User login
FDA clears endoscopic devices for sleeve gastroplasty, bariatric revision
The Food and Drug Administration has cleared for marketing the first devices indicated for endoscopic sleeve gastroplasty (ESG) and endoscopic bariatric revision, according to the manufacturer.
The Apollo ESG, Apollo ESG Sx, Apollo Revise, and Apollo Revise Sx systems made by Apollo Endosurgery were reviewed through the de novo premarket review pathway, a regulatory pathway for low- to moderate-risk devices of a new type.
“The Apollo ESG and Apollo Revise systems offer a compelling mix of effectiveness, safety, durability, and convenience for treatment of patients with obesity,” Chas McKhann, president and CEO of the company, said in a news release.
“The authorization of these new endoscopic systems represents a major step forward in addressing the global obesity epidemic,” Mr. McKhann added.
The Apollo ESG and Apollo ESG Sx systems are intended for use by trained gastroenterologists or surgeons to facilitate weight loss in adults with obesity who have failed to lose weight or maintain weight loss through more conservative measures, the company says.
The Apollo Revise and Apollo Revise Sx systems allow gastroenterologists or surgeons to perform transoral outlet reduction (TORe) as a revision to a previous bariatric procedure.
Studies have shown that 10 years after bariatric surgery, patients have regained an average of 20%-30% of weight they initially lost. Bariatric revision procedures are the fastest growing segment of the bariatric surgery market.
TORe is an endoscopic procedure performed to revise a previous gastric bypass and like ESG, can be performed as a same-day procedure without incisions or scars.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has cleared for marketing the first devices indicated for endoscopic sleeve gastroplasty (ESG) and endoscopic bariatric revision, according to the manufacturer.
The Apollo ESG, Apollo ESG Sx, Apollo Revise, and Apollo Revise Sx systems made by Apollo Endosurgery were reviewed through the de novo premarket review pathway, a regulatory pathway for low- to moderate-risk devices of a new type.
“The Apollo ESG and Apollo Revise systems offer a compelling mix of effectiveness, safety, durability, and convenience for treatment of patients with obesity,” Chas McKhann, president and CEO of the company, said in a news release.
“The authorization of these new endoscopic systems represents a major step forward in addressing the global obesity epidemic,” Mr. McKhann added.
The Apollo ESG and Apollo ESG Sx systems are intended for use by trained gastroenterologists or surgeons to facilitate weight loss in adults with obesity who have failed to lose weight or maintain weight loss through more conservative measures, the company says.
The Apollo Revise and Apollo Revise Sx systems allow gastroenterologists or surgeons to perform transoral outlet reduction (TORe) as a revision to a previous bariatric procedure.
Studies have shown that 10 years after bariatric surgery, patients have regained an average of 20%-30% of weight they initially lost. Bariatric revision procedures are the fastest growing segment of the bariatric surgery market.
TORe is an endoscopic procedure performed to revise a previous gastric bypass and like ESG, can be performed as a same-day procedure without incisions or scars.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has cleared for marketing the first devices indicated for endoscopic sleeve gastroplasty (ESG) and endoscopic bariatric revision, according to the manufacturer.
The Apollo ESG, Apollo ESG Sx, Apollo Revise, and Apollo Revise Sx systems made by Apollo Endosurgery were reviewed through the de novo premarket review pathway, a regulatory pathway for low- to moderate-risk devices of a new type.
“The Apollo ESG and Apollo Revise systems offer a compelling mix of effectiveness, safety, durability, and convenience for treatment of patients with obesity,” Chas McKhann, president and CEO of the company, said in a news release.
“The authorization of these new endoscopic systems represents a major step forward in addressing the global obesity epidemic,” Mr. McKhann added.
The Apollo ESG and Apollo ESG Sx systems are intended for use by trained gastroenterologists or surgeons to facilitate weight loss in adults with obesity who have failed to lose weight or maintain weight loss through more conservative measures, the company says.
The Apollo Revise and Apollo Revise Sx systems allow gastroenterologists or surgeons to perform transoral outlet reduction (TORe) as a revision to a previous bariatric procedure.
Studies have shown that 10 years after bariatric surgery, patients have regained an average of 20%-30% of weight they initially lost. Bariatric revision procedures are the fastest growing segment of the bariatric surgery market.
TORe is an endoscopic procedure performed to revise a previous gastric bypass and like ESG, can be performed as a same-day procedure without incisions or scars.
A version of this article first appeared on Medscape.com.
CDC warns about potentially deadly virus in infants
The potentially fatal parechovirus is now circulating in multiple states, causing fevers, seizures, and sepsis-like symptoms, including confusion and extreme pain, according to the CDC.
Human parechoviruses are common in children and most have been infected before they start kindergarten, the CDC said. Between 6 months and 5 years of age, symptoms include an upper respiratory tract infection, fever, and rash.
But infants younger than 3 months may have more serious, and possibly fatal, infections. They may get “sepsis-like illness, seizures, and meningitis or meningoencephalitis, particularly in infants younger than 1 month,” the CDC said. At least one newborn has reportedly died from the infection.
Parechovirus can spread like other common germs, from feces that are later ingested – likely due to poor handwashing – and through droplets sent airborne by coughing or sneezing. It can be transmitted by people both with and without symptoms of the infection.
The microbe can reproduce for 1-3 weeks in the upper respiratory tract and up to 6 months in the gastrointestinal tract, the CDC said.
Kristina Angel Bryant, MD, a pediatric infectious disease specialist at the University of Louisville Hospital, says parechoviruses often cause rashes on the hands and feet, which some experts refer to as “mittens and booties.”
The CDC is urging doctors to test for parechovirus if they recognize these symptoms in infants if there is no other explanation for what might be distressing them.
There is no specific treatment for parechovirus. And with no standard testing system in place, experts are unsure if the number of parechovirus cases is higher in 2022 than in previous years.
The message for parents, Dr. Bryant says, is: Don’t panic. “This is not a new virus.”
“One of the most common symptoms is fever, and in some kids, that is the only symptom,” she says. “Older infants and toddlers may have only cold symptoms, and some kids have no symptoms at all.”
Parents can take the usual steps to protect their child from the viral illness, including diligent handwashing and having less contact with people who are sick, Dr. Bryant says.
A version of this article first appeared on Medscape.com.
The potentially fatal parechovirus is now circulating in multiple states, causing fevers, seizures, and sepsis-like symptoms, including confusion and extreme pain, according to the CDC.
Human parechoviruses are common in children and most have been infected before they start kindergarten, the CDC said. Between 6 months and 5 years of age, symptoms include an upper respiratory tract infection, fever, and rash.
But infants younger than 3 months may have more serious, and possibly fatal, infections. They may get “sepsis-like illness, seizures, and meningitis or meningoencephalitis, particularly in infants younger than 1 month,” the CDC said. At least one newborn has reportedly died from the infection.
Parechovirus can spread like other common germs, from feces that are later ingested – likely due to poor handwashing – and through droplets sent airborne by coughing or sneezing. It can be transmitted by people both with and without symptoms of the infection.
The microbe can reproduce for 1-3 weeks in the upper respiratory tract and up to 6 months in the gastrointestinal tract, the CDC said.
Kristina Angel Bryant, MD, a pediatric infectious disease specialist at the University of Louisville Hospital, says parechoviruses often cause rashes on the hands and feet, which some experts refer to as “mittens and booties.”
The CDC is urging doctors to test for parechovirus if they recognize these symptoms in infants if there is no other explanation for what might be distressing them.
There is no specific treatment for parechovirus. And with no standard testing system in place, experts are unsure if the number of parechovirus cases is higher in 2022 than in previous years.
The message for parents, Dr. Bryant says, is: Don’t panic. “This is not a new virus.”
“One of the most common symptoms is fever, and in some kids, that is the only symptom,” she says. “Older infants and toddlers may have only cold symptoms, and some kids have no symptoms at all.”
Parents can take the usual steps to protect their child from the viral illness, including diligent handwashing and having less contact with people who are sick, Dr. Bryant says.
A version of this article first appeared on Medscape.com.
The potentially fatal parechovirus is now circulating in multiple states, causing fevers, seizures, and sepsis-like symptoms, including confusion and extreme pain, according to the CDC.
Human parechoviruses are common in children and most have been infected before they start kindergarten, the CDC said. Between 6 months and 5 years of age, symptoms include an upper respiratory tract infection, fever, and rash.
