User login
Climate change magnifies health effects of wildfire smoke in care deserts
DRESSLERVILLE, NEV. – Smoke began billowing into the skies of northwestern Nevada in September, clouding the mountains, dimming the sun – and quashing residents’ hopes that they would be spared from wildfires and the awful air quality the blazes produce.
The lung-irritating particles were blowing in from burning forests in California and settling in Douglas County, Nevada, home to nearly 50,000 people, prompting warnings that air quality had reached hazardous levels.
Those levels meant the air was very unhealthy, bad enough to raise alarms about people’s immediate health care needs and questions about whether worsening pollution could result in long-term health issues. People could increasingly face such risks as climate change makes wildfires, drought, dust storms, and floods more frequent across the United States and the world.
Some people simply feel powerless.
“There’s not much we could do about it,” said Serrell Smokey, chairman of the Washoe Tribe of Nevada and California. The tribe’s land straddles the border between California and Nevada near Lake Tahoe and extends into Douglas County, about 60 miles south of Reno.
Tribe members and other area residents are among millions of people nationwide who this year will experience poor air quality because of wildfires. In September, as smoke settled over Nevada, fire-related air quality alerts were dispatched in six other states: California, Idaho, Montana, Oregon, Washington, and Wyoming.
Yet, by one measure, people who live in Douglas County are better off than those in some other hard-hit areas. Douglas County residents must drive 30 minutes, on average, for medical care from lung specialists called pulmonologists. In other parts of the West and Upper Midwest, however, patients must drive an hour or more, according to data analyzed by GoodRx, a website that tracks prescription drug prices and conducts research.
Specifically, the research found that about 5.5 million Americans live in the 488 counties where drive times to pulmonologists are an hour or more. Much of Nevada and large parts of Montana fall into those gaps between specialists – places that have recently grappled with wildfires that fill the air with smoke and ash, which can cause lung problems or exacerbate existing ones.
Data from the Association of American Medical Colleges shows the number of pulmonary disease specialists in the United States dropped nearly 11% from 2014 to 2019. The group, which is based in Washington, D.C., and represents the academic medicine community, noted that the decline might not be as high as it appears because some physicians are opting to practice pulmonary critical care rather than just pulmonology. Many of those types of pulmonologists work in hospital intensive care units.
About 15,000 pulmonologists are practicing in the United States, according to the GoodRx report. Yet vast swaths of the country have few or none.
“New Mexico has one pulmonologist for the entire southeastern part of state, not counting Las Cruces, which is closer to El Paso,” said Victor Test, MD, a pulmonologist at Texas Tech Physicians.
Dr. Test, one of 13 pulmonologists in the Lubbock, Tex., region, said that his patients from within Texas sometimes drive 4 hours for an appointment and that other people travel from “New Mexico, Oklahoma, even far western Kansas.”
Increases in wildfires and their intensity will likely expand the need for pulmonologists.
“Climate change is going to affect lung disease,” said Nicholas Kenyon, MD, a professor of pulmonary, critical care, and sleep medicine at the University of California, Davis, where he and several other researchers are tracking the effects of wildfires. At his Sacramento practice, Dr. Kenyon said, he sees patients from far northern parts of California, including Eureka, a 5-hour drive from the state capital.
The short-term effects of breathing smoke are pretty well known. People show up in emergency rooms with asthma attacks, exacerbation of COPD, bronchitis, and even pneumonia, Dr. Kenyon said. Some have chest pain or other cardiac concerns.
“But we have very little understanding of what happens over the longer term,” he said. “If people get 2 or 3 weeks of wildfire exposure for 2 or 3 years, does that lead to worsening of asthma or COPD? We just don’t know.”
Fires release multiple pollutants, including carbon dioxide, carbon monoxide, and chemicals like benzene. All fires send particles into the air. Health researchers and air quality experts are most concerned about tiny pieces referred to as particulate matter 2.5. Far smaller than a human hair, the particles can lodge deep in the lungs and have been linked to heart and lung conditions.
Increases in those tiny particles are associated with a greater risk of death from all causes, excluding accidents, homicides, and other nonaccidental causes, for up to 4 days after a population is exposed, according to a 2020 New England Journal of Medicine overview.
The concentration of fine particulate matter is one of five gauges used to calculate the Air Quality Index, a numerical and color-coded index used to let the public know about local air pollution levels. Green denotes good air quality and is given if the total index is 50 or less. When the measurement exceeds 100, the air quality gets an orange label and may be bad for certain groups. Levels over 200 get a red label and are considered unhealthy for everyone.
Government agencies track those levels, as do people who use apps or websites to determine whether it’s safe to go outside.
When the AQI rises above 150, Farah Madhani-Lovely, MD, a pulmonologist, said, Renown Regional Medical Center in Reno shuts its outpatient pulmonary rehabilitation clinic because it doesn’t want to encourage patients to drive in. Some patients from Douglas County opt for care near home, about an hour away. “We don’t want these patients exposed outside because just 1 minute of exposure to the smoke can trigger an exacerbation of their chronic disease,” Dr. Madhani-Lovely said.
Mr. Smokey said connecting with pulmonologists can be difficult for Washoe Tribe members, particularly those who live on the California side of the reservation. “We cannot find providers for them,” he said. “We end up referring them out and sending them hundreds of miles out of their way just to get care that we should be able to provide here.”
Recruiting specialists to rural areas or smaller cities has long been difficult. For one thing, a specialist might be the only one for miles around, “so there’s a tremendous burden in terms of coverage and days off,” Dr. Test said.
Another concern is that physicians tend to train in larger cities and often want to practice in similar places. Even recruiting pulmonary physicians to Lubbock, a city of 260,000 in West Texas, is a challenge, Dr. Test said.
“I love Lubbock,” he said. “But I tell people who have never been here, I say, ‘It’s really flat.’ They don’t understand flat until they get here.”
In Nevada, on days when the air quality is bad, Washoe tribal members try to protect themselves with makeshift air purifiers created from fans, duct tape, and air filters, Mr. Smokey said.
Longer term, Mr. Smokey and other tribal leaders are pushing the Indian Health Service to establish a specialty care hospital in northern Nevada. The closest specialty care hospital for Washoe tribal members is more than 700 miles away, in Phoenix.
It’s difficult because “there’s a need we should be taking care of,” Mr. Smokey said. “But we have to fight for it. And sometimes that fight takes years, years, and years to accomplish.”
A version of this article first appeared on Medscape.com.
DRESSLERVILLE, NEV. – Smoke began billowing into the skies of northwestern Nevada in September, clouding the mountains, dimming the sun – and quashing residents’ hopes that they would be spared from wildfires and the awful air quality the blazes produce.
The lung-irritating particles were blowing in from burning forests in California and settling in Douglas County, Nevada, home to nearly 50,000 people, prompting warnings that air quality had reached hazardous levels.
Those levels meant the air was very unhealthy, bad enough to raise alarms about people’s immediate health care needs and questions about whether worsening pollution could result in long-term health issues. People could increasingly face such risks as climate change makes wildfires, drought, dust storms, and floods more frequent across the United States and the world.
Some people simply feel powerless.
“There’s not much we could do about it,” said Serrell Smokey, chairman of the Washoe Tribe of Nevada and California. The tribe’s land straddles the border between California and Nevada near Lake Tahoe and extends into Douglas County, about 60 miles south of Reno.
Tribe members and other area residents are among millions of people nationwide who this year will experience poor air quality because of wildfires. In September, as smoke settled over Nevada, fire-related air quality alerts were dispatched in six other states: California, Idaho, Montana, Oregon, Washington, and Wyoming.
Yet, by one measure, people who live in Douglas County are better off than those in some other hard-hit areas. Douglas County residents must drive 30 minutes, on average, for medical care from lung specialists called pulmonologists. In other parts of the West and Upper Midwest, however, patients must drive an hour or more, according to data analyzed by GoodRx, a website that tracks prescription drug prices and conducts research.
Specifically, the research found that about 5.5 million Americans live in the 488 counties where drive times to pulmonologists are an hour or more. Much of Nevada and large parts of Montana fall into those gaps between specialists – places that have recently grappled with wildfires that fill the air with smoke and ash, which can cause lung problems or exacerbate existing ones.
Data from the Association of American Medical Colleges shows the number of pulmonary disease specialists in the United States dropped nearly 11% from 2014 to 2019. The group, which is based in Washington, D.C., and represents the academic medicine community, noted that the decline might not be as high as it appears because some physicians are opting to practice pulmonary critical care rather than just pulmonology. Many of those types of pulmonologists work in hospital intensive care units.
About 15,000 pulmonologists are practicing in the United States, according to the GoodRx report. Yet vast swaths of the country have few or none.
“New Mexico has one pulmonologist for the entire southeastern part of state, not counting Las Cruces, which is closer to El Paso,” said Victor Test, MD, a pulmonologist at Texas Tech Physicians.
Dr. Test, one of 13 pulmonologists in the Lubbock, Tex., region, said that his patients from within Texas sometimes drive 4 hours for an appointment and that other people travel from “New Mexico, Oklahoma, even far western Kansas.”
Increases in wildfires and their intensity will likely expand the need for pulmonologists.
“Climate change is going to affect lung disease,” said Nicholas Kenyon, MD, a professor of pulmonary, critical care, and sleep medicine at the University of California, Davis, where he and several other researchers are tracking the effects of wildfires. At his Sacramento practice, Dr. Kenyon said, he sees patients from far northern parts of California, including Eureka, a 5-hour drive from the state capital.
The short-term effects of breathing smoke are pretty well known. People show up in emergency rooms with asthma attacks, exacerbation of COPD, bronchitis, and even pneumonia, Dr. Kenyon said. Some have chest pain or other cardiac concerns.
“But we have very little understanding of what happens over the longer term,” he said. “If people get 2 or 3 weeks of wildfire exposure for 2 or 3 years, does that lead to worsening of asthma or COPD? We just don’t know.”
Fires release multiple pollutants, including carbon dioxide, carbon monoxide, and chemicals like benzene. All fires send particles into the air. Health researchers and air quality experts are most concerned about tiny pieces referred to as particulate matter 2.5. Far smaller than a human hair, the particles can lodge deep in the lungs and have been linked to heart and lung conditions.
Increases in those tiny particles are associated with a greater risk of death from all causes, excluding accidents, homicides, and other nonaccidental causes, for up to 4 days after a population is exposed, according to a 2020 New England Journal of Medicine overview.
The concentration of fine particulate matter is one of five gauges used to calculate the Air Quality Index, a numerical and color-coded index used to let the public know about local air pollution levels. Green denotes good air quality and is given if the total index is 50 or less. When the measurement exceeds 100, the air quality gets an orange label and may be bad for certain groups. Levels over 200 get a red label and are considered unhealthy for everyone.
Government agencies track those levels, as do people who use apps or websites to determine whether it’s safe to go outside.
When the AQI rises above 150, Farah Madhani-Lovely, MD, a pulmonologist, said, Renown Regional Medical Center in Reno shuts its outpatient pulmonary rehabilitation clinic because it doesn’t want to encourage patients to drive in. Some patients from Douglas County opt for care near home, about an hour away. “We don’t want these patients exposed outside because just 1 minute of exposure to the smoke can trigger an exacerbation of their chronic disease,” Dr. Madhani-Lovely said.
Mr. Smokey said connecting with pulmonologists can be difficult for Washoe Tribe members, particularly those who live on the California side of the reservation. “We cannot find providers for them,” he said. “We end up referring them out and sending them hundreds of miles out of their way just to get care that we should be able to provide here.”
Recruiting specialists to rural areas or smaller cities has long been difficult. For one thing, a specialist might be the only one for miles around, “so there’s a tremendous burden in terms of coverage and days off,” Dr. Test said.
Another concern is that physicians tend to train in larger cities and often want to practice in similar places. Even recruiting pulmonary physicians to Lubbock, a city of 260,000 in West Texas, is a challenge, Dr. Test said.
“I love Lubbock,” he said. “But I tell people who have never been here, I say, ‘It’s really flat.’ They don’t understand flat until they get here.”
In Nevada, on days when the air quality is bad, Washoe tribal members try to protect themselves with makeshift air purifiers created from fans, duct tape, and air filters, Mr. Smokey said.
Longer term, Mr. Smokey and other tribal leaders are pushing the Indian Health Service to establish a specialty care hospital in northern Nevada. The closest specialty care hospital for Washoe tribal members is more than 700 miles away, in Phoenix.
It’s difficult because “there’s a need we should be taking care of,” Mr. Smokey said. “But we have to fight for it. And sometimes that fight takes years, years, and years to accomplish.”
A version of this article first appeared on Medscape.com.
DRESSLERVILLE, NEV. – Smoke began billowing into the skies of northwestern Nevada in September, clouding the mountains, dimming the sun – and quashing residents’ hopes that they would be spared from wildfires and the awful air quality the blazes produce.
The lung-irritating particles were blowing in from burning forests in California and settling in Douglas County, Nevada, home to nearly 50,000 people, prompting warnings that air quality had reached hazardous levels.
Those levels meant the air was very unhealthy, bad enough to raise alarms about people’s immediate health care needs and questions about whether worsening pollution could result in long-term health issues. People could increasingly face such risks as climate change makes wildfires, drought, dust storms, and floods more frequent across the United States and the world.
Some people simply feel powerless.
“There’s not much we could do about it,” said Serrell Smokey, chairman of the Washoe Tribe of Nevada and California. The tribe’s land straddles the border between California and Nevada near Lake Tahoe and extends into Douglas County, about 60 miles south of Reno.
Tribe members and other area residents are among millions of people nationwide who this year will experience poor air quality because of wildfires. In September, as smoke settled over Nevada, fire-related air quality alerts were dispatched in six other states: California, Idaho, Montana, Oregon, Washington, and Wyoming.
Yet, by one measure, people who live in Douglas County are better off than those in some other hard-hit areas. Douglas County residents must drive 30 minutes, on average, for medical care from lung specialists called pulmonologists. In other parts of the West and Upper Midwest, however, patients must drive an hour or more, according to data analyzed by GoodRx, a website that tracks prescription drug prices and conducts research.
Specifically, the research found that about 5.5 million Americans live in the 488 counties where drive times to pulmonologists are an hour or more. Much of Nevada and large parts of Montana fall into those gaps between specialists – places that have recently grappled with wildfires that fill the air with smoke and ash, which can cause lung problems or exacerbate existing ones.
Data from the Association of American Medical Colleges shows the number of pulmonary disease specialists in the United States dropped nearly 11% from 2014 to 2019. The group, which is based in Washington, D.C., and represents the academic medicine community, noted that the decline might not be as high as it appears because some physicians are opting to practice pulmonary critical care rather than just pulmonology. Many of those types of pulmonologists work in hospital intensive care units.
About 15,000 pulmonologists are practicing in the United States, according to the GoodRx report. Yet vast swaths of the country have few or none.
“New Mexico has one pulmonologist for the entire southeastern part of state, not counting Las Cruces, which is closer to El Paso,” said Victor Test, MD, a pulmonologist at Texas Tech Physicians.
Dr. Test, one of 13 pulmonologists in the Lubbock, Tex., region, said that his patients from within Texas sometimes drive 4 hours for an appointment and that other people travel from “New Mexico, Oklahoma, even far western Kansas.”
Increases in wildfires and their intensity will likely expand the need for pulmonologists.
“Climate change is going to affect lung disease,” said Nicholas Kenyon, MD, a professor of pulmonary, critical care, and sleep medicine at the University of California, Davis, where he and several other researchers are tracking the effects of wildfires. At his Sacramento practice, Dr. Kenyon said, he sees patients from far northern parts of California, including Eureka, a 5-hour drive from the state capital.
The short-term effects of breathing smoke are pretty well known. People show up in emergency rooms with asthma attacks, exacerbation of COPD, bronchitis, and even pneumonia, Dr. Kenyon said. Some have chest pain or other cardiac concerns.
“But we have very little understanding of what happens over the longer term,” he said. “If people get 2 or 3 weeks of wildfire exposure for 2 or 3 years, does that lead to worsening of asthma or COPD? We just don’t know.”
Fires release multiple pollutants, including carbon dioxide, carbon monoxide, and chemicals like benzene. All fires send particles into the air. Health researchers and air quality experts are most concerned about tiny pieces referred to as particulate matter 2.5. Far smaller than a human hair, the particles can lodge deep in the lungs and have been linked to heart and lung conditions.
Increases in those tiny particles are associated with a greater risk of death from all causes, excluding accidents, homicides, and other nonaccidental causes, for up to 4 days after a population is exposed, according to a 2020 New England Journal of Medicine overview.
The concentration of fine particulate matter is one of five gauges used to calculate the Air Quality Index, a numerical and color-coded index used to let the public know about local air pollution levels. Green denotes good air quality and is given if the total index is 50 or less. When the measurement exceeds 100, the air quality gets an orange label and may be bad for certain groups. Levels over 200 get a red label and are considered unhealthy for everyone.
