User login
FDA panel okays teprotumumab for thyroid eye disease
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
Influenza activity continues to be unusually high
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
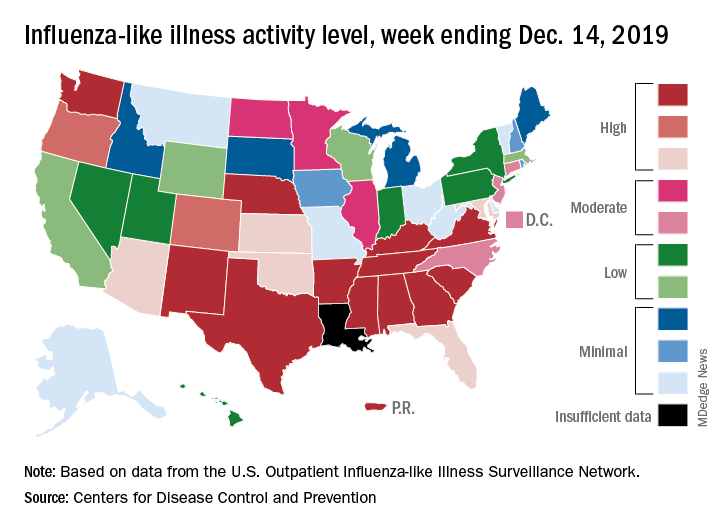
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
FDA warns gabapentin, pregabalin may cause serious breathing problems
Elderly patients who take these drugs also are at increased risk of breathing problems, the announcement said.
Gabapentin (marketed as Neurontin, Gralise, and Horizant) and pregabalin (Lyrica and Lyrica CR) are used to treat seizures, nerve pain, and restless legs syndrome. Physicians increasingly are prescribing these medications, and people are misusing and abusing these drugs more frequently, the agency said. Gabapentin and pregabalin often are combined with central nervous system depressants such as opioids, antianxiety medicines, antidepressants, and antihistamines, which increases the risk of respiratory depression.
Conditions that reduce lung function, including chronic obstructive pulmonary disease (COPD), also increase the likelihood of breathing problems when taking gabapentin and pregabalin.
“There is less evidence supporting the risk of serious breathing difficulties in healthy individuals taking gabapentinoids alone. We will continue to monitor these medicines as part of our routine monitoring of all FDA-approved drugs,” the announcement said.
The FDA is requiring new warnings about the risk of respiratory depression in the prescribing information of gabapentinoids. In addition, drug manufacturers must further assess the abuse potential of these drugs, particularly in combination with opioids.
Patients and caregivers should seek immediate medical attention for respiratory problems, which can be life threatening. Symptoms include confusion or disorientation; unusual dizziness or lightheadedness; extreme sleepiness or lethargy; slowed, shallow, or difficult breathing; unresponsiveness; and bluish-colored or tinted skin, especially on the lips, fingers, and toes.
Physicians should start gabapentinoids at the lowest dose and monitor patients for symptoms of respiratory depression and sedation when coprescribing these drugs with an opioid or other central nervous system depressant such as a benzodiazepine, according to the FDA.
The agency reviewed 49 case reports that were submitted between 2012 and 2017. Among these cases, 12 people died from respiratory depression with gabapentinoids. All of the patients who died had at least one risk factor.
Gabapentin first was approved in 1993, and pregabalin was approved in 2004. Drug adverse events and side effects can be reported online, the agency noted.
Elderly patients who take these drugs also are at increased risk of breathing problems, the announcement said.
Gabapentin (marketed as Neurontin, Gralise, and Horizant) and pregabalin (Lyrica and Lyrica CR) are used to treat seizures, nerve pain, and restless legs syndrome. Physicians increasingly are prescribing these medications, and people are misusing and abusing these drugs more frequently, the agency said. Gabapentin and pregabalin often are combined with central nervous system depressants such as opioids, antianxiety medicines, antidepressants, and antihistamines, which increases the risk of respiratory depression.
