN) 2A or 2B, and undifferentiated or anaplastic thyroid cancer are less common. The increasing incidence of thyroid cancer is primarily the result of an increase in nonclinically relevant DTC.
What is the diagnostic strategy? Thyroid cancer usually presents as a thyroid nodule found by the patient or incidentally on physical examination or imaging. Other presenting signs and symptoms include hoarseness, voice changes, and dysphagia.
Continue to: Thyroid US is the study of...
Thyroid US is the study of choice for initial evaluation of the size and features of a nodule; findings are used to make recommendations for further workup. If further evaluation is indicated, FNA biopsy is the test of choice.29
In 2016, the American Thyroid Association released updated guidelines for evaluating thyroid nodules (TABLE).30 The US Preventive Services Task Force recommends against screening for thyroid cancer by neck palpation or US in asymptomatic patients because evidence of significant mortality benefit is lacking.31

How is it treated? Treatment of thyroid cancer focuses on local excision of the nodule by partial or total thyroidectomy (depending on the size and type of cancer) and surgical removal of involved lymph nodes. Differentiated thyroid cancer is categorized as high-, medium-, or low-risk, depending on tumor extension, incomplete tumor resection, size of lymph nodes > 3 cm, and distant metastases. Adjuvant treatment with radioactive iodine can be considered for intermediate-risk DTC and is recommended for high-risk DTC.32
Following surgical treatment, thyroid-stimulating hormone suppression is recommended using levothyroxine.33 Patients at higher risk of recurrence should have longer and more intense suppression of thyroid-stimulating hormone.30 Levels of serum thyroglobulin and anti-thyroglobulin antibody should be followed postoperatively; rising values can indicate recurrent disease. The calcitonin level should be followed in patients with a history of MTC. Thyroid US should be performed 6 to 12 months postoperatively, then periodically, depending on determination of recurrence risk and any change in the thyroglobulin level.30
(Note: Glucagon-like peptide-1 [GLP-1] receptor agonists, used to treat type 2 diabetes mellitus, carry a black-box warning for their risk of MTC and are contraindicated in patients who have a personal or family history of MTC, MEN2A, or MEN2B.34)
Continue to: Anaplastic thyroid cancer...
Anaplastic thyroid cancer, a rare form of thyroid cancer, carries a high mortality rate, with a median survival of 5 months from diagnosis and 1-year survival of 20%. Patients require expeditious total thyroidectomy and neck dissection, followed by external-beam radiation with or without chemotherapy. If this strategy is not feasible, tracheostomy might be necessary to maintain a patent airway.2 Family physicians treating a patient who has anaplastic thyroid cancer can fulfill a crucial role by ensuring that an advance directive is established, a surrogate decision-maker is appointed, and goals of care are well defined.
Follow-up care for head and neck Ca
The risk of adverse effects after radiation therapy for head and neck cancer calls for close monitoring, appropriate treatment, and referral and counseling as needed. See “Follow-up care after treatment of head and neck cancer.” 35-39
SIDEBAR
Follow-up care after treatment of head and neck cancer35-39
Challenge: After radiation to the head and neck, as many as 53% of patients develop subclinical hypothyroidism and 33% develop clinical hypothyroidism.35Strategy: Measure the thyroid-stimulating hormone level within 1 year of the completion of radiotherapy and every 6 to 12 months thereafter.36
Challenge: Radiation to the head and neck can decrease the function of salivary glands, causing xerostomia in as many as 40% of patients. This condition can lead to problems with oral hygiene and difficulty with speech, eating, and swallowing.37Strategy:
- Treat xerostomia with artificial saliva, sugar-free candy and gum, or muscarinic cholinergic agonists, such as pilocarpine and cevimeline.
- Consider treatment with pilocarpine or cevimeline. Pilocarpine alleviates xerostomia in approximately 50% of patients who develop the condition, although its use can be limited by adverse cholinergic effects.3,7 Cevimeline causes fewer and less pronounced adverse effects than pilocarpine because it acts more specifically on receptors in the salivary glands.38
- Mention the possibility of acupuncture to your patients. There is evidence that it can stimulate salivary flow.39
Challenge: Patients who have had radiation to the head and neck have an increased risk of dental caries from xerostomia and the direct effect of radiation, which causes demineralization of teeth.
Strategy: Following radiation, instruct the patient about appropriate oral hygiene:
- regular flossing
- brushing and application of daily fluoride
- regular visits for dental care.39
Challenge: Trismus occurs in 5% to 25% of patients, depending on the type of radiation.36Strategy: Recommend exercise-based treatment, the treatment of choice. Surgery is indicated for severe cases.
Challenge: Dysphagia occurs in approximately 25% of patients treated with radiation.36Strategy: Provide a referral for swallowing exercises, which might be helpful. Some cases are severe enough to warrant placement of a feeding tube.37
Last, counsel all patients who have been treated for cancer of the head or neck, with any modality, about cessation of smoking and alcohol.
CORRESPONDENCE
Anne Mounsey, MD, Family Medicine Residency, The University of North Carolina at Chapel Hill, 590 Manning Dr., Chapel Hill, NC 27599; [email protected]
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
2. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104-1139.
3. Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489-501.
4. Seoane J, Alvarez-Novoa P, Gomez I, et al. Early oral cancer diagnosis: The Aarhus statement perspective. A systematic review and meta-analysis. Head Neck. 2016;38(suppl 1):E2182-E2189.
5. Franco J, Elghouche AN, Harris MS, et al Diagnostic delays and errors in head and neck cancer patients: opportunities for improvement. Am J Med Qual. 2017;32:330-335.
6. Pynnonen MA, Gillespie MB, Roman B, et al. Clinical practice guideline: evaluation of the neck mass in adults. Otolaryngol Head Neck Surg. 2017;157(suppl 2):S1-S30.
7. Bosetti C, Gallus S, Franceschi S, et al. Cancer of the larynx in non-smoking alcohol drinkers and in non-drinking tobacco smokers. Br J Cancer. 2002;87:516-518.
8. Hoffman HT, Porter K, Karnell LH, et al. Laryngeal cancer in the United States: changes in demographics, patterns of care, and survival. Laryngoscope. 2006;116(9 pt 2 suppl 111):1-13.
9. Schwartz SR, Cohen SM, Dailey SH, et al. Clinical practice guideline: hoarseness (dysphonia). Otolaryngol Head Neck Surg. 2009;141(3 suppl 2):S1-S31.
10. Steuer CE, El-Deiry M, Parks JR, et al. An update on larynx cancer. CA Cancer J Clin. 2017;67:31-50.
11. Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349:2091-2098.
12. Mendenhall WM, Werning JW, Hinerman RW, et al. Management of T1-T2 glottic carcinomas. Cancer. 2004;100:1786-1792.
13. Surveillance, Epidemiology, and End Results Unit. National Cancer Institute. Cancer stat facts: oral cavity and pharynx. https://seer.cancer.gov/statfacts/html/oralcav.html. Accessed October 18, 2019.
14. Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014;50:380-386.
15. Tarakji B, Gazal G, Al-Maweri SA, et al. Guideline for the diagnosis and treatment of recurrent aphthous stomatitis for dental practitioners. J Int Oral Health. 2015;7:74-80.
16. Siu A, Landon K, Ramos DM. Differential diagnosis and management of oral ulcers. Semin Cutan Med Surg. 2015;34:171-177.
17. US Preventive Services Task Force. Final recommendation statement: oral cancer: screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/oral-cancer-screening1. Updated November 2013. Accessed October 18, 2019.
18. Villa A, Woo SB. Leukoplakia—a diagnostic and management algorithm. J Oral Maxillofac Surg. 2017;75:723-734.
19. Yang S, Wu S, Zhou J, et al. Screening for nasopharyngeal cancer. Cochrane Database Syst Rev. 2015;(11):CD008423.
20. Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: a historical analysis of population-based data. Head Neck. 2012;34:877-885.
21. Ou SH, Zell JA, Ziogas A, et al. Epidemiology of nasopharyngeal carcinoma in the United States: improved survival of Chinese patients within the keratinizing squamous cell carcinoma histology. Ann Oncol. 2007;18:29-35.
22. Chang ET, Adami H-O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765-1777.
23. Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma. Lancet. 2016;387:1012-1024.
24. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg. 1986;8:177-184.
25. Lewis JS. Sinonasal squamous cell carcinoma: a review with emphasis on emerging histologic subtypes and the role of human papillomavirus. Head Neck Pathol. 2016;10:60-67.
26. Horn-Ross PL, Ljung BM, Morrow M. Environmental factors and the risk of salivary gland cancer. Epidemiology. 1997;8:414-419.
27. Colella G, Cannavale R, Flamminio F, et al. Fine-needle aspiration cytology of salivary gland lesions: a systematic review. J Oral Maxillofac Surg. 2010;68:2146-2153.
28. Berrino F, De Angelis R, Sant M, et al; EUROCARE Working Group. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995-99: results of the EUROCARE-4 study. Lancet Oncol. 2007;8:773-783.
