User login
Disseminated Intravascular Coagulation
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1). 
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8 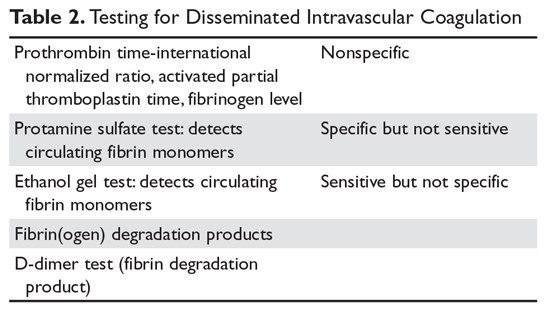
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12 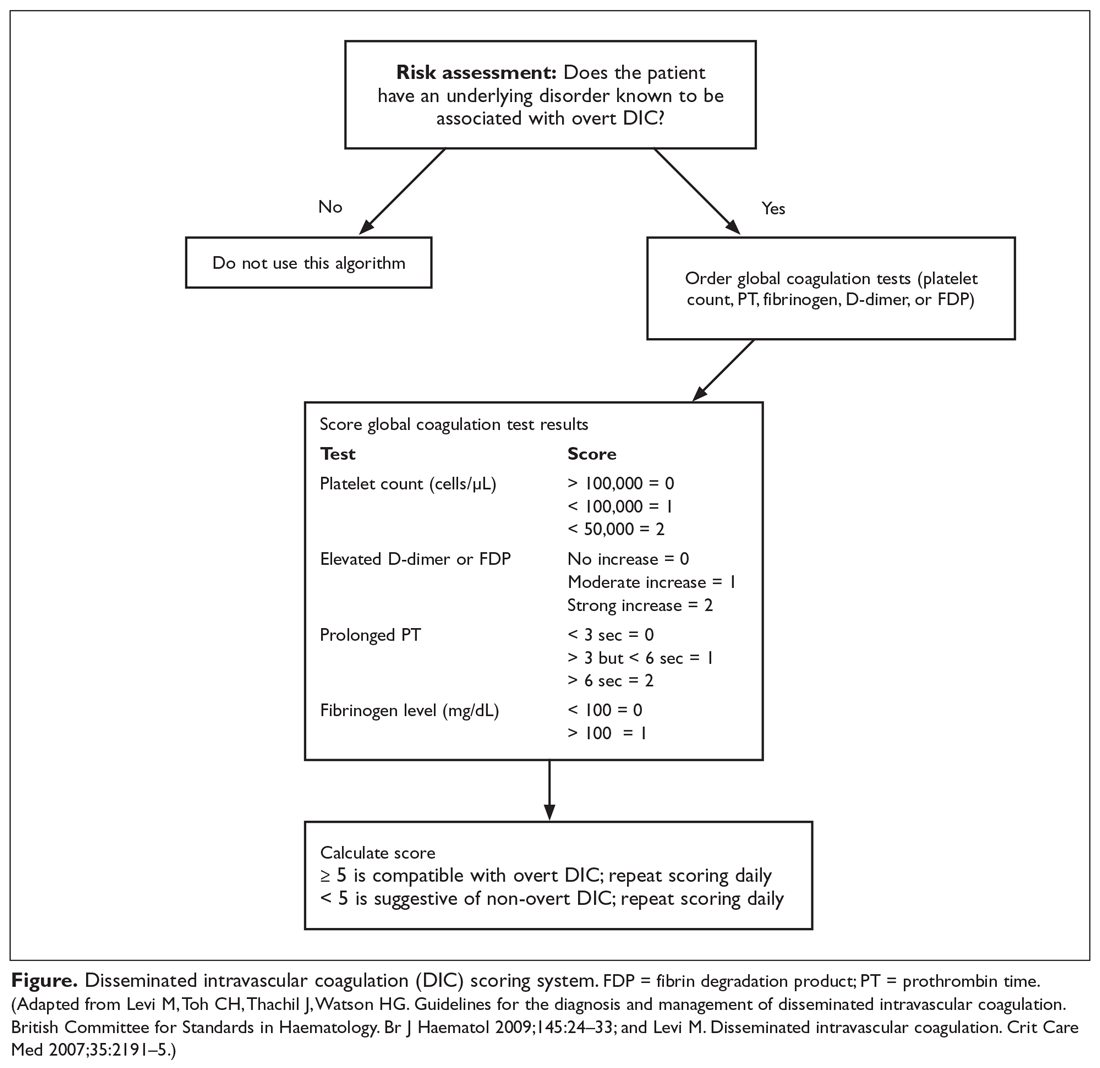
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3). 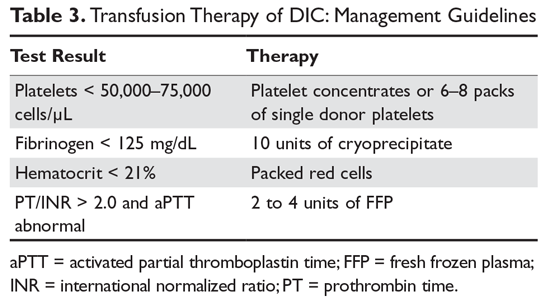
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
Reverse Shoulder Arthroplasty and Latissimus Dorsi Tendon Transfer
Take-Home Points
- CTA with loss of teres minor has been associated with worse clinical outcomes.
- Combined RSA and LDTT has been proposed and studied as a solution to this problem.
- LD tendon can be transferred to native teres minor insertion or lateral bicipital groove.
- Published studies have shown significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength.
- Overall complication rates appear similar to RSA alone, however rates of neuropraxia may be higher.
Reverse shoulder arthroplasty (RSA) is a proven procedure that typically improves pain and function in patients with rotator cuff tear arthropathy.1 Worse clinical outcomes are seen in patients with loss of teres minor function.2,3 The teres minor is often the last important external rotator of the shoulder left in cuff tear arthropathy. When its function is lost, the ability to achieve active external rotation may become diminished. This phenomenon was termed combined loss of active elevation and external rotation (CLEER) by Boileau and colleagues.4 Patients with CLEER typically exhibit weakness with external rotation of the shoulder—most pronounced with the arm in an abducted position. Clinical examination may reveal a positive Hornblower test, and magnetic resonance imaging (MRI) of the shoulder often shows atrophy in the teres minor muscle.5
Patients with CLEER often do not exhibit the same degree of clinical improvement after RSA, largely because the external rotation strength deficit remains unchanged, causing persistent difficulty in completing activities of daily living (eg, combing hair, brushing teeth, eating).6 One option for treating patients with CLEER is to combine RSA with latissimus dorsi tendon transfer (LDTT) with or without teres major (TM)tendon transfer. In 1934, L’Episcopo7 was the first to describe performing LDTT with TM tendon transfer in an attempt to restore external rotation in patients with brachial plexus palsy. This procedure typically is used for irreparable posterior-superior rotator cuff tears in younger patients.8 Although the transfer was originally popularized with use of 2 incisions,9 Boileau and colleagues4 described a modified technique that allows the transfer to be performed through a single deltopectoral approach during RSA.
Although several authors have described the outcomes of RSA with LDTT, the expected clinical outcomes and complication rates remain elusive because of the relatively small number of patients in each case series. In a systematic review, we critically examined and synthesized the results of individual studies on RSA with LDTT. We had 3 questions: What are the demographics of patients treated with RSA-LDTT? What outcomes are associated with this combined procedure? What are the associated complications, and how often do they occur?
Methods
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines were followed. PubMed and Scopus computerized literature databases were searched through July 2015. Articles were identified with keyword searches (Figure). In our review, we included only studies that were reported in English, that included a minimum of 10 patients at baseline, and that had follow-up of at least 12 months; we excluded review papers, case reports, and technique papers without patient data. Mr. Sheth performed the initial search, and he and Dr. Namdari reviewed the qualifying abstracts. If one of the authors selected a paper, it was moved to the next phase of the review process. At the final phase (full-text review), there were no disagreements about which articles ultimately would be included (Figure). 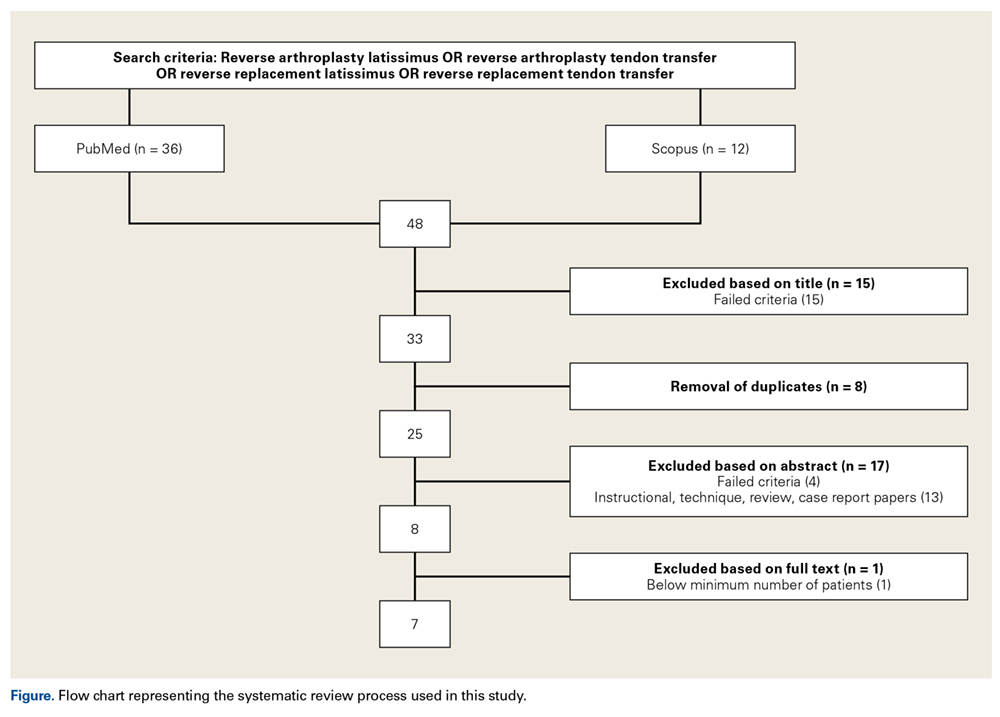
We obtained 36 articles from PubMed and 12 from Scopus (Figure). Of these 48 articles, 15 were removed on the basis of their titles (reviews or editorials), and 8 for being duplicates. The remaining 25 articles underwent abstract review, which eliminated 17: reviews, case reports, technique articles, instructional articles, and reports on small case series (<10 patients) or studies lacking the minimum 12-month follow-up. The remaining 8 articles underwent full-text review. Inclusion/exclusion criteria removed 1 article, leaving 7 qualifying articles for analysis.
None of the studies compared outcomes with those of a control (nonoperative) group or an alternative surgical treatment. One study reported outcomes of RSA with and without LDTT; in this instance, we included only the data specific to the RSA-with-LDTT cases. Data from the individual studies were compiled to obtain demographic statistics. In cases in which outcomes data were consistently reported between studies, results were pooled for calculation of percentages and frequency-weighted (FW) means. FW means and grouped standard deviations were used to generate P values, using the number of “subjects” as the number of studies. As a result, comparative statistics for each variable were reported as means that 95% of the studies would report.
Results
Seven studies met the inclusion/exclusion criteria and were included in this systematic review. Five were retrospective,10-14 and 2 were prospective.5,6 All were published between 2007 and 2015. Table 1 lists the full study characteristics between groups.
Demographics
All 7 studies reported number of patients at baseline (Table 1); 133 patients (study range, 11-40) underwent RSA with LDTT.5,6,10-14 All 7 studies reported patient ages; FW mean age was 69.5 years (range, 66-73 years).5,6,10-14 Six studies reported sex at follow-up; there were 36 men (33.6%) and 71 women (66.4%).5,6,10,12-14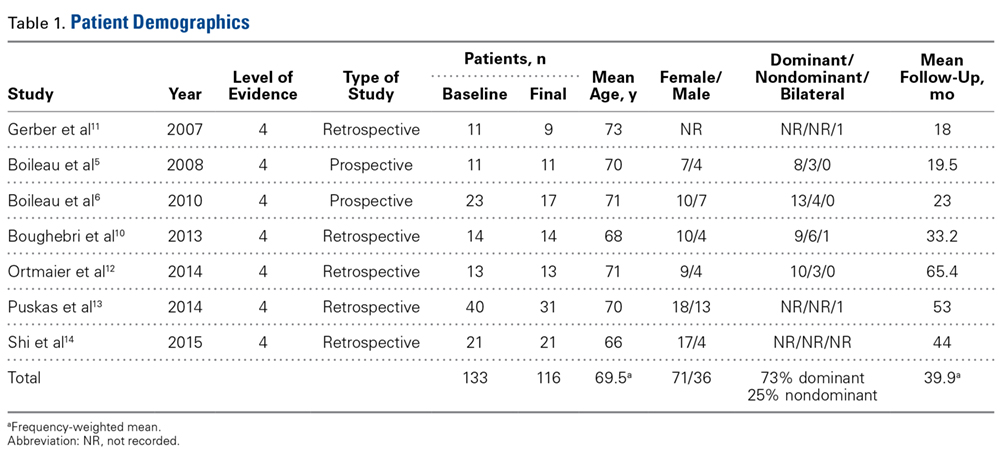
Surgical Indications and Technique
All patients underwent RSA with LDTT with or without TM tendon transfer for the indications of cuff tear arthropathy and CLEER. All 7 studies assessed loss of elevation as active forward elevation of <80° or <90° and loss of external rotation as active external rotation of <0°, inability to maintain abducted arm at 0°, or external rotation lag sign of >30°. All surgeries were performed with the deltopectoral approach. Combined LD/TM tendons were transferred in 6 studies5,6,10,12-14 and only the LD tendon in the seventh.11 Of the 6 studies that indicated tendon transfer location, 4 reported attaching to the posterolateral aspect of the greater tuberosity at the level of the original teres minor insertion5,6,11,12 and 2 reported attaching to the lateral aspect of the bicipital groove at the level of the LD insertion,10,14 . Six studies reported use of a sling or brace for 6 weeks after surgery.5,6,10-12,14
Outcomes
The 7 studies reported outcomes data for 116 (87%) of their 133 baseline patients (Table 2). Patients were followed up an FW mean of 39.9 months (range, 18-65 months). Six studies reported postoperative Constant scores; FW mean Constant score was 28.7 before surgery and 64.4 afterward (P = .0001).5,6,10-13 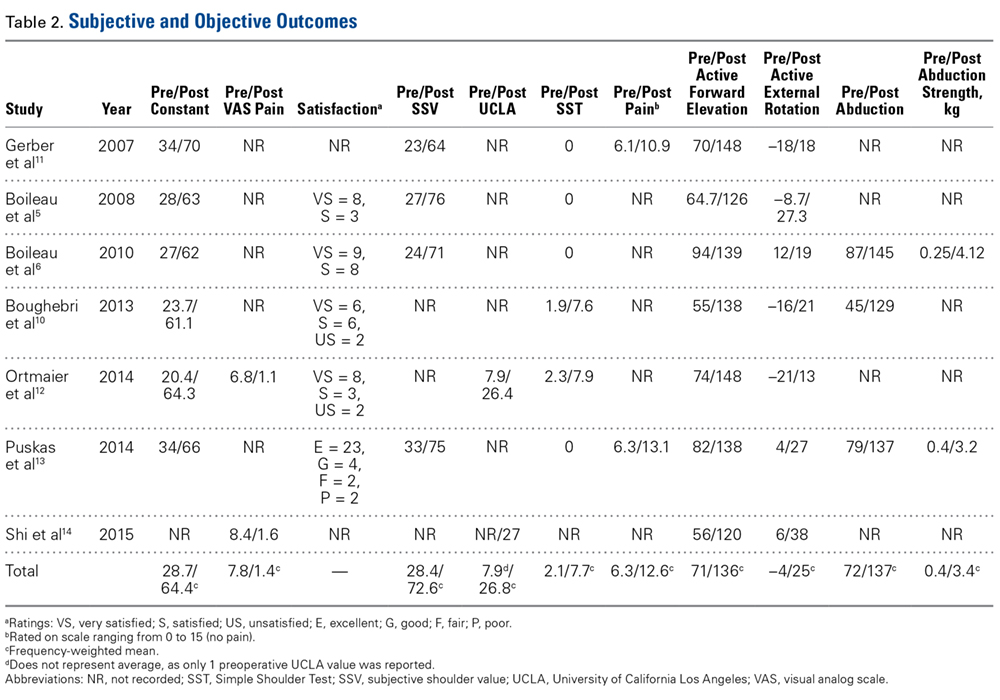
With regard to functional evaluation on physical examination, all 7 studies reported preoperative and postoperative active forward elevation and external rotation.5,6,10-14 Active forward elevation improved to an FW mean of 136°, from 71° (P < .0001), and external rotation improved to an FW mean of 25°, from –4° (P < .0001). Three studies reported preoperative and postoperative abduction; abduction improved to an FW mean of 137°, from 72° (P = .003).6,10,13
Complications and Reoperations
The 7 studies reported 31 complications, for an overall complication rate of 22.8% (31/126).5,6,10-14 There were 9 cases of neuropraxia (7.1%), 7 infections (6.0%), 4 dislocations or subluxations (3.4%), 2 cases of aseptic loosening (1.7%), 2 deltoid separations (1.7%), 2 periprosthetic fractures (1.7%), 1 acromion fracture (0.9%), 1 hematoma (0.9%), 1 LD/TM tendon rupture (0.9%), 1 intraoperative metaphyseal fracture (0.9%), and 1 painful baseplate screw (prominent where it penetrated the scapular spine)7 (0.9%).
The 7 studies also reported 19 reoperations, for an overall reoperation rate of 15.1% (19/126).5,6,10-14 There were 4 wound revisions, 3 revision RSAs, 3 open reduction and internal fixations, 2 deltoid repairs, 2 irrigation and débridements, 1 revision to hemiarthroplasty, 1 acromioclavicular resection, 1 procedure for a shoulder dislocation, 1 cerclage wire fixation to correct an intraoperative metaphyseal fracture, and 1 procedure to burr down a protruding baseplate screw.
Discussion
RSA with LDTT improves postoperative function in patients with cuff tear arthropathy associated with profound external rotation weakness caused by loss of a functional teres minor muscle. That statement is consistent with the findings of our systematic review, as all 7 reviewed studies found functional improvements, particularly in active external rotation (~30° improvement). In addition, there were consistent reductions in pain and improvements in forward elevation.
Our review found a mean patient age of 69.5 years, similar to the 72.7 years reported in a recent population-based study on RSA utilization.15 Likewise, our percentage of women who underwent RSA with LDTT, 66.4%, is similar to the overall rate of 63.6%.15 It appears that the RSA-with-LDTT population and the traditional RSA population are not dramatically different.
The improvements we found in subjective outcome scores and range of motion can be compared with those found in RSA-only treatment of rotator cuff tear arthropathy. Wall and colleagues16 found an approximate 44-point Constant score improvement, to 65.1 from 21.7, which is similar to our 36-point improvement for RSA with LDTT. They also found an approximate 10-point increase in pain relief; ours was about 6 points. Regarding range of motion, they found 66° improvement in active forward elevation and 2° in active external rotation, and we found 65° and 29° improvement, respectively. Thus, the outcomes of RSA with LDTT and RSA alone appear to be comparable. Simovitch and colleagues17 evaluated RSA outcomes as a function of teres minor muscle atrophy and found that, compared with patients with stage 3 or 4 fatty infiltration, patients with stage 0, 1, or 2 infiltration had significantly better ultimate Constant scores, significantly better SSVs, and significantly more preoperative-to-postoperative improvement. On average, Constant scores and SSVs increased 32% and 25%, respectively, in patients with more extensive fatty atrophy, and these patients experienced an average net loss of 7° in external rotation. It appears that, whereas RSA-with-LDTT outcomes are similar to outcomes in a nonspecific group of cuff tear arthropathy patients treated with RSA alone, adding LDTT to RSA may substantially improve outcomes in cases in which the teres minor is of poor quality.
We found no differences in implant types. However, with the exception of the Arrow prosthesis, which had 8.5 mm of lateralization, all implants had a traditional Grammont design. Greiner and colleagues2 recently found a trend toward improved external rotation in lateralized RSA designs, and a statistically significant improvement in external rotation in patients with an intact teres minor. The impact of LDTT with use of a lateralized design is unknown.
Our review found a relatively high rate of complications, 22.8%, and a reoperation rate of 15.1%. These are not dramatically different from the historical rates of complications (21%) and reoperations (13.4%).18 Although RSA with LDTT appears to have a higher rate of a specific complication, nerve-related injury, this is not necessarily surprising given the proximity of the axillary and radial nerves, the operative field, and the tendons transferred. This review’s rate of neuropraxia, 7.1%, is higher than the historical rate of 1.2% reported for RSA alone.18
This systematic review was limited by the quality of the studies available for inclusion. Although we followed PRISMA guidelines, none of the reviewed studies reported methods for controlling bias, confounding, and chance. In addition, the number of patients included and the relatively short follow-up period limit the impact of our findings. Finally, the individual studies used different outcome measures and did not report raw patient data, which limited our ability to perform more advanced statistical analysis.
Conclusion
This systematic review describes the demographics and outcomes of patients who underwent RSA with LDTT. Compiled data and FW means showed significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength. For RSA with LDTT and RSA alone, complication rates appear comparable, but the rate of neuropraxia may be higher for the combined procedure. Although this review provides valuable information on RSA with LDTT, its lack of a control comparison group and its relatively short follow-up period limited our ability to draw meaningful conclusions about the efficacy of the combined procedure in treating rotator cuff tear arthropathy in the absence of a functional teres minor.
1. Cuff D, Pupello D, Virani N, Levy J, Frankle M. Reverse shoulder arthroplasty for the treatment of rotator cuff deficiency. J Bone Joint Surg Am. 2008;90(6):1244-1251.
2. Greiner S, Schmidt C, Herrmann S, Pauly S, Perka C. Clinical performance of lateralized versus non-lateralized reverse shoulder arthroplasty: a prospective randomized study. J Shoulder Elbow Surg. 2015;24(9):1397-1404.
3. Young AA, Smith MM, Bacle G, Moraga C, Walch G. Early results of reverse shoulder arthroplasty in patients with rheumatoid arthritis. J Bone Joint Surg Am. 2011;93(20):1915-1923.
4. Boileau P, Chuinard C, Roussanne Y, Neyton L, Trojani C. Modified latissimus dorsi and teres major transfer through a single delto-pectoral approach for external rotation deficit of the shoulder: as an isolated procedure or with a reverse arthroplasty. J Shoulder Elbow Surg. 2007;16(6):671-682.
5. Boileau P, Chuinard C, Roussanne Y, Bicknell RT, Rochet N, Trojani C. Reverse shoulder arthroplasty combined with a modified latissimus dorsi and teres major tendon transfer for shoulder pseudoparalysis associated with dropping arm. Clin Orthop Relat Res. 2008;466(3):584-593.
6. Boileau P, Rumian AP, Zumstein MA. Reversed shoulder arthroplasty with modified L’Episcopo for combined loss of active elevation and external rotation. J Shoulder Elbow Surg. 2010;19(2 suppl):20-30.
7. L’Episcopo JB. Tendon transplantation in obstetrical paralysis. Am J Surg. 1934;25:122-125.
8. Namdari S, Voleti P, Baldwin K, Glaser D, Huffman GR. Latissimus dorsi tendon transfer for irreparable rotator cuff tears: a systematic review. J Bone Joint Surg Am. 2012;94(10):891-898.
9. Gerber C, Vinh TS, Hertel R, Hess CW. Latissimus dorsi transfer for the treatment of massive tears of the rotator cuff. A preliminary report. Clin Orthop Relat Res. 1988;(232):51-61.
10. Boughebri O, Kilinc A, Valenti P. Reverse shoulder arthroplasty combined with a latissimus dorsi and teres major transfer for a deficit of both active elevation and external rotation. Results of 15 cases with a minimum of 2-year follow-up. Orthop Traumatol Surg Res. 2013;99(2):131-137.
11. Gerber C, Pennington SD, Lingenfelter EJ, Sukthankar A. Reverse Delta-III total shoulder replacement combined with latissimus dorsi transfer. A preliminary report. J Bone Joint Surg Am. 2007;89(5):940-947.
12. Ortmaier R, Resch H, Hitzl W, et al. Reverse shoulder arthroplasty combined with latissimus dorsi transfer using the bone-chip technique. Int Orthop. 2014;38(3):553-559.
13. Puskas GJ, Catanzaro S, Gerber C. Clinical outcome of reverse total shoulder arthroplasty combined with latissimus dorsi transfer for the treatment of chronic combined pseudoparesis of elevation and external rotation of the shoulder. J Shoulder Elbow Surg. 2014;23(1):49-57.
14. Shi LL, Cahill KE, Ek ET, Tompson JD, Higgins LD, Warner JJ. Latissimus dorsi and teres major transfer with reverse shoulder arthroplasty restores active motion and reduces pain for posterosuperior cuff dysfunction. Clin Orthop Relat Res. 2015;473(10):3212-3217.
15. Schairer WW, Nwachukwu BU, Lyman S, Craig EV, Gulotta LV. National utilization of reverse total shoulder arthroplasty in the United States. J Shoulder Elbow Surg. 2015;24(1):91-97.
16. Wall B, Nove-Josserand L, O’Connor DP, Edwards TB, Walch G. Reverse total shoulder arthroplasty: a review of results according to etiology. J Bone Joint Surg Am. 2007;89(7):1476-1485.
17. Simovitch RW, Helmy N, Zumstein MA, Gerber C. Impact of fatty infiltration of the teres minor muscle on the outcome of reverse total shoulder arthroplasty. J Bone Joint Surg Am. 2007;89(5):934-939.
18. Zumstein MA, Pinedo M, Old J, Boileau P. Problems, complications, reoperations, and revisions in reverse total shoulder arthroplasty: a systematic review. J Shoulder Elbow Surg. 2011;20(1):146-157.
Take-Home Points
- CTA with loss of teres minor has been associated with worse clinical outcomes.
- Combined RSA and LDTT has been proposed and studied as a solution to this problem.
- LD tendon can be transferred to native teres minor insertion or lateral bicipital groove.
- Published studies have shown significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength.
- Overall complication rates appear similar to RSA alone, however rates of neuropraxia may be higher.
Reverse shoulder arthroplasty (RSA) is a proven procedure that typically improves pain and function in patients with rotator cuff tear arthropathy.1 Worse clinical outcomes are seen in patients with loss of teres minor function.2,3 The teres minor is often the last important external rotator of the shoulder left in cuff tear arthropathy. When its function is lost, the ability to achieve active external rotation may become diminished. This phenomenon was termed combined loss of active elevation and external rotation (CLEER) by Boileau and colleagues.4 Patients with CLEER typically exhibit weakness with external rotation of the shoulder—most pronounced with the arm in an abducted position. Clinical examination may reveal a positive Hornblower test, and magnetic resonance imaging (MRI) of the shoulder often shows atrophy in the teres minor muscle.5
Patients with CLEER often do not exhibit the same degree of clinical improvement after RSA, largely because the external rotation strength deficit remains unchanged, causing persistent difficulty in completing activities of daily living (eg, combing hair, brushing teeth, eating).6 One option for treating patients with CLEER is to combine RSA with latissimus dorsi tendon transfer (LDTT) with or without teres major (TM)tendon transfer. In 1934, L’Episcopo7 was the first to describe performing LDTT with TM tendon transfer in an attempt to restore external rotation in patients with brachial plexus palsy. This procedure typically is used for irreparable posterior-superior rotator cuff tears in younger patients.8 Although the transfer was originally popularized with use of 2 incisions,9 Boileau and colleagues4 described a modified technique that allows the transfer to be performed through a single deltopectoral approach during RSA.
Although several authors have described the outcomes of RSA with LDTT, the expected clinical outcomes and complication rates remain elusive because of the relatively small number of patients in each case series. In a systematic review, we critically examined and synthesized the results of individual studies on RSA with LDTT. We had 3 questions: What are the demographics of patients treated with RSA-LDTT? What outcomes are associated with this combined procedure? What are the associated complications, and how often do they occur?
Methods
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines were followed. PubMed and Scopus computerized literature databases were searched through July 2015. Articles were identified with keyword searches (Figure). In our review, we included only studies that were reported in English, that included a minimum of 10 patients at baseline, and that had follow-up of at least 12 months; we excluded review papers, case reports, and technique papers without patient data. Mr. Sheth performed the initial search, and he and Dr. Namdari reviewed the qualifying abstracts. If one of the authors selected a paper, it was moved to the next phase of the review process. At the final phase (full-text review), there were no disagreements about which articles ultimately would be included (Figure).
We obtained 36 articles from PubMed and 12 from Scopus (Figure). Of these 48 articles, 15 were removed on the basis of their titles (reviews or editorials), and 8 for being duplicates. The remaining 25 articles underwent abstract review, which eliminated 17: reviews, case reports, technique articles, instructional articles, and reports on small case series (<10 patients) or studies lacking the minimum 12-month follow-up. The remaining 8 articles underwent full-text review. Inclusion/exclusion criteria removed 1 article, leaving 7 qualifying articles for analysis.
None of the studies compared outcomes with those of a control (nonoperative) group or an alternative surgical treatment. One study reported outcomes of RSA with and without LDTT; in this instance, we included only the data specific to the RSA-with-LDTT cases. Data from the individual studies were compiled to obtain demographic statistics. In cases in which outcomes data were consistently reported between studies, results were pooled for calculation of percentages and frequency-weighted (FW) means. FW means and grouped standard deviations were used to generate P values, using the number of “subjects” as the number of studies. As a result, comparative statistics for each variable were reported as means that 95% of the studies would report.
Results
Seven studies met the inclusion/exclusion criteria and were included in this systematic review. Five were retrospective,10-14 and 2 were prospective.5,6 All were published between 2007 and 2015. Table 1 lists the full study characteristics between groups.
Demographics
All 7 studies reported number of patients at baseline (Table 1); 133 patients (study range, 11-40) underwent RSA with LDTT.5,6,10-14 All 7 studies reported patient ages; FW mean age was 69.5 years (range, 66-73 years).5,6,10-14 Six studies reported sex at follow-up; there were 36 men (33.6%) and 71 women (66.4%).5,6,10,12-14
Surgical Indications and Technique
All patients underwent RSA with LDTT with or without TM tendon transfer for the indications of cuff tear arthropathy and CLEER. All 7 studies assessed loss of elevation as active forward elevation of <80° or <90° and loss of external rotation as active external rotation of <0°, inability to maintain abducted arm at 0°, or external rotation lag sign of >30°. All surgeries were performed with the deltopectoral approach. Combined LD/TM tendons were transferred in 6 studies5,6,10,12-14 and only the LD tendon in the seventh.11 Of the 6 studies that indicated tendon transfer location, 4 reported attaching to the posterolateral aspect of the greater tuberosity at the level of the original teres minor insertion5,6,11,12 and 2 reported attaching to the lateral aspect of the bicipital groove at the level of the LD insertion,10,14 . Six studies reported use of a sling or brace for 6 weeks after surgery.5,6,10-12,14
Outcomes
The 7 studies reported outcomes data for 116 (87%) of their 133 baseline patients (Table 2). Patients were followed up an FW mean of 39.9 months (range, 18-65 months). Six studies reported postoperative Constant scores; FW mean Constant score was 28.7 before surgery and 64.4 afterward (P = .0001).5,6,10-13
With regard to functional evaluation on physical examination, all 7 studies reported preoperative and postoperative active forward elevation and external rotation.5,6,10-14 Active forward elevation improved to an FW mean of 136°, from 71° (P < .0001), and external rotation improved to an FW mean of 25°, from –4° (P < .0001). Three studies reported preoperative and postoperative abduction; abduction improved to an FW mean of 137°, from 72° (P = .003).6,10,13
Complications and Reoperations
The 7 studies reported 31 complications, for an overall complication rate of 22.8% (31/126).5,6,10-14 There were 9 cases of neuropraxia (7.1%), 7 infections (6.0%), 4 dislocations or subluxations (3.4%), 2 cases of aseptic loosening (1.7%), 2 deltoid separations (1.7%), 2 periprosthetic fractures (1.7%), 1 acromion fracture (0.9%), 1 hematoma (0.9%), 1 LD/TM tendon rupture (0.9%), 1 intraoperative metaphyseal fracture (0.9%), and 1 painful baseplate screw (prominent where it penetrated the scapular spine)7 (0.9%).
The 7 studies also reported 19 reoperations, for an overall reoperation rate of 15.1% (19/126).5,6,10-14 There were 4 wound revisions, 3 revision RSAs, 3 open reduction and internal fixations, 2 deltoid repairs, 2 irrigation and débridements, 1 revision to hemiarthroplasty, 1 acromioclavicular resection, 1 procedure for a shoulder dislocation, 1 cerclage wire fixation to correct an intraoperative metaphyseal fracture, and 1 procedure to burr down a protruding baseplate screw.
Discussion
RSA with LDTT improves postoperative function in patients with cuff tear arthropathy associated with profound external rotation weakness caused by loss of a functional teres minor muscle. That statement is consistent with the findings of our systematic review, as all 7 reviewed studies found functional improvements, particularly in active external rotation (~30° improvement). In addition, there were consistent reductions in pain and improvements in forward elevation.
Our review found a mean patient age of 69.5 years, similar to the 72.7 years reported in a recent population-based study on RSA utilization.15 Likewise, our percentage of women who underwent RSA with LDTT, 66.4%, is similar to the overall rate of 63.6%.15 It appears that the RSA-with-LDTT population and the traditional RSA population are not dramatically different.
The improvements we found in subjective outcome scores and range of motion can be compared with those found in RSA-only treatment of rotator cuff tear arthropathy. Wall and colleagues16 found an approximate 44-point Constant score improvement, to 65.1 from 21.7, which is similar to our 36-point improvement for RSA with LDTT. They also found an approximate 10-point increase in pain relief; ours was about 6 points. Regarding range of motion, they found 66° improvement in active forward elevation and 2° in active external rotation, and we found 65° and 29° improvement, respectively. Thus, the outcomes of RSA with LDTT and RSA alone appear to be comparable. Simovitch and colleagues17 evaluated RSA outcomes as a function of teres minor muscle atrophy and found that, compared with patients with stage 3 or 4 fatty infiltration, patients with stage 0, 1, or 2 infiltration had significantly better ultimate Constant scores, significantly better SSVs, and significantly more preoperative-to-postoperative improvement. On average, Constant scores and SSVs increased 32% and 25%, respectively, in patients with more extensive fatty atrophy, and these patients experienced an average net loss of 7° in external rotation. It appears that, whereas RSA-with-LDTT outcomes are similar to outcomes in a nonspecific group of cuff tear arthropathy patients treated with RSA alone, adding LDTT to RSA may substantially improve outcomes in cases in which the teres minor is of poor quality.
We found no differences in implant types. However, with the exception of the Arrow prosthesis, which had 8.5 mm of lateralization, all implants had a traditional Grammont design. Greiner and colleagues2 recently found a trend toward improved external rotation in lateralized RSA designs, and a statistically significant improvement in external rotation in patients with an intact teres minor. The impact of LDTT with use of a lateralized design is unknown.
Our review found a relatively high rate of complications, 22.8%, and a reoperation rate of 15.1%. These are not dramatically different from the historical rates of complications (21%) and reoperations (13.4%).18 Although RSA with LDTT appears to have a higher rate of a specific complication, nerve-related injury, this is not necessarily surprising given the proximity of the axillary and radial nerves, the operative field, and the tendons transferred. This review’s rate of neuropraxia, 7.1%, is higher than the historical rate of 1.2% reported for RSA alone.18
This systematic review was limited by the quality of the studies available for inclusion. Although we followed PRISMA guidelines, none of the reviewed studies reported methods for controlling bias, confounding, and chance. In addition, the number of patients included and the relatively short follow-up period limit the impact of our findings. Finally, the individual studies used different outcome measures and did not report raw patient data, which limited our ability to perform more advanced statistical analysis.
Conclusion
This systematic review describes the demographics and outcomes of patients who underwent RSA with LDTT. Compiled data and FW means showed significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength. For RSA with LDTT and RSA alone, complication rates appear comparable, but the rate of neuropraxia may be higher for the combined procedure. Although this review provides valuable information on RSA with LDTT, its lack of a control comparison group and its relatively short follow-up period limited our ability to draw meaningful conclusions about the efficacy of the combined procedure in treating rotator cuff tear arthropathy in the absence of a functional teres minor.
Take-Home Points
- CTA with loss of teres minor has been associated with worse clinical outcomes.
- Combined RSA and LDTT has been proposed and studied as a solution to this problem.
- LD tendon can be transferred to native teres minor insertion or lateral bicipital groove.
- Published studies have shown significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength.
- Overall complication rates appear similar to RSA alone, however rates of neuropraxia may be higher.
Reverse shoulder arthroplasty (RSA) is a proven procedure that typically improves pain and function in patients with rotator cuff tear arthropathy.1 Worse clinical outcomes are seen in patients with loss of teres minor function.2,3 The teres minor is often the last important external rotator of the shoulder left in cuff tear arthropathy. When its function is lost, the ability to achieve active external rotation may become diminished. This phenomenon was termed combined loss of active elevation and external rotation (CLEER) by Boileau and colleagues.4 Patients with CLEER typically exhibit weakness with external rotation of the shoulder—most pronounced with the arm in an abducted position. Clinical examination may reveal a positive Hornblower test, and magnetic resonance imaging (MRI) of the shoulder often shows atrophy in the teres minor muscle.5
Patients with CLEER often do not exhibit the same degree of clinical improvement after RSA, largely because the external rotation strength deficit remains unchanged, causing persistent difficulty in completing activities of daily living (eg, combing hair, brushing teeth, eating).6 One option for treating patients with CLEER is to combine RSA with latissimus dorsi tendon transfer (LDTT) with or without teres major (TM)tendon transfer. In 1934, L’Episcopo7 was the first to describe performing LDTT with TM tendon transfer in an attempt to restore external rotation in patients with brachial plexus palsy. This procedure typically is used for irreparable posterior-superior rotator cuff tears in younger patients.8 Although the transfer was originally popularized with use of 2 incisions,9 Boileau and colleagues4 described a modified technique that allows the transfer to be performed through a single deltopectoral approach during RSA.
Although several authors have described the outcomes of RSA with LDTT, the expected clinical outcomes and complication rates remain elusive because of the relatively small number of patients in each case series. In a systematic review, we critically examined and synthesized the results of individual studies on RSA with LDTT. We had 3 questions: What are the demographics of patients treated with RSA-LDTT? What outcomes are associated with this combined procedure? What are the associated complications, and how often do they occur?
Methods
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines were followed. PubMed and Scopus computerized literature databases were searched through July 2015. Articles were identified with keyword searches (Figure). In our review, we included only studies that were reported in English, that included a minimum of 10 patients at baseline, and that had follow-up of at least 12 months; we excluded review papers, case reports, and technique papers without patient data. Mr. Sheth performed the initial search, and he and Dr. Namdari reviewed the qualifying abstracts. If one of the authors selected a paper, it was moved to the next phase of the review process. At the final phase (full-text review), there were no disagreements about which articles ultimately would be included (Figure).
We obtained 36 articles from PubMed and 12 from Scopus (Figure). Of these 48 articles, 15 were removed on the basis of their titles (reviews or editorials), and 8 for being duplicates. The remaining 25 articles underwent abstract review, which eliminated 17: reviews, case reports, technique articles, instructional articles, and reports on small case series (<10 patients) or studies lacking the minimum 12-month follow-up. The remaining 8 articles underwent full-text review. Inclusion/exclusion criteria removed 1 article, leaving 7 qualifying articles for analysis.
None of the studies compared outcomes with those of a control (nonoperative) group or an alternative surgical treatment. One study reported outcomes of RSA with and without LDTT; in this instance, we included only the data specific to the RSA-with-LDTT cases. Data from the individual studies were compiled to obtain demographic statistics. In cases in which outcomes data were consistently reported between studies, results were pooled for calculation of percentages and frequency-weighted (FW) means. FW means and grouped standard deviations were used to generate P values, using the number of “subjects” as the number of studies. As a result, comparative statistics for each variable were reported as means that 95% of the studies would report.
