User login
Consider These Factors in an Academic Radiation Oncology Position
TOPLINE:
— and accept an offer if the practice is “great” in at least two of those areas and “good” in the third, experts say in a recent editorial.
METHODOLOGY:
- Many physicians choose to go into academic medicine because they want to stay involved in research and education while still treating patients.
- However, graduating radiation oncology residents often lack or have limited guidance on what to look for in a prospective job and how to assess their contract.
- This recent editorial provides guidance to radiation oncologists seeking academic positions. The authors advise prospective employees to evaluate three main factors — compensation, daily duties, and location — as well as provide tips for identifying red flags in each category.
TAKEAWAY:
- Compensation: Prospective faculty should assess both direct compensation, that is, salary, and indirect compensation, which typically includes retirement contributions and other perks. For direct compensation, what is the base salary? Is extra work compensated? How does the salary offer measure up to salary data reported by national agencies? Also: Don’t overlook uncompensated duties, such as time in tumor boards or in meetings, which may be time-consuming, and make sure compensation terms are clearly delineated in a contract and equitable among physicians in a specific rank.
- Daily duties: When it comes to daily life on the job, a prospective employee should consider many factors, including the cancer center’s excitement to hire you, the reputation of the faculty and leaders at the organization, employee turnover rates, diversity among faculty, and the time line of career advancement.
- Location: The location of the job encompasses the geography — such as distance from home to work, the number of practices covered, cost of living, and the area itself — as well as the atmosphere for conducting research and publishing.
- Finally, carefully review the job contract. All the key aspects of the job, including compensation and benefits, should be clearly stated in the contract to “improve communication of expectations.”
IN PRACTICE:
“A prospective faculty member can ask 100 questions, but they can’t make 100 demands; consideration of the three domains can help to focus negotiation efforts where the efforts are needed,” the authors noted.
SOURCE:
This editorial, led by Nicholas G. Zaorsky from the Department of Radiation Oncology, University Hospitals Seidman Cancer Center, Case Western Reserve School of Medicine, Cleveland, Ohio, was published online in Practical Radiation Oncology
DISCLOSURES:
The lead author declared being supported by the American Cancer Society and National Institutes of Health. He also reported having ties with many other sources.
A version of this article appeared on Medscape.com.
TOPLINE:
— and accept an offer if the practice is “great” in at least two of those areas and “good” in the third, experts say in a recent editorial.
METHODOLOGY:
- Many physicians choose to go into academic medicine because they want to stay involved in research and education while still treating patients.
- However, graduating radiation oncology residents often lack or have limited guidance on what to look for in a prospective job and how to assess their contract.
- This recent editorial provides guidance to radiation oncologists seeking academic positions. The authors advise prospective employees to evaluate three main factors — compensation, daily duties, and location — as well as provide tips for identifying red flags in each category.
TAKEAWAY:
- Compensation: Prospective faculty should assess both direct compensation, that is, salary, and indirect compensation, which typically includes retirement contributions and other perks. For direct compensation, what is the base salary? Is extra work compensated? How does the salary offer measure up to salary data reported by national agencies? Also: Don’t overlook uncompensated duties, such as time in tumor boards or in meetings, which may be time-consuming, and make sure compensation terms are clearly delineated in a contract and equitable among physicians in a specific rank.
- Daily duties: When it comes to daily life on the job, a prospective employee should consider many factors, including the cancer center’s excitement to hire you, the reputation of the faculty and leaders at the organization, employee turnover rates, diversity among faculty, and the time line of career advancement.
- Location: The location of the job encompasses the geography — such as distance from home to work, the number of practices covered, cost of living, and the area itself — as well as the atmosphere for conducting research and publishing.
- Finally, carefully review the job contract. All the key aspects of the job, including compensation and benefits, should be clearly stated in the contract to “improve communication of expectations.”
IN PRACTICE:
“A prospective faculty member can ask 100 questions, but they can’t make 100 demands; consideration of the three domains can help to focus negotiation efforts where the efforts are needed,” the authors noted.
SOURCE:
This editorial, led by Nicholas G. Zaorsky from the Department of Radiation Oncology, University Hospitals Seidman Cancer Center, Case Western Reserve School of Medicine, Cleveland, Ohio, was published online in Practical Radiation Oncology
DISCLOSURES:
The lead author declared being supported by the American Cancer Society and National Institutes of Health. He also reported having ties with many other sources.
A version of this article appeared on Medscape.com.
TOPLINE:
— and accept an offer if the practice is “great” in at least two of those areas and “good” in the third, experts say in a recent editorial.
METHODOLOGY:
- Many physicians choose to go into academic medicine because they want to stay involved in research and education while still treating patients.
- However, graduating radiation oncology residents often lack or have limited guidance on what to look for in a prospective job and how to assess their contract.
- This recent editorial provides guidance to radiation oncologists seeking academic positions. The authors advise prospective employees to evaluate three main factors — compensation, daily duties, and location — as well as provide tips for identifying red flags in each category.
TAKEAWAY:
- Compensation: Prospective faculty should assess both direct compensation, that is, salary, and indirect compensation, which typically includes retirement contributions and other perks. For direct compensation, what is the base salary? Is extra work compensated? How does the salary offer measure up to salary data reported by national agencies? Also: Don’t overlook uncompensated duties, such as time in tumor boards or in meetings, which may be time-consuming, and make sure compensation terms are clearly delineated in a contract and equitable among physicians in a specific rank.
- Daily duties: When it comes to daily life on the job, a prospective employee should consider many factors, including the cancer center’s excitement to hire you, the reputation of the faculty and leaders at the organization, employee turnover rates, diversity among faculty, and the time line of career advancement.
- Location: The location of the job encompasses the geography — such as distance from home to work, the number of practices covered, cost of living, and the area itself — as well as the atmosphere for conducting research and publishing.
- Finally, carefully review the job contract. All the key aspects of the job, including compensation and benefits, should be clearly stated in the contract to “improve communication of expectations.”
IN PRACTICE:
“A prospective faculty member can ask 100 questions, but they can’t make 100 demands; consideration of the three domains can help to focus negotiation efforts where the efforts are needed,” the authors noted.
SOURCE:
This editorial, led by Nicholas G. Zaorsky from the Department of Radiation Oncology, University Hospitals Seidman Cancer Center, Case Western Reserve School of Medicine, Cleveland, Ohio, was published online in Practical Radiation Oncology
DISCLOSURES:
The lead author declared being supported by the American Cancer Society and National Institutes of Health. He also reported having ties with many other sources.
A version of this article appeared on Medscape.com.
Look Beyond BMI: Metabolic Factors’ Link to Cancer Explained
The new research finds that adults with persistent metabolic syndrome that worsens over time are at increased risk for any type of cancer.
