User login
Flibanserin is poised for FDA approval. But why this drug? And why now?
Women’s sexual health took a step forward last month when an advisory panel to the US Food and Drug Administration (FDA) recommended approval of the drug flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in premenopausal women. The approval came with some reservations regarding safety (use with certain medications and alcohol). And it’s worthwhile to note that the FDA had on hand its own Drug Safety and Risk Management Committee during deliberations. However, assuming the agency follows the recommendations of the Bone, Reproductive, and Urologic Drugs Advisory Committee, women will soon have available the first agent for sexual dysfunction—aside from a medication for intercourse-associated pain—developed specifically for them.
Had the panel voted down the approval, it would have set a dangerous precedent that likely would have led to a standstill in new drug development in all therapeutic classes of women’s sexual health for the next decade.
Why do I say that—and state it so emphatically?
To answer that question, let’s review the approval process for flibanserin, as well as for the 2 testosterone products that preceded its appearance before the FDA.

Hypoactive sexual desire disorder (HSDD) is the most common sexual dysfunction in women. Few interventions have proven to be effective for the treatment of HSDD. Education, counseling, and psychotherapy may be helpful in some women. Exogenous testosterone has been reported to be effective in the treatment of low sexual desire in postmenopausal women taking estrogen, but this treatment is not approved by the US Food and Drug Administration (FDA), and the long-term safety of exogenous testosterone in women is not established. Clinicians who treat women with sexual desire disorders are frustrated by their inability to prescribe an effective treatment for this common problem. An FDA advisory panel recently voted to support the approval of flibanserin for the treatment of HSDD in premenopausal women. In this month’s editorial, Dr. James Simon provides a history of the FDA review process for medications designed to treat HSDD, including the decision to not approve testosterone and the vote of the FDA advisory panel to support the approval of flibanserin. Readers of OBG Management should be aware that Dr. Simon, as is apparent in this piece and fully disclosed, has served as Medical Director and an advisor to Sprout Pharmaceuticals, the company with the rights to flibanserin. As editors we have concluded that Dr. Simon’s unique perspective, knowledge, and insights about the history of the FDA review of treatments of HSDD, including testosterone, would be of great interest to our readers.
—Robert L. Barbieri, MD, Editor in Chief
—Lila O’Connor, Editor
A tale of 2 products: The testosterone story
In 2004, Procter & Gamble filed a new drug application (NDA) for a testosterone patch (Intrinsa) developed for the treatment of female sexual dysfunction. Specifically, the patch was created for the treatment of HSDD in surgically menopausal women (those who had undergone bilateral oophorectomy) who were receiving concomitant estrogen therapy.
Both the FDA and the advisory committee considering the NDA agreed that the patch was effective. The question was whether its efficacy outweighed its risks. Recall that this discussion was taking place only 2 years after the initial publication of findings from the Women’s Health Initiative (WHI) estrogen-progestin arm. That arm had been halted prematurely because the risk of breast cancer exceeded the prespecified stopping boundary. In addition, the global index summarizing the balance of risks and benefits showed that risks outweighed benefits in regard to breast cancer, coronary heart disease, stroke, and pulmonary embolism.1 As a result, the safety of all forms of hormone therapy was being closely scrutinized.
The FDA was also on “high alert” for safety as rofecoxib (Vioxx) had just been removed from the market due to unforeseen risks, with much media fanfare and large numbers of lawsuits.
After consideration of the NDA for the testosterone patch, the FDA advised Procter & Gamble to undertake an adequately designed and powered safety study to confirm that it would not increase the risks of coronary heart disease or breast cancer among users, since testosterone can be converted to estradiol in women.
Procter & Gamble ultimately withdrew its NDA. Such a safety study would have taken an additional 5 years to complete and cost upwards of $300 million. And because testosterone is not a patentable compound, a study that long and costly didn’t seem like a smart business proposition. Shortly after the NDA was withdrawn, the European Medicines Agency—the European counterpart of the FDA—approved the Intrinsa testosterone patch for the same indication as proposed in the United States.
Next up, BioSante Pharmaceuticals filed its NDA for LibiGel, a testosterone gel formulated specifically for postmenopausal women with HSDD. In its efficacy study, LibiGel failed to demonstrate superiority above and beyond placebo. The manufacturer, which was concurrently conducting a large, long-term safety study to satisfy the FDA’s concerns, ran out of money shortly thereafter, and that was the end of that.
Flibanserin’s focus: premenopausal women
In contrast to the 2 testosterone products, flibanserin was developed for premenopausal women. Although preliminary data on flibanserin use among postmenopausal women are available,2 the drug was studied primarily in premenopausal women with HSDD, the indication sought at this time.
In the premenopausal population, problems such as pain with intercourse or hypoestrogenism aren’t typically present, simplifying the identification of HSDD. (See the sidebar below, “What is HSDD and how is it diagnosed?”) In clinical trials of the drug, HSDD was secondary, generalized, and acquired—that is, it followed a period of normal sexual function. And it didn’t come and go but was present regardless of location and circumstance. Study participants had had a normal sex drive before their desire “turned off,” an occurrence they found distressing.3–6
What is hypoactive sexual desire disorderand how is it diagnosed?
In the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), hypoactive sexual desire disorder (HSDD) is described as having the following characteristics:
- persistently or recurrently deficient (or absent) sexual fantasies and desire for sexual activity
- marked distress or interpersonal difficulty in response to this deficiency
- lack of another explanation, such as another Axis I disorder or use of a substance known to affect sexual function.
In the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), published in 2013, HSDD was folded with female sexual arousal disorder into a new diagnosis, female sexual interest and arousal disorder. This is somewhat confusing in that the physiologies of these 2 disorders are quite separate and distinct.
In clinical practice, HSDD is easily identified using the Decreased Sexual Desire Screener (DSDS), a simple screening test that asks 4 yes/no questions:
- In the past, was your level of sexual desire or interest good and satisfying to you?
- Has there been a decrease in your level of sexual desire or interest?
- Are you bothered by your decreased level of sexual desire or interest?
- Would you like your level of sexual desire or interest to increase?
A “yes” response to each of these questions is required. In addition, a fifth question asks whether a number of conditions, drugs, or circumstances might be responsible for the decreased desire or interest:
- an operation, depression, injuries, or other medical condition
- medications, drugs, or alcohol you are currently taking
- pregnancy, recent childbirth, or menopausal symptoms
- other sexual issues you may be having (pain, decreased arousal or orgasm)
- your partner’s sexual problems
- dissatisfaction with your relationship or partner
- stress or fatigue.
Boehringer Ingelheim, a German concern, developed flibanserin and filed the initial NDA in 2009. In clinical trials, that company ran into problems because the electronic diary it was using to measure desire failed to demonstrate efficacy for flibanserin. It turns out that, if you ask women who are distressed about having low desire to report their level of desire every single day, they quickly get turned off by the question and eventually stop answering altogether. Such changes in behavior play havoc with the validity of the instrument to assess desire.
After flibanserin failed the electronic diary desire domain—one of the study’s endpoints—the company substituted a different measure, the Female Sexual Function Index (FSFI) desire domain. Although the FSFI showed statistically significantly greater efficacy for flibanserin than placebo, the FDA argued that it was unreasonable for the company to change the rules to fit the outcome. For that and other reasons, it turned down the NDA.
In response, Boehringer Ingelheim went back to the drawing board and undertook a new study intended to achieve several goals:
- substitute the FSFI desire score for the electronic diary desire score
- reduce the number of restricted medications to see if the results could be more generalizable to the normal population of women with HSDD
- determine whether there were any safety signals for drug-drug interaction that weren’t apparent in the first 3 trials, in which a large number of medications had been excluded.
About the time this study was drawing to a conclusion, the company got cold feet and decided to shelve its plans for the drug.
Sprout steps in
I was among the delegation of medical and pharmaceutical professionals who visited Boehringer Ingelheim in 2011 to explore the possibility of Sprout Pharmaceuticals acquiring flibanserin. Boehringer Ingelheim agreed to the deal, and Sprout took over drug development, resubmitting the NDA to the FDA in 2013 with the additional study and other data. The FDA again denied the application. In response, Sprout filed a request for a dispute resolution, a formal procedure convened when the sponsor of an NDA cannot reach agreement with the FDA. In the course of this procedure, the FDA asked for additional analyses, as well as some pharmacogenomics and a driving study.
Around this time, the FDA had determined that the sleep aid zolpidem (Ambien) is metabolized differently in women than in men and that, in some of these women, there is a significant cognitive deficit carried over to the next day when the drug is taken as prescribed at bedtime. Because flibanserin came on the heels of this determination and was known to cause drowsiness, the FDA requested the driving study. It also requested a drug-drug interaction study to determine whether flibanserin is metabolized differently in some women with genetically different medication metabolism.
Sprout conducted those studies, both of which came back “clean.” Armed with this new data, the FDA scheduled a meeting of its advisory committee on June 4, 2015. And the rest, as they say, is history.
Flibanserin vs placebo
During the advisory committee’s deliberations on June 4, discussion focused, in part, on how flibanserin performed in comparison with placebo. It was noted that flibanserin increased the number of satisfying sexual events (SSEs) by only 1 per month, compared with placebo. But that conclusion doesn’t accurately convey the findings of the efficacy studies.
First, even without flibanserin, women in the trials reported that they continued to have sex with their partners 2 to 3 times per month. That established a baseline number of SSEs of approximately 2.5. The consent form for the flibanserin trial contained a clause stating that the woman would agree to try and have sex at least 1 additional time per month. This requirement, independent of any treatment, was bound to increase the placebo effect because, regardless of the drug given (flibanserin or placebo), the participant was going to try to have sex at least 1 more time per month.
In the flibanserin trials, the placebo effect was 1.5 additional SSEs per month. Add that to the baseline number of SSEs and you have a total of 4 SSEs per month. Among flibanserin users, the number of SSEs per month was about 5. And even though that’s only 1 more time per month than the placebo group, those 5 events were more desired events. That means that the baseline of 2.5 SSEs, among flibanserin users, had a different character by the end of the study period.
There is also a ceiling effect in play. Consider that the participants in the flibanserin trials were women who had been married an average of 10 years, with an average age of approximately 36 years. How much more sex is likely even possible in this population?
This isn’t to say that women are incapable of having more sex. It is merely a reflection of the data, which show that, among married women aged 30 to 39 years, only roughly 25% have sex more often than weekly, and only 5.1% have sex 4 or more times per week.7 If women were shown to be having sex more than 5 times per month, a likely criticism would have been that the drug was making them hypersexual or even abnormal.
Also keep in mind that the drug doesn’t work in every woman, just as antidepressants are not effective in every person. So when the responders and nonresponders were lumped together, the magnitude of the drug’s response in the combined group was smaller. In reality, approximately 25% of women in the flibanserin trials experienced an increase of 4 or more SSEs per month, compared with 15% among placebo users.
Flibanserin dosing and side effects
Flibanserin is indicated for the treatment of hypoactive sexual desire disorder in premenopausal women. It is taken daily in a 100-mg tablet. Bedtime dosing is preferred to mitigate potential side effects such as drowsiness, hypotension, and syncope. These effects can occur with flibanserin alone, in combination with certain drugs, or in combination with alcohol.
Significant drug-drug interactions have been documented for flibanserin in combination with moderate and strong CYP3A4 inhibitors such as fluconazole and ketoconazole. Package labeling for flibanserin will detail this risk.
Why now?
As I stated earlier, a failure to approve flibanserin would set a dangerous precedent. Why? Because the company did everything the FDA asked it to do, and the results came out statistically significantly better than placebo—which was the desired endpoint. If the FDA were to deny approval of the drug, it would be saying, in effect, that it can change its mind in the middle of the argument—something it faulted Boehringer Ingelheim for in earlier deliberations (switching the insensitive electronic diary for the statistically significant FSFI).
In reality, the FDA is likely to say yes to approval, but with restrictions, as that is what its advisory committee recommended. What those restrictions will be remains to be determined, but they are likely to resemble those of other drugs in the class, such as selective serotonin reuptake inhibitors (SSRIs), including a warning to be careful using flibanserin with alcohol until the drug’s effects are clear.
Opposition to flibanserin misses the mark
During the public hearing portion of the advisory committee meeting, most of the testimony came from women seeking approval of the drug. However, there were some naysayers. Their arguments against approval boiled down to 4 perspectives:
- the view that development of flibanserin represents “medicalization” of a disorder that can be treated effectively with psychotherapy and education. This perspective is best embodied by an organization called the New View Campaign. Refuting this perspective, however, is research in animal models that clearly demonstrates that HSDD (or its equivalent in animals) is the result of an imbalance between dopamine and norepinephrine on the positive end and serotonin on the negative end. These findings are supported by functional magnetic resonance imaging (MRI) and positron emission tomography (PET) scans of the brains of women with HSDD who are shown erotic stimuli.8,9 The scans demonstrate that their brains respond differently from those of normal women. So if it’s all about education and counseling, why are the brains of women with HSDD functioning differently? I would argue that, if depression and HSDD are both abnormalities of the serotonergic system (flibanserin is essentially an SSRI), then how can depression be a biologically based disorder but HSDD can’t? In my opinion, the New View Campaign isn’t new at all.
- the view, represented by an organization called PharmedOUT, that marketing by pharmaceutical companies overly influences prescribers, ultimately medicalizing problems that don’t require medication or overselling medications for problems that may require drug treatment for a short time only. This organization is headed by an academic physician who has not seen patients in many years and has never treated women for HSDD.
- the view, represented by the Public Citizen Health Research Group, that the safety profile of flibanserin is lacking. This organization argues not just against flibanserin but against pretty much any drug. In its view, there are never enough safety data. I would argue that, when it comes to flibanserin, there are more safety and efficacy data than there are for almost any other women’s health drug. Most drug companies test their medications in 1,500 to 2,000 people, as the FDA requires. Sprout Pharmaceuticals tested flibanserin in almost 8,000 women. The total number of individuals who have been studied, in fact, exceeds 11,000.
- the view, represented by the National Women’s Health Network, that the drug’s risks outweigh its modest benefits. As I have pointed out, however, the benefits of flibanserin have been downplayed in data analysis, and the body of safety data for the drug is substantial.
There is also the sociological view that HSDD is a normal variant of healthy sexual function. Its adherents argue that women with HSDD feel distress because their male partners are forcing them to feel inadequate. But I have yet to hear a single critical voice from a physician who actually treats women and who can prescribe drugs. The opposition to flibanserin, such as it is, flows from people who don’t see patients and can’t prescribe medications.
These naysayers are negative in a theoretical vacuum. It’s very easy to fall into that trap when you don’t have to look across the consultation desk to a patient who is asking you for a remedy—a woman who’s been suffering for 25 years, say—and have to tell her you have nothing to offer. You might mention testosterone, explaining that it was approved for men but you’ll try to prescribe an appropriate dose. But be sure to include discussion of its many side effects.
A long and winding road
Flibanserin’s journey from inception to probable approval has been long and eventful, and you can be sure that the pharmaceutical industry has been paying attention. Hundreds of millions of dollars in funding for drug development hang in the balance. That women deserve remedies developed specifically for their needs and metabolism is a given. The approval of flibanserin will send a hopeful signal to them as well as to industry—that the FDA takes them seriously and seeks to identify effective remedies. In this case, it seems likely, the agency will end up on the right side of history.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333.
2. Simon JA, Kingsberg SA, Shumel B, Hanes V, Garcia M Jr, Sand M. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21(6):633–640.
3. DeRogatis LR, Komer L, Katz M, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074–1085.
4. Jayne C, Simon JA, Taylor LV, Kimura T, Lesko L; SUNFLOWER study investigators. Open-label extension study of flibanserin in women with hypoactive sexual desire disorder. J Sex Med. 2012;9(12):3180–3188.
5. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793–804.
6. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807–1815.
7. Kinsey Institute. Percentage of Women Reporting Frequency of Vaginal Sex, N = 2,393. http://www.kinseyinstitute.org/resources/FAQ.html. Accessed June 17, 2015.
8. Arnow BA, Millheiser L, Garrett A, et al. Women with hypoactive sexual desire disorder compared to normal females: a functional magnetic resonance imaging study. Neuroscience. 2009;158(2):484–502.
9. Bianchi-Demicheli F, Cojan Y, et al. Neural bases of hypoactive sexual desire disorder in women: an event-related FMRI study. J Sex Med. 2011;8(9):2546–2559.

James A. Simon, MD, CCD, NCMP, IF
Dr. Simon is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University, and Medical Director, Women’s Health & Research Consultants, Washington, DC. Dr. Simon is a North American Menopause Society past president, a certified menopause practitioner, and clinical densitometrist. Dr. Simon is an ISSWSH fellow and an AASECT certified sexuality counselor. He also serves on the OBG Management Board of Editors.
Dr. Simon reports that within the past 36 months, he has been a consultant to or served on the advisory boards of AbbVie, Actavis, Amgen, Amneal, Apotex, Ascend Therapeutics, BioSante, Depomed, Dr. Reddy Laboratories, Everett Laboratories, Intimina by Lelo, Lupin, Meda, Merck, Novartis, Noven, Novo Nordisk, Nuelle, Pfizer, Regeneron, Sanofi SA, Sermonix, Shionogi, Shippan Point Advisors, Sprout, Teva, TherapeuticsMD, Warner Chilcott, and Watson.
