User login
Pediatric, adolescent migraine treatment and prevention guidelines are updated
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
FROM NEUROLOGY
Fatal Drug-Resistant Invasive Pulmonary Aspergillus fumigatus in a 56-Year-Old Immunosuppressed Man (FULL)
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1). 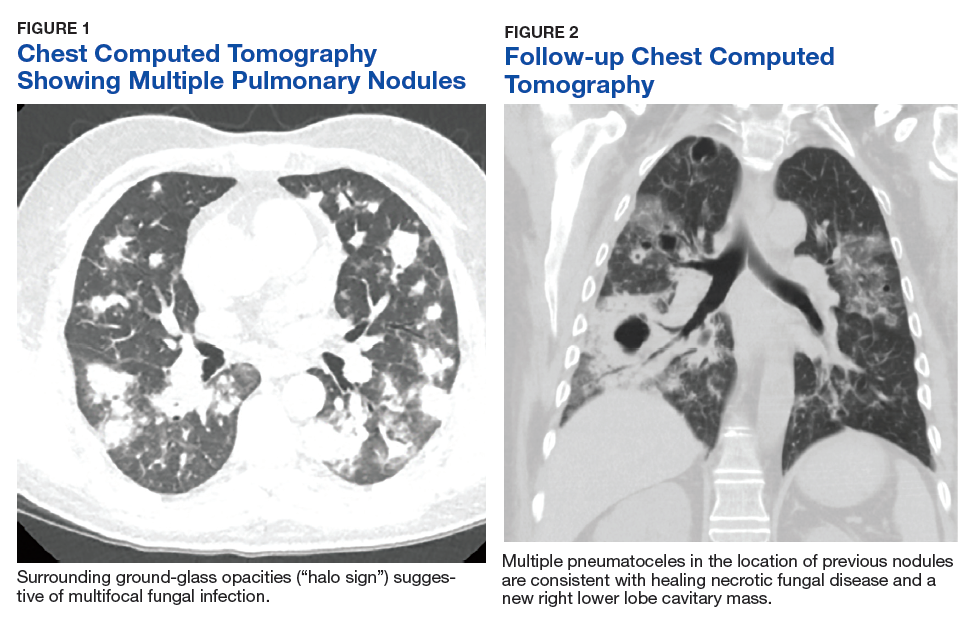
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4). 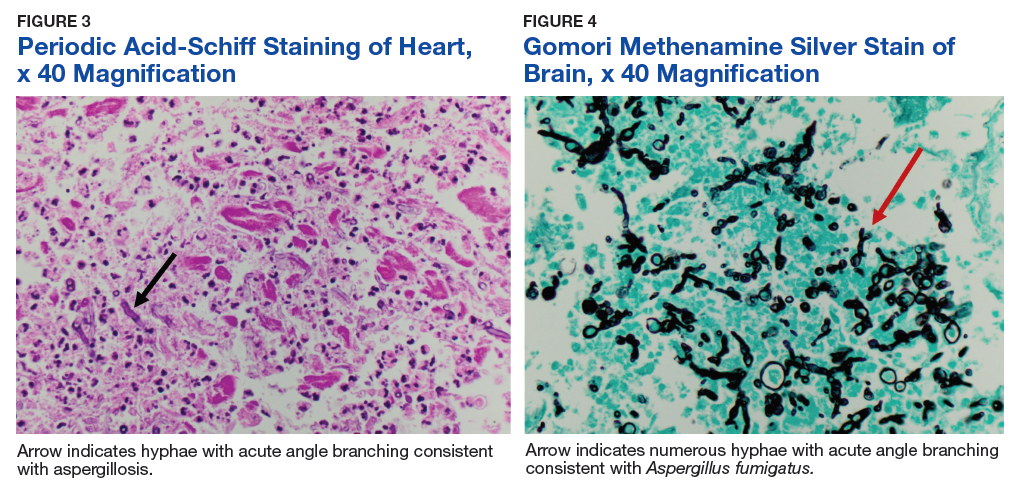
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Interview with Andrew Pachner, MD, about the molecular processes of multiple sclerosis
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.

What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.

Treatment Facility: An Important Prognostic Factor for Dedifferentiated Liposarcoma Survival (FULL)
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
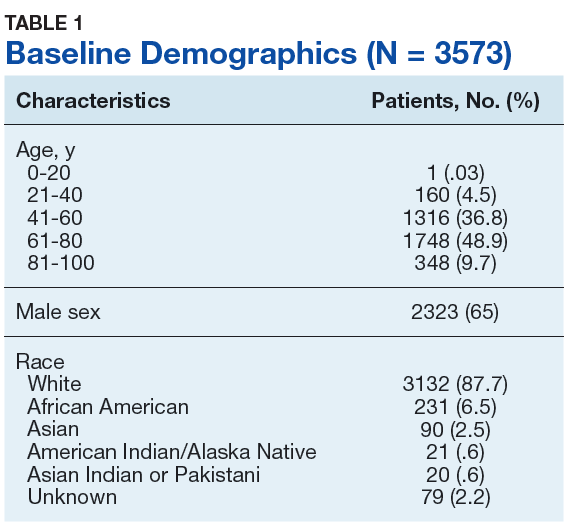
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
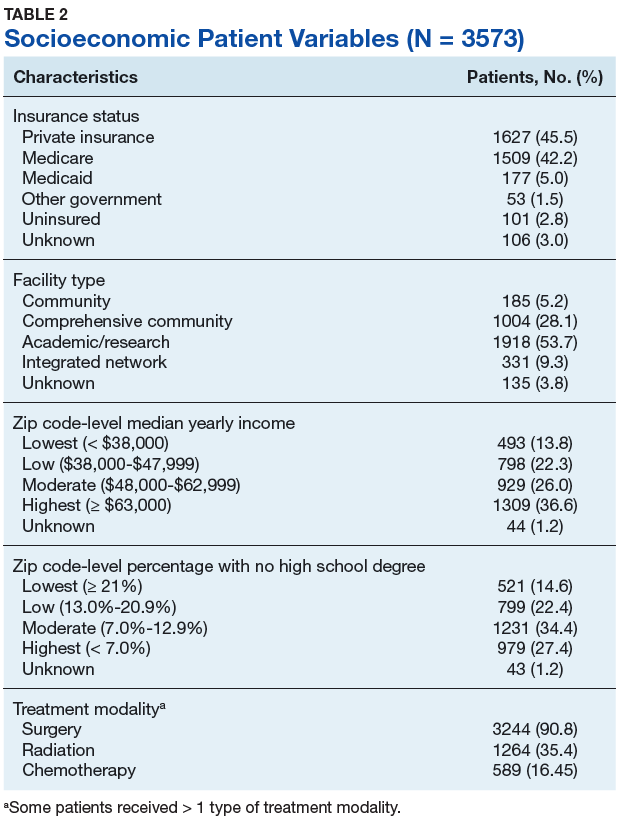
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
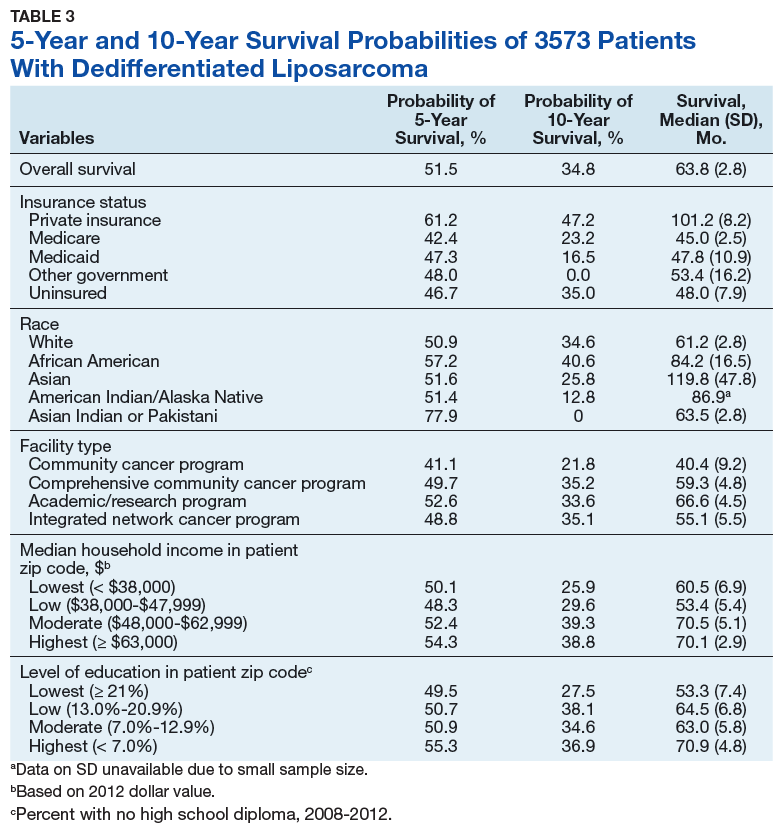
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
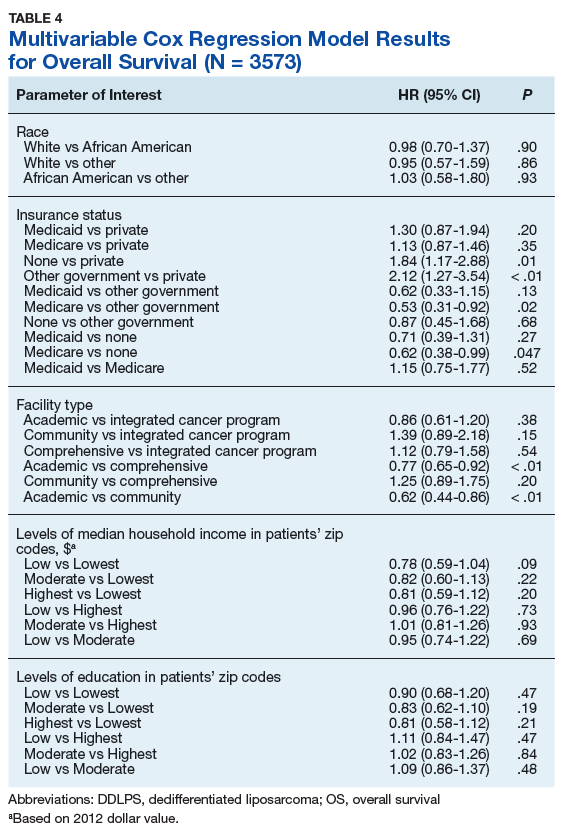
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Prevalence of Cancer in Thyroid Nodules In the Veteran Population (FULL)
Thyroid nodules are identified incidentally in 4% to 10% of the general population in the US.1,2 Clinicians and patients often are concerned about potential malignancy when thyroid nodules are identified because 5% to 15% of nodules will be cancerous.1 The most common form of cancer is papillary carcinoma followed by follicular carcinoma.2 Initially, serum thyroid-stimulating hormone (TSH) levels and thyroid ultrasound are used to evaluate a thyroid nodule because both tests can reveal vital information about malignancy potential.3 Ultrasound characteristics, such as macrocalcifications, hypoechogenicity, absence of halo, increased vascularity, and irregular nodular margins, increase suspicion for malignancy and warrant further investigation.3
Ultrasound-guided fine-needle aspiration (FNA) is the modality of choice for evaluation of thyroid nodules with sensitivity and specificity > 90%.2,4 Most patients receive a definitive diagnosis with this test; however, about 25% of cases are indeterminate based on the Bethesda System and require surgical investigation.3
Currently, it is well accepted clinical practice to refer all nodules > 4 cm for surgical intervention regardless of malignancy risk factors or the mass effect of the nodule.3-6 The preference for surgery—rather than FNA—is because of the notable false negative rate with FNA in larger nodules; studies have described false negative rates for FNA close to 10%.7,8 In contrast, Megwalu recently reported a FNA false negative rate of 0%.9
The risk of malignancy associated with nodule size has been researched for many years, but studies have produced conflicting results. In this retrospective cohort study, the authors compared malignancy rates between patients with nodules ≥ 3 cm and those with nodules < 3 cm.