Results
Seven studies met the inclusion/exclusion criteria and were included in this systematic review. Five were retrospective,10-14 and 2 were prospective.5,6 All were published between 2007 and 2015. Table 1 lists the full study characteristics between groups.
Demographics
All 7 studies reported number of patients at baseline (Table 1); 133 patients (study range, 11-40) underwent RSA with LDTT.5,6,10-14 All 7 studies reported patient ages; FW mean age was 69.5 years (range, 66-73 years).5,6,10-14 Six studies reported sex at follow-up; there were 36 men (33.6%) and 71 women (66.4%).5,6,10,12-14
Surgical Indications and Technique
All patients underwent RSA with LDTT with or without TM tendon transfer for the indications of cuff tear arthropathy and CLEER. All 7 studies assessed loss of elevation as active forward elevation of <80° or <90° and loss of external rotation as active external rotation of <0°, inability to maintain abducted arm at 0°, or external rotation lag sign of >30°. All surgeries were performed with the deltopectoral approach. Combined LD/TM tendons were transferred in 6 studies5,6,10,12-14 and only the LD tendon in the seventh.11 Of the 6 studies that indicated tendon transfer location, 4 reported attaching to the posterolateral aspect of the greater tuberosity at the level of the original teres minor insertion5,6,11,12 and 2 reported attaching to the lateral aspect of the bicipital groove at the level of the LD insertion,10,14 . Six studies reported use of a sling or brace for 6 weeks after surgery.5,6,10-12,14
Outcomes
The 7 studies reported outcomes data for 116 (87%) of their 133 baseline patients (Table 2). Patients were followed up an FW mean of 39.9 months (range, 18-65 months). Six studies reported postoperative Constant scores; FW mean Constant score was 28.7 before surgery and 64.4 afterward (P = .0001).5,6,10-13
With regard to functional evaluation on physical examination, all 7 studies reported preoperative and postoperative active forward elevation and external rotation.5,6,10-14 Active forward elevation improved to an FW mean of 136°, from 71° (P < .0001), and external rotation improved to an FW mean of 25°, from –4° (P < .0001). Three studies reported preoperative and postoperative abduction; abduction improved to an FW mean of 137°, from 72° (P = .003).6,10,13
Complications and Reoperations
The 7 studies reported 31 complications, for an overall complication rate of 22.8% (31/126).5,6,10-14 There were 9 cases of neuropraxia (7.1%), 7 infections (6.0%), 4 dislocations or subluxations (3.4%), 2 cases of aseptic loosening (1.7%), 2 deltoid separations (1.7%), 2 periprosthetic fractures (1.7%), 1 acromion fracture (0.9%), 1 hematoma (0.9%), 1 LD/TM tendon rupture (0.9%), 1 intraoperative metaphyseal fracture (0.9%), and 1 painful baseplate screw (prominent where it penetrated the scapular spine)7 (0.9%).
The 7 studies also reported 19 reoperations, for an overall reoperation rate of 15.1% (19/126).5,6,10-14 There were 4 wound revisions, 3 revision RSAs, 3 open reduction and internal fixations, 2 deltoid repairs, 2 irrigation and débridements, 1 revision to hemiarthroplasty, 1 acromioclavicular resection, 1 procedure for a shoulder dislocation, 1 cerclage wire fixation to correct an intraoperative metaphyseal fracture, and 1 procedure to burr down a protruding baseplate screw.
Discussion
RSA with LDTT improves postoperative function in patients with cuff tear arthropathy associated with profound external rotation weakness caused by loss of a functional teres minor muscle. That statement is consistent with the findings of our systematic review, as all 7 reviewed studies found functional improvements, particularly in active external rotation (~30° improvement). In addition, there were consistent reductions in pain and improvements in forward elevation.
Our review found a mean patient age of 69.5 years, similar to the 72.7 years reported in a recent population-based study on RSA utilization.15 Likewise, our percentage of women who underwent RSA with LDTT, 66.4%, is similar to the overall rate of 63.6%.15 It appears that the RSA-with-LDTT population and the traditional RSA population are not dramatically different.
The improvements we found in subjective outcome scores and range of motion can be compared with those found in RSA-only treatment of rotator cuff tear arthropathy. Wall and colleagues16 found an approximate 44-point Constant score improvement, to 65.1 from 21.7, which is similar to our 36-point improvement for RSA with LDTT. They also found an approximate 10-point increase in pain relief; ours was about 6 points. Regarding range of motion, they found 66° improvement in active forward elevation and 2° in active external rotation, and we found 65° and 29° improvement, respectively. Thus, the outcomes of RSA with LDTT and RSA alone appear to be comparable. Simovitch and colleagues17 evaluated RSA outcomes as a function of teres minor muscle atrophy and found that, compared with patients with stage 3 or 4 fatty infiltration, patients with stage 0, 1, or 2 infiltration had significantly better ultimate Constant scores, significantly better SSVs, and significantly more preoperative-to-postoperative improvement. On average, Constant scores and SSVs increased 32% and 25%, respectively, in patients with more extensive fatty atrophy, and these patients experienced an average net loss of 7° in external rotation. It appears that, whereas RSA-with-LDTT outcomes are similar to outcomes in a nonspecific group of cuff tear arthropathy patients treated with RSA alone, adding LDTT to RSA may substantially improve outcomes in cases in which the teres minor is of poor quality.
We found no differences in implant types. However, with the exception of the Arrow prosthesis, which had 8.5 mm of lateralization, all implants had a traditional Grammont design. Greiner and colleagues2 recently found a trend toward improved external rotation in lateralized RSA designs, and a statistically significant improvement in external rotation in patients with an intact teres minor. The impact of LDTT with use of a lateralized design is unknown.
Our review found a relatively high rate of complications, 22.8%, and a reoperation rate of 15.1%. These are not dramatically different from the historical rates of complications (21%) and reoperations (13.4%).18 Although RSA with LDTT appears to have a higher rate of a specific complication, nerve-related injury, this is not necessarily surprising given the proximity of the axillary and radial nerves, the operative field, and the tendons transferred. This review’s rate of neuropraxia, 7.1%, is higher than the historical rate of 1.2% reported for RSA alone.18
This systematic review was limited by the quality of the studies available for inclusion. Although we followed PRISMA guidelines, none of the reviewed studies reported methods for controlling bias, confounding, and chance. In addition, the number of patients included and the relatively short follow-up period limit the impact of our findings. Finally, the individual studies used different outcome measures and did not report raw patient data, which limited our ability to perform more advanced statistical analysis.
Conclusion
This systematic review describes the demographics and outcomes of patients who underwent RSA with LDTT. Compiled data and FW means showed significant improvements in various subjective values, active forward elevation, external rotation, and abduction strength. For RSA with LDTT and RSA alone, complication rates appear comparable, but the rate of neuropraxia may be higher for the combined procedure. Although this review provides valuable information on RSA with LDTT, its lack of a control comparison group and its relatively short follow-up period limited our ability to draw meaningful conclusions about the efficacy of the combined procedure in treating rotator cuff tear arthropathy in the absence of a functional teres minor.
1. Cuff D, Pupello D, Virani N, Levy J, Frankle M. Reverse shoulder arthroplasty for the treatment of rotator cuff deficiency. J Bone Joint Surg Am. 2008;90(6):1244-1251.
2. Greiner S, Schmidt C, Herrmann S, Pauly S, Perka C. Clinical performance of lateralized versus non-lateralized reverse shoulder arthroplasty: a prospective randomized study. J Shoulder Elbow Surg. 2015;24(9):1397-1404.
3. Young AA, Smith MM, Bacle G, Moraga C, Walch G. Early results of reverse shoulder arthroplasty in patients with rheumatoid arthritis. J Bone Joint Surg Am. 2011;93(20):1915-1923.
4. Boileau P, Chuinard C, Roussanne Y, Neyton L, Trojani C. Modified latissimus dorsi and teres major transfer through a single delto-pectoral approach for external rotation deficit of the shoulder: as an isolated procedure or with a reverse arthroplasty. J Shoulder Elbow Surg. 2007;16(6):671-682.
5. Boileau P, Chuinard C, Roussanne Y, Bicknell RT, Rochet N, Trojani C. Reverse shoulder arthroplasty combined with a modified latissimus dorsi and teres major tendon transfer for shoulder pseudoparalysis associated with dropping arm. Clin Orthop Relat Res. 2008;466(3):584-593.
6. Boileau P, Rumian AP, Zumstein MA. Reversed shoulder arthroplasty with modified L’Episcopo for combined loss of active elevation and external rotation. J Shoulder Elbow Surg. 2010;19(2 suppl):20-30.
7. L’Episcopo JB. Tendon transplantation in obstetrical paralysis. Am J Surg. 1934;25:122-125.
8. Namdari S, Voleti P, Baldwin K, Glaser D, Huffman GR. Latissimus dorsi tendon transfer for irreparable rotator cuff tears: a systematic review. J Bone Joint Surg Am. 2012;94(10):891-898.
9. Gerber C, Vinh TS, Hertel R, Hess CW. Latissimus dorsi transfer for the treatment of massive tears of the rotator cuff. A preliminary report. Clin Orthop Relat Res. 1988;(232):51-61.
10. Boughebri O, Kilinc A, Valenti P. Reverse shoulder arthroplasty combined with a latissimus dorsi and teres major transfer for a deficit of both active elevation and external rotation. Results of 15 cases with a minimum of 2-year follow-up. Orthop Traumatol Surg Res. 2013;99(2):131-137.
11. Gerber C, Pennington SD, Lingenfelter EJ, Sukthankar A. Reverse Delta-III total shoulder replacement combined with latissimus dorsi transfer. A preliminary report. J Bone Joint Surg Am. 2007;89(5):940-947.
12. Ortmaier R, Resch H, Hitzl W, et al. Reverse shoulder arthroplasty combined with latissimus dorsi transfer using the bone-chip technique. Int Orthop. 2014;38(3):553-559.
13. Puskas GJ, Catanzaro S, Gerber C. Clinical outcome of reverse total shoulder arthroplasty combined with latissimus dorsi transfer for the treatment of chronic combined pseudoparesis of elevation and external rotation of the shoulder. J Shoulder Elbow Surg. 2014;23(1):49-57.
14. Shi LL, Cahill KE, Ek ET, Tompson JD, Higgins LD, Warner JJ. Latissimus dorsi and teres major transfer with reverse shoulder arthroplasty restores active motion and reduces pain for posterosuperior cuff dysfunction. Clin Orthop Relat Res. 2015;473(10):3212-3217.
15. Schairer WW, Nwachukwu BU, Lyman S, Craig EV, Gulotta LV. National utilization of reverse total shoulder arthroplasty in the United States. J Shoulder Elbow Surg. 2015;24(1):91-97.
16. Wall B, Nove-Josserand L, O’Connor DP, Edwards TB, Walch G. Reverse total shoulder arthroplasty: a review of results according to etiology. J Bone Joint Surg Am. 2007;89(7):1476-1485.
17. Simovitch RW, Helmy N, Zumstein MA, Gerber C. Impact of fatty infiltration of the teres minor muscle on the outcome of reverse total shoulder arthroplasty. J Bone Joint Surg Am. 2007;89(5):934-939.
18. Zumstein MA, Pinedo M, Old J, Boileau P. Problems, complications, reoperations, and revisions in reverse total shoulder arthroplasty: a systematic review. J Shoulder Elbow Surg. 2011;20(1):146-157.
1. Cuff D, Pupello D, Virani N, Levy J, Frankle M. Reverse shoulder arthroplasty for the treatment of rotator cuff deficiency. J Bone Joint Surg Am. 2008;90(6):1244-1251.
2. Greiner S, Schmidt C, Herrmann S, Pauly S, Perka C. Clinical performance of lateralized versus non-lateralized reverse shoulder arthroplasty: a prospective randomized study. J Shoulder Elbow Surg. 2015;24(9):1397-1404.
3. Young AA, Smith MM, Bacle G, Moraga C, Walch G. Early results of reverse shoulder arthroplasty in patients with rheumatoid arthritis. J Bone Joint Surg Am. 2011;93(20):1915-1923.
4. Boileau P, Chuinard C, Roussanne Y, Neyton L, Trojani C. Modified latissimus dorsi and teres major transfer through a single delto-pectoral approach for external rotation deficit of the shoulder: as an isolated procedure or with a reverse arthroplasty. J Shoulder Elbow Surg. 2007;16(6):671-682.
5. Boileau P, Chuinard C, Roussanne Y, Bicknell RT, Rochet N, Trojani C. Reverse shoulder arthroplasty combined with a modified latissimus dorsi and teres major tendon transfer for shoulder pseudoparalysis associated with dropping arm. Clin Orthop Relat Res. 2008;466(3):584-593.
6. Boileau P, Rumian AP, Zumstein MA. Reversed shoulder arthroplasty with modified L’Episcopo for combined loss of active elevation and external rotation. J Shoulder Elbow Surg. 2010;19(2 suppl):20-30.
7. L’Episcopo JB. Tendon transplantation in obstetrical paralysis. Am J Surg. 1934;25:122-125.
8. Namdari S, Voleti P, Baldwin K, Glaser D, Huffman GR. Latissimus dorsi tendon transfer for irreparable rotator cuff tears: a systematic review. J Bone Joint Surg Am. 2012;94(10):891-898.
9. Gerber C, Vinh TS, Hertel R, Hess CW. Latissimus dorsi transfer for the treatment of massive tears of the rotator cuff. A preliminary report. Clin Orthop Relat Res. 1988;(232):51-61.
10. Boughebri O, Kilinc A, Valenti P. Reverse shoulder arthroplasty combined with a latissimus dorsi and teres major transfer for a deficit of both active elevation and external rotation. Results of 15 cases with a minimum of 2-year follow-up. Orthop Traumatol Surg Res. 2013;99(2):131-137.
11. Gerber C, Pennington SD, Lingenfelter EJ, Sukthankar A. Reverse Delta-III total shoulder replacement combined with latissimus dorsi transfer. A preliminary report. J Bone Joint Surg Am. 2007;89(5):940-947.
12. Ortmaier R, Resch H, Hitzl W, et al. Reverse shoulder arthroplasty combined with latissimus dorsi transfer using the bone-chip technique. Int Orthop. 2014;38(3):553-559.
13. Puskas GJ, Catanzaro S, Gerber C. Clinical outcome of reverse total shoulder arthroplasty combined with latissimus dorsi transfer for the treatment of chronic combined pseudoparesis of elevation and external rotation of the shoulder. J Shoulder Elbow Surg. 2014;23(1):49-57.
14. Shi LL, Cahill KE, Ek ET, Tompson JD, Higgins LD, Warner JJ. Latissimus dorsi and teres major transfer with reverse shoulder arthroplasty restores active motion and reduces pain for posterosuperior cuff dysfunction. Clin Orthop Relat Res. 2015;473(10):3212-3217.
15. Schairer WW, Nwachukwu BU, Lyman S, Craig EV, Gulotta LV. National utilization of reverse total shoulder arthroplasty in the United States. J Shoulder Elbow Surg. 2015;24(1):91-97.
16. Wall B, Nove-Josserand L, O’Connor DP, Edwards TB, Walch G. Reverse total shoulder arthroplasty: a review of results according to etiology. J Bone Joint Surg Am. 2007;89(7):1476-1485.
17. Simovitch RW, Helmy N, Zumstein MA, Gerber C. Impact of fatty infiltration of the teres minor muscle on the outcome of reverse total shoulder arthroplasty. J Bone Joint Surg Am. 2007;89(5):934-939.
18. Zumstein MA, Pinedo M, Old J, Boileau P. Problems, complications, reoperations, and revisions in reverse total shoulder arthroplasty: a systematic review. J Shoulder Elbow Surg. 2011;20(1):146-157.
Cancer-Related Fatigue: Approach to Assessment and Management
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
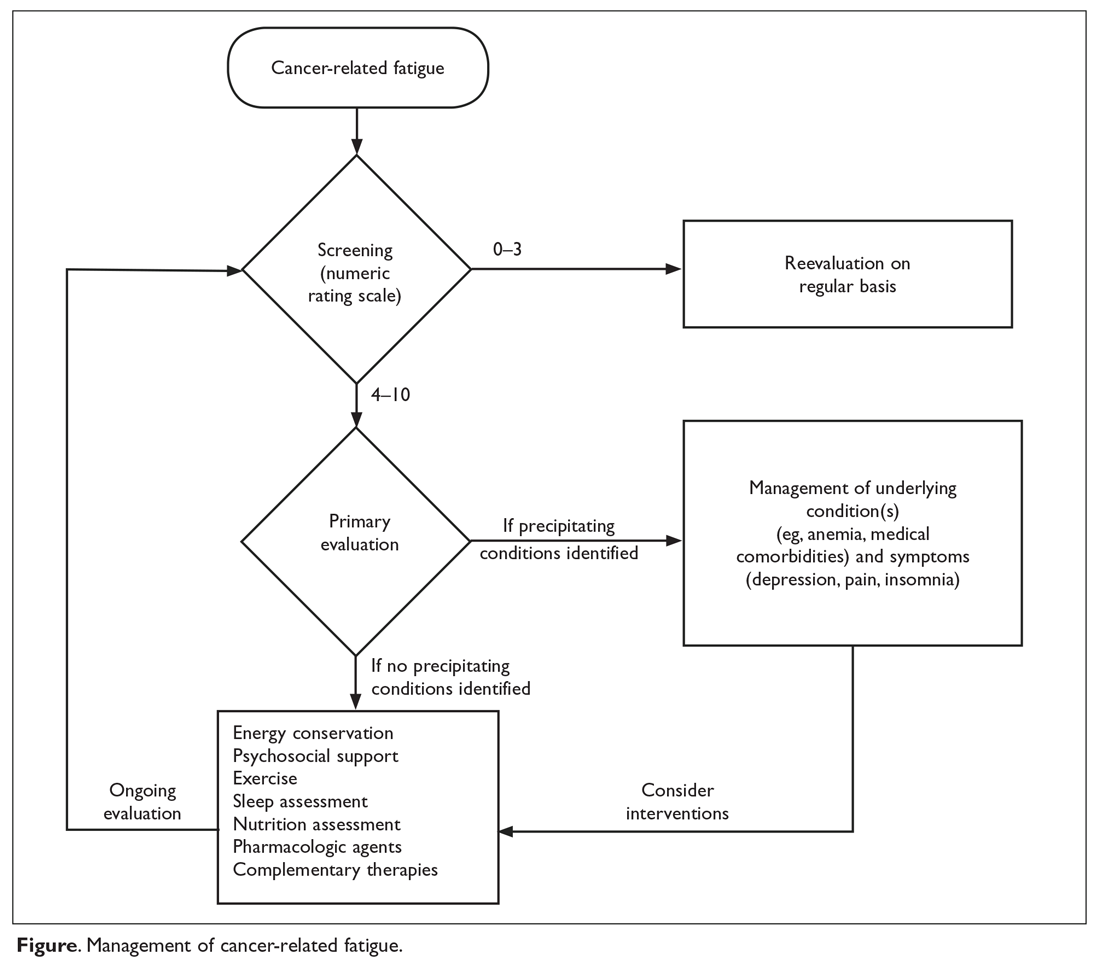
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below. 
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below.
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below.
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
Advance Care Planning: Making It Easier for Patients (and You)

With the number of aging Americans projected to grow dramatically in the next several years, the need for primary palliative care and advance care planning (ACP) is more important than ever. Patients and their families want and expect palliative care when needed, but initial conversations about ACP can be difficult for them. Appropriate timing in raising this subject and clear communication can give patients the opportunity, while they are still independent, to set their goals for medical care.
For the past several decades, political decisions and judicial cases have shaped palliative care as we know it today. And its shape is still evolving. In support of ACP, advocacy groups at a national level are developing models that practitioners can use to engage patients in setting goals. And Medicare is now reimbursing primary care providers for this work that they have been doing for years (although many still may not be billing for the service).
Finally, the busy primary care office may have its own set of challenges in addressing ACP. Our aim in this review is to identify the barriers we face and the solutions we can implement to make a difference in our patients’ end-of-life care planning.
LANDMARK EVENTS HAVE DEFINED ACP TODAY
In 1969, Luis Kutner, an Illinois attorney, proposed the idea of a “living will,” envisioned as a document specifying the types of treatment a person would be willing to receive were he or she unable at a later time to participate in making a decision.1 In 1976, California became the first state to give living wills the power of the law through the Natural Death Act.2
Throughout the 1970s and ‘80s, several high-profile court cases brought this idea into the national spotlight. In 1975, the New Jersey Supreme Court granted the parents of 21-year-old Karen Ann Quinlan the right to discontinue the treatment sustaining her in a persistent vegetative state. Ms. Quinlan was removed from the ventilator and lived nine more months before dying in a nursing home.
In 1983, age 25, Nancy Cruzan was involved in a motor vehicle accident that left her in a persistent vegetative state. She remained so until 1988, when her parents asked that her feeding tube be removed. The hospital refused, indicating that it would lead to her death. The family sued, and the case eventually went to the US Supreme Court in 1989.
In a 5-to-4 decision, the Supreme Court ruled that a state was legally able to require “clear and convincing evidence” of a patient’s wish for removal of life-sustaining therapies. Cruzan’s family was able to provide such evidence, and her artificial nutrition was withheld. She died 12 days later.
The Cruzan case was instrumental in furthering ACP, leading to the passage of the Patient Self Determination Act by Congress in 1990. All federally funded health care facilities were now required to educate patients of their rights in determining their medical care and to ask about advance directives.3 The ACP movement gained additional momentum from the landmark SUPPORT study that documented shortcomings in communication between physicians and patients/families about treatment preferences and end-of-life care in US hospitals.4
In the Terri Schiavo case, the patient’s husband disagreed with the life-sustaining decisions of his wife’s parents, given her persistent vegetative state and the fact that she had no chance of meaningful recovery. After a prolonged national debate, it was ultimately decided that the husband could elect to withhold artificial nutrition. (She died in 2005.) The Schiavo case, as well as the Institute of Medicine’s report on Dying in America, influenced Congress in 2016 to pass legislation funding ACP conversations.5
THE DEMONSTRATED BENEFITS OF ACP
When done comprehensively, ACP yields many benefits for patients and families and for the health care system. A systematic review demonstrated that, despite the few studies examining the economic cost of ACP, the process may lead to decreased health care costs in certain populations (nursing home residents, community-dwelling adults with dementia, and those living in high health care–spending regions) and at the very least does not increase health care costs.6 ACP has increased the number of do-not-resuscitate orders and has decreased hospitalizations, admissions to intensive care units, and rates of cardiopulmonary resuscitation, mechanical ventilation, and use of tube feeding.6-8
More noteworthy than the decrease in resource utilization and potential cost savings is the impact that ACP can have on a patient’s quality of life. Patients who receive aggressive care at the end of life tend to experience decreased quality of life compared with those receiving hospice care.7 Quality-of-life scores for patients in hospice improved with the length of enrollment in that care.7 When ACP discussions have taken place, the care patients receive at the end of life tends to conform more closely to their wishes and to increase family satisfaction.9-11
One reason that practitioners often give for not completing ACP is the fear of increasing patient or family anxiety or post-traumatic stress disorder (PTSD). However, studies have shown this concern to be unfounded.7,12 While ACP studies have not shown a decrease in rates of anxiety or PTSD, no study has shown an increase in these psychologic morbidities.8
Caveats to keep in mind. Not all studies have shown unambiguous benefits related to ACP. Among the systematic reviews previously noted, there was significant variability in quality of data. Additionally, some experts argue that the traditional view of ACP (ie, completion of a single advance directive/living will) is outdated and should be replaced with a method that prepares patients and families to anticipate “in-the-moment decision making.”13 While we still believe that completion of an advance directive is useful, the experts’ point is well taken, especially since many patients change their preferences over time (and typically toward more aggressive care).14,15 While the advance directive serves a role, it is more important to help patients recognize their goals and preferences and to facilitate ongoing discussions between the patient and his or her family/surrogate decision-maker and providers.
A SNAPSHOT OF PARTICIPATION IN ACP
Despite the ACP movement and the likely benefits associated with it, most individuals have not participated. Rates of completion do seem to be rising, but there is still room for improvement. Among all individuals older than 18, only 26.3% have an advance directive.16 In a cohort of older patients seen in an emergency department, only 40% had a living will, while nearly 54% had a designated health care power of attorney.17 Perhaps more alarming is the lack of ACP for those patients almost all providers would agree need it: the long-term care population. The National Center for Health Statistics has reported that only 28% of home health care patients, 65% of nursing home residents, and 88% of hospice patients have an advance directive on file.18
PROVIDER AND PATIENT BARRIERS TO ACP
If ACP can decrease resource utilization and improve caregiver compliance with a patient’s wishes for end of life, the obvious question is: Why isn’t it done more often? A longstanding barrier for providers has been that these types of discussions are time intensive and have not been billable. However, since January 1, 2016, we are now able to bill for these discussions. (More on this in a bit.) Providers do cite other barriers, though.
A recent systematic review showed that ACP is hindered by time constraints imposed by other clinical and administrative tasks that are heavily monitored.19 Barriers to engaging in ACP reported by patients include a reluctance to think about dying, a belief that family or providers will know what to do, difficulty understanding ACP forms, and the absence of a person who can serve as a surrogate decision-maker.20,21
NATIONAL MODELS TO HELP WITH IMPLEMENTATION
The percentage of individuals with an advance directive in the US has not increased significantly over the past decade.22 The lack of traction in completion and use of advance directives has led several authors to question the utility of this older model of ACP.22 Several experts in the field believe that more robust, ongoing goals-of-care conversations between patients, families, and providers are equally, or even more, important than the completion of actual advance directive documents.23,24
National models such as the POLST (Physician Orders for Life-Sustaining Treatment) paradigm have become popular in several states (www.polst.org). Intended for those with estimated life expectancy of less than one year, POLST is not an advance directive but a physician order for these seriously ill patients. Emergency medical service workers are legally able to follow a POLST document but not a living will or advance directive—a significant reason for those with end-stage illness to consider completing a POLST document with their health care provider. Programs such as “Respecting Choices” have incorporated POLST documentation as part of ongoing goals-of-care conversations between patients and health care providers (www.gundersenhealth.org/respecting-choices).
Many groups have developed products to encourage patients and their families to initiate conversations at home. An example is the Conversation Project, a free online resource available in multiple languages that can help break the ice for patients and get them talking about their wishes for end-of-life care (www.theconversationproject.org). It poses simple stimulating questions such as “What kind of role do you want to have in the decision-making process?” and “What are your concerns about treatment?”
HOW-TO TIPS FOR ACP IN OUTPATIENT SETTINGS
When approaching the topic of ACP with patients, it’s important to do so over time, starting as soon as possible with older patients and those with chronic illness that confers a high risk for significant morbidity or mortality. Assess each patient’s understanding of ACP and readiness to discuss the topic. Many patients think of ACP in the context of a document (eg, living will), so asking about the existence of a living will may help to start the conversation. Alternatively, consider inquiring about whether the patient has had experience with family or friends at the end of life or during a difficult medical situation, and whether the patient has thought about making personal plans for such a situation.25
When a patient is ready to have this conversation, your goal should be three-fold:26
- Help the patient articulate personal values, goals, and preferences.
- Ask the patient to formally assign health care power of attorney (POA) to a trusted individual or to name a surrogate decision-maker. Document this decision in the medical record.
- Help the patient translate expressed values into specific medical care plans, if applicable.
Because ACP conversations are often time consuming, it’s a good idea to schedule separate appointments to focus on this alone. If, however, a patient is unable to return for a dedicated ACP visit, a first step that can be completed in a reasonably short period would be choosing a surrogate decision-maker.
Helping a patient articulate personal values may be eased by asking such questions as, “Have you ever thought about what kind of care you would want if the time came when you could not make your own decisions?” or “What worries you the most about possibly not being able to make your own decisions?”27 If the patient is able to identify a surrogate decision-maker before the ACP appointment, ask that this person attend. A family member or close friend may remember instances in which the patient expressed health care preferences, and their presence can help to minimize gaps in communication.
Once the patient’s preferences are clear, document them in the medical record. Some preferences may be suitable for translation into POLST orders or an advance directive, but this is less important than the overall discussion. ACP should be an ongoing conversation, since a patient’s goals may change over time. And encourage the patient to share any desired change in plans with their surrogate decision-maker or update the POA document.
BE SURE TO BILL FOR ACP SERVICES
To encourage office-based providers to conduct ACP, the Centers for Medicare and Medicaid Services (CMS) implemented payment for CPT codes 99497 and 99498.
CPT code 99497 covers the first 30 minutes of face-to-face time with patients or their family members or medical decision-makers. This time can be used to discuss living wills or advance directives.
CPT code 99498 can be applied to each additional 30 minutes of ACP services. Typically, this billing code would be used as an add-on for a particular diagnosis, such as heart failure, chronic obstructive pulmonary disease, or pancreatic cancer.
CPT Code 99497 equates to 2.40 relative-value units (RVU) with an estimated payment of $85.99, while CPT code 99498 equates to 2.09 RVU with an estimated payment of $74.88.28
According to CMS, there is no annual limit to the number of times the ACP codes can be billed for a particular patient. And there are no restrictions regarding location of service, meaning a provider could perform this in an outpatient setting, an inpatient setting, or a long-term care facility. All health care providers are allowed to bill with this code. Also worth noting: You don’t need to complete any particular documentation for a visit to be billed as an ACP service. CMS provides a helpful Q & A at www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/FAQ-Advance-Care-Planning.pdf.
1. Kutner L. Due process of euthanasia: the living will, a proposal. Indiana Law J. 1969;44:539-554.
2. California Law Revision Commission. 2000 Health Care Decisions Law and Revised Power of Attorney Law. www.clrc.ca.gov/pub/Printed-Reports/Pub208.pdf. Accessed August 14, 2017.
3. H.R. 5067 - 101st Congress. Patient Self Determination Act of 1990. www.govtrack.us/congress/bills/101/hr5067. Accessed August 14, 2017.
4. The SUPPORT Principle Investigators. A controlled trial to improve care for seriously ill hospitalized patients. The Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments (SUPPORT). JAMA. 1995;274:1591-1598.
5. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
6. Dixon J, Matosevic T, Knapp M. The economic evidence for advance care planning: systematic review of evidence. Palliat Med. 2015;29:869-884.
7. Wright AA, Ray A, Mack JW, et al. Associations between end-of-life discussions, patient mental-health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
8. Brinkman-Stoppelenburg A, Rietjens JAC, van der Heide A. The effects of advance care planning on end-of-life care: a systematic review. Palliat Med. 2014;28:1000-1025.
9. Detering KM, Hancock AD, Reade MC, et al. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ. 2010;340:c1345.
10. Morrison RS, Chichin E, Carter J, et al. The effect of a social work intervention to enhance advance care planning documentation in the nursing home. J Am Geriatr Soc. 2005;53:290-294.
11. Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives. J Am Med Dir Assoc. 2006;7:339-344.
12. Walczak A, Butow PN, Bu S, et al. A systematic review of evidence for end-of-life communication interventions: who do they target, how are they structured and do they work? Patient Educ Couns. 2016;99:3-16.
13. Sudore RL, Fried TR. Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med. 2010;153:256-261.
14. Straton JB, Wang NY, Meoni LA, et al. Physical functioning, depression, and preferences for treatment at the end of life: the Johns Hopkins Precursors study. J Am Geriatr Soc. 2004;52:577-582.
15. Fried TR, Byers AL, Gallo WT, et al. Prospective study of health status preferences and changes in preferences over time in older adults. Arch Intern Med. 2006;166:890-895.
16. Rao JK, Anderson LA, Lin F, et al. Completion of advance directives among U.S. consumers. Am J Prev Med. 2014;46:65-70.
17. Grudzen CR, Buonocore P, Steinberg J, et al; AAHPM Research Committee Writing Group. Concordance of advance care plans with inpatient directives in the electronic medical record for older patients admitted from the emergency department. J Pain Symptom Manage. 2016;51:647-651.
18. Jones AL, Moss AJ, Harris-Kojetin LD. Use of advance directives in long-term care populations. NCHS Data Brief. 2011;(54):1-8.
19. Lund S, Richardson A, May C. Barriers to advance care planning at the end of life: an explanatory systematic review of implementation studies. PLoS One. 2015;10:e0116629.
20. Fried TR, Bullock K, Iannone L, et al. Understanding advance care planning as a process of health behavior change. J Am Geriatr Soc. 2009;57:1547-1555.
21. Schickedanz AD, Schillinger D, Landefeld CS, et al. A clinical framework for improving the advance care planning process: start with patients’ self-identified barriers. J Am Geriatr Soc. 2009;57:31-39.
22. Winter L, Parks SM, Diamond JJ. Ask a different question, get a different answer: why living wills are poor guides to care preferences at the end of life. J Palliat Med. 2010;13:567-572.
23. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. www.nap.edu/read/18748/chapter/1. Accessed August 14, 2017.
24. Sudore RL, Schickedanz AD, Landefeld CS, et al. Engagement in multiple steps of the advance care planning process: a descriptive study of diverse older adults. J Am Geriatr Soc. 2008;56:1006-1013.
25. McMahan RD, Knight SJ, Fried TR, et al. Advance care planning beyond advance directives: perspectives from patients and surrogates. J Pain Symptom Manage. 2013;46:355-365.
26. Lum HD, Sudore RL, Bekelman DB. Advance care planning in the elderly. Med Clin North Am. 2015;99:391-403.
27. Lum HD, Sudore RL. Advance care planning and goals of care communication in older adults with cardiovascular disease and multi-morbidity. Clin Geriatr Med. 2016;32:247-260.
28. American College of Physicians. Advanced Care Planning: Implementation for practices. www.acponline.org/system/files/documents/practice-resources/business-resources/payment/advance_care_planning_toolkit.pdf. Accessed August 14, 2017.
With the number of aging Americans projected to grow dramatically in the next several years, the need for primary palliative care and advance care planning (ACP) is more important than ever. Patients and their families want and expect palliative care when needed, but initial conversations about ACP can be difficult for them. Appropriate timing in raising this subject and clear communication can give patients the opportunity, while they are still independent, to set their goals for medical care.
For the past several decades, political decisions and judicial cases have shaped palliative care as we know it today. And its shape is still evolving. In support of ACP, advocacy groups at a national level are developing models that practitioners can use to engage patients in setting goals. And Medicare is now reimbursing primary care providers for this work that they have been doing for years (although many still may not be billing for the service).
Finally, the busy primary care office may have its own set of challenges in addressing ACP. Our aim in this review is to identify the barriers we face and the solutions we can implement to make a difference in our patients’ end-of-life care planning.
LANDMARK EVENTS HAVE DEFINED ACP TODAY
In 1969, Luis Kutner, an Illinois attorney, proposed the idea of a “living will,” envisioned as a document specifying the types of treatment a person would be willing to receive were he or she unable at a later time to participate in making a decision.1 In 1976, California became the first state to give living wills the power of the law through the Natural Death Act.2
Throughout the 1970s and ‘80s, several high-profile court cases brought this idea into the national spotlight. In 1975, the New Jersey Supreme Court granted the parents of 21-year-old Karen Ann Quinlan the right to discontinue the treatment sustaining her in a persistent vegetative state. Ms. Quinlan was removed from the ventilator and lived nine more months before dying in a nursing home.
In 1983, age 25, Nancy Cruzan was involved in a motor vehicle accident that left her in a persistent vegetative state. She remained so until 1988, when her parents asked that her feeding tube be removed. The hospital refused, indicating that it would lead to her death. The family sued, and the case eventually went to the US Supreme Court in 1989.
In a 5-to-4 decision, the Supreme Court ruled that a state was legally able to require “clear and convincing evidence” of a patient’s wish for removal of life-sustaining therapies. Cruzan’s family was able to provide such evidence, and her artificial nutrition was withheld. She died 12 days later.
The Cruzan case was instrumental in furthering ACP, leading to the passage of the Patient Self Determination Act by Congress in 1990. All federally funded health care facilities were now required to educate patients of their rights in determining their medical care and to ask about advance directives.3 The ACP movement gained additional momentum from the landmark SUPPORT study that documented shortcomings in communication between physicians and patients/families about treatment preferences and end-of-life care in US hospitals.4
In the Terri Schiavo case, the patient’s husband disagreed with the life-sustaining decisions of his wife’s parents, given her persistent vegetative state and the fact that she had no chance of meaningful recovery. After a prolonged national debate, it was ultimately decided that the husband could elect to withhold artificial nutrition. (She died in 2005.) The Schiavo case, as well as the Institute of Medicine’s report on Dying in America, influenced Congress in 2016 to pass legislation funding ACP conversations.5
THE DEMONSTRATED BENEFITS OF ACP
When done comprehensively, ACP yields many benefits for patients and families and for the health care system. A systematic review demonstrated that, despite the few studies examining the economic cost of ACP, the process may lead to decreased health care costs in certain populations (nursing home residents, community-dwelling adults with dementia, and those living in high health care–spending regions) and at the very least does not increase health care costs.6 ACP has increased the number of do-not-resuscitate orders and has decreased hospitalizations, admissions to intensive care units, and rates of cardiopulmonary resuscitation, mechanical ventilation, and use of tube feeding.6-8
More noteworthy than the decrease in resource utilization and potential cost savings is the impact that ACP can have on a patient’s quality of life. Patients who receive aggressive care at the end of life tend to experience decreased quality of life compared with those receiving hospice care.7 Quality-of-life scores for patients in hospice improved with the length of enrollment in that care.7 When ACP discussions have taken place, the care patients receive at the end of life tends to conform more closely to their wishes and to increase family satisfaction.9-11
One reason that practitioners often give for not completing ACP is the fear of increasing patient or family anxiety or post-traumatic stress disorder (PTSD). However, studies have shown this concern to be unfounded.7,12 While ACP studies have not shown a decrease in rates of anxiety or PTSD, no study has shown an increase in these psychologic morbidities.8
Caveats to keep in mind. Not all studies have shown unambiguous benefits related to ACP. Among the systematic reviews previously noted, there was significant variability in quality of data. Additionally, some experts argue that the traditional view of ACP (ie, completion of a single advance directive/living will) is outdated and should be replaced with a method that prepares patients and families to anticipate “in-the-moment decision making.”13 While we still believe that completion of an advance directive is useful, the experts’ point is well taken, especially since many patients change their preferences over time (and typically toward more aggressive care).14,15 While the advance directive serves a role, it is more important to help patients recognize their goals and preferences and to facilitate ongoing discussions between the patient and his or her family/surrogate decision-maker and providers.
A SNAPSHOT OF PARTICIPATION IN ACP
Despite the ACP movement and the likely benefits associated with it, most individuals have not participated. Rates of completion do seem to be rising, but there is still room for improvement. Among all individuals older than 18, only 26.3% have an advance directive.16 In a cohort of older patients seen in an emergency department, only 40% had a living will, while nearly 54% had a designated health care power of attorney.17 Perhaps more alarming is the lack of ACP for those patients almost all providers would agree need it: the long-term care population. The National Center for Health Statistics has reported that only 28% of home health care patients, 65% of nursing home residents, and 88% of hospice patients have an advance directive on file.18
PROVIDER AND PATIENT BARRIERS TO ACP
If ACP can decrease resource utilization and improve caregiver compliance with a patient’s wishes for end of life, the obvious question is: Why isn’t it done more often? A longstanding barrier for providers has been that these types of discussions are time intensive and have not been billable. However, since January 1, 2016, we are now able to bill for these discussions. (More on this in a bit.) Providers do cite other barriers, though.
A recent systematic review showed that ACP is hindered by time constraints imposed by other clinical and administrative tasks that are heavily monitored.19 Barriers to engaging in ACP reported by patients include a reluctance to think about dying, a belief that family or providers will know what to do, difficulty understanding ACP forms, and the absence of a person who can serve as a surrogate decision-maker.20,21
NATIONAL MODELS TO HELP WITH IMPLEMENTATION
The percentage of individuals with an advance directive in the US has not increased significantly over the past decade.22 The lack of traction in completion and use of advance directives has led several authors to question the utility of this older model of ACP.22 Several experts in the field believe that more robust, ongoing goals-of-care conversations between patients, families, and providers are equally, or even more, important than the completion of actual advance directive documents.23,24
National models such as the POLST (Physician Orders for Life-Sustaining Treatment) paradigm have become popular in several states (www.polst.org). Intended for those with estimated life expectancy of less than one year, POLST is not an advance directive but a physician order for these seriously ill patients. Emergency medical service workers are legally able to follow a POLST document but not a living will or advance directive—a significant reason for those with end-stage illness to consider completing a POLST document with their health care provider. Programs such as “Respecting Choices” have incorporated POLST documentation as part of ongoing goals-of-care conversations between patients and health care providers (www.gundersenhealth.org/respecting-choices).