Government agencies track those levels, as do people who use apps or websites to determine whether it’s safe to go outside.
When the AQI rises above 150, Farah Madhani-Lovely, MD, a pulmonologist, said, Renown Regional Medical Center in Reno shuts its outpatient pulmonary rehabilitation clinic because it doesn’t want to encourage patients to drive in. Some patients from Douglas County opt for care near home, about an hour away. “We don’t want these patients exposed outside because just 1 minute of exposure to the smoke can trigger an exacerbation of their chronic disease,” Dr. Madhani-Lovely said.
Mr. Smokey said connecting with pulmonologists can be difficult for Washoe Tribe members, particularly those who live on the California side of the reservation. “We cannot find providers for them,” he said. “We end up referring them out and sending them hundreds of miles out of their way just to get care that we should be able to provide here.”
Recruiting specialists to rural areas or smaller cities has long been difficult. For one thing, a specialist might be the only one for miles around, “so there’s a tremendous burden in terms of coverage and days off,” Dr. Test said.
Another concern is that physicians tend to train in larger cities and often want to practice in similar places. Even recruiting pulmonary physicians to Lubbock, a city of 260,000 in West Texas, is a challenge, Dr. Test said.
“I love Lubbock,” he said. “But I tell people who have never been here, I say, ‘It’s really flat.’ They don’t understand flat until they get here.”
In Nevada, on days when the air quality is bad, Washoe tribal members try to protect themselves with makeshift air purifiers created from fans, duct tape, and air filters, Mr. Smokey said.
Longer term, Mr. Smokey and other tribal leaders are pushing the Indian Health Service to establish a specialty care hospital in northern Nevada. The closest specialty care hospital for Washoe tribal members is more than 700 miles away, in Phoenix.
It’s difficult because “there’s a need we should be taking care of,” Mr. Smokey said. “But we have to fight for it. And sometimes that fight takes years, years, and years to accomplish.”
A version of this article first appeared on Medscape.com.
Hormone changes: The star of every stage in women’s sleep
MADRID – Because of the hormone changes that occur throughout their lives, women experience sleep problems that differ significantly from those experienced by men. Indeed, 75%-84% of pregnant women don’t sleep well during the third trimester, and up to 80% of women in menopause have symptoms that prevent them from getting a good night’s rest. For those seeking a precision medicine approach, the challenge is to identify the relationship between the different sex-related phenotypes and the sleep conditions.
Irene Cano, MD, PhD, is the coordinator of the sleep department at the Spanish Society of Pulmonology and Thoracic Surgery. She spoke with this news organization about the significant impact of hormones on sleep disorders in women.
“Reproductive hormones like estrogen and progesterone play a meaningful role in brain functions – not only those linked to the regulation of reproduction but also other physiological processes related to the regulation of circadian rhythms, cognitive performance, mood, and sleep. In addition, other hormones – for example, prolactin, growth hormone, cortisol, and melatonin – have sex-dependent effects on sleep,” Dr. Cano said.
Girls start puberty at a younger age than boys. As girls enter adolescence, they go to bed later and waking up earlier. So, girls are getting less than the 10 hours of sleep that they should be getting at this stage of life. The result is sleep debt, which gives rise to various problems: poor academic performance, ADHD, obesity, and metabolic problems, to name a few. As Ariadna Farré, RN, a sleep unit nurse, noted at SEPAR’s Joint Winter Meeting, “schools would have to start morning classes later to get adolescents to perform well academically. As the situation is now, half of the kids are falling asleep at their desks.”
Influencing sleep quality
Dr. Cano explained the issue as follows: “In adolescence, along with changes in young women’s hormone levels, we begin to see differences between the sexes. The changes in levels of estrogens and progesterone are what’s responsible for the changes that, to some extent, cause those disturbances in the quality of our sleep and in the stages of our sleep.”
Thus, sleep can be affected by the changes in hormone level that occur during a menstrual cycle. Estrogens, which increase during the follicular phase, are associated with REM sleep, while progesterone, which increases during the luteal phase, increases non-REM sleep. “In the 3-6 days prior to menstruation, it’s quite common for a woman to report difficulties falling asleep and staying asleep, in connection with a decline in the percentage of time she spends in REM sleep, in the context of premenstrual syndrome. In addition,” Dr. Cano pointed out, “menstrual bleeding, that loss of blood, is associated with a drop in iron levels, making it more likely that the woman will experience restless legs syndrome.”
Cardiovascular system
This news organization also spoke with Milagros Merino, MD, PhD, president of the Spanish Sleep Society. “The consequences that lack of sleep have on the cardiovascular system – we’re essentially talking about certain arrhythmias, high blood pressure, thrombosis in some cases, stroke, and heart attack. Lack of sleep also gives rise to endocrine and metabolic issues, like overweight and being at a greater risk of developing diabetes. And as for mental health, we see, among other things, attention and memory problems, emotional lability, and irascibility. Numerous studies have confirmed all of this.”
Sleep apnea also deserves mention, Dr. Merino added. “Although this disorder is more common in men, we’re seeing it more and more now in women, along with the cardiovascular issues that it brings about.”
Another cardiovascular risk factor is insomnia, said Dr. Merino. “This sleep disorder is more prevalent in women. As hormones constantly change, the ways women sleep constantly change, from one stage of life to the next. They sleep one way in childhood, another way in adolescence, and yet another way in menopause.”
Sleep in pregnancy
During pregnancy, hormone changes are much more pronounced. During the first trimester, progesterone levels increase, making the woman drowsy. On top of that, her sleep is interrupted by more frequent visits to the bathroom as well as greater general discomfort.
In the second trimester, sleep interruptions persist but are not as bad as they were during the first 3 months. In the third trimester, 75%-84% of pregnant women find it difficult to sleep because of aches and pains, the need to urinate during the night, cramps, and heartburn.
“Major physical changes are happening. When the bladder gets compressed, the woman has to get up and go to the bathroom. There’s an interruption in her sleep,” Ms. Farré explained. In addition, as the pregnancy progresses, the woman gains weight and her body mass index (BMI) increases, which can bring on obstructive sleep apnea, high blood pressure, preeclampsia, and diabetes, if not closely monitored.
Other factors include concomitant treatments, such as contraceptives, and the stages of life, such as pregnancy and lactation. “When a woman of childbearing age has restless legs syndrome, more often than not, this means that she has an iron deficiency that needs to be treated with oral iron supplements,” said Dr. Merino. “However, there are few medications that can be given to a pregnant woman – and RLS is relatively common during pregnancy. So, we have to turn to oral or intravenous iron supplements. Yet another matter is narcolepsy. In these cases, all medications have to be stopped during pregnancy and lactation, as they can be harmful to the baby.”
Sleep apnea
While one in five menopausal women are asymptomatic, the others experience mild to severe symptoms of apnea that frequently interrupt their sleep. In this stage of life, which begins around age 50 years, the hormones that had provided protection against sleep disruptions start to decrease. As a result, there is a rise in sleep problems, especially insomnia, breathing-related sleep disorders (for example, apnea), and restless legs syndrome.
The prevalence of breathing-related sleep disorders during menopause is attributable to weight gain, the drop in levels of estrogens, and the redistribution of adipose tissue in the body. Other factors also increase a woman’s risk of experiencing apnea. They range from stress, depression, and other psychological and psychiatric conditions to health status, medication use, and simply the fact of getting older. “Sleep apnea is more common in men than in premenopausal women. The numbers even out, though, when we compare men against menopausal women,” Dr. Cano noted.
In women, symptoms of sleep apnea are frequently attributed to menopause. There is some overlap: insomnia, headache, irritability, low mood, decreased libido, fatigue during the day, and feeling sleepy. Only much later is the woman’s condition correctly diagnosed as sleep apnea. So, even though presenting with the same complaints, a man will be diagnosed with sleep apnea sooner than a woman will – in some cases, around 10 years sooner.
“On the other hand, we’d always thought that, in menopause, insomnia was characterized by awakenings occurring throughout the second half of the night. But perhaps what happens more often is that women are regularly waking up repeatedly over the course of the entire night, as opposed to experiencing a wakefulness that starts early and lasts throughout the night or having a problem falling asleep to begin with,” said Dr. Merino. “The good news is that hormone replacement therapy can get things back to the way they were. And getting better sleep will help to overcome insomnia.”
Socioeconomic status
Insomnia is the most common sleep disorder. It affects 10%-20% of people, mostly women. “The fact that sleep problems are more prevalent in women can be explained by the fact that among women, there is a higher incidence of conditions that disrupt sleep, such as depression,” said Dr. Cano.
“Insomnia is much more common in adult women than adult men. And at menopause, women find that the insomnia only gets worse,” Dr. Merino added. “But around that same age, 50 years old, what we start to see more frequently in men is REM sleep behavior disorder, a type of parasomnia that’s a risk marker of degenerative nerve diseases.”
Dr. Cano emphasized one finding that, though basic, is not well known. “After adjusting for socioeconomic characteristics, the difference between the sexes in reporting sleep problems is cut in half. This suggests that an important factor that explains why there are differences in sleep problems between the sexes is that women’s socioeconomic status is generally lower than men’s.
“As for sleep apnea in particular,” Dr. Cano continued, “the kinds of symptoms that women have can be different from the classic ones seen in men – snoring, pauses in breathing, and daytime sleepiness; women are being underdiagnosed, and when they are diagnosed, that’s happening at a later age and at a higher BMI.”
So, it’s alarming that, as reported by SEPAR, 90% of women with obstructive sleep apnea are not being diagnosed.
Precision medicine approach
“The majority of research studies on sleep apnea have focused on men – given the prevalence of cases – and the results have been extrapolated to women. This is why there’s still a lot of work to be done in terms of better defining the characteristics specific to each sleep disorder and how they relate to each sex,” said Dr. Cano. “Being able to identify the relationship between the different sex-related phenotypes and each condition will allow us to take a precision medicine approach tailored to a patient’s particular characteristics.”
As Dr. Merino put it: “The approach to sleep disorders is always personalized. The patient’s sex, in and of itself, doesn’t have that great of an impact on this approach. What does have a great impact are women’s life stages. There are some subtle differences here and there, such as types of continuous positive airway pressure machines. The ones that are designed for women have masks that are better suited to their facial features, which differ from men’s.”
A precision medicine approach can be taken to treat any sleep disorder. For insomnia, the approach allows healthcare professionals to employ an appropriate cognitive-behavioral therapy plan or to determine which drugs would be more effective – all on the basis of symptoms and the characteristics of the particular case. Regarding sleep apnea, Dr. Cano explained, “taking into account the different anatomical characteristics or the higher prevalence of positional apnea will also allow us to offer different therapeutic alternatives to continuous positive airway pressure, such as mandibular advancement devices or positional therapy devices.”
Women should be encouraged to develop good sleep habits. These include taking circadian rhythms into account and aligning lifestyles accordingly. It also means going to bed earlier than the men in the household. For menopausal women, recommended sleep habits range from keeping their bedroom at an ideal temperature, following a diet rich in vegetables to avoid becoming overweight, and exercising daily. While this advice may be more applicable to teenagers, adults can benefit from it as well: Electronic devices should be turned off well before bedtime. Whether from a phone screen, a tablet screen, or a TV screen, the light emitted can keep one awake, which can be harmful to one’s health.
Dr. Cano and Dr. Merino disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com. This article was translated from the Medscape Spanish edition.
MADRID – Because of the hormone changes that occur throughout their lives, women experience sleep problems that differ significantly from those experienced by men. Indeed, 75%-84% of pregnant women don’t sleep well during the third trimester, and up to 80% of women in menopause have symptoms that prevent them from getting a good night’s rest. For those seeking a precision medicine approach, the challenge is to identify the relationship between the different sex-related phenotypes and the sleep conditions.
Irene Cano, MD, PhD, is the coordinator of the sleep department at the Spanish Society of Pulmonology and Thoracic Surgery. She spoke with this news organization about the significant impact of hormones on sleep disorders in women.
“Reproductive hormones like estrogen and progesterone play a meaningful role in brain functions – not only those linked to the regulation of reproduction but also other physiological processes related to the regulation of circadian rhythms, cognitive performance, mood, and sleep. In addition, other hormones – for example, prolactin, growth hormone, cortisol, and melatonin – have sex-dependent effects on sleep,” Dr. Cano said.
Girls start puberty at a younger age than boys. As girls enter adolescence, they go to bed later and waking up earlier. So, girls are getting less than the 10 hours of sleep that they should be getting at this stage of life. The result is sleep debt, which gives rise to various problems: poor academic performance, ADHD, obesity, and metabolic problems, to name a few. As Ariadna Farré, RN, a sleep unit nurse, noted at SEPAR’s Joint Winter Meeting, “schools would have to start morning classes later to get adolescents to perform well academically. As the situation is now, half of the kids are falling asleep at their desks.”
Influencing sleep quality
Dr. Cano explained the issue as follows: “In adolescence, along with changes in young women’s hormone levels, we begin to see differences between the sexes. The changes in levels of estrogens and progesterone are what’s responsible for the changes that, to some extent, cause those disturbances in the quality of our sleep and in the stages of our sleep.”
Thus, sleep can be affected by the changes in hormone level that occur during a menstrual cycle. Estrogens, which increase during the follicular phase, are associated with REM sleep, while progesterone, which increases during the luteal phase, increases non-REM sleep. “In the 3-6 days prior to menstruation, it’s quite common for a woman to report difficulties falling asleep and staying asleep, in connection with a decline in the percentage of time she spends in REM sleep, in the context of premenstrual syndrome. In addition,” Dr. Cano pointed out, “menstrual bleeding, that loss of blood, is associated with a drop in iron levels, making it more likely that the woman will experience restless legs syndrome.”
Cardiovascular system
This news organization also spoke with Milagros Merino, MD, PhD, president of the Spanish Sleep Society. “The consequences that lack of sleep have on the cardiovascular system – we’re essentially talking about certain arrhythmias, high blood pressure, thrombosis in some cases, stroke, and heart attack. Lack of sleep also gives rise to endocrine and metabolic issues, like overweight and being at a greater risk of developing diabetes. And as for mental health, we see, among other things, attention and memory problems, emotional lability, and irascibility. Numerous studies have confirmed all of this.”
Sleep apnea also deserves mention, Dr. Merino added. “Although this disorder is more common in men, we’re seeing it more and more now in women, along with the cardiovascular issues that it brings about.”
Another cardiovascular risk factor is insomnia, said Dr. Merino. “This sleep disorder is more prevalent in women. As hormones constantly change, the ways women sleep constantly change, from one stage of life to the next. They sleep one way in childhood, another way in adolescence, and yet another way in menopause.”
Sleep in pregnancy
During pregnancy, hormone changes are much more pronounced. During the first trimester, progesterone levels increase, making the woman drowsy. On top of that, her sleep is interrupted by more frequent visits to the bathroom as well as greater general discomfort.
In the second trimester, sleep interruptions persist but are not as bad as they were during the first 3 months. In the third trimester, 75%-84% of pregnant women find it difficult to sleep because of aches and pains, the need to urinate during the night, cramps, and heartburn.
“Major physical changes are happening. When the bladder gets compressed, the woman has to get up and go to the bathroom. There’s an interruption in her sleep,” Ms. Farré explained. In addition, as the pregnancy progresses, the woman gains weight and her body mass index (BMI) increases, which can bring on obstructive sleep apnea, high blood pressure, preeclampsia, and diabetes, if not closely monitored.
Other factors include concomitant treatments, such as contraceptives, and the stages of life, such as pregnancy and lactation. “When a woman of childbearing age has restless legs syndrome, more often than not, this means that she has an iron deficiency that needs to be treated with oral iron supplements,” said Dr. Merino. “However, there are few medications that can be given to a pregnant woman – and RLS is relatively common during pregnancy. So, we have to turn to oral or intravenous iron supplements. Yet another matter is narcolepsy. In these cases, all medications have to be stopped during pregnancy and lactation, as they can be harmful to the baby.”
Sleep apnea
While one in five menopausal women are asymptomatic, the others experience mild to severe symptoms of apnea that frequently interrupt their sleep. In this stage of life, which begins around age 50 years, the hormones that had provided protection against sleep disruptions start to decrease. As a result, there is a rise in sleep problems, especially insomnia, breathing-related sleep disorders (for example, apnea), and restless legs syndrome.
The prevalence of breathing-related sleep disorders during menopause is attributable to weight gain, the drop in levels of estrogens, and the redistribution of adipose tissue in the body. Other factors also increase a woman’s risk of experiencing apnea. They range from stress, depression, and other psychological and psychiatric conditions to health status, medication use, and simply the fact of getting older. “Sleep apnea is more common in men than in premenopausal women. The numbers even out, though, when we compare men against menopausal women,” Dr. Cano noted.
In women, symptoms of sleep apnea are frequently attributed to menopause. There is some overlap: insomnia, headache, irritability, low mood, decreased libido, fatigue during the day, and feeling sleepy. Only much later is the woman’s condition correctly diagnosed as sleep apnea. So, even though presenting with the same complaints, a man will be diagnosed with sleep apnea sooner than a woman will – in some cases, around 10 years sooner.