The conditions that make up metabolic syndrome (high blood pressure, high blood sugar, increased abdominal adiposity, and high cholesterol and triglycerides) have been associated with an increased risk of diseases, including heart disease, stroke, and type 2 diabetes, wrote Li Deng, PhD, of Capital Medical University, Beijing, and colleagues.
However, a single assessment of metabolic syndrome at one point in time is inadequate to show an association with cancer risk over time, they said. In the current study, the researchers used models to examine the association between trajectory patterns of metabolic syndrome over time and the risk of overall and specific cancer types. They also examined the impact of chronic inflammation concurrent with metabolic syndrome.
What We Know About Metabolic Syndrome and Cancer Risk
A systematic review and meta-analysis published in Diabetes Care in 2012 showed an association between the presence of metabolic syndrome and an increased risk of various cancers including liver, bladder, pancreatic, breast, and colorectal.
More recently, a 2020 study published in Diabetes showed evidence of increased risk for certain cancers (pancreatic, kidney, uterine, cervical) but no increased risk for cancer overall.
In addition, a 2022 study by some of the current study researchers of the same Chinese cohort focused on the role of inflammation in combination with metabolic syndrome on colorectal cancer specifically, and found an increased risk for cancer when both metabolic syndrome and inflammation were present.
However, the reasons for this association between metabolic syndrome and cancer remain unclear, and the effect of the fluctuating nature of metabolic syndrome over time on long-term cancer risk has not been explored, the researchers wrote.
“There is emerging evidence that even normal weight individuals who are metabolically unhealthy may be at an elevated cancer risk, and we need better metrics to define the underlying metabolic dysfunction in obesity,” Sheetal Hardikar, MBBS, PhD, MPH, an investigator at the Huntsman Cancer Institute, University of Utah, said in an interview.
Dr. Hardikar, who serves as assistant professor in the department of population health sciences at the University of Utah, was not involved in the current study. She and her colleagues published a research paper on data from the National Health and Nutrition Examination Survey in 2023 that showed an increased risk of obesity-related cancer.
What New Study Adds to Related Research
Previous studies have consistently reported an approximately 30% increased risk of cancer with metabolic syndrome, Dr. Hardikar said. “What is unique about this study is the examination of metabolic syndrome trajectories over four years, and not just the presence of metabolic syndrome at one point in time,” she said.
In the new study, published in Cancer on March 11 (doi: 10.1002/cncr.35235), 44,115 adults in China were separated into four trajectories based on metabolic syndrome scores for the period from 2006 to 2010. The scores were based on clinical evidence of metabolic syndrome, defined using the International Diabetes Federation criteria of central obesity and the presence of at least two other factors including increased triglycerides, decreased HDL cholesterol, high blood pressure (or treatment for previously diagnosed hypertension), and increased fasting plasma glucose (or previous diagnosis of type 2 diabetes).
The average age of the participants was 49 years; the mean body mass index ranged from approximately 22 kg/m2 in the low-stable group to approximately 28 kg/m2 in the elevated-increasing group.
The four trajectories of metabolic syndrome were low-stable (10.56% of participants), moderate-low (40.84%), moderate-high (41.46%), and elevated-increasing (7.14%), based on trends from the individuals’ initial physical exams on entering the study.
Over a median follow-up period of 9.4 years (from 2010 to 2021), 2,271 cancer diagnoses were reported in the study population. Those with an elevated-increasing metabolic syndrome trajectory had 1.3 times the risk of any cancer compared with those in the low-stable group. Risk for breast cancer, endometrial cancer, kidney cancer, colorectal cancer, and liver cancer in the highest trajectory group were 2.1, 3.3, 4.5, 2.5, and 1.6 times higher, respectively, compared to the lowest group. The increased risk in the elevated-trajectory group for all cancer types persisted when the low-stable, moderate-low, and moderate-high trajectory pattern groups were combined.
The researchers also examined the impact of chronic inflammation and found that individuals with persistently high metabolic syndrome scores and concurrent chronic inflammation had the highest risks of breast, endometrial, colon, and liver cancer. However, individuals with persistently high metabolic syndrome scores and no concurrent chronic inflammation had the highest risk of kidney cancer.
What Are the Limitations of This Research?
The researchers of the current study acknowledged the lack of information on other causes of cancer, including dietary habits, hepatitis C infection, and Helicobacter pylori infection. Other limitations include the focus only on individuals from a single community of mainly middle-aged men in China that may not generalize to other populations.
Also, the metabolic syndrome trajectories did not change much over time, which may be related to the short 4-year study period.
Using the International Diabetes Federation criteria was another limitation, because it prevented the assessment of cancer risk in normal weight individuals with metabolic dysfunction, Dr. Hardikar noted.
Does Metabolic Syndrome Cause Cancer?
“This research suggests that proactive and continuous management of metabolic syndrome may serve as an essential strategy in preventing cancer,” senior author Han-Ping Shi, MD, PhD, of Capital Medical University in Beijing, noted in a statement on the study.
More research is needed to assess the impact of these interventions on cancer risk. However, the data from the current study can guide future research that may lead to more targeted treatments and more effective preventive strategies, he continued.
“Current evidence based on this study and many other reports strongly suggests an increased risk for cancer associated with metabolic syndrome,” Dr. Hardikar said in an interview. The data serve as a reminder to clinicians to look beyond BMI as the only measure of obesity, and to consider metabolic factors together to identify individuals at increased risk for cancer, she said.
“We must continue to educate patients about obesity and all the chronic conditions it may lead to, but we cannot ignore this emerging phenotype of being of normal weight but metabolically unhealthy,” Dr. Hardikar emphasized.
What Additional Research is Needed?
Looking ahead, “we need well-designed interventions to test causality for metabolic syndrome and cancer risk, though the evidence from the observational studies is very strong,” Dr. Hardikar said.
In addition, a consensus is needed to better define metabolic dysfunction,and to explore cancer risk in normal weight but metabolically unhealthy individuals, she said.
The study was supported by the National Key Research and Development Program of China. The researchers and Dr. Hardikar had no financial conflicts to disclose.
The new research finds that adults with persistent metabolic syndrome that worsens over time are at increased risk for any type of cancer.
The conditions that make up metabolic syndrome (high blood pressure, high blood sugar, increased abdominal adiposity, and high cholesterol and triglycerides) have been associated with an increased risk of diseases, including heart disease, stroke, and type 2 diabetes, wrote Li Deng, PhD, of Capital Medical University, Beijing, and colleagues.
However, a single assessment of metabolic syndrome at one point in time is inadequate to show an association with cancer risk over time, they said. In the current study, the researchers used models to examine the association between trajectory patterns of metabolic syndrome over time and the risk of overall and specific cancer types. They also examined the impact of chronic inflammation concurrent with metabolic syndrome.