Many groups have developed products to encourage patients and their families to initiate conversations at home. An example is the Conversation Project, a free online resource available in multiple languages that can help break the ice for patients and get them talking about their wishes for end-of-life care (www.theconversationproject.org). It poses simple stimulating questions such as “What kind of role do you want to have in the decision-making process?” and “What are your concerns about treatment?”
HOW-TO TIPS FOR ACP IN OUTPATIENT SETTINGS
When approaching the topic of ACP with patients, it’s important to do so over time, starting as soon as possible with older patients and those with chronic illness that confers a high risk for significant morbidity or mortality. Assess each patient’s understanding of ACP and readiness to discuss the topic. Many patients think of ACP in the context of a document (eg, living will), so asking about the existence of a living will may help to start the conversation. Alternatively, consider inquiring about whether the patient has had experience with family or friends at the end of life or during a difficult medical situation, and whether the patient has thought about making personal plans for such a situation.25
When a patient is ready to have this conversation, your goal should be three-fold:26
- Help the patient articulate personal values, goals, and preferences.
- Ask the patient to formally assign health care power of attorney (POA) to a trusted individual or to name a surrogate decision-maker. Document this decision in the medical record.
- Help the patient translate expressed values into specific medical care plans, if applicable.
Because ACP conversations are often time consuming, it’s a good idea to schedule separate appointments to focus on this alone. If, however, a patient is unable to return for a dedicated ACP visit, a first step that can be completed in a reasonably short period would be choosing a surrogate decision-maker.
Helping a patient articulate personal values may be eased by asking such questions as, “Have you ever thought about what kind of care you would want if the time came when you could not make your own decisions?” or “What worries you the most about possibly not being able to make your own decisions?”27 If the patient is able to identify a surrogate decision-maker before the ACP appointment, ask that this person attend. A family member or close friend may remember instances in which the patient expressed health care preferences, and their presence can help to minimize gaps in communication.
Once the patient’s preferences are clear, document them in the medical record. Some preferences may be suitable for translation into POLST orders or an advance directive, but this is less important than the overall discussion. ACP should be an ongoing conversation, since a patient’s goals may change over time. And encourage the patient to share any desired change in plans with their surrogate decision-maker or update the POA document.
BE SURE TO BILL FOR ACP SERVICES
To encourage office-based providers to conduct ACP, the Centers for Medicare and Medicaid Services (CMS) implemented payment for CPT codes 99497 and 99498.
CPT code 99497 covers the first 30 minutes of face-to-face time with patients or their family members or medical decision-makers. This time can be used to discuss living wills or advance directives.
CPT code 99498 can be applied to each additional 30 minutes of ACP services. Typically, this billing code would be used as an add-on for a particular diagnosis, such as heart failure, chronic obstructive pulmonary disease, or pancreatic cancer.
CPT Code 99497 equates to 2.40 relative-value units (RVU) with an estimated payment of $85.99, while CPT code 99498 equates to 2.09 RVU with an estimated payment of $74.88.28
According to CMS, there is no annual limit to the number of times the ACP codes can be billed for a particular patient. And there are no restrictions regarding location of service, meaning a provider could perform this in an outpatient setting, an inpatient setting, or a long-term care facility. All health care providers are allowed to bill with this code. Also worth noting: You don’t need to complete any particular documentation for a visit to be billed as an ACP service. CMS provides a helpful Q & A at www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/FAQ-Advance-Care-Planning.pdf.
With the number of aging Americans projected to grow dramatically in the next several years, the need for primary palliative care and advance care planning (ACP) is more important than ever. Patients and their families want and expect palliative care when needed, but initial conversations about ACP can be difficult for them. Appropriate timing in raising this subject and clear communication can give patients the opportunity, while they are still independent, to set their goals for medical care.
For the past several decades, political decisions and judicial cases have shaped palliative care as we know it today. And its shape is still evolving. In support of ACP, advocacy groups at a national level are developing models that practitioners can use to engage patients in setting goals. And Medicare is now reimbursing primary care providers for this work that they have been doing for years (although many still may not be billing for the service).
Finally, the busy primary care office may have its own set of challenges in addressing ACP. Our aim in this review is to identify the barriers we face and the solutions we can implement to make a difference in our patients’ end-of-life care planning.
LANDMARK EVENTS HAVE DEFINED ACP TODAY
In 1969, Luis Kutner, an Illinois attorney, proposed the idea of a “living will,” envisioned as a document specifying the types of treatment a person would be willing to receive were he or she unable at a later time to participate in making a decision.1 In 1976, California became the first state to give living wills the power of the law through the Natural Death Act.2
Throughout the 1970s and ‘80s, several high-profile court cases brought this idea into the national spotlight. In 1975, the New Jersey Supreme Court granted the parents of 21-year-old Karen Ann Quinlan the right to discontinue the treatment sustaining her in a persistent vegetative state. Ms. Quinlan was removed from the ventilator and lived nine more months before dying in a nursing home.
In 1983, age 25, Nancy Cruzan was involved in a motor vehicle accident that left her in a persistent vegetative state. She remained so until 1988, when her parents asked that her feeding tube be removed. The hospital refused, indicating that it would lead to her death. The family sued, and the case eventually went to the US Supreme Court in 1989.
In a 5-to-4 decision, the Supreme Court ruled that a state was legally able to require “clear and convincing evidence” of a patient’s wish for removal of life-sustaining therapies. Cruzan’s family was able to provide such evidence, and her artificial nutrition was withheld. She died 12 days later.
The Cruzan case was instrumental in furthering ACP, leading to the passage of the Patient Self Determination Act by Congress in 1990. All federally funded health care facilities were now required to educate patients of their rights in determining their medical care and to ask about advance directives.3 The ACP movement gained additional momentum from the landmark SUPPORT study that documented shortcomings in communication between physicians and patients/families about treatment preferences and end-of-life care in US hospitals.4
In the Terri Schiavo case, the patient’s husband disagreed with the life-sustaining decisions of his wife’s parents, given her persistent vegetative state and the fact that she had no chance of meaningful recovery. After a prolonged national debate, it was ultimately decided that the husband could elect to withhold artificial nutrition. (She died in 2005.) The Schiavo case, as well as the Institute of Medicine’s report on Dying in America, influenced Congress in 2016 to pass legislation funding ACP conversations.5
THE DEMONSTRATED BENEFITS OF ACP
When done comprehensively, ACP yields many benefits for patients and families and for the health care system. A systematic review demonstrated that, despite the few studies examining the economic cost of ACP, the process may lead to decreased health care costs in certain populations (nursing home residents, community-dwelling adults with dementia, and those living in high health care–spending regions) and at the very least does not increase health care costs.6 ACP has increased the number of do-not-resuscitate orders and has decreased hospitalizations, admissions to intensive care units, and rates of cardiopulmonary resuscitation, mechanical ventilation, and use of tube feeding.6-8
More noteworthy than the decrease in resource utilization and potential cost savings is the impact that ACP can have on a patient’s quality of life. Patients who receive aggressive care at the end of life tend to experience decreased quality of life compared with those receiving hospice care.7 Quality-of-life scores for patients in hospice improved with the length of enrollment in that care.7 When ACP discussions have taken place, the care patients receive at the end of life tends to conform more closely to their wishes and to increase family satisfaction.9-11
One reason that practitioners often give for not completing ACP is the fear of increasing patient or family anxiety or post-traumatic stress disorder (PTSD). However, studies have shown this concern to be unfounded.7,12 While ACP studies have not shown a decrease in rates of anxiety or PTSD, no study has shown an increase in these psychologic morbidities.8
Caveats to keep in mind. Not all studies have shown unambiguous benefits related to ACP. Among the systematic reviews previously noted, there was significant variability in quality of data. Additionally, some experts argue that the traditional view of ACP (ie, completion of a single advance directive/living will) is outdated and should be replaced with a method that prepares patients and families to anticipate “in-the-moment decision making.”13 While we still believe that completion of an advance directive is useful, the experts’ point is well taken, especially since many patients change their preferences over time (and typically toward more aggressive care).14,15 While the advance directive serves a role, it is more important to help patients recognize their goals and preferences and to facilitate ongoing discussions between the patient and his or her family/surrogate decision-maker and providers.
A SNAPSHOT OF PARTICIPATION IN ACP
Despite the ACP movement and the likely benefits associated with it, most individuals have not participated. Rates of completion do seem to be rising, but there is still room for improvement. Among all individuals older than 18, only 26.3% have an advance directive.16 In a cohort of older patients seen in an emergency department, only 40% had a living will, while nearly 54% had a designated health care power of attorney.17 Perhaps more alarming is the lack of ACP for those patients almost all providers would agree need it: the long-term care population. The National Center for Health Statistics has reported that only 28% of home health care patients, 65% of nursing home residents, and 88% of hospice patients have an advance directive on file.18
PROVIDER AND PATIENT BARRIERS TO ACP
If ACP can decrease resource utilization and improve caregiver compliance with a patient’s wishes for end of life, the obvious question is: Why isn’t it done more often? A longstanding barrier for providers has been that these types of discussions are time intensive and have not been billable. However, since January 1, 2016, we are now able to bill for these discussions. (More on this in a bit.) Providers do cite other barriers, though.
A recent systematic review showed that ACP is hindered by time constraints imposed by other clinical and administrative tasks that are heavily monitored.19 Barriers to engaging in ACP reported by patients include a reluctance to think about dying, a belief that family or providers will know what to do, difficulty understanding ACP forms, and the absence of a person who can serve as a surrogate decision-maker.20,21
NATIONAL MODELS TO HELP WITH IMPLEMENTATION
The percentage of individuals with an advance directive in the US has not increased significantly over the past decade.22 The lack of traction in completion and use of advance directives has led several authors to question the utility of this older model of ACP.22 Several experts in the field believe that more robust, ongoing goals-of-care conversations between patients, families, and providers are equally, or even more, important than the completion of actual advance directive documents.23,24
National models such as the POLST (Physician Orders for Life-Sustaining Treatment) paradigm have become popular in several states (www.polst.org). Intended for those with estimated life expectancy of less than one year, POLST is not an advance directive but a physician order for these seriously ill patients. Emergency medical service workers are legally able to follow a POLST document but not a living will or advance directive—a significant reason for those with end-stage illness to consider completing a POLST document with their health care provider. Programs such as “Respecting Choices” have incorporated POLST documentation as part of ongoing goals-of-care conversations between patients and health care providers (www.gundersenhealth.org/respecting-choices).
Many groups have developed products to encourage patients and their families to initiate conversations at home. An example is the Conversation Project, a free online resource available in multiple languages that can help break the ice for patients and get them talking about their wishes for end-of-life care (www.theconversationproject.org). It poses simple stimulating questions such as “What kind of role do you want to have in the decision-making process?” and “What are your concerns about treatment?”
HOW-TO TIPS FOR ACP IN OUTPATIENT SETTINGS
When approaching the topic of ACP with patients, it’s important to do so over time, starting as soon as possible with older patients and those with chronic illness that confers a high risk for significant morbidity or mortality. Assess each patient’s understanding of ACP and readiness to discuss the topic. Many patients think of ACP in the context of a document (eg, living will), so asking about the existence of a living will may help to start the conversation. Alternatively, consider inquiring about whether the patient has had experience with family or friends at the end of life or during a difficult medical situation, and whether the patient has thought about making personal plans for such a situation.25
When a patient is ready to have this conversation, your goal should be three-fold:26
- Help the patient articulate personal values, goals, and preferences.
- Ask the patient to formally assign health care power of attorney (POA) to a trusted individual or to name a surrogate decision-maker. Document this decision in the medical record.
- Help the patient translate expressed values into specific medical care plans, if applicable.
Because ACP conversations are often time consuming, it’s a good idea to schedule separate appointments to focus on this alone. If, however, a patient is unable to return for a dedicated ACP visit, a first step that can be completed in a reasonably short period would be choosing a surrogate decision-maker.
Helping a patient articulate personal values may be eased by asking such questions as, “Have you ever thought about what kind of care you would want if the time came when you could not make your own decisions?” or “What worries you the most about possibly not being able to make your own decisions?”27 If the patient is able to identify a surrogate decision-maker before the ACP appointment, ask that this person attend. A family member or close friend may remember instances in which the patient expressed health care preferences, and their presence can help to minimize gaps in communication.
Once the patient’s preferences are clear, document them in the medical record. Some preferences may be suitable for translation into POLST orders or an advance directive, but this is less important than the overall discussion. ACP should be an ongoing conversation, since a patient’s goals may change over time. And encourage the patient to share any desired change in plans with their surrogate decision-maker or update the POA document.
BE SURE TO BILL FOR ACP SERVICES
To encourage office-based providers to conduct ACP, the Centers for Medicare and Medicaid Services (CMS) implemented payment for CPT codes 99497 and 99498.
CPT code 99497 covers the first 30 minutes of face-to-face time with patients or their family members or medical decision-makers. This time can be used to discuss living wills or advance directives.
CPT code 99498 can be applied to each additional 30 minutes of ACP services. Typically, this billing code would be used as an add-on for a particular diagnosis, such as heart failure, chronic obstructive pulmonary disease, or pancreatic cancer.
CPT Code 99497 equates to 2.40 relative-value units (RVU) with an estimated payment of $85.99, while CPT code 99498 equates to 2.09 RVU with an estimated payment of $74.88.28
According to CMS, there is no annual limit to the number of times the ACP codes can be billed for a particular patient. And there are no restrictions regarding location of service, meaning a provider could perform this in an outpatient setting, an inpatient setting, or a long-term care facility. All health care providers are allowed to bill with this code. Also worth noting: You don’t need to complete any particular documentation for a visit to be billed as an ACP service. CMS provides a helpful Q & A at www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/FAQ-Advance-Care-Planning.pdf.
1. Kutner L. Due process of euthanasia: the living will, a proposal. Indiana Law J. 1969;44:539-554.
2. California Law Revision Commission. 2000 Health Care Decisions Law and Revised Power of Attorney Law. www.clrc.ca.gov/pub/Printed-Reports/Pub208.pdf. Accessed August 14, 2017.
3. H.R. 5067 - 101st Congress. Patient Self Determination Act of 1990. www.govtrack.us/congress/bills/101/hr5067. Accessed August 14, 2017.
4. The SUPPORT Principle Investigators. A controlled trial to improve care for seriously ill hospitalized patients. The Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments (SUPPORT). JAMA. 1995;274:1591-1598.
5. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
6. Dixon J, Matosevic T, Knapp M. The economic evidence for advance care planning: systematic review of evidence. Palliat Med. 2015;29:869-884.
7. Wright AA, Ray A, Mack JW, et al. Associations between end-of-life discussions, patient mental-health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
8. Brinkman-Stoppelenburg A, Rietjens JAC, van der Heide A. The effects of advance care planning on end-of-life care: a systematic review. Palliat Med. 2014;28:1000-1025.
9. Detering KM, Hancock AD, Reade MC, et al. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ. 2010;340:c1345.
10. Morrison RS, Chichin E, Carter J, et al. The effect of a social work intervention to enhance advance care planning documentation in the nursing home. J Am Geriatr Soc. 2005;53:290-294.
11. Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives. J Am Med Dir Assoc. 2006;7:339-344.
12. Walczak A, Butow PN, Bu S, et al. A systematic review of evidence for end-of-life communication interventions: who do they target, how are they structured and do they work? Patient Educ Couns. 2016;99:3-16.
13. Sudore RL, Fried TR. Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med. 2010;153:256-261.
14. Straton JB, Wang NY, Meoni LA, et al. Physical functioning, depression, and preferences for treatment at the end of life: the Johns Hopkins Precursors study. J Am Geriatr Soc. 2004;52:577-582.
15. Fried TR, Byers AL, Gallo WT, et al. Prospective study of health status preferences and changes in preferences over time in older adults. Arch Intern Med. 2006;166:890-895.
16. Rao JK, Anderson LA, Lin F, et al. Completion of advance directives among U.S. consumers. Am J Prev Med. 2014;46:65-70.
17. Grudzen CR, Buonocore P, Steinberg J, et al; AAHPM Research Committee Writing Group. Concordance of advance care plans with inpatient directives in the electronic medical record for older patients admitted from the emergency department. J Pain Symptom Manage. 2016;51:647-651.
18. Jones AL, Moss AJ, Harris-Kojetin LD. Use of advance directives in long-term care populations. NCHS Data Brief. 2011;(54):1-8.
19. Lund S, Richardson A, May C. Barriers to advance care planning at the end of life: an explanatory systematic review of implementation studies. PLoS One. 2015;10:e0116629.
20. Fried TR, Bullock K, Iannone L, et al. Understanding advance care planning as a process of health behavior change. J Am Geriatr Soc. 2009;57:1547-1555.
21. Schickedanz AD, Schillinger D, Landefeld CS, et al. A clinical framework for improving the advance care planning process: start with patients’ self-identified barriers. J Am Geriatr Soc. 2009;57:31-39.
22. Winter L, Parks SM, Diamond JJ. Ask a different question, get a different answer: why living wills are poor guides to care preferences at the end of life. J Palliat Med. 2010;13:567-572.
23. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. www.nap.edu/read/18748/chapter/1. Accessed August 14, 2017.
24. Sudore RL, Schickedanz AD, Landefeld CS, et al. Engagement in multiple steps of the advance care planning process: a descriptive study of diverse older adults. J Am Geriatr Soc. 2008;56:1006-1013.
25. McMahan RD, Knight SJ, Fried TR, et al. Advance care planning beyond advance directives: perspectives from patients and surrogates. J Pain Symptom Manage. 2013;46:355-365.
26. Lum HD, Sudore RL, Bekelman DB. Advance care planning in the elderly. Med Clin North Am. 2015;99:391-403.
27. Lum HD, Sudore RL. Advance care planning and goals of care communication in older adults with cardiovascular disease and multi-morbidity. Clin Geriatr Med. 2016;32:247-260.
28. American College of Physicians. Advanced Care Planning: Implementation for practices. www.acponline.org/system/files/documents/practice-resources/business-resources/payment/advance_care_planning_toolkit.pdf. Accessed August 14, 2017.
1. Kutner L. Due process of euthanasia: the living will, a proposal. Indiana Law J. 1969;44:539-554.
2. California Law Revision Commission. 2000 Health Care Decisions Law and Revised Power of Attorney Law. www.clrc.ca.gov/pub/Printed-Reports/Pub208.pdf. Accessed August 14, 2017.
3. H.R. 5067 - 101st Congress. Patient Self Determination Act of 1990. www.govtrack.us/congress/bills/101/hr5067. Accessed August 14, 2017.
4. The SUPPORT Principle Investigators. A controlled trial to improve care for seriously ill hospitalized patients. The Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments (SUPPORT). JAMA. 1995;274:1591-1598.
5. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
6. Dixon J, Matosevic T, Knapp M. The economic evidence for advance care planning: systematic review of evidence. Palliat Med. 2015;29:869-884.
7. Wright AA, Ray A, Mack JW, et al. Associations between end-of-life discussions, patient mental-health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300:1665-1673.
8. Brinkman-Stoppelenburg A, Rietjens JAC, van der Heide A. The effects of advance care planning on end-of-life care: a systematic review. Palliat Med. 2014;28:1000-1025.
9. Detering KM, Hancock AD, Reade MC, et al. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ. 2010;340:c1345.
10. Morrison RS, Chichin E, Carter J, et al. The effect of a social work intervention to enhance advance care planning documentation in the nursing home. J Am Geriatr Soc. 2005;53:290-294.
11. Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives. J Am Med Dir Assoc. 2006;7:339-344.
12. Walczak A, Butow PN, Bu S, et al. A systematic review of evidence for end-of-life communication interventions: who do they target, how are they structured and do they work? Patient Educ Couns. 2016;99:3-16.
13. Sudore RL, Fried TR. Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med. 2010;153:256-261.
14. Straton JB, Wang NY, Meoni LA, et al. Physical functioning, depression, and preferences for treatment at the end of life: the Johns Hopkins Precursors study. J Am Geriatr Soc. 2004;52:577-582.
15. Fried TR, Byers AL, Gallo WT, et al. Prospective study of health status preferences and changes in preferences over time in older adults. Arch Intern Med. 2006;166:890-895.
16. Rao JK, Anderson LA, Lin F, et al. Completion of advance directives among U.S. consumers. Am J Prev Med. 2014;46:65-70.
17. Grudzen CR, Buonocore P, Steinberg J, et al; AAHPM Research Committee Writing Group. Concordance of advance care plans with inpatient directives in the electronic medical record for older patients admitted from the emergency department. J Pain Symptom Manage. 2016;51:647-651.
18. Jones AL, Moss AJ, Harris-Kojetin LD. Use of advance directives in long-term care populations. NCHS Data Brief. 2011;(54):1-8.
19. Lund S, Richardson A, May C. Barriers to advance care planning at the end of life: an explanatory systematic review of implementation studies. PLoS One. 2015;10:e0116629.
20. Fried TR, Bullock K, Iannone L, et al. Understanding advance care planning as a process of health behavior change. J Am Geriatr Soc. 2009;57:1547-1555.
21. Schickedanz AD, Schillinger D, Landefeld CS, et al. A clinical framework for improving the advance care planning process: start with patients’ self-identified barriers. J Am Geriatr Soc. 2009;57:31-39.
22. Winter L, Parks SM, Diamond JJ. Ask a different question, get a different answer: why living wills are poor guides to care preferences at the end of life. J Palliat Med. 2010;13:567-572.
23. Institute of Medicine. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. www.nap.edu/read/18748/chapter/1. Accessed August 14, 2017.
24. Sudore RL, Schickedanz AD, Landefeld CS, et al. Engagement in multiple steps of the advance care planning process: a descriptive study of diverse older adults. J Am Geriatr Soc. 2008;56:1006-1013.
25. McMahan RD, Knight SJ, Fried TR, et al. Advance care planning beyond advance directives: perspectives from patients and surrogates. J Pain Symptom Manage. 2013;46:355-365.
26. Lum HD, Sudore RL, Bekelman DB. Advance care planning in the elderly. Med Clin North Am. 2015;99:391-403.
27. Lum HD, Sudore RL. Advance care planning and goals of care communication in older adults with cardiovascular disease and multi-morbidity. Clin Geriatr Med. 2016;32:247-260.
28. American College of Physicians. Advanced Care Planning: Implementation for practices. www.acponline.org/system/files/documents/practice-resources/business-resources/payment/advance_care_planning_toolkit.pdf. Accessed August 14, 2017.
First EDition: ED Visits by Older Patients Increase in the Weeks After a Disaster
ED Visits by Older Patients Increase in the Weeks After a Disaster
BY KELLIE DESANTIS
Visits to an ED by adults ages 65 years and older increase significantly in the weeks following a disaster, according to a study published in Disaster Medicine and Public Health Preparedness.1
Older adults are vulnerable to the effects of disasters because of their diminished ability to adequately prepare for and respond to the effects of a disaster. Older adults suffering from visual, auditory, proprioceptive, and cognitive impairments are especially vulnerable and have the most difficulty complying with evacuation and preparatory warnings. Individuals with multiple chronic diseases, living in long-term care facilities or suffering from cognitive impairments are among the most vulnerable.
To better understand the impact of natural disasters on this vulnerable population, researchers examined the effects of the 2012 disaster, Hurricane Sandy, on older adults living in New York City (NYC) during the disaster. Researchers turned to the New York State Department of Health (NYSDOH) for data. The NYSDOH compiles a comprehensive database of claims from all ED visits in the Statewide Planning and Research Cooperative System (SPARCS), which is the most complete source for ED utilization in New York state, and includes primary and secondary diagnosis codes and patient addresses.
Researchers evaluated ED utilization by adults 65 years and older in the weeks immediately before and after the Hurricane Sandy landfall. They excluded patients who lived in a nursing home, were incarcerated, or visited an ED associated with a specialty hospital (surgical subspecialty, oncological, or Veterans Administration). By using geographic distribution information available from SPARCS and the NYC Office of Emergency Management evacuation zones, researchers were able to compare the ED utilization for older adults living in the evacuation zones before the landfall of Hurricane Sandy and in the weeks shortly after the storm.
The analysis revealed a significant increase in ED utilization for older adults living in the evacuation zones in the 3 weeks after the storm compared to ED use before the storm. The number of weekly ED visits by older adults from all evacuation zones was 9,852 in the weeks before Hurricane Sandy and increased in the first week after the storm to 10,073. Among the most severely impacted were older adults in evacuation zone one, where ED utilization increased from 552 visits to 1,111 visits. The number of ED visits remained elevated for 3 weeks after the storm but returned to normal by the fourth week.
Researchers suggested several reasons for this increase in ED visits, including seeking refuge in the ED as a result of homelessness due to the disaster, the interruption of ongoing care for chronic illness, environmental exposure, and the lack of preparation for the lasting effect of the disaster.
To improve the response to such a disaster in the future, a NYC Hurricane Sandy Assessment report2 recommended developing a door-to-door service task force for older adults to improve preparedness for this vulnerable population. The task force would be responsible for implementing an action plan to ensure that healthcare services, communication, and provisions for this population continue without interruption in the weeks following a disaster. Legal and regulatory changes would allow for Medicare recipients to be eligible for "early medication refill" and pre-storm "early dialysis" programs to improve the continuity of care of the chronically ill.
1. Malik S, Lee DC, Doran KM, et al. Vulnerability of older adults in disasters: emergency department utilization by geriatric patients after hurricane sandy. Disaster Med Public Health Prep. 2017:1-10. doi:10.1017/dmp.2017.44
2. The City of New York, Office of the Mayor. Hurricane Sandy After Action Report. Published May 2013. http://www.nyc.gov/html/recovery/downlaods/pdf/sandy_aar_5.2.13.pdf. Accessed September 1, 2017.
Digital Rectal Examination of ED Patients with Acute GI Bleeding Cuts Rates of Admissions, Pharmacotherapy, and Endoscopy
BY JEFF BAUER
Patients presenting to the ED with acute gastrointestinal (GI) bleeding who receive a digital rectal examination have significantly lower rates of admissions, pharmacotherapy, and endoscopies, according to a retrospective study published in The American Journal of Medicine. Digital rectal examinations are an established part of the physical examination of a patient with GI bleeding, but physicians often are reluctant to conduct such examinations. Previous studies have found that 10% to 35% of patients with acute GI bleeding do not receive digital rectal examinations.
In the current study, researchers analyzed data from the electronic health records (EHRs) of patients ages 18 years and older who presented to the ED of a single institution with acute GI bleeding, as identified by International Classification of Diseases, Ninth Edition codes. They collected patients’ medical histories, demographic information, and clinical and laboratory data. ED clinician notes were used to determine which patients received a digital rectal examination. The outcomes researchers assessed were hospital admission, intensive care unit (ICU) admission, initiation of medical therapy (a proton pump inhibitor or octreotide), inpatient endoscopy (upper endoscopy or colonoscopy), and packed red blood cell (RBC) transfusion.
Overall, 1237 patients presented with acute GI bleeding. Most patients were Caucasian (49.2%) or Hispanic (38.4%), 44.9% were female, and the median age was 53 years.
Slightly more than one-half of patients (55.6%) received a digital rectal examination. In total, 736 patients were admitted—including 222 admissions to the ICU; 751 were started on a proton pump inhibitor or octreotide, 274 underwent endoscopy, and 321 received an RBC transfusion.
Patients were more likely to receive a digital rectal examination if they were older, Hispanic, or receiving an anticoagulant. Patients were less likely to undergo such examinations if they presented with altered mental status or hematemesis. Compared to patients who did not receive a digital rectal examination, those who did were significantly less likely to be admitted to the hospital (P = .004), to be starting on medical therapy (P = .04), or to undergo endoscopy (P = .02). There were no significant differences between these two groups in terms of ICU admissions, gastroenterology consultations, or transfusions.
Researchers suggested that the 44% rate of patients with acute GI bleeding who did not receive digital rectal examinations was higher than had been reported in previous studies. The difference had been the result of relying solely on ED clinician notes for this data, without including notes from admitting or consulting clinicians. The authors also were unable to determine the reasons these examinations were not conducted.
Shrestha MP, Borgstrom M, Trowers E. Digital rectal examination reduces hospital admissions, endoscopies, and medical therapy in patients with acute gastrointestinal bleeding. Am J Med. 2017;130(7):819-825. doi: 10.1016/j.amjmed.2017.01.036.
ED Visits by Older Patients Increase in the Weeks After a Disaster
BY KELLIE DESANTIS
Visits to an ED by adults ages 65 years and older increase significantly in the weeks following a disaster, according to a study published in Disaster Medicine and Public Health Preparedness.1
Older adults are vulnerable to the effects of disasters because of their diminished ability to adequately prepare for and respond to the effects of a disaster. Older adults suffering from visual, auditory, proprioceptive, and cognitive impairments are especially vulnerable and have the most difficulty complying with evacuation and preparatory warnings. Individuals with multiple chronic diseases, living in long-term care facilities or suffering from cognitive impairments are among the most vulnerable.
To better understand the impact of natural disasters on this vulnerable population, researchers examined the effects of the 2012 disaster, Hurricane Sandy, on older adults living in New York City (NYC) during the disaster. Researchers turned to the New York State Department of Health (NYSDOH) for data. The NYSDOH compiles a comprehensive database of claims from all ED visits in the Statewide Planning and Research Cooperative System (SPARCS), which is the most complete source for ED utilization in New York state, and includes primary and secondary diagnosis codes and patient addresses.
Researchers evaluated ED utilization by adults 65 years and older in the weeks immediately before and after the Hurricane Sandy landfall. They excluded patients who lived in a nursing home, were incarcerated, or visited an ED associated with a specialty hospital (surgical subspecialty, oncological, or Veterans Administration). By using geographic distribution information available from SPARCS and the NYC Office of Emergency Management evacuation zones, researchers were able to compare the ED utilization for older adults living in the evacuation zones before the landfall of Hurricane Sandy and in the weeks shortly after the storm.
The analysis revealed a significant increase in ED utilization for older adults living in the evacuation zones in the 3 weeks after the storm compared to ED use before the storm. The number of weekly ED visits by older adults from all evacuation zones was 9,852 in the weeks before Hurricane Sandy and increased in the first week after the storm to 10,073. Among the most severely impacted were older adults in evacuation zone one, where ED utilization increased from 552 visits to 1,111 visits. The number of ED visits remained elevated for 3 weeks after the storm but returned to normal by the fourth week.
Researchers suggested several reasons for this increase in ED visits, including seeking refuge in the ED as a result of homelessness due to the disaster, the interruption of ongoing care for chronic illness, environmental exposure, and the lack of preparation for the lasting effect of the disaster.
To improve the response to such a disaster in the future, a NYC Hurricane Sandy Assessment report2 recommended developing a door-to-door service task force for older adults to improve preparedness for this vulnerable population. The task force would be responsible for implementing an action plan to ensure that healthcare services, communication, and provisions for this population continue without interruption in the weeks following a disaster. Legal and regulatory changes would allow for Medicare recipients to be eligible for "early medication refill" and pre-storm "early dialysis" programs to improve the continuity of care of the chronically ill.
1. Malik S, Lee DC, Doran KM, et al. Vulnerability of older adults in disasters: emergency department utilization by geriatric patients after hurricane sandy. Disaster Med Public Health Prep. 2017:1-10. doi:10.1017/dmp.2017.44
2. The City of New York, Office of the Mayor. Hurricane Sandy After Action Report. Published May 2013. http://www.nyc.gov/html/recovery/downlaods/pdf/sandy_aar_5.2.13.pdf. Accessed September 1, 2017.
Digital Rectal Examination of ED Patients with Acute GI Bleeding Cuts Rates of Admissions, Pharmacotherapy, and Endoscopy
BY JEFF BAUER
Patients presenting to the ED with acute gastrointestinal (GI) bleeding who receive a digital rectal examination have significantly lower rates of admissions, pharmacotherapy, and endoscopies, according to a retrospective study published in The American Journal of Medicine. Digital rectal examinations are an established part of the physical examination of a patient with GI bleeding, but physicians often are reluctant to conduct such examinations. Previous studies have found that 10% to 35% of patients with acute GI bleeding do not receive digital rectal examinations.
In the current study, researchers analyzed data from the electronic health records (EHRs) of patients ages 18 years and older who presented to the ED of a single institution with acute GI bleeding, as identified by International Classification of Diseases, Ninth Edition codes. They collected patients’ medical histories, demographic information, and clinical and laboratory data. ED clinician notes were used to determine which patients received a digital rectal examination. The outcomes researchers assessed were hospital admission, intensive care unit (ICU) admission, initiation of medical therapy (a proton pump inhibitor or octreotide), inpatient endoscopy (upper endoscopy or colonoscopy), and packed red blood cell (RBC) transfusion.
Overall, 1237 patients presented with acute GI bleeding. Most patients were Caucasian (49.2%) or Hispanic (38.4%), 44.9% were female, and the median age was 53 years.
Slightly more than one-half of patients (55.6%) received a digital rectal examination. In total, 736 patients were admitted—including 222 admissions to the ICU; 751 were started on a proton pump inhibitor or octreotide, 274 underwent endoscopy, and 321 received an RBC transfusion.
Patients were more likely to receive a digital rectal examination if they were older, Hispanic, or receiving an anticoagulant. Patients were less likely to undergo such examinations if they presented with altered mental status or hematemesis. Compared to patients who did not receive a digital rectal examination, those who did were significantly less likely to be admitted to the hospital (P = .004), to be starting on medical therapy (P = .04), or to undergo endoscopy (P = .02). There were no significant differences between these two groups in terms of ICU admissions, gastroenterology consultations, or transfusions.
Researchers suggested that the 44% rate of patients with acute GI bleeding who did not receive digital rectal examinations was higher than had been reported in previous studies. The difference had been the result of relying solely on ED clinician notes for this data, without including notes from admitting or consulting clinicians. The authors also were unable to determine the reasons these examinations were not conducted.
Shrestha MP, Borgstrom M, Trowers E. Digital rectal examination reduces hospital admissions, endoscopies, and medical therapy in patients with acute gastrointestinal bleeding. Am J Med. 2017;130(7):819-825. doi: 10.1016/j.amjmed.2017.01.036.
ED Visits by Older Patients Increase in the Weeks After a Disaster
BY KELLIE DESANTIS
Visits to an ED by adults ages 65 years and older increase significantly in the weeks following a disaster, according to a study published in Disaster Medicine and Public Health Preparedness.1
Older adults are vulnerable to the effects of disasters because of their diminished ability to adequately prepare for and respond to the effects of a disaster. Older adults suffering from visual, auditory, proprioceptive, and cognitive impairments are especially vulnerable and have the most difficulty complying with evacuation and preparatory warnings. Individuals with multiple chronic diseases, living in long-term care facilities or suffering from cognitive impairments are among the most vulnerable.
To better understand the impact of natural disasters on this vulnerable population, researchers examined the effects of the 2012 disaster, Hurricane Sandy, on older adults living in New York City (NYC) during the disaster. Researchers turned to the New York State Department of Health (NYSDOH) for data. The NYSDOH compiles a comprehensive database of claims from all ED visits in the Statewide Planning and Research Cooperative System (SPARCS), which is the most complete source for ED utilization in New York state, and includes primary and secondary diagnosis codes and patient addresses.
Researchers evaluated ED utilization by adults 65 years and older in the weeks immediately before and after the Hurricane Sandy landfall. They excluded patients who lived in a nursing home, were incarcerated, or visited an ED associated with a specialty hospital (surgical subspecialty, oncological, or Veterans Administration). By using geographic distribution information available from SPARCS and the NYC Office of Emergency Management evacuation zones, researchers were able to compare the ED utilization for older adults living in the evacuation zones before the landfall of Hurricane Sandy and in the weeks shortly after the storm.
The analysis revealed a significant increase in ED utilization for older adults living in the evacuation zones in the 3 weeks after the storm compared to ED use before the storm. The number of weekly ED visits by older adults from all evacuation zones was 9,852 in the weeks before Hurricane Sandy and increased in the first week after the storm to 10,073. Among the most severely impacted were older adults in evacuation zone one, where ED utilization increased from 552 visits to 1,111 visits. The number of ED visits remained elevated for 3 weeks after the storm but returned to normal by the fourth week.
Researchers suggested several reasons for this increase in ED visits, including seeking refuge in the ED as a result of homelessness due to the disaster, the interruption of ongoing care for chronic illness, environmental exposure, and the lack of preparation for the lasting effect of the disaster.
To improve the response to such a disaster in the future, a NYC Hurricane Sandy Assessment report2 recommended developing a door-to-door service task force for older adults to improve preparedness for this vulnerable population. The task force would be responsible for implementing an action plan to ensure that healthcare services, communication, and provisions for this population continue without interruption in the weeks following a disaster. Legal and regulatory changes would allow for Medicare recipients to be eligible for "early medication refill" and pre-storm "early dialysis" programs to improve the continuity of care of the chronically ill.
1. Malik S, Lee DC, Doran KM, et al. Vulnerability of older adults in disasters: emergency department utilization by geriatric patients after hurricane sandy. Disaster Med Public Health Prep. 2017:1-10. doi:10.1017/dmp.2017.44
2. The City of New York, Office of the Mayor. Hurricane Sandy After Action Report. Published May 2013. http://www.nyc.gov/html/recovery/downlaods/pdf/sandy_aar_5.2.13.pdf. Accessed September 1, 2017.
Digital Rectal Examination of ED Patients with Acute GI Bleeding Cuts Rates of Admissions, Pharmacotherapy, and Endoscopy
BY JEFF BAUER
Patients presenting to the ED with acute gastrointestinal (GI) bleeding who receive a digital rectal examination have significantly lower rates of admissions, pharmacotherapy, and endoscopies, according to a retrospective study published in The American Journal of Medicine. Digital rectal examinations are an established part of the physical examination of a patient with GI bleeding, but physicians often are reluctant to conduct such examinations. Previous studies have found that 10% to 35% of patients with acute GI bleeding do not receive digital rectal examinations.
In the current study, researchers analyzed data from the electronic health records (EHRs) of patients ages 18 years and older who presented to the ED of a single institution with acute GI bleeding, as identified by International Classification of Diseases, Ninth Edition codes. They collected patients’ medical histories, demographic information, and clinical and laboratory data. ED clinician notes were used to determine which patients received a digital rectal examination. The outcomes researchers assessed were hospital admission, intensive care unit (ICU) admission, initiation of medical therapy (a proton pump inhibitor or octreotide), inpatient endoscopy (upper endoscopy or colonoscopy), and packed red blood cell (RBC) transfusion.
Overall, 1237 patients presented with acute GI bleeding. Most patients were Caucasian (49.2%) or Hispanic (38.4%), 44.9% were female, and the median age was 53 years.
Slightly more than one-half of patients (55.6%) received a digital rectal examination. In total, 736 patients were admitted—including 222 admissions to the ICU; 751 were started on a proton pump inhibitor or octreotide, 274 underwent endoscopy, and 321 received an RBC transfusion.
Patients were more likely to receive a digital rectal examination if they were older, Hispanic, or receiving an anticoagulant. Patients were less likely to undergo such examinations if they presented with altered mental status or hematemesis. Compared to patients who did not receive a digital rectal examination, those who did were significantly less likely to be admitted to the hospital (P = .004), to be starting on medical therapy (P = .04), or to undergo endoscopy (P = .02). There were no significant differences between these two groups in terms of ICU admissions, gastroenterology consultations, or transfusions.
Researchers suggested that the 44% rate of patients with acute GI bleeding who did not receive digital rectal examinations was higher than had been reported in previous studies. The difference had been the result of relying solely on ED clinician notes for this data, without including notes from admitting or consulting clinicians. The authors also were unable to determine the reasons these examinations were not conducted.