“On the other hand, we’d always thought that, in menopause, insomnia was characterized by awakenings occurring throughout the second half of the night. But perhaps what happens more often is that women are regularly waking up repeatedly over the course of the entire night, as opposed to experiencing a wakefulness that starts early and lasts throughout the night or having a problem falling asleep to begin with,” said Dr. Merino. “The good news is that hormone replacement therapy can get things back to the way they were. And getting better sleep will help to overcome insomnia.”
Socioeconomic status
Insomnia is the most common sleep disorder. It affects 10%-20% of people, mostly women. “The fact that sleep problems are more prevalent in women can be explained by the fact that among women, there is a higher incidence of conditions that disrupt sleep, such as depression,” said Dr. Cano.
“Insomnia is much more common in adult women than adult men. And at menopause, women find that the insomnia only gets worse,” Dr. Merino added. “But around that same age, 50 years old, what we start to see more frequently in men is REM sleep behavior disorder, a type of parasomnia that’s a risk marker of degenerative nerve diseases.”
Dr. Cano emphasized one finding that, though basic, is not well known. “After adjusting for socioeconomic characteristics, the difference between the sexes in reporting sleep problems is cut in half. This suggests that an important factor that explains why there are differences in sleep problems between the sexes is that women’s socioeconomic status is generally lower than men’s.
“As for sleep apnea in particular,” Dr. Cano continued, “the kinds of symptoms that women have can be different from the classic ones seen in men – snoring, pauses in breathing, and daytime sleepiness; women are being underdiagnosed, and when they are diagnosed, that’s happening at a later age and at a higher BMI.”
So, it’s alarming that, as reported by SEPAR, 90% of women with obstructive sleep apnea are not being diagnosed.
Precision medicine approach
“The majority of research studies on sleep apnea have focused on men – given the prevalence of cases – and the results have been extrapolated to women. This is why there’s still a lot of work to be done in terms of better defining the characteristics specific to each sleep disorder and how they relate to each sex,” said Dr. Cano. “Being able to identify the relationship between the different sex-related phenotypes and each condition will allow us to take a precision medicine approach tailored to a patient’s particular characteristics.”
As Dr. Merino put it: “The approach to sleep disorders is always personalized. The patient’s sex, in and of itself, doesn’t have that great of an impact on this approach. What does have a great impact are women’s life stages. There are some subtle differences here and there, such as types of continuous positive airway pressure machines. The ones that are designed for women have masks that are better suited to their facial features, which differ from men’s.”
A precision medicine approach can be taken to treat any sleep disorder. For insomnia, the approach allows healthcare professionals to employ an appropriate cognitive-behavioral therapy plan or to determine which drugs would be more effective – all on the basis of symptoms and the characteristics of the particular case. Regarding sleep apnea, Dr. Cano explained, “taking into account the different anatomical characteristics or the higher prevalence of positional apnea will also allow us to offer different therapeutic alternatives to continuous positive airway pressure, such as mandibular advancement devices or positional therapy devices.”
Women should be encouraged to develop good sleep habits. These include taking circadian rhythms into account and aligning lifestyles accordingly. It also means going to bed earlier than the men in the household. For menopausal women, recommended sleep habits range from keeping their bedroom at an ideal temperature, following a diet rich in vegetables to avoid becoming overweight, and exercising daily. While this advice may be more applicable to teenagers, adults can benefit from it as well: Electronic devices should be turned off well before bedtime. Whether from a phone screen, a tablet screen, or a TV screen, the light emitted can keep one awake, which can be harmful to one’s health.
Dr. Cano and Dr. Merino disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com. This article was translated from the Medscape Spanish edition.
MADRID – Because of the hormone changes that occur throughout their lives, women experience sleep problems that differ significantly from those experienced by men. Indeed, 75%-84% of pregnant women don’t sleep well during the third trimester, and up to 80% of women in menopause have symptoms that prevent them from getting a good night’s rest. For those seeking a precision medicine approach, the challenge is to identify the relationship between the different sex-related phenotypes and the sleep conditions.
Irene Cano, MD, PhD, is the coordinator of the sleep department at the Spanish Society of Pulmonology and Thoracic Surgery. She spoke with this news organization about the significant impact of hormones on sleep disorders in women.
“Reproductive hormones like estrogen and progesterone play a meaningful role in brain functions – not only those linked to the regulation of reproduction but also other physiological processes related to the regulation of circadian rhythms, cognitive performance, mood, and sleep. In addition, other hormones – for example, prolactin, growth hormone, cortisol, and melatonin – have sex-dependent effects on sleep,” Dr. Cano said.
Girls start puberty at a younger age than boys. As girls enter adolescence, they go to bed later and waking up earlier. So, girls are getting less than the 10 hours of sleep that they should be getting at this stage of life. The result is sleep debt, which gives rise to various problems: poor academic performance, ADHD, obesity, and metabolic problems, to name a few. As Ariadna Farré, RN, a sleep unit nurse, noted at SEPAR’s Joint Winter Meeting, “schools would have to start morning classes later to get adolescents to perform well academically. As the situation is now, half of the kids are falling asleep at their desks.”
Influencing sleep quality
Dr. Cano explained the issue as follows: “In adolescence, along with changes in young women’s hormone levels, we begin to see differences between the sexes. The changes in levels of estrogens and progesterone are what’s responsible for the changes that, to some extent, cause those disturbances in the quality of our sleep and in the stages of our sleep.”
Thus, sleep can be affected by the changes in hormone level that occur during a menstrual cycle. Estrogens, which increase during the follicular phase, are associated with REM sleep, while progesterone, which increases during the luteal phase, increases non-REM sleep. “In the 3-6 days prior to menstruation, it’s quite common for a woman to report difficulties falling asleep and staying asleep, in connection with a decline in the percentage of time she spends in REM sleep, in the context of premenstrual syndrome. In addition,” Dr. Cano pointed out, “menstrual bleeding, that loss of blood, is associated with a drop in iron levels, making it more likely that the woman will experience restless legs syndrome.”
Cardiovascular system
This news organization also spoke with Milagros Merino, MD, PhD, president of the Spanish Sleep Society. “The consequences that lack of sleep have on the cardiovascular system – we’re essentially talking about certain arrhythmias, high blood pressure, thrombosis in some cases, stroke, and heart attack. Lack of sleep also gives rise to endocrine and metabolic issues, like overweight and being at a greater risk of developing diabetes. And as for mental health, we see, among other things, attention and memory problems, emotional lability, and irascibility. Numerous studies have confirmed all of this.”
Sleep apnea also deserves mention, Dr. Merino added. “Although this disorder is more common in men, we’re seeing it more and more now in women, along with the cardiovascular issues that it brings about.”
Another cardiovascular risk factor is insomnia, said Dr. Merino. “This sleep disorder is more prevalent in women. As hormones constantly change, the ways women sleep constantly change, from one stage of life to the next. They sleep one way in childhood, another way in adolescence, and yet another way in menopause.”
Sleep in pregnancy
During pregnancy, hormone changes are much more pronounced. During the first trimester, progesterone levels increase, making the woman drowsy. On top of that, her sleep is interrupted by more frequent visits to the bathroom as well as greater general discomfort.
In the second trimester, sleep interruptions persist but are not as bad as they were during the first 3 months. In the third trimester, 75%-84% of pregnant women find it difficult to sleep because of aches and pains, the need to urinate during the night, cramps, and heartburn.
“Major physical changes are happening. When the bladder gets compressed, the woman has to get up and go to the bathroom. There’s an interruption in her sleep,” Ms. Farré explained. In addition, as the pregnancy progresses, the woman gains weight and her body mass index (BMI) increases, which can bring on obstructive sleep apnea, high blood pressure, preeclampsia, and diabetes, if not closely monitored.
Other factors include concomitant treatments, such as contraceptives, and the stages of life, such as pregnancy and lactation. “When a woman of childbearing age has restless legs syndrome, more often than not, this means that she has an iron deficiency that needs to be treated with oral iron supplements,” said Dr. Merino. “However, there are few medications that can be given to a pregnant woman – and RLS is relatively common during pregnancy. So, we have to turn to oral or intravenous iron supplements. Yet another matter is narcolepsy. In these cases, all medications have to be stopped during pregnancy and lactation, as they can be harmful to the baby.”
Sleep apnea
While one in five menopausal women are asymptomatic, the others experience mild to severe symptoms of apnea that frequently interrupt their sleep. In this stage of life, which begins around age 50 years, the hormones that had provided protection against sleep disruptions start to decrease. As a result, there is a rise in sleep problems, especially insomnia, breathing-related sleep disorders (for example, apnea), and restless legs syndrome.
The prevalence of breathing-related sleep disorders during menopause is attributable to weight gain, the drop in levels of estrogens, and the redistribution of adipose tissue in the body. Other factors also increase a woman’s risk of experiencing apnea. They range from stress, depression, and other psychological and psychiatric conditions to health status, medication use, and simply the fact of getting older. “Sleep apnea is more common in men than in premenopausal women. The numbers even out, though, when we compare men against menopausal women,” Dr. Cano noted.
In women, symptoms of sleep apnea are frequently attributed to menopause. There is some overlap: insomnia, headache, irritability, low mood, decreased libido, fatigue during the day, and feeling sleepy. Only much later is the woman’s condition correctly diagnosed as sleep apnea. So, even though presenting with the same complaints, a man will be diagnosed with sleep apnea sooner than a woman will – in some cases, around 10 years sooner.
“On the other hand, we’d always thought that, in menopause, insomnia was characterized by awakenings occurring throughout the second half of the night. But perhaps what happens more often is that women are regularly waking up repeatedly over the course of the entire night, as opposed to experiencing a wakefulness that starts early and lasts throughout the night or having a problem falling asleep to begin with,” said Dr. Merino. “The good news is that hormone replacement therapy can get things back to the way they were. And getting better sleep will help to overcome insomnia.”
Socioeconomic status
Insomnia is the most common sleep disorder. It affects 10%-20% of people, mostly women. “The fact that sleep problems are more prevalent in women can be explained by the fact that among women, there is a higher incidence of conditions that disrupt sleep, such as depression,” said Dr. Cano.
“Insomnia is much more common in adult women than adult men. And at menopause, women find that the insomnia only gets worse,” Dr. Merino added. “But around that same age, 50 years old, what we start to see more frequently in men is REM sleep behavior disorder, a type of parasomnia that’s a risk marker of degenerative nerve diseases.”
Dr. Cano emphasized one finding that, though basic, is not well known. “After adjusting for socioeconomic characteristics, the difference between the sexes in reporting sleep problems is cut in half. This suggests that an important factor that explains why there are differences in sleep problems between the sexes is that women’s socioeconomic status is generally lower than men’s.
“As for sleep apnea in particular,” Dr. Cano continued, “the kinds of symptoms that women have can be different from the classic ones seen in men – snoring, pauses in breathing, and daytime sleepiness; women are being underdiagnosed, and when they are diagnosed, that’s happening at a later age and at a higher BMI.”
So, it’s alarming that, as reported by SEPAR, 90% of women with obstructive sleep apnea are not being diagnosed.
Precision medicine approach
“The majority of research studies on sleep apnea have focused on men – given the prevalence of cases – and the results have been extrapolated to women. This is why there’s still a lot of work to be done in terms of better defining the characteristics specific to each sleep disorder and how they relate to each sex,” said Dr. Cano. “Being able to identify the relationship between the different sex-related phenotypes and each condition will allow us to take a precision medicine approach tailored to a patient’s particular characteristics.”
As Dr. Merino put it: “The approach to sleep disorders is always personalized. The patient’s sex, in and of itself, doesn’t have that great of an impact on this approach. What does have a great impact are women’s life stages. There are some subtle differences here and there, such as types of continuous positive airway pressure machines. The ones that are designed for women have masks that are better suited to their facial features, which differ from men’s.”
A precision medicine approach can be taken to treat any sleep disorder. For insomnia, the approach allows healthcare professionals to employ an appropriate cognitive-behavioral therapy plan or to determine which drugs would be more effective – all on the basis of symptoms and the characteristics of the particular case. Regarding sleep apnea, Dr. Cano explained, “taking into account the different anatomical characteristics or the higher prevalence of positional apnea will also allow us to offer different therapeutic alternatives to continuous positive airway pressure, such as mandibular advancement devices or positional therapy devices.”
Women should be encouraged to develop good sleep habits. These include taking circadian rhythms into account and aligning lifestyles accordingly. It also means going to bed earlier than the men in the household. For menopausal women, recommended sleep habits range from keeping their bedroom at an ideal temperature, following a diet rich in vegetables to avoid becoming overweight, and exercising daily. While this advice may be more applicable to teenagers, adults can benefit from it as well: Electronic devices should be turned off well before bedtime. Whether from a phone screen, a tablet screen, or a TV screen, the light emitted can keep one awake, which can be harmful to one’s health.
Dr. Cano and Dr. Merino disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com. This article was translated from the Medscape Spanish edition.
‘Not in our lane’: Physicians rebel at idea they should discuss gun safety with patients
The latest NPR/PBS NewsHour/Marist poll shows that that the margin of public opinion in the United States is the widest that it has been during the past 10 years in favor of taking steps to control gun violence; 59% of U.S. adults said it’s more important to control gun violence than to protect gun rights, and 35% said the opposite.
Have physicians’ opinions about gun issues in our country shifted meaningfully during that period? That’s a complex question that can be informed with the basic snapshot provided by doctors› comments to New York University (and Medscape blogger) bioethicist Arthur L. Caplan’s four video blogs on whether physicians should discuss gun safety with their patients. Dr. Caplan’s video blogs appeared on the Medscape website in 2014, 2016, 2018, and 2022.
Hundreds of physicians have posted comments to Dr. Caplan’s arguments that doctors should bring up gun safety when talking to their patients. The great majority of comments opposed his position in 2014, and that remained the case through 2022, regardless of incidents of gun-related violence. Supportive comments have been a small minority that has grown only slightly over his four video blogs.
Physicians’ lack of qualifications
The most prevalent counterarguments expressed against Dr. Caplan’s position are that physicians lack the proper knowledge to discuss gun safety with patients; and the responsibility falls on family members, certified firearms instructors, teachers, and others – but not doctors – to educate people about firearm safety.
“Then there’s a third group that says, ‘I don’t want to do this because I am too busy trying to figure out what is wrong with the patient,’ ” Dr. Caplan says.
Here are a few on-point comments that were posted to his video blogs:
- “Unless physicians become certified firearms instructors like myself, they are not qualified to talk to patients on the subject and should advise patients to find a program and take a course.” – Dr. Ken Long, March 31, 2014
- “Gun safety should be taught in school, just like health and sex education.” – Patricia L., Feb. 11, 2016
- “None of my medical or surgical training or experience qualifies me as a policy expert on gun laws or regulations.” – Dr. Kelly Hyde, Dec. 23, 2018
- “I have the Constitution hanging in my office with an NRA plaque next to it. Most MDs can’t mow their own yard.” – Dr. Brian Anseeuw, June 21, 2022
Do mental health issues trump gun talks?
Another counterargument to discussing gun safety with patients involves mental health issues that many physicians may not be trained to address. Mental health entered comments to Dr. Caplan’s video blogs in 2016 and has shaped much of the discussion since.
- “First of all, two-thirds of gun deaths are suicides. It is foolish to talk about counseling patients about gun safety, etc, and ignore the mental health issues.” – Dr. Jeffrey Jennings, Jan. 25, 2016
- “Suicide victims and those committing mass shootings are mentally ill. ... Blame society, drugs, mental illness, easy access to illegal firearms, and poor recognition of SOS (signs of suicide).” – Dr. Alan DeCarlo, Dec. 24, 2018
- “Yes, we have gun violence, but what is the underlying problem? Bullying? Mental issues? Not enough parental supervision? These and others are the issues I feel need to be discussed.” – T. Deese, June 24, 2022
- “The causes of increased gun violence are mental health, problems with bullying, social media, and normalization of deviant behavior.” – Julie Johng, 2022
Added responsibility is too much
Another theme that has grown over time is that talks of gun safety just heap issues onto physicians’ treatment plates that are already too full.
- “Oh, for God’s sake, is there anything else I can do while I›m at it? Primary care has gotten to be more headache than it’s worth. Thanks for another reason to think about retiring.” – Dr. Kathleen Collins, March 31, 2014
- “THE JOB OF POLICE, COURTS, AND LAW-EDUCATED PROSECUTORS SHOULD NOT BE HANDLED BY PHYSICIANS.” – Dr. Sudarshan Singla, Jan. 25, 2016
- “This is a debate that only those at the academic/ivory tower–level of medicine even have time to lament. The frontline medical providers barely have enough time to adequately address the pertinent.” – Tobin Purslow, Jan. 15, 2016
Other ways to communicate
For his part, Dr. Caplan believes there is a variety of ways physicians can effectively discuss gun safety with patients to help minimize the potential of injury or death.
