User login
Painful Mouth Ulcers
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
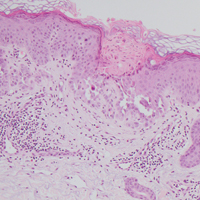
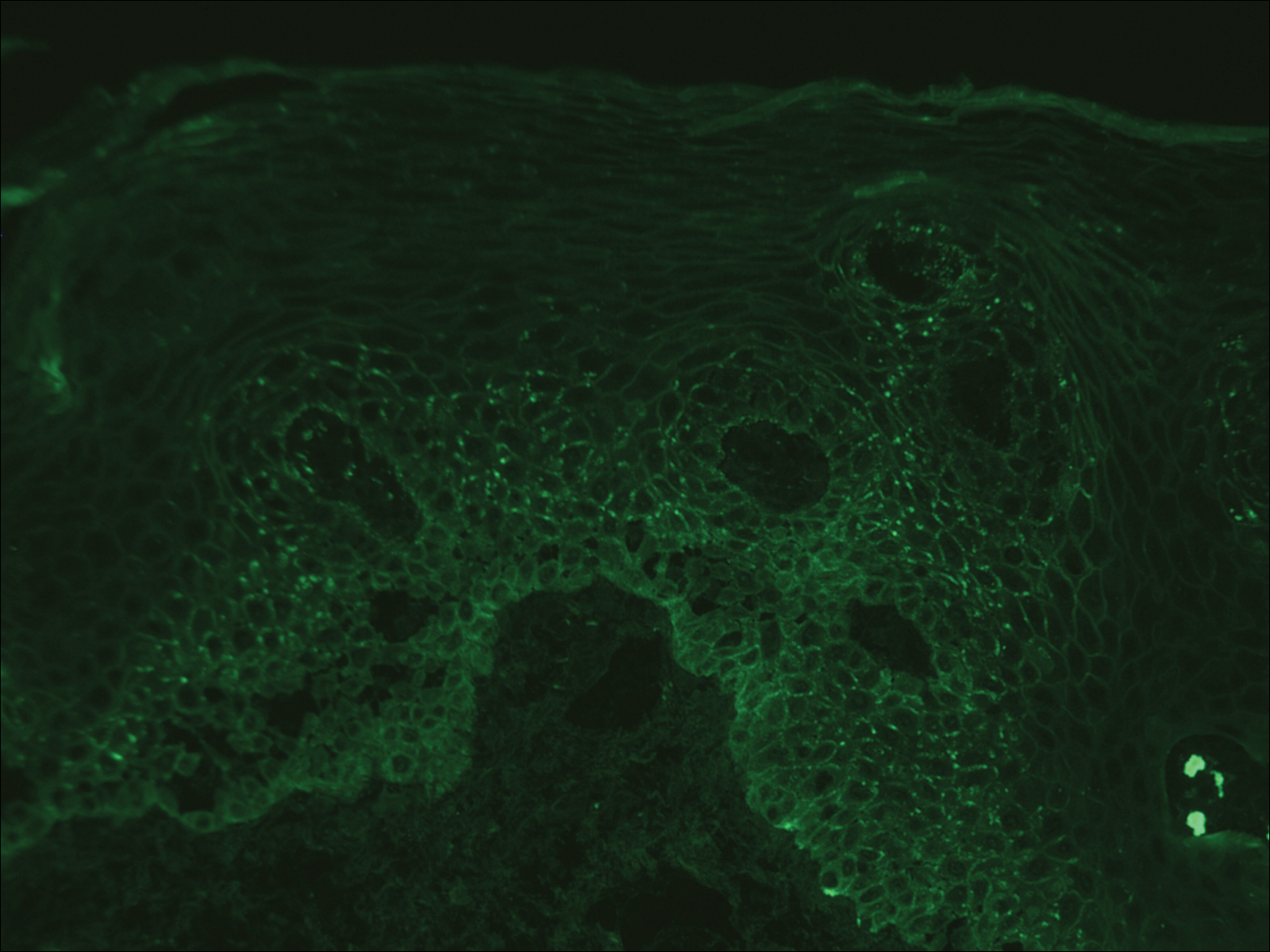
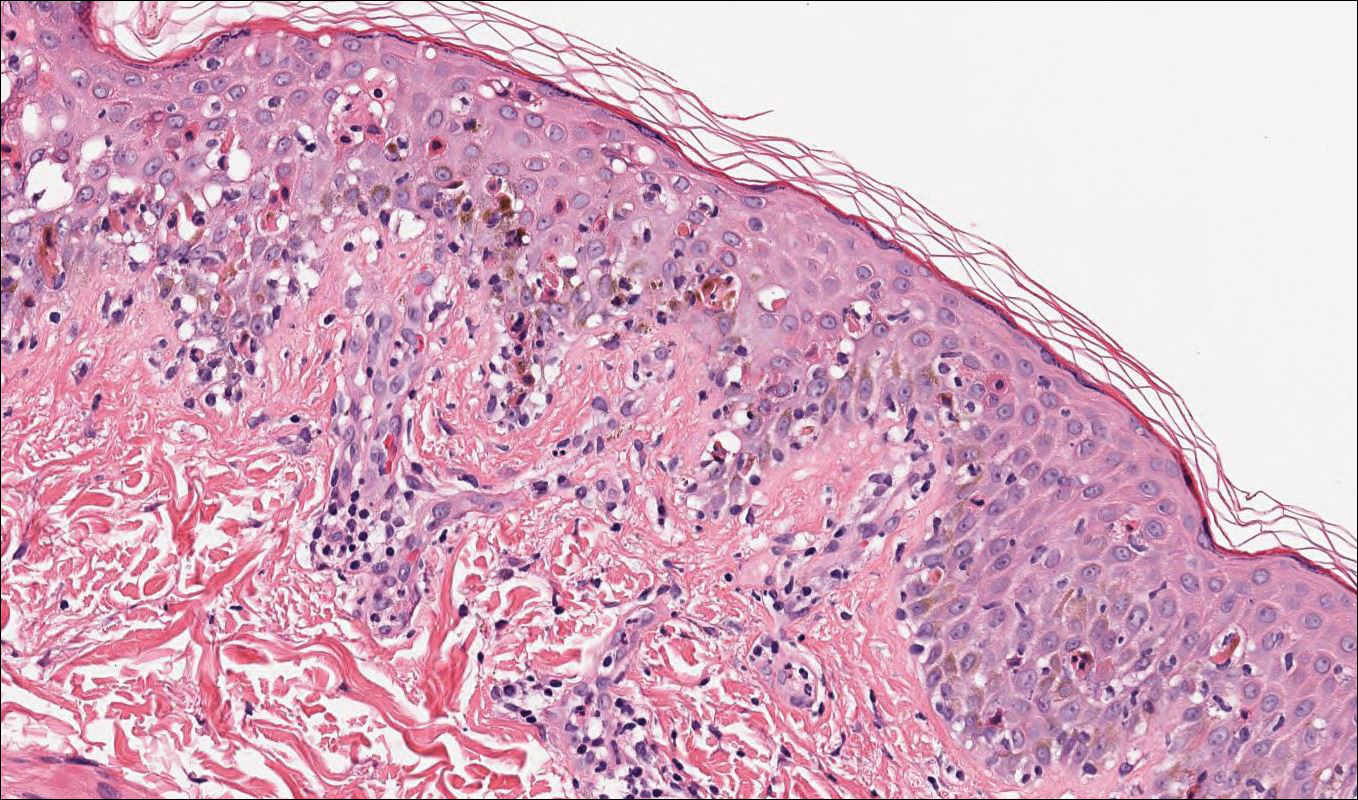
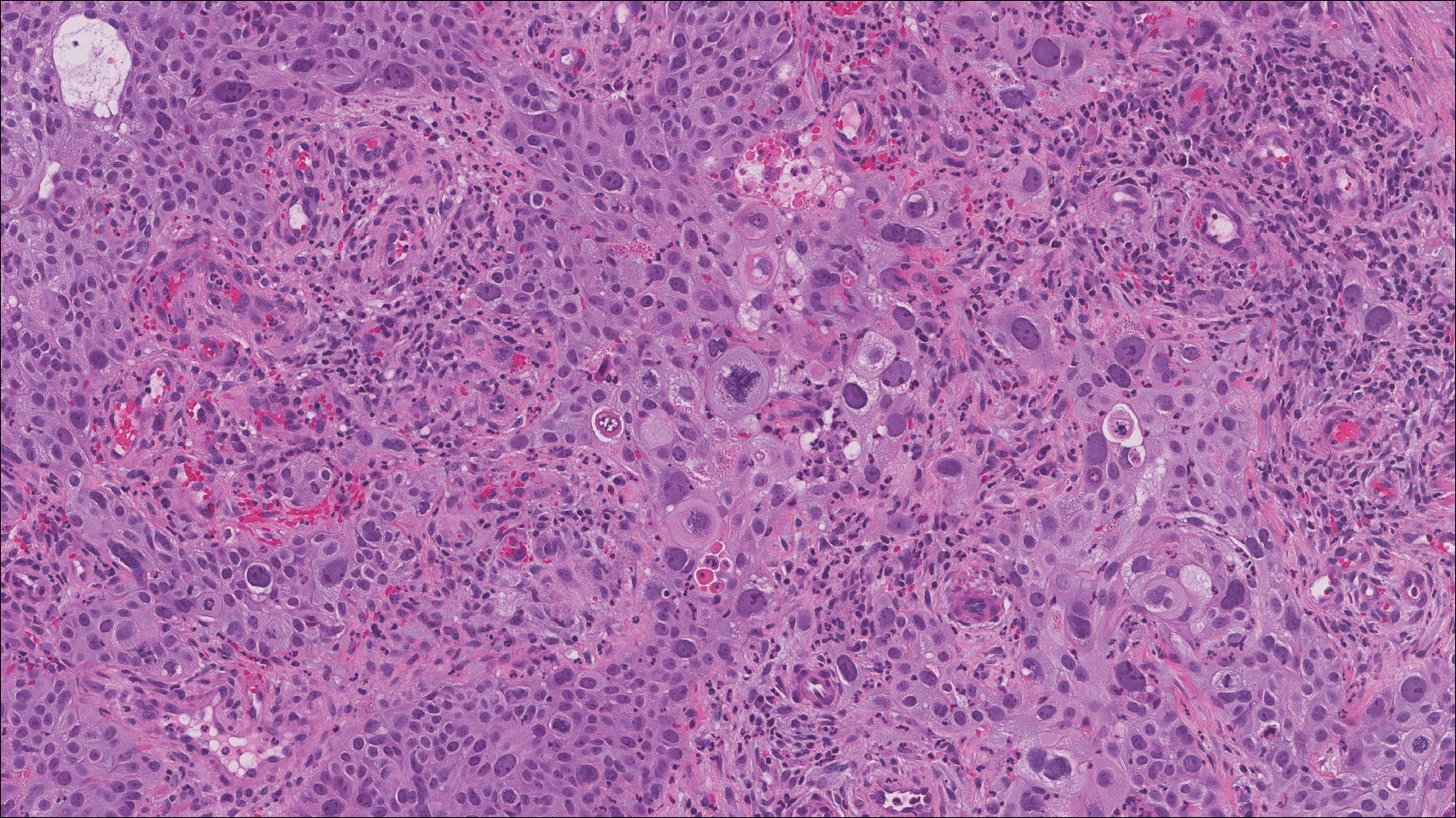
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6

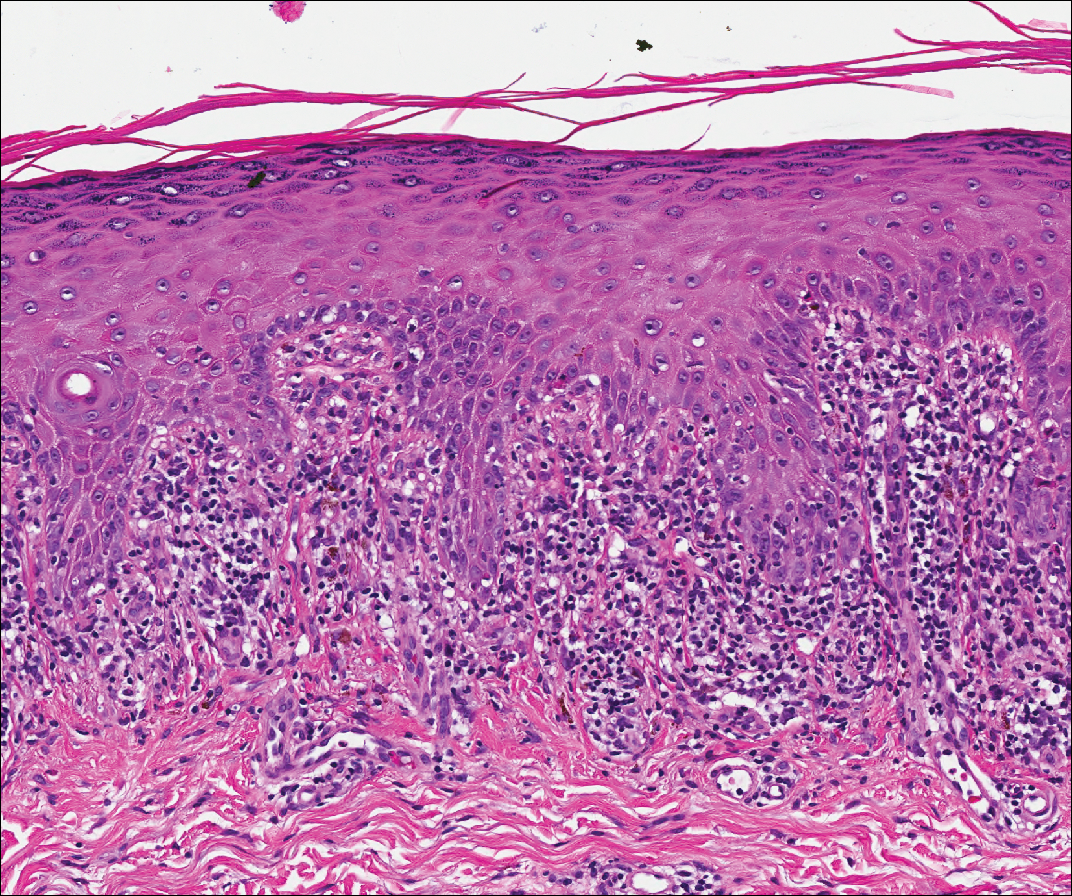
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
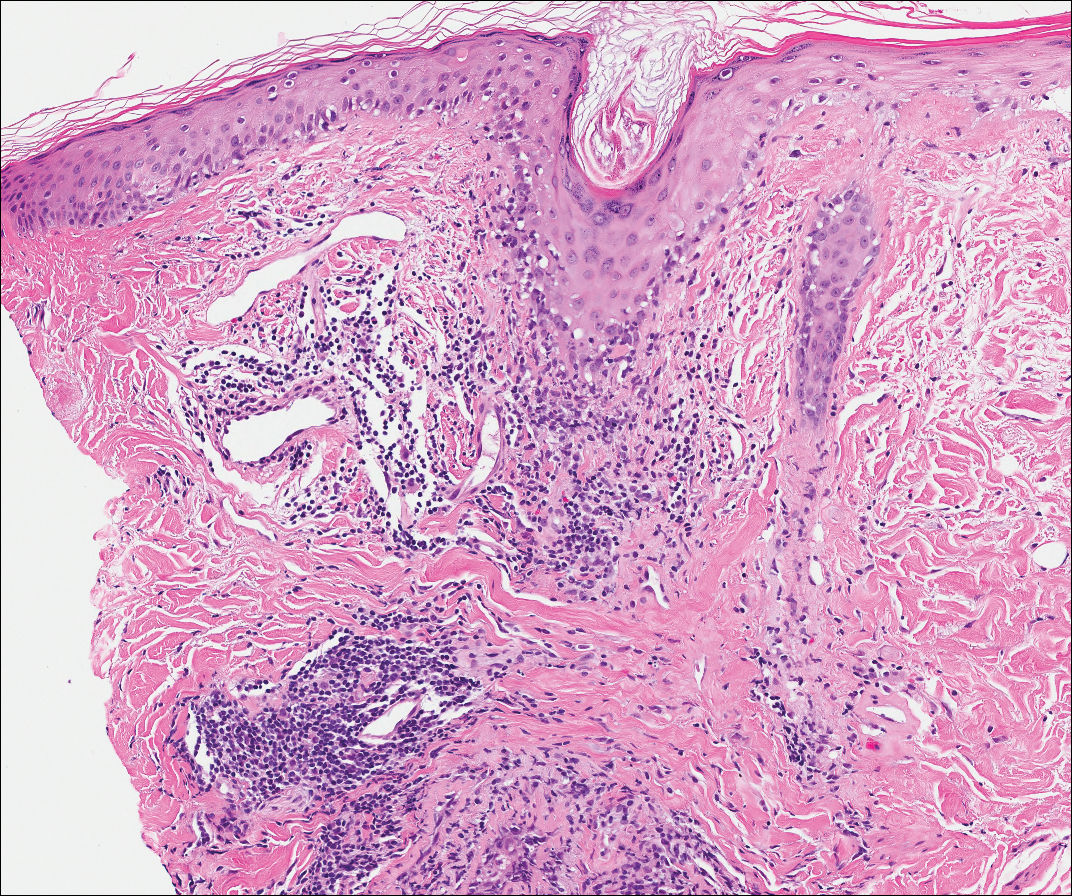
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
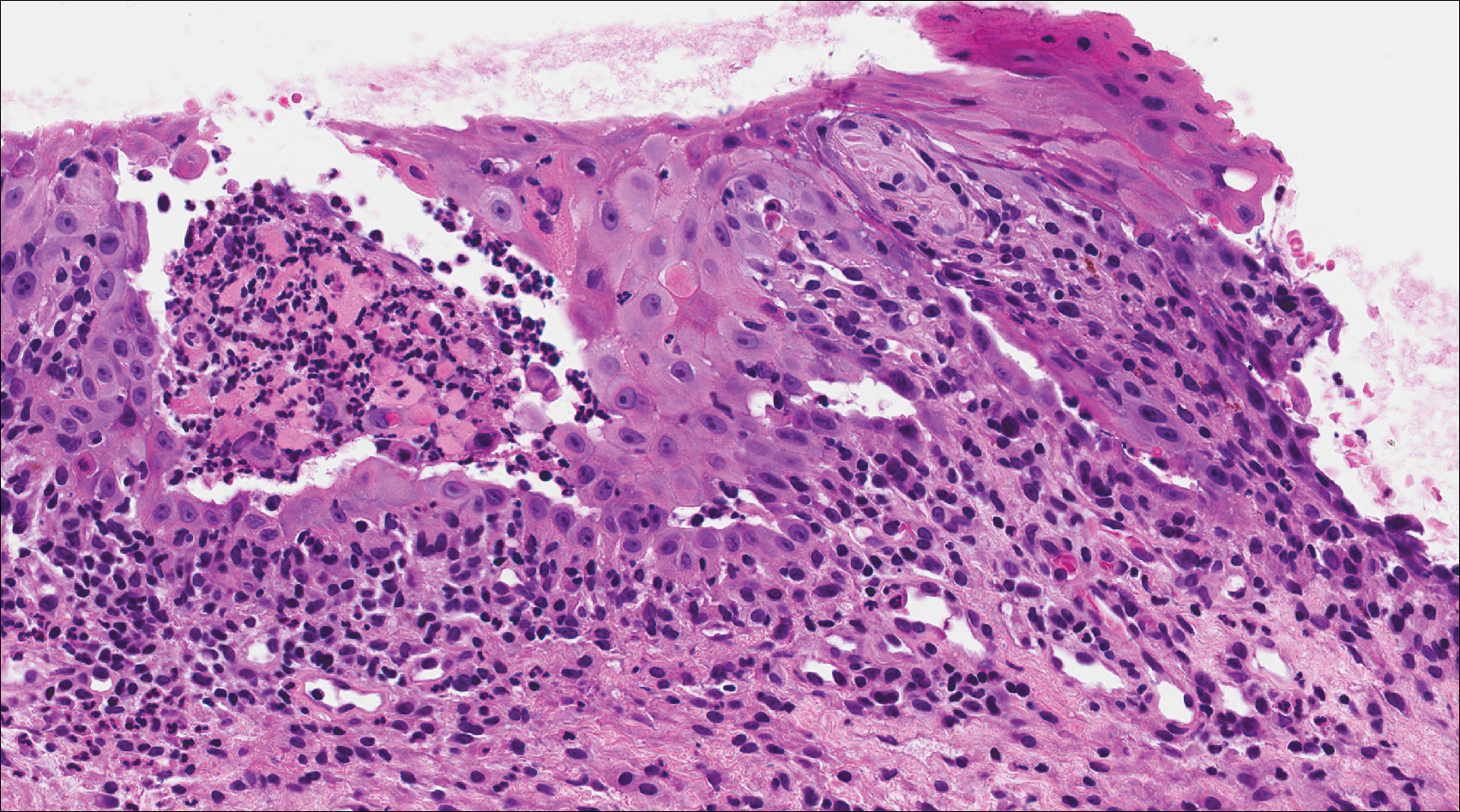
A 41-year-old woman presented with painful ulcers on the oral mucosa of 2 months' duration that were unresponsive to treatment with acyclovir. She had been diagnosed with a pelvic tumor a few weeks prior to the development of the mouth ulcers. Direct immunofluorescence of the perilesional mucosa showed positive IgG and complement C3 with an intercellular distribution. A biopsy of an oral lesion was performed.
