User login
To prevent depression recurrence, interpersonal psychotherapy is a first-line treatment with long-term benefits
Major depressive disorder (MDD) frequently is recurrent, with new episodes causing substantial social and economic impairment1 and increasing the likelihood of future episodes.2 For this reason, contemporary psychiatric practitioners think of depression treatment as long-term and plan thoughtfully for maintenance therapy.
Recognizing the importance of engaging depressed individuals beyond the initial response,3 American Psychiatric Association practice guidelines conceptualize depression treatment as 3 phases:
• acute treatment, with the aim of remission (symptom removal)
• continuation treatment, with the aim of preventing relapse (symptom return)
• maintenance treatment, with the aim of preventing recurrence (new episodes).4
Interpersonal psychotherapy (IPT) is an evidence-based psychosocial treatment that adheres to this model.5 As a time-limited, manual-driven6,7 approach, IPT focuses on interpersonal distresses as precipitating and perpetuating factors of depression.8
Acute IPT’s efficacy is well-established across >200 empirical studies—making it an evidence-based, first-line treatment for adult depression.4,9,10 Meta-analyses show that acute IPT is superior to placebo and no-treatment controls, and largely comparable to antidepressant medication and other active, first-line psychotherapies, such as cognitive-behavioral therapy (CBT).11,12
Although this review, as well as the literature, focuses largely on adult outpatients with depression, evidence of IPT’s general efficacy exists for adolescents,13 chronically depressed patients,11 and depressed inpatients.14 This article presents a case study to describe the structure of IPT when used to treat depressed adults. We also present evidence of IPT’s acute and long-term efficacy in preventing depression recurrence and data to guide its use in practice.
CASE REPORT
‘Safe’ but depressed
Timothy, age 18, is a first-year college student who presents for outpatient psychotherapy to address recurrent depression. He reports general unhappiness, loss of interest in things, low energy, sleep problems, poor academic and work functioning, and low self-esteem. He experienced at least 3 similar depressive episodes while in high school.
The therapist’s diagnostic and interpersonal assessment suggests that Timothy’s depression is interpersonally driven. Timothy longs for relational intimacy but fears he will fail or burden people with his needs. He has difficulty gauging appropriate levels of enmeshment with others and either becomes overdependent or stays at a distance. This “safe” approach to relationships contributes to boredom, loneliness, and isolation. His recent transition to college away from home and the failure of a romantic relationship have compounded these experiences.
Interpersonal model of IPT
IPT conceptualizes depression as involving predisposing, precipitating, and perpetuating biopsychosocial factors, including:
• underlying biological and social vulnerability, such as insecure attach ment (ie, tenuous and often negative views of self and others)
• current interpersonal life stressors
• inadequate social supports.15,16
For example, poor early attachment to caregivers can give rise to despair, isolation, and low mood. In turn, this can be exacerbated by poor social and communication skills that promote further rejection and withdrawal of social support and thus, intensified despair, isolation, and low mood. As in Timothy’s case, this vicious cycle underscores psychosocial stressors as a causal factor, maintaining factor, and result of depression. Specifically, IPT conceptualizes 4 main biopsychosocial problem domains:
• grief and loss
• interpersonal disputes
• role transitions
• interpersonal/communication deficits (often connected to isolation).
Working within 1 or 2 of the most salient problem domains, IPT centers on strategies for helping patients solve interpersonal problems based on the notion that modified relationships, revised interpersonal expectations, improved communications, and increased social support will lead to symptom reduction.15-17
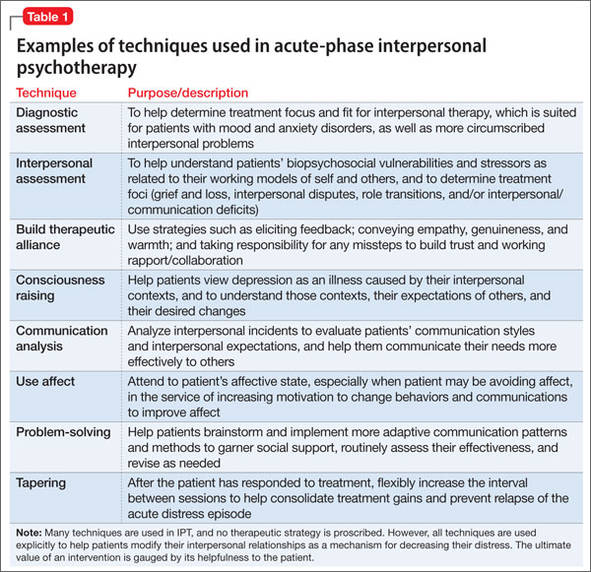
Many techniques are utilized in IPT (Table 1) to help patients modify their interpersonal relationships as a mechanism for decreasing their distress. IPT is problem-focused, aiming to improve patients’ relationships by drawing on their assets and helping to build skills around shortcomings. Therefore, IPT focuses on observable interpersonal patterns, as opposed to latent personality dynamics.
CASE CONTINUED
Setting goals
When the clinical explains in the non-technical terms the data supporting IPT’s efficacy for depression, including with young adults, Timothy agrees to teeatment with acute IPT. The therapist behins with consciousness-raising techniques to help Timothy adopt the “sick role” by viewing depressing as an illness to be cured. Collaboratively, they establish treatment goals that fit the IPT formulation of depression— ie, revising current relationships and expectations of them, increasing social support, improving communication skills, and solving problems within 1 or 2 of the IPT problem domains.
For Timothy, the most pressing psychosocial problems seem to be interpersonal deficits and role transitions. He appears to be insecurely attached to others, which is a risk factor for poor facilitation of, and boundaries around, good relationships. A transition to a new and intimidating interpersonal context—living on a college campus—compounded his vulnerabilities and increased his depression.
Acute treatment. The acute phase of IPT is time-limited—often, 12 to 16 sessions with gradual tapering toward the end (akin to a continuation phase). The time limit’s purpose is to focus both patient and therapist on the specific goal of removing the acute “illness” of depression. The IPT clinician takes an interpersonal inventory to learn about the patient’s most important relationships and hones in on the IPT domain foci. Working collaboratively, the therapist might help the patient mourn a loss, reconstruct a narrative with a deceased loved one, consider ways to increase social contact, develop assertiveness, label feelings and needs, resolve an impasse with a significant other, and so forth.
The IPT therapist is an advocate for the patient and adopts an active stance laced with empathy and warmth. However, the therapist is more than unconditionally accepting as depression is viewed as a problem to be actively resolved.
CASE CONTINUED
Creating new patterns
The therapist uses various IPT strategies to work collaboratively with Timothy. She attempts to develop a strong working alliance by building interpersonal safety and trust— which take time with an insecurely attached patient. She tries to provide a new model for how close relationships can develop, while also focusing on current relationships. She and Timothy address his romantic desire for a coworker and work on developing realistic expectations and effective methods for conveying his interest.
When Timothy approaches his coworker, she does not reject him—as he expected— but wants to pursue friendship before possibly dating. The therapist then works with Timothy’s emotional reaction and explores ways to effectively convey his emotions to this young woman. Drawing on communication analysis and problem-solving strategies, Timothy is able to sustain this friendship—a shift from his typical retreat when relationships have not gone as hoped or expected.
Timothy develops confidence to take more risks in initiating social encounters and starts to confide in his roommates when he feels upset. After 3 months of treatment, his expanded social network and improved interpersonal skills result in decreased depression. When Timothy suggests termination, he and the therapist agree to end acute IPT but—given his history of depression—to continue maintenance sessions.
Limited data exist on variables that relate to IPT’s acute success or conditions under which it works best. Although process research lags behind acute IPT outcome research, some findings can help guide the IPT practitioner. For example, variables shown to predict outcomes of acute IPT for depression include a positive therapeutic alliance, therapist warmth, and psycho psychotherapist use of exploratory techniques (Table 2).

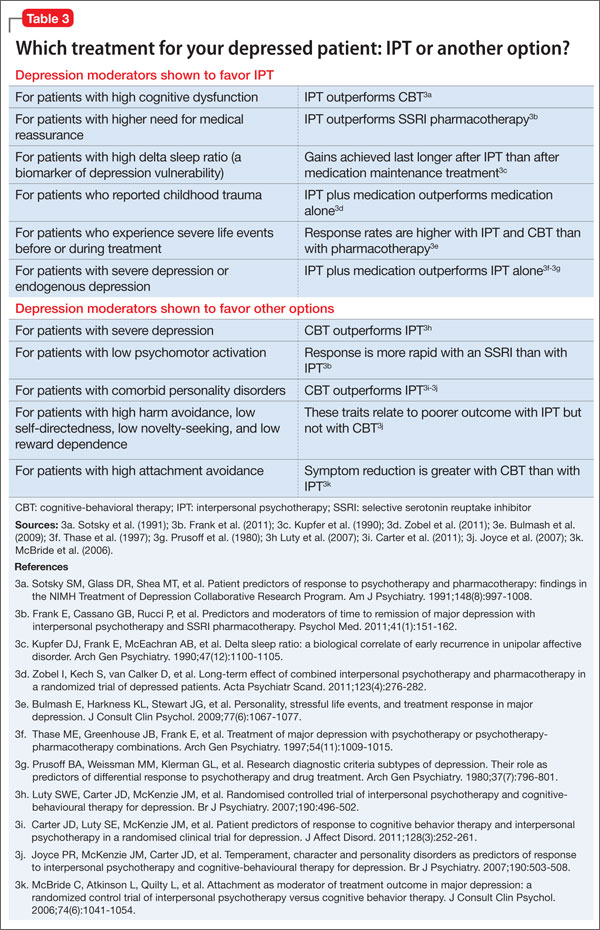
Similarly, IPT has been shown to be more effective in some patients than others, depending on various moderators of depression. For example:
• For patients with high cognitive dysfunction, IPT outperforms CBT.
• For patients with higher need for medical reassurance, IPT outperforms selective serotonin reuptake inhibitor (SSRI) pharmacotherapy.
• For patients with severe depression, CBT outperforms IPT.
• For patients with low psychomotor activation, response is more rapid with an SSRI than with IPT (Table 3).18
Durability of acute IPT
One way to understand recurrence prevention is to examine the durability of a treatment’s acute effect in the absence of a specific maintenance plan. In theory, patients will continue to apply the skills learned in acute IPT to maintain gains and prevent recurrences, even after they stop seeing the psychotherapist.
Initial findings. Some research speaks to IPT’s acute-phase durability. The inaugural clinical trial of IPT by Weissman et al19 included 4 months of acute treatment and a 1-year uncontrolled naturalistic follow-up assessment. At follow-up, depression and global clinical symptoms were the same, whether patients had been acutely treated with IPT alone, pharmacotherapy alone (amitriptyline), combined IPT and pharmacotherapy, or nonscheduled treatment with a psychiatrist.
Some patients continued to function well, whereas others did not fully maintain acute treatment gains. Patients who received IPT acutely, either singly or with medication, showed better social functioning at follow-up compared with patients who did not receive IPT. This long-term durability of social improvements was an obvious target of IPT.
Support from TDCRP. In the National Institute of Mental Health Treatment of Depression Collaborative Research Project (TDCRP),20 patients in the acute phase of depression were assigned to 16 weeks of IPT, CBT, pharmacotherapy (imipramine) and clinical management (CM), or placebo plus CM. Among those who recovered by acute treatment’s end, MDD relapse rates at 18-month naturalistic follow-up were 33% for IPT, 36% for CBT, 50% for imipramine, and 33% for placebo. Between-group differences were not statistically significant.
Because acute responders to different types of treatment might have different inherent relapse tendencies, these data do not support causal attributions about the enduring effects of acute-phase treatment. The relapse rates do suggest, however, that 16 weeks of acute treatment, irrespective of kind, was insufficient for some patients to achieve full recovery and lasting remission. Consistent with the initial IPT trial,19 IPT (and CBT) outperformed medi cation and placebo in maintaining relationship quality.21
Long-term benefits. A more recent trial by Zobel et al22 examined the durability of benefits from 5 weeks of acute IPT plus pharmacotherapy and pharmacotherapy plus CM for inpatients with MDD. Although caution is required in interpreting naturalistic follow-up studies, patients in both groups showed decreased depression from baseline to 5-year follow-up. Early symptom reduction was more rapid for patients in the IPT plus pharmacotherapy group, but no significant difference existed at 5 years. More IPT patients than CM patients showed sustained remission (28% vs 11%, respectively). These rates demonstrate a need for longer-term potency of acute treatments and more targeted maintenance treatments.
IPT-M for preventing recurrence
A second way to understand recurrence prevention is to examine the efficacy of a treatment’s maintenance protocol added to an acute treatment phase. IPT has been adapted as a maintenance treatment (IPT-M), with emphasis on keeping patients well. With this revised focus, IPT-M differs somewhat from acute IPT. Although treatment continues to center on interpersonal functioning, IPT-M favors:
• vigilance for possible triggers of new depressive episodes
• longer-term contact with a therapist
• reinforcing skills learned
• addressing an expanded number of interpersonal problem areas (given that such problems can be addressed more efficiently relative to acute treatment).
Efficacy of IPT-M. In the initial trial, Frank et al23 compared the efficacy of IPT-M with that of pharmacotherapy (imipramine) in preventing depressive relapse among patients with recurrent depression who had responded to ≥16 sessions of acute IPT and imipramine and remained well during a 17- week continuation phase. For maintenance, patients were assigned to IPT-M alone, imipramine alone, placebo alone, IPT-M plus imipramine, or IPT-M plus placebo. Maintenance imipramine was continued at the acute dosage (target 200 mg/d; up to 400 mg/d was allowed). Maintenance IPT was monthly sessions. Patients remained in the trial for 3 years or until depression recurred.
On its own, IPT-M showed some efficacy in preventing recurrence, as the mean time to recurrence was 82 weeks for IPT-M alone and 74 weeks for IPT-M plus placebo. The prophylactic effect of imipramine was stronger, however. The mean time to recurrence for imipramine with IPT was 131 weeks, and the mean time to recurrence for imipramine without IPT was 124 weeks. Therefore, whereas monthly IPT-M can certainly help prolong wellness and delay recurrence, IPT maintenance treatment with acute doses of imipramine might be even more effective— if the patient is willing to take medication. These findings must be considered with caution because of the inherent inequity between imipramine and IPT-M in regard to maintenance dosage strength.
Frequency of treatment. In another trial, Frank et al24 examined whether the frequency of maintenance IPT sessions played a role in its prophylactic effect. Adult women who had achieved depression remission with acute IPT (alone or in combination with SSRI pharmacotherapy) were randomized to weekly, bi-weekly, or monthly IPT-M alone for 2 years or until recurrence. Depression recurred during IPT-M in:
• 26% of patients who had received acute IPT alone
• 50% of those who had received acute IPT plus an SSRI.
Frequency of IPT-M sessions did not affect time to recurrence. Thus, for women who can achieve remission with IPT alone, varied frequencies of IPT-M can be good prophylaxis. For women who need an SSRI to augment acute IPT, IPT-M alone at varied dosages is less effective in preventing depression recurrence. Therefore, acute treatment response patterns can inform maintenance plans, with the most prudent maintenance strategy being to maintain the acute treatment strategy over a longer period.
IPT-M for late-life depression. A trial by Reynolds et al25 examined the efficacy of maintenance nortriptyline and IPT-M in preventing depression recurrence in patients age ≥59 who initially recovered after combined acute and continuation IPT plus nortriptyline. The 4 conditions (with their recurrence rates) were:
• monthly IPT-M with nortriptyline (20%)
• monthly IPT-M with placebo (64%)
• nortriptyline plus medication visits (43%)
• placebo plus medication visits (90%).
Clearly, the combined active treatments outperformed placebo and antidepressant alone in terms of delaying or preventing recurrence, which suggests an optimal maintenance strategy with this population.
IPT-M for later life. Another trial by the same group26 enrolled patients age ≥70 with MDD that responded to acute IPT plus paroxetine. The maintenance treatments to which they were randomly assigned (and recurrence rates within 2 years) were:
• paroxetine plus IPT-M (35%)
• placebo plus IPT-M (68%)
• paroxetine plus clinical management (37%)
• placebo plus clinical management (58%).
Recurrence rates were the same for patients receiving medication plus IPT-M and medication plus clinical management, and depression was 2.4 times more likely to recur in patients receiving placebo vs active medication. Therefore, for later life depression, the optimal maintenance strategy was the SSRI.
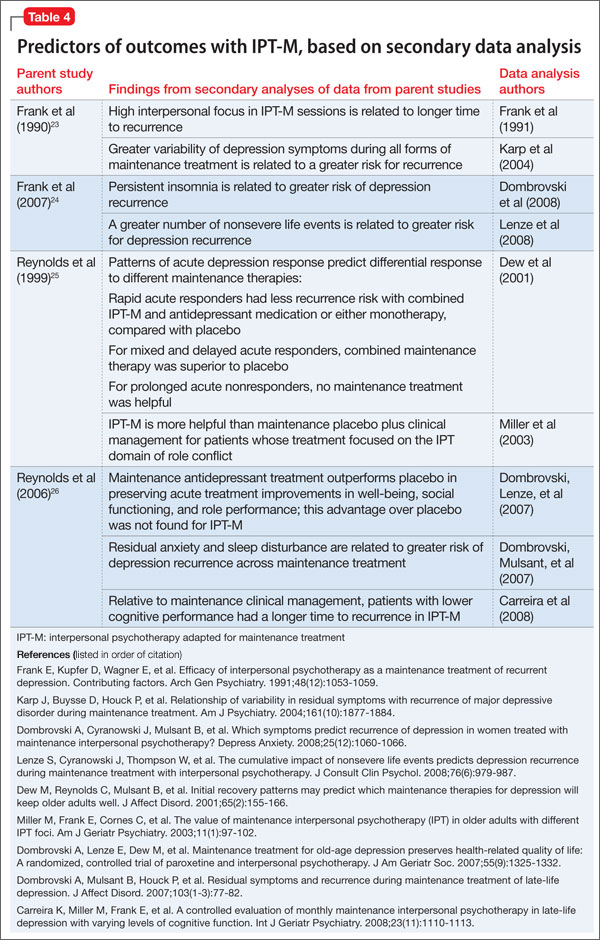
Secondary analyses of data from these seminal trials of IPT-M point to other predictors of how and for whom maintenance IPT may work (Table 4). For example:
• Greater variability of depression symptoms during all forms of maintenance treatment is related to a greater risk of recurrence.
• Persistent insomnia is related to greater risk of recurrent depression.
• High interpersonal focus in IPT-M sessions is related to longer time to recurrence.
Bottom Line
Interpersonal psychotherapy (IPT) is efficacious for acute depression and for preventing recurrences. Patients treated successfully with acute IPT alone benefit from varied doses of maintenance IPT. Combining IPT-M with antidepressant medication can be more potent than IPT-M alone. For late-life depression, medication appears to be most effective for maintenance treatment.
Related Resources
Media
• Video demonstration, role-play transcripts, lesson plans, and quizzes. In: Appendices in and DVD companion to Ravitz P, Watson P, Grigoriadas S. Interpersonal psychotherapy for depression. New York, NY: Norton; 2013.
• Video demonstration of IPT sessions. In: DVD companion to Dewan, M, Steenbarger, B, Greenberg, R, eds. The art and science of brief psychotherapies: An illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012.
Text
• Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London, United Kingdom: Taylor & Francis; 2012.
• Weissman MM, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
• Weissman M, Markowitz J, Klerman GL. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
Websites
• Interpersonal Psychotherapy Institute. http://iptinstitute.com.
• International Society for Interpersonal Psychotherapy. http://interpersonalpsychotherapy.org.
Drug Brand Names
Amitriptyline • Elavil Nortriptyline • Pamelor
Imipramine • Tofranil Paroxetine • Paxil
Acknowledgments
The authors are grateful to Samantha L. Bernecker, MS, and Nicholas R. Morrison for their assistance with the research review.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. ten Doesschate MC, Koeter MW, Bockting CL, et al. Health related quality of life in recurrent depression: a comparison with a general population sample. J Affect Disord. 2010; 120(1-3):126-132.
2. Hardeveld F, Spijker J, De Graaf R, et al. Prevalence and predictors of recurrence of major depressive disorder in the adult population. Acta Psychiatr Scand. 2010;122(3):184-91.
3. Arnow BA, Constantino MJ. Effectiveness of psychotherapy and combination treatment for chronic depression. J Clin Psychol. 2003;59(8):893-905.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2010.
5. Klerman GL, Weissman MM, Rounsaville BJ, et al. Interpersonal psychotherapy of depression. New York, NY: Basic Books; 1984.
6. Weissman MM, Markowitz JC, Klerman G. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
7. Weissman M, Markowitz J, Klerman G. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
8. Brakemeier EL, Frase L. Interpersonal psychotherapy (IPT) in major depressive disorder. Eur Arch Psychiatry Clin Neurosci. 2012;262(suppl 2):S117-1121.
9. Depression in adults (update): NICE guideline CG90). National Institute for Health and Care Excellence. (2009). http://www.nice.org.uk/cg90. Updated October 2009. Accessed March 5, 2014.
10. Depression. National Institutes of Mental Health. http://www.nimh.nih.gov/health/publications/depression/ index.shtml. Revised 2011. Accessed March 5, 2014.
11. Cuijpers P, van Straten A, Andersson G, et al. Psychotherapy for depression in adults: a meta-analysis of comparative outcome studies. J Consult Clin Psychol. 2008;76(6):909-922.
12. Cuijpers P, Geraedts AS, van Oppen P, et al. Interpersonal psychotherapy for depression: a meta-analysis [Erratum in: Am J Psychiatry. 2011;168(6):652]. Am J Psychiatry. 2011; 168(6):581-592.
13. Mufson L, Dorta K, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6): 577-584.
14. Schramm E, Schneider D, Zobel I, et al. Efficacy of interpersonal psychotherapy plus pharmacotherapy in chronically depressed inpatients. J Affect Disord. 2008; 109(1-2):65-73.
15. Bernecker SL. How and for whom does interpersonal psychotherapy work? Psychotherapy Bulletin. 2012;47(2):13-17.
16. Stuart S. Interpersonal psychotherapy. In: Dewan MJ, Steenbarger BN, Greenberg RP, eds. The art and science of brief psychotherapies: an illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012: 157-193.
17. Grigoriadas S, Watson P, Maunder R, eds. Psychotherapy essentials to go: Interpersonal psychotherapy for depression. New York, NY: W. W. Norton & Company, Inc.; 2013.
18. Bleiberg KL, Markowitz JC. Interpersonal psychotherapy for depression. In: Barlow D, ed. Clinical handbook of psychological disorders: a step-by-step treatment manual. New York, NY: The Guilford Press; 2008:306-327.
19. Weissman MM, Klerman GL, Prusoff BA, et al. Depressed outpatients. Results one year after treatment with drugs and/or interpersonal psychotherapy. Arch Gen Psychiatry. 1981;38(1):51-55.
20. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49(10):782-787.
21. Blatt S, Zuroff D, Bondi C, et al. Short- and long-term effect of medication and psychotherapy in the brief treatment of depression: further analyses of data from the NIMH TDCRP. Psychother Res. 2000;10(2):215-234.
22. Zobel I, Kech S, van Calker D, et al. Long-term effect of combined interpersonal psychotherapy and pharmacotherapy in a randomized trial of depressed patients. Acta Psychiatr Scand. 2011;123(4):276-282.
23. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry. 1990;47(12):1093-1099.
24. Frank E, Kupfer DJ, Buysse DJ, et al. Randomized trial of weekly, twice-monthly, and monthly interpersonal psychotherapy as maintenance treatment for women with recurrent depression. Am J Psychiatry. 2007;164(5): 761-767.
25. Reynolds CF 3rd, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA. 1999;281(1): 39-45.
26. Reynolds CF 3rd, Dew MA, Pollock BG, et al. Maintenance treatment of major depression in old age. N Engl J Med. 2006;354(11):1130-1138.
Major depressive disorder (MDD) frequently is recurrent, with new episodes causing substantial social and economic impairment1 and increasing the likelihood of future episodes.2 For this reason, contemporary psychiatric practitioners think of depression treatment as long-term and plan thoughtfully for maintenance therapy.
Recognizing the importance of engaging depressed individuals beyond the initial response,3 American Psychiatric Association practice guidelines conceptualize depression treatment as 3 phases:
• acute treatment, with the aim of remission (symptom removal)
• continuation treatment, with the aim of preventing relapse (symptom return)
• maintenance treatment, with the aim of preventing recurrence (new episodes).4
Interpersonal psychotherapy (IPT) is an evidence-based psychosocial treatment that adheres to this model.5 As a time-limited, manual-driven6,7 approach, IPT focuses on interpersonal distresses as precipitating and perpetuating factors of depression.8
Acute IPT’s efficacy is well-established across >200 empirical studies—making it an evidence-based, first-line treatment for adult depression.4,9,10 Meta-analyses show that acute IPT is superior to placebo and no-treatment controls, and largely comparable to antidepressant medication and other active, first-line psychotherapies, such as cognitive-behavioral therapy (CBT).11,12
Although this review, as well as the literature, focuses largely on adult outpatients with depression, evidence of IPT’s general efficacy exists for adolescents,13 chronically depressed patients,11 and depressed inpatients.14 This article presents a case study to describe the structure of IPT when used to treat depressed adults. We also present evidence of IPT’s acute and long-term efficacy in preventing depression recurrence and data to guide its use in practice.
CASE REPORT
‘Safe’ but depressed
Timothy, age 18, is a first-year college student who presents for outpatient psychotherapy to address recurrent depression. He reports general unhappiness, loss of interest in things, low energy, sleep problems, poor academic and work functioning, and low self-esteem. He experienced at least 3 similar depressive episodes while in high school.
The therapist’s diagnostic and interpersonal assessment suggests that Timothy’s depression is interpersonally driven. Timothy longs for relational intimacy but fears he will fail or burden people with his needs. He has difficulty gauging appropriate levels of enmeshment with others and either becomes overdependent or stays at a distance. This “safe” approach to relationships contributes to boredom, loneliness, and isolation. His recent transition to college away from home and the failure of a romantic relationship have compounded these experiences.
Interpersonal model of IPT
IPT conceptualizes depression as involving predisposing, precipitating, and perpetuating biopsychosocial factors, including:
• underlying biological and social vulnerability, such as insecure attach ment (ie, tenuous and often negative views of self and others)
• current interpersonal life stressors
• inadequate social supports.15,16
For example, poor early attachment to caregivers can give rise to despair, isolation, and low mood. In turn, this can be exacerbated by poor social and communication skills that promote further rejection and withdrawal of social support and thus, intensified despair, isolation, and low mood. As in Timothy’s case, this vicious cycle underscores psychosocial stressors as a causal factor, maintaining factor, and result of depression. Specifically, IPT conceptualizes 4 main biopsychosocial problem domains:
• grief and loss
• interpersonal disputes
• role transitions
• interpersonal/communication deficits (often connected to isolation).
Working within 1 or 2 of the most salient problem domains, IPT centers on strategies for helping patients solve interpersonal problems based on the notion that modified relationships, revised interpersonal expectations, improved communications, and increased social support will lead to symptom reduction.15-17
Many techniques are utilized in IPT (Table 1) to help patients modify their interpersonal relationships as a mechanism for decreasing their distress. IPT is problem-focused, aiming to improve patients’ relationships by drawing on their assets and helping to build skills around shortcomings. Therefore, IPT focuses on observable interpersonal patterns, as opposed to latent personality dynamics.
CASE CONTINUED
Setting goals
When the clinical explains in the non-technical terms the data supporting IPT’s efficacy for depression, including with young adults, Timothy agrees to teeatment with acute IPT. The therapist behins with consciousness-raising techniques to help Timothy adopt the “sick role” by viewing depressing as an illness to be cured. Collaboratively, they establish treatment goals that fit the IPT formulation of depression— ie, revising current relationships and expectations of them, increasing social support, improving communication skills, and solving problems within 1 or 2 of the IPT problem domains.
For Timothy, the most pressing psychosocial problems seem to be interpersonal deficits and role transitions. He appears to be insecurely attached to others, which is a risk factor for poor facilitation of, and boundaries around, good relationships. A transition to a new and intimidating interpersonal context—living on a college campus—compounded his vulnerabilities and increased his depression.
Acute treatment. The acute phase of IPT is time-limited—often, 12 to 16 sessions with gradual tapering toward the end (akin to a continuation phase). The time limit’s purpose is to focus both patient and therapist on the specific goal of removing the acute “illness” of depression. The IPT clinician takes an interpersonal inventory to learn about the patient’s most important relationships and hones in on the IPT domain foci. Working collaboratively, the therapist might help the patient mourn a loss, reconstruct a narrative with a deceased loved one, consider ways to increase social contact, develop assertiveness, label feelings and needs, resolve an impasse with a significant other, and so forth.
The IPT therapist is an advocate for the patient and adopts an active stance laced with empathy and warmth. However, the therapist is more than unconditionally accepting as depression is viewed as a problem to be actively resolved.
CASE CONTINUED
Creating new patterns
The therapist uses various IPT strategies to work collaboratively with Timothy. She attempts to develop a strong working alliance by building interpersonal safety and trust— which take time with an insecurely attached patient. She tries to provide a new model for how close relationships can develop, while also focusing on current relationships. She and Timothy address his romantic desire for a coworker and work on developing realistic expectations and effective methods for conveying his interest.
When Timothy approaches his coworker, she does not reject him—as he expected— but wants to pursue friendship before possibly dating. The therapist then works with Timothy’s emotional reaction and explores ways to effectively convey his emotions to this young woman. Drawing on communication analysis and problem-solving strategies, Timothy is able to sustain this friendship—a shift from his typical retreat when relationships have not gone as hoped or expected.
Timothy develops confidence to take more risks in initiating social encounters and starts to confide in his roommates when he feels upset. After 3 months of treatment, his expanded social network and improved interpersonal skills result in decreased depression. When Timothy suggests termination, he and the therapist agree to end acute IPT but—given his history of depression—to continue maintenance sessions.
Limited data exist on variables that relate to IPT’s acute success or conditions under which it works best. Although process research lags behind acute IPT outcome research, some findings can help guide the IPT practitioner. For example, variables shown to predict outcomes of acute IPT for depression include a positive therapeutic alliance, therapist warmth, and psycho psychotherapist use of exploratory techniques (Table 2).
Similarly, IPT has been shown to be more effective in some patients than others, depending on various moderators of depression. For example:
• For patients with high cognitive dysfunction, IPT outperforms CBT.
• For patients with higher need for medical reassurance, IPT outperforms selective serotonin reuptake inhibitor (SSRI) pharmacotherapy.
• For patients with severe depression, CBT outperforms IPT.
• For patients with low psychomotor activation, response is more rapid with an SSRI than with IPT (Table 3).18
Durability of acute IPT
One way to understand recurrence prevention is to examine the durability of a treatment’s acute effect in the absence of a specific maintenance plan. In theory, patients will continue to apply the skills learned in acute IPT to maintain gains and prevent recurrences, even after they stop seeing the psychotherapist.
Initial findings. Some research speaks to IPT’s acute-phase durability. The inaugural clinical trial of IPT by Weissman et al19 included 4 months of acute treatment and a 1-year uncontrolled naturalistic follow-up assessment. At follow-up, depression and global clinical symptoms were the same, whether patients had been acutely treated with IPT alone, pharmacotherapy alone (amitriptyline), combined IPT and pharmacotherapy, or nonscheduled treatment with a psychiatrist.
Some patients continued to function well, whereas others did not fully maintain acute treatment gains. Patients who received IPT acutely, either singly or with medication, showed better social functioning at follow-up compared with patients who did not receive IPT. This long-term durability of social improvements was an obvious target of IPT.
Support from TDCRP. In the National Institute of Mental Health Treatment of Depression Collaborative Research Project (TDCRP),20 patients in the acute phase of depression were assigned to 16 weeks of IPT, CBT, pharmacotherapy (imipramine) and clinical management (CM), or placebo plus CM. Among those who recovered by acute treatment’s end, MDD relapse rates at 18-month naturalistic follow-up were 33% for IPT, 36% for CBT, 50% for imipramine, and 33% for placebo. Between-group differences were not statistically significant.
Because acute responders to different types of treatment might have different inherent relapse tendencies, these data do not support causal attributions about the enduring effects of acute-phase treatment. The relapse rates do suggest, however, that 16 weeks of acute treatment, irrespective of kind, was insufficient for some patients to achieve full recovery and lasting remission. Consistent with the initial IPT trial,19 IPT (and CBT) outperformed medi cation and placebo in maintaining relationship quality.21
Long-term benefits. A more recent trial by Zobel et al22 examined the durability of benefits from 5 weeks of acute IPT plus pharmacotherapy and pharmacotherapy plus CM for inpatients with MDD. Although caution is required in interpreting naturalistic follow-up studies, patients in both groups showed decreased depression from baseline to 5-year follow-up. Early symptom reduction was more rapid for patients in the IPT plus pharmacotherapy group, but no significant difference existed at 5 years. More IPT patients than CM patients showed sustained remission (28% vs 11%, respectively). These rates demonstrate a need for longer-term potency of acute treatments and more targeted maintenance treatments.
IPT-M for preventing recurrence
A second way to understand recurrence prevention is to examine the efficacy of a treatment’s maintenance protocol added to an acute treatment phase. IPT has been adapted as a maintenance treatment (IPT-M), with emphasis on keeping patients well. With this revised focus, IPT-M differs somewhat from acute IPT. Although treatment continues to center on interpersonal functioning, IPT-M favors:
• vigilance for possible triggers of new depressive episodes
• longer-term contact with a therapist
• reinforcing skills learned
• addressing an expanded number of interpersonal problem areas (given that such problems can be addressed more efficiently relative to acute treatment).
Efficacy of IPT-M. In the initial trial, Frank et al23 compared the efficacy of IPT-M with that of pharmacotherapy (imipramine) in preventing depressive relapse among patients with recurrent depression who had responded to ≥16 sessions of acute IPT and imipramine and remained well during a 17- week continuation phase. For maintenance, patients were assigned to IPT-M alone, imipramine alone, placebo alone, IPT-M plus imipramine, or IPT-M plus placebo. Maintenance imipramine was continued at the acute dosage (target 200 mg/d; up to 400 mg/d was allowed). Maintenance IPT was monthly sessions. Patients remained in the trial for 3 years or until depression recurred.
On its own, IPT-M showed some efficacy in preventing recurrence, as the mean time to recurrence was 82 weeks for IPT-M alone and 74 weeks for IPT-M plus placebo. The prophylactic effect of imipramine was stronger, however. The mean time to recurrence for imipramine with IPT was 131 weeks, and the mean time to recurrence for imipramine without IPT was 124 weeks. Therefore, whereas monthly IPT-M can certainly help prolong wellness and delay recurrence, IPT maintenance treatment with acute doses of imipramine might be even more effective— if the patient is willing to take medication. These findings must be considered with caution because of the inherent inequity between imipramine and IPT-M in regard to maintenance dosage strength.
Frequency of treatment. In another trial, Frank et al24 examined whether the frequency of maintenance IPT sessions played a role in its prophylactic effect. Adult women who had achieved depression remission with acute IPT (alone or in combination with SSRI pharmacotherapy) were randomized to weekly, bi-weekly, or monthly IPT-M alone for 2 years or until recurrence. Depression recurred during IPT-M in:
• 26% of patients who had received acute IPT alone
• 50% of those who had received acute IPT plus an SSRI.
Frequency of IPT-M sessions did not affect time to recurrence. Thus, for women who can achieve remission with IPT alone, varied frequencies of IPT-M can be good prophylaxis. For women who need an SSRI to augment acute IPT, IPT-M alone at varied dosages is less effective in preventing depression recurrence. Therefore, acute treatment response patterns can inform maintenance plans, with the most prudent maintenance strategy being to maintain the acute treatment strategy over a longer period.
IPT-M for late-life depression. A trial by Reynolds et al25 examined the efficacy of maintenance nortriptyline and IPT-M in preventing depression recurrence in patients age ≥59 who initially recovered after combined acute and continuation IPT plus nortriptyline. The 4 conditions (with their recurrence rates) were:
• monthly IPT-M with nortriptyline (20%)
• monthly IPT-M with placebo (64%)
• nortriptyline plus medication visits (43%)
• placebo plus medication visits (90%).
Clearly, the combined active treatments outperformed placebo and antidepressant alone in terms of delaying or preventing recurrence, which suggests an optimal maintenance strategy with this population.
IPT-M for later life. Another trial by the same group26 enrolled patients age ≥70 with MDD that responded to acute IPT plus paroxetine. The maintenance treatments to which they were randomly assigned (and recurrence rates within 2 years) were:
• paroxetine plus IPT-M (35%)
• placebo plus IPT-M (68%)
• paroxetine plus clinical management (37%)
• placebo plus clinical management (58%).
Recurrence rates were the same for patients receiving medication plus IPT-M and medication plus clinical management, and depression was 2.4 times more likely to recur in patients receiving placebo vs active medication. Therefore, for later life depression, the optimal maintenance strategy was the SSRI.
Secondary analyses of data from these seminal trials of IPT-M point to other predictors of how and for whom maintenance IPT may work (Table 4). For example:
• Greater variability of depression symptoms during all forms of maintenance treatment is related to a greater risk of recurrence.
• Persistent insomnia is related to greater risk of recurrent depression.
• High interpersonal focus in IPT-M sessions is related to longer time to recurrence.
Bottom Line
Interpersonal psychotherapy (IPT) is efficacious for acute depression and for preventing recurrences. Patients treated successfully with acute IPT alone benefit from varied doses of maintenance IPT. Combining IPT-M with antidepressant medication can be more potent than IPT-M alone. For late-life depression, medication appears to be most effective for maintenance treatment.
Related Resources
Media
• Video demonstration, role-play transcripts, lesson plans, and quizzes. In: Appendices in and DVD companion to Ravitz P, Watson P, Grigoriadas S. Interpersonal psychotherapy for depression. New York, NY: Norton; 2013.
• Video demonstration of IPT sessions. In: DVD companion to Dewan, M, Steenbarger, B, Greenberg, R, eds. The art and science of brief psychotherapies: An illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012.
Text
• Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London, United Kingdom: Taylor & Francis; 2012.
• Weissman MM, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
• Weissman M, Markowitz J, Klerman GL. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
Websites
• Interpersonal Psychotherapy Institute. http://iptinstitute.com.
• International Society for Interpersonal Psychotherapy. http://interpersonalpsychotherapy.org.
Drug Brand Names
Amitriptyline • Elavil Nortriptyline • Pamelor
Imipramine • Tofranil Paroxetine • Paxil
Acknowledgments
The authors are grateful to Samantha L. Bernecker, MS, and Nicholas R. Morrison for their assistance with the research review.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Major depressive disorder (MDD) frequently is recurrent, with new episodes causing substantial social and economic impairment1 and increasing the likelihood of future episodes.2 For this reason, contemporary psychiatric practitioners think of depression treatment as long-term and plan thoughtfully for maintenance therapy.
Recognizing the importance of engaging depressed individuals beyond the initial response,3 American Psychiatric Association practice guidelines conceptualize depression treatment as 3 phases:
• acute treatment, with the aim of remission (symptom removal)
• continuation treatment, with the aim of preventing relapse (symptom return)
• maintenance treatment, with the aim of preventing recurrence (new episodes).4
Interpersonal psychotherapy (IPT) is an evidence-based psychosocial treatment that adheres to this model.5 As a time-limited, manual-driven6,7 approach, IPT focuses on interpersonal distresses as precipitating and perpetuating factors of depression.8
Acute IPT’s efficacy is well-established across >200 empirical studies—making it an evidence-based, first-line treatment for adult depression.4,9,10 Meta-analyses show that acute IPT is superior to placebo and no-treatment controls, and largely comparable to antidepressant medication and other active, first-line psychotherapies, such as cognitive-behavioral therapy (CBT).11,12
Although this review, as well as the literature, focuses largely on adult outpatients with depression, evidence of IPT’s general efficacy exists for adolescents,13 chronically depressed patients,11 and depressed inpatients.14 This article presents a case study to describe the structure of IPT when used to treat depressed adults. We also present evidence of IPT’s acute and long-term efficacy in preventing depression recurrence and data to guide its use in practice.
CASE REPORT
‘Safe’ but depressed
Timothy, age 18, is a first-year college student who presents for outpatient psychotherapy to address recurrent depression. He reports general unhappiness, loss of interest in things, low energy, sleep problems, poor academic and work functioning, and low self-esteem. He experienced at least 3 similar depressive episodes while in high school.
The therapist’s diagnostic and interpersonal assessment suggests that Timothy’s depression is interpersonally driven. Timothy longs for relational intimacy but fears he will fail or burden people with his needs. He has difficulty gauging appropriate levels of enmeshment with others and either becomes overdependent or stays at a distance. This “safe” approach to relationships contributes to boredom, loneliness, and isolation. His recent transition to college away from home and the failure of a romantic relationship have compounded these experiences.
Interpersonal model of IPT
IPT conceptualizes depression as involving predisposing, precipitating, and perpetuating biopsychosocial factors, including:
• underlying biological and social vulnerability, such as insecure attach ment (ie, tenuous and often negative views of self and others)
• current interpersonal life stressors
• inadequate social supports.15,16
For example, poor early attachment to caregivers can give rise to despair, isolation, and low mood. In turn, this can be exacerbated by poor social and communication skills that promote further rejection and withdrawal of social support and thus, intensified despair, isolation, and low mood. As in Timothy’s case, this vicious cycle underscores psychosocial stressors as a causal factor, maintaining factor, and result of depression. Specifically, IPT conceptualizes 4 main biopsychosocial problem domains:
• grief and loss
• interpersonal disputes
• role transitions
• interpersonal/communication deficits (often connected to isolation).
Working within 1 or 2 of the most salient problem domains, IPT centers on strategies for helping patients solve interpersonal problems based on the notion that modified relationships, revised interpersonal expectations, improved communications, and increased social support will lead to symptom reduction.15-17
Many techniques are utilized in IPT (Table 1) to help patients modify their interpersonal relationships as a mechanism for decreasing their distress. IPT is problem-focused, aiming to improve patients’ relationships by drawing on their assets and helping to build skills around shortcomings. Therefore, IPT focuses on observable interpersonal patterns, as opposed to latent personality dynamics.
CASE CONTINUED
Setting goals
When the clinical explains in the non-technical terms the data supporting IPT’s efficacy for depression, including with young adults, Timothy agrees to teeatment with acute IPT. The therapist behins with consciousness-raising techniques to help Timothy adopt the “sick role” by viewing depressing as an illness to be cured. Collaboratively, they establish treatment goals that fit the IPT formulation of depression— ie, revising current relationships and expectations of them, increasing social support, improving communication skills, and solving problems within 1 or 2 of the IPT problem domains.
For Timothy, the most pressing psychosocial problems seem to be interpersonal deficits and role transitions. He appears to be insecurely attached to others, which is a risk factor for poor facilitation of, and boundaries around, good relationships. A transition to a new and intimidating interpersonal context—living on a college campus—compounded his vulnerabilities and increased his depression.
Acute treatment. The acute phase of IPT is time-limited—often, 12 to 16 sessions with gradual tapering toward the end (akin to a continuation phase). The time limit’s purpose is to focus both patient and therapist on the specific goal of removing the acute “illness” of depression. The IPT clinician takes an interpersonal inventory to learn about the patient’s most important relationships and hones in on the IPT domain foci. Working collaboratively, the therapist might help the patient mourn a loss, reconstruct a narrative with a deceased loved one, consider ways to increase social contact, develop assertiveness, label feelings and needs, resolve an impasse with a significant other, and so forth.
The IPT therapist is an advocate for the patient and adopts an active stance laced with empathy and warmth. However, the therapist is more than unconditionally accepting as depression is viewed as a problem to be actively resolved.
CASE CONTINUED
Creating new patterns
The therapist uses various IPT strategies to work collaboratively with Timothy. She attempts to develop a strong working alliance by building interpersonal safety and trust— which take time with an insecurely attached patient. She tries to provide a new model for how close relationships can develop, while also focusing on current relationships. She and Timothy address his romantic desire for a coworker and work on developing realistic expectations and effective methods for conveying his interest.
When Timothy approaches his coworker, she does not reject him—as he expected— but wants to pursue friendship before possibly dating. The therapist then works with Timothy’s emotional reaction and explores ways to effectively convey his emotions to this young woman. Drawing on communication analysis and problem-solving strategies, Timothy is able to sustain this friendship—a shift from his typical retreat when relationships have not gone as hoped or expected.
Timothy develops confidence to take more risks in initiating social encounters and starts to confide in his roommates when he feels upset. After 3 months of treatment, his expanded social network and improved interpersonal skills result in decreased depression. When Timothy suggests termination, he and the therapist agree to end acute IPT but—given his history of depression—to continue maintenance sessions.
Limited data exist on variables that relate to IPT’s acute success or conditions under which it works best. Although process research lags behind acute IPT outcome research, some findings can help guide the IPT practitioner. For example, variables shown to predict outcomes of acute IPT for depression include a positive therapeutic alliance, therapist warmth, and psycho psychotherapist use of exploratory techniques (Table 2).
Similarly, IPT has been shown to be more effective in some patients than others, depending on various moderators of depression. For example:
• For patients with high cognitive dysfunction, IPT outperforms CBT.
• For patients with higher need for medical reassurance, IPT outperforms selective serotonin reuptake inhibitor (SSRI) pharmacotherapy.
• For patients with severe depression, CBT outperforms IPT.
• For patients with low psychomotor activation, response is more rapid with an SSRI than with IPT (Table 3).18
Durability of acute IPT
One way to understand recurrence prevention is to examine the durability of a treatment’s acute effect in the absence of a specific maintenance plan. In theory, patients will continue to apply the skills learned in acute IPT to maintain gains and prevent recurrences, even after they stop seeing the psychotherapist.
Initial findings. Some research speaks to IPT’s acute-phase durability. The inaugural clinical trial of IPT by Weissman et al19 included 4 months of acute treatment and a 1-year uncontrolled naturalistic follow-up assessment. At follow-up, depression and global clinical symptoms were the same, whether patients had been acutely treated with IPT alone, pharmacotherapy alone (amitriptyline), combined IPT and pharmacotherapy, or nonscheduled treatment with a psychiatrist.
Some patients continued to function well, whereas others did not fully maintain acute treatment gains. Patients who received IPT acutely, either singly or with medication, showed better social functioning at follow-up compared with patients who did not receive IPT. This long-term durability of social improvements was an obvious target of IPT.
Support from TDCRP. In the National Institute of Mental Health Treatment of Depression Collaborative Research Project (TDCRP),20 patients in the acute phase of depression were assigned to 16 weeks of IPT, CBT, pharmacotherapy (imipramine) and clinical management (CM), or placebo plus CM. Among those who recovered by acute treatment’s end, MDD relapse rates at 18-month naturalistic follow-up were 33% for IPT, 36% for CBT, 50% for imipramine, and 33% for placebo. Between-group differences were not statistically significant.
Because acute responders to different types of treatment might have different inherent relapse tendencies, these data do not support causal attributions about the enduring effects of acute-phase treatment. The relapse rates do suggest, however, that 16 weeks of acute treatment, irrespective of kind, was insufficient for some patients to achieve full recovery and lasting remission. Consistent with the initial IPT trial,19 IPT (and CBT) outperformed medi cation and placebo in maintaining relationship quality.21
Long-term benefits. A more recent trial by Zobel et al22 examined the durability of benefits from 5 weeks of acute IPT plus pharmacotherapy and pharmacotherapy plus CM for inpatients with MDD. Although caution is required in interpreting naturalistic follow-up studies, patients in both groups showed decreased depression from baseline to 5-year follow-up. Early symptom reduction was more rapid for patients in the IPT plus pharmacotherapy group, but no significant difference existed at 5 years. More IPT patients than CM patients showed sustained remission (28% vs 11%, respectively). These rates demonstrate a need for longer-term potency of acute treatments and more targeted maintenance treatments.
IPT-M for preventing recurrence
A second way to understand recurrence prevention is to examine the efficacy of a treatment’s maintenance protocol added to an acute treatment phase. IPT has been adapted as a maintenance treatment (IPT-M), with emphasis on keeping patients well. With this revised focus, IPT-M differs somewhat from acute IPT. Although treatment continues to center on interpersonal functioning, IPT-M favors:
• vigilance for possible triggers of new depressive episodes
• longer-term contact with a therapist
• reinforcing skills learned
• addressing an expanded number of interpersonal problem areas (given that such problems can be addressed more efficiently relative to acute treatment).
Efficacy of IPT-M. In the initial trial, Frank et al23 compared the efficacy of IPT-M with that of pharmacotherapy (imipramine) in preventing depressive relapse among patients with recurrent depression who had responded to ≥16 sessions of acute IPT and imipramine and remained well during a 17- week continuation phase. For maintenance, patients were assigned to IPT-M alone, imipramine alone, placebo alone, IPT-M plus imipramine, or IPT-M plus placebo. Maintenance imipramine was continued at the acute dosage (target 200 mg/d; up to 400 mg/d was allowed). Maintenance IPT was monthly sessions. Patients remained in the trial for 3 years or until depression recurred.
On its own, IPT-M showed some efficacy in preventing recurrence, as the mean time to recurrence was 82 weeks for IPT-M alone and 74 weeks for IPT-M plus placebo. The prophylactic effect of imipramine was stronger, however. The mean time to recurrence for imipramine with IPT was 131 weeks, and the mean time to recurrence for imipramine without IPT was 124 weeks. Therefore, whereas monthly IPT-M can certainly help prolong wellness and delay recurrence, IPT maintenance treatment with acute doses of imipramine might be even more effective— if the patient is willing to take medication. These findings must be considered with caution because of the inherent inequity between imipramine and IPT-M in regard to maintenance dosage strength.
Frequency of treatment. In another trial, Frank et al24 examined whether the frequency of maintenance IPT sessions played a role in its prophylactic effect. Adult women who had achieved depression remission with acute IPT (alone or in combination with SSRI pharmacotherapy) were randomized to weekly, bi-weekly, or monthly IPT-M alone for 2 years or until recurrence. Depression recurred during IPT-M in:
• 26% of patients who had received acute IPT alone
• 50% of those who had received acute IPT plus an SSRI.
Frequency of IPT-M sessions did not affect time to recurrence. Thus, for women who can achieve remission with IPT alone, varied frequencies of IPT-M can be good prophylaxis. For women who need an SSRI to augment acute IPT, IPT-M alone at varied dosages is less effective in preventing depression recurrence. Therefore, acute treatment response patterns can inform maintenance plans, with the most prudent maintenance strategy being to maintain the acute treatment strategy over a longer period.
IPT-M for late-life depression. A trial by Reynolds et al25 examined the efficacy of maintenance nortriptyline and IPT-M in preventing depression recurrence in patients age ≥59 who initially recovered after combined acute and continuation IPT plus nortriptyline. The 4 conditions (with their recurrence rates) were:
• monthly IPT-M with nortriptyline (20%)
• monthly IPT-M with placebo (64%)
• nortriptyline plus medication visits (43%)
• placebo plus medication visits (90%).
Clearly, the combined active treatments outperformed placebo and antidepressant alone in terms of delaying or preventing recurrence, which suggests an optimal maintenance strategy with this population.
IPT-M for later life. Another trial by the same group26 enrolled patients age ≥70 with MDD that responded to acute IPT plus paroxetine. The maintenance treatments to which they were randomly assigned (and recurrence rates within 2 years) were:
• paroxetine plus IPT-M (35%)
• placebo plus IPT-M (68%)
• paroxetine plus clinical management (37%)
• placebo plus clinical management (58%).
Recurrence rates were the same for patients receiving medication plus IPT-M and medication plus clinical management, and depression was 2.4 times more likely to recur in patients receiving placebo vs active medication. Therefore, for later life depression, the optimal maintenance strategy was the SSRI.
Secondary analyses of data from these seminal trials of IPT-M point to other predictors of how and for whom maintenance IPT may work (Table 4). For example:
• Greater variability of depression symptoms during all forms of maintenance treatment is related to a greater risk of recurrence.
• Persistent insomnia is related to greater risk of recurrent depression.
• High interpersonal focus in IPT-M sessions is related to longer time to recurrence.
Bottom Line
Interpersonal psychotherapy (IPT) is efficacious for acute depression and for preventing recurrences. Patients treated successfully with acute IPT alone benefit from varied doses of maintenance IPT. Combining IPT-M with antidepressant medication can be more potent than IPT-M alone. For late-life depression, medication appears to be most effective for maintenance treatment.
Related Resources
Media
• Video demonstration, role-play transcripts, lesson plans, and quizzes. In: Appendices in and DVD companion to Ravitz P, Watson P, Grigoriadas S. Interpersonal psychotherapy for depression. New York, NY: Norton; 2013.
• Video demonstration of IPT sessions. In: DVD companion to Dewan, M, Steenbarger, B, Greenberg, R, eds. The art and science of brief psychotherapies: An illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012.
Text
• Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London, United Kingdom: Taylor & Francis; 2012.
• Weissman MM, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
• Weissman M, Markowitz J, Klerman GL. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
Websites
• Interpersonal Psychotherapy Institute. http://iptinstitute.com.
• International Society for Interpersonal Psychotherapy. http://interpersonalpsychotherapy.org.
Drug Brand Names
Amitriptyline • Elavil Nortriptyline • Pamelor
Imipramine • Tofranil Paroxetine • Paxil
Acknowledgments
The authors are grateful to Samantha L. Bernecker, MS, and Nicholas R. Morrison for their assistance with the research review.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. ten Doesschate MC, Koeter MW, Bockting CL, et al. Health related quality of life in recurrent depression: a comparison with a general population sample. J Affect Disord. 2010; 120(1-3):126-132.
2. Hardeveld F, Spijker J, De Graaf R, et al. Prevalence and predictors of recurrence of major depressive disorder in the adult population. Acta Psychiatr Scand. 2010;122(3):184-91.
3. Arnow BA, Constantino MJ. Effectiveness of psychotherapy and combination treatment for chronic depression. J Clin Psychol. 2003;59(8):893-905.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2010.
5. Klerman GL, Weissman MM, Rounsaville BJ, et al. Interpersonal psychotherapy of depression. New York, NY: Basic Books; 1984.
6. Weissman MM, Markowitz JC, Klerman G. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
7. Weissman M, Markowitz J, Klerman G. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
8. Brakemeier EL, Frase L. Interpersonal psychotherapy (IPT) in major depressive disorder. Eur Arch Psychiatry Clin Neurosci. 2012;262(suppl 2):S117-1121.
9. Depression in adults (update): NICE guideline CG90). National Institute for Health and Care Excellence. (2009). http://www.nice.org.uk/cg90. Updated October 2009. Accessed March 5, 2014.
10. Depression. National Institutes of Mental Health. http://www.nimh.nih.gov/health/publications/depression/ index.shtml. Revised 2011. Accessed March 5, 2014.
11. Cuijpers P, van Straten A, Andersson G, et al. Psychotherapy for depression in adults: a meta-analysis of comparative outcome studies. J Consult Clin Psychol. 2008;76(6):909-922.
12. Cuijpers P, Geraedts AS, van Oppen P, et al. Interpersonal psychotherapy for depression: a meta-analysis [Erratum in: Am J Psychiatry. 2011;168(6):652]. Am J Psychiatry. 2011; 168(6):581-592.
13. Mufson L, Dorta K, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6): 577-584.
14. Schramm E, Schneider D, Zobel I, et al. Efficacy of interpersonal psychotherapy plus pharmacotherapy in chronically depressed inpatients. J Affect Disord. 2008; 109(1-2):65-73.
15. Bernecker SL. How and for whom does interpersonal psychotherapy work? Psychotherapy Bulletin. 2012;47(2):13-17.
16. Stuart S. Interpersonal psychotherapy. In: Dewan MJ, Steenbarger BN, Greenberg RP, eds. The art and science of brief psychotherapies: an illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012: 157-193.
17. Grigoriadas S, Watson P, Maunder R, eds. Psychotherapy essentials to go: Interpersonal psychotherapy for depression. New York, NY: W. W. Norton & Company, Inc.; 2013.
18. Bleiberg KL, Markowitz JC. Interpersonal psychotherapy for depression. In: Barlow D, ed. Clinical handbook of psychological disorders: a step-by-step treatment manual. New York, NY: The Guilford Press; 2008:306-327.
19. Weissman MM, Klerman GL, Prusoff BA, et al. Depressed outpatients. Results one year after treatment with drugs and/or interpersonal psychotherapy. Arch Gen Psychiatry. 1981;38(1):51-55.
20. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49(10):782-787.
21. Blatt S, Zuroff D, Bondi C, et al. Short- and long-term effect of medication and psychotherapy in the brief treatment of depression: further analyses of data from the NIMH TDCRP. Psychother Res. 2000;10(2):215-234.
22. Zobel I, Kech S, van Calker D, et al. Long-term effect of combined interpersonal psychotherapy and pharmacotherapy in a randomized trial of depressed patients. Acta Psychiatr Scand. 2011;123(4):276-282.
23. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry. 1990;47(12):1093-1099.
24. Frank E, Kupfer DJ, Buysse DJ, et al. Randomized trial of weekly, twice-monthly, and monthly interpersonal psychotherapy as maintenance treatment for women with recurrent depression. Am J Psychiatry. 2007;164(5): 761-767.
25. Reynolds CF 3rd, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA. 1999;281(1): 39-45.
26. Reynolds CF 3rd, Dew MA, Pollock BG, et al. Maintenance treatment of major depression in old age. N Engl J Med. 2006;354(11):1130-1138.
1. ten Doesschate MC, Koeter MW, Bockting CL, et al. Health related quality of life in recurrent depression: a comparison with a general population sample. J Affect Disord. 2010; 120(1-3):126-132.
2. Hardeveld F, Spijker J, De Graaf R, et al. Prevalence and predictors of recurrence of major depressive disorder in the adult population. Acta Psychiatr Scand. 2010;122(3):184-91.
3. Arnow BA, Constantino MJ. Effectiveness of psychotherapy and combination treatment for chronic depression. J Clin Psychol. 2003;59(8):893-905.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2010.
5. Klerman GL, Weissman MM, Rounsaville BJ, et al. Interpersonal psychotherapy of depression. New York, NY: Basic Books; 1984.
6. Weissman MM, Markowitz JC, Klerman G. Comprehensive guide to interpersonal psychotherapy. New York, NY: Basic Books; 2000.
7. Weissman M, Markowitz J, Klerman G. Clinician’s quick guide to interpersonal psychotherapy. New York, NY: Oxford University Press; 2007.
8. Brakemeier EL, Frase L. Interpersonal psychotherapy (IPT) in major depressive disorder. Eur Arch Psychiatry Clin Neurosci. 2012;262(suppl 2):S117-1121.
9. Depression in adults (update): NICE guideline CG90). National Institute for Health and Care Excellence. (2009). http://www.nice.org.uk/cg90. Updated October 2009. Accessed March 5, 2014.
10. Depression. National Institutes of Mental Health. http://www.nimh.nih.gov/health/publications/depression/ index.shtml. Revised 2011. Accessed March 5, 2014.
11. Cuijpers P, van Straten A, Andersson G, et al. Psychotherapy for depression in adults: a meta-analysis of comparative outcome studies. J Consult Clin Psychol. 2008;76(6):909-922.
12. Cuijpers P, Geraedts AS, van Oppen P, et al. Interpersonal psychotherapy for depression: a meta-analysis [Erratum in: Am J Psychiatry. 2011;168(6):652]. Am J Psychiatry. 2011; 168(6):581-592.
13. Mufson L, Dorta K, Wickramaratne P, et al. A randomized effectiveness trial of interpersonal psychotherapy for depressed adolescents. Arch Gen Psychiatry. 2004;61(6): 577-584.
14. Schramm E, Schneider D, Zobel I, et al. Efficacy of interpersonal psychotherapy plus pharmacotherapy in chronically depressed inpatients. J Affect Disord. 2008; 109(1-2):65-73.
15. Bernecker SL. How and for whom does interpersonal psychotherapy work? Psychotherapy Bulletin. 2012;47(2):13-17.
16. Stuart S. Interpersonal psychotherapy. In: Dewan MJ, Steenbarger BN, Greenberg RP, eds. The art and science of brief psychotherapies: an illustrated guide. 2nd ed. Arlington, VA: American Psychiatric Publishing; 2012: 157-193.
17. Grigoriadas S, Watson P, Maunder R, eds. Psychotherapy essentials to go: Interpersonal psychotherapy for depression. New York, NY: W. W. Norton & Company, Inc.; 2013.
18. Bleiberg KL, Markowitz JC. Interpersonal psychotherapy for depression. In: Barlow D, ed. Clinical handbook of psychological disorders: a step-by-step treatment manual. New York, NY: The Guilford Press; 2008:306-327.
19. Weissman MM, Klerman GL, Prusoff BA, et al. Depressed outpatients. Results one year after treatment with drugs and/or interpersonal psychotherapy. Arch Gen Psychiatry. 1981;38(1):51-55.
20. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49(10):782-787.
21. Blatt S, Zuroff D, Bondi C, et al. Short- and long-term effect of medication and psychotherapy in the brief treatment of depression: further analyses of data from the NIMH TDCRP. Psychother Res. 2000;10(2):215-234.
22. Zobel I, Kech S, van Calker D, et al. Long-term effect of combined interpersonal psychotherapy and pharmacotherapy in a randomized trial of depressed patients. Acta Psychiatr Scand. 2011;123(4):276-282.
23. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry. 1990;47(12):1093-1099.
24. Frank E, Kupfer DJ, Buysse DJ, et al. Randomized trial of weekly, twice-monthly, and monthly interpersonal psychotherapy as maintenance treatment for women with recurrent depression. Am J Psychiatry. 2007;164(5): 761-767.
25. Reynolds CF 3rd, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA. 1999;281(1): 39-45.
26. Reynolds CF 3rd, Dew MA, Pollock BG, et al. Maintenance treatment of major depression in old age. N Engl J Med. 2006;354(11):1130-1138.