But infants younger than 3 months may have more serious, and possibly fatal, infections. They may get “sepsis-like illness, seizures, and meningitis or meningoencephalitis, particularly in infants younger than 1 month,” the CDC said. At least one newborn has reportedly died from the infection.
Parechovirus can spread like other common germs, from feces that are later ingested – likely due to poor handwashing – and through droplets sent airborne by coughing or sneezing. It can be transmitted by people both with and without symptoms of the infection.
The microbe can reproduce for 1-3 weeks in the upper respiratory tract and up to 6 months in the gastrointestinal tract, the CDC said.
Kristina Angel Bryant, MD, a pediatric infectious disease specialist at the University of Louisville Hospital, says parechoviruses often cause rashes on the hands and feet, which some experts refer to as “mittens and booties.”
The CDC is urging doctors to test for parechovirus if they recognize these symptoms in infants if there is no other explanation for what might be distressing them.
There is no specific treatment for parechovirus. And with no standard testing system in place, experts are unsure if the number of parechovirus cases is higher in 2022 than in previous years.
The message for parents, Dr. Bryant says, is: Don’t panic. “This is not a new virus.”
“One of the most common symptoms is fever, and in some kids, that is the only symptom,” she says. “Older infants and toddlers may have only cold symptoms, and some kids have no symptoms at all.”
Parents can take the usual steps to protect their child from the viral illness, including diligent handwashing and having less contact with people who are sick, Dr. Bryant says.
A version of this article first appeared on Medscape.com.
FDA approves topical ruxolitinib for nonsegmental vitiligo
The on July 18. The treatment, which was approved for treating mild to moderate atopic dermatitis in September 2021, is a cream formulation of ruxolitinib, a Janus kinase 1 (JAK1)/JAK2 inhibitor.
Previously, no treatment was approved to repigment patients with vitiligo, says David Rosmarin, MD, vice chair for research and education in the department of dermatology at Tufts Medical Center, Boston. “It’s important to have options that we can give to patients that are both safe and effective to get them the desired results,” Dr. Rosmarin, the lead investigator of the phase 3 clinical trials of topical ruxolitinib, said in an interview. Vitiligo is “a disease that can really affect quality of life. Some people [with vitiligo] feel as if they’re being stared at or they’re being bullied; they don’t feel confident. It can affect relationships and intimacy.”
Approval was based on the results of two phase 3 trials (TruE-V1 and TruE-V2) in 674 patients with nonsegmental vitiligo aged 12 years or older. At 24 weeks, about 30% of the patients on treatment, applied twice a day, achieved at least a 75% improvement in the facial Vitiligo Area Scoring Index (F-VASI75), compared with about 8% and 13% among those in the vehicle groups in the two trials.
At 52 weeks, about 50% of the patients treated with topical ruxolitinib achieved F-VASI75.
Also, using self-reporting as measured by the Vitiligo Noticeability Scale, about 30%-40% of patients described their vitiligo as being “a lot less noticeable” or “no longer noticeable” at week 52. Dr. Rosmarin reported the 52-week results at the 2022 annual meeting of the American Academy of Dermatology.
The trial group used 1.5% ruxolitinib cream twice daily for the full year. The vehicle group began using ruxolitinib halfway through the trial. In this group, 26.8% and 29.6% achieved F-VASI 75 at 52 weeks in the two trials.
For treating vitiligo, patients are advised to apply a thin layer of topical ruxolitinib to affected areas twice a day, “up to 10% body surface area,” according to the prescribing information, which adds: “Satisfactory patient response may require treatment … for more than 24 weeks. If the patient does not find the repigmentation meaningful by 24 weeks, the patient should be reevaluated by the health care provider.”
The most common side effects during the vehicle-controlled part of the trials were development of acne and pruritus at the application site, headache, urinary tract infections, erythema at the application site, and pyrexia, according to the company.
The approved label for topical ruxolitinib includes a boxed warning about serious infections, mortality, cancer, major adverse cardiovascular events, and thrombosis – which, the warning notes, is based on reports in patients treated with oral JAK inhibitors for inflammatory conditions.
Dr. Rosmarin believes that using this drug with other therapies, like light treatment, might yield even better responses. The available data are in patients treated with ruxolitinib as monotherapy, without complementary therapies.
William Damsky, MD, PhD, professor of dermatology and dermatopathology at Yale University, New Haven, who was not involved in the trials, said what is most exciting about this drug is its novelty. Although some topical steroids are used off-label to treat vitiligo, their efficacy is far from what’s been observed in these trials of topical ruxolitinib, he told this news organization. “It’s huge for a number of reasons. … One very big reason is it just provides some hope” for the many patients with vitiligo who, over the years, have been told “that there’s nothing that could be done for their disease, and this really changes that.”
Dr. Rosmarin reports financial relationships with over 20 pharmaceutical companies. Dr. Damsky disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The on July 18. The treatment, which was approved for treating mild to moderate atopic dermatitis in September 2021, is a cream formulation of ruxolitinib, a Janus kinase 1 (JAK1)/JAK2 inhibitor.
Previously, no treatment was approved to repigment patients with vitiligo, says David Rosmarin, MD, vice chair for research and education in the department of dermatology at Tufts Medical Center, Boston. “It’s important to have options that we can give to patients that are both safe and effective to get them the desired results,” Dr. Rosmarin, the lead investigator of the phase 3 clinical trials of topical ruxolitinib, said in an interview. Vitiligo is “a disease that can really affect quality of life. Some people [with vitiligo] feel as if they’re being stared at or they’re being bullied; they don’t feel confident. It can affect relationships and intimacy.”
Approval was based on the results of two phase 3 trials (TruE-V1 and TruE-V2) in 674 patients with nonsegmental vitiligo aged 12 years or older. At 24 weeks, about 30% of the patients on treatment, applied twice a day, achieved at least a 75% improvement in the facial Vitiligo Area Scoring Index (F-VASI75), compared with about 8% and 13% among those in the vehicle groups in the two trials.
At 52 weeks, about 50% of the patients treated with topical ruxolitinib achieved F-VASI75.
Also, using self-reporting as measured by the Vitiligo Noticeability Scale, about 30%-40% of patients described their vitiligo as being “a lot less noticeable” or “no longer noticeable” at week 52. Dr. Rosmarin reported the 52-week results at the 2022 annual meeting of the American Academy of Dermatology.
The trial group used 1.5% ruxolitinib cream twice daily for the full year. The vehicle group began using ruxolitinib halfway through the trial. In this group, 26.8% and 29.6% achieved F-VASI 75 at 52 weeks in the two trials.
For treating vitiligo, patients are advised to apply a thin layer of topical ruxolitinib to affected areas twice a day, “up to 10% body surface area,” according to the prescribing information, which adds: “Satisfactory patient response may require treatment … for more than 24 weeks. If the patient does not find the repigmentation meaningful by 24 weeks, the patient should be reevaluated by the health care provider.”
The most common side effects during the vehicle-controlled part of the trials were development of acne and pruritus at the application site, headache, urinary tract infections, erythema at the application site, and pyrexia, according to the company.
The approved label for topical ruxolitinib includes a boxed warning about serious infections, mortality, cancer, major adverse cardiovascular events, and thrombosis – which, the warning notes, is based on reports in patients treated with oral JAK inhibitors for inflammatory conditions.
Dr. Rosmarin believes that using this drug with other therapies, like light treatment, might yield even better responses. The available data are in patients treated with ruxolitinib as monotherapy, without complementary therapies.
William Damsky, MD, PhD, professor of dermatology and dermatopathology at Yale University, New Haven, who was not involved in the trials, said what is most exciting about this drug is its novelty. Although some topical steroids are used off-label to treat vitiligo, their efficacy is far from what’s been observed in these trials of topical ruxolitinib, he told this news organization. “It’s huge for a number of reasons. … One very big reason is it just provides some hope” for the many patients with vitiligo who, over the years, have been told “that there’s nothing that could be done for their disease, and this really changes that.”
Dr. Rosmarin reports financial relationships with over 20 pharmaceutical companies. Dr. Damsky disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The on July 18. The treatment, which was approved for treating mild to moderate atopic dermatitis in September 2021, is a cream formulation of ruxolitinib, a Janus kinase 1 (JAK1)/JAK2 inhibitor.