What We Know About Metabolic Syndrome and Cancer Risk
A systematic review and meta-analysis published in Diabetes Care in 2012 showed an association between the presence of metabolic syndrome and an increased risk of various cancers including liver, bladder, pancreatic, breast, and colorectal.
More recently, a 2020 study published in Diabetes showed evidence of increased risk for certain cancers (pancreatic, kidney, uterine, cervical) but no increased risk for cancer overall.
In addition, a 2022 study by some of the current study researchers of the same Chinese cohort focused on the role of inflammation in combination with metabolic syndrome on colorectal cancer specifically, and found an increased risk for cancer when both metabolic syndrome and inflammation were present.
However, the reasons for this association between metabolic syndrome and cancer remain unclear, and the effect of the fluctuating nature of metabolic syndrome over time on long-term cancer risk has not been explored, the researchers wrote.
“There is emerging evidence that even normal weight individuals who are metabolically unhealthy may be at an elevated cancer risk, and we need better metrics to define the underlying metabolic dysfunction in obesity,” Sheetal Hardikar, MBBS, PhD, MPH, an investigator at the Huntsman Cancer Institute, University of Utah, said in an interview.
Dr. Hardikar, who serves as assistant professor in the department of population health sciences at the University of Utah, was not involved in the current study. She and her colleagues published a research paper on data from the National Health and Nutrition Examination Survey in 2023 that showed an increased risk of obesity-related cancer.
What New Study Adds to Related Research
Previous studies have consistently reported an approximately 30% increased risk of cancer with metabolic syndrome, Dr. Hardikar said. “What is unique about this study is the examination of metabolic syndrome trajectories over four years, and not just the presence of metabolic syndrome at one point in time,” she said.
In the new study, published in Cancer on March 11 (doi: 10.1002/cncr.35235), 44,115 adults in China were separated into four trajectories based on metabolic syndrome scores for the period from 2006 to 2010. The scores were based on clinical evidence of metabolic syndrome, defined using the International Diabetes Federation criteria of central obesity and the presence of at least two other factors including increased triglycerides, decreased HDL cholesterol, high blood pressure (or treatment for previously diagnosed hypertension), and increased fasting plasma glucose (or previous diagnosis of type 2 diabetes).
The average age of the participants was 49 years; the mean body mass index ranged from approximately 22 kg/m2 in the low-stable group to approximately 28 kg/m2 in the elevated-increasing group.
The four trajectories of metabolic syndrome were low-stable (10.56% of participants), moderate-low (40.84%), moderate-high (41.46%), and elevated-increasing (7.14%), based on trends from the individuals’ initial physical exams on entering the study.
Over a median follow-up period of 9.4 years (from 2010 to 2021), 2,271 cancer diagnoses were reported in the study population. Those with an elevated-increasing metabolic syndrome trajectory had 1.3 times the risk of any cancer compared with those in the low-stable group. Risk for breast cancer, endometrial cancer, kidney cancer, colorectal cancer, and liver cancer in the highest trajectory group were 2.1, 3.3, 4.5, 2.5, and 1.6 times higher, respectively, compared to the lowest group. The increased risk in the elevated-trajectory group for all cancer types persisted when the low-stable, moderate-low, and moderate-high trajectory pattern groups were combined.
The researchers also examined the impact of chronic inflammation and found that individuals with persistently high metabolic syndrome scores and concurrent chronic inflammation had the highest risks of breast, endometrial, colon, and liver cancer. However, individuals with persistently high metabolic syndrome scores and no concurrent chronic inflammation had the highest risk of kidney cancer.
What Are the Limitations of This Research?
The researchers of the current study acknowledged the lack of information on other causes of cancer, including dietary habits, hepatitis C infection, and Helicobacter pylori infection. Other limitations include the focus only on individuals from a single community of mainly middle-aged men in China that may not generalize to other populations.
Also, the metabolic syndrome trajectories did not change much over time, which may be related to the short 4-year study period.
Using the International Diabetes Federation criteria was another limitation, because it prevented the assessment of cancer risk in normal weight individuals with metabolic dysfunction, Dr. Hardikar noted.
Does Metabolic Syndrome Cause Cancer?
“This research suggests that proactive and continuous management of metabolic syndrome may serve as an essential strategy in preventing cancer,” senior author Han-Ping Shi, MD, PhD, of Capital Medical University in Beijing, noted in a statement on the study.
More research is needed to assess the impact of these interventions on cancer risk. However, the data from the current study can guide future research that may lead to more targeted treatments and more effective preventive strategies, he continued.
“Current evidence based on this study and many other reports strongly suggests an increased risk for cancer associated with metabolic syndrome,” Dr. Hardikar said in an interview. The data serve as a reminder to clinicians to look beyond BMI as the only measure of obesity, and to consider metabolic factors together to identify individuals at increased risk for cancer, she said.
“We must continue to educate patients about obesity and all the chronic conditions it may lead to, but we cannot ignore this emerging phenotype of being of normal weight but metabolically unhealthy,” Dr. Hardikar emphasized.
What Additional Research is Needed?
Looking ahead, “we need well-designed interventions to test causality for metabolic syndrome and cancer risk, though the evidence from the observational studies is very strong,” Dr. Hardikar said.
In addition, a consensus is needed to better define metabolic dysfunction,and to explore cancer risk in normal weight but metabolically unhealthy individuals, she said.
The study was supported by the National Key Research and Development Program of China. The researchers and Dr. Hardikar had no financial conflicts to disclose.
The new research finds that adults with persistent metabolic syndrome that worsens over time are at increased risk for any type of cancer.
The conditions that make up metabolic syndrome (high blood pressure, high blood sugar, increased abdominal adiposity, and high cholesterol and triglycerides) have been associated with an increased risk of diseases, including heart disease, stroke, and type 2 diabetes, wrote Li Deng, PhD, of Capital Medical University, Beijing, and colleagues.
However, a single assessment of metabolic syndrome at one point in time is inadequate to show an association with cancer risk over time, they said. In the current study, the researchers used models to examine the association between trajectory patterns of metabolic syndrome over time and the risk of overall and specific cancer types. They also examined the impact of chronic inflammation concurrent with metabolic syndrome.
What We Know About Metabolic Syndrome and Cancer Risk
A systematic review and meta-analysis published in Diabetes Care in 2012 showed an association between the presence of metabolic syndrome and an increased risk of various cancers including liver, bladder, pancreatic, breast, and colorectal.
More recently, a 2020 study published in Diabetes showed evidence of increased risk for certain cancers (pancreatic, kidney, uterine, cervical) but no increased risk for cancer overall.
In addition, a 2022 study by some of the current study researchers of the same Chinese cohort focused on the role of inflammation in combination with metabolic syndrome on colorectal cancer specifically, and found an increased risk for cancer when both metabolic syndrome and inflammation were present.