Taking the spice route: Psychoactive properties of culinary spices
Many substances that are not typically thought of as “substances of abuse” possess—when adequately dosed—clinically meaningful psychoactive properties. In addition to the more familiar effects of alcohol, psychostimulants, opioids, Cannabis, and hallucinogens, you may encounter psychiatric phenomena resulting from abuse of more obscure substances, including culinary spices.
The clinician treating a patient in an apparent intoxicated state who has a negative drug screen might ask that patient if he (she) abuses spices. This might be particularly relevant when treating patients thought to have limited access to illicit substances or those with ready access to large amounts of spices, such as prisoners, young patients, and those working in the food service industry.
Abuse of spices can be a problematic diagnosis
Patients may misuse culinary spices to achieve euphoria, or a “natural high.” They may present with medical or psychiatric symptoms, including acute altered mental status, but the psychoactive substances are not identified on routine toxicology studies. In addition, patients may not attribute their use of spices for psychoactive effect to “drugs,” because these materials are legal and readily available. This may lead to misdiagnosis of a systemic medical disorder or a primary psychiatric illness to explain the patient’s symptoms and initiating a psychotropic agent and other psychiatric services when a substance abuse program might be a more appropriate clinical intervention.
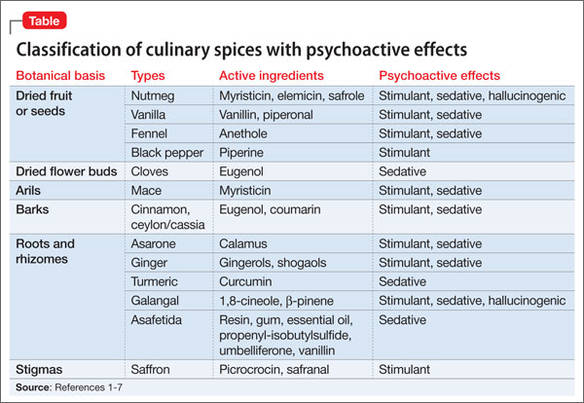
Some spices contain psychoactive compounds that can alter CNS function (Table1-7), might be abused for recreational purposes, and can be toxic in an excessive amount. Internet resources, including anonymous web-based communications, and anecdotal materials about non-traditional recreational drugs, are available to anyone with Internet access.8 However, little research has been conducted into the prevalence of abuse (Box)9 and spices’ psychoactive properties. The lack of toxicology detection of spices in the medical setting presents a diagnostic challenge.
The psychoactive plants used in “natural high” products mainly are psychoactively inactive in their natural form, but extracts or alkaloids obtained from them might induce 1 or more of 3 classifications of psychoactivity:
• stimulant
• sedative
• hallucinogenic.
Many of these substances are considered to be aphrodisiac, and some may be abused to increase sexual function.
The following is a review of common spices that have been reported to possess potential psychoactive properties.
Nutmeg
Nutmeg (Myristica fragrans) is a common and easily accessible means of reaching euphoria in adults.10 The aromatic oil of nutmeg contains myristicin, a psychoactive substance that is chemically similar to hallucinogenic compounds such as mescaline. Its psychoactive effects could be attributed to metabolic formation of amphetamine derivatives from its core ingredients, elemicin, myristicin, and safrole.11,12
Nutmeg and its active component, myristicin, produce central monoamine oxidase (MAO) inhibition as evidenced by the ability to lower the convulsive dose of IV tryptamine in mice and to increase brain 5-hydroxytryptamine concentrations.13,14 Although myristicin’s potency is not comparable to that of the more potent MAO inhibitors such as tranylcypromine and iproniazid (which is not available in the United States), it seems adequate when compared with its low toxicity.14 Nutmeg extract is associated with a significant antidepressant effect in mice, which seemed to be mediated by interaction with the adrenergic, dopaminergic, and serotonergic systems.13 Nutmeg is associated with sustained increase in sexual activity in animal studies, with no evidence of adverse effects and toxicity, suggesting that nutmeg possesses clinically significant aphrodisiac activity.15
Psychoactive effects can be achieved by ingesting 5 to 15 g of nutmeg.11 Acute nutmeg intoxication produces palpitations, dizziness, anxiety, and hallucinations, mostly resolving within 24 hours, while effects of chronic abuse are reported to be similar to Cannabis use, including euphoria, giddiness, anxiety, fear, sense of impending doom, detachment, confabulation, and hallucinations.11,16 Urine drug screens are negative unless other psychoactive substances have been ingested.17
Suspected nutmeg intoxication or poisoning should be treated with supportive treatment. Use sedatives with caution because of alternating periods of delirium and obtundation during nutmeg intoxication.17
In case reports, myristicin poisoning induced CNS neuromodulatory signs that mimicked an anticholinergic hyperstimulation state.12,18 Fatal myristicin poisoning is rare; 2 cases have been reported, 1 in combination with flunitrazepam (not available in the United States).19,20 Nutmeg also has sedative properties and can cause GI symptoms when ingesting excessive amounts.1,20,21 Grover et al21 described no harmful effects on blood pressure and electrocardiogram; however, Shah et al22 reported palpitations and dry mouth.
Vanilla
Vanilla (species of the genus Vanilla) contains piperonal, also known as heliotropin.1 Piperonal has aromatherapeutic qualities that might elevate mood and well-being. In the early 1990s, the Memorial Sloan- Kettering Cancer Center in New York City described heliotropin as a powerful aromatherapy tool. Patients who were undergoing an MRI in an environment scented with heliotropin demonstrated a 63% reduction in anxiety compared with those who were not exposed to fragrance.23 The Smell and Taste Treatment and Research Foundation in Chicago found that vanilla can promote sexual arousal.24
Short-term effects of vanillin—a major component of vanilla—include a feeling of relaxation and reduced stress; long-term use can produce an antidepressant effect.1 There are no reports of vanilla abuse to achieve these effects; however, patients might abuse vanilla extract because of its alcohol content (up to 35% ethanol).25
Fennel
The essential oil of fennel (Foeniculum vulgare) can be neurotoxic and epileptogenic. Skalli and colleagues recently reported a case of seizure induction in a young woman after ingesting cakes containing fennel oil.26 Fennel oil also has been reported to have significant interaction with the fluoroquinolone-type antibiotics. Be aware of adverse effects associated with fennel ingestion; question patients if atypical seizures or reactions to antibiotics occur.27
Spices such as fennel, dill, cinnamon, saffron, and anise also contain psychoactive substances that are chemically similar to myristicin, which can induce sedation, stimulation, or hallucinations.7
Black pepper
Piperine, which gives black pepper (Piper nigrum) its spiciness, enhances thermogenesis of lipid metabolism, accelerates energy metabolism, and increases serotonin and endorphin production in the brain.28 Black pepper is reported to potentiate γ-aminobutyric acid A receptor subtypes,29 and could present possible applications for treating insomnia, epilepsy, and anxiety disorders.
Cloves
Non-culinary uses of clove (Syzygium aromaticum, a tree in the myrtle family) include flavored cigarettes. However, in 2009 clove cigarettes were banned in the United States as part of a public policy to reduce the number of children who start smoking.30 Eugenol, which constitutes as much as 90% of the essential oil extracted from cloves (and is responsible for the aroma), can cause hepatotoxicity31 and palpitations32; it can be toxic in quantities as low as 5 mL.33 Eugenol is present in other spices, such as nutmeg and cinnamon, and has been reported to have sedative properties.1
Mace
Mace is made from the covering of nutmeg (Myristica fragrans) seeds. It has a strong aroma resembling that of nutmeg. Whole mace contains 4% to 14% of a volatile oil similar to that found in nutmeg. Because mace contains the same oils that make nutmeg psychoactive1 in excessive amounts—although nutmeg seeds are more potent—be aware of the psychoactive potential of mace.
CinnamonCassia cinnamon (Cinnamomum aromaticum) is spicier and tarter than Ceylon cinnamon (Cinnamomum zeylanicum), which has a more flowery aroma. The 2 types of cinnamon can be distinguished by their different chemical composition. Ceylon cinnamon contains eugenol and benzyl benzoate; cassia cinnamon contains coumarin.3 Eugenol is reported to have sedative effects.1 Coumarin is a precursor molecule in the synthesis of a number of synthetic anticoagulant pharmaceuticals, including coumadin. Because of the toxic component of coumarin, European health agencies have warned against consuming high amounts of cassia.34 There are no reports of side effects arising from the occasional use of cinnamon as a spice.
In a study by Frydman-Marom et al,35 cinnamon extract (CEppt) was found to act on the CNS by inhibiting development of Alzheimer’s disease in animal models.
Asarone
Asarone is found in the Asarum family of spices that includes Acorus calamus. Asarone is chemically similar to mescaline. Although anecdotal reports indicate that A. calamus is a hallucinogen, research shows no evidence that it contains hallucinogenic substances.36 Han et al37 reported an antidepressant effect with the essential oil and asarones for the rhizomes of Acorus tatarinowii. In animal studies, asarone was found to reduce spontaneous motor activity, and even in low doses, reduced anxiety without decreasing acuity of perception.38
Ginger
Ginger (Zingiber officinale) is regarded as a sedative, general stimulant, and aphrodisiac.1,4,5 Its main constituents are phenolic compounds such as gingerols and shogaols, and sesquiterpenes such as zingiberene.4 Ginger is an inhibitor of thromboxane synthetase, a property shared by tricyclic antidepressants.39
Research indicates that 9 compounds found in ginger may interact with the serotonin 5-HT1A receptor, suggesting a possible mechanism for reducing anxiety.40 A study by Nievergelt et al41 indicates that by binding to human serotonin receptors, ginger might influence GI function. Ginger extract contains a cholinergic and spasmogenic component, which provides a mechanistic insight for the prokinetic action of ginger.40
Turmeric
Turmeric (Curcuma longa) has been investigated for possible benefit in Alzheimer’s disease42; research into curcumin, the active substance of turmeric, is increasing. Although the original report was retracted after publication, curcumin was reported to selectively bind to human cannabinoid receptors type 1 (CB1) with nanomolar affinities and to function as an antagonist/inverse agonist.43 However, Gertsch et al44 found that curcumin did not interact functionally with the CB1 receptor, although this compound appears to share ability of the CB1 receptor inverse agonist.
Galangal
Major constituents identified in the galangal (or galanga) rhizome and leaf oil were 1,8-cineole, and β-pinene and camphor.6 Galangal, a member of the ginger (Zingiberaceae) family, interacts with MAO inhibitors, H2 receptor antagonists, and proton-pump inhibitors.1 Anxiolytic, hallucinogenic, and stimulant properties have been reported.1 An excessive amount can induce diarrhea, dizziness, nausea, and vomiting.1
Saffron
Stigma of saffron (a member of the family Iridaceae) was found to be significantly more effective than placebo and equally as efficacious as fluoxetine and imipramine in treating depression. Saffron petal was found to be significantly more effective than placebo and as effective as fluoxetine and saffron stigma in a recent systematic review.45-48
Asafetida
Asafetida (Ferula assa-foetida), when combined with valerian root, is used as a sedative to treat hyperactivity.2 The active ingredients of asafetida are the resin, endogenous gum, essential oil, propenyl-isobutylsulfide, umbelliferone, and vanillin. Several of the volatile constituents produce a sedative effect.2 Additive effects can occur between the hypotensive property of asafetida and dopamine receptor agonists such as bromocriptine mesylate. Use caution when combining asafetida in conjunction with a CNS depressant or a stimulant.2
Recommendations for treating spice-abusers
Patients may present to psychiatry services with psychological and physiological evidence of intoxication with culinary spices that may mimic 1) abuse of other substances, 2) primary psychiatric illness, and 3) primary medical illness. When you encounter a patient with a new psychiatric symptom, consider inquiring about the abuse of spices.
Patients might abuse more than 1 spice; a comprehensive screening approach might therefore be useful. Caution patients that ingesting these substance to excess can have harmful effects. Consider appropriate psychopharmacotherapy for underlying psychiatric symptoms to help patients who use spices maladaptively to self-medicate psychiatric symptoms.
Consider abuse of culinary spices in clinical presentations of psychiatric symptoms that do not seem adequate for a diagnosis of a primary anxiety, mood, or psychotic disorder, or in cases atypical psychiatric presentations that are—perhaps to your surprise—associated with negative toxicology studies for common, more familiar substances of abuse.
Physicians practicing in an environment where street drugs are difficult to obtain (eg, prisons) should consider monitoring for possible abuse of spices. Based on the available, albeit limited, literature, it appears that most culinary spice–associated intoxication can be managed:
• with an elevated level of clinical suspicion
• by ruling out other causes of intoxication
• using targeted, empirical psychopharmacotherapy to manage symptoms
• with supportive care that includes close psychiatric follow-up.
Consider comorbid abuse of other, more familiar substances of abuse in patients who misuse spices. As with inhalant abuse, the concept of “substance abuse” in clinical practice may need to be further expanded to include patients who abuse culinary spices. Patients could be screened for psychiatric illnesses known to increase the risk of substance abuse. These might include—but are not limited to:
• comorbid psychotic disorders
• mood disorders, particularly bipolar disorders
• trauma- and stressor-related disorders, particularly posttraumatic stress disorder
• personality disorders, particularly antisocial, borderline, and narcissistic personality disorders.
Pending the availability of population-based studies on abuse of culinary spices, the usual cautions regarding substance abuse seem to be appropriate when caring for these patients. Assessment for and management of comorbid psychiatric conditions is essential in the comprehensive psychiatric care of patients who abuse substances.
Last, general consideration of a 12-step recovery program appears warranted for these patients; the self-reflection and group support of such programs can be useful in helping patients control their use of these substances.
Bottom Line
Presentation of culinary spice intoxication can parallel that of other medical or psychiatric illnesses, or other drugs of abuse. Consideration and questioning for abuse of spices is necessary to ascertain the psychoactive effects of these substances when used surreptitiously. Management should follow substance abuse treatment protocols: inquiry into patterns of problematic use and readiness to change, assessment and management of psychiatric comorbidity, and referral to a recovery program.
Related Resources
• Srinivasan K. Role of spices beyond food flavoring: nutraceuticals with multiple health effects. Food Reviews International. 2005;21(2):167-188.
• Parthasarathi U, Hategan A, Bourgeois JA. Out of the cupboard and into the clinic: Nutmeg-induced mood disorder. Current Psychiatry. 2013;12(12):E1-E2.
Drug Brand Names
Bromocriptine mesylate • Parlodel Imipramine • Tofrani
Flunitrazepam • Rohypnol Iproniazid • Marsilid
Fluoxetine • Prozac Tranylcypromine • Parnate
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. O’Mahony Carey S. Psychoactive substances. A guide to ethnobotanical plants and herbs, synthetic chemicals, compounds and products. http://www.drugs.ie/ resourcesfiles/guides/Psychoactive_substances_low_res. pdf. Accessed March 4, 2014.
2. Asafetida. Applied Health. http://www.appliedhealth.com/index.php?option=com _content&view=article&id= 108207. Accessed March 4, 2014.
3. Jayatilaka A, Poole SK, Poole CF, et al. Simultaneous micro steam distillation/solvent extraction for the isolation of semivolatile flavor compounds from cinnamon and their separation by series coupled-column gas chromatography. Analytica Chimica Acta. 1995;302(2-3):147-162.
4. Spices. History & Special Collections UCLA Louise M. Darling Biomedical Library. http://unitproj.library.ucla. edu/biomed/spice/index.cfm?displayID=15. Accessed March 4, 2014.
5. Ginger action and uses. Ginger extract. Gingerols. MDidea Web site. http://www.mdidea.com/products/new/ new02108.html. Accessed March 4, 2014.
6. Raina VK, Srivastava SK, Syamasunder KV. The essential oil of ‘greater galangal’ [Alpinia galanga (L.) Willd.] from the lower Himalayan region of India. Flavour and Fragrance Journal. 2002;17(5):358-360.
7. Wenk G. Psychoactive spices - Bon appetite! http://www.psychologytoday.com/blog/your-brain-food/201008/ psychoactive-spices-bon-appetite. Published August 4, 2010. Accessed March 4, 2014.
8. Wax PM. Just a click away: recreational drug Web sites on the Internet. Pediatrics.2002;109(6):e96.
9. Forrester MB. Nutmeg intoxication in Texas, 1998-2004. Hum Exp Toxicol. 2005;24(11):563-566.
10. Abernethy MK, Becker LB. Acute nutmeg intoxication. Am J Emerg Med. 1992;10(5):429-430.
11. Brenner N, Frank OS, Knight E. Chronic nutmeg psychosis. J R Soc Med. 1993;86(3):179-180.
12. McKenna A, Nordt SP, Ryan J. Acute nutmeg poisoning. Eur J Emerg Med. 2004;11(4):240-241.
13. Dhingra D, Sharma A. Antidepressant-like activity of n-hexane extract of nutmeg (Myristica fragrans) seeds in mice. J Med Food. 2006;9(1):84-89.
14. Truitt EB Jr, Duritz G, Ebersberger EM. Evidence of monoamine oxidase inhibition by myristicin and nutmeg. Proc Soc Exp Biol Med. 1963;112:647-650.
15. Tajuddin, Ahmad S, Latif A, et al. An experimental study of sexual function improving effect of Myristica fragrans Houtt. (nutmeg). BMC Complement Altern Med. 2005;5:16.
16. Quin GI, Fanning NF, Plunkett PK. Nutmeg intoxication. J Accid Emerg Med. 1998;15(4):287-288.
17. Barceloux DG. Nutmeg (Myristica fragrans Houtt.) Dis Mon. 2009;55(6):373-379.
18. Demetriades AK, Wallman PD, McGuiness A, et al. Low cost, high risk: accidental nutmeg intoxication. Emerg Med J. 2005;22(3):223-225.
19. Weil A. The use of nutmeg as a psychotropic agent. Bull Narc. 1966;18(4):15-23. http://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1966-01-01_4_ page003.html. Accessed March 5, 2013.
20. Stein U, Greyer H, Hentschel H. Nutmeg (myristicin) poisoning - report on a fatal case and a series of cases recorded by a poison information centre. Forensic Sci Int. 2001;118(1):87-90.
21. Grover JK, Khandkar S, Vats V, et al. Pharmacological studies on Myristica fragrans—antidiarrheal, hypnotic, analgesic and hemodynamic (blood pressure) parameters. Methods Find Exp Clin Pharmacol. 2002;24(10):675-680.
22. Shah AM, Calello DP, Quintero-Solivan J, et al. The not-so-nice spice: a teenage girl with palpitations and dry mouth. Pediatr Emerg Care. 2011;27(12):1205-1207.
23. Heliotropin. Polarized light microscopy digital image gallery. http://micro.magnet.fsu.edu/primer/techniques/ polarized/gallery/pages/heliotropinsmall.html. Accessed March 5, 2014.
24. Gage E. Romancing the bean. Budget Travel. http://articles.cnn.com/2007-09-11/travel/vanilla_1_vanilla-orchid-totonaca?_s=PM:TRAVEL. Published September 11, 2007. Updated September 16, 2012. Accessed March 5, 2014.
25. Mazor S, DesLauriers CA, Mycyk MB. Adolescent ethanol intoxication from vanilla extract ingestion: a case report. The Internet Journal of Family Practice. 2005;4(1). doi: 10.5580/bc.
26. Skalli S, Soulaymani Bencheikh R. Epileptic seizure induced by fennel essential oil. Epileptic Disord. 2011;13(3):345-347.
27. Zhu M, Wong PY, Li RC. Effect of oral administration of fennel (Foeniculum vulgare) on ciprofloxacin absorption and disposition in the rat. J Pharm Pharmacol. 1999;51(12):1391-1396.
28. Malini T, Arunakaran J, Aruldhas MM, et al. Effects of piperine on the lipid composition and enzymes of the pyruvate-malate cycle in the testis of the rat in vivo. Biochem Mol Biol Int. 1999;47(3):537-545.
29. Zaugg J, Baburin I, Hering S, et al. Identifying GABAA receptor ligands in black pepper by activity profiling, LC-TOFMS, and offline microprobe NMR. Planta Med. 2009; 75(9):888-889. doi: 10.1055/s-0029-1234276.
30. Flavored tobacco. FDA.gov. http://www.fda.gov/TobaccoProducts/ProtectingKidsfromTobacco/ FlavoredTobacco/default.htm. Published September 22, 2009. Updated March 21, 2013. Accessed March 18, 2014.
31. Fujisawa S, Atsumi T, Kadoma Y, et al. Antioxidant and prooxidant action of eugenol-related compounds and their cytotoxicity. Toxicology. 2002;177(1):39-54.
32. Eugenol oil overdose. New York Times Health Guide. http://health.nytimes.com/health/guides/poison/ eugenol-oil-overdose/overview.html. Accessed March 5, 2014.
33. Hartnoll G, Moore D, Douek D. Near fatal ingestion of oil of cloves. Arch Dis Child. 1993;69(3):392-393.
34. Harris E. NPR. German Christmas cookies pose health danger. http://www.npr.org/templates/story/story.php? storyId=6672644. Published December 25, 2006. Accessed March 5, 2014.
35. Frydman-Marom A, Levin A, Farfara D, et al. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS One. 2011; 6(1):e16564. doi:10.1371/journal.pone.001656453.
36. Björnstad K, Helander A, Hultén P, et al. Bioanalytical investigation of asarone in connection with Acorus calamus oil intoxications. J Anal Toxicol. 2009;33(9):604-609.
37. Han P, Han T, Peng W, et al. Antidepressant-like effects of essential oil and asarone, a major essential oil component from the rhizome of Acorus tatarinowii. Pharm Biol. 2013;51(5):589-594.
38. Dandiya PC, Menon MK. Actions of asarone on behavior, stress, and hyperpyrexia, and its interaction with central stimulants. J Pharmacol Exp Ther. 1964;145:42-46.
39. Bockon J. Ginger: inhibition of thromboxane synthetase and stimulation of prostacyclin: relevance for medicine and psychiatry. Med Hypotheses. 1986;20(3):271-278.
40. Ghayur MN, Gilani AH. Pharmacological basis for the medicinal use of ginger in gastrointestinal disorders. Dig Dis Sci. 2005;50(10):1889-1897.
41. Nievergelt A, Huonker P, Schoop R, et al. Identification of serotonin 5-HT1A receptor partial agonists in ginger. Bioorg Med Chem. 2010;18(9):3345-3351.
42. Mishra A, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann Indian Acad Neurol. 2008;11(1):13-19.
43. Seely KA, Levi MS, Prather PL. The dietary polyphenols trans-resveratrol and curcumin selectively bind human CB1 cannabinoid receptors with nanomolar affinities and function as antagonists/inverse agonists [retracted in: J Pharmacol Exp Ther. 2009;331(3):1147]. J Pharmacol Exp Ther. 2009;330(1): 31-39.
44. Gertsch J, Pertwee RG, Di Marzo V. Phytocannabinoids beyond the Cannabis plant – do they exist? Br J Pharmacol. 2010;160(3):523-529.
45. Dwyer AV, Whitten DL, Hawrelak JA. Herbal medicines, other than St. John’s Wort, in the treatment of depression: a systematic review. Altern Med Rev. 2011;16(1):40-49.
46. Moshiri E, Basti AA, Noorbala AA, et al. Crocus sativus L. (petal) in the treatment of mild-to-moderate depression: a double-blind, randomized and placebo controlled trial. Phytomedicine. 2006;13(9-10):607-611.
47. Noorbala AA, Akhondzadeh S, Tahmacebi-Pour N, et al. Hydro-alcoholic extract of Crocus sativus L. versus fluoxetine in the treatment of mild to moderate depression: a double-blind, randomized pilot trial. J Ethnopharmacol. 2005;97(2):281-284.
48. Akhondzadeh S, Tahmacebi-Pour N, Noorbala AA, et al. Crocus sativus L. in the treatment of mild to moderate depression: a double-blind, randomized, and placebo-controlled trial. Phytother Res. 2005;19(2):148-151.
Many substances that are not typically thought of as “substances of abuse” possess—when adequately dosed—clinically meaningful psychoactive properties. In addition to the more familiar effects of alcohol, psychostimulants, opioids, Cannabis, and hallucinogens, you may encounter psychiatric phenomena resulting from abuse of more obscure substances, including culinary spices.
The clinician treating a patient in an apparent intoxicated state who has a negative drug screen might ask that patient if he (she) abuses spices. This might be particularly relevant when treating patients thought to have limited access to illicit substances or those with ready access to large amounts of spices, such as prisoners, young patients, and those working in the food service industry.
Abuse of spices can be a problematic diagnosis
Patients may misuse culinary spices to achieve euphoria, or a “natural high.” They may present with medical or psychiatric symptoms, including acute altered mental status, but the psychoactive substances are not identified on routine toxicology studies. In addition, patients may not attribute their use of spices for psychoactive effect to “drugs,” because these materials are legal and readily available. This may lead to misdiagnosis of a systemic medical disorder or a primary psychiatric illness to explain the patient’s symptoms and initiating a psychotropic agent and other psychiatric services when a substance abuse program might be a more appropriate clinical intervention.
Some spices contain psychoactive compounds that can alter CNS function (Table1-7), might be abused for recreational purposes, and can be toxic in an excessive amount. Internet resources, including anonymous web-based communications, and anecdotal materials about non-traditional recreational drugs, are available to anyone with Internet access.8 However, little research has been conducted into the prevalence of abuse (Box)9 and spices’ psychoactive properties. The lack of toxicology detection of spices in the medical setting presents a diagnostic challenge.
The psychoactive plants used in “natural high” products mainly are psychoactively inactive in their natural form, but extracts or alkaloids obtained from them might induce 1 or more of 3 classifications of psychoactivity:
• stimulant
• sedative
• hallucinogenic.
Many of these substances are considered to be aphrodisiac, and some may be abused to increase sexual function.
The following is a review of common spices that have been reported to possess potential psychoactive properties.
Nutmeg
Nutmeg (Myristica fragrans) is a common and easily accessible means of reaching euphoria in adults.10 The aromatic oil of nutmeg contains myristicin, a psychoactive substance that is chemically similar to hallucinogenic compounds such as mescaline. Its psychoactive effects could be attributed to metabolic formation of amphetamine derivatives from its core ingredients, elemicin, myristicin, and safrole.11,12
Nutmeg and its active component, myristicin, produce central monoamine oxidase (MAO) inhibition as evidenced by the ability to lower the convulsive dose of IV tryptamine in mice and to increase brain 5-hydroxytryptamine concentrations.13,14 Although myristicin’s potency is not comparable to that of the more potent MAO inhibitors such as tranylcypromine and iproniazid (which is not available in the United States), it seems adequate when compared with its low toxicity.14 Nutmeg extract is associated with a significant antidepressant effect in mice, which seemed to be mediated by interaction with the adrenergic, dopaminergic, and serotonergic systems.13 Nutmeg is associated with sustained increase in sexual activity in animal studies, with no evidence of adverse effects and toxicity, suggesting that nutmeg possesses clinically significant aphrodisiac activity.15
Psychoactive effects can be achieved by ingesting 5 to 15 g of nutmeg.11 Acute nutmeg intoxication produces palpitations, dizziness, anxiety, and hallucinations, mostly resolving within 24 hours, while effects of chronic abuse are reported to be similar to Cannabis use, including euphoria, giddiness, anxiety, fear, sense of impending doom, detachment, confabulation, and hallucinations.11,16 Urine drug screens are negative unless other psychoactive substances have been ingested.17
Suspected nutmeg intoxication or poisoning should be treated with supportive treatment. Use sedatives with caution because of alternating periods of delirium and obtundation during nutmeg intoxication.17
In case reports, myristicin poisoning induced CNS neuromodulatory signs that mimicked an anticholinergic hyperstimulation state.12,18 Fatal myristicin poisoning is rare; 2 cases have been reported, 1 in combination with flunitrazepam (not available in the United States).19,20 Nutmeg also has sedative properties and can cause GI symptoms when ingesting excessive amounts.1,20,21 Grover et al21 described no harmful effects on blood pressure and electrocardiogram; however, Shah et al22 reported palpitations and dry mouth.
Vanilla
Vanilla (species of the genus Vanilla) contains piperonal, also known as heliotropin.1 Piperonal has aromatherapeutic qualities that might elevate mood and well-being. In the early 1990s, the Memorial Sloan- Kettering Cancer Center in New York City described heliotropin as a powerful aromatherapy tool. Patients who were undergoing an MRI in an environment scented with heliotropin demonstrated a 63% reduction in anxiety compared with those who were not exposed to fragrance.23 The Smell and Taste Treatment and Research Foundation in Chicago found that vanilla can promote sexual arousal.24
Short-term effects of vanillin—a major component of vanilla—include a feeling of relaxation and reduced stress; long-term use can produce an antidepressant effect.1 There are no reports of vanilla abuse to achieve these effects; however, patients might abuse vanilla extract because of its alcohol content (up to 35% ethanol).25
Fennel
The essential oil of fennel (Foeniculum vulgare) can be neurotoxic and epileptogenic. Skalli and colleagues recently reported a case of seizure induction in a young woman after ingesting cakes containing fennel oil.26 Fennel oil also has been reported to have significant interaction with the fluoroquinolone-type antibiotics. Be aware of adverse effects associated with fennel ingestion; question patients if atypical seizures or reactions to antibiotics occur.27
Spices such as fennel, dill, cinnamon, saffron, and anise also contain psychoactive substances that are chemically similar to myristicin, which can induce sedation, stimulation, or hallucinations.7
Black pepper
Piperine, which gives black pepper (Piper nigrum) its spiciness, enhances thermogenesis of lipid metabolism, accelerates energy metabolism, and increases serotonin and endorphin production in the brain.28 Black pepper is reported to potentiate γ-aminobutyric acid A receptor subtypes,29 and could present possible applications for treating insomnia, epilepsy, and anxiety disorders.
Cloves
Non-culinary uses of clove (Syzygium aromaticum, a tree in the myrtle family) include flavored cigarettes. However, in 2009 clove cigarettes were banned in the United States as part of a public policy to reduce the number of children who start smoking.30 Eugenol, which constitutes as much as 90% of the essential oil extracted from cloves (and is responsible for the aroma), can cause hepatotoxicity31 and palpitations32; it can be toxic in quantities as low as 5 mL.33 Eugenol is present in other spices, such as nutmeg and cinnamon, and has been reported to have sedative properties.1
Mace
Mace is made from the covering of nutmeg (Myristica fragrans) seeds. It has a strong aroma resembling that of nutmeg. Whole mace contains 4% to 14% of a volatile oil similar to that found in nutmeg. Because mace contains the same oils that make nutmeg psychoactive1 in excessive amounts—although nutmeg seeds are more potent—be aware of the psychoactive potential of mace.
CinnamonCassia cinnamon (Cinnamomum aromaticum) is spicier and tarter than Ceylon cinnamon (Cinnamomum zeylanicum), which has a more flowery aroma. The 2 types of cinnamon can be distinguished by their different chemical composition. Ceylon cinnamon contains eugenol and benzyl benzoate; cassia cinnamon contains coumarin.3 Eugenol is reported to have sedative effects.1 Coumarin is a precursor molecule in the synthesis of a number of synthetic anticoagulant pharmaceuticals, including coumadin. Because of the toxic component of coumarin, European health agencies have warned against consuming high amounts of cassia.34 There are no reports of side effects arising from the occasional use of cinnamon as a spice.
In a study by Frydman-Marom et al,35 cinnamon extract (CEppt) was found to act on the CNS by inhibiting development of Alzheimer’s disease in animal models.
Asarone
Asarone is found in the Asarum family of spices that includes Acorus calamus. Asarone is chemically similar to mescaline. Although anecdotal reports indicate that A. calamus is a hallucinogen, research shows no evidence that it contains hallucinogenic substances.36 Han et al37 reported an antidepressant effect with the essential oil and asarones for the rhizomes of Acorus tatarinowii. In animal studies, asarone was found to reduce spontaneous motor activity, and even in low doses, reduced anxiety without decreasing acuity of perception.38
Ginger
Ginger (Zingiber officinale) is regarded as a sedative, general stimulant, and aphrodisiac.1,4,5 Its main constituents are phenolic compounds such as gingerols and shogaols, and sesquiterpenes such as zingiberene.4 Ginger is an inhibitor of thromboxane synthetase, a property shared by tricyclic antidepressants.39
Research indicates that 9 compounds found in ginger may interact with the serotonin 5-HT1A receptor, suggesting a possible mechanism for reducing anxiety.40 A study by Nievergelt et al41 indicates that by binding to human serotonin receptors, ginger might influence GI function. Ginger extract contains a cholinergic and spasmogenic component, which provides a mechanistic insight for the prokinetic action of ginger.40
Turmeric
Turmeric (Curcuma longa) has been investigated for possible benefit in Alzheimer’s disease42; research into curcumin, the active substance of turmeric, is increasing. Although the original report was retracted after publication, curcumin was reported to selectively bind to human cannabinoid receptors type 1 (CB1) with nanomolar affinities and to function as an antagonist/inverse agonist.43 However, Gertsch et al44 found that curcumin did not interact functionally with the CB1 receptor, although this compound appears to share ability of the CB1 receptor inverse agonist.
Galangal
Major constituents identified in the galangal (or galanga) rhizome and leaf oil were 1,8-cineole, and β-pinene and camphor.6 Galangal, a member of the ginger (Zingiberaceae) family, interacts with MAO inhibitors, H2 receptor antagonists, and proton-pump inhibitors.1 Anxiolytic, hallucinogenic, and stimulant properties have been reported.1 An excessive amount can induce diarrhea, dizziness, nausea, and vomiting.1
Saffron
Stigma of saffron (a member of the family Iridaceae) was found to be significantly more effective than placebo and equally as efficacious as fluoxetine and imipramine in treating depression. Saffron petal was found to be significantly more effective than placebo and as effective as fluoxetine and saffron stigma in a recent systematic review.45-48
Asafetida
Asafetida (Ferula assa-foetida), when combined with valerian root, is used as a sedative to treat hyperactivity.2 The active ingredients of asafetida are the resin, endogenous gum, essential oil, propenyl-isobutylsulfide, umbelliferone, and vanillin. Several of the volatile constituents produce a sedative effect.2 Additive effects can occur between the hypotensive property of asafetida and dopamine receptor agonists such as bromocriptine mesylate. Use caution when combining asafetida in conjunction with a CNS depressant or a stimulant.2