Previously, no treatment was approved to repigment patients with vitiligo, says David Rosmarin, MD, vice chair for research and education in the department of dermatology at Tufts Medical Center, Boston. “It’s important to have options that we can give to patients that are both safe and effective to get them the desired results,” Dr. Rosmarin, the lead investigator of the phase 3 clinical trials of topical ruxolitinib, said in an interview. Vitiligo is “a disease that can really affect quality of life. Some people [with vitiligo] feel as if they’re being stared at or they’re being bullied; they don’t feel confident. It can affect relationships and intimacy.”
Approval was based on the results of two phase 3 trials (TruE-V1 and TruE-V2) in 674 patients with nonsegmental vitiligo aged 12 years or older. At 24 weeks, about 30% of the patients on treatment, applied twice a day, achieved at least a 75% improvement in the facial Vitiligo Area Scoring Index (F-VASI75), compared with about 8% and 13% among those in the vehicle groups in the two trials.
At 52 weeks, about 50% of the patients treated with topical ruxolitinib achieved F-VASI75.
Also, using self-reporting as measured by the Vitiligo Noticeability Scale, about 30%-40% of patients described their vitiligo as being “a lot less noticeable” or “no longer noticeable” at week 52. Dr. Rosmarin reported the 52-week results at the 2022 annual meeting of the American Academy of Dermatology.
The trial group used 1.5% ruxolitinib cream twice daily for the full year. The vehicle group began using ruxolitinib halfway through the trial. In this group, 26.8% and 29.6% achieved F-VASI 75 at 52 weeks in the two trials.
For treating vitiligo, patients are advised to apply a thin layer of topical ruxolitinib to affected areas twice a day, “up to 10% body surface area,” according to the prescribing information, which adds: “Satisfactory patient response may require treatment … for more than 24 weeks. If the patient does not find the repigmentation meaningful by 24 weeks, the patient should be reevaluated by the health care provider.”
The most common side effects during the vehicle-controlled part of the trials were development of acne and pruritus at the application site, headache, urinary tract infections, erythema at the application site, and pyrexia, according to the company.
The approved label for topical ruxolitinib includes a boxed warning about serious infections, mortality, cancer, major adverse cardiovascular events, and thrombosis – which, the warning notes, is based on reports in patients treated with oral JAK inhibitors for inflammatory conditions.
Dr. Rosmarin believes that using this drug with other therapies, like light treatment, might yield even better responses. The available data are in patients treated with ruxolitinib as monotherapy, without complementary therapies.
William Damsky, MD, PhD, professor of dermatology and dermatopathology at Yale University, New Haven, who was not involved in the trials, said what is most exciting about this drug is its novelty. Although some topical steroids are used off-label to treat vitiligo, their efficacy is far from what’s been observed in these trials of topical ruxolitinib, he told this news organization. “It’s huge for a number of reasons. … One very big reason is it just provides some hope” for the many patients with vitiligo who, over the years, have been told “that there’s nothing that could be done for their disease, and this really changes that.”
Dr. Rosmarin reports financial relationships with over 20 pharmaceutical companies. Dr. Damsky disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA grants emergency authorization for Novavax COVID vaccine
on July 13.

The vaccine is authorized for adults only. Should the Centers for Disease Control and Prevention follow suit and approve its use, Novavax would join Moderna, Pfizer and Johnson & Johnson on the U.S. market. A CDC panel of advisors is expected to consider the new entry on July 19.
The Novavax vaccine is only for those who have not yet been vaccinated at all.
“Today’s authorization offers adults in the United States who have not yet received a COVID-19 vaccine another option that meets the FDA’s rigorous standards for safety, effectiveness and manufacturing quality needed to support emergency use authorization,” FDA Commissioner Robert Califf, MD, said in a statement. “COVID-19 vaccines remain the best preventive measure against severe disease caused by COVID-19 and I encourage anyone who is eligible for, but has not yet received a COVID-19 vaccine, to consider doing so.”
The Novavax vaccine is protein-based, making it different than mRNA vaccines from Pfizer and Moderna. It contains harmless elements of actual coronavirus spike protein and an ingredient known as a adjuvant that enhances the patient’s immune response.
Clinical trials found the vaccine to be 90.4% effective in preventing mild, moderate or severe COVID-19. Only 17 patients out of 17,200 developed COVID-19 after receiving both doses.
The FDA said, however, that Novavax’s vaccine did show evidence of increased risk of myocarditis – inflammation of the heart – and pericarditis, inflammation of tissue surrounding the heart. In most people both disorders began within 10 days.
A version of this article first appeared on WebMD.com.
on July 13.
The vaccine is authorized for adults only. Should the Centers for Disease Control and Prevention follow suit and approve its use, Novavax would join Moderna, Pfizer and Johnson & Johnson on the U.S. market. A CDC panel of advisors is expected to consider the new entry on July 19.
The Novavax vaccine is only for those who have not yet been vaccinated at all.
“Today’s authorization offers adults in the United States who have not yet received a COVID-19 vaccine another option that meets the FDA’s rigorous standards for safety, effectiveness and manufacturing quality needed to support emergency use authorization,” FDA Commissioner Robert Califf, MD, said in a statement. “COVID-19 vaccines remain the best preventive measure against severe disease caused by COVID-19 and I encourage anyone who is eligible for, but has not yet received a COVID-19 vaccine, to consider doing so.”
The Novavax vaccine is protein-based, making it different than mRNA vaccines from Pfizer and Moderna. It contains harmless elements of actual coronavirus spike protein and an ingredient known as a adjuvant that enhances the patient’s immune response.
Clinical trials found the vaccine to be 90.4% effective in preventing mild, moderate or severe COVID-19. Only 17 patients out of 17,200 developed COVID-19 after receiving both doses.
The FDA said, however, that Novavax’s vaccine did show evidence of increased risk of myocarditis – inflammation of the heart – and pericarditis, inflammation of tissue surrounding the heart. In most people both disorders began within 10 days.
A version of this article first appeared on WebMD.com.
on July 13.
The vaccine is authorized for adults only. Should the Centers for Disease Control and Prevention follow suit and approve its use, Novavax would join Moderna, Pfizer and Johnson & Johnson on the U.S. market. A CDC panel of advisors is expected to consider the new entry on July 19.
The Novavax vaccine is only for those who have not yet been vaccinated at all.
“Today’s authorization offers adults in the United States who have not yet received a COVID-19 vaccine another option that meets the FDA’s rigorous standards for safety, effectiveness and manufacturing quality needed to support emergency use authorization,” FDA Commissioner Robert Califf, MD, said in a statement. “COVID-19 vaccines remain the best preventive measure against severe disease caused by COVID-19 and I encourage anyone who is eligible for, but has not yet received a COVID-19 vaccine, to consider doing so.”
The Novavax vaccine is protein-based, making it different than mRNA vaccines from Pfizer and Moderna. It contains harmless elements of actual coronavirus spike protein and an ingredient known as a adjuvant that enhances the patient’s immune response.
Clinical trials found the vaccine to be 90.4% effective in preventing mild, moderate or severe COVID-19. Only 17 patients out of 17,200 developed COVID-19 after receiving both doses.
The FDA said, however, that Novavax’s vaccine did show evidence of increased risk of myocarditis – inflammation of the heart – and pericarditis, inflammation of tissue surrounding the heart. In most people both disorders began within 10 days.
A version of this article first appeared on WebMD.com.
FDA approves combination pegloticase and methotrexate for refractory gout

Pegloticase, which has been available for 12 years, is a pegylated uric acid specific enzyme that lowers sUA by converting it to allantoin.
Though pegloticase is effective in treating chronic gout in patients refractory to conventional treatment, approximately 92% of patients develop antibodies against the drug, resulting in reduced efficacy.
Based on the immunomodulatory effects of methotrexate, researchers of the randomized, placebo-controlled MIRROR trial sought to determine whether combination treatment of pegloticase with methotrexate (multiple brands) would prevent the development of anti-drug antibodies.
Findings from the phase 4 trial found that co-administration of pegloticase and methotrexate reduced the formation of new anti-PEG antibodies. In the group receiving methotrexate and pegloticase, 23.2% (22 out of 95) of patients had an increase in anti-PEG antibodies, compared with 50% (24 of 48) in the pegloticase plus placebo group, according to a recent company press release.
Nearly three-quarters (71%) of participants in the group pretreated with methotrexate, followed by combination pegloticase-methotrexate, had sUA levels that dopped to below 6 mg/dL during the 52-week study. By comparison, 38.5% of participants in the pegloticase and placebo group reached the endpoint. Though gout flare occurred in both groups, methotrexate did not appear to increase the risk for adverse events or gout flare.