Shrestha MP, Borgstrom M, Trowers E. Digital rectal examination reduces hospital admissions, endoscopies, and medical therapy in patients with acute gastrointestinal bleeding. Am J Med. 2017;130(7):819-825. doi: 10.1016/j.amjmed.2017.01.036.
Is Ketamine the New Wonder Drug for Treating Suicide?
In 2014 the suicide rate in the U.S. was 13/100,000, the highest recorded in 28 years.1 Suicide is now considered the 10th leading cause of death for all ages, and the rate has increased every year from 2000 to 2014 among both women and men and in every age group except those aged ≥ 75 years.1-3 For those aged 15 to 44 years, suicide is among the top 3 causes of death worldwide.4-6
Background
In 2013, more than 490,000 hospital visits related to suicide attempts were reported in the U.S.4 Health care expenditures related to suicide are estimated at $56.9 billion in combined medical and work loss costs annually and an unmeasurable cost to the affected families.7 The mental health care community is desperate for ways to address this epidemic, and the National Academies of Medicine (NAM) has declared that research that directly addresses comparative effectiveness of treatment strategies following a suicide attempt should be a national priority.8
The most recent reports from 2014 indicate that the suicide rates are higher for male veterans than for male nonveterans (32.1 vs 20.9 per 100,000, respectively) and are much higher for female veterans than for female nonveterans (28.7 vs 5.2 per 100,000, respectively).3 Suicide rates also may be associated with veteran-specific comorbidities, such as higher rates of depression, anxiety, posttraumatic stress disorder (PTSD), and war-related trauma.3 According to the VHA, the suicide rate for veterans aged > 30 years also is rapidly increasing, and VHA has echoed the calls from NAM to make suicide prevention research a national priority.3
The VA has tried to stem the tide of suicides in veterans by implementing many advances in suicide prevention, including hiring suicide prevention coordinators at every VA hospital, enhanced monitoring, and the availability of 24-hour crisis hotline services. Yet the suicide rates for veterans continue to rise and remain higher than the rates in the general population.3
About 90% of deaths by suicide are by persons who have a treatable psychiatric disorder, most commonly a mood disorder, such as depression.4 However, most studies show that antidepressant therapy does not provide rapid or significant relief of suicidal ideation (SI).4 Therefore, the current standard of care for the treatment of acutely suicidal patients includes a combination of hospitalization, cognitive behavioral therapy or psychotherapy, case management, antidepressant medications, and electroconvulsive therapy (ECT).4 Even though these therapies have become more widely available over the past decade, rates of suicide continue to increase.1,4 These interventions have limited effectiveness in acute settings. Although both intensive outpatient follow-up and routine outpatient care have been studied in relation to the decrease of suicidal behavior, neither intervention has been shown to immediately reduce suicidal behavior significantly in patients.
Suicidality Interventions
Therapy and case management require patients to be well enough to make office visits and follow through with care for periods as long as 1 year, which is often not possible for individuals with severe depression.5 One-third of patients who attended 6 months of outpatient therapy consistently still met the criteria for major depressive disorder (MDD), a major risk for suicide attempt.9 Antidepressant medications take a minimum of 4 weeks to reach full efficacy, and many patients stop taking the medications before that point because of concern that the medication is not helping or because of adverse effects (AEs), such as sleep disturbance, sexual dysfunction, or weight gain.9
Electroconvulsive therapy has been shown to be an effective treatment for patients with depression and suicidal behavior, but adherence with 12 weeks of recommended therapy has been a barrier for this intervention. Additionally, ECT may not provide reduction in SI for 1 to 2 weeks.4,10 A review of research studies showed that nearly 50% of patients with high-expressed SI did not complete the prescribed amount of ECT due to the length of time to complete the recommended 12 sessions.10 Therefore, current treatment barriers for suicidal patients include: (1) long periods in treatment for therapy, medication, and ECT before any relief of symptoms is noted; (2) high recidivism rates for MDD symptoms and risk of suicide following treatment; and (3) high treatment dropout rates.
Pharmacologic treatments currently used in suicidal patients have not fared much better. Many have received FDA approval for treatment of associated mental health diagnoses such as bipolar disorder, schizophrenia, or MDD, but there are no approved treatments that specifically target suicidal behavior. Lithium is approved for reducing the long-term risk of SI primarily because it reduces the risk of mood disorders associated with SI, but lithium has not been shown to be effective in acute settings.11 Clozapine is approved for reducing the long-term risk of recurrent suicide in patients with schizophrenia or schizoaffective disorder.4 Clozapine has not been shown to be effective in patients with mood disorders, which make up the majority of patients who attempt suicide.4 Additionally, both medications are plagued by the same barriers listed earlier, such as long time to effect (it takes an average 4 weeks to reach efficacy), lack of efficacy in acute settings, and AEs (eg, sleep problems, weight gain, and sexual dysfunction).9 Thus finding better pharmacologic interventions for suicidal patients is a priority for current research.
Ketamine
Recently, researchers have identified ketamine as a potential therapeutic option for depression and SI. A single ketamine infusion treatment has a rapid response, minimal AEs, and potentially long-lasting efficacy with SI, which would make it ideal for the treatment of acutely suicidal patients.4 Ketamine is an N-methyl-D-aspartate receptor (NMDAR) inhibitor that also has been found to be a weak μ- and κ-opioid receptor agonist and an inhibitor of the reuptake of serotonin, dopamine, and norepinephrine. Inhibition of the NMDAR results in analgesia, and ketamine is approved for the induction of anesthesia, pain relief, and sedation.12
Although AEs such as hallucinations and sedation create the potential for dangerous recreational use, ketamine is safely used in health care settings for a variety of indications. Effects are noted within 5 minutes of administration if given by infusion, and the main effects can last between 20 and 40 minutes.
Ketamine has a complex pharmacology and plays a role in other cell signaling mechanisms, but the significance of these additional mechanisms in the therapeutic effects of ketamine have only recently been elucidated. Preclinical studies indicate a probable NMDAR inhibition-independent mechanism responsible for the antidepressant response to ketamine.13,14 The complex associations with rapamycin signaling, eukaryotic elongation factor 2 dephosphorylation, increased synthesis of brain-derived neurotrophic factor, and activation of glutamatergic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors have been linked to its rapid antidepressant effect and ketamine’s induction of synaptogenesis within the limbic system.13,14
Clinical Research
Ketamine was studied as an adjunctive treatment to psychotherapy for addictions as far back as the 1970s.15 The available reports indicate a universally positive result, with increased rates of remission and decreased rates of relapse attributed to ketamine’s ability to alter one’s thought processes by reinforcing limbic-cortex interactions that facilitate the growth of more positive cognitive schemas and improved emotional attitudes about the self in support of the recovery process.15
Neurobiologic studies have shown that treatment with ketamine has a direct and immediate effect on neuronal pathways of the limbic system. It is known to regulate the mind’s reaction to positive stimuli by reversing the depressed subject’s blunted reaction to positive faces.16 This rapid normalization of the positive faces test is unique to ketamine infusion and is not seen in tests with traditional antidepressants.
In 2000, the first placebo-controlled trial using ketamine for treatment resistant depression (TRD) demonstrated the rapid antidepressant effects of a single dose of ketamine, but this study only looked at these effects for 1 week.17 In multiple double blind, placebo-controlled trials since then, IV infusion of ketamine was shown to be an effective intervention for TRD.13,18,19 More recently, a published investigation involving the treatment of MDD showed that ketamine in conjunction with a selective serotonin reuptake inhibitor (SSRI) accelerated and enhanced the effectiveness of the SSRI in reducing depressive symptoms.20
Based on the rapid resolution of depressive symptoms using ketamine, researchers have looked at its effect on suicidality as a secondary measure. A case study of a patient with severe depressive episodes and multiple previous suicidal attempts reported that the patient responded to a single dose of ketamine, described the experience as “being reborn,” and maintained complete remission of SI for the 6-month study period.21 In a larger study, 133 TRD patients received a single IV dose of ketamine with significant reductions in SI independent of depressive and anxiety symptoms.22
Depression Treatment
These results have led to an excitement for ketamine therapy as a novel treatment of depression, and off-label use by treatment centers now exists in several countries to aid those with TRD.23 This off-label use continues to be controversial, as research has yet to determine the safest most effective route and duration of treatment and whether the ketamine treatment AEs will exceed any accrued therapeutic benefit.13
The American Psychological Association Council of Research Task Force on Novel Biomarkers and Treatment critically examined the clinical evidence of ketamine use and has raised important concerns about the use of ketamine in the outpatient setting, administered in the absence of consensus therapeutic monitoring guidelines, and ambitiously marketed as a panacea for TRD.13,24 A study showed permanent impairment of brain function for both groups compared with monkeys treated with saline infusions.25 In 2016, the FDA gave fast-track approval for an intranasal ketamine that would make the treatment more easily available in the outpatient setting, but this could lead to certain patients developing a dependency on ketamine or engaging in its diversion for recreational use. There are case reports and anecdotes in the literature of patients and research subjects developing drug-seeking behaviors and overuse of ketamine.24 Additionally, the comorbidities associated with TRD and SI have not been fully evaluated. For instance, there is evidence that depressed patients with obsessive compulsive disorder may have worse outcomes that include delayed onset SI.26
There also is concern for the use of ketamine for chronic opioid users. The combination of ketamine with opioids may increase the response to the opioid in an otherwise drug tolerant patient, leading to risk of death by overdose in patients who have not increased their usual dose.27 However, this effect was noted only when ketamine and opioids were administered together, and the effect does not seem to last postinfusion.27
The challenges in treatment of TRD include finding an effective formulation—IV infusion of ketamine requires cardiovascular monitoring and is administered by anesthesiologists. The short duration of action for depression requires repeated infusions, and the frequency and quantity of infusions have not been determined. Efforts to find other NMDAR inhibitors (eg, memantine, nitrous oxide, D-cycloserine, and others) that match ketamine’s antidepressant efficacy but with easier delivery methods and fewer risks have thus far been unsuccessful.13 It is now believed that ketamine’s unique ability to activate intracellular signaling pathways linked to synaptic plasticity gives it the antidepressant function. Recent studies have further narrowed ketamine’s antidepressant function to the R- enantiomer of the ketamine metabolite, hydroxynorketamine.14 The nasal spray for ketamine is the S- enantiomer, which has better bioavailability but may have less antidepressant efficacy compared with the racemic mixture used in ketamine infusions.
Suicide Ideation Treatment
The many challenges faced by researchers and clinicians trying to develop ketamine treatment for TRD may not apply to the treatment of SI. Whereas repeated doses of ketamine cannot reliably produce sustained remission of depression, the few studies that have looked at the long-term effects of ketamine treatment on SI indicate the potential for long-term efficacy after a single IV infusion.21,22 Although treatment with IV infusions have additional costs and logistics, if it is found beneficial, it could be given in the emergency department (ED) prior to hospitalization and potentially lead to better outcomes.
In 2011, a small preliminary observational study of patients with depression and SI presenting to the ED indicated that SI was rapidly reduced following an infusion of ketamine.28 This study showed that both depressive symptoms and suicidality rapidly and significantly diminished within 40 minutes with no evidence of the recurrence of symptoms 10 days postadministration.
A more recent study used ketamine in a military field hospital to treat SI and also concluded that it could be effective and safe when administered in an ED setting. This preliminary study suggests that ketamine could be a safe and potentially effective medication for rapid reduction of depression and suicidality in a busy ED setting.29 These limited studies involving the use of ketamine in patients with SI show promise with long-term effectiveness. However, more research is needed to clarify whether the efficacy with SI will be similar to the clinical experience seen in TRD; a duration of effect limited to 2 weeks with recurrence after treatment discontinued.24
Conclusion
There has been a compelling accumulation of scientific data since 2000 to support the use of ketamine for the treatment of depression and SI. Ketamine use in patients with these diagnoses showed a rapid decrease of symptoms and minimal AEs among a significant number of patients.22,30
Although the initial findings involving the use of ketamine in suicidal patients are promising, the clinical use of ketamine needs further research, using larger sample sizes and exploring both the short-term and long-term effects of this medication. Researchers need to further establish the safe and effective route, point of care, and patient type that would best respond to this novel treatment. The initial evidence would suggest that health care providers have every right to be hopeful that ketamine will become the first pharmacologic treatment of acute SI in a majority of patients presenting to EDs, mental health clinics, community hospitals, and VA medical centers.
1. Curtin SC, Warner MA, Hedegaard H. Increase in suicide in the United States 199-2014. NCHS data brief, no. 241. https://www.cdc.gov/nchs/data/data -briefs/db241.pdf. Published April 2016. Accessed August 3, 2017.
2. Nock MK, Borges G, Bromet EJ, Cha CB, Kessler RC, Lee S. Suicide and suicidal behavior. Epidemiol Rev. 2008;30(1):133-154.
3. U.S. Department of Veteran Affairs Office of Suicide Prevention. Suicide among veterans and other Americans 2001-2014. https://www.mentalhealth .va.gov/docs/2016suicidedatareport.pdf Published August 3, 2016. Accessed August 11, 2017.
4. Wilkinson ST, Sanacora G. Ketamine: a potential rapid-acting antisuicidal agent? Depress Anxiety. 2016;33(8):711-717.
5. Aleman A, Denys D. Mental health: a road map for suicide research and prevention. Nature. 2014;509(7501):421-423.
6. Griffiths JJ, Zarate CA, Jr, Rasimas JJ. Existing and novel biological therapeutics in suicide prevention. Am J Prev Med. 2014;47(3)(suppl 2):S195-S203.
7. Centers for Disease Control and Prevention. Leading causes of death reports, 1981-2015. https://www.cdc.gov/injury/wisqars/leading_causes_death.html. Updated February 19, 2017. Accessed August 14, 2017.
8. Institute of Medicine of the National Academies; Board on Health Care Services; Committee on Comparative Effectiveness Research Prioritization. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: The National Academies Press; 2009.
9. Weinberger MI, Sirey JA, Bruce ML, Heo M, Papademetriou E, Meyers BS. Predictors of major depression six months after admission for outpatient treatment. Psychiatr Serv. 2008;59(10):1211-1215.
10. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
11. Lewitzka U, Jabs B, Fülle M, et al. Does lithium reduce acute suicidal ideation and behavior? A protocol for a randomized, placebo-controlled multicenter trial of lithium plus treatment as usual (TAU) in patients with suicidal major depressive episode. BMC Psychiatry. 2015;15:117.
12. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman RD, Kaye AD. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. 2016;32(3):298-306.
13. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
14. Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481-486.
15. Krupitsky EM, Grinenko AY. Ketamine psychedelic therapy (KPT): a review of the results of ten years of research. J Psychoactive Drugs. 1997;29(2):165-183.
16. Murrough JW, Collins KA, Fields J, et al. Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Transl Psychiatry. 2015;5:e509.
17. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351-354.
18. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
19. Zarate CA Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856-864.
20. Hu YD, Xiang YT, Fang JX, et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: results from a randomized, placebo-controlled 4-week study. Psychol Med. 2016;46(3):623-635.
21. Aligeti S, Quinones M, Salazar R. Rapid resolution of suicidal behavior and depression with single low-dose ketamine intravenous push even after 6 months of follow-up. J Clin Psychopharmacol. 2014;34(4):533-535.
22. Ballard ED, Ionescu DF, Vande Voort JL, et al. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J Psychiatr Res. 2014;58:161-166.
23. Henderson TA. Practical application of the neuroregenerative properties of ketamine: real world treatment experience. Neural Regen Res. 2016;11(2):195-200.
24. Newport DJ, Schatzberg AF, Nemeroff CB. Whither ketamine as an antidepressant: panacea or toxin? Depress Anxiety. 2016;33(8):685-688.
25. Sun L, Li Q, Li Q, et al. Chronic ketamine exposure induces permanent impairment of brain functions in adolescent cynomolgus monkeys. Addict Biol. 2014;19(2):185-194.
26. Niciu MJ, Grunschel BD, Corlett PR, Pittenger C, Bloch MH. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
27. Huxtable CA, Roberts LJ, Somogyi AA, MacIntyre PE. Acute pain management in opioid-tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39(5):804-823.
28. Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14(8):1127-1131.
29. Burger J, Capobianco M, Lovem R, et al. A double-blinded, randomized, placebo-controlled sub-dissociative dose ketamine pilot study in the treatment of acute depression and suicidality in a military emergency department setting. Mil Med. 2016;181(10):1195-1199.
30. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
In 2014 the suicide rate in the U.S. was 13/100,000, the highest recorded in 28 years.1 Suicide is now considered the 10th leading cause of death for all ages, and the rate has increased every year from 2000 to 2014 among both women and men and in every age group except those aged ≥ 75 years.1-3 For those aged 15 to 44 years, suicide is among the top 3 causes of death worldwide.4-6
Background
In 2013, more than 490,000 hospital visits related to suicide attempts were reported in the U.S.4 Health care expenditures related to suicide are estimated at $56.9 billion in combined medical and work loss costs annually and an unmeasurable cost to the affected families.7 The mental health care community is desperate for ways to address this epidemic, and the National Academies of Medicine (NAM) has declared that research that directly addresses comparative effectiveness of treatment strategies following a suicide attempt should be a national priority.8
The most recent reports from 2014 indicate that the suicide rates are higher for male veterans than for male nonveterans (32.1 vs 20.9 per 100,000, respectively) and are much higher for female veterans than for female nonveterans (28.7 vs 5.2 per 100,000, respectively).3 Suicide rates also may be associated with veteran-specific comorbidities, such as higher rates of depression, anxiety, posttraumatic stress disorder (PTSD), and war-related trauma.3 According to the VHA, the suicide rate for veterans aged > 30 years also is rapidly increasing, and VHA has echoed the calls from NAM to make suicide prevention research a national priority.3
The VA has tried to stem the tide of suicides in veterans by implementing many advances in suicide prevention, including hiring suicide prevention coordinators at every VA hospital, enhanced monitoring, and the availability of 24-hour crisis hotline services. Yet the suicide rates for veterans continue to rise and remain higher than the rates in the general population.3
About 90% of deaths by suicide are by persons who have a treatable psychiatric disorder, most commonly a mood disorder, such as depression.4 However, most studies show that antidepressant therapy does not provide rapid or significant relief of suicidal ideation (SI).4 Therefore, the current standard of care for the treatment of acutely suicidal patients includes a combination of hospitalization, cognitive behavioral therapy or psychotherapy, case management, antidepressant medications, and electroconvulsive therapy (ECT).4 Even though these therapies have become more widely available over the past decade, rates of suicide continue to increase.1,4 These interventions have limited effectiveness in acute settings. Although both intensive outpatient follow-up and routine outpatient care have been studied in relation to the decrease of suicidal behavior, neither intervention has been shown to immediately reduce suicidal behavior significantly in patients.
Suicidality Interventions
Therapy and case management require patients to be well enough to make office visits and follow through with care for periods as long as 1 year, which is often not possible for individuals with severe depression.5 One-third of patients who attended 6 months of outpatient therapy consistently still met the criteria for major depressive disorder (MDD), a major risk for suicide attempt.9 Antidepressant medications take a minimum of 4 weeks to reach full efficacy, and many patients stop taking the medications before that point because of concern that the medication is not helping or because of adverse effects (AEs), such as sleep disturbance, sexual dysfunction, or weight gain.9
Electroconvulsive therapy has been shown to be an effective treatment for patients with depression and suicidal behavior, but adherence with 12 weeks of recommended therapy has been a barrier for this intervention. Additionally, ECT may not provide reduction in SI for 1 to 2 weeks.4,10 A review of research studies showed that nearly 50% of patients with high-expressed SI did not complete the prescribed amount of ECT due to the length of time to complete the recommended 12 sessions.10 Therefore, current treatment barriers for suicidal patients include: (1) long periods in treatment for therapy, medication, and ECT before any relief of symptoms is noted; (2) high recidivism rates for MDD symptoms and risk of suicide following treatment; and (3) high treatment dropout rates.
Pharmacologic treatments currently used in suicidal patients have not fared much better. Many have received FDA approval for treatment of associated mental health diagnoses such as bipolar disorder, schizophrenia, or MDD, but there are no approved treatments that specifically target suicidal behavior. Lithium is approved for reducing the long-term risk of SI primarily because it reduces the risk of mood disorders associated with SI, but lithium has not been shown to be effective in acute settings.11 Clozapine is approved for reducing the long-term risk of recurrent suicide in patients with schizophrenia or schizoaffective disorder.4 Clozapine has not been shown to be effective in patients with mood disorders, which make up the majority of patients who attempt suicide.4 Additionally, both medications are plagued by the same barriers listed earlier, such as long time to effect (it takes an average 4 weeks to reach efficacy), lack of efficacy in acute settings, and AEs (eg, sleep problems, weight gain, and sexual dysfunction).9 Thus finding better pharmacologic interventions for suicidal patients is a priority for current research.
Ketamine
Recently, researchers have identified ketamine as a potential therapeutic option for depression and SI. A single ketamine infusion treatment has a rapid response, minimal AEs, and potentially long-lasting efficacy with SI, which would make it ideal for the treatment of acutely suicidal patients.4 Ketamine is an N-methyl-D-aspartate receptor (NMDAR) inhibitor that also has been found to be a weak μ- and κ-opioid receptor agonist and an inhibitor of the reuptake of serotonin, dopamine, and norepinephrine. Inhibition of the NMDAR results in analgesia, and ketamine is approved for the induction of anesthesia, pain relief, and sedation.12
Although AEs such as hallucinations and sedation create the potential for dangerous recreational use, ketamine is safely used in health care settings for a variety of indications. Effects are noted within 5 minutes of administration if given by infusion, and the main effects can last between 20 and 40 minutes.
Ketamine has a complex pharmacology and plays a role in other cell signaling mechanisms, but the significance of these additional mechanisms in the therapeutic effects of ketamine have only recently been elucidated. Preclinical studies indicate a probable NMDAR inhibition-independent mechanism responsible for the antidepressant response to ketamine.13,14 The complex associations with rapamycin signaling, eukaryotic elongation factor 2 dephosphorylation, increased synthesis of brain-derived neurotrophic factor, and activation of glutamatergic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors have been linked to its rapid antidepressant effect and ketamine’s induction of synaptogenesis within the limbic system.13,14
Clinical Research
Ketamine was studied as an adjunctive treatment to psychotherapy for addictions as far back as the 1970s.15 The available reports indicate a universally positive result, with increased rates of remission and decreased rates of relapse attributed to ketamine’s ability to alter one’s thought processes by reinforcing limbic-cortex interactions that facilitate the growth of more positive cognitive schemas and improved emotional attitudes about the self in support of the recovery process.15
Neurobiologic studies have shown that treatment with ketamine has a direct and immediate effect on neuronal pathways of the limbic system. It is known to regulate the mind’s reaction to positive stimuli by reversing the depressed subject’s blunted reaction to positive faces.16 This rapid normalization of the positive faces test is unique to ketamine infusion and is not seen in tests with traditional antidepressants.
In 2000, the first placebo-controlled trial using ketamine for treatment resistant depression (TRD) demonstrated the rapid antidepressant effects of a single dose of ketamine, but this study only looked at these effects for 1 week.17 In multiple double blind, placebo-controlled trials since then, IV infusion of ketamine was shown to be an effective intervention for TRD.13,18,19 More recently, a published investigation involving the treatment of MDD showed that ketamine in conjunction with a selective serotonin reuptake inhibitor (SSRI) accelerated and enhanced the effectiveness of the SSRI in reducing depressive symptoms.20
Based on the rapid resolution of depressive symptoms using ketamine, researchers have looked at its effect on suicidality as a secondary measure. A case study of a patient with severe depressive episodes and multiple previous suicidal attempts reported that the patient responded to a single dose of ketamine, described the experience as “being reborn,” and maintained complete remission of SI for the 6-month study period.21 In a larger study, 133 TRD patients received a single IV dose of ketamine with significant reductions in SI independent of depressive and anxiety symptoms.22
Depression Treatment
These results have led to an excitement for ketamine therapy as a novel treatment of depression, and off-label use by treatment centers now exists in several countries to aid those with TRD.23 This off-label use continues to be controversial, as research has yet to determine the safest most effective route and duration of treatment and whether the ketamine treatment AEs will exceed any accrued therapeutic benefit.13
The American Psychological Association Council of Research Task Force on Novel Biomarkers and Treatment critically examined the clinical evidence of ketamine use and has raised important concerns about the use of ketamine in the outpatient setting, administered in the absence of consensus therapeutic monitoring guidelines, and ambitiously marketed as a panacea for TRD.13,24 A study showed permanent impairment of brain function for both groups compared with monkeys treated with saline infusions.25 In 2016, the FDA gave fast-track approval for an intranasal ketamine that would make the treatment more easily available in the outpatient setting, but this could lead to certain patients developing a dependency on ketamine or engaging in its diversion for recreational use. There are case reports and anecdotes in the literature of patients and research subjects developing drug-seeking behaviors and overuse of ketamine.24 Additionally, the comorbidities associated with TRD and SI have not been fully evaluated. For instance, there is evidence that depressed patients with obsessive compulsive disorder may have worse outcomes that include delayed onset SI.26
There also is concern for the use of ketamine for chronic opioid users. The combination of ketamine with opioids may increase the response to the opioid in an otherwise drug tolerant patient, leading to risk of death by overdose in patients who have not increased their usual dose.27 However, this effect was noted only when ketamine and opioids were administered together, and the effect does not seem to last postinfusion.27
The challenges in treatment of TRD include finding an effective formulation—IV infusion of ketamine requires cardiovascular monitoring and is administered by anesthesiologists. The short duration of action for depression requires repeated infusions, and the frequency and quantity of infusions have not been determined. Efforts to find other NMDAR inhibitors (eg, memantine, nitrous oxide, D-cycloserine, and others) that match ketamine’s antidepressant efficacy but with easier delivery methods and fewer risks have thus far been unsuccessful.13 It is now believed that ketamine’s unique ability to activate intracellular signaling pathways linked to synaptic plasticity gives it the antidepressant function. Recent studies have further narrowed ketamine’s antidepressant function to the R- enantiomer of the ketamine metabolite, hydroxynorketamine.14 The nasal spray for ketamine is the S- enantiomer, which has better bioavailability but may have less antidepressant efficacy compared with the racemic mixture used in ketamine infusions.
Suicide Ideation Treatment
The many challenges faced by researchers and clinicians trying to develop ketamine treatment for TRD may not apply to the treatment of SI. Whereas repeated doses of ketamine cannot reliably produce sustained remission of depression, the few studies that have looked at the long-term effects of ketamine treatment on SI indicate the potential for long-term efficacy after a single IV infusion.21,22 Although treatment with IV infusions have additional costs and logistics, if it is found beneficial, it could be given in the emergency department (ED) prior to hospitalization and potentially lead to better outcomes.
In 2011, a small preliminary observational study of patients with depression and SI presenting to the ED indicated that SI was rapidly reduced following an infusion of ketamine.28 This study showed that both depressive symptoms and suicidality rapidly and significantly diminished within 40 minutes with no evidence of the recurrence of symptoms 10 days postadministration.
A more recent study used ketamine in a military field hospital to treat SI and also concluded that it could be effective and safe when administered in an ED setting. This preliminary study suggests that ketamine could be a safe and potentially effective medication for rapid reduction of depression and suicidality in a busy ED setting.29 These limited studies involving the use of ketamine in patients with SI show promise with long-term effectiveness. However, more research is needed to clarify whether the efficacy with SI will be similar to the clinical experience seen in TRD; a duration of effect limited to 2 weeks with recurrence after treatment discontinued.24
Conclusion
There has been a compelling accumulation of scientific data since 2000 to support the use of ketamine for the treatment of depression and SI. Ketamine use in patients with these diagnoses showed a rapid decrease of symptoms and minimal AEs among a significant number of patients.22,30
Although the initial findings involving the use of ketamine in suicidal patients are promising, the clinical use of ketamine needs further research, using larger sample sizes and exploring both the short-term and long-term effects of this medication. Researchers need to further establish the safe and effective route, point of care, and patient type that would best respond to this novel treatment. The initial evidence would suggest that health care providers have every right to be hopeful that ketamine will become the first pharmacologic treatment of acute SI in a majority of patients presenting to EDs, mental health clinics, community hospitals, and VA medical centers.
In 2014 the suicide rate in the U.S. was 13/100,000, the highest recorded in 28 years.1 Suicide is now considered the 10th leading cause of death for all ages, and the rate has increased every year from 2000 to 2014 among both women and men and in every age group except those aged ≥ 75 years.1-3 For those aged 15 to 44 years, suicide is among the top 3 causes of death worldwide.4-6
Background
In 2013, more than 490,000 hospital visits related to suicide attempts were reported in the U.S.4 Health care expenditures related to suicide are estimated at $56.9 billion in combined medical and work loss costs annually and an unmeasurable cost to the affected families.7 The mental health care community is desperate for ways to address this epidemic, and the National Academies of Medicine (NAM) has declared that research that directly addresses comparative effectiveness of treatment strategies following a suicide attempt should be a national priority.8
The most recent reports from 2014 indicate that the suicide rates are higher for male veterans than for male nonveterans (32.1 vs 20.9 per 100,000, respectively) and are much higher for female veterans than for female nonveterans (28.7 vs 5.2 per 100,000, respectively).3 Suicide rates also may be associated with veteran-specific comorbidities, such as higher rates of depression, anxiety, posttraumatic stress disorder (PTSD), and war-related trauma.3 According to the VHA, the suicide rate for veterans aged > 30 years also is rapidly increasing, and VHA has echoed the calls from NAM to make suicide prevention research a national priority.3
The VA has tried to stem the tide of suicides in veterans by implementing many advances in suicide prevention, including hiring suicide prevention coordinators at every VA hospital, enhanced monitoring, and the availability of 24-hour crisis hotline services. Yet the suicide rates for veterans continue to rise and remain higher than the rates in the general population.3
About 90% of deaths by suicide are by persons who have a treatable psychiatric disorder, most commonly a mood disorder, such as depression.4 However, most studies show that antidepressant therapy does not provide rapid or significant relief of suicidal ideation (SI).4 Therefore, the current standard of care for the treatment of acutely suicidal patients includes a combination of hospitalization, cognitive behavioral therapy or psychotherapy, case management, antidepressant medications, and electroconvulsive therapy (ECT).4 Even though these therapies have become more widely available over the past decade, rates of suicide continue to increase.1,4 These interventions have limited effectiveness in acute settings. Although both intensive outpatient follow-up and routine outpatient care have been studied in relation to the decrease of suicidal behavior, neither intervention has been shown to immediately reduce suicidal behavior significantly in patients.
Suicidality Interventions
Therapy and case management require patients to be well enough to make office visits and follow through with care for periods as long as 1 year, which is often not possible for individuals with severe depression.5 One-third of patients who attended 6 months of outpatient therapy consistently still met the criteria for major depressive disorder (MDD), a major risk for suicide attempt.9 Antidepressant medications take a minimum of 4 weeks to reach full efficacy, and many patients stop taking the medications before that point because of concern that the medication is not helping or because of adverse effects (AEs), such as sleep disturbance, sexual dysfunction, or weight gain.9
Electroconvulsive therapy has been shown to be an effective treatment for patients with depression and suicidal behavior, but adherence with 12 weeks of recommended therapy has been a barrier for this intervention. Additionally, ECT may not provide reduction in SI for 1 to 2 weeks.4,10 A review of research studies showed that nearly 50% of patients with high-expressed SI did not complete the prescribed amount of ECT due to the length of time to complete the recommended 12 sessions.10 Therefore, current treatment barriers for suicidal patients include: (1) long periods in treatment for therapy, medication, and ECT before any relief of symptoms is noted; (2) high recidivism rates for MDD symptoms and risk of suicide following treatment; and (3) high treatment dropout rates.
Pharmacologic treatments currently used in suicidal patients have not fared much better. Many have received FDA approval for treatment of associated mental health diagnoses such as bipolar disorder, schizophrenia, or MDD, but there are no approved treatments that specifically target suicidal behavior. Lithium is approved for reducing the long-term risk of SI primarily because it reduces the risk of mood disorders associated with SI, but lithium has not been shown to be effective in acute settings.11 Clozapine is approved for reducing the long-term risk of recurrent suicide in patients with schizophrenia or schizoaffective disorder.4 Clozapine has not been shown to be effective in patients with mood disorders, which make up the majority of patients who attempt suicide.4 Additionally, both medications are plagued by the same barriers listed earlier, such as long time to effect (it takes an average 4 weeks to reach efficacy), lack of efficacy in acute settings, and AEs (eg, sleep problems, weight gain, and sexual dysfunction).9 Thus finding better pharmacologic interventions for suicidal patients is a priority for current research.
Ketamine
Recently, researchers have identified ketamine as a potential therapeutic option for depression and SI. A single ketamine infusion treatment has a rapid response, minimal AEs, and potentially long-lasting efficacy with SI, which would make it ideal for the treatment of acutely suicidal patients.4 Ketamine is an N-methyl-D-aspartate receptor (NMDAR) inhibitor that also has been found to be a weak μ- and κ-opioid receptor agonist and an inhibitor of the reuptake of serotonin, dopamine, and norepinephrine. Inhibition of the NMDAR results in analgesia, and ketamine is approved for the induction of anesthesia, pain relief, and sedation.12
Although AEs such as hallucinations and sedation create the potential for dangerous recreational use, ketamine is safely used in health care settings for a variety of indications. Effects are noted within 5 minutes of administration if given by infusion, and the main effects can last between 20 and 40 minutes.
Ketamine has a complex pharmacology and plays a role in other cell signaling mechanisms, but the significance of these additional mechanisms in the therapeutic effects of ketamine have only recently been elucidated. Preclinical studies indicate a probable NMDAR inhibition-independent mechanism responsible for the antidepressant response to ketamine.13,14 The complex associations with rapamycin signaling, eukaryotic elongation factor 2 dephosphorylation, increased synthesis of brain-derived neurotrophic factor, and activation of glutamatergic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors have been linked to its rapid antidepressant effect and ketamine’s induction of synaptogenesis within the limbic system.13,14
Clinical Research
Ketamine was studied as an adjunctive treatment to psychotherapy for addictions as far back as the 1970s.15 The available reports indicate a universally positive result, with increased rates of remission and decreased rates of relapse attributed to ketamine’s ability to alter one’s thought processes by reinforcing limbic-cortex interactions that facilitate the growth of more positive cognitive schemas and improved emotional attitudes about the self in support of the recovery process.15
Neurobiologic studies have shown that treatment with ketamine has a direct and immediate effect on neuronal pathways of the limbic system. It is known to regulate the mind’s reaction to positive stimuli by reversing the depressed subject’s blunted reaction to positive faces.16 This rapid normalization of the positive faces test is unique to ketamine infusion and is not seen in tests with traditional antidepressants.
In 2000, the first placebo-controlled trial using ketamine for treatment resistant depression (TRD) demonstrated the rapid antidepressant effects of a single dose of ketamine, but this study only looked at these effects for 1 week.17 In multiple double blind, placebo-controlled trials since then, IV infusion of ketamine was shown to be an effective intervention for TRD.13,18,19 More recently, a published investigation involving the treatment of MDD showed that ketamine in conjunction with a selective serotonin reuptake inhibitor (SSRI) accelerated and enhanced the effectiveness of the SSRI in reducing depressive symptoms.20
Based on the rapid resolution of depressive symptoms using ketamine, researchers have looked at its effect on suicidality as a secondary measure. A case study of a patient with severe depressive episodes and multiple previous suicidal attempts reported that the patient responded to a single dose of ketamine, described the experience as “being reborn,” and maintained complete remission of SI for the 6-month study period.21 In a larger study, 133 TRD patients received a single IV dose of ketamine with significant reductions in SI independent of depressive and anxiety symptoms.22
Depression Treatment
These results have led to an excitement for ketamine therapy as a novel treatment of depression, and off-label use by treatment centers now exists in several countries to aid those with TRD.23 This off-label use continues to be controversial, as research has yet to determine the safest most effective route and duration of treatment and whether the ketamine treatment AEs will exceed any accrued therapeutic benefit.13
The American Psychological Association Council of Research Task Force on Novel Biomarkers and Treatment critically examined the clinical evidence of ketamine use and has raised important concerns about the use of ketamine in the outpatient setting, administered in the absence of consensus therapeutic monitoring guidelines, and ambitiously marketed as a panacea for TRD.13,24 A study showed permanent impairment of brain function for both groups compared with monkeys treated with saline infusions.25 In 2016, the FDA gave fast-track approval for an intranasal ketamine that would make the treatment more easily available in the outpatient setting, but this could lead to certain patients developing a dependency on ketamine or engaging in its diversion for recreational use. There are case reports and anecdotes in the literature of patients and research subjects developing drug-seeking behaviors and overuse of ketamine.24 Additionally, the comorbidities associated with TRD and SI have not been fully evaluated. For instance, there is evidence that depressed patients with obsessive compulsive disorder may have worse outcomes that include delayed onset SI.26
There also is concern for the use of ketamine for chronic opioid users. The combination of ketamine with opioids may increase the response to the opioid in an otherwise drug tolerant patient, leading to risk of death by overdose in patients who have not increased their usual dose.27 However, this effect was noted only when ketamine and opioids were administered together, and the effect does not seem to last postinfusion.27
The challenges in treatment of TRD include finding an effective formulation—IV infusion of ketamine requires cardiovascular monitoring and is administered by anesthesiologists. The short duration of action for depression requires repeated infusions, and the frequency and quantity of infusions have not been determined. Efforts to find other NMDAR inhibitors (eg, memantine, nitrous oxide, D-cycloserine, and others) that match ketamine’s antidepressant efficacy but with easier delivery methods and fewer risks have thus far been unsuccessful.13 It is now believed that ketamine’s unique ability to activate intracellular signaling pathways linked to synaptic plasticity gives it the antidepressant function. Recent studies have further narrowed ketamine’s antidepressant function to the R- enantiomer of the ketamine metabolite, hydroxynorketamine.14 The nasal spray for ketamine is the S- enantiomer, which has better bioavailability but may have less antidepressant efficacy compared with the racemic mixture used in ketamine infusions.
Suicide Ideation Treatment
The many challenges faced by researchers and clinicians trying to develop ketamine treatment for TRD may not apply to the treatment of SI. Whereas repeated doses of ketamine cannot reliably produce sustained remission of depression, the few studies that have looked at the long-term effects of ketamine treatment on SI indicate the potential for long-term efficacy after a single IV infusion.21,22 Although treatment with IV infusions have additional costs and logistics, if it is found beneficial, it could be given in the emergency department (ED) prior to hospitalization and potentially lead to better outcomes.
In 2011, a small preliminary observational study of patients with depression and SI presenting to the ED indicated that SI was rapidly reduced following an infusion of ketamine.28 This study showed that both depressive symptoms and suicidality rapidly and significantly diminished within 40 minutes with no evidence of the recurrence of symptoms 10 days postadministration.
A more recent study used ketamine in a military field hospital to treat SI and also concluded that it could be effective and safe when administered in an ED setting. This preliminary study suggests that ketamine could be a safe and potentially effective medication for rapid reduction of depression and suicidality in a busy ED setting.29 These limited studies involving the use of ketamine in patients with SI show promise with long-term effectiveness. However, more research is needed to clarify whether the efficacy with SI will be similar to the clinical experience seen in TRD; a duration of effect limited to 2 weeks with recurrence after treatment discontinued.24
Conclusion
There has been a compelling accumulation of scientific data since 2000 to support the use of ketamine for the treatment of depression and SI. Ketamine use in patients with these diagnoses showed a rapid decrease of symptoms and minimal AEs among a significant number of patients.22,30
Although the initial findings involving the use of ketamine in suicidal patients are promising, the clinical use of ketamine needs further research, using larger sample sizes and exploring both the short-term and long-term effects of this medication. Researchers need to further establish the safe and effective route, point of care, and patient type that would best respond to this novel treatment. The initial evidence would suggest that health care providers have every right to be hopeful that ketamine will become the first pharmacologic treatment of acute SI in a majority of patients presenting to EDs, mental health clinics, community hospitals, and VA medical centers.
1. Curtin SC, Warner MA, Hedegaard H. Increase in suicide in the United States 199-2014. NCHS data brief, no. 241. https://www.cdc.gov/nchs/data/data -briefs/db241.pdf. Published April 2016. Accessed August 3, 2017.
2. Nock MK, Borges G, Bromet EJ, Cha CB, Kessler RC, Lee S. Suicide and suicidal behavior. Epidemiol Rev. 2008;30(1):133-154.