Recommendations for treating spice-abusers
Patients may present to psychiatry services with psychological and physiological evidence of intoxication with culinary spices that may mimic 1) abuse of other substances, 2) primary psychiatric illness, and 3) primary medical illness. When you encounter a patient with a new psychiatric symptom, consider inquiring about the abuse of spices.
Patients might abuse more than 1 spice; a comprehensive screening approach might therefore be useful. Caution patients that ingesting these substance to excess can have harmful effects. Consider appropriate psychopharmacotherapy for underlying psychiatric symptoms to help patients who use spices maladaptively to self-medicate psychiatric symptoms.
Consider abuse of culinary spices in clinical presentations of psychiatric symptoms that do not seem adequate for a diagnosis of a primary anxiety, mood, or psychotic disorder, or in cases atypical psychiatric presentations that are—perhaps to your surprise—associated with negative toxicology studies for common, more familiar substances of abuse.
Physicians practicing in an environment where street drugs are difficult to obtain (eg, prisons) should consider monitoring for possible abuse of spices. Based on the available, albeit limited, literature, it appears that most culinary spice–associated intoxication can be managed:
• with an elevated level of clinical suspicion
• by ruling out other causes of intoxication
• using targeted, empirical psychopharmacotherapy to manage symptoms
• with supportive care that includes close psychiatric follow-up.
Consider comorbid abuse of other, more familiar substances of abuse in patients who misuse spices. As with inhalant abuse, the concept of “substance abuse” in clinical practice may need to be further expanded to include patients who abuse culinary spices. Patients could be screened for psychiatric illnesses known to increase the risk of substance abuse. These might include—but are not limited to:
• comorbid psychotic disorders
• mood disorders, particularly bipolar disorders
• trauma- and stressor-related disorders, particularly posttraumatic stress disorder
• personality disorders, particularly antisocial, borderline, and narcissistic personality disorders.
Pending the availability of population-based studies on abuse of culinary spices, the usual cautions regarding substance abuse seem to be appropriate when caring for these patients. Assessment for and management of comorbid psychiatric conditions is essential in the comprehensive psychiatric care of patients who abuse substances.
Last, general consideration of a 12-step recovery program appears warranted for these patients; the self-reflection and group support of such programs can be useful in helping patients control their use of these substances.
Bottom Line
Presentation of culinary spice intoxication can parallel that of other medical or psychiatric illnesses, or other drugs of abuse. Consideration and questioning for abuse of spices is necessary to ascertain the psychoactive effects of these substances when used surreptitiously. Management should follow substance abuse treatment protocols: inquiry into patterns of problematic use and readiness to change, assessment and management of psychiatric comorbidity, and referral to a recovery program.
Related Resources
• Srinivasan K. Role of spices beyond food flavoring: nutraceuticals with multiple health effects. Food Reviews International. 2005;21(2):167-188.
• Parthasarathi U, Hategan A, Bourgeois JA. Out of the cupboard and into the clinic: Nutmeg-induced mood disorder. Current Psychiatry. 2013;12(12):E1-E2.
Drug Brand Names
Bromocriptine mesylate • Parlodel Imipramine • Tofrani
Flunitrazepam • Rohypnol Iproniazid • Marsilid
Fluoxetine • Prozac Tranylcypromine • Parnate
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Many substances that are not typically thought of as “substances of abuse” possess—when adequately dosed—clinically meaningful psychoactive properties. In addition to the more familiar effects of alcohol, psychostimulants, opioids, Cannabis, and hallucinogens, you may encounter psychiatric phenomena resulting from abuse of more obscure substances, including culinary spices.
The clinician treating a patient in an apparent intoxicated state who has a negative drug screen might ask that patient if he (she) abuses spices. This might be particularly relevant when treating patients thought to have limited access to illicit substances or those with ready access to large amounts of spices, such as prisoners, young patients, and those working in the food service industry.
Abuse of spices can be a problematic diagnosis
Patients may misuse culinary spices to achieve euphoria, or a “natural high.” They may present with medical or psychiatric symptoms, including acute altered mental status, but the psychoactive substances are not identified on routine toxicology studies. In addition, patients may not attribute their use of spices for psychoactive effect to “drugs,” because these materials are legal and readily available. This may lead to misdiagnosis of a systemic medical disorder or a primary psychiatric illness to explain the patient’s symptoms and initiating a psychotropic agent and other psychiatric services when a substance abuse program might be a more appropriate clinical intervention.
Some spices contain psychoactive compounds that can alter CNS function (Table1-7), might be abused for recreational purposes, and can be toxic in an excessive amount. Internet resources, including anonymous web-based communications, and anecdotal materials about non-traditional recreational drugs, are available to anyone with Internet access.8 However, little research has been conducted into the prevalence of abuse (Box)9 and spices’ psychoactive properties. The lack of toxicology detection of spices in the medical setting presents a diagnostic challenge.
The psychoactive plants used in “natural high” products mainly are psychoactively inactive in their natural form, but extracts or alkaloids obtained from them might induce 1 or more of 3 classifications of psychoactivity:
• stimulant
• sedative
• hallucinogenic.
Many of these substances are considered to be aphrodisiac, and some may be abused to increase sexual function.
The following is a review of common spices that have been reported to possess potential psychoactive properties.
Nutmeg
Nutmeg (Myristica fragrans) is a common and easily accessible means of reaching euphoria in adults.10 The aromatic oil of nutmeg contains myristicin, a psychoactive substance that is chemically similar to hallucinogenic compounds such as mescaline. Its psychoactive effects could be attributed to metabolic formation of amphetamine derivatives from its core ingredients, elemicin, myristicin, and safrole.11,12
Nutmeg and its active component, myristicin, produce central monoamine oxidase (MAO) inhibition as evidenced by the ability to lower the convulsive dose of IV tryptamine in mice and to increase brain 5-hydroxytryptamine concentrations.13,14 Although myristicin’s potency is not comparable to that of the more potent MAO inhibitors such as tranylcypromine and iproniazid (which is not available in the United States), it seems adequate when compared with its low toxicity.14 Nutmeg extract is associated with a significant antidepressant effect in mice, which seemed to be mediated by interaction with the adrenergic, dopaminergic, and serotonergic systems.13 Nutmeg is associated with sustained increase in sexual activity in animal studies, with no evidence of adverse effects and toxicity, suggesting that nutmeg possesses clinically significant aphrodisiac activity.15
Psychoactive effects can be achieved by ingesting 5 to 15 g of nutmeg.11 Acute nutmeg intoxication produces palpitations, dizziness, anxiety, and hallucinations, mostly resolving within 24 hours, while effects of chronic abuse are reported to be similar to Cannabis use, including euphoria, giddiness, anxiety, fear, sense of impending doom, detachment, confabulation, and hallucinations.11,16 Urine drug screens are negative unless other psychoactive substances have been ingested.17
Suspected nutmeg intoxication or poisoning should be treated with supportive treatment. Use sedatives with caution because of alternating periods of delirium and obtundation during nutmeg intoxication.17
In case reports, myristicin poisoning induced CNS neuromodulatory signs that mimicked an anticholinergic hyperstimulation state.12,18 Fatal myristicin poisoning is rare; 2 cases have been reported, 1 in combination with flunitrazepam (not available in the United States).19,20 Nutmeg also has sedative properties and can cause GI symptoms when ingesting excessive amounts.1,20,21 Grover et al21 described no harmful effects on blood pressure and electrocardiogram; however, Shah et al22 reported palpitations and dry mouth.
Vanilla
Vanilla (species of the genus Vanilla) contains piperonal, also known as heliotropin.1 Piperonal has aromatherapeutic qualities that might elevate mood and well-being. In the early 1990s, the Memorial Sloan- Kettering Cancer Center in New York City described heliotropin as a powerful aromatherapy tool. Patients who were undergoing an MRI in an environment scented with heliotropin demonstrated a 63% reduction in anxiety compared with those who were not exposed to fragrance.23 The Smell and Taste Treatment and Research Foundation in Chicago found that vanilla can promote sexual arousal.24
Short-term effects of vanillin—a major component of vanilla—include a feeling of relaxation and reduced stress; long-term use can produce an antidepressant effect.1 There are no reports of vanilla abuse to achieve these effects; however, patients might abuse vanilla extract because of its alcohol content (up to 35% ethanol).25
Fennel
The essential oil of fennel (Foeniculum vulgare) can be neurotoxic and epileptogenic. Skalli and colleagues recently reported a case of seizure induction in a young woman after ingesting cakes containing fennel oil.26 Fennel oil also has been reported to have significant interaction with the fluoroquinolone-type antibiotics. Be aware of adverse effects associated with fennel ingestion; question patients if atypical seizures or reactions to antibiotics occur.27
Spices such as fennel, dill, cinnamon, saffron, and anise also contain psychoactive substances that are chemically similar to myristicin, which can induce sedation, stimulation, or hallucinations.7
Black pepper
Piperine, which gives black pepper (Piper nigrum) its spiciness, enhances thermogenesis of lipid metabolism, accelerates energy metabolism, and increases serotonin and endorphin production in the brain.28 Black pepper is reported to potentiate γ-aminobutyric acid A receptor subtypes,29 and could present possible applications for treating insomnia, epilepsy, and anxiety disorders.
Cloves
Non-culinary uses of clove (Syzygium aromaticum, a tree in the myrtle family) include flavored cigarettes. However, in 2009 clove cigarettes were banned in the United States as part of a public policy to reduce the number of children who start smoking.30 Eugenol, which constitutes as much as 90% of the essential oil extracted from cloves (and is responsible for the aroma), can cause hepatotoxicity31 and palpitations32; it can be toxic in quantities as low as 5 mL.33 Eugenol is present in other spices, such as nutmeg and cinnamon, and has been reported to have sedative properties.1
Mace
Mace is made from the covering of nutmeg (Myristica fragrans) seeds. It has a strong aroma resembling that of nutmeg. Whole mace contains 4% to 14% of a volatile oil similar to that found in nutmeg. Because mace contains the same oils that make nutmeg psychoactive1 in excessive amounts—although nutmeg seeds are more potent—be aware of the psychoactive potential of mace.
CinnamonCassia cinnamon (Cinnamomum aromaticum) is spicier and tarter than Ceylon cinnamon (Cinnamomum zeylanicum), which has a more flowery aroma. The 2 types of cinnamon can be distinguished by their different chemical composition. Ceylon cinnamon contains eugenol and benzyl benzoate; cassia cinnamon contains coumarin.3 Eugenol is reported to have sedative effects.1 Coumarin is a precursor molecule in the synthesis of a number of synthetic anticoagulant pharmaceuticals, including coumadin. Because of the toxic component of coumarin, European health agencies have warned against consuming high amounts of cassia.34 There are no reports of side effects arising from the occasional use of cinnamon as a spice.
In a study by Frydman-Marom et al,35 cinnamon extract (CEppt) was found to act on the CNS by inhibiting development of Alzheimer’s disease in animal models.
Asarone
Asarone is found in the Asarum family of spices that includes Acorus calamus. Asarone is chemically similar to mescaline. Although anecdotal reports indicate that A. calamus is a hallucinogen, research shows no evidence that it contains hallucinogenic substances.36 Han et al37 reported an antidepressant effect with the essential oil and asarones for the rhizomes of Acorus tatarinowii. In animal studies, asarone was found to reduce spontaneous motor activity, and even in low doses, reduced anxiety without decreasing acuity of perception.38
Ginger
Ginger (Zingiber officinale) is regarded as a sedative, general stimulant, and aphrodisiac.1,4,5 Its main constituents are phenolic compounds such as gingerols and shogaols, and sesquiterpenes such as zingiberene.4 Ginger is an inhibitor of thromboxane synthetase, a property shared by tricyclic antidepressants.39
Research indicates that 9 compounds found in ginger may interact with the serotonin 5-HT1A receptor, suggesting a possible mechanism for reducing anxiety.40 A study by Nievergelt et al41 indicates that by binding to human serotonin receptors, ginger might influence GI function. Ginger extract contains a cholinergic and spasmogenic component, which provides a mechanistic insight for the prokinetic action of ginger.40
Turmeric
Turmeric (Curcuma longa) has been investigated for possible benefit in Alzheimer’s disease42; research into curcumin, the active substance of turmeric, is increasing. Although the original report was retracted after publication, curcumin was reported to selectively bind to human cannabinoid receptors type 1 (CB1) with nanomolar affinities and to function as an antagonist/inverse agonist.43 However, Gertsch et al44 found that curcumin did not interact functionally with the CB1 receptor, although this compound appears to share ability of the CB1 receptor inverse agonist.
Galangal
Major constituents identified in the galangal (or galanga) rhizome and leaf oil were 1,8-cineole, and β-pinene and camphor.6 Galangal, a member of the ginger (Zingiberaceae) family, interacts with MAO inhibitors, H2 receptor antagonists, and proton-pump inhibitors.1 Anxiolytic, hallucinogenic, and stimulant properties have been reported.1 An excessive amount can induce diarrhea, dizziness, nausea, and vomiting.1
Saffron
Stigma of saffron (a member of the family Iridaceae) was found to be significantly more effective than placebo and equally as efficacious as fluoxetine and imipramine in treating depression. Saffron petal was found to be significantly more effective than placebo and as effective as fluoxetine and saffron stigma in a recent systematic review.45-48
Asafetida
Asafetida (Ferula assa-foetida), when combined with valerian root, is used as a sedative to treat hyperactivity.2 The active ingredients of asafetida are the resin, endogenous gum, essential oil, propenyl-isobutylsulfide, umbelliferone, and vanillin. Several of the volatile constituents produce a sedative effect.2 Additive effects can occur between the hypotensive property of asafetida and dopamine receptor agonists such as bromocriptine mesylate. Use caution when combining asafetida in conjunction with a CNS depressant or a stimulant.2
Recommendations for treating spice-abusers
Patients may present to psychiatry services with psychological and physiological evidence of intoxication with culinary spices that may mimic 1) abuse of other substances, 2) primary psychiatric illness, and 3) primary medical illness. When you encounter a patient with a new psychiatric symptom, consider inquiring about the abuse of spices.
Patients might abuse more than 1 spice; a comprehensive screening approach might therefore be useful. Caution patients that ingesting these substance to excess can have harmful effects. Consider appropriate psychopharmacotherapy for underlying psychiatric symptoms to help patients who use spices maladaptively to self-medicate psychiatric symptoms.
Consider abuse of culinary spices in clinical presentations of psychiatric symptoms that do not seem adequate for a diagnosis of a primary anxiety, mood, or psychotic disorder, or in cases atypical psychiatric presentations that are—perhaps to your surprise—associated with negative toxicology studies for common, more familiar substances of abuse.
Physicians practicing in an environment where street drugs are difficult to obtain (eg, prisons) should consider monitoring for possible abuse of spices. Based on the available, albeit limited, literature, it appears that most culinary spice–associated intoxication can be managed:
• with an elevated level of clinical suspicion
• by ruling out other causes of intoxication
• using targeted, empirical psychopharmacotherapy to manage symptoms
• with supportive care that includes close psychiatric follow-up.
Consider comorbid abuse of other, more familiar substances of abuse in patients who misuse spices. As with inhalant abuse, the concept of “substance abuse” in clinical practice may need to be further expanded to include patients who abuse culinary spices. Patients could be screened for psychiatric illnesses known to increase the risk of substance abuse. These might include—but are not limited to:
• comorbid psychotic disorders
• mood disorders, particularly bipolar disorders
• trauma- and stressor-related disorders, particularly posttraumatic stress disorder
• personality disorders, particularly antisocial, borderline, and narcissistic personality disorders.
Pending the availability of population-based studies on abuse of culinary spices, the usual cautions regarding substance abuse seem to be appropriate when caring for these patients. Assessment for and management of comorbid psychiatric conditions is essential in the comprehensive psychiatric care of patients who abuse substances.
Last, general consideration of a 12-step recovery program appears warranted for these patients; the self-reflection and group support of such programs can be useful in helping patients control their use of these substances.
Bottom Line
Presentation of culinary spice intoxication can parallel that of other medical or psychiatric illnesses, or other drugs of abuse. Consideration and questioning for abuse of spices is necessary to ascertain the psychoactive effects of these substances when used surreptitiously. Management should follow substance abuse treatment protocols: inquiry into patterns of problematic use and readiness to change, assessment and management of psychiatric comorbidity, and referral to a recovery program.
Related Resources
• Srinivasan K. Role of spices beyond food flavoring: nutraceuticals with multiple health effects. Food Reviews International. 2005;21(2):167-188.
• Parthasarathi U, Hategan A, Bourgeois JA. Out of the cupboard and into the clinic: Nutmeg-induced mood disorder. Current Psychiatry. 2013;12(12):E1-E2.
Drug Brand Names
Bromocriptine mesylate • Parlodel Imipramine • Tofrani
Flunitrazepam • Rohypnol Iproniazid • Marsilid
Fluoxetine • Prozac Tranylcypromine • Parnate
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. O’Mahony Carey S. Psychoactive substances. A guide to ethnobotanical plants and herbs, synthetic chemicals, compounds and products. http://www.drugs.ie/ resourcesfiles/guides/Psychoactive_substances_low_res. pdf. Accessed March 4, 2014.
2. Asafetida. Applied Health. http://www.appliedhealth.com/index.php?option=com _content&view=article&id= 108207. Accessed March 4, 2014.
3. Jayatilaka A, Poole SK, Poole CF, et al. Simultaneous micro steam distillation/solvent extraction for the isolation of semivolatile flavor compounds from cinnamon and their separation by series coupled-column gas chromatography. Analytica Chimica Acta. 1995;302(2-3):147-162.
4. Spices. History & Special Collections UCLA Louise M. Darling Biomedical Library. http://unitproj.library.ucla. edu/biomed/spice/index.cfm?displayID=15. Accessed March 4, 2014.
5. Ginger action and uses. Ginger extract. Gingerols. MDidea Web site. http://www.mdidea.com/products/new/ new02108.html. Accessed March 4, 2014.
6. Raina VK, Srivastava SK, Syamasunder KV. The essential oil of ‘greater galangal’ [Alpinia galanga (L.) Willd.] from the lower Himalayan region of India. Flavour and Fragrance Journal. 2002;17(5):358-360.
7. Wenk G. Psychoactive spices - Bon appetite! http://www.psychologytoday.com/blog/your-brain-food/201008/ psychoactive-spices-bon-appetite. Published August 4, 2010. Accessed March 4, 2014.
8. Wax PM. Just a click away: recreational drug Web sites on the Internet. Pediatrics.2002;109(6):e96.
9. Forrester MB. Nutmeg intoxication in Texas, 1998-2004. Hum Exp Toxicol. 2005;24(11):563-566.
10. Abernethy MK, Becker LB. Acute nutmeg intoxication. Am J Emerg Med. 1992;10(5):429-430.
11. Brenner N, Frank OS, Knight E. Chronic nutmeg psychosis. J R Soc Med. 1993;86(3):179-180.
12. McKenna A, Nordt SP, Ryan J. Acute nutmeg poisoning. Eur J Emerg Med. 2004;11(4):240-241.
13. Dhingra D, Sharma A. Antidepressant-like activity of n-hexane extract of nutmeg (Myristica fragrans) seeds in mice. J Med Food. 2006;9(1):84-89.
14. Truitt EB Jr, Duritz G, Ebersberger EM. Evidence of monoamine oxidase inhibition by myristicin and nutmeg. Proc Soc Exp Biol Med. 1963;112:647-650.
15. Tajuddin, Ahmad S, Latif A, et al. An experimental study of sexual function improving effect of Myristica fragrans Houtt. (nutmeg). BMC Complement Altern Med. 2005;5:16.
16. Quin GI, Fanning NF, Plunkett PK. Nutmeg intoxication. J Accid Emerg Med. 1998;15(4):287-288.
17. Barceloux DG. Nutmeg (Myristica fragrans Houtt.) Dis Mon. 2009;55(6):373-379.
18. Demetriades AK, Wallman PD, McGuiness A, et al. Low cost, high risk: accidental nutmeg intoxication. Emerg Med J. 2005;22(3):223-225.
19. Weil A. The use of nutmeg as a psychotropic agent. Bull Narc. 1966;18(4):15-23. http://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1966-01-01_4_ page003.html. Accessed March 5, 2013.
20. Stein U, Greyer H, Hentschel H. Nutmeg (myristicin) poisoning - report on a fatal case and a series of cases recorded by a poison information centre. Forensic Sci Int. 2001;118(1):87-90.
21. Grover JK, Khandkar S, Vats V, et al. Pharmacological studies on Myristica fragrans—antidiarrheal, hypnotic, analgesic and hemodynamic (blood pressure) parameters. Methods Find Exp Clin Pharmacol. 2002;24(10):675-680.
22. Shah AM, Calello DP, Quintero-Solivan J, et al. The not-so-nice spice: a teenage girl with palpitations and dry mouth. Pediatr Emerg Care. 2011;27(12):1205-1207.
23. Heliotropin. Polarized light microscopy digital image gallery. http://micro.magnet.fsu.edu/primer/techniques/ polarized/gallery/pages/heliotropinsmall.html. Accessed March 5, 2014.
24. Gage E. Romancing the bean. Budget Travel. http://articles.cnn.com/2007-09-11/travel/vanilla_1_vanilla-orchid-totonaca?_s=PM:TRAVEL. Published September 11, 2007. Updated September 16, 2012. Accessed March 5, 2014.
25. Mazor S, DesLauriers CA, Mycyk MB. Adolescent ethanol intoxication from vanilla extract ingestion: a case report. The Internet Journal of Family Practice. 2005;4(1). doi: 10.5580/bc.
26. Skalli S, Soulaymani Bencheikh R. Epileptic seizure induced by fennel essential oil. Epileptic Disord. 2011;13(3):345-347.
27. Zhu M, Wong PY, Li RC. Effect of oral administration of fennel (Foeniculum vulgare) on ciprofloxacin absorption and disposition in the rat. J Pharm Pharmacol. 1999;51(12):1391-1396.
28. Malini T, Arunakaran J, Aruldhas MM, et al. Effects of piperine on the lipid composition and enzymes of the pyruvate-malate cycle in the testis of the rat in vivo. Biochem Mol Biol Int. 1999;47(3):537-545.
29. Zaugg J, Baburin I, Hering S, et al. Identifying GABAA receptor ligands in black pepper by activity profiling, LC-TOFMS, and offline microprobe NMR. Planta Med. 2009; 75(9):888-889. doi: 10.1055/s-0029-1234276.
30. Flavored tobacco. FDA.gov. http://www.fda.gov/TobaccoProducts/ProtectingKidsfromTobacco/ FlavoredTobacco/default.htm. Published September 22, 2009. Updated March 21, 2013. Accessed March 18, 2014.
31. Fujisawa S, Atsumi T, Kadoma Y, et al. Antioxidant and prooxidant action of eugenol-related compounds and their cytotoxicity. Toxicology. 2002;177(1):39-54.
32. Eugenol oil overdose. New York Times Health Guide. http://health.nytimes.com/health/guides/poison/ eugenol-oil-overdose/overview.html. Accessed March 5, 2014.
33. Hartnoll G, Moore D, Douek D. Near fatal ingestion of oil of cloves. Arch Dis Child. 1993;69(3):392-393.
34. Harris E. NPR. German Christmas cookies pose health danger. http://www.npr.org/templates/story/story.php? storyId=6672644. Published December 25, 2006. Accessed March 5, 2014.
35. Frydman-Marom A, Levin A, Farfara D, et al. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS One. 2011; 6(1):e16564. doi:10.1371/journal.pone.001656453.
36. Björnstad K, Helander A, Hultén P, et al. Bioanalytical investigation of asarone in connection with Acorus calamus oil intoxications. J Anal Toxicol. 2009;33(9):604-609.
37. Han P, Han T, Peng W, et al. Antidepressant-like effects of essential oil and asarone, a major essential oil component from the rhizome of Acorus tatarinowii. Pharm Biol. 2013;51(5):589-594.
38. Dandiya PC, Menon MK. Actions of asarone on behavior, stress, and hyperpyrexia, and its interaction with central stimulants. J Pharmacol Exp Ther. 1964;145:42-46.
39. Bockon J. Ginger: inhibition of thromboxane synthetase and stimulation of prostacyclin: relevance for medicine and psychiatry. Med Hypotheses. 1986;20(3):271-278.
40. Ghayur MN, Gilani AH. Pharmacological basis for the medicinal use of ginger in gastrointestinal disorders. Dig Dis Sci. 2005;50(10):1889-1897.
41. Nievergelt A, Huonker P, Schoop R, et al. Identification of serotonin 5-HT1A receptor partial agonists in ginger. Bioorg Med Chem. 2010;18(9):3345-3351.
42. Mishra A, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann Indian Acad Neurol. 2008;11(1):13-19.
43. Seely KA, Levi MS, Prather PL. The dietary polyphenols trans-resveratrol and curcumin selectively bind human CB1 cannabinoid receptors with nanomolar affinities and function as antagonists/inverse agonists [retracted in: J Pharmacol Exp Ther. 2009;331(3):1147]. J Pharmacol Exp Ther. 2009;330(1): 31-39.
44. Gertsch J, Pertwee RG, Di Marzo V. Phytocannabinoids beyond the Cannabis plant – do they exist? Br J Pharmacol. 2010;160(3):523-529.
45. Dwyer AV, Whitten DL, Hawrelak JA. Herbal medicines, other than St. John’s Wort, in the treatment of depression: a systematic review. Altern Med Rev. 2011;16(1):40-49.
46. Moshiri E, Basti AA, Noorbala AA, et al. Crocus sativus L. (petal) in the treatment of mild-to-moderate depression: a double-blind, randomized and placebo controlled trial. Phytomedicine. 2006;13(9-10):607-611.
47. Noorbala AA, Akhondzadeh S, Tahmacebi-Pour N, et al. Hydro-alcoholic extract of Crocus sativus L. versus fluoxetine in the treatment of mild to moderate depression: a double-blind, randomized pilot trial. J Ethnopharmacol. 2005;97(2):281-284.
48. Akhondzadeh S, Tahmacebi-Pour N, Noorbala AA, et al. Crocus sativus L. in the treatment of mild to moderate depression: a double-blind, randomized, and placebo-controlled trial. Phytother Res. 2005;19(2):148-151.
1. O’Mahony Carey S. Psychoactive substances. A guide to ethnobotanical plants and herbs, synthetic chemicals, compounds and products. http://www.drugs.ie/ resourcesfiles/guides/Psychoactive_substances_low_res. pdf. Accessed March 4, 2014.
2. Asafetida. Applied Health. http://www.appliedhealth.com/index.php?option=com _content&view=article&id= 108207. Accessed March 4, 2014.
3. Jayatilaka A, Poole SK, Poole CF, et al. Simultaneous micro steam distillation/solvent extraction for the isolation of semivolatile flavor compounds from cinnamon and their separation by series coupled-column gas chromatography. Analytica Chimica Acta. 1995;302(2-3):147-162.
4. Spices. History & Special Collections UCLA Louise M. Darling Biomedical Library. http://unitproj.library.ucla. edu/biomed/spice/index.cfm?displayID=15. Accessed March 4, 2014.
5. Ginger action and uses. Ginger extract. Gingerols. MDidea Web site. http://www.mdidea.com/products/new/ new02108.html. Accessed March 4, 2014.
6. Raina VK, Srivastava SK, Syamasunder KV. The essential oil of ‘greater galangal’ [Alpinia galanga (L.) Willd.] from the lower Himalayan region of India. Flavour and Fragrance Journal. 2002;17(5):358-360.
7. Wenk G. Psychoactive spices - Bon appetite! http://www.psychologytoday.com/blog/your-brain-food/201008/ psychoactive-spices-bon-appetite. Published August 4, 2010. Accessed March 4, 2014.
8. Wax PM. Just a click away: recreational drug Web sites on the Internet. Pediatrics.2002;109(6):e96.
9. Forrester MB. Nutmeg intoxication in Texas, 1998-2004. Hum Exp Toxicol. 2005;24(11):563-566.
10. Abernethy MK, Becker LB. Acute nutmeg intoxication. Am J Emerg Med. 1992;10(5):429-430.
11. Brenner N, Frank OS, Knight E. Chronic nutmeg psychosis. J R Soc Med. 1993;86(3):179-180.
12. McKenna A, Nordt SP, Ryan J. Acute nutmeg poisoning. Eur J Emerg Med. 2004;11(4):240-241.
13. Dhingra D, Sharma A. Antidepressant-like activity of n-hexane extract of nutmeg (Myristica fragrans) seeds in mice. J Med Food. 2006;9(1):84-89.
14. Truitt EB Jr, Duritz G, Ebersberger EM. Evidence of monoamine oxidase inhibition by myristicin and nutmeg. Proc Soc Exp Biol Med. 1963;112:647-650.
15. Tajuddin, Ahmad S, Latif A, et al. An experimental study of sexual function improving effect of Myristica fragrans Houtt. (nutmeg). BMC Complement Altern Med. 2005;5:16.
16. Quin GI, Fanning NF, Plunkett PK. Nutmeg intoxication. J Accid Emerg Med. 1998;15(4):287-288.
17. Barceloux DG. Nutmeg (Myristica fragrans Houtt.) Dis Mon. 2009;55(6):373-379.
18. Demetriades AK, Wallman PD, McGuiness A, et al. Low cost, high risk: accidental nutmeg intoxication. Emerg Med J. 2005;22(3):223-225.
19. Weil A. The use of nutmeg as a psychotropic agent. Bull Narc. 1966;18(4):15-23. http://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1966-01-01_4_ page003.html. Accessed March 5, 2013.
20. Stein U, Greyer H, Hentschel H. Nutmeg (myristicin) poisoning - report on a fatal case and a series of cases recorded by a poison information centre. Forensic Sci Int. 2001;118(1):87-90.
21. Grover JK, Khandkar S, Vats V, et al. Pharmacological studies on Myristica fragrans—antidiarrheal, hypnotic, analgesic and hemodynamic (blood pressure) parameters. Methods Find Exp Clin Pharmacol. 2002;24(10):675-680.
22. Shah AM, Calello DP, Quintero-Solivan J, et al. The not-so-nice spice: a teenage girl with palpitations and dry mouth. Pediatr Emerg Care. 2011;27(12):1205-1207.
23. Heliotropin. Polarized light microscopy digital image gallery. http://micro.magnet.fsu.edu/primer/techniques/ polarized/gallery/pages/heliotropinsmall.html. Accessed March 5, 2014.
24. Gage E. Romancing the bean. Budget Travel. http://articles.cnn.com/2007-09-11/travel/vanilla_1_vanilla-orchid-totonaca?_s=PM:TRAVEL. Published September 11, 2007. Updated September 16, 2012. Accessed March 5, 2014.
25. Mazor S, DesLauriers CA, Mycyk MB. Adolescent ethanol intoxication from vanilla extract ingestion: a case report. The Internet Journal of Family Practice. 2005;4(1). doi: 10.5580/bc.
26. Skalli S, Soulaymani Bencheikh R. Epileptic seizure induced by fennel essential oil. Epileptic Disord. 2011;13(3):345-347.
27. Zhu M, Wong PY, Li RC. Effect of oral administration of fennel (Foeniculum vulgare) on ciprofloxacin absorption and disposition in the rat. J Pharm Pharmacol. 1999;51(12):1391-1396.
28. Malini T, Arunakaran J, Aruldhas MM, et al. Effects of piperine on the lipid composition and enzymes of the pyruvate-malate cycle in the testis of the rat in vivo. Biochem Mol Biol Int. 1999;47(3):537-545.
29. Zaugg J, Baburin I, Hering S, et al. Identifying GABAA receptor ligands in black pepper by activity profiling, LC-TOFMS, and offline microprobe NMR. Planta Med. 2009; 75(9):888-889. doi: 10.1055/s-0029-1234276.
30. Flavored tobacco. FDA.gov. http://www.fda.gov/TobaccoProducts/ProtectingKidsfromTobacco/ FlavoredTobacco/default.htm. Published September 22, 2009. Updated March 21, 2013. Accessed March 18, 2014.
31. Fujisawa S, Atsumi T, Kadoma Y, et al. Antioxidant and prooxidant action of eugenol-related compounds and their cytotoxicity. Toxicology. 2002;177(1):39-54.
32. Eugenol oil overdose. New York Times Health Guide. http://health.nytimes.com/health/guides/poison/ eugenol-oil-overdose/overview.html. Accessed March 5, 2014.
33. Hartnoll G, Moore D, Douek D. Near fatal ingestion of oil of cloves. Arch Dis Child. 1993;69(3):392-393.
34. Harris E. NPR. German Christmas cookies pose health danger. http://www.npr.org/templates/story/story.php? storyId=6672644. Published December 25, 2006. Accessed March 5, 2014.
35. Frydman-Marom A, Levin A, Farfara D, et al. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS One. 2011; 6(1):e16564. doi:10.1371/journal.pone.001656453.
36. Björnstad K, Helander A, Hultén P, et al. Bioanalytical investigation of asarone in connection with Acorus calamus oil intoxications. J Anal Toxicol. 2009;33(9):604-609.
37. Han P, Han T, Peng W, et al. Antidepressant-like effects of essential oil and asarone, a major essential oil component from the rhizome of Acorus tatarinowii. Pharm Biol. 2013;51(5):589-594.
38. Dandiya PC, Menon MK. Actions of asarone on behavior, stress, and hyperpyrexia, and its interaction with central stimulants. J Pharmacol Exp Ther. 1964;145:42-46.
39. Bockon J. Ginger: inhibition of thromboxane synthetase and stimulation of prostacyclin: relevance for medicine and psychiatry. Med Hypotheses. 1986;20(3):271-278.
40. Ghayur MN, Gilani AH. Pharmacological basis for the medicinal use of ginger in gastrointestinal disorders. Dig Dis Sci. 2005;50(10):1889-1897.
41. Nievergelt A, Huonker P, Schoop R, et al. Identification of serotonin 5-HT1A receptor partial agonists in ginger. Bioorg Med Chem. 2010;18(9):3345-3351.
42. Mishra A, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann Indian Acad Neurol. 2008;11(1):13-19.
43. Seely KA, Levi MS, Prather PL. The dietary polyphenols trans-resveratrol and curcumin selectively bind human CB1 cannabinoid receptors with nanomolar affinities and function as antagonists/inverse agonists [retracted in: J Pharmacol Exp Ther. 2009;331(3):1147]. J Pharmacol Exp Ther. 2009;330(1): 31-39.
44. Gertsch J, Pertwee RG, Di Marzo V. Phytocannabinoids beyond the Cannabis plant – do they exist? Br J Pharmacol. 2010;160(3):523-529.
45. Dwyer AV, Whitten DL, Hawrelak JA. Herbal medicines, other than St. John’s Wort, in the treatment of depression: a systematic review. Altern Med Rev. 2011;16(1):40-49.
46. Moshiri E, Basti AA, Noorbala AA, et al. Crocus sativus L. (petal) in the treatment of mild-to-moderate depression: a double-blind, randomized and placebo controlled trial. Phytomedicine. 2006;13(9-10):607-611.
47. Noorbala AA, Akhondzadeh S, Tahmacebi-Pour N, et al. Hydro-alcoholic extract of Crocus sativus L. versus fluoxetine in the treatment of mild to moderate depression: a double-blind, randomized pilot trial. J Ethnopharmacol. 2005;97(2):281-284.
48. Akhondzadeh S, Tahmacebi-Pour N, Noorbala AA, et al. Crocus sativus L. in the treatment of mild to moderate depression: a double-blind, randomized, and placebo-controlled trial. Phytother Res. 2005;19(2):148-151.