Perianal Condyloma Acuminatum-like Plaque
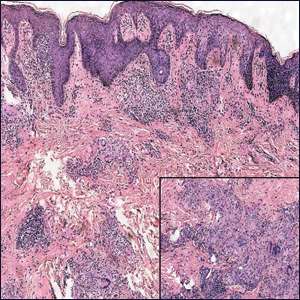
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
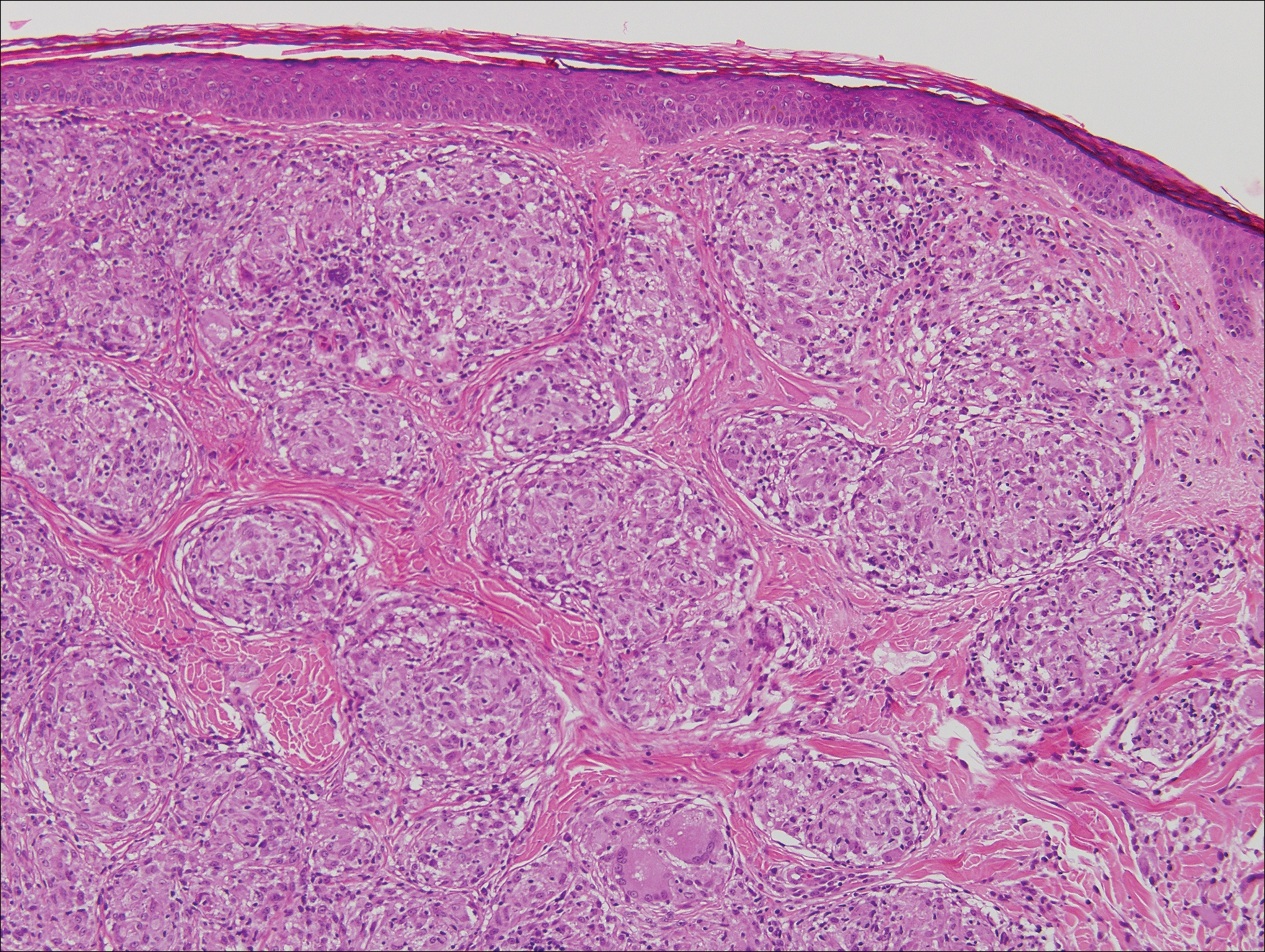
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
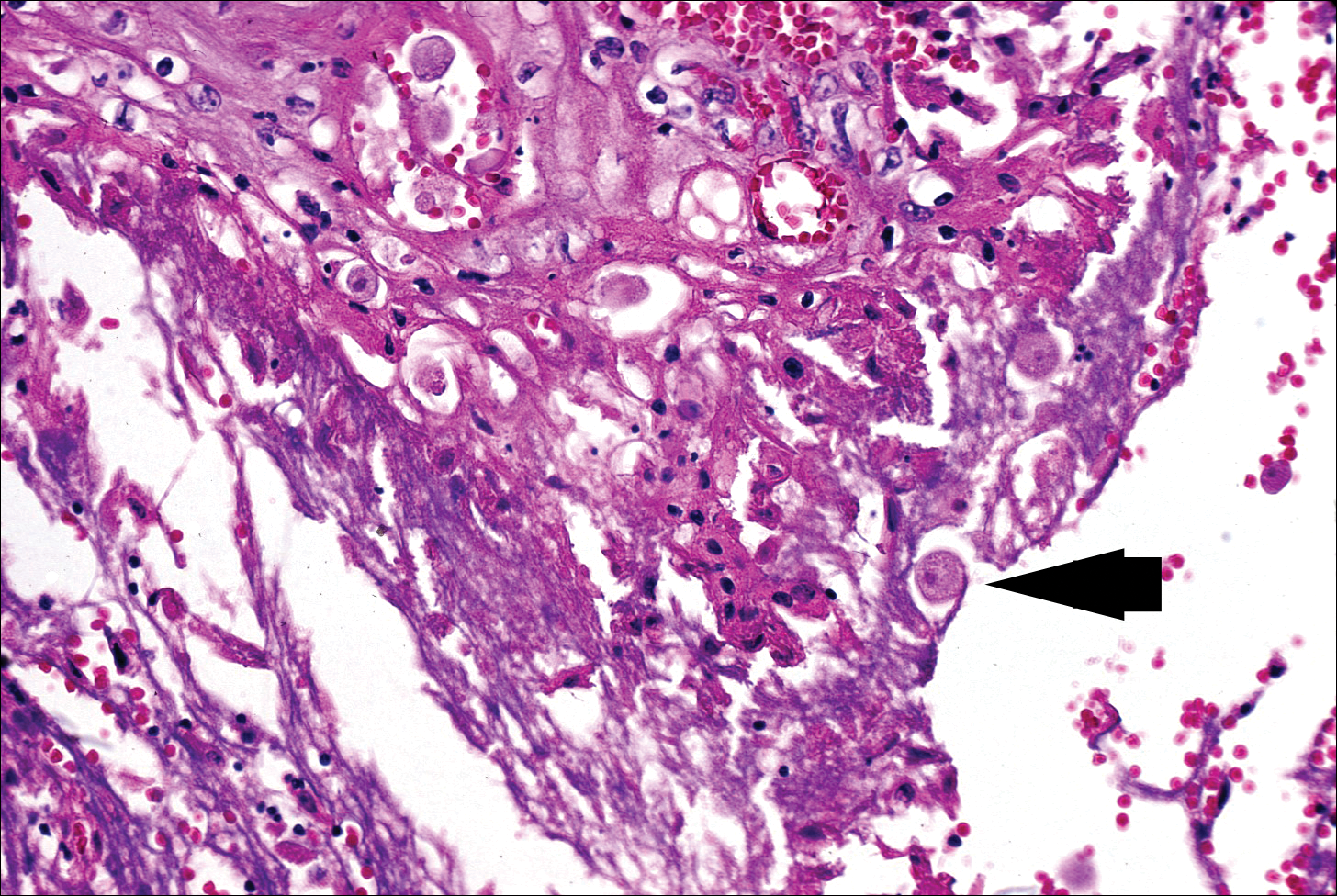
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
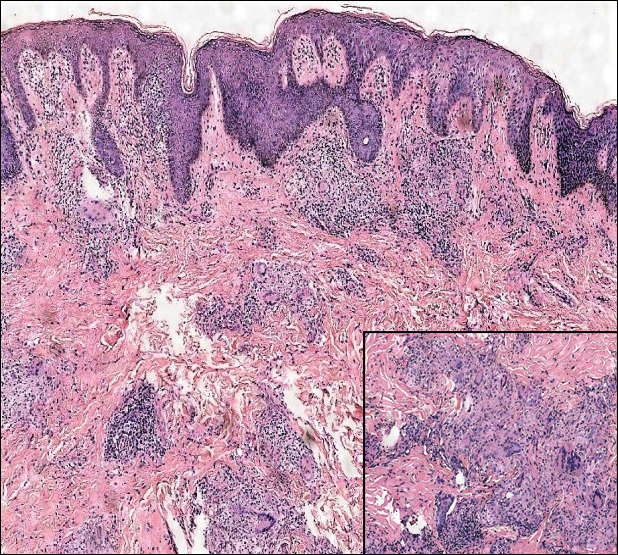
A 19-year-old man presented with a perianal condyloma acuminatum-like plaque of 2 years' duration and a 6-month history of diarrhea.
Asymptomatic Subcutaneous Nodule on the Cheek
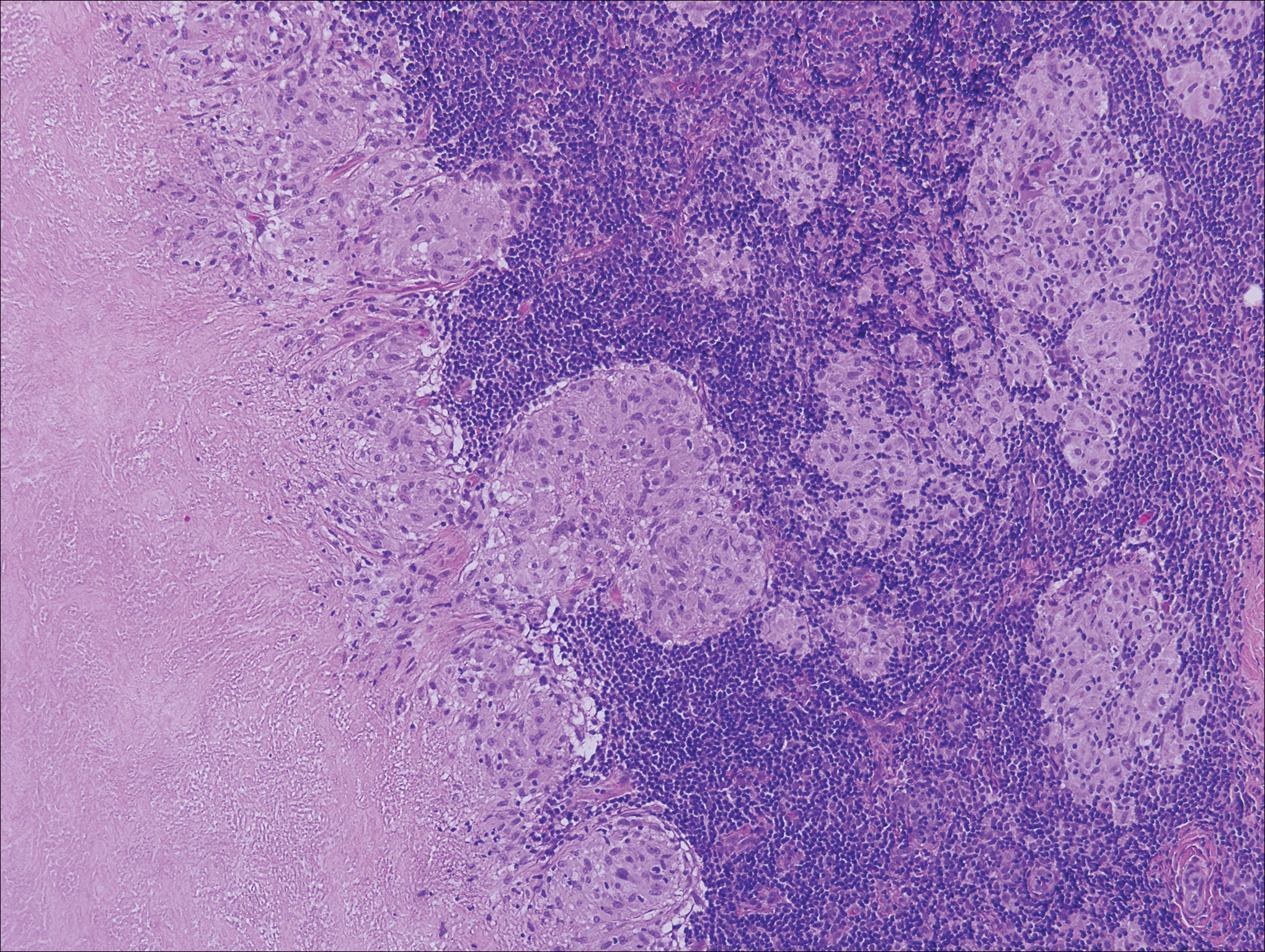
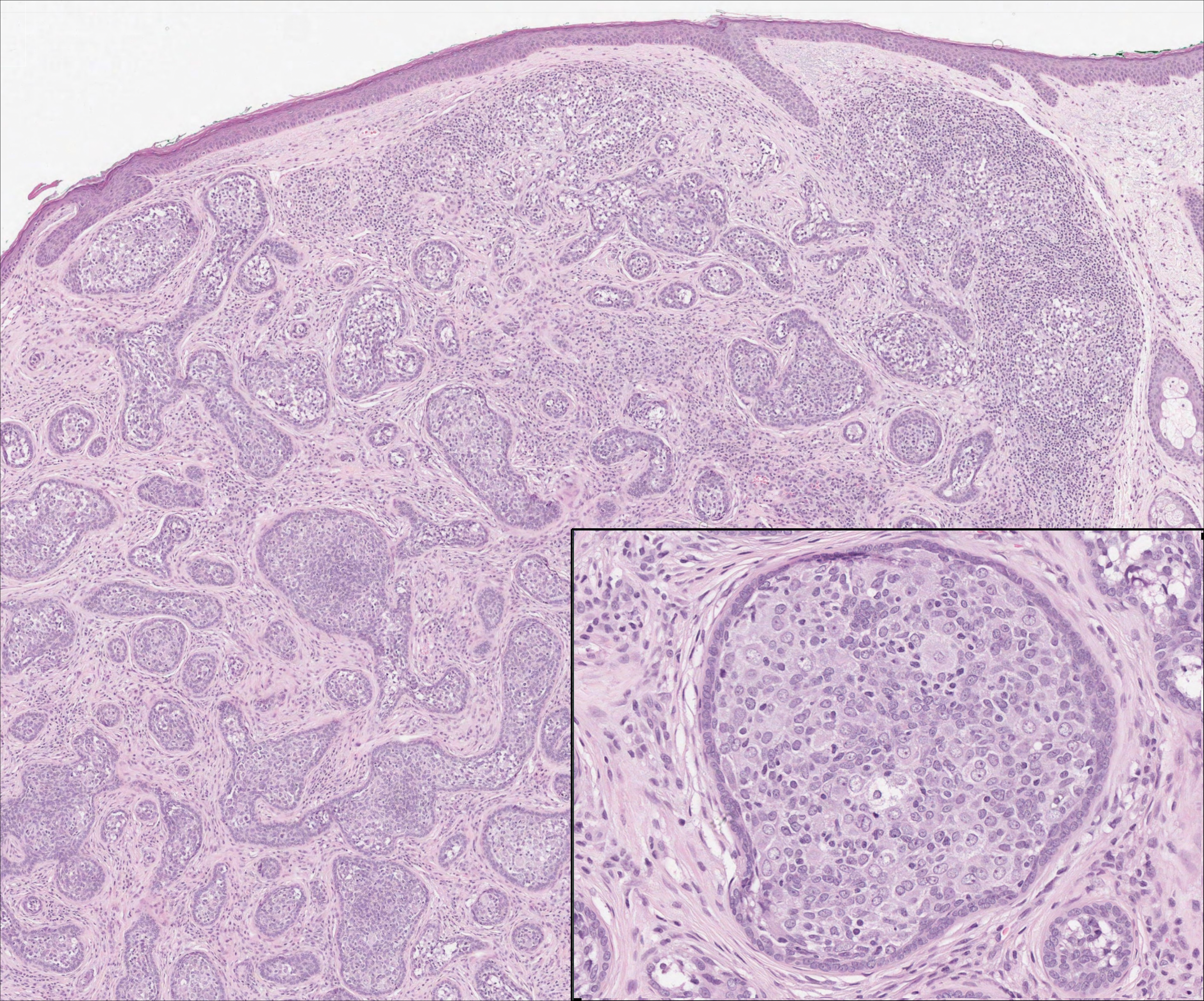
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
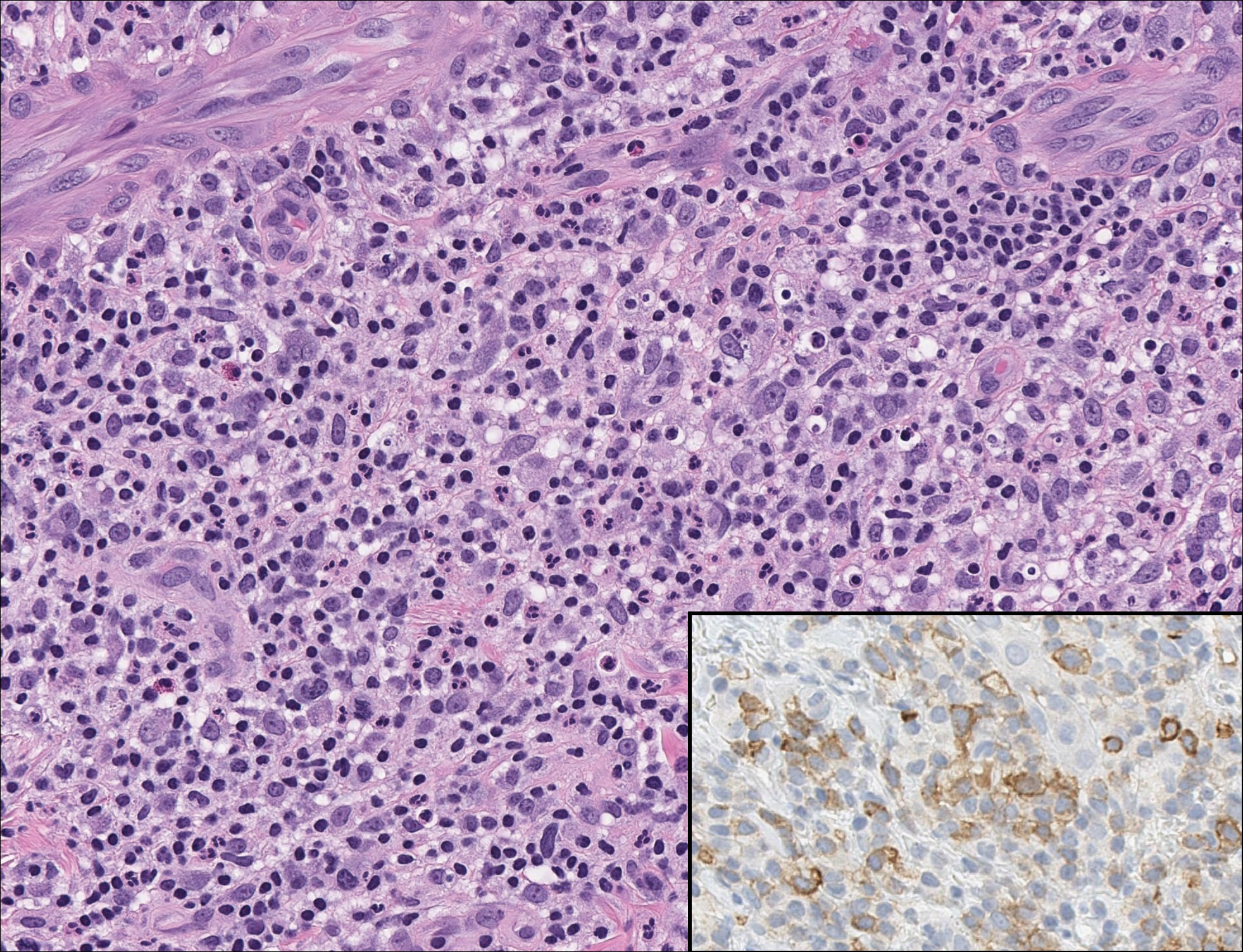
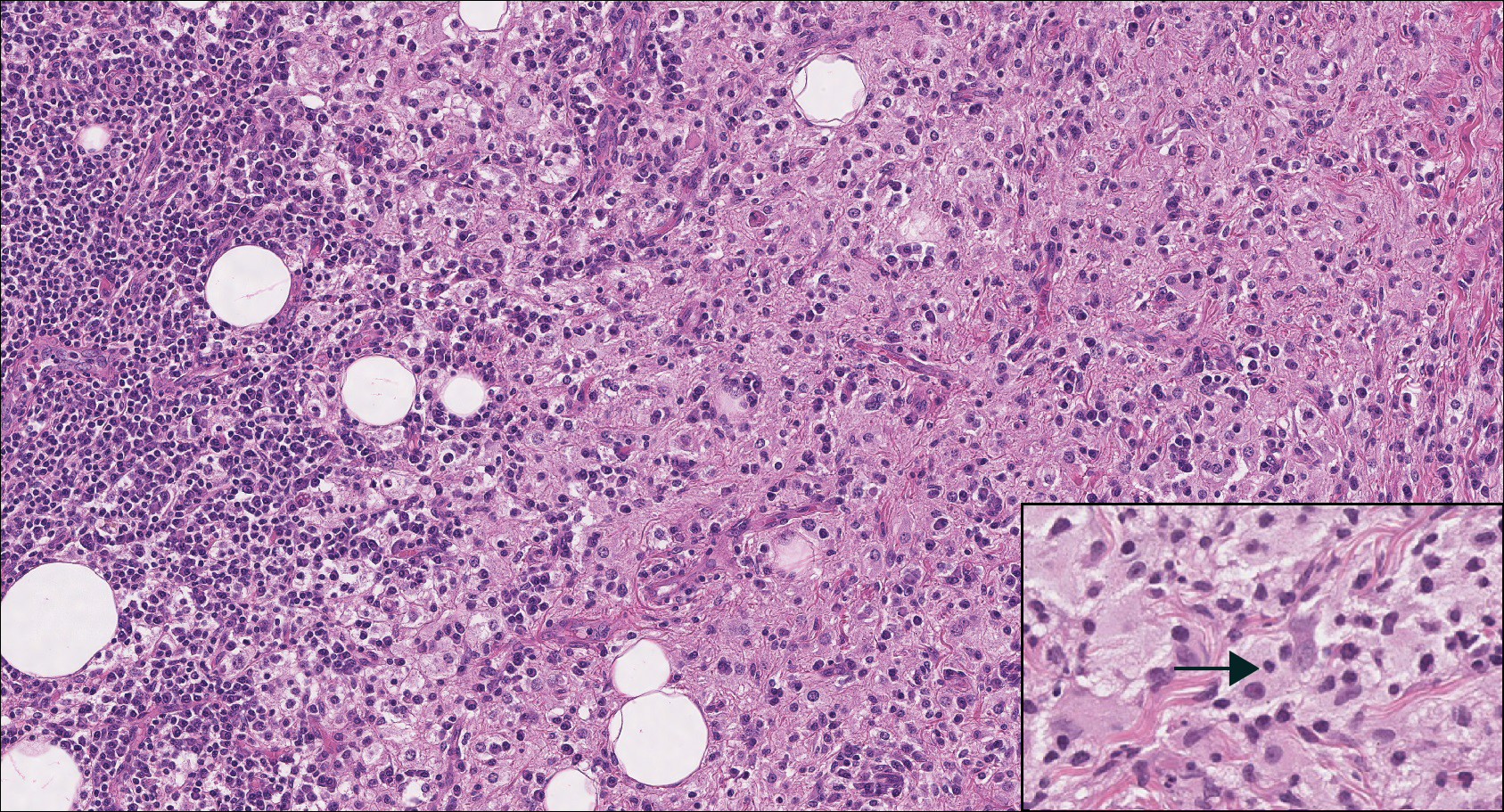
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
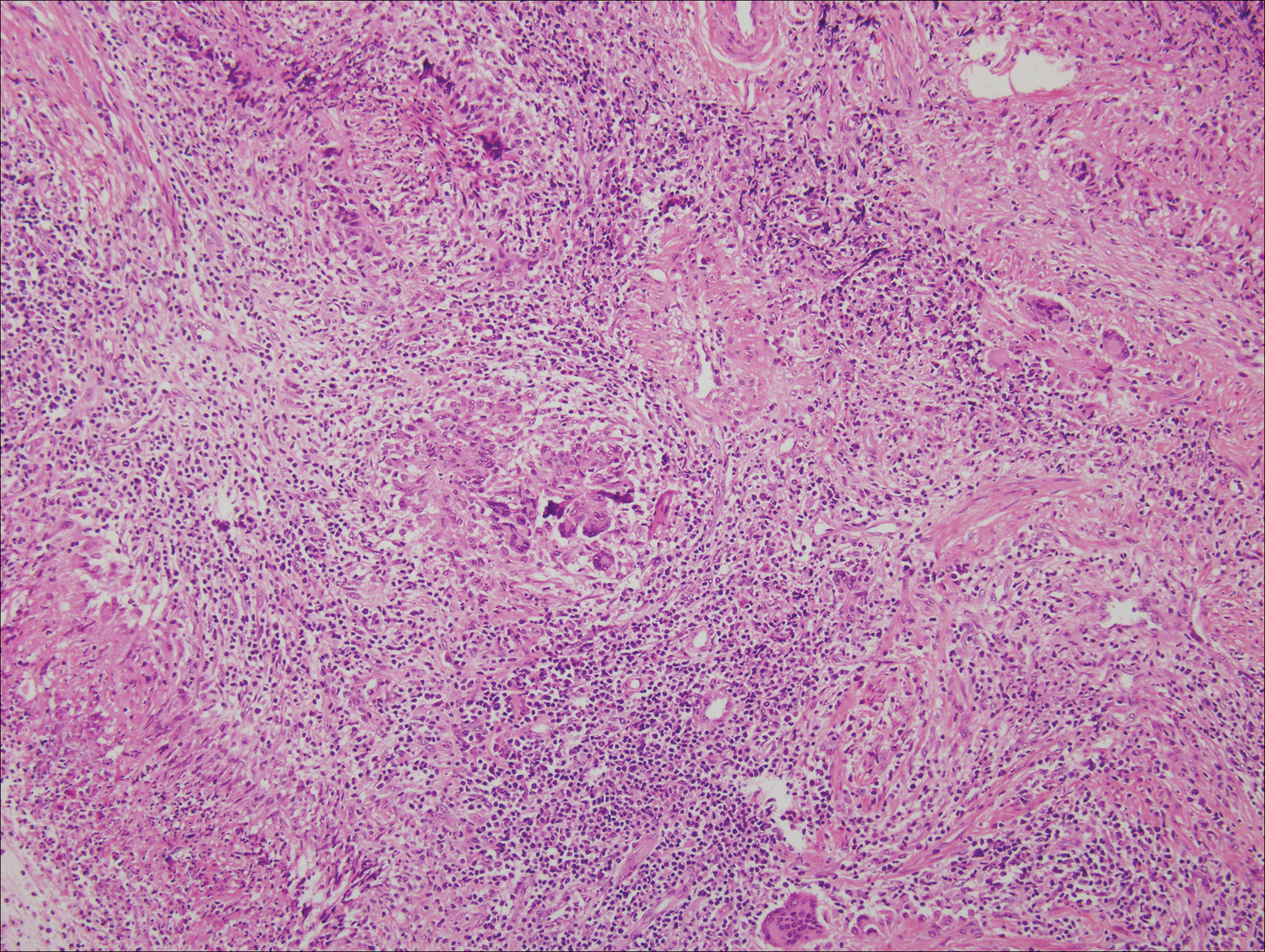
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
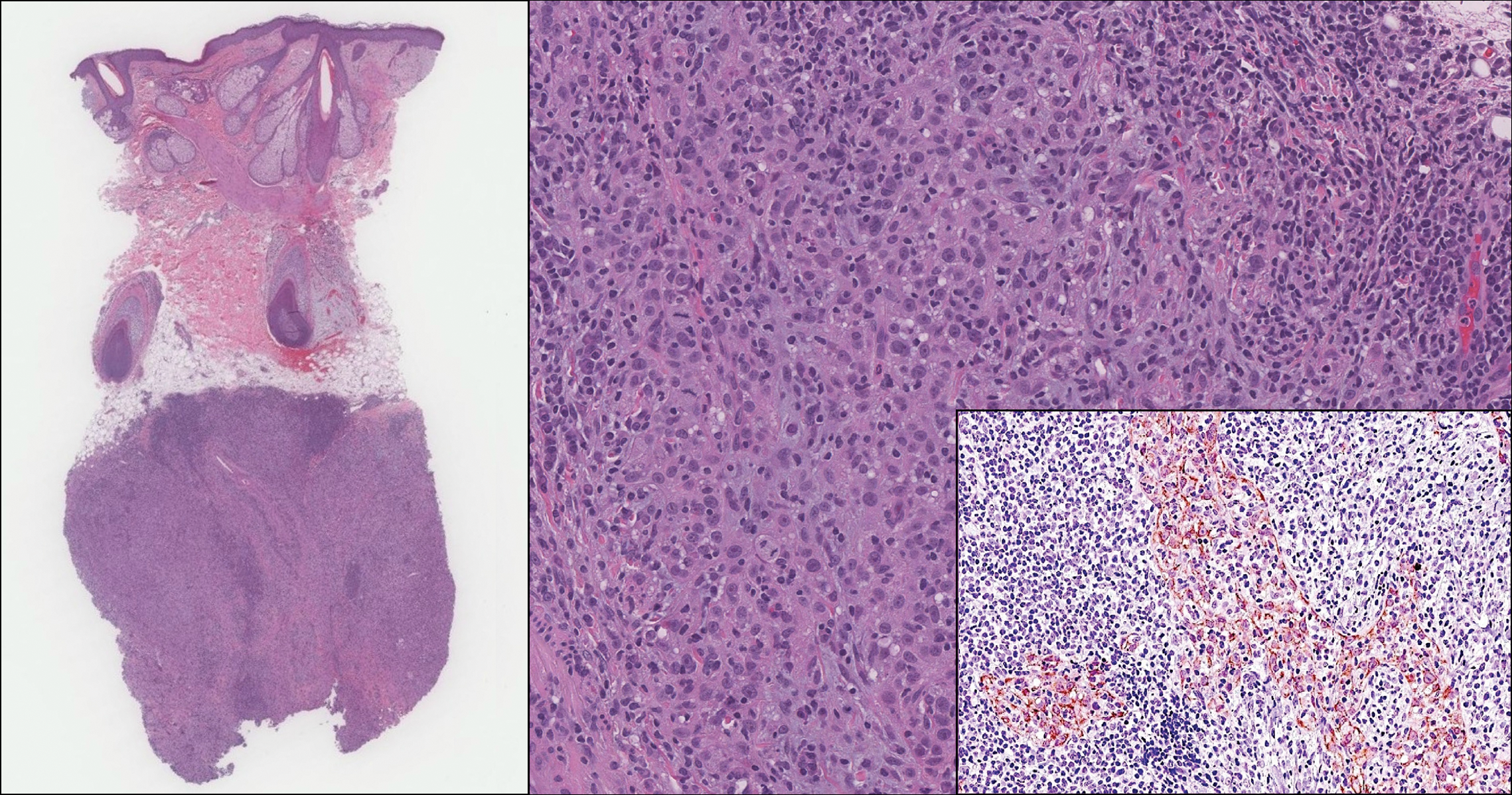
An 81-year-old man with history of melanoma and nonmelanoma skin cancer presented with a subcutaneous nodule on the left cheek of 3 months' duration. The lesion was reportedly asymptomatic and measured 2.6×2.9 cm. A punch biopsy of the lesion was obtained for histopathologic evaluation.