Conditions that reduce lung function, including chronic obstructive pulmonary disease (COPD), also increase the likelihood of breathing problems when taking gabapentin and pregabalin.
“There is less evidence supporting the risk of serious breathing difficulties in healthy individuals taking gabapentinoids alone. We will continue to monitor these medicines as part of our routine monitoring of all FDA-approved drugs,” the announcement said.
The FDA is requiring new warnings about the risk of respiratory depression in the prescribing information of gabapentinoids. In addition, drug manufacturers must further assess the abuse potential of these drugs, particularly in combination with opioids.
Patients and caregivers should seek immediate medical attention for respiratory problems, which can be life threatening. Symptoms include confusion or disorientation; unusual dizziness or lightheadedness; extreme sleepiness or lethargy; slowed, shallow, or difficult breathing; unresponsiveness; and bluish-colored or tinted skin, especially on the lips, fingers, and toes.
Physicians should start gabapentinoids at the lowest dose and monitor patients for symptoms of respiratory depression and sedation when coprescribing these drugs with an opioid or other central nervous system depressant such as a benzodiazepine, according to the FDA.
The agency reviewed 49 case reports that were submitted between 2012 and 2017. Among these cases, 12 people died from respiratory depression with gabapentinoids. All of the patients who died had at least one risk factor.
Gabapentin first was approved in 1993, and pregabalin was approved in 2004. Drug adverse events and side effects can be reported online, the agency noted.
Elderly patients who take these drugs also are at increased risk of breathing problems, the announcement said.
Gabapentin (marketed as Neurontin, Gralise, and Horizant) and pregabalin (Lyrica and Lyrica CR) are used to treat seizures, nerve pain, and restless legs syndrome. Physicians increasingly are prescribing these medications, and people are misusing and abusing these drugs more frequently, the agency said. Gabapentin and pregabalin often are combined with central nervous system depressants such as opioids, antianxiety medicines, antidepressants, and antihistamines, which increases the risk of respiratory depression.
Conditions that reduce lung function, including chronic obstructive pulmonary disease (COPD), also increase the likelihood of breathing problems when taking gabapentin and pregabalin.
“There is less evidence supporting the risk of serious breathing difficulties in healthy individuals taking gabapentinoids alone. We will continue to monitor these medicines as part of our routine monitoring of all FDA-approved drugs,” the announcement said.
The FDA is requiring new warnings about the risk of respiratory depression in the prescribing information of gabapentinoids. In addition, drug manufacturers must further assess the abuse potential of these drugs, particularly in combination with opioids.
Patients and caregivers should seek immediate medical attention for respiratory problems, which can be life threatening. Symptoms include confusion or disorientation; unusual dizziness or lightheadedness; extreme sleepiness or lethargy; slowed, shallow, or difficult breathing; unresponsiveness; and bluish-colored or tinted skin, especially on the lips, fingers, and toes.
Physicians should start gabapentinoids at the lowest dose and monitor patients for symptoms of respiratory depression and sedation when coprescribing these drugs with an opioid or other central nervous system depressant such as a benzodiazepine, according to the FDA.
The agency reviewed 49 case reports that were submitted between 2012 and 2017. Among these cases, 12 people died from respiratory depression with gabapentinoids. All of the patients who died had at least one risk factor.
Gabapentin first was approved in 1993, and pregabalin was approved in 2004. Drug adverse events and side effects can be reported online, the agency noted.
FDA approves antibody-drug conjugate for advanced urothelial cancer
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.

The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.
The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.
The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
FDA approves Vyondys 53 for Duchenne muscular dystrophy subtype
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.
FDA clears first fully disposable duodenoscope
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
Flu activity dropped in early December
according to the Centers for Disease Control and Prevention.