Answers to 7 questions about using neuropsychological testing in your practice
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.

3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
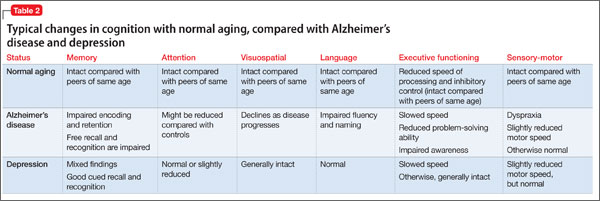
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.
3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.
3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.

Schizophrenia prodrome: An optimal approach
In studies of schizophrenia, one of the more striking findings is the delay in the initiation of treatment. That delay ranges from 1 to 2 years for patients experiencing psychotic symptoms to several years if the prodromal phase is taken into account.1 Yet duration of untreated psychosis has been found to be a critical factor in prognosis, including psychosocial functioning, in patients with schizophrenia.2,3 Identification of individuals in the prodromal phase not only offers an opportunity to intervene at an earlier symptomatic stage, but might be associated with a better response to antipsychotics and a better overall treatment outcome as well.
What’s in a name?
Several terms, including ultra high risk, clinical high risk, at-risk mental state, psychosis risk syndrome, and schizophrenia/psychosis prodrome, have been used to describe the prodromal phase of schizophrenia. The proposal to include attenuated psychosis syndrome (APS) in the DSM-5—originally intended to capture those with subthreshold delusions, hallucinations, or disorganized behavior, occurring at least once a week for the past month and worsening over the past year—generated a debate about the validity of such a diagnostic category4,5 that culminated in the inclusion of APS as a condition for further study but not as a term for clinical use.6 Its presence in the DSM-5 brings to the forefront the importance of early clinical intervention in patients at risk of developing psychotic illness.
Schizophrenia is not inevitable
The prodromal phase can be viewed as a sequence of evolving symptoms7 (Box 18,9), starting with subtle differences evident only to the person experiencing them and often progressing to brief limited intermittent psychosis (BLIPS) or attenuated psychosis.8

In fact, prodrome is a retrospective diagnosis. The predictive power of conversion to psychosis has been found to fluctuate from as low as 9% to as high as 76%,10 prompting ethical concerns about a high false-positive rate, the assumption of inevitability associated with the term “schizophrenia prodrome,”9 and the potential for overdiagnosis and misdiagnosis. Concerns about psychosocial stigma and exposure to antipsychotic medications have been expressed as well.11
A case for early engagement
In retrospect, patients who eventually progress to psychotic illness are commonly found to have been in the prodromal phase for several years. Yet many patients’ first contact with psychiatric services occurs during a florid episode of acute psychosis. Identifying patients in the early prodromal period offers the opportunity to more effectively engage them and form a therapeutic alliance.12 Any young adult who presents with a decline in academic or occupational function, social withdrawal, perplexity, and apparent distress or agitation (Table 113-16) without a clear precipitating factor should therefore be closely monitored, particularly if he (she) has a family history of psychosis.

Screening tools. A variety of interviews and rating scales (Table 28) have been developed to assess and monitor at-risk persons, a number of which have been designed to detect basic symptoms in the early phase of prodrome. In addition to the structured scales, several self-report tools—including the Prodromal Questionnaire-Brief (PQ-B), Youth Psychosis At Risk Questionnaire-Brief (YPARQ-B), Prime Screen-Revised, and PROD-screen (Screen for prodromal symptoms of psychosis)—have been found to be useful in screening a large sample to identify those who might need further evaluation.17
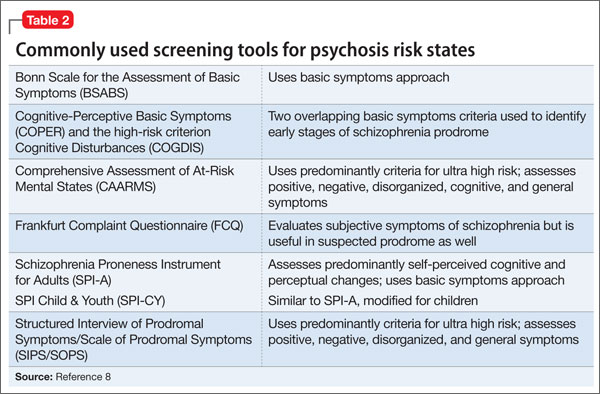
Increased risk of conversion. Several clinical factors are associated with an increased risk of conversion to psychotic illness.9 In addition to family history, these include:
• greater severity and longer duration of attenuated positive symptoms
• presence of bizarre thoughts and behavior
• paranoia
• decline in global assessment of functioning score over the previous year
• use of either Cannabis or amphetamines.
A history of childhood trauma, increased sensitivity to psychosocial stressors, and dysregulation of the hypothalamicpituitary axis also have been associated with progression to psychosis.18
Recent evidence suggests that the prodromal phase is a predictor not only for psychosis but also for other disabling psychiatric illnesses, such as bipolar disorder and obsessive-compulsive disorder.19
From a phenomenological standpoint, disturbance of the sense of self—characterized by features such as depersonalization, derealization, decreased reactivity to other people and the environment, and intense reflectivity to oneself or others—has been proposed as a critical marker for progression to psychosis.20 Another predictor is the perception of negativity of others toward oneself. Examples include heightened sensitivity to rejection or shame, which seems to emerge from a pattern of insecure attachment, and the outsider status experienced by immigrants faced with multiple social, cultural, and language barriers.21 The presence of obsessivecompulsive symptoms during the prodromal phase has been linked to significant impairment in functioning, an acute switch to psychosis, and an increased risk of suicide.22
Monitor or treat? An optimal approach
A key dilemma in the management of patients who exhibit signs and symptoms of schizophrenia prodrome is whether to simply monitor closely or to initiate treatment.
International clinical practice guidelines recommend several practical steps in the monitoring of patients in a prepsychotic state (Table 3),23 but caution against the use of antipsychotic agents unless the patient meets diagnostic criteria for a psychotic disorder.

CBT. Some evidence supports the initiation of cognitive-behavioral therapy (CBT) during the initial prodromal phase and the addition of alow-dose atypical antipsychotic if the patient progresses to a later phase, characterized by BLIPS/APS.24,25 Evidence also suggests that a combination of CBT and antipsychotic medication might delay, but not prevent, the progression to a psychotic episode.9 Any risk of adverse metabolic complications precludes use ofan atypical antipsychotic.One potential alternative is the use of omega-3 polyunsaturated fatty acids (Box 2).26,27
A clinically useful approach would be to view schizophrenia/psychosis prodrome not as a distinct diagnostic category but as a cluster of signs and symptoms associated with an increased risk of psychosis, with persons in this phase in need of close follow-up and, possibly, early initiation of an antipsychotic agent. It is important to engage the patient and his family at an early stage to educate them about the diagnostic uncertainty; to help them deal with the stigma; to manage risk factors; and, collaboratively, to decide on an intervention strategy.23,28

Bottom Line
Despite several drawbacks, the concept of schizophrenia/psychosis prodrome may
be viewed as a cluster of signs and symptoms (rather than a distinct diagnostic category) associated with increased risk for psychosis that need close follow up. Follow up may involve psychoeducational and psychotherapeutic interventions and, need be, early initiation of antipsychotics. In addition, such symptoms may be associated with other psychiatric disorders such as bipolar disorder and obsessive- compulsive disorder. Timely attention and early intervention may alter the course
and improve overall prognosis.
Related Resources
• Early intervention in psychosis. WPA Education Committee’s recommended roles of the psychiatrist. www.wpanet.org/uploads/Education/Educational_Resources/earlyintervention-psychosis.pdf.
• Early Psychosis Prevention and Intervention Centre, Melbourne, Australia. http://eppic.org.au/psychosis.
• International Early Psychosis Association Writing Group. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-124. http://bjp.rcpsych.org/content/187/48/s120.full.
Disclosures
Dr. Madaan is an employee of University of Virginia Health System. As an employee with the University of Virginia, Dr. Madaan has received research support from Eli Lilly and Company, Forest, Merck, Otsuka, Pfizer, Shire, and Sunovion. He also has served as a consultant for the NOW Coalition for Bipolar Disorder, and on the American Psychiatric Association’s Focus Self-Assessment editorial board. Drs. Bestha and Kolli report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Salokangas RK, McGlashan TH. Early detection and intervention of psychosis. A review. Nord J Psychiatry. 2008;62:92-105.
2. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10): 1785-1804.
3. Stefanopoulou E, Lafuente AR, Fonseca AS, et al. Global assessment of psychosocial functioning and predictors of outcome in schizophrenia. Int J Psychiatry Clin Pract. 2011;15(1):62-68.
4. Yung AR, Nelson B, Thompson AD, et al. Should a “Risk Syndrome for Psychosis” be included in the DSMV? Schizophr Res. 2010;120(1-3):7-15.
5. Corcoran CM, First MB, Cornblatt B. The psychosis risk syndrome and its proposed inclusion in the DSM-V: a risk-benefit analysis. Schizophr Res. 2010;120(1-3):16-22.
6. Diagnostic and statistical manual of mental disorders, 5th ed, text rev. Washington, DC: American Psychiatric Association; 2013.
7. Schultze-Lutter F, Ruhrmann S, Berning J, et al. Basic symptoms and ultrahigh risk criteria: symptom development in the initial prodromal state. Schizophr Bull. 2010;36(1):182-191.
8. Correll CU, Hauser M, Auther AM, et al. Research in people with psychosis risk syndrome: a review of the current evidence and future directions. J Child Psychol
Psychiatry. 2010;51(4):390-431.
9. Addington J, Heinssen R. Prediction and prevention of psychosis in youth at clinical high risk. Annu Rev Clin Psychol. 2012;8:269-289.
10. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
11. Singh F, Mirzakhanian H, Fusar-Poli P, et al. Ethical implications for clinical practice and future research in “at risk” individuals. Curr Pharm Des. 2012;18(4):606-612.
12. Bota RG, Munro JS, Ricci WF, et al. The dynamics of insight in the prodrome of schizophrenia. CNS Spectr. 2006;11(5):355-362.
13. Phillips LJ, Yung AR, McGorry PD. Identification of young people at risk of psychosis: validation of Personal Assessment and Crisis Evaluation Clinic intake criteria. Aust N Z J Psychiatry. 2000;34(suppl):S164-S169.
14. Miller TJ, McGlashan TH, Woods SW, et al. Symptom assessment in schizophrenic prodromal states. Psychiatr Q. 1999;70(4):273-287.
15. Schultze-Lutter F, Klosterkötter J, Picker H. Predicting first-episode psychosis by basic symptom criteria. Clinical Neuropsychiatry. 2007;4(1):11-22.
16. Schultze-Lutter F, Ruhrmann S, Picker H, et al. Basic symptoms in early psychotic and depressive disorders. Br J Psychiatry Suppl. 2007;51:s31-s37.
17. Kline E, Wilson C, Ereshefsky S, et al. Convergent and discriminant validity of attenuated psychosis screening tools. Schizophr Res. 2012;134(1):49-53.
18. Holtzman CW, Shapiro DI, Trotman HD, et al. Stress and the prodromal phase of psychosis. Curr Pharm Des. 2012;18(4):527-533.
19. Rössler W, Hengartner MP, Ajdacic-Gross V, et al. Subclinical psychosis symptoms in young adults are risk factors for subsequent common mental disorders. Schizophr Res. 2011;131(1-3):18-23.
20. Nelson B, Yung AR, Bechdolf A, et al. The phenomenological critique and self-disturbance: implications for ultra-high risk (“prodrome”) research. Schizophr Bull. 2008;34(2):381-392.
21. Salokangas RK, Heinimaa M, Svirskis T, et al. Perceived negative attitude of others as an early sign of psychosis. Eur Psychiatry. 2009;24(4):233-238.
22. Niendam TA, Berzak J, Cannon TD, et al. Obsessive compulsive symptoms in the psychosis prodrome:correlates of clinical and functional outcome. Schizophr
Res. 2009;108(1-3):170-175.
23. Addington J, Amminger GP, Barbato A. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-s124.
24. Klosterkötter J, Schultze-Lutter F, Bechdolf A, et al. Prediction and prevention of schizophrenia: what has been achieved and where to go next? World Psychiatry.
2011;10(3):165-174.
25. Stafford MR, Jackson H, Mayo-Wilson E, et al. Early interventions to prevent psychosis: systematic review and meta-analysis. BMJ. 2013;346:f185.
In studies of schizophrenia, one of the more striking findings is the delay in the initiation of treatment. That delay ranges from 1 to 2 years for patients experiencing psychotic symptoms to several years if the prodromal phase is taken into account.1 Yet duration of untreated psychosis has been found to be a critical factor in prognosis, including psychosocial functioning, in patients with schizophrenia.2,3 Identification of individuals in the prodromal phase not only offers an opportunity to intervene at an earlier symptomatic stage, but might be associated with a better response to antipsychotics and a better overall treatment outcome as well.
What’s in a name?
Several terms, including ultra high risk, clinical high risk, at-risk mental state, psychosis risk syndrome, and schizophrenia/psychosis prodrome, have been used to describe the prodromal phase of schizophrenia. The proposal to include attenuated psychosis syndrome (APS) in the DSM-5—originally intended to capture those with subthreshold delusions, hallucinations, or disorganized behavior, occurring at least once a week for the past month and worsening over the past year—generated a debate about the validity of such a diagnostic category4,5 that culminated in the inclusion of APS as a condition for further study but not as a term for clinical use.6 Its presence in the DSM-5 brings to the forefront the importance of early clinical intervention in patients at risk of developing psychotic illness.
Schizophrenia is not inevitable
The prodromal phase can be viewed as a sequence of evolving symptoms7 (Box 18,9), starting with subtle differences evident only to the person experiencing them and often progressing to brief limited intermittent psychosis (BLIPS) or attenuated psychosis.8
In fact, prodrome is a retrospective diagnosis. The predictive power of conversion to psychosis has been found to fluctuate from as low as 9% to as high as 76%,10 prompting ethical concerns about a high false-positive rate, the assumption of inevitability associated with the term “schizophrenia prodrome,”9 and the potential for overdiagnosis and misdiagnosis. Concerns about psychosocial stigma and exposure to antipsychotic medications have been expressed as well.11
A case for early engagement
In retrospect, patients who eventually progress to psychotic illness are commonly found to have been in the prodromal phase for several years. Yet many patients’ first contact with psychiatric services occurs during a florid episode of acute psychosis. Identifying patients in the early prodromal period offers the opportunity to more effectively engage them and form a therapeutic alliance.12 Any young adult who presents with a decline in academic or occupational function, social withdrawal, perplexity, and apparent distress or agitation (Table 113-16) without a clear precipitating factor should therefore be closely monitored, particularly if he (she) has a family history of psychosis.
Screening tools. A variety of interviews and rating scales (Table 28) have been developed to assess and monitor at-risk persons, a number of which have been designed to detect basic symptoms in the early phase of prodrome. In addition to the structured scales, several self-report tools—including the Prodromal Questionnaire-Brief (PQ-B), Youth Psychosis At Risk Questionnaire-Brief (YPARQ-B), Prime Screen-Revised, and PROD-screen (Screen for prodromal symptoms of psychosis)—have been found to be useful in screening a large sample to identify those who might need further evaluation.17
Increased risk of conversion. Several clinical factors are associated with an increased risk of conversion to psychotic illness.9 In addition to family history, these include:
• greater severity and longer duration of attenuated positive symptoms
• presence of bizarre thoughts and behavior
• paranoia
• decline in global assessment of functioning score over the previous year
• use of either Cannabis or amphetamines.
A history of childhood trauma, increased sensitivity to psychosocial stressors, and dysregulation of the hypothalamicpituitary axis also have been associated with progression to psychosis.18
Recent evidence suggests that the prodromal phase is a predictor not only for psychosis but also for other disabling psychiatric illnesses, such as bipolar disorder and obsessive-compulsive disorder.19
From a phenomenological standpoint, disturbance of the sense of self—characterized by features such as depersonalization, derealization, decreased reactivity to other people and the environment, and intense reflectivity to oneself or others—has been proposed as a critical marker for progression to psychosis.20 Another predictor is the perception of negativity of others toward oneself. Examples include heightened sensitivity to rejection or shame, which seems to emerge from a pattern of insecure attachment, and the outsider status experienced by immigrants faced with multiple social, cultural, and language barriers.21 The presence of obsessivecompulsive symptoms during the prodromal phase has been linked to significant impairment in functioning, an acute switch to psychosis, and an increased risk of suicide.22
Monitor or treat? An optimal approach
A key dilemma in the management of patients who exhibit signs and symptoms of schizophrenia prodrome is whether to simply monitor closely or to initiate treatment.
International clinical practice guidelines recommend several practical steps in the monitoring of patients in a prepsychotic state (Table 3),23 but caution against the use of antipsychotic agents unless the patient meets diagnostic criteria for a psychotic disorder.
CBT. Some evidence supports the initiation of cognitive-behavioral therapy (CBT) during the initial prodromal phase and the addition of alow-dose atypical antipsychotic if the patient progresses to a later phase, characterized by BLIPS/APS.24,25 Evidence also suggests that a combination of CBT and antipsychotic medication might delay, but not prevent, the progression to a psychotic episode.9 Any risk of adverse metabolic complications precludes use ofan atypical antipsychotic.One potential alternative is the use of omega-3 polyunsaturated fatty acids (Box 2).26,27
A clinically useful approach would be to view schizophrenia/psychosis prodrome not as a distinct diagnostic category but as a cluster of signs and symptoms associated with an increased risk of psychosis, with persons in this phase in need of close follow-up and, possibly, early initiation of an antipsychotic agent. It is important to engage the patient and his family at an early stage to educate them about the diagnostic uncertainty; to help them deal with the stigma; to manage risk factors; and, collaboratively, to decide on an intervention strategy.23,28
Bottom Line
Despite several drawbacks, the concept of schizophrenia/psychosis prodrome may
be viewed as a cluster of signs and symptoms (rather than a distinct diagnostic category) associated with increased risk for psychosis that need close follow up. Follow up may involve psychoeducational and psychotherapeutic interventions and, need be, early initiation of antipsychotics. In addition, such symptoms may be associated with other psychiatric disorders such as bipolar disorder and obsessive- compulsive disorder. Timely attention and early intervention may alter the course
and improve overall prognosis.
Related Resources
• Early intervention in psychosis. WPA Education Committee’s recommended roles of the psychiatrist. www.wpanet.org/uploads/Education/Educational_Resources/earlyintervention-psychosis.pdf.
• Early Psychosis Prevention and Intervention Centre, Melbourne, Australia. http://eppic.org.au/psychosis.
• International Early Psychosis Association Writing Group. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-124. http://bjp.rcpsych.org/content/187/48/s120.full.
Disclosures
Dr. Madaan is an employee of University of Virginia Health System. As an employee with the University of Virginia, Dr. Madaan has received research support from Eli Lilly and Company, Forest, Merck, Otsuka, Pfizer, Shire, and Sunovion. He also has served as a consultant for the NOW Coalition for Bipolar Disorder, and on the American Psychiatric Association’s Focus Self-Assessment editorial board. Drs. Bestha and Kolli report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
In studies of schizophrenia, one of the more striking findings is the delay in the initiation of treatment. That delay ranges from 1 to 2 years for patients experiencing psychotic symptoms to several years if the prodromal phase is taken into account.1 Yet duration of untreated psychosis has been found to be a critical factor in prognosis, including psychosocial functioning, in patients with schizophrenia.2,3 Identification of individuals in the prodromal phase not only offers an opportunity to intervene at an earlier symptomatic stage, but might be associated with a better response to antipsychotics and a better overall treatment outcome as well.
What’s in a name?
Several terms, including ultra high risk, clinical high risk, at-risk mental state, psychosis risk syndrome, and schizophrenia/psychosis prodrome, have been used to describe the prodromal phase of schizophrenia. The proposal to include attenuated psychosis syndrome (APS) in the DSM-5—originally intended to capture those with subthreshold delusions, hallucinations, or disorganized behavior, occurring at least once a week for the past month and worsening over the past year—generated a debate about the validity of such a diagnostic category4,5 that culminated in the inclusion of APS as a condition for further study but not as a term for clinical use.6 Its presence in the DSM-5 brings to the forefront the importance of early clinical intervention in patients at risk of developing psychotic illness.
Schizophrenia is not inevitable
The prodromal phase can be viewed as a sequence of evolving symptoms7 (Box 18,9), starting with subtle differences evident only to the person experiencing them and often progressing to brief limited intermittent psychosis (BLIPS) or attenuated psychosis.8
In fact, prodrome is a retrospective diagnosis. The predictive power of conversion to psychosis has been found to fluctuate from as low as 9% to as high as 76%,10 prompting ethical concerns about a high false-positive rate, the assumption of inevitability associated with the term “schizophrenia prodrome,”9 and the potential for overdiagnosis and misdiagnosis. Concerns about psychosocial stigma and exposure to antipsychotic medications have been expressed as well.11
A case for early engagement
In retrospect, patients who eventually progress to psychotic illness are commonly found to have been in the prodromal phase for several years. Yet many patients’ first contact with psychiatric services occurs during a florid episode of acute psychosis. Identifying patients in the early prodromal period offers the opportunity to more effectively engage them and form a therapeutic alliance.12 Any young adult who presents with a decline in academic or occupational function, social withdrawal, perplexity, and apparent distress or agitation (Table 113-16) without a clear precipitating factor should therefore be closely monitored, particularly if he (she) has a family history of psychosis.
Screening tools. A variety of interviews and rating scales (Table 28) have been developed to assess and monitor at-risk persons, a number of which have been designed to detect basic symptoms in the early phase of prodrome. In addition to the structured scales, several self-report tools—including the Prodromal Questionnaire-Brief (PQ-B), Youth Psychosis At Risk Questionnaire-Brief (YPARQ-B), Prime Screen-Revised, and PROD-screen (Screen for prodromal symptoms of psychosis)—have been found to be useful in screening a large sample to identify those who might need further evaluation.17
Increased risk of conversion. Several clinical factors are associated with an increased risk of conversion to psychotic illness.9 In addition to family history, these include:
• greater severity and longer duration of attenuated positive symptoms
• presence of bizarre thoughts and behavior
• paranoia
• decline in global assessment of functioning score over the previous year
• use of either Cannabis or amphetamines.
A history of childhood trauma, increased sensitivity to psychosocial stressors, and dysregulation of the hypothalamicpituitary axis also have been associated with progression to psychosis.18
Recent evidence suggests that the prodromal phase is a predictor not only for psychosis but also for other disabling psychiatric illnesses, such as bipolar disorder and obsessive-compulsive disorder.19
From a phenomenological standpoint, disturbance of the sense of self—characterized by features such as depersonalization, derealization, decreased reactivity to other people and the environment, and intense reflectivity to oneself or others—has been proposed as a critical marker for progression to psychosis.20 Another predictor is the perception of negativity of others toward oneself. Examples include heightened sensitivity to rejection or shame, which seems to emerge from a pattern of insecure attachment, and the outsider status experienced by immigrants faced with multiple social, cultural, and language barriers.21 The presence of obsessivecompulsive symptoms during the prodromal phase has been linked to significant impairment in functioning, an acute switch to psychosis, and an increased risk of suicide.22
Monitor or treat? An optimal approach
A key dilemma in the management of patients who exhibit signs and symptoms of schizophrenia prodrome is whether to simply monitor closely or to initiate treatment.
International clinical practice guidelines recommend several practical steps in the monitoring of patients in a prepsychotic state (Table 3),23 but caution against the use of antipsychotic agents unless the patient meets diagnostic criteria for a psychotic disorder.
CBT. Some evidence supports the initiation of cognitive-behavioral therapy (CBT) during the initial prodromal phase and the addition of alow-dose atypical antipsychotic if the patient progresses to a later phase, characterized by BLIPS/APS.24,25 Evidence also suggests that a combination of CBT and antipsychotic medication might delay, but not prevent, the progression to a psychotic episode.9 Any risk of adverse metabolic complications precludes use ofan atypical antipsychotic.One potential alternative is the use of omega-3 polyunsaturated fatty acids (Box 2).26,27
A clinically useful approach would be to view schizophrenia/psychosis prodrome not as a distinct diagnostic category but as a cluster of signs and symptoms associated with an increased risk of psychosis, with persons in this phase in need of close follow-up and, possibly, early initiation of an antipsychotic agent. It is important to engage the patient and his family at an early stage to educate them about the diagnostic uncertainty; to help them deal with the stigma; to manage risk factors; and, collaboratively, to decide on an intervention strategy.23,28
Bottom Line
Despite several drawbacks, the concept of schizophrenia/psychosis prodrome may
be viewed as a cluster of signs and symptoms (rather than a distinct diagnostic category) associated with increased risk for psychosis that need close follow up. Follow up may involve psychoeducational and psychotherapeutic interventions and, need be, early initiation of antipsychotics. In addition, such symptoms may be associated with other psychiatric disorders such as bipolar disorder and obsessive- compulsive disorder. Timely attention and early intervention may alter the course
and improve overall prognosis.
Related Resources
• Early intervention in psychosis. WPA Education Committee’s recommended roles of the psychiatrist. www.wpanet.org/uploads/Education/Educational_Resources/earlyintervention-psychosis.pdf.
• Early Psychosis Prevention and Intervention Centre, Melbourne, Australia. http://eppic.org.au/psychosis.
• International Early Psychosis Association Writing Group. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-124. http://bjp.rcpsych.org/content/187/48/s120.full.
Disclosures
Dr. Madaan is an employee of University of Virginia Health System. As an employee with the University of Virginia, Dr. Madaan has received research support from Eli Lilly and Company, Forest, Merck, Otsuka, Pfizer, Shire, and Sunovion. He also has served as a consultant for the NOW Coalition for Bipolar Disorder, and on the American Psychiatric Association’s Focus Self-Assessment editorial board. Drs. Bestha and Kolli report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Salokangas RK, McGlashan TH. Early detection and intervention of psychosis. A review. Nord J Psychiatry. 2008;62:92-105.
2. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10): 1785-1804.
3. Stefanopoulou E, Lafuente AR, Fonseca AS, et al. Global assessment of psychosocial functioning and predictors of outcome in schizophrenia. Int J Psychiatry Clin Pract. 2011;15(1):62-68.
4. Yung AR, Nelson B, Thompson AD, et al. Should a “Risk Syndrome for Psychosis” be included in the DSMV? Schizophr Res. 2010;120(1-3):7-15.
5. Corcoran CM, First MB, Cornblatt B. The psychosis risk syndrome and its proposed inclusion in the DSM-V: a risk-benefit analysis. Schizophr Res. 2010;120(1-3):16-22.
6. Diagnostic and statistical manual of mental disorders, 5th ed, text rev. Washington, DC: American Psychiatric Association; 2013.
7. Schultze-Lutter F, Ruhrmann S, Berning J, et al. Basic symptoms and ultrahigh risk criteria: symptom development in the initial prodromal state. Schizophr Bull. 2010;36(1):182-191.
8. Correll CU, Hauser M, Auther AM, et al. Research in people with psychosis risk syndrome: a review of the current evidence and future directions. J Child Psychol
Psychiatry. 2010;51(4):390-431.
9. Addington J, Heinssen R. Prediction and prevention of psychosis in youth at clinical high risk. Annu Rev Clin Psychol. 2012;8:269-289.
10. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
11. Singh F, Mirzakhanian H, Fusar-Poli P, et al. Ethical implications for clinical practice and future research in “at risk” individuals. Curr Pharm Des. 2012;18(4):606-612.
12. Bota RG, Munro JS, Ricci WF, et al. The dynamics of insight in the prodrome of schizophrenia. CNS Spectr. 2006;11(5):355-362.
13. Phillips LJ, Yung AR, McGorry PD. Identification of young people at risk of psychosis: validation of Personal Assessment and Crisis Evaluation Clinic intake criteria. Aust N Z J Psychiatry. 2000;34(suppl):S164-S169.
14. Miller TJ, McGlashan TH, Woods SW, et al. Symptom assessment in schizophrenic prodromal states. Psychiatr Q. 1999;70(4):273-287.
15. Schultze-Lutter F, Klosterkötter J, Picker H. Predicting first-episode psychosis by basic symptom criteria. Clinical Neuropsychiatry. 2007;4(1):11-22.
16. Schultze-Lutter F, Ruhrmann S, Picker H, et al. Basic symptoms in early psychotic and depressive disorders. Br J Psychiatry Suppl. 2007;51:s31-s37.
17. Kline E, Wilson C, Ereshefsky S, et al. Convergent and discriminant validity of attenuated psychosis screening tools. Schizophr Res. 2012;134(1):49-53.
18. Holtzman CW, Shapiro DI, Trotman HD, et al. Stress and the prodromal phase of psychosis. Curr Pharm Des. 2012;18(4):527-533.
19. Rössler W, Hengartner MP, Ajdacic-Gross V, et al. Subclinical psychosis symptoms in young adults are risk factors for subsequent common mental disorders. Schizophr Res. 2011;131(1-3):18-23.
20. Nelson B, Yung AR, Bechdolf A, et al. The phenomenological critique and self-disturbance: implications for ultra-high risk (“prodrome”) research. Schizophr Bull. 2008;34(2):381-392.
21. Salokangas RK, Heinimaa M, Svirskis T, et al. Perceived negative attitude of others as an early sign of psychosis. Eur Psychiatry. 2009;24(4):233-238.
22. Niendam TA, Berzak J, Cannon TD, et al. Obsessive compulsive symptoms in the psychosis prodrome:correlates of clinical and functional outcome. Schizophr
Res. 2009;108(1-3):170-175.
23. Addington J, Amminger GP, Barbato A. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-s124.
24. Klosterkötter J, Schultze-Lutter F, Bechdolf A, et al. Prediction and prevention of schizophrenia: what has been achieved and where to go next? World Psychiatry.
2011;10(3):165-174.
25. Stafford MR, Jackson H, Mayo-Wilson E, et al. Early interventions to prevent psychosis: systematic review and meta-analysis. BMJ. 2013;346:f185.
1. Salokangas RK, McGlashan TH. Early detection and intervention of psychosis. A review. Nord J Psychiatry. 2008;62:92-105.
2. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10): 1785-1804.
3. Stefanopoulou E, Lafuente AR, Fonseca AS, et al. Global assessment of psychosocial functioning and predictors of outcome in schizophrenia. Int J Psychiatry Clin Pract. 2011;15(1):62-68.
4. Yung AR, Nelson B, Thompson AD, et al. Should a “Risk Syndrome for Psychosis” be included in the DSMV? Schizophr Res. 2010;120(1-3):7-15.
5. Corcoran CM, First MB, Cornblatt B. The psychosis risk syndrome and its proposed inclusion in the DSM-V: a risk-benefit analysis. Schizophr Res. 2010;120(1-3):16-22.
6. Diagnostic and statistical manual of mental disorders, 5th ed, text rev. Washington, DC: American Psychiatric Association; 2013.
7. Schultze-Lutter F, Ruhrmann S, Berning J, et al. Basic symptoms and ultrahigh risk criteria: symptom development in the initial prodromal state. Schizophr Bull. 2010;36(1):182-191.
8. Correll CU, Hauser M, Auther AM, et al. Research in people with psychosis risk syndrome: a review of the current evidence and future directions. J Child Psychol
Psychiatry. 2010;51(4):390-431.
9. Addington J, Heinssen R. Prediction and prevention of psychosis in youth at clinical high risk. Annu Rev Clin Psychol. 2012;8:269-289.
10. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
11. Singh F, Mirzakhanian H, Fusar-Poli P, et al. Ethical implications for clinical practice and future research in “at risk” individuals. Curr Pharm Des. 2012;18(4):606-612.
12. Bota RG, Munro JS, Ricci WF, et al. The dynamics of insight in the prodrome of schizophrenia. CNS Spectr. 2006;11(5):355-362.
13. Phillips LJ, Yung AR, McGorry PD. Identification of young people at risk of psychosis: validation of Personal Assessment and Crisis Evaluation Clinic intake criteria. Aust N Z J Psychiatry. 2000;34(suppl):S164-S169.
14. Miller TJ, McGlashan TH, Woods SW, et al. Symptom assessment in schizophrenic prodromal states. Psychiatr Q. 1999;70(4):273-287.
15. Schultze-Lutter F, Klosterkötter J, Picker H. Predicting first-episode psychosis by basic symptom criteria. Clinical Neuropsychiatry. 2007;4(1):11-22.
16. Schultze-Lutter F, Ruhrmann S, Picker H, et al. Basic symptoms in early psychotic and depressive disorders. Br J Psychiatry Suppl. 2007;51:s31-s37.
17. Kline E, Wilson C, Ereshefsky S, et al. Convergent and discriminant validity of attenuated psychosis screening tools. Schizophr Res. 2012;134(1):49-53.
18. Holtzman CW, Shapiro DI, Trotman HD, et al. Stress and the prodromal phase of psychosis. Curr Pharm Des. 2012;18(4):527-533.
19. Rössler W, Hengartner MP, Ajdacic-Gross V, et al. Subclinical psychosis symptoms in young adults are risk factors for subsequent common mental disorders. Schizophr Res. 2011;131(1-3):18-23.
20. Nelson B, Yung AR, Bechdolf A, et al. The phenomenological critique and self-disturbance: implications for ultra-high risk (“prodrome”) research. Schizophr Bull. 2008;34(2):381-392.
21. Salokangas RK, Heinimaa M, Svirskis T, et al. Perceived negative attitude of others as an early sign of psychosis. Eur Psychiatry. 2009;24(4):233-238.
22. Niendam TA, Berzak J, Cannon TD, et al. Obsessive compulsive symptoms in the psychosis prodrome:correlates of clinical and functional outcome. Schizophr
Res. 2009;108(1-3):170-175.
23. Addington J, Amminger GP, Barbato A. International clinical practice guidelines for early psychosis. Br J Psychiatry. 2005;187:s120-s124.
24. Klosterkötter J, Schultze-Lutter F, Bechdolf A, et al. Prediction and prevention of schizophrenia: what has been achieved and where to go next? World Psychiatry.
2011;10(3):165-174.
25. Stafford MR, Jackson H, Mayo-Wilson E, et al. Early interventions to prevent psychosis: systematic review and meta-analysis. BMJ. 2013;346:f185.