However, the reasons for this association between metabolic syndrome and cancer remain unclear, and the effect of the fluctuating nature of metabolic syndrome over time on long-term cancer risk has not been explored, the researchers wrote.
“There is emerging evidence that even normal weight individuals who are metabolically unhealthy may be at an elevated cancer risk, and we need better metrics to define the underlying metabolic dysfunction in obesity,” Sheetal Hardikar, MBBS, PhD, MPH, an investigator at the Huntsman Cancer Institute, University of Utah, said in an interview.
Dr. Hardikar, who serves as assistant professor in the department of population health sciences at the University of Utah, was not involved in the current study. She and her colleagues published a research paper on data from the National Health and Nutrition Examination Survey in 2023 that showed an increased risk of obesity-related cancer.
What New Study Adds to Related Research
Previous studies have consistently reported an approximately 30% increased risk of cancer with metabolic syndrome, Dr. Hardikar said. “What is unique about this study is the examination of metabolic syndrome trajectories over four years, and not just the presence of metabolic syndrome at one point in time,” she said.
In the new study, published in Cancer on March 11 (doi: 10.1002/cncr.35235), 44,115 adults in China were separated into four trajectories based on metabolic syndrome scores for the period from 2006 to 2010. The scores were based on clinical evidence of metabolic syndrome, defined using the International Diabetes Federation criteria of central obesity and the presence of at least two other factors including increased triglycerides, decreased HDL cholesterol, high blood pressure (or treatment for previously diagnosed hypertension), and increased fasting plasma glucose (or previous diagnosis of type 2 diabetes).
The average age of the participants was 49 years; the mean body mass index ranged from approximately 22 kg/m2 in the low-stable group to approximately 28 kg/m2 in the elevated-increasing group.
The four trajectories of metabolic syndrome were low-stable (10.56% of participants), moderate-low (40.84%), moderate-high (41.46%), and elevated-increasing (7.14%), based on trends from the individuals’ initial physical exams on entering the study.
Over a median follow-up period of 9.4 years (from 2010 to 2021), 2,271 cancer diagnoses were reported in the study population. Those with an elevated-increasing metabolic syndrome trajectory had 1.3 times the risk of any cancer compared with those in the low-stable group. Risk for breast cancer, endometrial cancer, kidney cancer, colorectal cancer, and liver cancer in the highest trajectory group were 2.1, 3.3, 4.5, 2.5, and 1.6 times higher, respectively, compared to the lowest group. The increased risk in the elevated-trajectory group for all cancer types persisted when the low-stable, moderate-low, and moderate-high trajectory pattern groups were combined.
The researchers also examined the impact of chronic inflammation and found that individuals with persistently high metabolic syndrome scores and concurrent chronic inflammation had the highest risks of breast, endometrial, colon, and liver cancer. However, individuals with persistently high metabolic syndrome scores and no concurrent chronic inflammation had the highest risk of kidney cancer.
What Are the Limitations of This Research?
The researchers of the current study acknowledged the lack of information on other causes of cancer, including dietary habits, hepatitis C infection, and Helicobacter pylori infection. Other limitations include the focus only on individuals from a single community of mainly middle-aged men in China that may not generalize to other populations.
Also, the metabolic syndrome trajectories did not change much over time, which may be related to the short 4-year study period.
Using the International Diabetes Federation criteria was another limitation, because it prevented the assessment of cancer risk in normal weight individuals with metabolic dysfunction, Dr. Hardikar noted.
Does Metabolic Syndrome Cause Cancer?
“This research suggests that proactive and continuous management of metabolic syndrome may serve as an essential strategy in preventing cancer,” senior author Han-Ping Shi, MD, PhD, of Capital Medical University in Beijing, noted in a statement on the study.
More research is needed to assess the impact of these interventions on cancer risk. However, the data from the current study can guide future research that may lead to more targeted treatments and more effective preventive strategies, he continued.
“Current evidence based on this study and many other reports strongly suggests an increased risk for cancer associated with metabolic syndrome,” Dr. Hardikar said in an interview. The data serve as a reminder to clinicians to look beyond BMI as the only measure of obesity, and to consider metabolic factors together to identify individuals at increased risk for cancer, she said.
“We must continue to educate patients about obesity and all the chronic conditions it may lead to, but we cannot ignore this emerging phenotype of being of normal weight but metabolically unhealthy,” Dr. Hardikar emphasized.
What Additional Research is Needed?
Looking ahead, “we need well-designed interventions to test causality for metabolic syndrome and cancer risk, though the evidence from the observational studies is very strong,” Dr. Hardikar said.
In addition, a consensus is needed to better define metabolic dysfunction,and to explore cancer risk in normal weight but metabolically unhealthy individuals, she said.
The study was supported by the National Key Research and Development Program of China. The researchers and Dr. Hardikar had no financial conflicts to disclose.
FROM CANCER
ASTRO Pushes Return to Direct Supervision in RT: Needed or ‘Babysitting’?
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
FDA Removes Harmful Chemicals From Food Packaging
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
New Trials in Leukemia and Lymphoma: Could Your Patient Benefit?
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
Are Food Emulsifiers Associated With Increased Cancer Risk?