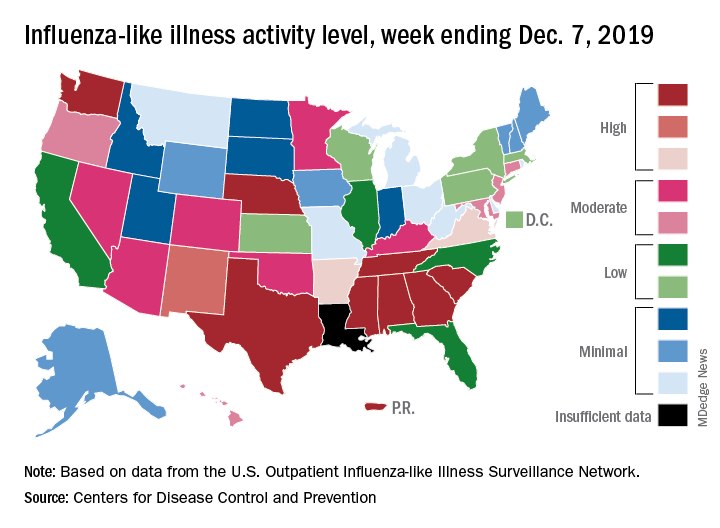
Nationally, 3.2% of outpatient visits were for influenza-like illness (ILI) during the week of Dec. 1-7, the CDC reported. That is down from 3.4% the week before, which was the highest November rate in 10 years. The national baseline rate is 2.4%, and the current 3.2% marks the fifth consecutive week that the outpatient ILI rate has been at or above the baseline level, the CDC report noted.
The drop in activity “may be influenced in part by a reduction in routine healthcare visits surrounding the Thanksgiving holiday. … as has occurred during previous seasons,” the CDC influenza division said Dec. 13 in its weekly flu report.
The early spike in “activity is being caused mostly by influenza B/Victoria viruses, which is unusual for this time of year,” the report said. Since the beginning of the 2019-2020 season a little over 2 months ago, almost 70% of specimens that have been positive for influenza have been identified as type B.
The nationwide decline in activity doesn’t, however, show up at the state level. For the week ending Dec. 7, there were eight states along with Puerto Rico at level 10 on the CDC’s 1-10 scale of flu activity, as there were the previous week. Washington state moved up from 9 to 10, but Louisiana, which was at level 10 last week, had insufficient data to be included this week, the CDC data show.
There were four flu-related pediatric deaths reported to the CDC during the week ending Dec. 7, all occurring in previous weeks, which brings the total to 10 for the season. In 2018-2019, there were 143 pediatric deaths caused by influenza, the CDC said.
according to the Centers for Disease Control and Prevention.
Nationally, 3.2% of outpatient visits were for influenza-like illness (ILI) during the week of Dec. 1-7, the CDC reported. That is down from 3.4% the week before, which was the highest November rate in 10 years. The national baseline rate is 2.4%, and the current 3.2% marks the fifth consecutive week that the outpatient ILI rate has been at or above the baseline level, the CDC report noted.
The drop in activity “may be influenced in part by a reduction in routine healthcare visits surrounding the Thanksgiving holiday. … as has occurred during previous seasons,” the CDC influenza division said Dec. 13 in its weekly flu report.
The early spike in “activity is being caused mostly by influenza B/Victoria viruses, which is unusual for this time of year,” the report said. Since the beginning of the 2019-2020 season a little over 2 months ago, almost 70% of specimens that have been positive for influenza have been identified as type B.
The nationwide decline in activity doesn’t, however, show up at the state level. For the week ending Dec. 7, there were eight states along with Puerto Rico at level 10 on the CDC’s 1-10 scale of flu activity, as there were the previous week. Washington state moved up from 9 to 10, but Louisiana, which was at level 10 last week, had insufficient data to be included this week, the CDC data show.
There were four flu-related pediatric deaths reported to the CDC during the week ending Dec. 7, all occurring in previous weeks, which brings the total to 10 for the season. In 2018-2019, there were 143 pediatric deaths caused by influenza, the CDC said.
according to the Centers for Disease Control and Prevention.