In the past 36 months, Dr. Simon has received grant/research support from AbbVie, Actavis, Agile Therapeutics, Bayer Healthcare, EndoCeutics, New England Research Institute, Novo Nordisk, Palatin Technologies, Teva, and TherapeuticsMD.
Within the past 36 months, Dr. Simon also has served on the speaker’s bureaus of Amgen, Eisai, Merck, Novartis, Noven, Novo Nordisk, Shionogi, Teva, and Warner Chilcott.
Dr. Simon served as Chief Medical Officer of Sprout Pharmaceuticals until April 1, 2013.
James A. Simon, MD, CCD, NCMP, IF
Dr. Simon is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University, and Medical Director, Women’s Health & Research Consultants, Washington, DC. Dr. Simon is a North American Menopause Society past president, a certified menopause practitioner, and clinical densitometrist. Dr. Simon is an ISSWSH fellow and an AASECT certified sexuality counselor. He also serves on the OBG Management Board of Editors.
Dr. Simon reports that within the past 36 months, he has been a consultant to or served on the advisory boards of AbbVie, Actavis, Amgen, Amneal, Apotex, Ascend Therapeutics, BioSante, Depomed, Dr. Reddy Laboratories, Everett Laboratories, Intimina by Lelo, Lupin, Meda, Merck, Novartis, Noven, Novo Nordisk, Nuelle, Pfizer, Regeneron, Sanofi SA, Sermonix, Shionogi, Shippan Point Advisors, Sprout, Teva, TherapeuticsMD, Warner Chilcott, and Watson.
In the past 36 months, Dr. Simon has received grant/research support from AbbVie, Actavis, Agile Therapeutics, Bayer Healthcare, EndoCeutics, New England Research Institute, Novo Nordisk, Palatin Technologies, Teva, and TherapeuticsMD.
Within the past 36 months, Dr. Simon also has served on the speaker’s bureaus of Amgen, Eisai, Merck, Novartis, Noven, Novo Nordisk, Shionogi, Teva, and Warner Chilcott.
Dr. Simon served as Chief Medical Officer of Sprout Pharmaceuticals until April 1, 2013.
James A. Simon, MD, CCD, NCMP, IF
Dr. Simon is Clinical Professor, Department of Obstetrics and Gynecology, George Washington University, and Medical Director, Women’s Health & Research Consultants, Washington, DC. Dr. Simon is a North American Menopause Society past president, a certified menopause practitioner, and clinical densitometrist. Dr. Simon is an ISSWSH fellow and an AASECT certified sexuality counselor. He also serves on the OBG Management Board of Editors.
Dr. Simon reports that within the past 36 months, he has been a consultant to or served on the advisory boards of AbbVie, Actavis, Amgen, Amneal, Apotex, Ascend Therapeutics, BioSante, Depomed, Dr. Reddy Laboratories, Everett Laboratories, Intimina by Lelo, Lupin, Meda, Merck, Novartis, Noven, Novo Nordisk, Nuelle, Pfizer, Regeneron, Sanofi SA, Sermonix, Shionogi, Shippan Point Advisors, Sprout, Teva, TherapeuticsMD, Warner Chilcott, and Watson.
In the past 36 months, Dr. Simon has received grant/research support from AbbVie, Actavis, Agile Therapeutics, Bayer Healthcare, EndoCeutics, New England Research Institute, Novo Nordisk, Palatin Technologies, Teva, and TherapeuticsMD.
Within the past 36 months, Dr. Simon also has served on the speaker’s bureaus of Amgen, Eisai, Merck, Novartis, Noven, Novo Nordisk, Shionogi, Teva, and Warner Chilcott.
Dr. Simon served as Chief Medical Officer of Sprout Pharmaceuticals until April 1, 2013.
Women’s sexual health took a step forward last month when an advisory panel to the US Food and Drug Administration (FDA) recommended approval of the drug flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in premenopausal women. The approval came with some reservations regarding safety (use with certain medications and alcohol). And it’s worthwhile to note that the FDA had on hand its own Drug Safety and Risk Management Committee during deliberations. However, assuming the agency follows the recommendations of the Bone, Reproductive, and Urologic Drugs Advisory Committee, women will soon have available the first agent for sexual dysfunction—aside from a medication for intercourse-associated pain—developed specifically for them.
Had the panel voted down the approval, it would have set a dangerous precedent that likely would have led to a standstill in new drug development in all therapeutic classes of women’s sexual health for the next decade.
Why do I say that—and state it so emphatically?
To answer that question, let’s review the approval process for flibanserin, as well as for the 2 testosterone products that preceded its appearance before the FDA.
Hypoactive sexual desire disorder (HSDD) is the most common sexual dysfunction in women. Few interventions have proven to be effective for the treatment of HSDD. Education, counseling, and psychotherapy may be helpful in some women. Exogenous testosterone has been reported to be effective in the treatment of low sexual desire in postmenopausal women taking estrogen, but this treatment is not approved by the US Food and Drug Administration (FDA), and the long-term safety of exogenous testosterone in women is not established. Clinicians who treat women with sexual desire disorders are frustrated by their inability to prescribe an effective treatment for this common problem. An FDA advisory panel recently voted to support the approval of flibanserin for the treatment of HSDD in premenopausal women. In this month’s editorial, Dr. James Simon provides a history of the FDA review process for medications designed to treat HSDD, including the decision to not approve testosterone and the vote of the FDA advisory panel to support the approval of flibanserin. Readers of OBG Management should be aware that Dr. Simon, as is apparent in this piece and fully disclosed, has served as Medical Director and an advisor to Sprout Pharmaceuticals, the company with the rights to flibanserin. As editors we have concluded that Dr. Simon’s unique perspective, knowledge, and insights about the history of the FDA review of treatments of HSDD, including testosterone, would be of great interest to our readers.
—Robert L. Barbieri, MD, Editor in Chief
—Lila O’Connor, Editor
A tale of 2 products: The testosterone story
In 2004, Procter & Gamble filed a new drug application (NDA) for a testosterone patch (Intrinsa) developed for the treatment of female sexual dysfunction. Specifically, the patch was created for the treatment of HSDD in surgically menopausal women (those who had undergone bilateral oophorectomy) who were receiving concomitant estrogen therapy.
Both the FDA and the advisory committee considering the NDA agreed that the patch was effective. The question was whether its efficacy outweighed its risks. Recall that this discussion was taking place only 2 years after the initial publication of findings from the Women’s Health Initiative (WHI) estrogen-progestin arm. That arm had been halted prematurely because the risk of breast cancer exceeded the prespecified stopping boundary. In addition, the global index summarizing the balance of risks and benefits showed that risks outweighed benefits in regard to breast cancer, coronary heart disease, stroke, and pulmonary embolism.1 As a result, the safety of all forms of hormone therapy was being closely scrutinized.
The FDA was also on “high alert” for safety as rofecoxib (Vioxx) had just been removed from the market due to unforeseen risks, with much media fanfare and large numbers of lawsuits.
After consideration of the NDA for the testosterone patch, the FDA advised Procter & Gamble to undertake an adequately designed and powered safety study to confirm that it would not increase the risks of coronary heart disease or breast cancer among users, since testosterone can be converted to estradiol in women.
Procter & Gamble ultimately withdrew its NDA. Such a safety study would have taken an additional 5 years to complete and cost upwards of $300 million. And because testosterone is not a patentable compound, a study that long and costly didn’t seem like a smart business proposition. Shortly after the NDA was withdrawn, the European Medicines Agency—the European counterpart of the FDA—approved the Intrinsa testosterone patch for the same indication as proposed in the United States.
Next up, BioSante Pharmaceuticals filed its NDA for LibiGel, a testosterone gel formulated specifically for postmenopausal women with HSDD. In its efficacy study, LibiGel failed to demonstrate superiority above and beyond placebo. The manufacturer, which was concurrently conducting a large, long-term safety study to satisfy the FDA’s concerns, ran out of money shortly thereafter, and that was the end of that.
Flibanserin’s focus: premenopausal women
In contrast to the 2 testosterone products, flibanserin was developed for premenopausal women. Although preliminary data on flibanserin use among postmenopausal women are available,2 the drug was studied primarily in premenopausal women with HSDD, the indication sought at this time.
In the premenopausal population, problems such as pain with intercourse or hypoestrogenism aren’t typically present, simplifying the identification of HSDD. (See the sidebar below, “What is HSDD and how is it diagnosed?”) In clinical trials of the drug, HSDD was secondary, generalized, and acquired—that is, it followed a period of normal sexual function. And it didn’t come and go but was present regardless of location and circumstance. Study participants had had a normal sex drive before their desire “turned off,” an occurrence they found distressing.3–6
What is hypoactive sexual desire disorderand how is it diagnosed?
In the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), hypoactive sexual desire disorder (HSDD) is described as having the following characteristics:
- persistently or recurrently deficient (or absent) sexual fantasies and desire for sexual activity
- marked distress or interpersonal difficulty in response to this deficiency
- lack of another explanation, such as another Axis I disorder or use of a substance known to affect sexual function.
In the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), published in 2013, HSDD was folded with female sexual arousal disorder into a new diagnosis, female sexual interest and arousal disorder. This is somewhat confusing in that the physiologies of these 2 disorders are quite separate and distinct.
In clinical practice, HSDD is easily identified using the Decreased Sexual Desire Screener (DSDS), a simple screening test that asks 4 yes/no questions:
- In the past, was your level of sexual desire or interest good and satisfying to you?
- Has there been a decrease in your level of sexual desire or interest?
- Are you bothered by your decreased level of sexual desire or interest?
- Would you like your level of sexual desire or interest to increase?
A “yes” response to each of these questions is required. In addition, a fifth question asks whether a number of conditions, drugs, or circumstances might be responsible for the decreased desire or interest:
- an operation, depression, injuries, or other medical condition
- medications, drugs, or alcohol you are currently taking
- pregnancy, recent childbirth, or menopausal symptoms
- other sexual issues you may be having (pain, decreased arousal or orgasm)
- your partner’s sexual problems
- dissatisfaction with your relationship or partner
- stress or fatigue.
Boehringer Ingelheim, a German concern, developed flibanserin and filed the initial NDA in 2009. In clinical trials, that company ran into problems because the electronic diary it was using to measure desire failed to demonstrate efficacy for flibanserin. It turns out that, if you ask women who are distressed about having low desire to report their level of desire every single day, they quickly get turned off by the question and eventually stop answering altogether. Such changes in behavior play havoc with the validity of the instrument to assess desire.
After flibanserin failed the electronic diary desire domain—one of the study’s endpoints—the company substituted a different measure, the Female Sexual Function Index (FSFI) desire domain. Although the FSFI showed statistically significantly greater efficacy for flibanserin than placebo, the FDA argued that it was unreasonable for the company to change the rules to fit the outcome. For that and other reasons, it turned down the NDA.
In response, Boehringer Ingelheim went back to the drawing board and undertook a new study intended to achieve several goals:
- substitute the FSFI desire score for the electronic diary desire score
- reduce the number of restricted medications to see if the results could be more generalizable to the normal population of women with HSDD
- determine whether there were any safety signals for drug-drug interaction that weren’t apparent in the first 3 trials, in which a large number of medications had been excluded.
About the time this study was drawing to a conclusion, the company got cold feet and decided to shelve its plans for the drug.
Sprout steps in
I was among the delegation of medical and pharmaceutical professionals who visited Boehringer Ingelheim in 2011 to explore the possibility of Sprout Pharmaceuticals acquiring flibanserin. Boehringer Ingelheim agreed to the deal, and Sprout took over drug development, resubmitting the NDA to the FDA in 2013 with the additional study and other data. The FDA again denied the application. In response, Sprout filed a request for a dispute resolution, a formal procedure convened when the sponsor of an NDA cannot reach agreement with the FDA. In the course of this procedure, the FDA asked for additional analyses, as well as some pharmacogenomics and a driving study.
Around this time, the FDA had determined that the sleep aid zolpidem (Ambien) is metabolized differently in women than in men and that, in some of these women, there is a significant cognitive deficit carried over to the next day when the drug is taken as prescribed at bedtime. Because flibanserin came on the heels of this determination and was known to cause drowsiness, the FDA requested the driving study. It also requested a drug-drug interaction study to determine whether flibanserin is metabolized differently in some women with genetically different medication metabolism.
Sprout conducted those studies, both of which came back “clean.” Armed with this new data, the FDA scheduled a meeting of its advisory committee on June 4, 2015. And the rest, as they say, is history.
Flibanserin vs placebo
During the advisory committee’s deliberations on June 4, discussion focused, in part, on how flibanserin performed in comparison with placebo. It was noted that flibanserin increased the number of satisfying sexual events (SSEs) by only 1 per month, compared with placebo. But that conclusion doesn’t accurately convey the findings of the efficacy studies.
First, even without flibanserin, women in the trials reported that they continued to have sex with their partners 2 to 3 times per month. That established a baseline number of SSEs of approximately 2.5. The consent form for the flibanserin trial contained a clause stating that the woman would agree to try and have sex at least 1 additional time per month. This requirement, independent of any treatment, was bound to increase the placebo effect because, regardless of the drug given (flibanserin or placebo), the participant was going to try to have sex at least 1 more time per month.
In the flibanserin trials, the placebo effect was 1.5 additional SSEs per month. Add that to the baseline number of SSEs and you have a total of 4 SSEs per month. Among flibanserin users, the number of SSEs per month was about 5. And even though that’s only 1 more time per month than the placebo group, those 5 events were more desired events. That means that the baseline of 2.5 SSEs, among flibanserin users, had a different character by the end of the study period.
There is also a ceiling effect in play. Consider that the participants in the flibanserin trials were women who had been married an average of 10 years, with an average age of approximately 36 years. How much more sex is likely even possible in this population?
This isn’t to say that women are incapable of having more sex. It is merely a reflection of the data, which show that, among married women aged 30 to 39 years, only roughly 25% have sex more often than weekly, and only 5.1% have sex 4 or more times per week.7 If women were shown to be having sex more than 5 times per month, a likely criticism would have been that the drug was making them hypersexual or even abnormal.
Also keep in mind that the drug doesn’t work in every woman, just as antidepressants are not effective in every person. So when the responders and nonresponders were lumped together, the magnitude of the drug’s response in the combined group was smaller. In reality, approximately 25% of women in the flibanserin trials experienced an increase of 4 or more SSEs per month, compared with 15% among placebo users.
Flibanserin dosing and side effects
Flibanserin is indicated for the treatment of hypoactive sexual desire disorder in premenopausal women. It is taken daily in a 100-mg tablet. Bedtime dosing is preferred to mitigate potential side effects such as drowsiness, hypotension, and syncope. These effects can occur with flibanserin alone, in combination with certain drugs, or in combination with alcohol.
Significant drug-drug interactions have been documented for flibanserin in combination with moderate and strong CYP3A4 inhibitors such as fluconazole and ketoconazole. Package labeling for flibanserin will detail this risk.
Why now?
As I stated earlier, a failure to approve flibanserin would set a dangerous precedent. Why? Because the company did everything the FDA asked it to do, and the results came out statistically significantly better than placebo—which was the desired endpoint. If the FDA were to deny approval of the drug, it would be saying, in effect, that it can change its mind in the middle of the argument—something it faulted Boehringer Ingelheim for in earlier deliberations (switching the insensitive electronic diary for the statistically significant FSFI).
In reality, the FDA is likely to say yes to approval, but with restrictions, as that is what its advisory committee recommended. What those restrictions will be remains to be determined, but they are likely to resemble those of other drugs in the class, such as selective serotonin reuptake inhibitors (SSRIs), including a warning to be careful using flibanserin with alcohol until the drug’s effects are clear.
Opposition to flibanserin misses the mark
During the public hearing portion of the advisory committee meeting, most of the testimony came from women seeking approval of the drug. However, there were some naysayers. Their arguments against approval boiled down to 4 perspectives:
- the view that development of flibanserin represents “medicalization” of a disorder that can be treated effectively with psychotherapy and education. This perspective is best embodied by an organization called the New View Campaign. Refuting this perspective, however, is research in animal models that clearly demonstrates that HSDD (or its equivalent in animals) is the result of an imbalance between dopamine and norepinephrine on the positive end and serotonin on the negative end. These findings are supported by functional magnetic resonance imaging (MRI) and positron emission tomography (PET) scans of the brains of women with HSDD who are shown erotic stimuli.8,9 The scans demonstrate that their brains respond differently from those of normal women. So if it’s all about education and counseling, why are the brains of women with HSDD functioning differently? I would argue that, if depression and HSDD are both abnormalities of the serotonergic system (flibanserin is essentially an SSRI), then how can depression be a biologically based disorder but HSDD can’t? In my opinion, the New View Campaign isn’t new at all.
- the view, represented by an organization called PharmedOUT, that marketing by pharmaceutical companies overly influences prescribers, ultimately medicalizing problems that don’t require medication or overselling medications for problems that may require drug treatment for a short time only. This organization is headed by an academic physician who has not seen patients in many years and has never treated women for HSDD.
- the view, represented by the Public Citizen Health Research Group, that the safety profile of flibanserin is lacking. This organization argues not just against flibanserin but against pretty much any drug. In its view, there are never enough safety data. I would argue that, when it comes to flibanserin, there are more safety and efficacy data than there are for almost any other women’s health drug. Most drug companies test their medications in 1,500 to 2,000 people, as the FDA requires. Sprout Pharmaceuticals tested flibanserin in almost 8,000 women. The total number of individuals who have been studied, in fact, exceeds 11,000.
