User login
First generics for Gilenya approved by FDA
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
FDA approves infliximab-axxq for numerous indications
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.

The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.
The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.
The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
2019-2020 flu season starts off full throttle
For the week ending Nov. 23, there were five states, along with Puerto Rico, at the highest level of the Centers for Disease Control and Prevention’s 1-10 scale of flu activity. That’s more than any year since 2012, including the pandemic season of 2017-2018, according to CDC data, and may suggest either an early peak or the beginning of a particularly bad winter.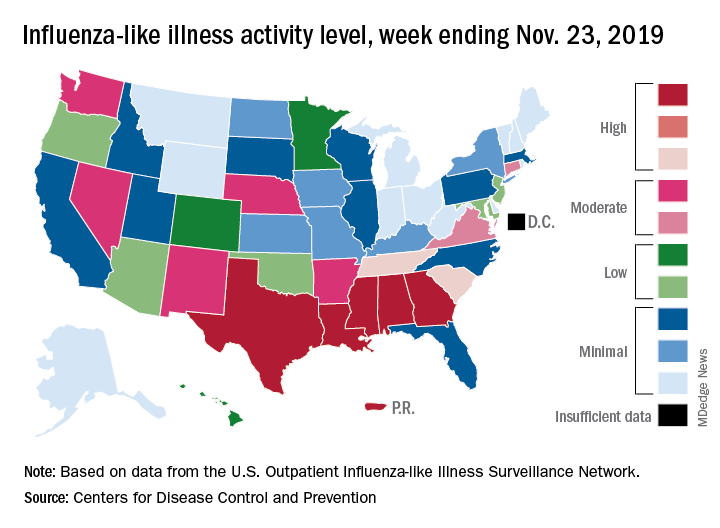
“Nationally, ILI [influenza-like illness] activity has been at or above baseline for 3 weeks; however, the amount of influenza activity across the country varies with the south and parts of the west seeing elevated activity while other parts of the country are still seeing low activity,” the CDC’s influenza division said in its weekly FluView report.
The five highest-activity states – Alabama, Georgia, Louisiana, Mississippi, and Texas – are all at level 10, and they join two others – South Carolina and Tennessee, which are at level 8 – in the “high” range from 8-10 on the ILI activity scale; Puerto Rico also is at level 10. ILI is defined as “fever (temperature of 100° F [37.8° C] or greater) and a cough and/or a sore throat without a known cause other than influenza,” the CDC said.
The activity scale is based on the percentage of outpatient visits for ILI in each state, which is reported to the CDC’s Outpatient Influenza-like Illness Surveillance Network (ILINet) each week. The national rate for the week ending Nov. 23 was 2.9%, which is above the new-for-this-season baseline rate of 2.4%. For the three previous flu seasons, the national baseline was 2.2%, having been raised from its previous level of 2.1% in 2015-2016, CDC data show.
The peak month of flu activity occurs most often in February – 15 times from 1982-1983 to 2017-2018 – but there were seven peaks in December and six each in January and March over that time period, along with one peak each in October and November, the CDC said. The October peak occurred during the H1N1 pandemic year of 2009, when the national outpatient ILI rate climbed to just over 7.7%.
For the week ending Nov. 23, there were five states, along with Puerto Rico, at the highest level of the Centers for Disease Control and Prevention’s 1-10 scale of flu activity. That’s more than any year since 2012, including the pandemic season of 2017-2018, according to CDC data, and may suggest either an early peak or the beginning of a particularly bad winter.
“Nationally, ILI [influenza-like illness] activity has been at or above baseline for 3 weeks; however, the amount of influenza activity across the country varies with the south and parts of the west seeing elevated activity while other parts of the country are still seeing low activity,” the CDC’s influenza division said in its weekly FluView report.
The five highest-activity states – Alabama, Georgia, Louisiana, Mississippi, and Texas – are all at level 10, and they join two others – South Carolina and Tennessee, which are at level 8 – in the “high” range from 8-10 on the ILI activity scale; Puerto Rico also is at level 10. ILI is defined as “fever (temperature of 100° F [37.8° C] or greater) and a cough and/or a sore throat without a known cause other than influenza,” the CDC said.
The activity scale is based on the percentage of outpatient visits for ILI in each state, which is reported to the CDC’s Outpatient Influenza-like Illness Surveillance Network (ILINet) each week. The national rate for the week ending Nov. 23 was 2.9%, which is above the new-for-this-season baseline rate of 2.4%. For the three previous flu seasons, the national baseline was 2.2%, having been raised from its previous level of 2.1% in 2015-2016, CDC data show.