Awakening to the dangers of obstructive sleep apnea
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1

The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.

Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12

Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.

Dissecting melancholia with evidence-based biomarker tools
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).

Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).


In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
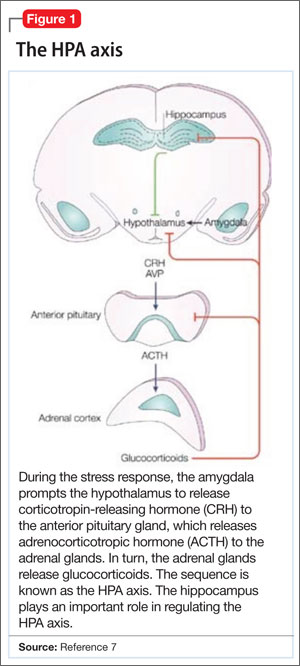
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
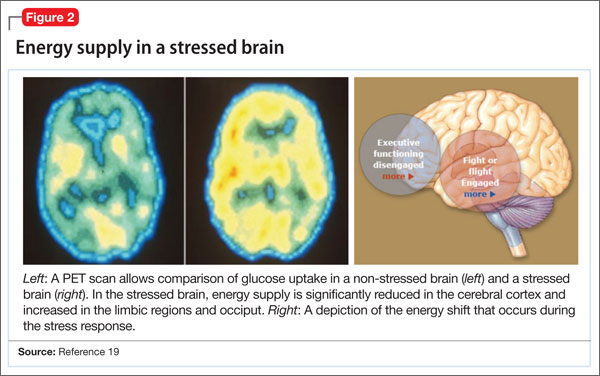
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
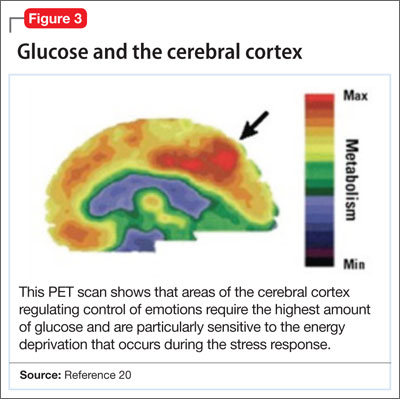
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
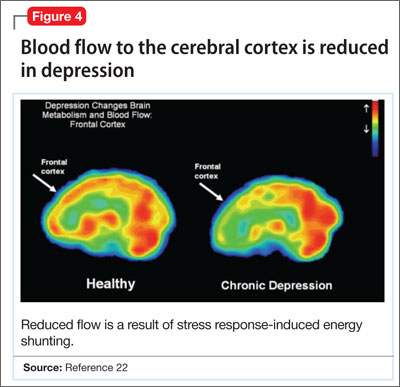
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).

Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
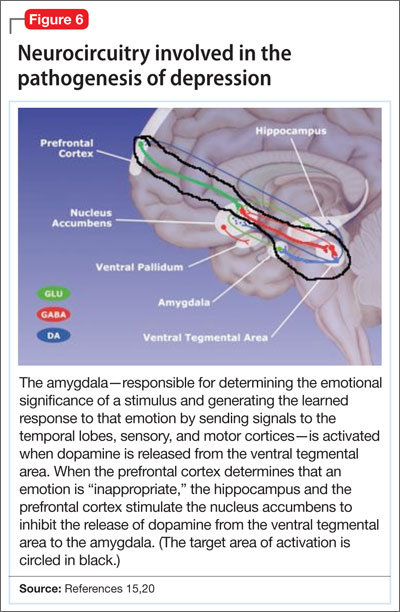
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
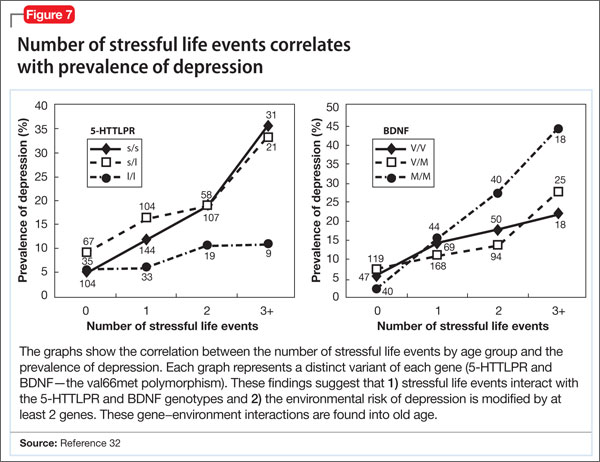
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.

Recovery on higher ground: Spirituality in the treatment of substance abuse
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.

The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
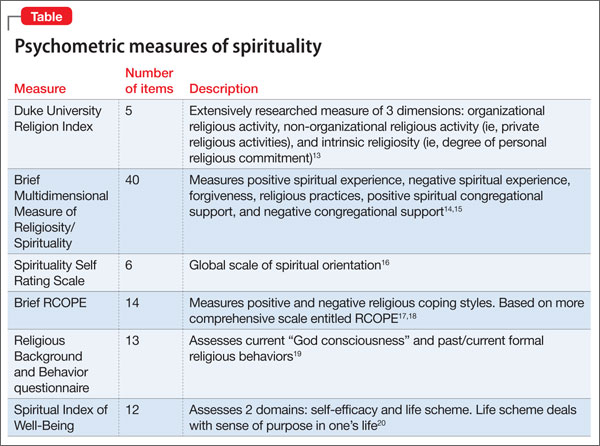
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.

Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.

Medication for alcohol use disorder: Which agents work best?
Historically, alcohol use disorder (AUD; classified as alcohol abuse or dependence in DSM-IV-TR) has been treated with psychosocial therapies, but many patients treated this way relapse into heavy drinking patterns and are unable to sustain sobriety (Box 11). Although vital for treating AUD, psychosocial methods have, to date, a modest success rate. Research has demonstrated that combining pharmacotherapy with psychosocial programs is effective for treating AUD.2

Patients and clinicians might associate AUD medications with so-called aversion therapy because, for many years, the only treatment was disulfiram, which causes unpleasant physical effects when consumed with alcohol. However, newer medications help patients maintain abstinence by targeting brain neurotransmitters relevant to addiction neurocircuitry, such as dopamine, serotonin, ϒ-aminobutyric acid (GABA), glutamate, and opioid.3 These medications may help patients with AUD achieve sobriety, avoid relapse, decrease heavy drinking days, and delay time to recurrent drinking.
In this article, we review FDA-approved medications (Table 1)4-6 and off-label agents (Table 2)3,7-18 and provide recommendations for treating patients with AUD (Box 2).19-21
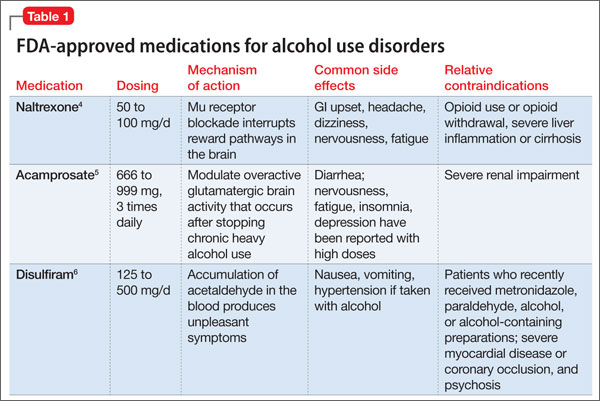
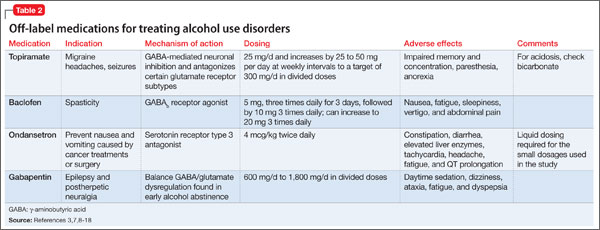