Nationally, 3.2% of outpatient visits were for influenza-like illness (ILI) during the week of Dec. 1-7, the CDC reported. That is down from 3.4% the week before, which was the highest November rate in 10 years. The national baseline rate is 2.4%, and the current 3.2% marks the fifth consecutive week that the outpatient ILI rate has been at or above the baseline level, the CDC report noted.
The drop in activity “may be influenced in part by a reduction in routine healthcare visits surrounding the Thanksgiving holiday. … as has occurred during previous seasons,” the CDC influenza division said Dec. 13 in its weekly flu report.
The early spike in “activity is being caused mostly by influenza B/Victoria viruses, which is unusual for this time of year,” the report said. Since the beginning of the 2019-2020 season a little over 2 months ago, almost 70% of specimens that have been positive for influenza have been identified as type B.
The nationwide decline in activity doesn’t, however, show up at the state level. For the week ending Dec. 7, there were eight states along with Puerto Rico at level 10 on the CDC’s 1-10 scale of flu activity, as there were the previous week. Washington state moved up from 9 to 10, but Louisiana, which was at level 10 last week, had insufficient data to be included this week, the CDC data show.
There were four flu-related pediatric deaths reported to the CDC during the week ending Dec. 7, all occurring in previous weeks, which brings the total to 10 for the season. In 2018-2019, there were 143 pediatric deaths caused by influenza, the CDC said.
Contacts with health care professionals increased among adults
Adults aged 18-64 years were more likely to see or talk to a health care professional in 2017-2018 than they were in 2012-2013, according to the Centers for Disease Control and Prevention.
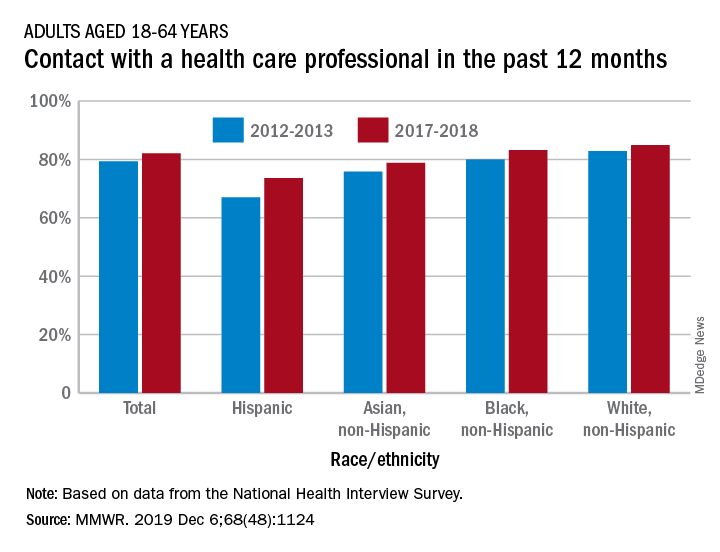
The percentage of American adults who had seen or talked to a health care professional in the past 12 months rose from 79.3% in 2012-2013 to 82.1% in 2017-2018, Michael E. Martinez, MPH, and Tainya C. Clarke, PhD, reported in the Morbidity and Mortality Weekly Report.
Analysis by race/ethnicity showed that Hispanic adults were still the least likely to have seen or talked to a health care professional in 2017-2018, even though they had the largest increase – more than six percentage points – between the two time periods, the CDC investigators reported.
White adults were the most likely to have seen or talked to a health care provider in both 2012-2013 and 2017-2018 but their 2.1-percentage-point increase over the course of the analysis was the smallest of the four groups included, based on data from the National Health Interview Survey.
SOURCE: Martinez ME, Clarke TC. MMWR. 2019 Dec 6;68(48):1124.
Adults aged 18-64 years were more likely to see or talk to a health care professional in 2017-2018 than they were in 2012-2013, according to the Centers for Disease Control and Prevention.
The percentage of American adults who had seen or talked to a health care professional in the past 12 months rose from 79.3% in 2012-2013 to 82.1% in 2017-2018, Michael E. Martinez, MPH, and Tainya C. Clarke, PhD, reported in the Morbidity and Mortality Weekly Report.