Acknowledging that other aspects of treatment are often more pressing, he suggested that the gun safety education could be done through educational videos that are shown in waiting rooms, through pamphlets available at the front desk, or throuigh a newsletter sent to patients.
“Everything doesn’t have to happen in conversation. The doctor’s office should become more of an educational site.
“I am 100% more passionate about this than when I first started down this road.”
A version of this article first appeared on Medscape.com.
The latest NPR/PBS NewsHour/Marist poll shows that that the margin of public opinion in the United States is the widest that it has been during the past 10 years in favor of taking steps to control gun violence; 59% of U.S. adults said it’s more important to control gun violence than to protect gun rights, and 35% said the opposite.
Have physicians’ opinions about gun issues in our country shifted meaningfully during that period? That’s a complex question that can be informed with the basic snapshot provided by doctors› comments to New York University (and Medscape blogger) bioethicist Arthur L. Caplan’s four video blogs on whether physicians should discuss gun safety with their patients. Dr. Caplan’s video blogs appeared on the Medscape website in 2014, 2016, 2018, and 2022.
Hundreds of physicians have posted comments to Dr. Caplan’s arguments that doctors should bring up gun safety when talking to their patients. The great majority of comments opposed his position in 2014, and that remained the case through 2022, regardless of incidents of gun-related violence. Supportive comments have been a small minority that has grown only slightly over his four video blogs.
Physicians’ lack of qualifications
The most prevalent counterarguments expressed against Dr. Caplan’s position are that physicians lack the proper knowledge to discuss gun safety with patients; and the responsibility falls on family members, certified firearms instructors, teachers, and others – but not doctors – to educate people about firearm safety.
“Then there’s a third group that says, ‘I don’t want to do this because I am too busy trying to figure out what is wrong with the patient,’ ” Dr. Caplan says.
Here are a few on-point comments that were posted to his video blogs:
- “Unless physicians become certified firearms instructors like myself, they are not qualified to talk to patients on the subject and should advise patients to find a program and take a course.” – Dr. Ken Long, March 31, 2014
- “Gun safety should be taught in school, just like health and sex education.” – Patricia L., Feb. 11, 2016
- “None of my medical or surgical training or experience qualifies me as a policy expert on gun laws or regulations.” – Dr. Kelly Hyde, Dec. 23, 2018
- “I have the Constitution hanging in my office with an NRA plaque next to it. Most MDs can’t mow their own yard.” – Dr. Brian Anseeuw, June 21, 2022
Do mental health issues trump gun talks?
Another counterargument to discussing gun safety with patients involves mental health issues that many physicians may not be trained to address. Mental health entered comments to Dr. Caplan’s video blogs in 2016 and has shaped much of the discussion since.
- “First of all, two-thirds of gun deaths are suicides. It is foolish to talk about counseling patients about gun safety, etc, and ignore the mental health issues.” – Dr. Jeffrey Jennings, Jan. 25, 2016
- “Suicide victims and those committing mass shootings are mentally ill. ... Blame society, drugs, mental illness, easy access to illegal firearms, and poor recognition of SOS (signs of suicide).” – Dr. Alan DeCarlo, Dec. 24, 2018
- “Yes, we have gun violence, but what is the underlying problem? Bullying? Mental issues? Not enough parental supervision? These and others are the issues I feel need to be discussed.” – T. Deese, June 24, 2022
- “The causes of increased gun violence are mental health, problems with bullying, social media, and normalization of deviant behavior.” – Julie Johng, 2022
Added responsibility is too much
Another theme that has grown over time is that talks of gun safety just heap issues onto physicians’ treatment plates that are already too full.
- “Oh, for God’s sake, is there anything else I can do while I›m at it? Primary care has gotten to be more headache than it’s worth. Thanks for another reason to think about retiring.” – Dr. Kathleen Collins, March 31, 2014
- “THE JOB OF POLICE, COURTS, AND LAW-EDUCATED PROSECUTORS SHOULD NOT BE HANDLED BY PHYSICIANS.” – Dr. Sudarshan Singla, Jan. 25, 2016
- “This is a debate that only those at the academic/ivory tower–level of medicine even have time to lament. The frontline medical providers barely have enough time to adequately address the pertinent.” – Tobin Purslow, Jan. 15, 2016
Other ways to communicate
For his part, Dr. Caplan believes there is a variety of ways physicians can effectively discuss gun safety with patients to help minimize the potential of injury or death.
Acknowledging that other aspects of treatment are often more pressing, he suggested that the gun safety education could be done through educational videos that are shown in waiting rooms, through pamphlets available at the front desk, or throuigh a newsletter sent to patients.
“Everything doesn’t have to happen in conversation. The doctor’s office should become more of an educational site.
“I am 100% more passionate about this than when I first started down this road.”
A version of this article first appeared on Medscape.com.
The latest NPR/PBS NewsHour/Marist poll shows that that the margin of public opinion in the United States is the widest that it has been during the past 10 years in favor of taking steps to control gun violence; 59% of U.S. adults said it’s more important to control gun violence than to protect gun rights, and 35% said the opposite.
Have physicians’ opinions about gun issues in our country shifted meaningfully during that period? That’s a complex question that can be informed with the basic snapshot provided by doctors› comments to New York University (and Medscape blogger) bioethicist Arthur L. Caplan’s four video blogs on whether physicians should discuss gun safety with their patients. Dr. Caplan’s video blogs appeared on the Medscape website in 2014, 2016, 2018, and 2022.
Hundreds of physicians have posted comments to Dr. Caplan’s arguments that doctors should bring up gun safety when talking to their patients. The great majority of comments opposed his position in 2014, and that remained the case through 2022, regardless of incidents of gun-related violence. Supportive comments have been a small minority that has grown only slightly over his four video blogs.
Physicians’ lack of qualifications
The most prevalent counterarguments expressed against Dr. Caplan’s position are that physicians lack the proper knowledge to discuss gun safety with patients; and the responsibility falls on family members, certified firearms instructors, teachers, and others – but not doctors – to educate people about firearm safety.
“Then there’s a third group that says, ‘I don’t want to do this because I am too busy trying to figure out what is wrong with the patient,’ ” Dr. Caplan says.
Here are a few on-point comments that were posted to his video blogs:
- “Unless physicians become certified firearms instructors like myself, they are not qualified to talk to patients on the subject and should advise patients to find a program and take a course.” – Dr. Ken Long, March 31, 2014
- “Gun safety should be taught in school, just like health and sex education.” – Patricia L., Feb. 11, 2016
- “None of my medical or surgical training or experience qualifies me as a policy expert on gun laws or regulations.” – Dr. Kelly Hyde, Dec. 23, 2018
- “I have the Constitution hanging in my office with an NRA plaque next to it. Most MDs can’t mow their own yard.” – Dr. Brian Anseeuw, June 21, 2022
Do mental health issues trump gun talks?
Another counterargument to discussing gun safety with patients involves mental health issues that many physicians may not be trained to address. Mental health entered comments to Dr. Caplan’s video blogs in 2016 and has shaped much of the discussion since.
- “First of all, two-thirds of gun deaths are suicides. It is foolish to talk about counseling patients about gun safety, etc, and ignore the mental health issues.” – Dr. Jeffrey Jennings, Jan. 25, 2016
- “Suicide victims and those committing mass shootings are mentally ill. ... Blame society, drugs, mental illness, easy access to illegal firearms, and poor recognition of SOS (signs of suicide).” – Dr. Alan DeCarlo, Dec. 24, 2018
- “Yes, we have gun violence, but what is the underlying problem? Bullying? Mental issues? Not enough parental supervision? These and others are the issues I feel need to be discussed.” – T. Deese, June 24, 2022
- “The causes of increased gun violence are mental health, problems with bullying, social media, and normalization of deviant behavior.” – Julie Johng, 2022
Added responsibility is too much
Another theme that has grown over time is that talks of gun safety just heap issues onto physicians’ treatment plates that are already too full.
- “Oh, for God’s sake, is there anything else I can do while I›m at it? Primary care has gotten to be more headache than it’s worth. Thanks for another reason to think about retiring.” – Dr. Kathleen Collins, March 31, 2014
- “THE JOB OF POLICE, COURTS, AND LAW-EDUCATED PROSECUTORS SHOULD NOT BE HANDLED BY PHYSICIANS.” – Dr. Sudarshan Singla, Jan. 25, 2016
- “This is a debate that only those at the academic/ivory tower–level of medicine even have time to lament. The frontline medical providers barely have enough time to adequately address the pertinent.” – Tobin Purslow, Jan. 15, 2016
Other ways to communicate
For his part, Dr. Caplan believes there is a variety of ways physicians can effectively discuss gun safety with patients to help minimize the potential of injury or death.
Acknowledging that other aspects of treatment are often more pressing, he suggested that the gun safety education could be done through educational videos that are shown in waiting rooms, through pamphlets available at the front desk, or throuigh a newsletter sent to patients.
“Everything doesn’t have to happen in conversation. The doctor’s office should become more of an educational site.
“I am 100% more passionate about this than when I first started down this road.”
A version of this article first appeared on Medscape.com.
You and the skeptical patient: Who’s the doctor here?
“I spoke to him on many occasions about the dangers of COVID, but he just didn’t believe me,” said Dr. Hood, an internist in Lexington, Ky. “He just didn’t give me enough time to help him. He waited to let me know he was ill with COVID and took days to pick up the medicine. Unfortunately, he then passed away.”
The rise of the skeptical patient
It can be extremely frustrating for doctors when patients question or disbelieve their physician’s medical advice and explanations. And many physicians resent the amount of time they spend trying to explain or make their case, especially during a busy day. But patients’ skepticism about the validity of some treatments seems to be increasing.
“Patients are now more likely to have their own medical explanation for their complaint than they used to, and that can be bad for their health,” Dr. Hood said.
Dr. Hood sees medical cynicism as part of Americans’ growing distrust of experts, leveraged by easy access to the internet. “When people Google, they tend to look for support of their opinions, rather than arrive at a fully educated decision.”
Only about half of patients believe their physicians “provide fair and accurate treatment information all or most of the time,” according to a 2019 survey by the Pew Research Center.
Patients’ distrust has become more obvious during the COVID-19 pandemic, said John Schumann, MD, an internist with Oak Street Health, a practice with more than 500 physicians and other providers in 20 states, treating almost exclusively Medicare patients.
“The skeptics became more entrenched during the pandemic,” said Dr. Schumann, who is based in Tulsa, Okla. “They may think the COVID vaccines were approved too quickly, or believe the pandemic itself is a hoax.”
“There’s a lot of antiscience rhetoric now,” Dr. Schumann added. “I’d say about half of my patients are comfortable with science-based decisions and the other half are not.”
What are patients mistrustful about?
Patients’ suspicions of certain therapies began long before the pandemic. In dermatology, for example, some patients refuse to take topical steroids, said Steven R. Feldman, MD, a dermatologist in Winston-Salem, N.C.
“Their distrust is usually based on anecdotal stories they read about,” he noted. “Patients in other specialties are dead set against vaccinations.”
In addition to refusing treatments and inoculations, some patients ask for questionable regimens mentioned in the news. “Some patients have demanded hydroxychloroquine or Noromectin, drugs that are unproven in the treatment of COVID,” Dr. Schumann said. “We refuse to prescribe them.”
Dr. Hood said patients’ reluctance to follow medical advice can often be based on cost. “I have a patient who was more willing to save $20 than to save his life. But when the progression of his test results fit my predictions, he became more willing to take treatments. I had to wait for the opportune moment to convince him.”
Many naysayer patients keep their views to themselves, and physicians may be unaware that the patients are stonewalling. A 2006 study estimated that about 10%-16% of primary care patients actively resist medical authority.
Dr. Schumann cited patients who don’t want to hear an upsetting diagnosis. “Some patients might refuse to take a biopsy to see if they have cancer because they don’t want to know,” he said. “In many cases, they simply won’t get the biopsy and won’t tell the doctor that they didn’t.”
Sometimes skeptics’ arguments have merit
Some patients’ concerns can be valid, such as when they refuse to go on statins, said Zain Hakeem, DO, a physician in Austin, Tex.
“In some cases, I feel that statins are not necessary,” he said. “The science on statins for primary prevention is not strong, although they should be used for exceedingly high-risk patients.”
Certain patients, especially those with chronic conditions, do a great deal of research, using legitimate sources on the Web, and their research is well supported.
However, these patients can be overconfident in their conclusions. Several studies have shown that with just a little experience, people can replace beginners’ caution with a false sense of competence.
For example, “Patients may not weigh the risks correctly,” Dr. Hakeem said. “They can be more concerned about the risk of having their colon perforated during a colonoscopy, while the risk of cancer if they don’t have a colonoscopy is much higher.”
Some highly successful people may be more likely to trust their own medical instincts. When Steve Jobs, the founder of Apple, was diagnosed with pancreatic cancer in 2003, he put off surgery for 9 months while he tried to cure his disease with a vegan diet, acupuncture, herbs, bowel cleansings, and other remedies he read about. He died in 2011. Some experts believe that delay hastened his death.
Of course, not all physicians’ diagnoses or treatments are correct. One study indicated doctors’ diagnostic error rate could be as high as 15%. And just as patients can be overconfident in their conclusions, so can doctors. Another study found that physicians’ stated confidence in their diagnosis was only slightly affected by the inaccuracy of that diagnosis or the difficulty of the case.
Best ways to deal with cynical patients
Patients’ skepticism can frustrate doctors, reduce the efficiency of care delivery, and interfere with recovery. What can doctors do to deal with these problems?
1. Build the patient’s trust in you. “Getting patients to adhere to your advice involves making sure they feel they have a caring doctor whom they trust,” Dr. Feldman said.
“I want to show patients that I am entirely focused on them,” he added. “For example, I may rush to the door of the exam room from my last appointment, but I open the door very slowly and deliberately, because I want the patient to see that I won’t hurry with them.”
2. Spend time with the patient. Familiarity builds trust. Dr. Schumann said doctors at Oak Street Health see their patients an average of six to eight times a year, an unusually high number. “The more patients see their physicians, the more likely they are to trust them.”
3. Keep up to date. “I make sure I’m up to date with the literature, and I try to present a truthful message,” Dr. Hood said. “For instance, my research showed that inflammation played a strong role in developing complications from COVID, so I wrote a detailed treatment protocol aimed at the inflammation and the immune response, which has been very effective.”
4. Confront patients tactfully. Patients who do research on the Web don’t want to be scolded, Dr. Feldman said. In fact, he praises them, even if he doesn’t agree with their findings. “I might say: ‘What a relief to finally find patients who’ve taken the time to educate themselves before coming here.’ ”
Dr. Feldman is careful not to dispute patients’ conclusions. “Debating the issues is not an effective approach to get patients to trust you. The last thing you want to tell a patient is: ‘Listen to me! I’m an expert.’ People just dig in.”
However, it does help to give patients feedback. “I’m a big fan of patients arguing with me,” Dr. Hakeem said. “It means you can straighten out misunderstandings and improve decision-making.”
5. Explain your reasoning. “You need to communicate clearly and show them your thinking,” Dr. Hood said. “For instance, I’ll explain why a patient has a strong risk for heart attack.”
6. Acknowledge uncertainties. “The doctor may present the science as far more certain than it is,” Dr. Hakeem said. “If you don’t acknowledge the uncertainties, you could break the patient’s trust in you.”
7. Don’t use a lot of numbers. “Data is not a good tool to convince patients,” Dr. Feldman said. “The human brain isn’t designed to work that way.”
If you want to use numbers to show clinical risk, Dr. Hakeem advisd using natural frequencies, such as 10 out of 10,000, which is less confusing to the patient than the equivalent percentage of 0.1%.
It can be helpful to refer to familiar concepts. One way to understand a risk is to compare it with risks in daily life, such as the dangers of driving or falling in the shower, Dr. Hakeem added.
Dr. Feldman often refers to another person’s experience when presenting his medical advice. “I might say to the patient: ‘You remind me of another patient I had. They were sitting in the same chair you’re sitting in. They did really well on this drug, and I think it’s probably the best choice for you, too.’ ”
8. Adopt shared decision-making. This approach involves empowering the patient to become an equal partner in medical decisions. The patient is given information through portals and is encouraged to do research. Critics, however, say that most patients don’t want this degree of empowerment and would rather depend on the doctor’s advice.
Conclusion
It’s often impossible to get through to a skeptical patient, which can be disheartening for doctors. “Physicians want to do what is best for the patient, so when the patient doesn’t listen, they may take it personally,” Dr. Hood said. “But you always have to remember, the patient is the one with disease, and it’s up to the patient to open the door.”
Still, some skeptical patients ultimately change their minds. Dr. Schumann said patients who initially declined the COVID vaccine eventually decided to get it. “It often took them more than a year. but it’s never too late.”
A version of this article first appeared on Medscape.com.