3. U.S. Department of Veteran Affairs Office of Suicide Prevention. Suicide among veterans and other Americans 2001-2014. https://www.mentalhealth .va.gov/docs/2016suicidedatareport.pdf Published August 3, 2016. Accessed August 11, 2017.
4. Wilkinson ST, Sanacora G. Ketamine: a potential rapid-acting antisuicidal agent? Depress Anxiety. 2016;33(8):711-717.
5. Aleman A, Denys D. Mental health: a road map for suicide research and prevention. Nature. 2014;509(7501):421-423.
6. Griffiths JJ, Zarate CA, Jr, Rasimas JJ. Existing and novel biological therapeutics in suicide prevention. Am J Prev Med. 2014;47(3)(suppl 2):S195-S203.
7. Centers for Disease Control and Prevention. Leading causes of death reports, 1981-2015. https://www.cdc.gov/injury/wisqars/leading_causes_death.html. Updated February 19, 2017. Accessed August 14, 2017.
8. Institute of Medicine of the National Academies; Board on Health Care Services; Committee on Comparative Effectiveness Research Prioritization. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: The National Academies Press; 2009.
9. Weinberger MI, Sirey JA, Bruce ML, Heo M, Papademetriou E, Meyers BS. Predictors of major depression six months after admission for outpatient treatment. Psychiatr Serv. 2008;59(10):1211-1215.
10. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
11. Lewitzka U, Jabs B, Fülle M, et al. Does lithium reduce acute suicidal ideation and behavior? A protocol for a randomized, placebo-controlled multicenter trial of lithium plus treatment as usual (TAU) in patients with suicidal major depressive episode. BMC Psychiatry. 2015;15:117.
12. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman RD, Kaye AD. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. 2016;32(3):298-306.
13. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
14. Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481-486.
15. Krupitsky EM, Grinenko AY. Ketamine psychedelic therapy (KPT): a review of the results of ten years of research. J Psychoactive Drugs. 1997;29(2):165-183.
16. Murrough JW, Collins KA, Fields J, et al. Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Transl Psychiatry. 2015;5:e509.
17. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351-354.
18. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
19. Zarate CA Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856-864.
20. Hu YD, Xiang YT, Fang JX, et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: results from a randomized, placebo-controlled 4-week study. Psychol Med. 2016;46(3):623-635.
21. Aligeti S, Quinones M, Salazar R. Rapid resolution of suicidal behavior and depression with single low-dose ketamine intravenous push even after 6 months of follow-up. J Clin Psychopharmacol. 2014;34(4):533-535.
22. Ballard ED, Ionescu DF, Vande Voort JL, et al. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J Psychiatr Res. 2014;58:161-166.
23. Henderson TA. Practical application of the neuroregenerative properties of ketamine: real world treatment experience. Neural Regen Res. 2016;11(2):195-200.
24. Newport DJ, Schatzberg AF, Nemeroff CB. Whither ketamine as an antidepressant: panacea or toxin? Depress Anxiety. 2016;33(8):685-688.
25. Sun L, Li Q, Li Q, et al. Chronic ketamine exposure induces permanent impairment of brain functions in adolescent cynomolgus monkeys. Addict Biol. 2014;19(2):185-194.
26. Niciu MJ, Grunschel BD, Corlett PR, Pittenger C, Bloch MH. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
27. Huxtable CA, Roberts LJ, Somogyi AA, MacIntyre PE. Acute pain management in opioid-tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39(5):804-823.
28. Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14(8):1127-1131.
29. Burger J, Capobianco M, Lovem R, et al. A double-blinded, randomized, placebo-controlled sub-dissociative dose ketamine pilot study in the treatment of acute depression and suicidality in a military emergency department setting. Mil Med. 2016;181(10):1195-1199.
30. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
1. Curtin SC, Warner MA, Hedegaard H. Increase in suicide in the United States 199-2014. NCHS data brief, no. 241. https://www.cdc.gov/nchs/data/data -briefs/db241.pdf. Published April 2016. Accessed August 3, 2017.
2. Nock MK, Borges G, Bromet EJ, Cha CB, Kessler RC, Lee S. Suicide and suicidal behavior. Epidemiol Rev. 2008;30(1):133-154.
3. U.S. Department of Veteran Affairs Office of Suicide Prevention. Suicide among veterans and other Americans 2001-2014. https://www.mentalhealth .va.gov/docs/2016suicidedatareport.pdf Published August 3, 2016. Accessed August 11, 2017.
4. Wilkinson ST, Sanacora G. Ketamine: a potential rapid-acting antisuicidal agent? Depress Anxiety. 2016;33(8):711-717.
5. Aleman A, Denys D. Mental health: a road map for suicide research and prevention. Nature. 2014;509(7501):421-423.
6. Griffiths JJ, Zarate CA, Jr, Rasimas JJ. Existing and novel biological therapeutics in suicide prevention. Am J Prev Med. 2014;47(3)(suppl 2):S195-S203.
7. Centers for Disease Control and Prevention. Leading causes of death reports, 1981-2015. https://www.cdc.gov/injury/wisqars/leading_causes_death.html. Updated February 19, 2017. Accessed August 14, 2017.
8. Institute of Medicine of the National Academies; Board on Health Care Services; Committee on Comparative Effectiveness Research Prioritization. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: The National Academies Press; 2009.
9. Weinberger MI, Sirey JA, Bruce ML, Heo M, Papademetriou E, Meyers BS. Predictors of major depression six months after admission for outpatient treatment. Psychiatr Serv. 2008;59(10):1211-1215.
10. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
11. Lewitzka U, Jabs B, Fülle M, et al. Does lithium reduce acute suicidal ideation and behavior? A protocol for a randomized, placebo-controlled multicenter trial of lithium plus treatment as usual (TAU) in patients with suicidal major depressive episode. BMC Psychiatry. 2015;15:117.
12. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman RD, Kaye AD. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. 2016;32(3):298-306.
13. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
14. Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481-486.
15. Krupitsky EM, Grinenko AY. Ketamine psychedelic therapy (KPT): a review of the results of ten years of research. J Psychoactive Drugs. 1997;29(2):165-183.
16. Murrough JW, Collins KA, Fields J, et al. Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Transl Psychiatry. 2015;5:e509.
17. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351-354.
18. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
19. Zarate CA Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856-864.
20. Hu YD, Xiang YT, Fang JX, et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: results from a randomized, placebo-controlled 4-week study. Psychol Med. 2016;46(3):623-635.
21. Aligeti S, Quinones M, Salazar R. Rapid resolution of suicidal behavior and depression with single low-dose ketamine intravenous push even after 6 months of follow-up. J Clin Psychopharmacol. 2014;34(4):533-535.
22. Ballard ED, Ionescu DF, Vande Voort JL, et al. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J Psychiatr Res. 2014;58:161-166.
23. Henderson TA. Practical application of the neuroregenerative properties of ketamine: real world treatment experience. Neural Regen Res. 2016;11(2):195-200.
24. Newport DJ, Schatzberg AF, Nemeroff CB. Whither ketamine as an antidepressant: panacea or toxin? Depress Anxiety. 2016;33(8):685-688.
25. Sun L, Li Q, Li Q, et al. Chronic ketamine exposure induces permanent impairment of brain functions in adolescent cynomolgus monkeys. Addict Biol. 2014;19(2):185-194.
26. Niciu MJ, Grunschel BD, Corlett PR, Pittenger C, Bloch MH. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
27. Huxtable CA, Roberts LJ, Somogyi AA, MacIntyre PE. Acute pain management in opioid-tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39(5):804-823.
28. Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14(8):1127-1131.
29. Burger J, Capobianco M, Lovem R, et al. A double-blinded, randomized, placebo-controlled sub-dissociative dose ketamine pilot study in the treatment of acute depression and suicidality in a military emergency department setting. Mil Med. 2016;181(10):1195-1199.
30. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
Behavioral Health: Using Rating Scales in a Clinical Setting
In the current health care environment, there is an increasing demand for objective assessment of disease states.1 This is particularly apparent in the realm of behavioral health, where documentation of outcomes lags that of other areas of medicine.
In 2012, the additional health care costs incurred by persons with mental health diagnoses were estimated to be $293 billion among commercially insured, Medicaid, and Medicare beneficiaries in the United States—a figure that is 273% higher than the cost for those without psychiatric diagnoses.2 Psychiatric and medical illnesses can be so tightly linked that accurate diagnosis and treatment of psychiatric disorders becomes essential to control medical illnesses. It is not surprising that there is increased scrutiny to the ways in which behavioral health care can be objectively assessed and monitored, and payers such as the Centers for Medicare and Medicaid Services increasingly require objective documentation of disease state improvement for payment.3
Support for objective assessment of disease derives from the collaborative care model. This model is designed to better integrate mental health and primary care (among other practices) by establishing the Patient-Centered Medical Home and emphasizing screening and monitoring patient-reported outcomes over time to assess treatment response.4 This approach, which is endorsed by the American Psychiatric Association, is associated with significant improvements in outcomes compared with usual care.5 It tracks patient progress using validated clinical rating scales and other screening tools (eg, Patient Health Questionnaire [PHQ-9] for depression), an approach that is analogous to how patients with type 2 diabetes are monitored by A1C lab tests.6 An extensive body of research supports the impact of this approach on treatment. A 2012 Cochrane review associated collaborative care with significant improvements in depression and anxiety outcomes compared with usual treatment.7
Despite these findings, a recent Kennedy Forum brief asserts that behavioral health is characterized by a "lack of systematic measurement to determine whether patients are responding to treatment."8 That same brief points to the many validated, easy-to-administer rating scales and screening tools that can reliably measure the frequency and severity of psychiatric symptoms over time, and likens the lack of their use to "treating high blood pressure without using a blood pressure cuff to measure if a patient's blood pressure is improving."8 In fact, it is estimated that only 18% of psychiatrists and 11% of psychologists use rating scales routinely.9,10 This lack of use denies clinicians important information that can help detect deterioration or lack of improvement in their patients; implementing these scales in primary care can help early detection of behavioral health problems.
Behavioral health is replete with rating scales and screening tools, and the number of competing scales can make choosing a measure difficult.1 Nonetheless, not all scales are appropriate for clinical use; many are designed for research, for instance, and are lengthy and difficult to administer.
Let's review a number of rating scales that are brief, useful, and easy to administer. A framework for the screening tools addressed in this article is available on the federally funded Center for Integrated Health Solutions website (www.integration.samhsa.gov). This site promotes the use of tools designed to assist in screening and monitoring for depression, anxiety, bipolar disorder, substance use, and suicidality.11
QUALITY CRITERIA FOR RATING SCALES
The quality of a rating scale is determined by the following attributes.
Objectivity. The ability of a scale to obtain the same results, regardless of who administers, analyzes, or interprets it.
Reliability. The ability of a scale to convey consistent and reproducible information across time, patients, and raters.
Validity. The degree to which the scale measures what it is supposed to measure (eg, depressive symptoms). Sensitivity and specificity are measures of validity and provide additional information about the rating scale; namely, whether the scale can detect the presence of a disease (sensitivity) and whether it detects only that disease or condition and not another (specificity).
Establishment of norms. Whether a scale provides reference values for different clinical groups.
Practicability. The resources required to administer the assessment instrument in terms of time, staff, and material.12
In addition to meeting these quality criteria, selection of a scale can be based on whether it is self-rated or observer-rated. Advantages to self-rated scales, such as the PHQ-9, Mood Disorder Questionnaire (MDQ), and Generalized Anxiety Disorder 7-item (GAD-7) scale, are their practicability—they are easy to administer and don't require much time—and their use in evaluating and raising awareness of subjective states.
However, reliability may be a concern, as some patients may lack insight or exaggerate or mask symptoms when completing such scales.13 Both observer- and self-rated scales can be used together to minimize bias, identify symptoms that might have been missed/not addressed in the clinical interview, and drive clinical decision-making. Both can also help patients communicate with their providers and make them feel more involved in clinical decision-making.8
ENDORSED RATING SCALES
The following scales have met many of the quality criteria described here and are endorsed by the government payer system. They can easily be incorporated into clinical practice and will provide useful clinical information that can assist in diagnosis and monitoring patient outcomes.
Patient Health Questionnaire
PHQ-9 is a nine-item self-report questionnaire that can help to detect depression and supplement a thorough mental health interview. It scores the nine DSM-IV criteria for depression on a scale of 0 (not at all) to 3 (nearly every day). It is a public resource that is easy to find online, available without cost in several languages, and takes just a few minutes to complete.14
PHQ-9 has shown excellent test-retest reliability in screening for depression, and normative data on the instrument's use are available in various clinical populations.15 Research has shown that as PHQ-9 depression scores increase, functional status decreases, while depressive symptoms, sick days, and health care utilization increase.15 In one study, a PHQ-9 score of ≥ 10 had 88% sensitivity and specificity for detecting depression, with scores of 5, 10, 15, and 20 indicating mild, moderate, moderately severe, and severe depression, respectively.16 In addition to its use as a screening tool, PHQ-9 is a responsive and reliable measure of depression treatment outcomes.17
Mood Disorder Questionnaire
MDQ is another brief, self-report questionnaire that is available online. It is designed to identify and monitor patients who are likely to meet diagnostic criteria for bipolar disorder.18,19
The first question on the MDQ asks if the patient has experienced any of 13 common mood and behavior symptoms. The second question asks if these symptoms have ever occurred at the same time, and the third asks the degree to which the patient finds the symptoms to be problematic. The remaining two questions provide additional clinical information, addressing family history of manic-depressive illness or bipolar disorder and whether a diagnosis of either disorder has been made.
The MDQ has shown validity in assessing bipolar disorder symptoms in a general population, although recent research suggests that imprecise recall bias may limit its reliability in detecting hypomanic episodes earlier in life.20,21 Nonetheless, its specificity of > 97% means that it will effectively screen out just about all true negatives.18
Generalized Anxiety Disorder 7-item scale
The GAD-7 scale is a brief, self-administered questionnaire for screening and measuring severity of GAD.22 It asks patients to rate seven items that represent problems with general anxiety and scores each item on a scale of 0 (not at all) to 3 (nearly every day). Similar to the other measures, it is easily accessible online.
Research evidence supports the reliability and validity of GAD-7 as a measure of anxiety in the general population. Sensitivity and specificity are 89% and 82%, respectively. Normative data for age- and sex-specific subgroups support its use across age groups and in both males and females.23 The GAD-7 performs well for detecting and monitoring not only GAD but also panic disorder, social anxiety disorder, and posttraumatic stress disorder.24
CAGE questionnaire for detection of substance use
The CAGE questionnaire is a widely used screening tool that was originally developed to detect alcohol abuse but has been adapted to assess other substance abuse.25,26 The omission of substance abuse from diagnostic consideration can have a major effect on quality of care, because substance abuse can be the underlying cause of other diseases. Therefore, routine administration of this instrument in clinical practice can lead to better understanding and monitoring of patient health.27
Similar to other instruments, CAGE is free and available online.27 It contains four simple questions, with 1 point assigned to each positive answer (see Table); the simple mnemonic makes the questions easy to remember and to administer in a clinical setting.

CAGE has demonstrated validity, with one study determining that scores ≥ 2 had a specificity and sensitivity of 76% and 93%, respectively, for identifying excessive drinking, and a specificity and sensitivity of 77% and 91%, respectively, for identifying alcohol abuse.28
Columbia Suicide Severity Rating Scale (C-SSRS)
C-SSRS was developed by researchers at Columbia University to assess the severity of and track changes over time in suicidal ideation and behavior. C-SSRS is two pages and takes only a few minutes to administer; however, it also may be completed as a self-report measure. The questions are phrased in an interview format, and while clinicians are encouraged to receive training prior to its administration, specific training in mental health is not required.
The "Lifetime/Recent" version allows practitioners to gather lifetime history of suicidality as well as any recent suicidal ideation and/or behavior, whereas the "Since Last Visit" version of the scale assesses suicidality in patients who have completed at least one Lifetime/Recent C-SSRS assessment. A truncated, six-item "Screener" version is typically used in emergency situations. A risk assessment can be added to either the Full or Screener version to summarize the answers from C-SSRS and document risk and protective factors.29
Several studies have found C-SSRS to be reliable and valid for identifying suicide risk in children and adults.30,31USA Today reported that an individual exhibiting even a single behavior identified by the scale is eight to 10 times more likely to complete suicide.32 In addition, the C-SSRS has helped reduce the suicide rate by 65% in one of the largest providers of community-based behavioral health care in the United States.32
USING SCALES TO AUGMENT CARE
Each of the scales described in this article can easily be incorporated into clinical practice. The information the scales provide can be used to track progression of symptoms and effectiveness of treatment. Although rating scales should never be used alone to establish a diagnosis or clinical treatment plan, they can and should be used to augment information from the clinician's assessment and follow-up interviews.5
1. McDowell I. Measuring Health: A Guide to Rating Scales and Questionnaires. 3rd ed. New York, NY: Oxford University Press; 2006.
2. Kennedy Forum. Fixing behavioral health care in America: a national call for integrating and coordinating specialty behavioral health care with the medical system. http://thekennedyforum-dot-org.s3.amazonaws.com/documents/KennedyForum-BehavioralHealth_FINAL_3.pdf. Accessed August 14, 2017.
3. The Office of the National Coordinator for Health Information Technology. Behavioral health (BH) Clinical Quality Measures (CQMs) Program initiatives. www.healthit.gov/sites/default/files/pdf/2012-09-27-behavioral-health-clinical-quality-measures-program-initiatives-public-forum.pdf. Accessed August 14, 2017.
4. Unutzer J, Harbin H, Schoenbaum M. The collaborative care model: an approach for integrating physical and mental health care in Medicaid health homes. www.medicaid.gov/State-Resource-Center/Medicaid-State-Technical-Assistance/Health-Homes-Technical-Assistance/Downloads/HH-IRC-Collaborative-5-13.pdf. Accessed August 14, 2017.
5. World Group On Psychiatric Evaluation; American Psychiatric Association Steering Committee On Practice Guidelines. Practice guideline for the psychiatric evaluation of adults. 2nd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/psychevaladults.pdf. Accessed August 14, 2017.
6. Melek S, Norris D, Paulus J. Economic Impact of Integrated Medical-Behavioral Healthcare: Implications for Psychiatry. Denver, CO: Milliman, Inc; 2014.
7. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
8. Kennedy Forum. Fixing behavioral health care in America: a national call for measurement-based care. www.thekennedyforum.org/a-national-call-for-measurement-based-care/. Accessed August 14, 2017.
9. Zimmerman M, McGlinchey JB. Why don't psychiatrists use scales to measure outcome when treating depressed patients? J Clin Psychiatry. 2008;69(12):1916-1919.
10. Hatfield D, McCullough L, Frantz SH, et al. Do we know when our clients get worse? An investigation of therapists' ability to detect negative client change. Clin Psychol Psychother. 2010;17(1):25-32.
11. SAMHSA-HRSA Center for Integrated Solutions. Screening tools. www.integration.samhsa.gov/clinical-practice/screening-tools. Accessed August 14, 2017.
12. Moller HJ. Standardised rating scales in psychiatry: methodological basis, their possibilities and limitations and descriptions of important rating scales. World J Biol Psychiatry. 2009;10(1):6-26.
13. Sajatovic M, Ramirez LF. Rating Scales in Mental Health. 2nd ed. Hudson, OH: Lexi-Comp; 2003.
14. Patient Health Questionnaire-9 (PHQ-9). www.agencymeddirectors.wa.gov/files/AssessmentTools/14-PHQ-9%20overview.pdf. Accessed August 14, 2017.
15. Patient Health Questionnaire-9 (PHQ-9). Rehab Measures Web site. www.rehabmeasures.org/Lists/RehabMeasures/DispForm.aspx?ID=954. Accessed August 14, 2017.
16. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
17. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
18. Ketter TA. Strategies for monitoring outcomes in patients with bipolar disorder. Prim Care Companion J Clin Psychiatry. 2010;12(suppl 1):10-16.
19. The Mood Disorder Questionnaire. University of Texas Medical Branch. www.dbsalliance.org/pdfs/MDQ.pdf. Accessed August 14, 2017.
20. Hirschfeld RM, Holzer C, Calabrese JR, et al. Validity of the Mood Disorder Questionnaire: a general population study. Am J Psychiatry. 2003;160(1):178-180.
21. Boschloo L, Nolen WA, Spijker AT, et al. The Mood Disorder Questionnaire (MDQ) for detecting (hypo) manic episodes: its validity and impact of recall bias. J Affect Disord. 2013;151(1):203-208.
22. Spitzer RL, Kroenke K, Williams JB, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092-1097.
23. Lowe B, Decker O, Müller S, et al. Validation and standardization of the Generalized Anxiety Disorder Screener (GAD-7) in the general population. Med Care. 2008;46(3):266-274.
24. Kroenke K, Spitzer RL, Williams JB, et al. Anxiety disorders in primary care: prevalence, impairment, comorbidity, and detection. Ann Intern Med. 2007;146(5):317-325.
25. Ewing JA. Detecting alcoholism. The CAGE Questionnaire. JAMA. 1984;252(14):1905-1907.
26. CAGE substance abuse screening tool. Johns Hopkins Medicine. www.hopkinsmedicine.org/johns_hopkins_healthcare/downloads/cage%20substance%20screening%20tool.pdf. Accessed August 14, 2017.
27. O'Brien CP. The CAGE questionnaire for detection of alcoholism: a remarkably useful but simple tool. JAMA. 2008;300(17):2054-2056.
28. Bernadt MW, Mumford J, Taylor C, et al. Comparison of questionnaire and laboratory tests in the detection of excessive drinking and alcoholism. Lancet. 1982;1(8267):325-328.
29. Columbia Suicide-Severity Rating Scale (CS-SRS). http://cssrs.columbia.edu/the-columbia-scale-c-ssrs/cssrs-for-communities-and-healthcare/#filter=.general-use.english. Accessed August 14, 2017.
30. Mundt JC, Greist JH, Jefferson JW, et al. Prediction of suicidal behavior in clinical research by lifetime suicidal ideation and behavior ascertained by the electronic Columbia-Suicide Severity Rating Scale. J Clin Psychiatry. 2013;74(9):887-893.
31. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277.
32. Esposito L. Suicide checklist spots people at highest risk. USA Today. http://usatoday30.usatoday.com/news/health/story/health/story/2011-11-09/Suicide-checklist-spots-peo ple-at-highest-risk/51135944/1. Accessed August 14, 2017.
In the current health care environment, there is an increasing demand for objective assessment of disease states.1 This is particularly apparent in the realm of behavioral health, where documentation of outcomes lags that of other areas of medicine.
In 2012, the additional health care costs incurred by persons with mental health diagnoses were estimated to be $293 billion among commercially insured, Medicaid, and Medicare beneficiaries in the United States—a figure that is 273% higher than the cost for those without psychiatric diagnoses.2 Psychiatric and medical illnesses can be so tightly linked that accurate diagnosis and treatment of psychiatric disorders becomes essential to control medical illnesses. It is not surprising that there is increased scrutiny to the ways in which behavioral health care can be objectively assessed and monitored, and payers such as the Centers for Medicare and Medicaid Services increasingly require objective documentation of disease state improvement for payment.3
Support for objective assessment of disease derives from the collaborative care model. This model is designed to better integrate mental health and primary care (among other practices) by establishing the Patient-Centered Medical Home and emphasizing screening and monitoring patient-reported outcomes over time to assess treatment response.4 This approach, which is endorsed by the American Psychiatric Association, is associated with significant improvements in outcomes compared with usual care.5 It tracks patient progress using validated clinical rating scales and other screening tools (eg, Patient Health Questionnaire [PHQ-9] for depression), an approach that is analogous to how patients with type 2 diabetes are monitored by A1C lab tests.6 An extensive body of research supports the impact of this approach on treatment. A 2012 Cochrane review associated collaborative care with significant improvements in depression and anxiety outcomes compared with usual treatment.7
Despite these findings, a recent Kennedy Forum brief asserts that behavioral health is characterized by a "lack of systematic measurement to determine whether patients are responding to treatment."8 That same brief points to the many validated, easy-to-administer rating scales and screening tools that can reliably measure the frequency and severity of psychiatric symptoms over time, and likens the lack of their use to "treating high blood pressure without using a blood pressure cuff to measure if a patient's blood pressure is improving."8 In fact, it is estimated that only 18% of psychiatrists and 11% of psychologists use rating scales routinely.9,10 This lack of use denies clinicians important information that can help detect deterioration or lack of improvement in their patients; implementing these scales in primary care can help early detection of behavioral health problems.
Behavioral health is replete with rating scales and screening tools, and the number of competing scales can make choosing a measure difficult.1 Nonetheless, not all scales are appropriate for clinical use; many are designed for research, for instance, and are lengthy and difficult to administer.
Let's review a number of rating scales that are brief, useful, and easy to administer. A framework for the screening tools addressed in this article is available on the federally funded Center for Integrated Health Solutions website (www.integration.samhsa.gov). This site promotes the use of tools designed to assist in screening and monitoring for depression, anxiety, bipolar disorder, substance use, and suicidality.11
QUALITY CRITERIA FOR RATING SCALES
The quality of a rating scale is determined by the following attributes.
Objectivity. The ability of a scale to obtain the same results, regardless of who administers, analyzes, or interprets it.
Reliability. The ability of a scale to convey consistent and reproducible information across time, patients, and raters.
Validity. The degree to which the scale measures what it is supposed to measure (eg, depressive symptoms). Sensitivity and specificity are measures of validity and provide additional information about the rating scale; namely, whether the scale can detect the presence of a disease (sensitivity) and whether it detects only that disease or condition and not another (specificity).
Establishment of norms. Whether a scale provides reference values for different clinical groups.
Practicability. The resources required to administer the assessment instrument in terms of time, staff, and material.12
In addition to meeting these quality criteria, selection of a scale can be based on whether it is self-rated or observer-rated. Advantages to self-rated scales, such as the PHQ-9, Mood Disorder Questionnaire (MDQ), and Generalized Anxiety Disorder 7-item (GAD-7) scale, are their practicability—they are easy to administer and don't require much time—and their use in evaluating and raising awareness of subjective states.
However, reliability may be a concern, as some patients may lack insight or exaggerate or mask symptoms when completing such scales.13 Both observer- and self-rated scales can be used together to minimize bias, identify symptoms that might have been missed/not addressed in the clinical interview, and drive clinical decision-making. Both can also help patients communicate with their providers and make them feel more involved in clinical decision-making.8
ENDORSED RATING SCALES
The following scales have met many of the quality criteria described here and are endorsed by the government payer system. They can easily be incorporated into clinical practice and will provide useful clinical information that can assist in diagnosis and monitoring patient outcomes.
Patient Health Questionnaire
PHQ-9 is a nine-item self-report questionnaire that can help to detect depression and supplement a thorough mental health interview. It scores the nine DSM-IV criteria for depression on a scale of 0 (not at all) to 3 (nearly every day). It is a public resource that is easy to find online, available without cost in several languages, and takes just a few minutes to complete.14
PHQ-9 has shown excellent test-retest reliability in screening for depression, and normative data on the instrument's use are available in various clinical populations.15 Research has shown that as PHQ-9 depression scores increase, functional status decreases, while depressive symptoms, sick days, and health care utilization increase.15 In one study, a PHQ-9 score of ≥ 10 had 88% sensitivity and specificity for detecting depression, with scores of 5, 10, 15, and 20 indicating mild, moderate, moderately severe, and severe depression, respectively.16 In addition to its use as a screening tool, PHQ-9 is a responsive and reliable measure of depression treatment outcomes.17
Mood Disorder Questionnaire
MDQ is another brief, self-report questionnaire that is available online. It is designed to identify and monitor patients who are likely to meet diagnostic criteria for bipolar disorder.18,19
The first question on the MDQ asks if the patient has experienced any of 13 common mood and behavior symptoms. The second question asks if these symptoms have ever occurred at the same time, and the third asks the degree to which the patient finds the symptoms to be problematic. The remaining two questions provide additional clinical information, addressing family history of manic-depressive illness or bipolar disorder and whether a diagnosis of either disorder has been made.
The MDQ has shown validity in assessing bipolar disorder symptoms in a general population, although recent research suggests that imprecise recall bias may limit its reliability in detecting hypomanic episodes earlier in life.20,21 Nonetheless, its specificity of > 97% means that it will effectively screen out just about all true negatives.18
Generalized Anxiety Disorder 7-item scale
The GAD-7 scale is a brief, self-administered questionnaire for screening and measuring severity of GAD.22 It asks patients to rate seven items that represent problems with general anxiety and scores each item on a scale of 0 (not at all) to 3 (nearly every day). Similar to the other measures, it is easily accessible online.
Research evidence supports the reliability and validity of GAD-7 as a measure of anxiety in the general population. Sensitivity and specificity are 89% and 82%, respectively. Normative data for age- and sex-specific subgroups support its use across age groups and in both males and females.23 The GAD-7 performs well for detecting and monitoring not only GAD but also panic disorder, social anxiety disorder, and posttraumatic stress disorder.24
CAGE questionnaire for detection of substance use
The CAGE questionnaire is a widely used screening tool that was originally developed to detect alcohol abuse but has been adapted to assess other substance abuse.25,26 The omission of substance abuse from diagnostic consideration can have a major effect on quality of care, because substance abuse can be the underlying cause of other diseases. Therefore, routine administration of this instrument in clinical practice can lead to better understanding and monitoring of patient health.27
Similar to other instruments, CAGE is free and available online.27 It contains four simple questions, with 1 point assigned to each positive answer (see Table); the simple mnemonic makes the questions easy to remember and to administer in a clinical setting.
CAGE has demonstrated validity, with one study determining that scores ≥ 2 had a specificity and sensitivity of 76% and 93%, respectively, for identifying excessive drinking, and a specificity and sensitivity of 77% and 91%, respectively, for identifying alcohol abuse.28
Columbia Suicide Severity Rating Scale (C-SSRS)
C-SSRS was developed by researchers at Columbia University to assess the severity of and track changes over time in suicidal ideation and behavior. C-SSRS is two pages and takes only a few minutes to administer; however, it also may be completed as a self-report measure. The questions are phrased in an interview format, and while clinicians are encouraged to receive training prior to its administration, specific training in mental health is not required.
The "Lifetime/Recent" version allows practitioners to gather lifetime history of suicidality as well as any recent suicidal ideation and/or behavior, whereas the "Since Last Visit" version of the scale assesses suicidality in patients who have completed at least one Lifetime/Recent C-SSRS assessment. A truncated, six-item "Screener" version is typically used in emergency situations. A risk assessment can be added to either the Full or Screener version to summarize the answers from C-SSRS and document risk and protective factors.29
Several studies have found C-SSRS to be reliable and valid for identifying suicide risk in children and adults.30,31USA Today reported that an individual exhibiting even a single behavior identified by the scale is eight to 10 times more likely to complete suicide.32 In addition, the C-SSRS has helped reduce the suicide rate by 65% in one of the largest providers of community-based behavioral health care in the United States.32
USING SCALES TO AUGMENT CARE
Each of the scales described in this article can easily be incorporated into clinical practice. The information the scales provide can be used to track progression of symptoms and effectiveness of treatment. Although rating scales should never be used alone to establish a diagnosis or clinical treatment plan, they can and should be used to augment information from the clinician's assessment and follow-up interviews.5
In the current health care environment, there is an increasing demand for objective assessment of disease states.1 This is particularly apparent in the realm of behavioral health, where documentation of outcomes lags that of other areas of medicine.
In 2012, the additional health care costs incurred by persons with mental health diagnoses were estimated to be $293 billion among commercially insured, Medicaid, and Medicare beneficiaries in the United States—a figure that is 273% higher than the cost for those without psychiatric diagnoses.2 Psychiatric and medical illnesses can be so tightly linked that accurate diagnosis and treatment of psychiatric disorders becomes essential to control medical illnesses. It is not surprising that there is increased scrutiny to the ways in which behavioral health care can be objectively assessed and monitored, and payers such as the Centers for Medicare and Medicaid Services increasingly require objective documentation of disease state improvement for payment.3
Support for objective assessment of disease derives from the collaborative care model. This model is designed to better integrate mental health and primary care (among other practices) by establishing the Patient-Centered Medical Home and emphasizing screening and monitoring patient-reported outcomes over time to assess treatment response.4 This approach, which is endorsed by the American Psychiatric Association, is associated with significant improvements in outcomes compared with usual care.5 It tracks patient progress using validated clinical rating scales and other screening tools (eg, Patient Health Questionnaire [PHQ-9] for depression), an approach that is analogous to how patients with type 2 diabetes are monitored by A1C lab tests.6 An extensive body of research supports the impact of this approach on treatment. A 2012 Cochrane review associated collaborative care with significant improvements in depression and anxiety outcomes compared with usual treatment.7
Despite these findings, a recent Kennedy Forum brief asserts that behavioral health is characterized by a "lack of systematic measurement to determine whether patients are responding to treatment."8 That same brief points to the many validated, easy-to-administer rating scales and screening tools that can reliably measure the frequency and severity of psychiatric symptoms over time, and likens the lack of their use to "treating high blood pressure without using a blood pressure cuff to measure if a patient's blood pressure is improving."8 In fact, it is estimated that only 18% of psychiatrists and 11% of psychologists use rating scales routinely.9,10 This lack of use denies clinicians important information that can help detect deterioration or lack of improvement in their patients; implementing these scales in primary care can help early detection of behavioral health problems.
Behavioral health is replete with rating scales and screening tools, and the number of competing scales can make choosing a measure difficult.1 Nonetheless, not all scales are appropriate for clinical use; many are designed for research, for instance, and are lengthy and difficult to administer.
Let's review a number of rating scales that are brief, useful, and easy to administer. A framework for the screening tools addressed in this article is available on the federally funded Center for Integrated Health Solutions website (www.integration.samhsa.gov). This site promotes the use of tools designed to assist in screening and monitoring for depression, anxiety, bipolar disorder, substance use, and suicidality.11
QUALITY CRITERIA FOR RATING SCALES
The quality of a rating scale is determined by the following attributes.
Objectivity. The ability of a scale to obtain the same results, regardless of who administers, analyzes, or interprets it.
Reliability. The ability of a scale to convey consistent and reproducible information across time, patients, and raters.
Validity. The degree to which the scale measures what it is supposed to measure (eg, depressive symptoms). Sensitivity and specificity are measures of validity and provide additional information about the rating scale; namely, whether the scale can detect the presence of a disease (sensitivity) and whether it detects only that disease or condition and not another (specificity).
Establishment of norms. Whether a scale provides reference values for different clinical groups.
Practicability. The resources required to administer the assessment instrument in terms of time, staff, and material.12
In addition to meeting these quality criteria, selection of a scale can be based on whether it is self-rated or observer-rated. Advantages to self-rated scales, such as the PHQ-9, Mood Disorder Questionnaire (MDQ), and Generalized Anxiety Disorder 7-item (GAD-7) scale, are their practicability—they are easy to administer and don't require much time—and their use in evaluating and raising awareness of subjective states.
However, reliability may be a concern, as some patients may lack insight or exaggerate or mask symptoms when completing such scales.13 Both observer- and self-rated scales can be used together to minimize bias, identify symptoms that might have been missed/not addressed in the clinical interview, and drive clinical decision-making. Both can also help patients communicate with their providers and make them feel more involved in clinical decision-making.8
ENDORSED RATING SCALES
The following scales have met many of the quality criteria described here and are endorsed by the government payer system. They can easily be incorporated into clinical practice and will provide useful clinical information that can assist in diagnosis and monitoring patient outcomes.
Patient Health Questionnaire
PHQ-9 is a nine-item self-report questionnaire that can help to detect depression and supplement a thorough mental health interview. It scores the nine DSM-IV criteria for depression on a scale of 0 (not at all) to 3 (nearly every day). It is a public resource that is easy to find online, available without cost in several languages, and takes just a few minutes to complete.14
PHQ-9 has shown excellent test-retest reliability in screening for depression, and normative data on the instrument's use are available in various clinical populations.15 Research has shown that as PHQ-9 depression scores increase, functional status decreases, while depressive symptoms, sick days, and health care utilization increase.15 In one study, a PHQ-9 score of ≥ 10 had 88% sensitivity and specificity for detecting depression, with scores of 5, 10, 15, and 20 indicating mild, moderate, moderately severe, and severe depression, respectively.16 In addition to its use as a screening tool, PHQ-9 is a responsive and reliable measure of depression treatment outcomes.17
Mood Disorder Questionnaire
MDQ is another brief, self-report questionnaire that is available online. It is designed to identify and monitor patients who are likely to meet diagnostic criteria for bipolar disorder.18,19
The first question on the MDQ asks if the patient has experienced any of 13 common mood and behavior symptoms. The second question asks if these symptoms have ever occurred at the same time, and the third asks the degree to which the patient finds the symptoms to be problematic. The remaining two questions provide additional clinical information, addressing family history of manic-depressive illness or bipolar disorder and whether a diagnosis of either disorder has been made.
The MDQ has shown validity in assessing bipolar disorder symptoms in a general population, although recent research suggests that imprecise recall bias may limit its reliability in detecting hypomanic episodes earlier in life.20,21 Nonetheless, its specificity of > 97% means that it will effectively screen out just about all true negatives.18
Generalized Anxiety Disorder 7-item scale
The GAD-7 scale is a brief, self-administered questionnaire for screening and measuring severity of GAD.22 It asks patients to rate seven items that represent problems with general anxiety and scores each item on a scale of 0 (not at all) to 3 (nearly every day). Similar to the other measures, it is easily accessible online.
Research evidence supports the reliability and validity of GAD-7 as a measure of anxiety in the general population. Sensitivity and specificity are 89% and 82%, respectively. Normative data for age- and sex-specific subgroups support its use across age groups and in both males and females.23 The GAD-7 performs well for detecting and monitoring not only GAD but also panic disorder, social anxiety disorder, and posttraumatic stress disorder.24
CAGE questionnaire for detection of substance use
The CAGE questionnaire is a widely used screening tool that was originally developed to detect alcohol abuse but has been adapted to assess other substance abuse.25,26 The omission of substance abuse from diagnostic consideration can have a major effect on quality of care, because substance abuse can be the underlying cause of other diseases. Therefore, routine administration of this instrument in clinical practice can lead to better understanding and monitoring of patient health.27
Similar to other instruments, CAGE is free and available online.27 It contains four simple questions, with 1 point assigned to each positive answer (see Table); the simple mnemonic makes the questions easy to remember and to administer in a clinical setting.
CAGE has demonstrated validity, with one study determining that scores ≥ 2 had a specificity and sensitivity of 76% and 93%, respectively, for identifying excessive drinking, and a specificity and sensitivity of 77% and 91%, respectively, for identifying alcohol abuse.28
Columbia Suicide Severity Rating Scale (C-SSRS)
C-SSRS was developed by researchers at Columbia University to assess the severity of and track changes over time in suicidal ideation and behavior. C-SSRS is two pages and takes only a few minutes to administer; however, it also may be completed as a self-report measure. The questions are phrased in an interview format, and while clinicians are encouraged to receive training prior to its administration, specific training in mental health is not required.
The "Lifetime/Recent" version allows practitioners to gather lifetime history of suicidality as well as any recent suicidal ideation and/or behavior, whereas the "Since Last Visit" version of the scale assesses suicidality in patients who have completed at least one Lifetime/Recent C-SSRS assessment. A truncated, six-item "Screener" version is typically used in emergency situations. A risk assessment can be added to either the Full or Screener version to summarize the answers from C-SSRS and document risk and protective factors.29
Several studies have found C-SSRS to be reliable and valid for identifying suicide risk in children and adults.30,31USA Today reported that an individual exhibiting even a single behavior identified by the scale is eight to 10 times more likely to complete suicide.32 In addition, the C-SSRS has helped reduce the suicide rate by 65% in one of the largest providers of community-based behavioral health care in the United States.32
USING SCALES TO AUGMENT CARE
Each of the scales described in this article can easily be incorporated into clinical practice. The information the scales provide can be used to track progression of symptoms and effectiveness of treatment. Although rating scales should never be used alone to establish a diagnosis or clinical treatment plan, they can and should be used to augment information from the clinician's assessment and follow-up interviews.5
1. McDowell I. Measuring Health: A Guide to Rating Scales and Questionnaires. 3rd ed. New York, NY: Oxford University Press; 2006.