- the view, represented by the National Women’s Health Network, that the drug’s risks outweigh its modest benefits. As I have pointed out, however, the benefits of flibanserin have been downplayed in data analysis, and the body of safety data for the drug is substantial.
There is also the sociological view that HSDD is a normal variant of healthy sexual function. Its adherents argue that women with HSDD feel distress because their male partners are forcing them to feel inadequate. But I have yet to hear a single critical voice from a physician who actually treats women and who can prescribe drugs. The opposition to flibanserin, such as it is, flows from people who don’t see patients and can’t prescribe medications.
These naysayers are negative in a theoretical vacuum. It’s very easy to fall into that trap when you don’t have to look across the consultation desk to a patient who is asking you for a remedy—a woman who’s been suffering for 25 years, say—and have to tell her you have nothing to offer. You might mention testosterone, explaining that it was approved for men but you’ll try to prescribe an appropriate dose. But be sure to include discussion of its many side effects.
A long and winding road
Flibanserin’s journey from inception to probable approval has been long and eventful, and you can be sure that the pharmaceutical industry has been paying attention. Hundreds of millions of dollars in funding for drug development hang in the balance. That women deserve remedies developed specifically for their needs and metabolism is a given. The approval of flibanserin will send a hopeful signal to them as well as to industry—that the FDA takes them seriously and seeks to identify effective remedies. In this case, it seems likely, the agency will end up on the right side of history.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Women’s sexual health took a step forward last month when an advisory panel to the US Food and Drug Administration (FDA) recommended approval of the drug flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in premenopausal women. The approval came with some reservations regarding safety (use with certain medications and alcohol). And it’s worthwhile to note that the FDA had on hand its own Drug Safety and Risk Management Committee during deliberations. However, assuming the agency follows the recommendations of the Bone, Reproductive, and Urologic Drugs Advisory Committee, women will soon have available the first agent for sexual dysfunction—aside from a medication for intercourse-associated pain—developed specifically for them.
Had the panel voted down the approval, it would have set a dangerous precedent that likely would have led to a standstill in new drug development in all therapeutic classes of women’s sexual health for the next decade.
Why do I say that—and state it so emphatically?
To answer that question, let’s review the approval process for flibanserin, as well as for the 2 testosterone products that preceded its appearance before the FDA.
Hypoactive sexual desire disorder (HSDD) is the most common sexual dysfunction in women. Few interventions have proven to be effective for the treatment of HSDD. Education, counseling, and psychotherapy may be helpful in some women. Exogenous testosterone has been reported to be effective in the treatment of low sexual desire in postmenopausal women taking estrogen, but this treatment is not approved by the US Food and Drug Administration (FDA), and the long-term safety of exogenous testosterone in women is not established. Clinicians who treat women with sexual desire disorders are frustrated by their inability to prescribe an effective treatment for this common problem. An FDA advisory panel recently voted to support the approval of flibanserin for the treatment of HSDD in premenopausal women. In this month’s editorial, Dr. James Simon provides a history of the FDA review process for medications designed to treat HSDD, including the decision to not approve testosterone and the vote of the FDA advisory panel to support the approval of flibanserin. Readers of OBG Management should be aware that Dr. Simon, as is apparent in this piece and fully disclosed, has served as Medical Director and an advisor to Sprout Pharmaceuticals, the company with the rights to flibanserin. As editors we have concluded that Dr. Simon’s unique perspective, knowledge, and insights about the history of the FDA review of treatments of HSDD, including testosterone, would be of great interest to our readers.
—Robert L. Barbieri, MD, Editor in Chief
—Lila O’Connor, Editor
A tale of 2 products: The testosterone story
In 2004, Procter & Gamble filed a new drug application (NDA) for a testosterone patch (Intrinsa) developed for the treatment of female sexual dysfunction. Specifically, the patch was created for the treatment of HSDD in surgically menopausal women (those who had undergone bilateral oophorectomy) who were receiving concomitant estrogen therapy.
Both the FDA and the advisory committee considering the NDA agreed that the patch was effective. The question was whether its efficacy outweighed its risks. Recall that this discussion was taking place only 2 years after the initial publication of findings from the Women’s Health Initiative (WHI) estrogen-progestin arm. That arm had been halted prematurely because the risk of breast cancer exceeded the prespecified stopping boundary. In addition, the global index summarizing the balance of risks and benefits showed that risks outweighed benefits in regard to breast cancer, coronary heart disease, stroke, and pulmonary embolism.1 As a result, the safety of all forms of hormone therapy was being closely scrutinized.
The FDA was also on “high alert” for safety as rofecoxib (Vioxx) had just been removed from the market due to unforeseen risks, with much media fanfare and large numbers of lawsuits.
After consideration of the NDA for the testosterone patch, the FDA advised Procter & Gamble to undertake an adequately designed and powered safety study to confirm that it would not increase the risks of coronary heart disease or breast cancer among users, since testosterone can be converted to estradiol in women.
Procter & Gamble ultimately withdrew its NDA. Such a safety study would have taken an additional 5 years to complete and cost upwards of $300 million. And because testosterone is not a patentable compound, a study that long and costly didn’t seem like a smart business proposition. Shortly after the NDA was withdrawn, the European Medicines Agency—the European counterpart of the FDA—approved the Intrinsa testosterone patch for the same indication as proposed in the United States.
Next up, BioSante Pharmaceuticals filed its NDA for LibiGel, a testosterone gel formulated specifically for postmenopausal women with HSDD. In its efficacy study, LibiGel failed to demonstrate superiority above and beyond placebo. The manufacturer, which was concurrently conducting a large, long-term safety study to satisfy the FDA’s concerns, ran out of money shortly thereafter, and that was the end of that.
Flibanserin’s focus: premenopausal women
In contrast to the 2 testosterone products, flibanserin was developed for premenopausal women. Although preliminary data on flibanserin use among postmenopausal women are available,2 the drug was studied primarily in premenopausal women with HSDD, the indication sought at this time.
In the premenopausal population, problems such as pain with intercourse or hypoestrogenism aren’t typically present, simplifying the identification of HSDD. (See the sidebar below, “What is HSDD and how is it diagnosed?”) In clinical trials of the drug, HSDD was secondary, generalized, and acquired—that is, it followed a period of normal sexual function. And it didn’t come and go but was present regardless of location and circumstance. Study participants had had a normal sex drive before their desire “turned off,” an occurrence they found distressing.3–6
What is hypoactive sexual desire disorderand how is it diagnosed?
In the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), hypoactive sexual desire disorder (HSDD) is described as having the following characteristics:
- persistently or recurrently deficient (or absent) sexual fantasies and desire for sexual activity
- marked distress or interpersonal difficulty in response to this deficiency
- lack of another explanation, such as another Axis I disorder or use of a substance known to affect sexual function.
In the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), published in 2013, HSDD was folded with female sexual arousal disorder into a new diagnosis, female sexual interest and arousal disorder. This is somewhat confusing in that the physiologies of these 2 disorders are quite separate and distinct.
In clinical practice, HSDD is easily identified using the Decreased Sexual Desire Screener (DSDS), a simple screening test that asks 4 yes/no questions:
- In the past, was your level of sexual desire or interest good and satisfying to you?
- Has there been a decrease in your level of sexual desire or interest?
- Are you bothered by your decreased level of sexual desire or interest?
- Would you like your level of sexual desire or interest to increase?
A “yes” response to each of these questions is required. In addition, a fifth question asks whether a number of conditions, drugs, or circumstances might be responsible for the decreased desire or interest:
- an operation, depression, injuries, or other medical condition
- medications, drugs, or alcohol you are currently taking
- pregnancy, recent childbirth, or menopausal symptoms
- other sexual issues you may be having (pain, decreased arousal or orgasm)
- your partner’s sexual problems
- dissatisfaction with your relationship or partner
- stress or fatigue.
Boehringer Ingelheim, a German concern, developed flibanserin and filed the initial NDA in 2009. In clinical trials, that company ran into problems because the electronic diary it was using to measure desire failed to demonstrate efficacy for flibanserin. It turns out that, if you ask women who are distressed about having low desire to report their level of desire every single day, they quickly get turned off by the question and eventually stop answering altogether. Such changes in behavior play havoc with the validity of the instrument to assess desire.
After flibanserin failed the electronic diary desire domain—one of the study’s endpoints—the company substituted a different measure, the Female Sexual Function Index (FSFI) desire domain. Although the FSFI showed statistically significantly greater efficacy for flibanserin than placebo, the FDA argued that it was unreasonable for the company to change the rules to fit the outcome. For that and other reasons, it turned down the NDA.
In response, Boehringer Ingelheim went back to the drawing board and undertook a new study intended to achieve several goals:
- substitute the FSFI desire score for the electronic diary desire score
- reduce the number of restricted medications to see if the results could be more generalizable to the normal population of women with HSDD
- determine whether there were any safety signals for drug-drug interaction that weren’t apparent in the first 3 trials, in which a large number of medications had been excluded.
About the time this study was drawing to a conclusion, the company got cold feet and decided to shelve its plans for the drug.
Sprout steps in
I was among the delegation of medical and pharmaceutical professionals who visited Boehringer Ingelheim in 2011 to explore the possibility of Sprout Pharmaceuticals acquiring flibanserin. Boehringer Ingelheim agreed to the deal, and Sprout took over drug development, resubmitting the NDA to the FDA in 2013 with the additional study and other data. The FDA again denied the application. In response, Sprout filed a request for a dispute resolution, a formal procedure convened when the sponsor of an NDA cannot reach agreement with the FDA. In the course of this procedure, the FDA asked for additional analyses, as well as some pharmacogenomics and a driving study.
Around this time, the FDA had determined that the sleep aid zolpidem (Ambien) is metabolized differently in women than in men and that, in some of these women, there is a significant cognitive deficit carried over to the next day when the drug is taken as prescribed at bedtime. Because flibanserin came on the heels of this determination and was known to cause drowsiness, the FDA requested the driving study. It also requested a drug-drug interaction study to determine whether flibanserin is metabolized differently in some women with genetically different medication metabolism.
Sprout conducted those studies, both of which came back “clean.” Armed with this new data, the FDA scheduled a meeting of its advisory committee on June 4, 2015. And the rest, as they say, is history.
Flibanserin vs placebo
During the advisory committee’s deliberations on June 4, discussion focused, in part, on how flibanserin performed in comparison with placebo. It was noted that flibanserin increased the number of satisfying sexual events (SSEs) by only 1 per month, compared with placebo. But that conclusion doesn’t accurately convey the findings of the efficacy studies.
First, even without flibanserin, women in the trials reported that they continued to have sex with their partners 2 to 3 times per month. That established a baseline number of SSEs of approximately 2.5. The consent form for the flibanserin trial contained a clause stating that the woman would agree to try and have sex at least 1 additional time per month. This requirement, independent of any treatment, was bound to increase the placebo effect because, regardless of the drug given (flibanserin or placebo), the participant was going to try to have sex at least 1 more time per month.
In the flibanserin trials, the placebo effect was 1.5 additional SSEs per month. Add that to the baseline number of SSEs and you have a total of 4 SSEs per month. Among flibanserin users, the number of SSEs per month was about 5. And even though that’s only 1 more time per month than the placebo group, those 5 events were more desired events. That means that the baseline of 2.5 SSEs, among flibanserin users, had a different character by the end of the study period.
There is also a ceiling effect in play. Consider that the participants in the flibanserin trials were women who had been married an average of 10 years, with an average age of approximately 36 years. How much more sex is likely even possible in this population?
This isn’t to say that women are incapable of having more sex. It is merely a reflection of the data, which show that, among married women aged 30 to 39 years, only roughly 25% have sex more often than weekly, and only 5.1% have sex 4 or more times per week.7 If women were shown to be having sex more than 5 times per month, a likely criticism would have been that the drug was making them hypersexual or even abnormal.
Also keep in mind that the drug doesn’t work in every woman, just as antidepressants are not effective in every person. So when the responders and nonresponders were lumped together, the magnitude of the drug’s response in the combined group was smaller. In reality, approximately 25% of women in the flibanserin trials experienced an increase of 4 or more SSEs per month, compared with 15% among placebo users.
Flibanserin dosing and side effects
Flibanserin is indicated for the treatment of hypoactive sexual desire disorder in premenopausal women. It is taken daily in a 100-mg tablet. Bedtime dosing is preferred to mitigate potential side effects such as drowsiness, hypotension, and syncope. These effects can occur with flibanserin alone, in combination with certain drugs, or in combination with alcohol.
Significant drug-drug interactions have been documented for flibanserin in combination with moderate and strong CYP3A4 inhibitors such as fluconazole and ketoconazole. Package labeling for flibanserin will detail this risk.
Why now?
As I stated earlier, a failure to approve flibanserin would set a dangerous precedent. Why? Because the company did everything the FDA asked it to do, and the results came out statistically significantly better than placebo—which was the desired endpoint. If the FDA were to deny approval of the drug, it would be saying, in effect, that it can change its mind in the middle of the argument—something it faulted Boehringer Ingelheim for in earlier deliberations (switching the insensitive electronic diary for the statistically significant FSFI).
In reality, the FDA is likely to say yes to approval, but with restrictions, as that is what its advisory committee recommended. What those restrictions will be remains to be determined, but they are likely to resemble those of other drugs in the class, such as selective serotonin reuptake inhibitors (SSRIs), including a warning to be careful using flibanserin with alcohol until the drug’s effects are clear.
Opposition to flibanserin misses the mark
During the public hearing portion of the advisory committee meeting, most of the testimony came from women seeking approval of the drug. However, there were some naysayers. Their arguments against approval boiled down to 4 perspectives:
- the view that development of flibanserin represents “medicalization” of a disorder that can be treated effectively with psychotherapy and education. This perspective is best embodied by an organization called the New View Campaign. Refuting this perspective, however, is research in animal models that clearly demonstrates that HSDD (or its equivalent in animals) is the result of an imbalance between dopamine and norepinephrine on the positive end and serotonin on the negative end. These findings are supported by functional magnetic resonance imaging (MRI) and positron emission tomography (PET) scans of the brains of women with HSDD who are shown erotic stimuli.8,9 The scans demonstrate that their brains respond differently from those of normal women. So if it’s all about education and counseling, why are the brains of women with HSDD functioning differently? I would argue that, if depression and HSDD are both abnormalities of the serotonergic system (flibanserin is essentially an SSRI), then how can depression be a biologically based disorder but HSDD can’t? In my opinion, the New View Campaign isn’t new at all.
- the view, represented by an organization called PharmedOUT, that marketing by pharmaceutical companies overly influences prescribers, ultimately medicalizing problems that don’t require medication or overselling medications for problems that may require drug treatment for a short time only. This organization is headed by an academic physician who has not seen patients in many years and has never treated women for HSDD.
- the view, represented by the Public Citizen Health Research Group, that the safety profile of flibanserin is lacking. This organization argues not just against flibanserin but against pretty much any drug. In its view, there are never enough safety data. I would argue that, when it comes to flibanserin, there are more safety and efficacy data than there are for almost any other women’s health drug. Most drug companies test their medications in 1,500 to 2,000 people, as the FDA requires. Sprout Pharmaceuticals tested flibanserin in almost 8,000 women. The total number of individuals who have been studied, in fact, exceeds 11,000.
- the view, represented by the National Women’s Health Network, that the drug’s risks outweigh its modest benefits. As I have pointed out, however, the benefits of flibanserin have been downplayed in data analysis, and the body of safety data for the drug is substantial.
There is also the sociological view that HSDD is a normal variant of healthy sexual function. Its adherents argue that women with HSDD feel distress because their male partners are forcing them to feel inadequate. But I have yet to hear a single critical voice from a physician who actually treats women and who can prescribe drugs. The opposition to flibanserin, such as it is, flows from people who don’t see patients and can’t prescribe medications.
These naysayers are negative in a theoretical vacuum. It’s very easy to fall into that trap when you don’t have to look across the consultation desk to a patient who is asking you for a remedy—a woman who’s been suffering for 25 years, say—and have to tell her you have nothing to offer. You might mention testosterone, explaining that it was approved for men but you’ll try to prescribe an appropriate dose. But be sure to include discussion of its many side effects.
A long and winding road
Flibanserin’s journey from inception to probable approval has been long and eventful, and you can be sure that the pharmaceutical industry has been paying attention. Hundreds of millions of dollars in funding for drug development hang in the balance. That women deserve remedies developed specifically for their needs and metabolism is a given. The approval of flibanserin will send a hopeful signal to them as well as to industry—that the FDA takes them seriously and seeks to identify effective remedies. In this case, it seems likely, the agency will end up on the right side of history.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333.
2. Simon JA, Kingsberg SA, Shumel B, Hanes V, Garcia M Jr, Sand M. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21(6):633–640.
3. DeRogatis LR, Komer L, Katz M, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074–1085.
4. Jayne C, Simon JA, Taylor LV, Kimura T, Lesko L; SUNFLOWER study investigators. Open-label extension study of flibanserin in women with hypoactive sexual desire disorder. J Sex Med. 2012;9(12):3180–3188.
5. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793–804.
6. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807–1815.
7. Kinsey Institute. Percentage of Women Reporting Frequency of Vaginal Sex, N = 2,393. http://www.kinseyinstitute.org/resources/FAQ.html. Accessed June 17, 2015.
8. Arnow BA, Millheiser L, Garrett A, et al. Women with hypoactive sexual desire disorder compared to normal females: a functional magnetic resonance imaging study. Neuroscience. 2009;158(2):484–502.