Growing Nodule on the Arm
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
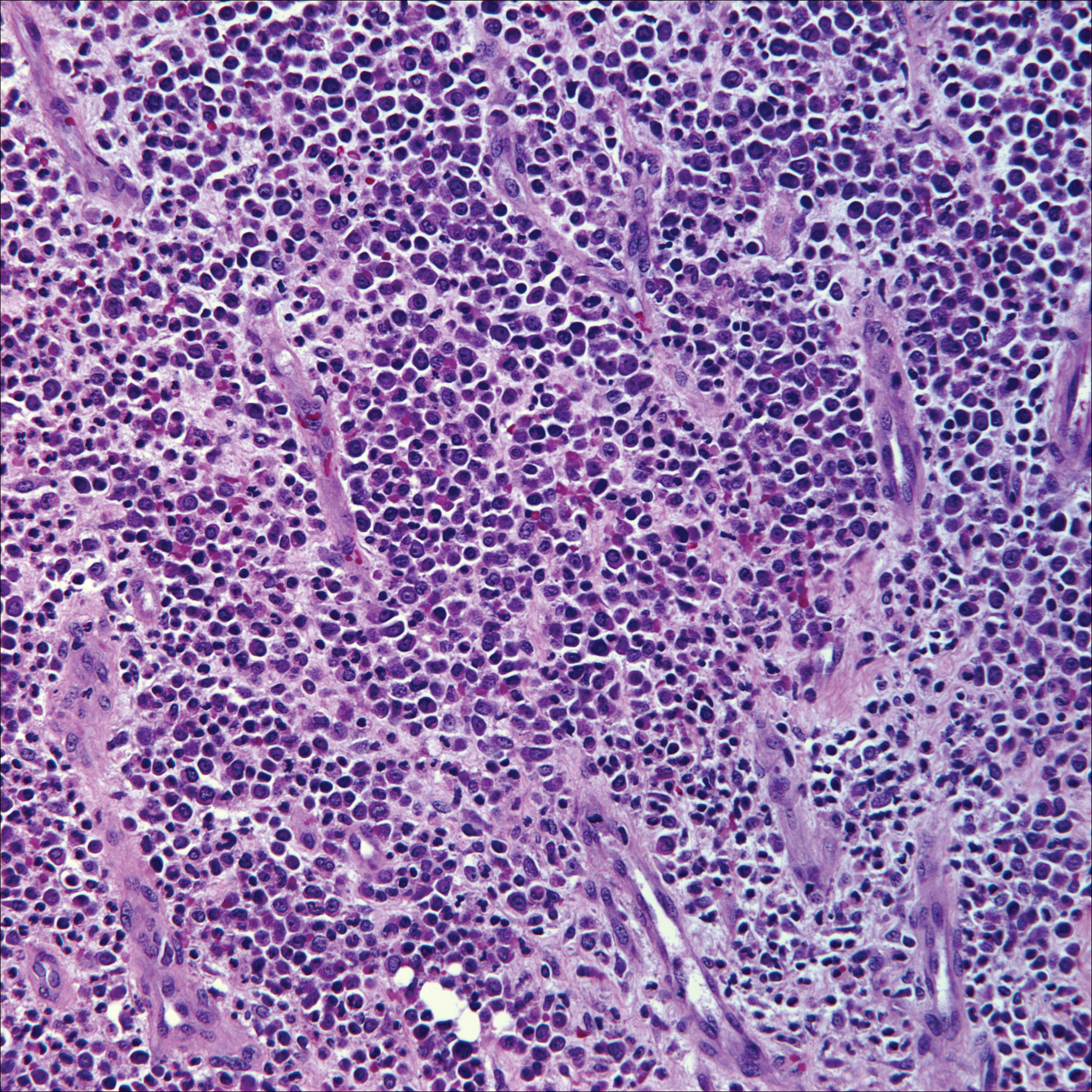
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
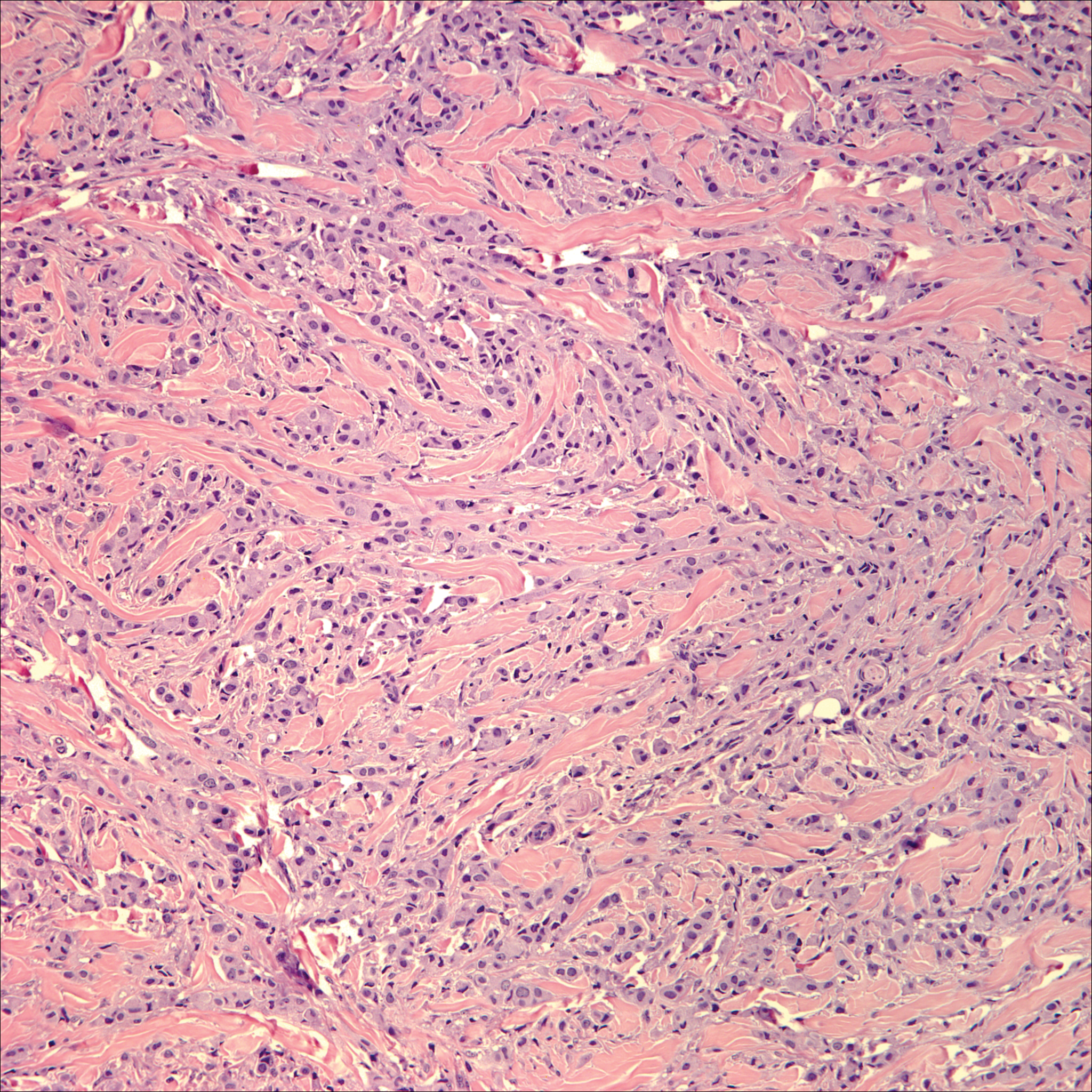
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
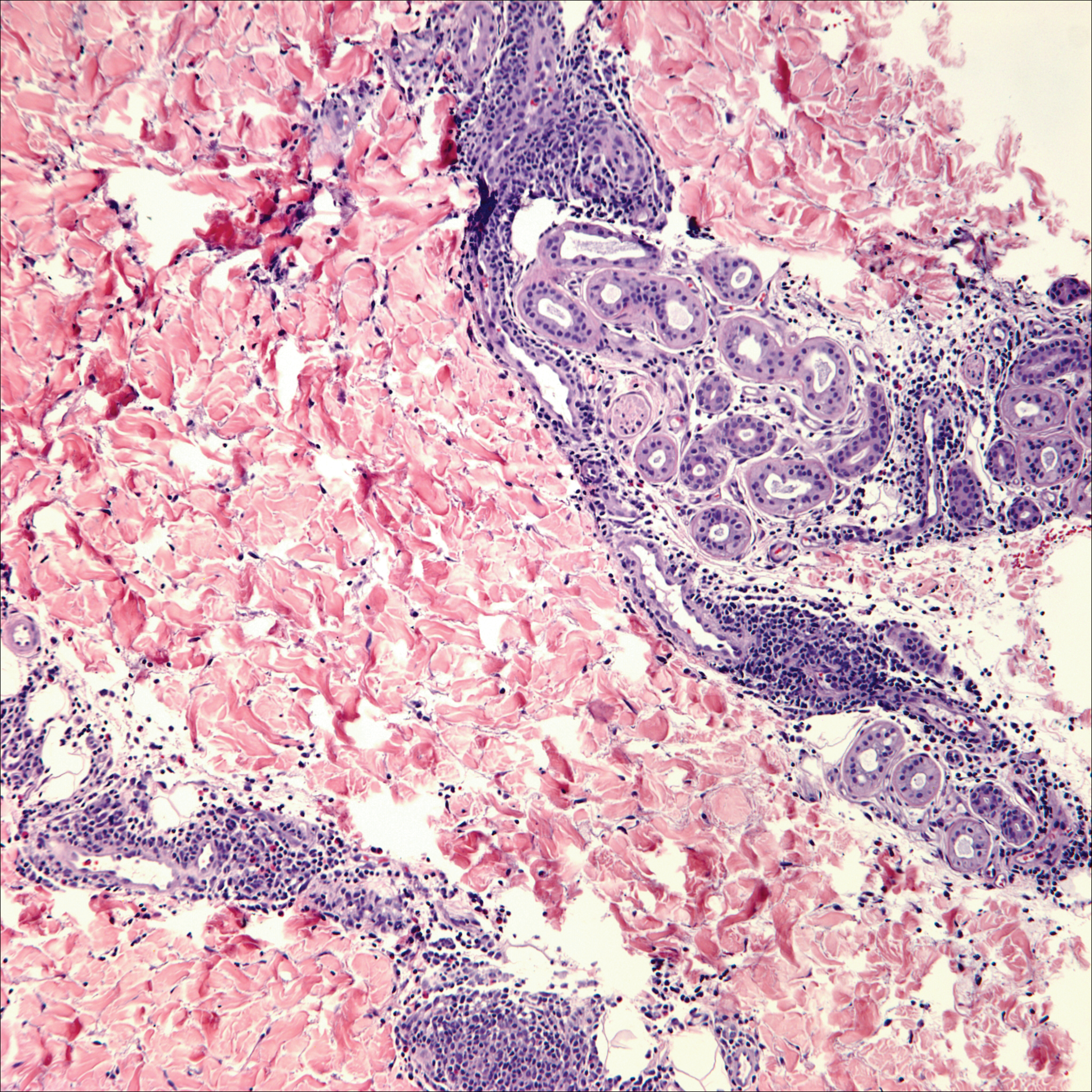
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
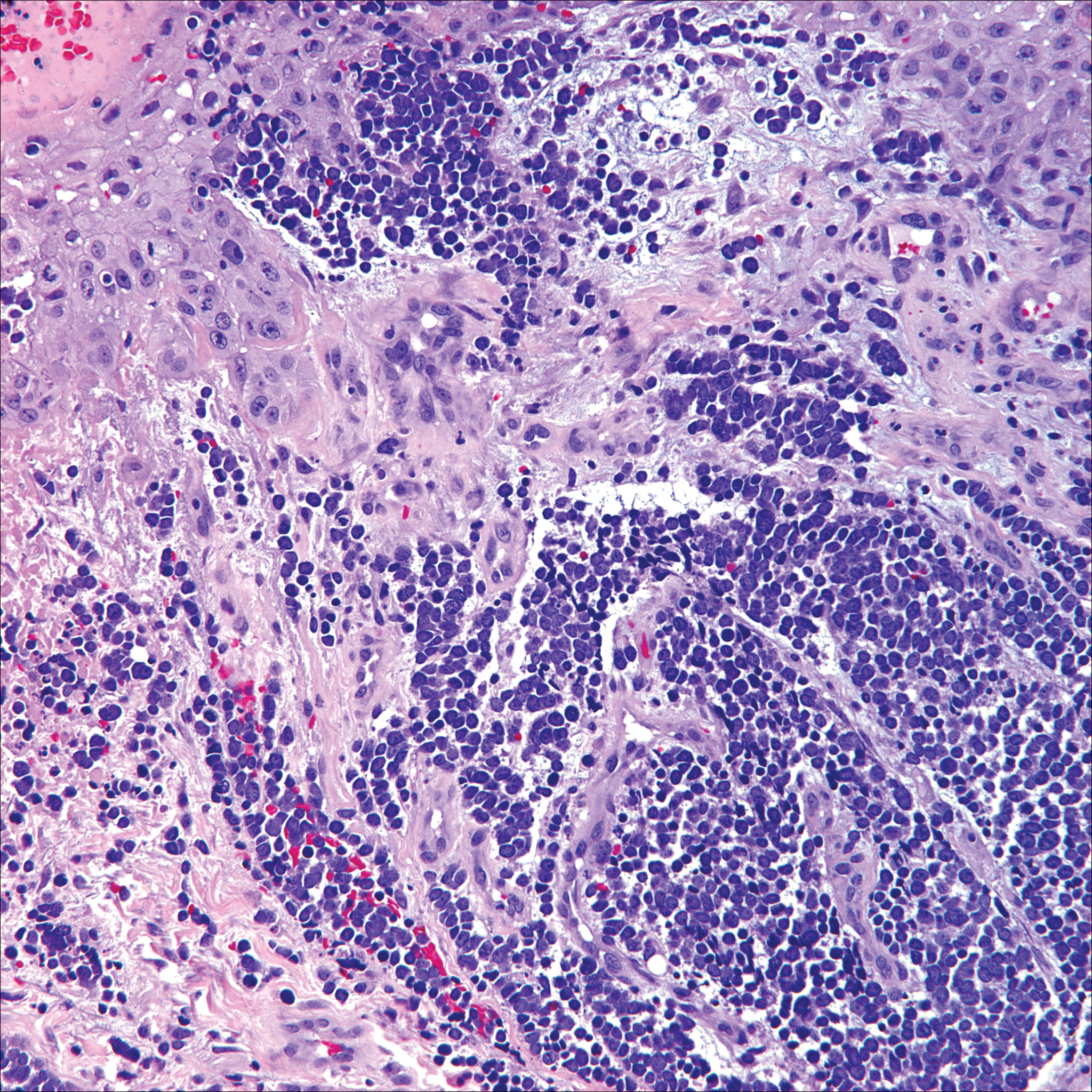
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
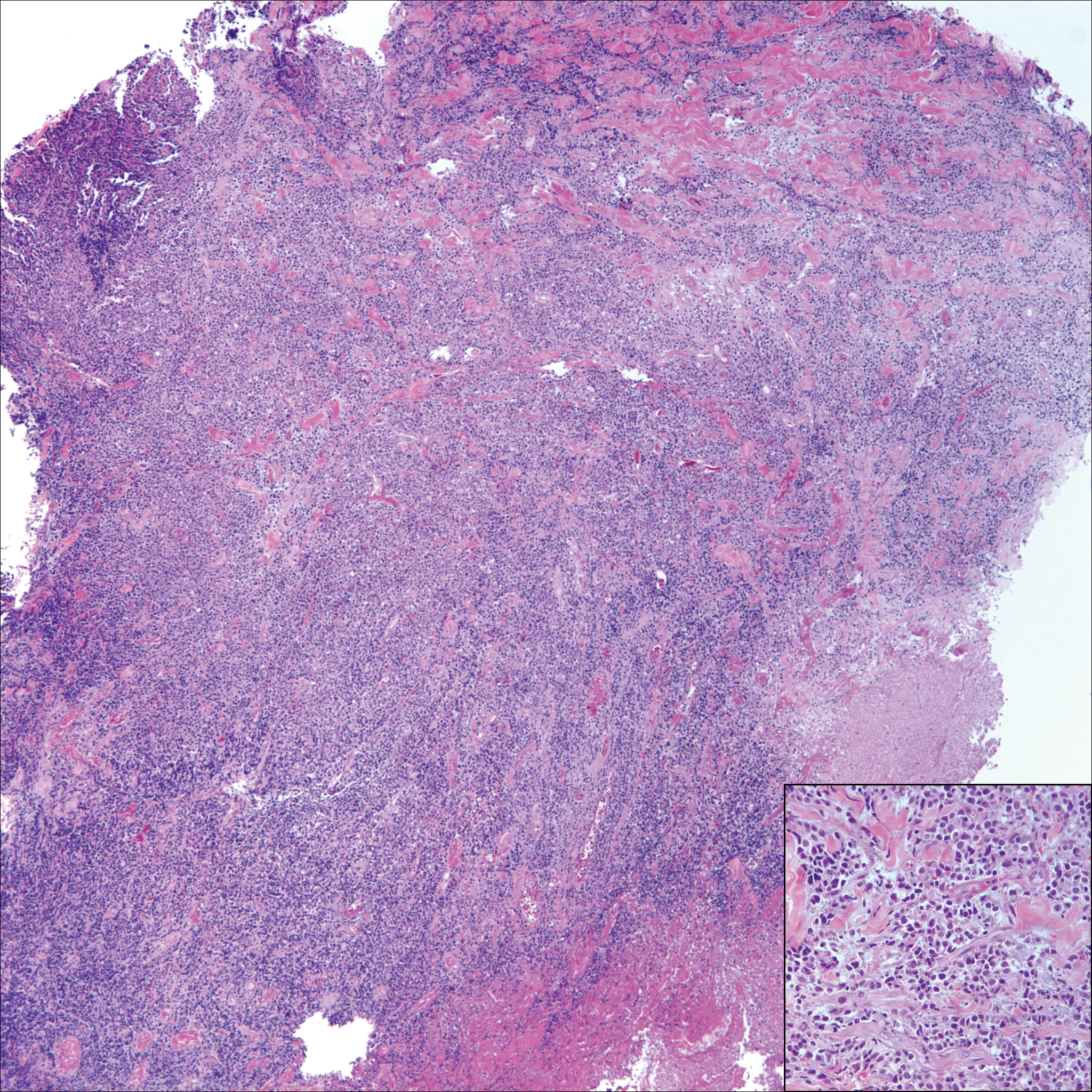
A 65-year-old white woman presented with an asymptomatic bump on the left upper arm of 4 months' duration that arose following a cat scratch. Physical examination was notable for a 35×30-mm, firm, ulcerated, exophytic nodule. Histologic examination demonstrated an ulcerated epidermis and a dense basophilic infiltrate occupying the entire dermis and extending to the subcutaneous tissue. Higher magnification (inset) demonstrated a pleomorphic population of medium- to large-sized discohesive round cells containing variable amounts of slightly eosinophilic cytoplasm, irregular nuclear contours, and prominent nucleoli. Scattered atypical mitotic figures were identified. CD30, CD4, leukocyte common antigen, and Ki-67 immunostains were strongly and diffusely positive. Notable negative stains included anaplastic lymphoma kinase, synaptophysin, epithelial membrane antigen, neuron-specific enolase, CD20, and S-100.