29. Baloch ZW, LiVolsi VA, Asa SL, et al. Diagnostic terminology and morphologic criteria for cytologic diagnosis of thyroid lesions: a synopsis of the National Cancer Institute Thyroid Fine-Needle Aspiration State of the Science Conference. Diagn Cytopathol. 2008;36:425-437.
30. Haugen BR, Alexander EK, Bible KC, et al; The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133.
31. US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for thyroid Cancer: US Preventive Services Task Force recommendation statement. JAMA. 2017;317:1882-1887.
32. Jonklaas J, Cooper DS, Ain KB, et al; National Thyroid Cancer Treatment Cooperative Study Group. Radioiodine therapy in patients with stage I differentiated thyroid cancer. Thyroid. 2010;20:1423-1424.
33. Cooper DS, Specker B, Ho M, et al. Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. Thyroid. 1998;8:737-744.
34. US Food and Drug Administration. Highlight of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125431s020lbl.pdf. Updated December 2017. Accessed October 30, 1019.
35. Boomsma MJ, Bijl HP, Langendijk JA. Radiation-induced hypothyroidism in head and neck cancer patients: a systematic review. Radiother Oncol. 2011;99:1-5.
36. The development of quality of care measures for oral cavity cancer. Arch Otolaryngol Head Neck Surg. 2008;134:672.
37. Strojan P, Hutcheson KA, Eisbruch A, et al. Treatment of late sequelae after radiotherapy for head and neck cancer. Cancer Treat Rev. 2017;59:79-92.
38. Chambers MS, Posner M, Jones CU, et al. Cevimeline for the treatment of postirradiation xerostomia in patients with head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;68:1102-1109.
39. Gupta N, Pal M, Rawat S, et al. Radiation-induced dental caries, prevention and treatment - a systematic review. Natl J Maxillofac Surg. 2015;6:160-166.
The statistics reveal a serious problem: This year, an estimated 63,030 Americans will be given a diagnosis of head and neck cancer (which includes laryngeal, oropharyngeal, sinonasal, nasopharyngeal, and salivary gland cancer1); approximately 13,360 of them will die. Furthermore, thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an estimated 56,870 cases in 2017.1,2 Major risk factors for head and neck cancer are tobacco and alcohol exposure and infection with Epstein-Barr virus and human papillomavirus (HPV).3
In this article, we review the background for each of the principal types of head and neck cancer with which you should be familiar. We also discuss how to evaluate signs and symptoms that raise suspicion of these neoplasms; outline the diagnostic strategy in the face of such suspicion; and summarize accepted therapeutic approaches. Last, we describe the important role that you, the family physician, play in providing posttreatment care for these patients, especially prevention and management of late adverse effects of radiation therapy.
General characterizationsof these cancers
Approximately one-half of patients with head and neck cancer present initially with a nonspecific, persistent neck mass that should be deemed malignant until proven otherwise, because a delay in diagnosis is associated with a worse outcome.4 In a series of 100 patients with head and neck cancer, for example, delay in diagnosis occurred in nearly 25%—most often because of time spent providing inappropriate antibiotic treatment.5 Guidelines for management of neck masses recommend against the use of antibiotics in patients who do not have evidence of infection.6
Patients with a neck mass that has been present for longer than 2 weeks or that is ulcerated, fixed to underlying tissues, of firm consistency, or > 1.5 cm should have a physical examination that includes visualization of the base of tongue, pharynx, and larynx. The mass should be evaluated with fine-needle aspiration (FNA) biopsy, which has a positive predictive value of 96% and negative predictive value of 90% for the diagnosis of a head and neck mass. (Note: Anticoagulation therapy is not an absolute contraindication to FNA, which is not associated with an increased risk of bleeding.6)
Laryngeal cancer
What you need to know. More than 90% of laryngeal cancers are squamous cell carcinoma (SCC). Smoking or heavy drinking (> 8 drinks/d), compared to neither behavior, is associated with an increased risk of laryngeal cancer (odds ratio, 9.4 and 2.5, respectively).7 The risk of cancer is directly proportional to the degree of tobacco exposure.
Laryngeal cancer occurs in the supraglottic region in one-third of patients; in the glottic region in one-half; and in the subglottic region in a very few.8 Glottic cancer presents earlier than supraglottic cancer with hoarseness, whereas supraglottic cancer presents with more advanced disease, causing stridor, dysphagia, and throat pain. (Note: Guidelines recommend against prescribing acid suppressants in patients with hoarseness who do not have symptoms of reflux.9)
Stage 1 and Stage 2 laryngeal cancers are localized; Stages 3-4B are locally advanced or involve lymph nodes, or both; Stage 4C is metastatic disease. Overall, 60% of patients have Stage 3 or Stage 4 disease at diagnosis.10
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Laryngoscopy should be performed before computed tomography (CT) or magnetic resonance imaging is considered in a patient with hoarseness that does not resolve after 3 months—or sooner, if there is suspicion of malignancy.
How is it treated? Most patients presenting with Stage 1 or Stage 2 cancer can be treated with local radiation or, less commonly, larynx-preserving surgery. Patients with Stage 3 or Stage 4 disease can be treated with a combination of radiation and chemotherapy, which, compared to radiation alone, confers a decreased risk of local recurrence and increased laryngectomy-free survival.11 Patients whose vocal cords are destroyed or who have recurrence following radiation and chemotherapy might need total laryngectomy and formation of a tracheostomy and prosthetic for voice creation.
Five-year overall survival for Stage 1 and Stage 2 supraglottic and glottic cancers is 80%—lower, however, for later-presenting subglottic cancers.12
Oropharyngeal cancer
What you need to know. The lifetime risk for cancer of the oropharynx is approximately 1%.13 SCC is responsible for approximately 90% of these cancers. Early detection is important: The 5-year survival rate is more than twice as high for localized disease (83%) than it is for metastatic disease (39%) at detection.13
At any given time, 7% of the US population has HPV infection of the oropharynx. Most of these cases clear spontaneously, but persistent high-risk HPV infection led to a 225% increase in HPV-positive oropharyngeal SCC from 1988 to 2004.14 The representative case of HPV-positive oropharyngeal SCC is a middle-aged (40- to 59-year-old) white male with a history of multiple sexual partners and with little or no tobacco exposure and low alcohol consumption.
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Oral cancers present with a lesion, often ulcerative, that should be examined by palpation with a gloved finger to describe the presence, color, and number of lesions; any tenderness; tissue consistency (soft, firm, hard); and fixation to underlying structures.15 The oropharynx should be examined without protrusion of the tongue, which obscures the oropharynx and can make it harder to depress the posterior part of the tongue.
A finding of leukoplakia (white plaques) and erythroplakia (red plaques) of the oropharynx might reflect benign hyperkeratosis or premalignant lesions; the plaques do not wipe off on examination. Referral to a dentist or otorhinolaryngologist for biopsy is indicated for all erythroplakia and leukoplakia, and for ulcers that persist longer than 2 weeks.16
(Note: Evidence is insufficient to support screening asymptomatic patients for oral and oropharyngeal cancers by physical examination. There is no US Food and Drug Administration-approved screening test for oral HPV infection.17)
How is it treated? A diagnosis of moderate dysplasia or carcinoma in situ should be treated with surgical excision to clear margins followed by routine monitoring every 3 to 6 months, for life.18 Topical medication, electrocautery, laser ablation, and cryosurgery are management options for less severe dysplasia.
Sinonasal cancer
What you need to know. Worldwide, sinonasal cancer accounts for approximately 0.7% of all new cancers but demonstrates strong genetic and regional associations, particularly among the Cantonese population of southern China.19 One-half of new sinonasal malignancies are SCC; the rest are adenocarcinoma, lymphoepithelial carcinoma, and rare subtypes.20
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Presentation tends to mimic common, nonmalignant conditions, such as sinusitis, until invasion into adjacent structures. When sinonasal passages are involved, the history might include epistaxis or nasal discharge; facial or dental pain; unilateral nasal obstruction with unexplained onset later in life; and failure to respond to treatment of presumed rhinosinusitis. Physical examination should include assessment of cranial nerves, palpation of the sinuses, and anterior rhinoscopy.
Thin-cut CT of the paranasal sinuses is the first-line imaging study. Sinonasal endoscopy, with targeted biopsy of suspicious lesions, is the evaluation of choice when malignancy is suspected.
How is it treated? Surgery is the treatment of choice, with postoperative radiation for patients at higher risk of recurrence because of more extensive disase.12 Five-year survival for advanced disease is poor (35%); only 15% of cases are diagnosed at a localized stage because presenting symptoms are nonspecific.21
Nasopharyngeal cancer
What you need to know. Nasopharyngeal cancer is rare in the United States and Europe, compared with China, where it is endemic (and where a variety of risk factors, including intake of salt-preserved fish, have been proposed22). Epstein-Barr virus infection and a history of smoking increase the risk.