9. Bianchi-Demicheli F, Cojan Y, et al. Neural bases of hypoactive sexual desire disorder in women: an event-related FMRI study. J Sex Med. 2011;8(9):2546–2559.
1. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333.
2. Simon JA, Kingsberg SA, Shumel B, Hanes V, Garcia M Jr, Sand M. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21(6):633–640.
3. DeRogatis LR, Komer L, Katz M, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074–1085.
4. Jayne C, Simon JA, Taylor LV, Kimura T, Lesko L; SUNFLOWER study investigators. Open-label extension study of flibanserin in women with hypoactive sexual desire disorder. J Sex Med. 2012;9(12):3180–3188.
5. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793–804.
6. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807–1815.
7. Kinsey Institute. Percentage of Women Reporting Frequency of Vaginal Sex, N = 2,393. http://www.kinseyinstitute.org/resources/FAQ.html. Accessed June 17, 2015.
8. Arnow BA, Millheiser L, Garrett A, et al. Women with hypoactive sexual desire disorder compared to normal females: a functional magnetic resonance imaging study. Neuroscience. 2009;158(2):484–502.
9. Bianchi-Demicheli F, Cojan Y, et al. Neural bases of hypoactive sexual desire disorder in women: an event-related FMRI study. J Sex Med. 2011;8(9):2546–2559.

Based on interval cancer rates, which women with dense breasts are most likely to benefit from supplemental imaging?
The number of states that require notification to a woman who is identified on mammography as having heterogeneous (Breast Imaging-Reporting and Data System [BI-RADS] C) or extremely dense breasts (BI-RADS D) is growing. In fact, Michigan became the 22nd state to require such notification when its law went into effect on June 1, 2015. How do we advise our patients who come to us wondering what they should do with the new-found information?
Since supplemental imaging after normal mammography findings can result in false positives and potentially unnecessary biopsies or treatment, Kerlikowski and colleagues investigated for which patients supplemental screening could be beneficial. In other words, which patients are at highest risk for interval cancer (invasive cancer diagnosed within 12 months of a normal mammogram), as these women would be most likely to benefit from supplemental imaging that could potentially detect a tumor not identified on digital screening mammography.
Details of the study
The researchers included 831,455 digital screening mammography examinations performed among 365,426 women aged 40 to 74 years who did not have a history of breast cancer or breast implants and had complete information on demographic and breast health history. To calculate breast cancer risk, they used the Breast Cancer Surveillance Consortium (BCSC) 5-year risk model, which requires 5 risk factors: first-degree relatives with history of breast cancer, history of breast biopsy, BI-RADS breast density, and race/ethnicity. They used breast density to stratify women by risk for interval cancer within the next year and to identify women at increased 5-year risk for breast cancer.
In which patient populations are cases of interval cancer highest?
The authors found the interval cancer rates to exceed 1 case per 1,000 mammography examinations among:
- women aged 70 to 74 years with heterogeneously dense breasts
- women aged 50 to 74 years with extremely dense breasts
- women with breast cancer risk of 1.67% or greater and extremely dense breasts (47.5% of women with extremely dense breasts)
- women with breast cancer risk of 2.50% or greater and heterogeneously dense breasts (19.5% of women with heterogeneously dense breasts).
The authors point out that, together, these 2 latter groups represent 24% of women aged 40 to 74 years with dense breasts, or 12% of women having screening mammography.
For women aged 40 to 49 years, interval cancer rates were less than 1 case per 1,000 examinations for all density categories. For 51% of these women with heterogeneously dense breasts, the 5-year risk of breast cancer was low to average (0% to 1.66%), with interval cancer rates of 0.58 to 0.63 per 1,000 examinations. For 52.5% of 40- to 49-year-old women with extremely dense breasts, the 5-year risk of breast cancer was low to average, with interval cancer rates of 0.72 to 0.89 cases per 1,000 examinations.
The interval cancer rate for women with scattered fibroglandular densities (BI-RADS B) and 5-year risk of 2.50% or greater was 0.90 cases per 1,000 mammography examinations.
Kerlikowski and colleagues conclude that breast density should not be the sole criterion for deciding whether supplemental imaging is justified because not all women with dense breasts have high interval cancer rates.
What this evidence means for practice
BCSC 5-year risk combined with BI-RADS breast density can identify women at high risk for interval cancer to inform patient–provider discussions about alternative screening strategies, as the study authors state. However, there remains a huge gap in our knowledge about whether supplemental imaging in any of these risk groups improved stage of diagnosis or breast cancer–specific mortality.
Nearly all national guidelines groups (US Preventive Services Task Force,1 American College of Obstetricians and Gynecologists,2 National Comprehensive Cancer Network,3 and the American Cancer Society4) concur that supplemental breast imaging should not be performed on women with dense breasts until there are reasonable data that demonstrate an improvement in stage of diagnosis or breast cancer mortality.
—Mark D. Pearlman, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Calonge N, Petitti DB, DeWitt TG, et al. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151(10):716–726.
2. American College of Obstetricians and Gynecologists. Practice Bulletin No. 122: Breast cancer screening. Obstet Gynecol. 2011;118(2 Part 1):372–382.
3. Bevers TB, Anderson BO, Bonaccio E, et al; National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: breast cancer screening and diagnosis. J Natl Compr Canc Netw. 2009;7(10):1060–1096.
4. American Cancer Society. Breast Cancer Screening Guidelines. http://www.cancer.org/healthy/informationforhealthcareprofessionals/acsguidelines/breastcancerscreeningguidelines/index. Accessed June 17, 2015.
The number of states that require notification to a woman who is identified on mammography as having heterogeneous (Breast Imaging-Reporting and Data System [BI-RADS] C) or extremely dense breasts (BI-RADS D) is growing. In fact, Michigan became the 22nd state to require such notification when its law went into effect on June 1, 2015. How do we advise our patients who come to us wondering what they should do with the new-found information?
Since supplemental imaging after normal mammography findings can result in false positives and potentially unnecessary biopsies or treatment, Kerlikowski and colleagues investigated for which patients supplemental screening could be beneficial. In other words, which patients are at highest risk for interval cancer (invasive cancer diagnosed within 12 months of a normal mammogram), as these women would be most likely to benefit from supplemental imaging that could potentially detect a tumor not identified on digital screening mammography.
Details of the study
The researchers included 831,455 digital screening mammography examinations performed among 365,426 women aged 40 to 74 years who did not have a history of breast cancer or breast implants and had complete information on demographic and breast health history. To calculate breast cancer risk, they used the Breast Cancer Surveillance Consortium (BCSC) 5-year risk model, which requires 5 risk factors: first-degree relatives with history of breast cancer, history of breast biopsy, BI-RADS breast density, and race/ethnicity. They used breast density to stratify women by risk for interval cancer within the next year and to identify women at increased 5-year risk for breast cancer.
In which patient populations are cases of interval cancer highest?
The authors found the interval cancer rates to exceed 1 case per 1,000 mammography examinations among:
- women aged 70 to 74 years with heterogeneously dense breasts
- women aged 50 to 74 years with extremely dense breasts
- women with breast cancer risk of 1.67% or greater and extremely dense breasts (47.5% of women with extremely dense breasts)
- women with breast cancer risk of 2.50% or greater and heterogeneously dense breasts (19.5% of women with heterogeneously dense breasts).
The authors point out that, together, these 2 latter groups represent 24% of women aged 40 to 74 years with dense breasts, or 12% of women having screening mammography.
For women aged 40 to 49 years, interval cancer rates were less than 1 case per 1,000 examinations for all density categories. For 51% of these women with heterogeneously dense breasts, the 5-year risk of breast cancer was low to average (0% to 1.66%), with interval cancer rates of 0.58 to 0.63 per 1,000 examinations. For 52.5% of 40- to 49-year-old women with extremely dense breasts, the 5-year risk of breast cancer was low to average, with interval cancer rates of 0.72 to 0.89 cases per 1,000 examinations.
The interval cancer rate for women with scattered fibroglandular densities (BI-RADS B) and 5-year risk of 2.50% or greater was 0.90 cases per 1,000 mammography examinations.
Kerlikowski and colleagues conclude that breast density should not be the sole criterion for deciding whether supplemental imaging is justified because not all women with dense breasts have high interval cancer rates.
What this evidence means for practice
BCSC 5-year risk combined with BI-RADS breast density can identify women at high risk for interval cancer to inform patient–provider discussions about alternative screening strategies, as the study authors state. However, there remains a huge gap in our knowledge about whether supplemental imaging in any of these risk groups improved stage of diagnosis or breast cancer–specific mortality.
Nearly all national guidelines groups (US Preventive Services Task Force,1 American College of Obstetricians and Gynecologists,2 National Comprehensive Cancer Network,3 and the American Cancer Society4) concur that supplemental breast imaging should not be performed on women with dense breasts until there are reasonable data that demonstrate an improvement in stage of diagnosis or breast cancer mortality.
—Mark D. Pearlman, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The number of states that require notification to a woman who is identified on mammography as having heterogeneous (Breast Imaging-Reporting and Data System [BI-RADS] C) or extremely dense breasts (BI-RADS D) is growing. In fact, Michigan became the 22nd state to require such notification when its law went into effect on June 1, 2015. How do we advise our patients who come to us wondering what they should do with the new-found information?
Since supplemental imaging after normal mammography findings can result in false positives and potentially unnecessary biopsies or treatment, Kerlikowski and colleagues investigated for which patients supplemental screening could be beneficial. In other words, which patients are at highest risk for interval cancer (invasive cancer diagnosed within 12 months of a normal mammogram), as these women would be most likely to benefit from supplemental imaging that could potentially detect a tumor not identified on digital screening mammography.
Details of the study
The researchers included 831,455 digital screening mammography examinations performed among 365,426 women aged 40 to 74 years who did not have a history of breast cancer or breast implants and had complete information on demographic and breast health history. To calculate breast cancer risk, they used the Breast Cancer Surveillance Consortium (BCSC) 5-year risk model, which requires 5 risk factors: first-degree relatives with history of breast cancer, history of breast biopsy, BI-RADS breast density, and race/ethnicity. They used breast density to stratify women by risk for interval cancer within the next year and to identify women at increased 5-year risk for breast cancer.
In which patient populations are cases of interval cancer highest?
The authors found the interval cancer rates to exceed 1 case per 1,000 mammography examinations among:
- women aged 70 to 74 years with heterogeneously dense breasts
- women aged 50 to 74 years with extremely dense breasts
- women with breast cancer risk of 1.67% or greater and extremely dense breasts (47.5% of women with extremely dense breasts)
- women with breast cancer risk of 2.50% or greater and heterogeneously dense breasts (19.5% of women with heterogeneously dense breasts).
The authors point out that, together, these 2 latter groups represent 24% of women aged 40 to 74 years with dense breasts, or 12% of women having screening mammography.
For women aged 40 to 49 years, interval cancer rates were less than 1 case per 1,000 examinations for all density categories. For 51% of these women with heterogeneously dense breasts, the 5-year risk of breast cancer was low to average (0% to 1.66%), with interval cancer rates of 0.58 to 0.63 per 1,000 examinations. For 52.5% of 40- to 49-year-old women with extremely dense breasts, the 5-year risk of breast cancer was low to average, with interval cancer rates of 0.72 to 0.89 cases per 1,000 examinations.
The interval cancer rate for women with scattered fibroglandular densities (BI-RADS B) and 5-year risk of 2.50% or greater was 0.90 cases per 1,000 mammography examinations.
Kerlikowski and colleagues conclude that breast density should not be the sole criterion for deciding whether supplemental imaging is justified because not all women with dense breasts have high interval cancer rates.
What this evidence means for practice
BCSC 5-year risk combined with BI-RADS breast density can identify women at high risk for interval cancer to inform patient–provider discussions about alternative screening strategies, as the study authors state. However, there remains a huge gap in our knowledge about whether supplemental imaging in any of these risk groups improved stage of diagnosis or breast cancer–specific mortality.
Nearly all national guidelines groups (US Preventive Services Task Force,1 American College of Obstetricians and Gynecologists,2 National Comprehensive Cancer Network,3 and the American Cancer Society4) concur that supplemental breast imaging should not be performed on women with dense breasts until there are reasonable data that demonstrate an improvement in stage of diagnosis or breast cancer mortality.
—Mark D. Pearlman, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Calonge N, Petitti DB, DeWitt TG, et al. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151(10):716–726.
2. American College of Obstetricians and Gynecologists. Practice Bulletin No. 122: Breast cancer screening. Obstet Gynecol. 2011;118(2 Part 1):372–382.
3. Bevers TB, Anderson BO, Bonaccio E, et al; National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: breast cancer screening and diagnosis. J Natl Compr Canc Netw. 2009;7(10):1060–1096.
4. American Cancer Society. Breast Cancer Screening Guidelines. http://www.cancer.org/healthy/informationforhealthcareprofessionals/acsguidelines/breastcancerscreeningguidelines/index. Accessed June 17, 2015.
1. Calonge N, Petitti DB, DeWitt TG, et al. Screening for breast cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151(10):716–726.
2. American College of Obstetricians and Gynecologists. Practice Bulletin No. 122: Breast cancer screening. Obstet Gynecol. 2011;118(2 Part 1):372–382.
3. Bevers TB, Anderson BO, Bonaccio E, et al; National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: breast cancer screening and diagnosis. J Natl Compr Canc Netw. 2009;7(10):1060–1096.
4. American Cancer Society. Breast Cancer Screening Guidelines. http://www.cancer.org/healthy/informationforhealthcareprofessionals/acsguidelines/breastcancerscreeningguidelines/index. Accessed June 17, 2015.
Does adjuvant oophorectomy improve survival in BRCA1 or BRCA2 mutation carriers with breast cancer?
Although bilateral salpingo-oophorectomy is known to prevent breast and ovarian cancer in BRCA mutation carriers,1 published reports also have suggested that, among mutation carriers with breast cancer, oophorectomy improves survival. In this retrospective analysis, investigators focused on women with stage I or II breast cancer and a BRCA1 or BRCA2 mutation, observing them for as long as 20 years after diagnosis. Survival rates were compared between women who did and did not undergo oophorectomy.
Details of the trial
Metcalfe and colleagues followed women with a BRCA1 or BRCA2 mutation and intact ovaries who were diagnosed with breast cancer at age 65 or younger between 1975 and 2008, tracking them for a mean of 12.5 years. Of 676 women, 345 underwent oophorectomy, usually with the intent of preventing ovarian cancer.
Overall, oophorectomy was associated with a 56% reduction in the risk of breast cancer-specific mortality (P = .005). Among breast cancer survivors with a BRCA1 mutation, oophorectomy was associated with a significant 62% reduction in breast cancer mortality. Among BRCA2 carriers, the observed 43% reduction in breast cancer mortality did not achieve statistical significance (P = .23).
Full impact of oophorectomy may be difficult to tease out
As Metcalfe and colleagues point out, recent improvements in breast imaging that have led to earlier diagnosis, as well as improvements in the treatment of breast cancer, might attenuate the mortality benefits observed with oophorectomy.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This important report underscores the importance of testing all women with early-stage breast cancer for BRCA mutations, and informs the management of known BRCA1 carriers with breast cancer.
—Andrew M. Kaunitz, MD
Reference
1. Finch APM, Lubinski J, Moller P, et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. JCO. 2014;32(15):1547–1553.
Although bilateral salpingo-oophorectomy is known to prevent breast and ovarian cancer in BRCA mutation carriers,1 published reports also have suggested that, among mutation carriers with breast cancer, oophorectomy improves survival. In this retrospective analysis, investigators focused on women with stage I or II breast cancer and a BRCA1 or BRCA2 mutation, observing them for as long as 20 years after diagnosis. Survival rates were compared between women who did and did not undergo oophorectomy.
Details of the trial
Metcalfe and colleagues followed women with a BRCA1 or BRCA2 mutation and intact ovaries who were diagnosed with breast cancer at age 65 or younger between 1975 and 2008, tracking them for a mean of 12.5 years. Of 676 women, 345 underwent oophorectomy, usually with the intent of preventing ovarian cancer.
Overall, oophorectomy was associated with a 56% reduction in the risk of breast cancer-specific mortality (P = .005). Among breast cancer survivors with a BRCA1 mutation, oophorectomy was associated with a significant 62% reduction in breast cancer mortality. Among BRCA2 carriers, the observed 43% reduction in breast cancer mortality did not achieve statistical significance (P = .23).
Full impact of oophorectomy may be difficult to tease out
As Metcalfe and colleagues point out, recent improvements in breast imaging that have led to earlier diagnosis, as well as improvements in the treatment of breast cancer, might attenuate the mortality benefits observed with oophorectomy.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This important report underscores the importance of testing all women with early-stage breast cancer for BRCA mutations, and informs the management of known BRCA1 carriers with breast cancer.
—Andrew M. Kaunitz, MD
Although bilateral salpingo-oophorectomy is known to prevent breast and ovarian cancer in BRCA mutation carriers,1 published reports also have suggested that, among mutation carriers with breast cancer, oophorectomy improves survival. In this retrospective analysis, investigators focused on women with stage I or II breast cancer and a BRCA1 or BRCA2 mutation, observing them for as long as 20 years after diagnosis. Survival rates were compared between women who did and did not undergo oophorectomy.
Details of the trial
Metcalfe and colleagues followed women with a BRCA1 or BRCA2 mutation and intact ovaries who were diagnosed with breast cancer at age 65 or younger between 1975 and 2008, tracking them for a mean of 12.5 years. Of 676 women, 345 underwent oophorectomy, usually with the intent of preventing ovarian cancer.
