User login
Sudden hypoxia during knee surgery
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1. What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
- Fat embolism
- Intraoperative pneumonia
- Venous thromboembolism with pulmonary embolism
- Acute myocardial infarction
- Acute pulmonary edema
- Excessive sedation
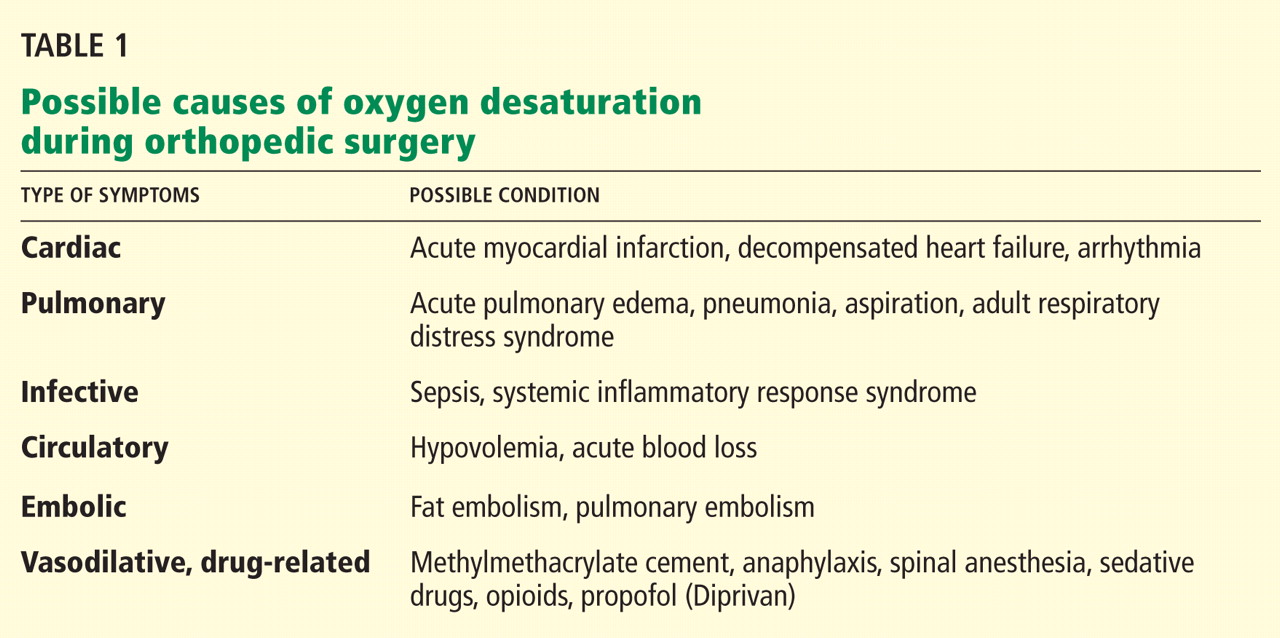
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2. Which consequence of fat embolism is most likely at this time in this patient?
- Coexisting sepsis
- Fat embolism syndrome
- Acute cardioembolic stroke
- Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3. Where on the body is the rash associated with fat embolism syndrome usually seen?
- Face
- Near a site of fracture or surgery
- Chest, axilla, conjunctiva
- Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27

Body systems affected by fat embolism syndrome are summarized in Table 2.
4. How many hours after injury does fat embolism syndrome typically manifest?
- 1 to 2 hours
- 6 to 12 hours
- 12 to 20 hours
- 24 to 48 hours
- 72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.



Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
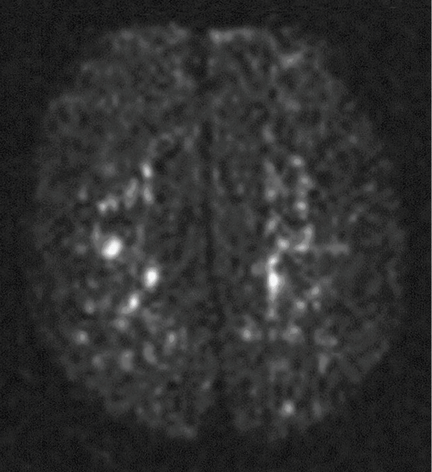
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome (Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5. Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
- Intravenous ethanol
- Steroids
- Heparin
- Dextran
- Aspirin
- None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
- Akhtar S. Fat embolism. Anesthesiol Clin 2009; 27:533–550.
- Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras 2005; 13:196–208.
- ten Duis HJ. The fat embolism syndrome. Injury 1997; 28:77–85.
- Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res 1969; 66:241–253.
- Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res 1982; 165:68–82.
- Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics 2003; 26:523–527.
- Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br 2003; 85:90–94.
- Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br 1995; 77:450–455.
- Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev 1993; 22:567–571.
- Mellor A, Soni N. Fat embolism. Anaesthesia 2001; 56:145–154.
- Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today 2007; 37:5–8.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br 1987; 69:128–131.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br 1974; 56B:408–416.
- Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res 1993; 291:208–214.
- Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg 1997; 85:441–443.
- Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot 1997; 83:9–21.
- Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty 2001; 16:730–739.
- Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res 1989; 248:112–118.
- Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res 1994; 309:94–101.
- Gauss H. The pathology of fat embolism. Arch Surg 1924; 9:593–605.
- Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg 1927; 14:621–662.
- Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics 1996; 19:41–48.
- Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev 1993; 20:165–170.
- Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology 2002; 96:1027–1029.
- Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med 1994; 150:1416–1422.
- Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir 1911; 112:192.
- Hagley SR. The fulminant fat embolism syndrome. Anaesth Intensive Care 1983; 11:167–170.
- Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med 1991; 8:233–239.
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg 1997; 132:435–439.
- King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest 1971; 59:524–530.
- Talbot M, Schemitsch EH. Fat embolism syndrome: history, definition, epidemiology. Injury 2006; 37(suppl 4):S3–S7.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med 1983; 99:438–443.
- Tetzlaff J, Massoli K. Fat embolism. In:Tetzlaff J, editor. Clinical Orthopedic Anesthesia. Boston, MA: Butterworth-Heinemann; 1995:341–349.
- Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am 1993; 11:25–54.
- Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci 1997; 33:654–658.
- Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest 1998; 113:178–181.
- Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol 1998; 8:1590–1593.
- Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma 1999; 46:324–327.
- Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke 2001; 32:2942–2944.
- Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury 2006; 37(suppl 4):S68–S73.
- Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J 1958; 1:1160–1161.
- Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am 1977; 59:878–880.
- Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J 1964; 2:101–102.
- Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med 2008; 36:565–571.
- Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma 1987; 27:1173–1176.
- Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res 1979; 143:211–221.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg 2009; 52:386–393.
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1. What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
- Fat embolism
- Intraoperative pneumonia
- Venous thromboembolism with pulmonary embolism
- Acute myocardial infarction
- Acute pulmonary edema
- Excessive sedation
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2. Which consequence of fat embolism is most likely at this time in this patient?
- Coexisting sepsis
- Fat embolism syndrome
- Acute cardioembolic stroke
- Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3. Where on the body is the rash associated with fat embolism syndrome usually seen?
- Face
- Near a site of fracture or surgery
- Chest, axilla, conjunctiva
- Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27
Body systems affected by fat embolism syndrome are summarized in Table 2.
4. How many hours after injury does fat embolism syndrome typically manifest?
- 1 to 2 hours
- 6 to 12 hours
- 12 to 20 hours
- 24 to 48 hours
- 72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.
Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome (Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5. Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
- Intravenous ethanol
- Steroids
- Heparin
- Dextran
- Aspirin
- None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
A 75-year-old man with type 2 diabetes and hypothyroidism underwent bilateral total knee replacement at our hospital.
His functional capacity had been moderately limited by knee pain, but he could easily climb one flight of stairs without symptoms. His medications at that time included levothyroxine (Synthroid) and metformin (Glucophage). He had no known cardiac or pulmonary disease. The preoperative evaluation, including laboratory tests and electrocardiography, was within normal limits.
Spinal anesthesia was used for surgery, and he was given 2 mg of midazolam (Versed) intravenously for sedation. No additional sedation was given. He was given oxygen via nasal cannula at 2 L/min.
All vital signs were stable at the start of the procedure. However, about halfway through, when the thigh tourniquet was released, his oxygen saturation dropped abruptly from 100% to 92%. All other vital signs remained stable, and he was asymptomatic, was oriented to person, time, and place, was conversing freely, and was in no distress. The oxygen flow was increased to 6 L/min, his oxygen saturation improved, and the procedure was then completed as planned.
At the conclusion of the surgery, before the patient was transported to the postanesthesia care unit (PACU) and while his oxygen flow rate was still 6 L/min, his oxygen saturation again dropped to 92%. A simple face mask was placed, and the oxygen flow rate was increased to 10 L/min. His oxygen saturation stayed low, near 90%.
Bleeding during surgery had been nominal. He had received 2 L of lactated Ringer’s solution and 500 mL of hetastarch (Hextend) during surgery. He continued to be asymptomatic in the PACU.
1. What is the most likely cause of oxygen desaturation during bilateral total knee arthroplasty?
- Fat embolism
- Intraoperative pneumonia
- Venous thromboembolism with pulmonary embolism
- Acute myocardial infarction
- Acute pulmonary edema
- Excessive sedation
The differential diagnosis of oxygen desaturation during orthopedic procedures is listed in Table 1.
Fat embolism is the most likely cause, particularly given the greater fatty embolic load that occurs with bilateral total knee arthroplasty than with unilateral total knee arthroplasty.
At what point the maximal showering of fat emboli occurs is not known. Fat may be released into the circulation with pressurization of the medullary canal during surgery or with manipulation of a fracture. The emboli may collect in the leg veins and then be released in a shower when the thigh tourniquet is released. Vasoactive mediators and methylmethacrylate cement released into the circulatory system after tourniquet deflation may also cause vasodilation, hypotension, and increased dead-space ventilation, resulting in hypoxia and a drop in end-tidal CO2.
Pneumonia during surgery is rare without an apparent aspiration event.
Venous thromboembolism is possible but is more likely later in the postoperative period after major orthopedic surgery.
Acute myocardial infarction could present with hypoxia, particularly in a diabetic patient, who may not experience chest pain. However, intraoperative electrocardiographic changes would likely be seen. If myocardial infarction is suspected, postoperative serial electrocardiograms and measuring troponin and cardiac enzyme levels aid in the diagnosis.
Acute pulmonary edema is possible but not as highly suspected, as the patient had no history of congestive heart failure and received an appropriate amount of fluid for this type of surgery.
Excessive sedation could cause hypoventilation and, thus, oxygen desaturatation. However, this patient’s oxygen desaturatation began more than an hour after the midazolam was given. Midazolam is a short-acting benzodiazepine. It is unlikely that the patient would show signs of hypoventilation and oversedation an hour after the drug was given. Our patient also did not show any signs of excessive sedation, as he was awake and conversing during the surgery.
Fat emboli vs fat embolism syndrome
Fat embolism is the presence of fat drops within the systemic and pulmonary microcirculation, with or without clinical sequelae.1 Fat embolism syndrome, on the other hand, is defined as injury to and dysfunction of one or more organs as a result of the embolization of fat, usually within 24 hours of injury or orthopedic surgery.2
Fat embolism syndrome is an unpredictable condition with a varied presentation. Fat droplets are thought to embolize via the venous circulation into the pulmonary arteries, occluding small blood vessels in the lung. However, they also get into the arterial circulation and occlude arteries in the brain, kidney, heart, and liver (more on this phenomenon below).
Fat embolism is reported to originate primarily from fractures of the femur, tibia, and pelvis.2,3 As many as 90% of trauma patients have been shown to have evidence of fat embolism on autopsy.4 However, only a small number of patients develop the classic fat embolism syndrome,2,3,5 Why some develop the syndrome and others do not is still unknown.
Orthopedic procedures associated with fat embolization include knee arthroplasty and hip arthroplasty, particularly if it involves intramedullary manipulation or medullary fixation.6 It has also been reported during spinal procedures in which pedicular screws are used.7 The syndrome occurs in 0.25% to 30% of patients following multiple fractures and in 0.1% to 12% of patients during or following knee or hip arthroplasty.
One study8 showed evidence of fat on transesophageal echocardiography in 88% of patients undergoing medullary reaming of lower-extremity fractures and hip hemiarthroplasty. Blood sampling from the right atrium confirmed that fat was responsible for the echocardiographic abnormalities. The study also showed that the severity of the embolic showering correlated with the severity of hypoxia and the decrease in end-tidal CO2.8
CASE CONTINUED
On arrival at the PACU, our patient’s oxygen saturation was 94% while he was breathing oxygen via a simple face mask at a flow rate of 10 L/min. His heart rate was 60 bpm, blood pressure 110/60 mm Hg, and temperature 37.5°C (96.3°F). Chest sounds were normal on auscultation.
However, 3 hours later, his mental status rapidly deteriorated. He was oriented only to person, and he was drowsy. He had escalating respiratory distress with a rapid respiratory rate and decreasing oxygen saturation. At this point, auscultation of his chest wall revealed bilateral crackles and rales.
He was promptly intubated. Profuse fluid and secretions were noted to be coming from his lungs, filling the endotracheal tube. Arterial blood gas measurement showed a pH of 7.22, Pao2 64 mm Hg, and Paco2 56 mm Hg on 100% fraction of inspired oxygen, with no increased anion gap.
2. Which consequence of fat embolism is most likely at this time in this patient?
- Coexisting sepsis
- Fat embolism syndrome
- Acute cardioembolic stroke
- Anaphylaxis
Fat embolism syndrome should be highly suspected in this patient. As mentioned, it can affect many different organs. It is the most serious condition resulting from fat embolization after surgery or trauma.
Sepsis was unlikely in our patient, since he presented for his surgery in good health and with no preexisting signs or symptoms of infection. Acute cardioembolic stroke could have caused the neurologic signs, but this would not necessarily explain the coexisting hypoxia. An anaphylactic reaction to drugs or surgical cement would most likely present intraoperatively, shortly after exposure occurred, rather than several hours after surgery.
How common is fat embolism syndrome?
The occurrence rate of fat embolism syndrome has been reported to be 0.25% to 30% after multiple fractures and 0.1% to 12% after knee and hip joint surgery, with a mortality rate of 13% to 36%.2,9–14 The rate of occurrence after unilateral total knee joint replacement has been reported to be 1.8% to 5%, and 4% to 12% after bilateral total knee replacement.15–19
The syndrome is relatively more common with traumatic fractures of the lower extremities. However, it has also been reported with liposuction, total parenteral nutrition, bone marrow harvest and transplantation, burns, and acute pancreatitis, to mention a few.10
The broad range of reported incidence rates can be attributed to the fact that many studies were in patients with multiple trauma, whose concomitant injuries may have made it difficult to clearly define the contribution of fat embolism syndrome to the overall rates of morbidity and mortality. Also, different studies used different criteria to define the syndrome.
How does fat embolism syndrome occur?
Two hypotheses for how this syndrome occurs were proposed nearly a century ago.20,21
The “mechanical” theory is that fat emboli are formed as a result of trauma and disruption of adipose tissue and other cells in the bone marrow. Increases in intramedullary pressure force the fat emboli through damaged medullary venous channels in the bone and into the circulation of the lower extremities. This embolization of fat causes an initial mechanical pulmonary obstruction. Mechanical obstruction by fat emboli in the pulmonary system leads to increased pulmonary pressures and an increase in right heart outflow pressure. The right heart becomes strained, leading to a decreased right-sided cardiac output. As a result, the left heart filling pressures diminish and hypotension ensues.20
The “biochemical” theory, on the other hand, is that chylomicrons within the vascular system are modified and their stability is compromised as a result of stress. These traumatized chylomicrons then coalesce to form droplets of fat that accumulate in the pulmonary circulation and produce a mechanical obstruction. This would explain why nontraumatic, nonorthopedic insults can produce this syndrome.
Autopsy studies show that there is little correlation between the presence and quantity of intravascular fat and the severity of clinical symptoms, thus implying that the syndrome is caused by more than just mechanical obstruction. The biochemical theory postulates that fat globules within the circulatory system then cause the release of lipase from the pulmonary alveolar cells, which then hydrolyses the fat into free fatty acids. These free fatty acids cause an inflammatory reaction, complementmediated leukocyte aggregation, chemotoxin release, and subsequently endothelial damage. These vasoactive substances damage type 2 pneumocytes and lead to an increased permeability of the pulmonary capillary beds. Acute respiratory distress syndrome (ARDS) may ensue. Disseminated intravascular coagulation may occur as a result of the formation of microthrombi involving lipids, platelets, and fibrin.21,22
Embolization of fat to the central nervous system can occur as fat globules cross into the systemic circulation via a patent foramen ovale, an atrioventricular shunt, or the pulmonary capillaries. This can then result in cerebral ischemia.23
Although patent foramen ovale may seem the most direct route for cerebral embolization, the neurologic impairment and signs of cerebral emboli in fat embolism syndrome may occur in the absence of patent foramen ovale.24,25 The fat globules may actually go through the lung capillaries, being flexible and forced through by increased pulmonary pressure.
But whether the cause of fat embolism syndrome is occlusion by globules, the release of biochemical mediators, or a combination of both is unknown. Both mechanisms are likely responsible. We can only suspect that the degree of fat load and intrinsic metabolic differences between individuals account for the variation in susceptibility.
FAT EMBOLI AFFECT THE LUNGS, SKIN, AND BRAIN
3. Where on the body is the rash associated with fat embolism syndrome usually seen?
- Face
- Near a site of fracture or surgery
- Chest, axilla, conjunctiva
- Distal extremities
Petechiae are part of the classic presenting triad of fat embolism syndrome, which also includes pulmonary and cerebral dysfunction.
Petechiae usually appear on the 2nd to 4th day after injury.26 They are usually found across the chest, the anterior axillary folds, and the neck, as well as on the oral mucosa and the conjunctiva. The rash is caused by occlusion of dermal capillaries by fat, which increases their fragility.10
Pulmonary changes usually begin with tachypnea, dyspnea, and a drop in oxygen saturation, leading to generalized hypoxia. Respiratory symptoms are present in 100% of cases.2 Respiratory symptoms can acutely develop with the sudden manipulation of a fracture, reaming of bone, or release of a limb tourniquet.27
Body systems affected by fat embolism syndrome are summarized in Table 2.
4. How many hours after injury does fat embolism syndrome typically manifest?
- 1 to 2 hours
- 6 to 12 hours
- 12 to 20 hours
- 24 to 48 hours
- 72 to 84 hours
Most patients develop signs and symptoms 24 to 48 hours after injury. Patients presenting earlier than 12 hours usually have a more fulminant course.29
The time between fat embolization and the development of fat embolism syndrome is thought to be related to the time required for the metabolic conversion of fat to free fatty acids.30 We suspect that the early desaturation seen in our patient was the result of a heavy showering of fat intraoperatively. However, this could only be concluded after we had ruled out other causes of acute hypoxia and hypotension.
Fat embolism syndrome is a diagnosis of exclusion and is based on clinical criteria. No specific sign, symptom, or test is pathognomonic. It may often be confused with other conditions such as systemic inflammatory response syndrome or sepsis. However, the triad of respiratory and neurologic symptoms and petechiae coupled with the clinical picture of recent trauma or orthopedic surgery almost assures the diagnosis.
Fat embolism syndrome can range from subclinical to fulminating, with the more fulminating course attributable to a huge load of fat emboli, which leads to acute cor pulmonale.
Regardless of the criteria used, one must have a high index of suspicion for fat embolization syndrome in patients undergoing orthopedic procedures, particularly hip and knee surgery, and in patients with fractures, especially fractures of the femur, tibia, or pelvis and multiple, concomitant fractures.
CASE CONTINUED
Our patient was given furosemide (Lasix) empirically for diuresis and to improve oxygenation. However, his oxygen saturation remained low.
Chest radiography 4 hours after surgery showed bilateral pulmonary infiltrates. Serial electrocardiography showed no acute changes. Levels of cardiac enzymes and troponins were normal. Transthoracic echocardiography showed no left ventricular dysfunction, a normal right ventricle, and no evidence of valvular lesions. Urine and blood fat stains were negative, but the sputum stain was positive for copious extracellular fat. The patient became comatose 5 hours postoperatively. Computed tomography of the brain was normal. He was transferred to the surgical intensive care unit.