Methods
The authors performed a retrospective chart review of the medical records of 329 patients presenting for thyroid nodule evaluation found on physical exam or incidentally identified with imaging at the Dayton Veteran Affairs Medical Center from January 2000 to May 2016. Data collection included sex, age, race, personal history of neck radiation treatment, family history of thyroid cancer, personal history of thyroid cancer, hot nodules/Graves disease, abnormal neck lymph nodes, and serum TSH levels. The authors looked for an association between TSH level and cancer. Hot thyroid nodules are known to have low risk of malignancy.
All patients aged 18 to 99 years with a thyroid nodule evaluated with FNA were included in the study. Patients were divided into 2 groups, those with nodules ≥ 3 cm and those with nodules < 3 cm. For nodules requiring subsequent biopsies, only the initial nodule biopsy was included in our study. The 3-cm cutoff was selected based on previous studies.1,5,10 Patients who did not undergo a FNA study were excluded. Indications for surgery were positive FNA results, suspicious imaging, size of nodule, or patient preference.
Means and standard deviations are reported for continuous variables and counts and percentages for categorical variables. We used the Mann-Whitney test for comparisons involving continuous variables with 2 groups and the Kruskal-Wallis test for 4 groups. The chi-square test—corrected for continuity if necessary—was used to compare 2 categorical variables. We used multiple logistic regression to adjust for demographic and clinical variables other than nodule size that were related to malignancy. Inferences were made at the 0.05 level of significance.
Results
A total of 329 patients with thyroid nodules were identified: 236 were < 3 cm and 93 were ≥ 3 cm. The 2 groups differed on race, with more white patients in the < 3-cm nodule group (78% vs 67%, P = .036) (Table 1). 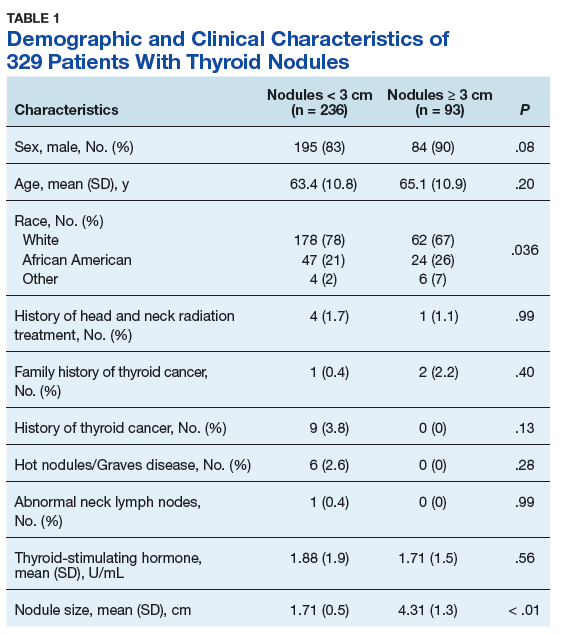
Prevalence of cancer based on FNA in nodules < 3 cm was 6.4% (95% CI, 3.6%–10.3%) and nodules ≥ 3 cm was 8.6% (95% CI, 3.8%–16.2%; P = .23) (Table 2). 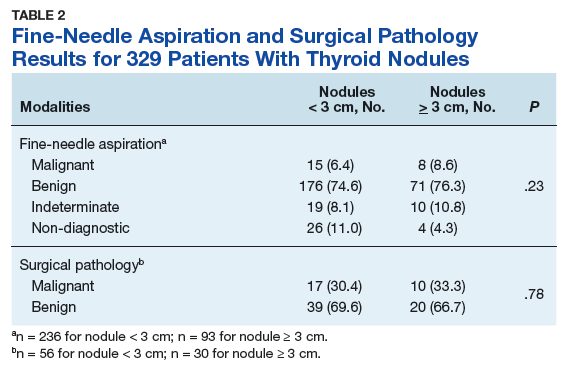
When divided into 4 subgroups, cancer using FNA was found in 35.1% of nodules < 2 cm, 21.1% of nodules 2 cm to < 3 cm, 42.1% of nodules 3 cm to 4 cm, and 18.2% of nodules > 4 cm (P = .32) (Table 3). 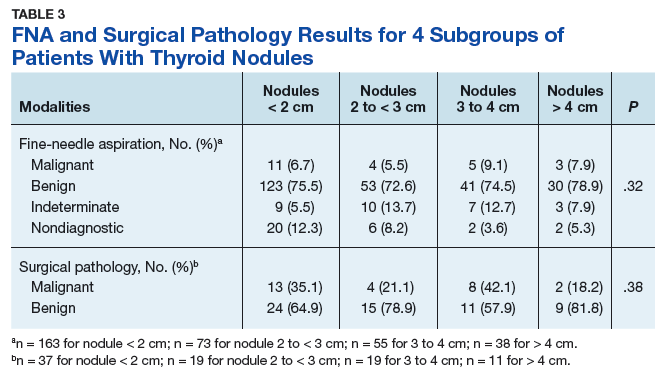
Surgical pathology results showed 17 cases of papillary carcinoma in nodules < 3 cm, whereas there were 9 cases of papillary carcinoma and 1 case of follicular carcinoma in nodules > 3 cm. When correlated with the cytology results, 10 cases were reported as benign, 11 were malignant, and 6 samples were non-diagnostic.
There were 30 nondiagnostic FNA samples: 7 patients had surgery, 19 were monitored with serial imaging, 2 were lost to follow-up, and 2 expired for other reasons. Of the 19 patients who were monitored with serial imaging, the nodules were stable and did not require repeat sampling.
Discussion
The authors found no relationship between thyroid nodule size and malignancy over a 16-year period in a veteran population, either with FNA or surgical pathology. The lack of relationship persists when adjusted for the only nonthyroid variable on which the 2 groups differed (race).
The finding of no relationship between larger thyroid nodule size and cancer is consistent with other studies. In a 10-year chart review of 695 patients at Walter Reed Army Medical Center, Burch and colleagues found a malignancy rate of 18.6% but no association between thyroid nodule size and malignancy.11 They concluded that nodules ≥ 4 cm did not increase malignancy risk. In a 3-year retrospective study of 326 patients, Mangister and colleagues reported that the malignancy rate was higher in nodules < 3 cm (48.4%) compared with nodules ≥ 3 cm (33.3%).10 This study concluded that the malignancy potential of thyroid nodules peaked at 2 cm and decreased at > 3 cm. Kamran and colleagues reported a nonlinear relationship between nodule size and malignancy with a threshold of 2 cm, beyond which there was no increased risk of malignancy.1
Conversely, in a prospective study Kuru and colleagues followed 571 patients who had undergone thyroidectomy and found that nodules ≥ 4 cm were associated with increased malignancy risk compared with nodules < 4 cm. However, with a cutoff of 3 cm there was no relationship.5 Discrepancies among studies might be because of variability in patient demographics and the prevalence of thyroid cancer in a specific institution. Although the majority of thyroid nodules are seen in females, the current study’s population was predominantly male and entirely veteran. Consequently, interpretation of these studies highlight the need to individualize clinical decision-making for each patient.
Limitations
This study has several limitations. It was conducted at a single institution with a group of veterans, which limits the ability to generalize its results to the general population. Second, data omissions are likely in retrospective chart reviews, and ensuring accuracy of data collection could be challenging. Third, all thyroid nodules found to be benign with cytology did not undergo surgical intervention to confirm the diagnosis; therefore, only 93 of 329 nodules were evaluated with the definitive diagnostic test. Therefore, selection bias was introduced into the nodule size comparisons when surgical intervention was used to measure the outcome. However, because false negative rates for FNA is low, likely few malignant nodules were missed. In addition, all patients with thyroid nodules are not referred for surgery because of potential complications.
Conclusion
This study strongly suggests there is no increased or decreased cancer risk for thyroid nodules ≥ 3 cm compared with those < 3 cm. Current clinical practice is to refer patients with larger nodules for surgical evaluation. In a large systemic review, Shin and colleagues reported higher pretest probability of malignancy in larger nodules and recommended consideration of surgical intervention for nodules > 3 cm because of false negatives and concerns for diagnostic inaccuracy with FNA.8 Although data were mixed, Shin and colleagues reported higher incidence of false negative FNA results in larger nodules.8 Given the authors’ findings and earlier conflicting results, the decision for surgical intervention cannot be made solely on nodule size and requires consideration of additional factors including FNA results, nodule characteristics, patient risk factors, and patient preference.
1. Kamran SC, Marqusee E, Kim MI, et al. Thyroid nodule size and prediction of cancer. J Clin Endocrinol Metab. 2013;98(2):564-570.
2. Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association Management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26(1):1-33.
3. Popoveniuc G, Jonklaas J. Thyroid nodules. Med Clin North Am. 2012;96(2):329-349.
4. Amrikachi M, Ramzy I, Rubenfeld S, Wheeler TM. Accuracy of fine needle aspiration of thyroid. Arch Pathol Lab Med. 2001;125(4):484-488.
5. Kuru B, Gulcelik NE, Gulcelik MA, Dincer H. Predictive index for carcinoma of thyroid nodules and its integration with fine-needle aspiration cytology. Head Neck. 2009;31(7):856-866.
6. Kim JH, Kim NK, Oh YL, et al. The validity of ultrasonography-guided fine needle aspiration biopsy in thyroid nodules 4 cm or larger depends on ultrasound characteristics. Endocrinol Metab (Seoul). 2014;29(4):545-552.
7. Wharry LI, McCoy KL, Stang MT, et al. Thyroid nodules (≥4 cm): can ultrasound and cytology reliably exclude cancer? World J Surg. 2014;38(3):614-621.
8. Pinchot SN, Al-Wagih H, Schaefer S, Sippel R, Chen H. Accuracy of fine needle aspiration biopsy for predicting neoplasm or carcinoma in thyroid nodules 4 cm or larger. Arch Surg. 2009;144(7):649-655.
9. Megwalu UC. Risk of malignancy in thyroid nodules 4 cm or larger. Endocrinol Metab (Seoul). 2017;32(1):77-82.
10. Magister MJ, Chaikhoutdinov I, Schaefer E, et al. Association of thyroid nodule size and Bethesda class with rate of malignant disease. JAMA Otolaryngol Head Neck Surg. 2015;141(12):1089-1095.
11. Shrestha M, Crothers BA, Burch HB. The impact of thyroid nodule size on the risk of malignancy and accuracy of fine needle aspiration: a 10-year study from a single institution. Thyroid. 2012;22(12):1251-1256.
Thyroid nodules are identified incidentally in 4% to 10% of the general population in the US.1,2 Clinicians and patients often are concerned about potential malignancy when thyroid nodules are identified because 5% to 15% of nodules will be cancerous.1 The most common form of cancer is papillary carcinoma followed by follicular carcinoma.2 Initially, serum thyroid-stimulating hormone (TSH) levels and thyroid ultrasound are used to evaluate a thyroid nodule because both tests can reveal vital information about malignancy potential.3 Ultrasound characteristics, such as macrocalcifications, hypoechogenicity, absence of halo, increased vascularity, and irregular nodular margins, increase suspicion for malignancy and warrant further investigation.3
Ultrasound-guided fine-needle aspiration (FNA) is the modality of choice for evaluation of thyroid nodules with sensitivity and specificity > 90%.2,4 Most patients receive a definitive diagnosis with this test; however, about 25% of cases are indeterminate based on the Bethesda System and require surgical investigation.3
Currently, it is well accepted clinical practice to refer all nodules > 4 cm for surgical intervention regardless of malignancy risk factors or the mass effect of the nodule.3-6 The preference for surgery—rather than FNA—is because of the notable false negative rate with FNA in larger nodules; studies have described false negative rates for FNA close to 10%.7,8 In contrast, Megwalu recently reported a FNA false negative rate of 0%.9
The risk of malignancy associated with nodule size has been researched for many years, but studies have produced conflicting results. In this retrospective cohort study, the authors compared malignancy rates between patients with nodules ≥ 3 cm and those with nodules < 3 cm.