Overall, oophorectomy was associated with a 56% reduction in the risk of breast cancer-specific mortality (P = .005). Among breast cancer survivors with a BRCA1 mutation, oophorectomy was associated with a significant 62% reduction in breast cancer mortality. Among BRCA2 carriers, the observed 43% reduction in breast cancer mortality did not achieve statistical significance (P = .23).
Full impact of oophorectomy may be difficult to tease out
As Metcalfe and colleagues point out, recent improvements in breast imaging that have led to earlier diagnosis, as well as improvements in the treatment of breast cancer, might attenuate the mortality benefits observed with oophorectomy.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This important report underscores the importance of testing all women with early-stage breast cancer for BRCA mutations, and informs the management of known BRCA1 carriers with breast cancer.
—Andrew M. Kaunitz, MD
Reference
1. Finch APM, Lubinski J, Moller P, et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. JCO. 2014;32(15):1547–1553.
Reference
1. Finch APM, Lubinski J, Moller P, et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. JCO. 2014;32(15):1547–1553.
IHCC: Childhood migraine hurts school performance
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.

In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.
In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.
In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
EXPERT ANALYSIS FROM IHC 2015

AAS: New approach underway to ID teens at high suicide risk
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.

“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.
“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.
“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
EXPERT ANALYSIS FROM THE ANNUAL AAS CONFERENCE

Bundled Payment and Hospital Medicine, Pt. 2
Editor’s note: Second in a two-part series examining bundled payments and hospital medicine. In full disclosure, Dr. Whitcomb works for a company that is an Awardee Convener in the CMS Bundled Payments for Care Improvement (BPCI) Initiative.
In part one of this series, we discussed the basics of the BPCI program. Now we will delve into specific roles and opportunities for hospitalists in bundled payment programs in general, and the BPCI program in particular.
The bundled payment model can be hard to explain. One example that might make it clearer is that of LASIK vision correction surgery, where a single bundled payment covers the fees of the ophthalmologist, the operating facility, and any other services (like optometry) and medications (like eye drops). Another example is the diagnosis-related group (DRG) payment for hospital care, in which all facility costs are bundled together into a single payment.
A simplistic way to differentiate bundled payment from accountable care organization (ACOs) is that the former is typically initiated by an acute medical or surgical event and concludes after a recovery period—often 30, 60, or 90 days. Conversely, the latter generally covers the care of individuals within a population over time, often focusing on the management of chronic conditions.
The Opportunity
Two major opportunities for hospitalists to improve value (quality/cost) present themselves through the BPCI initiative. One is in post-acute facility utilization, and the other is in reducing readmissions. Figure 1 shows that for 30-day episodes starting with a hospitalization for five common conditions, payments for post-acute care are surprisingly close in amount to those for the preceding hospitalization.1
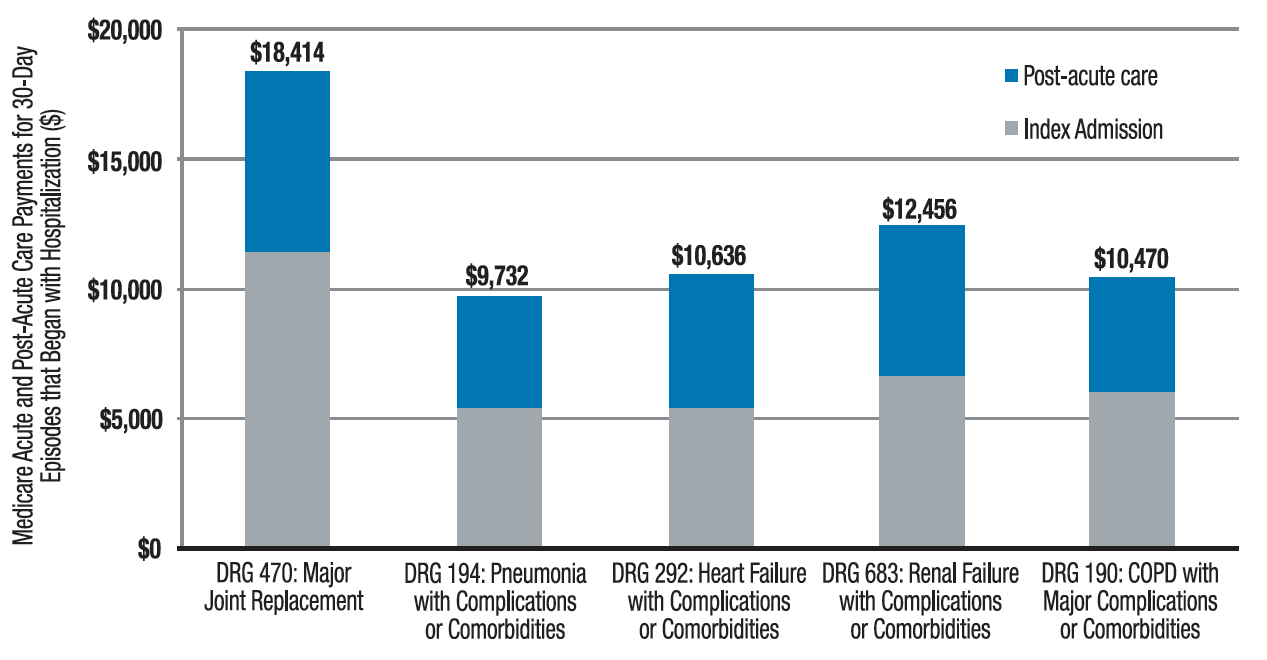
Much of the cost of post-acute care comes from skilled nursing facilities (SNFs) and, to a lesser degree, inpatient rehabilitation facilities. A broad range of research studies has demonstrated that inpatient care managed by hospitalists—compared with the traditional model—is associated with a decrease in inpatient costs; however, recent research indicates that the hospital cost savings generated by hospitalists are offset by more spending in the 30 days post discharge, specifically on more SNF care and increased readmissions.2 As another indicator that post-acute care needs a closer look, a 2013 Institute of Medicine report concluded that spending on post-acute care was responsible for the majority of Medicare’s overall regional variation in spending.1,3
Of course, success in a bundled payment model will also be derived from reducing costs in the hospital setting, such as those stemming from unnecessary or duplicative testing and imaging, injudicious use of consultants, and practices identified in programs such as Choosing Wisely.
How Your Practice Can Drive Bundled Payment Success
The aforementioned observations point to the need to improve the value of post-acute care by optimizing post-acute spending—driven mostly by SNF costs—and minimizing avoidable readmissions. I offer the following inpatient interventions to achieve these goals:
- Speak with patients early and often regarding expectations for recovery post discharge. When possible, set a goal of home discharge with the needed support.
- Write orders for early ambulation. Develop an early ambulation program with nursing and physical therapy.
- Address goals of care during the patient/family meeting. For appropriate patients with life-limiting illness, involve the palliative care service or equivalent and discuss the role of future aggressive interventions, including hospitalization, so that the course set is consistent with the patients’ goals and wishes.
- Lead the in-hospital team, instead of defaulting to others, like case management, in making an informed decision about ideal post-discharge location by factoring in caregiver availability, independence, and SNF needs. Marshal resources to enable a home recovery (i.e., home health evaluation), whether or not there is an intervening SNF stay. If patients go to a SNF, set expectations for length of stay in the facility.
- Adhere to best practices for care transitions, such as those in Project BOOST, including thorough medication reconciliation.
Beyond the Four Walls
As you aim for a high-value (high quality and affordable) discharge, your hospital medicine practice may consider new approaches to filling longstanding gaps in post-acute care. Forward-looking hospitalist groups have implemented the following approaches:
- Establish a post-discharge clinic where patients are seen after discharge, in the interim before they have an opportunity for primary care follow-up;
- Send teams to work in SNFs;
- Call patients after discharge to ensure they are following their plan of care;
- Leverage newer current procedural terminology (CPT) codes, like the Transitional Care Management or Chronic Care Management codes, to support your transitional care services;
- Provide home visits for high-risk patients; and
- Access waivers for G-codes for home visits and/or telemedicine outside of rural areas. These waivers exist under the BPCI initiative.
Shift from ‘Traditional’ Hospitalist to ‘Value’ Hospitalist
If some of the changes in practice needed to succeed in a bundled payment world seem daunting to you, it may be helpful to realize that with the challenge comes an opportunity. This opportunity for hospitalists parallels that of the early days of the specialty, when reducing length of stay created substantial support from hospital leaders and was a factor leading to the rapid growth in the number of hospitalists. In January, the U.S. Secretary of Health and Human Services set a goal to tie 50% of Medicare payments to ‘alternative payment models’ like bundled payments by 2018. In April, as part of the sustainable growth rate fix, Medicare announced it would create substantial new bonuses for physicians who have at least 25% of their revenue in such models.1
As healthcare policy aligns behind ‘alternative payment models,’ bundled payment programs are likely to be a potent driver of an evolving hospitalist specialty. Next-generation hospitalists will be asked to take a leadership role in addressing ‘value’ with responsibility for improving care coordination and affordability over an episode of illness.
Now may be the time to take to heart the words of computer scientist Alan Kay: “The best way to predict the future is to invent it.”

References
- Mechanic R. Post-acute care–the next frontier for controlling Medicare spending. N Engl J Med. 2014;370(8):692-694.
- Kuo YF, Goodwin JS. Association of hospitalist care with medical utilization after discharge: evidence of cost shift from a cohort study. Ann Intern Med. 2011;155(3):152-159.
- Newhouse JP, Garber AM. Geographic variation in Medicare services. N Engl J Med. 2013;368(16):1465-1468.
Editor’s note: Second in a two-part series examining bundled payments and hospital medicine. In full disclosure, Dr. Whitcomb works for a company that is an Awardee Convener in the CMS Bundled Payments for Care Improvement (BPCI) Initiative.
In part one of this series, we discussed the basics of the BPCI program. Now we will delve into specific roles and opportunities for hospitalists in bundled payment programs in general, and the BPCI program in particular.
The bundled payment model can be hard to explain. One example that might make it clearer is that of LASIK vision correction surgery, where a single bundled payment covers the fees of the ophthalmologist, the operating facility, and any other services (like optometry) and medications (like eye drops). Another example is the diagnosis-related group (DRG) payment for hospital care, in which all facility costs are bundled together into a single payment.
A simplistic way to differentiate bundled payment from accountable care organization (ACOs) is that the former is typically initiated by an acute medical or surgical event and concludes after a recovery period—often 30, 60, or 90 days. Conversely, the latter generally covers the care of individuals within a population over time, often focusing on the management of chronic conditions.
The Opportunity
Two major opportunities for hospitalists to improve value (quality/cost) present themselves through the BPCI initiative. One is in post-acute facility utilization, and the other is in reducing readmissions. Figure 1 shows that for 30-day episodes starting with a hospitalization for five common conditions, payments for post-acute care are surprisingly close in amount to those for the preceding hospitalization.1
Much of the cost of post-acute care comes from skilled nursing facilities (SNFs) and, to a lesser degree, inpatient rehabilitation facilities. A broad range of research studies has demonstrated that inpatient care managed by hospitalists—compared with the traditional model—is associated with a decrease in inpatient costs; however, recent research indicates that the hospital cost savings generated by hospitalists are offset by more spending in the 30 days post discharge, specifically on more SNF care and increased readmissions.2 As another indicator that post-acute care needs a closer look, a 2013 Institute of Medicine report concluded that spending on post-acute care was responsible for the majority of Medicare’s overall regional variation in spending.1,3
Of course, success in a bundled payment model will also be derived from reducing costs in the hospital setting, such as those stemming from unnecessary or duplicative testing and imaging, injudicious use of consultants, and practices identified in programs such as Choosing Wisely.
How Your Practice Can Drive Bundled Payment Success
The aforementioned observations point to the need to improve the value of post-acute care by optimizing post-acute spending—driven mostly by SNF costs—and minimizing avoidable readmissions. I offer the following inpatient interventions to achieve these goals:
- Speak with patients early and often regarding expectations for recovery post discharge. When possible, set a goal of home discharge with the needed support.
- Write orders for early ambulation. Develop an early ambulation program with nursing and physical therapy.
- Address goals of care during the patient/family meeting. For appropriate patients with life-limiting illness, involve the palliative care service or equivalent and discuss the role of future aggressive interventions, including hospitalization, so that the course set is consistent with the patients’ goals and wishes.
- Lead the in-hospital team, instead of defaulting to others, like case management, in making an informed decision about ideal post-discharge location by factoring in caregiver availability, independence, and SNF needs. Marshal resources to enable a home recovery (i.e., home health evaluation), whether or not there is an intervening SNF stay. If patients go to a SNF, set expectations for length of stay in the facility.
- Adhere to best practices for care transitions, such as those in Project BOOST, including thorough medication reconciliation.
Beyond the Four Walls
As you aim for a high-value (high quality and affordable) discharge, your hospital medicine practice may consider new approaches to filling longstanding gaps in post-acute care. Forward-looking hospitalist groups have implemented the following approaches:
- Establish a post-discharge clinic where patients are seen after discharge, in the interim before they have an opportunity for primary care follow-up;
- Send teams to work in SNFs;
- Call patients after discharge to ensure they are following their plan of care;
- Leverage newer current procedural terminology (CPT) codes, like the Transitional Care Management or Chronic Care Management codes, to support your transitional care services;
- Provide home visits for high-risk patients; and
- Access waivers for G-codes for home visits and/or telemedicine outside of rural areas. These waivers exist under the BPCI initiative.
Shift from ‘Traditional’ Hospitalist to ‘Value’ Hospitalist
If some of the changes in practice needed to succeed in a bundled payment world seem daunting to you, it may be helpful to realize that with the challenge comes an opportunity. This opportunity for hospitalists parallels that of the early days of the specialty, when reducing length of stay created substantial support from hospital leaders and was a factor leading to the rapid growth in the number of hospitalists. In January, the U.S. Secretary of Health and Human Services set a goal to tie 50% of Medicare payments to ‘alternative payment models’ like bundled payments by 2018. In April, as part of the sustainable growth rate fix, Medicare announced it would create substantial new bonuses for physicians who have at least 25% of their revenue in such models.1
As healthcare policy aligns behind ‘alternative payment models,’ bundled payment programs are likely to be a potent driver of an evolving hospitalist specialty. Next-generation hospitalists will be asked to take a leadership role in addressing ‘value’ with responsibility for improving care coordination and affordability over an episode of illness.
Now may be the time to take to heart the words of computer scientist Alan Kay: “The best way to predict the future is to invent it.”
References
- Mechanic R. Post-acute care–the next frontier for controlling Medicare spending. N Engl J Med. 2014;370(8):692-694.
- Kuo YF, Goodwin JS. Association of hospitalist care with medical utilization after discharge: evidence of cost shift from a cohort study. Ann Intern Med. 2011;155(3):152-159.
- Newhouse JP, Garber AM. Geographic variation in Medicare services. N Engl J Med. 2013;368(16):1465-1468.
Editor’s note: Second in a two-part series examining bundled payments and hospital medicine. In full disclosure, Dr. Whitcomb works for a company that is an Awardee Convener in the CMS Bundled Payments for Care Improvement (BPCI) Initiative.
In part one of this series, we discussed the basics of the BPCI program. Now we will delve into specific roles and opportunities for hospitalists in bundled payment programs in general, and the BPCI program in particular.
The bundled payment model can be hard to explain. One example that might make it clearer is that of LASIK vision correction surgery, where a single bundled payment covers the fees of the ophthalmologist, the operating facility, and any other services (like optometry) and medications (like eye drops). Another example is the diagnosis-related group (DRG) payment for hospital care, in which all facility costs are bundled together into a single payment.
A simplistic way to differentiate bundled payment from accountable care organization (ACOs) is that the former is typically initiated by an acute medical or surgical event and concludes after a recovery period—often 30, 60, or 90 days. Conversely, the latter generally covers the care of individuals within a population over time, often focusing on the management of chronic conditions.
The Opportunity
Two major opportunities for hospitalists to improve value (quality/cost) present themselves through the BPCI initiative. One is in post-acute facility utilization, and the other is in reducing readmissions. Figure 1 shows that for 30-day episodes starting with a hospitalization for five common conditions, payments for post-acute care are surprisingly close in amount to those for the preceding hospitalization.1
Much of the cost of post-acute care comes from skilled nursing facilities (SNFs) and, to a lesser degree, inpatient rehabilitation facilities. A broad range of research studies has demonstrated that inpatient care managed by hospitalists—compared with the traditional model—is associated with a decrease in inpatient costs; however, recent research indicates that the hospital cost savings generated by hospitalists are offset by more spending in the 30 days post discharge, specifically on more SNF care and increased readmissions.2 As another indicator that post-acute care needs a closer look, a 2013 Institute of Medicine report concluded that spending on post-acute care was responsible for the majority of Medicare’s overall regional variation in spending.1,3
Of course, success in a bundled payment model will also be derived from reducing costs in the hospital setting, such as those stemming from unnecessary or duplicative testing and imaging, injudicious use of consultants, and practices identified in programs such as Choosing Wisely.
How Your Practice Can Drive Bundled Payment Success
The aforementioned observations point to the need to improve the value of post-acute care by optimizing post-acute spending—driven mostly by SNF costs—and minimizing avoidable readmissions. I offer the following inpatient interventions to achieve these goals:
- Speak with patients early and often regarding expectations for recovery post discharge. When possible, set a goal of home discharge with the needed support.