The study, led by John Botson, MD, RPh, CCD, a rheumatologist in Anchorage, Alaska, concluded that these measurements demonstrated a significant improvement from traditional pegloticase-only treatment of gout. “This trial confirms not only improved efficacy but improved safety in patients treated with pegloticase in combination with methotrexate 15 mg orally once weekly,” Dr. Botson said last month in an interview with this news organization.
The study was funded by Horizon. Dr. Botson reports receiving research support from Horizon and Radius Health and speaker fees from AbbVie, Amgen, Aurinia, ChemoCentryx, Horizon, Eli Lilly, and Novartis.
A version of this article first appeared on Medscape.com.
Pegloticase, which has been available for 12 years, is a pegylated uric acid specific enzyme that lowers sUA by converting it to allantoin.
Though pegloticase is effective in treating chronic gout in patients refractory to conventional treatment, approximately 92% of patients develop antibodies against the drug, resulting in reduced efficacy.
Based on the immunomodulatory effects of methotrexate, researchers of the randomized, placebo-controlled MIRROR trial sought to determine whether combination treatment of pegloticase with methotrexate (multiple brands) would prevent the development of anti-drug antibodies.
Findings from the phase 4 trial found that co-administration of pegloticase and methotrexate reduced the formation of new anti-PEG antibodies. In the group receiving methotrexate and pegloticase, 23.2% (22 out of 95) of patients had an increase in anti-PEG antibodies, compared with 50% (24 of 48) in the pegloticase plus placebo group, according to a recent company press release.
Nearly three-quarters (71%) of participants in the group pretreated with methotrexate, followed by combination pegloticase-methotrexate, had sUA levels that dopped to below 6 mg/dL during the 52-week study. By comparison, 38.5% of participants in the pegloticase and placebo group reached the endpoint. Though gout flare occurred in both groups, methotrexate did not appear to increase the risk for adverse events or gout flare.
The study, led by John Botson, MD, RPh, CCD, a rheumatologist in Anchorage, Alaska, concluded that these measurements demonstrated a significant improvement from traditional pegloticase-only treatment of gout. “This trial confirms not only improved efficacy but improved safety in patients treated with pegloticase in combination with methotrexate 15 mg orally once weekly,” Dr. Botson said last month in an interview with this news organization.
The study was funded by Horizon. Dr. Botson reports receiving research support from Horizon and Radius Health and speaker fees from AbbVie, Amgen, Aurinia, ChemoCentryx, Horizon, Eli Lilly, and Novartis.
A version of this article first appeared on Medscape.com.
Pegloticase, which has been available for 12 years, is a pegylated uric acid specific enzyme that lowers sUA by converting it to allantoin.
Though pegloticase is effective in treating chronic gout in patients refractory to conventional treatment, approximately 92% of patients develop antibodies against the drug, resulting in reduced efficacy.
Based on the immunomodulatory effects of methotrexate, researchers of the randomized, placebo-controlled MIRROR trial sought to determine whether combination treatment of pegloticase with methotrexate (multiple brands) would prevent the development of anti-drug antibodies.
Findings from the phase 4 trial found that co-administration of pegloticase and methotrexate reduced the formation of new anti-PEG antibodies. In the group receiving methotrexate and pegloticase, 23.2% (22 out of 95) of patients had an increase in anti-PEG antibodies, compared with 50% (24 of 48) in the pegloticase plus placebo group, according to a recent company press release.
Nearly three-quarters (71%) of participants in the group pretreated with methotrexate, followed by combination pegloticase-methotrexate, had sUA levels that dopped to below 6 mg/dL during the 52-week study. By comparison, 38.5% of participants in the pegloticase and placebo group reached the endpoint. Though gout flare occurred in both groups, methotrexate did not appear to increase the risk for adverse events or gout flare.
The study, led by John Botson, MD, RPh, CCD, a rheumatologist in Anchorage, Alaska, concluded that these measurements demonstrated a significant improvement from traditional pegloticase-only treatment of gout. “This trial confirms not only improved efficacy but improved safety in patients treated with pegloticase in combination with methotrexate 15 mg orally once weekly,” Dr. Botson said last month in an interview with this news organization.
The study was funded by Horizon. Dr. Botson reports receiving research support from Horizon and Radius Health and speaker fees from AbbVie, Amgen, Aurinia, ChemoCentryx, Horizon, Eli Lilly, and Novartis.
A version of this article first appeared on Medscape.com.
Neck floats may not be right for certain babies, FDA warns
The FDA is warning that parents should avoid using neck floats for infants with special needs or developmental delays.
According to the agency, companies have been advertising the products as having health benefits for children with physical and developmental problems, despite a lack of evidence for such claims. The companies, which the FDA did not name, claimed that water therapy with floats could help babies with special needs – like those with spina bifida – to increase muscle tone, boost flexibility and range of motion, and build lung capacity, among other benefits.
But used improperly, neck floats can lead to serious injury and death. At least one baby has died, and one was hospitalized, after using the floats, FDA officials said.
The inflatable plastic rings are worn around a baby’s neck, allowing them to float freely in water. Some of these products are being marketed for infants as young as 2 weeks old, as well as for premature babies. But the FDA said the safety and effectiveness of the products for these children have not been proven.
The floats “have not been evaluated by the FDA, and we are not aware of any demonstrated benefit with the use of neck floats for water therapy interventions,” the agency said in the June 28 statement.
While injuries and deaths from neck floats are rare, the FDA said families and caregivers should be aware that these incidents can and do occur.
People who have problems with the neck floats are encouraged to report them through MedWatch, the FDA Safety Information and Adverse Event Reporting Program. Health care personnel employed by the FDA are required to file new reports with the FDA.
A version of this article first appeared on WebMD.com.
The FDA is warning that parents should avoid using neck floats for infants with special needs or developmental delays.
According to the agency, companies have been advertising the products as having health benefits for children with physical and developmental problems, despite a lack of evidence for such claims. The companies, which the FDA did not name, claimed that water therapy with floats could help babies with special needs – like those with spina bifida – to increase muscle tone, boost flexibility and range of motion, and build lung capacity, among other benefits.
But used improperly, neck floats can lead to serious injury and death. At least one baby has died, and one was hospitalized, after using the floats, FDA officials said.
The inflatable plastic rings are worn around a baby’s neck, allowing them to float freely in water. Some of these products are being marketed for infants as young as 2 weeks old, as well as for premature babies. But the FDA said the safety and effectiveness of the products for these children have not been proven.
The floats “have not been evaluated by the FDA, and we are not aware of any demonstrated benefit with the use of neck floats for water therapy interventions,” the agency said in the June 28 statement.
While injuries and deaths from neck floats are rare, the FDA said families and caregivers should be aware that these incidents can and do occur.
People who have problems with the neck floats are encouraged to report them through MedWatch, the FDA Safety Information and Adverse Event Reporting Program. Health care personnel employed by the FDA are required to file new reports with the FDA.
A version of this article first appeared on WebMD.com.
The FDA is warning that parents should avoid using neck floats for infants with special needs or developmental delays.
According to the agency, companies have been advertising the products as having health benefits for children with physical and developmental problems, despite a lack of evidence for such claims. The companies, which the FDA did not name, claimed that water therapy with floats could help babies with special needs – like those with spina bifida – to increase muscle tone, boost flexibility and range of motion, and build lung capacity, among other benefits.
But used improperly, neck floats can lead to serious injury and death. At least one baby has died, and one was hospitalized, after using the floats, FDA officials said.
The inflatable plastic rings are worn around a baby’s neck, allowing them to float freely in water. Some of these products are being marketed for infants as young as 2 weeks old, as well as for premature babies. But the FDA said the safety and effectiveness of the products for these children have not been proven.
The floats “have not been evaluated by the FDA, and we are not aware of any demonstrated benefit with the use of neck floats for water therapy interventions,” the agency said in the June 28 statement.
While injuries and deaths from neck floats are rare, the FDA said families and caregivers should be aware that these incidents can and do occur.
People who have problems with the neck floats are encouraged to report them through MedWatch, the FDA Safety Information and Adverse Event Reporting Program. Health care personnel employed by the FDA are required to file new reports with the FDA.
A version of this article first appeared on WebMD.com.
Baricitinib’s approval for alopecia areata: Considerations for starting patients on treatment
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.

Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
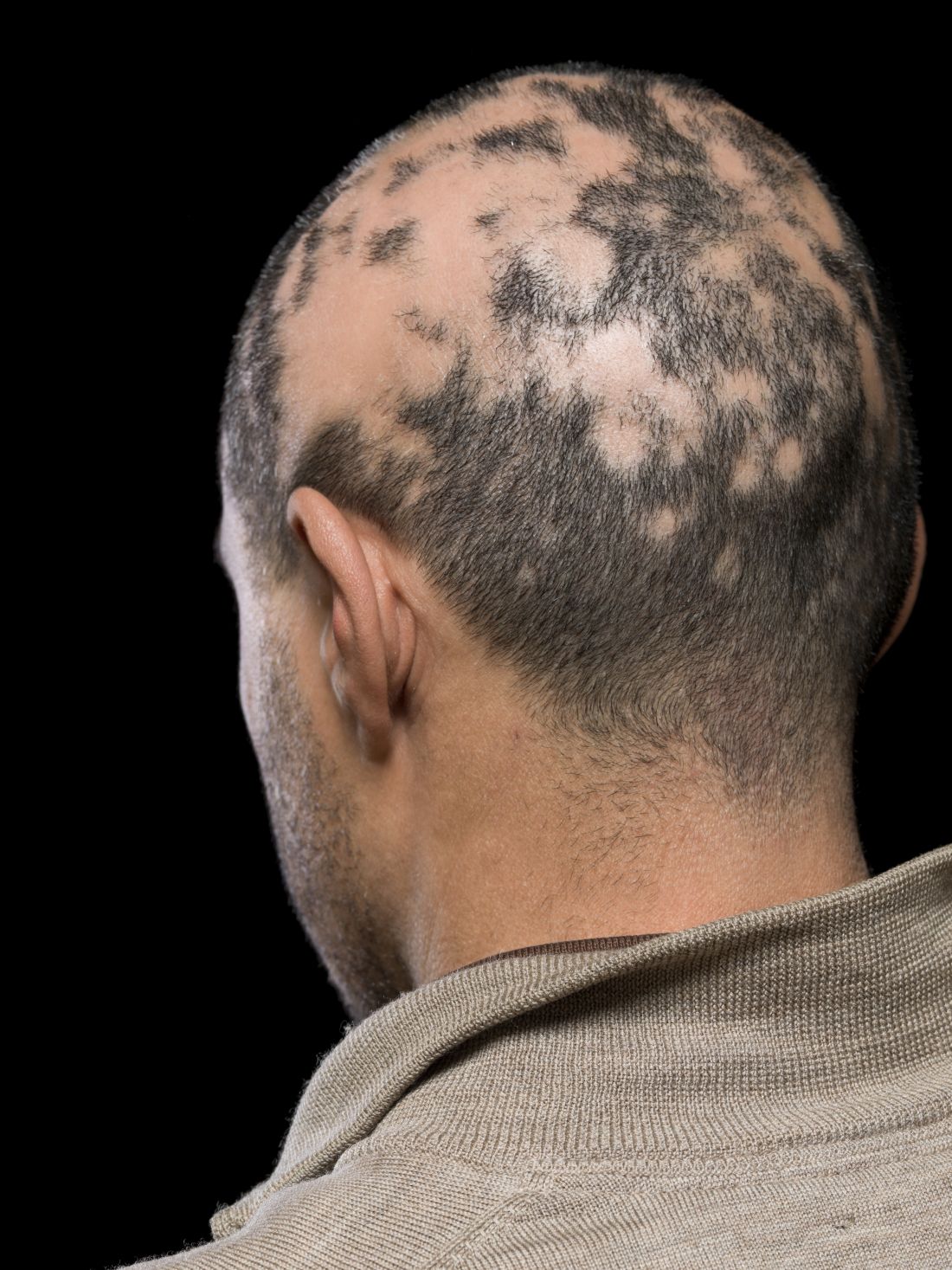
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.

“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.

Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.
Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.
“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.
Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.
Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.
“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.
Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Will the headache field embrace rofecoxib?
In June, the Concord, Mass.–based company Tremeau Pharmaceuticals announced that the Food and Drug Administration was letting it proceed with a phase 3 clinical trial to test rofecoxib, the once-bestselling painkiller known as Vioxx, in patients with migraine.
The anti-inflammatory drug, a cyclooxygenase-2 (COX-2) inhibitor, received its first FDA approval in 1999 and became widely prescribed for arthritis and acute pain. In 2004 it was withdrawn by its manufacturer, Merck, after being shown to raise the risk of cardiovascular events.
In clinical trials and in real-world epidemiological studies, rofecoxib was associated with elevated heart attack, stroke, and related deaths; one 2005 study estimated that it had been responsible for some 38,000 excess deaths in the United States before being withdrawn. In 2007 Merck, beset with allegations that it had suppressed and mischaracterized rofecoxib’s safety data, paid out nearly $5 billion to settle thousands of lawsuits filed by patients and their families.
, an indication for which it received an orphan drug designation in 2017 and the agency’s green light for trials in 2020.
Brad Sippy, Tremeau’s chief executive officer, said that his company chose the two indications in part because both patient populations have low cardiovascular risk. Migraine patients are generally younger than the arthritis populations formerly treated with rofecoxib and are unlikely to take the drug for more than a day or 2 at time, avoiding the risks associated with extended exposure.
A crowded market
The past several years have seen the emergence of a cornucopia of new migraine treatments, including monoclonal antibodies such as erenumab (Aimovig, Amgen), which help prevent attacks by blocking the vasodilator calcitonin gene-related peptide, or CGRP. In addition to the standard arsenal of triptans and nonsteroidal anti-inflammatory drugs for acute pain relief, migraine patients can now choose among serotonin-blocking agents such as lasmiditan (Reyvow, Eli Lilly), known as “ditans,” and small-molecule CGRP antagonists such as ubrogepant (Ubrelvy, Abbie), known as “gepants.” Some NSAIDs, including one COX inhibitor, have been formulated into rapidly absorbed powders or liquids for migraine.
Mr. Sippy said he sees a role for rofecoxib even in this crowded space. “Migraine as you know is a multimodal situation – few people say that only one drug works for them,” he said. “We think this is an option that would basically be like a high dose of ibuprofen,” but with less frequent dosing and lower gastrointestinal and platelet effects compared with ibuprofen and other NSAIDs.
An improved formulation
Rofecoxib “crosses the blood brain barrier very readily – better than other COX inhibitors on the market,” Mr. Sippy added. “It was well absorbed in its original formulation, and our product is even better absorbed than the original – we estimate it’s probably an hour quicker to [peak concentration].” In addition, he said, “our formulation is more efficient at delivering the drug so we don’t need as much active ingredient – our 17.5 milligrams gets you the same systemic exposure as 25 milligrams of the old product.”
A different mechanism of action
Neurologist Alan M. Rapoport, MD, editor-in-chief of Neurology Reviews and professor of neurology at the University of California, Los Angeles, said that he was “cautiously optimistic” that “if used correctly and not too frequently, [rofecoxib] will find its niche in migraine treatment.”
“Patients liked Vioxx,” said Dr. Rapoport, past president of the International Headache Society. Even people currently on prevention “need to have an acute care drug handy.” While some patients on monoclonal antibodies have had success with gepants for acute care, “these both target the same pathway. It’s always nice to have options with a different mechanism of action.”
One of the arguments Tremeau has cited for reintroducing rofecoxib has been an urgent need for alternatives to opioid painkillers. Indeed some analysts have linked the demise of Vioxx with a subsequent increase in opioid prescribing.
Dr. Rapoport noted that he never prescribes opioids or butalbital, a barbiturate, for migraine, and that most headache specialists avoid them in clinical practice. But in the emergency setting, he said, patients receive them all too frequently.
Mr. Sippy said that opioid prescribing, while not unknown in migraine, was a bigger problem in hemophilic arthropathy, the first indication his company has pursued for rofecoxib. People with hemophilia “have a kind of arthritis that would respond well to an anti-inflammatory drug but they can’t take NSAIDs due to bleeding risk. This is why so many end up on opioids. Rofecoxib, as a COX-2 inhibitor, doesn’t have any effect on platelet aggregation, which would make it another option.”
No unique risks at prescribed doses
The migraine indication originally started out narrower: Patients with both migraine and bleeding disorders. “But in talking with the FDA, they encouraged us to develop it for migraine,” Mr. Sippy said. The company is considering pursuing a third indication: menstrual pain co-occurring with migraine. Tremeau has not ruled out seeking an indication in patients with arthritis who cannot take other painkillers, whether opioids or NSAIDs.
