User login
Index finger plaque

The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.
The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.

Does early introduction of peanuts to an infant’s diet reduce the risk for peanut allergy?
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
EVIDENCE-BASED ANSWER:
Probably not, unless the child has severe eczema or egg allergy. In a general pediatric population, introducing peanuts early (at age 3 to 6 months) doesn’t appear to alter rates of subsequent peanut allergy compared with introduction after age 6 months (strength of recommendation [SOR]: B, randomized clinical trial [RCT] using multiple potential food allergens).
In children with severe eczema, egg allergy, or both, however, the risk for a peanut allergy is 12% to 24% lower when peanut-containing foods are introduced at age 4 to 11 months than after age 1 year. Early introduction of peanuts is associated with about 1 additional mild virus-associated syndrome (upper respiratory infection [URI], exanthem, conjunctivitis, or gastroenteritis) per patient (SOR: B, RCT).
Introducing peanuts before age 1 year is recommended for atopic children without evidence of pre-existing peanut allergy; an earlier start, at age 4 to 6 months, is advised for infants with severe eczema or egg allergy (SOR: C, expert opinion).

Hidradenitis Suppurativa in the Military
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Practice Points
- Hidradenitis suppurativa (HS) can be more difficult to treat in physically active military servicemembers (SMs).
- Patient education and primary care physician awareness of HS is critical to initial diagnosis and long-term management.
- Primary care physician knowledge of HS as well as an understanding of the capabilities at local military medical facilities is important for optimal treatment of HS in military SMs.

Neurofibromatosis type 1: More than skin deep
Neurofibromatosis type 1 (NF1) is an autosomal dominant inherited disorder that is estimated to occur in 1:2500 births and to have a prevalence of 1:2000 to 1:4000.1,2 It was first described in 1882 by Friedrich Daniel Von Recklinghausen, who identified patients and their relatives with signs of neuroectodermal abnormalities (café-au-lait macules [CALMs], axillary and inguinal freckling, and neurofibromas).
NF1 may begin insidiously in childhood and evolves as the patient ages. It is associated with intracranial, intraspinal, and intraorbital neoplasms, although other organs and tissues can also be involved.
The family physician might be the first one to recognize the signs of this condition during a well-child exam and is in a unique position to coordinate a multidisciplinary approach to care.
A mutated allele and early manifestations on the skin
NF1 has been attributed to genetic mosaicism and is classified as segmental, generalized, or (less frequently) gonadal. The disorder results from germline mutations in the NF1 tumor-suppressor gene on chromosome 17, known to codify the cytoplasmic protein called neurofibromin.3 The penetrance of NF1 is complete, which means that 100% of patients with the mutated allele will develop the disease.
Patients typically have symptoms by the third decade of life, although many will show signs of the disease in early childhood. CALMs are the earliest expression of NF1. They manifest in the first 2 years of life and are found in almost all affected patients. The lesions are well defined and measure 10 to 40 mm. They are typically light brown, although they may darken with sun exposure.
Histologically, the lesions will show macromelanosomes and high concentrations of melanin but do not represent an increased risk for malignancy.4 Not all isolated CALMs are a sign of NF1. While children younger than 29 months with 6 or more CALMs have a high risk for NF1 (80.4%; 95% confidence interval [CI], 74.6% to 86.2%), those who are older than 29 months with at least 1 atypical CALM or fewer than 6 CALMs have just a 0.9% (95% CI, 0% to 2.6%) risk for constitutional NF1.5
Freckles are also observed in 90% of patients with NF1; these tend to develop after the third year of life. The breast and trunk are the most commonly affected areas in adults. The pathophysiology is unknown, but this freckling is believed to be related to skin friction, high humidity, and ambient temperature.6
Continue to: Neurofibromas are benign...