Patients with nasopharyngeal cancer can present with epistaxis, nasal obstruction, and auditory symptoms, such as serous otitis media. Direct extension of the tumor can lead to cranial-nerve palsy, most commonly III, V, VI, and XII.23
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Three-quarters of patients present with a neck mass from lymph-node metastases. Patients with the risk factors for nasopharyngeal cancer noted above who present with concerning symptoms should have nasoendoscopy with biopsy.
How is it treated? Radiation is the primary treatment, which is combined with chemotherapy for more advanced disease.23 Screening high-risk populations for antibodies to Epstein-Barr virus and performing nasopharyngeal endoscopy on patients who screen positive increases the detection rate of nasopharyngeal cancer; however, this strategy has not been shown to improve survival.9
Salivary gland tumors
What you need to know. Salivary gland neoplasms are a rare and heterogeneous entity, comprising 6% to 8% of head and neck cancers.24 More than 70% of these tumors are located in the parotid gland; 8%, in the submandibular glands; 1%, in the sublingual glands; and the rest, in the minor salivary glands. Most salivary gland tumors are benign; the most prevalent malignant tumors are mucoepidermoid carcinoma (30%) and adenoid cystic carcinoma (10%).25 Additional identified risk factors for a salivary gland tumor include irradiation, prior head and neck cancer, and environmental exposures, including hairdressing, rubber manufacturing, and exposure to nickel compounds.26
What is the diagnostic strategy? The history and physical exam are essential to distinguish a salivary gland tumor from an infectious cause and sialolithiasis. Parotid tumors most commonly present as asymptomatic parotid swelling, although pain can be present in as many as 40% of malignant parotid tumors.25 Facial nerve weakness is found in 25% of parotid tumors; although the differential diagnosis of facial nerve palsy is broad, suspicion of malignancy should be raised in the presence of a parotid mass, progressive unilateral symptoms, hemifacial spasm progressing to weakness, and a history of skin cancer on the face or scalp. Additional characteristics that favor a neoplastic cause are trismus and nontender lymphadenopathy.25
In contrast, sialolithiasis is associated with intermittent pain caused by eating and is more common in the settings of dehydration and poor dental hygiene. Sialadenitis should be suspected when the presentation is fever, increased pain and swelling, erythema, and expression of pus from the salivary gland.
Continue to: If malignancy is suspected...
If malignancy is suspected, the initial diagnostic evaluation should include ultrasonography (US); concurrent FNA biopsy should be performed if a mass is detected.27 US-guided FNA has a sensitivity of 73% to 86% for salivary neoplasm.7 CT and magnetic resonance imaging are useful for further characterization of tumors and can be advantageous for surgical planning.
How is it treated? Treatment of a salivary gland tumor involves surgical resection, followed by radiotherapy for patients in whom disease is more extensive or who exhibit high-risk pathology. Primary radiotherapy can be used in patients with an unresectable tumor. Typically, chemotherapy is used only for palliative purposes in relapsing disease, when a tumor is not amenable to radiotherapy, and in metastatic disease.25
Prognosis varies by histotype but is generally favorable. The survival rates for a malignant salivary gland tumor are 83% at 1 year, 69% at 3 years, and 65% at 5 years.28 Distant metastases are the most common cause of death, occurring primarily in the lungs (80%), bone (15%), and liver.27 Factors that indicate poor prognosis include facial nerve involvement, trismus, a tumor > 4 cm, bone involvement, nodal spread, and recurrence.25
Thyroid cancer
What you need to know. Thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an annual incidence of 4.5%.1 In the United States, most thyroid cancers are differentiated thyroid cancer (DTC), which includes papillary and follicular cancers. Less-differentiated medullary thyroid cancer (MTC), typically associated with multiple endocrine neoplasia (MEN) 2A or 2B, and undifferentiated or anaplastic thyroid cancer are less common. The increasing incidence of thyroid cancer is primarily the result of an increase in nonclinically relevant DTC.
What is the diagnostic strategy? Thyroid cancer usually presents as a thyroid nodule found by the patient or incidentally on physical examination or imaging. Other presenting signs and symptoms include hoarseness, voice changes, and dysphagia.
Continue to: Thyroid US is the study of...
Thyroid US is the study of choice for initial evaluation of the size and features of a nodule; findings are used to make recommendations for further workup. If further evaluation is indicated, FNA biopsy is the test of choice.29
In 2016, the American Thyroid Association released updated guidelines for evaluating thyroid nodules (TABLE).30 The US Preventive Services Task Force recommends against screening for thyroid cancer by neck palpation or US in asymptomatic patients because evidence of significant mortality benefit is lacking.31
How is it treated? Treatment of thyroid cancer focuses on local excision of the nodule by partial or total thyroidectomy (depending on the size and type of cancer) and surgical removal of involved lymph nodes. Differentiated thyroid cancer is categorized as high-, medium-, or low-risk, depending on tumor extension, incomplete tumor resection, size of lymph nodes > 3 cm, and distant metastases. Adjuvant treatment with radioactive iodine can be considered for intermediate-risk DTC and is recommended for high-risk DTC.32
Following surgical treatment, thyroid-stimulating hormone suppression is recommended using levothyroxine.33 Patients at higher risk of recurrence should have longer and more intense suppression of thyroid-stimulating hormone.30 Levels of serum thyroglobulin and anti-thyroglobulin antibody should be followed postoperatively; rising values can indicate recurrent disease. The calcitonin level should be followed in patients with a history of MTC. Thyroid US should be performed 6 to 12 months postoperatively, then periodically, depending on determination of recurrence risk and any change in the thyroglobulin level.30
(Note: Glucagon-like peptide-1 [GLP-1] receptor agonists, used to treat type 2 diabetes mellitus, carry a black-box warning for their risk of MTC and are contraindicated in patients who have a personal or family history of MTC, MEN2A, or MEN2B.34)
Continue to: Anaplastic thyroid cancer...
Anaplastic thyroid cancer, a rare form of thyroid cancer, carries a high mortality rate, with a median survival of 5 months from diagnosis and 1-year survival of 20%. Patients require expeditious total thyroidectomy and neck dissection, followed by external-beam radiation with or without chemotherapy. If this strategy is not feasible, tracheostomy might be necessary to maintain a patent airway.2 Family physicians treating a patient who has anaplastic thyroid cancer can fulfill a crucial role by ensuring that an advance directive is established, a surrogate decision-maker is appointed, and goals of care are well defined.
Follow-up care for head and neck Ca
The risk of adverse effects after radiation therapy for head and neck cancer calls for close monitoring, appropriate treatment, and referral and counseling as needed. See “Follow-up care after treatment of head and neck cancer.” 35-39
SIDEBAR
Follow-up care after treatment of head and neck cancer35-39
Challenge: After radiation to the head and neck, as many as 53% of patients develop subclinical hypothyroidism and 33% develop clinical hypothyroidism.35Strategy: Measure the thyroid-stimulating hormone level within 1 year of the completion of radiotherapy and every 6 to 12 months thereafter.36
Challenge: Radiation to the head and neck can decrease the function of salivary glands, causing xerostomia in as many as 40% of patients. This condition can lead to problems with oral hygiene and difficulty with speech, eating, and swallowing.37Strategy:
- Treat xerostomia with artificial saliva, sugar-free candy and gum, or muscarinic cholinergic agonists, such as pilocarpine and cevimeline.
- Consider treatment with pilocarpine or cevimeline. Pilocarpine alleviates xerostomia in approximately 50% of patients who develop the condition, although its use can be limited by adverse cholinergic effects.3,7 Cevimeline causes fewer and less pronounced adverse effects than pilocarpine because it acts more specifically on receptors in the salivary glands.38
- Mention the possibility of acupuncture to your patients. There is evidence that it can stimulate salivary flow.39
Challenge: Patients who have had radiation to the head and neck have an increased risk of dental caries from xerostomia and the direct effect of radiation, which causes demineralization of teeth.
Strategy: Following radiation, instruct the patient about appropriate oral hygiene:
- regular flossing
- brushing and application of daily fluoride
- regular visits for dental care.39
Challenge: Trismus occurs in 5% to 25% of patients, depending on the type of radiation.36Strategy: Recommend exercise-based treatment, the treatment of choice. Surgery is indicated for severe cases.
Challenge: Dysphagia occurs in approximately 25% of patients treated with radiation.36Strategy: Provide a referral for swallowing exercises, which might be helpful. Some cases are severe enough to warrant placement of a feeding tube.37
Last, counsel all patients who have been treated for cancer of the head or neck, with any modality, about cessation of smoking and alcohol.