Methods
The authors performed a retrospective chart review of the medical records of 329 patients presenting for thyroid nodule evaluation found on physical exam or incidentally identified with imaging at the Dayton Veteran Affairs Medical Center from January 2000 to May 2016. Data collection included sex, age, race, personal history of neck radiation treatment, family history of thyroid cancer, personal history of thyroid cancer, hot nodules/Graves disease, abnormal neck lymph nodes, and serum TSH levels. The authors looked for an association between TSH level and cancer. Hot thyroid nodules are known to have low risk of malignancy.
All patients aged 18 to 99 years with a thyroid nodule evaluated with FNA were included in the study. Patients were divided into 2 groups, those with nodules ≥ 3 cm and those with nodules < 3 cm. For nodules requiring subsequent biopsies, only the initial nodule biopsy was included in our study. The 3-cm cutoff was selected based on previous studies.1,5,10 Patients who did not undergo a FNA study were excluded. Indications for surgery were positive FNA results, suspicious imaging, size of nodule, or patient preference.
Means and standard deviations are reported for continuous variables and counts and percentages for categorical variables. We used the Mann-Whitney test for comparisons involving continuous variables with 2 groups and the Kruskal-Wallis test for 4 groups. The chi-square test—corrected for continuity if necessary—was used to compare 2 categorical variables. We used multiple logistic regression to adjust for demographic and clinical variables other than nodule size that were related to malignancy. Inferences were made at the 0.05 level of significance.
Results
A total of 329 patients with thyroid nodules were identified: 236 were < 3 cm and 93 were ≥ 3 cm. The 2 groups differed on race, with more white patients in the < 3-cm nodule group (78% vs 67%, P = .036) (Table 1).
Prevalence of cancer based on FNA in nodules < 3 cm was 6.4% (95% CI, 3.6%–10.3%) and nodules ≥ 3 cm was 8.6% (95% CI, 3.8%–16.2%; P = .23) (Table 2).
When divided into 4 subgroups, cancer using FNA was found in 35.1% of nodules < 2 cm, 21.1% of nodules 2 cm to < 3 cm, 42.1% of nodules 3 cm to 4 cm, and 18.2% of nodules > 4 cm (P = .32) (Table 3).
Surgical pathology results showed 17 cases of papillary carcinoma in nodules < 3 cm, whereas there were 9 cases of papillary carcinoma and 1 case of follicular carcinoma in nodules > 3 cm. When correlated with the cytology results, 10 cases were reported as benign, 11 were malignant, and 6 samples were non-diagnostic.
There were 30 nondiagnostic FNA samples: 7 patients had surgery, 19 were monitored with serial imaging, 2 were lost to follow-up, and 2 expired for other reasons. Of the 19 patients who were monitored with serial imaging, the nodules were stable and did not require repeat sampling.
Discussion
The authors found no relationship between thyroid nodule size and malignancy over a 16-year period in a veteran population, either with FNA or surgical pathology. The lack of relationship persists when adjusted for the only nonthyroid variable on which the 2 groups differed (race).
The finding of no relationship between larger thyroid nodule size and cancer is consistent with other studies. In a 10-year chart review of 695 patients at Walter Reed Army Medical Center, Burch and colleagues found a malignancy rate of 18.6% but no association between thyroid nodule size and malignancy.11 They concluded that nodules ≥ 4 cm did not increase malignancy risk. In a 3-year retrospective study of 326 patients, Mangister and colleagues reported that the malignancy rate was higher in nodules < 3 cm (48.4%) compared with nodules ≥ 3 cm (33.3%).10 This study concluded that the malignancy potential of thyroid nodules peaked at 2 cm and decreased at > 3 cm. Kamran and colleagues reported a nonlinear relationship between nodule size and malignancy with a threshold of 2 cm, beyond which there was no increased risk of malignancy.1
Conversely, in a prospective study Kuru and colleagues followed 571 patients who had undergone thyroidectomy and found that nodules ≥ 4 cm were associated with increased malignancy risk compared with nodules < 4 cm. However, with a cutoff of 3 cm there was no relationship.5 Discrepancies among studies might be because of variability in patient demographics and the prevalence of thyroid cancer in a specific institution. Although the majority of thyroid nodules are seen in females, the current study’s population was predominantly male and entirely veteran. Consequently, interpretation of these studies highlight the need to individualize clinical decision-making for each patient.
Limitations
This study has several limitations. It was conducted at a single institution with a group of veterans, which limits the ability to generalize its results to the general population. Second, data omissions are likely in retrospective chart reviews, and ensuring accuracy of data collection could be challenging. Third, all thyroid nodules found to be benign with cytology did not undergo surgical intervention to confirm the diagnosis; therefore, only 93 of 329 nodules were evaluated with the definitive diagnostic test. Therefore, selection bias was introduced into the nodule size comparisons when surgical intervention was used to measure the outcome. However, because false negative rates for FNA is low, likely few malignant nodules were missed. In addition, all patients with thyroid nodules are not referred for surgery because of potential complications.
Conclusion
This study strongly suggests there is no increased or decreased cancer risk for thyroid nodules ≥ 3 cm compared with those < 3 cm. Current clinical practice is to refer patients with larger nodules for surgical evaluation. In a large systemic review, Shin and colleagues reported higher pretest probability of malignancy in larger nodules and recommended consideration of surgical intervention for nodules > 3 cm because of false negatives and concerns for diagnostic inaccuracy with FNA.8 Although data were mixed, Shin and colleagues reported higher incidence of false negative FNA results in larger nodules.8 Given the authors’ findings and earlier conflicting results, the decision for surgical intervention cannot be made solely on nodule size and requires consideration of additional factors including FNA results, nodule characteristics, patient risk factors, and patient preference.
Thyroid nodules are identified incidentally in 4% to 10% of the general population in the US.1,2 Clinicians and patients often are concerned about potential malignancy when thyroid nodules are identified because 5% to 15% of nodules will be cancerous.1 The most common form of cancer is papillary carcinoma followed by follicular carcinoma.2 Initially, serum thyroid-stimulating hormone (TSH) levels and thyroid ultrasound are used to evaluate a thyroid nodule because both tests can reveal vital information about malignancy potential.3 Ultrasound characteristics, such as macrocalcifications, hypoechogenicity, absence of halo, increased vascularity, and irregular nodular margins, increase suspicion for malignancy and warrant further investigation.3
Ultrasound-guided fine-needle aspiration (FNA) is the modality of choice for evaluation of thyroid nodules with sensitivity and specificity > 90%.2,4 Most patients receive a definitive diagnosis with this test; however, about 25% of cases are indeterminate based on the Bethesda System and require surgical investigation.3
Currently, it is well accepted clinical practice to refer all nodules > 4 cm for surgical intervention regardless of malignancy risk factors or the mass effect of the nodule.3-6 The preference for surgery—rather than FNA—is because of the notable false negative rate with FNA in larger nodules; studies have described false negative rates for FNA close to 10%.7,8 In contrast, Megwalu recently reported a FNA false negative rate of 0%.9
The risk of malignancy associated with nodule size has been researched for many years, but studies have produced conflicting results. In this retrospective cohort study, the authors compared malignancy rates between patients with nodules ≥ 3 cm and those with nodules < 3 cm.
Methods
The authors performed a retrospective chart review of the medical records of 329 patients presenting for thyroid nodule evaluation found on physical exam or incidentally identified with imaging at the Dayton Veteran Affairs Medical Center from January 2000 to May 2016. Data collection included sex, age, race, personal history of neck radiation treatment, family history of thyroid cancer, personal history of thyroid cancer, hot nodules/Graves disease, abnormal neck lymph nodes, and serum TSH levels. The authors looked for an association between TSH level and cancer. Hot thyroid nodules are known to have low risk of malignancy.
All patients aged 18 to 99 years with a thyroid nodule evaluated with FNA were included in the study. Patients were divided into 2 groups, those with nodules ≥ 3 cm and those with nodules < 3 cm. For nodules requiring subsequent biopsies, only the initial nodule biopsy was included in our study. The 3-cm cutoff was selected based on previous studies.1,5,10 Patients who did not undergo a FNA study were excluded. Indications for surgery were positive FNA results, suspicious imaging, size of nodule, or patient preference.
Means and standard deviations are reported for continuous variables and counts and percentages for categorical variables. We used the Mann-Whitney test for comparisons involving continuous variables with 2 groups and the Kruskal-Wallis test for 4 groups. The chi-square test—corrected for continuity if necessary—was used to compare 2 categorical variables. We used multiple logistic regression to adjust for demographic and clinical variables other than nodule size that were related to malignancy. Inferences were made at the 0.05 level of significance.
Results
A total of 329 patients with thyroid nodules were identified: 236 were < 3 cm and 93 were ≥ 3 cm. The 2 groups differed on race, with more white patients in the < 3-cm nodule group (78% vs 67%, P = .036) (Table 1).
Prevalence of cancer based on FNA in nodules < 3 cm was 6.4% (95% CI, 3.6%–10.3%) and nodules ≥ 3 cm was 8.6% (95% CI, 3.8%–16.2%; P = .23) (Table 2).
When divided into 4 subgroups, cancer using FNA was found in 35.1% of nodules < 2 cm, 21.1% of nodules 2 cm to < 3 cm, 42.1% of nodules 3 cm to 4 cm, and 18.2% of nodules > 4 cm (P = .32) (Table 3).
Surgical pathology results showed 17 cases of papillary carcinoma in nodules < 3 cm, whereas there were 9 cases of papillary carcinoma and 1 case of follicular carcinoma in nodules > 3 cm. When correlated with the cytology results, 10 cases were reported as benign, 11 were malignant, and 6 samples were non-diagnostic.
There were 30 nondiagnostic FNA samples: 7 patients had surgery, 19 were monitored with serial imaging, 2 were lost to follow-up, and 2 expired for other reasons. Of the 19 patients who were monitored with serial imaging, the nodules were stable and did not require repeat sampling.
Discussion
The authors found no relationship between thyroid nodule size and malignancy over a 16-year period in a veteran population, either with FNA or surgical pathology. The lack of relationship persists when adjusted for the only nonthyroid variable on which the 2 groups differed (race).
The finding of no relationship between larger thyroid nodule size and cancer is consistent with other studies. In a 10-year chart review of 695 patients at Walter Reed Army Medical Center, Burch and colleagues found a malignancy rate of 18.6% but no association between thyroid nodule size and malignancy.11 They concluded that nodules ≥ 4 cm did not increase malignancy risk. In a 3-year retrospective study of 326 patients, Mangister and colleagues reported that the malignancy rate was higher in nodules < 3 cm (48.4%) compared with nodules ≥ 3 cm (33.3%).10 This study concluded that the malignancy potential of thyroid nodules peaked at 2 cm and decreased at > 3 cm. Kamran and colleagues reported a nonlinear relationship between nodule size and malignancy with a threshold of 2 cm, beyond which there was no increased risk of malignancy.1
Conversely, in a prospective study Kuru and colleagues followed 571 patients who had undergone thyroidectomy and found that nodules ≥ 4 cm were associated with increased malignancy risk compared with nodules < 4 cm. However, with a cutoff of 3 cm there was no relationship.5 Discrepancies among studies might be because of variability in patient demographics and the prevalence of thyroid cancer in a specific institution. Although the majority of thyroid nodules are seen in females, the current study’s population was predominantly male and entirely veteran. Consequently, interpretation of these studies highlight the need to individualize clinical decision-making for each patient.
Limitations
This study has several limitations. It was conducted at a single institution with a group of veterans, which limits the ability to generalize its results to the general population. Second, data omissions are likely in retrospective chart reviews, and ensuring accuracy of data collection could be challenging. Third, all thyroid nodules found to be benign with cytology did not undergo surgical intervention to confirm the diagnosis; therefore, only 93 of 329 nodules were evaluated with the definitive diagnostic test. Therefore, selection bias was introduced into the nodule size comparisons when surgical intervention was used to measure the outcome. However, because false negative rates for FNA is low, likely few malignant nodules were missed. In addition, all patients with thyroid nodules are not referred for surgery because of potential complications.