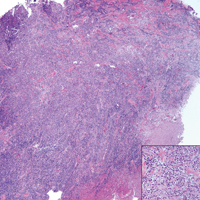
Purpuric Macule of the Right Axilla
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
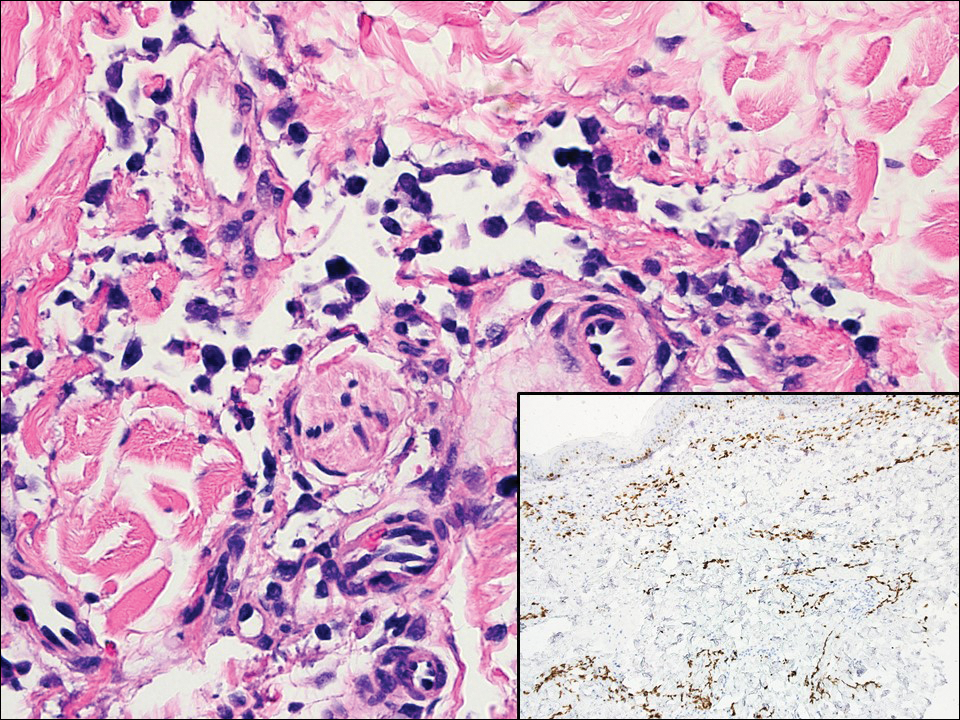
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
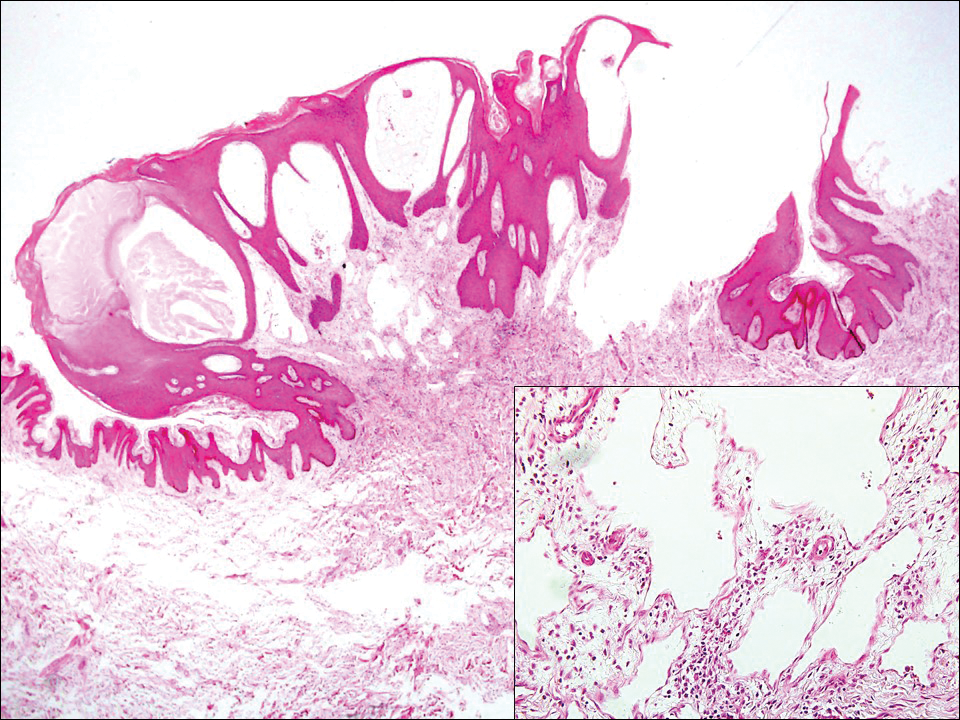
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
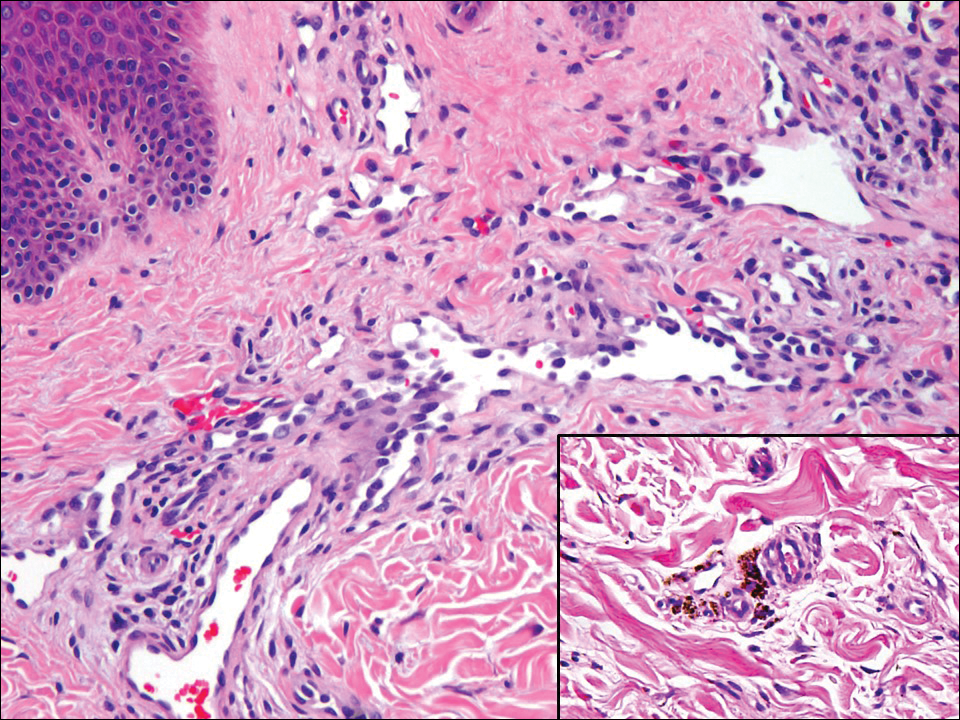
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
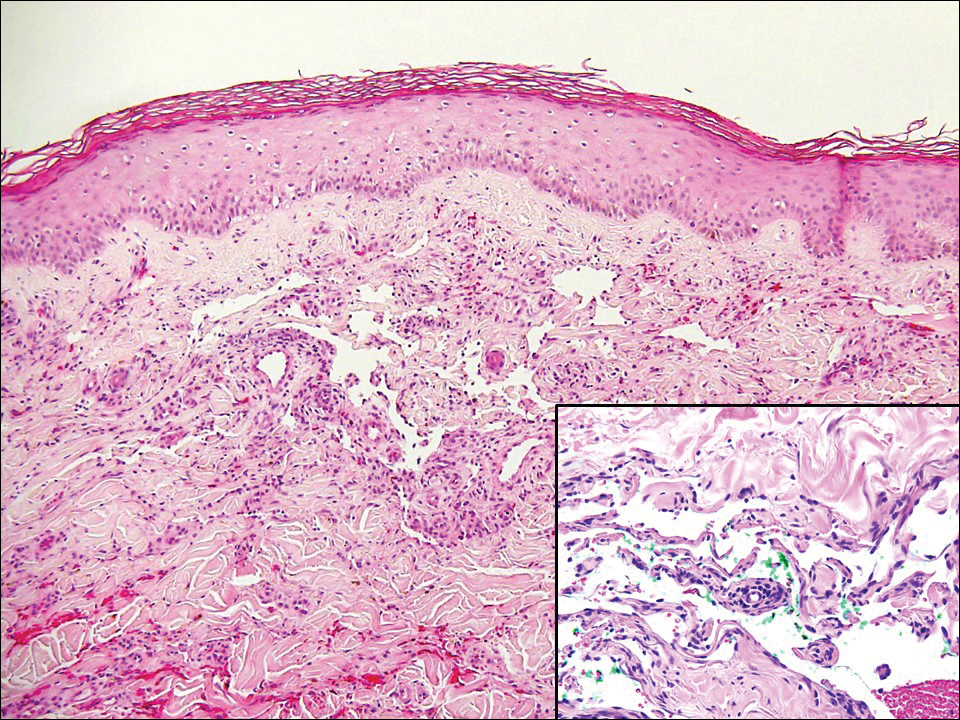
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
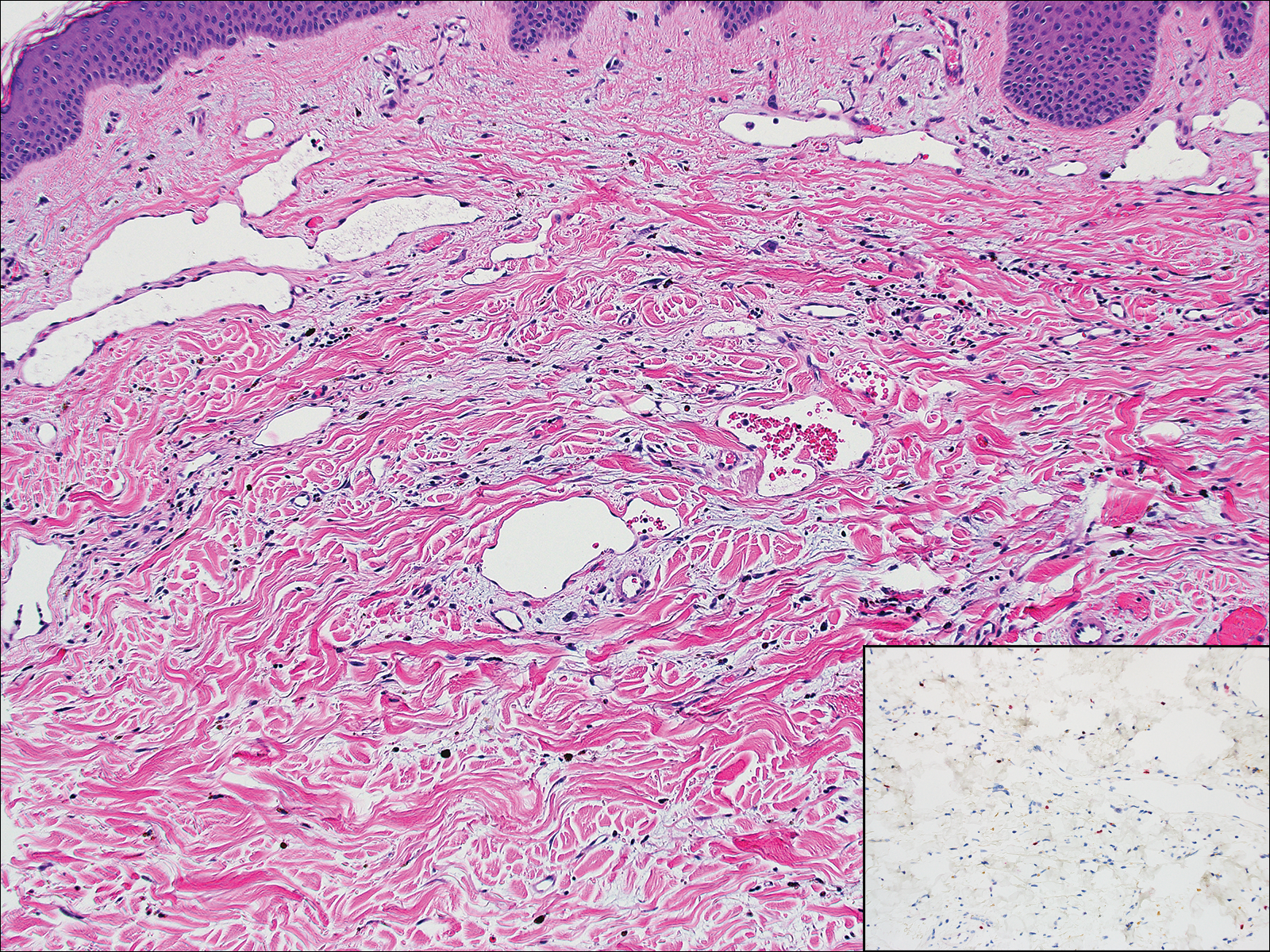
A 67-year-old woman presented with a lesion on the medial aspect of the right axilla of 2 weeks' duration. The patient had a history of cancer of the right breast treated with a mastectomy and adjuvant radiation. She denied pain, bleeding, pruritus, or rapid growth, as well as any changes in medication or recent trauma. Physical examination revealed a 5-mm purpuric macule of the right axilla. A punch biopsy was performed. Amplification for the C-MYC gene was negative by fluorescence in situ hybridization.