2. Kennedy Forum. Fixing behavioral health care in America: a national call for integrating and coordinating specialty behavioral health care with the medical system. http://thekennedyforum-dot-org.s3.amazonaws.com/documents/KennedyForum-BehavioralHealth_FINAL_3.pdf. Accessed August 14, 2017.
3. The Office of the National Coordinator for Health Information Technology. Behavioral health (BH) Clinical Quality Measures (CQMs) Program initiatives. www.healthit.gov/sites/default/files/pdf/2012-09-27-behavioral-health-clinical-quality-measures-program-initiatives-public-forum.pdf. Accessed August 14, 2017.
4. Unutzer J, Harbin H, Schoenbaum M. The collaborative care model: an approach for integrating physical and mental health care in Medicaid health homes. www.medicaid.gov/State-Resource-Center/Medicaid-State-Technical-Assistance/Health-Homes-Technical-Assistance/Downloads/HH-IRC-Collaborative-5-13.pdf. Accessed August 14, 2017.
5. World Group On Psychiatric Evaluation; American Psychiatric Association Steering Committee On Practice Guidelines. Practice guideline for the psychiatric evaluation of adults. 2nd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/psychevaladults.pdf. Accessed August 14, 2017.
6. Melek S, Norris D, Paulus J. Economic Impact of Integrated Medical-Behavioral Healthcare: Implications for Psychiatry. Denver, CO: Milliman, Inc; 2014.
7. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
8. Kennedy Forum. Fixing behavioral health care in America: a national call for measurement-based care. www.thekennedyforum.org/a-national-call-for-measurement-based-care/. Accessed August 14, 2017.
9. Zimmerman M, McGlinchey JB. Why don't psychiatrists use scales to measure outcome when treating depressed patients? J Clin Psychiatry. 2008;69(12):1916-1919.
10. Hatfield D, McCullough L, Frantz SH, et al. Do we know when our clients get worse? An investigation of therapists' ability to detect negative client change. Clin Psychol Psychother. 2010;17(1):25-32.
11. SAMHSA-HRSA Center for Integrated Solutions. Screening tools. www.integration.samhsa.gov/clinical-practice/screening-tools. Accessed August 14, 2017.
12. Moller HJ. Standardised rating scales in psychiatry: methodological basis, their possibilities and limitations and descriptions of important rating scales. World J Biol Psychiatry. 2009;10(1):6-26.
13. Sajatovic M, Ramirez LF. Rating Scales in Mental Health. 2nd ed. Hudson, OH: Lexi-Comp; 2003.
14. Patient Health Questionnaire-9 (PHQ-9). www.agencymeddirectors.wa.gov/files/AssessmentTools/14-PHQ-9%20overview.pdf. Accessed August 14, 2017.
15. Patient Health Questionnaire-9 (PHQ-9). Rehab Measures Web site. www.rehabmeasures.org/Lists/RehabMeasures/DispForm.aspx?ID=954. Accessed August 14, 2017.
16. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
17. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
18. Ketter TA. Strategies for monitoring outcomes in patients with bipolar disorder. Prim Care Companion J Clin Psychiatry. 2010;12(suppl 1):10-16.
19. The Mood Disorder Questionnaire. University of Texas Medical Branch. www.dbsalliance.org/pdfs/MDQ.pdf. Accessed August 14, 2017.
20. Hirschfeld RM, Holzer C, Calabrese JR, et al. Validity of the Mood Disorder Questionnaire: a general population study. Am J Psychiatry. 2003;160(1):178-180.
21. Boschloo L, Nolen WA, Spijker AT, et al. The Mood Disorder Questionnaire (MDQ) for detecting (hypo) manic episodes: its validity and impact of recall bias. J Affect Disord. 2013;151(1):203-208.
22. Spitzer RL, Kroenke K, Williams JB, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092-1097.
23. Lowe B, Decker O, Müller S, et al. Validation and standardization of the Generalized Anxiety Disorder Screener (GAD-7) in the general population. Med Care. 2008;46(3):266-274.
24. Kroenke K, Spitzer RL, Williams JB, et al. Anxiety disorders in primary care: prevalence, impairment, comorbidity, and detection. Ann Intern Med. 2007;146(5):317-325.
25. Ewing JA. Detecting alcoholism. The CAGE Questionnaire. JAMA. 1984;252(14):1905-1907.
26. CAGE substance abuse screening tool. Johns Hopkins Medicine. www.hopkinsmedicine.org/johns_hopkins_healthcare/downloads/cage%20substance%20screening%20tool.pdf. Accessed August 14, 2017.
27. O'Brien CP. The CAGE questionnaire for detection of alcoholism: a remarkably useful but simple tool. JAMA. 2008;300(17):2054-2056.
28. Bernadt MW, Mumford J, Taylor C, et al. Comparison of questionnaire and laboratory tests in the detection of excessive drinking and alcoholism. Lancet. 1982;1(8267):325-328.
29. Columbia Suicide-Severity Rating Scale (CS-SRS). http://cssrs.columbia.edu/the-columbia-scale-c-ssrs/cssrs-for-communities-and-healthcare/#filter=.general-use.english. Accessed August 14, 2017.
30. Mundt JC, Greist JH, Jefferson JW, et al. Prediction of suicidal behavior in clinical research by lifetime suicidal ideation and behavior ascertained by the electronic Columbia-Suicide Severity Rating Scale. J Clin Psychiatry. 2013;74(9):887-893.
31. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277.
32. Esposito L. Suicide checklist spots people at highest risk. USA Today. http://usatoday30.usatoday.com/news/health/story/health/story/2011-11-09/Suicide-checklist-spots-peo ple-at-highest-risk/51135944/1. Accessed August 14, 2017.
1. McDowell I. Measuring Health: A Guide to Rating Scales and Questionnaires. 3rd ed. New York, NY: Oxford University Press; 2006.
2. Kennedy Forum. Fixing behavioral health care in America: a national call for integrating and coordinating specialty behavioral health care with the medical system. http://thekennedyforum-dot-org.s3.amazonaws.com/documents/KennedyForum-BehavioralHealth_FINAL_3.pdf. Accessed August 14, 2017.
3. The Office of the National Coordinator for Health Information Technology. Behavioral health (BH) Clinical Quality Measures (CQMs) Program initiatives. www.healthit.gov/sites/default/files/pdf/2012-09-27-behavioral-health-clinical-quality-measures-program-initiatives-public-forum.pdf. Accessed August 14, 2017.
4. Unutzer J, Harbin H, Schoenbaum M. The collaborative care model: an approach for integrating physical and mental health care in Medicaid health homes. www.medicaid.gov/State-Resource-Center/Medicaid-State-Technical-Assistance/Health-Homes-Technical-Assistance/Downloads/HH-IRC-Collaborative-5-13.pdf. Accessed August 14, 2017.
5. World Group On Psychiatric Evaluation; American Psychiatric Association Steering Committee On Practice Guidelines. Practice guideline for the psychiatric evaluation of adults. 2nd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/psychevaladults.pdf. Accessed August 14, 2017.
6. Melek S, Norris D, Paulus J. Economic Impact of Integrated Medical-Behavioral Healthcare: Implications for Psychiatry. Denver, CO: Milliman, Inc; 2014.
7. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525.
8. Kennedy Forum. Fixing behavioral health care in America: a national call for measurement-based care. www.thekennedyforum.org/a-national-call-for-measurement-based-care/. Accessed August 14, 2017.
9. Zimmerman M, McGlinchey JB. Why don't psychiatrists use scales to measure outcome when treating depressed patients? J Clin Psychiatry. 2008;69(12):1916-1919.
10. Hatfield D, McCullough L, Frantz SH, et al. Do we know when our clients get worse? An investigation of therapists' ability to detect negative client change. Clin Psychol Psychother. 2010;17(1):25-32.
11. SAMHSA-HRSA Center for Integrated Solutions. Screening tools. www.integration.samhsa.gov/clinical-practice/screening-tools. Accessed August 14, 2017.
12. Moller HJ. Standardised rating scales in psychiatry: methodological basis, their possibilities and limitations and descriptions of important rating scales. World J Biol Psychiatry. 2009;10(1):6-26.
13. Sajatovic M, Ramirez LF. Rating Scales in Mental Health. 2nd ed. Hudson, OH: Lexi-Comp; 2003.
14. Patient Health Questionnaire-9 (PHQ-9). www.agencymeddirectors.wa.gov/files/AssessmentTools/14-PHQ-9%20overview.pdf. Accessed August 14, 2017.
15. Patient Health Questionnaire-9 (PHQ-9). Rehab Measures Web site. www.rehabmeasures.org/Lists/RehabMeasures/DispForm.aspx?ID=954. Accessed August 14, 2017.
16. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606-613.
17. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
18. Ketter TA. Strategies for monitoring outcomes in patients with bipolar disorder. Prim Care Companion J Clin Psychiatry. 2010;12(suppl 1):10-16.
19. The Mood Disorder Questionnaire. University of Texas Medical Branch. www.dbsalliance.org/pdfs/MDQ.pdf. Accessed August 14, 2017.
20. Hirschfeld RM, Holzer C, Calabrese JR, et al. Validity of the Mood Disorder Questionnaire: a general population study. Am J Psychiatry. 2003;160(1):178-180.
21. Boschloo L, Nolen WA, Spijker AT, et al. The Mood Disorder Questionnaire (MDQ) for detecting (hypo) manic episodes: its validity and impact of recall bias. J Affect Disord. 2013;151(1):203-208.
22. Spitzer RL, Kroenke K, Williams JB, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092-1097.
23. Lowe B, Decker O, Müller S, et al. Validation and standardization of the Generalized Anxiety Disorder Screener (GAD-7) in the general population. Med Care. 2008;46(3):266-274.
24. Kroenke K, Spitzer RL, Williams JB, et al. Anxiety disorders in primary care: prevalence, impairment, comorbidity, and detection. Ann Intern Med. 2007;146(5):317-325.
25. Ewing JA. Detecting alcoholism. The CAGE Questionnaire. JAMA. 1984;252(14):1905-1907.
26. CAGE substance abuse screening tool. Johns Hopkins Medicine. www.hopkinsmedicine.org/johns_hopkins_healthcare/downloads/cage%20substance%20screening%20tool.pdf. Accessed August 14, 2017.
27. O'Brien CP. The CAGE questionnaire for detection of alcoholism: a remarkably useful but simple tool. JAMA. 2008;300(17):2054-2056.
28. Bernadt MW, Mumford J, Taylor C, et al. Comparison of questionnaire and laboratory tests in the detection of excessive drinking and alcoholism. Lancet. 1982;1(8267):325-328.
29. Columbia Suicide-Severity Rating Scale (CS-SRS). http://cssrs.columbia.edu/the-columbia-scale-c-ssrs/cssrs-for-communities-and-healthcare/#filter=.general-use.english. Accessed August 14, 2017.
30. Mundt JC, Greist JH, Jefferson JW, et al. Prediction of suicidal behavior in clinical research by lifetime suicidal ideation and behavior ascertained by the electronic Columbia-Suicide Severity Rating Scale. J Clin Psychiatry. 2013;74(9):887-893.
31. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277.
32. Esposito L. Suicide checklist spots people at highest risk. USA Today. http://usatoday30.usatoday.com/news/health/story/health/story/2011-11-09/Suicide-checklist-spots-peo ple-at-highest-risk/51135944/1. Accessed August 14, 2017.

Atopic Dermatitis Prevention and Treatment
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
Practice Points
- Prevention of atopic dermatitis is desired in high-risk settings (ie, 1 or more relatives with atopy).
- Emollient therapy from early infancy has been described as one method.
- Other forms of disease prevention have not yet been adequately developed.
2017 Update on female sexual dysfunction
Sexual function is a complex, multifaceted process mediated by neurologic functions, hormonal regulation, and ps
As it turns out, quite a lot. Female sexual dysfunction is a common, vastly undertreated sexual health problem that can have wide-reaching effects on a woman’s life. These effects may include impaired body image, self-confidence, and self-worth. Sexual dysfunction also can contribute to relationship dissatisfaction and leave one feeling less connected with her partner.1,2 Studies have shown women with sexual dysfunction have higher health care expenditures3 and that depression and fatigue are common comorbidities, as is frequently seen in other chronic conditions such as diabetes and back pain.4
Understanding the pathogenesis of female sexual dysfunction helps to guide our approach to its management. Indeed, increased understanding of its pathology has helped to usher in new and emerging treatment options, as well as a personalized, biopsychosocial approach to its management.
Related article:
2016 Update on female sexual dysfunction
In this Update, I discuss the interplay of physiologic and psychological factors that affect female sexual function as well as the latest options for its management. I have also assembled a panel of experts to discuss 2 cases representative of sexual dysfunction that you may encounter in your clinical practice and how prescribing decisions are made for their management.
Read about factors that impact sexual function and agents to help manage dysfunction.
Multiple transmitters in the brain can increase or decrease sexual desire and function
Neurotransmitters involved in sexual excitation include brain dopamine, melanocortin, oxytocin, vasopressin, and norepinephrine, whereas brain opioids, serotonin, prolactin, and endocannabinoids function as sexual inhibitors. Inhibitory transmitters are activated normally during sexual refractoriness but also from primary aversion or secondary avoidance disorders.1 Drugs or conditions that reduce brain dopamine levels, increase the action of brain serotonin, or enhance brain opioid pathways have been shown to inhibit sexual desire, while those that increase hypothalamic and mesolimbic dopamine or decrease serotonin release have been shown to stimulate sexual desire.1
Estradiol and progesterone can impact sexual function and desire
In addition to the neurotransmitters, hormones are important modulators of female sexual function. Decreasing levels of circulating estrogen after menopause lead to physiologic, biologic, and clinical changes in the urogenital tissues, such as decreased elastin, thinning of the epithelium, reduced vaginal blood flow, diminished lubrication, and decreased flexibility and elasticity. These changes result in the symptoms of genitourinary syndrome of menopause (GSM), which affects as many as half of all menopausal women.5,6 In clinical trials, dyspareunia and vaginal dryness are the most bothersome GSM symptoms reported.7
The role of hormonal regulation in sexual dysfunction among premenopausal women is not yet fully understood, but we do know that estradiol has been shown to improve sexual desire, progesterone tends to dampen sexual desire, and that testosterone at physiological levels has been shown in most studies to have a neutral effect on sexual desire in a well-estrogenized patient.8
Related article:
Focus on treating genital atrophy symptoms
Experience and behavior modulate or reinforce sexual dysfunction
The most common psychological factors that trigger or amplify female sexual dysfunction are depression, anxiety, distraction, negative body image, sexual abuse, and emotional neglect.9 Contextual or sociocultural factors, such as relationship discord, life-stage stressors (the empty nest syndrome or anxiety and sleep deprivation from a new baby), as well as cultural or religious values that suppress sexuality, also should be considered.9 Experience-based neuroplasticity (changes in brain pathways that become solidified by negative or positive experiences) may elucidate how a multimodal approach, utilizing medical and psychological treatment, can be beneficial for patients, particularly those with hypoactive sexual desire disorder (HSDD).1
New and emerging approaches to managing female sexual dysfunction
Three agents, one of which has been available for prescription for some time, one that is newly available, and one in the pipeline, are or may soon be in the gynecologist's armamentarium.
Flibanserin
Medications that target excitatory pathways or blunt inhibitory pathways are in development, and one, flibanserin (Addyi), has been US Food and Drug Administration (FDA)-approved for the treatment of acquired, generalized HSDD in premenopausal women.1,10 Flibanserin is a nonhormonal, centrally acting, postsynaptic serotonin 1A receptor agonist and a serotonin 2A receptor antagonist that is taken daily at bedtime (100 mg); several weeks are usually needed before any effects are noted.1 It is not approved for postmenopausal women and has a boxed warning about the risks of hypotension and syncope; its use is contraindicated in women who drink alcohol, in those who have hepatic impairment, and with the use of moderate or strong CYP3A4 inhibitors.11
Also keep in mind that flibanserin is only available through a Risk Evaluation and Mitigation Strategy program, so clinicians who wish to prescribe it must enroll in and complete training to become certified providers.9
Related article:
What you need to know (and do) to prescribe the new drug flibanserin
Prasterone
Prasterone (Intrarosa), a once-daily intravaginal dehydroepiandrosterone (DHEA) product, is a prohormone that increases local estrogen and testosterone and has the advantage of improved sexual function, desire, arousal, lubrication, orgasm, satisfaction, as well as pain at sexual activity.12 It was approved by the FDA in November 2016 to treat moderate to severe dyspareunia and has been available for prescribing since July 2017. Its cost is comparable to topical estrogen products, with a $25 copay program.
Because prasterone is not an estrogen, it does not have the boxed warning that all estrogen products are mandated by the FDA to have. This may make it more acceptable to patients, who often decline to use an estrogen product after seeing the boxed warning on the package. The Centers for Medicare and Medicaid Services (CMS) does not have prasterone on its list of potentially hazardous drugs for the elderly. However, keep in mind that because its label is for dyspareunia and not specifically for GSM, CMS considers it a drug of choice--in other words, like sildenafil (Viagra), a lifestyle choice and not for treatment of a medical condition. As such, at the present time, Medicare does not cover it.
Bremelanotide
Late-stage trials of bremelanotide, a melanocortin receptor agonist, are underway. Its mechanism of action is somewhat like that of flibanserin in that both drugs increase dopamine and norepinephrine levels. The advantage of bremelanotide is that it is used as needed. It is dosed subcutaneously (1.75 mg) and it can be used as often as a woman would like to use it. The FDA is expected to consider it for approval in about a year. Unpublished data from poster sessions at recent meetings show that, in a phase 3 study of 1,247 premenopausal women with HSDD (who had already been screened for depression and were found to have a physiologic condition), improvements in desire, arousal, lubrication, and orgasm were shown with bremelanotide. About 18% of women stopped using the drug because of adverse effects (nausea, vomiting, flushing, or headache) versus 2% for placebo. Like flibanserin, it is expected to be approved for premenopausal women only.
Read how 3 experts would manage differing GSM symptoms.
What would you prescribe for these patients?
CASE Genitourinary syndrome of menopause (GSM) in a 55-year-old woman
A 55-year-old widow is beginning a new relationship. She has not had partnered sexual activity for several years, but she recently has begun a relationship. She describes pain with attempted penetration with her new partner. Her last menstrual period was 3 years ago and she has experienced very minor menopausal symptoms, which are not bothersome. On examination, the vulva and vagina are pale, with thin epithelium and absent rugae. The tissue lacks elasticity. A virginal speculum is needed to visualize the cervix.
How would you go about deciding which of the many options for management of GSM you will recommend for this patient? What do you weigh as you consider DHEA versus estrogen and topical versus oral therapy?
JoAnn V. Pinkerton, MD: Vulvovaginal atrophy (VVA), part of GSM, is associated with postmenopausal estrogen deficiency and includes the signs and symptoms seen on this patient's physical exam: vaginal narrowing, pallor, loss of elasticity, as well as pain with intercourse.6 Estrogen therapy is the most effective treatment for vaginal atrophy.13 Since she does not have significant menopausal symptoms, low-dose vaginal estrogen preparations are effective and generally safe treatments for VVA; these include creams, tablets containing estradiol or conjugated equine estrogen (CEE), and a low-dose vaginal estradiol ring--all available at doses that result in minimal systemic absorption.
Choice is usually made based on patient desire and likely adherence. If the patient prefers nonestrogen therapies that improve VVA and have been approved for relief of dyspareunia in postmenopausal women, I would discuss with the patient the oral selective estrogen receptor modulator ospemifene,14 and the new intravaginal DHEA suppositories, prasterone.15 Ospemifene is taken daily as an oral tablet, has a small risk of blood clots, and is my choice for women who do not need systemic hormone therapy and prefer to avoid vaginal therapy.
Andrew M. Kaunitz, MD: GSM is prevalent in menopausal women and, if not treated, causes progressive vaginal dryness and sexual discomfort. When the main indication for hormonal management in a menopausal woman is GSM (as opposed to treatment of vasomotor symptoms or prevention of osteoporosis), the treatment of choice is low-dose local vaginal estrogen, ospemifene, or prasterone (DHEA). Prasterone is a vaginally administered nonestrogen steroid that was approved by the FDA to treat dyspareunia associated with GSM. DHEA is an endogenous inactive steroid that is converted locally into androgens and estrogens; one vaginal insert is placed nightly.16,17
This 55-year-old widow has not been sexually active for some time. The facts that attempted penetration was painful and only an ultrathin (virginal) speculum could be used for examination indicate that contraction of the pelvic floor muscles is likely present. Simply starting medical management may not lead to comfortable/successful penetrative sex for this woman. In addition to medical management, she would likely benefit from referral for physical therapy. Using dilators and other strategies, along with the positive impact that medical management will have on the vaginal mucosa, a woman's physical therapist can work with this patient to help the pelvic floor muscles relax and facilitate comfortable penetrative sex.
James A. Simon, MD: With only minor vasomotor symptoms, I would assess the other potential benefits of a systemic therapy. These might include cardiovascular risk reduction (systemic estrogens or estrogens/progesterone in some), breast cancer risk reduction (some data suggesting ospemifene can accomplish this), osteoporosis prevention (systemic estrogens and estrogen/androgens), etc. If there is an option for a treatment to address more than one symptom, in this case GSM, assessing the risks/benefits of each of these therapies should be estimated for this specific patient.
If there are no systemic benefits to be had, then any of the local treatments should be helpful. As there are no head-to-head comparisons available, local estrogen cream, tablets, rings, local DHEA, or systemic ospemifene each should be considered possible treatments. I also feel this patient may benefit from supplementary self-dilation and/or physical therapy.
Related article:
2017 Update on menopause
CASE Dyspareunia and vasomotor symptoms in a 42-year-old breast cancer survivor
A 42-year-old woman with a BRCA1 mutation has undergone prophylactic mastectomies as well as hysterectomy with bilateral salpingo-oophorectomy. She reports mild to moderate hot flashes and bothersome vaginal dryness and dyspareunia. Examination confirms GSM.
Would you advise systemic hormone therapy for this patient? What would your recommendation be for management of her GSM symptoms?
Dr. Simon: While one's gut reaction would be to avoid systemic estrogen therapy in a patient with a BRCA1 mutation, the scientific information confirming this fear is lacking.18 Such patients may benefit significantly from systemic estrogen therapy (reduced risk of cardiovascular disease and cognitive decline, etc.), and with both breasts and both ovaries removed, estrogen's breast cancer risks, if any in this population, are largely avoided. The patient also may benefit from additional local therapy with either estrogens or DHEA.
Dr. Kaunitz: Due to her high lifetime risk of breast and ovarian cancer, this woman has proceeded with risk-reducing breast and gynecologic surgery. As more BRCA mutation carriers are being identified and undergo risk-reducing bilateral mastectomy (usually with reconstruction) and salpingo-oophorectomy, clinicians and mutation carriers more frequently face decisions regarding use of systemic hormone therapy.
Mutation carriers who have undergone bilateral risk-reducing mastectomy experience a very low baseline future risk for breast cancer; accordingly, concerns regarding this disease should not prevent use of systemic hormone therapy. Furthermore, without hormone replacement, induced menopause in women this age is associated with an elevated risk of osteoporosis, persistent vasomotor symptoms, cardiovascular disease, stroke, mood changes, dementia, Parkinson disease, and overall mortality. Recognizing the safety of estrogen therapy in this setting, this 42-year-old BRCA1 mutation carrier can initiate estrogen therapy. Standard dose estrogen therapy refers to oral estradiol 1.0 mg, conjugated equine estrogen 0.625 mg,or transdermal estradiol 0.05 mg. In younger women like this 42-year-old with surgically induced menopause, higher than standard replacement doses of estrogen are often appropriate.17
Due to concerns the hormone therapy might further increase future risk of breast cancer, some mutation carriers may delay or avoid risk-reducing bilateral salpingo-oophorectomy, a potentially lifesaving surgery which reduces not only future risk of ovarian cancer but also future risk for breast cancer.
Among mutation carriers with intact breasts, several studies address risk of breast cancer with use of systemic hormone therapy. Although limited in numbers of participants and years of follow-up, in aggregate, these studies provide reassurance that short-term use of systemic hormone therapy does not increase breast cancer risk in women with BRCA1 or BRCA2 mutations and intact breasts.19
Dr. Pinkerton: For this woman with early surgical menopause and hysterectomy, estrogen therapy could improve her vasomotor symptoms and decrease her risk of bone loss and GSM.17 In the Women's Health Initiative trial, there were 7 fewer breast cancers per 10,000 women-years in the estrogen-onlyarm.20 Observational studies suggest that hormone therapy, when given to the average age of menopause, decreases the risks of heart disease, Parkinson disease, and dementia.21 Limited observational evidence suggests that hormone therapy use does not further increase risk of breast cancer in women following oophorectomy for BRCA1 or BRCA2 gene mutation.22
The absolute risks observed with hormone therapy tended to be small, especially in younger, healthy women. Systemic hormone therapy could treat her hot flashes and her GSM symptoms and potentially decrease health risks associated with premature estrogen deficiency. Nonestrogen therapies for hot flashes include low-dose antidepressants, gabapentin, and mind-body options, such as cognitive behavioral therapy and hypnosis, but these would not decrease her health risks or treat her GSM.
If she only requests treatment of her GSM symptoms, she would be a candidate for low-dose vaginal estrogen therapy, given as a cream, tablet, or ring depending on her choice. I would not choose ospemifene as my first choice as she is having hot flashes, and there are no data yet on the drug's health benefits in early menopause. If she prefers nonestrogen vaginal therapy, the new intravaginal DHEA might be a good choice as both estrogen and testosterone are increased locally in the vagina while hormone levels remain in the postmenopausal range. There is no boxed warning on the patient insert, although safety in women with breast cancer or in those with elevated risk of breast cancer has not been tested.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review. Mayo Clin Proc. 2017;92(1):114–128.
- Kingsberg SA. Attitudinal survey of women living with low sexual desire. J Womens Health (Larchmt). 2014;23(10):817–823.
- Foley K, Foley D, Johnson BH. Healthcare resource utilization and expenditures of women diagnosed with hypoactive sexual desire disorder. J Med Econ. 2010;13(4):583–590.
- Biddle AK, West SL, D’Aloisio AA, Wheeler SB, Borisov NN, Thorp J. Hypoactive sexual desire disorder in postmenopausal women: quality of life and health burden. Value Health. 2009;12(5):763–772.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;21(10):1063–1068.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause. 2013;20(9):888–902.
- Ettinger B, Hait H, Reape KZ, Shu H. Measuring symptom relief in studies of vaginal and vulvar atrophy: the most bothersome symptom approach. Menopause. 2008;15(5):885–889.
- Dennerstein L, Randolph J, Taffe J, Dudley E, Burger H. Hormones, mood, sexuality, and the menopausal transition. Fertil Steril. 2002;77(suppl 4):S42–S48.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women’s sexual desire and arousal disorders [published correction appears in J Sex Med. 2010;7(2 pt 1):856]. J Sex Med. 2010;7(1 pt 2):586–614.
- US Food and Drug Administration website. FDA approves first treatment for sexual desire disorder. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm458734.htm. Accessed August 14, 2017.
- Addyi (flibanserin) [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America, LLC; 2016.
- Labrie F, Derogatis L, Archer DF, et al; Members of the VVA Prasterone Research Group. Effect of intravaginal prasterone on sexual dysfunction in postmenopausal women with vulvovaginal atrophy. J Sex Med. 2015;12(12):2401–2412.
- Lethaby A, Ayeleke RO, Roberts H. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2016;8:CD001500.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause. 2013;20(6):623–630.
- Labrie F, Archer DF, Koltun, W, et al; VVA Prasterone Research Group. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause. 2016;23(3):243–256.
- Kaunitz AM. Focus on treating genital atrophy symptoms. OBG Manag. 2017;29(1):14, 16–17.
- The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24(7):728–753.
- Crandall CJ, Hovey KM, Andrews CA, et al. Breast cancer, endometrial cancer, and cardiovascular events in participants who used vaginal estrogen in the Women’s Health Initiative Observational Study. Menopause. August 14, 2017. doi:10.1097/GME.0000000000000956.
- Domchek S, Kaunitz AM. Use of systemic hormone therapy in BRCA mutation carriers. Menopause. 2016;23(9):1026–1027.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–1712.
- Faubion SS, Kuhle CL, Shuster LT, Rocca WA. Long-term health consequences of premature or early menopause and considerations for management. Climacteric. 2015;18(4):483–491.
- Gabriel CA, Tigges-Cardwell J, Stopfer J, Erlichman J, Nathanson K, Domchek SM. Use of total abdominal hysterectomy and hormone replacement therapy in BRCA1 and BRCA2 mutation carriers undergoing risk-reducing salpingo-oophorectomy. Fam Cancer. 2009;8(1):23-28.

Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.

Andrew M. Kaunitz, MD, NCMP, is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women's Health Specialists at Emerson, Jacksonville, Florida.

JoAnn V. Pinkerton, MD, NCMP, is Professor, Department of Obstetrics and Gynecology, and Director, Midlife Health, University of Virginia Health System, Charlottesville, Virginia; Executive Director, The North American Menopause Society, Pepper Pike, Ohio.

James A. Simon, MD, CCD, IF, NCMP, is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University; Medical Director, Women's Health & Research Consultants, Washington, DC.
Dr. Kaunitz reports that he receives grant or research support from Bayer, Endoceutics, and TherapeuticsMD and is a consultant to Bayer Healthcare, AMAG Pharmaceuticals, Allergan, Pfizer, and Shionogi. Dr. Kaunitz is a member of the OBG Management Board of Editors.
Dr. Pinkerton reports that she receives grant or research support from Grants/Research at TherapeuticsMD (fees go to the University of Virginia). She is a member of the OBG Management Board of Editors.
Dr. Simon reports he has served (within the last year) or is currently serving as a consultant to or on the advisory boards of: AbbVie, Allergan, AMAG Pharmaceuticals, Amgen, Ascend Therapeutics, Azure Biotech, Bayer Healthcare Pharmaceuticals, CEEK Enterprises, Covance, Millendo Therapeutics, Mitsubishi Tanabe Pharma Development America, ObsEva, Radius Health, Sanofi, Sebela Pharmaceuticals, Sermonix Pharmaceuticals, Shionogi, Symbiotec Pharmalab, TherapeuticsMD, and Valeant Pharmaceuticals. He has also served (within the last year) or is currently serving on the speaker's bureaus of: Duchesnay USA, Novo Nordisk, Shionogi, and Valeant Pharmaceuticals. In the last year, he has received or is currently receiving grant/research support from: AbbVie, Allergan, Agile Therapeutics, Bayer Healthcare, New England Research Institute, ObsEva, Palatin Technologies, Symbio Research, and TherapeuticsMD. He is a stockholder (direct purchase) in Sermonix Pharmaceuticals. Dr. Simon is a member of the OBG Management Board of Editors.
Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.
Andrew M. Kaunitz, MD, NCMP, is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women's Health Specialists at Emerson, Jacksonville, Florida.
JoAnn V. Pinkerton, MD, NCMP, is Professor, Department of Obstetrics and Gynecology, and Director, Midlife Health, University of Virginia Health System, Charlottesville, Virginia; Executive Director, The North American Menopause Society, Pepper Pike, Ohio.
James A. Simon, MD, CCD, IF, NCMP, is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University; Medical Director, Women's Health & Research Consultants, Washington, DC.
Dr. Kaunitz reports that he receives grant or research support from Bayer, Endoceutics, and TherapeuticsMD and is a consultant to Bayer Healthcare, AMAG Pharmaceuticals, Allergan, Pfizer, and Shionogi. Dr. Kaunitz is a member of the OBG Management Board of Editors.
Dr. Pinkerton reports that she receives grant or research support from Grants/Research at TherapeuticsMD (fees go to the University of Virginia). She is a member of the OBG Management Board of Editors.
Dr. Simon reports he has served (within the last year) or is currently serving as a consultant to or on the advisory boards of: AbbVie, Allergan, AMAG Pharmaceuticals, Amgen, Ascend Therapeutics, Azure Biotech, Bayer Healthcare Pharmaceuticals, CEEK Enterprises, Covance, Millendo Therapeutics, Mitsubishi Tanabe Pharma Development America, ObsEva, Radius Health, Sanofi, Sebela Pharmaceuticals, Sermonix Pharmaceuticals, Shionogi, Symbiotec Pharmalab, TherapeuticsMD, and Valeant Pharmaceuticals. He has also served (within the last year) or is currently serving on the speaker's bureaus of: Duchesnay USA, Novo Nordisk, Shionogi, and Valeant Pharmaceuticals. In the last year, he has received or is currently receiving grant/research support from: AbbVie, Allergan, Agile Therapeutics, Bayer Healthcare, New England Research Institute, ObsEva, Palatin Technologies, Symbio Research, and TherapeuticsMD. He is a stockholder (direct purchase) in Sermonix Pharmaceuticals. Dr. Simon is a member of the OBG Management Board of Editors.
Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.
Andrew M. Kaunitz, MD, NCMP, is University of Florida Term Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine-Jacksonville; Medical Director and Director of Menopause and Gynecologic Ultrasound Services, UF Women's Health Specialists at Emerson, Jacksonville, Florida.
JoAnn V. Pinkerton, MD, NCMP, is Professor, Department of Obstetrics and Gynecology, and Director, Midlife Health, University of Virginia Health System, Charlottesville, Virginia; Executive Director, The North American Menopause Society, Pepper Pike, Ohio.
James A. Simon, MD, CCD, IF, NCMP, is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University; Medical Director, Women's Health & Research Consultants, Washington, DC.
Dr. Kaunitz reports that he receives grant or research support from Bayer, Endoceutics, and TherapeuticsMD and is a consultant to Bayer Healthcare, AMAG Pharmaceuticals, Allergan, Pfizer, and Shionogi. Dr. Kaunitz is a member of the OBG Management Board of Editors.
Dr. Pinkerton reports that she receives grant or research support from Grants/Research at TherapeuticsMD (fees go to the University of Virginia). She is a member of the OBG Management Board of Editors.
Dr. Simon reports he has served (within the last year) or is currently serving as a consultant to or on the advisory boards of: AbbVie, Allergan, AMAG Pharmaceuticals, Amgen, Ascend Therapeutics, Azure Biotech, Bayer Healthcare Pharmaceuticals, CEEK Enterprises, Covance, Millendo Therapeutics, Mitsubishi Tanabe Pharma Development America, ObsEva, Radius Health, Sanofi, Sebela Pharmaceuticals, Sermonix Pharmaceuticals, Shionogi, Symbiotec Pharmalab, TherapeuticsMD, and Valeant Pharmaceuticals. He has also served (within the last year) or is currently serving on the speaker's bureaus of: Duchesnay USA, Novo Nordisk, Shionogi, and Valeant Pharmaceuticals. In the last year, he has received or is currently receiving grant/research support from: AbbVie, Allergan, Agile Therapeutics, Bayer Healthcare, New England Research Institute, ObsEva, Palatin Technologies, Symbio Research, and TherapeuticsMD. He is a stockholder (direct purchase) in Sermonix Pharmaceuticals. Dr. Simon is a member of the OBG Management Board of Editors.
Sexual function is a complex, multifaceted process mediated by neurologic functions, hormonal regulation, and ps
As it turns out, quite a lot. Female sexual dysfunction is a common, vastly undertreated sexual health problem that can have wide-reaching effects on a woman’s life. These effects may include impaired body image, self-confidence, and self-worth. Sexual dysfunction also can contribute to relationship dissatisfaction and leave one feeling less connected with her partner.1,2 Studies have shown women with sexual dysfunction have higher health care expenditures3 and that depression and fatigue are common comorbidities, as is frequently seen in other chronic conditions such as diabetes and back pain.4
Understanding the pathogenesis of female sexual dysfunction helps to guide our approach to its management. Indeed, increased understanding of its pathology has helped to usher in new and emerging treatment options, as well as a personalized, biopsychosocial approach to its management.
Related article:
2016 Update on female sexual dysfunction
In this Update, I discuss the interplay of physiologic and psychological factors that affect female sexual function as well as the latest options for its management. I have also assembled a panel of experts to discuss 2 cases representative of sexual dysfunction that you may encounter in your clinical practice and how prescribing decisions are made for their management.
Read about factors that impact sexual function and agents to help manage dysfunction.
Multiple transmitters in the brain can increase or decrease sexual desire and function
Neurotransmitters involved in sexual excitation include brain dopamine, melanocortin, oxytocin, vasopressin, and norepinephrine, whereas brain opioids, serotonin, prolactin, and endocannabinoids function as sexual inhibitors. Inhibitory transmitters are activated normally during sexual refractoriness but also from primary aversion or secondary avoidance disorders.1 Drugs or conditions that reduce brain dopamine levels, increase the action of brain serotonin, or enhance brain opioid pathways have been shown to inhibit sexual desire, while those that increase hypothalamic and mesolimbic dopamine or decrease serotonin release have been shown to stimulate sexual desire.1
Estradiol and progesterone can impact sexual function and desire
In addition to the neurotransmitters, hormones are important modulators of female sexual function. Decreasing levels of circulating estrogen after menopause lead to physiologic, biologic, and clinical changes in the urogenital tissues, such as decreased elastin, thinning of the epithelium, reduced vaginal blood flow, diminished lubrication, and decreased flexibility and elasticity. These changes result in the symptoms of genitourinary syndrome of menopause (GSM), which affects as many as half of all menopausal women.5,6 In clinical trials, dyspareunia and vaginal dryness are the most bothersome GSM symptoms reported.7
The role of hormonal regulation in sexual dysfunction among premenopausal women is not yet fully understood, but we do know that estradiol has been shown to improve sexual desire, progesterone tends to dampen sexual desire, and that testosterone at physiological levels has been shown in most studies to have a neutral effect on sexual desire in a well-estrogenized patient.8
Related article:
Focus on treating genital atrophy symptoms
Experience and behavior modulate or reinforce sexual dysfunction
The most common psychological factors that trigger or amplify female sexual dysfunction are depression, anxiety, distraction, negative body image, sexual abuse, and emotional neglect.9 Contextual or sociocultural factors, such as relationship discord, life-stage stressors (the empty nest syndrome or anxiety and sleep deprivation from a new baby), as well as cultural or religious values that suppress sexuality, also should be considered.9 Experience-based neuroplasticity (changes in brain pathways that become solidified by negative or positive experiences) may elucidate how a multimodal approach, utilizing medical and psychological treatment, can be beneficial for patients, particularly those with hypoactive sexual desire disorder (HSDD).1
New and emerging approaches to managing female sexual dysfunction
Three agents, one of which has been available for prescription for some time, one that is newly available, and one in the pipeline, are or may soon be in the gynecologist's armamentarium.
Flibanserin
Medications that target excitatory pathways or blunt inhibitory pathways are in development, and one, flibanserin (Addyi), has been US Food and Drug Administration (FDA)-approved for the treatment of acquired, generalized HSDD in premenopausal women.1,10 Flibanserin is a nonhormonal, centrally acting, postsynaptic serotonin 1A receptor agonist and a serotonin 2A receptor antagonist that is taken daily at bedtime (100 mg); several weeks are usually needed before any effects are noted.1 It is not approved for postmenopausal women and has a boxed warning about the risks of hypotension and syncope; its use is contraindicated in women who drink alcohol, in those who have hepatic impairment, and with the use of moderate or strong CYP3A4 inhibitors.11
Also keep in mind that flibanserin is only available through a Risk Evaluation and Mitigation Strategy program, so clinicians who wish to prescribe it must enroll in and complete training to become certified providers.9
Related article:
What you need to know (and do) to prescribe the new drug flibanserin
Prasterone
Prasterone (Intrarosa), a once-daily intravaginal dehydroepiandrosterone (DHEA) product, is a prohormone that increases local estrogen and testosterone and has the advantage of improved sexual function, desire, arousal, lubrication, orgasm, satisfaction, as well as pain at sexual activity.12 It was approved by the FDA in November 2016 to treat moderate to severe dyspareunia and has been available for prescribing since July 2017. Its cost is comparable to topical estrogen products, with a $25 copay program.