CORRESPONDENCE
Anne Mounsey, MD, Family Medicine Residency, The University of North Carolina at Chapel Hill, 590 Manning Dr., Chapel Hill, NC 27599; [email protected]
The statistics reveal a serious problem: This year, an estimated 63,030 Americans will be given a diagnosis of head and neck cancer (which includes laryngeal, oropharyngeal, sinonasal, nasopharyngeal, and salivary gland cancer1); approximately 13,360 of them will die. Furthermore, thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an estimated 56,870 cases in 2017.1,2 Major risk factors for head and neck cancer are tobacco and alcohol exposure and infection with Epstein-Barr virus and human papillomavirus (HPV).3
In this article, we review the background for each of the principal types of head and neck cancer with which you should be familiar. We also discuss how to evaluate signs and symptoms that raise suspicion of these neoplasms; outline the diagnostic strategy in the face of such suspicion; and summarize accepted therapeutic approaches. Last, we describe the important role that you, the family physician, play in providing posttreatment care for these patients, especially prevention and management of late adverse effects of radiation therapy.
General characterizationsof these cancers
Approximately one-half of patients with head and neck cancer present initially with a nonspecific, persistent neck mass that should be deemed malignant until proven otherwise, because a delay in diagnosis is associated with a worse outcome.4 In a series of 100 patients with head and neck cancer, for example, delay in diagnosis occurred in nearly 25%—most often because of time spent providing inappropriate antibiotic treatment.5 Guidelines for management of neck masses recommend against the use of antibiotics in patients who do not have evidence of infection.6
Patients with a neck mass that has been present for longer than 2 weeks or that is ulcerated, fixed to underlying tissues, of firm consistency, or > 1.5 cm should have a physical examination that includes visualization of the base of tongue, pharynx, and larynx. The mass should be evaluated with fine-needle aspiration (FNA) biopsy, which has a positive predictive value of 96% and negative predictive value of 90% for the diagnosis of a head and neck mass. (Note: Anticoagulation therapy is not an absolute contraindication to FNA, which is not associated with an increased risk of bleeding.6)
Laryngeal cancer
What you need to know. More than 90% of laryngeal cancers are squamous cell carcinoma (SCC). Smoking or heavy drinking (> 8 drinks/d), compared to neither behavior, is associated with an increased risk of laryngeal cancer (odds ratio, 9.4 and 2.5, respectively).7 The risk of cancer is directly proportional to the degree of tobacco exposure.
Laryngeal cancer occurs in the supraglottic region in one-third of patients; in the glottic region in one-half; and in the subglottic region in a very few.8 Glottic cancer presents earlier than supraglottic cancer with hoarseness, whereas supraglottic cancer presents with more advanced disease, causing stridor, dysphagia, and throat pain. (Note: Guidelines recommend against prescribing acid suppressants in patients with hoarseness who do not have symptoms of reflux.9)
Stage 1 and Stage 2 laryngeal cancers are localized; Stages 3-4B are locally advanced or involve lymph nodes, or both; Stage 4C is metastatic disease. Overall, 60% of patients have Stage 3 or Stage 4 disease at diagnosis.10
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Laryngoscopy should be performed before computed tomography (CT) or magnetic resonance imaging is considered in a patient with hoarseness that does not resolve after 3 months—or sooner, if there is suspicion of malignancy.
How is it treated? Most patients presenting with Stage 1 or Stage 2 cancer can be treated with local radiation or, less commonly, larynx-preserving surgery. Patients with Stage 3 or Stage 4 disease can be treated with a combination of radiation and chemotherapy, which, compared to radiation alone, confers a decreased risk of local recurrence and increased laryngectomy-free survival.11 Patients whose vocal cords are destroyed or who have recurrence following radiation and chemotherapy might need total laryngectomy and formation of a tracheostomy and prosthetic for voice creation.
Five-year overall survival for Stage 1 and Stage 2 supraglottic and glottic cancers is 80%—lower, however, for later-presenting subglottic cancers.12
Oropharyngeal cancer
What you need to know. The lifetime risk for cancer of the oropharynx is approximately 1%.13 SCC is responsible for approximately 90% of these cancers. Early detection is important: The 5-year survival rate is more than twice as high for localized disease (83%) than it is for metastatic disease (39%) at detection.13
At any given time, 7% of the US population has HPV infection of the oropharynx. Most of these cases clear spontaneously, but persistent high-risk HPV infection led to a 225% increase in HPV-positive oropharyngeal SCC from 1988 to 2004.14 The representative case of HPV-positive oropharyngeal SCC is a middle-aged (40- to 59-year-old) white male with a history of multiple sexual partners and with little or no tobacco exposure and low alcohol consumption.
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Oral cancers present with a lesion, often ulcerative, that should be examined by palpation with a gloved finger to describe the presence, color, and number of lesions; any tenderness; tissue consistency (soft, firm, hard); and fixation to underlying structures.15 The oropharynx should be examined without protrusion of the tongue, which obscures the oropharynx and can make it harder to depress the posterior part of the tongue.
A finding of leukoplakia (white plaques) and erythroplakia (red plaques) of the oropharynx might reflect benign hyperkeratosis or premalignant lesions; the plaques do not wipe off on examination. Referral to a dentist or otorhinolaryngologist for biopsy is indicated for all erythroplakia and leukoplakia, and for ulcers that persist longer than 2 weeks.16
(Note: Evidence is insufficient to support screening asymptomatic patients for oral and oropharyngeal cancers by physical examination. There is no US Food and Drug Administration-approved screening test for oral HPV infection.17)
How is it treated? A diagnosis of moderate dysplasia or carcinoma in situ should be treated with surgical excision to clear margins followed by routine monitoring every 3 to 6 months, for life.18 Topical medication, electrocautery, laser ablation, and cryosurgery are management options for less severe dysplasia.
Sinonasal cancer
What you need to know. Worldwide, sinonasal cancer accounts for approximately 0.7% of all new cancers but demonstrates strong genetic and regional associations, particularly among the Cantonese population of southern China.19 One-half of new sinonasal malignancies are SCC; the rest are adenocarcinoma, lymphoepithelial carcinoma, and rare subtypes.20
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Presentation tends to mimic common, nonmalignant conditions, such as sinusitis, until invasion into adjacent structures. When sinonasal passages are involved, the history might include epistaxis or nasal discharge; facial or dental pain; unilateral nasal obstruction with unexplained onset later in life; and failure to respond to treatment of presumed rhinosinusitis. Physical examination should include assessment of cranial nerves, palpation of the sinuses, and anterior rhinoscopy.
Thin-cut CT of the paranasal sinuses is the first-line imaging study. Sinonasal endoscopy, with targeted biopsy of suspicious lesions, is the evaluation of choice when malignancy is suspected.
How is it treated? Surgery is the treatment of choice, with postoperative radiation for patients at higher risk of recurrence because of more extensive disase.12 Five-year survival for advanced disease is poor (35%); only 15% of cases are diagnosed at a localized stage because presenting symptoms are nonspecific.21
Nasopharyngeal cancer
What you need to know. Nasopharyngeal cancer is rare in the United States and Europe, compared with China, where it is endemic (and where a variety of risk factors, including intake of salt-preserved fish, have been proposed22). Epstein-Barr virus infection and a history of smoking increase the risk.
Patients with nasopharyngeal cancer can present with epistaxis, nasal obstruction, and auditory symptoms, such as serous otitis media. Direct extension of the tumor can lead to cranial-nerve palsy, most commonly III, V, VI, and XII.23
Continue to: What is the diagnostic strategy?
What is the diagnostic strategy? Three-quarters of patients present with a neck mass from lymph-node metastases. Patients with the risk factors for nasopharyngeal cancer noted above who present with concerning symptoms should have nasoendoscopy with biopsy.
How is it treated? Radiation is the primary treatment, which is combined with chemotherapy for more advanced disease.23 Screening high-risk populations for antibodies to Epstein-Barr virus and performing nasopharyngeal endoscopy on patients who screen positive increases the detection rate of nasopharyngeal cancer; however, this strategy has not been shown to improve survival.9
Salivary gland tumors
What you need to know. Salivary gland neoplasms are a rare and heterogeneous entity, comprising 6% to 8% of head and neck cancers.24 More than 70% of these tumors are located in the parotid gland; 8%, in the submandibular glands; 1%, in the sublingual glands; and the rest, in the minor salivary glands. Most salivary gland tumors are benign; the most prevalent malignant tumors are mucoepidermoid carcinoma (30%) and adenoid cystic carcinoma (10%).25 Additional identified risk factors for a salivary gland tumor include irradiation, prior head and neck cancer, and environmental exposures, including hairdressing, rubber manufacturing, and exposure to nickel compounds.26
What is the diagnostic strategy? The history and physical exam are essential to distinguish a salivary gland tumor from an infectious cause and sialolithiasis. Parotid tumors most commonly present as asymptomatic parotid swelling, although pain can be present in as many as 40% of malignant parotid tumors.25 Facial nerve weakness is found in 25% of parotid tumors; although the differential diagnosis of facial nerve palsy is broad, suspicion of malignancy should be raised in the presence of a parotid mass, progressive unilateral symptoms, hemifacial spasm progressing to weakness, and a history of skin cancer on the face or scalp. Additional characteristics that favor a neoplastic cause are trismus and nontender lymphadenopathy.25
In contrast, sialolithiasis is associated with intermittent pain caused by eating and is more common in the settings of dehydration and poor dental hygiene. Sialadenitis should be suspected when the presentation is fever, increased pain and swelling, erythema, and expression of pus from the salivary gland.