The peak month of flu activity occurs most often in February – 15 times from 1982-1983 to 2017-2018 – but there were seven peaks in December and six each in January and March over that time period, along with one peak each in October and November, the CDC said. The October peak occurred during the H1N1 pandemic year of 2009, when the national outpatient ILI rate climbed to just over 7.7%.
For the week ending Nov. 23, there were five states, along with Puerto Rico, at the highest level of the Centers for Disease Control and Prevention’s 1-10 scale of flu activity. That’s more than any year since 2012, including the pandemic season of 2017-2018, according to CDC data, and may suggest either an early peak or the beginning of a particularly bad winter.
“Nationally, ILI [influenza-like illness] activity has been at or above baseline for 3 weeks; however, the amount of influenza activity across the country varies with the south and parts of the west seeing elevated activity while other parts of the country are still seeing low activity,” the CDC’s influenza division said in its weekly FluView report.
The five highest-activity states – Alabama, Georgia, Louisiana, Mississippi, and Texas – are all at level 10, and they join two others – South Carolina and Tennessee, which are at level 8 – in the “high” range from 8-10 on the ILI activity scale; Puerto Rico also is at level 10. ILI is defined as “fever (temperature of 100° F [37.8° C] or greater) and a cough and/or a sore throat without a known cause other than influenza,” the CDC said.
The activity scale is based on the percentage of outpatient visits for ILI in each state, which is reported to the CDC’s Outpatient Influenza-like Illness Surveillance Network (ILINet) each week. The national rate for the week ending Nov. 23 was 2.9%, which is above the new-for-this-season baseline rate of 2.4%. For the three previous flu seasons, the national baseline was 2.2%, having been raised from its previous level of 2.1% in 2015-2016, CDC data show.
The peak month of flu activity occurs most often in February – 15 times from 1982-1983 to 2017-2018 – but there were seven peaks in December and six each in January and March over that time period, along with one peak each in October and November, the CDC said. The October peak occurred during the H1N1 pandemic year of 2009, when the national outpatient ILI rate climbed to just over 7.7%.
FDA approves Oxbryta for sickle cell disease treatment
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.

Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
FDA: Two repackagers issue voluntary ranitidine recall
The ranitidine recall saga continues as two more companies have issued voluntary recalls of their repackaged ranitidine products because of possibly unacceptable levels of N-nitrosodimethylamine (NDMA), according to the Food and Drug Administration.
Golden State Medical Supply has recalled 150-mg and 300-mg ranitidine capsules, manufactured by Novitium, and Precision Dose has recalled a ranitidine oral solution at 150 mg/mL, manufactured by Amneal Pharmaceuticals.
“FDA has advised companies to recall their ranitidine if testing shows levels of NDMA above the acceptable daily intake (96 ng/day or 0.32 parts per million for ranitidine),” the FDA said. The agency has “posted the results of its testing of ranitidine samples and has asked companies to conduct their own laboratory testing.”
The FDA added that consumers taking over-the-counter ranitidine can consider alternatives such as famotidine (Pepcid), cimetidine (Tagamet), esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
Find the full press release and more information on other ranitidine recalls on the FDA website.
The ranitidine recall saga continues as two more companies have issued voluntary recalls of their repackaged ranitidine products because of possibly unacceptable levels of N-nitrosodimethylamine (NDMA), according to the Food and Drug Administration.
Golden State Medical Supply has recalled 150-mg and 300-mg ranitidine capsules, manufactured by Novitium, and Precision Dose has recalled a ranitidine oral solution at 150 mg/mL, manufactured by Amneal Pharmaceuticals.
“FDA has advised companies to recall their ranitidine if testing shows levels of NDMA above the acceptable daily intake (96 ng/day or 0.32 parts per million for ranitidine),” the FDA said. The agency has “posted the results of its testing of ranitidine samples and has asked companies to conduct their own laboratory testing.”
The FDA added that consumers taking over-the-counter ranitidine can consider alternatives such as famotidine (Pepcid), cimetidine (Tagamet), esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
Find the full press release and more information on other ranitidine recalls on the FDA website.
The ranitidine recall saga continues as two more companies have issued voluntary recalls of their repackaged ranitidine products because of possibly unacceptable levels of N-nitrosodimethylamine (NDMA), according to the Food and Drug Administration.