Because prasterone is not an estrogen, it does not have the boxed warning that all estrogen products are mandated by the FDA to have. This may make it more acceptable to patients, who often decline to use an estrogen product after seeing the boxed warning on the package. The Centers for Medicare and Medicaid Services (CMS) does not have prasterone on its list of potentially hazardous drugs for the elderly. However, keep in mind that because its label is for dyspareunia and not specifically for GSM, CMS considers it a drug of choice--in other words, like sildenafil (Viagra), a lifestyle choice and not for treatment of a medical condition. As such, at the present time, Medicare does not cover it.
Bremelanotide
Late-stage trials of bremelanotide, a melanocortin receptor agonist, are underway. Its mechanism of action is somewhat like that of flibanserin in that both drugs increase dopamine and norepinephrine levels. The advantage of bremelanotide is that it is used as needed. It is dosed subcutaneously (1.75 mg) and it can be used as often as a woman would like to use it. The FDA is expected to consider it for approval in about a year. Unpublished data from poster sessions at recent meetings show that, in a phase 3 study of 1,247 premenopausal women with HSDD (who had already been screened for depression and were found to have a physiologic condition), improvements in desire, arousal, lubrication, and orgasm were shown with bremelanotide. About 18% of women stopped using the drug because of adverse effects (nausea, vomiting, flushing, or headache) versus 2% for placebo. Like flibanserin, it is expected to be approved for premenopausal women only.
Read how 3 experts would manage differing GSM symptoms.
What would you prescribe for these patients?
CASE Genitourinary syndrome of menopause (GSM) in a 55-year-old woman
A 55-year-old widow is beginning a new relationship. She has not had partnered sexual activity for several years, but she recently has begun a relationship. She describes pain with attempted penetration with her new partner. Her last menstrual period was 3 years ago and she has experienced very minor menopausal symptoms, which are not bothersome. On examination, the vulva and vagina are pale, with thin epithelium and absent rugae. The tissue lacks elasticity. A virginal speculum is needed to visualize the cervix.
How would you go about deciding which of the many options for management of GSM you will recommend for this patient? What do you weigh as you consider DHEA versus estrogen and topical versus oral therapy?
JoAnn V. Pinkerton, MD: Vulvovaginal atrophy (VVA), part of GSM, is associated with postmenopausal estrogen deficiency and includes the signs and symptoms seen on this patient's physical exam: vaginal narrowing, pallor, loss of elasticity, as well as pain with intercourse.6 Estrogen therapy is the most effective treatment for vaginal atrophy.13 Since she does not have significant menopausal symptoms, low-dose vaginal estrogen preparations are effective and generally safe treatments for VVA; these include creams, tablets containing estradiol or conjugated equine estrogen (CEE), and a low-dose vaginal estradiol ring--all available at doses that result in minimal systemic absorption.
Choice is usually made based on patient desire and likely adherence. If the patient prefers nonestrogen therapies that improve VVA and have been approved for relief of dyspareunia in postmenopausal women, I would discuss with the patient the oral selective estrogen receptor modulator ospemifene,14 and the new intravaginal DHEA suppositories, prasterone.15 Ospemifene is taken daily as an oral tablet, has a small risk of blood clots, and is my choice for women who do not need systemic hormone therapy and prefer to avoid vaginal therapy.
Andrew M. Kaunitz, MD: GSM is prevalent in menopausal women and, if not treated, causes progressive vaginal dryness and sexual discomfort. When the main indication for hormonal management in a menopausal woman is GSM (as opposed to treatment of vasomotor symptoms or prevention of osteoporosis), the treatment of choice is low-dose local vaginal estrogen, ospemifene, or prasterone (DHEA). Prasterone is a vaginally administered nonestrogen steroid that was approved by the FDA to treat dyspareunia associated with GSM. DHEA is an endogenous inactive steroid that is converted locally into androgens and estrogens; one vaginal insert is placed nightly.16,17
This 55-year-old widow has not been sexually active for some time. The facts that attempted penetration was painful and only an ultrathin (virginal) speculum could be used for examination indicate that contraction of the pelvic floor muscles is likely present. Simply starting medical management may not lead to comfortable/successful penetrative sex for this woman. In addition to medical management, she would likely benefit from referral for physical therapy. Using dilators and other strategies, along with the positive impact that medical management will have on the vaginal mucosa, a woman's physical therapist can work with this patient to help the pelvic floor muscles relax and facilitate comfortable penetrative sex.
James A. Simon, MD: With only minor vasomotor symptoms, I would assess the other potential benefits of a systemic therapy. These might include cardiovascular risk reduction (systemic estrogens or estrogens/progesterone in some), breast cancer risk reduction (some data suggesting ospemifene can accomplish this), osteoporosis prevention (systemic estrogens and estrogen/androgens), etc. If there is an option for a treatment to address more than one symptom, in this case GSM, assessing the risks/benefits of each of these therapies should be estimated for this specific patient.
If there are no systemic benefits to be had, then any of the local treatments should be helpful. As there are no head-to-head comparisons available, local estrogen cream, tablets, rings, local DHEA, or systemic ospemifene each should be considered possible treatments. I also feel this patient may benefit from supplementary self-dilation and/or physical therapy.
Related article:
2017 Update on menopause
CASE Dyspareunia and vasomotor symptoms in a 42-year-old breast cancer survivor
A 42-year-old woman with a BRCA1 mutation has undergone prophylactic mastectomies as well as hysterectomy with bilateral salpingo-oophorectomy. She reports mild to moderate hot flashes and bothersome vaginal dryness and dyspareunia. Examination confirms GSM.
Would you advise systemic hormone therapy for this patient? What would your recommendation be for management of her GSM symptoms?
Dr. Simon: While one's gut reaction would be to avoid systemic estrogen therapy in a patient with a BRCA1 mutation, the scientific information confirming this fear is lacking.18 Such patients may benefit significantly from systemic estrogen therapy (reduced risk of cardiovascular disease and cognitive decline, etc.), and with both breasts and both ovaries removed, estrogen's breast cancer risks, if any in this population, are largely avoided. The patient also may benefit from additional local therapy with either estrogens or DHEA.
Dr. Kaunitz: Due to her high lifetime risk of breast and ovarian cancer, this woman has proceeded with risk-reducing breast and gynecologic surgery. As more BRCA mutation carriers are being identified and undergo risk-reducing bilateral mastectomy (usually with reconstruction) and salpingo-oophorectomy, clinicians and mutation carriers more frequently face decisions regarding use of systemic hormone therapy.
Mutation carriers who have undergone bilateral risk-reducing mastectomy experience a very low baseline future risk for breast cancer; accordingly, concerns regarding this disease should not prevent use of systemic hormone therapy. Furthermore, without hormone replacement, induced menopause in women this age is associated with an elevated risk of osteoporosis, persistent vasomotor symptoms, cardiovascular disease, stroke, mood changes, dementia, Parkinson disease, and overall mortality. Recognizing the safety of estrogen therapy in this setting, this 42-year-old BRCA1 mutation carrier can initiate estrogen therapy. Standard dose estrogen therapy refers to oral estradiol 1.0 mg, conjugated equine estrogen 0.625 mg,or transdermal estradiol 0.05 mg. In younger women like this 42-year-old with surgically induced menopause, higher than standard replacement doses of estrogen are often appropriate.17
Due to concerns the hormone therapy might further increase future risk of breast cancer, some mutation carriers may delay or avoid risk-reducing bilateral salpingo-oophorectomy, a potentially lifesaving surgery which reduces not only future risk of ovarian cancer but also future risk for breast cancer.
Among mutation carriers with intact breasts, several studies address risk of breast cancer with use of systemic hormone therapy. Although limited in numbers of participants and years of follow-up, in aggregate, these studies provide reassurance that short-term use of systemic hormone therapy does not increase breast cancer risk in women with BRCA1 or BRCA2 mutations and intact breasts.19
Dr. Pinkerton: For this woman with early surgical menopause and hysterectomy, estrogen therapy could improve her vasomotor symptoms and decrease her risk of bone loss and GSM.17 In the Women's Health Initiative trial, there were 7 fewer breast cancers per 10,000 women-years in the estrogen-onlyarm.20 Observational studies suggest that hormone therapy, when given to the average age of menopause, decreases the risks of heart disease, Parkinson disease, and dementia.21 Limited observational evidence suggests that hormone therapy use does not further increase risk of breast cancer in women following oophorectomy for BRCA1 or BRCA2 gene mutation.22
The absolute risks observed with hormone therapy tended to be small, especially in younger, healthy women. Systemic hormone therapy could treat her hot flashes and her GSM symptoms and potentially decrease health risks associated with premature estrogen deficiency. Nonestrogen therapies for hot flashes include low-dose antidepressants, gabapentin, and mind-body options, such as cognitive behavioral therapy and hypnosis, but these would not decrease her health risks or treat her GSM.
If she only requests treatment of her GSM symptoms, she would be a candidate for low-dose vaginal estrogen therapy, given as a cream, tablet, or ring depending on her choice. I would not choose ospemifene as my first choice as she is having hot flashes, and there are no data yet on the drug's health benefits in early menopause. If she prefers nonestrogen vaginal therapy, the new intravaginal DHEA might be a good choice as both estrogen and testosterone are increased locally in the vagina while hormone levels remain in the postmenopausal range. There is no boxed warning on the patient insert, although safety in women with breast cancer or in those with elevated risk of breast cancer has not been tested.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Sexual function is a complex, multifaceted process mediated by neurologic functions, hormonal regulation, and ps
As it turns out, quite a lot. Female sexual dysfunction is a common, vastly undertreated sexual health problem that can have wide-reaching effects on a woman’s life. These effects may include impaired body image, self-confidence, and self-worth. Sexual dysfunction also can contribute to relationship dissatisfaction and leave one feeling less connected with her partner.1,2 Studies have shown women with sexual dysfunction have higher health care expenditures3 and that depression and fatigue are common comorbidities, as is frequently seen in other chronic conditions such as diabetes and back pain.4
Understanding the pathogenesis of female sexual dysfunction helps to guide our approach to its management. Indeed, increased understanding of its pathology has helped to usher in new and emerging treatment options, as well as a personalized, biopsychosocial approach to its management.
Related article:
2016 Update on female sexual dysfunction
In this Update, I discuss the interplay of physiologic and psychological factors that affect female sexual function as well as the latest options for its management. I have also assembled a panel of experts to discuss 2 cases representative of sexual dysfunction that you may encounter in your clinical practice and how prescribing decisions are made for their management.
Read about factors that impact sexual function and agents to help manage dysfunction.
Multiple transmitters in the brain can increase or decrease sexual desire and function
Neurotransmitters involved in sexual excitation include brain dopamine, melanocortin, oxytocin, vasopressin, and norepinephrine, whereas brain opioids, serotonin, prolactin, and endocannabinoids function as sexual inhibitors. Inhibitory transmitters are activated normally during sexual refractoriness but also from primary aversion or secondary avoidance disorders.1 Drugs or conditions that reduce brain dopamine levels, increase the action of brain serotonin, or enhance brain opioid pathways have been shown to inhibit sexual desire, while those that increase hypothalamic and mesolimbic dopamine or decrease serotonin release have been shown to stimulate sexual desire.1
Estradiol and progesterone can impact sexual function and desire
In addition to the neurotransmitters, hormones are important modulators of female sexual function. Decreasing levels of circulating estrogen after menopause lead to physiologic, biologic, and clinical changes in the urogenital tissues, such as decreased elastin, thinning of the epithelium, reduced vaginal blood flow, diminished lubrication, and decreased flexibility and elasticity. These changes result in the symptoms of genitourinary syndrome of menopause (GSM), which affects as many as half of all menopausal women.5,6 In clinical trials, dyspareunia and vaginal dryness are the most bothersome GSM symptoms reported.7
The role of hormonal regulation in sexual dysfunction among premenopausal women is not yet fully understood, but we do know that estradiol has been shown to improve sexual desire, progesterone tends to dampen sexual desire, and that testosterone at physiological levels has been shown in most studies to have a neutral effect on sexual desire in a well-estrogenized patient.8
Related article:
Focus on treating genital atrophy symptoms
Experience and behavior modulate or reinforce sexual dysfunction
The most common psychological factors that trigger or amplify female sexual dysfunction are depression, anxiety, distraction, negative body image, sexual abuse, and emotional neglect.9 Contextual or sociocultural factors, such as relationship discord, life-stage stressors (the empty nest syndrome or anxiety and sleep deprivation from a new baby), as well as cultural or religious values that suppress sexuality, also should be considered.9 Experience-based neuroplasticity (changes in brain pathways that become solidified by negative or positive experiences) may elucidate how a multimodal approach, utilizing medical and psychological treatment, can be beneficial for patients, particularly those with hypoactive sexual desire disorder (HSDD).1
New and emerging approaches to managing female sexual dysfunction
Three agents, one of which has been available for prescription for some time, one that is newly available, and one in the pipeline, are or may soon be in the gynecologist's armamentarium.
Flibanserin
Medications that target excitatory pathways or blunt inhibitory pathways are in development, and one, flibanserin (Addyi), has been US Food and Drug Administration (FDA)-approved for the treatment of acquired, generalized HSDD in premenopausal women.1,10 Flibanserin is a nonhormonal, centrally acting, postsynaptic serotonin 1A receptor agonist and a serotonin 2A receptor antagonist that is taken daily at bedtime (100 mg); several weeks are usually needed before any effects are noted.1 It is not approved for postmenopausal women and has a boxed warning about the risks of hypotension and syncope; its use is contraindicated in women who drink alcohol, in those who have hepatic impairment, and with the use of moderate or strong CYP3A4 inhibitors.11
Also keep in mind that flibanserin is only available through a Risk Evaluation and Mitigation Strategy program, so clinicians who wish to prescribe it must enroll in and complete training to become certified providers.9
Related article:
What you need to know (and do) to prescribe the new drug flibanserin
Prasterone
Prasterone (Intrarosa), a once-daily intravaginal dehydroepiandrosterone (DHEA) product, is a prohormone that increases local estrogen and testosterone and has the advantage of improved sexual function, desire, arousal, lubrication, orgasm, satisfaction, as well as pain at sexual activity.12 It was approved by the FDA in November 2016 to treat moderate to severe dyspareunia and has been available for prescribing since July 2017. Its cost is comparable to topical estrogen products, with a $25 copay program.
Because prasterone is not an estrogen, it does not have the boxed warning that all estrogen products are mandated by the FDA to have. This may make it more acceptable to patients, who often decline to use an estrogen product after seeing the boxed warning on the package. The Centers for Medicare and Medicaid Services (CMS) does not have prasterone on its list of potentially hazardous drugs for the elderly. However, keep in mind that because its label is for dyspareunia and not specifically for GSM, CMS considers it a drug of choice--in other words, like sildenafil (Viagra), a lifestyle choice and not for treatment of a medical condition. As such, at the present time, Medicare does not cover it.
Bremelanotide
Late-stage trials of bremelanotide, a melanocortin receptor agonist, are underway. Its mechanism of action is somewhat like that of flibanserin in that both drugs increase dopamine and norepinephrine levels. The advantage of bremelanotide is that it is used as needed. It is dosed subcutaneously (1.75 mg) and it can be used as often as a woman would like to use it. The FDA is expected to consider it for approval in about a year. Unpublished data from poster sessions at recent meetings show that, in a phase 3 study of 1,247 premenopausal women with HSDD (who had already been screened for depression and were found to have a physiologic condition), improvements in desire, arousal, lubrication, and orgasm were shown with bremelanotide. About 18% of women stopped using the drug because of adverse effects (nausea, vomiting, flushing, or headache) versus 2% for placebo. Like flibanserin, it is expected to be approved for premenopausal women only.
Read how 3 experts would manage differing GSM symptoms.
What would you prescribe for these patients?
CASE Genitourinary syndrome of menopause (GSM) in a 55-year-old woman
A 55-year-old widow is beginning a new relationship. She has not had partnered sexual activity for several years, but she recently has begun a relationship. She describes pain with attempted penetration with her new partner. Her last menstrual period was 3 years ago and she has experienced very minor menopausal symptoms, which are not bothersome. On examination, the vulva and vagina are pale, with thin epithelium and absent rugae. The tissue lacks elasticity. A virginal speculum is needed to visualize the cervix.
How would you go about deciding which of the many options for management of GSM you will recommend for this patient? What do you weigh as you consider DHEA versus estrogen and topical versus oral therapy?
JoAnn V. Pinkerton, MD: Vulvovaginal atrophy (VVA), part of GSM, is associated with postmenopausal estrogen deficiency and includes the signs and symptoms seen on this patient's physical exam: vaginal narrowing, pallor, loss of elasticity, as well as pain with intercourse.6 Estrogen therapy is the most effective treatment for vaginal atrophy.13 Since she does not have significant menopausal symptoms, low-dose vaginal estrogen preparations are effective and generally safe treatments for VVA; these include creams, tablets containing estradiol or conjugated equine estrogen (CEE), and a low-dose vaginal estradiol ring--all available at doses that result in minimal systemic absorption.
Choice is usually made based on patient desire and likely adherence. If the patient prefers nonestrogen therapies that improve VVA and have been approved for relief of dyspareunia in postmenopausal women, I would discuss with the patient the oral selective estrogen receptor modulator ospemifene,14 and the new intravaginal DHEA suppositories, prasterone.15 Ospemifene is taken daily as an oral tablet, has a small risk of blood clots, and is my choice for women who do not need systemic hormone therapy and prefer to avoid vaginal therapy.
Andrew M. Kaunitz, MD: GSM is prevalent in menopausal women and, if not treated, causes progressive vaginal dryness and sexual discomfort. When the main indication for hormonal management in a menopausal woman is GSM (as opposed to treatment of vasomotor symptoms or prevention of osteoporosis), the treatment of choice is low-dose local vaginal estrogen, ospemifene, or prasterone (DHEA). Prasterone is a vaginally administered nonestrogen steroid that was approved by the FDA to treat dyspareunia associated with GSM. DHEA is an endogenous inactive steroid that is converted locally into androgens and estrogens; one vaginal insert is placed nightly.16,17
This 55-year-old widow has not been sexually active for some time. The facts that attempted penetration was painful and only an ultrathin (virginal) speculum could be used for examination indicate that contraction of the pelvic floor muscles is likely present. Simply starting medical management may not lead to comfortable/successful penetrative sex for this woman. In addition to medical management, she would likely benefit from referral for physical therapy. Using dilators and other strategies, along with the positive impact that medical management will have on the vaginal mucosa, a woman's physical therapist can work with this patient to help the pelvic floor muscles relax and facilitate comfortable penetrative sex.
James A. Simon, MD: With only minor vasomotor symptoms, I would assess the other potential benefits of a systemic therapy. These might include cardiovascular risk reduction (systemic estrogens or estrogens/progesterone in some), breast cancer risk reduction (some data suggesting ospemifene can accomplish this), osteoporosis prevention (systemic estrogens and estrogen/androgens), etc. If there is an option for a treatment to address more than one symptom, in this case GSM, assessing the risks/benefits of each of these therapies should be estimated for this specific patient.
If there are no systemic benefits to be had, then any of the local treatments should be helpful. As there are no head-to-head comparisons available, local estrogen cream, tablets, rings, local DHEA, or systemic ospemifene each should be considered possible treatments. I also feel this patient may benefit from supplementary self-dilation and/or physical therapy.
Related article:
2017 Update on menopause
CASE Dyspareunia and vasomotor symptoms in a 42-year-old breast cancer survivor
A 42-year-old woman with a BRCA1 mutation has undergone prophylactic mastectomies as well as hysterectomy with bilateral salpingo-oophorectomy. She reports mild to moderate hot flashes and bothersome vaginal dryness and dyspareunia. Examination confirms GSM.
Would you advise systemic hormone therapy for this patient? What would your recommendation be for management of her GSM symptoms?
Dr. Simon: While one's gut reaction would be to avoid systemic estrogen therapy in a patient with a BRCA1 mutation, the scientific information confirming this fear is lacking.18 Such patients may benefit significantly from systemic estrogen therapy (reduced risk of cardiovascular disease and cognitive decline, etc.), and with both breasts and both ovaries removed, estrogen's breast cancer risks, if any in this population, are largely avoided. The patient also may benefit from additional local therapy with either estrogens or DHEA.
Dr. Kaunitz: Due to her high lifetime risk of breast and ovarian cancer, this woman has proceeded with risk-reducing breast and gynecologic surgery. As more BRCA mutation carriers are being identified and undergo risk-reducing bilateral mastectomy (usually with reconstruction) and salpingo-oophorectomy, clinicians and mutation carriers more frequently face decisions regarding use of systemic hormone therapy.
Mutation carriers who have undergone bilateral risk-reducing mastectomy experience a very low baseline future risk for breast cancer; accordingly, concerns regarding this disease should not prevent use of systemic hormone therapy. Furthermore, without hormone replacement, induced menopause in women this age is associated with an elevated risk of osteoporosis, persistent vasomotor symptoms, cardiovascular disease, stroke, mood changes, dementia, Parkinson disease, and overall mortality. Recognizing the safety of estrogen therapy in this setting, this 42-year-old BRCA1 mutation carrier can initiate estrogen therapy. Standard dose estrogen therapy refers to oral estradiol 1.0 mg, conjugated equine estrogen 0.625 mg,or transdermal estradiol 0.05 mg. In younger women like this 42-year-old with surgically induced menopause, higher than standard replacement doses of estrogen are often appropriate.17
Due to concerns the hormone therapy might further increase future risk of breast cancer, some mutation carriers may delay or avoid risk-reducing bilateral salpingo-oophorectomy, a potentially lifesaving surgery which reduces not only future risk of ovarian cancer but also future risk for breast cancer.
Among mutation carriers with intact breasts, several studies address risk of breast cancer with use of systemic hormone therapy. Although limited in numbers of participants and years of follow-up, in aggregate, these studies provide reassurance that short-term use of systemic hormone therapy does not increase breast cancer risk in women with BRCA1 or BRCA2 mutations and intact breasts.19
Dr. Pinkerton: For this woman with early surgical menopause and hysterectomy, estrogen therapy could improve her vasomotor symptoms and decrease her risk of bone loss and GSM.17 In the Women's Health Initiative trial, there were 7 fewer breast cancers per 10,000 women-years in the estrogen-onlyarm.20 Observational studies suggest that hormone therapy, when given to the average age of menopause, decreases the risks of heart disease, Parkinson disease, and dementia.21 Limited observational evidence suggests that hormone therapy use does not further increase risk of breast cancer in women following oophorectomy for BRCA1 or BRCA2 gene mutation.22
The absolute risks observed with hormone therapy tended to be small, especially in younger, healthy women. Systemic hormone therapy could treat her hot flashes and her GSM symptoms and potentially decrease health risks associated with premature estrogen deficiency. Nonestrogen therapies for hot flashes include low-dose antidepressants, gabapentin, and mind-body options, such as cognitive behavioral therapy and hypnosis, but these would not decrease her health risks or treat her GSM.
If she only requests treatment of her GSM symptoms, she would be a candidate for low-dose vaginal estrogen therapy, given as a cream, tablet, or ring depending on her choice. I would not choose ospemifene as my first choice as she is having hot flashes, and there are no data yet on the drug's health benefits in early menopause. If she prefers nonestrogen vaginal therapy, the new intravaginal DHEA might be a good choice as both estrogen and testosterone are increased locally in the vagina while hormone levels remain in the postmenopausal range. There is no boxed warning on the patient insert, although safety in women with breast cancer or in those with elevated risk of breast cancer has not been tested.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review. Mayo Clin Proc. 2017;92(1):114–128.
- Kingsberg SA. Attitudinal survey of women living with low sexual desire. J Womens Health (Larchmt). 2014;23(10):817–823.
- Foley K, Foley D, Johnson BH. Healthcare resource utilization and expenditures of women diagnosed with hypoactive sexual desire disorder. J Med Econ. 2010;13(4):583–590.
- Biddle AK, West SL, D’Aloisio AA, Wheeler SB, Borisov NN, Thorp J. Hypoactive sexual desire disorder in postmenopausal women: quality of life and health burden. Value Health. 2009;12(5):763–772.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;21(10):1063–1068.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause. 2013;20(9):888–902.
- Ettinger B, Hait H, Reape KZ, Shu H. Measuring symptom relief in studies of vaginal and vulvar atrophy: the most bothersome symptom approach. Menopause. 2008;15(5):885–889.
- Dennerstein L, Randolph J, Taffe J, Dudley E, Burger H. Hormones, mood, sexuality, and the menopausal transition. Fertil Steril. 2002;77(suppl 4):S42–S48.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women’s sexual desire and arousal disorders [published correction appears in J Sex Med. 2010;7(2 pt 1):856]. J Sex Med. 2010;7(1 pt 2):586–614.
- US Food and Drug Administration website. FDA approves first treatment for sexual desire disorder. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm458734.htm. Accessed August 14, 2017.
- Addyi (flibanserin) [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America, LLC; 2016.
- Labrie F, Derogatis L, Archer DF, et al; Members of the VVA Prasterone Research Group. Effect of intravaginal prasterone on sexual dysfunction in postmenopausal women with vulvovaginal atrophy. J Sex Med. 2015;12(12):2401–2412.
- Lethaby A, Ayeleke RO, Roberts H. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2016;8:CD001500.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause. 2013;20(6):623–630.
- Labrie F, Archer DF, Koltun, W, et al; VVA Prasterone Research Group. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause. 2016;23(3):243–256.
- Kaunitz AM. Focus on treating genital atrophy symptoms. OBG Manag. 2017;29(1):14, 16–17.
- The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24(7):728–753.
- Crandall CJ, Hovey KM, Andrews CA, et al. Breast cancer, endometrial cancer, and cardiovascular events in participants who used vaginal estrogen in the Women’s Health Initiative Observational Study. Menopause. August 14, 2017. doi:10.1097/GME.0000000000000956.
- Domchek S, Kaunitz AM. Use of systemic hormone therapy in BRCA mutation carriers. Menopause. 2016;23(9):1026–1027.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–1712.
- Faubion SS, Kuhle CL, Shuster LT, Rocca WA. Long-term health consequences of premature or early menopause and considerations for management. Climacteric. 2015;18(4):483–491.
- Gabriel CA, Tigges-Cardwell J, Stopfer J, Erlichman J, Nathanson K, Domchek SM. Use of total abdominal hysterectomy and hormone replacement therapy in BRCA1 and BRCA2 mutation carriers undergoing risk-reducing salpingo-oophorectomy. Fam Cancer. 2009;8(1):23-28.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review. Mayo Clin Proc. 2017;92(1):114–128.
- Kingsberg SA. Attitudinal survey of women living with low sexual desire. J Womens Health (Larchmt). 2014;23(10):817–823.
- Foley K, Foley D, Johnson BH. Healthcare resource utilization and expenditures of women diagnosed with hypoactive sexual desire disorder. J Med Econ. 2010;13(4):583–590.
- Biddle AK, West SL, D’Aloisio AA, Wheeler SB, Borisov NN, Thorp J. Hypoactive sexual desire disorder in postmenopausal women: quality of life and health burden. Value Health. 2009;12(5):763–772.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;21(10):1063–1068.
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause. 2013;20(9):888–902.
- Ettinger B, Hait H, Reape KZ, Shu H. Measuring symptom relief in studies of vaginal and vulvar atrophy: the most bothersome symptom approach. Menopause. 2008;15(5):885–889.
- Dennerstein L, Randolph J, Taffe J, Dudley E, Burger H. Hormones, mood, sexuality, and the menopausal transition. Fertil Steril. 2002;77(suppl 4):S42–S48.
- Brotto LA, Bitzer J, Laan E, Leiblum S, Luria M. Women’s sexual desire and arousal disorders [published correction appears in J Sex Med. 2010;7(2 pt 1):856]. J Sex Med. 2010;7(1 pt 2):586–614.
- US Food and Drug Administration website. FDA approves first treatment for sexual desire disorder. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm458734.htm. Accessed August 14, 2017.
- Addyi (flibanserin) [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals North America, LLC; 2016.
- Labrie F, Derogatis L, Archer DF, et al; Members of the VVA Prasterone Research Group. Effect of intravaginal prasterone on sexual dysfunction in postmenopausal women with vulvovaginal atrophy. J Sex Med. 2015;12(12):2401–2412.
- Lethaby A, Ayeleke RO, Roberts H. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2016;8:CD001500.
- Portman DJ, Bachmann GA, Simon JA; Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause. 2013;20(6):623–630.
- Labrie F, Archer DF, Koltun, W, et al; VVA Prasterone Research Group. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause. 2016;23(3):243–256.
- Kaunitz AM. Focus on treating genital atrophy symptoms. OBG Manag. 2017;29(1):14, 16–17.
- The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24(7):728–753.
- Crandall CJ, Hovey KM, Andrews CA, et al. Breast cancer, endometrial cancer, and cardiovascular events in participants who used vaginal estrogen in the Women’s Health Initiative Observational Study. Menopause. August 14, 2017. doi:10.1097/GME.0000000000000956.
- Domchek S, Kaunitz AM. Use of systemic hormone therapy in BRCA mutation carriers. Menopause. 2016;23(9):1026–1027.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–1712.
- Faubion SS, Kuhle CL, Shuster LT, Rocca WA. Long-term health consequences of premature or early menopause and considerations for management. Climacteric. 2015;18(4):483–491.
- Gabriel CA, Tigges-Cardwell J, Stopfer J, Erlichman J, Nathanson K, Domchek SM. Use of total abdominal hysterectomy and hormone replacement therapy in BRCA1 and BRCA2 mutation carriers undergoing risk-reducing salpingo-oophorectomy. Fam Cancer. 2009;8(1):23-28.
Fast Tracks
- Although not fully understood how, estradiol can improve sexual desire, progesterone tends to dampen sexual desire, and testosterone has a neutral effect in premenopausal women
- Newly available since July 2017, prasterone is a once-daily intravaginal agent that treats moderate to severe dyspareunia and has costs similar to topical estrogens
- Estrogen therapy may be considered in a breast cancer mutation carrier who has undergone prophylactic mastectomies and bilateral salpingo-oophorectomy
5 Points on Stiff Elbow
Take-Home Points
- Proper patient selection is critical as extensive postoperative rehabilitation is required to obtain an excellent outcome.
- Open and arthroscopic approaches are effective treatment options for elbow contractures.
- Elbow stability must be restored to obtain a successful outcome.
- Knowledge of neurovascular anatomy is essential to prevent neurologic complications.
- Prophylactic ulnar nerve release should be considered, especially in patients with limited flexion.
Elbow stiffness has several etiologies, posttraumatic being the most common. Elbow stiffness can have debilitating functional effects necessitating treatment. In a biomechanical study of normal elbow function, Morrey and colleagues1 determined that a flexion extension arc of 100° (30°-130°) and a forearm rotation arc of 100° (50° pronation-50° supination) are required in 90% of activities of daily living. Similarly, elbow flexion of <105° was poorly tolerated, whereas patients could easier adapt to flexion contractures up to 40°.2
The goal of initial evaluation should be to establish the cause of the contracture and the patient’s functional demands and ability to cooperate in the extensive postoperative rehabilitation that is essential in achieving an excellent functional outcome. In a thorough clinical examination, the clinician must note skin, range of motion (ROM), ligamentous stability, and neurovascular structures and give special attention to ulnar nerve function and symptoms. Mid-arc pain suggests additional intra-articular pathology, as stiffness typically causes pain only at the limits of motion as osteophytes impinge and soft tissue is under maximal tension. Routine elbow radiographs are required in all cases, and computed tomography (CT) can be useful in evaluating osseous sources of contracture. Suspected ligamentous instability and cartilaginous defects particularly in the setting of mid-arc pain are best evaluated with magnetic resonance imaging.3
In this 5-point review, we evaluate treatment options as well as rehabilitation protocols in the management of elbow stiffness.
1 Anatomy of Contracture: The Usual Suspects
The cause of elbow stiffness is incompletely understood. Several posited contributing factors include biology, complex intra-articular anatomy, capsular distention favoring a flexed position, and tenuous postoperative fixation necessitating prolonged immobilization. Identifying intrinsic and extrinsic anatomical sources of stiffness can help guide treatment.4 Intrinsic pathology includes intra-articular malunion, osteophytes, loose bodies, and adhesions; extrinsic pathology includes soft-tissue contracture, heterotopic ossification, and extra-articular malunion.
Compared with the normal elbow, the capsule becomes thickened and fibrotic and thereby prevents motion. Severe contractures, and extension contractures in particular, may require release of the posterior medial capsule and the posterior medial collateral ligament (MCL) to regain motion. In a series of 42 patients with flexion <100°, Park and colleagues5 noted that all patients required release of the posterior band of the MCL to regain flexion. Other muscular impediments to motion include contracture of the brachialis and scarring of the triceps to the posterior humerus. Scarring of the triceps to the humerus can limit flexion.
In the posttrauma setting, intra-articular and extra-articular malunion must be considered. Extension malunion of the distal humerus can reduce flexion,6 and shortening with compromise of the olecranon and coronoid fossae can limit both flexion and extension.
Last, heterotopic ossification and osteophytes should be assessed as potential causes of limited ROM. Both the coronoid process and the olecranon can develop osteophytes, and their respective fossae should be assessed with CT. Posterior impingement is rare at the tip of the olecranon; it occurs because of "widening" of the olecranon by "Mickey Mouse ear" osteophytes and bony encroachment along the medial and lateral columns. Thus, the olecranon must be narrowed and the fossa widened and deepened.
In case of concomitant ligament instability, we prefer to reconstruct the ligament first, and then perform contracture release as a staged procedure. We favor a staged approach because the rehabilitation regimens for instability and contracture release are diametrically opposed: Instability requires immobilization, and contracture release requires immediate motion. Last, incision placement and ulnar nerve management are crucial in minimizing the potential complications of the second procedure.
2 Nonoperative Treatment
In the absence of significant bony impediments to motion—such as heterotopic ossification or malunion—initial treatment should commence with nonoperative therapy. Therapy should be initiated as soon as concern for stiffness arises in order to prevent contracture. Initial nonoperative treatment can also serve as an important litmus test of postoperative adherence. Adequate patient relaxation is crucial in avoiding co-contracture resisting stretching forces. Passive ROM exercises use sustained force to allow time-dependent stress relaxation to increase tissue length as well as fatigue antagonist muscles. In addition, hold-and-relax techniques apply isometric resistance to induce relaxation of antagonist muscles.7 Active ROM should emphasize triceps isolation and elbow extension to prevent scarring of the triceps to the posterior humerus.
Corrective splinting can be an effective adjuvant to physiotherapy. Static progressive turnbuckle splints was described as an effective treatment for both elbow flexion and extension contractures, effecting an average 43° increase in elbow motion in a series of 15 patients.8 Similarly, Gelinas and colleagues9 noted improvement among 22 patients treated with turnbuckle splinting for an average of 4.5 months. In addition, serial extension splints may be used in the treatment of elbow flexion contractures.
3 Open Contacture Release and Surgical Approach
When nonoperative therapies fail to restore the functional arc of motion, patients with flexion contractures or extension contractures of >30° may be indicated for contracture release. Surgical approach should be determined by meticulous preoperative planning that notes prior incisions and CT findings. It can be helpful to organize common offending structures and their effects on flexion and extension (Table). 

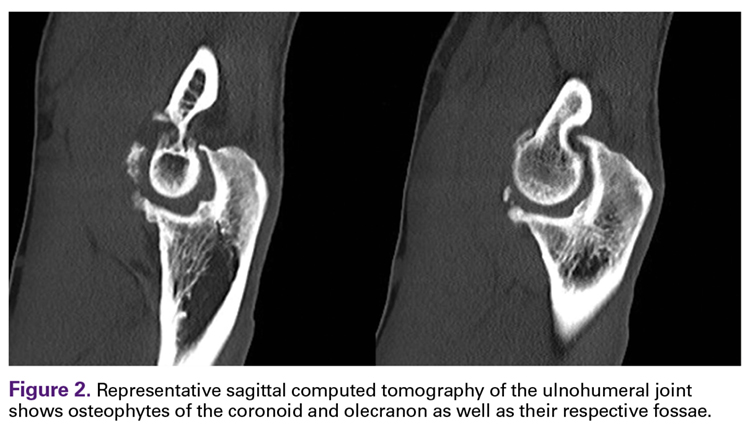
A medial over-the-top approach uses the medial supracondylar ridge as a landmark, subperiosteally reflecting the brachialis anteriorly.10 The ulnar nerve is neurolyzed and protected posteriorly. The flexor-pronator mass is split distally and elevated along with the brachialis as a single sleeve of muscle. The coronal plane of dissection should be the anterior half of the lateral epicondyle to avoid injury to the MCL. Large Bennett or Hohmann retractors can hinge on the lateral border of the humerus and provide clear visualization of the anterior capsule and the ulnohumeral joint. Exposure of the radiocapitellar joint is possible, but this joint is very deep in the operative field, and caution should be taken excising the anterolateral capsule because of the risk of radial nerve injury. The ulnar nerve can be temporarily transposed anteriorly to dissect posteriorly along the supracondylar ridge of the humerus. The triceps is reflected off the distal humerus. Occasionally, the posterior band of the MCL must be resected in severe extension contractures. If possible, the anterior bundle should be preserved. With this approach, the anterior capsule, distal humerus, coronoid process, posterior MCL, posterior capsule, and triceps can be addressed. The zone anterior to the radial head and the anterolateral and posterolateral capsule cannot be safely exposed with a medial approach. As described by Wada and colleagues,11 a primarily medial approach resulted in an average 64° increase in arc of motion.
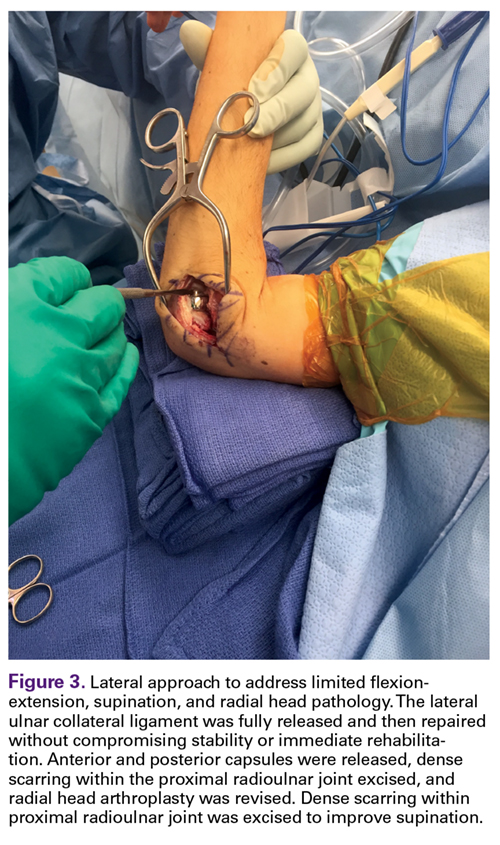
an internal joint stabilizer (Skeletal Dynamics) (Figure 4) and to initiate motion therapy immediately. External fixation (hinged or unhinged is rarely used in our practice. 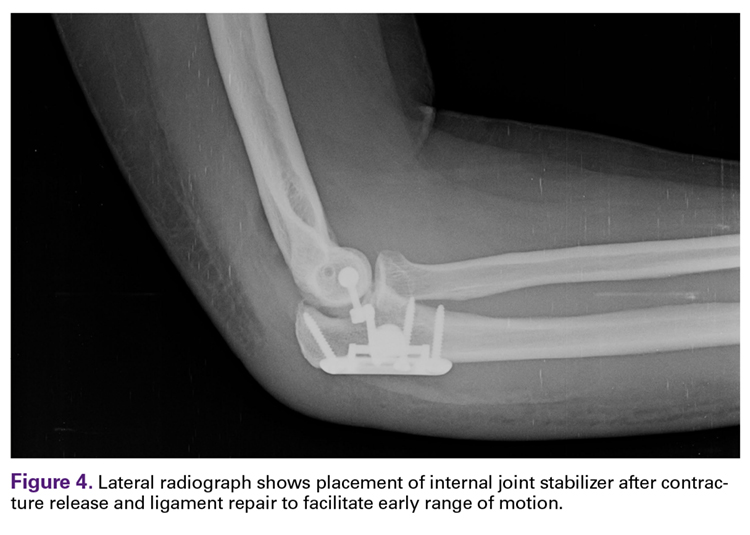
4 Arthroscopic Contracture Release and Technique
Recently, arthroscopic elbow contracture release, a technically demanding but effective treatment option, has gained popularity. Knowledge of neurovascular anatomy is a prerequisite to the prevention of devastating neurologic complications (ulnar, median, and radial nerve transections have been described14,15). Relative contraindications include extensive heterotopic ossification, ulnar nerve transposition, and limited arthroscopic experience. Functional improvements as well as average 26° to 42° increases in arc of motion have been described with arthroscopic release.16-18 In thin-framed patients with dense elbow capsular scarring (severe loss of elbow motion with hard block) and small joint space, arthroscopic release and particularly arthroscope insertion are notoriously difficult.