Neurofibromas are benign subcutaneous palpable lesions that grow within peripheral nerve tissue, including spinal, subcutaneous, plexiform, or dermal encapsulated nerves. Originating in Schwann cells, they are composed of fibroblasts, mast cells, macrophages, endothelial cells, and other perineural cells. Some patients show disfiguration when hundreds of these masses are present (FIGURE). These tumors increase in number as the patient ages or during pregnancy, which is thought to be secondary to hormonal changes.7 They are sometimes painful and can be pruritic. Their appearance can also cause patient distress.

The diagnosis is a clinical one
Suspicion for NF1 should be high in patients presenting with the dermatologic findings described, although CALMs and freckling are not exclusive to NF1. Diagnostic criteria for NF1, which distinguish it from other conditions, were first outlined in a National Institutes of Health Consensus Development Conference Statement in 1987.8 The list of criteria has subsequently been expanded.
While the presence of at least 2 criteria is required for diagnosis,2 NF1 should be suspected in individuals who have any of the following findings8,9:
- the presence of at least 6 CALMs that are > 5 mm in prepubertal children and > 15 mm in adults
- 2 or more neurofibromas of any type, or at least one plexiform neurofibroma
- axillary or groin freckling
- optic pathway glioma
- 2 or more Lisch nodules (iris hamartomas seen on slit-lamp examination)
- bony dysplasia (sphenoid wing dysplasia, bowing of long bone ± pseudarthrosis)
- first-degree relative with NF1.
What you’ll see as the disease progresses
NF1 can affect a variety of systems, and potential complications of the disease are numerous and varied (see TABLE9). Here is some of what you may see as the patient’s disease progresses to various organ systems:
Learning disabilities and other cognitive and behavioral problems, such as attention-deficit/hyperactivity disorder, may affect up to 70% of children with NF1. Additionally, children with NF1 have visual/spatial problems, impaired visual motor integration, and language deficits.10 The etiology of cognitive impairment in NF1 is unknown.11

Continue to: Hypertension
Hypertension is common and may contribute to premature death in patients with NF1. Up to 27% of patients will have significant cardiovascular anomalies, including pulmonary valve stenosis, hypertrophic cardiomyopathy in patients with complete deletions of the NF1 gene, intracardiac neurofibromas, renal artery stenosis, coarctation of the aorta, and cerebral infarctions.12 Renal artery stenosis occurs in approximately 2% of the NF1 population, and the diagnosis should be considered in hypertensive children, young adults, pregnant women, older individuals with refractory hypertension, and those with an abdominal bruit.13
Psychological issues. The disfigurement caused by neurofibromas and the uncertainty of an unpredictable disease course can cause psychological manifestations for patients with NF1. Anxiety and depression are common. Not surprisingly, patients with more severe disease report more adverse psychological effects.
Orthopedic deformities. Spinal deformities are the most common skeletal manifestation of NF1, with an incidence estimated from 10% to 25% in various studies. Bone mineral density, as measured by age- and gender-adjusted Z-scores, is significantly lower in NF1 patients than in the general population.14 Children may develop bowing of the long bones, particularly the tibia, and pseudarthrosis, a false joint in a long bone. Children with NF1 need yearly assessment of the spine. Patients with clinical evidence of scoliosis should be referred to Orthopedics for further evaluation.