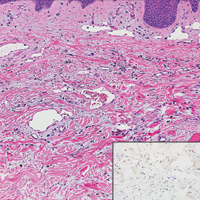
Indurated Plaque on the Eyebrow
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).

Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
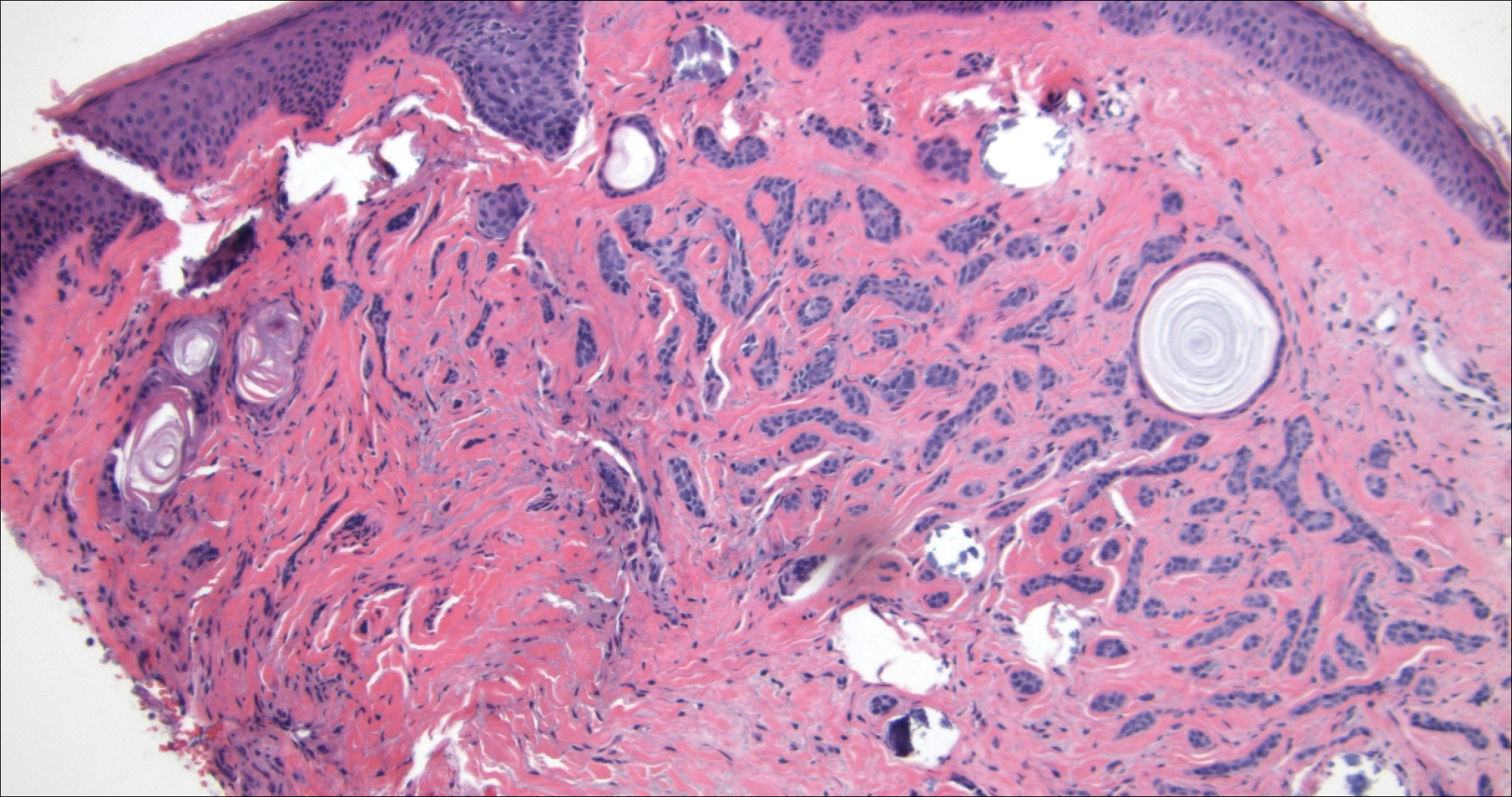
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
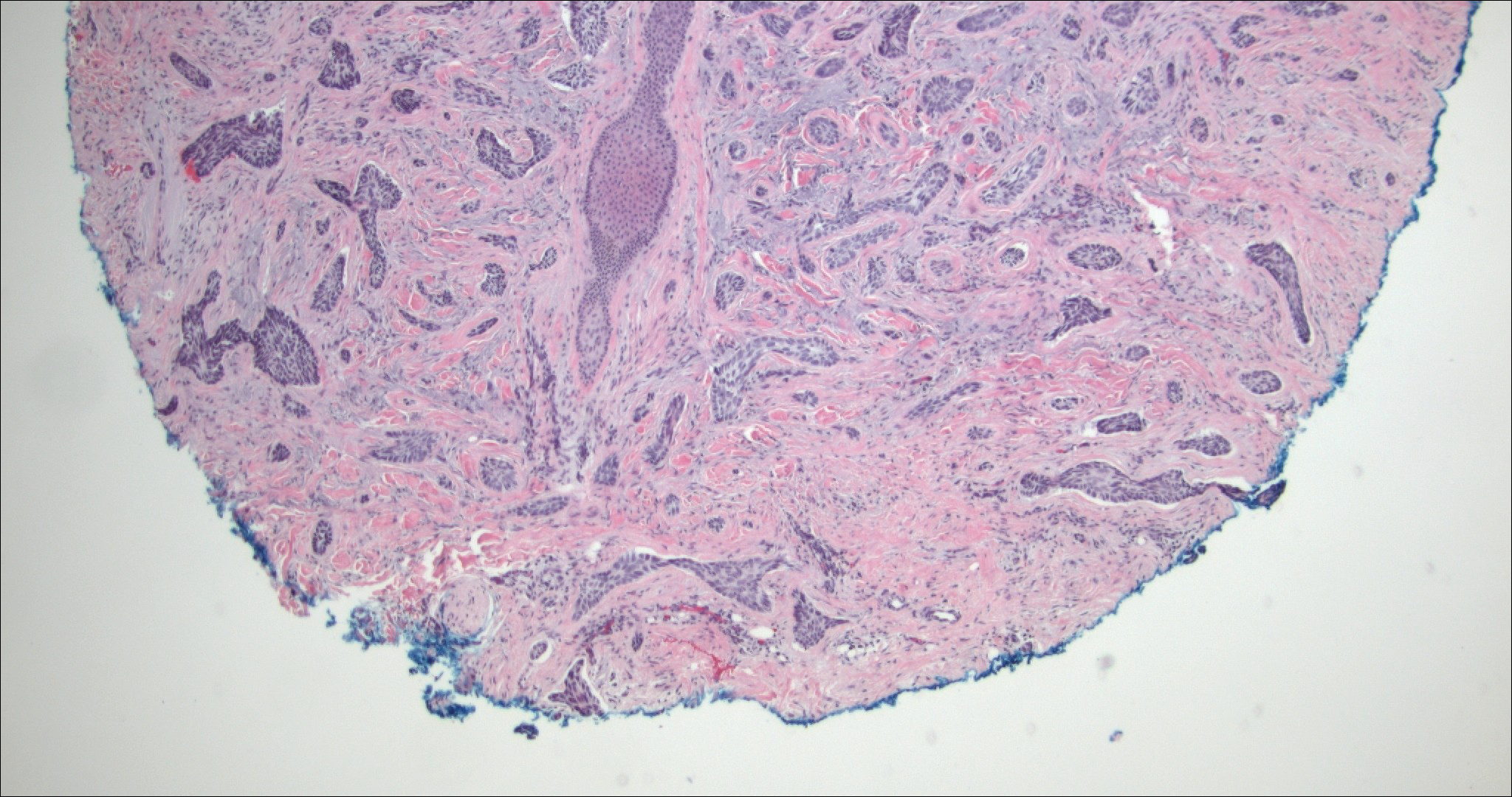
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
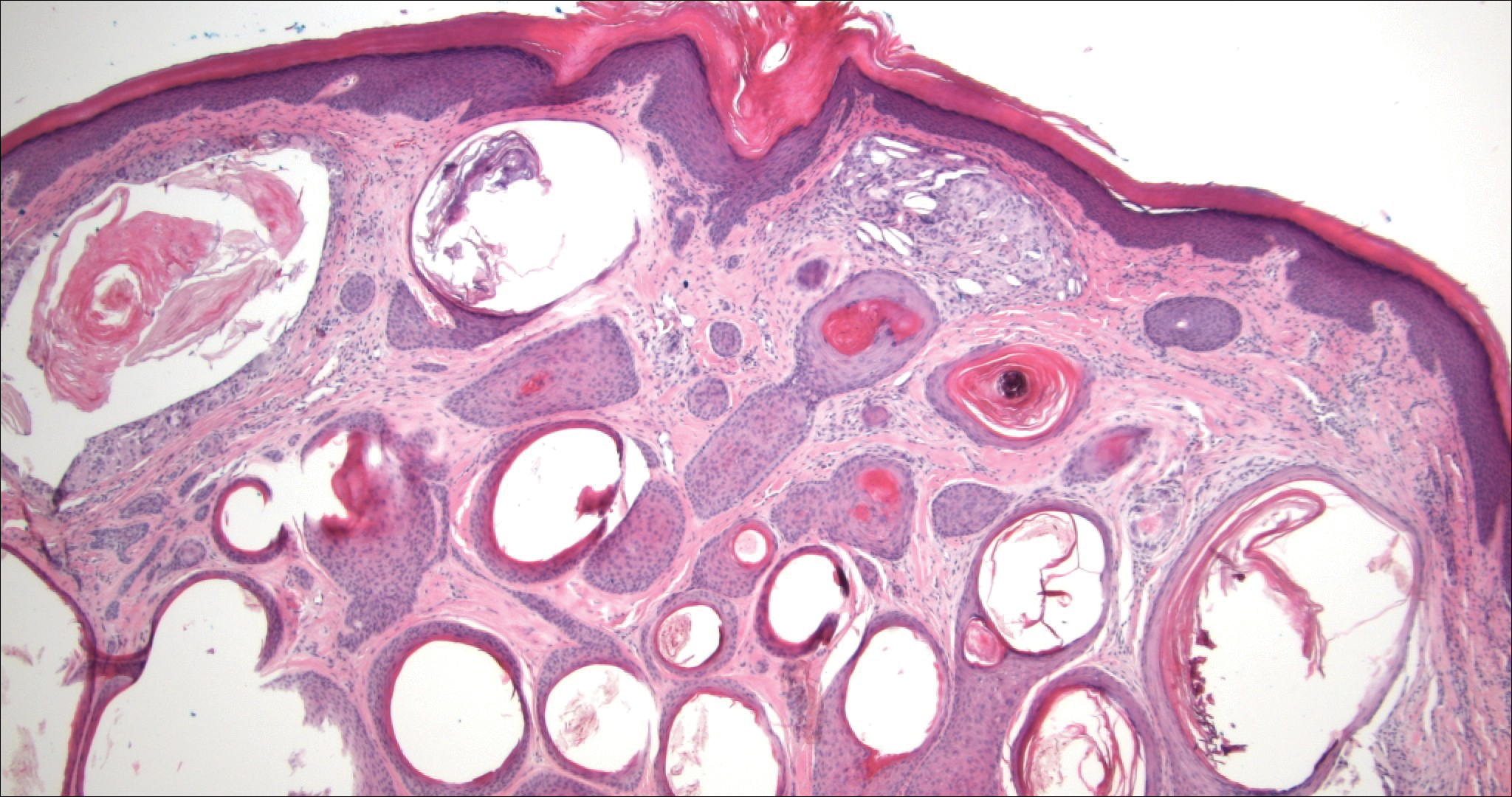
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
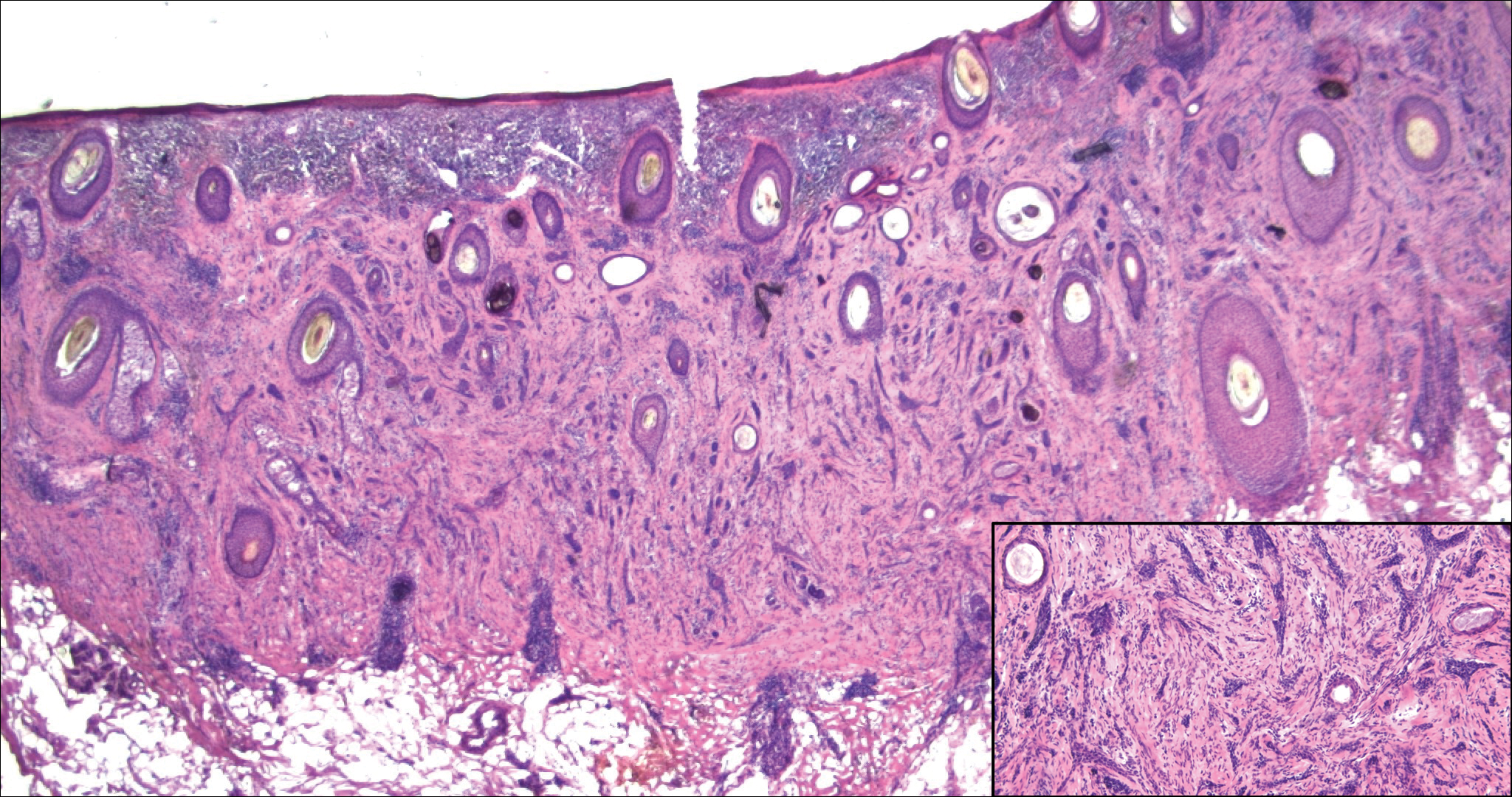
A 52-year-old woman presented with an indurated plaque on the right lateral eyebrow that had been slowly enlarging over the last 4 months.
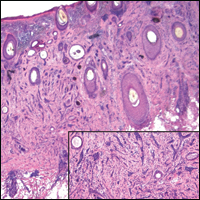
Solitary Tender Nodule on the Back
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
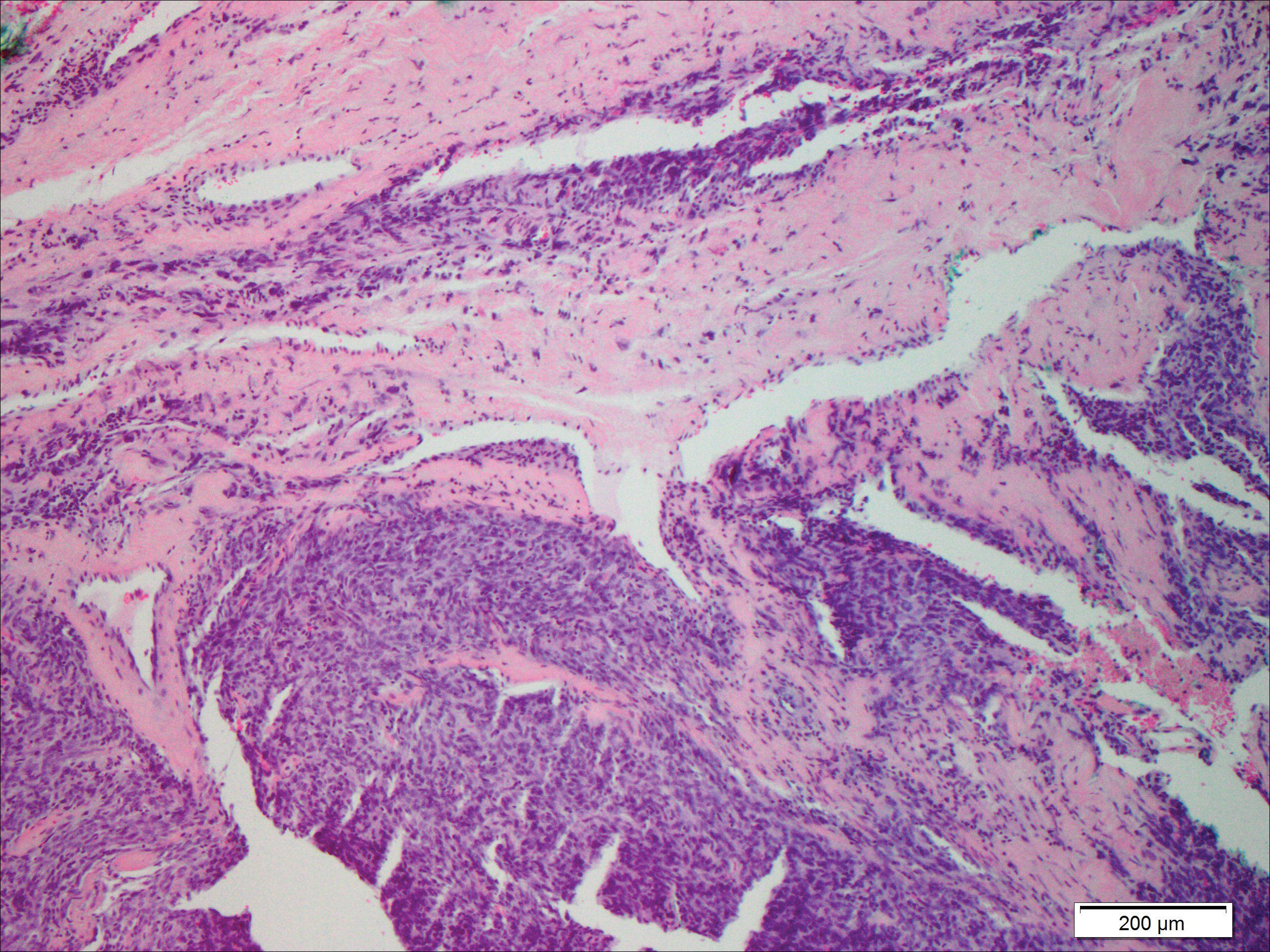
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.

Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
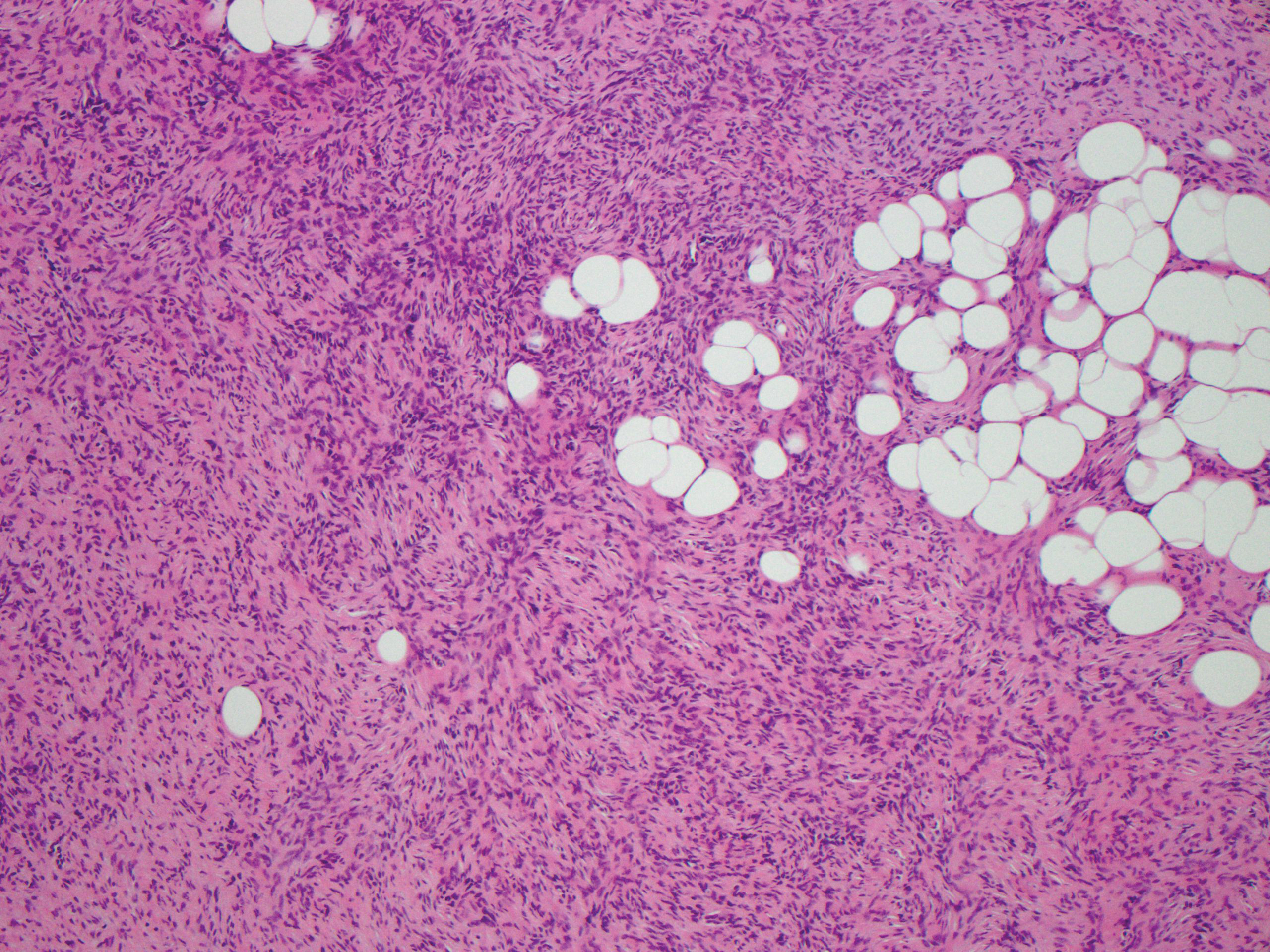
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
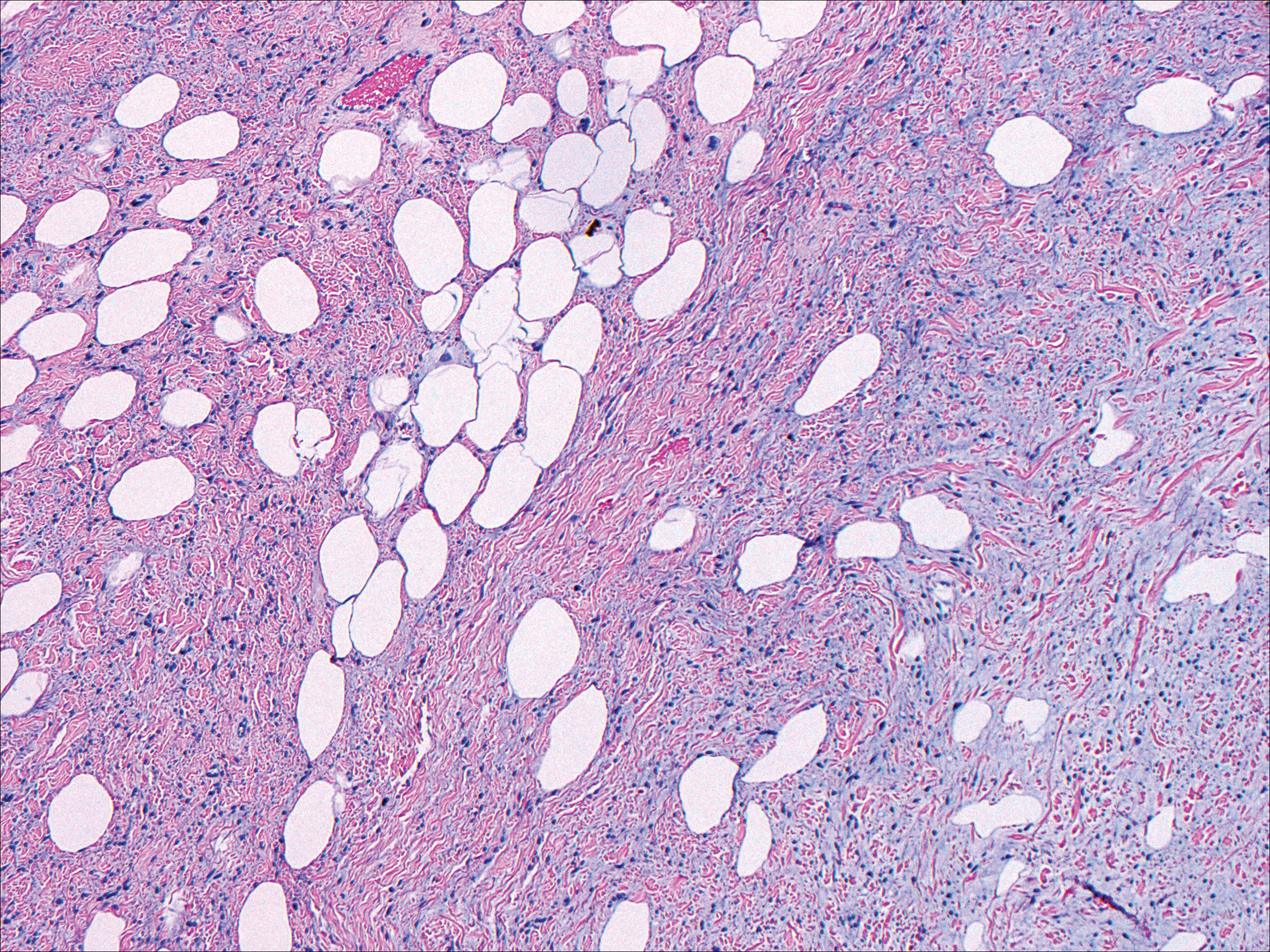
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
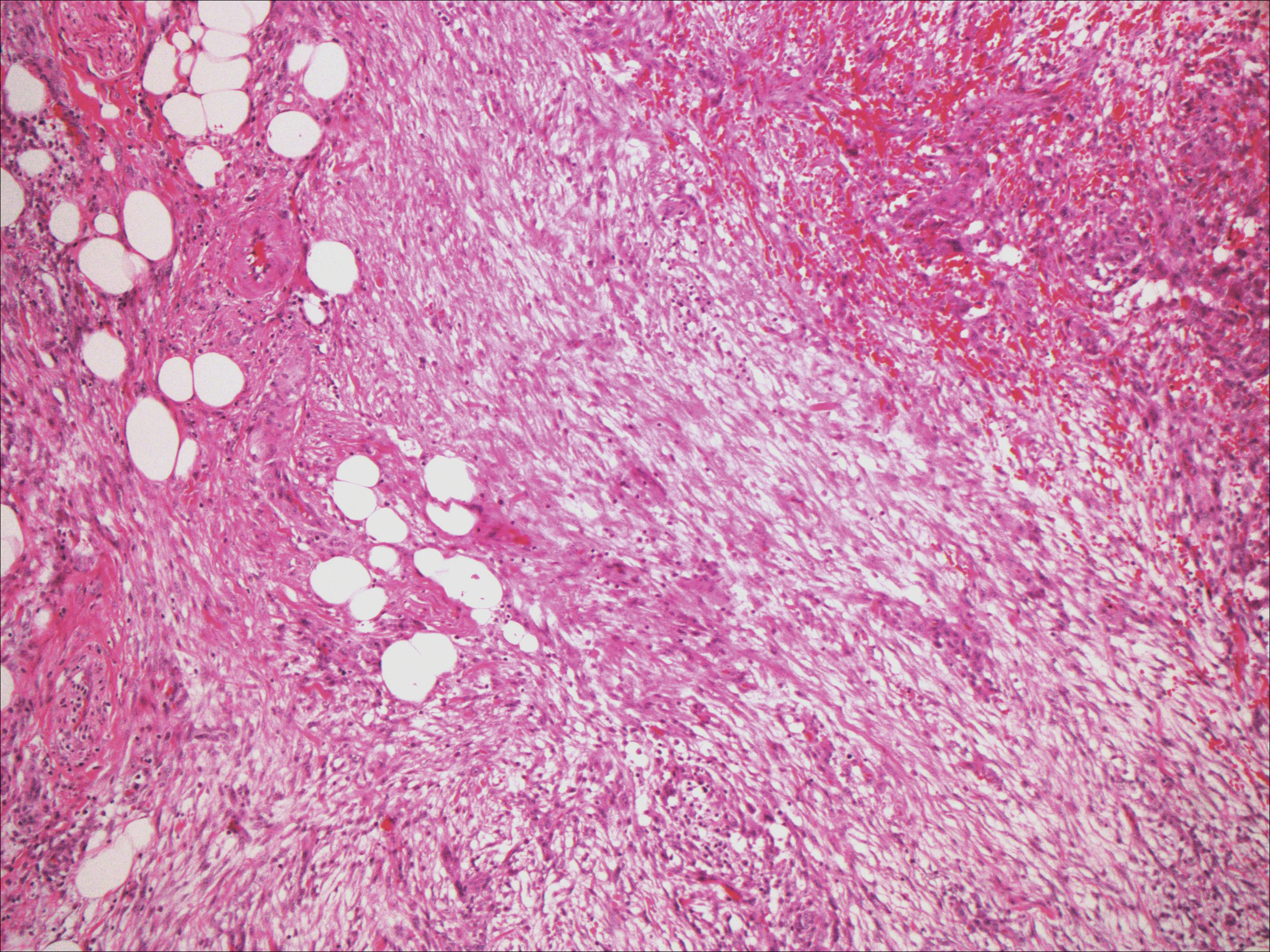
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
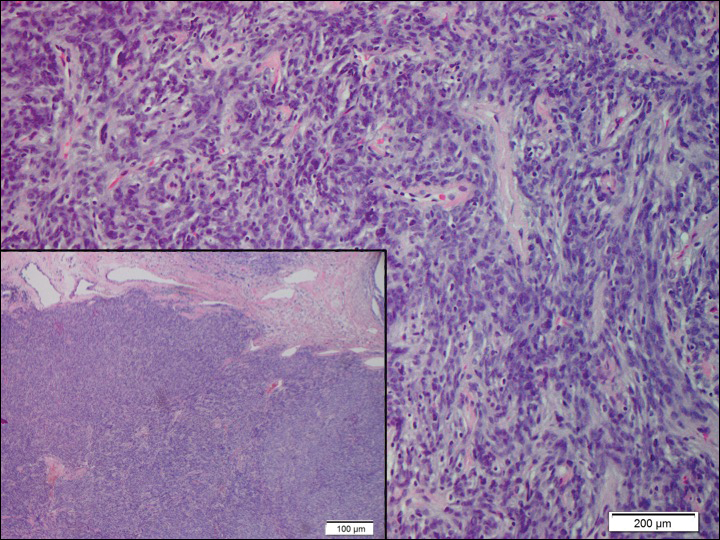
A 73-year-old man presented with a tender nodule on the back that had recently increased in size. On physical examination, a solitary 4-cm nodule was noted in the right trapezius region. The patient denied any personal or family history of similar lesions or a penchant for cysts. Due to the symptomatic nature of the lesion, surgical excision was performed.
Verrucoid Lesion on the Eyelid
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.

Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11

Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).

Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
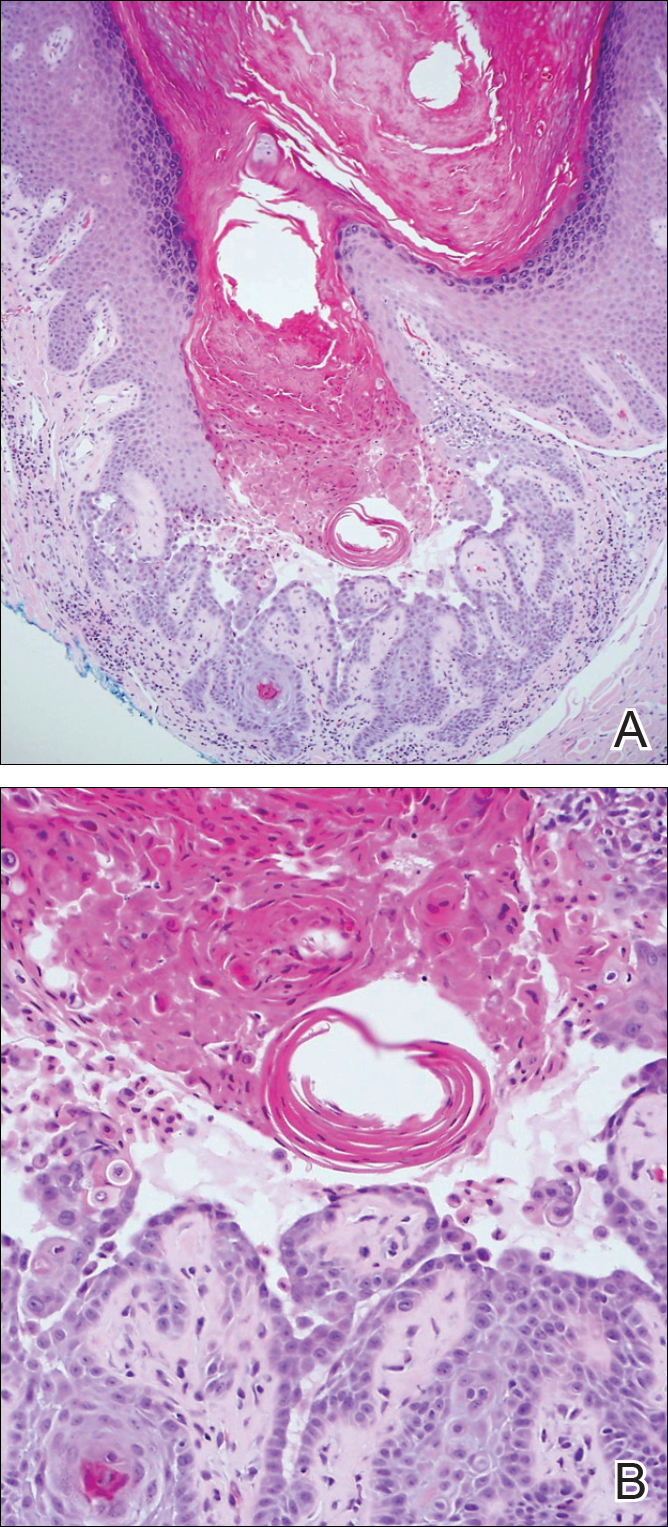
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
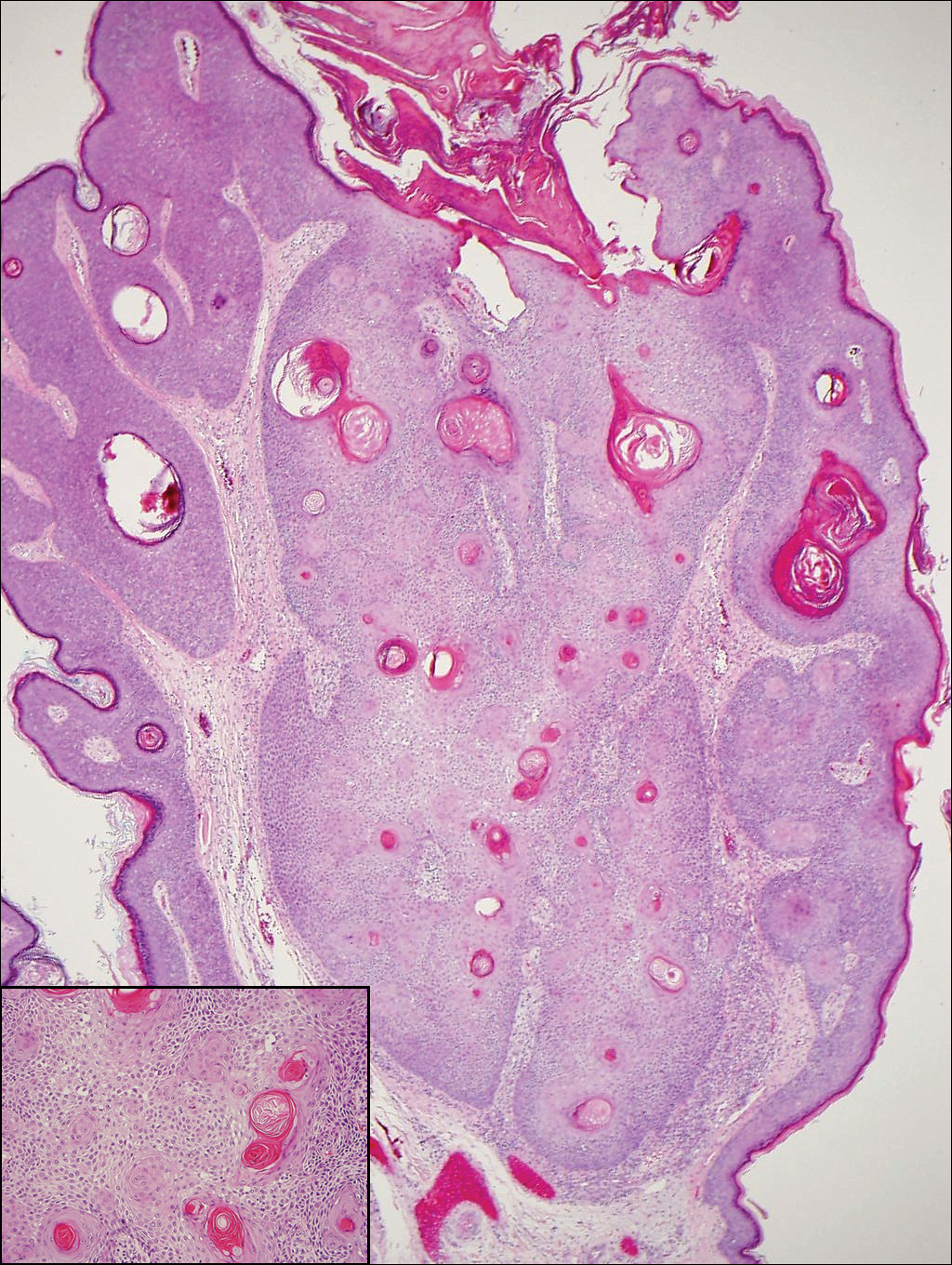
A 60-year-old man presented with a 3-mm verrucous papule on the right upper eyelid of 2 years' duration.