FDA-approved treatments
Naltrexone is an opiate antagonist that blocks the mu receptor and is believed to interrupt the dopamine reward pathway in the brain for alcohol. A meta-analysis of 2,861 patients in 24 combined randomized controlled trials (RCTs) demonstrated naltrexone to be an effective short-term (12 weeks) treatment for alcoholism, significantly decreasing relapses.22 The large multisite COMBINE study (N = 1,383) showed that naltrexone, 100 mg/d, and medical treatment without behavioral treatment over 16 weeks was more effective than placebo in increasing percentage of days abstinent (80.6% vs 75.1%, respectively) and reducing the percentage of patients experiencing heavy drinking days (66.2% vs 73.1%, respectively).2 Patients who have a family history of AUD or strong cravings, or both, may benefit most from naltrexone.3,7,23 Despite evidence of the effectiveness of naltrexone for AUD, not all studies have yielded positive results.24
Common side effects of naltrexone, if present, appear early in treatment and include GI upset (eg, nausea, vomiting, abdominal pain), headache, and fatigue.2,3,22,23 Hepatotoxicity has been reported with dosages of 100 to 300 mg/d, but lab values typically normalize when naltrexone is discontinued.2,3,23 Monitor markers of liver function including ϒ-glutamyltransferase, aspartate aminotransferase, alanine aminotransferase, and bilirubin before and during naltrexone treatment (we check patients 1 to 3 months after starting treatment and yearly thereafter). Obtain a negative urine drug screen for opioids before administering naltrexone, because if opioids were consumed recently naltrexone could precipitate withdrawal. Because of the unknown teratogenicity of naltrexone, women of childbearing age should undergo pregnancy testing before and periodically during naltrexone therapy.
Naltrexone also is available in a once-monthly, 380-mg injectable formulation. Studies show that, similar to its oral counterpart, injectable naltrexone effectively reduces heavy drinking days and number of drinks a day compared with placebo.25,26 Advantages of injectable naltrexone are its extended steady release of medication and its efficacy for patients who do not adhere to oral dosing.7,27 Side effects are similar to oral naltrexone, except for injection site reactions and pain.
Contraindications to naltrexone include current opioid use because its antagonistic effects on opioid receptors render opioid analgesia ineffective. Patients who have used opioids within 7 to 10 days or who may be surreptitiously using opioids should not take naltrexone because it may cause opioid withdrawal. Some patients may try to override the opioid receptor blockade of naltrexone with higher opioid doses, which could result in overdose.
Naltrexone is approved for treating opioid use disorder and may be useful for persons with comorbid opioid use disorder and AUD, if the patient has been adequately detoxified from opioids and intends to abstain from these drugs. Patients who have extensive liver damage secondary to acute hepatitis or uncompensated cirrhosis would not be good candidates for naltrexone because of a risk of hepatotoxicity.2,3,22
Because of ease of dosing, we recommend naltrexone as a first-line treatment for AUD, unless the patient requires opioids or has severe liver disease. We recommend increasing naltrexone from 50 mg to 100 mg before switching to acamprosate, based on European studies.
Acamprosate is a glutamate antagonist that is thought to modulate overactive glutamatergic brain activity that occurs after stopping chronic heavy alcohol use. In a meta-analysis of 17 studies (N = 4,087), continuous abstinence rates at 6 months were significantly higher in acamprosate-treated patients (36.1%) than in patients receiving placebo (23.4%).28 In a review of European studies, acamprosate benefited patients who have increased anxiety, physiological dependence, negative family history of AUD, and late age of onset (age >25) of alcohol dependence.7 However, in the COMBINE trial acamprosate was no more effective than placebo.2
We consider acamprosate an effective option for patients who do not respond to naltrexone or have a contraindication. Dosages of 333 mg to 666 mg, 3 times a day, are recommended, although dosages up to 3 g/d have been studied; titration is not required.2,7,28 We recommend advising patients to continue treatment even if they relapse, because these medications may mitigate relapse severity. Adherence to multiple daily doses can be problematic for some patients, but pairing medications with meals or bedtime may improve adherence.
Diarrhea is the most common side effect of acamprosate; nervousness, fatigue, insomnia, and depression have been reported with high dosages.2,3,7,28 Acamprosate is excreted through the kidney and is safe for patients with liver disorders such as acute hepatitis or cirrhosis. The drug is contraindicated in patients with acute or chronic renal failure with creatinine clearance <30 mL/min; those with less severe renal insufficiency might need a lower dosage. Obtain baseline renal function before starting acamprosate; women of childbearing age should undergo a pregnancy test.
Disulfiram inhibits alcohol metabolism, resulting in acetaldehyde accumulation, which causes unpleasant physical effects such as nausea, vomiting, and hypotension. This creates a negative rather than a positive experience with drinking. A US Veterans Administration Cooperative Study randomized 605 participants to riboflavin, disulfiram, 1 mg/d (an inactive dose), or disulfiram, 250 mg/d (standard dose). There was no difference in percentage of patients remaining abstinent or time to first drink.8 Participants receiving disulfiram, 250 mg/d, had fewer drinking days after relapse compared with the other groups.8
Adverse physical effects produced when disulfiram and alcohol interact include tremor, diaphoresis, unstable blood pressure, and severe diarrhea and vomiting. Disulfiram can cause medically serious reactions in a small percentage of patients, especially those with significant medical comorbidity or advanced age. Patients with severe hypertension, diabetes mellitus, heart disease, a history of stroke, peripheral neuropathy, epilepsy, or renal or hepatic insufficiency should not use disulfiram.3 Patients taking disulfiram should avoid casual exposures to food, aftershave, mouthwash, and hand sanitizer that might contain alcohol. Disulfiram has no significant effect on alcohol craving. Social support to help oversee dosing may enhance adherence.7
Off-label medications
Topiramate is FDA-approved to treat migraine headaches and some seizure disorders. Topiramate facilitates GABA-mediated neuronal inhibition and antagonizes certain glutamate receptor subtypes. In an RCT (N = 150), topiramate, up to 300 mg/d, was more effective than placebo at reducing heavy drinking days and number of drinks per day, increasing days abstinent, and alleviating cravings.9 In a 12-week, double-blind RCT (N = 150), topiramate increased “safe drinking”—defined as ≤1 standard drink per day for women and ≤2 per day for men—vs placebo.10 Dosages were 75 mg to 300 mg/d in twice daily divided doses. Dosing starts at 25 mg/d and increases by 25 to 50 mg a day at weekly intervals. We recommend reserving topiramate for persons who do not respond to or cannot tolerate naltrexone and acamprosate because of the slow titration needed to prevent side effects. Although not studied, it may seem that topiramate’s antiepileptic actions could prevent seizures during alcohol withdrawal, but the protracted titration would limit its utility.
Side effects of topiramate include impaired memory and concentration, paresthesia, and anorexia and are more likely to present during rapid titration or with a high dosage.7,8 Rare reports of spontaneous myopia, angle-closure glaucoma, increased intraocular pressure, ocular pain, and blurry vision have been reported, but these complications often resolve with discontinuation of topiramate.7,8
Topiramate primarily is excreted through the kidney, and its action in the renal tubules can lead to metabolic acidosis or nephrolithiasis.8 Relative contraindications include acute or chronic kidney disease, including kidney stones. Consider slower titration and a 50% reduction in dosing if creatinine clearance is <70 mL/min. Obtain renal function tests before starting topiramate and consider monitoring serum bicarbonate for metabolic acidosis (we test at 3 and 6 months, then every 6 months). Because of teratogenic effects of topiramate (eg, cleft lip and palate), rule out pregnancy in all women of childbearing age.
Baclofen is a GABAb receptor agonist that is FDA approved for treating spasticity. Because GABA transmission is down-regulated in chronic AUD, it is a commonly targeted neurotransmitter when developing medications for AUD. GABAa receptors are fast-acting inhibitory ion channels, and its agonists (eg, benzodiazepines) have a significant abuse and cross-addiction liability. GABAb receptors, however, are slow-acting through a complex cascade of intracellular signals, and therefore GABAb agonists such as baclofen have been studied for treating addiction.
In a randomized double-blind, placebo-controlled trial (N = 39), baclofen was superior to placebo in suppressing obsessive aspects of cravings and decreasing state anxiety.11 Baclofen, 10 mg 3 times daily, in another randomized double-blind, placebo-controlled trial (n = 42) reduced the number of drinks per day by 53% vs placebo; 20 mg 3 times a day resulted in a 68% reduction in drinks per day vs placebo.12 However, a placebo-controlled RCT (n = 80) reported that baclofen, 10 mg 3 times daily, was not superior to placebo for primary outcomes related to alcohol consumption, although it did significantly decrease cravings and anxiety among persons with AUD.13 Evidence suggests that baclofen might be effective for promoting abstinence, reducing the risk of relapse, and alleviating cravings and anxiety in persons with AUD, although further investigation is needed.
In studies for AUD, the side-effect profile for baclofen was relatively benign.11-13 Nausea, fatigue, sleepiness, vertigo, and abdominal pain were reported; overall, baclofen was found to be safe and to have no abuse liability.7,10,12 The addictive potential of other muscle relaxers may have dissuaded providers from using baclofen for AUD, but we consider it a reasonable alternative when FDA-approved treatments fail.
Because baclofen is primarily eliminated by the kidneys, it may be safe for people with cirrhosis or severe liver disease.12 Baseline renal labs should be performed before administering baclofen and a negative pregnancy test obtained for women of childbearing age.
Ondansetron is a serotonin receptor type 3 (5-HT3) antagonist that has shown promising results for AUD.3,7 Research suggests that 5-HT3 receptors are an action site for alcohol in the brain and are thought to play a role in its rewarding effects.7 Ondansetron may be more effective for early-onset alcoholism (EOA) than late-onset alcoholism (LOA).14,15 EOA (age ≤25) is characterized by strong family history of AUD and prominent antisocial traits. A randomized double-blind, placebo-controlled trial (n = 271) reported that ondansetron, 4 mcg/kg twice daily, was superior to placebo in reducing number of drinks per day, increasing days abstinent, and reducing cravings in patients with EOA.14 Among persons with EOA, ondansetron, 16 mcg/kg twice daily, significantly reduced the severity of symptoms of fatigue, confusion, and overall mood disturbance such as depression, anxiety, and hostility compared with placebo in an RCT (n = 321).15 The lowest available oral dosage of ondansetron is 4 mg tablets or 4 mg/5 mL solution. We have used 4 mg twice daily for patients who have failed naltrexone and acamprosate or when these agents are contraindicated.
Common side effects of ondansetron include constipation, diarrhea, elevated liver enzymes, tachycardia, headache, and fatigue. Contraindications include congenital long QT syndrome, QTc prolongation risk, or significant hepatic impairment. We suggest evaluating baseline electrocardiogram and liver function tests. Women should undergo a pregnancy test before receiving medications.
Gabapentin is an anticonvulsant that is FDA-approved for treating epilepsy and postherpetic neuralgia. Gabapentin is related structurally to GABA and may potentiate central nervous system GABA activity, inhibit glutamate activity, and reduce norepinephrine and dopamine release.16 Gabapentin is thought to balance the GABA/glutamate dysregulation found in early alcohol abstinence and reduce risk for alcohol relapse.16A randomized, double-blind, placebo-controlled trial (N = 60) demonstrated that gabapentin, 600 mg/d, significantly reduced number of drinks per day and heavy drinking days and increased days of abstinence compared with placebo over 28 days.17 Another double-blind, randomized, placebo-controlled trial of 150 people used gabapentin, 900 or 1,800 mg/d; there was a linear dose response for increased days abstinent and no heavy drinking days in favor of gabapentin.18
An RCT (N = 150) evaluated adding gabapentin, up to 1200 mg/d, to naltrexone, 50 mg/d, vs naltrexone with placebo or double placebo over 6 weeks of treatment. The combined gabapentin-naltrexone group outperformed the other 2 groups on time to heavy drinking, number of heavy drinking days, and number of drinks per day. Gabapentin’s positive effects on sleep may have mediated some of its beneficial effects.29 In an open-label pilot study, gabapentin was more effective than trazodone for insomnia during early alcohol abstinence.30 Of note, gabapentin is a safe alternative to benzodiazepines for alcohol detoxification in patients with severe hepatic disease or those at risk of interacting with alcohol (eg, outpatients at high risk to drink during detoxification).31 Gabapentin, 400 mg/d to 1,600 mg/d, generally is safe and well tolerated and has some support for improving cravings, reducing alcohol consumption, delaying relapse, and improving sleep in patients with AUD.
Side effects of gabapentin include daytime sedation, dizziness, ataxia, fatigue, and dyspepsia. Using 3 divided doses might enhance tolerability. To reduce daytime sedation, we recommend administering most of the dose at night, which also may relieve insomnia. Gabapentin is excreted through the kidney; baseline renal function tests should be performed before initiating treatment, because the dosage might need to be adjusted in people with renal insufficiency.
Bottom Line
FDA-approved (acamprosate, naltrexone, and disulfiram) and off-label (baclofen, gabapentin, ondansetron, and topiramate) agents can help patients with alcohol use disorder achieve abstinence, reduce heavy drinking days, prevent relapse, and maintain sobriety. Research supports the use of pharmacotherapy combined with psychosocial modalities, such as 12-step programs, motivational interviewing, and cognitive-behavioral therapy.
Related Resources
- National Institute on Drug Abuse. Principles of drug addiction treatment: A research-based guide (third edition). www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/evidence-based-approaches-to-drug-addiction-treatment/pharmacotherapi-1.
- Substance Abuse and Mental Health Services Administration. Incorporating alcohol pharmacotherapies into medical practice. http://162.99.3.213/products/manuals/tips/pdf/TIP49.pdf.
- Pettinati HM, Mattson ME. Medical management treatment manual: A clinical guide for researchers and clinicians providing pharmacotherapy for alcohol dependence. http://pubs.niaaa.nih.gov/publications/MedicalManual/MMManual.pdf.
Drug Brand Names
Acamprosate • Campral Naltrexone • Vivitrol, ReVia
Baclofen • Lioresal Ondansetron • Zofran
Disulfiram • Antabuse Topiramate • Topamax
Gabapentin • Neurontin Trazodone • Desyrel, Oleptro
Metronidazole • Flagyl
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. World Health Organization. Alcohol fact sheet. http://www.who.int/mediacentre/factsheets/fs349/en/index.html. Published February 2011. Accessed April 30, 2013.
2. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence. The COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
3. Mann K. Pharmacotherapy of alcohol dependence a review of the clinical data. CNS Drugs. 2004;18(8):485-504.
4. ReVia [package insert]. Pomona, NY: Barr Pharmaceuticals; 2009.
5. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2004.
6. Antabuse [package insert]. Pomona, NY: Barr Pharmaceuticals; 2010.
7. Johnson BA. Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem Pharmacol. 2008;75(1):34-56.
8. Fuller RK, Branchey L, Brightwell DR, et al. Disulfiram treatment of alcoholism: a Veterans Administration cooperative study. JAMA. 1986;256(11):1449-1454.
9. Johnson BA, Ait-Daoud N, Bowden CL, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003;361(9370):
1677-1685.
10. Ma JZ, Ait-Daoud N, Johnson BA. Topiramate reduces the harm of excessive drinking: implications for public health and primary care. Addiction. 2006;101:1561-1568.
11. Addolorato G, Caputo F, Capristo E, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002;37(5):504-508.
12. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
13. Garbutt JC, Kampov-Polevoy AB, Gallop R, et al. Efficacy and safety of baclofen for alcohol dependence: a randomized, double-blind placebo-controlled trial. Alcohol Clin Exp Res. 2010;34(11):1849-1857.
14. Johnson BA, Roache JD, Javors MA, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA. 2000;284(8):963-971.
15. Johnson BA, Ait-Daoud N, Ma JZ, et al. Ondansetron reduces mood disturbance among biologically predisposed, alcohol-dependent individuals. Alcohol Clin Exp Res. 2003;27(11):1773-1779.
16. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
17. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68:1691-1700.
18. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial [published online November 4, 2013]. JAMA Intern Med. doi: 10.1001/jamainternmed.2013.11950.
19. The Management of Substance Use Disorders Working Group. VA/DoD clinical practice guideline for management of substance use disorders (SUD). http://www.healthquality.va.gov/sud/sud_full_601f.pdf. Published August 2009. Accessed November 22, 2013.
20. Mark TL, Kranzler HR, Song X, et al. Physicians’ opinions about medication to treat alcoholism. Addiction. 2003;98(5):617-626.
21. Thomas CP, Wallack SS, Lee S, et al. Research to practice: adoption of naltrexone in alcoholism treatment. J Subst Abuse Treat. 2003;24(1):1-11.
22. Srisurapanont M, Jarusuraisin N. Naltrexone for the treatment of alcoholism: a meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2005;8:267-280.
23. Anton RF. Naltrexone for the management of alcohol dependence. N Engl J Med. 2008;359(7):715-721.
24. Gueorguieva R, Wu R, Pittman B, et al. New insights into the efficacy of naltrexone based on trajectory-based reanalysis of two negative clinical trials. Biol Psychiatry. 2007;61(11): 1290-1295.
25. Lapham S, Forman R, Alexander M, et al. The effects of extended-release naltrexone on holiday drinking in alcohol-dependent patients. J Subst Abuse. 2009;36(1):1-6.
26. Ciraulo DA, Dong Q, Silverman BL, et al. Early treatment response in alcohol dependence with extended-release naltrexone. J Clin Psychiatry. 2008;69(2):190-195.
27. Mark TL, Montejano LB, Kranzler HR, et al. Comparison of healthcare utilization among patients treated with alcoholism medications. Am J Managed Care. 2010;16(12): 879-888.
28. Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28(1):51-63.
29. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
30. Karam-Hage M, Brower KJ. Open pilot study of gabapentin versus trazodone to treat insomnia in alcoholic outpatients. Psychiatry Clin Neurosci. 2003;57(5):542-544.
31. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.
Historically, alcohol use disorder (AUD; classified as alcohol abuse or dependence in DSM-IV-TR) has been treated with psychosocial therapies, but many patients treated this way relapse into heavy drinking patterns and are unable to sustain sobriety (Box 11). Although vital for treating AUD, psychosocial methods have, to date, a modest success rate. Research has demonstrated that combining pharmacotherapy with psychosocial programs is effective for treating AUD.2
Patients and clinicians might associate AUD medications with so-called aversion therapy because, for many years, the only treatment was disulfiram, which causes unpleasant physical effects when consumed with alcohol. However, newer medications help patients maintain abstinence by targeting brain neurotransmitters relevant to addiction neurocircuitry, such as dopamine, serotonin, ϒ-aminobutyric acid (GABA), glutamate, and opioid.3 These medications may help patients with AUD achieve sobriety, avoid relapse, decrease heavy drinking days, and delay time to recurrent drinking.
In this article, we review FDA-approved medications (Table 1)4-6 and off-label agents (Table 2)3,7-18 and provide recommendations for treating patients with AUD (Box 2).19-21
FDA-approved treatments
Naltrexone is an opiate antagonist that blocks the mu receptor and is believed to interrupt the dopamine reward pathway in the brain for alcohol. A meta-analysis of 2,861 patients in 24 combined randomized controlled trials (RCTs) demonstrated naltrexone to be an effective short-term (12 weeks) treatment for alcoholism, significantly decreasing relapses.22 The large multisite COMBINE study (N = 1,383) showed that naltrexone, 100 mg/d, and medical treatment without behavioral treatment over 16 weeks was more effective than placebo in increasing percentage of days abstinent (80.6% vs 75.1%, respectively) and reducing the percentage of patients experiencing heavy drinking days (66.2% vs 73.1%, respectively).2 Patients who have a family history of AUD or strong cravings, or both, may benefit most from naltrexone.3,7,23 Despite evidence of the effectiveness of naltrexone for AUD, not all studies have yielded positive results.24
Common side effects of naltrexone, if present, appear early in treatment and include GI upset (eg, nausea, vomiting, abdominal pain), headache, and fatigue.2,3,22,23 Hepatotoxicity has been reported with dosages of 100 to 300 mg/d, but lab values typically normalize when naltrexone is discontinued.2,3,23 Monitor markers of liver function including ϒ-glutamyltransferase, aspartate aminotransferase, alanine aminotransferase, and bilirubin before and during naltrexone treatment (we check patients 1 to 3 months after starting treatment and yearly thereafter). Obtain a negative urine drug screen for opioids before administering naltrexone, because if opioids were consumed recently naltrexone could precipitate withdrawal. Because of the unknown teratogenicity of naltrexone, women of childbearing age should undergo pregnancy testing before and periodically during naltrexone therapy.
Naltrexone also is available in a once-monthly, 380-mg injectable formulation. Studies show that, similar to its oral counterpart, injectable naltrexone effectively reduces heavy drinking days and number of drinks a day compared with placebo.25,26 Advantages of injectable naltrexone are its extended steady release of medication and its efficacy for patients who do not adhere to oral dosing.7,27 Side effects are similar to oral naltrexone, except for injection site reactions and pain.
Contraindications to naltrexone include current opioid use because its antagonistic effects on opioid receptors render opioid analgesia ineffective. Patients who have used opioids within 7 to 10 days or who may be surreptitiously using opioids should not take naltrexone because it may cause opioid withdrawal. Some patients may try to override the opioid receptor blockade of naltrexone with higher opioid doses, which could result in overdose.
Naltrexone is approved for treating opioid use disorder and may be useful for persons with comorbid opioid use disorder and AUD, if the patient has been adequately detoxified from opioids and intends to abstain from these drugs. Patients who have extensive liver damage secondary to acute hepatitis or uncompensated cirrhosis would not be good candidates for naltrexone because of a risk of hepatotoxicity.2,3,22
Because of ease of dosing, we recommend naltrexone as a first-line treatment for AUD, unless the patient requires opioids or has severe liver disease. We recommend increasing naltrexone from 50 mg to 100 mg before switching to acamprosate, based on European studies.
Acamprosate is a glutamate antagonist that is thought to modulate overactive glutamatergic brain activity that occurs after stopping chronic heavy alcohol use. In a meta-analysis of 17 studies (N = 4,087), continuous abstinence rates at 6 months were significantly higher in acamprosate-treated patients (36.1%) than in patients receiving placebo (23.4%).28 In a review of European studies, acamprosate benefited patients who have increased anxiety, physiological dependence, negative family history of AUD, and late age of onset (age >25) of alcohol dependence.7 However, in the COMBINE trial acamprosate was no more effective than placebo.2
We consider acamprosate an effective option for patients who do not respond to naltrexone or have a contraindication. Dosages of 333 mg to 666 mg, 3 times a day, are recommended, although dosages up to 3 g/d have been studied; titration is not required.2,7,28 We recommend advising patients to continue treatment even if they relapse, because these medications may mitigate relapse severity. Adherence to multiple daily doses can be problematic for some patients, but pairing medications with meals or bedtime may improve adherence.
Diarrhea is the most common side effect of acamprosate; nervousness, fatigue, insomnia, and depression have been reported with high dosages.2,3,7,28 Acamprosate is excreted through the kidney and is safe for patients with liver disorders such as acute hepatitis or cirrhosis. The drug is contraindicated in patients with acute or chronic renal failure with creatinine clearance <30 mL/min; those with less severe renal insufficiency might need a lower dosage. Obtain baseline renal function before starting acamprosate; women of childbearing age should undergo a pregnancy test.
Disulfiram inhibits alcohol metabolism, resulting in acetaldehyde accumulation, which causes unpleasant physical effects such as nausea, vomiting, and hypotension. This creates a negative rather than a positive experience with drinking. A US Veterans Administration Cooperative Study randomized 605 participants to riboflavin, disulfiram, 1 mg/d (an inactive dose), or disulfiram, 250 mg/d (standard dose). There was no difference in percentage of patients remaining abstinent or time to first drink.8 Participants receiving disulfiram, 250 mg/d, had fewer drinking days after relapse compared with the other groups.8
Adverse physical effects produced when disulfiram and alcohol interact include tremor, diaphoresis, unstable blood pressure, and severe diarrhea and vomiting. Disulfiram can cause medically serious reactions in a small percentage of patients, especially those with significant medical comorbidity or advanced age. Patients with severe hypertension, diabetes mellitus, heart disease, a history of stroke, peripheral neuropathy, epilepsy, or renal or hepatic insufficiency should not use disulfiram.3 Patients taking disulfiram should avoid casual exposures to food, aftershave, mouthwash, and hand sanitizer that might contain alcohol. Disulfiram has no significant effect on alcohol craving. Social support to help oversee dosing may enhance adherence.7
Off-label medications
Topiramate is FDA-approved to treat migraine headaches and some seizure disorders. Topiramate facilitates GABA-mediated neuronal inhibition and antagonizes certain glutamate receptor subtypes. In an RCT (N = 150), topiramate, up to 300 mg/d, was more effective than placebo at reducing heavy drinking days and number of drinks per day, increasing days abstinent, and alleviating cravings.9 In a 12-week, double-blind RCT (N = 150), topiramate increased “safe drinking”—defined as ≤1 standard drink per day for women and ≤2 per day for men—vs placebo.10 Dosages were 75 mg to 300 mg/d in twice daily divided doses. Dosing starts at 25 mg/d and increases by 25 to 50 mg a day at weekly intervals. We recommend reserving topiramate for persons who do not respond to or cannot tolerate naltrexone and acamprosate because of the slow titration needed to prevent side effects. Although not studied, it may seem that topiramate’s antiepileptic actions could prevent seizures during alcohol withdrawal, but the protracted titration would limit its utility.
Side effects of topiramate include impaired memory and concentration, paresthesia, and anorexia and are more likely to present during rapid titration or with a high dosage.7,8 Rare reports of spontaneous myopia, angle-closure glaucoma, increased intraocular pressure, ocular pain, and blurry vision have been reported, but these complications often resolve with discontinuation of topiramate.7,8
Topiramate primarily is excreted through the kidney, and its action in the renal tubules can lead to metabolic acidosis or nephrolithiasis.8 Relative contraindications include acute or chronic kidney disease, including kidney stones. Consider slower titration and a 50% reduction in dosing if creatinine clearance is <70 mL/min. Obtain renal function tests before starting topiramate and consider monitoring serum bicarbonate for metabolic acidosis (we test at 3 and 6 months, then every 6 months). Because of teratogenic effects of topiramate (eg, cleft lip and palate), rule out pregnancy in all women of childbearing age.
Baclofen is a GABAb receptor agonist that is FDA approved for treating spasticity. Because GABA transmission is down-regulated in chronic AUD, it is a commonly targeted neurotransmitter when developing medications for AUD. GABAa receptors are fast-acting inhibitory ion channels, and its agonists (eg, benzodiazepines) have a significant abuse and cross-addiction liability. GABAb receptors, however, are slow-acting through a complex cascade of intracellular signals, and therefore GABAb agonists such as baclofen have been studied for treating addiction.
In a randomized double-blind, placebo-controlled trial (N = 39), baclofen was superior to placebo in suppressing obsessive aspects of cravings and decreasing state anxiety.11 Baclofen, 10 mg 3 times daily, in another randomized double-blind, placebo-controlled trial (n = 42) reduced the number of drinks per day by 53% vs placebo; 20 mg 3 times a day resulted in a 68% reduction in drinks per day vs placebo.12 However, a placebo-controlled RCT (n = 80) reported that baclofen, 10 mg 3 times daily, was not superior to placebo for primary outcomes related to alcohol consumption, although it did significantly decrease cravings and anxiety among persons with AUD.13 Evidence suggests that baclofen might be effective for promoting abstinence, reducing the risk of relapse, and alleviating cravings and anxiety in persons with AUD, although further investigation is needed.
In studies for AUD, the side-effect profile for baclofen was relatively benign.11-13 Nausea, fatigue, sleepiness, vertigo, and abdominal pain were reported; overall, baclofen was found to be safe and to have no abuse liability.7,10,12 The addictive potential of other muscle relaxers may have dissuaded providers from using baclofen for AUD, but we consider it a reasonable alternative when FDA-approved treatments fail.
Because baclofen is primarily eliminated by the kidneys, it may be safe for people with cirrhosis or severe liver disease.12 Baseline renal labs should be performed before administering baclofen and a negative pregnancy test obtained for women of childbearing age.
Ondansetron is a serotonin receptor type 3 (5-HT3) antagonist that has shown promising results for AUD.3,7 Research suggests that 5-HT3 receptors are an action site for alcohol in the brain and are thought to play a role in its rewarding effects.7 Ondansetron may be more effective for early-onset alcoholism (EOA) than late-onset alcoholism (LOA).14,15 EOA (age ≤25) is characterized by strong family history of AUD and prominent antisocial traits. A randomized double-blind, placebo-controlled trial (n = 271) reported that ondansetron, 4 mcg/kg twice daily, was superior to placebo in reducing number of drinks per day, increasing days abstinent, and reducing cravings in patients with EOA.14 Among persons with EOA, ondansetron, 16 mcg/kg twice daily, significantly reduced the severity of symptoms of fatigue, confusion, and overall mood disturbance such as depression, anxiety, and hostility compared with placebo in an RCT (n = 321).15 The lowest available oral dosage of ondansetron is 4 mg tablets or 4 mg/5 mL solution. We have used 4 mg twice daily for patients who have failed naltrexone and acamprosate or when these agents are contraindicated.
Common side effects of ondansetron include constipation, diarrhea, elevated liver enzymes, tachycardia, headache, and fatigue. Contraindications include congenital long QT syndrome, QTc prolongation risk, or significant hepatic impairment. We suggest evaluating baseline electrocardiogram and liver function tests. Women should undergo a pregnancy test before receiving medications.
Gabapentin is an anticonvulsant that is FDA-approved for treating epilepsy and postherpetic neuralgia. Gabapentin is related structurally to GABA and may potentiate central nervous system GABA activity, inhibit glutamate activity, and reduce norepinephrine and dopamine release.16 Gabapentin is thought to balance the GABA/glutamate dysregulation found in early alcohol abstinence and reduce risk for alcohol relapse.16A randomized, double-blind, placebo-controlled trial (N = 60) demonstrated that gabapentin, 600 mg/d, significantly reduced number of drinks per day and heavy drinking days and increased days of abstinence compared with placebo over 28 days.17 Another double-blind, randomized, placebo-controlled trial of 150 people used gabapentin, 900 or 1,800 mg/d; there was a linear dose response for increased days abstinent and no heavy drinking days in favor of gabapentin.18
An RCT (N = 150) evaluated adding gabapentin, up to 1200 mg/d, to naltrexone, 50 mg/d, vs naltrexone with placebo or double placebo over 6 weeks of treatment. The combined gabapentin-naltrexone group outperformed the other 2 groups on time to heavy drinking, number of heavy drinking days, and number of drinks per day. Gabapentin’s positive effects on sleep may have mediated some of its beneficial effects.29 In an open-label pilot study, gabapentin was more effective than trazodone for insomnia during early alcohol abstinence.30 Of note, gabapentin is a safe alternative to benzodiazepines for alcohol detoxification in patients with severe hepatic disease or those at risk of interacting with alcohol (eg, outpatients at high risk to drink during detoxification).31 Gabapentin, 400 mg/d to 1,600 mg/d, generally is safe and well tolerated and has some support for improving cravings, reducing alcohol consumption, delaying relapse, and improving sleep in patients with AUD.
Side effects of gabapentin include daytime sedation, dizziness, ataxia, fatigue, and dyspepsia. Using 3 divided doses might enhance tolerability. To reduce daytime sedation, we recommend administering most of the dose at night, which also may relieve insomnia. Gabapentin is excreted through the kidney; baseline renal function tests should be performed before initiating treatment, because the dosage might need to be adjusted in people with renal insufficiency.
Bottom Line
FDA-approved (acamprosate, naltrexone, and disulfiram) and off-label (baclofen, gabapentin, ondansetron, and topiramate) agents can help patients with alcohol use disorder achieve abstinence, reduce heavy drinking days, prevent relapse, and maintain sobriety. Research supports the use of pharmacotherapy combined with psychosocial modalities, such as 12-step programs, motivational interviewing, and cognitive-behavioral therapy.
Related Resources
- National Institute on Drug Abuse. Principles of drug addiction treatment: A research-based guide (third edition). www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/evidence-based-approaches-to-drug-addiction-treatment/pharmacotherapi-1.
- Substance Abuse and Mental Health Services Administration. Incorporating alcohol pharmacotherapies into medical practice. http://162.99.3.213/products/manuals/tips/pdf/TIP49.pdf.
- Pettinati HM, Mattson ME. Medical management treatment manual: A clinical guide for researchers and clinicians providing pharmacotherapy for alcohol dependence. http://pubs.niaaa.nih.gov/publications/MedicalManual/MMManual.pdf.
Drug Brand Names
Acamprosate • Campral Naltrexone • Vivitrol, ReVia
Baclofen • Lioresal Ondansetron • Zofran
Disulfiram • Antabuse Topiramate • Topamax
Gabapentin • Neurontin Trazodone • Desyrel, Oleptro
Metronidazole • Flagyl
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Historically, alcohol use disorder (AUD; classified as alcohol abuse or dependence in DSM-IV-TR) has been treated with psychosocial therapies, but many patients treated this way relapse into heavy drinking patterns and are unable to sustain sobriety (Box 11). Although vital for treating AUD, psychosocial methods have, to date, a modest success rate. Research has demonstrated that combining pharmacotherapy with psychosocial programs is effective for treating AUD.2
Patients and clinicians might associate AUD medications with so-called aversion therapy because, for many years, the only treatment was disulfiram, which causes unpleasant physical effects when consumed with alcohol. However, newer medications help patients maintain abstinence by targeting brain neurotransmitters relevant to addiction neurocircuitry, such as dopamine, serotonin, ϒ-aminobutyric acid (GABA), glutamate, and opioid.3 These medications may help patients with AUD achieve sobriety, avoid relapse, decrease heavy drinking days, and delay time to recurrent drinking.
In this article, we review FDA-approved medications (Table 1)4-6 and off-label agents (Table 2)3,7-18 and provide recommendations for treating patients with AUD (Box 2).19-21
FDA-approved treatments
Naltrexone is an opiate antagonist that blocks the mu receptor and is believed to interrupt the dopamine reward pathway in the brain for alcohol. A meta-analysis of 2,861 patients in 24 combined randomized controlled trials (RCTs) demonstrated naltrexone to be an effective short-term (12 weeks) treatment for alcoholism, significantly decreasing relapses.22 The large multisite COMBINE study (N = 1,383) showed that naltrexone, 100 mg/d, and medical treatment without behavioral treatment over 16 weeks was more effective than placebo in increasing percentage of days abstinent (80.6% vs 75.1%, respectively) and reducing the percentage of patients experiencing heavy drinking days (66.2% vs 73.1%, respectively).2 Patients who have a family history of AUD or strong cravings, or both, may benefit most from naltrexone.3,7,23 Despite evidence of the effectiveness of naltrexone for AUD, not all studies have yielded positive results.24
Common side effects of naltrexone, if present, appear early in treatment and include GI upset (eg, nausea, vomiting, abdominal pain), headache, and fatigue.2,3,22,23 Hepatotoxicity has been reported with dosages of 100 to 300 mg/d, but lab values typically normalize when naltrexone is discontinued.2,3,23 Monitor markers of liver function including ϒ-glutamyltransferase, aspartate aminotransferase, alanine aminotransferase, and bilirubin before and during naltrexone treatment (we check patients 1 to 3 months after starting treatment and yearly thereafter). Obtain a negative urine drug screen for opioids before administering naltrexone, because if opioids were consumed recently naltrexone could precipitate withdrawal. Because of the unknown teratogenicity of naltrexone, women of childbearing age should undergo pregnancy testing before and periodically during naltrexone therapy.
Naltrexone also is available in a once-monthly, 380-mg injectable formulation. Studies show that, similar to its oral counterpart, injectable naltrexone effectively reduces heavy drinking days and number of drinks a day compared with placebo.25,26 Advantages of injectable naltrexone are its extended steady release of medication and its efficacy for patients who do not adhere to oral dosing.7,27 Side effects are similar to oral naltrexone, except for injection site reactions and pain.
Contraindications to naltrexone include current opioid use because its antagonistic effects on opioid receptors render opioid analgesia ineffective. Patients who have used opioids within 7 to 10 days or who may be surreptitiously using opioids should not take naltrexone because it may cause opioid withdrawal. Some patients may try to override the opioid receptor blockade of naltrexone with higher opioid doses, which could result in overdose.
Naltrexone is approved for treating opioid use disorder and may be useful for persons with comorbid opioid use disorder and AUD, if the patient has been adequately detoxified from opioids and intends to abstain from these drugs. Patients who have extensive liver damage secondary to acute hepatitis or uncompensated cirrhosis would not be good candidates for naltrexone because of a risk of hepatotoxicity.2,3,22
Because of ease of dosing, we recommend naltrexone as a first-line treatment for AUD, unless the patient requires opioids or has severe liver disease. We recommend increasing naltrexone from 50 mg to 100 mg before switching to acamprosate, based on European studies.
Acamprosate is a glutamate antagonist that is thought to modulate overactive glutamatergic brain activity that occurs after stopping chronic heavy alcohol use. In a meta-analysis of 17 studies (N = 4,087), continuous abstinence rates at 6 months were significantly higher in acamprosate-treated patients (36.1%) than in patients receiving placebo (23.4%).28 In a review of European studies, acamprosate benefited patients who have increased anxiety, physiological dependence, negative family history of AUD, and late age of onset (age >25) of alcohol dependence.7 However, in the COMBINE trial acamprosate was no more effective than placebo.2
We consider acamprosate an effective option for patients who do not respond to naltrexone or have a contraindication. Dosages of 333 mg to 666 mg, 3 times a day, are recommended, although dosages up to 3 g/d have been studied; titration is not required.2,7,28 We recommend advising patients to continue treatment even if they relapse, because these medications may mitigate relapse severity. Adherence to multiple daily doses can be problematic for some patients, but pairing medications with meals or bedtime may improve adherence.
Diarrhea is the most common side effect of acamprosate; nervousness, fatigue, insomnia, and depression have been reported with high dosages.2,3,7,28 Acamprosate is excreted through the kidney and is safe for patients with liver disorders such as acute hepatitis or cirrhosis. The drug is contraindicated in patients with acute or chronic renal failure with creatinine clearance <30 mL/min; those with less severe renal insufficiency might need a lower dosage. Obtain baseline renal function before starting acamprosate; women of childbearing age should undergo a pregnancy test.
Disulfiram inhibits alcohol metabolism, resulting in acetaldehyde accumulation, which causes unpleasant physical effects such as nausea, vomiting, and hypotension. This creates a negative rather than a positive experience with drinking. A US Veterans Administration Cooperative Study randomized 605 participants to riboflavin, disulfiram, 1 mg/d (an inactive dose), or disulfiram, 250 mg/d (standard dose). There was no difference in percentage of patients remaining abstinent or time to first drink.8 Participants receiving disulfiram, 250 mg/d, had fewer drinking days after relapse compared with the other groups.8
Adverse physical effects produced when disulfiram and alcohol interact include tremor, diaphoresis, unstable blood pressure, and severe diarrhea and vomiting. Disulfiram can cause medically serious reactions in a small percentage of patients, especially those with significant medical comorbidity or advanced age. Patients with severe hypertension, diabetes mellitus, heart disease, a history of stroke, peripheral neuropathy, epilepsy, or renal or hepatic insufficiency should not use disulfiram.3 Patients taking disulfiram should avoid casual exposures to food, aftershave, mouthwash, and hand sanitizer that might contain alcohol. Disulfiram has no significant effect on alcohol craving. Social support to help oversee dosing may enhance adherence.7
Off-label medications
Topiramate is FDA-approved to treat migraine headaches and some seizure disorders. Topiramate facilitates GABA-mediated neuronal inhibition and antagonizes certain glutamate receptor subtypes. In an RCT (N = 150), topiramate, up to 300 mg/d, was more effective than placebo at reducing heavy drinking days and number of drinks per day, increasing days abstinent, and alleviating cravings.9 In a 12-week, double-blind RCT (N = 150), topiramate increased “safe drinking”—defined as ≤1 standard drink per day for women and ≤2 per day for men—vs placebo.10 Dosages were 75 mg to 300 mg/d in twice daily divided doses. Dosing starts at 25 mg/d and increases by 25 to 50 mg a day at weekly intervals. We recommend reserving topiramate for persons who do not respond to or cannot tolerate naltrexone and acamprosate because of the slow titration needed to prevent side effects. Although not studied, it may seem that topiramate’s antiepileptic actions could prevent seizures during alcohol withdrawal, but the protracted titration would limit its utility.
Side effects of topiramate include impaired memory and concentration, paresthesia, and anorexia and are more likely to present during rapid titration or with a high dosage.7,8 Rare reports of spontaneous myopia, angle-closure glaucoma, increased intraocular pressure, ocular pain, and blurry vision have been reported, but these complications often resolve with discontinuation of topiramate.7,8
Topiramate primarily is excreted through the kidney, and its action in the renal tubules can lead to metabolic acidosis or nephrolithiasis.8 Relative contraindications include acute or chronic kidney disease, including kidney stones. Consider slower titration and a 50% reduction in dosing if creatinine clearance is <70 mL/min. Obtain renal function tests before starting topiramate and consider monitoring serum bicarbonate for metabolic acidosis (we test at 3 and 6 months, then every 6 months). Because of teratogenic effects of topiramate (eg, cleft lip and palate), rule out pregnancy in all women of childbearing age.
Baclofen is a GABAb receptor agonist that is FDA approved for treating spasticity. Because GABA transmission is down-regulated in chronic AUD, it is a commonly targeted neurotransmitter when developing medications for AUD. GABAa receptors are fast-acting inhibitory ion channels, and its agonists (eg, benzodiazepines) have a significant abuse and cross-addiction liability. GABAb receptors, however, are slow-acting through a complex cascade of intracellular signals, and therefore GABAb agonists such as baclofen have been studied for treating addiction.
In a randomized double-blind, placebo-controlled trial (N = 39), baclofen was superior to placebo in suppressing obsessive aspects of cravings and decreasing state anxiety.11 Baclofen, 10 mg 3 times daily, in another randomized double-blind, placebo-controlled trial (n = 42) reduced the number of drinks per day by 53% vs placebo; 20 mg 3 times a day resulted in a 68% reduction in drinks per day vs placebo.12 However, a placebo-controlled RCT (n = 80) reported that baclofen, 10 mg 3 times daily, was not superior to placebo for primary outcomes related to alcohol consumption, although it did significantly decrease cravings and anxiety among persons with AUD.13 Evidence suggests that baclofen might be effective for promoting abstinence, reducing the risk of relapse, and alleviating cravings and anxiety in persons with AUD, although further investigation is needed.
In studies for AUD, the side-effect profile for baclofen was relatively benign.11-13 Nausea, fatigue, sleepiness, vertigo, and abdominal pain were reported; overall, baclofen was found to be safe and to have no abuse liability.7,10,12 The addictive potential of other muscle relaxers may have dissuaded providers from using baclofen for AUD, but we consider it a reasonable alternative when FDA-approved treatments fail.
Because baclofen is primarily eliminated by the kidneys, it may be safe for people with cirrhosis or severe liver disease.12 Baseline renal labs should be performed before administering baclofen and a negative pregnancy test obtained for women of childbearing age.
Ondansetron is a serotonin receptor type 3 (5-HT3) antagonist that has shown promising results for AUD.3,7 Research suggests that 5-HT3 receptors are an action site for alcohol in the brain and are thought to play a role in its rewarding effects.7 Ondansetron may be more effective for early-onset alcoholism (EOA) than late-onset alcoholism (LOA).14,15 EOA (age ≤25) is characterized by strong family history of AUD and prominent antisocial traits. A randomized double-blind, placebo-controlled trial (n = 271) reported that ondansetron, 4 mcg/kg twice daily, was superior to placebo in reducing number of drinks per day, increasing days abstinent, and reducing cravings in patients with EOA.14 Among persons with EOA, ondansetron, 16 mcg/kg twice daily, significantly reduced the severity of symptoms of fatigue, confusion, and overall mood disturbance such as depression, anxiety, and hostility compared with placebo in an RCT (n = 321).15 The lowest available oral dosage of ondansetron is 4 mg tablets or 4 mg/5 mL solution. We have used 4 mg twice daily for patients who have failed naltrexone and acamprosate or when these agents are contraindicated.
Common side effects of ondansetron include constipation, diarrhea, elevated liver enzymes, tachycardia, headache, and fatigue. Contraindications include congenital long QT syndrome, QTc prolongation risk, or significant hepatic impairment. We suggest evaluating baseline electrocardiogram and liver function tests. Women should undergo a pregnancy test before receiving medications.
Gabapentin is an anticonvulsant that is FDA-approved for treating epilepsy and postherpetic neuralgia. Gabapentin is related structurally to GABA and may potentiate central nervous system GABA activity, inhibit glutamate activity, and reduce norepinephrine and dopamine release.16 Gabapentin is thought to balance the GABA/glutamate dysregulation found in early alcohol abstinence and reduce risk for alcohol relapse.16A randomized, double-blind, placebo-controlled trial (N = 60) demonstrated that gabapentin, 600 mg/d, significantly reduced number of drinks per day and heavy drinking days and increased days of abstinence compared with placebo over 28 days.17 Another double-blind, randomized, placebo-controlled trial of 150 people used gabapentin, 900 or 1,800 mg/d; there was a linear dose response for increased days abstinent and no heavy drinking days in favor of gabapentin.18
An RCT (N = 150) evaluated adding gabapentin, up to 1200 mg/d, to naltrexone, 50 mg/d, vs naltrexone with placebo or double placebo over 6 weeks of treatment. The combined gabapentin-naltrexone group outperformed the other 2 groups on time to heavy drinking, number of heavy drinking days, and number of drinks per day. Gabapentin’s positive effects on sleep may have mediated some of its beneficial effects.29 In an open-label pilot study, gabapentin was more effective than trazodone for insomnia during early alcohol abstinence.30 Of note, gabapentin is a safe alternative to benzodiazepines for alcohol detoxification in patients with severe hepatic disease or those at risk of interacting with alcohol (eg, outpatients at high risk to drink during detoxification).31 Gabapentin, 400 mg/d to 1,600 mg/d, generally is safe and well tolerated and has some support for improving cravings, reducing alcohol consumption, delaying relapse, and improving sleep in patients with AUD.
Side effects of gabapentin include daytime sedation, dizziness, ataxia, fatigue, and dyspepsia. Using 3 divided doses might enhance tolerability. To reduce daytime sedation, we recommend administering most of the dose at night, which also may relieve insomnia. Gabapentin is excreted through the kidney; baseline renal function tests should be performed before initiating treatment, because the dosage might need to be adjusted in people with renal insufficiency.
Bottom Line
FDA-approved (acamprosate, naltrexone, and disulfiram) and off-label (baclofen, gabapentin, ondansetron, and topiramate) agents can help patients with alcohol use disorder achieve abstinence, reduce heavy drinking days, prevent relapse, and maintain sobriety. Research supports the use of pharmacotherapy combined with psychosocial modalities, such as 12-step programs, motivational interviewing, and cognitive-behavioral therapy.
Related Resources
- National Institute on Drug Abuse. Principles of drug addiction treatment: A research-based guide (third edition). www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/evidence-based-approaches-to-drug-addiction-treatment/pharmacotherapi-1.
- Substance Abuse and Mental Health Services Administration. Incorporating alcohol pharmacotherapies into medical practice. http://162.99.3.213/products/manuals/tips/pdf/TIP49.pdf.
- Pettinati HM, Mattson ME. Medical management treatment manual: A clinical guide for researchers and clinicians providing pharmacotherapy for alcohol dependence. http://pubs.niaaa.nih.gov/publications/MedicalManual/MMManual.pdf.
Drug Brand Names
Acamprosate • Campral Naltrexone • Vivitrol, ReVia
Baclofen • Lioresal Ondansetron • Zofran
Disulfiram • Antabuse Topiramate • Topamax
Gabapentin • Neurontin Trazodone • Desyrel, Oleptro
Metronidazole • Flagyl
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. World Health Organization. Alcohol fact sheet. http://www.who.int/mediacentre/factsheets/fs349/en/index.html. Published February 2011. Accessed April 30, 2013.
2. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence. The COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
3. Mann K. Pharmacotherapy of alcohol dependence a review of the clinical data. CNS Drugs. 2004;18(8):485-504.
4. ReVia [package insert]. Pomona, NY: Barr Pharmaceuticals; 2009.
5. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2004.
6. Antabuse [package insert]. Pomona, NY: Barr Pharmaceuticals; 2010.
7. Johnson BA. Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem Pharmacol. 2008;75(1):34-56.
8. Fuller RK, Branchey L, Brightwell DR, et al. Disulfiram treatment of alcoholism: a Veterans Administration cooperative study. JAMA. 1986;256(11):1449-1454.
9. Johnson BA, Ait-Daoud N, Bowden CL, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003;361(9370):
1677-1685.
10. Ma JZ, Ait-Daoud N, Johnson BA. Topiramate reduces the harm of excessive drinking: implications for public health and primary care. Addiction. 2006;101:1561-1568.
11. Addolorato G, Caputo F, Capristo E, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002;37(5):504-508.
12. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
13. Garbutt JC, Kampov-Polevoy AB, Gallop R, et al. Efficacy and safety of baclofen for alcohol dependence: a randomized, double-blind placebo-controlled trial. Alcohol Clin Exp Res. 2010;34(11):1849-1857.
14. Johnson BA, Roache JD, Javors MA, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA. 2000;284(8):963-971.
15. Johnson BA, Ait-Daoud N, Ma JZ, et al. Ondansetron reduces mood disturbance among biologically predisposed, alcohol-dependent individuals. Alcohol Clin Exp Res. 2003;27(11):1773-1779.
16. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
17. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68:1691-1700.
18. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial [published online November 4, 2013]. JAMA Intern Med. doi: 10.1001/jamainternmed.2013.11950.
19. The Management of Substance Use Disorders Working Group. VA/DoD clinical practice guideline for management of substance use disorders (SUD). http://www.healthquality.va.gov/sud/sud_full_601f.pdf. Published August 2009. Accessed November 22, 2013.
20. Mark TL, Kranzler HR, Song X, et al. Physicians’ opinions about medication to treat alcoholism. Addiction. 2003;98(5):617-626.
21. Thomas CP, Wallack SS, Lee S, et al. Research to practice: adoption of naltrexone in alcoholism treatment. J Subst Abuse Treat. 2003;24(1):1-11.
22. Srisurapanont M, Jarusuraisin N. Naltrexone for the treatment of alcoholism: a meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2005;8:267-280.
23. Anton RF. Naltrexone for the management of alcohol dependence. N Engl J Med. 2008;359(7):715-721.
24. Gueorguieva R, Wu R, Pittman B, et al. New insights into the efficacy of naltrexone based on trajectory-based reanalysis of two negative clinical trials. Biol Psychiatry. 2007;61(11): 1290-1295.
25. Lapham S, Forman R, Alexander M, et al. The effects of extended-release naltrexone on holiday drinking in alcohol-dependent patients. J Subst Abuse. 2009;36(1):1-6.
26. Ciraulo DA, Dong Q, Silverman BL, et al. Early treatment response in alcohol dependence with extended-release naltrexone. J Clin Psychiatry. 2008;69(2):190-195.
27. Mark TL, Montejano LB, Kranzler HR, et al. Comparison of healthcare utilization among patients treated with alcoholism medications. Am J Managed Care. 2010;16(12): 879-888.
28. Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28(1):51-63.
29. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
30. Karam-Hage M, Brower KJ. Open pilot study of gabapentin versus trazodone to treat insomnia in alcoholic outpatients. Psychiatry Clin Neurosci. 2003;57(5):542-544.
31. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.
1. World Health Organization. Alcohol fact sheet. http://www.who.int/mediacentre/factsheets/fs349/en/index.html. Published February 2011. Accessed April 30, 2013.
2. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence. The COMBINE study: a randomized controlled trial. JAMA. 2006;295(17):2003-2017.
3. Mann K. Pharmacotherapy of alcohol dependence a review of the clinical data. CNS Drugs. 2004;18(8):485-504.
4. ReVia [package insert]. Pomona, NY: Barr Pharmaceuticals; 2009.
5. Campral [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2004.
6. Antabuse [package insert]. Pomona, NY: Barr Pharmaceuticals; 2010.
7. Johnson BA. Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem Pharmacol. 2008;75(1):34-56.
8. Fuller RK, Branchey L, Brightwell DR, et al. Disulfiram treatment of alcoholism: a Veterans Administration cooperative study. JAMA. 1986;256(11):1449-1454.
9. Johnson BA, Ait-Daoud N, Bowden CL, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003;361(9370):
1677-1685.
10. Ma JZ, Ait-Daoud N, Johnson BA. Topiramate reduces the harm of excessive drinking: implications for public health and primary care. Addiction. 2006;101:1561-1568.
11. Addolorato G, Caputo F, Capristo E, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002;37(5):504-508.
12. Addolorato G, Leggio L, Ferrulli A, et al. Dose-response effect of baclofen in reducing daily alcohol intake in alcohol dependence: secondary analysis of a randomized, double-blind, placebo-controlled trial. Alcohol Alcohol. 2011;46(3):312-317.
13. Garbutt JC, Kampov-Polevoy AB, Gallop R, et al. Efficacy and safety of baclofen for alcohol dependence: a randomized, double-blind placebo-controlled trial. Alcohol Clin Exp Res. 2010;34(11):1849-1857.
14. Johnson BA, Roache JD, Javors MA, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA. 2000;284(8):963-971.
15. Johnson BA, Ait-Daoud N, Ma JZ, et al. Ondansetron reduces mood disturbance among biologically predisposed, alcohol-dependent individuals. Alcohol Clin Exp Res. 2003;27(11):1773-1779.
16. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
17. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68:1691-1700.
18. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial [published online November 4, 2013]. JAMA Intern Med. doi: 10.1001/jamainternmed.2013.11950.
19. The Management of Substance Use Disorders Working Group. VA/DoD clinical practice guideline for management of substance use disorders (SUD). http://www.healthquality.va.gov/sud/sud_full_601f.pdf. Published August 2009. Accessed November 22, 2013.
20. Mark TL, Kranzler HR, Song X, et al. Physicians’ opinions about medication to treat alcoholism. Addiction. 2003;98(5):617-626.
21. Thomas CP, Wallack SS, Lee S, et al. Research to practice: adoption of naltrexone in alcoholism treatment. J Subst Abuse Treat. 2003;24(1):1-11.
22. Srisurapanont M, Jarusuraisin N. Naltrexone for the treatment of alcoholism: a meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2005;8:267-280.
23. Anton RF. Naltrexone for the management of alcohol dependence. N Engl J Med. 2008;359(7):715-721.
24. Gueorguieva R, Wu R, Pittman B, et al. New insights into the efficacy of naltrexone based on trajectory-based reanalysis of two negative clinical trials. Biol Psychiatry. 2007;61(11): 1290-1295.
25. Lapham S, Forman R, Alexander M, et al. The effects of extended-release naltrexone on holiday drinking in alcohol-dependent patients. J Subst Abuse. 2009;36(1):1-6.
26. Ciraulo DA, Dong Q, Silverman BL, et al. Early treatment response in alcohol dependence with extended-release naltrexone. J Clin Psychiatry. 2008;69(2):190-195.
27. Mark TL, Montejano LB, Kranzler HR, et al. Comparison of healthcare utilization among patients treated with alcoholism medications. Am J Managed Care. 2010;16(12): 879-888.
28. Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28(1):51-63.
29. Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168(7):709-717.
30. Karam-Hage M, Brower KJ. Open pilot study of gabapentin versus trazodone to treat insomnia in alcoholic outpatients. Psychiatry Clin Neurosci. 2003;57(5):542-544.
31. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.

Performing capacity evaluations: What’s expected from your consult
One of the most common reasons medical colleagues seek consultation with a psychiatrist is to address the question of capacity. Indeed, this referral question often is posed as, “Is the patient competent?”
This referral question is incomplete and incorrectly phrased. The question should include the domain in which capacity is being questioned—for example, “Is the patient competent to refuse surgery?” Specifically identifying the area in which competency is questioned is necessary because a person might be competent in one area and incompetent in another (Box 1).