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Democratic Lawmakers Press Pfizer on Chemotherapy Drug Shortages
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
Unleashing Our Immune Response to Quash Cancer
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
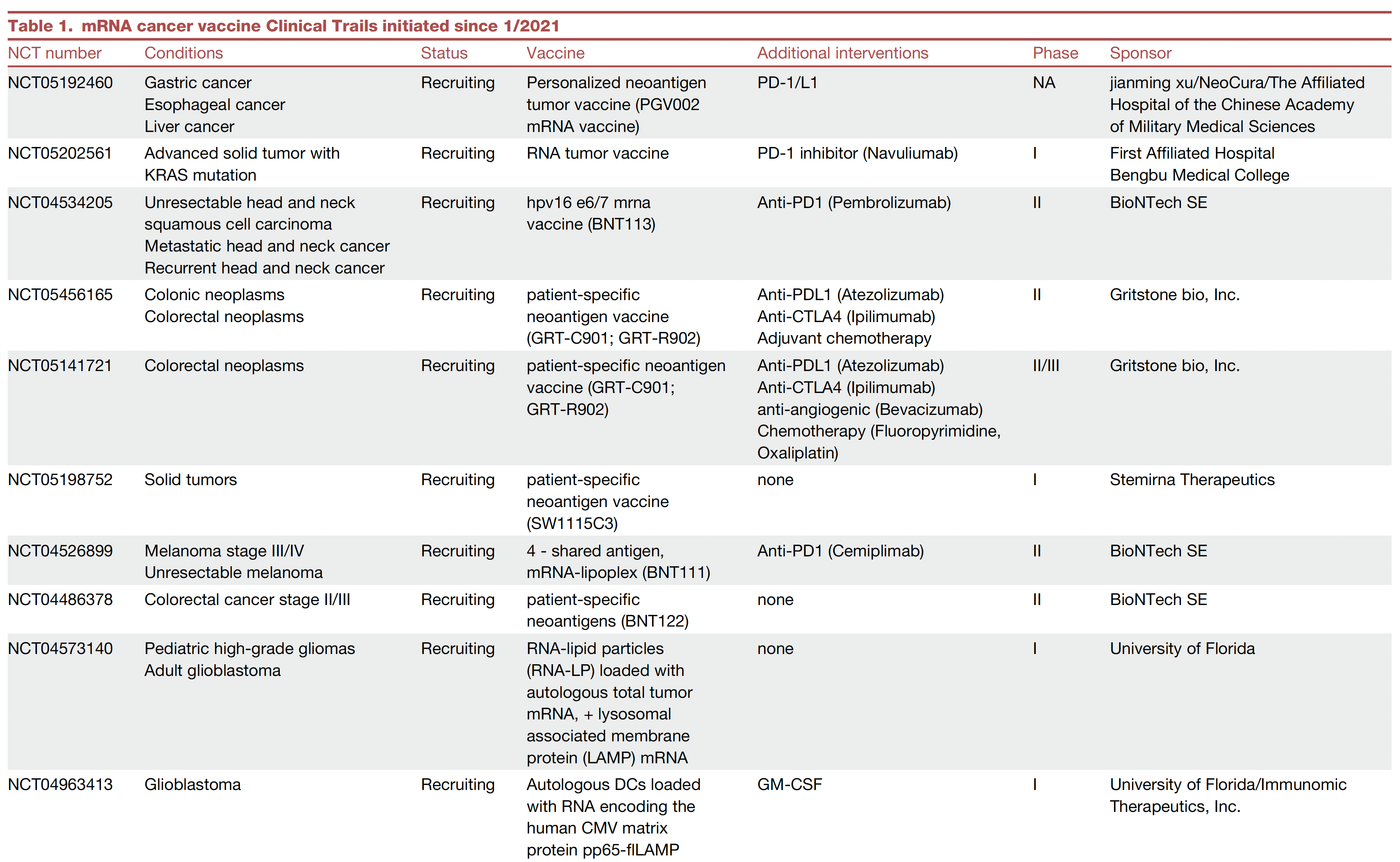{kind=link}
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
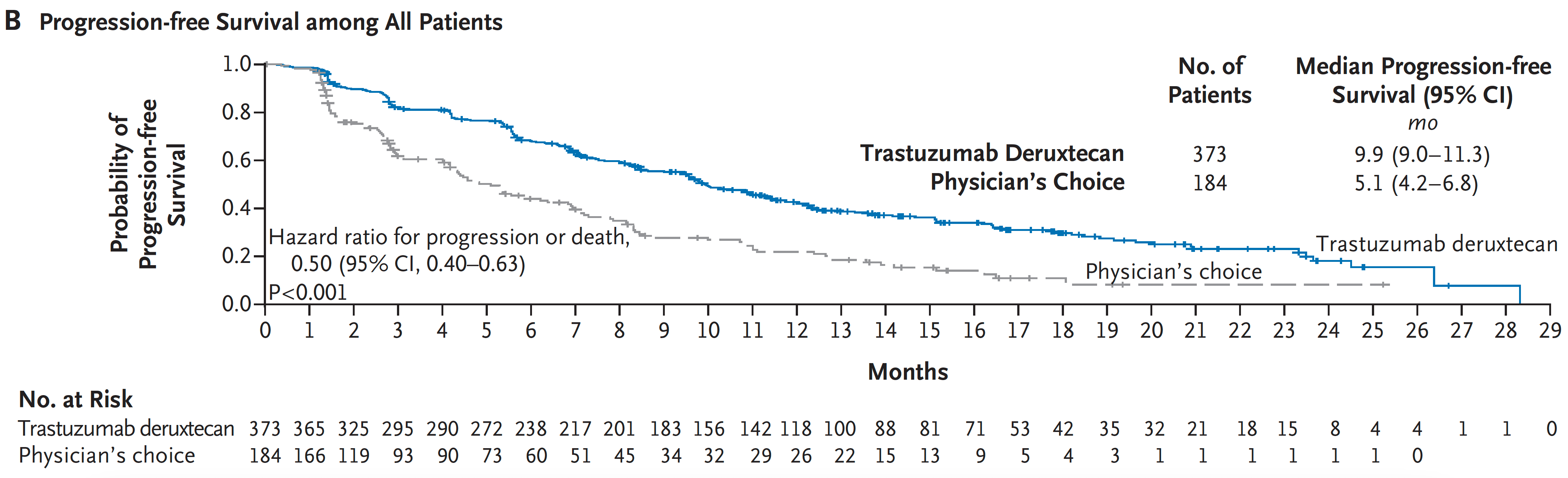{kind=link}
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
Measurable residual disease–guided therapy promising for CLL
The targeted therapy combination was also associated with better progression-free survival among patients with CLL with worse prognostic features, including immunoglobulin heavy chain variable (IGHV) unmutated disease and cytogenetic abnormalities, such as the 11q deletion, trisomy 12, and 13q deletion.
Personalizing treatment duration of ibrutinib-venetoclax, as determined by measurable residual disease (MRD), allowed more than half of patients assigned to the combination therapy to stop therapy by 3 years because they had achieved MRD negativity, reported Peter Hillmen, MBChB, PhD, from the Leeds Institute of Medical Research at St James’s University Hospital in Leeds, United Kingdom.
The shorter course of therapy could help to ameliorate toxicities and lower the risk for the development of drug-resistant disease, he said.
“This is the first trial to show that an MRD-guided approach with treatment beyond [MRD] negativity has a significant advantage over chemoimmunotherapy, both in terms of [progression-free] and overall survival. Over 90% of patients achieve an MRD-negative in this combination in the peripheral blood,” said Dr. Hillmen in a media briefing prior to his presentation of the data in an oral abstract session here at the American Society of Hematology annual meeting.
The study results were also published online in The New England Journal of Medicine to coincide with the presentation.
Adaptive Trial
The FLAIR study is a phase 3 open-label platform trial that initially compared ibrutinib-rituximab with fludarabine-cyclophosphamide-rituximab (FCR) in patients with untreated CLL. However, in 2017 the trial was adapted to include both an ibrutinib monotherapy and an ibrutinib-venetoclax arm with therapy duration determined by MRD.
At ASH 2023, Dr. Hillmen presented data from an interim analysis of 523 patients comparing ibrutinib-venetoclax with FCR.