- Write orders for early ambulation. Develop an early ambulation program with nursing and physical therapy.
- Address goals of care during the patient/family meeting. For appropriate patients with life-limiting illness, involve the palliative care service or equivalent and discuss the role of future aggressive interventions, including hospitalization, so that the course set is consistent with the patients’ goals and wishes.
- Lead the in-hospital team, instead of defaulting to others, like case management, in making an informed decision about ideal post-discharge location by factoring in caregiver availability, independence, and SNF needs. Marshal resources to enable a home recovery (i.e., home health evaluation), whether or not there is an intervening SNF stay. If patients go to a SNF, set expectations for length of stay in the facility.
- Adhere to best practices for care transitions, such as those in Project BOOST, including thorough medication reconciliation.
Beyond the Four Walls
As you aim for a high-value (high quality and affordable) discharge, your hospital medicine practice may consider new approaches to filling longstanding gaps in post-acute care. Forward-looking hospitalist groups have implemented the following approaches:
- Establish a post-discharge clinic where patients are seen after discharge, in the interim before they have an opportunity for primary care follow-up;
- Send teams to work in SNFs;
- Call patients after discharge to ensure they are following their plan of care;
- Leverage newer current procedural terminology (CPT) codes, like the Transitional Care Management or Chronic Care Management codes, to support your transitional care services;
- Provide home visits for high-risk patients; and
- Access waivers for G-codes for home visits and/or telemedicine outside of rural areas. These waivers exist under the BPCI initiative.
Shift from ‘Traditional’ Hospitalist to ‘Value’ Hospitalist
If some of the changes in practice needed to succeed in a bundled payment world seem daunting to you, it may be helpful to realize that with the challenge comes an opportunity. This opportunity for hospitalists parallels that of the early days of the specialty, when reducing length of stay created substantial support from hospital leaders and was a factor leading to the rapid growth in the number of hospitalists. In January, the U.S. Secretary of Health and Human Services set a goal to tie 50% of Medicare payments to ‘alternative payment models’ like bundled payments by 2018. In April, as part of the sustainable growth rate fix, Medicare announced it would create substantial new bonuses for physicians who have at least 25% of their revenue in such models.1
As healthcare policy aligns behind ‘alternative payment models,’ bundled payment programs are likely to be a potent driver of an evolving hospitalist specialty. Next-generation hospitalists will be asked to take a leadership role in addressing ‘value’ with responsibility for improving care coordination and affordability over an episode of illness.
Now may be the time to take to heart the words of computer scientist Alan Kay: “The best way to predict the future is to invent it.”
References
- Mechanic R. Post-acute care–the next frontier for controlling Medicare spending. N Engl J Med. 2014;370(8):692-694.
- Kuo YF, Goodwin JS. Association of hospitalist care with medical utilization after discharge: evidence of cost shift from a cohort study. Ann Intern Med. 2011;155(3):152-159.
- Newhouse JP, Garber AM. Geographic variation in Medicare services. N Engl J Med. 2013;368(16):1465-1468.
Standard Text Messaging for Smartphones Not HIPAA Compliant

Doctors were the first to begin using pagers and, along with drug dealers, appear to be the last to give them up. But we really need to get rid of them.
Sadly, for the foreseeable future, we will need a pager replacement, but, in the longer term, I’m hopeful that we can:
- Reduce the frequency of electronic interruptions—all forms of interruptions—and the adverse effects that reliably accompany them, and
- Ensure that each interruption has value—that is, reduce or eliminate the many low value and non-urgent messages we all get (e.g. the ones informing you of a lab result you’ve already seen).
Death to the Pager
I can’t imagine anyone who will be more pleased than I will if pagers go the way of now rare hospital-wide PA announcements. Some hospitals have eliminated these announcements entirely, and even critical messages like “code blue” announcements are sent directly to each responder via a pager or other personal device.
Around the time the first iPhone was born, hospital signs banning cell phones began coming down. It seems the fear that they would disrupt hospital electronics, such as telemetry and other monitoring devices, has proven largely unfounded (though, along with things like computer keyboards and stethoscopes, pagers and cell phones can serve as dangerous repositories of bacteria).
Now nearly everyone, from staff to patients, keeps a cell phone with them while in the hospital. I think that is the most important step toward getting rid of pagers. Many doctors already are using the standard text messaging apps that come with the phone to communicate with one another efficiently.
“Regular” Texting Won’t Cut It
Unfortunately, the standard text messaging that comes with every smartphone is not HIPAA compliant. Though I certainly don’t know how anyone would do it, it is apparently too easy for another person to intercept the message. So, if you’re texting information related to your clinical work, you need to make sure it doesn’t include anything that could be considered protected health information. It isn’t enough just to leave the patient’s name off the message. If you’re in the habit of regularly texting doctors, nurses, and other healthcare personnel about patient care, you are at high risk of violating HIPAA, even if you try hard to avoid it.
Another big drawback is that there isn’t a good way to turn off work-related texting when you’re off duty, while leaving your texting app open for communication with your friends and family. Hospital staff will sometimes fail to check whether you’re on duty before texting, and that will lead to your personal time being interrupted by work reminders.
I think these shortcomings mean that none of us should rely on the standard text messaging apps that come with our phones.
But in order for a different app or service to be of any value, we will need to ensure that most providers associated with our hospital are on the same messaging system. That is a tall order, but fortunately there are a lot of companies trying to produce an attractive product that makes it as easy as possible to attract a critical mass of users at your institution.
HIPAA-Compliant Texting Vendors
Many healthcare tech companies provide secure messaging, usually at no additional cost, as an add-on to their main products, such as charge capture software (e.g. IngeniousMed), or physician social networking (e.g. Doximity). Something like 30 companies now offer a dedicated HIPAA-compliant texting option, including IM Your Doc, Voalte, Telmediq, PerfectServe, Vocera, and TigerText. There are so many that it is awfully tough to understand all of their strengths and shortcomings in detail, but I’m having fun trying to do just that. And I anticipate there will be significant consolidation in vendors within the next two to three years.
The dedicated HIPAA-compliant texting services range in price from free for basic features to a monthly fee per user that varies depending on the features you choose to enable. Some offer integration with the hospital’s EHR, which can let a message sender who only knows the patient’s name to see which doctor, nurse, or other caregiver is currently responsible for the patient. Some offer integration with a call schedule and answering service, or even replace an answering service.
No pager replacement will be viable if there are sites in the hospital or elsewhere where it is out of contact; a solution that works on both cellular networks and Wi-Fi is essential. Some vendors offer the ability for messages not delivered to or acknowledged by the recipient to escalate to other forms of delivery after a specified period of time.
I would love to see a feature that I don’t think any vendor offers yet. It would be great if all messages the sender hasn’t marked “stat” or “urgent” first went to a queue in the EHR rather than immediately interrupting the recipient. That way a doctor or other caregiver could see messages while already working in the EHR, rather than glancing at each new message as it arrives, something that all too often needlessly interrupts another important task such as talking with a patient.
And, since most work in EHRs is done in front of a larger device with a full keyboard, it would be easier to type a quick reply message than it would be to rely on a smartphone keyboard for return messaging. Protocols could be established such that messages waiting in the EHR without a reply or dismissal after a specified time would then be sent to the recipient’s personal device.
A Texting Ecosystem
In nearly every case, the hospital will select the text messaging vendor, though hospitalists and nurses, who will typically be among the highest-volume users, should participate in the decision. But the real value of the system hinges on ensuring its wide adoption by most, or nearly all, hospital caregivers and affiliated ambulatory providers.
I would enjoy hearing from those who are already using a HIPAA-secure texting and pager replacement service now, as well as those still researching their options. This has the potential to meaningfully change the way hospitalists and others do their work.

Doctors were the first to begin using pagers and, along with drug dealers, appear to be the last to give them up. But we really need to get rid of them.
Sadly, for the foreseeable future, we will need a pager replacement, but, in the longer term, I’m hopeful that we can:
- Reduce the frequency of electronic interruptions—all forms of interruptions—and the adverse effects that reliably accompany them, and
- Ensure that each interruption has value—that is, reduce or eliminate the many low value and non-urgent messages we all get (e.g. the ones informing you of a lab result you’ve already seen).
Death to the Pager
I can’t imagine anyone who will be more pleased than I will if pagers go the way of now rare hospital-wide PA announcements. Some hospitals have eliminated these announcements entirely, and even critical messages like “code blue” announcements are sent directly to each responder via a pager or other personal device.
Around the time the first iPhone was born, hospital signs banning cell phones began coming down. It seems the fear that they would disrupt hospital electronics, such as telemetry and other monitoring devices, has proven largely unfounded (though, along with things like computer keyboards and stethoscopes, pagers and cell phones can serve as dangerous repositories of bacteria).
Now nearly everyone, from staff to patients, keeps a cell phone with them while in the hospital. I think that is the most important step toward getting rid of pagers. Many doctors already are using the standard text messaging apps that come with the phone to communicate with one another efficiently.
“Regular” Texting Won’t Cut It
Unfortunately, the standard text messaging that comes with every smartphone is not HIPAA compliant. Though I certainly don’t know how anyone would do it, it is apparently too easy for another person to intercept the message. So, if you’re texting information related to your clinical work, you need to make sure it doesn’t include anything that could be considered protected health information. It isn’t enough just to leave the patient’s name off the message. If you’re in the habit of regularly texting doctors, nurses, and other healthcare personnel about patient care, you are at high risk of violating HIPAA, even if you try hard to avoid it.
Another big drawback is that there isn’t a good way to turn off work-related texting when you’re off duty, while leaving your texting app open for communication with your friends and family. Hospital staff will sometimes fail to check whether you’re on duty before texting, and that will lead to your personal time being interrupted by work reminders.
I think these shortcomings mean that none of us should rely on the standard text messaging apps that come with our phones.
But in order for a different app or service to be of any value, we will need to ensure that most providers associated with our hospital are on the same messaging system. That is a tall order, but fortunately there are a lot of companies trying to produce an attractive product that makes it as easy as possible to attract a critical mass of users at your institution.
HIPAA-Compliant Texting Vendors
Many healthcare tech companies provide secure messaging, usually at no additional cost, as an add-on to their main products, such as charge capture software (e.g. IngeniousMed), or physician social networking (e.g. Doximity). Something like 30 companies now offer a dedicated HIPAA-compliant texting option, including IM Your Doc, Voalte, Telmediq, PerfectServe, Vocera, and TigerText. There are so many that it is awfully tough to understand all of their strengths and shortcomings in detail, but I’m having fun trying to do just that. And I anticipate there will be significant consolidation in vendors within the next two to three years.
The dedicated HIPAA-compliant texting services range in price from free for basic features to a monthly fee per user that varies depending on the features you choose to enable. Some offer integration with the hospital’s EHR, which can let a message sender who only knows the patient’s name to see which doctor, nurse, or other caregiver is currently responsible for the patient. Some offer integration with a call schedule and answering service, or even replace an answering service.
No pager replacement will be viable if there are sites in the hospital or elsewhere where it is out of contact; a solution that works on both cellular networks and Wi-Fi is essential. Some vendors offer the ability for messages not delivered to or acknowledged by the recipient to escalate to other forms of delivery after a specified period of time.
I would love to see a feature that I don’t think any vendor offers yet. It would be great if all messages the sender hasn’t marked “stat” or “urgent” first went to a queue in the EHR rather than immediately interrupting the recipient. That way a doctor or other caregiver could see messages while already working in the EHR, rather than glancing at each new message as it arrives, something that all too often needlessly interrupts another important task such as talking with a patient.
And, since most work in EHRs is done in front of a larger device with a full keyboard, it would be easier to type a quick reply message than it would be to rely on a smartphone keyboard for return messaging. Protocols could be established such that messages waiting in the EHR without a reply or dismissal after a specified time would then be sent to the recipient’s personal device.
A Texting Ecosystem
In nearly every case, the hospital will select the text messaging vendor, though hospitalists and nurses, who will typically be among the highest-volume users, should participate in the decision. But the real value of the system hinges on ensuring its wide adoption by most, or nearly all, hospital caregivers and affiliated ambulatory providers.
I would enjoy hearing from those who are already using a HIPAA-secure texting and pager replacement service now, as well as those still researching their options. This has the potential to meaningfully change the way hospitalists and others do their work.
Doctors were the first to begin using pagers and, along with drug dealers, appear to be the last to give them up. But we really need to get rid of them.
Sadly, for the foreseeable future, we will need a pager replacement, but, in the longer term, I’m hopeful that we can:
- Reduce the frequency of electronic interruptions—all forms of interruptions—and the adverse effects that reliably accompany them, and
- Ensure that each interruption has value—that is, reduce or eliminate the many low value and non-urgent messages we all get (e.g. the ones informing you of a lab result you’ve already seen).
Death to the Pager
I can’t imagine anyone who will be more pleased than I will if pagers go the way of now rare hospital-wide PA announcements. Some hospitals have eliminated these announcements entirely, and even critical messages like “code blue” announcements are sent directly to each responder via a pager or other personal device.
Around the time the first iPhone was born, hospital signs banning cell phones began coming down. It seems the fear that they would disrupt hospital electronics, such as telemetry and other monitoring devices, has proven largely unfounded (though, along with things like computer keyboards and stethoscopes, pagers and cell phones can serve as dangerous repositories of bacteria).
Now nearly everyone, from staff to patients, keeps a cell phone with them while in the hospital. I think that is the most important step toward getting rid of pagers. Many doctors already are using the standard text messaging apps that come with the phone to communicate with one another efficiently.
“Regular” Texting Won’t Cut It
Unfortunately, the standard text messaging that comes with every smartphone is not HIPAA compliant. Though I certainly don’t know how anyone would do it, it is apparently too easy for another person to intercept the message. So, if you’re texting information related to your clinical work, you need to make sure it doesn’t include anything that could be considered protected health information. It isn’t enough just to leave the patient’s name off the message. If you’re in the habit of regularly texting doctors, nurses, and other healthcare personnel about patient care, you are at high risk of violating HIPAA, even if you try hard to avoid it.
Another big drawback is that there isn’t a good way to turn off work-related texting when you’re off duty, while leaving your texting app open for communication with your friends and family. Hospital staff will sometimes fail to check whether you’re on duty before texting, and that will lead to your personal time being interrupted by work reminders.
I think these shortcomings mean that none of us should rely on the standard text messaging apps that come with our phones.
But in order for a different app or service to be of any value, we will need to ensure that most providers associated with our hospital are on the same messaging system. That is a tall order, but fortunately there are a lot of companies trying to produce an attractive product that makes it as easy as possible to attract a critical mass of users at your institution.
HIPAA-Compliant Texting Vendors
Many healthcare tech companies provide secure messaging, usually at no additional cost, as an add-on to their main products, such as charge capture software (e.g. IngeniousMed), or physician social networking (e.g. Doximity). Something like 30 companies now offer a dedicated HIPAA-compliant texting option, including IM Your Doc, Voalte, Telmediq, PerfectServe, Vocera, and TigerText. There are so many that it is awfully tough to understand all of their strengths and shortcomings in detail, but I’m having fun trying to do just that. And I anticipate there will be significant consolidation in vendors within the next two to three years.
The dedicated HIPAA-compliant texting services range in price from free for basic features to a monthly fee per user that varies depending on the features you choose to enable. Some offer integration with the hospital’s EHR, which can let a message sender who only knows the patient’s name to see which doctor, nurse, or other caregiver is currently responsible for the patient. Some offer integration with a call schedule and answering service, or even replace an answering service.
No pager replacement will be viable if there are sites in the hospital or elsewhere where it is out of contact; a solution that works on both cellular networks and Wi-Fi is essential. Some vendors offer the ability for messages not delivered to or acknowledged by the recipient to escalate to other forms of delivery after a specified period of time.
I would love to see a feature that I don’t think any vendor offers yet. It would be great if all messages the sender hasn’t marked “stat” or “urgent” first went to a queue in the EHR rather than immediately interrupting the recipient. That way a doctor or other caregiver could see messages while already working in the EHR, rather than glancing at each new message as it arrives, something that all too often needlessly interrupts another important task such as talking with a patient.
And, since most work in EHRs is done in front of a larger device with a full keyboard, it would be easier to type a quick reply message than it would be to rely on a smartphone keyboard for return messaging. Protocols could be established such that messages waiting in the EHR without a reply or dismissal after a specified time would then be sent to the recipient’s personal device.
A Texting Ecosystem
In nearly every case, the hospital will select the text messaging vendor, though hospitalists and nurses, who will typically be among the highest-volume users, should participate in the decision. But the real value of the system hinges on ensuring its wide adoption by most, or nearly all, hospital caregivers and affiliated ambulatory providers.
I would enjoy hearing from those who are already using a HIPAA-secure texting and pager replacement service now, as well as those still researching their options. This has the potential to meaningfully change the way hospitalists and others do their work.
AAS: Acute suicidal affective disturbance proposed as new diagnosis
ATLANTA – Among all deaths by suicide, 15%-20% are attributable to a previously undescribed entity that now has a name: acute suicidal affective disturbance, Thomas Joiner, Ph.D., said at the annual conference of the American Association of Suicidology.
“It’s real, it’s fairly common, and it’s extremely dangerous. We think it ranks up there among the most serious of conditions – more likely to result in death than the manic phase of bipolar disorder or schizophrenia, for example,” said Dr. Joiner, who first identified and characterized the condition, named it, plays a pivotal role in ongoing multicenter collaborative research efforts targeting it, and vows to see acute suicidal affective disturbance, or ASAD, included in the DSM-6.