The clinical course was marked by hemodynamic instability requiring norepinephrine (Levophed) and vasopressin (Pitressin) for hypotension. Right ventricular filling pressures via central venous pressure monitoring showed no evidence of hypovolemia. The hemoglobin concentration and the hematocrit were stable, with no evidence of acute or ongoing bleeding. Blood, urine, and sputum cultures remained negative. Acute myocardial infarction was ruled out by serial electrocardiography, cardiac enzyme testing, and troponin testing.
Magnetic resonance imaging (MRI) of the brain on postoperative day 2 showed foci of acute ischemia suggestive of embolic phenomena consistent with fat embolism syndrome (Figure 1). Transthoracic echocardiography was repeated but again showed no evidence of a patent foramen ovale. Electroencephalography on postoperative day 4 showed severe, diffuse encephalopathy. There was no petechial skin rash. Other laboratory studies showed progressive thrombocytopenia with a platelet count of 53 × 199/L on postoperative day 3.
TESTS THAT AID THE CLINICAL DIAGNOSIS
Although no single laboratory test is pathognomonic for fat embolism syndrome, several tests may help raise suspicion of it, especially in the setting of fracture or an orthopedic surgical procedure.
Arterial blood gases must be measured. A Pao2 of less than 60 mm Hg with no other obvious lung pathology in an orthopedic surgery patient is highly suspicious.12 An alveolar-arterial gradient of greater than 100 mm Hg may further increase suspicion.
Tests for fat. The blood and urine may be examined for fat, although positive findings are not specific for fat embolism syndrome.33 Fat in the urine indicates the occurrence of massive fat embolism, but this is not always accompanied by the syndrome.34 Gurd and Wilson13 found fat globules larger than 8 μm circulating in the serum in all documented cases. They stated that, even though the relationship of large fat globules to the pathogenesis of the clinical picture remains obscure, the demonstration of their presence can be helpful in the diagnosis.13
Also, samples obtained with bronchoalveolar lavage may be examined for fat. The macrophages may be stained for fat using the oil red O stain. Again, this is a nonspecific marker, as fat-stained macrophages are seen in trauma patients,35 but the finding has a very high negative predictive value.36 Anemia, thrombocytopenia, hypofibrinogenemia, an elevated lipase level, and a high erythrocyte sedimentation rate may be found in fat embolism syndrome.13
Chest radiography may show bilateral infiltrates, as in ARDS, but this is not diagnostic for fat embolism syndrome.
Electrocardiography may show changes in ST and T waves and signs of right heart strain.
Transesophageal echocardiography may show increased right heart and pulmonary artery pressures.
Computed tomography is often negative,37,38 but T2-weighted MRI is useful in the diagnosis of cerebral fat embolism syndrome, as it can show intracerebral microinfarcts as early as 4 hours after the onset of neurologic symptoms, and these findings correlate well with the clinical severity of brain injury.
Diffusion-weighted MRI may enhance the sensitivity and specificity of the neuroradiologic diagnosis. Diffusion-weighted MRI typically shows multiple nonconfluent areas of high-intensity signals or bright spots on a dark background, known as a “starfield pattern.” This pattern has been suggested to be pathognomonic of acute cerebral microinfarction. The abnormalities presumably reflect foci of cytotoxic edema that develops immediately, unlike vasogenic edema, seen in T2-weighted images, which may take up to several days to develop. Although these images are not necessarily specific for fat emboli, they are useful in helping make the diagnosis. Thus, diffusionweighted MRI should be done if fat embolism syndrome is suspected.38,39
CASE CONCLUDED
The patient’s course in the intensive care unit was further complicated by gastrointestinal bleeding and renal failure. His neurologic status did not improve. Repeated MRI of the brain showed evolving bilateral watershed infarction throughout the cortices. The neurologic consult service diagnosed the patient as having severe encephalopathy with a very poor prognosis. The decision was made to withdraw care. He was placed under palliative care and died on postoperative day 22.
DRUG TREATMENT OF FAT EMBOLISM SYNDROME
5. Which of the following drugs has been proven to be effective in treating fat embolism syndrome?
- Intravenous ethanol
- Steroids
- Heparin
- Dextran
- Aspirin
- None of the above
None of the above has been proven to be effective in treating this disorder. The management is largely supportive. Thus, prevention, early diagnosis, and symptom management are vital.
Pulmonary and hemodynamic support are the cornerstones of successful treatment. Aggressive respiratory support is often needed. Management of acute lung injury and ARDS focuses on achieving acceptable gas exchange while preventing ventilator-associated lung injury. Intravascular volume must be supported. Inotropes and pulmonary vasodilators may be required to maintain hemodynamics. Exacerbation of central nervous system ischemia from hypotension or hypoxia should be avoided.
If the thrombocytopenia leads to clinical bleeding, platelet transfusions may be warranted.
Supportive care should include prophylaxis of deep venous thrombosis and of gastrointestinal bleeding, and maintenance of nutrition.40 Patients who receive supportive care generally have a favorable outcome, with a mortality rate of less than 10%.28
Drug studies have been inconclusive
Drugs suggested in the treatment of fat embolism syndrome include heparin, aspirin, dextran, hypertonic glucose, and alcohol, but the results have been inconclusive.3,11,23,40–43
Heparin stimulates lipase activity, consequently decreasing the concentration of circulating fat globules. However, the increase in levels of free fatty acids may actually worsen the clinical picture. For this reason, and because of anticoagulation concerns and evidence of increased mortality rates, heparin is now contraindicated in the treatment of fat embolism syndrome.2,41,43
Alcohol. Patients with a higher blood alcohol level at the time of injury have been reported to have a lower incidence of fat embolism syndrome. Alcohol inhibits lipase, suppressing the rise of free fatty acids. In experimental studies, the incidence of fat embolism syndrome was lower when the blood alcohol level was maintained at 20 mg/dL. However, no prospective randomized trial has been done to determine the clinical efficacy of ethanol as a treatment for this condition.5,42
Dextran has been advocated, owing to its ability to improve small-vessel perfusion, but bleeding risk and acute renal failure associated with this drug have limited its use.5
N-acetylcysteine has been shown to attenuate fat-induced lung injury in a study of rats with induced fat embolism syndrome.44
Corticosteroid treatment for this condition is controversial. Studies in patients with femoral and tibial fractures show that steroids reduce the incidence of fat embolism syndrome when given prophylactically, and those treated with steroids had a higher Pao2 than controls. Doses of methylprednisolone in these studies ranged between 9 mg/kg to 90 mg/kg. A drawback of these studies is their small number of patients.12,32,45,46
A meta-analysis47 of randomized trials of corticosteroids to prevent fat embolism syndrome in patients with long-bone fractures identified 104 such studies. Only 7 of the 104 were considered adequate. In 389 patients with long-bone fractures, prophylactic corticosteroids reduced the risk of fat embolism syndrome by 78% (95% confidence interval 43%–92%) and corticosteroids also significantly reduced the risk of hypoxia with no difference in rates of infection or death. However, the overall quality of the trials was poor, and the authors of the meta-analysis concluded that more study is needed before corticosteroids could be formally recommended.47
There is no evidence that steroids improve the overall clinical course of already established fat embolism syndrome.12,32,45 The dosing and optimal timing of administration have also not been established. High doses pose a risk of septic complications, which may be devastating for the posttrauma or postoperative patient.
- Akhtar S. Fat embolism. Anesthesiol Clin 2009; 27:533–550.
- Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras 2005; 13:196–208.
- ten Duis HJ. The fat embolism syndrome. Injury 1997; 28:77–85.
- Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res 1969; 66:241–253.
- Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res 1982; 165:68–82.
- Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics 2003; 26:523–527.
- Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br 2003; 85:90–94.
- Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br 1995; 77:450–455.
- Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev 1993; 22:567–571.
- Mellor A, Soni N. Fat embolism. Anaesthesia 2001; 56:145–154.
- Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today 2007; 37:5–8.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br 1987; 69:128–131.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br 1974; 56B:408–416.
- Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res 1993; 291:208–214.
- Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg 1997; 85:441–443.
- Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot 1997; 83:9–21.
- Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty 2001; 16:730–739.
- Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res 1989; 248:112–118.
- Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res 1994; 309:94–101.
- Gauss H. The pathology of fat embolism. Arch Surg 1924; 9:593–605.
- Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg 1927; 14:621–662.
- Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics 1996; 19:41–48.
- Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev 1993; 20:165–170.
- Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology 2002; 96:1027–1029.
- Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med 1994; 150:1416–1422.
- Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir 1911; 112:192.
- Hagley SR. The fulminant fat embolism syndrome. Anaesth Intensive Care 1983; 11:167–170.
- Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med 1991; 8:233–239.
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg 1997; 132:435–439.
- King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest 1971; 59:524–530.
- Talbot M, Schemitsch EH. Fat embolism syndrome: history, definition, epidemiology. Injury 2006; 37(suppl 4):S3–S7.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med 1983; 99:438–443.
- Tetzlaff J, Massoli K. Fat embolism. In:Tetzlaff J, editor. Clinical Orthopedic Anesthesia. Boston, MA: Butterworth-Heinemann; 1995:341–349.
- Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am 1993; 11:25–54.
- Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci 1997; 33:654–658.
- Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest 1998; 113:178–181.
- Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol 1998; 8:1590–1593.
- Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma 1999; 46:324–327.
- Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke 2001; 32:2942–2944.
- Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury 2006; 37(suppl 4):S68–S73.
- Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J 1958; 1:1160–1161.
- Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am 1977; 59:878–880.
- Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J 1964; 2:101–102.
- Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med 2008; 36:565–571.
- Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma 1987; 27:1173–1176.
- Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res 1979; 143:211–221.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg 2009; 52:386–393.
- Akhtar S. Fat embolism. Anesthesiol Clin 2009; 27:533–550.
- Filomeno LT, Carelli CR, Silva NC, Filho TE, Amatuzzi MM. Fat embolism: a review for current orthopaedics practice. Acta Ortop Bras 2005; 13:196–208.
- ten Duis HJ. The fat embolism syndrome. Injury 1997; 28:77–85.
- Peltier LF. Fat embolism. A current concept. Clin Orthop Relat Res 1969; 66:241–253.
- Gossling HR, Pellegrini VD. Fat embolism syndrome: a review of the pathophysiology and physiological basis of treatment. Clin Orthop Relat Res 1982; 165:68–82.
- Papagelopoulos PJ, Apostolou CD, Karachalios TS, Themistocleous GS, Giannakopoulos CK, Ioannidis TT. Pulmonary fat embolism after total hip and total knee arthroplasty. Orthopedics 2003; 26:523–527.
- Takahashi S, Kitagawa H, Ishii T. Intraoperative pulmonary embolism during spinal instrumentation surgery. A prospective study using transoesophageal echocardiography. J Bone Joint Surg Br 2003; 85:90–94.
- Christie J, Robinson CM, Pell AC, McBirnie J, Burnett R. Transcardiac echocardiography during invasive intramedullary procedures. J Bone Joint Surg Br 1995; 77:450–455.
- Robert JH, Hoffmeyer P, Broquet PE, Cerutti P, Vasey H. Fat embolism syndrome. Orthop Rev 1993; 22:567–571.
- Mellor A, Soni N. Fat embolism. Anaesthesia 2001; 56:145–154.
- Taviloglu K, Yanar H. Fat embolism syndrome. Surg Today 2007; 37:5–8.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br 1987; 69:128–131.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br 1974; 56B:408–416.
- Ganong RB. Fat emboli syndrome in isolated fractures of the tibia and femur. Clin Orthop Relat Res 1993; 291:208–214.
- Djelouah I, Lefèvre G, Ozier Y, Rosencher N, Tallet F. Fat embolism in orthopedic surgery: role of bone marrow fatty acid. Anesth Analg 1997; 85:441–443.
- Barre J, Lepouse C, Segal P. Embolism and intramedullary femoral surgery. Rev Chir Orthop Reparatrice Appar Mot 1997; 83:9–21.
- Kim YH. Incidence of fat embolism syndrome after cemented or cementless bilateral simultaneous and unilateral total knee arthroplasty. J Arthroplasty 2001; 16:730–739.
- Dorr LD, Merkel C, Mellman MF, Klein I. Fat emboli in bilateral total knee arthroplasty. Predictive factors for neurologic manifestations. Clin Orthop Relat Res 1989; 248:112–118.
- Jankiewicz JJ, Sculco TP, Ranawat CS, Behr C, Tarrentino S. Onestage versus 2-stage bilateral total knee arthroplasty. Clin Orthop Relat Res 1994; 309:94–101.
- Gauss H. The pathology of fat embolism. Arch Surg 1924; 9:593–605.
- Lehman EP, Moore RM. Fat embolism, including experimental production without trauma. Arch Surg 1927; 14:621–662.
- Johnson MJ, Lucas GL. Fat embolism syndrome. Orthopedics 1996; 19:41–48.
- Benson KT. Diagnosis and treatment of fat embolism syndrome. Anesthesiology Rev 1993; 20:165–170.
- Colonna DM, Kilgus D, Brown W, Challa V, Stump DA, Moody DM. Acute brain fat embolization occurring after total hip arthroplasty in the absence of a patent foramen ovale. Anesthesiology 2002; 96:1027–1029.
- Byrick RJ, Mullen JB, Mazer CD, Guest CB. Transpulmonary systemic fat embolism. Studies in mongrel dogs after cemented arthroplasty. Am J Respir Crit Care Med 1994; 150:1416–1422.
- Benestad G. Drei Fälle von Fettembolie mit punktförmigen Blutungen in der Haut. Deutsche Ztschr f Chir 1911; 112:192.
- Hagley SR. The fulminant fat embolism syndrome. Anaesth Intensive Care 1983; 11:167–170.
- Fulde GW, Harrison P. Fat embolism—a review. Arch Emerg Med 1991; 8:233–239.
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg 1997; 132:435–439.
- King EG, Wagner WW, Ashbaugh DG, Latham LP, Halsey DR. Alterations in pulmonary microanatomy after fat embolism. In vivo observations via thoracic window of the oleic acid-embolized canine lung. Chest 1971; 59:524–530.
- Talbot M, Schemitsch EH. Fat embolism syndrome: history, definition, epidemiology. Injury 2006; 37(suppl 4):S3–S7.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med 1983; 99:438–443.
- Tetzlaff J, Massoli K. Fat embolism. In:Tetzlaff J, editor. Clinical Orthopedic Anesthesia. Boston, MA: Butterworth-Heinemann; 1995:341–349.
- Capan LM, Miller SM, Patel KP. Fat embolism. Anesthesiol Clin North Am 1993; 11:25–54.
- Reider E, Sherman Y, Weiss Y, Liebergall M, Pizov R. Alveolar macrophages fat stain in early diagnosis of fat embolism syndrome. Isr J Med Sci 1997; 33:654–658.
- Aoki N, Soma K, Shindo M, Kurosawa T, Ohwada T. Evaluation of potential fat emboli during placement of intramedullary nails after orthopedic fractures. Chest 1998; 113:178–181.
- Stoeger A, Daniaux M, Felber S, Stockhammer G, Aichner F, zur Nedden D. MRI findings in cerebral fat embolism. Eur Radiol 1998; 8:1590–1593.
- Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma 1999; 46:324–327.
- Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke 2001; 32:2942–2944.
- Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury 2006; 37(suppl 4):S68–S73.
- Sage RH, Tudor RW. Treatment of fat embolism with heparin. Br Med J 1958; 1:1160–1161.
- Myers R, Taljaard JJ. Blood alcohol and fat embolism syndrome. J Bone Joint Surg Am 1977; 59:878–880.
- Denman EE, Cairnes CS, Holmes CM. Case of severe fat embolism treated by intermittent positive-pressure respiration. Br Med J 1964; 2:101–102.
- Liu DD, Kao SJ, Chen HI. N-Acetylcysteine attenuates acute lung injury induced by fat embolism. Crit Care Med 2008; 36:565–571.
- Kallenbach J, Lewis M, Zaltzman M, Feldman C, Orford A, Zwi S. ‘Low-dose’ corticosteroid prophylaxis against fat embolism. J Trauma 1987; 27:1173–1176.
- Stoltenberg JJ, Gustilo RB. The use of methylprednisolone and hypertonic glucose in the prophylaxis of fat embolism syndrome. Clin Orthop Relat Res 1979; 143:211–221.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg 2009; 52:386–393.
A 37-year-old man with a chronic cough
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B

Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B
Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B
Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
A 54-year-old woman with pancytopenia
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
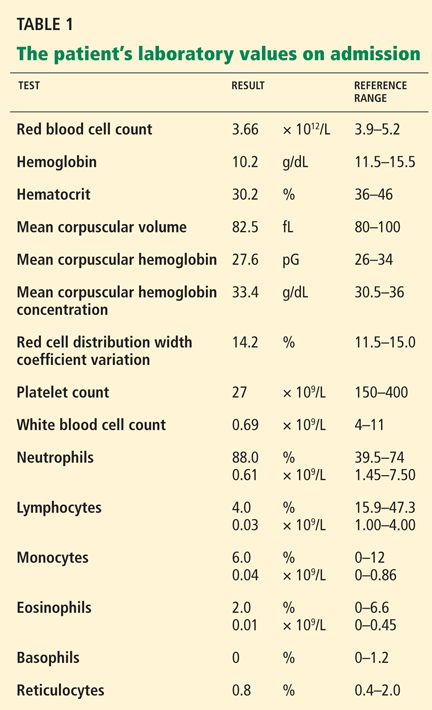
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
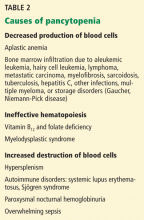
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other

Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
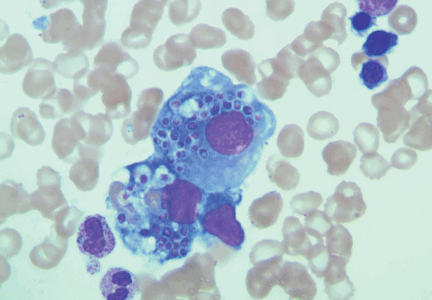
A 25-year-old man with very high alkaline phosphatase
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
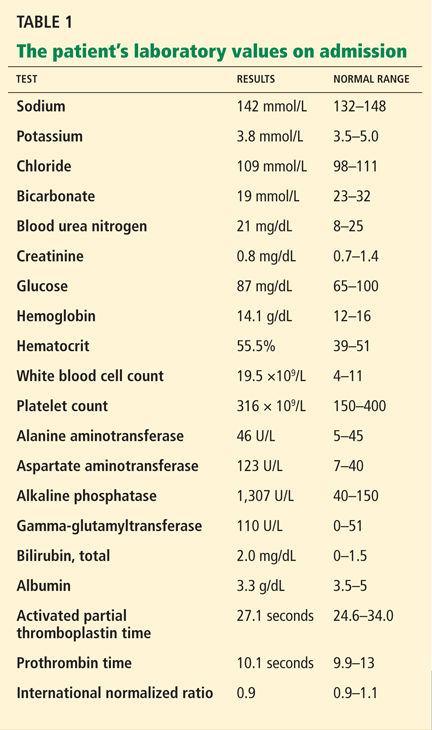
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
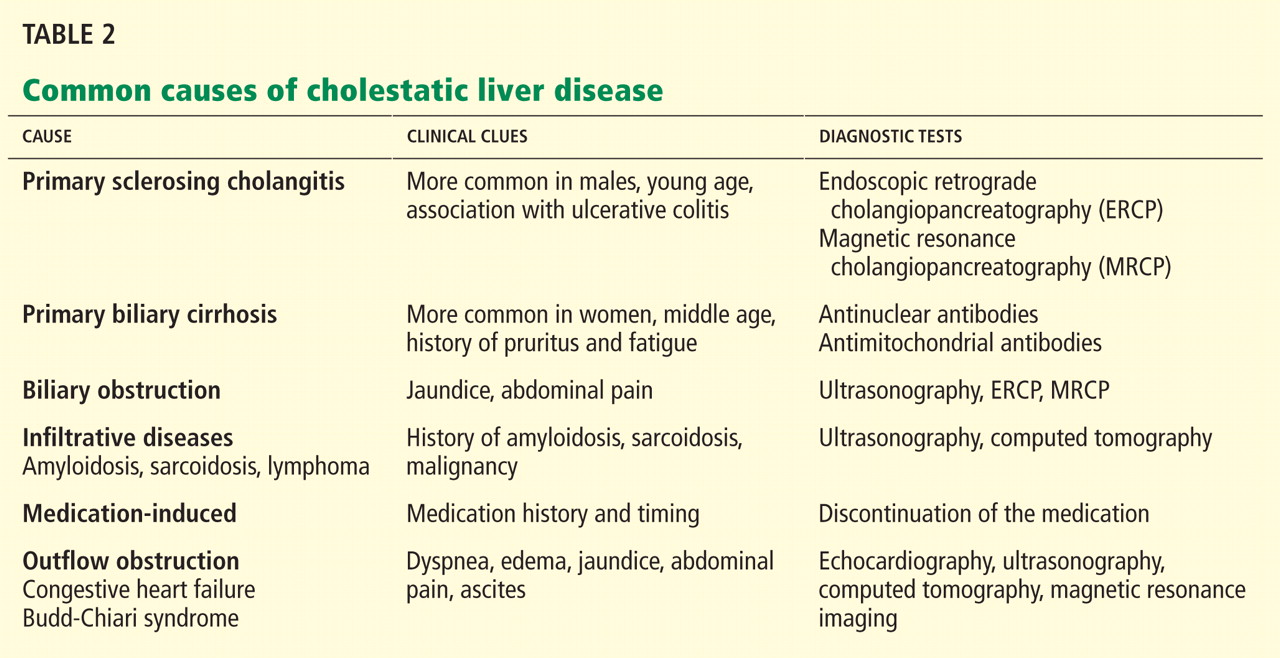
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations

Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 49-year-old woman with a persistent cough
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
A 49-year-old woman presents with a cough that has persisted for 3 weeks.