Five years ago, when Tremeau first announced its plans to bring rofecoxib back – indeed the company was set up for that purpose and has only this and another COX-2 inhibitor in development – some experts warned that there is little to prevent the drug from being used off-label, whether in higher doses or for other diseases.
“That’s something else we’re seeking to solve in addition to going for younger populations,” said Mr. Sippy, who worked at Merck during the Vioxx crisis and later headed neurology at Sunovion before starting his own company.
“We’re going for the former middle dose as our high dose and now we know that you don’t want to take more than the prescribed amount. If it doesn’t work you get off it; you don’t want to dose-creep on it. That’s been a key insight: At the appropriate dose, this product has no unique risk relative to the drug class and potentially some unique benefits,” he said.
Risk versus benefit
Joseph Ross, MD, a health policy researcher at Yale University in New Haven, Conn., who in a 2018 editorial expressed concerns about rofecoxib’s revival, said in an email that he felt its use in migraine could be justified, with caveats.
During Vioxx’s original approval and time on the market, “there was a cardiovascular risk associated with use that was not being transparently and clearly reported to patients and clinicians,” Dr. Ross said.
“In terms of testing the product for use in patients with migraine – a population of generally younger patients at lower risk of cardiovascular disease – my only concern is that the risk is clearly communicated and that there is adequate postmarket safety surveillance,” he said. “If patients are making fully informed decisions, the potential benefit of the drug with respect to pain control may be worth the risks.”
Dr. Rapoport serves as an adviser for AbbVie, Amgen, Biohaven, Cala Health, Collegium Pharmaceutical, Satsuma, Teva, Theranica and Xoc; he is on the speakers bureau of AbbVie, Amgen, Biohaven, Impel, Lundbeck, and Teva. Dr. Ross disclosed research support from Johnson and Johnson, the Medical Device Innovation Consortium, and the Laura and John Arnold Foundation, along with government grants; he is also an expert witness in a lawsuit against Biogen.
In June, the Concord, Mass.–based company Tremeau Pharmaceuticals announced that the Food and Drug Administration was letting it proceed with a phase 3 clinical trial to test rofecoxib, the once-bestselling painkiller known as Vioxx, in patients with migraine.
The anti-inflammatory drug, a cyclooxygenase-2 (COX-2) inhibitor, received its first FDA approval in 1999 and became widely prescribed for arthritis and acute pain. In 2004 it was withdrawn by its manufacturer, Merck, after being shown to raise the risk of cardiovascular events.
In clinical trials and in real-world epidemiological studies, rofecoxib was associated with elevated heart attack, stroke, and related deaths; one 2005 study estimated that it had been responsible for some 38,000 excess deaths in the United States before being withdrawn. In 2007 Merck, beset with allegations that it had suppressed and mischaracterized rofecoxib’s safety data, paid out nearly $5 billion to settle thousands of lawsuits filed by patients and their families.
, an indication for which it received an orphan drug designation in 2017 and the agency’s green light for trials in 2020.
Brad Sippy, Tremeau’s chief executive officer, said that his company chose the two indications in part because both patient populations have low cardiovascular risk. Migraine patients are generally younger than the arthritis populations formerly treated with rofecoxib and are unlikely to take the drug for more than a day or 2 at time, avoiding the risks associated with extended exposure.
A crowded market
The past several years have seen the emergence of a cornucopia of new migraine treatments, including monoclonal antibodies such as erenumab (Aimovig, Amgen), which help prevent attacks by blocking the vasodilator calcitonin gene-related peptide, or CGRP. In addition to the standard arsenal of triptans and nonsteroidal anti-inflammatory drugs for acute pain relief, migraine patients can now choose among serotonin-blocking agents such as lasmiditan (Reyvow, Eli Lilly), known as “ditans,” and small-molecule CGRP antagonists such as ubrogepant (Ubrelvy, Abbie), known as “gepants.” Some NSAIDs, including one COX inhibitor, have been formulated into rapidly absorbed powders or liquids for migraine.
Mr. Sippy said he sees a role for rofecoxib even in this crowded space. “Migraine as you know is a multimodal situation – few people say that only one drug works for them,” he said. “We think this is an option that would basically be like a high dose of ibuprofen,” but with less frequent dosing and lower gastrointestinal and platelet effects compared with ibuprofen and other NSAIDs.
An improved formulation
Rofecoxib “crosses the blood brain barrier very readily – better than other COX inhibitors on the market,” Mr. Sippy added. “It was well absorbed in its original formulation, and our product is even better absorbed than the original – we estimate it’s probably an hour quicker to [peak concentration].” In addition, he said, “our formulation is more efficient at delivering the drug so we don’t need as much active ingredient – our 17.5 milligrams gets you the same systemic exposure as 25 milligrams of the old product.”
A different mechanism of action
Neurologist Alan M. Rapoport, MD, editor-in-chief of Neurology Reviews and professor of neurology at the University of California, Los Angeles, said that he was “cautiously optimistic” that “if used correctly and not too frequently, [rofecoxib] will find its niche in migraine treatment.”
“Patients liked Vioxx,” said Dr. Rapoport, past president of the International Headache Society. Even people currently on prevention “need to have an acute care drug handy.” While some patients on monoclonal antibodies have had success with gepants for acute care, “these both target the same pathway. It’s always nice to have options with a different mechanism of action.”
One of the arguments Tremeau has cited for reintroducing rofecoxib has been an urgent need for alternatives to opioid painkillers. Indeed some analysts have linked the demise of Vioxx with a subsequent increase in opioid prescribing.
Dr. Rapoport noted that he never prescribes opioids or butalbital, a barbiturate, for migraine, and that most headache specialists avoid them in clinical practice. But in the emergency setting, he said, patients receive them all too frequently.
Mr. Sippy said that opioid prescribing, while not unknown in migraine, was a bigger problem in hemophilic arthropathy, the first indication his company has pursued for rofecoxib. People with hemophilia “have a kind of arthritis that would respond well to an anti-inflammatory drug but they can’t take NSAIDs due to bleeding risk. This is why so many end up on opioids. Rofecoxib, as a COX-2 inhibitor, doesn’t have any effect on platelet aggregation, which would make it another option.”
No unique risks at prescribed doses
The migraine indication originally started out narrower: Patients with both migraine and bleeding disorders. “But in talking with the FDA, they encouraged us to develop it for migraine,” Mr. Sippy said. The company is considering pursuing a third indication: menstrual pain co-occurring with migraine. Tremeau has not ruled out seeking an indication in patients with arthritis who cannot take other painkillers, whether opioids or NSAIDs.
Five years ago, when Tremeau first announced its plans to bring rofecoxib back – indeed the company was set up for that purpose and has only this and another COX-2 inhibitor in development – some experts warned that there is little to prevent the drug from being used off-label, whether in higher doses or for other diseases.
“That’s something else we’re seeking to solve in addition to going for younger populations,” said Mr. Sippy, who worked at Merck during the Vioxx crisis and later headed neurology at Sunovion before starting his own company.
“We’re going for the former middle dose as our high dose and now we know that you don’t want to take more than the prescribed amount. If it doesn’t work you get off it; you don’t want to dose-creep on it. That’s been a key insight: At the appropriate dose, this product has no unique risk relative to the drug class and potentially some unique benefits,” he said.
Risk versus benefit
Joseph Ross, MD, a health policy researcher at Yale University in New Haven, Conn., who in a 2018 editorial expressed concerns about rofecoxib’s revival, said in an email that he felt its use in migraine could be justified, with caveats.
During Vioxx’s original approval and time on the market, “there was a cardiovascular risk associated with use that was not being transparently and clearly reported to patients and clinicians,” Dr. Ross said.
“In terms of testing the product for use in patients with migraine – a population of generally younger patients at lower risk of cardiovascular disease – my only concern is that the risk is clearly communicated and that there is adequate postmarket safety surveillance,” he said. “If patients are making fully informed decisions, the potential benefit of the drug with respect to pain control may be worth the risks.”
Dr. Rapoport serves as an adviser for AbbVie, Amgen, Biohaven, Cala Health, Collegium Pharmaceutical, Satsuma, Teva, Theranica and Xoc; he is on the speakers bureau of AbbVie, Amgen, Biohaven, Impel, Lundbeck, and Teva. Dr. Ross disclosed research support from Johnson and Johnson, the Medical Device Innovation Consortium, and the Laura and John Arnold Foundation, along with government grants; he is also an expert witness in a lawsuit against Biogen.