“I spoke to him on many occasions about the dangers of COVID, but he just didn’t believe me,” said Dr. Hood, an internist in Lexington, Ky. “He just didn’t give me enough time to help him. He waited to let me know he was ill with COVID and took days to pick up the medicine. Unfortunately, he then passed away.”
The rise of the skeptical patient
It can be extremely frustrating for doctors when patients question or disbelieve their physician’s medical advice and explanations. And many physicians resent the amount of time they spend trying to explain or make their case, especially during a busy day. But patients’ skepticism about the validity of some treatments seems to be increasing.
“Patients are now more likely to have their own medical explanation for their complaint than they used to, and that can be bad for their health,” Dr. Hood said.
Dr. Hood sees medical cynicism as part of Americans’ growing distrust of experts, leveraged by easy access to the internet. “When people Google, they tend to look for support of their opinions, rather than arrive at a fully educated decision.”
Only about half of patients believe their physicians “provide fair and accurate treatment information all or most of the time,” according to a 2019 survey by the Pew Research Center.
Patients’ distrust has become more obvious during the COVID-19 pandemic, said John Schumann, MD, an internist with Oak Street Health, a practice with more than 500 physicians and other providers in 20 states, treating almost exclusively Medicare patients.
“The skeptics became more entrenched during the pandemic,” said Dr. Schumann, who is based in Tulsa, Okla. “They may think the COVID vaccines were approved too quickly, or believe the pandemic itself is a hoax.”
“There’s a lot of antiscience rhetoric now,” Dr. Schumann added. “I’d say about half of my patients are comfortable with science-based decisions and the other half are not.”
What are patients mistrustful about?
Patients’ suspicions of certain therapies began long before the pandemic. In dermatology, for example, some patients refuse to take topical steroids, said Steven R. Feldman, MD, a dermatologist in Winston-Salem, N.C.
“Their distrust is usually based on anecdotal stories they read about,” he noted. “Patients in other specialties are dead set against vaccinations.”
In addition to refusing treatments and inoculations, some patients ask for questionable regimens mentioned in the news. “Some patients have demanded hydroxychloroquine or Noromectin, drugs that are unproven in the treatment of COVID,” Dr. Schumann said. “We refuse to prescribe them.”
Dr. Hood said patients’ reluctance to follow medical advice can often be based on cost. “I have a patient who was more willing to save $20 than to save his life. But when the progression of his test results fit my predictions, he became more willing to take treatments. I had to wait for the opportune moment to convince him.”
Many naysayer patients keep their views to themselves, and physicians may be unaware that the patients are stonewalling. A 2006 study estimated that about 10%-16% of primary care patients actively resist medical authority.
Dr. Schumann cited patients who don’t want to hear an upsetting diagnosis. “Some patients might refuse to take a biopsy to see if they have cancer because they don’t want to know,” he said. “In many cases, they simply won’t get the biopsy and won’t tell the doctor that they didn’t.”
Sometimes skeptics’ arguments have merit
Some patients’ concerns can be valid, such as when they refuse to go on statins, said Zain Hakeem, DO, a physician in Austin, Tex.
“In some cases, I feel that statins are not necessary,” he said. “The science on statins for primary prevention is not strong, although they should be used for exceedingly high-risk patients.”
Certain patients, especially those with chronic conditions, do a great deal of research, using legitimate sources on the Web, and their research is well supported.
However, these patients can be overconfident in their conclusions. Several studies have shown that with just a little experience, people can replace beginners’ caution with a false sense of competence.
For example, “Patients may not weigh the risks correctly,” Dr. Hakeem said. “They can be more concerned about the risk of having their colon perforated during a colonoscopy, while the risk of cancer if they don’t have a colonoscopy is much higher.”
Some highly successful people may be more likely to trust their own medical instincts. When Steve Jobs, the founder of Apple, was diagnosed with pancreatic cancer in 2003, he put off surgery for 9 months while he tried to cure his disease with a vegan diet, acupuncture, herbs, bowel cleansings, and other remedies he read about. He died in 2011. Some experts believe that delay hastened his death.
Of course, not all physicians’ diagnoses or treatments are correct. One study indicated doctors’ diagnostic error rate could be as high as 15%. And just as patients can be overconfident in their conclusions, so can doctors. Another study found that physicians’ stated confidence in their diagnosis was only slightly affected by the inaccuracy of that diagnosis or the difficulty of the case.
Best ways to deal with cynical patients
Patients’ skepticism can frustrate doctors, reduce the efficiency of care delivery, and interfere with recovery. What can doctors do to deal with these problems?
1. Build the patient’s trust in you. “Getting patients to adhere to your advice involves making sure they feel they have a caring doctor whom they trust,” Dr. Feldman said.
“I want to show patients that I am entirely focused on them,” he added. “For example, I may rush to the door of the exam room from my last appointment, but I open the door very slowly and deliberately, because I want the patient to see that I won’t hurry with them.”
2. Spend time with the patient. Familiarity builds trust. Dr. Schumann said doctors at Oak Street Health see their patients an average of six to eight times a year, an unusually high number. “The more patients see their physicians, the more likely they are to trust them.”
3. Keep up to date. “I make sure I’m up to date with the literature, and I try to present a truthful message,” Dr. Hood said. “For instance, my research showed that inflammation played a strong role in developing complications from COVID, so I wrote a detailed treatment protocol aimed at the inflammation and the immune response, which has been very effective.”
4. Confront patients tactfully. Patients who do research on the Web don’t want to be scolded, Dr. Feldman said. In fact, he praises them, even if he doesn’t agree with their findings. “I might say: ‘What a relief to finally find patients who’ve taken the time to educate themselves before coming here.’ ”
Dr. Feldman is careful not to dispute patients’ conclusions. “Debating the issues is not an effective approach to get patients to trust you. The last thing you want to tell a patient is: ‘Listen to me! I’m an expert.’ People just dig in.”
However, it does help to give patients feedback. “I’m a big fan of patients arguing with me,” Dr. Hakeem said. “It means you can straighten out misunderstandings and improve decision-making.”
5. Explain your reasoning. “You need to communicate clearly and show them your thinking,” Dr. Hood said. “For instance, I’ll explain why a patient has a strong risk for heart attack.”
6. Acknowledge uncertainties. “The doctor may present the science as far more certain than it is,” Dr. Hakeem said. “If you don’t acknowledge the uncertainties, you could break the patient’s trust in you.”
7. Don’t use a lot of numbers. “Data is not a good tool to convince patients,” Dr. Feldman said. “The human brain isn’t designed to work that way.”
If you want to use numbers to show clinical risk, Dr. Hakeem advisd using natural frequencies, such as 10 out of 10,000, which is less confusing to the patient than the equivalent percentage of 0.1%.
It can be helpful to refer to familiar concepts. One way to understand a risk is to compare it with risks in daily life, such as the dangers of driving or falling in the shower, Dr. Hakeem added.
Dr. Feldman often refers to another person’s experience when presenting his medical advice. “I might say to the patient: ‘You remind me of another patient I had. They were sitting in the same chair you’re sitting in. They did really well on this drug, and I think it’s probably the best choice for you, too.’ ”
8. Adopt shared decision-making. This approach involves empowering the patient to become an equal partner in medical decisions. The patient is given information through portals and is encouraged to do research. Critics, however, say that most patients don’t want this degree of empowerment and would rather depend on the doctor’s advice.
Conclusion
It’s often impossible to get through to a skeptical patient, which can be disheartening for doctors. “Physicians want to do what is best for the patient, so when the patient doesn’t listen, they may take it personally,” Dr. Hood said. “But you always have to remember, the patient is the one with disease, and it’s up to the patient to open the door.”
Still, some skeptical patients ultimately change their minds. Dr. Schumann said patients who initially declined the COVID vaccine eventually decided to get it. “It often took them more than a year. but it’s never too late.”
A version of this article first appeared on Medscape.com.
“I spoke to him on many occasions about the dangers of COVID, but he just didn’t believe me,” said Dr. Hood, an internist in Lexington, Ky. “He just didn’t give me enough time to help him. He waited to let me know he was ill with COVID and took days to pick up the medicine. Unfortunately, he then passed away.”
The rise of the skeptical patient
It can be extremely frustrating for doctors when patients question or disbelieve their physician’s medical advice and explanations. And many physicians resent the amount of time they spend trying to explain or make their case, especially during a busy day. But patients’ skepticism about the validity of some treatments seems to be increasing.
“Patients are now more likely to have their own medical explanation for their complaint than they used to, and that can be bad for their health,” Dr. Hood said.
Dr. Hood sees medical cynicism as part of Americans’ growing distrust of experts, leveraged by easy access to the internet. “When people Google, they tend to look for support of their opinions, rather than arrive at a fully educated decision.”
Only about half of patients believe their physicians “provide fair and accurate treatment information all or most of the time,” according to a 2019 survey by the Pew Research Center.
Patients’ distrust has become more obvious during the COVID-19 pandemic, said John Schumann, MD, an internist with Oak Street Health, a practice with more than 500 physicians and other providers in 20 states, treating almost exclusively Medicare patients.
“The skeptics became more entrenched during the pandemic,” said Dr. Schumann, who is based in Tulsa, Okla. “They may think the COVID vaccines were approved too quickly, or believe the pandemic itself is a hoax.”
“There’s a lot of antiscience rhetoric now,” Dr. Schumann added. “I’d say about half of my patients are comfortable with science-based decisions and the other half are not.”
What are patients mistrustful about?
Patients’ suspicions of certain therapies began long before the pandemic. In dermatology, for example, some patients refuse to take topical steroids, said Steven R. Feldman, MD, a dermatologist in Winston-Salem, N.C.
“Their distrust is usually based on anecdotal stories they read about,” he noted. “Patients in other specialties are dead set against vaccinations.”
In addition to refusing treatments and inoculations, some patients ask for questionable regimens mentioned in the news. “Some patients have demanded hydroxychloroquine or Noromectin, drugs that are unproven in the treatment of COVID,” Dr. Schumann said. “We refuse to prescribe them.”
Dr. Hood said patients’ reluctance to follow medical advice can often be based on cost. “I have a patient who was more willing to save $20 than to save his life. But when the progression of his test results fit my predictions, he became more willing to take treatments. I had to wait for the opportune moment to convince him.”
Many naysayer patients keep their views to themselves, and physicians may be unaware that the patients are stonewalling. A 2006 study estimated that about 10%-16% of primary care patients actively resist medical authority.
Dr. Schumann cited patients who don’t want to hear an upsetting diagnosis. “Some patients might refuse to take a biopsy to see if they have cancer because they don’t want to know,” he said. “In many cases, they simply won’t get the biopsy and won’t tell the doctor that they didn’t.”
Sometimes skeptics’ arguments have merit
Some patients’ concerns can be valid, such as when they refuse to go on statins, said Zain Hakeem, DO, a physician in Austin, Tex.
“In some cases, I feel that statins are not necessary,” he said. “The science on statins for primary prevention is not strong, although they should be used for exceedingly high-risk patients.”
Certain patients, especially those with chronic conditions, do a great deal of research, using legitimate sources on the Web, and their research is well supported.
However, these patients can be overconfident in their conclusions. Several studies have shown that with just a little experience, people can replace beginners’ caution with a false sense of competence.
For example, “Patients may not weigh the risks correctly,” Dr. Hakeem said. “They can be more concerned about the risk of having their colon perforated during a colonoscopy, while the risk of cancer if they don’t have a colonoscopy is much higher.”
Some highly successful people may be more likely to trust their own medical instincts. When Steve Jobs, the founder of Apple, was diagnosed with pancreatic cancer in 2003, he put off surgery for 9 months while he tried to cure his disease with a vegan diet, acupuncture, herbs, bowel cleansings, and other remedies he read about. He died in 2011. Some experts believe that delay hastened his death.
Of course, not all physicians’ diagnoses or treatments are correct. One study indicated doctors’ diagnostic error rate could be as high as 15%. And just as patients can be overconfident in their conclusions, so can doctors. Another study found that physicians’ stated confidence in their diagnosis was only slightly affected by the inaccuracy of that diagnosis or the difficulty of the case.
Best ways to deal with cynical patients
Patients’ skepticism can frustrate doctors, reduce the efficiency of care delivery, and interfere with recovery. What can doctors do to deal with these problems?
1. Build the patient’s trust in you. “Getting patients to adhere to your advice involves making sure they feel they have a caring doctor whom they trust,” Dr. Feldman said.
“I want to show patients that I am entirely focused on them,” he added. “For example, I may rush to the door of the exam room from my last appointment, but I open the door very slowly and deliberately, because I want the patient to see that I won’t hurry with them.”
2. Spend time with the patient. Familiarity builds trust. Dr. Schumann said doctors at Oak Street Health see their patients an average of six to eight times a year, an unusually high number. “The more patients see their physicians, the more likely they are to trust them.”
3. Keep up to date. “I make sure I’m up to date with the literature, and I try to present a truthful message,” Dr. Hood said. “For instance, my research showed that inflammation played a strong role in developing complications from COVID, so I wrote a detailed treatment protocol aimed at the inflammation and the immune response, which has been very effective.”
4. Confront patients tactfully. Patients who do research on the Web don’t want to be scolded, Dr. Feldman said. In fact, he praises them, even if he doesn’t agree with their findings. “I might say: ‘What a relief to finally find patients who’ve taken the time to educate themselves before coming here.’ ”
Dr. Feldman is careful not to dispute patients’ conclusions. “Debating the issues is not an effective approach to get patients to trust you. The last thing you want to tell a patient is: ‘Listen to me! I’m an expert.’ People just dig in.”
However, it does help to give patients feedback. “I’m a big fan of patients arguing with me,” Dr. Hakeem said. “It means you can straighten out misunderstandings and improve decision-making.”
5. Explain your reasoning. “You need to communicate clearly and show them your thinking,” Dr. Hood said. “For instance, I’ll explain why a patient has a strong risk for heart attack.”
6. Acknowledge uncertainties. “The doctor may present the science as far more certain than it is,” Dr. Hakeem said. “If you don’t acknowledge the uncertainties, you could break the patient’s trust in you.”
7. Don’t use a lot of numbers. “Data is not a good tool to convince patients,” Dr. Feldman said. “The human brain isn’t designed to work that way.”
If you want to use numbers to show clinical risk, Dr. Hakeem advisd using natural frequencies, such as 10 out of 10,000, which is less confusing to the patient than the equivalent percentage of 0.1%.
It can be helpful to refer to familiar concepts. One way to understand a risk is to compare it with risks in daily life, such as the dangers of driving or falling in the shower, Dr. Hakeem added.
Dr. Feldman often refers to another person’s experience when presenting his medical advice. “I might say to the patient: ‘You remind me of another patient I had. They were sitting in the same chair you’re sitting in. They did really well on this drug, and I think it’s probably the best choice for you, too.’ ”
8. Adopt shared decision-making. This approach involves empowering the patient to become an equal partner in medical decisions. The patient is given information through portals and is encouraged to do research. Critics, however, say that most patients don’t want this degree of empowerment and would rather depend on the doctor’s advice.
Conclusion
It’s often impossible to get through to a skeptical patient, which can be disheartening for doctors. “Physicians want to do what is best for the patient, so when the patient doesn’t listen, they may take it personally,” Dr. Hood said. “But you always have to remember, the patient is the one with disease, and it’s up to the patient to open the door.”
Still, some skeptical patients ultimately change their minds. Dr. Schumann said patients who initially declined the COVID vaccine eventually decided to get it. “It often took them more than a year. but it’s never too late.”
A version of this article first appeared on Medscape.com.
Teens with diagnosed and undiagnosed ADHD report similar quality of life
The results align with findings from other studies suggesting lower quality of life (QOL) in teens with ADHD, but the current study is the first known to focus on the association between ADHD diagnosis itself vs. ADHD symptoms, and QOL, the researchers wrote. The findings show that at least some of the reduced QOL is associated with the diagnosis itself, they explained.
The researchers directly compared 393 teens with a childhood ADHD diagnosis to 393 matched teens with no ADHD diagnosis but who had hyperactive/inattentive behaviors.
The researchers reviewed self-reports from individuals who were enrolled in a population-based prospective study in Australia. The primary outcome was quality of life at age 14-15, which was measured with Child Health Utility 9D (CHU9D), a validated quality of life measure.
Study results
Overall, teens with and without an ADHD diagnosis reported similar levels of overall quality of life; the mean difference in the primary outcome CHU9D score was –0.03 (P = .10). Teens with and without an ADHD diagnosis also showed similar scores on measures of general health, happiness, and peer trust, the researchers noted.
The researchers also reviewed eight other prespecified, self-reported measures: academic self-concept, global health, negative social behaviors, overall happiness, peer trust, psychological sense of school membership, self-efficacy, and self-harm.
Teens diagnosed with ADHD in childhood were more than twice as likely to report self-harm (odds ratio 2.53, P less than .001) and displayed significantly more negative social behaviors (mean difference 1.56, P = .002), compared with teens without an ADHD diagnosis.