Two weeks ago, she was seen in the outpatient clinic for a nonproductive cough, rhinorrhea, sneezing, and a sore throat. At that time, she described coughing spells that were occasionally accompanied by posttussive chest pain and vomiting. The cough was worse at night and was occasionally associated with wheezing. She reported no fevers, chills, rigors, night sweats, or dyspnea. She said she has tried over-the-counter cough suppressants, antihistamines, and decongestants, but they provided no relief. Since she had a history of well-controlled asthma, she was diagnosed with an asthma exacerbation and was given prednisone 20 mg to take orally every day for 5 days, to be followed by an inhaled corticosteroid until her symptoms resolved.
Now, she has returned because her symptoms have persisted despite treatment, and she is seeking a second medical opinion. Her paroxysmal cough has become more frequent and more severe.
In addition to asthma, she has a history of allergic rhinitis. Her current medications include the over-the-counter histamine H1 antagonist cetirizine (Zyrtec), a fluticasone-salmeterol inhaler (Advair), and an albuterol inhaler (Proventil HFA). She reports having had mild asthma exacerbations in the past during the winter, which were managed well with her albuterol inhaler.
She has never smoked; she drinks alcohol socially. She has not traveled outside the United States during the past several months. She is married and has two children, ages 25 and 23. She lives at home with only her husband, and he has not been sick. However, she works at a greeting card store, and two of her coworkers have similar upper respiratory symptoms, although they have only a mild cough.
Her immunizations are not up-to-date. She last received the tetanus-diphtheria toxoid (Td) vaccine 12 years ago, and she never received the pediatric tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. She generally receives the influenza vaccine annually, and she received it about 6 weeks before this presentation.
She is not in distress, but she has paroxysms of severe coughing throughout her examination. Her pulse is 100 beats per minute, respiratory rate 18, and blood pressure 130/86 mm Hg. Her oropharynx is clear. The pulmonary examination reveals poor inspiratory effort due to coughing but is otherwise normal. The rest of the examination is normal, as is her chest radiograph.
WHAT DOES SHE HAVE?
1. Which of the following would best explain her symptoms?
- Asthma
- Postviral cough
- Pertussis
- Chronic bronchitis
- Pneumonia
- Gastroesophageal reflux disease
Asthma is a reasonable consideration, given her medical history, her occasional wheezing, and her nonproductive cough that is worse at night. However, asthma typically responds well to corticosteroid therapy. She has already received a course of prednisone, but her symptoms have not improved.
Postviral cough could also be considered in this patient. However, postviral cough does not typically occur in paroxysms, nor does it lead to posttussive vomiting. It is also generally regarded as a diagnosis of exclusion.
Pertussis (whooping cough) should be suspected in this patient, given the time course of her symptoms, the paroxysmal cough, and the posttussive vomiting. In addition, at her job she interacts with hundreds of people a day, increasing her risk of exposure to respiratory tract pathogens, including Bordetella pertussis.
Chronic bronchitis is defined by cough (typically productive) lasting at least 3 months per year for at least 2 consecutive years, which does not fit the time course for this patient. It is vastly more common in smokers.
Pneumonia typically presents with a cough that can be productive or nonproductive, but also with fever, chills, and radiologic evidence of a pulmonary infiltrate or consolidation. This woman has none of these.
Gastroesophageal reflux disease is one of the most common causes of chronic cough, with symptoms typically worse at night. However, it is generally associated with symptoms such as heartburn, a sour taste in the mouth, or regurgitation, which our patient did not report.
Thus, pertussis is the most likely diagnosis.
PERTUSSIS IS ON THE RISE
Pertussis is an acute and highly contagious disease caused by infection of the respiratory tract by B pertussis, a small, aerobic, gramnegative, pleomorphic coccobacillus that produces a number of antigenic and biologically active products, including pertussis toxin, filamentous hemagglutinin, agglutinogens, and tracheal cytotoxin. Transmitted by aerosolized droplets, it attaches to the ciliated epithelial cells of the lower respiratory tract, paralyzes the cilia via toxins, and causes inflammation, thus interfering with the clearing of respiratory secretions.
The incidence of pertussis is on the rise. In 2005, 25,827 cases were reported in the United States, the highest number since 1959.1 Pertussis is now epidemic in California. At the time of this writing, the number of confirmed, probable, and suspected cases in California was 9,477 (including 10 infant deaths) for the year 2010—the most cases reported in the past 65 years.2,3
In 2010, outbreaks were also reported in Michigan, Texas, Ohio, upstate New York, and Arizona.4 The overall incidence of pertussis is likely even higher than what is reported, since many cases go unrecognized or unreported.
Highly contagious
Pertussis is transmitted person-to-person, primarily through aerosolized droplets from coughing or sneezing or by direct contact with secretions from the respiratory tract of infected persons. It is highly contagious, with secondary attack rates of up to 80% in susceptible people.
A three-stage clinical course
The clinical definition of pertussis used by the US Centers for Disease Control and Prevention (CDC) and the Council of State and Territorial Epidemiologists is an acute cough illness lasting at least 2 weeks, with paroxysms of coughing, an inspiratory “whoop,” or posttussive vomiting without another apparent cause.5
The clinical course of the illness is traditionally divided into three stages:
The catarrhal phase typically lasts 1 to 2 weeks and is clinically indistinguishable from a viral upper respiratory infection. It is characterized by the insidious onset of malaise, coryza, sneezing, low-grade fever, and a mild cough that gradually becomes severe.6
The paroxysmal phase normally lasts 1 to 6 weeks but may persist for up to 10 weeks. The diagnosis of pertussis is usually suspected during this phase. The classic features of this phase are bursts or paroxysms of numerous, rapid coughs. These are followed by a long inspiratory effort usually accompanied by a characteristic high-pitched whoop, most notably observed in infants and children. Infants and children may appear very ill and distressed during this time and may become cyanotic, but cyanosis is uncommon in adults and adolescents. The paroxysms may also be followed by exhaustion and posttussive vomiting. In some cases, the cough is not paroxysmal, but rather simply persistent. The coughing attacks tend to occur more often at night, with an average of 15 attacks per 24 hours. During the first 1 to 2 weeks of this stage, the attacks generally increase in frequency, remain at the same intensity level for 2 to 3 weeks, and then gradually decrease over 1 to 2 weeks.1,7
The convalescent phase can have a variable course, ranging from weeks to months, with an average duration of 2 to 3 weeks. During this stage, the paroxysms of coughing become less frequent and gradually resolve. Paroxysms often recur with subsequent respiratory infections.
In infants and young children, pertussis tends to follow these stages in a predictable sequence. Adolescents and adults, however, tend to go through the stages without being as ill and typically do not exhibit the characteristic whoop.
TESTING FOR PERTUSSIS
2. Which would be the test of choice to confirm pertussis in this patient?
- Bacterial culture of nasopharyngeal secretions
- Polymerase chain reaction (PCR) testing of nasopharyngeal secretions
- Direct fluorescent antibody testing of nasopharyngeal secretions
- Enzyme-linked immunosorbent assay (ELISA) serologic testing
Establishing the diagnosis of pertussis is often rather challenging.
Bacterial culture: Very specific, but slow and not so sensitive
Bacterial culture is still the gold standard for diagnosing pertussis, as a positive culture for B pertussis is 100% specific.5
However, this test has drawbacks. Its sensitivity has a wide range (15% to 80%) and depends very much on the time from the onset of symptoms to the time the culture specimen is collected. The yield drops off significantly after 1 week, and after 3 weeks the test has a sensitivity of only 1% to 3%.8 Therefore, for our patient, who has had symptoms for 3 weeks already, bacterial culture would not be the best test. In addition, the results are usually not known for 7 to 14 days, which is too slow to be useful in managing acute cases.
For swabbing, a Dacron swab is inserted through the nostril to the posterior pharynx and is left in place for 10 seconds to maximize the yield of the specimen. Recovery rates for B pertussis are low if the throat or the anterior nasal passage is swabbed instead of the posterior pharynx.9
Nasopharyngeal aspiration is a more complicated procedure, requiring a suction device to trap the mucus, but it may provide higher yields than swabbing.10 In this method, the specimen is obtained by inserting a small tube (eg, an infant feeding tube) connected to a mucus trap into the nostril back to the posterior pharynx.
Often, direct inoculation of medium for B pertussis is not possible. In such cases, clinical specimens are placed in Regan Lowe transport medium (half-strength charcoal agar supplemented with horse blood and cephalexin).11,12
Polymerase chain reaction testing: Faster, more sensitive, but less specific
PCR testing of nasopharyngeal specimens is now being used instead of bacterial culture to diagnose pertussis in many situations. Alternatively, nasopharyngeal aspirate (or secretions collected with two Dacron swabs) can be obtained and divided at the time of collection and the specimens sent for both culture and PCR testing. Because bacterial culture is time-consuming and has poor sensitivity, the CDC states that a positive PCR test, along with the clinical symptoms and epidemiologic information, is sufficient for diagnosis.5
PCR testing can detect B pertussis with greater sensitivity and more rapidly than bacterial culture.12–14 Its sensitivity ranges from 61% to 99%, its specificity ranges from 88% to 98%,12,15,16 and its results can be available in 2 to 24 hours.12
PCR testing’s advantage in terms of sensitivity is especially pronounced in the later stages of the disease (as in our patient), when clinical suspicion of pertussis typically arises. It can be used effectively for up to 4 weeks from the onset of cough.14 Our patient, who presented nearly 3 weeks after the onset of symptoms, underwent nasopharyngeal sampling for PCR testing.
However, PCR testing is not as specific for B pertussis as is bacterial culture, since other Bordetella species can cause positive results on PCR testing. Also, as with culture, a negative test does not reliably rule out the disease, especially if the sample is collected late in the course.
Therefore, basing the diagnosis on PCR testing alone without the proper clinical context is not advised: pertussis outbreaks have been mistakenly declared on the basis of false-positive PCR test results. Three so-called “pertussis outbreaks” in three different states from 2004 to 200617 were largely the result of overdiagnosis based on equivocal or false-positive PCR test results without the appropriate clinical circumstances. Retrospective review of these pseudo-outbreaks revealed that few cases actually met the CDC’s diagnostic criteria.17 Many patients were not tested (by any method) for pertussis and were treated as having probable cases of pertussis on the basis of their symptoms. Patients who were tested and who had a positive PCR test did not meet the clinical definition of pertussis according to the Council of State and Territorial Epidemiologists.17
Since PCR testing varies in sensitivity and specificity, obtaining culture confirmation of pertussis for at least one suspicious case is recommended any time an outbreak is suspected. This is necessary for monitoring for continued presence of the agent among cases of disease, recruitment of isolates for epidemiologic studies, and surveillance for antibiotic resistance.
Direct fluorescence antibody testing
The CDC does not recommend direct fluorescence antibody testing to diagnose pertussis. This test is commercially available and is sometimes used to screen patients for B pertussis infection, but it lacks sensitivity and specificity for this organism. Cross-reaction with normal nasopharyngeal flora can lead to a false-positive result.18 In addition, the interpretation of the test is subjective, so the sensitivity and specificity are quite variable: the sensitivity is reported as 52% to 65%, while the specificity can vary from 15% to 99%.
Enzyme-linked immunosorbent assay
ELISA testing has been used in epidemiologic studies to measure serum antibodies to B pertussis. Many serologic tests exist, but none is commercially available. Many of these tests are used by the CDC and state health departments to help confirm the diagnosis, especially during outbreaks. Generally, serologic tests are more useful for diagnosis in later phases of the disease. Currently used ELISA tests use both paired and single serology techniques measuring elevated immunoglobulin G serum antibody concentrations against an array of antigens, including pertussis toxin, filamentous hemagglutinin, pertactin, and fimbrae. As a result, a range of sensitivities (33%–95%) and specificities (72%–100%) has been reported.12,14,19
TREATING PERTUSSIS
Our patient’s PCR test result comes back positive. In view of her symptoms and this result, we decide to treat her empirically for pertussis, even though she has had no known contact with anyone with the disease and there is currently no outbreak of it in the community.
3. According to the most recent evidence, which of the following would be the treatment of choice for pertussis in this patient?
- Azithromycin (Zithromax)
- Amoxicillin (Moxatag)
- Levofloxacin (Levaquin)
- Sulfamethoxazole-trimethoprim (Bactrim)
- Supportive measures (hydration, humidifier, antitussives, antihistamines, decongestants)
Azithromycin and the other macrolide antibiotics erythromycin and clarithromycin are first-line therapies for pertussis in adolescents and adults. If given during the catarrhal phase, they can reduce the duration and severity of symptoms and lessen the period of communicability.20,21 After the catarrhal phase, however, it is uncertain whether antibiotics change the clinical course of pertussis, as the data are conflicting.20–22
Factors to consider when selecting a macrolide antibiotic are tolerability, the potential for adverse events and drug interactions, ease of compliance, and cost. All three macrolides are equally effective against pertussis, but azithromycin and clarithromycin are generally better tolerated and are associated with milder and less frequent side effects than erythromycin, including lower rates of gastrointestinal side effects.
Erythromycin and clarithromycin inhibit the cytochrome P450 enzyme system, specifically CYP3A4, and can interact with a great many commonly prescribed drugs metabolized by this enzyme. Therefore, azithromycin may be a better choice for patients already taking other medications, like our patient.
Azithromycin and clarithromycin have longer half-lives and achieve higher tissue concentrations than erythromycin, allowing for less-frequent dosing (daily for azithromycin and twice daily for clarithromycin) and shorter treatment duration (5 days for azithromycin and 7 days for clarithromycin).
An advantage of erythromycin, though, is its lower cost. The cost of a recommended course of erythromycin treatment for pertussis (ie, 500 mg every 6 hours for 14 days) is roughly $20, compared with $75 for azithromycin.
Amoxicillin is not effective in clearing B pertussis from the nasopharynx and thus is not a reasonable option for the treatment of pertussis.23
Levofloxacin is also not recommended for the treatment of pertussis.
Sulfamethoxazole-trimethoprim is a second-line agent for pertussis. It is effective in eradicating B pertussis from the nasopharynx20 and is generally used as an alternative to the macrolide agents in patients who cannot tolerate or have contraindications to macrolides. Sulfamethoxazole-trimethoprim can also be an option for patients infected with rare macrolide-resistant strains of B pertussis.
Supportive measures by themselves are reasonable for patients with pertussis beyond the catarrhal phase, since antibiotics are typically not effective at that stage of the disease.
From 80% to 90% of patients with untreated pertussis spontaneously clear the bacteria from the nasopharynx within 3 to 4 weeks from the onset of cough symptoms.20 However, supportive measures, including antitussives (both over-the-counter and prescription), tend to have very little effect on the severity or duration of the illness, especially when used past the early stage of the illness.
POSTEXPOSURE CHEMOPROPHYLAXIS FOR CLOSE CONTACTS
Postexposure chemoprophylaxis should be given to close contacts of patients who have pertussis to help prevent secondary cases.22 The CDC defines a close contact as someone who has had face-to-face exposure within 3 feet of a symptomatic patient within 21 days after the onset of symptoms in the patient. Close contacts should be treated with antibiotic regimens similar to those used in confirmed cases of pertussis.
In our patient’s case, the diagnosis of pertussis was reported to the Ohio Department of Health. Shortly afterward, the department contacted the patient and obtained information about her close contacts. These people were then contacted and encouraged to complete a course of antibiotics for postexposure chemoprophylaxis, given the high secondary attack rates.
PERTUSSIS VACCINES
4. Which of the following vaccines could have reduced our patient’s chance of contracting the disease or reduced the severity or time course of the illness?
- DTaP
- Tdap
- Whole-cell pertussis vaccine
- No vaccine would have reduced her risk
It is important to prevent pertussis, given its associated morbidities and its generally poor response to drug therapy. Continued vigilance is imperative to maintain high levels of vaccine coverage, including the timely completion of the pertussis vaccination schedule.
The two vaccines in current use in the United States to produce immunity to pertussis—DTaP and Tdap—also confer immunity to diphtheria and tetanus. DTaP is used for children under 7 years of age, and Tdap is for ages 10 to 64. Thus, our patient should have received a series of DTaP injections as an infant and small child, and a Tdap booster at age 11 or 12 years and every 10 years after that.
The upper case “D,” “T,” and “P” in the abbreviations signifies full-strength doses and the lower case “d,” “t,” and “p” indicate that the doses of those components have been reduced. The “a” in both vaccines stands for “acellular”: ie, the pertussis component does not contain cellular elements.
DTaP for initial pertussis vaccination
The current recommendation for initial pertussis vaccination consists of a primary series of DTaP. DTaP vaccination is recommended for infants at 2 months of age, then again at 4 months of age, and again at 6 months of age. A fourth dose is given between the ages of 15 and 18 months, and a fifth dose is given between the ages of 4 to 6 years. If the fourth dose was given after age 4, then no fifth dose is needed.20
Tdap as a booster
The booster vaccine for adolescents and adults is Tdap. In 2005, two Tdap vaccines were licensed in the United States: Adacel for people ages 11 to 64 years, and Boostrix for people ages 10 to 18 years.
The CDC’s Advisory Committee on Immunization Practices (ACIP) recommends a booster dose of Tdap at age 11 or 12 years. Every 10 years thereafter, a booster of tetanus and diphtheria toxoid (Td) vaccine is recommended, except that one of the Td doses can be replaced by Tdap if the patient hasn’t received Tdap yet.
For adults ages 19 to 64, the ACIP currently recommends routine use of a single booster dose of Tdap to replace a single dose of Td if they received the last dose of toxoid vaccine 10 or more years earlier. If the previous dose of Td was given within the past 10 years, a single dose of Tdap is appropriate to protect patients against pertussis. This is especially true for patients at increased risk of pertussis or its complications, as well as for health care professionals and adults who have close contact with infants, such as new parents, grandparents, and child-care providers. The minimum interval since the last Td vaccination is ideally 2 years, although shorter intervals can be used for control of pertussis outbreaks and for those who have close contact with infants.24
In 2010, the ACIP decided that, for those ages 65 and older, a single dose of Tdap vaccine may be given in place of Td if the patient has not previously received Tdap, regardless of how much time has elapsed since the last vaccination with a Td-containing vaccine.25 Data from the Vaccine Adverse Event Reporting System suggest that Tdap vaccine in this age group is as safe as the Td vaccine.25
Subsequent tetanus vaccine doses, in the form of Td, should be given at 10-year intervals throughout adulthood. Administration of Tdap at 10-year intervals appears to be highly immunogenic and well tolerated,25 suggesting that it is possible that Tdap will become part of routine booster dosing instead of Td, pending further study.
Tdap is not contraindicated in pregnant women. Ideally, women should be vaccinated with Tdap before becoming pregnant if they have not previously received it. If the pregnant woman is not at risk of acquiring or transmitting pertussis during pregnancy, the ACIP recommends deferring Tdap vaccination until the immediate postpartum period.