In the ibrutinib-venetoclax group, patients received oral ibrutinib 420 mg daily, with venetoclax added after 2 months, beginning with a 20-mg dose ramped up to 400 mg in a weekly dose-escalation schedule. The combination could be given for 2-6 years, depending on MRD responses. FCR was delivered in up to six cycles of 28 days each. Two thirds of patients assigned to FCR completed all six cycles.
After a median follow-up of 43.7 months, 12 patients (4.6%) randomly assigned to ibrutinib-venetoclax had disease progression or died compared with 75 patients (28.5%) assigned to FCR. The estimated 3-year progression-free survival with ibrutinib-venetoclax was 97.2%, compared with 76.8% with FCR, translating into a hazard ratio (HR) for progression or death with the targeted therapy combination of 0.13 (P <.001).
Among patients with unmutated IGHV, the combination led to improved progression-free survival compared with FCR (hazard ratio [HR] for progression or death, 0.07); for patients with mutated IGHV, however, the combination did not improve progression-free survival (HR, 0.54; 95% CI, 0.21-1.38).
In all, eight patients (3.5%) assigned to ibrutinib-venetoclax and 23 assigned to FCR (9.5%) died.
The 3-year overall survival rates were 98% in the targeted therapy group vs 93% in the FCR group (HR for progression or death, 0.31).
At 2 years, 52.4% of patients assigned to ibrutinib-venetoclax had undetectable MRD in bone marrow compared with 49.8% with FCR. At 5 years, the respective percentages for MRD in bone marrow were 65.9% vs 49.8% and 92.7% vs 67.9% for MRD in peripheral blood.
The safety analysis showed higher rates of blood and lymphatic system disorders with FCR, whereas cardiac, metabolic/nutrition disorders, and eye disorders occurred more frequent with ibrutinib-venetoclax.
A total of 24 secondary cancers were diagnosed in 17 patients randomly assigned to ibrutinib-venetoclax and 45 secondary cancers among 34 patients randomly assigned to FCR. One patient assigned to ibrutinib-venetoclax developed myelodysplastic syndrome/acute myeloid leukemia (AML), as did eight patients assigned to FCR. One patient in the ibrutinib-venetoclax arm and four patients in the FCR arm had Richter’s transformation.
The incidence rate for other cancers was 2.6 per 100 person-years with ibrutinib-venetoclax compared with 5.4 per 100 person-years with FCR.
The most frequently occurring cancers in each arm were basal cell or squamous cell carcinomas. The incidence of myelodysplastic syndromes, AML lymphoma, and prostate/urologic cancers was higher among patients on FCR.
This research “unequivocally shows the superiority of targeted therapy over traditional cytotoxic chemotherapy,” commented briefing moderator Mikkael A. Sekeres, MD, from the University of Miami Miller School of Medicine.
The ibrutinib-venetoclax combination has the potential to reduce the incidence of myelodysplastic syndromes secondary to CLL therapy, Dr. Sekeres suggested.
“As someone who specializes in leukemia and myelodysplastic syndromes, I have the feeling I won’t be seeing these CLL patients in my clinic — years after being treated for CLL — much longer,” he said.
In an interview with this news organization, Lee Greenberger, PhD, said that “I think that duration-adapted therapy is a great story. Using MRD negativity is a perfectly justified way, I think, to go about a new combination that’s going to be really potent for CLL patients and probably give them many years of treatment and then to get off the drug, because ultimately the goal is to get cures.”
This combination, though highly efficacious, is unlikely to be curative; however, because even when MRD is undetectable, “it will come back,” said Dr. Greenberger, chief scientific officer for the Leukemia & Lymphoma Society.
Dr. Greenberger added that MRD testing of bone marrow, which provides a more detailed picture of MRD status than testing of peripheral blood, is feasible in academic medical centers but may be a barrier to MRD-adapted therapy in community oncology practices.
The FLAIR study is supported by grants from Cancer Research UK, Janssen, Pharmacyclics, and AbbVie. Dr. Hillmen disclosed employment and equity participation with Apellis Pharmaceuticals. Dr. Sekeres disclosed board activities for Geron, Novartis, and Bristol-Myers Squibb and owner of stock options Kurome. Dr. Greenberger reported no relevant financial disclosures.
A version of this article appeared on Medscape.com.
The targeted therapy combination was also associated with better progression-free survival among patients with CLL with worse prognostic features, including immunoglobulin heavy chain variable (IGHV) unmutated disease and cytogenetic abnormalities, such as the 11q deletion, trisomy 12, and 13q deletion.
Personalizing treatment duration of ibrutinib-venetoclax, as determined by measurable residual disease (MRD), allowed more than half of patients assigned to the combination therapy to stop therapy by 3 years because they had achieved MRD negativity, reported Peter Hillmen, MBChB, PhD, from the Leeds Institute of Medical Research at St James’s University Hospital in Leeds, United Kingdom.
The shorter course of therapy could help to ameliorate toxicities and lower the risk for the development of drug-resistant disease, he said.
“This is the first trial to show that an MRD-guided approach with treatment beyond [MRD] negativity has a significant advantage over chemoimmunotherapy, both in terms of [progression-free] and overall survival. Over 90% of patients achieve an MRD-negative in this combination in the peripheral blood,” said Dr. Hillmen in a media briefing prior to his presentation of the data in an oral abstract session here at the American Society of Hematology annual meeting.
The study results were also published online in The New England Journal of Medicine to coincide with the presentation.
Adaptive Trial
The FLAIR study is a phase 3 open-label platform trial that initially compared ibrutinib-rituximab with fludarabine-cyclophosphamide-rituximab (FCR) in patients with untreated CLL. However, in 2017 the trial was adapted to include both an ibrutinib monotherapy and an ibrutinib-venetoclax arm with therapy duration determined by MRD.
At ASH 2023, Dr. Hillmen presented data from an interim analysis of 523 patients comparing ibrutinib-venetoclax with FCR.
In the ibrutinib-venetoclax group, patients received oral ibrutinib 420 mg daily, with venetoclax added after 2 months, beginning with a 20-mg dose ramped up to 400 mg in a weekly dose-escalation schedule. The combination could be given for 2-6 years, depending on MRD responses. FCR was delivered in up to six cycles of 28 days each. Two thirds of patients assigned to FCR completed all six cycles.
After a median follow-up of 43.7 months, 12 patients (4.6%) randomly assigned to ibrutinib-venetoclax had disease progression or died compared with 75 patients (28.5%) assigned to FCR. The estimated 3-year progression-free survival with ibrutinib-venetoclax was 97.2%, compared with 76.8% with FCR, translating into a hazard ratio (HR) for progression or death with the targeted therapy combination of 0.13 (P <.001).
Among patients with unmutated IGHV, the combination led to improved progression-free survival compared with FCR (hazard ratio [HR] for progression or death, 0.07); for patients with mutated IGHV, however, the combination did not improve progression-free survival (HR, 0.54; 95% CI, 0.21-1.38).