Continue to: If malignancy is suspected...
If malignancy is suspected, the initial diagnostic evaluation should include ultrasonography (US); concurrent FNA biopsy should be performed if a mass is detected.27 US-guided FNA has a sensitivity of 73% to 86% for salivary neoplasm.7 CT and magnetic resonance imaging are useful for further characterization of tumors and can be advantageous for surgical planning.
How is it treated? Treatment of a salivary gland tumor involves surgical resection, followed by radiotherapy for patients in whom disease is more extensive or who exhibit high-risk pathology. Primary radiotherapy can be used in patients with an unresectable tumor. Typically, chemotherapy is used only for palliative purposes in relapsing disease, when a tumor is not amenable to radiotherapy, and in metastatic disease.25
Prognosis varies by histotype but is generally favorable. The survival rates for a malignant salivary gland tumor are 83% at 1 year, 69% at 3 years, and 65% at 5 years.28 Distant metastases are the most common cause of death, occurring primarily in the lungs (80%), bone (15%), and liver.27 Factors that indicate poor prognosis include facial nerve involvement, trismus, a tumor > 4 cm, bone involvement, nodal spread, and recurrence.25
Thyroid cancer
What you need to know. Thyroid cancer is the most rapidly increasing cancer diagnosis in the United States, with an annual incidence of 4.5%.1 In the United States, most thyroid cancers are differentiated thyroid cancer (DTC), which includes papillary and follicular cancers. Less-differentiated medullary thyroid cancer (MTC), typically associated with multiple endocrine neoplasia (MEN) 2A or 2B, and undifferentiated or anaplastic thyroid cancer are less common. The increasing incidence of thyroid cancer is primarily the result of an increase in nonclinically relevant DTC.
What is the diagnostic strategy? Thyroid cancer usually presents as a thyroid nodule found by the patient or incidentally on physical examination or imaging. Other presenting signs and symptoms include hoarseness, voice changes, and dysphagia.
Continue to: Thyroid US is the study of...
Thyroid US is the study of choice for initial evaluation of the size and features of a nodule; findings are used to make recommendations for further workup. If further evaluation is indicated, FNA biopsy is the test of choice.29
In 2016, the American Thyroid Association released updated guidelines for evaluating thyroid nodules (TABLE).30 The US Preventive Services Task Force recommends against screening for thyroid cancer by neck palpation or US in asymptomatic patients because evidence of significant mortality benefit is lacking.31
How is it treated? Treatment of thyroid cancer focuses on local excision of the nodule by partial or total thyroidectomy (depending on the size and type of cancer) and surgical removal of involved lymph nodes. Differentiated thyroid cancer is categorized as high-, medium-, or low-risk, depending on tumor extension, incomplete tumor resection, size of lymph nodes > 3 cm, and distant metastases. Adjuvant treatment with radioactive iodine can be considered for intermediate-risk DTC and is recommended for high-risk DTC.32
Following surgical treatment, thyroid-stimulating hormone suppression is recommended using levothyroxine.33 Patients at higher risk of recurrence should have longer and more intense suppression of thyroid-stimulating hormone.30 Levels of serum thyroglobulin and anti-thyroglobulin antibody should be followed postoperatively; rising values can indicate recurrent disease. The calcitonin level should be followed in patients with a history of MTC. Thyroid US should be performed 6 to 12 months postoperatively, then periodically, depending on determination of recurrence risk and any change in the thyroglobulin level.30
(Note: Glucagon-like peptide-1 [GLP-1] receptor agonists, used to treat type 2 diabetes mellitus, carry a black-box warning for their risk of MTC and are contraindicated in patients who have a personal or family history of MTC, MEN2A, or MEN2B.34)
Continue to: Anaplastic thyroid cancer...
Anaplastic thyroid cancer, a rare form of thyroid cancer, carries a high mortality rate, with a median survival of 5 months from diagnosis and 1-year survival of 20%. Patients require expeditious total thyroidectomy and neck dissection, followed by external-beam radiation with or without chemotherapy. If this strategy is not feasible, tracheostomy might be necessary to maintain a patent airway.2 Family physicians treating a patient who has anaplastic thyroid cancer can fulfill a crucial role by ensuring that an advance directive is established, a surrogate decision-maker is appointed, and goals of care are well defined.
Follow-up care for head and neck Ca
The risk of adverse effects after radiation therapy for head and neck cancer calls for close monitoring, appropriate treatment, and referral and counseling as needed. See “Follow-up care after treatment of head and neck cancer.” 35-39
SIDEBAR
Follow-up care after treatment of head and neck cancer35-39
Challenge: After radiation to the head and neck, as many as 53% of patients develop subclinical hypothyroidism and 33% develop clinical hypothyroidism.35Strategy: Measure the thyroid-stimulating hormone level within 1 year of the completion of radiotherapy and every 6 to 12 months thereafter.36
Challenge: Radiation to the head and neck can decrease the function of salivary glands, causing xerostomia in as many as 40% of patients. This condition can lead to problems with oral hygiene and difficulty with speech, eating, and swallowing.37Strategy:
- Treat xerostomia with artificial saliva, sugar-free candy and gum, or muscarinic cholinergic agonists, such as pilocarpine and cevimeline.
- Consider treatment with pilocarpine or cevimeline. Pilocarpine alleviates xerostomia in approximately 50% of patients who develop the condition, although its use can be limited by adverse cholinergic effects.3,7 Cevimeline causes fewer and less pronounced adverse effects than pilocarpine because it acts more specifically on receptors in the salivary glands.38
- Mention the possibility of acupuncture to your patients. There is evidence that it can stimulate salivary flow.39
Challenge: Patients who have had radiation to the head and neck have an increased risk of dental caries from xerostomia and the direct effect of radiation, which causes demineralization of teeth.
Strategy: Following radiation, instruct the patient about appropriate oral hygiene:
- regular flossing
- brushing and application of daily fluoride
- regular visits for dental care.39
Challenge: Trismus occurs in 5% to 25% of patients, depending on the type of radiation.36Strategy: Recommend exercise-based treatment, the treatment of choice. Surgery is indicated for severe cases.
Challenge: Dysphagia occurs in approximately 25% of patients treated with radiation.36Strategy: Provide a referral for swallowing exercises, which might be helpful. Some cases are severe enough to warrant placement of a feeding tube.37
Last, counsel all patients who have been treated for cancer of the head or neck, with any modality, about cessation of smoking and alcohol.
CORRESPONDENCE
Anne Mounsey, MD, Family Medicine Residency, The University of North Carolina at Chapel Hill, 590 Manning Dr., Chapel Hill, NC 27599; [email protected]
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
2. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104-1139.
3. Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489-501.
4. Seoane J, Alvarez-Novoa P, Gomez I, et al. Early oral cancer diagnosis: The Aarhus statement perspective. A systematic review and meta-analysis. Head Neck. 2016;38(suppl 1):E2182-E2189.
5. Franco J, Elghouche AN, Harris MS, et al Diagnostic delays and errors in head and neck cancer patients: opportunities for improvement. Am J Med Qual. 2017;32:330-335.
6. Pynnonen MA, Gillespie MB, Roman B, et al. Clinical practice guideline: evaluation of the neck mass in adults. Otolaryngol Head Neck Surg. 2017;157(suppl 2):S1-S30.
7. Bosetti C, Gallus S, Franceschi S, et al. Cancer of the larynx in non-smoking alcohol drinkers and in non-drinking tobacco smokers. Br J Cancer. 2002;87:516-518.
8. Hoffman HT, Porter K, Karnell LH, et al. Laryngeal cancer in the United States: changes in demographics, patterns of care, and survival. Laryngoscope. 2006;116(9 pt 2 suppl 111):1-13.
9. Schwartz SR, Cohen SM, Dailey SH, et al. Clinical practice guideline: hoarseness (dysphonia). Otolaryngol Head Neck Surg. 2009;141(3 suppl 2):S1-S31.
10. Steuer CE, El-Deiry M, Parks JR, et al. An update on larynx cancer. CA Cancer J Clin. 2017;67:31-50.
11. Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349:2091-2098.
12. Mendenhall WM, Werning JW, Hinerman RW, et al. Management of T1-T2 glottic carcinomas. Cancer. 2004;100:1786-1792.
13. Surveillance, Epidemiology, and End Results Unit. National Cancer Institute. Cancer stat facts: oral cavity and pharynx. https://seer.cancer.gov/statfacts/html/oralcav.html. Accessed October 18, 2019.
14. Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014;50:380-386.
15. Tarakji B, Gazal G, Al-Maweri SA, et al. Guideline for the diagnosis and treatment of recurrent aphthous stomatitis for dental practitioners. J Int Oral Health. 2015;7:74-80.