Eye issues. A majority of adult patients develop neurofibroma-like nodules in the iris known as Lisch nodules. The nodules are not thought to cause any ophthalmologic complications. Patients may also develop palpebral neurofibroma, which may become large and sporadically show malignant transformation. Optic nerve glioma may cause strabismus and proptosis, and a large number of patients will also develop glaucoma and globe enlargement.15
Gastrointestinal lesions and cancer. Neurofibromas can grow in the stomach, liver, mesentery, retroperitoneum, and bowel. Adenocarcinoma developed in 23% of patients.16 Gastrointestinal tract bleeding, pseudo-obstruction, and protein-losing enteropathy also may occur.17
Continue to: Central nervous system manifestations
Central nervous system manifestations. Neurological manifestations have been observed in 55% of patients with NF1.18 These include headache, hydrocephalus, epilepsy, lacunar stroke, white matter disease, intraspinal neurofibroma, facial palsy, radiculopathy, and polyneuropathy. Tumors include optic pathway tumors, meningioma, and cerebral glioma. Glioma is the predominant tumor type in NF1 and occurs in all parts of the nervous system, with a predilection for the optic pathways, brainstem, and cerebellum.18
Malignant peripheral nerve sheath tumors. There is an 8% to 13% lifetime risk for malignant peripheral nerve sheath tumors (MPNST), predominantly in individuals between the ages of 20 and 35.19,20 Any change in neurofibroma from soft to hard, or a rapid increase in the size, is suspicious for MPNST. Other symptoms include persistent pain lasting for longer than a month, pain that disturbs sleep, and new neurological deficits. These cancers can be hard to detect, leading to poor prognosis secondary to metastasis.19,20 The greatest risk factors for MPNST are pain associated with a mass and the presence of cutaneous and subcutaneous neurofibromas.21
Treatment is symptom based, but there is a new option
Treatment is individualized to the patient’s symptoms. Neurofibromas that are disfiguring, disruptive, or malignant may be surgically removed.
In April 2020, the US Food and Drug Administration (FDA) approved selumetinib (Koselugo) for the treatment of pediatric patients (ages ≥ 2 years) with NF1 who have symptomatic, inoperable plexiform neurofibromas (PNs).22 In a clinical trial, patients received selumetinib 25 mg/m2 orally twice a day until they demonstrated disease progression or experienced “unacceptable” adverse events.22,23 The overall response rate was 66%, defined as “the percentage of patients with a complete response and those who experienced more than a 20% reduction in PN volume on MRI that was confirmed on a subsequent MRI within 3 to 6 months.”22
Of note, all patients had a partial, not complete, response. Common adverse effects included vomiting, rash, abdominal pain, diarrhea, and nausea.23 Selumetinib may also cause more serious adverse effects, including cardiomyopathy and ocular toxicity. Prior to treatment initiation and at regular intervals during treatment, patients should undergo cardiac and ophthalmic evaluation.22,23 Selumetinib was granted priority review and orphan drug status by the FDA.22
Continue to: You play a key role in ongoing monitoring
You play a key role in ongoing monitoring
In light of the condition’s heterogeneity, the goals of care include early recognition and treatment of complications, especially neoplasms; optimization of quality of life; and identification and treatment of comorbidities. Family physicians are well positioned to monitor patients with NF1 for age-specific disease manifestations and potential complications.9 All patients require:
- an annual physical examination by a physician who is familiar with the individual and with the disease
- annual ophthalmologic examination in early childhood; less frequent examination in older children and adults
- regular blood pressure monitoring
- other studies (eg, MRI) only as indicated on the basis of clinically apparent signs or symptoms
- monitoring by an appropriate specialist if there are abnormalities of the central nervous, skeletal, or cardiovascular systems
- referral to a neurologist for any unexplained neurological signs and symptoms. Referral should be urgent if there are acute symptoms of progressive sensory disturbance, motor deficit and incoordination, or sphincter disturbances since these might indicate an intracranial lesion or spinal cord compression. Headaches on waking, morning vomiting, and altered consciousness are suggestive of raised intracranial pressure.
Children with NF1 benefit from coordinated care between the FP and a pediatrician or other specialist familiar with the disease. In addition to providing usual well care, perform regular assessment of development and school performance. Pay careful attention to the cardiovascular system (particularly blood pressure) and evaluate for scoliosis.
Young adults should be continually monitored for all complications, especially hypertension. This population requires continued education about NF1 and its possible complications and may benefit from counseling about disease inheritance. Screen for anxiety and depression; offer psychological support.
Adults require monitoring based on patient preference and disease severity. For this population, blood pressure should be measured annually, or more frequently if the patient’s values indicate borderline hypertension. Provide education about complications, especially MPNSTs and spinal cord compression. Patients who have abnormalities of the central nervous, skeletal, or cardiovascular systems should be monitored by an appropriate specialist. If desired, the patient may be referred to a geneticist, especially if he or she expresses concern about inheritance. Cutaneous neurofibromas can be removed if they cause discomfort, although removal occasionally results in neurological deficit.