Teens diagnosed with ADHD in childhood also scored significantly worse on measures of sense of school membership (mean difference −2.58, P less than .001), academic self-concept (mean difference, −0.14; P = .02), and self-efficacy (mean difference −0.20; P = .007), compared to teens without an ADHD diagnosis.
The average age at ADHD diagnosis was 10 years, and 72% of the ADHD-diagnosed group were boys. No significant differences were noted for levels of hyperactive/inattentive behaviors and between girls and boys, but girls overall and children with the highest levels of hyperactive and inattentive behaviors reported generally worse outcomes, regardless of ADHD diagnosis, the researchers noted.
Don’t rush to diagnosis
Although rates of ADHD diagnosis in children continue to rise, the prevalence of hyperactivity and inattentive behaviors appears stable, which suggests a problem with diagnosis, senior author Alexandra Barratt, MBBS, MPH, PhD, professor of public health at the University of Sydney, Australia, said in an interview.
“Our hypothesis was that children who had been diagnosed, and we assume treated for, ADHD would have better outcomes, compared to children matched for hyperactivity/inattention behaviors who were left undiagnosed and untreated, but we were surprised to find that, at best, outcomes were unchanged, and for some outcomes, worse,” Dr. Barratt said.
“Our study provides evidence that diagnosing ADHD may lead, inadvertently, to long-term harms, particularly for children with mild or borderline hyperactivity and inattention behaviors,” she emphasized.
“We can’t say from this study what to do instead, but previously one of our team has looked at stepped diagnosis as an alternative option for children with mild or borderline hyperactivity and inattention behaviors,” she said.
The stepped diagnosis includes such actions as gathering behavior data from multiple sources, and conducting a period of watchful waiting without presumption of a diagnosis or active treatment.
Given the findings of the new study, “I would ask that health professionals considering a child who may have ADHD be aware that there is an evidence gap around the long-term impact of an ADHD diagnosis on children, and to proceed cautiously,” Dr. Barratt said. As for additional research, independent, high-quality, randomized controlled trials of ADHD diagnosis in children with mild or borderline hyperactivity/inattention behaviors are urgently needed, with long-term, patient-centered outcomes including quality of life she noted.
ADHD screening needs improvement
The incidence and prevalence of ADHD is on the rise, but much of the perceived increase in ADHD may be due to overdiagnosis, “and a lack of robust thorough psychological testing as standard of care for diagnosis,” Peter Loper, MD, a pediatrician and psychiatrist at the University of South Carolina, Columbia, said in an interview.
The current study “reinforces the necessity of consistent screening for comorbid mental health problems, and specifically for thoughts of self-harm, in those children who are diagnosed with ADHD,” he said.
Expressing his lack of astonishment about the study findings, Dr. Loper said: “Previous data indicates that while following initial diagnosis of a medical or mental health problem, patients may experience a sense of relief; however, this is followed shortly thereafter by feelings of insufficiency or anxiety related to their specific diagnosis.”
“As it stands now, ADHD is often diagnosed in children and adolescents using basic screening questionnaires,” said Dr. Loper. “The findings of this study may bolster calls for more robust and thorough psychological testing for supporting the diagnosis of ADHD,” he said.
Individuals diagnosed with ADHD can sometimes have difficulty with social skills and relating to others, said Dr. Loper. “They may be more prone to internalize their poor school performance as due to being ‘stupid’ or ‘dumb,’ ” he said. Children and teens with ADHD should, whenever possible, be involved in extracurricular activities that support the development of social skills, he said. Parents’ praise of the process/effort, rather than focusing only on outcomes such as grades, is very important for the esteem of children and teens with ADHD, he added.
The study limitations included the use of observational data vs. data from randomized trials, and the potential for confounding factors in propensity scoring, the researchers wrote. Additional limitations include the size of the sample, which may have been too small to detect additional differences between diagnosed teens and matched controls, they noted.
“As the study authors appropriately cite, a large, randomized trial would be very helpful in supporting additional understanding of this issue,” Dr. Loper added.
The study was supported by the National Health and Medical Research Council The researchers and Dr. Loper had no financial conflicts to disclose.
The results align with findings from other studies suggesting lower quality of life (QOL) in teens with ADHD, but the current study is the first known to focus on the association between ADHD diagnosis itself vs. ADHD symptoms, and QOL, the researchers wrote. The findings show that at least some of the reduced QOL is associated with the diagnosis itself, they explained.
The researchers directly compared 393 teens with a childhood ADHD diagnosis to 393 matched teens with no ADHD diagnosis but who had hyperactive/inattentive behaviors.
The researchers reviewed self-reports from individuals who were enrolled in a population-based prospective study in Australia. The primary outcome was quality of life at age 14-15, which was measured with Child Health Utility 9D (CHU9D), a validated quality of life measure.
Study results
Overall, teens with and without an ADHD diagnosis reported similar levels of overall quality of life; the mean difference in the primary outcome CHU9D score was –0.03 (P = .10). Teens with and without an ADHD diagnosis also showed similar scores on measures of general health, happiness, and peer trust, the researchers noted.
The researchers also reviewed eight other prespecified, self-reported measures: academic self-concept, global health, negative social behaviors, overall happiness, peer trust, psychological sense of school membership, self-efficacy, and self-harm.
Teens diagnosed with ADHD in childhood were more than twice as likely to report self-harm (odds ratio 2.53, P less than .001) and displayed significantly more negative social behaviors (mean difference 1.56, P = .002), compared with teens without an ADHD diagnosis.
Teens diagnosed with ADHD in childhood also scored significantly worse on measures of sense of school membership (mean difference −2.58, P less than .001), academic self-concept (mean difference, −0.14; P = .02), and self-efficacy (mean difference −0.20; P = .007), compared to teens without an ADHD diagnosis.
The average age at ADHD diagnosis was 10 years, and 72% of the ADHD-diagnosed group were boys. No significant differences were noted for levels of hyperactive/inattentive behaviors and between girls and boys, but girls overall and children with the highest levels of hyperactive and inattentive behaviors reported generally worse outcomes, regardless of ADHD diagnosis, the researchers noted.
Don’t rush to diagnosis
Although rates of ADHD diagnosis in children continue to rise, the prevalence of hyperactivity and inattentive behaviors appears stable, which suggests a problem with diagnosis, senior author Alexandra Barratt, MBBS, MPH, PhD, professor of public health at the University of Sydney, Australia, said in an interview.
“Our hypothesis was that children who had been diagnosed, and we assume treated for, ADHD would have better outcomes, compared to children matched for hyperactivity/inattention behaviors who were left undiagnosed and untreated, but we were surprised to find that, at best, outcomes were unchanged, and for some outcomes, worse,” Dr. Barratt said.
“Our study provides evidence that diagnosing ADHD may lead, inadvertently, to long-term harms, particularly for children with mild or borderline hyperactivity and inattention behaviors,” she emphasized.
“We can’t say from this study what to do instead, but previously one of our team has looked at stepped diagnosis as an alternative option for children with mild or borderline hyperactivity and inattention behaviors,” she said.
The stepped diagnosis includes such actions as gathering behavior data from multiple sources, and conducting a period of watchful waiting without presumption of a diagnosis or active treatment.
Given the findings of the new study, “I would ask that health professionals considering a child who may have ADHD be aware that there is an evidence gap around the long-term impact of an ADHD diagnosis on children, and to proceed cautiously,” Dr. Barratt said. As for additional research, independent, high-quality, randomized controlled trials of ADHD diagnosis in children with mild or borderline hyperactivity/inattention behaviors are urgently needed, with long-term, patient-centered outcomes including quality of life she noted.
ADHD screening needs improvement
The incidence and prevalence of ADHD is on the rise, but much of the perceived increase in ADHD may be due to overdiagnosis, “and a lack of robust thorough psychological testing as standard of care for diagnosis,” Peter Loper, MD, a pediatrician and psychiatrist at the University of South Carolina, Columbia, said in an interview.
The current study “reinforces the necessity of consistent screening for comorbid mental health problems, and specifically for thoughts of self-harm, in those children who are diagnosed with ADHD,” he said.
Expressing his lack of astonishment about the study findings, Dr. Loper said: “Previous data indicates that while following initial diagnosis of a medical or mental health problem, patients may experience a sense of relief; however, this is followed shortly thereafter by feelings of insufficiency or anxiety related to their specific diagnosis.”
“As it stands now, ADHD is often diagnosed in children and adolescents using basic screening questionnaires,” said Dr. Loper. “The findings of this study may bolster calls for more robust and thorough psychological testing for supporting the diagnosis of ADHD,” he said.
Individuals diagnosed with ADHD can sometimes have difficulty with social skills and relating to others, said Dr. Loper. “They may be more prone to internalize their poor school performance as due to being ‘stupid’ or ‘dumb,’ ” he said. Children and teens with ADHD should, whenever possible, be involved in extracurricular activities that support the development of social skills, he said. Parents’ praise of the process/effort, rather than focusing only on outcomes such as grades, is very important for the esteem of children and teens with ADHD, he added.
The study limitations included the use of observational data vs. data from randomized trials, and the potential for confounding factors in propensity scoring, the researchers wrote. Additional limitations include the size of the sample, which may have been too small to detect additional differences between diagnosed teens and matched controls, they noted.
“As the study authors appropriately cite, a large, randomized trial would be very helpful in supporting additional understanding of this issue,” Dr. Loper added.
The study was supported by the National Health and Medical Research Council The researchers and Dr. Loper had no financial conflicts to disclose.
The results align with findings from other studies suggesting lower quality of life (QOL) in teens with ADHD, but the current study is the first known to focus on the association between ADHD diagnosis itself vs. ADHD symptoms, and QOL, the researchers wrote. The findings show that at least some of the reduced QOL is associated with the diagnosis itself, they explained.
The researchers directly compared 393 teens with a childhood ADHD diagnosis to 393 matched teens with no ADHD diagnosis but who had hyperactive/inattentive behaviors.
The researchers reviewed self-reports from individuals who were enrolled in a population-based prospective study in Australia. The primary outcome was quality of life at age 14-15, which was measured with Child Health Utility 9D (CHU9D), a validated quality of life measure.
Study results
Overall, teens with and without an ADHD diagnosis reported similar levels of overall quality of life; the mean difference in the primary outcome CHU9D score was –0.03 (P = .10). Teens with and without an ADHD diagnosis also showed similar scores on measures of general health, happiness, and peer trust, the researchers noted.
The researchers also reviewed eight other prespecified, self-reported measures: academic self-concept, global health, negative social behaviors, overall happiness, peer trust, psychological sense of school membership, self-efficacy, and self-harm.
Teens diagnosed with ADHD in childhood were more than twice as likely to report self-harm (odds ratio 2.53, P less than .001) and displayed significantly more negative social behaviors (mean difference 1.56, P = .002), compared with teens without an ADHD diagnosis.
Teens diagnosed with ADHD in childhood also scored significantly worse on measures of sense of school membership (mean difference −2.58, P less than .001), academic self-concept (mean difference, −0.14; P = .02), and self-efficacy (mean difference −0.20; P = .007), compared to teens without an ADHD diagnosis.
The average age at ADHD diagnosis was 10 years, and 72% of the ADHD-diagnosed group were boys. No significant differences were noted for levels of hyperactive/inattentive behaviors and between girls and boys, but girls overall and children with the highest levels of hyperactive and inattentive behaviors reported generally worse outcomes, regardless of ADHD diagnosis, the researchers noted.
Don’t rush to diagnosis
Although rates of ADHD diagnosis in children continue to rise, the prevalence of hyperactivity and inattentive behaviors appears stable, which suggests a problem with diagnosis, senior author Alexandra Barratt, MBBS, MPH, PhD, professor of public health at the University of Sydney, Australia, said in an interview.
“Our hypothesis was that children who had been diagnosed, and we assume treated for, ADHD would have better outcomes, compared to children matched for hyperactivity/inattention behaviors who were left undiagnosed and untreated, but we were surprised to find that, at best, outcomes were unchanged, and for some outcomes, worse,” Dr. Barratt said.
“Our study provides evidence that diagnosing ADHD may lead, inadvertently, to long-term harms, particularly for children with mild or borderline hyperactivity and inattention behaviors,” she emphasized.
“We can’t say from this study what to do instead, but previously one of our team has looked at stepped diagnosis as an alternative option for children with mild or borderline hyperactivity and inattention behaviors,” she said.
The stepped diagnosis includes such actions as gathering behavior data from multiple sources, and conducting a period of watchful waiting without presumption of a diagnosis or active treatment.
Given the findings of the new study, “I would ask that health professionals considering a child who may have ADHD be aware that there is an evidence gap around the long-term impact of an ADHD diagnosis on children, and to proceed cautiously,” Dr. Barratt said. As for additional research, independent, high-quality, randomized controlled trials of ADHD diagnosis in children with mild or borderline hyperactivity/inattention behaviors are urgently needed, with long-term, patient-centered outcomes including quality of life she noted.
ADHD screening needs improvement
The incidence and prevalence of ADHD is on the rise, but much of the perceived increase in ADHD may be due to overdiagnosis, “and a lack of robust thorough psychological testing as standard of care for diagnosis,” Peter Loper, MD, a pediatrician and psychiatrist at the University of South Carolina, Columbia, said in an interview.
The current study “reinforces the necessity of consistent screening for comorbid mental health problems, and specifically for thoughts of self-harm, in those children who are diagnosed with ADHD,” he said.
Expressing his lack of astonishment about the study findings, Dr. Loper said: “Previous data indicates that while following initial diagnosis of a medical or mental health problem, patients may experience a sense of relief; however, this is followed shortly thereafter by feelings of insufficiency or anxiety related to their specific diagnosis.”
“As it stands now, ADHD is often diagnosed in children and adolescents using basic screening questionnaires,” said Dr. Loper. “The findings of this study may bolster calls for more robust and thorough psychological testing for supporting the diagnosis of ADHD,” he said.
Individuals diagnosed with ADHD can sometimes have difficulty with social skills and relating to others, said Dr. Loper. “They may be more prone to internalize their poor school performance as due to being ‘stupid’ or ‘dumb,’ ” he said. Children and teens with ADHD should, whenever possible, be involved in extracurricular activities that support the development of social skills, he said. Parents’ praise of the process/effort, rather than focusing only on outcomes such as grades, is very important for the esteem of children and teens with ADHD, he added.
The study limitations included the use of observational data vs. data from randomized trials, and the potential for confounding factors in propensity scoring, the researchers wrote. Additional limitations include the size of the sample, which may have been too small to detect additional differences between diagnosed teens and matched controls, they noted.
“As the study authors appropriately cite, a large, randomized trial would be very helpful in supporting additional understanding of this issue,” Dr. Loper added.
The study was supported by the National Health and Medical Research Council The researchers and Dr. Loper had no financial conflicts to disclose.
FROM JAMA NETWORK OPEN
Dementia signs detected years before diagnosis
offering hope for interventions to reduce the risk of the disease developing.
To date it has been unclear whether it might be possible to detect changes in brain function before the onset of symptoms, so researchers at the University of Cambridge and Cambridge University Hospitals NHS Foundation Trust set out to determine whether people who developed a range of neurodegenerative diagnoses demonstrated reduced cognitive function at their baseline assessment.
The authors explained: “The pathophysiological processes of neurodegenerative diseases begin years before diagnosis. However, prediagnostic changes in cognition and physical function are poorly understood, especially in sporadic neurodegenerative disease.”
Prediagnostic cognitive and functional impairment identified
The researchers analyzed data from the UK Biobank and compared cognitive and functional measures, including problem solving, memory, reaction times and grip strength, as well as data on weight loss and gain and on the number of falls, in individuals who subsequently developed a number of dementia-related diseases (Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, progressive supranuclear palsy, dementia with Lewy bodies, and multiple system atrophy), with those who did not have a neurodegenerative diagnosis. After adjustment for the effects of age, the same measures were regressed against time to diagnosis. The study was published in Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association.
The researchers found evidence of prediagnostic cognitive impairment and decline with time, particularly in Alzheimer’s disease where those who went on to develop the disease scored more poorly compared with healthy individuals when it came to problem solving tasks, reaction times, remembering lists of numbers, prospective memory, and pair matching. This was also the case for people who developed frontotemporal dementia, the authors said.
Nol Swaddiwudhipong, MB, of the University of Cambridge, and first author, said: “When we looked back at patients’ histories, it became clear that they were showing some cognitive impairment several years before their symptoms became obvious enough to prompt a diagnosis. The impairments were often subtle, but across a number of aspects of cognition.”
Prediagnostic functional impairment and decline was also observed in multiple diseases, the authors said. People who went on to develop Alzheimer’s disease were more likely than were healthy adults to have had a fall in the previous 12 months, with those patients who went on to develop progressive supranuclear palsy (PSP) being more than twice as likely as healthy individuals to have had a fall.
The time between baseline assessment and diagnosis varied between 4.7 years for dementia with Lewy bodies and 8.3 years for Alzheimer’s disease.
“For every condition studied – including Parkinson’s disease and dementia with Lewy bodies – patients reported poorer overall health at baseline,” said the authors.