The question of competency should be modified as follows: “Does the patient have capacity to refuse surgery?” Competency is the degree of mental soundness necessary to make decisions about a specific issue or to carry out a specific act. Capacity is a person’s ability to make an informed decision. A determination of competency is a judicial finding made by the court. A physician can opine about a patient’s capacity, but cannot determine competency.
Adults are presumed to have capacity unless determined otherwise by the court. A person who lacks capacity to make an informed decision or give consent might need to be referred for a competency hearing or have a guardian appointed. Psychiatrists often are called on to provide an opinion to the court regarding a person’s capacity. Psychiatrists are particularly skilled at accessing a person’s mental status and gauging its potential for interfering with specific areas of functioning, but, in fact, any physician can make a determination of capacity.1
In this article, I:
- outline the components of a capacity evaluation
- describe the tools used in the determination of capacity
- review the typical features of patients and psychiatrists who perform capacity evaluations.
What constitutes a capacity evaluation?
The components of a capacity evaluation are comprehension, free choice, and reliability.
Comprehension refers to a patient’s factual understanding of his (her) medical condition—for example, including the risks and benefits of treatment and reasonable alternatives. The patient should show an understanding of 1) the situation as it relates to his condition, and 2) the consequences of his decisions. He also should demonstrate a rational manipulation of the information presented, applying a coherent and logical thought process to analyze possible courses of action.2
To determine if the patient has the requisite knowledge regarding his condition, the physician must be familiar with the patient’s clinical status. This might require consultation with the treating physician. Communication is a key component of capacity evaluations. Barriers to good communication can lead to the evaluating physician’s perception that the patient lacks capacity. If a patient does not understand his condition or the proposed treatments, the psychiatrist should educate him. It might be useful to arrange a meeting with the treating physician to facilitate communication.
Free choice. The patient’s decision to accept or reject a proposed treatment should be voluntary and free of coercion. In assessing a patient’s capacity, the psychiatrist should determine whether choices have been rendered impossible because of unrealistic fears or expectations about treatment, or because of impaired mental processes.
Reliability refers to a patient’s ability to provide a consistent choice over time. A patient who vacillates or is inconsistent does not have capacity to make decisions.
Features of patients referred for evaluation, and their evaluators
The most common reason for a capacity evaluation is a patient’s refusal of medical treatment. Between 3% and 25% of requests for psychiatric consultation in hospital settings involve questions about patients’ competence to make a treatment-related decision.3 Approximately 25% of adult medicine inpatients lack capacity for medical decision-making.4
Decision-making capacity is a functional evaluation. Decision-making capacity does not relate specifically to a person’s psychiatric diagnosis. In other words, the presence of a mental disorder does not render a person incapable of making decisions. However, people with Alzheimer’s disease or dementia have a high rate of impaired capacity for making treatment decisions.
Schizophrenia has been found to have the highest rate of impaired decision-making among psychiatric disorders; depression is second and bipolar disorder, third. The strongest predictor of incapacity in psychiatric patients is lack of insight.5 Positive symptoms, negative symptoms, severity of symptoms, involuntary admission, lack of insight, and treatment refusal were strong predictors of incapacity in a sample of psychiatric patients.6
The neuronal basis of decision-making is unknown. Studies have implicated functioning of the medial and lateral prefrontal cortex as an important correlate of decision-making capacity.7 As a result of these findings, a brain-based criterion could be added to the conceptual criteria of capacity. The specific neuropsychological components necessary for decision-making capacity are unknown. Some studies suggest that poor executive functioning and limited learning ability correlate with impaired decision-making capacity.8 Little is known about the relationship between emotion and capacity. Supady et al demonstrated that higher cognitive empathy and good emotion recognition were associated with increased decision-making capacity and higher rates of refusal to give informed consent.9
Physician bias has been identified in capacity evaluations. See Box 2.4,10-12

Tools used in capacity evaluations
Most capacity evaluations are conducted by clinical interview (Box 3). The reliability of physicians’ unstructured judgments of capacity has been poor.13 In a study of 5 physicians who made a determination of capacity after watching a videotape of capacity assessments, the rate of agreement among the subjects was no better than that of chance.14

There is no specific, simple, quick test to assess capacity.
Folstein Mini-Mental State Examination. The MMSE has not been found to be predictive of decision-making capacity. It has been found to correlate with clinical judgments of incapacity, and may be used to identify patients at the high and low ends of the range of capacity, especially among older persons who exhibit cognitive impairment.15 Patients who have severe dementia (MMSE score <16) have a high likelihood of being unable to consent to treatment.16
MacArthur Competence Assessment Tool-Treatment. The MacCAT-T is a structured interviewing tool used to evaluate a patient’s decision-making ability. It is the most commonly used screening tool to evaluate decision-making capacity. Advantages of the MacCAT-T include a higher inter-rater agreement and—unlike other assessment instruments—its ability to incorporate information specific to a patient’s decision-making situation.17 The MacCAT-T requires training and experience to administer.
Bottom Line
Physicians make decisions about a patient’s decision-making capacity. Courts determine competence by a formal judicial proceeding. The psychiatric consultant’s role in capacity evaluations is to determine if the patient 1) possesses the requisite knowledge about the specific referral issue and 2) demonstrates a voluntary and reliable decision.
Related Resources
- The MacArthur Treatment Competence Study. www.macarthur.virginia.edu/treatment.html.
- Resnick P, Sorrentino R: Forensic considerations (chapter. 8). In: Psychosomatic medicine. Blumenfield M, Strain JJ, eds. Baltimore, MD: Lippincott Williams & Wilkins; 2006:91-106.
- Appelbaum PS. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.
Disclosure
Dr. Sorrentino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Grisson T, Appelbaum PS. Assessing competence to consent to treatment—a guide for physicians and other health professionals. New York, NY: Oxford University Press; 1998.
2. Cohen LM, McCue JD, Green GM. Do clinical and formal assessments of capacity of patients in the intensive care unit to make decisions agree? Arch Intern Med. 1993;153(21): 2841-2845.
3. Farnsworth MG. Competency evaluations in a general hospital. Psychosomatics. 1990;31(1):60-66.
4. Sessums LL, Zembrzuska H, Jackson JL. Does this patient have medical decision-making capacity? JAMA. 2011; 306(4):420-427.
5. Cairns R, Maddock C, Buchanan A, et al. Prevalence and predictors of mental incapacity in psychiatric in-patients. Br J Psychiatry. 2005;187:379-385.
6. Candia PC, Barba AC. Mental capacity and consent to treatment in psychiatric patients: the state of the research. Curr Opin Psychiatry. 2011;24(5):442-446.
7. Duncan J. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci. 2000;23(10):475-483.
8. Mandarelli G, Parmigiani G, Tarsitani L, et al. The relationship between executive functions and capacity to consent to treatment in acute psychiatric hospitalization. J Empir Res Hum Res Ethics. 2012;7(5):63-70.
9. Supady A, Voelkel A, Witzel J, et al. How is informed consent related to emotions and empathy? An exploratory neuroethical investigation. J Med Ethics. 2011;37(5):311-317.
10. Jeste DV, Depp CA, Palmer BW. Magnitude of impairment in decisional capacity in people with schizophrenia compared to normal subjects: an overview. Schizophr Bull. 2006;32(1):121-128.
11. Feldman-Stewart D, Brundage MD. Challenges for designing and implementing decision aids. Patient Educ Couns. 2004;54(3):265-273.
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
13. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
14. Marson DC, McIntruff B, Hawkins L, et al. Consistency of physician judgments of capacity to consent to mild Alzheimer’s disease. J Am Geriatr Soc. 1997;45(4):453-457.
15. Kim SY, Caine ED. Utility and limits of the mini mental status examination in evaluating consent capacity in Alzheimer’s disease. Psychiatr Serv. 2002;53(10):1322-1324.
16. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
17. Grisso T, Appelbaum PS. MacArthur competence assessment tool for treatment (MacCAT-T). Sarasota, FL: Professional Resource Press; 1998.
One of the most common reasons medical colleagues seek consultation with a psychiatrist is to address the question of capacity. Indeed, this referral question often is posed as, “Is the patient competent?”
This referral question is incomplete and incorrectly phrased. The question should include the domain in which capacity is being questioned—for example, “Is the patient competent to refuse surgery?” Specifically identifying the area in which competency is questioned is necessary because a person might be competent in one area and incompetent in another (Box 1).
The question of competency should be modified as follows: “Does the patient have capacity to refuse surgery?” Competency is the degree of mental soundness necessary to make decisions about a specific issue or to carry out a specific act. Capacity is a person’s ability to make an informed decision. A determination of competency is a judicial finding made by the court. A physician can opine about a patient’s capacity, but cannot determine competency.
Adults are presumed to have capacity unless determined otherwise by the court. A person who lacks capacity to make an informed decision or give consent might need to be referred for a competency hearing or have a guardian appointed. Psychiatrists often are called on to provide an opinion to the court regarding a person’s capacity. Psychiatrists are particularly skilled at accessing a person’s mental status and gauging its potential for interfering with specific areas of functioning, but, in fact, any physician can make a determination of capacity.1
In this article, I:
- outline the components of a capacity evaluation
- describe the tools used in the determination of capacity
- review the typical features of patients and psychiatrists who perform capacity evaluations.
What constitutes a capacity evaluation?
The components of a capacity evaluation are comprehension, free choice, and reliability.
Comprehension refers to a patient’s factual understanding of his (her) medical condition—for example, including the risks and benefits of treatment and reasonable alternatives. The patient should show an understanding of 1) the situation as it relates to his condition, and 2) the consequences of his decisions. He also should demonstrate a rational manipulation of the information presented, applying a coherent and logical thought process to analyze possible courses of action.2
To determine if the patient has the requisite knowledge regarding his condition, the physician must be familiar with the patient’s clinical status. This might require consultation with the treating physician. Communication is a key component of capacity evaluations. Barriers to good communication can lead to the evaluating physician’s perception that the patient lacks capacity. If a patient does not understand his condition or the proposed treatments, the psychiatrist should educate him. It might be useful to arrange a meeting with the treating physician to facilitate communication.
Free choice. The patient’s decision to accept or reject a proposed treatment should be voluntary and free of coercion. In assessing a patient’s capacity, the psychiatrist should determine whether choices have been rendered impossible because of unrealistic fears or expectations about treatment, or because of impaired mental processes.
Reliability refers to a patient’s ability to provide a consistent choice over time. A patient who vacillates or is inconsistent does not have capacity to make decisions.
Features of patients referred for evaluation, and their evaluators
The most common reason for a capacity evaluation is a patient’s refusal of medical treatment. Between 3% and 25% of requests for psychiatric consultation in hospital settings involve questions about patients’ competence to make a treatment-related decision.3 Approximately 25% of adult medicine inpatients lack capacity for medical decision-making.4
Decision-making capacity is a functional evaluation. Decision-making capacity does not relate specifically to a person’s psychiatric diagnosis. In other words, the presence of a mental disorder does not render a person incapable of making decisions. However, people with Alzheimer’s disease or dementia have a high rate of impaired capacity for making treatment decisions.
Schizophrenia has been found to have the highest rate of impaired decision-making among psychiatric disorders; depression is second and bipolar disorder, third. The strongest predictor of incapacity in psychiatric patients is lack of insight.5 Positive symptoms, negative symptoms, severity of symptoms, involuntary admission, lack of insight, and treatment refusal were strong predictors of incapacity in a sample of psychiatric patients.6
The neuronal basis of decision-making is unknown. Studies have implicated functioning of the medial and lateral prefrontal cortex as an important correlate of decision-making capacity.7 As a result of these findings, a brain-based criterion could be added to the conceptual criteria of capacity. The specific neuropsychological components necessary for decision-making capacity are unknown. Some studies suggest that poor executive functioning and limited learning ability correlate with impaired decision-making capacity.8 Little is known about the relationship between emotion and capacity. Supady et al demonstrated that higher cognitive empathy and good emotion recognition were associated with increased decision-making capacity and higher rates of refusal to give informed consent.9
Physician bias has been identified in capacity evaluations. See Box 2.4,10-12
Tools used in capacity evaluations
Most capacity evaluations are conducted by clinical interview (Box 3). The reliability of physicians’ unstructured judgments of capacity has been poor.13 In a study of 5 physicians who made a determination of capacity after watching a videotape of capacity assessments, the rate of agreement among the subjects was no better than that of chance.14
There is no specific, simple, quick test to assess capacity.
Folstein Mini-Mental State Examination. The MMSE has not been found to be predictive of decision-making capacity. It has been found to correlate with clinical judgments of incapacity, and may be used to identify patients at the high and low ends of the range of capacity, especially among older persons who exhibit cognitive impairment.15 Patients who have severe dementia (MMSE score <16) have a high likelihood of being unable to consent to treatment.16
MacArthur Competence Assessment Tool-Treatment. The MacCAT-T is a structured interviewing tool used to evaluate a patient’s decision-making ability. It is the most commonly used screening tool to evaluate decision-making capacity. Advantages of the MacCAT-T include a higher inter-rater agreement and—unlike other assessment instruments—its ability to incorporate information specific to a patient’s decision-making situation.17 The MacCAT-T requires training and experience to administer.
Bottom Line
Physicians make decisions about a patient’s decision-making capacity. Courts determine competence by a formal judicial proceeding. The psychiatric consultant’s role in capacity evaluations is to determine if the patient 1) possesses the requisite knowledge about the specific referral issue and 2) demonstrates a voluntary and reliable decision.
Related Resources
- The MacArthur Treatment Competence Study. www.macarthur.virginia.edu/treatment.html.
- Resnick P, Sorrentino R: Forensic considerations (chapter. 8). In: Psychosomatic medicine. Blumenfield M, Strain JJ, eds. Baltimore, MD: Lippincott Williams & Wilkins; 2006:91-106.
- Appelbaum PS. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.
Disclosure
Dr. Sorrentino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
One of the most common reasons medical colleagues seek consultation with a psychiatrist is to address the question of capacity. Indeed, this referral question often is posed as, “Is the patient competent?”
This referral question is incomplete and incorrectly phrased. The question should include the domain in which capacity is being questioned—for example, “Is the patient competent to refuse surgery?” Specifically identifying the area in which competency is questioned is necessary because a person might be competent in one area and incompetent in another (Box 1).
The question of competency should be modified as follows: “Does the patient have capacity to refuse surgery?” Competency is the degree of mental soundness necessary to make decisions about a specific issue or to carry out a specific act. Capacity is a person’s ability to make an informed decision. A determination of competency is a judicial finding made by the court. A physician can opine about a patient’s capacity, but cannot determine competency.
Adults are presumed to have capacity unless determined otherwise by the court. A person who lacks capacity to make an informed decision or give consent might need to be referred for a competency hearing or have a guardian appointed. Psychiatrists often are called on to provide an opinion to the court regarding a person’s capacity. Psychiatrists are particularly skilled at accessing a person’s mental status and gauging its potential for interfering with specific areas of functioning, but, in fact, any physician can make a determination of capacity.1
In this article, I:
- outline the components of a capacity evaluation
- describe the tools used in the determination of capacity
- review the typical features of patients and psychiatrists who perform capacity evaluations.
What constitutes a capacity evaluation?
The components of a capacity evaluation are comprehension, free choice, and reliability.
Comprehension refers to a patient’s factual understanding of his (her) medical condition—for example, including the risks and benefits of treatment and reasonable alternatives. The patient should show an understanding of 1) the situation as it relates to his condition, and 2) the consequences of his decisions. He also should demonstrate a rational manipulation of the information presented, applying a coherent and logical thought process to analyze possible courses of action.2
To determine if the patient has the requisite knowledge regarding his condition, the physician must be familiar with the patient’s clinical status. This might require consultation with the treating physician. Communication is a key component of capacity evaluations. Barriers to good communication can lead to the evaluating physician’s perception that the patient lacks capacity. If a patient does not understand his condition or the proposed treatments, the psychiatrist should educate him. It might be useful to arrange a meeting with the treating physician to facilitate communication.
Free choice. The patient’s decision to accept or reject a proposed treatment should be voluntary and free of coercion. In assessing a patient’s capacity, the psychiatrist should determine whether choices have been rendered impossible because of unrealistic fears or expectations about treatment, or because of impaired mental processes.
Reliability refers to a patient’s ability to provide a consistent choice over time. A patient who vacillates or is inconsistent does not have capacity to make decisions.
Features of patients referred for evaluation, and their evaluators
The most common reason for a capacity evaluation is a patient’s refusal of medical treatment. Between 3% and 25% of requests for psychiatric consultation in hospital settings involve questions about patients’ competence to make a treatment-related decision.3 Approximately 25% of adult medicine inpatients lack capacity for medical decision-making.4
Decision-making capacity is a functional evaluation. Decision-making capacity does not relate specifically to a person’s psychiatric diagnosis. In other words, the presence of a mental disorder does not render a person incapable of making decisions. However, people with Alzheimer’s disease or dementia have a high rate of impaired capacity for making treatment decisions.
Schizophrenia has been found to have the highest rate of impaired decision-making among psychiatric disorders; depression is second and bipolar disorder, third. The strongest predictor of incapacity in psychiatric patients is lack of insight.5 Positive symptoms, negative symptoms, severity of symptoms, involuntary admission, lack of insight, and treatment refusal were strong predictors of incapacity in a sample of psychiatric patients.6
The neuronal basis of decision-making is unknown. Studies have implicated functioning of the medial and lateral prefrontal cortex as an important correlate of decision-making capacity.7 As a result of these findings, a brain-based criterion could be added to the conceptual criteria of capacity. The specific neuropsychological components necessary for decision-making capacity are unknown. Some studies suggest that poor executive functioning and limited learning ability correlate with impaired decision-making capacity.8 Little is known about the relationship between emotion and capacity. Supady et al demonstrated that higher cognitive empathy and good emotion recognition were associated with increased decision-making capacity and higher rates of refusal to give informed consent.9
Physician bias has been identified in capacity evaluations. See Box 2.4,10-12
Tools used in capacity evaluations
Most capacity evaluations are conducted by clinical interview (Box 3). The reliability of physicians’ unstructured judgments of capacity has been poor.13 In a study of 5 physicians who made a determination of capacity after watching a videotape of capacity assessments, the rate of agreement among the subjects was no better than that of chance.14
There is no specific, simple, quick test to assess capacity.
Folstein Mini-Mental State Examination. The MMSE has not been found to be predictive of decision-making capacity. It has been found to correlate with clinical judgments of incapacity, and may be used to identify patients at the high and low ends of the range of capacity, especially among older persons who exhibit cognitive impairment.15 Patients who have severe dementia (MMSE score <16) have a high likelihood of being unable to consent to treatment.16
MacArthur Competence Assessment Tool-Treatment. The MacCAT-T is a structured interviewing tool used to evaluate a patient’s decision-making ability. It is the most commonly used screening tool to evaluate decision-making capacity. Advantages of the MacCAT-T include a higher inter-rater agreement and—unlike other assessment instruments—its ability to incorporate information specific to a patient’s decision-making situation.17 The MacCAT-T requires training and experience to administer.
Bottom Line
Physicians make decisions about a patient’s decision-making capacity. Courts determine competence by a formal judicial proceeding. The psychiatric consultant’s role in capacity evaluations is to determine if the patient 1) possesses the requisite knowledge about the specific referral issue and 2) demonstrates a voluntary and reliable decision.
Related Resources
- The MacArthur Treatment Competence Study. www.macarthur.virginia.edu/treatment.html.
- Resnick P, Sorrentino R: Forensic considerations (chapter. 8). In: Psychosomatic medicine. Blumenfield M, Strain JJ, eds. Baltimore, MD: Lippincott Williams & Wilkins; 2006:91-106.
- Appelbaum PS. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.
Disclosure
Dr. Sorrentino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Grisson T, Appelbaum PS. Assessing competence to consent to treatment—a guide for physicians and other health professionals. New York, NY: Oxford University Press; 1998.
2. Cohen LM, McCue JD, Green GM. Do clinical and formal assessments of capacity of patients in the intensive care unit to make decisions agree? Arch Intern Med. 1993;153(21): 2841-2845.
3. Farnsworth MG. Competency evaluations in a general hospital. Psychosomatics. 1990;31(1):60-66.
4. Sessums LL, Zembrzuska H, Jackson JL. Does this patient have medical decision-making capacity? JAMA. 2011; 306(4):420-427.
5. Cairns R, Maddock C, Buchanan A, et al. Prevalence and predictors of mental incapacity in psychiatric in-patients. Br J Psychiatry. 2005;187:379-385.
6. Candia PC, Barba AC. Mental capacity and consent to treatment in psychiatric patients: the state of the research. Curr Opin Psychiatry. 2011;24(5):442-446.
7. Duncan J. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci. 2000;23(10):475-483.
8. Mandarelli G, Parmigiani G, Tarsitani L, et al. The relationship between executive functions and capacity to consent to treatment in acute psychiatric hospitalization. J Empir Res Hum Res Ethics. 2012;7(5):63-70.
9. Supady A, Voelkel A, Witzel J, et al. How is informed consent related to emotions and empathy? An exploratory neuroethical investigation. J Med Ethics. 2011;37(5):311-317.
10. Jeste DV, Depp CA, Palmer BW. Magnitude of impairment in decisional capacity in people with schizophrenia compared to normal subjects: an overview. Schizophr Bull. 2006;32(1):121-128.
11. Feldman-Stewart D, Brundage MD. Challenges for designing and implementing decision aids. Patient Educ Couns. 2004;54(3):265-273.
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
13. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
14. Marson DC, McIntruff B, Hawkins L, et al. Consistency of physician judgments of capacity to consent to mild Alzheimer’s disease. J Am Geriatr Soc. 1997;45(4):453-457.
15. Kim SY, Caine ED. Utility and limits of the mini mental status examination in evaluating consent capacity in Alzheimer’s disease. Psychiatr Serv. 2002;53(10):1322-1324.
16. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
17. Grisso T, Appelbaum PS. MacArthur competence assessment tool for treatment (MacCAT-T). Sarasota, FL: Professional Resource Press; 1998.
1. Grisson T, Appelbaum PS. Assessing competence to consent to treatment—a guide for physicians and other health professionals. New York, NY: Oxford University Press; 1998.
2. Cohen LM, McCue JD, Green GM. Do clinical and formal assessments of capacity of patients in the intensive care unit to make decisions agree? Arch Intern Med. 1993;153(21): 2841-2845.
3. Farnsworth MG. Competency evaluations in a general hospital. Psychosomatics. 1990;31(1):60-66.
4. Sessums LL, Zembrzuska H, Jackson JL. Does this patient have medical decision-making capacity? JAMA. 2011; 306(4):420-427.
5. Cairns R, Maddock C, Buchanan A, et al. Prevalence and predictors of mental incapacity in psychiatric in-patients. Br J Psychiatry. 2005;187:379-385.
6. Candia PC, Barba AC. Mental capacity and consent to treatment in psychiatric patients: the state of the research. Curr Opin Psychiatry. 2011;24(5):442-446.
7. Duncan J. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci. 2000;23(10):475-483.
8. Mandarelli G, Parmigiani G, Tarsitani L, et al. The relationship between executive functions and capacity to consent to treatment in acute psychiatric hospitalization. J Empir Res Hum Res Ethics. 2012;7(5):63-70.
9. Supady A, Voelkel A, Witzel J, et al. How is informed consent related to emotions and empathy? An exploratory neuroethical investigation. J Med Ethics. 2011;37(5):311-317.
10. Jeste DV, Depp CA, Palmer BW. Magnitude of impairment in decisional capacity in people with schizophrenia compared to normal subjects: an overview. Schizophr Bull. 2006;32(1):121-128.
11. Feldman-Stewart D, Brundage MD. Challenges for designing and implementing decision aids. Patient Educ Couns. 2004;54(3):265-273.
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
13. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
14. Marson DC, McIntruff B, Hawkins L, et al. Consistency of physician judgments of capacity to consent to mild Alzheimer’s disease. J Am Geriatr Soc. 1997;45(4):453-457.
15. Kim SY, Caine ED. Utility and limits of the mini mental status examination in evaluating consent capacity in Alzheimer’s disease. Psychiatr Serv. 2002;53(10):1322-1324.
16. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
17. Grisso T, Appelbaum PS. MacArthur competence assessment tool for treatment (MacCAT-T). Sarasota, FL: Professional Resource Press; 1998.

Expanding medication options for pediatric ADHD
Molly, age 9, is diagnosed with attention-deficit/hyperactivity disorder (ADHD) by her psychiatrist, who prescribes a long-acting methylphenidate formulation at 1 mg/kg. She tolerates the medication without side effects and shows significant improvement in her academic performance and on-task behavior in school. Molly takes methylphenidate before school at 7:00 am; this dose usually wears off at approximately 3:30 pm.
Molly and her parents are pleased with her response to methylphenidate, but report that she has difficulty getting ready for school because of distractibility. In the evenings Molly has trouble staying seated to do homework and often interrupts and argues with family members, but cannot tolerate afternoon dosing of immediate-release methylphenidate because of insomnia.
ADHD, the most common childhood neurobehavioral disorder, is characterized by difficulties with attention, impulse control, and modulating activity level. The pathophysiology of ADHD is thought to involve dysregulation of brain dopamine and norepinephrine systems.1 Managing ADHD includes pharmacotherapeutic and nonpharmacotherapeutic—ie, behavioral and psychoeducational—interventions.2,3
In this article, we provide an overview of the efficacy, side effects, and dosing for the 3 classes of ADHD medication—psychostimulants, atomoxetine, and α2 adrenergic agonists—including guidance on medication choice and combination treatment. We also discuss the effects of psychostimulants on tics, cardiovascular concerns, and substance abuse potential.
Psychostimulants
Methylphenidates and amphetamines are first-line agents for ADHD. Their primary mechanism of action involves blocking dopamine transporters, with additional effects including blockade of norepinephrine transporters, dampening action of monoamine oxidase (which slows dopamine and norepinephrine degradation), and enhanced release of dopamine into the synaptic space.1
Efficacy and response rates are similar for methylphenidate and amphetamine medications, although as many as 25% of patients may respond to only 1 agent.1 More than 90% of patients will have a positive response to one of the psychostimulants.1 The beneficial effects of psychostimulants on inattention, hyperactivity, and impulsivity are well documented.2Improvements in noncompliance, aggression, social interactions, and academic productivity also have been observed.4,5
Because of increased recognition of pervasive ADHD-related impairments, which can affect functioning in social, family, and extracurricular settings, practitioners have shifted to long-acting psychostimulants to reduce the need for in-school dosing, improve compliance, and obtain more after-school treatment effects. Long-acting formulations produce a slower rise and fall of psychostimulant levels in the brain, which may decrease side effects and potential for later drug abuse.6 See Table 12,7-9 and Table 22,7,9 for titration, dosing, and duration of action of psychostimulants.
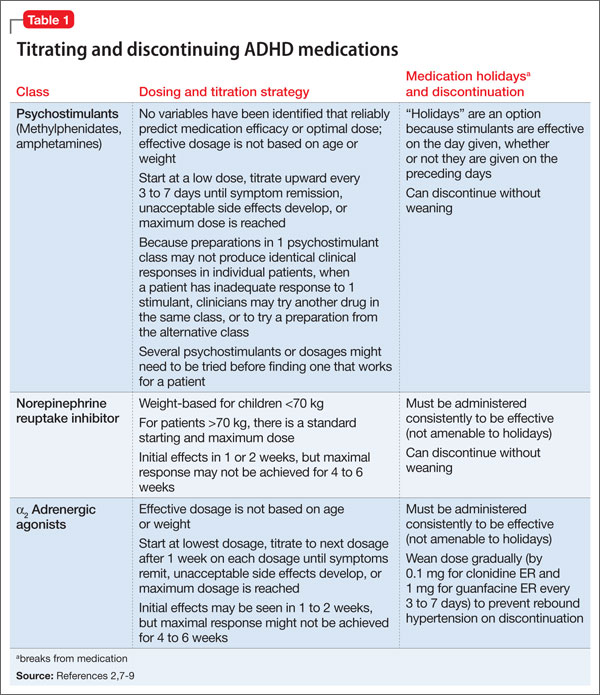
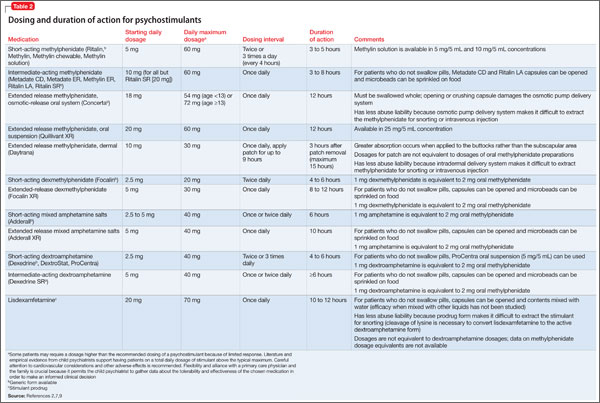
The most common side effects of psychostimulants are appetite loss, abdominal pain, headaches, and sleep disturbances.2 Emotional symptoms—irritability and nervousness—may be observed with psychostimulant use, but these behaviors may improve, rather than become worse, with treatment.5 Methylphenidates and amphetamines share many of the same side effects,2 with many studies indicating no differences between their side-effect profiles.1 Other studies indicate that sleep and emotional side effects may be more prominent with amphetamines than methylphenidates,10 although response varies by individual.
There is little evidence that methylphenidate, low-dose amphetamine, or low-dose dextroamphetamine makes tics worse in most children who have them, although significant tic exacerbation has been observed with higher-dose dextroamphetamine.11,12 In patients with comorbid ADHD and tic disorders, a trial of psychostimulants with monitoring for worsening tics is appropriate.
Changes in heart rate and blood pressure generally are not clinically significant in patients taking psychostimulants (average increases: 1 or 2 beats per minute and 1 to 4 mm Hg for systolic and diastolic blood pressures).12 However, psychostimulants may be associated with more substantial increases in heart rate and blood pressure in a subset of individuals (5% to 15%).12 Large studies of children and adults in the general population have not found an association between psychostimulant use and severe cardiovascular events (sudden cardiac death, myocardial infarction, stroke).12-14 Because of reports of sudden cardiac death in children with underlying heart disease who take a psychostimulant,15 clinicians are advised to screen patients and consider an electrocardiogram or evaluation by a cardiologist before starting a psychostimulant in a patient who has a personal or family history of specific cardiovascular risk factors (see Perrin et al16 and Cortese et al12 for screening questions and conditions).
Modest reductions in height (1 or 2 cm after 3 years of psychostimulant treatment) appear to be dose-dependent, and are similar across the methylphenidate and amphetamine classes. Some studies have shown reversal of growth deficits after treatment is stopped treatment and no adverse effects on final adult height.12,17 More study is needed to clarify the effects of continuous psychostimulant treatment from childhood to adulthood on growth.
Studies have failed to show an increased risk of substance abuse in persons with ADHD who were treated with psychostimulants during childhood. Some studies document a lower rate of later substance abuse in youths who received ADHD medications, although other reports show no effect of psychostimulant treatment on subsequent substance use disorder risk.12 Be aware that psychostimulants can be misused (eg, to get “high,” for performance enhancement, to suppress appetite, etc.). Misuse of psychostimulants is most common with short-acting preparations, and generally more difficult with long-acting preparations because extracting the active ingredients for snorting is difficult.2,12 Monitor refill requests and patient behavior for signs of misuse, and be alert for signs of illegal drug use in the patient’s family.
Psychotic symptoms—including hallucinations, delusions, mania, and extreme agitation—with psychostimulant treatment are rare, occurring at a rate of 1.5%.12
Atomoxetine
Approved by the FDA in 2002 for ADHD, atomoxetine is effective and generally well tolerated, although it is not as effective as psychostimulants.2 Atomoxetine is a potent norepinephrine reuptake inhibitor18 that does not produce euphoria, does not have potential for abuse, and has not been linked to increased tic onset or severity.19 Atomoxetine treatment is associated with a lower rate of sleep initiation difficulty compared with psychostimulants.18 Some studies suggest that atomoxetine may have mild beneficial effects on anxiety disorders,18 making it a reasonable choice for patients with significant anxiety or insomnia during psychostimulant treatment. Table 12,7-9 and Table 32,7,9 include information on dosing and duration of action for atomoxetine.
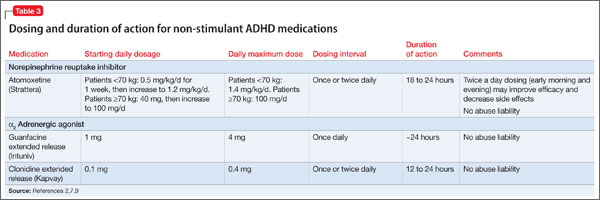
Common side effects of atomoxetine include sedation and fatigue, upset stomach, nausea and vomiting, reduced appetite, headache, and irritability.18 Inform patients that atomoxetine carries an FDA black-box warning for suicide risk; a review of 14 studies showed suicidal ideation was more common with atomoxetine than placebo, although no suicides occurred in any trials.20
Hepatotoxicity is rare with atomoxetine.21 Although routine liver enzyme testing is not required, discontinue atomoxetine if jaundice develops or elevated levels of liver enzymes are noted. Other rare but potentially serious side effects include changes in heart rate (≥20 beats per min) or blood pressure that occur in 5% to 10% of patients taking atomoxetine.22 The risk of serious cardiovascular events and sudden cardiac death with atomoxetine is extremely low, but patients should be screened for a personal and family history of cardiovascular risk factors and, if any of these are present, evaluated further before starting atomoxetine. Routine heart rate and blood pressure monitoring is recommended for all patients.12-14,16
Last, atomoxetine has been linked to growth delays in the first 1 or 2 years of treatment, with a return to expected measurements after an average 2 or 3 years of treatment; persistent decreases in growth rate were observed in patients who were taller or heavier than average before treatment.23
α2 Adrenergic agonists
Guanfacine ER and clonidine ER, the extended release (ER) formulations of α2 adrenergic agonists, were FDA-approved for treating ADHD in 2009 and 2010, respectively. Short-acting guanfacine and clonidine also are used for treating ADHD.24 Their mechanism of action involves stimulation of the pre-synaptic and post-synapic α2 adrenergic receptors, which control the release of norepinephrine and the rate of cell firing.25 The α2 agonists are considered a second-line treatment for ADHD because their efficacy and response rate for core ADHD symptoms lags behind those of psychostimulants.25 In addition to treating core ADHD symptoms, guanfacine and clonidine are used to treat tics and oppositional/aggressive behavior comorbid with ADHD.24,26 Clonidine, which is more sedating than guanfacine, can be used to treat comorbid ADHD and sleep disorders.24 The α2 agonists do not produce euphoria and do not have drug abuse potential.2Table 12,7-9 and Table 32,7,9 provide guidelines for prescribing guanfacine ER and clonidine ER.
The most common adverse effect is drowsiness; other common side effects include dizziness, irritability, headache, and abdominal pain.24 Short-term studies of α2 agonist treatment of ADHD have shown small, non-clinically significant reductions in heart rate and blood pressure; α2 agonist-associated bradycardia, increased QT interval, and cardiac arrhythmias have been reported,7,24,27 as well as rebound hypertension with abrupt discontinuation.24 Screen patients for a personal and family history of cardiovascular risk factors and, if present, evaluate further before initiating α2 agonists.
Combining ADHD medication classes
Combination therapy with >1 ADHD medications is employed when 1 class does not provide adequate symptom coverage or produces problematic side effects.8,24 Psychostimulants can be combined with low-dose atomoxetine (0.5 to 1.0 mg/kg/d) when atomoxetine does not adequately cover ADHD symptoms in school, or when psychostimulants do not adequately cover evening symptoms or patients experience problems with evening psychostimulant rebound.8 To date, prospective data on the safety and efficacy of combining atomoxetine and psychostimulants are limited, but what evidence is available suggests improved symptom control for some, but not all, patients, and a lack of serious adverse events.28
Psychostimulants have been combined with α2agonists when children have an inadequate response to psychostimulants alone, or in cases of ADHD comorbid with aggression or tics.24 Although early case reports raised concern about the safety of combining psychostimulants and α2 agonists, subsequent studies suggest that clonidine and guanfacine generally are well-tolerated when co-administered with psychostimulants.24,27,29
Case continued
Molly has derived substantial benefit from long-acting methylphenidate during the school day, but continues to have significant ADHD-related impairment in the mornings and evenings. Her physician tried afternoon dosing of immediate-release methylphenidate to address evening difficulties, but Molly experienced insomnia. It would be reasonable to consider adjunctive therapy with a non-stimulant medication. A medication that can provide round-the-clock ADHD symptom coverage—such as atomoxetine, guanfacine ER, or clonidine ER—could be added to her current day-time psychostimulant treatment, potentially improving her functioning at home before school and in the evenings.
Additional considerations
Combining medication and behavior therapy offers greater improvements on academic, conduct, and family satisfaction measures than either treatment alone.2 Clinicians can choose to employ behavior therapy alone, particularly if parents feel uncomfortable with—or children have not tolerated—medication.2,3 Evidence-based behavioral parent training and classroom management strategies (implemented by teachers) have shown the strongest and most consistent effects among nonpharmacotherapeutic interventions for ADHD.2 Most studies comparing behavior therapy to psychostimulants have found a stronger effect on core ADHD symptoms from psychostimulants than from behavior therapy.
When a patient does not respond adequately to FDA-approved ADHD medications alone or in combination, consider bupropion, an antidepressant with indirect dopamine and noradrenergic effects. Off-label bupropion has been shown to be effective for ADHD in controlled trials of both children and adults.30
Clinicians often encounter children who meet criteria for ADHD and an anxiety or mood disorder. Table 48,31 summarizes treatment recommendations for these patients.