Adults who require a vaccine containing tetanus toxoid for wound management should receive Tdap instead of Td if they have never received Tdap. Adults who have never received vaccine containing tetanus and diphtheria toxoid should receive a series of three vaccinations. The preferred schedule is a dose of Tdap, followed by a dose of Td more than 4 weeks later, and a second dose of Td 6 to 12 months later, though Tdap can be substituted for Td for any one of the three doses in the series. Adults with a history of pertussis generally should receive Tdap according to routine recommendations.
Tdap is contraindicated in people with a history of serious allergic reaction to any component of the Tdap vaccine or with a history of encephalopathy not attributable to an identifiable cause within 7 days of receiving a pertussis vaccine. Tdap is relatively contraindicated and should be deferred in people with current moderate to severe acute illness, current unstable neurologic condition, or a history of Arthus hypersensitivity reaction to a tetanus-toxoid-containing vaccine within the past 10 years, and in people who have developed Guillain-Barré syndrome, within 6 weeks of receiving a tetanus-toxoid–containing vaccine.
Tdap is generally well tolerated. Adverse effects are typically mild and may include localized pain, redness, and swelling; low-grade fever; headache; fatigue; and, less commonly, gastrointestinal upset, myalgia, arthralgia, rash, and swollen glands.
Whole-cell pertussis vaccine is no longer available in the United States
Whole-cell pertussis vaccine provides good protection against pertussis, with 70% to 90% efficacy after three doses. It is less expensive-than acellular formulations and therefore is used in many parts of the world where cost is an issue. It is no longer available in the United States, however, due to high rates of local reactions such as redness, swelling, and pain at the injection site.
The importance of staying up-to-date with booster shots
Booster vaccination for pertussis in adolescents and adults is critical, since the largest recent outbreaks have occurred in these groups.21 The high rate of outbreaks is presumably the result of waning immunity from childhood immunizations and of high interpersonal contact rates. Vaccination has been shown to reduce the chance of contracting the disease and to reduce the severity and time course of the illness.21
Adolescents and adults are an important reservoir for potentially serious infections in infants who are either unvaccinated or whose vaccination schedule has not been completed. These infants are at risk of severe illness, including pneumonia, seizures, encephalopathy, and apnea, or even death. Adults and teens can also suffer complications from pertussis, although these tend to be less serious, especially in those who have been vaccinated. Complications in teens and adults are often caused by malaise and the cough itself, including weight loss (33%), urinary stress incontinence (28%), syncope (6%), rib fractures from severe coughing (4%), and pneumonia (2%).26 Thus, it is important that adolescents and adults stay up-to-date with pertussis vaccination.
CASE CONTINUED
Our patient was treated with a short (5-day) course of azithromycin 500 mg daily. It did not improve her symptoms very much, but this was not unexpected, given her late presentation and duration of symptoms. Her cough persisted for about 2 months afterwards, but it improved with time and with supportive care at home.
CONTINUED CHALLENGES
Pertussis is a reemerging disease with an increased incidence over the past 30 years, and even more so over the past 10 years. Unfortunately, treatments are not very effective, especially since the disease is often diagnosed late in the course.
We are fortunate to have a vaccine that can prevent pertussis, yet pertussis persists, in large part because of waning immunity from childhood vaccination. The duration of immunity from childhood vaccination is not yet clear. Many adolescents and adults do not follow up on these booster vaccines, thus increasing their susceptibility to pertussis. Consequently, they can transmit the disease to children who are not fully immunized. Prevention by maintaining active immunity is the key to controlling this terrible disease.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
- Centers for Disease Control and Prevention. Pertussis. National Immunization Program, 2005. http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/pert.pdf. Accessed July 6, 2011.
- California Department of Public Health. Pertussis report. www.cdph.ca.gov/programs/immunize/Documents/PertussisReport2011-01-07.pdf. Accessed July 6, 2011.
- Centers for Disease Control and Prevention. Pertussis (whooping cough). www.cdc.gov/pertussis/outbreaks.html. Accessed July 3, 2011.
- Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep 2010; 59:847–861. http://www.cdc.gov/mmwr/PDF/wk/mm5927.pdf. Accessed July 1, 2011.
- Centers for Disease Control and Prevention. Pertussis. Vaccines and preventable diseases: pertussis (whooping cough) vaccination, 2010. http://www.cdc.gov/vaccines/vpd-vac/pertussis/default.htm. Accessed July 6, 2011.
- Hewlett EL, Edwards KM. Clinical practice. Pertussis—not just for kids. N Engl J Med 2005; 352:1215–1222.
- Hewlett E. Bordetella species. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed, Philadelphia, PA: Churchill Livingstone; 2000:2701.
- Viljanen MK, Ruuskanen O, Granberg C, Salmi TT. Serological diagnosis of pertussis: IgM, IgA and IgG antibodies against Bordetella pertussis measured by enzyme-linked immunosorbent assay (ELISA). Scand J Infect Dis 1982; 14:117–122.
- Bejuk D, Begovac J, Bace A, Kuzmanovic-Sterk N, Aleraj B. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995; 14:64–65.
- Hallander HO, Reizenstein E, Renemar B, Rasmuson G, Mardin L, Olin P. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993; 31:50–52.
- Regan J, Lowe F. Enrichment medium for the isolation of Bordetella. J Clin Microbiol 1977; 6:303–309.
- World Health Organization. Laboratory manual for the diagnosis of whooping cough caused by Bordetella pertussis/Bordetella para-pertussis. Department of Immunization, Vaccines and Biologicals. Printed 2004. Revised 2007. www.who.int/vaccines-documents/. Accessed July 6, 2011.
- Meade BD, Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994; 41:51–55.
- Wendelboe AM, Van Rie A. Diagnosis of pertussis: a historical review and recent developments. Expert Rev Mol Diagn 2006; 6:857–864.
- Knorr L, Fox JD, Tilley PA, Ahmed-Bentley J. Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. BMC Infect Dis 2006; 6:62.
- Sotir MJ, Cappozzo DL, Warshauer DM, et al. Evaluation of polymerase chain reaction and culture for diagnosis of pertussis in the control of a county-wide outbreak focused among adolescents and adults. Clin Infect Dis 2007; 44:1216–1219.
- Centers for Disease Control and Prevention (CDC). Outbreaks of respiratory illness mistakenly attributed to pertussis—New Hampshire, Massachusetts, and Tennessee, 2004–2006. MMWR Morb Mortal Wkly Rep 2007; 56:837–842.
- Ewanowich CA, Chui LW, Paranchych MG, Peppler MS, Marusyk RG, Albritton WL. Major outbreak of pertussis in northern Alberta, Canada: analysis of discrepant direct fluorescent-antibody and culture results by using polymerase chain reaction methodology. J Clin Microbiol 1993; 31:1715–1725.
- Müller FM, Hoppe JE, Wirsing von König CH. Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 1997; 35:2435–2443.
- Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep 2005; 54:1–16.
- Wirsing von König CH, Postels-Multani S, Bock HL, Schmitt HJ. Pertussis in adults: frequency of transmission after household exposure. Lancet 1995; 346:1326–1329.
- von König CH. Use of antibiotics in the prevention and treatment of pertussis. Pediatr Infect Dis J 2005; 24(suppl 5):S66–S68.
- Trollfors B. Effect of erythromycin and amoxycillin on Bordetella pertussis in the nasopharynx. Infection 1978; 6:228–230.
- Broder KR, Cortese MM, Iskander JK, et al; Advisory Committee on Immunization Practices (ACIP). Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–34.
- Centers for Disease Control and Prevention. Recommendations and Guidelines. ACIP presentation slides: October 2010 meeting. http://www.cdc.gov/vaccines/recs/acip/slides-oct10.htm. Accessed July 6, 2011.
- Cortese MM, Bisgard KM. Pertussis. In:Wallace RB, Kohatsu N, Last JM, editors. Wallace/Maxcy-Rosenau-Last Public Health & Preventive Medicine. 15th ed. New York, NY: McGraw-Hill Medical, 2008:111–114.
Progressive muscle weakness: More there than meets the eye
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.

Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
Ulcerative colitis and an abnormal cholangiogram
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
- Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
- White blood cell count 6.7 × 109/L (3.5–10.5)
- Platelet count 279 × 109/L (150–450)
- Alkaline phosphatase 387 U/L (45–115)
- Total bilirubin 0.9 mg/dL (0.1–1.0)
- Aspartate aminotransferase (AST) 35 U/L (35–48)
- Alanine aminotransferase (ALT) 30 U/L (7–55).
WHAT IS THE DIAGNOSIS?
1. Based on this information, which of the following is the most likely diagnosis?
- Autoimmune hepatitis
- Primary sclerosing cholangitis
- Primary biliary cirrhosis
- Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
- Characteristic cholangiographic abnormalities of the biliary tree
- Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
- Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
- Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
- Antimitochondrial antibody
- Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
- Crohn disease of the small bowel is the most common form
- Liver disease often precedes the bowel disease
- Treating the underlying bowel disease improves the long-term prognosis for the liver condition
- Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3. Which is the only life-prolonging therapy for primary sclerosing cholangitis?
- Methotrexate (Trexall)
- Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
- UDCA at a high dosage (20–30 mg/kg/day)
- Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4. In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
- Colon cancer
- Cholangiocarcinoma
- Osteoporosis
- Fat-soluble vitamin deficiency
- All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5. In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
- ERCP
- MRCP
- Abdominal ultrasonography
- Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
- Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
- This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
- Cholangiocarcinoma and colon cancer are dreaded complications.
- Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
- Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010; 51:660–678.
- Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol 2008; 14:3338–3349.
- Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med 1995; 332:924–933.
- Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis 2005; 11:62–72.
- Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003; 125:1364–1369.
- Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology 2004; 126:1929–1930.
- Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2006; 4:514–520.
- Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology 2004; 40:39–45.
- Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology 1981; 1:632–640.
- Krawitt EL. Autoimmune hepatitis. N Engl J Med 2006; 354:54–66.
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009; 50:291–308.
- Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol 1988; 7:193–199.
- Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc 1998; 73:285–291.
- Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis 1991; 11:31–39.
- Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology 1998; 27:685–690.
- Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology 1996; 110:432–440.
- Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology 1989; 96:790–794.
- Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995; 22:1404–1408.
- Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut 1997; 41:522–525.
- Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol 2009; 50:158–164.
- Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med 1997; 336:691–695.
- Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology 1995; 108:1199–1203.
- LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology 1988; 95:1036–1042.
- Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001; 121:900–907.
- Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol 2008; 48:792–800.
- Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology 2005; 129:1464–1472.
- Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009; 50:808–814.
- Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med 2001; 134:89–95.
- Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg 1996; 2(suppl 1):99–108..
- Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int 2007; 27:86–94.
- Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl 2006; 12:1813–1824.
- Rosen CB, Nagorney DM, Wiesner RH, Coffey RJ, LaRusso NF. Cholangiocarcinoma complicating primary sclerosing cholangitis. Ann Surg 1991; 213:21–25.
- Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology 2005; 128:1655–1667.
- Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology 2009; 136:2180–2186.
- Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology 1991; 14:257–261.
- Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol 2003; 98:2268–2274.
- Jorgensen RA, Lindor KD, Sartin JS, LaRusso NF, Wiesner RH. Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis. J Clin Gastroenterol 1995; 20:215–219.
- Saik RP, Greenburg AG, Farris JM, Peskin GW. Spectrum of cholangitis. Am J Surg 1975; 130:143–150.
- Bangarulingam SY, Gossard AA, Petersen BT, Ott BJ, Lindor KD. Complications of endoscopic retrograde cholangiopancreatography in primary sclerosing cholangitis. Am J Gastroenterol 2009; 104:855–860.
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
- Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
- White blood cell count 6.7 × 109/L (3.5–10.5)
- Platelet count 279 × 109/L (150–450)
- Alkaline phosphatase 387 U/L (45–115)
- Total bilirubin 0.9 mg/dL (0.1–1.0)
- Aspartate aminotransferase (AST) 35 U/L (35–48)
- Alanine aminotransferase (ALT) 30 U/L (7–55).
WHAT IS THE DIAGNOSIS?
1. Based on this information, which of the following is the most likely diagnosis?
- Autoimmune hepatitis
- Primary sclerosing cholangitis
- Primary biliary cirrhosis
- Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
- Characteristic cholangiographic abnormalities of the biliary tree
- Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
- Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
- Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
- Antimitochondrial antibody
- Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
- Crohn disease of the small bowel is the most common form
- Liver disease often precedes the bowel disease
- Treating the underlying bowel disease improves the long-term prognosis for the liver condition
- Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3. Which is the only life-prolonging therapy for primary sclerosing cholangitis?
- Methotrexate (Trexall)
- Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
- UDCA at a high dosage (20–30 mg/kg/day)
- Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4. In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
- Colon cancer
- Cholangiocarcinoma
- Osteoporosis
- Fat-soluble vitamin deficiency
- All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5. In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
- ERCP
- MRCP
- Abdominal ultrasonography
- Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
- Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
- This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
- Cholangiocarcinoma and colon cancer are dreaded complications.
- Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
- Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
- White blood cell count 6.7 × 109/L (3.5–10.5)
- Platelet count 279 × 109/L (150–450)
- Alkaline phosphatase 387 U/L (45–115)
- Total bilirubin 0.9 mg/dL (0.1–1.0)
- Aspartate aminotransferase (AST) 35 U/L (35–48)
- Alanine aminotransferase (ALT) 30 U/L (7–55).
WHAT IS THE DIAGNOSIS?
1. Based on this information, which of the following is the most likely diagnosis?
- Autoimmune hepatitis
- Primary sclerosing cholangitis
- Primary biliary cirrhosis
- Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
- Characteristic cholangiographic abnormalities of the biliary tree
- Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
- Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
- Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
- Antimitochondrial antibody
- Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
- Crohn disease of the small bowel is the most common form
- Liver disease often precedes the bowel disease
- Treating the underlying bowel disease improves the long-term prognosis for the liver condition
- Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3. Which is the only life-prolonging therapy for primary sclerosing cholangitis?
- Methotrexate (Trexall)
- Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
- UDCA at a high dosage (20–30 mg/kg/day)
- Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4. In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
- Colon cancer
- Cholangiocarcinoma
- Osteoporosis
- Fat-soluble vitamin deficiency
- All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5. In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
- ERCP
- MRCP
- Abdominal ultrasonography
- Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
- Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
- This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
- Cholangiocarcinoma and colon cancer are dreaded complications.
- Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
- Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010; 51:660–678.
- Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol 2008; 14:3338–3349.
- Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med 1995; 332:924–933.
- Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis 2005; 11:62–72.
- Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003; 125:1364–1369.
- Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology 2004; 126:1929–1930.
- Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2006; 4:514–520.
- Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology 2004; 40:39–45.
- Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology 1981; 1:632–640.
- Krawitt EL. Autoimmune hepatitis. N Engl J Med 2006; 354:54–66.
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009; 50:291–308.
- Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol 1988; 7:193–199.
- Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc 1998; 73:285–291.
- Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis 1991; 11:31–39.
- Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology 1998; 27:685–690.
- Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology 1996; 110:432–440.
- Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology 1989; 96:790–794.
- Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995; 22:1404–1408.
- Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut 1997; 41:522–525.
- Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol 2009; 50:158–164.
- Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med 1997; 336:691–695.
- Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology 1995; 108:1199–1203.
- LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology 1988; 95:1036–1042.
- Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001; 121:900–907.
- Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol 2008; 48:792–800.
- Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology 2005; 129:1464–1472.
- Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009; 50:808–814.
- Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med 2001; 134:89–95.
- Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg 1996; 2(suppl 1):99–108..
- Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int 2007; 27:86–94.
- Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl 2006; 12:1813–1824.
- Rosen CB, Nagorney DM, Wiesner RH, Coffey RJ, LaRusso NF. Cholangiocarcinoma complicating primary sclerosing cholangitis. Ann Surg 1991; 213:21–25.
- Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology 2005; 128:1655–1667.
- Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology 2009; 136:2180–2186.
- Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology 1991; 14:257–261.
- Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol 2003; 98:2268–2274.
- Jorgensen RA, Lindor KD, Sartin JS, LaRusso NF, Wiesner RH. Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis. J Clin Gastroenterol 1995; 20:215–219.
- Saik RP, Greenburg AG, Farris JM, Peskin GW. Spectrum of cholangitis. Am J Surg 1975; 130:143–150.
- Bangarulingam SY, Gossard AA, Petersen BT, Ott BJ, Lindor KD. Complications of endoscopic retrograde cholangiopancreatography in primary sclerosing cholangitis. Am J Gastroenterol 2009; 104:855–860.
- Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010; 51:660–678.
- Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol 2008; 14:3338–3349.
- Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med 1995; 332:924–933.
- Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis 2005; 11:62–72.
- Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003; 125:1364–1369.
- Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology 2004; 126:1929–1930.
- Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2006; 4:514–520.
- Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology 2004; 40:39–45.
- Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology 1981; 1:632–640.
- Krawitt EL. Autoimmune hepatitis. N Engl J Med 2006; 354:54–66.
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009; 50:291–308.
- Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol 1988; 7:193–199.
- Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc 1998; 73:285–291.
- Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis 1991; 11:31–39.
- Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology 1998; 27:685–690.
- Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology 1996; 110:432–440.
- Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology 1989; 96:790–794.
- Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995; 22:1404–1408.
- Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut 1997; 41:522–525.
- Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol 2009; 50:158–164.
- Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med 1997; 336:691–695.
- Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology 1995; 108:1199–1203.
- LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology 1988; 95:1036–1042.
- Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001; 121:900–907.
- Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol 2008; 48:792–800.
- Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology 2005; 129:1464–1472.
- Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009; 50:808–814.
- Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med 2001; 134:89–95.
- Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg 1996; 2(suppl 1):99–108..
- Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int 2007; 27:86–94.
- Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl 2006; 12:1813–1824.
- Rosen CB, Nagorney DM, Wiesner RH, Coffey RJ, LaRusso NF. Cholangiocarcinoma complicating primary sclerosing cholangitis. Ann Surg 1991; 213:21–25.
- Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology 2005; 128:1655–1667.
- Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology 2009; 136:2180–2186.
- Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology 1991; 14:257–261.
- Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol 2003; 98:2268–2274.
- Jorgensen RA, Lindor KD, Sartin JS, LaRusso NF, Wiesner RH. Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis. J Clin Gastroenterol 1995; 20:215–219.
- Saik RP, Greenburg AG, Farris JM, Peskin GW. Spectrum of cholangitis. Am J Surg 1975; 130:143–150.
- Bangarulingam SY, Gossard AA, Petersen BT, Ott BJ, Lindor KD. Complications of endoscopic retrograde cholangiopancreatography in primary sclerosing cholangitis. Am J Gastroenterol 2009; 104:855–860.
Nausea, vomiting, and panic attacks in a 50-year-old woman
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid

Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
Recurrent abdominal pain after laparoscopic cholecystectomy
Four months after undergoing laparoscopic cholecystectomy for symptomatic gallstones, an otherwise healthy 26-year-old woman begins to have episodes of epigastric and back pain similar to what she experienced before the surgery. The surgery was without complications, and her classic biliary colic disappeared afterward. Histologic evaluation of the surgical specimen revealed chronic cholecystitis with multiple small, mixed gallstones.
Now she describes a burning pain in her epigastrium and mid to upper back, starting about 30 minutes after a meal and lasting up to 4 hours. Sometimes it awakens her at night. She avoids eating for fear of inducing the pain. She has occasional chills but no fever, nausea, vomiting, jaundice, or changes in urine or stool color.
Three years ago she was diagnosed with a gastric ulcer induced by taking a nonsteroidal anti-inflammatory drug (NSAID). The ulcer was treated with a proton pump inhibitor for 1 month. She says the ulcer pain was dull and aching, different from her current pain.
Upper endoscopy 4 months ago (ie, before her laparoscopic cholecystectomy) showed no evidence of esophagitis or peptic ulcer disease.
Apart from her gallbladder operation, she has had no other surgery. According to the surgeon’s notes, intraoperative cholangiography was not performed, and no macroscopic changes of acute cholecystitis or difficult biliary anatomy were noted.
The patient does not smoke, does not drink alcohol, is not currently taking any medications, including NSAIDs or over-the-counter medications, and has not taken any recently. Her mother also had symptomatic gallstones requiring cholecystectomy.
On physical examination, only fever
On examination, her temperature is 101.2°F (38.4°C), blood pressure 117/80 mm Hg, heart rate 82 beats per minute, and blood oxygen saturation 99% on room air. Her weight is 138 lb (62.6 kg), height 5 feet 6 inches (168 cm).