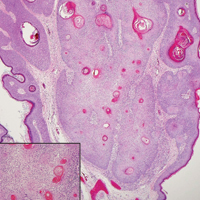
Orange Nodules on the Scalp
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
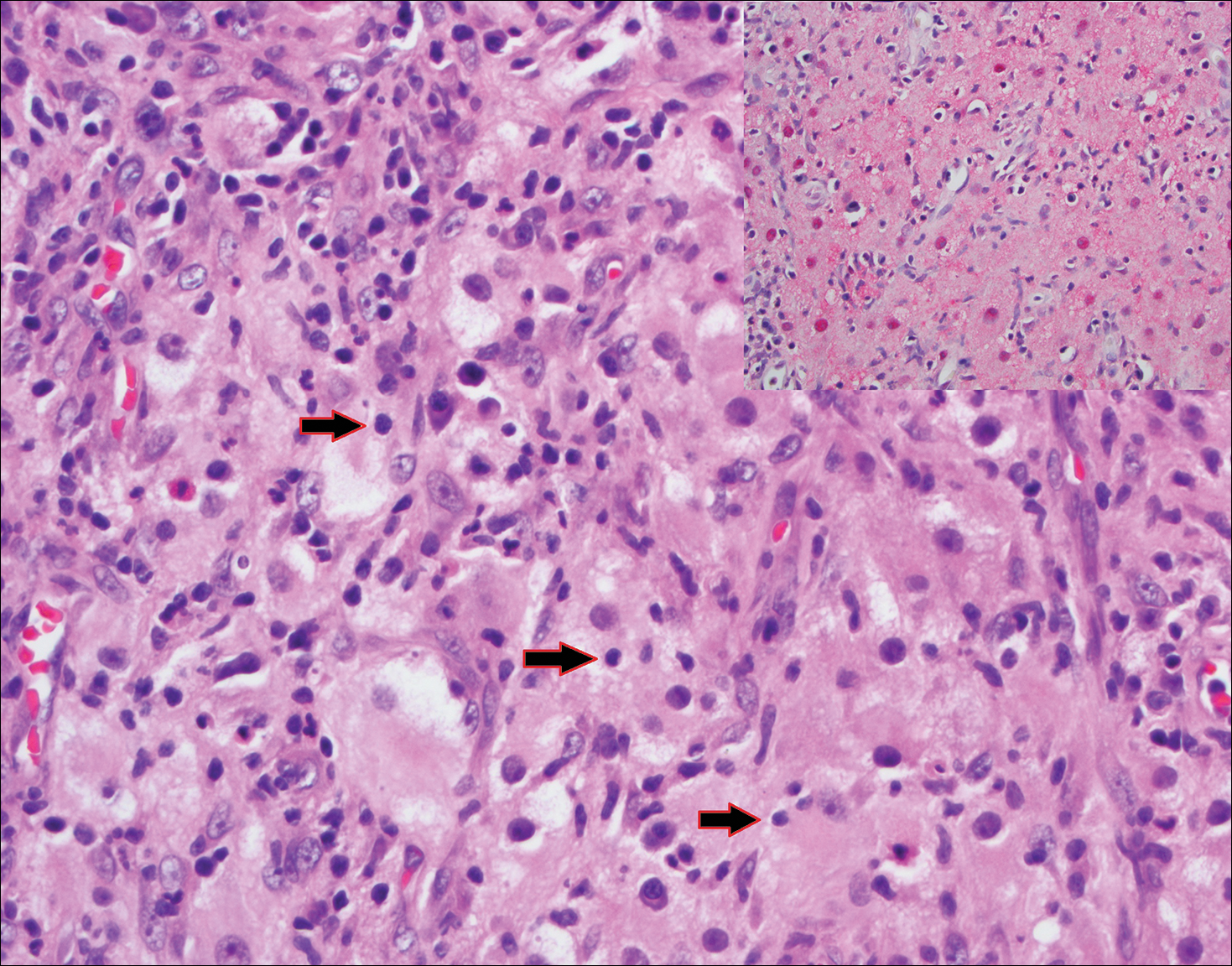
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
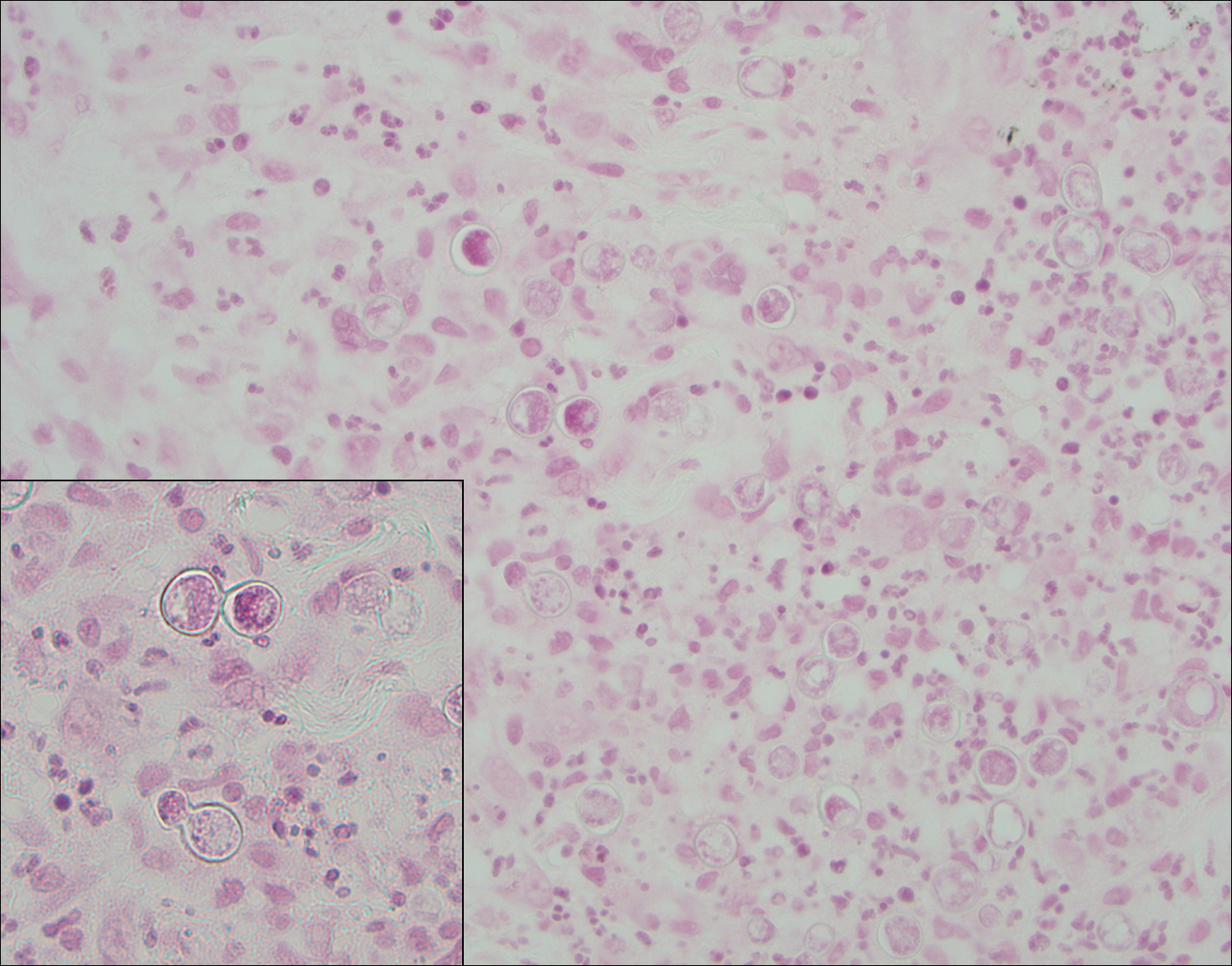
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
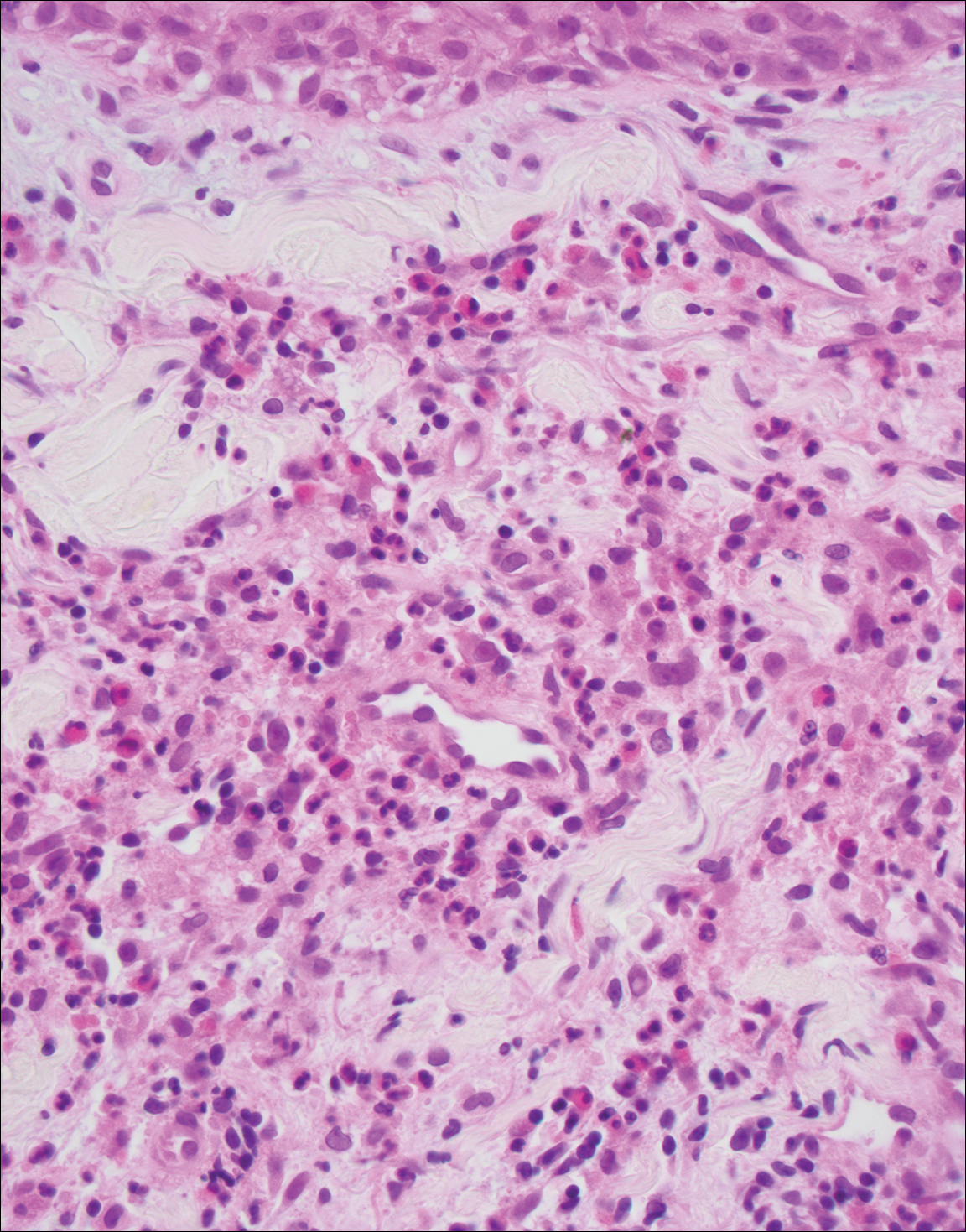
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
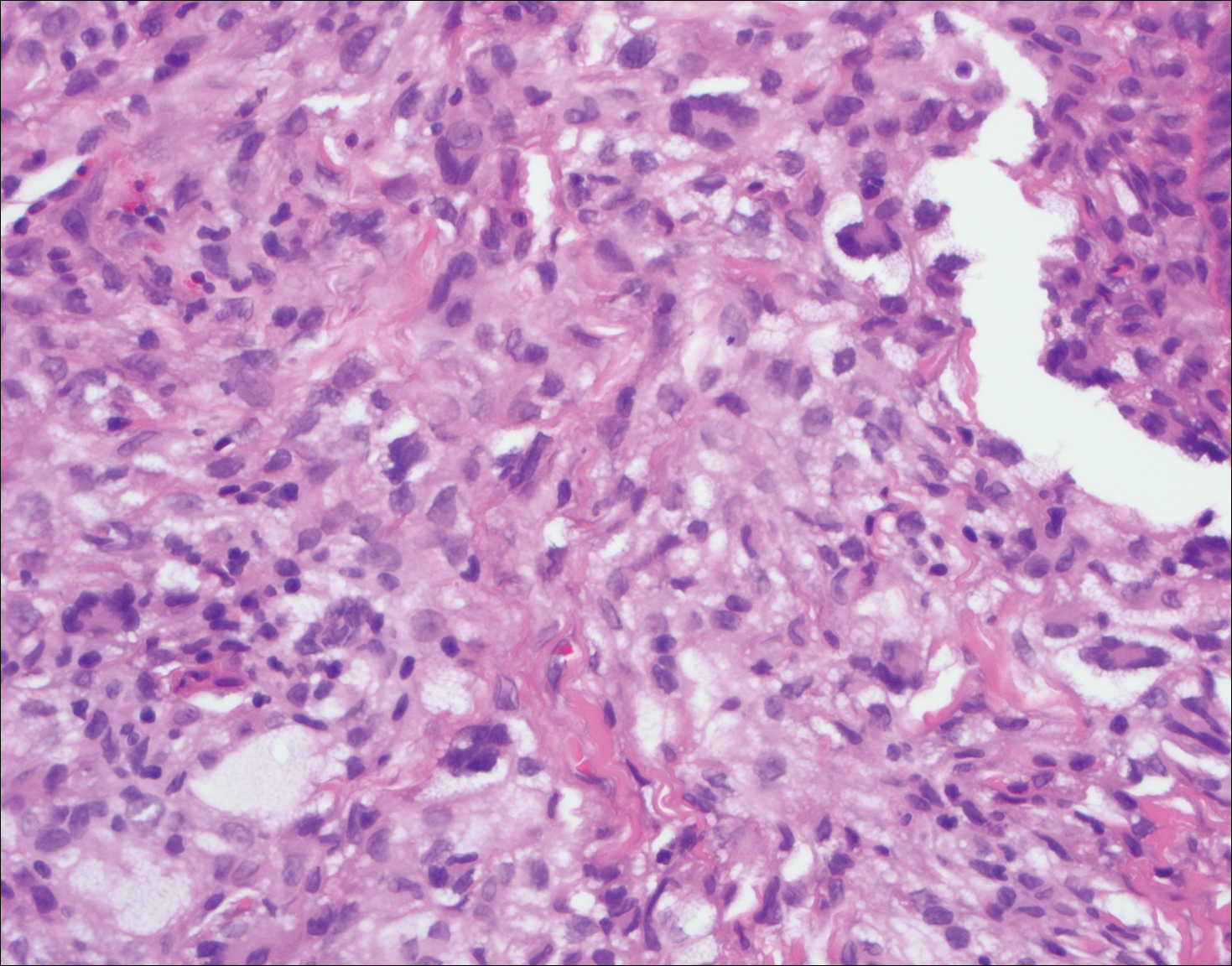
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
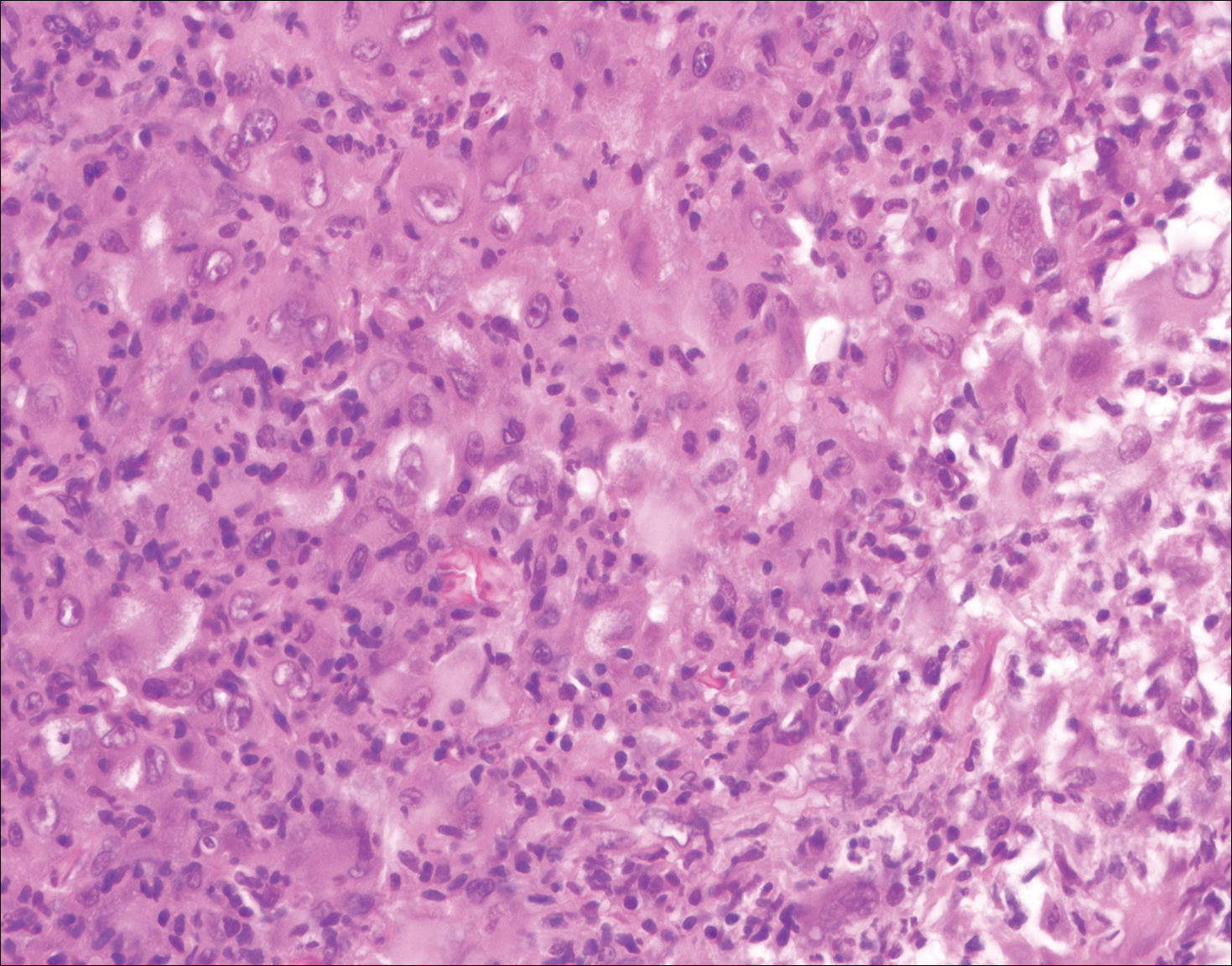
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
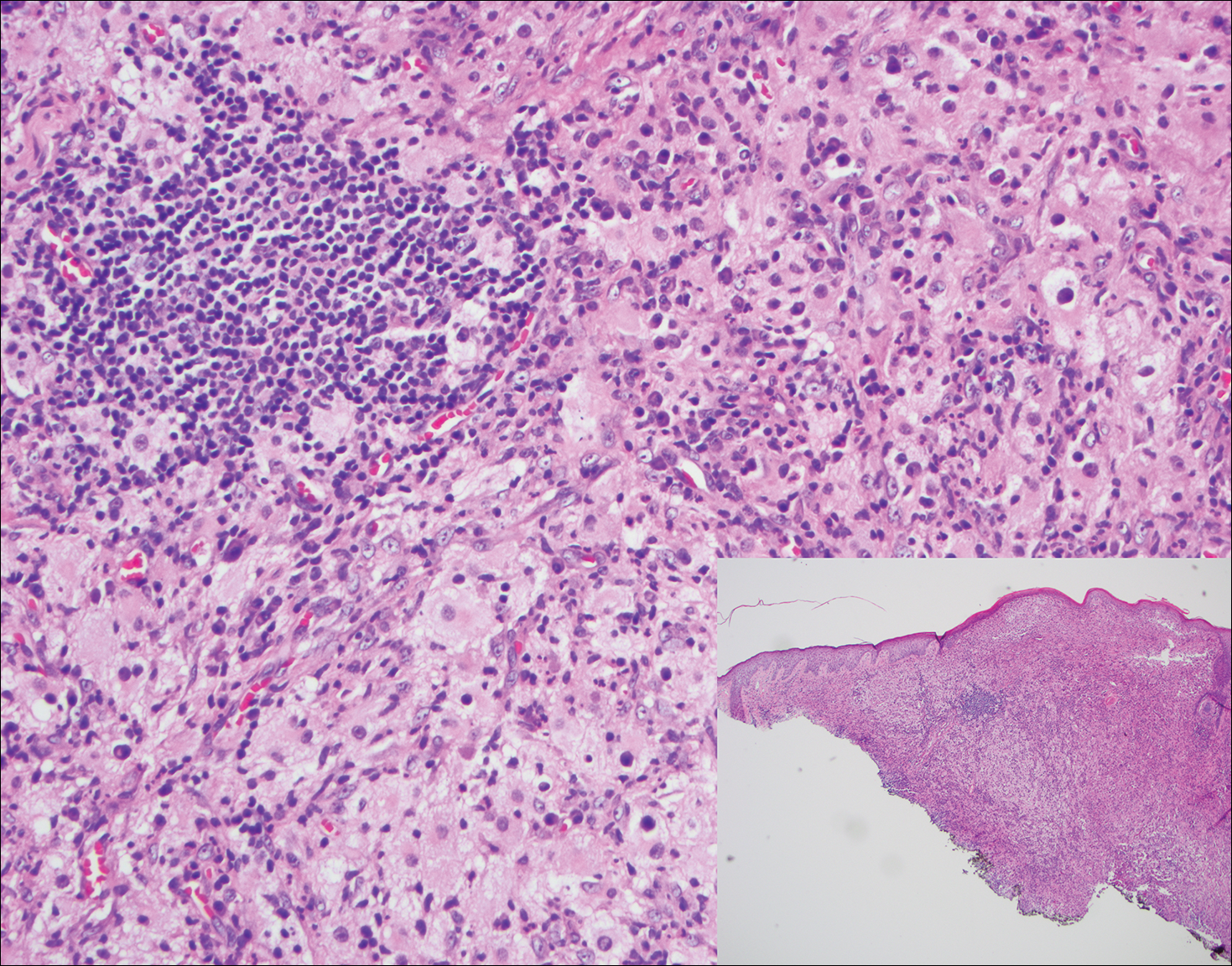
A 59-year-old man presented with itchy and mildly painful nodules on the head and neck of 7 months' duration. The patient denied fever, chills, unintentional weight loss, night sweats, and other systemic symptoms. Physical examination revealed multiple firm pink-orange nodules of varying sizes distributed on the scalp, face, and neck. Right-sided, painless, bulky cervical lymphadenopathy also was noted. An incisional biopsy was performed.