Conclusion
This study strongly suggests there is no increased or decreased cancer risk for thyroid nodules ≥ 3 cm compared with those < 3 cm. Current clinical practice is to refer patients with larger nodules for surgical evaluation. In a large systemic review, Shin and colleagues reported higher pretest probability of malignancy in larger nodules and recommended consideration of surgical intervention for nodules > 3 cm because of false negatives and concerns for diagnostic inaccuracy with FNA.8 Although data were mixed, Shin and colleagues reported higher incidence of false negative FNA results in larger nodules.8 Given the authors’ findings and earlier conflicting results, the decision for surgical intervention cannot be made solely on nodule size and requires consideration of additional factors including FNA results, nodule characteristics, patient risk factors, and patient preference.
1. Kamran SC, Marqusee E, Kim MI, et al. Thyroid nodule size and prediction of cancer. J Clin Endocrinol Metab. 2013;98(2):564-570.
2. Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association Management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26(1):1-33.
3. Popoveniuc G, Jonklaas J. Thyroid nodules. Med Clin North Am. 2012;96(2):329-349.
4. Amrikachi M, Ramzy I, Rubenfeld S, Wheeler TM. Accuracy of fine needle aspiration of thyroid. Arch Pathol Lab Med. 2001;125(4):484-488.
5. Kuru B, Gulcelik NE, Gulcelik MA, Dincer H. Predictive index for carcinoma of thyroid nodules and its integration with fine-needle aspiration cytology. Head Neck. 2009;31(7):856-866.
6. Kim JH, Kim NK, Oh YL, et al. The validity of ultrasonography-guided fine needle aspiration biopsy in thyroid nodules 4 cm or larger depends on ultrasound characteristics. Endocrinol Metab (Seoul). 2014;29(4):545-552.
7. Wharry LI, McCoy KL, Stang MT, et al. Thyroid nodules (≥4 cm): can ultrasound and cytology reliably exclude cancer? World J Surg. 2014;38(3):614-621.
8. Pinchot SN, Al-Wagih H, Schaefer S, Sippel R, Chen H. Accuracy of fine needle aspiration biopsy for predicting neoplasm or carcinoma in thyroid nodules 4 cm or larger. Arch Surg. 2009;144(7):649-655.
9. Megwalu UC. Risk of malignancy in thyroid nodules 4 cm or larger. Endocrinol Metab (Seoul). 2017;32(1):77-82.
10. Magister MJ, Chaikhoutdinov I, Schaefer E, et al. Association of thyroid nodule size and Bethesda class with rate of malignant disease. JAMA Otolaryngol Head Neck Surg. 2015;141(12):1089-1095.
11. Shrestha M, Crothers BA, Burch HB. The impact of thyroid nodule size on the risk of malignancy and accuracy of fine needle aspiration: a 10-year study from a single institution. Thyroid. 2012;22(12):1251-1256.
1. Kamran SC, Marqusee E, Kim MI, et al. Thyroid nodule size and prediction of cancer. J Clin Endocrinol Metab. 2013;98(2):564-570.
2. Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association Management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26(1):1-33.
3. Popoveniuc G, Jonklaas J. Thyroid nodules. Med Clin North Am. 2012;96(2):329-349.
4. Amrikachi M, Ramzy I, Rubenfeld S, Wheeler TM. Accuracy of fine needle aspiration of thyroid. Arch Pathol Lab Med. 2001;125(4):484-488.
5. Kuru B, Gulcelik NE, Gulcelik MA, Dincer H. Predictive index for carcinoma of thyroid nodules and its integration with fine-needle aspiration cytology. Head Neck. 2009;31(7):856-866.
6. Kim JH, Kim NK, Oh YL, et al. The validity of ultrasonography-guided fine needle aspiration biopsy in thyroid nodules 4 cm or larger depends on ultrasound characteristics. Endocrinol Metab (Seoul). 2014;29(4):545-552.
7. Wharry LI, McCoy KL, Stang MT, et al. Thyroid nodules (≥4 cm): can ultrasound and cytology reliably exclude cancer? World J Surg. 2014;38(3):614-621.
8. Pinchot SN, Al-Wagih H, Schaefer S, Sippel R, Chen H. Accuracy of fine needle aspiration biopsy for predicting neoplasm or carcinoma in thyroid nodules 4 cm or larger. Arch Surg. 2009;144(7):649-655.
9. Megwalu UC. Risk of malignancy in thyroid nodules 4 cm or larger. Endocrinol Metab (Seoul). 2017;32(1):77-82.
10. Magister MJ, Chaikhoutdinov I, Schaefer E, et al. Association of thyroid nodule size and Bethesda class with rate of malignant disease. JAMA Otolaryngol Head Neck Surg. 2015;141(12):1089-1095.
11. Shrestha M, Crothers BA, Burch HB. The impact of thyroid nodule size on the risk of malignancy and accuracy of fine needle aspiration: a 10-year study from a single institution. Thyroid. 2012;22(12):1251-1256.
Review of Radiologic Considerations in an Immunocompetent Patient With Primary Central Nervous System Lymphoma (FULL)
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
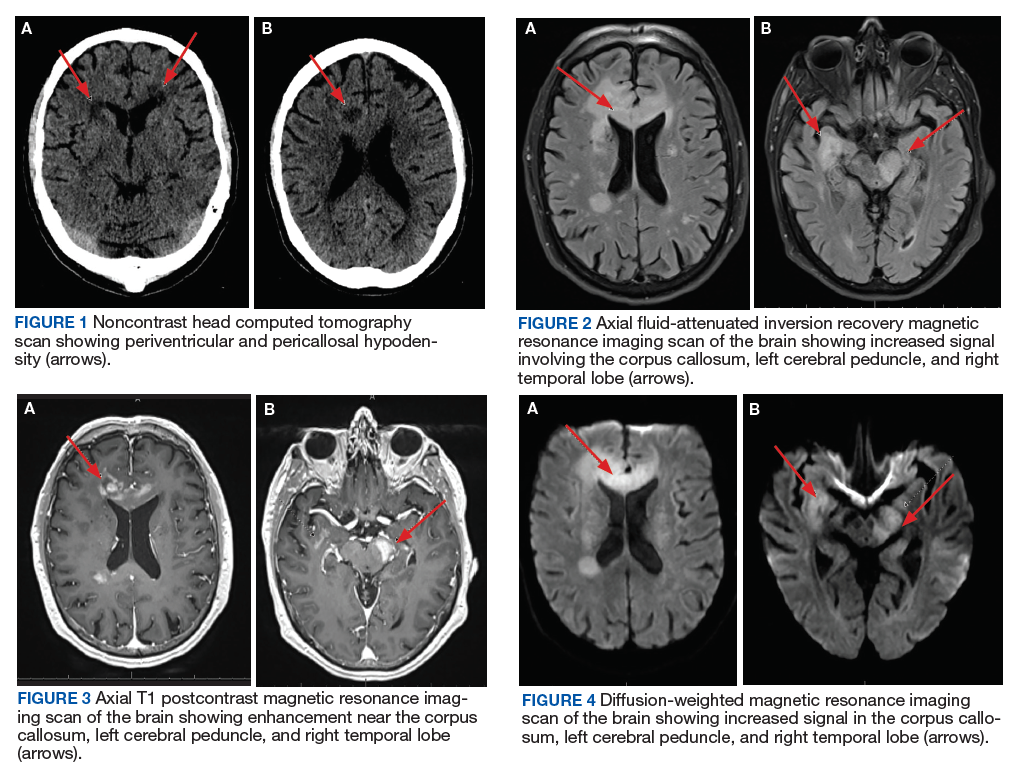
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Accuracy of Endoscopic Ultrasound in Staging of Early Rectal Cancer (FULL)
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
Vaccination is not associated with increased risk of MS
(MS), according to an analysis published July 30 in Neurology. Although the results suggest that vaccination is associated with a lower likelihood of incident MS within the following 5 years, “these data alone do not allow for any conclusion regarding a possible protective effect of vaccinations regarding the development of MS,” wrote Alexander Hapfelmeier, PhD, of the Technical University of Munich and colleagues.

In recent years, researchers have proposed and investigated various potential environmental risk factors for the development of MS. Vaccination is one proposed environmental risk factor, but case reports and small studies have yielded conflicting results about its association with incident MS.
To examine this question more closely, Dr. Hapfelmeier and colleagues performed a systematic retrospective analysis of ambulatory claims data held by the Bavarian Association of Statutory Health Insurance Physicians. They reviewed the data to identify patients with new-onset MS and at least two ICD-10 diagnoses of the disorder. They next identified two control cohorts of participants diagnosed with other autoimmune diseases: Crohn’s disease and psoriasis. Finally, they randomly selected a third control cohort of patients without any of these diagnoses and matched them by age, sex, and district to patients with MS in a 5:1 ratio. Eligible participants were younger than 70 years.
Dr. Hapfelmeier and colleagues reviewed the incidence and frequency of vaccinations (such as those targeting tick-borne encephalitis, human papillomavirus, and influenza virus) in all cohorts. They created unconditional logistic regression models to assess the association between vaccination and MS. They also created separate models to contrast the MS cohort with each of the control cohorts.
The researchers included 12,262 patients with MS, 19,296 patients with Crohn’s disease, 112,292 patients with psoriasis, and 79,185 participants without these autoimmune diseases in their analysis. They found 456 participants with Crohn’s disease and psoriasis, 216 participants with MS and psoriasis, 48 participants with Crohn’s disease and MS, and 2 participants with Crohn’s disease, psoriasis, and MS. Dr. Hapfelmeier and colleagues allocated these participants to each of the respective cohorts and did not analyze them differently because of the comparatively small sample sizes.
The investigators analyzed the occurrence of vaccination in all participants during the 5 years before first diagnosis. Among patients who received vaccination, the odds ratio of MS was 0.870 in participants without autoimmune disease, 0.919 in participants with Crohn’s disease, and 0.973 in participants with psoriasis. Decreased risk of MS was most notable for vaccinations against influenza and tick-borne encephalitis. The results were consistent regardless of time frame, control cohort, and definition of MS.
The subjective definition of the MS cohort was a limitation of the study, but the authors addressed it by also using several strict definitions of that cohort. Another limitation is that the source data may reflect entry errors and incorrect coding.
A grant from the German Federal Ministry of Education and Research Competence Network MS supported the study. The authors had no conflicts that were relevant to the topic of the study.
SOURCE: Hapfelmeier A et al. Neurology. 2019 Jul 30. doi: 10.1212/WNL.0000000000008012.
The analysis by Hapfelmeier et al. provides important evidence that vaccinations are not associated with multiple sclerosis (MS), said E. Ann Yeh, MD, a neurologist at the Hospital for Sick Children in Toronto, and Jennifer Graves, MD, PhD, a neurologist at the University of California, San Diego, in an accompanying editorial. On the contrary, the evidence supports a potential protective effect of vaccines on the risk of developing MS, they said.
“The reasons for this [finding] cannot be gleaned from this study and may range from biological to sociocultural/demographic reasons,” the authors added. “Infection, rather than vaccination, may be an MS trigger, or individuals obtaining vaccinations may be practicing other healthy behaviors protective for MS. These possibilities should be the subject of future studies.”
Until future studies are completed and their results published, the findings of Hapfelmeier et al. offer “strong evidence to share with worried patients and families when faced with the question of whether a vaccine in the recent or relatively distant past triggered the individual’s MS,” said Dr. Yeh and Dr. Graves.
The authors had various relationships with industry, including serving on advisory boards for and receiving funding from pharmaceutical companies.
The analysis by Hapfelmeier et al. provides important evidence that vaccinations are not associated with multiple sclerosis (MS), said E. Ann Yeh, MD, a neurologist at the Hospital for Sick Children in Toronto, and Jennifer Graves, MD, PhD, a neurologist at the University of California, San Diego, in an accompanying editorial. On the contrary, the evidence supports a potential protective effect of vaccines on the risk of developing MS, they said.