There is no jaundice or pallor. Her heart and lung examinations are normal.
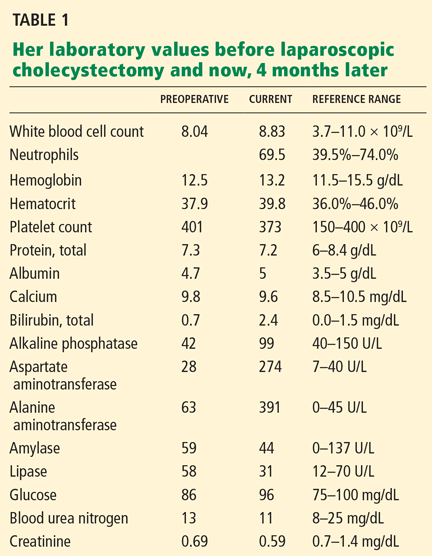
No costovertebral angle or spinal tenderness can be elicited.
Her laboratory values are shown in Table 1.
POSTCHOLECYSTECTOMY SYNDROME
1. After cholecystectomy, preoperative symptoms recur in what percentage of patients?
- 10% to 40%
- 50%
- 60%
- 80%
Postcholecystectomy syndrome—the recurrence of symptoms similar to those before the procedure—occurs in 10% to 40% of patients. The time to the onset of symptoms can range from 2 days to up to 25 years.1–4 Women may be at higher risk, with symptoms recurring in 43% vs 28% in men.5
Postcholecystectomy syndrome can have a biliary or a nonbiliary cause. Biliary causes include strictures, retained calculi, dropped calculi, tumors, sphincter of Oddi dysfunction, and calculi in the cystic duct remnant. Nonbiliary causes include functional and organic disorders such as peptic ulcer disease, gastroesophageal reflux, pancreatic disease, hepatocellular disorders, coronary artery disease, irritable bowel syndrome, and intercostal neuritis.
WHAT IS THE NEXT STEP?
2. Which is the most appropriate next step in the workup of this patient?
- Ultrasonography of the right upper quadrant
- Magnetic resonance cholangiopancreatography (MRCP)
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Observation and reassurance
- Review the operative record and consult with the surgeon
Although the patient is presenting with pain and fever, two features of the classic Charcot triad (pain, fever, jaundice) seen in cholangitis (infection of a bile duct), and although cholangitis almost confirms the diagnosis of common bile duct stones in a patient with gallstones (before or after cholecystectomy), other diagnoses to consider are bile duct injury, bile leak, and biloma.
Biloma can be detected with ultrasonography. Bile duct injuries are identified intraoperatively in up to 25% of patients. For those with an unrecognized injury, the clinical presentation is variable and depends on the type of injury. If a bile leak is present, patients present early, at a median of 3 days postoperatively. However, our patient presented with symptoms 4 months after her surgery. Patients with bile duct strictures without bile leak have a longer symptom-free interval and usually present with signs of biliary obstruction. Ultrasonography can then detect biliary dilatation.6
It would be very helpful to review the operative record and to talk to the surgeon to confirm that intraoperative cholangiography had not been done and to determine the level of difficulty of the surgery. (Intraoperative cholangiography involves the introduction of contrast dye into the biliary system by cannulation of the cystic duct or by direct injection into the common bile duct. An intraoperative cholangiogram is considered normal if the entire intrahepatic and extrahepatic biliary tree is seen to be filled with contrast.) A normal cholangiogram has a negative predictive value of 99.8% for the detection of ductal stones. Thus, a normal intraoperative cholangiogram can prevent unnecessary postoperative ECRP, since it almost always indicates a clean bile duct.7
Ultrasonography of the right upper quadrant has a low sensitivity (< 50%) for detecting common bile duct stones. However, it is highly operator-dependent, and it may be twice as sensitive if done by expert radiologists than by less experienced ones. Its limitations include poor visualization of the distal portion of the duct and low sensitivity in patients in whom the common bile duct is minimally dilated and also in patients with small stones. In most studies, however, it had a very high specificity—ie, greater than 95%.8
MRCP has a sensitivity of 82.6% and a specificity of 97.5% in detecting stones in the common bile duct.9 Therefore, normal results on abdominal ultrasonography and MRCP do not completely rule out stones.
Although this patient has a high pretest probability of having common bile duct stones, ERCP should be done only after a thorough review of the previous operative procedure.
Observation and reassurance are not appropriate in a patient with cholangitis, such as this patient, because waiting increases the risk of septicemia.
The patient undergoes ERCP with stone removal
Review of the operative report and discussion with the surgeon confirm that the laparoscopic procedure was uneventful and that intraoperative cholangiography was not done.
Therefore, the patient undergoes ERCP. The major papilla is normal. Cholangiography reveals nondilated common bile and intrahepatic ducts, with faint filling defects in the mid to distal common bile duct. Endoscopic sphincterotomy is performed, and three small stones are extracted from the common bile duct. Repeat balloon-occlusion cholangiography is normal.
The patient tolerates the procedure well and resumes a normal diet and normal activities.
Her pain persists, prompting an emergency room visit
Five days after her ERCP procedure, however, the same burning epigastric pain returns. As before, the pain occurs after eating and does not occur with fasting. At this time, she has no fever or chills.
WHAT IS CAUSING HER PAIN?
3. Which is the most likely cause of her persistent pain?
- Acute pancreatitis after ERCP
- Peptic ulcer disease
- Sphincter of Oddi dysfunction
- Biliary stones
The most likely cause is persistent biliary stones. The common bile duct was recently explored and stones were removed, but she may still have stones in the intrahepatic ducts or in the cystic duct remnant, both of which were unopacified during the ERCP procedure, indicating that either the test was incomplete or a stone is obstructing the passage of contrast. Her persistent symptoms warrant repeating her liver function tests.
Acute pancreatitis is the most common and feared complication of ERCP, and it should be suspected in any patient who develops abdominal pain within 6 hours of the procedure. It is much less likely to develop after 12 hours, however. Risk factors for post-ERCP pancreatitis include patient factors (young age, female sex, history of recurrent pancreatitis), procedural factors (difficult cannulation, minor papilla sphincterotomy), and, less likely, operator-related factors.10–13 In general, the more likely a patient is to have an abnormal and irregular common bile duct or pancreatic duct, the lower the risk of post-ERCP pancreatitis. The importance of operator-dependent factors is not yet clear.10–13
Despite the postprandial pattern of our patient’s pain and her history of gastric ulcer, peptic ulcer disease is unlikely in view of a normal esophagogastroduodenoscopic examination done 4 months earlier, and since she has no recent exposure to NSAIDs.
Sphincter of Oddi dysfunction may explain her symptoms, but she recently underwent endoscopic sphincterotomy, which is regarded as the most definitive treatment.14
WHAT SHOULD BE DONE NEXT?
4. What would be the best next step in her management?
- Repeat ERCP
- MRCP
- Endoscopic ultrasonography
- Observation and reassurance
MRCP is the most appropriate next step, given her recurrent symptoms. Repeat ERCP is not appropriate, since there is no evidence of cholangitis, and since her liver function tests had completely normalized.
A recent systematic review of endoscopic ultrasonography and MRCP for diagnosing choledocholithiasis found both tests to be highly accurate, with no statistically significant differences in sensitivity or specificity between the two.15 However, MRCP has the advantage of being noninvasive and of being able to show intrahepatic stones.
Park et al,16 in a prospective study of 66 patients with primary intrahepatic stones, concluded that MRCP findings were comparable to those of percutaneous transhepatic cholangioscopy, the reference standard for locating intrahepatic stones. The sensitivity, specificity, and accuracy of MRCP for detecting and locating intrahepatic stones were high (97%, 99%, and 98%, respectively).16 However, after sphincterotomy, pneumobilia may create an appearance that can be mistaken for intraductal stones.
She undergoes MRCP
The patient continues to have pain, and she has lost 5 pounds because she is still avoiding eating. At this point, she is beginning to wonder if her symptoms are psychogenic, since all the test results have been normal.
ERCP, MRCP, ULTRASONOGRAPHY?
5. What would be the best next step?
- Reassurance
- Referral to a psychiatrist
- Referral to a pain management clinic
- Endoscopic ultrasonography
- Repeat ERCP
Endoscopic ultrasonography is needed to look for cystic duct stones. Although several tests have shown normal results, the patient’s pain continues as in the previous episodes, making stone disease the most likely cause.
Although no stones were seen on MRCP and ultrasonography, a detailed evaluation for stones in a cystic duct or retained gallbladder remnant was not done satisfactorily.
Reassurance and referral to a psychiatrist or pain management clinic are not appropriate, since an organic cause of her pain has not been completely ruled out.
Findings on endoscopic ultrasonography
Endoscopic ultrasonography is performed and reveals a large (7-mm) stone in the area of the cystic duct remnant or gallbladder remnant (Figure 3). The common bile duct is normal.
CAUSES OF RETAINED GALLBLADDER AND CYSTIC DUCT REMNANT
6. What may have predisposed this patient to a retained gallbladder or cystic duct remnant after her surgery?
- Laparoscopic cholecystectomy
- Not doing intraoperative cholangiography
- Cholecystectomy for acute cholecystitis
- All of the above
All of the above may have contributed.
Postcholecystectomy syndrome can pose a diagnostic and therapeutic challenge, as in our patient. Although it has been reported since the advent of the operation, it is more common after laparoscopic cholecystectomy than after open surgery. One possible cause is stones in a cystic duct remnant, ie, a stub longer than 1 cm.
During open cholecystectomy, the cystic duct is ligated and cut as close to the common bile duct as possible, leaving only a small remnant. In laparoscopic cholecystectomy, it is divided closer to the gallbladder to avoid iatrogenic injury to the common bile duct, leaving a longer remnant. A long cystic duct remnant can be prevented by accurately locating the junction of the gallbladder and the cystic duct during cholecystectomy and by routinely doing intraoperative cholangiography. The presence of stones in a cystic duct or retained gallbladder remnant is a rare cause of postcholecystectomy syndrome, and suspicion is required to make the diagnosis.17–19
We should note that stones may also lurk in the short cystic duct remnant left after open cholecystectomy. In fact, the first case of cystic duct remnant, the so-called reformed gallbladder containing stones, was described in 1912 by Flörcken.20
Intraoperative cholangiography was introduced in 1931 by Mirizzi,21 who recommended its routine use. Since the advent of laparoscopic cholecystectomy in 1988, the routine use of intraoperative cholangiography has been debated. Advocates point to its ability to detect unsuspected calculi and to delineate the biliary anatomy, thus reducing the risk of biliary duct injury.7,22–25 Those who argue against its routine use emphasize the low reported rates of unsuspected stones in the common bile duct (2% to 3%), a longer operative time, the additional cost, and false-positive results that may lead to unnecessary common bile duct exploration. Another argument against its routine use is that most small ductal stones pass spontaneously without significant sequelae.26–28 Surgeons who use intraoperative cholangiography only selectively use it in patients with unclear biliary anatomy and preoperative biochemical or radiologic evidence of choledocholithiasis.
Case continued: She undergoes repeat ERCP
IF STONES ARE DIFFICULT TO EXTRACT
7. If the cystic duct stone were not amenable to endoscopic extraction, what would be the best alternative?
- Extracorporeal shock-wave lithotripsy (ESWL)
- Endoscopic biliary laser lithotripsy
- Repeat laparoscopic cholecystectomy
- All of the above
All of the above are alternatives.
A symptomatic stone in a cystic duct remnant is uncommon and is mentioned in the literature only in case series and case reports.
ESWL is effective for treating bile duct calculi.29 In a cohort of 239 patients with bile duct stones treated by ESWL, Benninger et al30 concluded that endoscopy plus ESWL was a definitive treatment for all patients except one, who subsequently underwent cholecystectomy. Once fragmented, the stones are extracted endoscopically.
Another fragmentation technique that can be offered to patients with stones in the cystic duct that are difficult to extract is contact fragmentation with a holmium laser placed in a transpapillary position under visual guidance.17
Repeat cholecystectomy with removal of stones in the cystic duct remnant (and removal of retained gallbladder remnants and reduction of the cystic duct remnant) has good postoperative results.17,18,31,32
After incomplete cholecystectomy, the cystic duct remnant and the Calot (cystohepatic) triangle are surrounded by inflamed scar tissue, and this was thought to make laparoscopic reoperation difficult.33 However, with advances in surgical technique and increasing experience of surgeons, repeat cholecystectomy can be done laparoscopically. It has now been suggested that laparoscopic exploration to remove the gallbladder remnants is safe and feasible in such patients.34,35
Discharge and follow-up
The patient is discharged home after the procedure. She is still free of symptoms 31 months later.
LESSONS LEARNED
Remnant cystic duct stones are uncommon
The estimated incidence of a retained calculus within the cystic duct remnant after cholecystectomy is less than 2.5%.2,36 In a series of 322 patients who underwent repeat surgery because of postcholecystectomy syndrome, Rogy et al36 found only 8 who had a stone in the cystic duct or gallbladder remnant, and in a series of 371 patients, Zhou el al2 found 4 who had a stone in the cystic duct remnant.
Stones in the cystic duct remnant are difficult to diagnose
Diagnosing stones in surgical remnants of the cystic duct or gallbladder can be difficult. The sensitivity of abdominal ultrasonography in detecting cystic duct stones is low—only 27% in one study, with a specificity of 100% and an accuracy of 75%.37 Ultrasonography may occasionally suggest cystic duct stones by showing an acoustic shadow in the anatomic region of the cystic duct. However, the results should be interpreted with caution.
Determining the accuracy of ERCP and MRCP in detecting cystic duct remnant stones is also difficult, as few cases have been reported and data may be conflicting. In a review of seven patients confirmed to have retained stones in a surgical remnant, Walsh et al17 found that ERCP correctly diagnosed the retained stone in only four out of six patients; MRCP was done in one patient, and it was read as normal.
In three cases of stones in a postsurgical gallbladder remnant, Hassan and Vilmann38 reported that ERCP and MRCP failed to identify the gallbladder remnant in two out of three cases, likely because the remaining structures are small. The diagnosis was finally made by endoscopic ultrasonography, which the authors concluded was a valuable method to visualize a small gallbladder remnant with stones.
Greater suspicion is needed in patients with typical biliary colic after cholecystectomy
Retained gallbladder remnant is described in the literature as a latent complication. The main problem is not the remnant itself but the chance that it harbors retained stones, which can lead to dilatation and inflammation of the remnant.
The patient can develop symptoms of acute cholecystitis or even acute cholangitis if the stone migrates to the common bile duct. Symptoms can develop as early as 2 weeks or as late as 25 years after laparoscopic cholecystectomy.
Endoscopic ultrasonography may be the best way to look for these remnant stones and to evaluate the bile duct and pancreas. Therefore, it should be part of the diagnostic algorithm in the evaluation of postcholecystectomy pain.
Mixed results with ERCP for extracting cystic duct stones
In case reports of cystic duct calculi after cholecystectomy, ERCP by itself has had mixed results. This traditional means of removing stones may succeed, as in our case. However, the success rate depends largely on anatomic factors such as the position of the stone in the cystic duct, the degree of stone impaction, the diameter of the cystic duct, and the number of valves in the duct.17
Stones in the cystic duct that cannot be extracted with ERCP may benefit from fragmentation techniques in situ via holmium laser followed by endoscopic extraction.
Repeat cholecystectomy is generally advised for any residual gallbladder, and it can be done laparoscopically.
- Lehman GA, Sherman S. Sphincter of Oddi dysfunction (postcholecystectomy syndrome). In:Yamada T, editor. Textbook of Gastroenterology. 2nd ed. Philadelphia: Lippincott; 1995:2251–2262.
- Zhou PH, Liu FL, Yao LQ, Qin XY. Endoscopic diagnosis and treatment of post-cholecystectomy syndrome. Hepatobiliary Pancreat Dis Int 2003; 2:117–120.
- Mergener K, Clavien PA, Branch MS, Baillie J. A stone in a grossly dilated cystic duct stump: a rare cause of postcholecystectomy pain. Am J Gastroenterol 1999; 94:229–231.
- Goenka MK, Kochhar R, Nagi B, Bhasin DK, Chowdhury A, Singh K. Endoscopic retrograde cholangiopancreatography in postcholecystectomy syndrome. J Assoc Physicians India 1996; 44:119–122.
- Bodvall B, Overgaard B. Cystic duct remnant after cholecystectomy: incidence studied by cholegraphy in 500 cases, and significance in 103 reoperations. Ann Surg 1966; 163:382–390.
- Bergman JJ, van den Brink GR, Rauws EA, et al. Treatment of bile duct lesions after laparoscopic cholecystectomy. Gut 1996; 38:141–147.
- Nickkholgh A, Soltaniyekta S, Kalbasi H. Routine versus selective intraoperative cholangiography during laparoscopic cholecystectomy: a survey of 2,130 patients undergoing laparoscopic cholecystectomy. Surg Endosc 2006; 20:868–874.
- Gandolfi L, Torresan F, Solmi L, Puccetti A. The role of ultrasound in biliary and pancreatic diseases. Eur J Ultrasound 2003; 16:141–159.
- Al Samaraee A, Khan U, Almashta Z, Yiannakou Y. Preoperative diagnosis of choledocholithiasis: the role of MRCP. Br J Hosp Med (Lond) 2009; 70:339–343.
- Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: a prospective, multicenter study. Gastrointest Endosc 2001; 54:425–434.
- Cheng CL, Sherman S, Watkins JL, et al. Risk factors for post-ERCP pancreatitis: a prospective multicenter study. Am J Gastroenterol 2006; 101:139–147.
- Mehta SN, Pavone E, Barkun JS, Bouchard S, Barkun AN. Predictors of post-ERCP complications in patients with suspected choledocholithiasis. Endoscopy 1998; 30:457–463.
- Badalov N, Tenner S, Baillie J. The prevention, recognition and treatment of post-ERCP pancreatitis. JOP 2009; 10:88–97.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64:248–254.
- Park DH, Kim MH, Lee SS, et al. Accuracy of magnetic resonance cholangiopancreatography for locating hepatolithiasis and detecting accompanying biliary strictures. Endoscopy 2004; 36:987–992.
- Walsh RM, Ponsky JL, Dumot J. Retained gallbladder/cystic duct remnant calculi as a cause of postcholecystectomy pain. Surg Endosc 2002; 16:981–984.
- Tantia O, Jain M, Khanna S, Sen B. Post cholecystectomy syndrome: role of cystic duct stump and re-intervention by laparoscopic surgery. J Minim Access Surg 2008; 4:71–75.
- Palanivelu C, Rangarajan M, Jategaonkar PA, Madankumar MV, Anand NV. Laparoscopic management of remnant cystic duct calculi: a retrospective study. Ann R Coll Surg Engl 2009; 91:25–29.
- Flörcken H. Gallenblasenregeneration mit Steinrecidiv nach Cholecystectomie. Deutsch Z Chir 1912; 113:604.
- Mirizzi PL. La colangiografía durante las operaciones de las vias biliares. Bol Soc Cirug Buenos Aires 1932; 16:1113.
- Soper NJ, Brunt LM. The case for routine operative cholangiography during laparoscopic cholecystectomy. Surg Clin North Am 1994; 74:953–959.
- Cuschieri A, Shimi S, Banting S, Nathanson LK, Pietrabissa A. Intraoperative cholangiography during laparoscopic cholecystectomy. Routine vs selective policy. Surg Endosc 1994; 8:302–305.
- Woods MS, Traverso LW, Kozarek RA, et al. Biliary tract complications of laparoscopic cholecystectomy are detected more frequently with routine intraoperative cholangiography. Surg Endosc 1995; 9:1076–1080.
- Vezakis A, Davides D, Ammori BJ, Martin IG, Larvin M, McMahon MJ. Intraoperative cholangiography during laparoscopic cholecystectomy. Surg Endosc 2000; 14:1118–1122.
- Ladocsi LT, Benitez LD, Filippone DR, Nance FC. Intraoperative cholangiography in laparoscopic cholecystectomy: a review of 734 consecutive cases. Am Surg 1997; 63:150–156.
- Clair DG, Brooks DC. Laparoscopic cholangiography. The case for a selective approach. Surg Clin North Am 1994; 74:961–966.
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239:28–33.