ASAD is a concept compatible with Dr. Joiner’s “Interpersonal Theory of Suicide,” which has taken the field of suicidology by storm. But ASAD , he stressed, is not a theoretical construct.
“ASAD exists as an entity, as a true object in nature that I worry over because it kills people and we can’t diagnose it currently because there’s a gap in the nomenclature. We’re proposing this condition to fill that gap,” explained Dr. Joiner, professor of psychology at Florida State University, Tallahassee.
Roughly 80% of all deaths by suicide can be attributed to a recognized mental health disorder, such as major depressive disorder, the depressive phase of bipolar disorder, anorexia nervosa, schizophrenia, or borderline personality disorder. Dr. Joiner presented highlights of three separate studies now in press – one conducted in Veterans Affairs outpatients, another in high-risk university undergraduates, and a third involving nearly 7,700 Mayo Clinic psychiatric inpatients and more than 3,400 U.S. Army high-risk patients – all of which support the construct validity of ASAD as a distinct entity that correlates negatively with impulsivity and is unrelated to alcohol use disorders or mood disorders.
In addition, Dr. Joiner continued, three different research teams – his own; the Military Suicide Research Consortium, which he codirects; and the U.S. Army STARRS group – have identified subgroups of patients at very high suicide risk who are characterized by ASAD symptoms that are discernible from those of established clinical entities (see list for proposed ASAD criteria).
“We intend to put ASAD into DSM-6. We know that’s going to be a battle, and we’re ready for it. We’re determined to fight it,” the psychologist said.
If ASAD were to eventually become a diagnosable condition, the clinical implications would be far-reaching, he observed. It would be a billable entity. Also, its existence in the formal psychiatric nomenclature would alert clinicians to be vigilant regarding ASAD; that’s crucial because recurrences can be lethal, and recognition of the prodromal overarousal symptoms may head off a full-blown crisis.
“If we’re right about ASAD, it would imply that mood disorders and ASAD are distinct, that their onset and remission patterns are different, thus supporting the rationale of suicide-specific treatments like CAMS [collaborative assessment and management of suicidality] and DBT [dialectical behavior therapy]. It would suggest the need for intensive and long-lasting therapies, because a recurrence can cost people their lives,” he continued.
Should ASAD become legitimized as a diagnosis, it would shore up the public health argument in favor of suicide-means restriction as a strategy for forestalling suicidal action.
“ASAD is a time-limited arousal state. You can’t sustain it for more than an hour or a few hours. It’ll abate with the passage of time,” according to Dr. Joiner.
In response to an audience question, he said it’s his clinical impression – not yet supported by data – that the most common comorbid conditions in patients with ASAD are anxiety disorders and personality disorders. Also, ASAD doesn’t appear to be age dependent: “We think it can emerge at any point in the lifespan.”
Dr. Joiner emphasized that many questions regarding ASAD remain unanswered.
“We have a long way to go,” he conceded. “And I’m not talking about years here, I’m talking about decades. The effort is going to be primarily research oriented. It’s a scientific effort. We’ve got to clear the hurdle scientifically, and we haven’t done that yet, although we’ve gotten a start.”
He reported having no financial conflicts regarding his presentation.
Proposed criteria for ASAD
1. A sudden surge in suicidal intent occurring over the course of minutes, hours, or days rather than weeks or months.
2. One or both of two alienation criteria: a) severe social withdrawal defined by extreme disgust with others or perceived liability to others; b) marked self-alienation manifested as self-disgust or the view that one’s selfhood is an onerous burden.
3. The perception that numbers 1 and 2 are hopelessly intractable.
4. At least two of the following manifestations of overarousal: insomnia, nightmares, irritability, agitation.
5. Exclusion criteria: The above is not due to exacerbation of a mood disorder or ingestion of alcohol or another substance.
Source: Dr. Joiner
ATLANTA – Among all deaths by suicide, 15%-20% are attributable to a previously undescribed entity that now has a name: acute suicidal affective disturbance, Thomas Joiner, Ph.D., said at the annual conference of the American Association of Suicidology.
“It’s real, it’s fairly common, and it’s extremely dangerous. We think it ranks up there among the most serious of conditions – more likely to result in death than the manic phase of bipolar disorder or schizophrenia, for example,” said Dr. Joiner, who first identified and characterized the condition, named it, plays a pivotal role in ongoing multicenter collaborative research efforts targeting it, and vows to see acute suicidal affective disturbance, or ASAD, included in the DSM-6.
ASAD is a concept compatible with Dr. Joiner’s “Interpersonal Theory of Suicide,” which has taken the field of suicidology by storm. But ASAD , he stressed, is not a theoretical construct.
“ASAD exists as an entity, as a true object in nature that I worry over because it kills people and we can’t diagnose it currently because there’s a gap in the nomenclature. We’re proposing this condition to fill that gap,” explained Dr. Joiner, professor of psychology at Florida State University, Tallahassee.
Roughly 80% of all deaths by suicide can be attributed to a recognized mental health disorder, such as major depressive disorder, the depressive phase of bipolar disorder, anorexia nervosa, schizophrenia, or borderline personality disorder. Dr. Joiner presented highlights of three separate studies now in press – one conducted in Veterans Affairs outpatients, another in high-risk university undergraduates, and a third involving nearly 7,700 Mayo Clinic psychiatric inpatients and more than 3,400 U.S. Army high-risk patients – all of which support the construct validity of ASAD as a distinct entity that correlates negatively with impulsivity and is unrelated to alcohol use disorders or mood disorders.
In addition, Dr. Joiner continued, three different research teams – his own; the Military Suicide Research Consortium, which he codirects; and the U.S. Army STARRS group – have identified subgroups of patients at very high suicide risk who are characterized by ASAD symptoms that are discernible from those of established clinical entities (see list for proposed ASAD criteria).
“We intend to put ASAD into DSM-6. We know that’s going to be a battle, and we’re ready for it. We’re determined to fight it,” the psychologist said.
If ASAD were to eventually become a diagnosable condition, the clinical implications would be far-reaching, he observed. It would be a billable entity. Also, its existence in the formal psychiatric nomenclature would alert clinicians to be vigilant regarding ASAD; that’s crucial because recurrences can be lethal, and recognition of the prodromal overarousal symptoms may head off a full-blown crisis.
“If we’re right about ASAD, it would imply that mood disorders and ASAD are distinct, that their onset and remission patterns are different, thus supporting the rationale of suicide-specific treatments like CAMS [collaborative assessment and management of suicidality] and DBT [dialectical behavior therapy]. It would suggest the need for intensive and long-lasting therapies, because a recurrence can cost people their lives,” he continued.
Should ASAD become legitimized as a diagnosis, it would shore up the public health argument in favor of suicide-means restriction as a strategy for forestalling suicidal action.
“ASAD is a time-limited arousal state. You can’t sustain it for more than an hour or a few hours. It’ll abate with the passage of time,” according to Dr. Joiner.
In response to an audience question, he said it’s his clinical impression – not yet supported by data – that the most common comorbid conditions in patients with ASAD are anxiety disorders and personality disorders. Also, ASAD doesn’t appear to be age dependent: “We think it can emerge at any point in the lifespan.”
Dr. Joiner emphasized that many questions regarding ASAD remain unanswered.
“We have a long way to go,” he conceded. “And I’m not talking about years here, I’m talking about decades. The effort is going to be primarily research oriented. It’s a scientific effort. We’ve got to clear the hurdle scientifically, and we haven’t done that yet, although we’ve gotten a start.”
He reported having no financial conflicts regarding his presentation.
Proposed criteria for ASAD
1. A sudden surge in suicidal intent occurring over the course of minutes, hours, or days rather than weeks or months.
2. One or both of two alienation criteria: a) severe social withdrawal defined by extreme disgust with others or perceived liability to others; b) marked self-alienation manifested as self-disgust or the view that one’s selfhood is an onerous burden.
3. The perception that numbers 1 and 2 are hopelessly intractable.
4. At least two of the following manifestations of overarousal: insomnia, nightmares, irritability, agitation.
5. Exclusion criteria: The above is not due to exacerbation of a mood disorder or ingestion of alcohol or another substance.
Source: Dr. Joiner
ATLANTA – Among all deaths by suicide, 15%-20% are attributable to a previously undescribed entity that now has a name: acute suicidal affective disturbance, Thomas Joiner, Ph.D., said at the annual conference of the American Association of Suicidology.
“It’s real, it’s fairly common, and it’s extremely dangerous. We think it ranks up there among the most serious of conditions – more likely to result in death than the manic phase of bipolar disorder or schizophrenia, for example,” said Dr. Joiner, who first identified and characterized the condition, named it, plays a pivotal role in ongoing multicenter collaborative research efforts targeting it, and vows to see acute suicidal affective disturbance, or ASAD, included in the DSM-6.
ASAD is a concept compatible with Dr. Joiner’s “Interpersonal Theory of Suicide,” which has taken the field of suicidology by storm. But ASAD , he stressed, is not a theoretical construct.
“ASAD exists as an entity, as a true object in nature that I worry over because it kills people and we can’t diagnose it currently because there’s a gap in the nomenclature. We’re proposing this condition to fill that gap,” explained Dr. Joiner, professor of psychology at Florida State University, Tallahassee.
Roughly 80% of all deaths by suicide can be attributed to a recognized mental health disorder, such as major depressive disorder, the depressive phase of bipolar disorder, anorexia nervosa, schizophrenia, or borderline personality disorder. Dr. Joiner presented highlights of three separate studies now in press – one conducted in Veterans Affairs outpatients, another in high-risk university undergraduates, and a third involving nearly 7,700 Mayo Clinic psychiatric inpatients and more than 3,400 U.S. Army high-risk patients – all of which support the construct validity of ASAD as a distinct entity that correlates negatively with impulsivity and is unrelated to alcohol use disorders or mood disorders.
In addition, Dr. Joiner continued, three different research teams – his own; the Military Suicide Research Consortium, which he codirects; and the U.S. Army STARRS group – have identified subgroups of patients at very high suicide risk who are characterized by ASAD symptoms that are discernible from those of established clinical entities (see list for proposed ASAD criteria).
“We intend to put ASAD into DSM-6. We know that’s going to be a battle, and we’re ready for it. We’re determined to fight it,” the psychologist said.
If ASAD were to eventually become a diagnosable condition, the clinical implications would be far-reaching, he observed. It would be a billable entity. Also, its existence in the formal psychiatric nomenclature would alert clinicians to be vigilant regarding ASAD; that’s crucial because recurrences can be lethal, and recognition of the prodromal overarousal symptoms may head off a full-blown crisis.
“If we’re right about ASAD, it would imply that mood disorders and ASAD are distinct, that their onset and remission patterns are different, thus supporting the rationale of suicide-specific treatments like CAMS [collaborative assessment and management of suicidality] and DBT [dialectical behavior therapy]. It would suggest the need for intensive and long-lasting therapies, because a recurrence can cost people their lives,” he continued.
Should ASAD become legitimized as a diagnosis, it would shore up the public health argument in favor of suicide-means restriction as a strategy for forestalling suicidal action.
“ASAD is a time-limited arousal state. You can’t sustain it for more than an hour or a few hours. It’ll abate with the passage of time,” according to Dr. Joiner.
In response to an audience question, he said it’s his clinical impression – not yet supported by data – that the most common comorbid conditions in patients with ASAD are anxiety disorders and personality disorders. Also, ASAD doesn’t appear to be age dependent: “We think it can emerge at any point in the lifespan.”
Dr. Joiner emphasized that many questions regarding ASAD remain unanswered.
“We have a long way to go,” he conceded. “And I’m not talking about years here, I’m talking about decades. The effort is going to be primarily research oriented. It’s a scientific effort. We’ve got to clear the hurdle scientifically, and we haven’t done that yet, although we’ve gotten a start.”
He reported having no financial conflicts regarding his presentation.
Proposed criteria for ASAD
1. A sudden surge in suicidal intent occurring over the course of minutes, hours, or days rather than weeks or months.
2. One or both of two alienation criteria: a) severe social withdrawal defined by extreme disgust with others or perceived liability to others; b) marked self-alienation manifested as self-disgust or the view that one’s selfhood is an onerous burden.
3. The perception that numbers 1 and 2 are hopelessly intractable.
4. At least two of the following manifestations of overarousal: insomnia, nightmares, irritability, agitation.
5. Exclusion criteria: The above is not due to exacerbation of a mood disorder or ingestion of alcohol or another substance.
Source: Dr. Joiner
EXPERT ANALYSIS FROM THE ANNUAL AAS CONFERENCE

How can electronic fetal heart-rate monitoring best improve neonatal outcomes during induction of labor?
In this prospective cohort study of women undergoing induction of labor of a singleton fetus at term (≥37 weeks), Clark and colleagues examined each patient chart for adherence to 6 clinical practices:
- fetal weight estimated prior to induction
- clinical assessment of pelvic adequacy prior to induction
- completion of a safety checklist prior to induction
- completion of a safety checklist every 30 minutes during induction
- oxytocin infusion rate decreased if the fetal heart rate did not meet defined requirements
- oxytocin infusion rate decreased if uterine activity did not meet defined requirements.
Defined requirements for fetal heart rate included at least 1 acceleration of 15 bpm x 15 seconds in 30 minutes, or adequate variability for 10 of the previous 30 minutes. There should have been no more than 1 late deceleration in the previous 30 minutes. And there should have been no more than 2 variable decelerations exceeding 60 seconds and decreasing more than 60 bpm from baseline in the previous 30 minutes.
Defined requirements for uterine activity included no more than 5 contractions in 10 minutes for any 20-minute interval. There should have been no 2 contractions longer than 120 seconds in duration. In addition, the uterus should have been soft upon palpation between contractions. If an intrauterine pressure catheter was in place, measurement in Montevideo units should have been less than 300 mm Hg, with baseline resting tone of less than 25 mm Hg.
Outcome measures included admission to a neonatal intensive care unit (NICU), 1- and 5-minute Apgar scores of less than 7, and primary cesarean delivery.
Study findings underscore value of a checklist
The study found that completion of a safety checklist every 30 minutes during labor was associated with a significantly reduced rate of NICU admission and cesarean delivery. When the clinician stopped or reduced oxytocin for failure to meet specific uterine activity requirements, the rate of NICU admission also was reduced, but there were no differences in other outcomes. When the clinician stopped or reduced oxytocin for a failure to meet specific fetal heart-rate requirements, the rate of NICU admission was significantly reduced, as was the rate of low Apgar scores at birth, but there was a significantly higher rate of cesarean delivery (26.6% vs 17.5%).
Strengths include size of the study
This is a large study in a population with demographics that likely reflect those of the general US population. The study size permitted detection of relatively small differences in outcome.
The data clearly support the conclusions that “electronic fetal heart-rate monitoring improves neonatal outcomes when unambiguous definitions of abnormal fetal heart rate and tachysystole are coupled with specific interventions” and that “utilization of a checklist for oxytocin monitoring is associated with improved neonatal outcomes.” However, data were conflicting regarding the impact of a standard oxytocin checklist on the rate of cesarean delivery.
What this evidence means for practice
Implementation of a conservative intrapartum checklist has been shown to improve specific measures of newborn outcome. Further study is needed to define the impact on the rate of cesarean delivery.
—David A. Miller, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In this prospective cohort study of women undergoing induction of labor of a singleton fetus at term (≥37 weeks), Clark and colleagues examined each patient chart for adherence to 6 clinical practices:
- fetal weight estimated prior to induction
- clinical assessment of pelvic adequacy prior to induction
- completion of a safety checklist prior to induction
- completion of a safety checklist every 30 minutes during induction
- oxytocin infusion rate decreased if the fetal heart rate did not meet defined requirements
- oxytocin infusion rate decreased if uterine activity did not meet defined requirements.
Defined requirements for fetal heart rate included at least 1 acceleration of 15 bpm x 15 seconds in 30 minutes, or adequate variability for 10 of the previous 30 minutes. There should have been no more than 1 late deceleration in the previous 30 minutes. And there should have been no more than 2 variable decelerations exceeding 60 seconds and decreasing more than 60 bpm from baseline in the previous 30 minutes.
Defined requirements for uterine activity included no more than 5 contractions in 10 minutes for any 20-minute interval. There should have been no 2 contractions longer than 120 seconds in duration. In addition, the uterus should have been soft upon palpation between contractions. If an intrauterine pressure catheter was in place, measurement in Montevideo units should have been less than 300 mm Hg, with baseline resting tone of less than 25 mm Hg.
Outcome measures included admission to a neonatal intensive care unit (NICU), 1- and 5-minute Apgar scores of less than 7, and primary cesarean delivery.
Study findings underscore value of a checklist
The study found that completion of a safety checklist every 30 minutes during labor was associated with a significantly reduced rate of NICU admission and cesarean delivery. When the clinician stopped or reduced oxytocin for failure to meet specific uterine activity requirements, the rate of NICU admission also was reduced, but there were no differences in other outcomes. When the clinician stopped or reduced oxytocin for a failure to meet specific fetal heart-rate requirements, the rate of NICU admission was significantly reduced, as was the rate of low Apgar scores at birth, but there was a significantly higher rate of cesarean delivery (26.6% vs 17.5%).
Strengths include size of the study
This is a large study in a population with demographics that likely reflect those of the general US population. The study size permitted detection of relatively small differences in outcome.