“The reasons for this [finding] cannot be gleaned from this study and may range from biological to sociocultural/demographic reasons,” the authors added. “Infection, rather than vaccination, may be an MS trigger, or individuals obtaining vaccinations may be practicing other healthy behaviors protective for MS. These possibilities should be the subject of future studies.”
Until future studies are completed and their results published, the findings of Hapfelmeier et al. offer “strong evidence to share with worried patients and families when faced with the question of whether a vaccine in the recent or relatively distant past triggered the individual’s MS,” said Dr. Yeh and Dr. Graves.
The authors had various relationships with industry, including serving on advisory boards for and receiving funding from pharmaceutical companies.
The analysis by Hapfelmeier et al. provides important evidence that vaccinations are not associated with multiple sclerosis (MS), said E. Ann Yeh, MD, a neurologist at the Hospital for Sick Children in Toronto, and Jennifer Graves, MD, PhD, a neurologist at the University of California, San Diego, in an accompanying editorial. On the contrary, the evidence supports a potential protective effect of vaccines on the risk of developing MS, they said.
“The reasons for this [finding] cannot be gleaned from this study and may range from biological to sociocultural/demographic reasons,” the authors added. “Infection, rather than vaccination, may be an MS trigger, or individuals obtaining vaccinations may be practicing other healthy behaviors protective for MS. These possibilities should be the subject of future studies.”
Until future studies are completed and their results published, the findings of Hapfelmeier et al. offer “strong evidence to share with worried patients and families when faced with the question of whether a vaccine in the recent or relatively distant past triggered the individual’s MS,” said Dr. Yeh and Dr. Graves.
The authors had various relationships with industry, including serving on advisory boards for and receiving funding from pharmaceutical companies.
(MS), according to an analysis published July 30 in Neurology. Although the results suggest that vaccination is associated with a lower likelihood of incident MS within the following 5 years, “these data alone do not allow for any conclusion regarding a possible protective effect of vaccinations regarding the development of MS,” wrote Alexander Hapfelmeier, PhD, of the Technical University of Munich and colleagues.
In recent years, researchers have proposed and investigated various potential environmental risk factors for the development of MS. Vaccination is one proposed environmental risk factor, but case reports and small studies have yielded conflicting results about its association with incident MS.
To examine this question more closely, Dr. Hapfelmeier and colleagues performed a systematic retrospective analysis of ambulatory claims data held by the Bavarian Association of Statutory Health Insurance Physicians. They reviewed the data to identify patients with new-onset MS and at least two ICD-10 diagnoses of the disorder. They next identified two control cohorts of participants diagnosed with other autoimmune diseases: Crohn’s disease and psoriasis. Finally, they randomly selected a third control cohort of patients without any of these diagnoses and matched them by age, sex, and district to patients with MS in a 5:1 ratio. Eligible participants were younger than 70 years.
Dr. Hapfelmeier and colleagues reviewed the incidence and frequency of vaccinations (such as those targeting tick-borne encephalitis, human papillomavirus, and influenza virus) in all cohorts. They created unconditional logistic regression models to assess the association between vaccination and MS. They also created separate models to contrast the MS cohort with each of the control cohorts.
The researchers included 12,262 patients with MS, 19,296 patients with Crohn’s disease, 112,292 patients with psoriasis, and 79,185 participants without these autoimmune diseases in their analysis. They found 456 participants with Crohn’s disease and psoriasis, 216 participants with MS and psoriasis, 48 participants with Crohn’s disease and MS, and 2 participants with Crohn’s disease, psoriasis, and MS. Dr. Hapfelmeier and colleagues allocated these participants to each of the respective cohorts and did not analyze them differently because of the comparatively small sample sizes.
The investigators analyzed the occurrence of vaccination in all participants during the 5 years before first diagnosis. Among patients who received vaccination, the odds ratio of MS was 0.870 in participants without autoimmune disease, 0.919 in participants with Crohn’s disease, and 0.973 in participants with psoriasis. Decreased risk of MS was most notable for vaccinations against influenza and tick-borne encephalitis. The results were consistent regardless of time frame, control cohort, and definition of MS.
The subjective definition of the MS cohort was a limitation of the study, but the authors addressed it by also using several strict definitions of that cohort. Another limitation is that the source data may reflect entry errors and incorrect coding.
A grant from the German Federal Ministry of Education and Research Competence Network MS supported the study. The authors had no conflicts that were relevant to the topic of the study.
SOURCE: Hapfelmeier A et al. Neurology. 2019 Jul 30. doi: 10.1212/WNL.0000000000008012.
(MS), according to an analysis published July 30 in Neurology. Although the results suggest that vaccination is associated with a lower likelihood of incident MS within the following 5 years, “these data alone do not allow for any conclusion regarding a possible protective effect of vaccinations regarding the development of MS,” wrote Alexander Hapfelmeier, PhD, of the Technical University of Munich and colleagues.
In recent years, researchers have proposed and investigated various potential environmental risk factors for the development of MS. Vaccination is one proposed environmental risk factor, but case reports and small studies have yielded conflicting results about its association with incident MS.
To examine this question more closely, Dr. Hapfelmeier and colleagues performed a systematic retrospective analysis of ambulatory claims data held by the Bavarian Association of Statutory Health Insurance Physicians. They reviewed the data to identify patients with new-onset MS and at least two ICD-10 diagnoses of the disorder. They next identified two control cohorts of participants diagnosed with other autoimmune diseases: Crohn’s disease and psoriasis. Finally, they randomly selected a third control cohort of patients without any of these diagnoses and matched them by age, sex, and district to patients with MS in a 5:1 ratio. Eligible participants were younger than 70 years.
Dr. Hapfelmeier and colleagues reviewed the incidence and frequency of vaccinations (such as those targeting tick-borne encephalitis, human papillomavirus, and influenza virus) in all cohorts. They created unconditional logistic regression models to assess the association between vaccination and MS. They also created separate models to contrast the MS cohort with each of the control cohorts.
The researchers included 12,262 patients with MS, 19,296 patients with Crohn’s disease, 112,292 patients with psoriasis, and 79,185 participants without these autoimmune diseases in their analysis. They found 456 participants with Crohn’s disease and psoriasis, 216 participants with MS and psoriasis, 48 participants with Crohn’s disease and MS, and 2 participants with Crohn’s disease, psoriasis, and MS. Dr. Hapfelmeier and colleagues allocated these participants to each of the respective cohorts and did not analyze them differently because of the comparatively small sample sizes.
The investigators analyzed the occurrence of vaccination in all participants during the 5 years before first diagnosis. Among patients who received vaccination, the odds ratio of MS was 0.870 in participants without autoimmune disease, 0.919 in participants with Crohn’s disease, and 0.973 in participants with psoriasis. Decreased risk of MS was most notable for vaccinations against influenza and tick-borne encephalitis. The results were consistent regardless of time frame, control cohort, and definition of MS.
The subjective definition of the MS cohort was a limitation of the study, but the authors addressed it by also using several strict definitions of that cohort. Another limitation is that the source data may reflect entry errors and incorrect coding.
A grant from the German Federal Ministry of Education and Research Competence Network MS supported the study. The authors had no conflicts that were relevant to the topic of the study.
SOURCE: Hapfelmeier A et al. Neurology. 2019 Jul 30. doi: 10.1212/WNL.0000000000008012.
FROM NEUROLOGY
VHA Practice Guideline Recommendations for Diffuse Gliomas (FULL)
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C). 
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C). 
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C). 
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points. 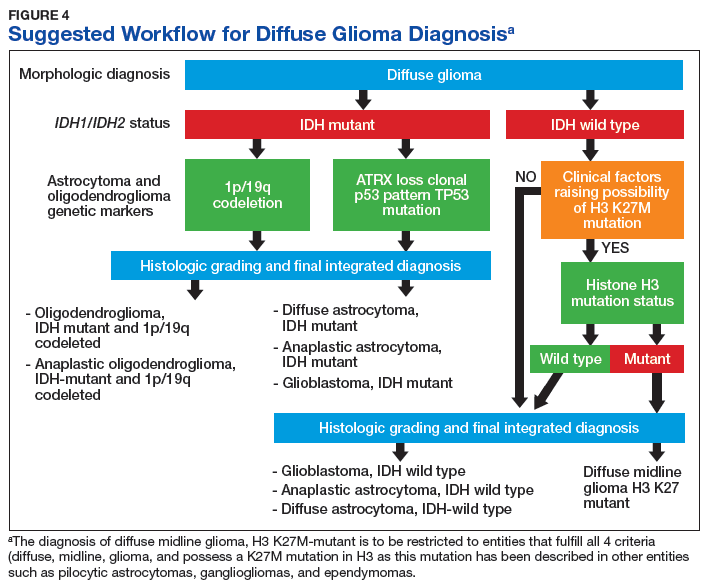
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1). 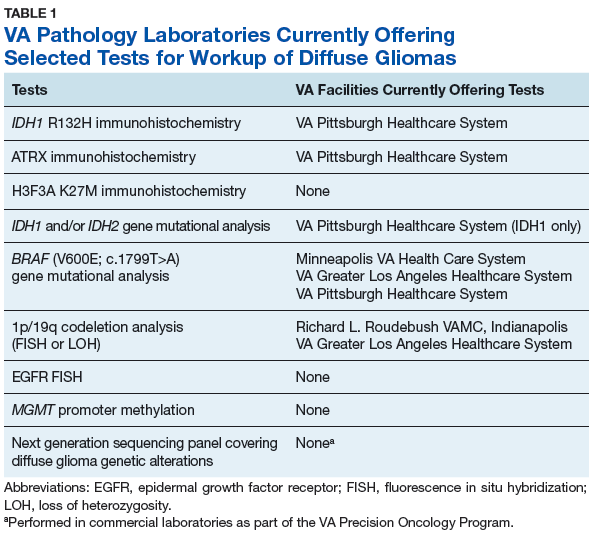
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas. 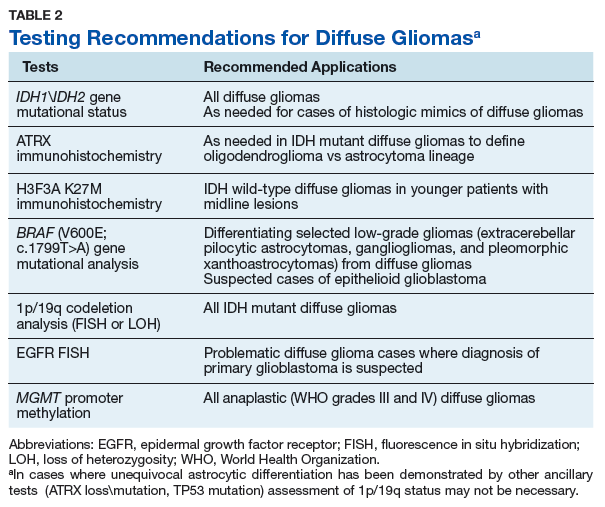

Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
Improving Comorbidities With Psoriasis Treatment
Psoriasis is a common immune-mediated inflammatory skin disorder that affects up to 3.2% of adults in the United States.1 It is a TH1, TH17, and TH22 inflammatory disease resulting in increased levels of cytokines in the skin, including IFN-γ, tumor necrosis factor (TNF), IL-17, and IL-22. Dendritic antigen-presenting cells also are increased in the skin of patients with psoriasis resulting in increased levels of IL-23.2 Although skin disease often is its most prominent and sometimes its only documented manifestation, an understanding of psoriasis as a chronic multisystem inflammatory disorder is essential to optimize outcomes.1,3 Multiple comorbidities that may affect treatment selection often are associated with psoriasis, including psoriatic arthritis, cardiovascular disease, depression, obesity, metabolic syndrome, cardiovascular disease (CVD), cerebrovascular disease, and peripheral vascular disease.