16. Siu A, Landon K, Ramos DM. Differential diagnosis and management of oral ulcers. Semin Cutan Med Surg. 2015;34:171-177.
17. US Preventive Services Task Force. Final recommendation statement: oral cancer: screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/oral-cancer-screening1. Updated November 2013. Accessed October 18, 2019.
18. Villa A, Woo SB. Leukoplakia—a diagnostic and management algorithm. J Oral Maxillofac Surg. 2017;75:723-734.
19. Yang S, Wu S, Zhou J, et al. Screening for nasopharyngeal cancer. Cochrane Database Syst Rev. 2015;(11):CD008423.
20. Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: a historical analysis of population-based data. Head Neck. 2012;34:877-885.
21. Ou SH, Zell JA, Ziogas A, et al. Epidemiology of nasopharyngeal carcinoma in the United States: improved survival of Chinese patients within the keratinizing squamous cell carcinoma histology. Ann Oncol. 2007;18:29-35.
22. Chang ET, Adami H-O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765-1777.
23. Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma. Lancet. 2016;387:1012-1024.
24. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg. 1986;8:177-184.
25. Lewis JS. Sinonasal squamous cell carcinoma: a review with emphasis on emerging histologic subtypes and the role of human papillomavirus. Head Neck Pathol. 2016;10:60-67.
26. Horn-Ross PL, Ljung BM, Morrow M. Environmental factors and the risk of salivary gland cancer. Epidemiology. 1997;8:414-419.
27. Colella G, Cannavale R, Flamminio F, et al. Fine-needle aspiration cytology of salivary gland lesions: a systematic review. J Oral Maxillofac Surg. 2010;68:2146-2153.
28. Berrino F, De Angelis R, Sant M, et al; EUROCARE Working Group. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995-99: results of the EUROCARE-4 study. Lancet Oncol. 2007;8:773-783.
29. Baloch ZW, LiVolsi VA, Asa SL, et al. Diagnostic terminology and morphologic criteria for cytologic diagnosis of thyroid lesions: a synopsis of the National Cancer Institute Thyroid Fine-Needle Aspiration State of the Science Conference. Diagn Cytopathol. 2008;36:425-437.
30. Haugen BR, Alexander EK, Bible KC, et al; The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133.
31. US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for thyroid Cancer: US Preventive Services Task Force recommendation statement. JAMA. 2017;317:1882-1887.
32. Jonklaas J, Cooper DS, Ain KB, et al; National Thyroid Cancer Treatment Cooperative Study Group. Radioiodine therapy in patients with stage I differentiated thyroid cancer. Thyroid. 2010;20:1423-1424.
33. Cooper DS, Specker B, Ho M, et al. Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. Thyroid. 1998;8:737-744.
34. US Food and Drug Administration. Highlight of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125431s020lbl.pdf. Updated December 2017. Accessed October 30, 1019.
35. Boomsma MJ, Bijl HP, Langendijk JA. Radiation-induced hypothyroidism in head and neck cancer patients: a systematic review. Radiother Oncol. 2011;99:1-5.
36. The development of quality of care measures for oral cavity cancer. Arch Otolaryngol Head Neck Surg. 2008;134:672.
37. Strojan P, Hutcheson KA, Eisbruch A, et al. Treatment of late sequelae after radiotherapy for head and neck cancer. Cancer Treat Rev. 2017;59:79-92.
38. Chambers MS, Posner M, Jones CU, et al. Cevimeline for the treatment of postirradiation xerostomia in patients with head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;68:1102-1109.
39. Gupta N, Pal M, Rawat S, et al. Radiation-induced dental caries, prevention and treatment - a systematic review. Natl J Maxillofac Surg. 2015;6:160-166.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30.
2. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104-1139.
3. Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489-501.
4. Seoane J, Alvarez-Novoa P, Gomez I, et al. Early oral cancer diagnosis: The Aarhus statement perspective. A systematic review and meta-analysis. Head Neck. 2016;38(suppl 1):E2182-E2189.
5. Franco J, Elghouche AN, Harris MS, et al Diagnostic delays and errors in head and neck cancer patients: opportunities for improvement. Am J Med Qual. 2017;32:330-335.
6. Pynnonen MA, Gillespie MB, Roman B, et al. Clinical practice guideline: evaluation of the neck mass in adults. Otolaryngol Head Neck Surg. 2017;157(suppl 2):S1-S30.
7. Bosetti C, Gallus S, Franceschi S, et al. Cancer of the larynx in non-smoking alcohol drinkers and in non-drinking tobacco smokers. Br J Cancer. 2002;87:516-518.
8. Hoffman HT, Porter K, Karnell LH, et al. Laryngeal cancer in the United States: changes in demographics, patterns of care, and survival. Laryngoscope. 2006;116(9 pt 2 suppl 111):1-13.
9. Schwartz SR, Cohen SM, Dailey SH, et al. Clinical practice guideline: hoarseness (dysphonia). Otolaryngol Head Neck Surg. 2009;141(3 suppl 2):S1-S31.
10. Steuer CE, El-Deiry M, Parks JR, et al. An update on larynx cancer. CA Cancer J Clin. 2017;67:31-50.
11. Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349:2091-2098.
12. Mendenhall WM, Werning JW, Hinerman RW, et al. Management of T1-T2 glottic carcinomas. Cancer. 2004;100:1786-1792.
13. Surveillance, Epidemiology, and End Results Unit. National Cancer Institute. Cancer stat facts: oral cavity and pharynx. https://seer.cancer.gov/statfacts/html/oralcav.html. Accessed October 18, 2019.
14. Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014;50:380-386.
15. Tarakji B, Gazal G, Al-Maweri SA, et al. Guideline for the diagnosis and treatment of recurrent aphthous stomatitis for dental practitioners. J Int Oral Health. 2015;7:74-80.
16. Siu A, Landon K, Ramos DM. Differential diagnosis and management of oral ulcers. Semin Cutan Med Surg. 2015;34:171-177.
17. US Preventive Services Task Force. Final recommendation statement: oral cancer: screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/oral-cancer-screening1. Updated November 2013. Accessed October 18, 2019.
18. Villa A, Woo SB. Leukoplakia—a diagnostic and management algorithm. J Oral Maxillofac Surg. 2017;75:723-734.
19. Yang S, Wu S, Zhou J, et al. Screening for nasopharyngeal cancer. Cochrane Database Syst Rev. 2015;(11):CD008423.
20. Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: a historical analysis of population-based data. Head Neck. 2012;34:877-885.
21. Ou SH, Zell JA, Ziogas A, et al. Epidemiology of nasopharyngeal carcinoma in the United States: improved survival of Chinese patients within the keratinizing squamous cell carcinoma histology. Ann Oncol. 2007;18:29-35.
22. Chang ET, Adami H-O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765-1777.
23. Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma. Lancet. 2016;387:1012-1024.
24. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg. 1986;8:177-184.
25. Lewis JS. Sinonasal squamous cell carcinoma: a review with emphasis on emerging histologic subtypes and the role of human papillomavirus. Head Neck Pathol. 2016;10:60-67.
26. Horn-Ross PL, Ljung BM, Morrow M. Environmental factors and the risk of salivary gland cancer. Epidemiology. 1997;8:414-419.
27. Colella G, Cannavale R, Flamminio F, et al. Fine-needle aspiration cytology of salivary gland lesions: a systematic review. J Oral Maxillofac Surg. 2010;68:2146-2153.
28. Berrino F, De Angelis R, Sant M, et al; EUROCARE Working Group. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995-99: results of the EUROCARE-4 study. Lancet Oncol. 2007;8:773-783.
29. Baloch ZW, LiVolsi VA, Asa SL, et al. Diagnostic terminology and morphologic criteria for cytologic diagnosis of thyroid lesions: a synopsis of the National Cancer Institute Thyroid Fine-Needle Aspiration State of the Science Conference. Diagn Cytopathol. 2008;36:425-437.
30. Haugen BR, Alexander EK, Bible KC, et al; The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133.
31. US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for thyroid Cancer: US Preventive Services Task Force recommendation statement. JAMA. 2017;317:1882-1887.
32. Jonklaas J, Cooper DS, Ain KB, et al; National Thyroid Cancer Treatment Cooperative Study Group. Radioiodine therapy in patients with stage I differentiated thyroid cancer. Thyroid. 2010;20:1423-1424.
33. Cooper DS, Specker B, Ho M, et al. Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. Thyroid. 1998;8:737-744.
34. US Food and Drug Administration. Highlight of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125431s020lbl.pdf. Updated December 2017. Accessed October 30, 1019.
35. Boomsma MJ, Bijl HP, Langendijk JA. Radiation-induced hypothyroidism in head and neck cancer patients: a systematic review. Radiother Oncol. 2011;99:1-5.
36. The development of quality of care measures for oral cavity cancer. Arch Otolaryngol Head Neck Surg. 2008;134:672.
37. Strojan P, Hutcheson KA, Eisbruch A, et al. Treatment of late sequelae after radiotherapy for head and neck cancer. Cancer Treat Rev. 2017;59:79-92.