- Ponsky LE, Geisinger MA, Ponsky JL, Streem SB. Contemporary ‘urologic’ intervention in the pancreaticobiliary tree. Urology 2001; 57:21–25.
- Benninger J, Rabenstein T, Farnbacher M, Keppler J, Hahn EG, Schneider HT. Extracorporeal shockwave lithotripsy of gallstones in cystic duct remnants and Mirizzi syndrome. Gastrointest Endosc 2004; 60:454–459.
- Demetriades H, Pramateftakis MG, Kanellos I, Angelopoulos S, Mantzoros I, Betsis D. Retained gallbladder remnant after laparoscopic cholecystectomy. J Laparoendosc Adv Surg Tech A 2008; 18:276–279.
- Shaw C, O’Hanlon DM, Fenlon HM, McEntee GP. Cystic duct remnant and the ‘post-cholecystectomy syndrome. ’ Hepatogastroenterology 2004; 51:36–38.
- Rozsos I, Magyaródi Z, Orbán P. Cystic duct syndrome and minimally invasive surgery. [Hungarian] Orv Hetil 1997; 138:2397–2401.
- Chowbey PK, Bandyopadhyay SK, Sharma A, Khullar R, Soni V, Baijal M. Laparoscopic reintervention for residual gallstone disease. Surg Laparosc Endosc Percutan Tech 2003; 13:31–35.
- Clemente G, Giuliante F, Cadeddu F, Nuzzo G. Laparoscopic removal of gallbladder remnant and long cystic stump. Endoscopy 2001; 33:814–815.
- Rogy MA, Függer R, Herbst F, Schulz F. Reoperation after cholecystectomy. The role of the cystic duct stump. HPB Surg 1991; 4:129–134.
- Laing FC, Jeffrey RB. Choledocholithiasis and cystic duct obstruction: difficult ultrasonographic diagnosis. Radiology 1983; 146:475–479.
- Hassan H, Vilmann P. Insufficient cholecystectomy diagnosed by endoscopic ultrasonography. Endoscopy 2004; 36:236–238.
Four months after undergoing laparoscopic cholecystectomy for symptomatic gallstones, an otherwise healthy 26-year-old woman begins to have episodes of epigastric and back pain similar to what she experienced before the surgery. The surgery was without complications, and her classic biliary colic disappeared afterward. Histologic evaluation of the surgical specimen revealed chronic cholecystitis with multiple small, mixed gallstones.
Now she describes a burning pain in her epigastrium and mid to upper back, starting about 30 minutes after a meal and lasting up to 4 hours. Sometimes it awakens her at night. She avoids eating for fear of inducing the pain. She has occasional chills but no fever, nausea, vomiting, jaundice, or changes in urine or stool color.
Three years ago she was diagnosed with a gastric ulcer induced by taking a nonsteroidal anti-inflammatory drug (NSAID). The ulcer was treated with a proton pump inhibitor for 1 month. She says the ulcer pain was dull and aching, different from her current pain.
Upper endoscopy 4 months ago (ie, before her laparoscopic cholecystectomy) showed no evidence of esophagitis or peptic ulcer disease.
Apart from her gallbladder operation, she has had no other surgery. According to the surgeon’s notes, intraoperative cholangiography was not performed, and no macroscopic changes of acute cholecystitis or difficult biliary anatomy were noted.
The patient does not smoke, does not drink alcohol, is not currently taking any medications, including NSAIDs or over-the-counter medications, and has not taken any recently. Her mother also had symptomatic gallstones requiring cholecystectomy.
On physical examination, only fever
On examination, her temperature is 101.2°F (38.4°C), blood pressure 117/80 mm Hg, heart rate 82 beats per minute, and blood oxygen saturation 99% on room air. Her weight is 138 lb (62.6 kg), height 5 feet 6 inches (168 cm).
There is no jaundice or pallor. Her heart and lung examinations are normal.
No costovertebral angle or spinal tenderness can be elicited.
Her laboratory values are shown in Table 1.
POSTCHOLECYSTECTOMY SYNDROME
1. After cholecystectomy, preoperative symptoms recur in what percentage of patients?
- 10% to 40%
- 50%
- 60%
- 80%
Postcholecystectomy syndrome—the recurrence of symptoms similar to those before the procedure—occurs in 10% to 40% of patients. The time to the onset of symptoms can range from 2 days to up to 25 years.1–4 Women may be at higher risk, with symptoms recurring in 43% vs 28% in men.5
Postcholecystectomy syndrome can have a biliary or a nonbiliary cause. Biliary causes include strictures, retained calculi, dropped calculi, tumors, sphincter of Oddi dysfunction, and calculi in the cystic duct remnant. Nonbiliary causes include functional and organic disorders such as peptic ulcer disease, gastroesophageal reflux, pancreatic disease, hepatocellular disorders, coronary artery disease, irritable bowel syndrome, and intercostal neuritis.
WHAT IS THE NEXT STEP?
2. Which is the most appropriate next step in the workup of this patient?
- Ultrasonography of the right upper quadrant
- Magnetic resonance cholangiopancreatography (MRCP)
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Observation and reassurance
- Review the operative record and consult with the surgeon
Although the patient is presenting with pain and fever, two features of the classic Charcot triad (pain, fever, jaundice) seen in cholangitis (infection of a bile duct), and although cholangitis almost confirms the diagnosis of common bile duct stones in a patient with gallstones (before or after cholecystectomy), other diagnoses to consider are bile duct injury, bile leak, and biloma.
Biloma can be detected with ultrasonography. Bile duct injuries are identified intraoperatively in up to 25% of patients. For those with an unrecognized injury, the clinical presentation is variable and depends on the type of injury. If a bile leak is present, patients present early, at a median of 3 days postoperatively. However, our patient presented with symptoms 4 months after her surgery. Patients with bile duct strictures without bile leak have a longer symptom-free interval and usually present with signs of biliary obstruction. Ultrasonography can then detect biliary dilatation.6
It would be very helpful to review the operative record and to talk to the surgeon to confirm that intraoperative cholangiography had not been done and to determine the level of difficulty of the surgery. (Intraoperative cholangiography involves the introduction of contrast dye into the biliary system by cannulation of the cystic duct or by direct injection into the common bile duct. An intraoperative cholangiogram is considered normal if the entire intrahepatic and extrahepatic biliary tree is seen to be filled with contrast.) A normal cholangiogram has a negative predictive value of 99.8% for the detection of ductal stones. Thus, a normal intraoperative cholangiogram can prevent unnecessary postoperative ECRP, since it almost always indicates a clean bile duct.7
Ultrasonography of the right upper quadrant has a low sensitivity (< 50%) for detecting common bile duct stones. However, it is highly operator-dependent, and it may be twice as sensitive if done by expert radiologists than by less experienced ones. Its limitations include poor visualization of the distal portion of the duct and low sensitivity in patients in whom the common bile duct is minimally dilated and also in patients with small stones. In most studies, however, it had a very high specificity—ie, greater than 95%.8
MRCP has a sensitivity of 82.6% and a specificity of 97.5% in detecting stones in the common bile duct.9 Therefore, normal results on abdominal ultrasonography and MRCP do not completely rule out stones.
Although this patient has a high pretest probability of having common bile duct stones, ERCP should be done only after a thorough review of the previous operative procedure.
Observation and reassurance are not appropriate in a patient with cholangitis, such as this patient, because waiting increases the risk of septicemia.
The patient undergoes ERCP with stone removal
Review of the operative report and discussion with the surgeon confirm that the laparoscopic procedure was uneventful and that intraoperative cholangiography was not done.
Therefore, the patient undergoes ERCP. The major papilla is normal. Cholangiography reveals nondilated common bile and intrahepatic ducts, with faint filling defects in the mid to distal common bile duct. Endoscopic sphincterotomy is performed, and three small stones are extracted from the common bile duct. Repeat balloon-occlusion cholangiography is normal.
The patient tolerates the procedure well and resumes a normal diet and normal activities.
Her pain persists, prompting an emergency room visit
Five days after her ERCP procedure, however, the same burning epigastric pain returns. As before, the pain occurs after eating and does not occur with fasting. At this time, she has no fever or chills.
WHAT IS CAUSING HER PAIN?
3. Which is the most likely cause of her persistent pain?
- Acute pancreatitis after ERCP
- Peptic ulcer disease
- Sphincter of Oddi dysfunction
- Biliary stones
The most likely cause is persistent biliary stones. The common bile duct was recently explored and stones were removed, but she may still have stones in the intrahepatic ducts or in the cystic duct remnant, both of which were unopacified during the ERCP procedure, indicating that either the test was incomplete or a stone is obstructing the passage of contrast. Her persistent symptoms warrant repeating her liver function tests.
Acute pancreatitis is the most common and feared complication of ERCP, and it should be suspected in any patient who develops abdominal pain within 6 hours of the procedure. It is much less likely to develop after 12 hours, however. Risk factors for post-ERCP pancreatitis include patient factors (young age, female sex, history of recurrent pancreatitis), procedural factors (difficult cannulation, minor papilla sphincterotomy), and, less likely, operator-related factors.10–13 In general, the more likely a patient is to have an abnormal and irregular common bile duct or pancreatic duct, the lower the risk of post-ERCP pancreatitis. The importance of operator-dependent factors is not yet clear.10–13
Despite the postprandial pattern of our patient’s pain and her history of gastric ulcer, peptic ulcer disease is unlikely in view of a normal esophagogastroduodenoscopic examination done 4 months earlier, and since she has no recent exposure to NSAIDs.
Sphincter of Oddi dysfunction may explain her symptoms, but she recently underwent endoscopic sphincterotomy, which is regarded as the most definitive treatment.14
WHAT SHOULD BE DONE NEXT?
4. What would be the best next step in her management?
- Repeat ERCP
- MRCP
- Endoscopic ultrasonography
- Observation and reassurance
MRCP is the most appropriate next step, given her recurrent symptoms. Repeat ERCP is not appropriate, since there is no evidence of cholangitis, and since her liver function tests had completely normalized.
A recent systematic review of endoscopic ultrasonography and MRCP for diagnosing choledocholithiasis found both tests to be highly accurate, with no statistically significant differences in sensitivity or specificity between the two.15 However, MRCP has the advantage of being noninvasive and of being able to show intrahepatic stones.
Park et al,16 in a prospective study of 66 patients with primary intrahepatic stones, concluded that MRCP findings were comparable to those of percutaneous transhepatic cholangioscopy, the reference standard for locating intrahepatic stones. The sensitivity, specificity, and accuracy of MRCP for detecting and locating intrahepatic stones were high (97%, 99%, and 98%, respectively).16 However, after sphincterotomy, pneumobilia may create an appearance that can be mistaken for intraductal stones.
She undergoes MRCP
The patient continues to have pain, and she has lost 5 pounds because she is still avoiding eating. At this point, she is beginning to wonder if her symptoms are psychogenic, since all the test results have been normal.
ERCP, MRCP, ULTRASONOGRAPHY?
5. What would be the best next step?
- Reassurance
- Referral to a psychiatrist
- Referral to a pain management clinic
- Endoscopic ultrasonography
- Repeat ERCP
Endoscopic ultrasonography is needed to look for cystic duct stones. Although several tests have shown normal results, the patient’s pain continues as in the previous episodes, making stone disease the most likely cause.
Although no stones were seen on MRCP and ultrasonography, a detailed evaluation for stones in a cystic duct or retained gallbladder remnant was not done satisfactorily.
Reassurance and referral to a psychiatrist or pain management clinic are not appropriate, since an organic cause of her pain has not been completely ruled out.
Findings on endoscopic ultrasonography
Endoscopic ultrasonography is performed and reveals a large (7-mm) stone in the area of the cystic duct remnant or gallbladder remnant (Figure 3). The common bile duct is normal.
CAUSES OF RETAINED GALLBLADDER AND CYSTIC DUCT REMNANT
6. What may have predisposed this patient to a retained gallbladder or cystic duct remnant after her surgery?
- Laparoscopic cholecystectomy
- Not doing intraoperative cholangiography
- Cholecystectomy for acute cholecystitis
- All of the above
All of the above may have contributed.
Postcholecystectomy syndrome can pose a diagnostic and therapeutic challenge, as in our patient. Although it has been reported since the advent of the operation, it is more common after laparoscopic cholecystectomy than after open surgery. One possible cause is stones in a cystic duct remnant, ie, a stub longer than 1 cm.
During open cholecystectomy, the cystic duct is ligated and cut as close to the common bile duct as possible, leaving only a small remnant. In laparoscopic cholecystectomy, it is divided closer to the gallbladder to avoid iatrogenic injury to the common bile duct, leaving a longer remnant. A long cystic duct remnant can be prevented by accurately locating the junction of the gallbladder and the cystic duct during cholecystectomy and by routinely doing intraoperative cholangiography. The presence of stones in a cystic duct or retained gallbladder remnant is a rare cause of postcholecystectomy syndrome, and suspicion is required to make the diagnosis.17–19
We should note that stones may also lurk in the short cystic duct remnant left after open cholecystectomy. In fact, the first case of cystic duct remnant, the so-called reformed gallbladder containing stones, was described in 1912 by Flörcken.20
Intraoperative cholangiography was introduced in 1931 by Mirizzi,21 who recommended its routine use. Since the advent of laparoscopic cholecystectomy in 1988, the routine use of intraoperative cholangiography has been debated. Advocates point to its ability to detect unsuspected calculi and to delineate the biliary anatomy, thus reducing the risk of biliary duct injury.7,22–25 Those who argue against its routine use emphasize the low reported rates of unsuspected stones in the common bile duct (2% to 3%), a longer operative time, the additional cost, and false-positive results that may lead to unnecessary common bile duct exploration. Another argument against its routine use is that most small ductal stones pass spontaneously without significant sequelae.26–28 Surgeons who use intraoperative cholangiography only selectively use it in patients with unclear biliary anatomy and preoperative biochemical or radiologic evidence of choledocholithiasis.
Case continued: She undergoes repeat ERCP
IF STONES ARE DIFFICULT TO EXTRACT
7. If the cystic duct stone were not amenable to endoscopic extraction, what would be the best alternative?
- Extracorporeal shock-wave lithotripsy (ESWL)
- Endoscopic biliary laser lithotripsy
- Repeat laparoscopic cholecystectomy
- All of the above
All of the above are alternatives.
A symptomatic stone in a cystic duct remnant is uncommon and is mentioned in the literature only in case series and case reports.
ESWL is effective for treating bile duct calculi.29 In a cohort of 239 patients with bile duct stones treated by ESWL, Benninger et al30 concluded that endoscopy plus ESWL was a definitive treatment for all patients except one, who subsequently underwent cholecystectomy. Once fragmented, the stones are extracted endoscopically.
Another fragmentation technique that can be offered to patients with stones in the cystic duct that are difficult to extract is contact fragmentation with a holmium laser placed in a transpapillary position under visual guidance.17
Repeat cholecystectomy with removal of stones in the cystic duct remnant (and removal of retained gallbladder remnants and reduction of the cystic duct remnant) has good postoperative results.17,18,31,32
After incomplete cholecystectomy, the cystic duct remnant and the Calot (cystohepatic) triangle are surrounded by inflamed scar tissue, and this was thought to make laparoscopic reoperation difficult.33 However, with advances in surgical technique and increasing experience of surgeons, repeat cholecystectomy can be done laparoscopically. It has now been suggested that laparoscopic exploration to remove the gallbladder remnants is safe and feasible in such patients.34,35
Discharge and follow-up
The patient is discharged home after the procedure. She is still free of symptoms 31 months later.
LESSONS LEARNED
Remnant cystic duct stones are uncommon
The estimated incidence of a retained calculus within the cystic duct remnant after cholecystectomy is less than 2.5%.2,36 In a series of 322 patients who underwent repeat surgery because of postcholecystectomy syndrome, Rogy et al36 found only 8 who had a stone in the cystic duct or gallbladder remnant, and in a series of 371 patients, Zhou el al2 found 4 who had a stone in the cystic duct remnant.
Stones in the cystic duct remnant are difficult to diagnose
Diagnosing stones in surgical remnants of the cystic duct or gallbladder can be difficult. The sensitivity of abdominal ultrasonography in detecting cystic duct stones is low—only 27% in one study, with a specificity of 100% and an accuracy of 75%.37 Ultrasonography may occasionally suggest cystic duct stones by showing an acoustic shadow in the anatomic region of the cystic duct. However, the results should be interpreted with caution.
Determining the accuracy of ERCP and MRCP in detecting cystic duct remnant stones is also difficult, as few cases have been reported and data may be conflicting. In a review of seven patients confirmed to have retained stones in a surgical remnant, Walsh et al17 found that ERCP correctly diagnosed the retained stone in only four out of six patients; MRCP was done in one patient, and it was read as normal.
In three cases of stones in a postsurgical gallbladder remnant, Hassan and Vilmann38 reported that ERCP and MRCP failed to identify the gallbladder remnant in two out of three cases, likely because the remaining structures are small. The diagnosis was finally made by endoscopic ultrasonography, which the authors concluded was a valuable method to visualize a small gallbladder remnant with stones.
Greater suspicion is needed in patients with typical biliary colic after cholecystectomy
Retained gallbladder remnant is described in the literature as a latent complication. The main problem is not the remnant itself but the chance that it harbors retained stones, which can lead to dilatation and inflammation of the remnant.
The patient can develop symptoms of acute cholecystitis or even acute cholangitis if the stone migrates to the common bile duct. Symptoms can develop as early as 2 weeks or as late as 25 years after laparoscopic cholecystectomy.
Endoscopic ultrasonography may be the best way to look for these remnant stones and to evaluate the bile duct and pancreas. Therefore, it should be part of the diagnostic algorithm in the evaluation of postcholecystectomy pain.
Mixed results with ERCP for extracting cystic duct stones
In case reports of cystic duct calculi after cholecystectomy, ERCP by itself has had mixed results. This traditional means of removing stones may succeed, as in our case. However, the success rate depends largely on anatomic factors such as the position of the stone in the cystic duct, the degree of stone impaction, the diameter of the cystic duct, and the number of valves in the duct.17
Stones in the cystic duct that cannot be extracted with ERCP may benefit from fragmentation techniques in situ via holmium laser followed by endoscopic extraction.
Repeat cholecystectomy is generally advised for any residual gallbladder, and it can be done laparoscopically.
Four months after undergoing laparoscopic cholecystectomy for symptomatic gallstones, an otherwise healthy 26-year-old woman begins to have episodes of epigastric and back pain similar to what she experienced before the surgery. The surgery was without complications, and her classic biliary colic disappeared afterward. Histologic evaluation of the surgical specimen revealed chronic cholecystitis with multiple small, mixed gallstones.
Now she describes a burning pain in her epigastrium and mid to upper back, starting about 30 minutes after a meal and lasting up to 4 hours. Sometimes it awakens her at night. She avoids eating for fear of inducing the pain. She has occasional chills but no fever, nausea, vomiting, jaundice, or changes in urine or stool color.
Three years ago she was diagnosed with a gastric ulcer induced by taking a nonsteroidal anti-inflammatory drug (NSAID). The ulcer was treated with a proton pump inhibitor for 1 month. She says the ulcer pain was dull and aching, different from her current pain.
Upper endoscopy 4 months ago (ie, before her laparoscopic cholecystectomy) showed no evidence of esophagitis or peptic ulcer disease.
Apart from her gallbladder operation, she has had no other surgery. According to the surgeon’s notes, intraoperative cholangiography was not performed, and no macroscopic changes of acute cholecystitis or difficult biliary anatomy were noted.
The patient does not smoke, does not drink alcohol, is not currently taking any medications, including NSAIDs or over-the-counter medications, and has not taken any recently. Her mother also had symptomatic gallstones requiring cholecystectomy.
On physical examination, only fever
On examination, her temperature is 101.2°F (38.4°C), blood pressure 117/80 mm Hg, heart rate 82 beats per minute, and blood oxygen saturation 99% on room air. Her weight is 138 lb (62.6 kg), height 5 feet 6 inches (168 cm).
There is no jaundice or pallor. Her heart and lung examinations are normal.
No costovertebral angle or spinal tenderness can be elicited.
Her laboratory values are shown in Table 1.