As with other immune-mediated inflammatory diseases, it has been hypothesized that psoriasis may influence comorbidities through shared genetic risks, environmental factors, and pathogenic factors or inflammatory pathways.2-4 For example, emerging evidence suggests that comorbidities such as metabolic syndrome may be related to the chronic inflammation that accompanies psoriasis, a finding that has important clinical implications.3
The interplay and dependence or interdependence of psoriasis and its comorbidities is complex, and it is an area deserving of vigorous research.1 At present, observational and epidemiological data such as the present case suggest that effective treatment of psoriasis could lead to benefits “beyond the skin” and potentially even prevent future disease-associated comorbidity.1-3
Metabolic Comorbidities and Psoriasis Treatment
Although the prevalence of CVD and CVD risk factors is increased in patients with psoriasis, studies suggest that the suppression of systemic inflammation that accompanies adequate psoriasis treatment, particularly in patients with moderate to severe disease, may decrease the risk for cardiovascular comorbidities.5 For example, a number of studies have found treatment of psoriasis with methotrexate may decrease the risk for cardiovascular events, including ischemic heart disease, stroke, and cardiovascular death.6-10 Low-dose methotrexate has been shown to be particularly advantageous for decreasing CVD in patients with psoriasis.5,8
Tumor necrosis factor α inhibitors, which are frequently used for moderate to severe plaque psoriasis, also may notably decrease cardiovascular risk.5 One study showed a significant decrease in the risk for myocardial infarction in patients with psoriasis who were treated with TNF-α inhibitors (hazard ratio, 0.50; 95% CI, 0.32-0.79)11; other studies have confirmed this benefit.12-17 Moreover, the reduction in cardiovascular events may be greater with TNF-α inhibitors than with methotrexate when the former is used for psoriasis treatment, with longer duration of TNF-α inhibition leading to greater risk reduction.18,19
In patients with severe psoriasis, treatment with TNF-α inhibitors has been associated with improvements in subclinical CVD (abnormalities in echocardiogram), improved coronary microvascular function (determined by transthoracic Doppler echocardiography), and reduced progression in coronary artery disease (assessed by coronary computed tomography).20-22 Improvement in endothelial function (brachial artery reactivity) and carotid arterial stiffness also has been reported following 6 months of treatment with adalimumab for moderate to severe psoriasis.21
Data concerning potential cardiovascular risk reduction with treatment of psoriasis utilizing newer agents are continuing to emerge. To date, no increase in the incidence of major adverse cardiovascular events has been shown in patients with psoriasis treated with anti–IL-17 agents, such as secukinumab; however, additional long-term studies are needed.18,23-25
Apremilast, an oral phosphodiesterase 4 inhibitor, is another addition to the psoriasis armamentarium.26 No increase in the risk for major cardiac events has been shown in randomized controlled trials of patients with psoriasis receiving apremilast for up to 156 weeks.27,28 As with secukinumab, additional long-term, large-scale studies are needed to determine the effects of apremilast on cardiovascular risk in patients with psoriasis.5
Other Comorbidities
Effective treatment of psoriasis also appears to benefit various other comorbidities. Numerous studies have shown an increased incidence of depression in patients with psoriasis vs controls and a concurrent improvement in psychiatric symptoms with psoriasis disease control.1 For instance, a multicenter, randomized, open-label study of 352 patients with psoriasis showed treatment with etanercept, a TNF inhibitor, significantly improved scores for concomitant depression and anxiety (P<.05).29 Similarly, a double-blind, randomized clinical trial of patients with psoriasis found significant improvement in depression at 12 weeks in patients treated with adalimumab vs placebo (P<.001).30 Likewise, a multicenter phase 3 trial of more than 600 psoriatic patients showed improved Beck depression inventory and Hamilton depression rating scale scores at 12 weeks in patients with psoriasis treated with etanercept compared to placebo.31
A much larger analysis of 7490 patients with psoriasis compared the rates of depression among patients receiving biologic therapy, phototherapy, and conventional systemic therapy. The greatest impact on depression symptoms was seen with biologic therapy (incidence rate, 3.01/100 patient-years), followed by conventional systemic therapy (5.70/100 patient-years), and phototherapy (5.85/100 patient-years).32
Uveitis, or inflammation of the middle layer of the eye (the uvea), frequently is seen in patients with psoriasis. In a cohort study of 60,000 patients with mild psoriasis and more than 7000 patients with severe psoriasis, the incidence of uveitis in patients was significantly increased in both patients with severe disease and those with mild disease (P<.001 for both).33 In a case series of 8 patients with concomitant psoriasis and uveitis, 4 patients were treated with infliximab and 4 with adalimumab; 7 patients treated achieved remission of their uveitis.34
Role of the TNF-α Blockade in Sickle Cell Disease
Presently, no reported human studies have shown TNF-α blockade as a possible treatment of sickle cell disease.35 However, increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and to the severity of sickle cell disease due to their integral role in the development of vascular wall dysfunction and ischemia.35,36 Studies have shown that TNF-α is released in homozygous sickle cell anemia (HbSS) disease and impedes blood flow during sickle cell crisis, resulting in worsening ischemia and painful infarction.35,36 Moreover, cytokine analysis has shown significantly (P<.05) elevated levels of TNF-α during sickle cell crises and at baseline in patients with HbSS vs healthy controls, suggesting a possible role of TNF-α in the pathogenesis of sickle cell crisis.36
The case patient reported a 50% reduction in pain level and the use of pain medications that overlapped with the initiation of adalimumab for treatment of her psoriasis. Moreover, although radiographs showed possible psoriatic changes of the distal metatarsal row, she described sickle cell pain and pain crises that were uncharacteristic of psoriatic arthralgia.35 Although these findings are observational in nature and limited to one patient, they do suggest an interesting hypothesis. If a common inflammatory mediator is the culprit, it is possible that TNF-α inhibitors could be the preferred treatment option for patients with psoriasis and comorbid HbSS or HbSC disease. Further studies are needed to analyze the role of TNF-α inhibition in sickle cell disease.
Bottom Line
Psoriasis may influence comorbidities through shared genetic risks, environmental factors, or inflammatory pathways. Improvement in metabolic and other comorbidities have been shown with the effective treatment of psoriasis. The case described here showed improvement in sickle cell crises and pain with treatment of psoriasis with adalimumab. Tumor necrosis factor inhibitors may be an optimal choice for patients with both psoriasis and sickle cell disease.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Davidovici BB, Sattar N, Prinz J, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol. 2010;130:1785-1796.
- Oliveira Mde F, Rocha Bde O, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90:9-20.
- Shah K, Mellars L, Changolkar A, Feldman SR. Real-world burden of comorbidities in US patients with psoriasis. J Am Acad Dermatol. 2017;77:287-292.
- Hu SC, Lan CE. Psoriasis and cardiovascular comorbidities: focusing on severe vascular events, cardiovascular risk factors and implications for treatment [published online October 21, 2017]. Int J Mol Sci. doi:10.3390/ijms18102211.
- Hugh J, Van Voorhees AS, Nijhawan RI, et al. From the Medical Board of The National Psoriasis Foundation: the risk of cardiovascular disease in individuals with psoriasis and the potential impact of current therapies. J Am Acad Dermatol. 2014;70:168-177.
- Churton S, Brown L, Shin TM, et al. Does treatment of psoriasis reduce the risk of cardiovascular disease? Drugs. 2014;74:169-182.
- Prodanovich S, Ma F, Taylor J, et al. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52:262-226.
- Gulliver WP, Young HM, Bachelez H, et al. Psoriasis patients treated with biologics and methotrexate have a reduced rate of myocardial infarction: a collaborative analysis using international cohorts. J Cutan Med Surg. 2016;20:550-554.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273:197-204.
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250.
- Wu JJ, Poon KY. Association of ethnicity, tumor necrosis factor inhibitor therapy, and myocardial infarction risk in patients with psoriasis. J Am Acad Dermatol. 2013;69:167-168.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Wu JJ, Poon KY, Bebchuk JD. Tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis, psoriatic arthritis, or both. J Drugs Dermatol. 2014;13:932-934.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular co-morbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Nguyen T, Wu JJ. Relationship between tumor necrosis factor-alpha inhibitors and cardiovascular disease in psoriasis: a review. Perm J. 2014;18:49-54.
- Shaaban D, Al-Mutairi N. The effect of tumour necrosis factor inhibitor therapy on the incidence of myocardial infarction in patients with psoriasis: a retrospective study [published online November 17, 2017]. J Dermatol Treat. doi:10.1080/09546634.2016.1254145.
- Wu D, Hou SY, Zhao S, et al. Efficacy and safety of interleukin-17 antagonists in patients with plaque psoriasis: A meta-analysis from phase 3 randomized controlled trials. J Eur Acad Dermatol Venereol. 2017;31:992-100.
- Yang ZS, Lin NN, Li L, et al. The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin Rev Allergy Immunol. 2016;51:240-247.
- Heredi E, Vegh J, Pogacsas L, et al. Subclinical cardiovascular disease and it’s improvement after long-term TNF-alpha inhibitor therapy in severe psoriatic patients. J Eur Acad Dermatol Venereol. 2016;30:1531-1536.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Piaserico S, Osto E, Famoso G, et al. Treatment with tumor necrosis factor inhibitors restores coronary microvascular function in young patients with severe psoriasis. Atherosclerosis. 2016;251:25-30.
- Van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Torres T, Raposo I, Selores M. IL-17 blockade in psoriasis: friend or foe in cardiovascular risk? Am J Clin Dermatol. 2016;17:107-112.
- Deeks ED. Apremilast: a review in psoriasis and psoriatic arthritis. Drugs. 2015;75:1393-1403.
- Crowley J, Thaci D, Joly P, et al. Long-term safety and tolerability of apremilast in patients with psoriasis: pooled safety analysis for >/= 156 weeks from 2 phase 3, randomized, controlled trials (ESTEEM 1 and 2). J Am Acad Dermatol. 2017;77:310-317.
- Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73:1020-1026.
- Daudén E, Griffiths CE, Ortonne JP, et al. Improvements in patient-reported outcomes in moderate-to-severe psoriasis patients receiving continuous or paused etanercept treatment over 54 weeks: the CRYSTEL study. J Eur Acad Dermatol Venereol. 2009;23:1374-1382.
- Menter A, Augustin M, Signorovitch J, et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J Am Acad Dermatol. 2010;62:812-818.
- Tyring S, Gottlieb A, Papp K, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29-35.
- Strober B, Gooderham M, de Jong EMGJ, et al. Depressive symptoms, depression, and the effect of biologic therapy among patients in Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Am Acad Dermatol. 2018;78:70-80.
- Egeberg A, Khalid U, Gislason GH, et al. Association of psoriatic disease with uveitis: a Danish nationwide cohort study. JAMA Dermatol. 2015;151:1200-1205.
- Huynh N, Cervantes-Castaneda RA, Bhat P, et al. Biologic response modifier therapy for psoriatic ocular inflammatory disease. Ocul Immunol Inflamm. 2008;16:89-93.
- Pulusani S, McMurray SL, Jensen K, et al. Psoriasis treatment in patients with sickle cell disease Cutis. 2019;103:93-94.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
Psoriasis is a common immune-mediated inflammatory skin disorder that affects up to 3.2% of adults in the United States.1 It is a TH1, TH17, and TH22 inflammatory disease resulting in increased levels of cytokines in the skin, including IFN-γ, tumor necrosis factor (TNF), IL-17, and IL-22. Dendritic antigen-presenting cells also are increased in the skin of patients with psoriasis resulting in increased levels of IL-23.2 Although skin disease often is its most prominent and sometimes its only documented manifestation, an understanding of psoriasis as a chronic multisystem inflammatory disorder is essential to optimize outcomes.1,3 Multiple comorbidities that may affect treatment selection often are associated with psoriasis, including psoriatic arthritis, cardiovascular disease, depression, obesity, metabolic syndrome, cardiovascular disease (CVD), cerebrovascular disease, and peripheral vascular disease.