38. Chambers MS, Posner M, Jones CU, et al. Cevimeline for the treatment of postirradiation xerostomia in patients with head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;68:1102-1109.
39. Gupta N, Pal M, Rawat S, et al. Radiation-induced dental caries, prevention and treatment - a systematic review. Natl J Maxillofac Surg. 2015;6:160-166.
PRACTICE RECOMMENDATIONS
› Do not treat a neck mass with antibiotics unless it has features consistent with infection. C
› Order laryngoscopy for all patients with hoarseness that does not resolve after 3 months—or sooner, if malignancy is suspected. C
› Order ultrasonography-guided fine-needle aspiration for diagnostic evaluation of salivary gland masses. B
› Manage a thyroid nodule based on its sonographic features, including size, consistency, and the presence of concerning features. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

How to use type 2 diabetes meds to lower CV disease risk
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4

Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).

Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32

See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.

ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; [email protected].
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4
Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).
Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32
See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.
ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; [email protected].
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4
Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).
Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32
See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.
ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; [email protected].
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
PRACTICE RECOMMENDATIONS
› Consider American Diabetes Association (ADA) guidance and prescribe a sodium–glucose cotransporter-2 (SGLT-2) inhibitor or glucagon-like peptide- 1 (GLP-1) receptor agonist that has demonstrated cardiovascular (CV) disease benefit for your patients who have type 2 diabetes (T2D) and established atherosclerotic CV disease. A
› Consider ADA’s recommendation for preferred therapy and prescribe an SGLT-2 inhibitor for your patients with T2D who have atherosclerotic CV disease and are at high risk of heart failure or in whom heart failure coexists. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Lipoprotein(a) Elevation: A New Diagnostic Code with Relevance to Service Members and Veterans (FULL)