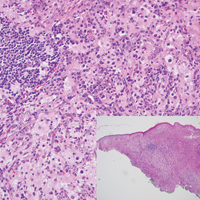
Pruritic Eruption on the Chest
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
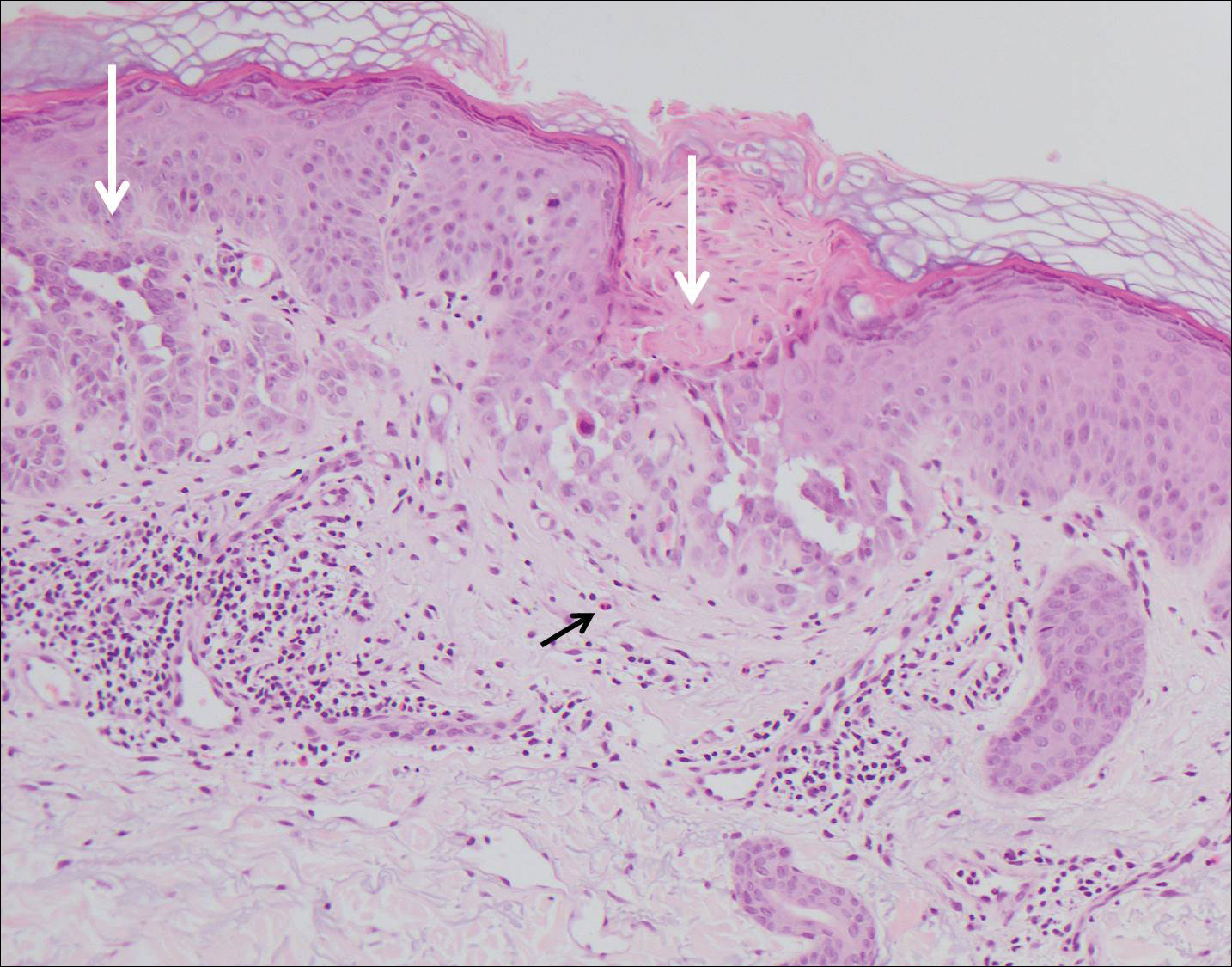
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
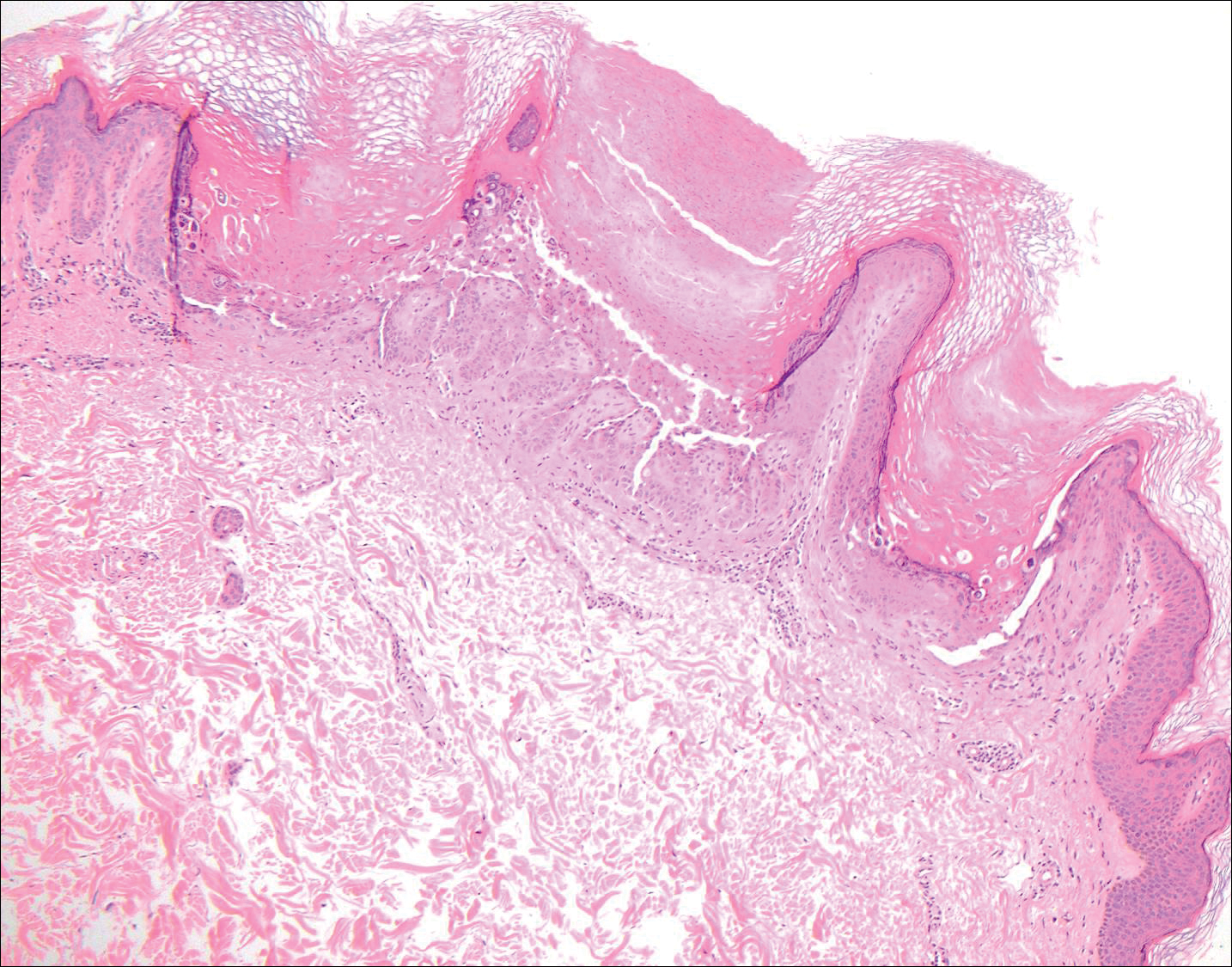
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
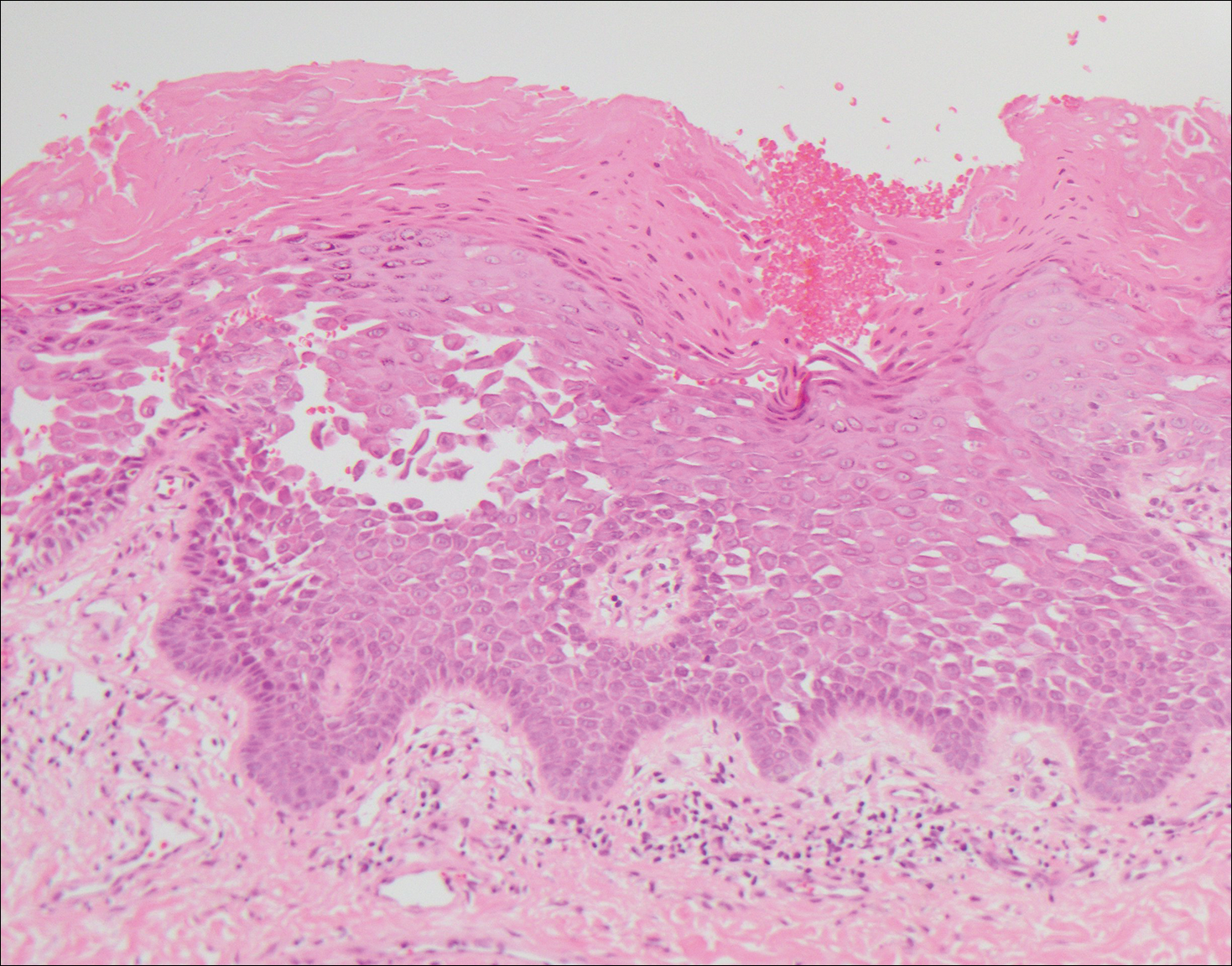
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
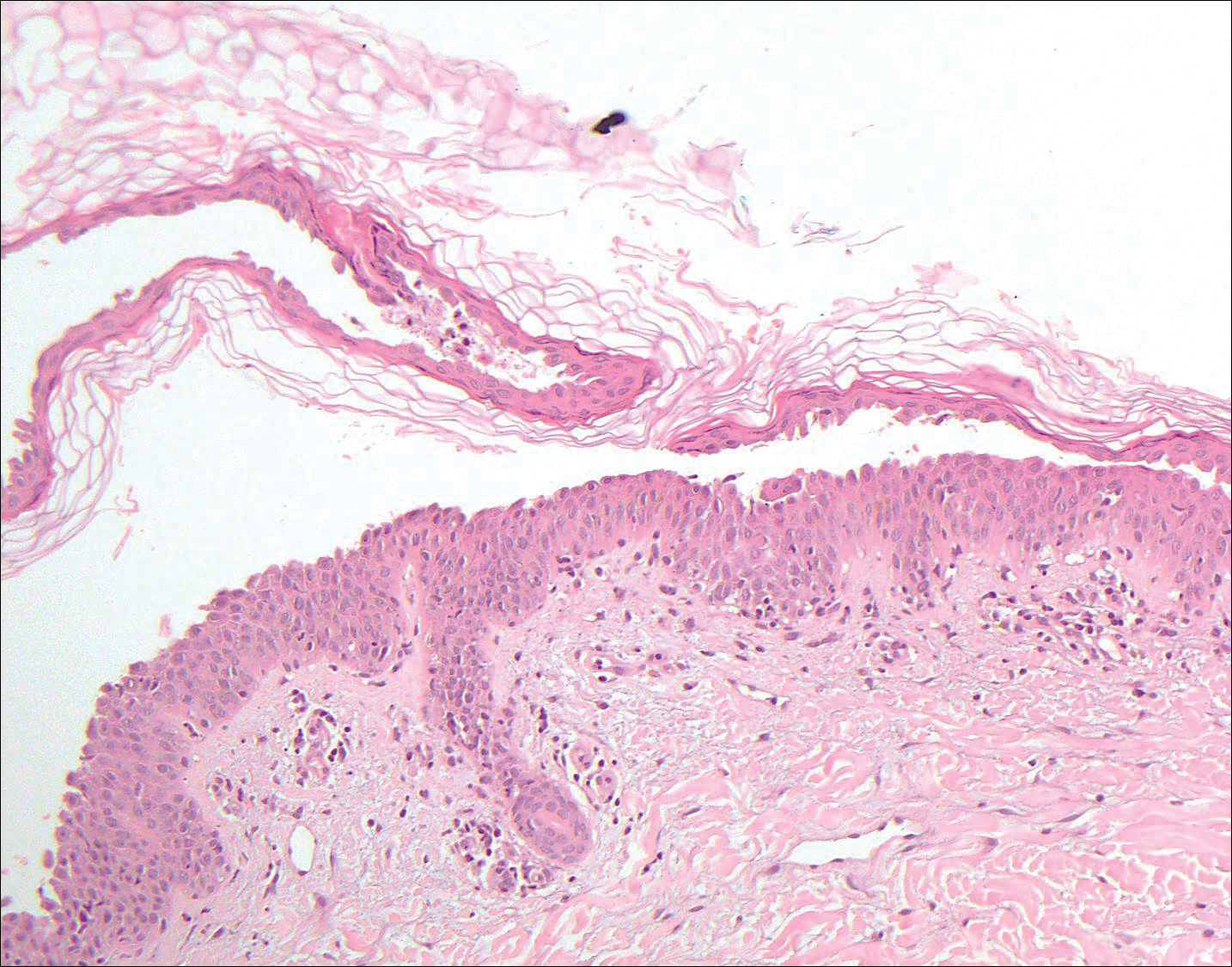
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
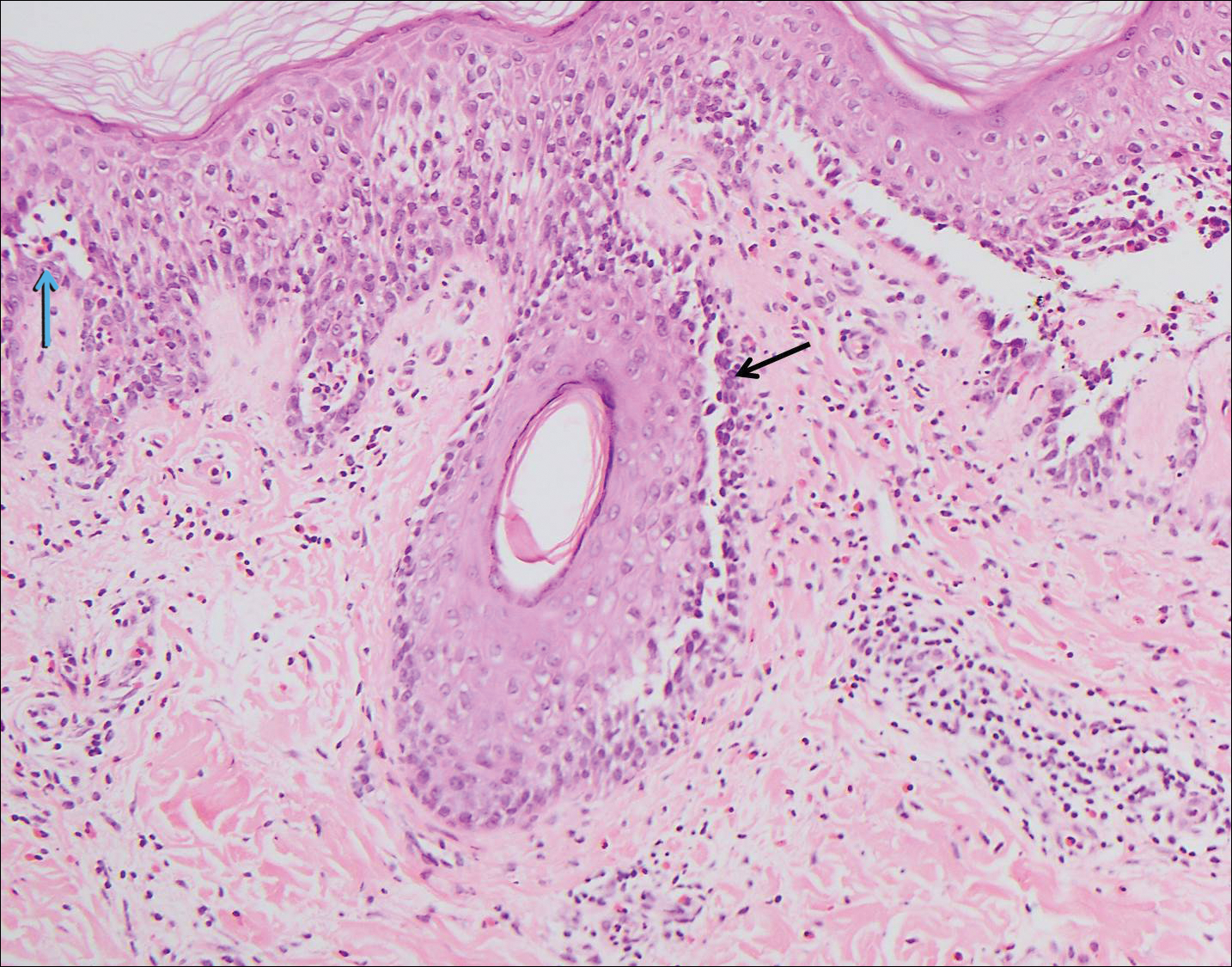
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
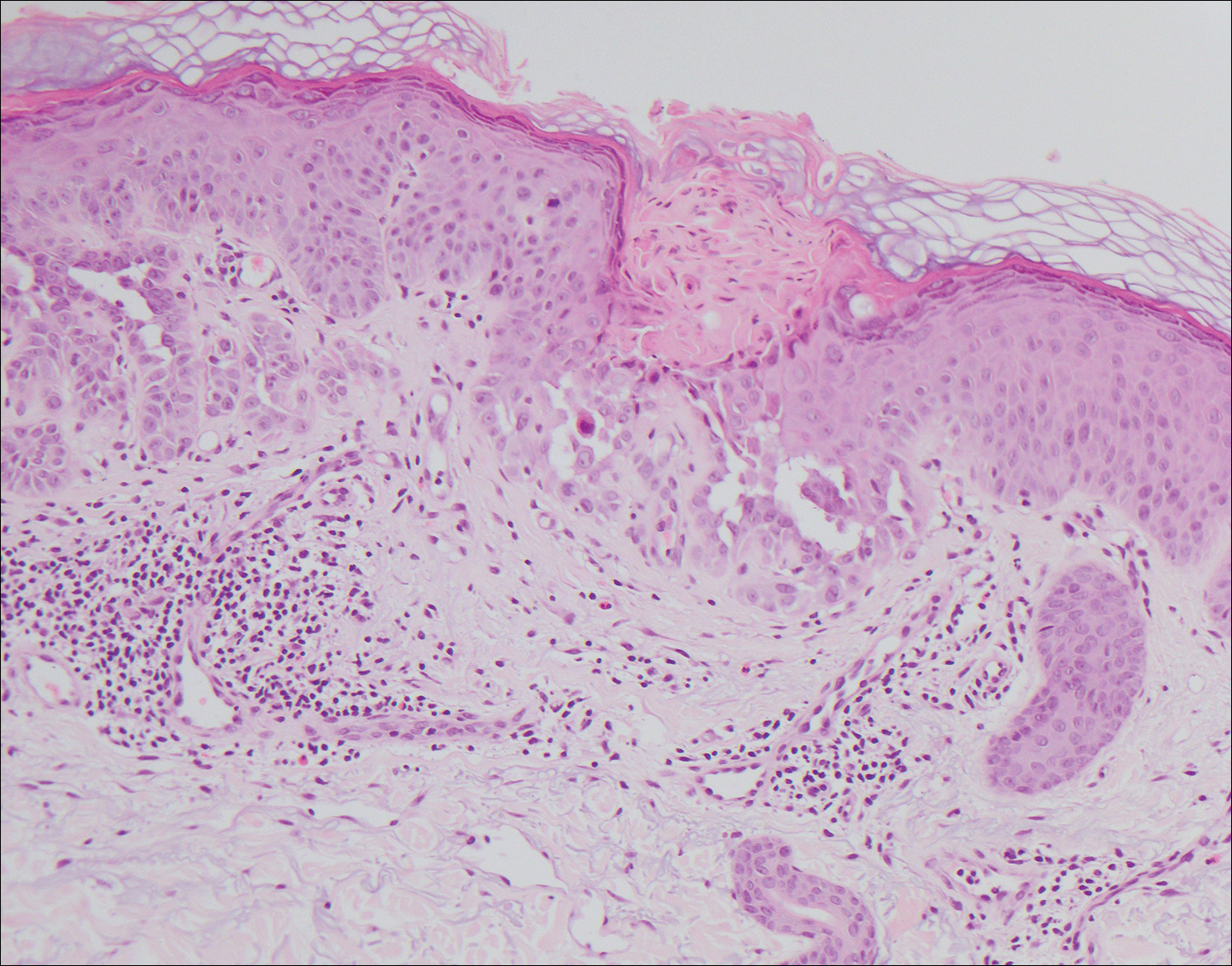
A 55-year-old man presented with small, erythematous, nonfollicular, pruritic papules on the mid chest.