The data clearly support the conclusions that “electronic fetal heart-rate monitoring improves neonatal outcomes when unambiguous definitions of abnormal fetal heart rate and tachysystole are coupled with specific interventions” and that “utilization of a checklist for oxytocin monitoring is associated with improved neonatal outcomes.” However, data were conflicting regarding the impact of a standard oxytocin checklist on the rate of cesarean delivery.
What this evidence means for practice
Implementation of a conservative intrapartum checklist has been shown to improve specific measures of newborn outcome. Further study is needed to define the impact on the rate of cesarean delivery.
—David A. Miller, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In this prospective cohort study of women undergoing induction of labor of a singleton fetus at term (≥37 weeks), Clark and colleagues examined each patient chart for adherence to 6 clinical practices:
- fetal weight estimated prior to induction
- clinical assessment of pelvic adequacy prior to induction
- completion of a safety checklist prior to induction
- completion of a safety checklist every 30 minutes during induction
- oxytocin infusion rate decreased if the fetal heart rate did not meet defined requirements
- oxytocin infusion rate decreased if uterine activity did not meet defined requirements.
Defined requirements for fetal heart rate included at least 1 acceleration of 15 bpm x 15 seconds in 30 minutes, or adequate variability for 10 of the previous 30 minutes. There should have been no more than 1 late deceleration in the previous 30 minutes. And there should have been no more than 2 variable decelerations exceeding 60 seconds and decreasing more than 60 bpm from baseline in the previous 30 minutes.
Defined requirements for uterine activity included no more than 5 contractions in 10 minutes for any 20-minute interval. There should have been no 2 contractions longer than 120 seconds in duration. In addition, the uterus should have been soft upon palpation between contractions. If an intrauterine pressure catheter was in place, measurement in Montevideo units should have been less than 300 mm Hg, with baseline resting tone of less than 25 mm Hg.
Outcome measures included admission to a neonatal intensive care unit (NICU), 1- and 5-minute Apgar scores of less than 7, and primary cesarean delivery.
Study findings underscore value of a checklist
The study found that completion of a safety checklist every 30 minutes during labor was associated with a significantly reduced rate of NICU admission and cesarean delivery. When the clinician stopped or reduced oxytocin for failure to meet specific uterine activity requirements, the rate of NICU admission also was reduced, but there were no differences in other outcomes. When the clinician stopped or reduced oxytocin for a failure to meet specific fetal heart-rate requirements, the rate of NICU admission was significantly reduced, as was the rate of low Apgar scores at birth, but there was a significantly higher rate of cesarean delivery (26.6% vs 17.5%).
Strengths include size of the study
This is a large study in a population with demographics that likely reflect those of the general US population. The study size permitted detection of relatively small differences in outcome.
The data clearly support the conclusions that “electronic fetal heart-rate monitoring improves neonatal outcomes when unambiguous definitions of abnormal fetal heart rate and tachysystole are coupled with specific interventions” and that “utilization of a checklist for oxytocin monitoring is associated with improved neonatal outcomes.” However, data were conflicting regarding the impact of a standard oxytocin checklist on the rate of cesarean delivery.
What this evidence means for practice
Implementation of a conservative intrapartum checklist has been shown to improve specific measures of newborn outcome. Further study is needed to define the impact on the rate of cesarean delivery.
—David A. Miller, MD
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
NCCI agrees to change recent edits regarding reconstructive procedures done at the time of vaginal hysterectomy
On October 1, 2014, the Centers for Medicare and Medicaid Services (CMS) and the National Correct Coding Initiative (NCCI) implemented a number of new coding pair edits that significantly restricted the types of surgical procedures that could be billed at the same time as vaginal hysterectomy. Most importantly, the new edits did not allow for combined anterior and posterior (AP) vaginal repair (57260) and apical vaginal suspensions (57282, 57283) to be separately billed and reimbursed when performed by the same surgeon and done in the same surgical session as a vaginal or laparoscopic hysterectomy. I initially reported on these changes in the January 2015 issue of OBG Management (“Recent NCCI edits have significantly impacted billing and reimbursement for vaginal hysterectomy”).
AUGS, ACOG, and others responded, and were heard
As a result of an intensive effort, the American Urogynecologic Society (AUGS), in conjunction with the American College of Obstetricians and Gynecologists (ACOG), the Society of Gynecologic Surgeons (SGS), and other professional societies, NCCI has agreed to change or modify a significant number of these pair edits.
As described in my earlier article, NCCI periodically reviews multiple surgical procedures performed in the same setting by a single surgeon to determine whether there is a significant overlap in services and therefore, in their opinion, redundant payment. Most significantly, CMS agreed with the argument made by the AUGS task force that it was inappropriate to bundle combined colporrhaphy with the various vaginal hysterectomy codes. These pair edits will no longer exist, and it is possible to bill separately for an AP repair performed in the same session as a vaginal hysterectomy—and expect additional payment for this procedure (subject to the multiple procedure reduction modifier -51).
Don’t miss out on reimbursement for your prior surgeries
It is important to recognize that this decision will be retroactive to October 1, 2014, which means that surgeons can resubmit charges for procedures performed between October 1, 2014, and March 31, 2015.
An exception to this rule is when the surgeon performs, and bills for, a vaginal hysterectomy code that includes enterocele repair, such as 58263. In these circumstances, the combined colporrhaphy remains bundled with the vaginal hysterectomy, unless a special modifier is used.
When can a modifier be used?
With regard to the majority of the other pair edits, CMS still contends that the edits are appropriate; however, they have modified the edits to allow for these procedures to be billed separately when the surgeon feels that substantial additional work has been performed. For example, CMS holds the position that all vaginal hysterectomies include some type of apical fixation to the surrounding tissues—such as fixation of the vaginal cuff at the time of closure to the distal uterosacral ligaments. However, it will allow for the additional billing for a more extensive vaginal apical suspension (such as a high uterosacral suspension or sacrospinous ligament suspension) as long as the surgeon documents the additional work performed and submits the claim using one of the NCCI-approved surgical modifiers. In this particular circumstance, the -59 modifier is the appropriate choice.
The AUGS task force also had objected to the bundling of a group of codes that, in the society’s opinion, were distinctly unrelated, such as a laparoscopic hysterectomy or vaginal hysterectomy combined with a sacral colpopexy performed through an open abdominal incision. CMS recognizes that these codes are used infrequently together and has therefore assumed that they may be billed in error. AUGS, however, has pointed out that, on occasion, a laparoscopic hysterectomy with an intended laparoscopic colpopexy may be converted to an open procedure due to a complication or technical difficulty, in which case the use of both codes would be appropriate.
The NCCI response was to keep the code pairs bundled but allow for billing with the -59 modifier.
We have cause for cheer, but bundling edits continue
Overall, these changes in pair edits represent a significant improvement in reimbursement for gynecologic surgeons, relative to the changes that went into effect on October 1. A more detailed explanation and list of the codes affected can be found on the AUGS Web site (http://www.augs.org). I emphasize again that these changes not only are in effect as of April 1, 2015, but also are retroactive to October 1, 2014.
Unfortunately, while AUGS and ACOG were advocating for these changes on behalf of gynecologists, NCCI recently has approved 6 additional pair edits that will go into effect on July 1. These changes specifically involve related procedures done at the time of vaginal hysterectomy with enterocele repair (58263, 58270 and 58294, 58292) and posterior vaginal repair (57250).
While these codes will be considered bundled on that date, NCCI will allow the use of the -59 modifier to override the bundle in clinically appropriate cases. The other code pairs affected are the 2 vaginal hysterectomy codes with colpourethropexy (58267, 58293) with anterior repair (57240). Again, these code pairs will be bundled, and it will require the use of a modifier to override this bundle.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Marc R. Toglia, MD
Dr. Toglia is a board-certified subspecialist in Female Pelvic Medicine and Reconstructive Surgery who serves as vice chair of the Coding Committee for the American Urogynecologic Society. He is Associate Professor of Obstetrics and Gynecology at Sidney Kimmel Medical College (formerly Jefferson Medical College) and practices at Main Line Health System in the Philadelphia, Pennsylvania, suburbs.
The author reports no financial relationships relevant to this article.
Marc R. Toglia, MD
Dr. Toglia is a board-certified subspecialist in Female Pelvic Medicine and Reconstructive Surgery who serves as vice chair of the Coding Committee for the American Urogynecologic Society. He is Associate Professor of Obstetrics and Gynecology at Sidney Kimmel Medical College (formerly Jefferson Medical College) and practices at Main Line Health System in the Philadelphia, Pennsylvania, suburbs.
The author reports no financial relationships relevant to this article.
Marc R. Toglia, MD
Dr. Toglia is a board-certified subspecialist in Female Pelvic Medicine and Reconstructive Surgery who serves as vice chair of the Coding Committee for the American Urogynecologic Society. He is Associate Professor of Obstetrics and Gynecology at Sidney Kimmel Medical College (formerly Jefferson Medical College) and practices at Main Line Health System in the Philadelphia, Pennsylvania, suburbs.
The author reports no financial relationships relevant to this article.
On October 1, 2014, the Centers for Medicare and Medicaid Services (CMS) and the National Correct Coding Initiative (NCCI) implemented a number of new coding pair edits that significantly restricted the types of surgical procedures that could be billed at the same time as vaginal hysterectomy. Most importantly, the new edits did not allow for combined anterior and posterior (AP) vaginal repair (57260) and apical vaginal suspensions (57282, 57283) to be separately billed and reimbursed when performed by the same surgeon and done in the same surgical session as a vaginal or laparoscopic hysterectomy. I initially reported on these changes in the January 2015 issue of OBG Management (“Recent NCCI edits have significantly impacted billing and reimbursement for vaginal hysterectomy”).
AUGS, ACOG, and others responded, and were heard
As a result of an intensive effort, the American Urogynecologic Society (AUGS), in conjunction with the American College of Obstetricians and Gynecologists (ACOG), the Society of Gynecologic Surgeons (SGS), and other professional societies, NCCI has agreed to change or modify a significant number of these pair edits.
As described in my earlier article, NCCI periodically reviews multiple surgical procedures performed in the same setting by a single surgeon to determine whether there is a significant overlap in services and therefore, in their opinion, redundant payment. Most significantly, CMS agreed with the argument made by the AUGS task force that it was inappropriate to bundle combined colporrhaphy with the various vaginal hysterectomy codes. These pair edits will no longer exist, and it is possible to bill separately for an AP repair performed in the same session as a vaginal hysterectomy—and expect additional payment for this procedure (subject to the multiple procedure reduction modifier -51).
Don’t miss out on reimbursement for your prior surgeries
It is important to recognize that this decision will be retroactive to October 1, 2014, which means that surgeons can resubmit charges for procedures performed between October 1, 2014, and March 31, 2015.
An exception to this rule is when the surgeon performs, and bills for, a vaginal hysterectomy code that includes enterocele repair, such as 58263. In these circumstances, the combined colporrhaphy remains bundled with the vaginal hysterectomy, unless a special modifier is used.
When can a modifier be used?
With regard to the majority of the other pair edits, CMS still contends that the edits are appropriate; however, they have modified the edits to allow for these procedures to be billed separately when the surgeon feels that substantial additional work has been performed. For example, CMS holds the position that all vaginal hysterectomies include some type of apical fixation to the surrounding tissues—such as fixation of the vaginal cuff at the time of closure to the distal uterosacral ligaments. However, it will allow for the additional billing for a more extensive vaginal apical suspension (such as a high uterosacral suspension or sacrospinous ligament suspension) as long as the surgeon documents the additional work performed and submits the claim using one of the NCCI-approved surgical modifiers. In this particular circumstance, the -59 modifier is the appropriate choice.
The AUGS task force also had objected to the bundling of a group of codes that, in the society’s opinion, were distinctly unrelated, such as a laparoscopic hysterectomy or vaginal hysterectomy combined with a sacral colpopexy performed through an open abdominal incision. CMS recognizes that these codes are used infrequently together and has therefore assumed that they may be billed in error. AUGS, however, has pointed out that, on occasion, a laparoscopic hysterectomy with an intended laparoscopic colpopexy may be converted to an open procedure due to a complication or technical difficulty, in which case the use of both codes would be appropriate.
The NCCI response was to keep the code pairs bundled but allow for billing with the -59 modifier.
We have cause for cheer, but bundling edits continue
Overall, these changes in pair edits represent a significant improvement in reimbursement for gynecologic surgeons, relative to the changes that went into effect on October 1. A more detailed explanation and list of the codes affected can be found on the AUGS Web site (http://www.augs.org). I emphasize again that these changes not only are in effect as of April 1, 2015, but also are retroactive to October 1, 2014.
Unfortunately, while AUGS and ACOG were advocating for these changes on behalf of gynecologists, NCCI recently has approved 6 additional pair edits that will go into effect on July 1. These changes specifically involve related procedures done at the time of vaginal hysterectomy with enterocele repair (58263, 58270 and 58294, 58292) and posterior vaginal repair (57250).
While these codes will be considered bundled on that date, NCCI will allow the use of the -59 modifier to override the bundle in clinically appropriate cases. The other code pairs affected are the 2 vaginal hysterectomy codes with colpourethropexy (58267, 58293) with anterior repair (57240). Again, these code pairs will be bundled, and it will require the use of a modifier to override this bundle.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
On October 1, 2014, the Centers for Medicare and Medicaid Services (CMS) and the National Correct Coding Initiative (NCCI) implemented a number of new coding pair edits that significantly restricted the types of surgical procedures that could be billed at the same time as vaginal hysterectomy. Most importantly, the new edits did not allow for combined anterior and posterior (AP) vaginal repair (57260) and apical vaginal suspensions (57282, 57283) to be separately billed and reimbursed when performed by the same surgeon and done in the same surgical session as a vaginal or laparoscopic hysterectomy. I initially reported on these changes in the January 2015 issue of OBG Management (“Recent NCCI edits have significantly impacted billing and reimbursement for vaginal hysterectomy”).
AUGS, ACOG, and others responded, and were heard
As a result of an intensive effort, the American Urogynecologic Society (AUGS), in conjunction with the American College of Obstetricians and Gynecologists (ACOG), the Society of Gynecologic Surgeons (SGS), and other professional societies, NCCI has agreed to change or modify a significant number of these pair edits.
As described in my earlier article, NCCI periodically reviews multiple surgical procedures performed in the same setting by a single surgeon to determine whether there is a significant overlap in services and therefore, in their opinion, redundant payment. Most significantly, CMS agreed with the argument made by the AUGS task force that it was inappropriate to bundle combined colporrhaphy with the various vaginal hysterectomy codes. These pair edits will no longer exist, and it is possible to bill separately for an AP repair performed in the same session as a vaginal hysterectomy—and expect additional payment for this procedure (subject to the multiple procedure reduction modifier -51).
Don’t miss out on reimbursement for your prior surgeries
It is important to recognize that this decision will be retroactive to October 1, 2014, which means that surgeons can resubmit charges for procedures performed between October 1, 2014, and March 31, 2015.
An exception to this rule is when the surgeon performs, and bills for, a vaginal hysterectomy code that includes enterocele repair, such as 58263. In these circumstances, the combined colporrhaphy remains bundled with the vaginal hysterectomy, unless a special modifier is used.
When can a modifier be used?
With regard to the majority of the other pair edits, CMS still contends that the edits are appropriate; however, they have modified the edits to allow for these procedures to be billed separately when the surgeon feels that substantial additional work has been performed. For example, CMS holds the position that all vaginal hysterectomies include some type of apical fixation to the surrounding tissues—such as fixation of the vaginal cuff at the time of closure to the distal uterosacral ligaments. However, it will allow for the additional billing for a more extensive vaginal apical suspension (such as a high uterosacral suspension or sacrospinous ligament suspension) as long as the surgeon documents the additional work performed and submits the claim using one of the NCCI-approved surgical modifiers. In this particular circumstance, the -59 modifier is the appropriate choice.
The AUGS task force also had objected to the bundling of a group of codes that, in the society’s opinion, were distinctly unrelated, such as a laparoscopic hysterectomy or vaginal hysterectomy combined with a sacral colpopexy performed through an open abdominal incision. CMS recognizes that these codes are used infrequently together and has therefore assumed that they may be billed in error. AUGS, however, has pointed out that, on occasion, a laparoscopic hysterectomy with an intended laparoscopic colpopexy may be converted to an open procedure due to a complication or technical difficulty, in which case the use of both codes would be appropriate.
The NCCI response was to keep the code pairs bundled but allow for billing with the -59 modifier.
We have cause for cheer, but bundling edits continue
Overall, these changes in pair edits represent a significant improvement in reimbursement for gynecologic surgeons, relative to the changes that went into effect on October 1. A more detailed explanation and list of the codes affected can be found on the AUGS Web site (http://www.augs.org). I emphasize again that these changes not only are in effect as of April 1, 2015, but also are retroactive to October 1, 2014.
Unfortunately, while AUGS and ACOG were advocating for these changes on behalf of gynecologists, NCCI recently has approved 6 additional pair edits that will go into effect on July 1. These changes specifically involve related procedures done at the time of vaginal hysterectomy with enterocele repair (58263, 58270 and 58294, 58292) and posterior vaginal repair (57250).
While these codes will be considered bundled on that date, NCCI will allow the use of the -59 modifier to override the bundle in clinically appropriate cases. The other code pairs affected are the 2 vaginal hysterectomy codes with colpourethropexy (58267, 58293) with anterior repair (57240). Again, these code pairs will be bundled, and it will require the use of a modifier to override this bundle.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