Analysis by race/ethnicity showed that Hispanic adults were still the least likely to have seen or talked to a health care professional in 2017-2018, even though they had the largest increase – more than six percentage points – between the two time periods, the CDC investigators reported.
White adults were the most likely to have seen or talked to a health care provider in both 2012-2013 and 2017-2018 but their 2.1-percentage-point increase over the course of the analysis was the smallest of the four groups included, based on data from the National Health Interview Survey.
SOURCE: Martinez ME, Clarke TC. MMWR. 2019 Dec 6;68(48):1124.
Adults aged 18-64 years were more likely to see or talk to a health care professional in 2017-2018 than they were in 2012-2013, according to the Centers for Disease Control and Prevention.
The percentage of American adults who had seen or talked to a health care professional in the past 12 months rose from 79.3% in 2012-2013 to 82.1% in 2017-2018, Michael E. Martinez, MPH, and Tainya C. Clarke, PhD, reported in the Morbidity and Mortality Weekly Report.
Analysis by race/ethnicity showed that Hispanic adults were still the least likely to have seen or talked to a health care professional in 2017-2018, even though they had the largest increase – more than six percentage points – between the two time periods, the CDC investigators reported.
White adults were the most likely to have seen or talked to a health care provider in both 2012-2013 and 2017-2018 but their 2.1-percentage-point increase over the course of the analysis was the smallest of the four groups included, based on data from the National Health Interview Survey.
SOURCE: Martinez ME, Clarke TC. MMWR. 2019 Dec 6;68(48):1124.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
EVALI outbreak ongoing, but new cases decline
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.

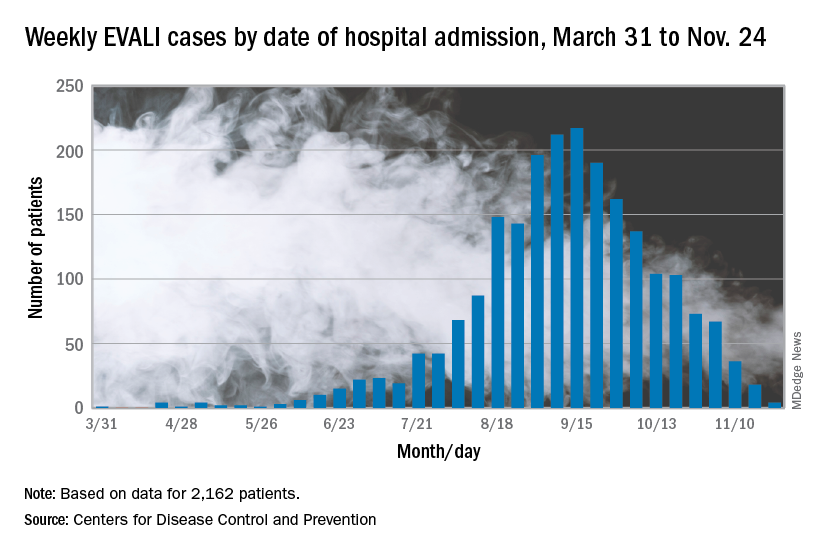
In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.
In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.
In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
FROM THE MMWR
Influenza already in midseason form
It’s been a decade since flu activity levels were this high this early in the season.