Golden State Medical Supply has recalled 150-mg and 300-mg ranitidine capsules, manufactured by Novitium, and Precision Dose has recalled a ranitidine oral solution at 150 mg/mL, manufactured by Amneal Pharmaceuticals.
“FDA has advised companies to recall their ranitidine if testing shows levels of NDMA above the acceptable daily intake (96 ng/day or 0.32 parts per million for ranitidine),” the FDA said. The agency has “posted the results of its testing of ranitidine samples and has asked companies to conduct their own laboratory testing.”
The FDA added that consumers taking over-the-counter ranitidine can consider alternatives such as famotidine (Pepcid), cimetidine (Tagamet), esomeprazole (Nexium), lansoprazole (Prevacid), and omeprazole (Prilosec).
Find the full press release and more information on other ranitidine recalls on the FDA website.
FDA approves acalabrutinib for CLL, SLL treatment
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
FDA approves Givlaari for treatment of acute hepatic porphyria
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
Ask about vaping in patients with respiratory symptoms, CDC says
“Do you vape?” may be one of the most important questions health care can providers can ask patients who present with respiratory symptoms this winter.

according to the Centers for Disease Control and Prevention.
Accordingly, providers need to ask patients with respiratory, gastrointestinal, or constitutional symptoms about their use of e-cigarette or vaping products, according to one several new CDC recommendations that appear in the Morbidity and Mortality Weekly Review.
“E-cigarette or vaping product use–associated lung injury (EVALI) remains a diagnosis of exclusion because, at present, no specific test or marker exists for its diagnosis, and evaluation should be guided by clinical judgment,” the CDC report reads.
As of Nov. 13, there have been 2,172 cases of EVALI reported to CDC, of which 42 (1.9%) have been fatal. Most of the patients with EVALI have been white (79%), male (68%), and under the age of 35 years (77%), according to CDC data.
Although vitamin E acetate was recently implicated as a potential cause of EVALI, the agency said evidence is “not sufficient” at this point in their investigation to rule out other chemicals of potential concern.
“Many different substances and product sources are still under investigation, and it might be that there is more than one cause of this outbreak,” CDC said.
Further recommendations
Beyond asking about vape use, providers should evaluate suspected EVALI with pulse oximetry and chest imaging, and should consider outpatient management for patients who are clinically stable, according to the recommendations.
The agency said influenza testing should be “strongly considered,” especially during influenza season, given that EVALI is a diagnosis of exclusion and that it may co-occur with other respiratory illnesses. Antimicrobials (including antivirals) should be given as warranted, they added.
Corticosteroids may be helpful in treating EVALI, but may worsen respiratory infections typically seen in outpatients, and so should be prescribed with caution in the outpatient setting, the CDC recommended.
Behavioral counseling, addiction treatment services, and Food and Drug Administration–approved cessation medications are recommended to help patients quit vaping or e-cigarette products, CDC said.
Health care providers should emphasize the importance of an annual flu shot for all patients 6 months of age or older, including those who use e-cigarette or vaping products, according to the agency.
“It is not known whether patients with EVALI are at higher risk for severe complications of influenza or other respiratory infections,” the report reads.
Blame it on vitamin E? THC? Other?
The report details how, as previously reported, vitamin E acetate was detected in bronchoalveolar lavage fluid samples from 29 patients with EVALI. Although other chemicals could contribute to EVALI, that finding provided “direct evidence” of vitamin E acetate at the primary site of injury, according to CDC.
Most patients with EVALI, 83%, have reported using a tetrahydrocannabinol (THC)-containing e-cigarette or vaping product, according to CDC, while 61% reported using a nicotine-containing product.
Based on that, CDC recommended that people avoid using THC-containing products. However, the agency cautioned that the specific cause or causes of EVALI remain to be elucidated.
“The only way for persons to assure that they are not at risk is to consider refraining from use of all e-cigarette, or vaping, products while this investigation continues,” CDC said in the report.
The need for this additional clinical guidance was assessed in anticipation of the seasonal uptick in influenza and other respiratory infections, according to the CDC, which said the recommendations were based in part on individual clinical perspectives from nine national experts who participated in a previously published clinical guidance on managing patients with EVALI.
SOURCES: Jatlaoui TC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e2; Chatham-Stephens K et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e1.
“Do you vape?” may be one of the most important questions health care can providers can ask patients who present with respiratory symptoms this winter.
according to the Centers for Disease Control and Prevention.
Accordingly, providers need to ask patients with respiratory, gastrointestinal, or constitutional symptoms about their use of e-cigarette or vaping products, according to one several new CDC recommendations that appear in the Morbidity and Mortality Weekly Review.