CORRESPONDENCE
T. Grant Phillips, MD, Associate Director, UPMC Altoona Family Physicians Residency, 501 Howard Avenue, Altoona, PA 16601-4899; [email protected]
1. Ly KI, Blakeley JO. The diagnosis and management of neurofibromatosis type 1. Med Clin North Am. 2019;103:1035-1054.
2. Miller DT, Freedenberg D, Schorry E, et al; Council on Genetics, American College of Medical Genetics and Genomics. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019;143:e20190660.
3. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 200l;61:1-14.
4. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834-844.
5. Ben-Shachar S, Dubov T, Toledano-Alhadef H, et al. Predicting neurofibromatosis type 1 risk among children with isolated café-au-lait macules. J Am Acad Dermatol. 2017;76:1077-1083.e3.
6. Friedman JM. Neurofibromatosis 1. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1109. Accessed Septemeber 28, 2020.
7. Roth TM, Petty EM, Barald KF. The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A. 2008;146A:1624-1633.
8. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, MD, July 13-15, 1987. Neurofibromatosis. 1988;1:172-178. https://consensus.nih.gov/1987/1987Neurofibramatosis064html.htm. Accessed Septemeber 28, 2020.
9. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81-88.
10. Koth CW, Cutting LE, Denckla MB. The association of neurofibromatosis type 1 and attention deficit hyperactivity disorder. Child Neuropsychol. 2000;6:185-194.
11. North KN, Riccardi VM, Samango‐Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121-1127.
12. Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
13. Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: a report of the NF1 Cardiovascular Task Force. Genet Med. 2003;4:105-111.
14. Lammert M, Kappler M, Mautner VF, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int. 2005;16:1161-1166.
15. Abdolrahimzadeh B, Piraino DC, Albanese G, et al. Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography. Clin Ophthalmol. 2016;10:851-860.
16. Bakker JR, Haber MM, Garcia FU. Gastrointestinal neurofibromatosis: an unusual cause of gastric outlet obstruction. Am Surg. 2005;71:100-105.
17. Rastogi R. Intra-abdominal manifestations of von Recklinghausen’s neurofibromatosis. Saudi J Gastroenterol. 2008;14:80-82.
18. Créange A, Zeller J, Rostaing-Rigattieri S, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122(pt 3):473-481.
19. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumours in neurofibromatosis 1. Cancer Res. 2002;62:1573-1577.
20. Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Med Genet. 2002;39:311-314.
21. King AA, Debaun MR, Riccardi VM, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am J Med Genet. 2000;93:388-392.
22. US Food and Drug Administration. FDA approves first therapy for children with debilitating and disfiguring rare disease [news release]. April 10, 2020. www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-children-debilitating-and-disfiguring-rare-disease. Accessed September 28, 2020.
23. Koselugo (selumetinib) [product information]. Wilmington, DC: AstraZeneca Pharmaceuticals LP; April 2020. www.accessdata.fda.gov/drugsatfda_docs/label/2020/213756s000lbl.pdf. Accessed September 24, 2020.
Neurofibromatosis type 1 (NF1) is an autosomal dominant inherited disorder that is estimated to occur in 1:2500 births and to have a prevalence of 1:2000 to 1:4000.1,2 It was first described in 1882 by Friedrich Daniel Von Recklinghausen, who identified patients and their relatives with signs of neuroectodermal abnormalities (café-au-lait macules [CALMs], axillary and inguinal freckling, and neurofibromas).
NF1 may begin insidiously in childhood and evolves as the patient ages. It is associated with intracranial, intraspinal, and intraorbital neoplasms, although other organs and tissues can also be involved.
The family physician might be the first one to recognize the signs of this condition during a well-child exam and is in a unique position to coordinate a multidisciplinary approach to care.