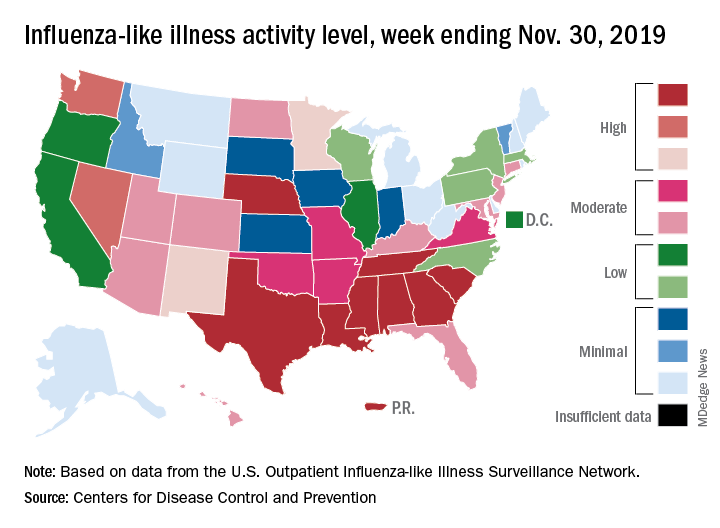
For the week ending Nov. 30, outpatient visits for influenza-like illness reached 3.5% of all visits to health care providers, the Centers for Disease Control and Prevention reported Dec. 6. That is the highest pre-December rate since the pandemic of 2009-2010, when the rate peaked at 7.7% in mid-October, CDC data show.
For the last week of November, eight states and Puerto Rico reported activity levels at the high point of the CDC’s 1-10 scale, which is at least five more states than any of the past five flu seasons. Three of the last five seasons had no states at level 10 this early in the season.
Another 4 states at levels 8 and 9 put a total of 13 jurisdictions in the “high” range of flu activity, with another 14 states in the “moderate” range of levels 6 and 7. Geographically speaking, 24 jurisdictions are experiencing regional or widespread activity, which is up from the 15 reported last week, the CDC’s influenza division said.
The hospitalization rate to date for the 2019-2020 season – 2.7 per 100,000 population – is “similar to what has been seen at this time during other recent seasons,” the CDC said.
One influenza-related pediatric death was reported during the week ending Nov. 30, which brings the total for the season to six, according to the CDC report.
It’s been a decade since flu activity levels were this high this early in the season.
For the week ending Nov. 30, outpatient visits for influenza-like illness reached 3.5% of all visits to health care providers, the Centers for Disease Control and Prevention reported Dec. 6. That is the highest pre-December rate since the pandemic of 2009-2010, when the rate peaked at 7.7% in mid-October, CDC data show.
For the last week of November, eight states and Puerto Rico reported activity levels at the high point of the CDC’s 1-10 scale, which is at least five more states than any of the past five flu seasons. Three of the last five seasons had no states at level 10 this early in the season.
Another 4 states at levels 8 and 9 put a total of 13 jurisdictions in the “high” range of flu activity, with another 14 states in the “moderate” range of levels 6 and 7. Geographically speaking, 24 jurisdictions are experiencing regional or widespread activity, which is up from the 15 reported last week, the CDC’s influenza division said.
The hospitalization rate to date for the 2019-2020 season – 2.7 per 100,000 population – is “similar to what has been seen at this time during other recent seasons,” the CDC said.
One influenza-related pediatric death was reported during the week ending Nov. 30, which brings the total for the season to six, according to the CDC report.
It’s been a decade since flu activity levels were this high this early in the season.
For the week ending Nov. 30, outpatient visits for influenza-like illness reached 3.5% of all visits to health care providers, the Centers for Disease Control and Prevention reported Dec. 6. That is the highest pre-December rate since the pandemic of 2009-2010, when the rate peaked at 7.7% in mid-October, CDC data show.
For the last week of November, eight states and Puerto Rico reported activity levels at the high point of the CDC’s 1-10 scale, which is at least five more states than any of the past five flu seasons. Three of the last five seasons had no states at level 10 this early in the season.
Another 4 states at levels 8 and 9 put a total of 13 jurisdictions in the “high” range of flu activity, with another 14 states in the “moderate” range of levels 6 and 7. Geographically speaking, 24 jurisdictions are experiencing regional or widespread activity, which is up from the 15 reported last week, the CDC’s influenza division said.
The hospitalization rate to date for the 2019-2020 season – 2.7 per 100,000 population – is “similar to what has been seen at this time during other recent seasons,” the CDC said.
One influenza-related pediatric death was reported during the week ending Nov. 30, which brings the total for the season to six, according to the CDC report.