POSTCHOLECYSTECTOMY SYNDROME
1. After cholecystectomy, preoperative symptoms recur in what percentage of patients?
- 10% to 40%
- 50%
- 60%
- 80%
Postcholecystectomy syndrome—the recurrence of symptoms similar to those before the procedure—occurs in 10% to 40% of patients. The time to the onset of symptoms can range from 2 days to up to 25 years.1–4 Women may be at higher risk, with symptoms recurring in 43% vs 28% in men.5
Postcholecystectomy syndrome can have a biliary or a nonbiliary cause. Biliary causes include strictures, retained calculi, dropped calculi, tumors, sphincter of Oddi dysfunction, and calculi in the cystic duct remnant. Nonbiliary causes include functional and organic disorders such as peptic ulcer disease, gastroesophageal reflux, pancreatic disease, hepatocellular disorders, coronary artery disease, irritable bowel syndrome, and intercostal neuritis.
WHAT IS THE NEXT STEP?
2. Which is the most appropriate next step in the workup of this patient?
- Ultrasonography of the right upper quadrant
- Magnetic resonance cholangiopancreatography (MRCP)
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Observation and reassurance
- Review the operative record and consult with the surgeon
Although the patient is presenting with pain and fever, two features of the classic Charcot triad (pain, fever, jaundice) seen in cholangitis (infection of a bile duct), and although cholangitis almost confirms the diagnosis of common bile duct stones in a patient with gallstones (before or after cholecystectomy), other diagnoses to consider are bile duct injury, bile leak, and biloma.
Biloma can be detected with ultrasonography. Bile duct injuries are identified intraoperatively in up to 25% of patients. For those with an unrecognized injury, the clinical presentation is variable and depends on the type of injury. If a bile leak is present, patients present early, at a median of 3 days postoperatively. However, our patient presented with symptoms 4 months after her surgery. Patients with bile duct strictures without bile leak have a longer symptom-free interval and usually present with signs of biliary obstruction. Ultrasonography can then detect biliary dilatation.6
It would be very helpful to review the operative record and to talk to the surgeon to confirm that intraoperative cholangiography had not been done and to determine the level of difficulty of the surgery. (Intraoperative cholangiography involves the introduction of contrast dye into the biliary system by cannulation of the cystic duct or by direct injection into the common bile duct. An intraoperative cholangiogram is considered normal if the entire intrahepatic and extrahepatic biliary tree is seen to be filled with contrast.) A normal cholangiogram has a negative predictive value of 99.8% for the detection of ductal stones. Thus, a normal intraoperative cholangiogram can prevent unnecessary postoperative ECRP, since it almost always indicates a clean bile duct.7
Ultrasonography of the right upper quadrant has a low sensitivity (< 50%) for detecting common bile duct stones. However, it is highly operator-dependent, and it may be twice as sensitive if done by expert radiologists than by less experienced ones. Its limitations include poor visualization of the distal portion of the duct and low sensitivity in patients in whom the common bile duct is minimally dilated and also in patients with small stones. In most studies, however, it had a very high specificity—ie, greater than 95%.8
MRCP has a sensitivity of 82.6% and a specificity of 97.5% in detecting stones in the common bile duct.9 Therefore, normal results on abdominal ultrasonography and MRCP do not completely rule out stones.
Although this patient has a high pretest probability of having common bile duct stones, ERCP should be done only after a thorough review of the previous operative procedure.
Observation and reassurance are not appropriate in a patient with cholangitis, such as this patient, because waiting increases the risk of septicemia.
The patient undergoes ERCP with stone removal
Review of the operative report and discussion with the surgeon confirm that the laparoscopic procedure was uneventful and that intraoperative cholangiography was not done.
Therefore, the patient undergoes ERCP. The major papilla is normal. Cholangiography reveals nondilated common bile and intrahepatic ducts, with faint filling defects in the mid to distal common bile duct. Endoscopic sphincterotomy is performed, and three small stones are extracted from the common bile duct. Repeat balloon-occlusion cholangiography is normal.
The patient tolerates the procedure well and resumes a normal diet and normal activities.
Her pain persists, prompting an emergency room visit
Five days after her ERCP procedure, however, the same burning epigastric pain returns. As before, the pain occurs after eating and does not occur with fasting. At this time, she has no fever or chills.
WHAT IS CAUSING HER PAIN?
3. Which is the most likely cause of her persistent pain?
- Acute pancreatitis after ERCP
- Peptic ulcer disease
- Sphincter of Oddi dysfunction
- Biliary stones
The most likely cause is persistent biliary stones. The common bile duct was recently explored and stones were removed, but she may still have stones in the intrahepatic ducts or in the cystic duct remnant, both of which were unopacified during the ERCP procedure, indicating that either the test was incomplete or a stone is obstructing the passage of contrast. Her persistent symptoms warrant repeating her liver function tests.
Acute pancreatitis is the most common and feared complication of ERCP, and it should be suspected in any patient who develops abdominal pain within 6 hours of the procedure. It is much less likely to develop after 12 hours, however. Risk factors for post-ERCP pancreatitis include patient factors (young age, female sex, history of recurrent pancreatitis), procedural factors (difficult cannulation, minor papilla sphincterotomy), and, less likely, operator-related factors.10–13 In general, the more likely a patient is to have an abnormal and irregular common bile duct or pancreatic duct, the lower the risk of post-ERCP pancreatitis. The importance of operator-dependent factors is not yet clear.10–13
Despite the postprandial pattern of our patient’s pain and her history of gastric ulcer, peptic ulcer disease is unlikely in view of a normal esophagogastroduodenoscopic examination done 4 months earlier, and since she has no recent exposure to NSAIDs.
Sphincter of Oddi dysfunction may explain her symptoms, but she recently underwent endoscopic sphincterotomy, which is regarded as the most definitive treatment.14
WHAT SHOULD BE DONE NEXT?
4. What would be the best next step in her management?
- Repeat ERCP
- MRCP
- Endoscopic ultrasonography
- Observation and reassurance
MRCP is the most appropriate next step, given her recurrent symptoms. Repeat ERCP is not appropriate, since there is no evidence of cholangitis, and since her liver function tests had completely normalized.
A recent systematic review of endoscopic ultrasonography and MRCP for diagnosing choledocholithiasis found both tests to be highly accurate, with no statistically significant differences in sensitivity or specificity between the two.15 However, MRCP has the advantage of being noninvasive and of being able to show intrahepatic stones.
Park et al,16 in a prospective study of 66 patients with primary intrahepatic stones, concluded that MRCP findings were comparable to those of percutaneous transhepatic cholangioscopy, the reference standard for locating intrahepatic stones. The sensitivity, specificity, and accuracy of MRCP for detecting and locating intrahepatic stones were high (97%, 99%, and 98%, respectively).16 However, after sphincterotomy, pneumobilia may create an appearance that can be mistaken for intraductal stones.
She undergoes MRCP
The patient continues to have pain, and she has lost 5 pounds because she is still avoiding eating. At this point, she is beginning to wonder if her symptoms are psychogenic, since all the test results have been normal.
ERCP, MRCP, ULTRASONOGRAPHY?
5. What would be the best next step?
- Reassurance
- Referral to a psychiatrist
- Referral to a pain management clinic
- Endoscopic ultrasonography
- Repeat ERCP
Endoscopic ultrasonography is needed to look for cystic duct stones. Although several tests have shown normal results, the patient’s pain continues as in the previous episodes, making stone disease the most likely cause.
Although no stones were seen on MRCP and ultrasonography, a detailed evaluation for stones in a cystic duct or retained gallbladder remnant was not done satisfactorily.
Reassurance and referral to a psychiatrist or pain management clinic are not appropriate, since an organic cause of her pain has not been completely ruled out.
Findings on endoscopic ultrasonography
Endoscopic ultrasonography is performed and reveals a large (7-mm) stone in the area of the cystic duct remnant or gallbladder remnant (Figure 3). The common bile duct is normal.
CAUSES OF RETAINED GALLBLADDER AND CYSTIC DUCT REMNANT
6. What may have predisposed this patient to a retained gallbladder or cystic duct remnant after her surgery?
- Laparoscopic cholecystectomy
- Not doing intraoperative cholangiography
- Cholecystectomy for acute cholecystitis
- All of the above
All of the above may have contributed.
Postcholecystectomy syndrome can pose a diagnostic and therapeutic challenge, as in our patient. Although it has been reported since the advent of the operation, it is more common after laparoscopic cholecystectomy than after open surgery. One possible cause is stones in a cystic duct remnant, ie, a stub longer than 1 cm.
During open cholecystectomy, the cystic duct is ligated and cut as close to the common bile duct as possible, leaving only a small remnant. In laparoscopic cholecystectomy, it is divided closer to the gallbladder to avoid iatrogenic injury to the common bile duct, leaving a longer remnant. A long cystic duct remnant can be prevented by accurately locating the junction of the gallbladder and the cystic duct during cholecystectomy and by routinely doing intraoperative cholangiography. The presence of stones in a cystic duct or retained gallbladder remnant is a rare cause of postcholecystectomy syndrome, and suspicion is required to make the diagnosis.17–19
We should note that stones may also lurk in the short cystic duct remnant left after open cholecystectomy. In fact, the first case of cystic duct remnant, the so-called reformed gallbladder containing stones, was described in 1912 by Flörcken.20
Intraoperative cholangiography was introduced in 1931 by Mirizzi,21 who recommended its routine use. Since the advent of laparoscopic cholecystectomy in 1988, the routine use of intraoperative cholangiography has been debated. Advocates point to its ability to detect unsuspected calculi and to delineate the biliary anatomy, thus reducing the risk of biliary duct injury.7,22–25 Those who argue against its routine use emphasize the low reported rates of unsuspected stones in the common bile duct (2% to 3%), a longer operative time, the additional cost, and false-positive results that may lead to unnecessary common bile duct exploration. Another argument against its routine use is that most small ductal stones pass spontaneously without significant sequelae.26–28 Surgeons who use intraoperative cholangiography only selectively use it in patients with unclear biliary anatomy and preoperative biochemical or radiologic evidence of choledocholithiasis.
Case continued: She undergoes repeat ERCP
IF STONES ARE DIFFICULT TO EXTRACT
7. If the cystic duct stone were not amenable to endoscopic extraction, what would be the best alternative?
- Extracorporeal shock-wave lithotripsy (ESWL)
- Endoscopic biliary laser lithotripsy
- Repeat laparoscopic cholecystectomy
- All of the above
All of the above are alternatives.
A symptomatic stone in a cystic duct remnant is uncommon and is mentioned in the literature only in case series and case reports.
ESWL is effective for treating bile duct calculi.29 In a cohort of 239 patients with bile duct stones treated by ESWL, Benninger et al30 concluded that endoscopy plus ESWL was a definitive treatment for all patients except one, who subsequently underwent cholecystectomy. Once fragmented, the stones are extracted endoscopically.
Another fragmentation technique that can be offered to patients with stones in the cystic duct that are difficult to extract is contact fragmentation with a holmium laser placed in a transpapillary position under visual guidance.17
Repeat cholecystectomy with removal of stones in the cystic duct remnant (and removal of retained gallbladder remnants and reduction of the cystic duct remnant) has good postoperative results.17,18,31,32
After incomplete cholecystectomy, the cystic duct remnant and the Calot (cystohepatic) triangle are surrounded by inflamed scar tissue, and this was thought to make laparoscopic reoperation difficult.33 However, with advances in surgical technique and increasing experience of surgeons, repeat cholecystectomy can be done laparoscopically. It has now been suggested that laparoscopic exploration to remove the gallbladder remnants is safe and feasible in such patients.34,35
Discharge and follow-up
The patient is discharged home after the procedure. She is still free of symptoms 31 months later.
LESSONS LEARNED
Remnant cystic duct stones are uncommon
The estimated incidence of a retained calculus within the cystic duct remnant after cholecystectomy is less than 2.5%.2,36 In a series of 322 patients who underwent repeat surgery because of postcholecystectomy syndrome, Rogy et al36 found only 8 who had a stone in the cystic duct or gallbladder remnant, and in a series of 371 patients, Zhou el al2 found 4 who had a stone in the cystic duct remnant.
Stones in the cystic duct remnant are difficult to diagnose
Diagnosing stones in surgical remnants of the cystic duct or gallbladder can be difficult. The sensitivity of abdominal ultrasonography in detecting cystic duct stones is low—only 27% in one study, with a specificity of 100% and an accuracy of 75%.37 Ultrasonography may occasionally suggest cystic duct stones by showing an acoustic shadow in the anatomic region of the cystic duct. However, the results should be interpreted with caution.
Determining the accuracy of ERCP and MRCP in detecting cystic duct remnant stones is also difficult, as few cases have been reported and data may be conflicting. In a review of seven patients confirmed to have retained stones in a surgical remnant, Walsh et al17 found that ERCP correctly diagnosed the retained stone in only four out of six patients; MRCP was done in one patient, and it was read as normal.
In three cases of stones in a postsurgical gallbladder remnant, Hassan and Vilmann38 reported that ERCP and MRCP failed to identify the gallbladder remnant in two out of three cases, likely because the remaining structures are small. The diagnosis was finally made by endoscopic ultrasonography, which the authors concluded was a valuable method to visualize a small gallbladder remnant with stones.
Greater suspicion is needed in patients with typical biliary colic after cholecystectomy
Retained gallbladder remnant is described in the literature as a latent complication. The main problem is not the remnant itself but the chance that it harbors retained stones, which can lead to dilatation and inflammation of the remnant.
The patient can develop symptoms of acute cholecystitis or even acute cholangitis if the stone migrates to the common bile duct. Symptoms can develop as early as 2 weeks or as late as 25 years after laparoscopic cholecystectomy.
Endoscopic ultrasonography may be the best way to look for these remnant stones and to evaluate the bile duct and pancreas. Therefore, it should be part of the diagnostic algorithm in the evaluation of postcholecystectomy pain.
Mixed results with ERCP for extracting cystic duct stones
In case reports of cystic duct calculi after cholecystectomy, ERCP by itself has had mixed results. This traditional means of removing stones may succeed, as in our case. However, the success rate depends largely on anatomic factors such as the position of the stone in the cystic duct, the degree of stone impaction, the diameter of the cystic duct, and the number of valves in the duct.17
Stones in the cystic duct that cannot be extracted with ERCP may benefit from fragmentation techniques in situ via holmium laser followed by endoscopic extraction.
Repeat cholecystectomy is generally advised for any residual gallbladder, and it can be done laparoscopically.
- Lehman GA, Sherman S. Sphincter of Oddi dysfunction (postcholecystectomy syndrome). In:Yamada T, editor. Textbook of Gastroenterology. 2nd ed. Philadelphia: Lippincott; 1995:2251–2262.
- Zhou PH, Liu FL, Yao LQ, Qin XY. Endoscopic diagnosis and treatment of post-cholecystectomy syndrome. Hepatobiliary Pancreat Dis Int 2003; 2:117–120.
- Mergener K, Clavien PA, Branch MS, Baillie J. A stone in a grossly dilated cystic duct stump: a rare cause of postcholecystectomy pain. Am J Gastroenterol 1999; 94:229–231.
- Goenka MK, Kochhar R, Nagi B, Bhasin DK, Chowdhury A, Singh K. Endoscopic retrograde cholangiopancreatography in postcholecystectomy syndrome. J Assoc Physicians India 1996; 44:119–122.
- Bodvall B, Overgaard B. Cystic duct remnant after cholecystectomy: incidence studied by cholegraphy in 500 cases, and significance in 103 reoperations. Ann Surg 1966; 163:382–390.
- Bergman JJ, van den Brink GR, Rauws EA, et al. Treatment of bile duct lesions after laparoscopic cholecystectomy. Gut 1996; 38:141–147.
- Nickkholgh A, Soltaniyekta S, Kalbasi H. Routine versus selective intraoperative cholangiography during laparoscopic cholecystectomy: a survey of 2,130 patients undergoing laparoscopic cholecystectomy. Surg Endosc 2006; 20:868–874.
- Gandolfi L, Torresan F, Solmi L, Puccetti A. The role of ultrasound in biliary and pancreatic diseases. Eur J Ultrasound 2003; 16:141–159.
- Al Samaraee A, Khan U, Almashta Z, Yiannakou Y. Preoperative diagnosis of choledocholithiasis: the role of MRCP. Br J Hosp Med (Lond) 2009; 70:339–343.
- Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: a prospective, multicenter study. Gastrointest Endosc 2001; 54:425–434.
- Cheng CL, Sherman S, Watkins JL, et al. Risk factors for post-ERCP pancreatitis: a prospective multicenter study. Am J Gastroenterol 2006; 101:139–147.
- Mehta SN, Pavone E, Barkun JS, Bouchard S, Barkun AN. Predictors of post-ERCP complications in patients with suspected choledocholithiasis. Endoscopy 1998; 30:457–463.
- Badalov N, Tenner S, Baillie J. The prevention, recognition and treatment of post-ERCP pancreatitis. JOP 2009; 10:88–97.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64:248–254.
- Park DH, Kim MH, Lee SS, et al. Accuracy of magnetic resonance cholangiopancreatography for locating hepatolithiasis and detecting accompanying biliary strictures. Endoscopy 2004; 36:987–992.
- Walsh RM, Ponsky JL, Dumot J. Retained gallbladder/cystic duct remnant calculi as a cause of postcholecystectomy pain. Surg Endosc 2002; 16:981–984.
- Tantia O, Jain M, Khanna S, Sen B. Post cholecystectomy syndrome: role of cystic duct stump and re-intervention by laparoscopic surgery. J Minim Access Surg 2008; 4:71–75.
- Palanivelu C, Rangarajan M, Jategaonkar PA, Madankumar MV, Anand NV. Laparoscopic management of remnant cystic duct calculi: a retrospective study. Ann R Coll Surg Engl 2009; 91:25–29.
- Flörcken H. Gallenblasenregeneration mit Steinrecidiv nach Cholecystectomie. Deutsch Z Chir 1912; 113:604.
- Mirizzi PL. La colangiografía durante las operaciones de las vias biliares. Bol Soc Cirug Buenos Aires 1932; 16:1113.
- Soper NJ, Brunt LM. The case for routine operative cholangiography during laparoscopic cholecystectomy. Surg Clin North Am 1994; 74:953–959.
- Cuschieri A, Shimi S, Banting S, Nathanson LK, Pietrabissa A. Intraoperative cholangiography during laparoscopic cholecystectomy. Routine vs selective policy. Surg Endosc 1994; 8:302–305.
- Woods MS, Traverso LW, Kozarek RA, et al. Biliary tract complications of laparoscopic cholecystectomy are detected more frequently with routine intraoperative cholangiography. Surg Endosc 1995; 9:1076–1080.
- Vezakis A, Davides D, Ammori BJ, Martin IG, Larvin M, McMahon MJ. Intraoperative cholangiography during laparoscopic cholecystectomy. Surg Endosc 2000; 14:1118–1122.
- Ladocsi LT, Benitez LD, Filippone DR, Nance FC. Intraoperative cholangiography in laparoscopic cholecystectomy: a review of 734 consecutive cases. Am Surg 1997; 63:150–156.
- Clair DG, Brooks DC. Laparoscopic cholangiography. The case for a selective approach. Surg Clin North Am 1994; 74:961–966.
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239:28–33.
- Ponsky LE, Geisinger MA, Ponsky JL, Streem SB. Contemporary ‘urologic’ intervention in the pancreaticobiliary tree. Urology 2001; 57:21–25.
- Benninger J, Rabenstein T, Farnbacher M, Keppler J, Hahn EG, Schneider HT. Extracorporeal shockwave lithotripsy of gallstones in cystic duct remnants and Mirizzi syndrome. Gastrointest Endosc 2004; 60:454–459.
- Demetriades H, Pramateftakis MG, Kanellos I, Angelopoulos S, Mantzoros I, Betsis D. Retained gallbladder remnant after laparoscopic cholecystectomy. J Laparoendosc Adv Surg Tech A 2008; 18:276–279.
- Shaw C, O’Hanlon DM, Fenlon HM, McEntee GP. Cystic duct remnant and the ‘post-cholecystectomy syndrome. ’ Hepatogastroenterology 2004; 51:36–38.
- Rozsos I, Magyaródi Z, Orbán P. Cystic duct syndrome and minimally invasive surgery. [Hungarian] Orv Hetil 1997; 138:2397–2401.
- Chowbey PK, Bandyopadhyay SK, Sharma A, Khullar R, Soni V, Baijal M. Laparoscopic reintervention for residual gallstone disease. Surg Laparosc Endosc Percutan Tech 2003; 13:31–35.