Clinical considerations
- Begin treatment with a psychostimulant at a low dosage, and titrate gradually until symptoms are controlled or side effects develop.
- Keep in mind that an effective dosage of a psychostimulant is not closely correlated with age, weight, or severity of symptoms.
- Monitor refill requests and patient behavior for signs of psychostimulant misuse. Be alert for signs of illegal drug use in patient family members.
- Lisdexamfetamine, dermal methylphenidate, and osmotic release oral system methylphenidate are the formulations least likely to be misused because their delivery systems make it difficult to extract the active ingredient for snorting or intravenous injection.
- Psychostimulants have not been shown to exacerbate tics in most children who have comorbid ADHD and a tic disorder. When a stimulant is associated with an exacerbation of tics, switching treatment to atomoxetine or α2 agonists is reasonable.
- For patients whose use of a stimulant is limited by an adverse effect on sleep, consider atomoxetine and α2 adrenergic agonists as alternative or adjunctive treatments.
- All 3 classes of FDA-approved ADHD medications (psychostimulants, atomoxetine, and adrenergic agonists) have been associated with adverse cardiac events in children who have underlying cardiovascular conditions. Before initiating treatment, screen patients for a personal or family history of cardiovascular risk factors, and undertake further evaluation as indicated.
Bottom Line
In general, the evidence supports psychostimulants as initial pharmacotherapy for ADHD, with additional options including atomoxetine and α2 agonists. When one medication class does not provide adequate coverage for ADHD symptoms, combining medication classes can be beneficial.
Related Resources
- National Institute of Mental Health. What is attention deficit hyperactivity disorder (ADHD, ADD)?” www.nimh.nih.gov/health/topics/attention-deficit-hyperactivity-disorder-adhd/index.shtml.
- National Resource Center on AD/HD. Managing medication for children and adolescents with ADHD. www.help4adhd.org/en/treatment/medication/WWK3.
Drug Brand Names
Atomoxetine • Strattera
Lisdexamfetamine • Vyvanse
Bupropion • Wellbutrin, Zyban
Clonidine extended release • Kapvay
Guanfacine extended release • Intuniv
Dexmethylphenidate • Focalin, Focalin XR
Mixed amphetamine salts • Adderall, Adderall XR
Dextroamphetamine • Dexedrine, Dexedrine SR, DextroStat, ProCentra
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Disclosures
Dr. Froehlich receives support from the National Institute of Mental Health Grant K23 MH083881. Dr. Delgado has received research support from Pfizer, Inc. Dr. Anixt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998; 94(1):127-152.
2. Subcommittee on Attention-Deficit/Hyperactivity Disorder; Steering Committee on Quality Improvement and Management; Wolraich M, Brown L, Brown RT, et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007-1022.
3. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
4. Zametkin AJ, Ernst M. Problems in the management of attention-deficit-hyperactivity disorder. N Engl J Med. 1999;340(1):40-46.
5. Goldman LS, Genel M, Bezman RJ, et al. Diagnosis and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Council on Scientific Affairs, American Medical Association. JAMA. 1998;279(14):1100-1107.
6. Swanson J, Gupta S, Lam A, et al. Development of a new once-a-day formulation of methylphenidate for the treatment of attention-deficit/hyperactivity disorder: proof-of-concept and proof-of-product studies. Arch Gen Psychiatry. 2003;60(2):204-211.
7. Vaughan B, Kratochvil CJ. Pharmacotherapy of pediatric attention-deficit/hyperactivity disorder. Child Adolesc Psychiatr Clin N Am. 2012;21(4):941-955.
8. Pliszka SR, Crismon ML, Hughes CW, et al; Texas Consensus Conference Panel on Pharmacotherapy of Childhood Attention Deficit Hyperactivity Disorder. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2006;45(6):642-657.
9. Antshel KM, Hargrave TM, Simonescu M, et al. Advances in understanding and treating ADHD. BMC Med. 2011;9:72.
10. Efron D, Jarman F, Barker M. Side effects of methylphenidate and dexamphetamine in children with attention deficit hyperactivity disorder: a double-blind, crossover trial. Pediatrics. 1997;100(4):662-666.
11. Pringsheim T, Steeves T. Pharmacological treatment for attention deficit hyperactivity disorder (ADHD) in children with comorbid tic disorders. Cochrane Database Syst Rev. 2011(4):CD007990.
12. Cortese S, Holtmann M, Banaschewski T, et al. Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry. 2013; 54(3):227-246.
13. Cooper WO, Habel LA, Sox CM, et al. ADHD drugs and serious cardiovascular events in children and young adults. N Engl J Med. 2011;365(20):1896-1904.
14. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
15. Vetter VL, Elia J, Erickson C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing. Circulation. 2008;117(18):2407-2423.
16. Perrin JM, Friedman RA, Knilans TK; Black Box Working Group; Section on Cardiology and Cardiac Surgery. Cardiovascular monitoring and stimulant drugs for attention-deficit/hyperactivity disorder. Pediatrics. 2008;122(2):451-453.
17. Faraone SV, Biederman J, Morley CP, et al. Effect of stimulants on height and weight: a review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47(9):994-1009.
18. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
19. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
20. Bangs ME, Tauscher-Wisniewski S, Polzer J, et al. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J Am Acad Child Adolesc Psychiatry. 2008;47(2):209-218.
21. Bangs ME, Jin L, Zhang S, et al. Hepatic events associated with atomoxetine treatment for attention-deficit hyperactivity disorder. Drug Saf. 2008;31(4):345-354.
22. U.S. Food and Drug Administration. Strattera (atomoxetine hydrochloride) capsule. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm223889.htm. Published August 2013. Accessed October 31, 2013.
23. Spencer TJ, Kratochvil CJ, Sangal RB, et al. Effects of atomoxetine on growth in children with attention-deficit/hyperactivity disorder following up to five years of treatment. J Child Adolesc Psychopharmacol. 2007;17(5):689-700.
24. Connor DF. Other medications. In: Barkley RA, ed. Attention-deficit/hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:658-677.
25. May DE, Kratochvil CJ. Attention-deficit hyperactivity disorder: recent advances in paediatric pharmacotherapy. Drugs. 2010;70(1):15-40.
26. Connor DF, Findling RL, Kollins SH, et al. Effects of guanfacine extended release on oppositional symptoms in children aged 6-12 years with attention-deficit hyperactivity disorder and oppositional symptoms: a randomized, double-blind, placebo-controlled trial. CNS Drugs. 2010; 24(9):755-768.
27. Croxtall JD. Clonidine extended-release: in attention-deficit hyperactivity disorder. Paediatr Drugs. 2011;13(5):329-336.
28. Treuer T, Gau SS, Mendez L, et al. A systematic review of combination therapy with stimulants and atomoxetine for attention-deficit/hyperactivity disorder, including patient characteristics, treatment strategies, effectiveness, and tolerability. J Child Adolesc Psychopharmacol. 2013;23(3):179-193.
29. Sallee FR. The role of alpha2-adrenergic agonists in attention-deficit/hyperactivity disorder. Postgrad Med. 2010;122(5):78-87.
30. Spencer TJ. Antidepressant and specific norepinephrine reuptake inhibitor treatments. In: Barkley RA, ed. Attention-deficit hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:648-657.
31. Singh MK, DelBello MP, Kowatch RA, et al. Co-occurrence of bipolar and attention-deficit hyperactivity disorders in children. Bipolar Disord. 2006;8(6):710-720.
Molly, age 9, is diagnosed with attention-deficit/hyperactivity disorder (ADHD) by her psychiatrist, who prescribes a long-acting methylphenidate formulation at 1 mg/kg. She tolerates the medication without side effects and shows significant improvement in her academic performance and on-task behavior in school. Molly takes methylphenidate before school at 7:00 am; this dose usually wears off at approximately 3:30 pm.
Molly and her parents are pleased with her response to methylphenidate, but report that she has difficulty getting ready for school because of distractibility. In the evenings Molly has trouble staying seated to do homework and often interrupts and argues with family members, but cannot tolerate afternoon dosing of immediate-release methylphenidate because of insomnia.
ADHD, the most common childhood neurobehavioral disorder, is characterized by difficulties with attention, impulse control, and modulating activity level. The pathophysiology of ADHD is thought to involve dysregulation of brain dopamine and norepinephrine systems.1 Managing ADHD includes pharmacotherapeutic and nonpharmacotherapeutic—ie, behavioral and psychoeducational—interventions.2,3
In this article, we provide an overview of the efficacy, side effects, and dosing for the 3 classes of ADHD medication—psychostimulants, atomoxetine, and α2 adrenergic agonists—including guidance on medication choice and combination treatment. We also discuss the effects of psychostimulants on tics, cardiovascular concerns, and substance abuse potential.
Psychostimulants
Methylphenidates and amphetamines are first-line agents for ADHD. Their primary mechanism of action involves blocking dopamine transporters, with additional effects including blockade of norepinephrine transporters, dampening action of monoamine oxidase (which slows dopamine and norepinephrine degradation), and enhanced release of dopamine into the synaptic space.1
Efficacy and response rates are similar for methylphenidate and amphetamine medications, although as many as 25% of patients may respond to only 1 agent.1 More than 90% of patients will have a positive response to one of the psychostimulants.1 The beneficial effects of psychostimulants on inattention, hyperactivity, and impulsivity are well documented.2Improvements in noncompliance, aggression, social interactions, and academic productivity also have been observed.4,5
Because of increased recognition of pervasive ADHD-related impairments, which can affect functioning in social, family, and extracurricular settings, practitioners have shifted to long-acting psychostimulants to reduce the need for in-school dosing, improve compliance, and obtain more after-school treatment effects. Long-acting formulations produce a slower rise and fall of psychostimulant levels in the brain, which may decrease side effects and potential for later drug abuse.6 See Table 12,7-9 and Table 22,7,9 for titration, dosing, and duration of action of psychostimulants.
The most common side effects of psychostimulants are appetite loss, abdominal pain, headaches, and sleep disturbances.2 Emotional symptoms—irritability and nervousness—may be observed with psychostimulant use, but these behaviors may improve, rather than become worse, with treatment.5 Methylphenidates and amphetamines share many of the same side effects,2 with many studies indicating no differences between their side-effect profiles.1 Other studies indicate that sleep and emotional side effects may be more prominent with amphetamines than methylphenidates,10 although response varies by individual.
There is little evidence that methylphenidate, low-dose amphetamine, or low-dose dextroamphetamine makes tics worse in most children who have them, although significant tic exacerbation has been observed with higher-dose dextroamphetamine.11,12 In patients with comorbid ADHD and tic disorders, a trial of psychostimulants with monitoring for worsening tics is appropriate.
Changes in heart rate and blood pressure generally are not clinically significant in patients taking psychostimulants (average increases: 1 or 2 beats per minute and 1 to 4 mm Hg for systolic and diastolic blood pressures).12 However, psychostimulants may be associated with more substantial increases in heart rate and blood pressure in a subset of individuals (5% to 15%).12 Large studies of children and adults in the general population have not found an association between psychostimulant use and severe cardiovascular events (sudden cardiac death, myocardial infarction, stroke).12-14 Because of reports of sudden cardiac death in children with underlying heart disease who take a psychostimulant,15 clinicians are advised to screen patients and consider an electrocardiogram or evaluation by a cardiologist before starting a psychostimulant in a patient who has a personal or family history of specific cardiovascular risk factors (see Perrin et al16 and Cortese et al12 for screening questions and conditions).
Modest reductions in height (1 or 2 cm after 3 years of psychostimulant treatment) appear to be dose-dependent, and are similar across the methylphenidate and amphetamine classes. Some studies have shown reversal of growth deficits after treatment is stopped treatment and no adverse effects on final adult height.12,17 More study is needed to clarify the effects of continuous psychostimulant treatment from childhood to adulthood on growth.
Studies have failed to show an increased risk of substance abuse in persons with ADHD who were treated with psychostimulants during childhood. Some studies document a lower rate of later substance abuse in youths who received ADHD medications, although other reports show no effect of psychostimulant treatment on subsequent substance use disorder risk.12 Be aware that psychostimulants can be misused (eg, to get “high,” for performance enhancement, to suppress appetite, etc.). Misuse of psychostimulants is most common with short-acting preparations, and generally more difficult with long-acting preparations because extracting the active ingredients for snorting is difficult.2,12 Monitor refill requests and patient behavior for signs of misuse, and be alert for signs of illegal drug use in the patient’s family.
Psychotic symptoms—including hallucinations, delusions, mania, and extreme agitation—with psychostimulant treatment are rare, occurring at a rate of 1.5%.12
Atomoxetine
Approved by the FDA in 2002 for ADHD, atomoxetine is effective and generally well tolerated, although it is not as effective as psychostimulants.2 Atomoxetine is a potent norepinephrine reuptake inhibitor18 that does not produce euphoria, does not have potential for abuse, and has not been linked to increased tic onset or severity.19 Atomoxetine treatment is associated with a lower rate of sleep initiation difficulty compared with psychostimulants.18 Some studies suggest that atomoxetine may have mild beneficial effects on anxiety disorders,18 making it a reasonable choice for patients with significant anxiety or insomnia during psychostimulant treatment. Table 12,7-9 and Table 32,7,9 include information on dosing and duration of action for atomoxetine.
Common side effects of atomoxetine include sedation and fatigue, upset stomach, nausea and vomiting, reduced appetite, headache, and irritability.18 Inform patients that atomoxetine carries an FDA black-box warning for suicide risk; a review of 14 studies showed suicidal ideation was more common with atomoxetine than placebo, although no suicides occurred in any trials.20
Hepatotoxicity is rare with atomoxetine.21 Although routine liver enzyme testing is not required, discontinue atomoxetine if jaundice develops or elevated levels of liver enzymes are noted. Other rare but potentially serious side effects include changes in heart rate (≥20 beats per min) or blood pressure that occur in 5% to 10% of patients taking atomoxetine.22 The risk of serious cardiovascular events and sudden cardiac death with atomoxetine is extremely low, but patients should be screened for a personal and family history of cardiovascular risk factors and, if any of these are present, evaluated further before starting atomoxetine. Routine heart rate and blood pressure monitoring is recommended for all patients.12-14,16
Last, atomoxetine has been linked to growth delays in the first 1 or 2 years of treatment, with a return to expected measurements after an average 2 or 3 years of treatment; persistent decreases in growth rate were observed in patients who were taller or heavier than average before treatment.23
α2 Adrenergic agonists
Guanfacine ER and clonidine ER, the extended release (ER) formulations of α2 adrenergic agonists, were FDA-approved for treating ADHD in 2009 and 2010, respectively. Short-acting guanfacine and clonidine also are used for treating ADHD.24 Their mechanism of action involves stimulation of the pre-synaptic and post-synapic α2 adrenergic receptors, which control the release of norepinephrine and the rate of cell firing.25 The α2 agonists are considered a second-line treatment for ADHD because their efficacy and response rate for core ADHD symptoms lags behind those of psychostimulants.25 In addition to treating core ADHD symptoms, guanfacine and clonidine are used to treat tics and oppositional/aggressive behavior comorbid with ADHD.24,26 Clonidine, which is more sedating than guanfacine, can be used to treat comorbid ADHD and sleep disorders.24 The α2 agonists do not produce euphoria and do not have drug abuse potential.2Table 12,7-9 and Table 32,7,9 provide guidelines for prescribing guanfacine ER and clonidine ER.
The most common adverse effect is drowsiness; other common side effects include dizziness, irritability, headache, and abdominal pain.24 Short-term studies of α2 agonist treatment of ADHD have shown small, non-clinically significant reductions in heart rate and blood pressure; α2 agonist-associated bradycardia, increased QT interval, and cardiac arrhythmias have been reported,7,24,27 as well as rebound hypertension with abrupt discontinuation.24 Screen patients for a personal and family history of cardiovascular risk factors and, if present, evaluate further before initiating α2 agonists.
Combining ADHD medication classes
Combination therapy with >1 ADHD medications is employed when 1 class does not provide adequate symptom coverage or produces problematic side effects.8,24 Psychostimulants can be combined with low-dose atomoxetine (0.5 to 1.0 mg/kg/d) when atomoxetine does not adequately cover ADHD symptoms in school, or when psychostimulants do not adequately cover evening symptoms or patients experience problems with evening psychostimulant rebound.8 To date, prospective data on the safety and efficacy of combining atomoxetine and psychostimulants are limited, but what evidence is available suggests improved symptom control for some, but not all, patients, and a lack of serious adverse events.28
Psychostimulants have been combined with α2agonists when children have an inadequate response to psychostimulants alone, or in cases of ADHD comorbid with aggression or tics.24 Although early case reports raised concern about the safety of combining psychostimulants and α2 agonists, subsequent studies suggest that clonidine and guanfacine generally are well-tolerated when co-administered with psychostimulants.24,27,29
Case continued
Molly has derived substantial benefit from long-acting methylphenidate during the school day, but continues to have significant ADHD-related impairment in the mornings and evenings. Her physician tried afternoon dosing of immediate-release methylphenidate to address evening difficulties, but Molly experienced insomnia. It would be reasonable to consider adjunctive therapy with a non-stimulant medication. A medication that can provide round-the-clock ADHD symptom coverage—such as atomoxetine, guanfacine ER, or clonidine ER—could be added to her current day-time psychostimulant treatment, potentially improving her functioning at home before school and in the evenings.
Additional considerations
Combining medication and behavior therapy offers greater improvements on academic, conduct, and family satisfaction measures than either treatment alone.2 Clinicians can choose to employ behavior therapy alone, particularly if parents feel uncomfortable with—or children have not tolerated—medication.2,3 Evidence-based behavioral parent training and classroom management strategies (implemented by teachers) have shown the strongest and most consistent effects among nonpharmacotherapeutic interventions for ADHD.2 Most studies comparing behavior therapy to psychostimulants have found a stronger effect on core ADHD symptoms from psychostimulants than from behavior therapy.
When a patient does not respond adequately to FDA-approved ADHD medications alone or in combination, consider bupropion, an antidepressant with indirect dopamine and noradrenergic effects. Off-label bupropion has been shown to be effective for ADHD in controlled trials of both children and adults.30
Clinicians often encounter children who meet criteria for ADHD and an anxiety or mood disorder. Table 48,31 summarizes treatment recommendations for these patients.
Clinical considerations
- Begin treatment with a psychostimulant at a low dosage, and titrate gradually until symptoms are controlled or side effects develop.
- Keep in mind that an effective dosage of a psychostimulant is not closely correlated with age, weight, or severity of symptoms.
- Monitor refill requests and patient behavior for signs of psychostimulant misuse. Be alert for signs of illegal drug use in patient family members.
- Lisdexamfetamine, dermal methylphenidate, and osmotic release oral system methylphenidate are the formulations least likely to be misused because their delivery systems make it difficult to extract the active ingredient for snorting or intravenous injection.
- Psychostimulants have not been shown to exacerbate tics in most children who have comorbid ADHD and a tic disorder. When a stimulant is associated with an exacerbation of tics, switching treatment to atomoxetine or α2 agonists is reasonable.
- For patients whose use of a stimulant is limited by an adverse effect on sleep, consider atomoxetine and α2 adrenergic agonists as alternative or adjunctive treatments.
- All 3 classes of FDA-approved ADHD medications (psychostimulants, atomoxetine, and adrenergic agonists) have been associated with adverse cardiac events in children who have underlying cardiovascular conditions. Before initiating treatment, screen patients for a personal or family history of cardiovascular risk factors, and undertake further evaluation as indicated.
Bottom Line
In general, the evidence supports psychostimulants as initial pharmacotherapy for ADHD, with additional options including atomoxetine and α2 agonists. When one medication class does not provide adequate coverage for ADHD symptoms, combining medication classes can be beneficial.
Related Resources
- National Institute of Mental Health. What is attention deficit hyperactivity disorder (ADHD, ADD)?” www.nimh.nih.gov/health/topics/attention-deficit-hyperactivity-disorder-adhd/index.shtml.
- National Resource Center on AD/HD. Managing medication for children and adolescents with ADHD. www.help4adhd.org/en/treatment/medication/WWK3.
Drug Brand Names
Atomoxetine • Strattera
Lisdexamfetamine • Vyvanse
Bupropion • Wellbutrin, Zyban
Clonidine extended release • Kapvay
Guanfacine extended release • Intuniv
Dexmethylphenidate • Focalin, Focalin XR
Mixed amphetamine salts • Adderall, Adderall XR
Dextroamphetamine • Dexedrine, Dexedrine SR, DextroStat, ProCentra
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Disclosures
Dr. Froehlich receives support from the National Institute of Mental Health Grant K23 MH083881. Dr. Delgado has received research support from Pfizer, Inc. Dr. Anixt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Molly, age 9, is diagnosed with attention-deficit/hyperactivity disorder (ADHD) by her psychiatrist, who prescribes a long-acting methylphenidate formulation at 1 mg/kg. She tolerates the medication without side effects and shows significant improvement in her academic performance and on-task behavior in school. Molly takes methylphenidate before school at 7:00 am; this dose usually wears off at approximately 3:30 pm.
Molly and her parents are pleased with her response to methylphenidate, but report that she has difficulty getting ready for school because of distractibility. In the evenings Molly has trouble staying seated to do homework and often interrupts and argues with family members, but cannot tolerate afternoon dosing of immediate-release methylphenidate because of insomnia.
ADHD, the most common childhood neurobehavioral disorder, is characterized by difficulties with attention, impulse control, and modulating activity level. The pathophysiology of ADHD is thought to involve dysregulation of brain dopamine and norepinephrine systems.1 Managing ADHD includes pharmacotherapeutic and nonpharmacotherapeutic—ie, behavioral and psychoeducational—interventions.2,3
In this article, we provide an overview of the efficacy, side effects, and dosing for the 3 classes of ADHD medication—psychostimulants, atomoxetine, and α2 adrenergic agonists—including guidance on medication choice and combination treatment. We also discuss the effects of psychostimulants on tics, cardiovascular concerns, and substance abuse potential.
Psychostimulants
Methylphenidates and amphetamines are first-line agents for ADHD. Their primary mechanism of action involves blocking dopamine transporters, with additional effects including blockade of norepinephrine transporters, dampening action of monoamine oxidase (which slows dopamine and norepinephrine degradation), and enhanced release of dopamine into the synaptic space.1
Efficacy and response rates are similar for methylphenidate and amphetamine medications, although as many as 25% of patients may respond to only 1 agent.1 More than 90% of patients will have a positive response to one of the psychostimulants.1 The beneficial effects of psychostimulants on inattention, hyperactivity, and impulsivity are well documented.2Improvements in noncompliance, aggression, social interactions, and academic productivity also have been observed.4,5
Because of increased recognition of pervasive ADHD-related impairments, which can affect functioning in social, family, and extracurricular settings, practitioners have shifted to long-acting psychostimulants to reduce the need for in-school dosing, improve compliance, and obtain more after-school treatment effects. Long-acting formulations produce a slower rise and fall of psychostimulant levels in the brain, which may decrease side effects and potential for later drug abuse.6 See Table 12,7-9 and Table 22,7,9 for titration, dosing, and duration of action of psychostimulants.
The most common side effects of psychostimulants are appetite loss, abdominal pain, headaches, and sleep disturbances.2 Emotional symptoms—irritability and nervousness—may be observed with psychostimulant use, but these behaviors may improve, rather than become worse, with treatment.5 Methylphenidates and amphetamines share many of the same side effects,2 with many studies indicating no differences between their side-effect profiles.1 Other studies indicate that sleep and emotional side effects may be more prominent with amphetamines than methylphenidates,10 although response varies by individual.
There is little evidence that methylphenidate, low-dose amphetamine, or low-dose dextroamphetamine makes tics worse in most children who have them, although significant tic exacerbation has been observed with higher-dose dextroamphetamine.11,12 In patients with comorbid ADHD and tic disorders, a trial of psychostimulants with monitoring for worsening tics is appropriate.
Changes in heart rate and blood pressure generally are not clinically significant in patients taking psychostimulants (average increases: 1 or 2 beats per minute and 1 to 4 mm Hg for systolic and diastolic blood pressures).12 However, psychostimulants may be associated with more substantial increases in heart rate and blood pressure in a subset of individuals (5% to 15%).12 Large studies of children and adults in the general population have not found an association between psychostimulant use and severe cardiovascular events (sudden cardiac death, myocardial infarction, stroke).12-14 Because of reports of sudden cardiac death in children with underlying heart disease who take a psychostimulant,15 clinicians are advised to screen patients and consider an electrocardiogram or evaluation by a cardiologist before starting a psychostimulant in a patient who has a personal or family history of specific cardiovascular risk factors (see Perrin et al16 and Cortese et al12 for screening questions and conditions).
Modest reductions in height (1 or 2 cm after 3 years of psychostimulant treatment) appear to be dose-dependent, and are similar across the methylphenidate and amphetamine classes. Some studies have shown reversal of growth deficits after treatment is stopped treatment and no adverse effects on final adult height.12,17 More study is needed to clarify the effects of continuous psychostimulant treatment from childhood to adulthood on growth.
Studies have failed to show an increased risk of substance abuse in persons with ADHD who were treated with psychostimulants during childhood. Some studies document a lower rate of later substance abuse in youths who received ADHD medications, although other reports show no effect of psychostimulant treatment on subsequent substance use disorder risk.12 Be aware that psychostimulants can be misused (eg, to get “high,” for performance enhancement, to suppress appetite, etc.). Misuse of psychostimulants is most common with short-acting preparations, and generally more difficult with long-acting preparations because extracting the active ingredients for snorting is difficult.2,12 Monitor refill requests and patient behavior for signs of misuse, and be alert for signs of illegal drug use in the patient’s family.
Psychotic symptoms—including hallucinations, delusions, mania, and extreme agitation—with psychostimulant treatment are rare, occurring at a rate of 1.5%.12
Atomoxetine
Approved by the FDA in 2002 for ADHD, atomoxetine is effective and generally well tolerated, although it is not as effective as psychostimulants.2 Atomoxetine is a potent norepinephrine reuptake inhibitor18 that does not produce euphoria, does not have potential for abuse, and has not been linked to increased tic onset or severity.19 Atomoxetine treatment is associated with a lower rate of sleep initiation difficulty compared with psychostimulants.18 Some studies suggest that atomoxetine may have mild beneficial effects on anxiety disorders,18 making it a reasonable choice for patients with significant anxiety or insomnia during psychostimulant treatment. Table 12,7-9 and Table 32,7,9 include information on dosing and duration of action for atomoxetine.
Common side effects of atomoxetine include sedation and fatigue, upset stomach, nausea and vomiting, reduced appetite, headache, and irritability.18 Inform patients that atomoxetine carries an FDA black-box warning for suicide risk; a review of 14 studies showed suicidal ideation was more common with atomoxetine than placebo, although no suicides occurred in any trials.20
Hepatotoxicity is rare with atomoxetine.21 Although routine liver enzyme testing is not required, discontinue atomoxetine if jaundice develops or elevated levels of liver enzymes are noted. Other rare but potentially serious side effects include changes in heart rate (≥20 beats per min) or blood pressure that occur in 5% to 10% of patients taking atomoxetine.22 The risk of serious cardiovascular events and sudden cardiac death with atomoxetine is extremely low, but patients should be screened for a personal and family history of cardiovascular risk factors and, if any of these are present, evaluated further before starting atomoxetine. Routine heart rate and blood pressure monitoring is recommended for all patients.12-14,16
Last, atomoxetine has been linked to growth delays in the first 1 or 2 years of treatment, with a return to expected measurements after an average 2 or 3 years of treatment; persistent decreases in growth rate were observed in patients who were taller or heavier than average before treatment.23
α2 Adrenergic agonists
Guanfacine ER and clonidine ER, the extended release (ER) formulations of α2 adrenergic agonists, were FDA-approved for treating ADHD in 2009 and 2010, respectively. Short-acting guanfacine and clonidine also are used for treating ADHD.24 Their mechanism of action involves stimulation of the pre-synaptic and post-synapic α2 adrenergic receptors, which control the release of norepinephrine and the rate of cell firing.25 The α2 agonists are considered a second-line treatment for ADHD because their efficacy and response rate for core ADHD symptoms lags behind those of psychostimulants.25 In addition to treating core ADHD symptoms, guanfacine and clonidine are used to treat tics and oppositional/aggressive behavior comorbid with ADHD.24,26 Clonidine, which is more sedating than guanfacine, can be used to treat comorbid ADHD and sleep disorders.24 The α2 agonists do not produce euphoria and do not have drug abuse potential.2Table 12,7-9 and Table 32,7,9 provide guidelines for prescribing guanfacine ER and clonidine ER.
The most common adverse effect is drowsiness; other common side effects include dizziness, irritability, headache, and abdominal pain.24 Short-term studies of α2 agonist treatment of ADHD have shown small, non-clinically significant reductions in heart rate and blood pressure; α2 agonist-associated bradycardia, increased QT interval, and cardiac arrhythmias have been reported,7,24,27 as well as rebound hypertension with abrupt discontinuation.24 Screen patients for a personal and family history of cardiovascular risk factors and, if present, evaluate further before initiating α2 agonists.
Combining ADHD medication classes
Combination therapy with >1 ADHD medications is employed when 1 class does not provide adequate symptom coverage or produces problematic side effects.8,24 Psychostimulants can be combined with low-dose atomoxetine (0.5 to 1.0 mg/kg/d) when atomoxetine does not adequately cover ADHD symptoms in school, or when psychostimulants do not adequately cover evening symptoms or patients experience problems with evening psychostimulant rebound.8 To date, prospective data on the safety and efficacy of combining atomoxetine and psychostimulants are limited, but what evidence is available suggests improved symptom control for some, but not all, patients, and a lack of serious adverse events.28
Psychostimulants have been combined with α2agonists when children have an inadequate response to psychostimulants alone, or in cases of ADHD comorbid with aggression or tics.24 Although early case reports raised concern about the safety of combining psychostimulants and α2 agonists, subsequent studies suggest that clonidine and guanfacine generally are well-tolerated when co-administered with psychostimulants.24,27,29
Case continued
Molly has derived substantial benefit from long-acting methylphenidate during the school day, but continues to have significant ADHD-related impairment in the mornings and evenings. Her physician tried afternoon dosing of immediate-release methylphenidate to address evening difficulties, but Molly experienced insomnia. It would be reasonable to consider adjunctive therapy with a non-stimulant medication. A medication that can provide round-the-clock ADHD symptom coverage—such as atomoxetine, guanfacine ER, or clonidine ER—could be added to her current day-time psychostimulant treatment, potentially improving her functioning at home before school and in the evenings.
Additional considerations
Combining medication and behavior therapy offers greater improvements on academic, conduct, and family satisfaction measures than either treatment alone.2 Clinicians can choose to employ behavior therapy alone, particularly if parents feel uncomfortable with—or children have not tolerated—medication.2,3 Evidence-based behavioral parent training and classroom management strategies (implemented by teachers) have shown the strongest and most consistent effects among nonpharmacotherapeutic interventions for ADHD.2 Most studies comparing behavior therapy to psychostimulants have found a stronger effect on core ADHD symptoms from psychostimulants than from behavior therapy.
When a patient does not respond adequately to FDA-approved ADHD medications alone or in combination, consider bupropion, an antidepressant with indirect dopamine and noradrenergic effects. Off-label bupropion has been shown to be effective for ADHD in controlled trials of both children and adults.30
Clinicians often encounter children who meet criteria for ADHD and an anxiety or mood disorder. Table 48,31 summarizes treatment recommendations for these patients.
Clinical considerations
- Begin treatment with a psychostimulant at a low dosage, and titrate gradually until symptoms are controlled or side effects develop.
- Keep in mind that an effective dosage of a psychostimulant is not closely correlated with age, weight, or severity of symptoms.
- Monitor refill requests and patient behavior for signs of psychostimulant misuse. Be alert for signs of illegal drug use in patient family members.
- Lisdexamfetamine, dermal methylphenidate, and osmotic release oral system methylphenidate are the formulations least likely to be misused because their delivery systems make it difficult to extract the active ingredient for snorting or intravenous injection.
- Psychostimulants have not been shown to exacerbate tics in most children who have comorbid ADHD and a tic disorder. When a stimulant is associated with an exacerbation of tics, switching treatment to atomoxetine or α2 agonists is reasonable.
- For patients whose use of a stimulant is limited by an adverse effect on sleep, consider atomoxetine and α2 adrenergic agonists as alternative or adjunctive treatments.
- All 3 classes of FDA-approved ADHD medications (psychostimulants, atomoxetine, and adrenergic agonists) have been associated with adverse cardiac events in children who have underlying cardiovascular conditions. Before initiating treatment, screen patients for a personal or family history of cardiovascular risk factors, and undertake further evaluation as indicated.
Bottom Line
In general, the evidence supports psychostimulants as initial pharmacotherapy for ADHD, with additional options including atomoxetine and α2 agonists. When one medication class does not provide adequate coverage for ADHD symptoms, combining medication classes can be beneficial.
Related Resources
- National Institute of Mental Health. What is attention deficit hyperactivity disorder (ADHD, ADD)?” www.nimh.nih.gov/health/topics/attention-deficit-hyperactivity-disorder-adhd/index.shtml.
- National Resource Center on AD/HD. Managing medication for children and adolescents with ADHD. www.help4adhd.org/en/treatment/medication/WWK3.
Drug Brand Names
Atomoxetine • Strattera
Lisdexamfetamine • Vyvanse
Bupropion • Wellbutrin, Zyban
Clonidine extended release • Kapvay
Guanfacine extended release • Intuniv
Dexmethylphenidate • Focalin, Focalin XR
Mixed amphetamine salts • Adderall, Adderall XR
Dextroamphetamine • Dexedrine, Dexedrine SR, DextroStat, ProCentra
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Disclosures
Dr. Froehlich receives support from the National Institute of Mental Health Grant K23 MH083881. Dr. Delgado has received research support from Pfizer, Inc. Dr. Anixt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998; 94(1):127-152.
2. Subcommittee on Attention-Deficit/Hyperactivity Disorder; Steering Committee on Quality Improvement and Management; Wolraich M, Brown L, Brown RT, et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007-1022.
3. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
4. Zametkin AJ, Ernst M. Problems in the management of attention-deficit-hyperactivity disorder. N Engl J Med. 1999;340(1):40-46.
5. Goldman LS, Genel M, Bezman RJ, et al. Diagnosis and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Council on Scientific Affairs, American Medical Association. JAMA. 1998;279(14):1100-1107.
6. Swanson J, Gupta S, Lam A, et al. Development of a new once-a-day formulation of methylphenidate for the treatment of attention-deficit/hyperactivity disorder: proof-of-concept and proof-of-product studies. Arch Gen Psychiatry. 2003;60(2):204-211.
7. Vaughan B, Kratochvil CJ. Pharmacotherapy of pediatric attention-deficit/hyperactivity disorder. Child Adolesc Psychiatr Clin N Am. 2012;21(4):941-955.
8. Pliszka SR, Crismon ML, Hughes CW, et al; Texas Consensus Conference Panel on Pharmacotherapy of Childhood Attention Deficit Hyperactivity Disorder. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2006;45(6):642-657.
9. Antshel KM, Hargrave TM, Simonescu M, et al. Advances in understanding and treating ADHD. BMC Med. 2011;9:72.
10. Efron D, Jarman F, Barker M. Side effects of methylphenidate and dexamphetamine in children with attention deficit hyperactivity disorder: a double-blind, crossover trial. Pediatrics. 1997;100(4):662-666.
11. Pringsheim T, Steeves T. Pharmacological treatment for attention deficit hyperactivity disorder (ADHD) in children with comorbid tic disorders. Cochrane Database Syst Rev. 2011(4):CD007990.
12. Cortese S, Holtmann M, Banaschewski T, et al. Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry. 2013; 54(3):227-246.
13. Cooper WO, Habel LA, Sox CM, et al. ADHD drugs and serious cardiovascular events in children and young adults. N Engl J Med. 2011;365(20):1896-1904.
14. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
15. Vetter VL, Elia J, Erickson C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing. Circulation. 2008;117(18):2407-2423.
16. Perrin JM, Friedman RA, Knilans TK; Black Box Working Group; Section on Cardiology and Cardiac Surgery. Cardiovascular monitoring and stimulant drugs for attention-deficit/hyperactivity disorder. Pediatrics. 2008;122(2):451-453.
17. Faraone SV, Biederman J, Morley CP, et al. Effect of stimulants on height and weight: a review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47(9):994-1009.
18. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
19. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
20. Bangs ME, Tauscher-Wisniewski S, Polzer J, et al. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J Am Acad Child Adolesc Psychiatry. 2008;47(2):209-218.
21. Bangs ME, Jin L, Zhang S, et al. Hepatic events associated with atomoxetine treatment for attention-deficit hyperactivity disorder. Drug Saf. 2008;31(4):345-354.
22. U.S. Food and Drug Administration. Strattera (atomoxetine hydrochloride) capsule. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm223889.htm. Published August 2013. Accessed October 31, 2013.
23. Spencer TJ, Kratochvil CJ, Sangal RB, et al. Effects of atomoxetine on growth in children with attention-deficit/hyperactivity disorder following up to five years of treatment. J Child Adolesc Psychopharmacol. 2007;17(5):689-700.
24. Connor DF. Other medications. In: Barkley RA, ed. Attention-deficit/hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:658-677.
25. May DE, Kratochvil CJ. Attention-deficit hyperactivity disorder: recent advances in paediatric pharmacotherapy. Drugs. 2010;70(1):15-40.
26. Connor DF, Findling RL, Kollins SH, et al. Effects of guanfacine extended release on oppositional symptoms in children aged 6-12 years with attention-deficit hyperactivity disorder and oppositional symptoms: a randomized, double-blind, placebo-controlled trial. CNS Drugs. 2010; 24(9):755-768.
27. Croxtall JD. Clonidine extended-release: in attention-deficit hyperactivity disorder. Paediatr Drugs. 2011;13(5):329-336.
28. Treuer T, Gau SS, Mendez L, et al. A systematic review of combination therapy with stimulants and atomoxetine for attention-deficit/hyperactivity disorder, including patient characteristics, treatment strategies, effectiveness, and tolerability. J Child Adolesc Psychopharmacol. 2013;23(3):179-193.
29. Sallee FR. The role of alpha2-adrenergic agonists in attention-deficit/hyperactivity disorder. Postgrad Med. 2010;122(5):78-87.
30. Spencer TJ. Antidepressant and specific norepinephrine reuptake inhibitor treatments. In: Barkley RA, ed. Attention-deficit hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:648-657.
31. Singh MK, DelBello MP, Kowatch RA, et al. Co-occurrence of bipolar and attention-deficit hyperactivity disorders in children. Bipolar Disord. 2006;8(6):710-720.
1. Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998; 94(1):127-152.
2. Subcommittee on Attention-Deficit/Hyperactivity Disorder; Steering Committee on Quality Improvement and Management; Wolraich M, Brown L, Brown RT, et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007-1022.
3. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
4. Zametkin AJ, Ernst M. Problems in the management of attention-deficit-hyperactivity disorder. N Engl J Med. 1999;340(1):40-46.
5. Goldman LS, Genel M, Bezman RJ, et al. Diagnosis and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Council on Scientific Affairs, American Medical Association. JAMA. 1998;279(14):1100-1107.
6. Swanson J, Gupta S, Lam A, et al. Development of a new once-a-day formulation of methylphenidate for the treatment of attention-deficit/hyperactivity disorder: proof-of-concept and proof-of-product studies. Arch Gen Psychiatry. 2003;60(2):204-211.
7. Vaughan B, Kratochvil CJ. Pharmacotherapy of pediatric attention-deficit/hyperactivity disorder. Child Adolesc Psychiatr Clin N Am. 2012;21(4):941-955.
8. Pliszka SR, Crismon ML, Hughes CW, et al; Texas Consensus Conference Panel on Pharmacotherapy of Childhood Attention Deficit Hyperactivity Disorder. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2006;45(6):642-657.
9. Antshel KM, Hargrave TM, Simonescu M, et al. Advances in understanding and treating ADHD. BMC Med. 2011;9:72.
10. Efron D, Jarman F, Barker M. Side effects of methylphenidate and dexamphetamine in children with attention deficit hyperactivity disorder: a double-blind, crossover trial. Pediatrics. 1997;100(4):662-666.
11. Pringsheim T, Steeves T. Pharmacological treatment for attention deficit hyperactivity disorder (ADHD) in children with comorbid tic disorders. Cochrane Database Syst Rev. 2011(4):CD007990.
12. Cortese S, Holtmann M, Banaschewski T, et al. Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry. 2013; 54(3):227-246.
13. Cooper WO, Habel LA, Sox CM, et al. ADHD drugs and serious cardiovascular events in children and young adults. N Engl J Med. 2011;365(20):1896-1904.
14. Martinez-Raga J, Knecht C, Szerman N, et al. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15-30.
15. Vetter VL, Elia J, Erickson C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing. Circulation. 2008;117(18):2407-2423.
16. Perrin JM, Friedman RA, Knilans TK; Black Box Working Group; Section on Cardiology and Cardiac Surgery. Cardiovascular monitoring and stimulant drugs for attention-deficit/hyperactivity disorder. Pediatrics. 2008;122(2):451-453.
17. Faraone SV, Biederman J, Morley CP, et al. Effect of stimulants on height and weight: a review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47(9):994-1009.
18. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11(3):203-226.
19. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
20. Bangs ME, Tauscher-Wisniewski S, Polzer J, et al. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J Am Acad Child Adolesc Psychiatry. 2008;47(2):209-218.
21. Bangs ME, Jin L, Zhang S, et al. Hepatic events associated with atomoxetine treatment for attention-deficit hyperactivity disorder. Drug Saf. 2008;31(4):345-354.
22. U.S. Food and Drug Administration. Strattera (atomoxetine hydrochloride) capsule. http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm223889.htm. Published August 2013. Accessed October 31, 2013.
23. Spencer TJ, Kratochvil CJ, Sangal RB, et al. Effects of atomoxetine on growth in children with attention-deficit/hyperactivity disorder following up to five years of treatment. J Child Adolesc Psychopharmacol. 2007;17(5):689-700.
24. Connor DF. Other medications. In: Barkley RA, ed. Attention-deficit/hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:658-677.
25. May DE, Kratochvil CJ. Attention-deficit hyperactivity disorder: recent advances in paediatric pharmacotherapy. Drugs. 2010;70(1):15-40.
26. Connor DF, Findling RL, Kollins SH, et al. Effects of guanfacine extended release on oppositional symptoms in children aged 6-12 years with attention-deficit hyperactivity disorder and oppositional symptoms: a randomized, double-blind, placebo-controlled trial. CNS Drugs. 2010; 24(9):755-768.
27. Croxtall JD. Clonidine extended-release: in attention-deficit hyperactivity disorder. Paediatr Drugs. 2011;13(5):329-336.
28. Treuer T, Gau SS, Mendez L, et al. A systematic review of combination therapy with stimulants and atomoxetine for attention-deficit/hyperactivity disorder, including patient characteristics, treatment strategies, effectiveness, and tolerability. J Child Adolesc Psychopharmacol. 2013;23(3):179-193.
29. Sallee FR. The role of alpha2-adrenergic agonists in attention-deficit/hyperactivity disorder. Postgrad Med. 2010;122(5):78-87.
30. Spencer TJ. Antidepressant and specific norepinephrine reuptake inhibitor treatments. In: Barkley RA, ed. Attention-deficit hyperactivity disorder: a handbook for diagnosis and treatment. 3rd ed. New York, NY: The Guilford Press; 2006:648-657.
31. Singh MK, DelBello MP, Kowatch RA, et al. Co-occurrence of bipolar and attention-deficit hyperactivity disorders in children. Bipolar Disord. 2006;8(6):710-720.