In June, the Concord, Mass.–based company Tremeau Pharmaceuticals announced that the Food and Drug Administration was letting it proceed with a phase 3 clinical trial to test rofecoxib, the once-bestselling painkiller known as Vioxx, in patients with migraine.
The anti-inflammatory drug, a cyclooxygenase-2 (COX-2) inhibitor, received its first FDA approval in 1999 and became widely prescribed for arthritis and acute pain. In 2004 it was withdrawn by its manufacturer, Merck, after being shown to raise the risk of cardiovascular events.
In clinical trials and in real-world epidemiological studies, rofecoxib was associated with elevated heart attack, stroke, and related deaths; one 2005 study estimated that it had been responsible for some 38,000 excess deaths in the United States before being withdrawn. In 2007 Merck, beset with allegations that it had suppressed and mischaracterized rofecoxib’s safety data, paid out nearly $5 billion to settle thousands of lawsuits filed by patients and their families.
, an indication for which it received an orphan drug designation in 2017 and the agency’s green light for trials in 2020.
Brad Sippy, Tremeau’s chief executive officer, said that his company chose the two indications in part because both patient populations have low cardiovascular risk. Migraine patients are generally younger than the arthritis populations formerly treated with rofecoxib and are unlikely to take the drug for more than a day or 2 at time, avoiding the risks associated with extended exposure.
A crowded market
The past several years have seen the emergence of a cornucopia of new migraine treatments, including monoclonal antibodies such as erenumab (Aimovig, Amgen), which help prevent attacks by blocking the vasodilator calcitonin gene-related peptide, or CGRP. In addition to the standard arsenal of triptans and nonsteroidal anti-inflammatory drugs for acute pain relief, migraine patients can now choose among serotonin-blocking agents such as lasmiditan (Reyvow, Eli Lilly), known as “ditans,” and small-molecule CGRP antagonists such as ubrogepant (Ubrelvy, Abbie), known as “gepants.” Some NSAIDs, including one COX inhibitor, have been formulated into rapidly absorbed powders or liquids for migraine.
Mr. Sippy said he sees a role for rofecoxib even in this crowded space. “Migraine as you know is a multimodal situation – few people say that only one drug works for them,” he said. “We think this is an option that would basically be like a high dose of ibuprofen,” but with less frequent dosing and lower gastrointestinal and platelet effects compared with ibuprofen and other NSAIDs.
An improved formulation
Rofecoxib “crosses the blood brain barrier very readily – better than other COX inhibitors on the market,” Mr. Sippy added. “It was well absorbed in its original formulation, and our product is even better absorbed than the original – we estimate it’s probably an hour quicker to [peak concentration].” In addition, he said, “our formulation is more efficient at delivering the drug so we don’t need as much active ingredient – our 17.5 milligrams gets you the same systemic exposure as 25 milligrams of the old product.”
A different mechanism of action
Neurologist Alan M. Rapoport, MD, editor-in-chief of Neurology Reviews and professor of neurology at the University of California, Los Angeles, said that he was “cautiously optimistic” that “if used correctly and not too frequently, [rofecoxib] will find its niche in migraine treatment.”
“Patients liked Vioxx,” said Dr. Rapoport, past president of the International Headache Society. Even people currently on prevention “need to have an acute care drug handy.” While some patients on monoclonal antibodies have had success with gepants for acute care, “these both target the same pathway. It’s always nice to have options with a different mechanism of action.”
One of the arguments Tremeau has cited for reintroducing rofecoxib has been an urgent need for alternatives to opioid painkillers. Indeed some analysts have linked the demise of Vioxx with a subsequent increase in opioid prescribing.
Dr. Rapoport noted that he never prescribes opioids or butalbital, a barbiturate, for migraine, and that most headache specialists avoid them in clinical practice. But in the emergency setting, he said, patients receive them all too frequently.
Mr. Sippy said that opioid prescribing, while not unknown in migraine, was a bigger problem in hemophilic arthropathy, the first indication his company has pursued for rofecoxib. People with hemophilia “have a kind of arthritis that would respond well to an anti-inflammatory drug but they can’t take NSAIDs due to bleeding risk. This is why so many end up on opioids. Rofecoxib, as a COX-2 inhibitor, doesn’t have any effect on platelet aggregation, which would make it another option.”
No unique risks at prescribed doses
The migraine indication originally started out narrower: Patients with both migraine and bleeding disorders. “But in talking with the FDA, they encouraged us to develop it for migraine,” Mr. Sippy said. The company is considering pursuing a third indication: menstrual pain co-occurring with migraine. Tremeau has not ruled out seeking an indication in patients with arthritis who cannot take other painkillers, whether opioids or NSAIDs.
Five years ago, when Tremeau first announced its plans to bring rofecoxib back – indeed the company was set up for that purpose and has only this and another COX-2 inhibitor in development – some experts warned that there is little to prevent the drug from being used off-label, whether in higher doses or for other diseases.
“That’s something else we’re seeking to solve in addition to going for younger populations,” said Mr. Sippy, who worked at Merck during the Vioxx crisis and later headed neurology at Sunovion before starting his own company.
“We’re going for the former middle dose as our high dose and now we know that you don’t want to take more than the prescribed amount. If it doesn’t work you get off it; you don’t want to dose-creep on it. That’s been a key insight: At the appropriate dose, this product has no unique risk relative to the drug class and potentially some unique benefits,” he said.
Risk versus benefit
Joseph Ross, MD, a health policy researcher at Yale University in New Haven, Conn., who in a 2018 editorial expressed concerns about rofecoxib’s revival, said in an email that he felt its use in migraine could be justified, with caveats.
During Vioxx’s original approval and time on the market, “there was a cardiovascular risk associated with use that was not being transparently and clearly reported to patients and clinicians,” Dr. Ross said.
“In terms of testing the product for use in patients with migraine – a population of generally younger patients at lower risk of cardiovascular disease – my only concern is that the risk is clearly communicated and that there is adequate postmarket safety surveillance,” he said. “If patients are making fully informed decisions, the potential benefit of the drug with respect to pain control may be worth the risks.”
Dr. Rapoport serves as an adviser for AbbVie, Amgen, Biohaven, Cala Health, Collegium Pharmaceutical, Satsuma, Teva, Theranica and Xoc; he is on the speakers bureau of AbbVie, Amgen, Biohaven, Impel, Lundbeck, and Teva. Dr. Ross disclosed research support from Johnson and Johnson, the Medical Device Innovation Consortium, and the Laura and John Arnold Foundation, along with government grants; he is also an expert witness in a lawsuit against Biogen.
Irritable bowel syndrome therapy removed from market (again)
Zelnorm (tegaserod), an oral short-term treatment of irritable bowel syndrome and constipation (IBS-C), is being removed from the U.S. market effective June 30, according to the manufacturer, Alfasigma.
The Italian pharmaceutical company said the drug is being removed for business purposes, not because of any concern involving its safety or efficacy, nor has it been recalled.
The drug has been through a teeter totter of regulations since its inception.
When it was first introduced in 2002, Zelnorm was a first-of-its-kind drug and was intended to treat all women with IBS-C in the short term. But it was removed from the market 5 years later following concerns about cardiovascular side effects. Clinical data showed an increased incidence of stroke and angina in women taking Zelnorm.
Despite these concerns, the U.S. Food and Drug Administration voted to reintroduce the drug into the market in 2019, but only for women without a history of heart health problems.
Though Alfasigma will stop making the drug, a company news release said current users can continue use for a while.
“Patients will continue to have access to Zelnorm (tegaserod) for as long as the existing supply of product remains in the trade channel,” Alfasigma said in a news release about the drug removal. The company urged its customers to discuss alternative IBS medications with their doctor.
Zelnorm is a serotonin agonist, meaning it binds to receptors and stops the release of serotonin into the system. These sorts of drugs can decrease the pain associated with IBS and help increase gut motility in order to pass stool. Other drugs besides Zelnorm that use this mechanism include alosetron and cilansetron.
A version of this article first appeared on Medscape.com.
Zelnorm (tegaserod), an oral short-term treatment of irritable bowel syndrome and constipation (IBS-C), is being removed from the U.S. market effective June 30, according to the manufacturer, Alfasigma.
The Italian pharmaceutical company said the drug is being removed for business purposes, not because of any concern involving its safety or efficacy, nor has it been recalled.