As with other immune-mediated inflammatory diseases, it has been hypothesized that psoriasis may influence comorbidities through shared genetic risks, environmental factors, and pathogenic factors or inflammatory pathways.2-4 For example, emerging evidence suggests that comorbidities such as metabolic syndrome may be related to the chronic inflammation that accompanies psoriasis, a finding that has important clinical implications.3
The interplay and dependence or interdependence of psoriasis and its comorbidities is complex, and it is an area deserving of vigorous research.1 At present, observational and epidemiological data such as the present case suggest that effective treatment of psoriasis could lead to benefits “beyond the skin” and potentially even prevent future disease-associated comorbidity.1-3
Metabolic Comorbidities and Psoriasis Treatment
Although the prevalence of CVD and CVD risk factors is increased in patients with psoriasis, studies suggest that the suppression of systemic inflammation that accompanies adequate psoriasis treatment, particularly in patients with moderate to severe disease, may decrease the risk for cardiovascular comorbidities.5 For example, a number of studies have found treatment of psoriasis with methotrexate may decrease the risk for cardiovascular events, including ischemic heart disease, stroke, and cardiovascular death.6-10 Low-dose methotrexate has been shown to be particularly advantageous for decreasing CVD in patients with psoriasis.5,8
Tumor necrosis factor α inhibitors, which are frequently used for moderate to severe plaque psoriasis, also may notably decrease cardiovascular risk.5 One study showed a significant decrease in the risk for myocardial infarction in patients with psoriasis who were treated with TNF-α inhibitors (hazard ratio, 0.50; 95% CI, 0.32-0.79)11; other studies have confirmed this benefit.12-17 Moreover, the reduction in cardiovascular events may be greater with TNF-α inhibitors than with methotrexate when the former is used for psoriasis treatment, with longer duration of TNF-α inhibition leading to greater risk reduction.18,19
In patients with severe psoriasis, treatment with TNF-α inhibitors has been associated with improvements in subclinical CVD (abnormalities in echocardiogram), improved coronary microvascular function (determined by transthoracic Doppler echocardiography), and reduced progression in coronary artery disease (assessed by coronary computed tomography).20-22 Improvement in endothelial function (brachial artery reactivity) and carotid arterial stiffness also has been reported following 6 months of treatment with adalimumab for moderate to severe psoriasis.21
Data concerning potential cardiovascular risk reduction with treatment of psoriasis utilizing newer agents are continuing to emerge. To date, no increase in the incidence of major adverse cardiovascular events has been shown in patients with psoriasis treated with anti–IL-17 agents, such as secukinumab; however, additional long-term studies are needed.18,23-25
Apremilast, an oral phosphodiesterase 4 inhibitor, is another addition to the psoriasis armamentarium.26 No increase in the risk for major cardiac events has been shown in randomized controlled trials of patients with psoriasis receiving apremilast for up to 156 weeks.27,28 As with secukinumab, additional long-term, large-scale studies are needed to determine the effects of apremilast on cardiovascular risk in patients with psoriasis.5
Other Comorbidities
Effective treatment of psoriasis also appears to benefit various other comorbidities. Numerous studies have shown an increased incidence of depression in patients with psoriasis vs controls and a concurrent improvement in psychiatric symptoms with psoriasis disease control.1 For instance, a multicenter, randomized, open-label study of 352 patients with psoriasis showed treatment with etanercept, a TNF inhibitor, significantly improved scores for concomitant depression and anxiety (P<.05).29 Similarly, a double-blind, randomized clinical trial of patients with psoriasis found significant improvement in depression at 12 weeks in patients treated with adalimumab vs placebo (P<.001).30 Likewise, a multicenter phase 3 trial of more than 600 psoriatic patients showed improved Beck depression inventory and Hamilton depression rating scale scores at 12 weeks in patients with psoriasis treated with etanercept compared to placebo.31
A much larger analysis of 7490 patients with psoriasis compared the rates of depression among patients receiving biologic therapy, phototherapy, and conventional systemic therapy. The greatest impact on depression symptoms was seen with biologic therapy (incidence rate, 3.01/100 patient-years), followed by conventional systemic therapy (5.70/100 patient-years), and phototherapy (5.85/100 patient-years).32
Uveitis, or inflammation of the middle layer of the eye (the uvea), frequently is seen in patients with psoriasis. In a cohort study of 60,000 patients with mild psoriasis and more than 7000 patients with severe psoriasis, the incidence of uveitis in patients was significantly increased in both patients with severe disease and those with mild disease (P<.001 for both).33 In a case series of 8 patients with concomitant psoriasis and uveitis, 4 patients were treated with infliximab and 4 with adalimumab; 7 patients treated achieved remission of their uveitis.34
Role of the TNF-α Blockade in Sickle Cell Disease
Presently, no reported human studies have shown TNF-α blockade as a possible treatment of sickle cell disease.35 However, increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and to the severity of sickle cell disease due to their integral role in the development of vascular wall dysfunction and ischemia.35,36 Studies have shown that TNF-α is released in homozygous sickle cell anemia (HbSS) disease and impedes blood flow during sickle cell crisis, resulting in worsening ischemia and painful infarction.35,36 Moreover, cytokine analysis has shown significantly (P<.05) elevated levels of TNF-α during sickle cell crises and at baseline in patients with HbSS vs healthy controls, suggesting a possible role of TNF-α in the pathogenesis of sickle cell crisis.36
The case patient reported a 50% reduction in pain level and the use of pain medications that overlapped with the initiation of adalimumab for treatment of her psoriasis. Moreover, although radiographs showed possible psoriatic changes of the distal metatarsal row, she described sickle cell pain and pain crises that were uncharacteristic of psoriatic arthralgia.35 Although these findings are observational in nature and limited to one patient, they do suggest an interesting hypothesis. If a common inflammatory mediator is the culprit, it is possible that TNF-α inhibitors could be the preferred treatment option for patients with psoriasis and comorbid HbSS or HbSC disease. Further studies are needed to analyze the role of TNF-α inhibition in sickle cell disease.
Bottom Line
Psoriasis may influence comorbidities through shared genetic risks, environmental factors, or inflammatory pathways. Improvement in metabolic and other comorbidities have been shown with the effective treatment of psoriasis. The case described here showed improvement in sickle cell crises and pain with treatment of psoriasis with adalimumab. Tumor necrosis factor inhibitors may be an optimal choice for patients with both psoriasis and sickle cell disease.
Psoriasis is a common immune-mediated inflammatory skin disorder that affects up to 3.2% of adults in the United States.1 It is a TH1, TH17, and TH22 inflammatory disease resulting in increased levels of cytokines in the skin, including IFN-γ, tumor necrosis factor (TNF), IL-17, and IL-22. Dendritic antigen-presenting cells also are increased in the skin of patients with psoriasis resulting in increased levels of IL-23.2 Although skin disease often is its most prominent and sometimes its only documented manifestation, an understanding of psoriasis as a chronic multisystem inflammatory disorder is essential to optimize outcomes.1,3 Multiple comorbidities that may affect treatment selection often are associated with psoriasis, including psoriatic arthritis, cardiovascular disease, depression, obesity, metabolic syndrome, cardiovascular disease (CVD), cerebrovascular disease, and peripheral vascular disease.
As with other immune-mediated inflammatory diseases, it has been hypothesized that psoriasis may influence comorbidities through shared genetic risks, environmental factors, and pathogenic factors or inflammatory pathways.2-4 For example, emerging evidence suggests that comorbidities such as metabolic syndrome may be related to the chronic inflammation that accompanies psoriasis, a finding that has important clinical implications.3
The interplay and dependence or interdependence of psoriasis and its comorbidities is complex, and it is an area deserving of vigorous research.1 At present, observational and epidemiological data such as the present case suggest that effective treatment of psoriasis could lead to benefits “beyond the skin” and potentially even prevent future disease-associated comorbidity.1-3
Metabolic Comorbidities and Psoriasis Treatment
Although the prevalence of CVD and CVD risk factors is increased in patients with psoriasis, studies suggest that the suppression of systemic inflammation that accompanies adequate psoriasis treatment, particularly in patients with moderate to severe disease, may decrease the risk for cardiovascular comorbidities.5 For example, a number of studies have found treatment of psoriasis with methotrexate may decrease the risk for cardiovascular events, including ischemic heart disease, stroke, and cardiovascular death.6-10 Low-dose methotrexate has been shown to be particularly advantageous for decreasing CVD in patients with psoriasis.5,8
Tumor necrosis factor α inhibitors, which are frequently used for moderate to severe plaque psoriasis, also may notably decrease cardiovascular risk.5 One study showed a significant decrease in the risk for myocardial infarction in patients with psoriasis who were treated with TNF-α inhibitors (hazard ratio, 0.50; 95% CI, 0.32-0.79)11; other studies have confirmed this benefit.12-17 Moreover, the reduction in cardiovascular events may be greater with TNF-α inhibitors than with methotrexate when the former is used for psoriasis treatment, with longer duration of TNF-α inhibition leading to greater risk reduction.18,19
In patients with severe psoriasis, treatment with TNF-α inhibitors has been associated with improvements in subclinical CVD (abnormalities in echocardiogram), improved coronary microvascular function (determined by transthoracic Doppler echocardiography), and reduced progression in coronary artery disease (assessed by coronary computed tomography).20-22 Improvement in endothelial function (brachial artery reactivity) and carotid arterial stiffness also has been reported following 6 months of treatment with adalimumab for moderate to severe psoriasis.21
Data concerning potential cardiovascular risk reduction with treatment of psoriasis utilizing newer agents are continuing to emerge. To date, no increase in the incidence of major adverse cardiovascular events has been shown in patients with psoriasis treated with anti–IL-17 agents, such as secukinumab; however, additional long-term studies are needed.18,23-25
Apremilast, an oral phosphodiesterase 4 inhibitor, is another addition to the psoriasis armamentarium.26 No increase in the risk for major cardiac events has been shown in randomized controlled trials of patients with psoriasis receiving apremilast for up to 156 weeks.27,28 As with secukinumab, additional long-term, large-scale studies are needed to determine the effects of apremilast on cardiovascular risk in patients with psoriasis.5
Other Comorbidities
Effective treatment of psoriasis also appears to benefit various other comorbidities. Numerous studies have shown an increased incidence of depression in patients with psoriasis vs controls and a concurrent improvement in psychiatric symptoms with psoriasis disease control.1 For instance, a multicenter, randomized, open-label study of 352 patients with psoriasis showed treatment with etanercept, a TNF inhibitor, significantly improved scores for concomitant depression and anxiety (P<.05).29 Similarly, a double-blind, randomized clinical trial of patients with psoriasis found significant improvement in depression at 12 weeks in patients treated with adalimumab vs placebo (P<.001).30 Likewise, a multicenter phase 3 trial of more than 600 psoriatic patients showed improved Beck depression inventory and Hamilton depression rating scale scores at 12 weeks in patients with psoriasis treated with etanercept compared to placebo.31
A much larger analysis of 7490 patients with psoriasis compared the rates of depression among patients receiving biologic therapy, phototherapy, and conventional systemic therapy. The greatest impact on depression symptoms was seen with biologic therapy (incidence rate, 3.01/100 patient-years), followed by conventional systemic therapy (5.70/100 patient-years), and phototherapy (5.85/100 patient-years).32
Uveitis, or inflammation of the middle layer of the eye (the uvea), frequently is seen in patients with psoriasis. In a cohort study of 60,000 patients with mild psoriasis and more than 7000 patients with severe psoriasis, the incidence of uveitis in patients was significantly increased in both patients with severe disease and those with mild disease (P<.001 for both).33 In a case series of 8 patients with concomitant psoriasis and uveitis, 4 patients were treated with infliximab and 4 with adalimumab; 7 patients treated achieved remission of their uveitis.34
Role of the TNF-α Blockade in Sickle Cell Disease
Presently, no reported human studies have shown TNF-α blockade as a possible treatment of sickle cell disease.35 However, increased levels of TNF-α have been shown to contribute to the onset of sickle cell crises and to the severity of sickle cell disease due to their integral role in the development of vascular wall dysfunction and ischemia.35,36 Studies have shown that TNF-α is released in homozygous sickle cell anemia (HbSS) disease and impedes blood flow during sickle cell crisis, resulting in worsening ischemia and painful infarction.35,36 Moreover, cytokine analysis has shown significantly (P<.05) elevated levels of TNF-α during sickle cell crises and at baseline in patients with HbSS vs healthy controls, suggesting a possible role of TNF-α in the pathogenesis of sickle cell crisis.36
The case patient reported a 50% reduction in pain level and the use of pain medications that overlapped with the initiation of adalimumab for treatment of her psoriasis. Moreover, although radiographs showed possible psoriatic changes of the distal metatarsal row, she described sickle cell pain and pain crises that were uncharacteristic of psoriatic arthralgia.35 Although these findings are observational in nature and limited to one patient, they do suggest an interesting hypothesis. If a common inflammatory mediator is the culprit, it is possible that TNF-α inhibitors could be the preferred treatment option for patients with psoriasis and comorbid HbSS or HbSC disease. Further studies are needed to analyze the role of TNF-α inhibition in sickle cell disease.