“E-cigarette or vaping product use–associated lung injury (EVALI) remains a diagnosis of exclusion because, at present, no specific test or marker exists for its diagnosis, and evaluation should be guided by clinical judgment,” the CDC report reads.
As of Nov. 13, there have been 2,172 cases of EVALI reported to CDC, of which 42 (1.9%) have been fatal. Most of the patients with EVALI have been white (79%), male (68%), and under the age of 35 years (77%), according to CDC data.
Although vitamin E acetate was recently implicated as a potential cause of EVALI, the agency said evidence is “not sufficient” at this point in their investigation to rule out other chemicals of potential concern.
“Many different substances and product sources are still under investigation, and it might be that there is more than one cause of this outbreak,” CDC said.
Further recommendations
Beyond asking about vape use, providers should evaluate suspected EVALI with pulse oximetry and chest imaging, and should consider outpatient management for patients who are clinically stable, according to the recommendations.
The agency said influenza testing should be “strongly considered,” especially during influenza season, given that EVALI is a diagnosis of exclusion and that it may co-occur with other respiratory illnesses. Antimicrobials (including antivirals) should be given as warranted, they added.
Corticosteroids may be helpful in treating EVALI, but may worsen respiratory infections typically seen in outpatients, and so should be prescribed with caution in the outpatient setting, the CDC recommended.
Behavioral counseling, addiction treatment services, and Food and Drug Administration–approved cessation medications are recommended to help patients quit vaping or e-cigarette products, CDC said.
Health care providers should emphasize the importance of an annual flu shot for all patients 6 months of age or older, including those who use e-cigarette or vaping products, according to the agency.
“It is not known whether patients with EVALI are at higher risk for severe complications of influenza or other respiratory infections,” the report reads.
Blame it on vitamin E? THC? Other?
The report details how, as previously reported, vitamin E acetate was detected in bronchoalveolar lavage fluid samples from 29 patients with EVALI. Although other chemicals could contribute to EVALI, that finding provided “direct evidence” of vitamin E acetate at the primary site of injury, according to CDC.
Most patients with EVALI, 83%, have reported using a tetrahydrocannabinol (THC)-containing e-cigarette or vaping product, according to CDC, while 61% reported using a nicotine-containing product.
Based on that, CDC recommended that people avoid using THC-containing products. However, the agency cautioned that the specific cause or causes of EVALI remain to be elucidated.
“The only way for persons to assure that they are not at risk is to consider refraining from use of all e-cigarette, or vaping, products while this investigation continues,” CDC said in the report.
The need for this additional clinical guidance was assessed in anticipation of the seasonal uptick in influenza and other respiratory infections, according to the CDC, which said the recommendations were based in part on individual clinical perspectives from nine national experts who participated in a previously published clinical guidance on managing patients with EVALI.
SOURCES: Jatlaoui TC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e2; Chatham-Stephens K et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e1.
“Do you vape?” may be one of the most important questions health care can providers can ask patients who present with respiratory symptoms this winter.
according to the Centers for Disease Control and Prevention.
Accordingly, providers need to ask patients with respiratory, gastrointestinal, or constitutional symptoms about their use of e-cigarette or vaping products, according to one several new CDC recommendations that appear in the Morbidity and Mortality Weekly Review.
“E-cigarette or vaping product use–associated lung injury (EVALI) remains a diagnosis of exclusion because, at present, no specific test or marker exists for its diagnosis, and evaluation should be guided by clinical judgment,” the CDC report reads.
As of Nov. 13, there have been 2,172 cases of EVALI reported to CDC, of which 42 (1.9%) have been fatal. Most of the patients with EVALI have been white (79%), male (68%), and under the age of 35 years (77%), according to CDC data.
Although vitamin E acetate was recently implicated as a potential cause of EVALI, the agency said evidence is “not sufficient” at this point in their investigation to rule out other chemicals of potential concern.
“Many different substances and product sources are still under investigation, and it might be that there is more than one cause of this outbreak,” CDC said.
Further recommendations
Beyond asking about vape use, providers should evaluate suspected EVALI with pulse oximetry and chest imaging, and should consider outpatient management for patients who are clinically stable, according to the recommendations.
The agency said influenza testing should be “strongly considered,” especially during influenza season, given that EVALI is a diagnosis of exclusion and that it may co-occur with other respiratory illnesses. Antimicrobials (including antivirals) should be given as warranted, they added.