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
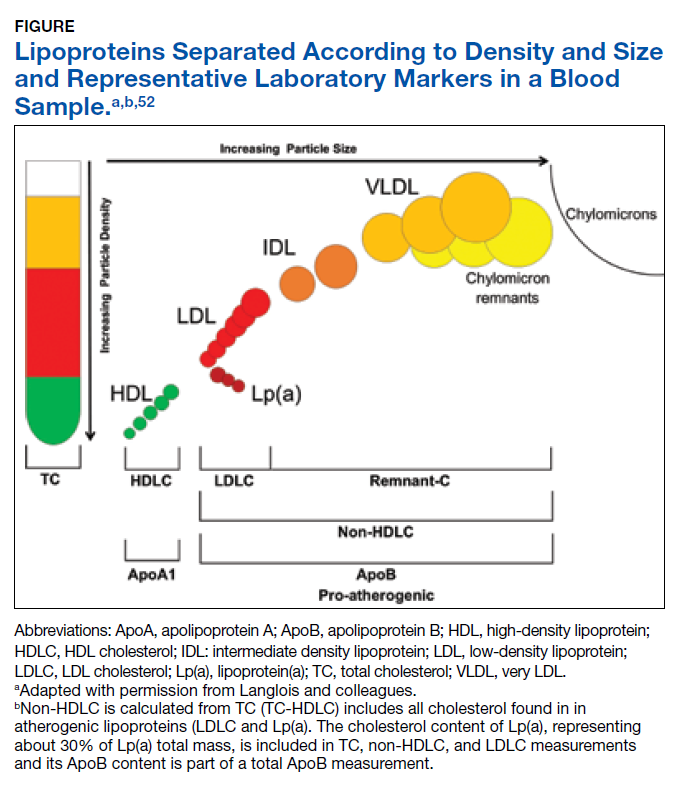
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
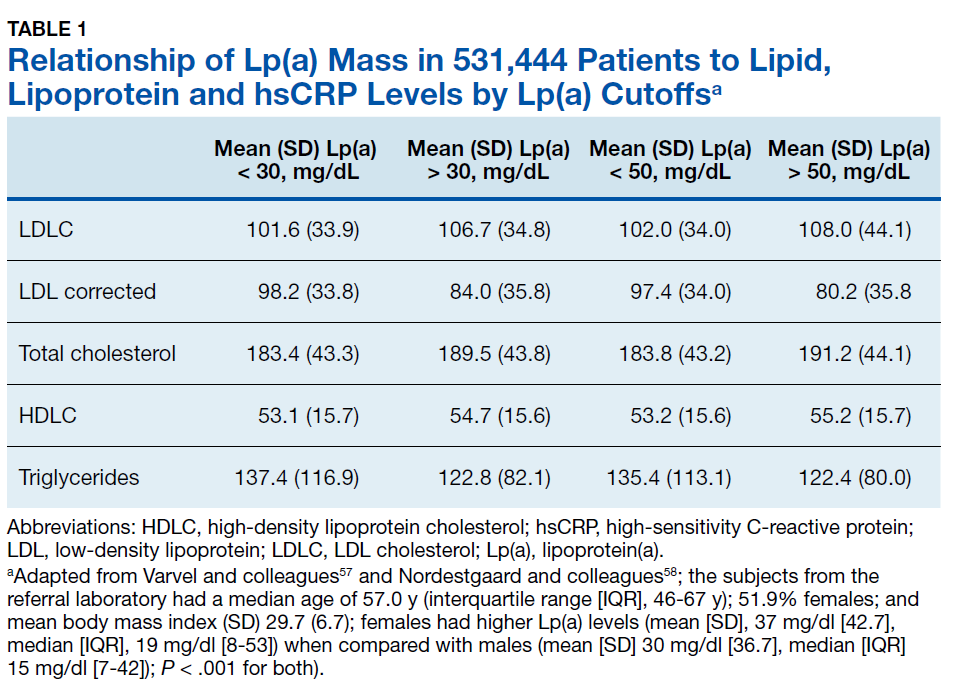
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
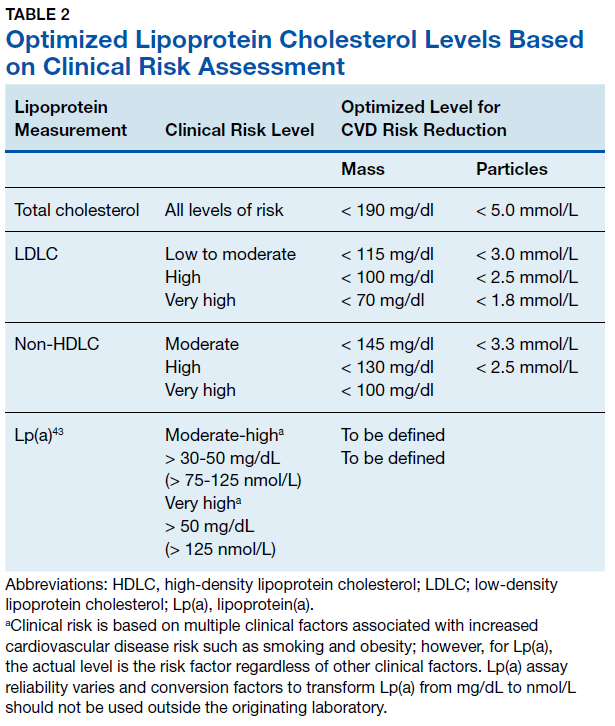
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
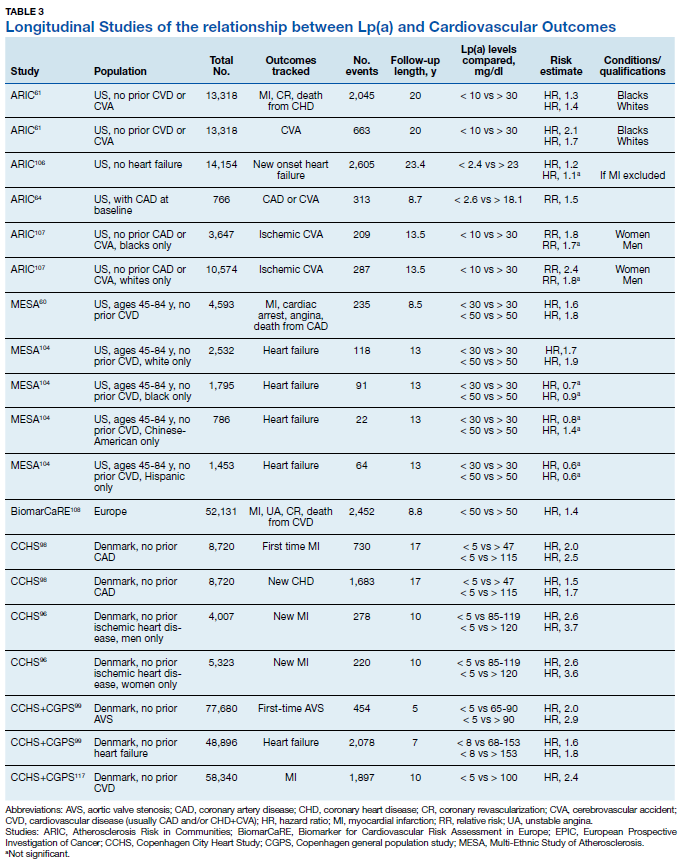
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
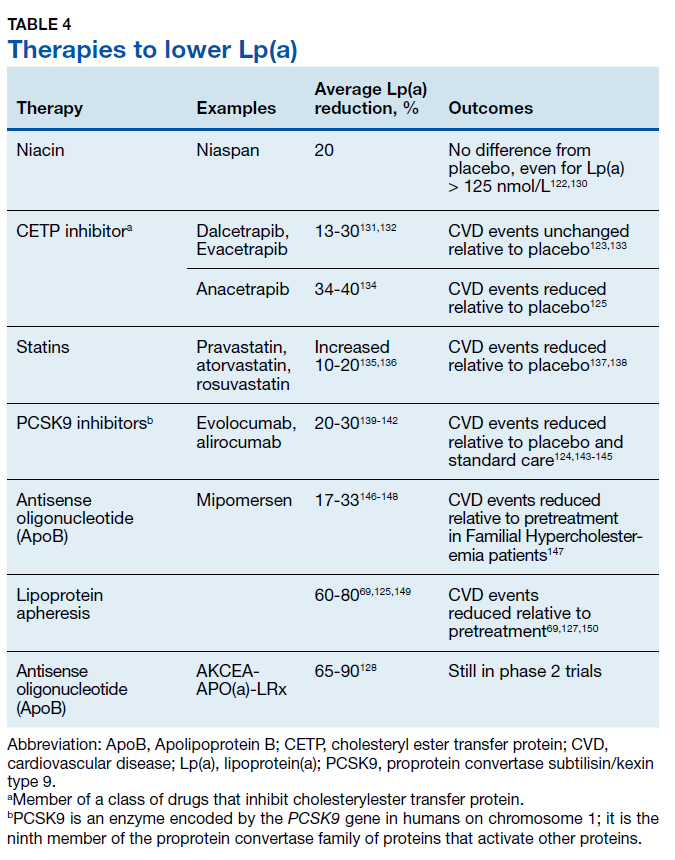
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
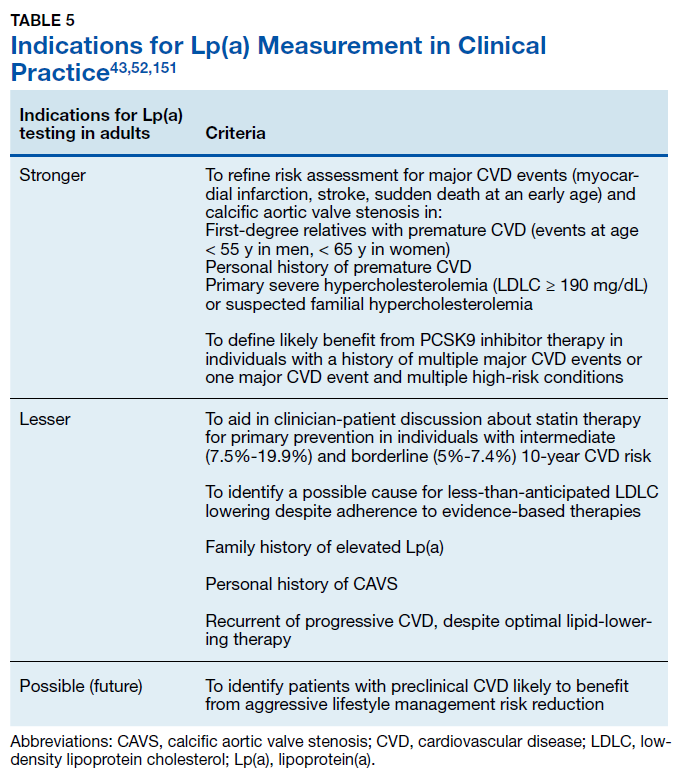
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
Systemic Medications Linked to an Increased Risk for Skin Malignancy
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Practice Points
- Patients should be educated about the increased risk for skin malignancy while undergoing treatment with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors.
- For BRAF inhibitors, sonic hedgehog–inhibiting agents, and JAK inhibitors, the increased risk for skin cancer warrants regular surveillance; however, given the indications for these medications, many patients will already be receiving regular skin screenings.
- The association between PDE-5 inhibitors and melanoma as well as nonmelanoma skin cancer remains questionable, and increased skin surveillance is not recommended at this time, unless patients have other risk factors for cutaneous malignancy.

Pediatric Molluscum: An Update
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38


Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39

Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.

Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
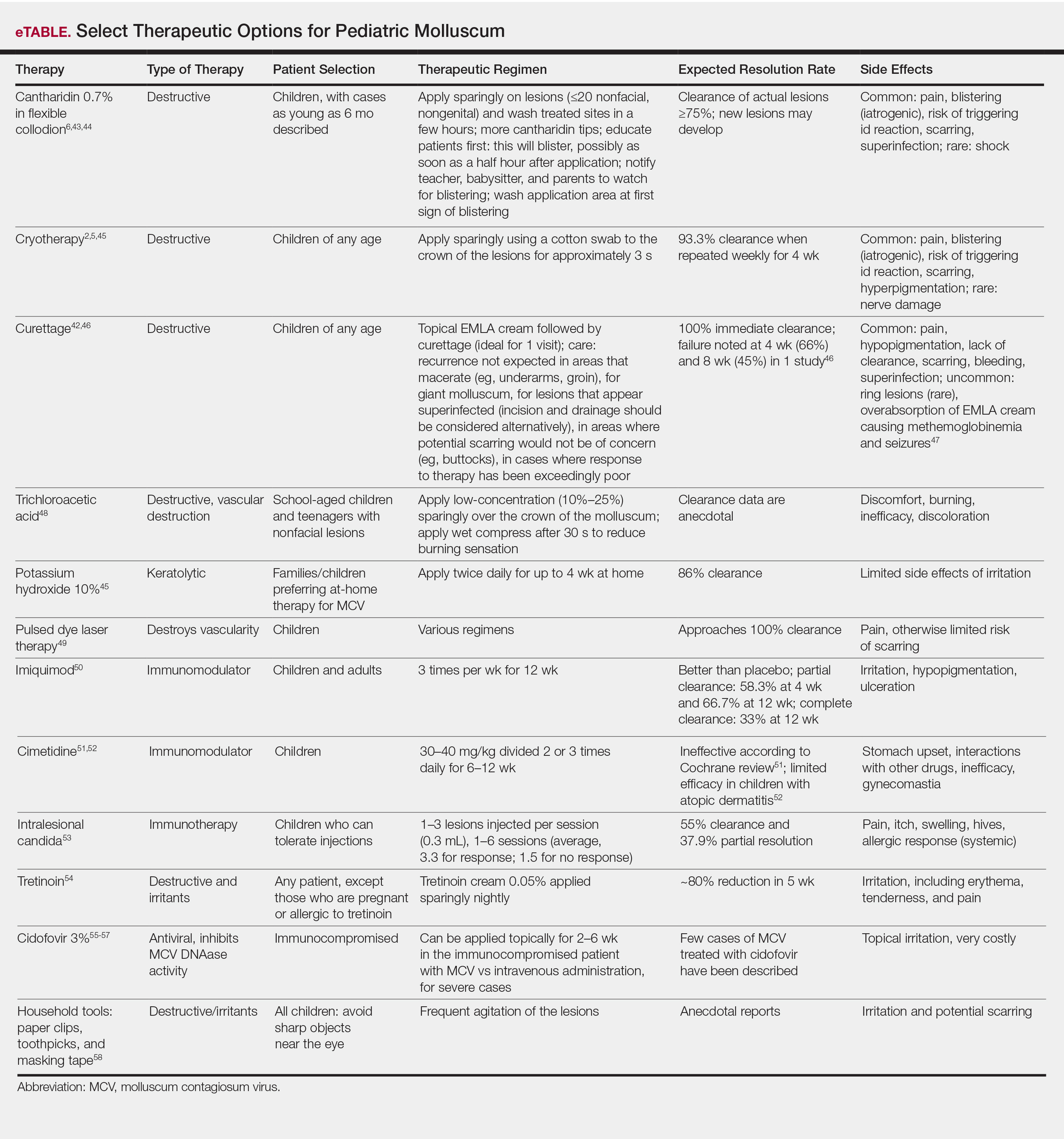
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38
Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39
Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.
Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38
Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39
Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.
Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
Practice Points
- Molluscum appears as pearly papules with a central dell (ie, umbilicated).
- Caused by a poxvirus, the disease is very contagious and transferred via skin-to-skin contact or fomites.
- One-third of children with molluscum will develop symptoms of local erythema, swelling, or pruritus.
- Diagnosis usually is clinical.
- Children are primarily managed through observation; however, cantharidin, cryotherapy, or curettage can be used for symptomatic or cosmetically concerning lesions.