A mutated allele and early manifestations on the skin
NF1 has been attributed to genetic mosaicism and is classified as segmental, generalized, or (less frequently) gonadal. The disorder results from germline mutations in the NF1 tumor-suppressor gene on chromosome 17, known to codify the cytoplasmic protein called neurofibromin.3 The penetrance of NF1 is complete, which means that 100% of patients with the mutated allele will develop the disease.
Patients typically have symptoms by the third decade of life, although many will show signs of the disease in early childhood. CALMs are the earliest expression of NF1. They manifest in the first 2 years of life and are found in almost all affected patients. The lesions are well defined and measure 10 to 40 mm. They are typically light brown, although they may darken with sun exposure.
Histologically, the lesions will show macromelanosomes and high concentrations of melanin but do not represent an increased risk for malignancy.4 Not all isolated CALMs are a sign of NF1. While children younger than 29 months with 6 or more CALMs have a high risk for NF1 (80.4%; 95% confidence interval [CI], 74.6% to 86.2%), those who are older than 29 months with at least 1 atypical CALM or fewer than 6 CALMs have just a 0.9% (95% CI, 0% to 2.6%) risk for constitutional NF1.5
Freckles are also observed in 90% of patients with NF1; these tend to develop after the third year of life. The breast and trunk are the most commonly affected areas in adults. The pathophysiology is unknown, but this freckling is believed to be related to skin friction, high humidity, and ambient temperature.6
Continue to: Neurofibromas are benign...
Neurofibromas are benign subcutaneous palpable lesions that grow within peripheral nerve tissue, including spinal, subcutaneous, plexiform, or dermal encapsulated nerves. Originating in Schwann cells, they are composed of fibroblasts, mast cells, macrophages, endothelial cells, and other perineural cells. Some patients show disfiguration when hundreds of these masses are present (FIGURE). These tumors increase in number as the patient ages or during pregnancy, which is thought to be secondary to hormonal changes.7 They are sometimes painful and can be pruritic. Their appearance can also cause patient distress.
The diagnosis is a clinical one
Suspicion for NF1 should be high in patients presenting with the dermatologic findings described, although CALMs and freckling are not exclusive to NF1. Diagnostic criteria for NF1, which distinguish it from other conditions, were first outlined in a National Institutes of Health Consensus Development Conference Statement in 1987.8 The list of criteria has subsequently been expanded.
While the presence of at least 2 criteria is required for diagnosis,2 NF1 should be suspected in individuals who have any of the following findings8,9:
- the presence of at least 6 CALMs that are > 5 mm in prepubertal children and > 15 mm in adults
- 2 or more neurofibromas of any type, or at least one plexiform neurofibroma
- axillary or groin freckling
- optic pathway glioma
- 2 or more Lisch nodules (iris hamartomas seen on slit-lamp examination)
- bony dysplasia (sphenoid wing dysplasia, bowing of long bone ± pseudarthrosis)
- first-degree relative with NF1.
What you’ll see as the disease progresses
NF1 can affect a variety of systems, and potential complications of the disease are numerous and varied (see TABLE9). Here is some of what you may see as the patient’s disease progresses to various organ systems:
Learning disabilities and other cognitive and behavioral problems, such as attention-deficit/hyperactivity disorder, may affect up to 70% of children with NF1. Additionally, children with NF1 have visual/spatial problems, impaired visual motor integration, and language deficits.10 The etiology of cognitive impairment in NF1 is unknown.11
Continue to: Hypertension
Hypertension is common and may contribute to premature death in patients with NF1. Up to 27% of patients will have significant cardiovascular anomalies, including pulmonary valve stenosis, hypertrophic cardiomyopathy in patients with complete deletions of the NF1 gene, intracardiac neurofibromas, renal artery stenosis, coarctation of the aorta, and cerebral infarctions.12 Renal artery stenosis occurs in approximately 2% of the NF1 population, and the diagnosis should be considered in hypertensive children, young adults, pregnant women, older individuals with refractory hypertension, and those with an abdominal bruit.13
Psychological issues. The disfigurement caused by neurofibromas and the uncertainty of an unpredictable disease course can cause psychological manifestations for patients with NF1. Anxiety and depression are common. Not surprisingly, patients with more severe disease report more adverse psychological effects.