Corticosteroids may be helpful in treating EVALI, but may worsen respiratory infections typically seen in outpatients, and so should be prescribed with caution in the outpatient setting, the CDC recommended.
Behavioral counseling, addiction treatment services, and Food and Drug Administration–approved cessation medications are recommended to help patients quit vaping or e-cigarette products, CDC said.
Health care providers should emphasize the importance of an annual flu shot for all patients 6 months of age or older, including those who use e-cigarette or vaping products, according to the agency.
“It is not known whether patients with EVALI are at higher risk for severe complications of influenza or other respiratory infections,” the report reads.
Blame it on vitamin E? THC? Other?
The report details how, as previously reported, vitamin E acetate was detected in bronchoalveolar lavage fluid samples from 29 patients with EVALI. Although other chemicals could contribute to EVALI, that finding provided “direct evidence” of vitamin E acetate at the primary site of injury, according to CDC.
Most patients with EVALI, 83%, have reported using a tetrahydrocannabinol (THC)-containing e-cigarette or vaping product, according to CDC, while 61% reported using a nicotine-containing product.
Based on that, CDC recommended that people avoid using THC-containing products. However, the agency cautioned that the specific cause or causes of EVALI remain to be elucidated.
“The only way for persons to assure that they are not at risk is to consider refraining from use of all e-cigarette, or vaping, products while this investigation continues,” CDC said in the report.
The need for this additional clinical guidance was assessed in anticipation of the seasonal uptick in influenza and other respiratory infections, according to the CDC, which said the recommendations were based in part on individual clinical perspectives from nine national experts who participated in a previously published clinical guidance on managing patients with EVALI.
SOURCES: Jatlaoui TC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e2; Chatham-Stephens K et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 19. doi. 10.15585/mmwr.mm6846e1.
FROM MMWR
FDA announces approval of fifth adalimumab biosimilar, Abrilada
The Food and Drug Administration has cleared adalimumab-afzb (Abrilada) as the fifth approved Humira biosimilar and the 25th approved biosimilar drug overall, the agency said in a Nov. 15 announcement.
According to a press release from Pfizer, approval for Abrilada was based on review of a comprehensive data package demonstrating biosimilarity of the drug to the reference product. This included data from a clinical comparative study, which found no clinically meaningful difference between Abrilada and the reference in terms of efficacy, safety, and immunogenicity in patients with moderate to severe rheumatoid arthritis (RA). In addition to RA, Abrilada is indicated for juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis.
Common adverse events in adalimumab clinical trials included infection, injection-site reactions, headache, and rash.
Pfizer said that it “is working to make Abrilada available to U.S. patients as soon as feasible based on the terms of our agreement with AbbVie [the manufacturer of Humira]. Our current plans are to launch in 2023.”
The Food and Drug Administration has cleared adalimumab-afzb (Abrilada) as the fifth approved Humira biosimilar and the 25th approved biosimilar drug overall, the agency said in a Nov. 15 announcement.
According to a press release from Pfizer, approval for Abrilada was based on review of a comprehensive data package demonstrating biosimilarity of the drug to the reference product. This included data from a clinical comparative study, which found no clinically meaningful difference between Abrilada and the reference in terms of efficacy, safety, and immunogenicity in patients with moderate to severe rheumatoid arthritis (RA). In addition to RA, Abrilada is indicated for juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis.
Common adverse events in adalimumab clinical trials included infection, injection-site reactions, headache, and rash.
Pfizer said that it “is working to make Abrilada available to U.S. patients as soon as feasible based on the terms of our agreement with AbbVie [the manufacturer of Humira]. Our current plans are to launch in 2023.”
The Food and Drug Administration has cleared adalimumab-afzb (Abrilada) as the fifth approved Humira biosimilar and the 25th approved biosimilar drug overall, the agency said in a Nov. 15 announcement.
According to a press release from Pfizer, approval for Abrilada was based on review of a comprehensive data package demonstrating biosimilarity of the drug to the reference product. This included data from a clinical comparative study, which found no clinically meaningful difference between Abrilada and the reference in terms of efficacy, safety, and immunogenicity in patients with moderate to severe rheumatoid arthritis (RA). In addition to RA, Abrilada is indicated for juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis.
Common adverse events in adalimumab clinical trials included infection, injection-site reactions, headache, and rash.
Pfizer said that it “is working to make Abrilada available to U.S. patients as soon as feasible based on the terms of our agreement with AbbVie [the manufacturer of Humira]. Our current plans are to launch in 2023.”
FDA approves treatment for sickle cell pain crises
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.