Potential for new treatments
The study findings that cognitive and functional decline occurs “years before symptoms become obvious” in multiple neurodegenerative diseases, raises the possibility that in the future at-risk patients could be screened to help select those who would benefit from interventions to reduce their risk of developing one of the conditions, or to help identify patients suitable for recruitment to clinical trials for new treatments.
Dr Swaddiwudhipong emphasized: “This is a step towards us being able to screen people who are at greatest risk – for example, people over 50 or those who have high blood pressure or do not do enough exercise – and intervene at an earlier stage to help them reduce their risk.”
There are currently very few effective treatments for dementia or other forms of neurodegeneration, the authors pointed out, in part because these conditions are often only diagnosed once symptoms appear, whereas the underlying neurodegeneration may have “begun years, even decades, earlier.” This means that by the time patients take part in clinical trials, it may already be too late in the disease process to alter its course, they explained.
Timothy Rittman, BMBS, PhD, department of clinical neurosciences, University of Cambridge, and senior author, explained that the findings could also help identify people who can participate in clinical trials for potential new treatments. “The problem with clinical trials is that by necessity they often recruit patients with a diagnosis, but we know that by this point they are already some way down the road and their condition cannot be stopped. If we can find these individuals early enough, we’ll have a better chance of seeing if the drugs are effective,” he emphasized.
Commenting on the new research, Richard Oakley, PhD, associate director of research at Alzheimer’s Society, said: “Studies like this show the importance in continued investment in dementia research to revolutionize diagnosis and drive new treatments, so one day we will beat dementia.”
The research was funded by the Medical Research Council with support from the NIHR Cambridge Biomedical Research Centre. The authors reported no conflicts of interest.
A version of this article first appeared on Medscape UK.
offering hope for interventions to reduce the risk of the disease developing.
To date it has been unclear whether it might be possible to detect changes in brain function before the onset of symptoms, so researchers at the University of Cambridge and Cambridge University Hospitals NHS Foundation Trust set out to determine whether people who developed a range of neurodegenerative diagnoses demonstrated reduced cognitive function at their baseline assessment.
The authors explained: “The pathophysiological processes of neurodegenerative diseases begin years before diagnosis. However, prediagnostic changes in cognition and physical function are poorly understood, especially in sporadic neurodegenerative disease.”
Prediagnostic cognitive and functional impairment identified
The researchers analyzed data from the UK Biobank and compared cognitive and functional measures, including problem solving, memory, reaction times and grip strength, as well as data on weight loss and gain and on the number of falls, in individuals who subsequently developed a number of dementia-related diseases (Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, progressive supranuclear palsy, dementia with Lewy bodies, and multiple system atrophy), with those who did not have a neurodegenerative diagnosis. After adjustment for the effects of age, the same measures were regressed against time to diagnosis. The study was published in Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association.
The researchers found evidence of prediagnostic cognitive impairment and decline with time, particularly in Alzheimer’s disease where those who went on to develop the disease scored more poorly compared with healthy individuals when it came to problem solving tasks, reaction times, remembering lists of numbers, prospective memory, and pair matching. This was also the case for people who developed frontotemporal dementia, the authors said.
Nol Swaddiwudhipong, MB, of the University of Cambridge, and first author, said: “When we looked back at patients’ histories, it became clear that they were showing some cognitive impairment several years before their symptoms became obvious enough to prompt a diagnosis. The impairments were often subtle, but across a number of aspects of cognition.”
Prediagnostic functional impairment and decline was also observed in multiple diseases, the authors said. People who went on to develop Alzheimer’s disease were more likely than were healthy adults to have had a fall in the previous 12 months, with those patients who went on to develop progressive supranuclear palsy (PSP) being more than twice as likely as healthy individuals to have had a fall.
The time between baseline assessment and diagnosis varied between 4.7 years for dementia with Lewy bodies and 8.3 years for Alzheimer’s disease.
“For every condition studied – including Parkinson’s disease and dementia with Lewy bodies – patients reported poorer overall health at baseline,” said the authors.
Potential for new treatments
The study findings that cognitive and functional decline occurs “years before symptoms become obvious” in multiple neurodegenerative diseases, raises the possibility that in the future at-risk patients could be screened to help select those who would benefit from interventions to reduce their risk of developing one of the conditions, or to help identify patients suitable for recruitment to clinical trials for new treatments.
Dr Swaddiwudhipong emphasized: “This is a step towards us being able to screen people who are at greatest risk – for example, people over 50 or those who have high blood pressure or do not do enough exercise – and intervene at an earlier stage to help them reduce their risk.”
There are currently very few effective treatments for dementia or other forms of neurodegeneration, the authors pointed out, in part because these conditions are often only diagnosed once symptoms appear, whereas the underlying neurodegeneration may have “begun years, even decades, earlier.” This means that by the time patients take part in clinical trials, it may already be too late in the disease process to alter its course, they explained.
Timothy Rittman, BMBS, PhD, department of clinical neurosciences, University of Cambridge, and senior author, explained that the findings could also help identify people who can participate in clinical trials for potential new treatments. “The problem with clinical trials is that by necessity they often recruit patients with a diagnosis, but we know that by this point they are already some way down the road and their condition cannot be stopped. If we can find these individuals early enough, we’ll have a better chance of seeing if the drugs are effective,” he emphasized.
Commenting on the new research, Richard Oakley, PhD, associate director of research at Alzheimer’s Society, said: “Studies like this show the importance in continued investment in dementia research to revolutionize diagnosis and drive new treatments, so one day we will beat dementia.”
The research was funded by the Medical Research Council with support from the NIHR Cambridge Biomedical Research Centre. The authors reported no conflicts of interest.
A version of this article first appeared on Medscape UK.
offering hope for interventions to reduce the risk of the disease developing.
To date it has been unclear whether it might be possible to detect changes in brain function before the onset of symptoms, so researchers at the University of Cambridge and Cambridge University Hospitals NHS Foundation Trust set out to determine whether people who developed a range of neurodegenerative diagnoses demonstrated reduced cognitive function at their baseline assessment.
The authors explained: “The pathophysiological processes of neurodegenerative diseases begin years before diagnosis. However, prediagnostic changes in cognition and physical function are poorly understood, especially in sporadic neurodegenerative disease.”
Prediagnostic cognitive and functional impairment identified
The researchers analyzed data from the UK Biobank and compared cognitive and functional measures, including problem solving, memory, reaction times and grip strength, as well as data on weight loss and gain and on the number of falls, in individuals who subsequently developed a number of dementia-related diseases (Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, progressive supranuclear palsy, dementia with Lewy bodies, and multiple system atrophy), with those who did not have a neurodegenerative diagnosis. After adjustment for the effects of age, the same measures were regressed against time to diagnosis. The study was published in Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association.
The researchers found evidence of prediagnostic cognitive impairment and decline with time, particularly in Alzheimer’s disease where those who went on to develop the disease scored more poorly compared with healthy individuals when it came to problem solving tasks, reaction times, remembering lists of numbers, prospective memory, and pair matching. This was also the case for people who developed frontotemporal dementia, the authors said.
Nol Swaddiwudhipong, MB, of the University of Cambridge, and first author, said: “When we looked back at patients’ histories, it became clear that they were showing some cognitive impairment several years before their symptoms became obvious enough to prompt a diagnosis. The impairments were often subtle, but across a number of aspects of cognition.”
Prediagnostic functional impairment and decline was also observed in multiple diseases, the authors said. People who went on to develop Alzheimer’s disease were more likely than were healthy adults to have had a fall in the previous 12 months, with those patients who went on to develop progressive supranuclear palsy (PSP) being more than twice as likely as healthy individuals to have had a fall.
The time between baseline assessment and diagnosis varied between 4.7 years for dementia with Lewy bodies and 8.3 years for Alzheimer’s disease.
“For every condition studied – including Parkinson’s disease and dementia with Lewy bodies – patients reported poorer overall health at baseline,” said the authors.
Potential for new treatments
The study findings that cognitive and functional decline occurs “years before symptoms become obvious” in multiple neurodegenerative diseases, raises the possibility that in the future at-risk patients could be screened to help select those who would benefit from interventions to reduce their risk of developing one of the conditions, or to help identify patients suitable for recruitment to clinical trials for new treatments.
Dr Swaddiwudhipong emphasized: “This is a step towards us being able to screen people who are at greatest risk – for example, people over 50 or those who have high blood pressure or do not do enough exercise – and intervene at an earlier stage to help them reduce their risk.”
There are currently very few effective treatments for dementia or other forms of neurodegeneration, the authors pointed out, in part because these conditions are often only diagnosed once symptoms appear, whereas the underlying neurodegeneration may have “begun years, even decades, earlier.” This means that by the time patients take part in clinical trials, it may already be too late in the disease process to alter its course, they explained.
Timothy Rittman, BMBS, PhD, department of clinical neurosciences, University of Cambridge, and senior author, explained that the findings could also help identify people who can participate in clinical trials for potential new treatments. “The problem with clinical trials is that by necessity they often recruit patients with a diagnosis, but we know that by this point they are already some way down the road and their condition cannot be stopped. If we can find these individuals early enough, we’ll have a better chance of seeing if the drugs are effective,” he emphasized.
Commenting on the new research, Richard Oakley, PhD, associate director of research at Alzheimer’s Society, said: “Studies like this show the importance in continued investment in dementia research to revolutionize diagnosis and drive new treatments, so one day we will beat dementia.”
The research was funded by the Medical Research Council with support from the NIHR Cambridge Biomedical Research Centre. The authors reported no conflicts of interest.
A version of this article first appeared on Medscape UK.
FROM ALZHEIMER’S & DEMENTIA
Nurse accused of murdering babies in her neonatal unit
Lucy Letby, 32, who worked at the Countess of Chester Hospital, is accused of multiple baby murders in the hospital’s neonatal unit from June 2015 to June 2016. She denies all charges.
Manchester Crown Court heard how Ms. Letby allegedly attempted to kill the children by injecting them with air, milk, or insulin, including two brothers from a set of triplets and one premature baby girl, who was only 98 minutes old.
Prosecutor Nicholas Johnson KC said the circumstances of the girl’s death were “an extreme example even by the standards of this case.”
“There were four separate occasions on which we allege Lucy Letby tried to kill her,” he said. “But ultimately at the fourth attempt, Lucy Letby succeeded in killing her.”
Attempts to murder the child ‘cold-blooded’ and ‘calculated’, says prosecutor
In the first alleged attempt, Ms. Letby injected the girl, identified for legal reasons as Child I, with air, but she was “resilient,” said Mr. Johnson. After the second attempt, Ms. Letby had stood in the doorway of Child I’s darkened room and commented that she looked pale. The designated nurse then approached and turned on the light, noticing that the child wasn’t breathing. After a third attempt the child was found to have excess air in her stomach, which had affected her breathing. Child I was then transferred to Arrowe Park Hospital, where she was stabilized before she was returned to Chester.
After the fourth attempt, Child I’s medical alarm rang, leading a nurse to spot Ms. Letby by the child’s incubator. Child I died that morning, said Mr. Johnson, who described the nurse’s attacks as premeditated. “It was persistent, it was calculated, and it was cold-blooded.”
The judge, Mr. Justice Goss, and jury heard how shortly after the parents were told of their child’s death, Ms. Letby approached the mother, who testified that the nurse was “smiling and kept going on about how she was present at the baby’s first bath and how much the baby had loved it.” She also sent a sympathy card to the parents, and the prosecutor says she kept an image of the card on her phone.
Doctor interrupted another alleged attempt
Dr. Ravi Jayaram, a paediatric consultant, had become suspicious of Ms. Letby in a number of unexplained child deaths. He later interrupted her as she allegedly tried to kill another baby, identified as Child K. He noticed that the nurse was alone with the baby and walked into the room, seeing Ms. Letby standing over the child’s incubator. He was “uncomfortable” as he had “started to notice a coincidence between unexplained deaths, serious collapses, and the presence of Lucy Letby,” said the prosecutor.
“Dr. Jayaram could see from the monitor on the wall that Child K’s oxygen saturation level was falling dangerously low, to somewhere in the 80s,” said Mr. Johnson. “He said an alarm should have been sounding as Child K’s oxygen levels were falling.” Despite this, the nurse had not called for assistance.
“We allege she was trying to kill Child K when Dr. Jayaram walked in,” Mr. Johnson said, adding that the child’s breathing tube was found dislodged. The prosecutor said it was possible for this to happen in an active baby, but Child K was very premature and had been sedated.
Despite his concerns, Dr. Jayaram did not make a note of his suspicions. Later that morning, Ms. Letby was again at Child K’s incubator calling for help. The nurse was assisting the baby with her breathing and the breathing tube was found to have slipped too far into her throat. The child was transferred to another hospital but later died. Ms. Letby is not accused of Child K’s murder.
However, after the death of Child K, Ms. Letby was moved to day shifts “because the consultants were concerned about the correlation between her presence and unexpected deaths and life-threatening episodes on the night shifts,” said Mr. Johnson. She was removed from the neonatal ward in June 2016 and moved to clerical duties where she would not come into contact with children.
Post-it note: Admission or anguish?
At the end of the prosecution’s presentation, Mr. Johnson mentioned a Post-it on which Ms. Letby had written, “I AM EVIL I DID THIS.” In the defense’s opening statements, Ben Myers KC, said the note was an “anguished outpouring of a young woman in fear and despair when she realises the enormity of what’s being said about her, in a moment to herself.”
He added that the nurse was dealing with employment issues at the time it was written, including a grievance procedure with the NHS Trust where she worked. Another note was shown on screens to the jury, which read: “Not good enough. I’m an awful person. I will never have children or marry. Despair.” and “I haven’t done anything wrong.”
Mr. Myers said that Ms. Letby was the type of person who often scribbles things down and the note was “nothing more extraordinary than that.”
In presenting the defense case, Mr. Myers argued that there was no evidence of Letby hurting the children, and that the prosecution’s case was “driven by the assumption that someone was doing deliberate harm” and that this was combined with “coincidence on certain occasions of Miss Letby’s presence.”
“What it isn’t driven by is evidence of Miss Letby actually doing what is alleged against her,” he added.
“There is a real danger that people will simply accept the prosecution theory of guilt, and that’s all we have so far,” Mr. Myers said. “A theory of guilt based firmly on coincidence – if anything can be based firmly on coincidence.”
A version of this article first appeared on Medscape UK.
Lucy Letby, 32, who worked at the Countess of Chester Hospital, is accused of multiple baby murders in the hospital’s neonatal unit from June 2015 to June 2016. She denies all charges.
Manchester Crown Court heard how Ms. Letby allegedly attempted to kill the children by injecting them with air, milk, or insulin, including two brothers from a set of triplets and one premature baby girl, who was only 98 minutes old.
Prosecutor Nicholas Johnson KC said the circumstances of the girl’s death were “an extreme example even by the standards of this case.”
“There were four separate occasions on which we allege Lucy Letby tried to kill her,” he said. “But ultimately at the fourth attempt, Lucy Letby succeeded in killing her.”
Attempts to murder the child ‘cold-blooded’ and ‘calculated’, says prosecutor
In the first alleged attempt, Ms. Letby injected the girl, identified for legal reasons as Child I, with air, but she was “resilient,” said Mr. Johnson. After the second attempt, Ms. Letby had stood in the doorway of Child I’s darkened room and commented that she looked pale. The designated nurse then approached and turned on the light, noticing that the child wasn’t breathing. After a third attempt the child was found to have excess air in her stomach, which had affected her breathing. Child I was then transferred to Arrowe Park Hospital, where she was stabilized before she was returned to Chester.
After the fourth attempt, Child I’s medical alarm rang, leading a nurse to spot Ms. Letby by the child’s incubator. Child I died that morning, said Mr. Johnson, who described the nurse’s attacks as premeditated. “It was persistent, it was calculated, and it was cold-blooded.”
The judge, Mr. Justice Goss, and jury heard how shortly after the parents were told of their child’s death, Ms. Letby approached the mother, who testified that the nurse was “smiling and kept going on about how she was present at the baby’s first bath and how much the baby had loved it.” She also sent a sympathy card to the parents, and the prosecutor says she kept an image of the card on her phone.
Doctor interrupted another alleged attempt
Dr. Ravi Jayaram, a paediatric consultant, had become suspicious of Ms. Letby in a number of unexplained child deaths. He later interrupted her as she allegedly tried to kill another baby, identified as Child K. He noticed that the nurse was alone with the baby and walked into the room, seeing Ms. Letby standing over the child’s incubator. He was “uncomfortable” as he had “started to notice a coincidence between unexplained deaths, serious collapses, and the presence of Lucy Letby,” said the prosecutor.
“Dr. Jayaram could see from the monitor on the wall that Child K’s oxygen saturation level was falling dangerously low, to somewhere in the 80s,” said Mr. Johnson. “He said an alarm should have been sounding as Child K’s oxygen levels were falling.” Despite this, the nurse had not called for assistance.