In all, eight patients (3.5%) assigned to ibrutinib-venetoclax and 23 assigned to FCR (9.5%) died.
The 3-year overall survival rates were 98% in the targeted therapy group vs 93% in the FCR group (HR for progression or death, 0.31).
At 2 years, 52.4% of patients assigned to ibrutinib-venetoclax had undetectable MRD in bone marrow compared with 49.8% with FCR. At 5 years, the respective percentages for MRD in bone marrow were 65.9% vs 49.8% and 92.7% vs 67.9% for MRD in peripheral blood.
The safety analysis showed higher rates of blood and lymphatic system disorders with FCR, whereas cardiac, metabolic/nutrition disorders, and eye disorders occurred more frequent with ibrutinib-venetoclax.
A total of 24 secondary cancers were diagnosed in 17 patients randomly assigned to ibrutinib-venetoclax and 45 secondary cancers among 34 patients randomly assigned to FCR. One patient assigned to ibrutinib-venetoclax developed myelodysplastic syndrome/acute myeloid leukemia (AML), as did eight patients assigned to FCR. One patient in the ibrutinib-venetoclax arm and four patients in the FCR arm had Richter’s transformation.
The incidence rate for other cancers was 2.6 per 100 person-years with ibrutinib-venetoclax compared with 5.4 per 100 person-years with FCR.
The most frequently occurring cancers in each arm were basal cell or squamous cell carcinomas. The incidence of myelodysplastic syndromes, AML lymphoma, and prostate/urologic cancers was higher among patients on FCR.
This research “unequivocally shows the superiority of targeted therapy over traditional cytotoxic chemotherapy,” commented briefing moderator Mikkael A. Sekeres, MD, from the University of Miami Miller School of Medicine.
The ibrutinib-venetoclax combination has the potential to reduce the incidence of myelodysplastic syndromes secondary to CLL therapy, Dr. Sekeres suggested.
“As someone who specializes in leukemia and myelodysplastic syndromes, I have the feeling I won’t be seeing these CLL patients in my clinic — years after being treated for CLL — much longer,” he said.
In an interview with this news organization, Lee Greenberger, PhD, said that “I think that duration-adapted therapy is a great story. Using MRD negativity is a perfectly justified way, I think, to go about a new combination that’s going to be really potent for CLL patients and probably give them many years of treatment and then to get off the drug, because ultimately the goal is to get cures.”
This combination, though highly efficacious, is unlikely to be curative; however, because even when MRD is undetectable, “it will come back,” said Dr. Greenberger, chief scientific officer for the Leukemia & Lymphoma Society.
Dr. Greenberger added that MRD testing of bone marrow, which provides a more detailed picture of MRD status than testing of peripheral blood, is feasible in academic medical centers but may be a barrier to MRD-adapted therapy in community oncology practices.
The FLAIR study is supported by grants from Cancer Research UK, Janssen, Pharmacyclics, and AbbVie. Dr. Hillmen disclosed employment and equity participation with Apellis Pharmaceuticals. Dr. Sekeres disclosed board activities for Geron, Novartis, and Bristol-Myers Squibb and owner of stock options Kurome. Dr. Greenberger reported no relevant financial disclosures.
A version of this article appeared on Medscape.com.
The targeted therapy combination was also associated with better progression-free survival among patients with CLL with worse prognostic features, including immunoglobulin heavy chain variable (IGHV) unmutated disease and cytogenetic abnormalities, such as the 11q deletion, trisomy 12, and 13q deletion.
Personalizing treatment duration of ibrutinib-venetoclax, as determined by measurable residual disease (MRD), allowed more than half of patients assigned to the combination therapy to stop therapy by 3 years because they had achieved MRD negativity, reported Peter Hillmen, MBChB, PhD, from the Leeds Institute of Medical Research at St James’s University Hospital in Leeds, United Kingdom.
The shorter course of therapy could help to ameliorate toxicities and lower the risk for the development of drug-resistant disease, he said.
“This is the first trial to show that an MRD-guided approach with treatment beyond [MRD] negativity has a significant advantage over chemoimmunotherapy, both in terms of [progression-free] and overall survival. Over 90% of patients achieve an MRD-negative in this combination in the peripheral blood,” said Dr. Hillmen in a media briefing prior to his presentation of the data in an oral abstract session here at the American Society of Hematology annual meeting.
The study results were also published online in The New England Journal of Medicine to coincide with the presentation.
Adaptive Trial
The FLAIR study is a phase 3 open-label platform trial that initially compared ibrutinib-rituximab with fludarabine-cyclophosphamide-rituximab (FCR) in patients with untreated CLL. However, in 2017 the trial was adapted to include both an ibrutinib monotherapy and an ibrutinib-venetoclax arm with therapy duration determined by MRD.
At ASH 2023, Dr. Hillmen presented data from an interim analysis of 523 patients comparing ibrutinib-venetoclax with FCR.
In the ibrutinib-venetoclax group, patients received oral ibrutinib 420 mg daily, with venetoclax added after 2 months, beginning with a 20-mg dose ramped up to 400 mg in a weekly dose-escalation schedule. The combination could be given for 2-6 years, depending on MRD responses. FCR was delivered in up to six cycles of 28 days each. Two thirds of patients assigned to FCR completed all six cycles.
After a median follow-up of 43.7 months, 12 patients (4.6%) randomly assigned to ibrutinib-venetoclax had disease progression or died compared with 75 patients (28.5%) assigned to FCR. The estimated 3-year progression-free survival with ibrutinib-venetoclax was 97.2%, compared with 76.8% with FCR, translating into a hazard ratio (HR) for progression or death with the targeted therapy combination of 0.13 (P <.001).
Among patients with unmutated IGHV, the combination led to improved progression-free survival compared with FCR (hazard ratio [HR] for progression or death, 0.07); for patients with mutated IGHV, however, the combination did not improve progression-free survival (HR, 0.54; 95% CI, 0.21-1.38).
In all, eight patients (3.5%) assigned to ibrutinib-venetoclax and 23 assigned to FCR (9.5%) died.
The 3-year overall survival rates were 98% in the targeted therapy group vs 93% in the FCR group (HR for progression or death, 0.31).
At 2 years, 52.4% of patients assigned to ibrutinib-venetoclax had undetectable MRD in bone marrow compared with 49.8% with FCR. At 5 years, the respective percentages for MRD in bone marrow were 65.9% vs 49.8% and 92.7% vs 67.9% for MRD in peripheral blood.
The safety analysis showed higher rates of blood and lymphatic system disorders with FCR, whereas cardiac, metabolic/nutrition disorders, and eye disorders occurred more frequent with ibrutinib-venetoclax.