Bottom Line
Psoriasis may influence comorbidities through shared genetic risks, environmental factors, or inflammatory pathways. Improvement in metabolic and other comorbidities have been shown with the effective treatment of psoriasis. The case described here showed improvement in sickle cell crises and pain with treatment of psoriasis with adalimumab. Tumor necrosis factor inhibitors may be an optimal choice for patients with both psoriasis and sickle cell disease.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Davidovici BB, Sattar N, Prinz J, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol. 2010;130:1785-1796.
- Oliveira Mde F, Rocha Bde O, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90:9-20.
- Shah K, Mellars L, Changolkar A, Feldman SR. Real-world burden of comorbidities in US patients with psoriasis. J Am Acad Dermatol. 2017;77:287-292.
- Hu SC, Lan CE. Psoriasis and cardiovascular comorbidities: focusing on severe vascular events, cardiovascular risk factors and implications for treatment [published online October 21, 2017]. Int J Mol Sci. doi:10.3390/ijms18102211.
- Hugh J, Van Voorhees AS, Nijhawan RI, et al. From the Medical Board of The National Psoriasis Foundation: the risk of cardiovascular disease in individuals with psoriasis and the potential impact of current therapies. J Am Acad Dermatol. 2014;70:168-177.
- Churton S, Brown L, Shin TM, et al. Does treatment of psoriasis reduce the risk of cardiovascular disease? Drugs. 2014;74:169-182.
- Prodanovich S, Ma F, Taylor J, et al. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52:262-226.
- Gulliver WP, Young HM, Bachelez H, et al. Psoriasis patients treated with biologics and methotrexate have a reduced rate of myocardial infarction: a collaborative analysis using international cohorts. J Cutan Med Surg. 2016;20:550-554.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273:197-204.
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250.
- Wu JJ, Poon KY. Association of ethnicity, tumor necrosis factor inhibitor therapy, and myocardial infarction risk in patients with psoriasis. J Am Acad Dermatol. 2013;69:167-168.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Wu JJ, Poon KY, Bebchuk JD. Tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis, psoriatic arthritis, or both. J Drugs Dermatol. 2014;13:932-934.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular co-morbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Nguyen T, Wu JJ. Relationship between tumor necrosis factor-alpha inhibitors and cardiovascular disease in psoriasis: a review. Perm J. 2014;18:49-54.
- Shaaban D, Al-Mutairi N. The effect of tumour necrosis factor inhibitor therapy on the incidence of myocardial infarction in patients with psoriasis: a retrospective study [published online November 17, 2017]. J Dermatol Treat. doi:10.1080/09546634.2016.1254145.
- Wu D, Hou SY, Zhao S, et al. Efficacy and safety of interleukin-17 antagonists in patients with plaque psoriasis: A meta-analysis from phase 3 randomized controlled trials. J Eur Acad Dermatol Venereol. 2017;31:992-100.
- Yang ZS, Lin NN, Li L, et al. The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin Rev Allergy Immunol. 2016;51:240-247.
- Heredi E, Vegh J, Pogacsas L, et al. Subclinical cardiovascular disease and it’s improvement after long-term TNF-alpha inhibitor therapy in severe psoriatic patients. J Eur Acad Dermatol Venereol. 2016;30:1531-1536.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Piaserico S, Osto E, Famoso G, et al. Treatment with tumor necrosis factor inhibitors restores coronary microvascular function in young patients with severe psoriasis. Atherosclerosis. 2016;251:25-30.
- Van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Torres T, Raposo I, Selores M. IL-17 blockade in psoriasis: friend or foe in cardiovascular risk? Am J Clin Dermatol. 2016;17:107-112.
- Deeks ED. Apremilast: a review in psoriasis and psoriatic arthritis. Drugs. 2015;75:1393-1403.
- Crowley J, Thaci D, Joly P, et al. Long-term safety and tolerability of apremilast in patients with psoriasis: pooled safety analysis for >/= 156 weeks from 2 phase 3, randomized, controlled trials (ESTEEM 1 and 2). J Am Acad Dermatol. 2017;77:310-317.
- Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73:1020-1026.
- Daudén E, Griffiths CE, Ortonne JP, et al. Improvements in patient-reported outcomes in moderate-to-severe psoriasis patients receiving continuous or paused etanercept treatment over 54 weeks: the CRYSTEL study. J Eur Acad Dermatol Venereol. 2009;23:1374-1382.
- Menter A, Augustin M, Signorovitch J, et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J Am Acad Dermatol. 2010;62:812-818.
- Tyring S, Gottlieb A, Papp K, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29-35.
- Strober B, Gooderham M, de Jong EMGJ, et al. Depressive symptoms, depression, and the effect of biologic therapy among patients in Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Am Acad Dermatol. 2018;78:70-80.
- Egeberg A, Khalid U, Gislason GH, et al. Association of psoriatic disease with uveitis: a Danish nationwide cohort study. JAMA Dermatol. 2015;151:1200-1205.
- Huynh N, Cervantes-Castaneda RA, Bhat P, et al. Biologic response modifier therapy for psoriatic ocular inflammatory disease. Ocul Immunol Inflamm. 2008;16:89-93.
- Pulusani S, McMurray SL, Jensen K, et al. Psoriasis treatment in patients with sickle cell disease Cutis. 2019;103:93-94.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Davidovici BB, Sattar N, Prinz J, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol. 2010;130:1785-1796.
- Oliveira Mde F, Rocha Bde O, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90:9-20.
- Shah K, Mellars L, Changolkar A, Feldman SR. Real-world burden of comorbidities in US patients with psoriasis. J Am Acad Dermatol. 2017;77:287-292.
- Hu SC, Lan CE. Psoriasis and cardiovascular comorbidities: focusing on severe vascular events, cardiovascular risk factors and implications for treatment [published online October 21, 2017]. Int J Mol Sci. doi:10.3390/ijms18102211.
- Hugh J, Van Voorhees AS, Nijhawan RI, et al. From the Medical Board of The National Psoriasis Foundation: the risk of cardiovascular disease in individuals with psoriasis and the potential impact of current therapies. J Am Acad Dermatol. 2014;70:168-177.
- Churton S, Brown L, Shin TM, et al. Does treatment of psoriasis reduce the risk of cardiovascular disease? Drugs. 2014;74:169-182.
- Prodanovich S, Ma F, Taylor J, et al. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52:262-226.
- Gulliver WP, Young HM, Bachelez H, et al. Psoriasis patients treated with biologics and methotrexate have a reduced rate of myocardial infarction: a collaborative analysis using international cohorts. J Cutan Med Surg. 2016;20:550-554.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273:197-204.
- Wu JJ, Poon KY, Channual JC, et al. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148:1244-1250.
- Wu JJ, Poon KY. Association of ethnicity, tumor necrosis factor inhibitor therapy, and myocardial infarction risk in patients with psoriasis. J Am Acad Dermatol. 2013;69:167-168.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Wu JJ, Poon KY, Bebchuk JD. Tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis, psoriatic arthritis, or both. J Drugs Dermatol. 2014;13:932-934.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular co-morbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Nguyen T, Wu JJ. Relationship between tumor necrosis factor-alpha inhibitors and cardiovascular disease in psoriasis: a review. Perm J. 2014;18:49-54.
- Shaaban D, Al-Mutairi N. The effect of tumour necrosis factor inhibitor therapy on the incidence of myocardial infarction in patients with psoriasis: a retrospective study [published online November 17, 2017]. J Dermatol Treat. doi:10.1080/09546634.2016.1254145.
- Wu D, Hou SY, Zhao S, et al. Efficacy and safety of interleukin-17 antagonists in patients with plaque psoriasis: A meta-analysis from phase 3 randomized controlled trials. J Eur Acad Dermatol Venereol. 2017;31:992-100.
- Yang ZS, Lin NN, Li L, et al. The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin Rev Allergy Immunol. 2016;51:240-247.
- Heredi E, Vegh J, Pogacsas L, et al. Subclinical cardiovascular disease and it’s improvement after long-term TNF-alpha inhibitor therapy in severe psoriatic patients. J Eur Acad Dermatol Venereol. 2016;30:1531-1536.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Piaserico S, Osto E, Famoso G, et al. Treatment with tumor necrosis factor inhibitors restores coronary microvascular function in young patients with severe psoriasis. Atherosclerosis. 2016;251:25-30.
- Van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Wu JJ, Guerin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Torres T, Raposo I, Selores M. IL-17 blockade in psoriasis: friend or foe in cardiovascular risk? Am J Clin Dermatol. 2016;17:107-112.
- Deeks ED. Apremilast: a review in psoriasis and psoriatic arthritis. Drugs. 2015;75:1393-1403.
- Crowley J, Thaci D, Joly P, et al. Long-term safety and tolerability of apremilast in patients with psoriasis: pooled safety analysis for >/= 156 weeks from 2 phase 3, randomized, controlled trials (ESTEEM 1 and 2). J Am Acad Dermatol. 2017;77:310-317.
- Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73:1020-1026.
- Daudén E, Griffiths CE, Ortonne JP, et al. Improvements in patient-reported outcomes in moderate-to-severe psoriasis patients receiving continuous or paused etanercept treatment over 54 weeks: the CRYSTEL study. J Eur Acad Dermatol Venereol. 2009;23:1374-1382.
- Menter A, Augustin M, Signorovitch J, et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J Am Acad Dermatol. 2010;62:812-818.
- Tyring S, Gottlieb A, Papp K, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29-35.
- Strober B, Gooderham M, de Jong EMGJ, et al. Depressive symptoms, depression, and the effect of biologic therapy among patients in Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Am Acad Dermatol. 2018;78:70-80.
- Egeberg A, Khalid U, Gislason GH, et al. Association of psoriatic disease with uveitis: a Danish nationwide cohort study. JAMA Dermatol. 2015;151:1200-1205.
- Huynh N, Cervantes-Castaneda RA, Bhat P, et al. Biologic response modifier therapy for psoriatic ocular inflammatory disease. Ocul Immunol Inflamm. 2008;16:89-93.
- Pulusani S, McMurray SL, Jensen K, et al. Psoriasis treatment in patients with sickle cell disease Cutis. 2019;103:93-94.
- Nnodim J, Meludu SC, Dioka CE, et al. Cytokine expression in homozygous sickle cell anaemia. JKIMSU. 2015;4:34-37.
A 31-year-old woman presented with moderate to severe plaque psoriasis (70% body surface area). The patient’s medical history was positive for sickle cell disease, specifically hemoglobin SC disease (HbSC). She reported chronic dull arthralgia in the ankles that was worse at night. She was being treated by hematology with ibuprofen and ketorolac. Radiographs of the feet and ankles showed erosive changes of the distal tarsal row and metatarsal bases. At the current presentation, her HbSC pain was 8/10 on a visual analog scale. She described her sickle cell pain crises as sharp 10/10 pain in the back, elbows, and ankles, associated with mild edema lasting 1 to 2 days. Radiographs of the spine, hands, and ankles were unremarkable.
Adalimumab was chosen as a systemic therapy for psoriasis based on its potential for improvement in HbSC symptoms as well as psoriasis.
Within 17 weeks of starting adalimumab, the psoriasis body surface area decreased from 70% to 40%, and she reported a decrease in her HbSC pain from 8/10 to 4/10 at 8-week follow-up and to 0/10 at 17-week follow-up. She also reported decreased use of pain medication with rare sickle cell pain crises following initiation of adalimumab.