The patient may be placed in the prone, lateral decubitus, or supine position, depending on surgeon preference (Figure 5). Before surgery, portals and the ulnar nerve should be carefully outlined.19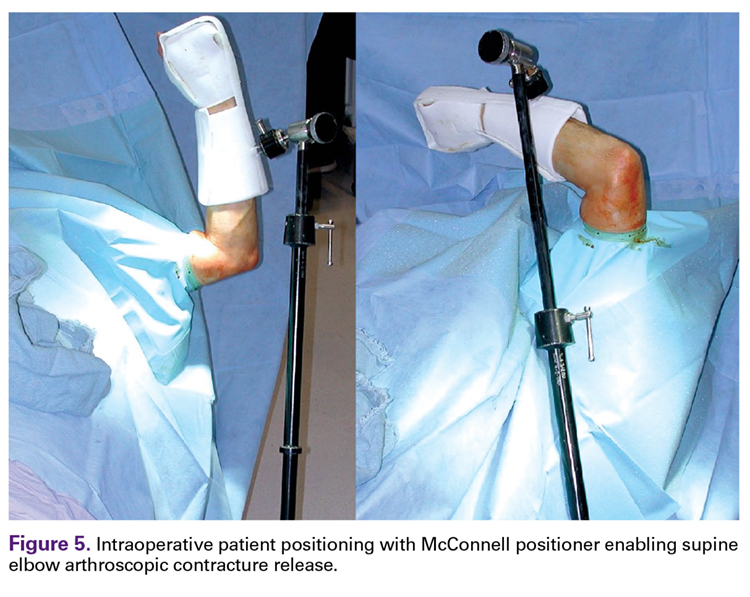
We prefer to start by entering the posterior compartment and using the shaver to create a working space. All bone work and resectioning should be performed before capsular resection. After the joint and the olecranon fossa are identified, soft-tissue and bony débridement of the olecranon and the fossa can be performed. Care should be taken to protect the ulnar nerve when the posteromedial corner or medial gutter is approached. 
5 Additional Considerations
After surgery, the elbow is immobilized in maximal extension and supination with an anterior splint, and therapy is initiated either immediately or after temporary immobilization.16,19,20 Regional anesthesia is crucial in obtaining adequate pain control and establishing an immediate postoperative therapy program. The utility of continuous passive motion (CPM) in postoperative protocols is controversial. A retrospective case-control study of 32 patients matched on age, diagnosis, and contraction severity found no benefit of CPM use, and increased costs and hospital length of stay, leading the authors to recommend against CPM use.20
Neurovascular risks are associated with both open and arthroscopic elbow contracture release. Particularly concerning is the risk of traction ulnar neuropathy, described in upward of 20% of patients.21 Anatomical studies have found decreases in cubital tunnel and ulnar nerve area as elbow flexion increases with corresponding increased intraneural pressure,22 leading some authors to recommend prophylactic ulnar nerve release with limited preoperative flexion.15 Nevertheless, despite transposition, ulnar nerve symptoms were noted in 8 of 40 patients who underwent open contracture release for posttraumatic loss of elbow flexion.5 In a retrospective review of 164 open and arthroscopic elbow contracture releases, Williams and colleagues21 noted an 8.1% rate of postoperative new-onset ulnar nerve symptoms. The rate of ulnar neuropathy was nonsignificantly elevated among patients with preoperative flexion of <100° (15.2% vs 3.7%; P = .057). Recently, a retrospective review of 564 consecutive arthroscopic contracture releases found a significantly higher rate of delayed-onset ulnar neuritis among patients without prophylactic ulnar nerve decompression or transposition (11% vs 3%; P < .001).23 Further analysis revealed that, compared with decompression, ulnar nerve transposition did not offer additional benefit but was associated with a significantly higher rate of wound complications (19% vs 4%; P = .03). We favor prophylactic release, particularly in the setting of preoperative extension contracture. For open contracture release from the lateral approach, however, we do not routinely release the ulnar nerve unless there were preoperative symptoms.
Although open and arthroscopic contracture releases can provide durable outcomes in the setting of painless elbow stiffness, options are more limited in the treatment of the painful stiff elbow. Total elbow arthroplasty remains an option in low-demand elderly patients but is not without significant risk of complications.24 In addition, durability concerns and postoperative restrictions make total elbow arthroplasty less attractive to younger patients. Interposition arthroplasty may be indicated as a salvage procedure in the treatment of a young or high-demand patient with a stiff painful elbow.25 Elbow stability is crucial in obtaining a successful outcome, and data on optimal graft choices are limited.
Conclusion
Elbow stiffness, a common complication of trauma, significantly impairs activities of daily living. Early after trauma, therapy should be initiated to prevent contracture. In the absence of symptomatic arthritis, both open and arthroscopic contracture releases are effective surgical treatments in properly selected and motivated patients. Although more research is needed to establish the optimal surgical approach, severity and anatomical cause of contracture should guide decisions as to which approach to use. Having a thorough understanding of neurovascular anatomy and of prophylactic ulnar nerve decompression in the setting of limited preoperative flexion can mitigate complications.
1. Morrey BF, Askew LJ, Chao EY. A biomechanical study of normal functional elbow motion. J Bone Joint Surg Am. 1981;63(6):872-877.
2. Hotchkiss RN. Elbow contracture. In: Green DP, Rotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 5th ed. New York, NY: Churchill-Livingstone; 2005:667-682.
3. Van Zeeland NL, Yamaguchi K. Arthroscopic capsular release of the elbow. J Shoulder Elbow Surg. 2010;19(2):13-19.
4. Morrey BF. Post-traumatic contracture of the elbow. Operative treatment, including distraction arthroplasty. J Bone Joint Surg Am. 1990;72(4):601-618.
5. Park MJ, Chang MJ, Lee YB, Kang HJ. Surgical release for posttraumatic loss of elbow flexion. J Bone Joint Surg Am. 2010;92(16):2692-2699.
6. Brouwer KM, Lindenhovius AL, Ring D. Loss of anterior translation of the distal humeral articular surface is associated with decreased elbow flexion. J Hand Surg Am. 2009;34(7):
1256-1260.
7. Taylor DC, Dalton JD, Seaber AV, Garrett WE. Viscoelastic properties of muscle-tendon units: the biomechanical effects of stretching. Am J Sports Med. 1990;18(3):300-309.
8. Green DP, McCoy H. Turnbuckle orthotic correction of elbow-flexion contractures after acute injuries. J Bone Joint Surg Am. 1979;61(7):1092-1095.
9. Gelinas JJ, Faber KJ, Patterson SD, King GJ. The effectiveness of turnbuckle splinting for elbow contractures. J Bone Joint Surg Br. 2000;82(1):74-78.
10. Hotchkiss RN, Kasparyan GN. The medial "over the top" approach to the elbow. Tech Orthop. 2000;15(2):105-112.
11. Wada T, Ishii S, Usui M, Miyano S. The medial approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Br. 2000;82(1):68-73.
12. Husband JB, Hastings H. The lateral approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Am. 1990;72(9):1353-1358.
13. Mansat P, Morrey BF. The column procedure: a limited lateral approach for extrinsic contracture of the elbow. J Bone Joint Surg Am. 1998;80(11):1603-1605.
14. Haapaniemi T, Berggren M, Adolfsson L. Complete transection of the median and radial nerves during arthroscopic release of post-traumatic elbow contracture. Arthroscopy. 1999;15(7):784-787.
15. Kelly EW, Morrey BF, O’Driscoll SW. Complications of elbow arthroscopy. J Bone Joint Surg Am. 2001;83(1):25-34.
16. Ball CM, Meunier M, Galatz LM, Calfee R, Yamaguchi K. Arthroscopic treatment of post-traumatic elbow contracture. J Shoulder Elbow Surg. 2002;11(6):624-629.
17. Ćefo I, Eygendaal D. Arthroscopic arthrolysis for posttraumatic elbow stiffness. J Shoulder Elbow Surg. 2011;20(3):434-439.
18. Nguyen D, Proper SI, MacDermid JC, King GJ, Faber KJ. Functional outcomes of arthroscopic capsular release of the elbow. Arthroscopy. 2006;22(8):842-849.
19. Sahajpal D, Choi T, Wright TW. Arthroscopic release of the stiff elbow. J Hand Surg. 2009;34(3):540-544.
20. Lindenhovius AL, Jupiter JB. The posttraumatic stiff elbow: a review of the literature. J Hand Surg. 2007;32(10):1605-1623.
21. Williams BG, Sotereanos DG, Baratz ME, Jarrett CD, Venouziou AI, Miller MC. The contracted elbow: is ulnar nerve release necessary? J Shoulder Elbow Surg. 2012;21(12):
1632-1636.
22. Gelberman RH, Yamaguchi K, Hollstien SB, et al. Changes in interstitial pressure and cross-sectional area of the cubital tunnel and of the ulnar nerve with flexion of the elbow. an experimental study in human cadavera. J Bone Joint Surg Am. 1998;80(4):492-501.
23. Blonna D, O’Driscoll SW. Delayed-onset ulnar neuritis after release of elbow contracture: preventive strategies derived from a study of 563 cases. Arthroscopy. 2014;30(8):947-956.
24. Mansat P, Morrey BF. Semiconstrained total elbow arthroplasty for ankylosed and stiff elbows. J Bone Joint Surg. 2000;82(9):1260-1268.
25. Hausman MR, Birnbaum PS. Interposition elbow arthroplasty. Tech Hand Up Extrem Surg. 2004;8(3):181-188.
Take-Home Points
- Proper patient selection is critical as extensive postoperative rehabilitation is required to obtain an excellent outcome.
- Open and arthroscopic approaches are effective treatment options for elbow contractures.
- Elbow stability must be restored to obtain a successful outcome.
- Knowledge of neurovascular anatomy is essential to prevent neurologic complications.
- Prophylactic ulnar nerve release should be considered, especially in patients with limited flexion.
Elbow stiffness has several etiologies, posttraumatic being the most common. Elbow stiffness can have debilitating functional effects necessitating treatment. In a biomechanical study of normal elbow function, Morrey and colleagues1 determined that a flexion extension arc of 100° (30°-130°) and a forearm rotation arc of 100° (50° pronation-50° supination) are required in 90% of activities of daily living. Similarly, elbow flexion of <105° was poorly tolerated, whereas patients could easier adapt to flexion contractures up to 40°.2
The goal of initial evaluation should be to establish the cause of the contracture and the patient’s functional demands and ability to cooperate in the extensive postoperative rehabilitation that is essential in achieving an excellent functional outcome. In a thorough clinical examination, the clinician must note skin, range of motion (ROM), ligamentous stability, and neurovascular structures and give special attention to ulnar nerve function and symptoms. Mid-arc pain suggests additional intra-articular pathology, as stiffness typically causes pain only at the limits of motion as osteophytes impinge and soft tissue is under maximal tension. Routine elbow radiographs are required in all cases, and computed tomography (CT) can be useful in evaluating osseous sources of contracture. Suspected ligamentous instability and cartilaginous defects particularly in the setting of mid-arc pain are best evaluated with magnetic resonance imaging.3
In this 5-point review, we evaluate treatment options as well as rehabilitation protocols in the management of elbow stiffness.
1 Anatomy of Contracture: The Usual Suspects
The cause of elbow stiffness is incompletely understood. Several posited contributing factors include biology, complex intra-articular anatomy, capsular distention favoring a flexed position, and tenuous postoperative fixation necessitating prolonged immobilization. Identifying intrinsic and extrinsic anatomical sources of stiffness can help guide treatment.4 Intrinsic pathology includes intra-articular malunion, osteophytes, loose bodies, and adhesions; extrinsic pathology includes soft-tissue contracture, heterotopic ossification, and extra-articular malunion.
Compared with the normal elbow, the capsule becomes thickened and fibrotic and thereby prevents motion. Severe contractures, and extension contractures in particular, may require release of the posterior medial capsule and the posterior medial collateral ligament (MCL) to regain motion. In a series of 42 patients with flexion <100°, Park and colleagues5 noted that all patients required release of the posterior band of the MCL to regain flexion. Other muscular impediments to motion include contracture of the brachialis and scarring of the triceps to the posterior humerus. Scarring of the triceps to the humerus can limit flexion.
In the posttrauma setting, intra-articular and extra-articular malunion must be considered. Extension malunion of the distal humerus can reduce flexion,6 and shortening with compromise of the olecranon and coronoid fossae can limit both flexion and extension.
Last, heterotopic ossification and osteophytes should be assessed as potential causes of limited ROM. Both the coronoid process and the olecranon can develop osteophytes, and their respective fossae should be assessed with CT. Posterior impingement is rare at the tip of the olecranon; it occurs because of "widening" of the olecranon by "Mickey Mouse ear" osteophytes and bony encroachment along the medial and lateral columns. Thus, the olecranon must be narrowed and the fossa widened and deepened.
In case of concomitant ligament instability, we prefer to reconstruct the ligament first, and then perform contracture release as a staged procedure. We favor a staged approach because the rehabilitation regimens for instability and contracture release are diametrically opposed: Instability requires immobilization, and contracture release requires immediate motion. Last, incision placement and ulnar nerve management are crucial in minimizing the potential complications of the second procedure.
2 Nonoperative Treatment
In the absence of significant bony impediments to motion—such as heterotopic ossification or malunion—initial treatment should commence with nonoperative therapy. Therapy should be initiated as soon as concern for stiffness arises in order to prevent contracture. Initial nonoperative treatment can also serve as an important litmus test of postoperative adherence. Adequate patient relaxation is crucial in avoiding co-contracture resisting stretching forces. Passive ROM exercises use sustained force to allow time-dependent stress relaxation to increase tissue length as well as fatigue antagonist muscles. In addition, hold-and-relax techniques apply isometric resistance to induce relaxation of antagonist muscles.7 Active ROM should emphasize triceps isolation and elbow extension to prevent scarring of the triceps to the posterior humerus.
Corrective splinting can be an effective adjuvant to physiotherapy. Static progressive turnbuckle splints was described as an effective treatment for both elbow flexion and extension contractures, effecting an average 43° increase in elbow motion in a series of 15 patients.8 Similarly, Gelinas and colleagues9 noted improvement among 22 patients treated with turnbuckle splinting for an average of 4.5 months. In addition, serial extension splints may be used in the treatment of elbow flexion contractures.
3 Open Contacture Release and Surgical Approach
When nonoperative therapies fail to restore the functional arc of motion, patients with flexion contractures or extension contractures of >30° may be indicated for contracture release. Surgical approach should be determined by meticulous preoperative planning that notes prior incisions and CT findings. It can be helpful to organize common offending structures and their effects on flexion and extension (Table).
A medial over-the-top approach uses the medial supracondylar ridge as a landmark, subperiosteally reflecting the brachialis anteriorly.10 The ulnar nerve is neurolyzed and protected posteriorly. The flexor-pronator mass is split distally and elevated along with the brachialis as a single sleeve of muscle. The coronal plane of dissection should be the anterior half of the lateral epicondyle to avoid injury to the MCL. Large Bennett or Hohmann retractors can hinge on the lateral border of the humerus and provide clear visualization of the anterior capsule and the ulnohumeral joint. Exposure of the radiocapitellar joint is possible, but this joint is very deep in the operative field, and caution should be taken excising the anterolateral capsule because of the risk of radial nerve injury. The ulnar nerve can be temporarily transposed anteriorly to dissect posteriorly along the supracondylar ridge of the humerus. The triceps is reflected off the distal humerus. Occasionally, the posterior band of the MCL must be resected in severe extension contractures. If possible, the anterior bundle should be preserved. With this approach, the anterior capsule, distal humerus, coronoid process, posterior MCL, posterior capsule, and triceps can be addressed. The zone anterior to the radial head and the anterolateral and posterolateral capsule cannot be safely exposed with a medial approach. As described by Wada and colleagues,11 a primarily medial approach resulted in an average 64° increase in arc of motion.
an internal joint stabilizer (Skeletal Dynamics) (Figure 4) and to initiate motion therapy immediately. External fixation (hinged or unhinged is rarely used in our practice.
4 Arthroscopic Contracture Release and Technique
Recently, arthroscopic elbow contracture release, a technically demanding but effective treatment option, has gained popularity. Knowledge of neurovascular anatomy is a prerequisite to the prevention of devastating neurologic complications (ulnar, median, and radial nerve transections have been described14,15). Relative contraindications include extensive heterotopic ossification, ulnar nerve transposition, and limited arthroscopic experience. Functional improvements as well as average 26° to 42° increases in arc of motion have been described with arthroscopic release.16-18 In thin-framed patients with dense elbow capsular scarring (severe loss of elbow motion with hard block) and small joint space, arthroscopic release and particularly arthroscope insertion are notoriously difficult.
The patient may be placed in the prone, lateral decubitus, or supine position, depending on surgeon preference (Figure 5). Before surgery, portals and the ulnar nerve should be carefully outlined.19
We prefer to start by entering the posterior compartment and using the shaver to create a working space. All bone work and resectioning should be performed before capsular resection. After the joint and the olecranon fossa are identified, soft-tissue and bony débridement of the olecranon and the fossa can be performed. Care should be taken to protect the ulnar nerve when the posteromedial corner or medial gutter is approached.
5 Additional Considerations
After surgery, the elbow is immobilized in maximal extension and supination with an anterior splint, and therapy is initiated either immediately or after temporary immobilization.16,19,20 Regional anesthesia is crucial in obtaining adequate pain control and establishing an immediate postoperative therapy program. The utility of continuous passive motion (CPM) in postoperative protocols is controversial. A retrospective case-control study of 32 patients matched on age, diagnosis, and contraction severity found no benefit of CPM use, and increased costs and hospital length of stay, leading the authors to recommend against CPM use.20
Neurovascular risks are associated with both open and arthroscopic elbow contracture release. Particularly concerning is the risk of traction ulnar neuropathy, described in upward of 20% of patients.21 Anatomical studies have found decreases in cubital tunnel and ulnar nerve area as elbow flexion increases with corresponding increased intraneural pressure,22 leading some authors to recommend prophylactic ulnar nerve release with limited preoperative flexion.15 Nevertheless, despite transposition, ulnar nerve symptoms were noted in 8 of 40 patients who underwent open contracture release for posttraumatic loss of elbow flexion.5 In a retrospective review of 164 open and arthroscopic elbow contracture releases, Williams and colleagues21 noted an 8.1% rate of postoperative new-onset ulnar nerve symptoms. The rate of ulnar neuropathy was nonsignificantly elevated among patients with preoperative flexion of <100° (15.2% vs 3.7%; P = .057). Recently, a retrospective review of 564 consecutive arthroscopic contracture releases found a significantly higher rate of delayed-onset ulnar neuritis among patients without prophylactic ulnar nerve decompression or transposition (11% vs 3%; P < .001).23 Further analysis revealed that, compared with decompression, ulnar nerve transposition did not offer additional benefit but was associated with a significantly higher rate of wound complications (19% vs 4%; P = .03). We favor prophylactic release, particularly in the setting of preoperative extension contracture. For open contracture release from the lateral approach, however, we do not routinely release the ulnar nerve unless there were preoperative symptoms.
Although open and arthroscopic contracture releases can provide durable outcomes in the setting of painless elbow stiffness, options are more limited in the treatment of the painful stiff elbow. Total elbow arthroplasty remains an option in low-demand elderly patients but is not without significant risk of complications.24 In addition, durability concerns and postoperative restrictions make total elbow arthroplasty less attractive to younger patients. Interposition arthroplasty may be indicated as a salvage procedure in the treatment of a young or high-demand patient with a stiff painful elbow.25 Elbow stability is crucial in obtaining a successful outcome, and data on optimal graft choices are limited.
Conclusion
Elbow stiffness, a common complication of trauma, significantly impairs activities of daily living. Early after trauma, therapy should be initiated to prevent contracture. In the absence of symptomatic arthritis, both open and arthroscopic contracture releases are effective surgical treatments in properly selected and motivated patients. Although more research is needed to establish the optimal surgical approach, severity and anatomical cause of contracture should guide decisions as to which approach to use. Having a thorough understanding of neurovascular anatomy and of prophylactic ulnar nerve decompression in the setting of limited preoperative flexion can mitigate complications.
Take-Home Points
- Proper patient selection is critical as extensive postoperative rehabilitation is required to obtain an excellent outcome.
- Open and arthroscopic approaches are effective treatment options for elbow contractures.
- Elbow stability must be restored to obtain a successful outcome.
- Knowledge of neurovascular anatomy is essential to prevent neurologic complications.
- Prophylactic ulnar nerve release should be considered, especially in patients with limited flexion.
Elbow stiffness has several etiologies, posttraumatic being the most common. Elbow stiffness can have debilitating functional effects necessitating treatment. In a biomechanical study of normal elbow function, Morrey and colleagues1 determined that a flexion extension arc of 100° (30°-130°) and a forearm rotation arc of 100° (50° pronation-50° supination) are required in 90% of activities of daily living. Similarly, elbow flexion of <105° was poorly tolerated, whereas patients could easier adapt to flexion contractures up to 40°.2
The goal of initial evaluation should be to establish the cause of the contracture and the patient’s functional demands and ability to cooperate in the extensive postoperative rehabilitation that is essential in achieving an excellent functional outcome. In a thorough clinical examination, the clinician must note skin, range of motion (ROM), ligamentous stability, and neurovascular structures and give special attention to ulnar nerve function and symptoms. Mid-arc pain suggests additional intra-articular pathology, as stiffness typically causes pain only at the limits of motion as osteophytes impinge and soft tissue is under maximal tension. Routine elbow radiographs are required in all cases, and computed tomography (CT) can be useful in evaluating osseous sources of contracture. Suspected ligamentous instability and cartilaginous defects particularly in the setting of mid-arc pain are best evaluated with magnetic resonance imaging.3
In this 5-point review, we evaluate treatment options as well as rehabilitation protocols in the management of elbow stiffness.
1 Anatomy of Contracture: The Usual Suspects
The cause of elbow stiffness is incompletely understood. Several posited contributing factors include biology, complex intra-articular anatomy, capsular distention favoring a flexed position, and tenuous postoperative fixation necessitating prolonged immobilization. Identifying intrinsic and extrinsic anatomical sources of stiffness can help guide treatment.4 Intrinsic pathology includes intra-articular malunion, osteophytes, loose bodies, and adhesions; extrinsic pathology includes soft-tissue contracture, heterotopic ossification, and extra-articular malunion.
Compared with the normal elbow, the capsule becomes thickened and fibrotic and thereby prevents motion. Severe contractures, and extension contractures in particular, may require release of the posterior medial capsule and the posterior medial collateral ligament (MCL) to regain motion. In a series of 42 patients with flexion <100°, Park and colleagues5 noted that all patients required release of the posterior band of the MCL to regain flexion. Other muscular impediments to motion include contracture of the brachialis and scarring of the triceps to the posterior humerus. Scarring of the triceps to the humerus can limit flexion.
In the posttrauma setting, intra-articular and extra-articular malunion must be considered. Extension malunion of the distal humerus can reduce flexion,6 and shortening with compromise of the olecranon and coronoid fossae can limit both flexion and extension.
Last, heterotopic ossification and osteophytes should be assessed as potential causes of limited ROM. Both the coronoid process and the olecranon can develop osteophytes, and their respective fossae should be assessed with CT. Posterior impingement is rare at the tip of the olecranon; it occurs because of "widening" of the olecranon by "Mickey Mouse ear" osteophytes and bony encroachment along the medial and lateral columns. Thus, the olecranon must be narrowed and the fossa widened and deepened.
In case of concomitant ligament instability, we prefer to reconstruct the ligament first, and then perform contracture release as a staged procedure. We favor a staged approach because the rehabilitation regimens for instability and contracture release are diametrically opposed: Instability requires immobilization, and contracture release requires immediate motion. Last, incision placement and ulnar nerve management are crucial in minimizing the potential complications of the second procedure.
2 Nonoperative Treatment
In the absence of significant bony impediments to motion—such as heterotopic ossification or malunion—initial treatment should commence with nonoperative therapy. Therapy should be initiated as soon as concern for stiffness arises in order to prevent contracture. Initial nonoperative treatment can also serve as an important litmus test of postoperative adherence. Adequate patient relaxation is crucial in avoiding co-contracture resisting stretching forces. Passive ROM exercises use sustained force to allow time-dependent stress relaxation to increase tissue length as well as fatigue antagonist muscles. In addition, hold-and-relax techniques apply isometric resistance to induce relaxation of antagonist muscles.7 Active ROM should emphasize triceps isolation and elbow extension to prevent scarring of the triceps to the posterior humerus.
Corrective splinting can be an effective adjuvant to physiotherapy. Static progressive turnbuckle splints was described as an effective treatment for both elbow flexion and extension contractures, effecting an average 43° increase in elbow motion in a series of 15 patients.8 Similarly, Gelinas and colleagues9 noted improvement among 22 patients treated with turnbuckle splinting for an average of 4.5 months. In addition, serial extension splints may be used in the treatment of elbow flexion contractures.
3 Open Contacture Release and Surgical Approach
When nonoperative therapies fail to restore the functional arc of motion, patients with flexion contractures or extension contractures of >30° may be indicated for contracture release. Surgical approach should be determined by meticulous preoperative planning that notes prior incisions and CT findings. It can be helpful to organize common offending structures and their effects on flexion and extension (Table).
A medial over-the-top approach uses the medial supracondylar ridge as a landmark, subperiosteally reflecting the brachialis anteriorly.10 The ulnar nerve is neurolyzed and protected posteriorly. The flexor-pronator mass is split distally and elevated along with the brachialis as a single sleeve of muscle. The coronal plane of dissection should be the anterior half of the lateral epicondyle to avoid injury to the MCL. Large Bennett or Hohmann retractors can hinge on the lateral border of the humerus and provide clear visualization of the anterior capsule and the ulnohumeral joint. Exposure of the radiocapitellar joint is possible, but this joint is very deep in the operative field, and caution should be taken excising the anterolateral capsule because of the risk of radial nerve injury. The ulnar nerve can be temporarily transposed anteriorly to dissect posteriorly along the supracondylar ridge of the humerus. The triceps is reflected off the distal humerus. Occasionally, the posterior band of the MCL must be resected in severe extension contractures. If possible, the anterior bundle should be preserved. With this approach, the anterior capsule, distal humerus, coronoid process, posterior MCL, posterior capsule, and triceps can be addressed. The zone anterior to the radial head and the anterolateral and posterolateral capsule cannot be safely exposed with a medial approach. As described by Wada and colleagues,11 a primarily medial approach resulted in an average 64° increase in arc of motion.
an internal joint stabilizer (Skeletal Dynamics) (Figure 4) and to initiate motion therapy immediately. External fixation (hinged or unhinged is rarely used in our practice.
4 Arthroscopic Contracture Release and Technique
Recently, arthroscopic elbow contracture release, a technically demanding but effective treatment option, has gained popularity. Knowledge of neurovascular anatomy is a prerequisite to the prevention of devastating neurologic complications (ulnar, median, and radial nerve transections have been described14,15). Relative contraindications include extensive heterotopic ossification, ulnar nerve transposition, and limited arthroscopic experience. Functional improvements as well as average 26° to 42° increases in arc of motion have been described with arthroscopic release.16-18 In thin-framed patients with dense elbow capsular scarring (severe loss of elbow motion with hard block) and small joint space, arthroscopic release and particularly arthroscope insertion are notoriously difficult.
The patient may be placed in the prone, lateral decubitus, or supine position, depending on surgeon preference (Figure 5). Before surgery, portals and the ulnar nerve should be carefully outlined.19
We prefer to start by entering the posterior compartment and using the shaver to create a working space. All bone work and resectioning should be performed before capsular resection. After the joint and the olecranon fossa are identified, soft-tissue and bony débridement of the olecranon and the fossa can be performed. Care should be taken to protect the ulnar nerve when the posteromedial corner or medial gutter is approached.
5 Additional Considerations
After surgery, the elbow is immobilized in maximal extension and supination with an anterior splint, and therapy is initiated either immediately or after temporary immobilization.16,19,20 Regional anesthesia is crucial in obtaining adequate pain control and establishing an immediate postoperative therapy program. The utility of continuous passive motion (CPM) in postoperative protocols is controversial. A retrospective case-control study of 32 patients matched on age, diagnosis, and contraction severity found no benefit of CPM use, and increased costs and hospital length of stay, leading the authors to recommend against CPM use.20
Neurovascular risks are associated with both open and arthroscopic elbow contracture release. Particularly concerning is the risk of traction ulnar neuropathy, described in upward of 20% of patients.21 Anatomical studies have found decreases in cubital tunnel and ulnar nerve area as elbow flexion increases with corresponding increased intraneural pressure,22 leading some authors to recommend prophylactic ulnar nerve release with limited preoperative flexion.15 Nevertheless, despite transposition, ulnar nerve symptoms were noted in 8 of 40 patients who underwent open contracture release for posttraumatic loss of elbow flexion.5 In a retrospective review of 164 open and arthroscopic elbow contracture releases, Williams and colleagues21 noted an 8.1% rate of postoperative new-onset ulnar nerve symptoms. The rate of ulnar neuropathy was nonsignificantly elevated among patients with preoperative flexion of <100° (15.2% vs 3.7%; P = .057). Recently, a retrospective review of 564 consecutive arthroscopic contracture releases found a significantly higher rate of delayed-onset ulnar neuritis among patients without prophylactic ulnar nerve decompression or transposition (11% vs 3%; P < .001).23 Further analysis revealed that, compared with decompression, ulnar nerve transposition did not offer additional benefit but was associated with a significantly higher rate of wound complications (19% vs 4%; P = .03). We favor prophylactic release, particularly in the setting of preoperative extension contracture. For open contracture release from the lateral approach, however, we do not routinely release the ulnar nerve unless there were preoperative symptoms.
Although open and arthroscopic contracture releases can provide durable outcomes in the setting of painless elbow stiffness, options are more limited in the treatment of the painful stiff elbow. Total elbow arthroplasty remains an option in low-demand elderly patients but is not without significant risk of complications.24 In addition, durability concerns and postoperative restrictions make total elbow arthroplasty less attractive to younger patients. Interposition arthroplasty may be indicated as a salvage procedure in the treatment of a young or high-demand patient with a stiff painful elbow.25 Elbow stability is crucial in obtaining a successful outcome, and data on optimal graft choices are limited.
Conclusion
Elbow stiffness, a common complication of trauma, significantly impairs activities of daily living. Early after trauma, therapy should be initiated to prevent contracture. In the absence of symptomatic arthritis, both open and arthroscopic contracture releases are effective surgical treatments in properly selected and motivated patients. Although more research is needed to establish the optimal surgical approach, severity and anatomical cause of contracture should guide decisions as to which approach to use. Having a thorough understanding of neurovascular anatomy and of prophylactic ulnar nerve decompression in the setting of limited preoperative flexion can mitigate complications.
1. Morrey BF, Askew LJ, Chao EY. A biomechanical study of normal functional elbow motion. J Bone Joint Surg Am. 1981;63(6):872-877.
2. Hotchkiss RN. Elbow contracture. In: Green DP, Rotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 5th ed. New York, NY: Churchill-Livingstone; 2005:667-682.
3. Van Zeeland NL, Yamaguchi K. Arthroscopic capsular release of the elbow. J Shoulder Elbow Surg. 2010;19(2):13-19.
4. Morrey BF. Post-traumatic contracture of the elbow. Operative treatment, including distraction arthroplasty. J Bone Joint Surg Am. 1990;72(4):601-618.
5. Park MJ, Chang MJ, Lee YB, Kang HJ. Surgical release for posttraumatic loss of elbow flexion. J Bone Joint Surg Am. 2010;92(16):2692-2699.
6. Brouwer KM, Lindenhovius AL, Ring D. Loss of anterior translation of the distal humeral articular surface is associated with decreased elbow flexion. J Hand Surg Am. 2009;34(7):
1256-1260.
7. Taylor DC, Dalton JD, Seaber AV, Garrett WE. Viscoelastic properties of muscle-tendon units: the biomechanical effects of stretching. Am J Sports Med. 1990;18(3):300-309.
8. Green DP, McCoy H. Turnbuckle orthotic correction of elbow-flexion contractures after acute injuries. J Bone Joint Surg Am. 1979;61(7):1092-1095.
9. Gelinas JJ, Faber KJ, Patterson SD, King GJ. The effectiveness of turnbuckle splinting for elbow contractures. J Bone Joint Surg Br. 2000;82(1):74-78.
10. Hotchkiss RN, Kasparyan GN. The medial "over the top" approach to the elbow. Tech Orthop. 2000;15(2):105-112.
11. Wada T, Ishii S, Usui M, Miyano S. The medial approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Br. 2000;82(1):68-73.
12. Husband JB, Hastings H. The lateral approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Am. 1990;72(9):1353-1358.
13. Mansat P, Morrey BF. The column procedure: a limited lateral approach for extrinsic contracture of the elbow. J Bone Joint Surg Am. 1998;80(11):1603-1605.
14. Haapaniemi T, Berggren M, Adolfsson L. Complete transection of the median and radial nerves during arthroscopic release of post-traumatic elbow contracture. Arthroscopy. 1999;15(7):784-787.
15. Kelly EW, Morrey BF, O’Driscoll SW. Complications of elbow arthroscopy. J Bone Joint Surg Am. 2001;83(1):25-34.
16. Ball CM, Meunier M, Galatz LM, Calfee R, Yamaguchi K. Arthroscopic treatment of post-traumatic elbow contracture. J Shoulder Elbow Surg. 2002;11(6):624-629.
17. Ćefo I, Eygendaal D. Arthroscopic arthrolysis for posttraumatic elbow stiffness. J Shoulder Elbow Surg. 2011;20(3):434-439.
18. Nguyen D, Proper SI, MacDermid JC, King GJ, Faber KJ. Functional outcomes of arthroscopic capsular release of the elbow. Arthroscopy. 2006;22(8):842-849.
19. Sahajpal D, Choi T, Wright TW. Arthroscopic release of the stiff elbow. J Hand Surg. 2009;34(3):540-544.
20. Lindenhovius AL, Jupiter JB. The posttraumatic stiff elbow: a review of the literature. J Hand Surg. 2007;32(10):1605-1623.
21. Williams BG, Sotereanos DG, Baratz ME, Jarrett CD, Venouziou AI, Miller MC. The contracted elbow: is ulnar nerve release necessary? J Shoulder Elbow Surg. 2012;21(12):
1632-1636.
22. Gelberman RH, Yamaguchi K, Hollstien SB, et al. Changes in interstitial pressure and cross-sectional area of the cubital tunnel and of the ulnar nerve with flexion of the elbow. an experimental study in human cadavera. J Bone Joint Surg Am. 1998;80(4):492-501.
23. Blonna D, O’Driscoll SW. Delayed-onset ulnar neuritis after release of elbow contracture: preventive strategies derived from a study of 563 cases. Arthroscopy. 2014;30(8):947-956.
24. Mansat P, Morrey BF. Semiconstrained total elbow arthroplasty for ankylosed and stiff elbows. J Bone Joint Surg. 2000;82(9):1260-1268.
25. Hausman MR, Birnbaum PS. Interposition elbow arthroplasty. Tech Hand Up Extrem Surg. 2004;8(3):181-188.
1. Morrey BF, Askew LJ, Chao EY. A biomechanical study of normal functional elbow motion. J Bone Joint Surg Am. 1981;63(6):872-877.
2. Hotchkiss RN. Elbow contracture. In: Green DP, Rotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 5th ed. New York, NY: Churchill-Livingstone; 2005:667-682.
3. Van Zeeland NL, Yamaguchi K. Arthroscopic capsular release of the elbow. J Shoulder Elbow Surg. 2010;19(2):13-19.
4. Morrey BF. Post-traumatic contracture of the elbow. Operative treatment, including distraction arthroplasty. J Bone Joint Surg Am. 1990;72(4):601-618.
5. Park MJ, Chang MJ, Lee YB, Kang HJ. Surgical release for posttraumatic loss of elbow flexion. J Bone Joint Surg Am. 2010;92(16):2692-2699.
6. Brouwer KM, Lindenhovius AL, Ring D. Loss of anterior translation of the distal humeral articular surface is associated with decreased elbow flexion. J Hand Surg Am. 2009;34(7):
1256-1260.
7. Taylor DC, Dalton JD, Seaber AV, Garrett WE. Viscoelastic properties of muscle-tendon units: the biomechanical effects of stretching. Am J Sports Med. 1990;18(3):300-309.
8. Green DP, McCoy H. Turnbuckle orthotic correction of elbow-flexion contractures after acute injuries. J Bone Joint Surg Am. 1979;61(7):1092-1095.
9. Gelinas JJ, Faber KJ, Patterson SD, King GJ. The effectiveness of turnbuckle splinting for elbow contractures. J Bone Joint Surg Br. 2000;82(1):74-78.
10. Hotchkiss RN, Kasparyan GN. The medial "over the top" approach to the elbow. Tech Orthop. 2000;15(2):105-112.
11. Wada T, Ishii S, Usui M, Miyano S. The medial approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Br. 2000;82(1):68-73.
12. Husband JB, Hastings H. The lateral approach for operative release of post-traumatic contracture of the elbow. J Bone Joint Surg Am. 1990;72(9):1353-1358.
13. Mansat P, Morrey BF. The column procedure: a limited lateral approach for extrinsic contracture of the elbow. J Bone Joint Surg Am. 1998;80(11):1603-1605.
14. Haapaniemi T, Berggren M, Adolfsson L. Complete transection of the median and radial nerves during arthroscopic release of post-traumatic elbow contracture. Arthroscopy. 1999;15(7):784-787.
15. Kelly EW, Morrey BF, O’Driscoll SW. Complications of elbow arthroscopy. J Bone Joint Surg Am. 2001;83(1):25-34.
16. Ball CM, Meunier M, Galatz LM, Calfee R, Yamaguchi K. Arthroscopic treatment of post-traumatic elbow contracture. J Shoulder Elbow Surg. 2002;11(6):624-629.
17. Ćefo I, Eygendaal D. Arthroscopic arthrolysis for posttraumatic elbow stiffness. J Shoulder Elbow Surg. 2011;20(3):434-439.
18. Nguyen D, Proper SI, MacDermid JC, King GJ, Faber KJ. Functional outcomes of arthroscopic capsular release of the elbow. Arthroscopy. 2006;22(8):842-849.
19. Sahajpal D, Choi T, Wright TW. Arthroscopic release of the stiff elbow. J Hand Surg. 2009;34(3):540-544.
20. Lindenhovius AL, Jupiter JB. The posttraumatic stiff elbow: a review of the literature. J Hand Surg. 2007;32(10):1605-1623.
21. Williams BG, Sotereanos DG, Baratz ME, Jarrett CD, Venouziou AI, Miller MC. The contracted elbow: is ulnar nerve release necessary? J Shoulder Elbow Surg. 2012;21(12):
1632-1636.
22. Gelberman RH, Yamaguchi K, Hollstien SB, et al. Changes in interstitial pressure and cross-sectional area of the cubital tunnel and of the ulnar nerve with flexion of the elbow. an experimental study in human cadavera. J Bone Joint Surg Am. 1998;80(4):492-501.
23. Blonna D, O’Driscoll SW. Delayed-onset ulnar neuritis after release of elbow contracture: preventive strategies derived from a study of 563 cases. Arthroscopy. 2014;30(8):947-956.
24. Mansat P, Morrey BF. Semiconstrained total elbow arthroplasty for ankylosed and stiff elbows. J Bone Joint Surg. 2000;82(9):1260-1268.
25. Hausman MR, Birnbaum PS. Interposition elbow arthroplasty. Tech Hand Up Extrem Surg. 2004;8(3):181-188.