A total of 24 secondary cancers were diagnosed in 17 patients randomly assigned to ibrutinib-venetoclax and 45 secondary cancers among 34 patients randomly assigned to FCR. One patient assigned to ibrutinib-venetoclax developed myelodysplastic syndrome/acute myeloid leukemia (AML), as did eight patients assigned to FCR. One patient in the ibrutinib-venetoclax arm and four patients in the FCR arm had Richter’s transformation.
The incidence rate for other cancers was 2.6 per 100 person-years with ibrutinib-venetoclax compared with 5.4 per 100 person-years with FCR.
The most frequently occurring cancers in each arm were basal cell or squamous cell carcinomas. The incidence of myelodysplastic syndromes, AML lymphoma, and prostate/urologic cancers was higher among patients on FCR.
This research “unequivocally shows the superiority of targeted therapy over traditional cytotoxic chemotherapy,” commented briefing moderator Mikkael A. Sekeres, MD, from the University of Miami Miller School of Medicine.
The ibrutinib-venetoclax combination has the potential to reduce the incidence of myelodysplastic syndromes secondary to CLL therapy, Dr. Sekeres suggested.
“As someone who specializes in leukemia and myelodysplastic syndromes, I have the feeling I won’t be seeing these CLL patients in my clinic — years after being treated for CLL — much longer,” he said.
In an interview with this news organization, Lee Greenberger, PhD, said that “I think that duration-adapted therapy is a great story. Using MRD negativity is a perfectly justified way, I think, to go about a new combination that’s going to be really potent for CLL patients and probably give them many years of treatment and then to get off the drug, because ultimately the goal is to get cures.”
This combination, though highly efficacious, is unlikely to be curative; however, because even when MRD is undetectable, “it will come back,” said Dr. Greenberger, chief scientific officer for the Leukemia & Lymphoma Society.
Dr. Greenberger added that MRD testing of bone marrow, which provides a more detailed picture of MRD status than testing of peripheral blood, is feasible in academic medical centers but may be a barrier to MRD-adapted therapy in community oncology practices.
The FLAIR study is supported by grants from Cancer Research UK, Janssen, Pharmacyclics, and AbbVie. Dr. Hillmen disclosed employment and equity participation with Apellis Pharmaceuticals. Dr. Sekeres disclosed board activities for Geron, Novartis, and Bristol-Myers Squibb and owner of stock options Kurome. Dr. Greenberger reported no relevant financial disclosures.
A version of this article appeared on Medscape.com.
FROM ASH 2023
FDA approves pirtobrutinib for previously treated CLL/SLL

The agent was initially approved in January 2023 for patients with mantle cell lymphoma who had previously received a BTK inhibitor.
Like the mantle cell approval, the CLL/SLL approval was based on findings from the open-label, single-arm, phase 1/2 BRUIN study that included adults with at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor.
The trial included 108 patients with either CLL or SLL. Overall, patients demonstrated an overall response rate of 72%, all of which were partial responses, and median duration of response of 12.2 months.
Before starting pirtobrutinib, 77% of patients with CLL or SLL had discontinued their last BTK inhibitor for refractory or progressive disease.
“Once patients with CLL or SLL have progressed on covalent BTK inhibitor and BCL-2 inhibitor therapies, treatments are limited and outcomes can be poor, making the approval of Jaypirca a meaningful advance and much-needed new treatment option for these patients,” William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston, said in an Eli Lilly press release.
Treatment during the study included the recommended dose of 200 mg given orally once daily until disease progression or unacceptable toxicity. Common adverse reactions that occurred in at least 20% of patients included fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache. Grade 3 or 4 laboratory abnormalities occurring in more than 10% of patients included decreased neutrophil counts, anemia, and decreased platelet counts.
Serious infections occurred in 32% of patients, including fatal infections in 10% of patients. The prescribing information for pirtobrutinib includes warnings about infections, hemorrhage, cytopenias, cardiac arrhythmias, and secondary primary malignancies.
A version of this article first appeared on Medscape.com.
The agent was initially approved in January 2023 for patients with mantle cell lymphoma who had previously received a BTK inhibitor.
Like the mantle cell approval, the CLL/SLL approval was based on findings from the open-label, single-arm, phase 1/2 BRUIN study that included adults with at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor.
The trial included 108 patients with either CLL or SLL. Overall, patients demonstrated an overall response rate of 72%, all of which were partial responses, and median duration of response of 12.2 months.
Before starting pirtobrutinib, 77% of patients with CLL or SLL had discontinued their last BTK inhibitor for refractory or progressive disease.
“Once patients with CLL or SLL have progressed on covalent BTK inhibitor and BCL-2 inhibitor therapies, treatments are limited and outcomes can be poor, making the approval of Jaypirca a meaningful advance and much-needed new treatment option for these patients,” William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston, said in an Eli Lilly press release.
Treatment during the study included the recommended dose of 200 mg given orally once daily until disease progression or unacceptable toxicity. Common adverse reactions that occurred in at least 20% of patients included fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache. Grade 3 or 4 laboratory abnormalities occurring in more than 10% of patients included decreased neutrophil counts, anemia, and decreased platelet counts.
Serious infections occurred in 32% of patients, including fatal infections in 10% of patients. The prescribing information for pirtobrutinib includes warnings about infections, hemorrhage, cytopenias, cardiac arrhythmias, and secondary primary malignancies.
A version of this article first appeared on Medscape.com.
The agent was initially approved in January 2023 for patients with mantle cell lymphoma who had previously received a BTK inhibitor.
Like the mantle cell approval, the CLL/SLL approval was based on findings from the open-label, single-arm, phase 1/2 BRUIN study that included adults with at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor.
The trial included 108 patients with either CLL or SLL. Overall, patients demonstrated an overall response rate of 72%, all of which were partial responses, and median duration of response of 12.2 months.
Before starting pirtobrutinib, 77% of patients with CLL or SLL had discontinued their last BTK inhibitor for refractory or progressive disease.
“Once patients with CLL or SLL have progressed on covalent BTK inhibitor and BCL-2 inhibitor therapies, treatments are limited and outcomes can be poor, making the approval of Jaypirca a meaningful advance and much-needed new treatment option for these patients,” William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston, said in an Eli Lilly press release.
Treatment during the study included the recommended dose of 200 mg given orally once daily until disease progression or unacceptable toxicity. Common adverse reactions that occurred in at least 20% of patients included fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache. Grade 3 or 4 laboratory abnormalities occurring in more than 10% of patients included decreased neutrophil counts, anemia, and decreased platelet counts.
Serious infections occurred in 32% of patients, including fatal infections in 10% of patients. The prescribing information for pirtobrutinib includes warnings about infections, hemorrhage, cytopenias, cardiac arrhythmias, and secondary primary malignancies.
A version of this article first appeared on Medscape.com.