“We allege she was trying to kill Child K when Dr. Jayaram walked in,” Mr. Johnson said, adding that the child’s breathing tube was found dislodged. The prosecutor said it was possible for this to happen in an active baby, but Child K was very premature and had been sedated.
Despite his concerns, Dr. Jayaram did not make a note of his suspicions. Later that morning, Ms. Letby was again at Child K’s incubator calling for help. The nurse was assisting the baby with her breathing and the breathing tube was found to have slipped too far into her throat. The child was transferred to another hospital but later died. Ms. Letby is not accused of Child K’s murder.
However, after the death of Child K, Ms. Letby was moved to day shifts “because the consultants were concerned about the correlation between her presence and unexpected deaths and life-threatening episodes on the night shifts,” said Mr. Johnson. She was removed from the neonatal ward in June 2016 and moved to clerical duties where she would not come into contact with children.
Post-it note: Admission or anguish?
At the end of the prosecution’s presentation, Mr. Johnson mentioned a Post-it on which Ms. Letby had written, “I AM EVIL I DID THIS.” In the defense’s opening statements, Ben Myers KC, said the note was an “anguished outpouring of a young woman in fear and despair when she realises the enormity of what’s being said about her, in a moment to herself.”
He added that the nurse was dealing with employment issues at the time it was written, including a grievance procedure with the NHS Trust where she worked. Another note was shown on screens to the jury, which read: “Not good enough. I’m an awful person. I will never have children or marry. Despair.” and “I haven’t done anything wrong.”
Mr. Myers said that Ms. Letby was the type of person who often scribbles things down and the note was “nothing more extraordinary than that.”
In presenting the defense case, Mr. Myers argued that there was no evidence of Letby hurting the children, and that the prosecution’s case was “driven by the assumption that someone was doing deliberate harm” and that this was combined with “coincidence on certain occasions of Miss Letby’s presence.”
“What it isn’t driven by is evidence of Miss Letby actually doing what is alleged against her,” he added.
“There is a real danger that people will simply accept the prosecution theory of guilt, and that’s all we have so far,” Mr. Myers said. “A theory of guilt based firmly on coincidence – if anything can be based firmly on coincidence.”
A version of this article first appeared on Medscape UK.
Lucy Letby, 32, who worked at the Countess of Chester Hospital, is accused of multiple baby murders in the hospital’s neonatal unit from June 2015 to June 2016. She denies all charges.
Manchester Crown Court heard how Ms. Letby allegedly attempted to kill the children by injecting them with air, milk, or insulin, including two brothers from a set of triplets and one premature baby girl, who was only 98 minutes old.
Prosecutor Nicholas Johnson KC said the circumstances of the girl’s death were “an extreme example even by the standards of this case.”
“There were four separate occasions on which we allege Lucy Letby tried to kill her,” he said. “But ultimately at the fourth attempt, Lucy Letby succeeded in killing her.”
Attempts to murder the child ‘cold-blooded’ and ‘calculated’, says prosecutor
In the first alleged attempt, Ms. Letby injected the girl, identified for legal reasons as Child I, with air, but she was “resilient,” said Mr. Johnson. After the second attempt, Ms. Letby had stood in the doorway of Child I’s darkened room and commented that she looked pale. The designated nurse then approached and turned on the light, noticing that the child wasn’t breathing. After a third attempt the child was found to have excess air in her stomach, which had affected her breathing. Child I was then transferred to Arrowe Park Hospital, where she was stabilized before she was returned to Chester.
After the fourth attempt, Child I’s medical alarm rang, leading a nurse to spot Ms. Letby by the child’s incubator. Child I died that morning, said Mr. Johnson, who described the nurse’s attacks as premeditated. “It was persistent, it was calculated, and it was cold-blooded.”
The judge, Mr. Justice Goss, and jury heard how shortly after the parents were told of their child’s death, Ms. Letby approached the mother, who testified that the nurse was “smiling and kept going on about how she was present at the baby’s first bath and how much the baby had loved it.” She also sent a sympathy card to the parents, and the prosecutor says she kept an image of the card on her phone.
Doctor interrupted another alleged attempt
Dr. Ravi Jayaram, a paediatric consultant, had become suspicious of Ms. Letby in a number of unexplained child deaths. He later interrupted her as she allegedly tried to kill another baby, identified as Child K. He noticed that the nurse was alone with the baby and walked into the room, seeing Ms. Letby standing over the child’s incubator. He was “uncomfortable” as he had “started to notice a coincidence between unexplained deaths, serious collapses, and the presence of Lucy Letby,” said the prosecutor.
“Dr. Jayaram could see from the monitor on the wall that Child K’s oxygen saturation level was falling dangerously low, to somewhere in the 80s,” said Mr. Johnson. “He said an alarm should have been sounding as Child K’s oxygen levels were falling.” Despite this, the nurse had not called for assistance.
“We allege she was trying to kill Child K when Dr. Jayaram walked in,” Mr. Johnson said, adding that the child’s breathing tube was found dislodged. The prosecutor said it was possible for this to happen in an active baby, but Child K was very premature and had been sedated.
Despite his concerns, Dr. Jayaram did not make a note of his suspicions. Later that morning, Ms. Letby was again at Child K’s incubator calling for help. The nurse was assisting the baby with her breathing and the breathing tube was found to have slipped too far into her throat. The child was transferred to another hospital but later died. Ms. Letby is not accused of Child K’s murder.
However, after the death of Child K, Ms. Letby was moved to day shifts “because the consultants were concerned about the correlation between her presence and unexpected deaths and life-threatening episodes on the night shifts,” said Mr. Johnson. She was removed from the neonatal ward in June 2016 and moved to clerical duties where she would not come into contact with children.
Post-it note: Admission or anguish?
At the end of the prosecution’s presentation, Mr. Johnson mentioned a Post-it on which Ms. Letby had written, “I AM EVIL I DID THIS.” In the defense’s opening statements, Ben Myers KC, said the note was an “anguished outpouring of a young woman in fear and despair when she realises the enormity of what’s being said about her, in a moment to herself.”
He added that the nurse was dealing with employment issues at the time it was written, including a grievance procedure with the NHS Trust where she worked. Another note was shown on screens to the jury, which read: “Not good enough. I’m an awful person. I will never have children or marry. Despair.” and “I haven’t done anything wrong.”
Mr. Myers said that Ms. Letby was the type of person who often scribbles things down and the note was “nothing more extraordinary than that.”
In presenting the defense case, Mr. Myers argued that there was no evidence of Letby hurting the children, and that the prosecution’s case was “driven by the assumption that someone was doing deliberate harm” and that this was combined with “coincidence on certain occasions of Miss Letby’s presence.”
“What it isn’t driven by is evidence of Miss Letby actually doing what is alleged against her,” he added.
“There is a real danger that people will simply accept the prosecution theory of guilt, and that’s all we have so far,” Mr. Myers said. “A theory of guilt based firmly on coincidence – if anything can be based firmly on coincidence.”
A version of this article first appeared on Medscape UK.
The urgent need to diagnose Sanfilippo syndrome at an early age
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.

Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8

“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.

“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.
Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8
“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.
“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.
Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8
“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.
“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.

Novel gene-based therapies for neuromuscular diseases
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5

Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.

Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
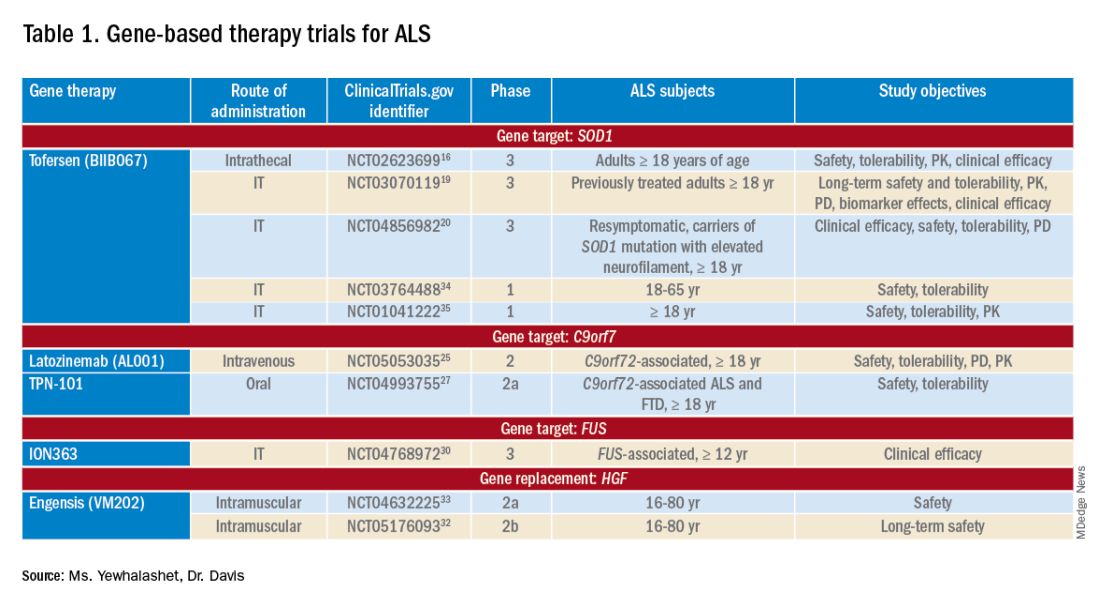
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
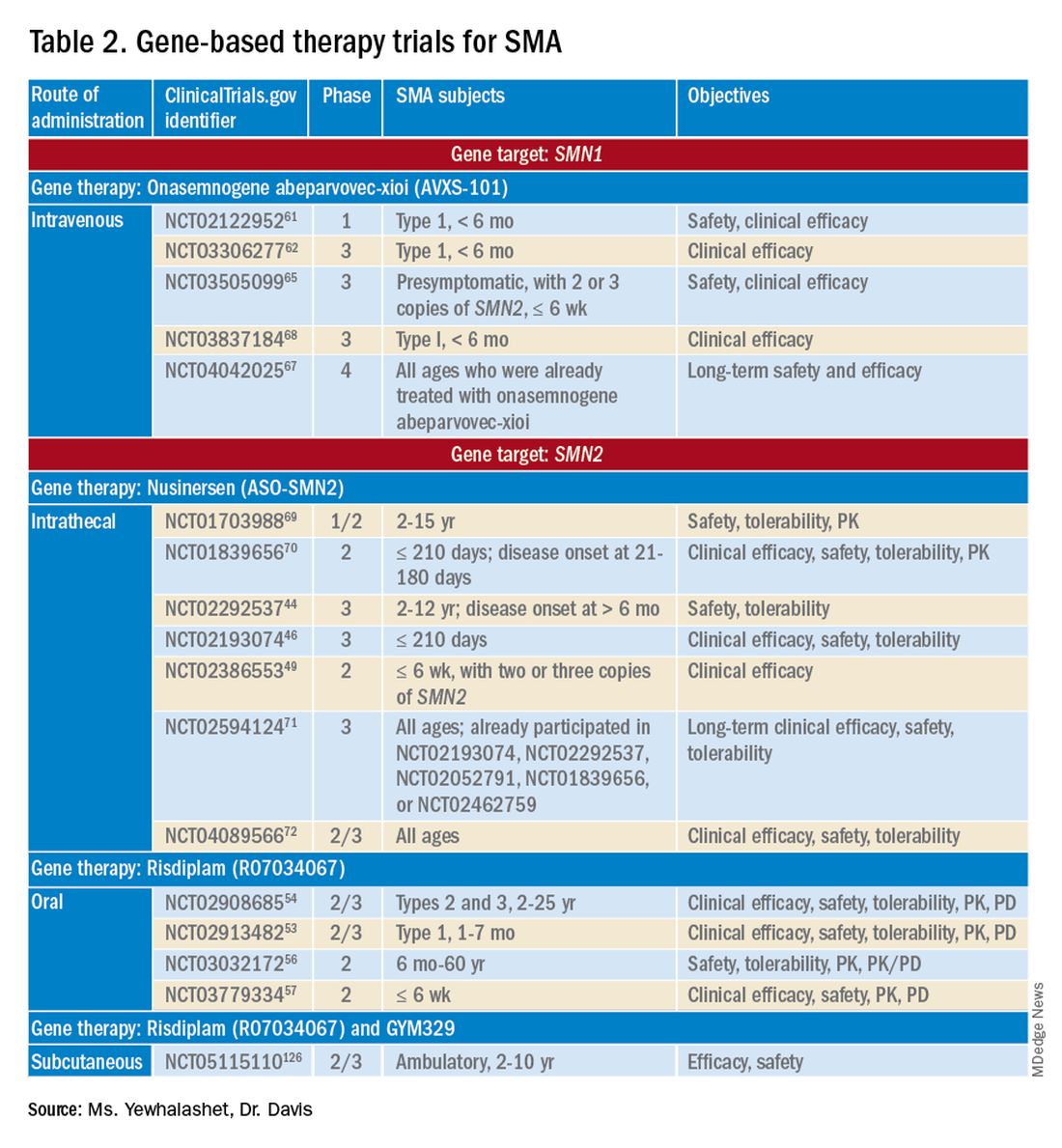
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
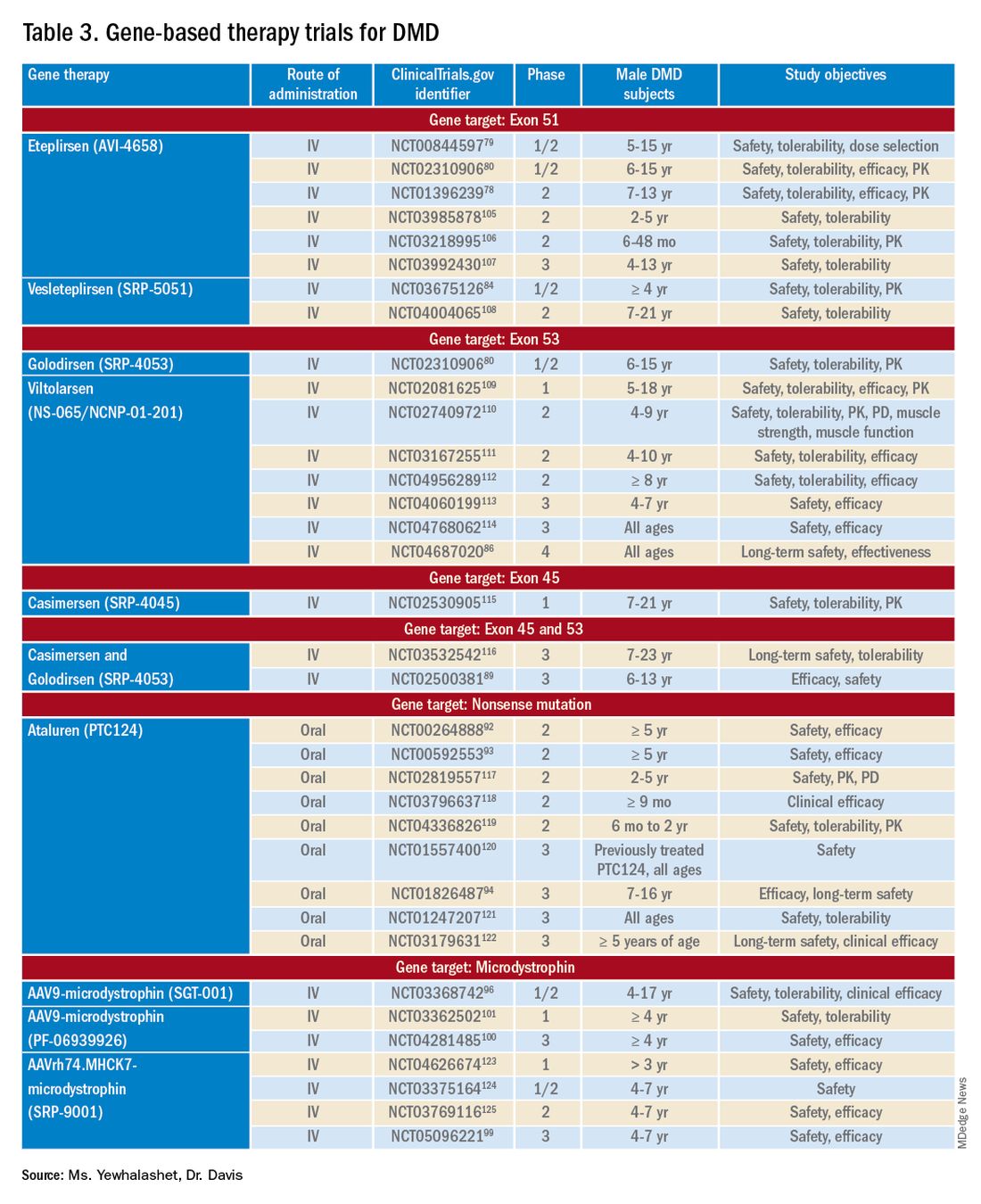
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Staying alert for patients with narcolepsy
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.

What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.

Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.

Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.
What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.
Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.
Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.
What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.
Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.
Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.