The drug has been through a teeter totter of regulations since its inception.
When it was first introduced in 2002, Zelnorm was a first-of-its-kind drug and was intended to treat all women with IBS-C in the short term. But it was removed from the market 5 years later following concerns about cardiovascular side effects. Clinical data showed an increased incidence of stroke and angina in women taking Zelnorm.
Despite these concerns, the U.S. Food and Drug Administration voted to reintroduce the drug into the market in 2019, but only for women without a history of heart health problems.
Though Alfasigma will stop making the drug, a company news release said current users can continue use for a while.
“Patients will continue to have access to Zelnorm (tegaserod) for as long as the existing supply of product remains in the trade channel,” Alfasigma said in a news release about the drug removal. The company urged its customers to discuss alternative IBS medications with their doctor.
Zelnorm is a serotonin agonist, meaning it binds to receptors and stops the release of serotonin into the system. These sorts of drugs can decrease the pain associated with IBS and help increase gut motility in order to pass stool. Other drugs besides Zelnorm that use this mechanism include alosetron and cilansetron.
A version of this article first appeared on Medscape.com.
Zelnorm (tegaserod), an oral short-term treatment of irritable bowel syndrome and constipation (IBS-C), is being removed from the U.S. market effective June 30, according to the manufacturer, Alfasigma.
The Italian pharmaceutical company said the drug is being removed for business purposes, not because of any concern involving its safety or efficacy, nor has it been recalled.
The drug has been through a teeter totter of regulations since its inception.
When it was first introduced in 2002, Zelnorm was a first-of-its-kind drug and was intended to treat all women with IBS-C in the short term. But it was removed from the market 5 years later following concerns about cardiovascular side effects. Clinical data showed an increased incidence of stroke and angina in women taking Zelnorm.
Despite these concerns, the U.S. Food and Drug Administration voted to reintroduce the drug into the market in 2019, but only for women without a history of heart health problems.
Though Alfasigma will stop making the drug, a company news release said current users can continue use for a while.
“Patients will continue to have access to Zelnorm (tegaserod) for as long as the existing supply of product remains in the trade channel,” Alfasigma said in a news release about the drug removal. The company urged its customers to discuss alternative IBS medications with their doctor.
Zelnorm is a serotonin agonist, meaning it binds to receptors and stops the release of serotonin into the system. These sorts of drugs can decrease the pain associated with IBS and help increase gut motility in order to pass stool. Other drugs besides Zelnorm that use this mechanism include alosetron and cilansetron.
A version of this article first appeared on Medscape.com.
FDA warning: Lymphoma drug heightens risk of death
The U.S. Food and Drug Administration issued a warning today that the cancer drug duvelisib (Copiktra, Verastem), a PI3 kinase inhibitor, may increase the risk of death and serious side effects.
Duvelisib was approved in 2018 to treat adults with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) who had received at least two prior therapies that did not work or stopped working.
However, more recent 5-year overall survival results from the randomized phase 3 DUO clinical trial found a possible increased risk of death with duvelisib, compared with another drug used to treat leukemia and lymphoma, according to an FDA Drug Safety Communication.
“The trial also found Copiktra was associated with a higher risk of serious side effects, including infections, diarrhea, inflammation of the intestines and lungs, skin reactions, and high liver enzyme levels in the blood,” states the warning, which advises prescribers to weigh the risks and benefits of continued use versus use of other treatments.
More specifically, median 5-year overall survival among 319 patients with CLL or SLL in the DUO trial was 52.3 months with duvelisib versus 63.3 months with the monoclonal antibody ofatumumab (hazard ratio, 1.09 overall and 1.06 among patients who received at least two prior lines of therapy).
Serious adverse events of grade 3 or higher were also more common in those treated with duvelisib.
Of note, in April, the FDA also announced that it was withdrawing approval of the relapsed or refractory follicular lymphoma indication for duvelisib following a voluntary request by the drug manufacturer Secura Bio.
A public meeting will be scheduled to discuss the findings of the trial and whether the drug should continue to be prescribed.
This FDA warning follows the agency’s June 1 withdrawal of approval for umbralisib (Ukoniq), another PI3 kinase inhibitor, following an investigation into a “possible increased risk of death.”
As reported by this news organization, umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
“These safety findings were similar for other medicines in the same PI3 kinase inhibitor class, which were discussed at an advisory committee meeting of non-FDA experts in April 2022,” according to the FDA warning.
The FDA urges patients and health care professionals to report side effects involving duvelisib or other medicines to the FDA MedWatch program.
A version of this article first appeared on Medscape.com
The U.S. Food and Drug Administration issued a warning today that the cancer drug duvelisib (Copiktra, Verastem), a PI3 kinase inhibitor, may increase the risk of death and serious side effects.
Duvelisib was approved in 2018 to treat adults with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) who had received at least two prior therapies that did not work or stopped working.
However, more recent 5-year overall survival results from the randomized phase 3 DUO clinical trial found a possible increased risk of death with duvelisib, compared with another drug used to treat leukemia and lymphoma, according to an FDA Drug Safety Communication.
“The trial also found Copiktra was associated with a higher risk of serious side effects, including infections, diarrhea, inflammation of the intestines and lungs, skin reactions, and high liver enzyme levels in the blood,” states the warning, which advises prescribers to weigh the risks and benefits of continued use versus use of other treatments.
More specifically, median 5-year overall survival among 319 patients with CLL or SLL in the DUO trial was 52.3 months with duvelisib versus 63.3 months with the monoclonal antibody ofatumumab (hazard ratio, 1.09 overall and 1.06 among patients who received at least two prior lines of therapy).
Serious adverse events of grade 3 or higher were also more common in those treated with duvelisib.
Of note, in April, the FDA also announced that it was withdrawing approval of the relapsed or refractory follicular lymphoma indication for duvelisib following a voluntary request by the drug manufacturer Secura Bio.
A public meeting will be scheduled to discuss the findings of the trial and whether the drug should continue to be prescribed.
This FDA warning follows the agency’s June 1 withdrawal of approval for umbralisib (Ukoniq), another PI3 kinase inhibitor, following an investigation into a “possible increased risk of death.”
As reported by this news organization, umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
“These safety findings were similar for other medicines in the same PI3 kinase inhibitor class, which were discussed at an advisory committee meeting of non-FDA experts in April 2022,” according to the FDA warning.
The FDA urges patients and health care professionals to report side effects involving duvelisib or other medicines to the FDA MedWatch program.
A version of this article first appeared on Medscape.com
The U.S. Food and Drug Administration issued a warning today that the cancer drug duvelisib (Copiktra, Verastem), a PI3 kinase inhibitor, may increase the risk of death and serious side effects.
Duvelisib was approved in 2018 to treat adults with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) who had received at least two prior therapies that did not work or stopped working.
However, more recent 5-year overall survival results from the randomized phase 3 DUO clinical trial found a possible increased risk of death with duvelisib, compared with another drug used to treat leukemia and lymphoma, according to an FDA Drug Safety Communication.
“The trial also found Copiktra was associated with a higher risk of serious side effects, including infections, diarrhea, inflammation of the intestines and lungs, skin reactions, and high liver enzyme levels in the blood,” states the warning, which advises prescribers to weigh the risks and benefits of continued use versus use of other treatments.
More specifically, median 5-year overall survival among 319 patients with CLL or SLL in the DUO trial was 52.3 months with duvelisib versus 63.3 months with the monoclonal antibody ofatumumab (hazard ratio, 1.09 overall and 1.06 among patients who received at least two prior lines of therapy).
Serious adverse events of grade 3 or higher were also more common in those treated with duvelisib.
Of note, in April, the FDA also announced that it was withdrawing approval of the relapsed or refractory follicular lymphoma indication for duvelisib following a voluntary request by the drug manufacturer Secura Bio.
A public meeting will be scheduled to discuss the findings of the trial and whether the drug should continue to be prescribed.
This FDA warning follows the agency’s June 1 withdrawal of approval for umbralisib (Ukoniq), another PI3 kinase inhibitor, following an investigation into a “possible increased risk of death.”
As reported by this news organization, umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
“These safety findings were similar for other medicines in the same PI3 kinase inhibitor class, which were discussed at an advisory committee meeting of non-FDA experts in April 2022,” according to the FDA warning.
The FDA urges patients and health care professionals to report side effects involving duvelisib or other medicines to the FDA MedWatch program.
A version of this article first appeared on Medscape.com