- Clemente G, Giuliante F, Cadeddu F, Nuzzo G. Laparoscopic removal of gallbladder remnant and long cystic stump. Endoscopy 2001; 33:814–815.
- Rogy MA, Függer R, Herbst F, Schulz F. Reoperation after cholecystectomy. The role of the cystic duct stump. HPB Surg 1991; 4:129–134.
- Laing FC, Jeffrey RB. Choledocholithiasis and cystic duct obstruction: difficult ultrasonographic diagnosis. Radiology 1983; 146:475–479.
- Hassan H, Vilmann P. Insufficient cholecystectomy diagnosed by endoscopic ultrasonography. Endoscopy 2004; 36:236–238.
- Lehman GA, Sherman S. Sphincter of Oddi dysfunction (postcholecystectomy syndrome). In:Yamada T, editor. Textbook of Gastroenterology. 2nd ed. Philadelphia: Lippincott; 1995:2251–2262.
- Zhou PH, Liu FL, Yao LQ, Qin XY. Endoscopic diagnosis and treatment of post-cholecystectomy syndrome. Hepatobiliary Pancreat Dis Int 2003; 2:117–120.
- Mergener K, Clavien PA, Branch MS, Baillie J. A stone in a grossly dilated cystic duct stump: a rare cause of postcholecystectomy pain. Am J Gastroenterol 1999; 94:229–231.
- Goenka MK, Kochhar R, Nagi B, Bhasin DK, Chowdhury A, Singh K. Endoscopic retrograde cholangiopancreatography in postcholecystectomy syndrome. J Assoc Physicians India 1996; 44:119–122.
- Bodvall B, Overgaard B. Cystic duct remnant after cholecystectomy: incidence studied by cholegraphy in 500 cases, and significance in 103 reoperations. Ann Surg 1966; 163:382–390.
- Bergman JJ, van den Brink GR, Rauws EA, et al. Treatment of bile duct lesions after laparoscopic cholecystectomy. Gut 1996; 38:141–147.
- Nickkholgh A, Soltaniyekta S, Kalbasi H. Routine versus selective intraoperative cholangiography during laparoscopic cholecystectomy: a survey of 2,130 patients undergoing laparoscopic cholecystectomy. Surg Endosc 2006; 20:868–874.
- Gandolfi L, Torresan F, Solmi L, Puccetti A. The role of ultrasound in biliary and pancreatic diseases. Eur J Ultrasound 2003; 16:141–159.
- Al Samaraee A, Khan U, Almashta Z, Yiannakou Y. Preoperative diagnosis of choledocholithiasis: the role of MRCP. Br J Hosp Med (Lond) 2009; 70:339–343.
- Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: a prospective, multicenter study. Gastrointest Endosc 2001; 54:425–434.
- Cheng CL, Sherman S, Watkins JL, et al. Risk factors for post-ERCP pancreatitis: a prospective multicenter study. Am J Gastroenterol 2006; 101:139–147.
- Mehta SN, Pavone E, Barkun JS, Bouchard S, Barkun AN. Predictors of post-ERCP complications in patients with suspected choledocholithiasis. Endoscopy 1998; 30:457–463.
- Badalov N, Tenner S, Baillie J. The prevention, recognition and treatment of post-ERCP pancreatitis. JOP 2009; 10:88–97.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64:248–254.
- Park DH, Kim MH, Lee SS, et al. Accuracy of magnetic resonance cholangiopancreatography for locating hepatolithiasis and detecting accompanying biliary strictures. Endoscopy 2004; 36:987–992.
- Walsh RM, Ponsky JL, Dumot J. Retained gallbladder/cystic duct remnant calculi as a cause of postcholecystectomy pain. Surg Endosc 2002; 16:981–984.
- Tantia O, Jain M, Khanna S, Sen B. Post cholecystectomy syndrome: role of cystic duct stump and re-intervention by laparoscopic surgery. J Minim Access Surg 2008; 4:71–75.
- Palanivelu C, Rangarajan M, Jategaonkar PA, Madankumar MV, Anand NV. Laparoscopic management of remnant cystic duct calculi: a retrospective study. Ann R Coll Surg Engl 2009; 91:25–29.
- Flörcken H. Gallenblasenregeneration mit Steinrecidiv nach Cholecystectomie. Deutsch Z Chir 1912; 113:604.
- Mirizzi PL. La colangiografía durante las operaciones de las vias biliares. Bol Soc Cirug Buenos Aires 1932; 16:1113.
- Soper NJ, Brunt LM. The case for routine operative cholangiography during laparoscopic cholecystectomy. Surg Clin North Am 1994; 74:953–959.
- Cuschieri A, Shimi S, Banting S, Nathanson LK, Pietrabissa A. Intraoperative cholangiography during laparoscopic cholecystectomy. Routine vs selective policy. Surg Endosc 1994; 8:302–305.
- Woods MS, Traverso LW, Kozarek RA, et al. Biliary tract complications of laparoscopic cholecystectomy are detected more frequently with routine intraoperative cholangiography. Surg Endosc 1995; 9:1076–1080.
- Vezakis A, Davides D, Ammori BJ, Martin IG, Larvin M, McMahon MJ. Intraoperative cholangiography during laparoscopic cholecystectomy. Surg Endosc 2000; 14:1118–1122.
- Ladocsi LT, Benitez LD, Filippone DR, Nance FC. Intraoperative cholangiography in laparoscopic cholecystectomy: a review of 734 consecutive cases. Am Surg 1997; 63:150–156.
- Clair DG, Brooks DC. Laparoscopic cholangiography. The case for a selective approach. Surg Clin North Am 1994; 74:961–966.
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239:28–33.
- Ponsky LE, Geisinger MA, Ponsky JL, Streem SB. Contemporary ‘urologic’ intervention in the pancreaticobiliary tree. Urology 2001; 57:21–25.
- Benninger J, Rabenstein T, Farnbacher M, Keppler J, Hahn EG, Schneider HT. Extracorporeal shockwave lithotripsy of gallstones in cystic duct remnants and Mirizzi syndrome. Gastrointest Endosc 2004; 60:454–459.
- Demetriades H, Pramateftakis MG, Kanellos I, Angelopoulos S, Mantzoros I, Betsis D. Retained gallbladder remnant after laparoscopic cholecystectomy. J Laparoendosc Adv Surg Tech A 2008; 18:276–279.
- Shaw C, O’Hanlon DM, Fenlon HM, McEntee GP. Cystic duct remnant and the ‘post-cholecystectomy syndrome. ’ Hepatogastroenterology 2004; 51:36–38.
- Rozsos I, Magyaródi Z, Orbán P. Cystic duct syndrome and minimally invasive surgery. [Hungarian] Orv Hetil 1997; 138:2397–2401.
- Chowbey PK, Bandyopadhyay SK, Sharma A, Khullar R, Soni V, Baijal M. Laparoscopic reintervention for residual gallstone disease. Surg Laparosc Endosc Percutan Tech 2003; 13:31–35.
- Clemente G, Giuliante F, Cadeddu F, Nuzzo G. Laparoscopic removal of gallbladder remnant and long cystic stump. Endoscopy 2001; 33:814–815.
- Rogy MA, Függer R, Herbst F, Schulz F. Reoperation after cholecystectomy. The role of the cystic duct stump. HPB Surg 1991; 4:129–134.
- Laing FC, Jeffrey RB. Choledocholithiasis and cystic duct obstruction: difficult ultrasonographic diagnosis. Radiology 1983; 146:475–479.
- Hassan H, Vilmann P. Insufficient cholecystectomy diagnosed by endoscopic ultrasonography. Endoscopy 2004; 36:236–238.
A 31-year-old man with abdominal pain and a rectal nodule
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
A 31-year-old man presents to the emergency department with abdominal pain and diarrhea, which began 4 days ago. The pain is in both of the lower quadrants, is crampy and persistent, and is relieved with bowel movements. He has been having watery stools five to six times per day, without frank blood.
He reports no fevers, chills, nausea, or vomiting, and he has never travelled outside the country. He underwent laparotomy 6 months ago for a gunshot wound. He takes no prescription drugs. He smokes and he drinks alcohol, and he says he has used heroin and oxycodone recreationally.
His blood pressure is 134/74 mm Hg, and he is afebrile. An abdominal examination reveals no mass or tenderness.
Results of a complete blood count, serum chemistry panel, and serum amylase level are normal. His lipase level is slightly elevated at 80 U/L (reference range 12–70). His stool is negative for Clostridium difficile toxin on enzyme immunoassay.
Computed tomography of the abdomen reveals diffuse pericolonic hyperemia and possible thickening of the rectosigmoid colon, raising the concern that he might have infectious or inflammatory colitis. The patient is admitted for further evaluation.
Colonoscopy to evaluate the abnormalities on computed tomography finds only a 5-mm submucosal nodule in the rectum (Figure 1). Biopsy of the nodule shows it to be a well-differentiated neuroendocrine neoplasm (carcinoid tumor). Random colon biopsy samples are normal.
The patient’s symptoms resolve over the next 24 hours without any treatment.
WHAT EXPLAINS THE PATIENT’S SYMPTOMS?
1. Which of the following best explains the patient’s clinical presentation?
- Narcotic withdrawal
- Carcinoid syndrome
- Viral gastroenteritis
- Acute pancreatitis
Viral gastroenteritis is common and affects people of all ages. The very young and the elderly are at higher risk of adverse outcomes, but few people die of it in the United States.
Our patient’s symptoms were consistent with viral gastroenteritis that resolved spontaneously while he received only supportive care.
Narcotic withdrawal can also cause watery stools and abdominal pain. However, this patient lacked other signs and symptoms of withdrawal, and his symptoms improved without any detoxification or maintenance treatment.
Pancreatitis. Although the patient had a mildly elevated lipase level, his lack of nausea and vomiting and the location of the pain were not consistent with acute pancreatitis.
Carcinoid syndrome. Carcinoid tumors are rare, typically indolent neuroendocrine neoplasms. The carcinoid syndrome consists of cutaneous flushing, gut hypermotility with diarrhea, and bronchospasm.1–5 Our patient did not have the full range of these symptoms. However, the presentation of carcinoid tumors varies broadly depending on the location, morphology, or biology of the tumor.6 Although our patient had diarrhea, his symptoms improved without any specific treatment. Rectal carcinoid tumors rarely cause diarrhea, and therefore the tumor noted on colonoscopy was almost certainly an incidental finding unrelated to his clinical presentation.
The classic symptoms are caused by production of 5-hydroxyindoleacetic acid, typically by a carcinoid tumor of the small bowel. Rectal carcinoids do not produce the 5-hydroxyindoleacetic acid responsible for this “malignant” serotonin-driven syndrome and are typically asymptomatic. When rectal carcinoid tumors are symptomatic, patients may have symptoms of local irritation or obstruction, such as hematochezia, constipation, other changes in bowel habits, rectal pain, pruritis ani, or weight loss.2,7
Nearly 50% of rectal carcinoid tumors are discovered incidentally. The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry database documented a 10-fold increase in the incidence of rectal carcinoids in the last 35 years, attributed in part to an increase in screening colonoscopy.8 Furthermore, although studies of large national or multicenter databases have found that 65% to 80% of all rectal carcinoid tumors are smaller than 1.0 cm, 93.3% to 100% of those discovered on screening endoscopy were 1.0 cm or smaller.8
Rectal carcinoid tumors have a characteristic feel on digital examination, with a hard, “buckshot” consistency, and are freely mobile.5 They have also been described as firm, nodular, rubbery, yellow, submucosal, and polypoid.8
WHERE DO CARCINOID TUMORS TEND TO ARISE?
2. Which of the following sites is the most commonly recognized site of a primary carcinoid tumor?
- Small bowel
- Lung
- Liver
- Pancreas
- Rectum
The small bowel is the most common site.
Carcinoid tumors derive from neoplastic proliferation of cells of the diffuse neuroendocrine system. Therefore, they can be found anywhere neuroendocrine cells are present, commonly in the gastrointestinal tract, urogenital tract, and the bronchial epithelium.
Traditionally, neuroendocrine tumors were classified by their embryologic origin: foregut (including the respiratory tract, thymus, stomach, and pancreas), midgut (including the small intestine, appendix, and right colon), and hindgut (including the transverse, descending, and sigmoid colon and rectum). Functionally, this was sensible, as each class of tumors presented similarly due to the similar hormonal secretory products.2,3,9
A 2004 population-based review of the SEER database10 classified incidence rates of carcinoid tumors and their distribution throughout the body. Most (54.5%) were discovered in the gastrointestinal tract, and of these, 44.7% were in the small intestine, 19.6% were in the rectum, 16.7% were in the appendix, 10.6% were in the colon, 7.2% were in the stomach, and the remaining 1.2% were at other gastrointestinal sites. Nongastrointestinal sites included the lungs and bronchi (30.1%), pancreas (2.3%), female reproductive tract and ovaries (1.2%), biliary system (1.1%), and head and neck (0.4%).10
The incidence rates have increased and the distribution of sites in the body has changed over time. For example, the appendix was once considered the site of highest incidence, with tumors often discovered incidentally during surgical resection. However, these data were based on anecdotal or single-institution reports and so may have been subject to reporting bias. According to the SEER data, the small intestine is now the leading site, perhaps because of increased awareness or improved diagnostic technology and imaging.10,11
The liver is a common site of metastasis, but it is an exceptionally rare location for a primary tumor.
HOW SHOULD THIS PATIENT BE MANAGED?
3. What is the appropriate management of rectal carcinoid in this patient?
- Since the nodule is 1.0 cm or smaller, watchful waiting is acceptable
- Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up is required
- Because all carcinoid tumors are potentially malignant, radical resection (eg, abdominal perineal resection) is appropriate
- Because all carcinoid tumors are potentially malignant, radical resection with chemotherapy with 5-fluorouracil (Adrucil) and doxorubicin (Adriamycin) is required
Since the nodule is 1.0 cm or smaller, local excision is appropriate, and no follow-up will be required. Rectal carcinoid tumors generally have a favorable prognosis, with a 5-year survival rate of 87.5%.10
PROGNOSIS DEPENDS ON TUMOR SIZE, OTHER FACTORS
Many studies have examined risk factors contributing to poor prognosis, and this is an area of active study. Early research categorized rectal carcinoid risk in terms of tumor diameter, and this is still widely used to guide management. As early as 1959, Hanley et al5 recognized that tumors that were likely to metastasize were often larger than 1 cm, had infiltrated the muscularis, or were ulcerated. Today, it is understood that only 3% to 10% of rectal carcinoids smaller than 1 cm metastasize, whereas 17% to 42% of those 1 to 2 cm and 60% to 80% of those larger than 2 cm do.2,8,12,13
However, size is not the only consideration. Wang et al12 showed that muscular invasion is an independent risk factor for survival, and that tumor diameter is a significant predictor of invasion and metastasis. Similarly, a metaanalysis by Mani et al13 recognized tumor size and muscularis invasion as the most important predictors of malignancy in these neoplasms.
To aid in predicting prognosis, staging systems have been developed from institutional or national registries. Landry et al14 developed a TNM (tumor, node, metastasis) staging system for rectal carcinoids, in which the T value was based on tumor size and degree of invasion. A group at Memorial-Sloan Kettering Cancer Center15 developed a system for risk stratification of carcinoid of the rectum that is based on tumor size, muscularis invasion, lymphovascular invasion, and the mitotic rate.
TREATMENT IS BY EXCISION
Despite these new prognostic systems, there is no new guidance on therapeutic management. Surgical therapy is still largely guided by tumor size.
Lesions smaller than 1 cm are resected endoscopically or by another local transanal technique.2,3,15,16 Standard endoscopic mucosal resection is performed, and recent studies have suggested that endoscopic submucosal dissection is as effective17 or even preferred, because it resects to the deeper submucosa (as the name suggests).18 This en bloc technique may be appropriate for lesions with evidence of local invasion.18 Other situations may call for deeper resection, such as transanal resection for higher lesions and full-thickness mucosal-muscularis resection.
Tumors 1 to 2 cm are currently evaluated for other factors such as ulceration and umbilication, which influence the choice of local vs radical resection. Otherwise, there is little guidance for tumors of 1 to 2 cm.
Tumors larger than 2 cm have a high risk of muscularis invasion and metastasis, and hence they are resected with wide margins and imaging is then used to evaluate for metastasis.8,19 In cases of metastasis, local resection is often palliative, providing local symptom relief.19
AN INCIDENTALLY DISCOVERED CASE; PATIENT LOST TO FOLLOW-UP
Our patient’s case is typical of rectal carcinoid in that it was discovered incidentally during colonoscopy. His clinical presentation was likely unrelated to his carcinoid tumor, and he improved without specific treatment. His symptoms resolved within 24 hours with supportive treatment and he was discharged.
Pathologic confirmation of carcinoid tumor occurred after his discharge. Despite persistent attempts to contact the patient, he never returned for a follow-up appointment.
TAKE-HOME POINTS
- Carcinoid tumors are rare neoplasms of neuroendocrine origin.
- Rectal carcinoids are the third most common carcinoid of the gastrointestinal tract.
- Most rectal carcinoids are asymptomatic.
- Diagnosis is most often incidental and histologic.
- Treatment is by excision.
- Prognosis is favorable for smaller carcinoids and depends on size (and therefore, invasion).
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.
- Thorson A, Biorck G, Bjorkman G, Waldenstrom J. Malignant carcinoid of the small intestine with metastases to the liver, valvular disease of the right side of the heart (pulmonary stenosis and tricuspid regurgitation without septal defects), peripheral vasomotor symptoms, bronchoconstriction, and an unusual type of cyanosis; a clinical and pathologic syndrome. Am Heart J 1954; 47:795–817.
- Wang AY, Ahmad NA. Rectal carcinoids. Curr Opin Gastroenterol 2006; 22:529–535.
- Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology 2005; 128:1717–1751.
- Aggarwal G, Obideen K, Wehbi M. Carcinoid tumors: what should increase our suspicion? Cleve Clin J Med 2008; 75:849–855.
- Hanley PH, Hines MO, Ray J, Armstrong R. Carcinoid tumors of the rectum. Experience with 26 cases. Proc R Soc Med 1959; 52(suppl):113–117.
- Pasieka JL. Carcinoid tumors. Surg Clin North Am 2009; 89:1123–1137.
- Jetmore AB, Ray JE, Gathright JB, McMullen KM, Hicks TC, Timmcke AE. Rectal carcinoids: the most frequent carcinoid tumor. Dis Colon Rectum 1992; 35:717–725.
- Scherübl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009; 41:162–165.
- Wilander E, Lundqvist M, Oberg K. Gastrointestinal carcinoid tumours. Histogenetic, histochemical, immunohistochemical, clinical and therapeutic aspects. Prog Histochem Cytochem 1989; 19:1–88.
- Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg 2004; 240:117–122.
- Modlin IM, Sandor A. An analyisis of 8,305 cases of carcinoid tumors. Cancer 1997; 79:813–829.
- Wang M, Peng J, Yang W, Chen W, Mo S, Cai S. Prognostic analysis for carcinoid tumors of the rectum: a single institutional analysis of 106 cases. Colorectal Dis 2009; Epub ahead of print.
- Mani S, Modlin IM, Ballantyne G, Ahlman H, West B. Carcinoids of the rectum. J Am Coll Surg 1994; 179:231–248.
- Landry CS, Brock G, Scoggins CR, McMasters KM, Martin RC. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008; 144:460–466.
- Fahy BN, Tang LH, Klimstra D, et al. Carcinoid of the rectum risk stratification (CaRRs): a strategy for preoperative outcome assessment. Ann Surg Oncol 2007; 14:1735–1743.
- Shirouzu K, Isomoto H, Kakegawa T, Morimatsu M. Treatment of rectal carcinoid tumors. Am J Surg 1990; 160:262–265.
- Baek IH. Endoscopic submucosal dissection or conventional endoscopic mucosal resection is an effective and safe treatment for rectal carcinoid tumors: a retrospective study. J Laparoendosc Adv Surg Tech A 2010; 20:329–331.
- Yamaguchi N, Isomoto H, Nishiyama H, et al. Endoscopic submucosal dissection for rectal carcinoid tumors. Surg Endosc 2010; 24:504–508.
- Ramage JK, Goretzki PE, Manfredi R, et al; Frascati Consensus Conference participants. Consensus guidelines for the management of patients with digestive neuroendocrine tumours: well-differentiated colon and rectum tumour/carcinoma. Neuroendocrinology 2008; 87:31–39.