Orthopedic deformities. Spinal deformities are the most common skeletal manifestation of NF1, with an incidence estimated from 10% to 25% in various studies. Bone mineral density, as measured by age- and gender-adjusted Z-scores, is significantly lower in NF1 patients than in the general population.14 Children may develop bowing of the long bones, particularly the tibia, and pseudarthrosis, a false joint in a long bone. Children with NF1 need yearly assessment of the spine. Patients with clinical evidence of scoliosis should be referred to Orthopedics for further evaluation.
Eye issues. A majority of adult patients develop neurofibroma-like nodules in the iris known as Lisch nodules. The nodules are not thought to cause any ophthalmologic complications. Patients may also develop palpebral neurofibroma, which may become large and sporadically show malignant transformation. Optic nerve glioma may cause strabismus and proptosis, and a large number of patients will also develop glaucoma and globe enlargement.15
Gastrointestinal lesions and cancer. Neurofibromas can grow in the stomach, liver, mesentery, retroperitoneum, and bowel. Adenocarcinoma developed in 23% of patients.16 Gastrointestinal tract bleeding, pseudo-obstruction, and protein-losing enteropathy also may occur.17
Continue to: Central nervous system manifestations
Central nervous system manifestations. Neurological manifestations have been observed in 55% of patients with NF1.18 These include headache, hydrocephalus, epilepsy, lacunar stroke, white matter disease, intraspinal neurofibroma, facial palsy, radiculopathy, and polyneuropathy. Tumors include optic pathway tumors, meningioma, and cerebral glioma. Glioma is the predominant tumor type in NF1 and occurs in all parts of the nervous system, with a predilection for the optic pathways, brainstem, and cerebellum.18
Malignant peripheral nerve sheath tumors. There is an 8% to 13% lifetime risk for malignant peripheral nerve sheath tumors (MPNST), predominantly in individuals between the ages of 20 and 35.19,20 Any change in neurofibroma from soft to hard, or a rapid increase in the size, is suspicious for MPNST. Other symptoms include persistent pain lasting for longer than a month, pain that disturbs sleep, and new neurological deficits. These cancers can be hard to detect, leading to poor prognosis secondary to metastasis.19,20 The greatest risk factors for MPNST are pain associated with a mass and the presence of cutaneous and subcutaneous neurofibromas.21
Treatment is symptom based, but there is a new option
Treatment is individualized to the patient’s symptoms. Neurofibromas that are disfiguring, disruptive, or malignant may be surgically removed.
In April 2020, the US Food and Drug Administration (FDA) approved selumetinib (Koselugo) for the treatment of pediatric patients (ages ≥ 2 years) with NF1 who have symptomatic, inoperable plexiform neurofibromas (PNs).22 In a clinical trial, patients received selumetinib 25 mg/m2 orally twice a day until they demonstrated disease progression or experienced “unacceptable” adverse events.22,23 The overall response rate was 66%, defined as “the percentage of patients with a complete response and those who experienced more than a 20% reduction in PN volume on MRI that was confirmed on a subsequent MRI within 3 to 6 months.”22
Of note, all patients had a partial, not complete, response. Common adverse effects included vomiting, rash, abdominal pain, diarrhea, and nausea.23 Selumetinib may also cause more serious adverse effects, including cardiomyopathy and ocular toxicity. Prior to treatment initiation and at regular intervals during treatment, patients should undergo cardiac and ophthalmic evaluation.22,23 Selumetinib was granted priority review and orphan drug status by the FDA.22
Continue to: You play a key role in ongoing monitoring
You play a key role in ongoing monitoring
In light of the condition’s heterogeneity, the goals of care include early recognition and treatment of complications, especially neoplasms; optimization of quality of life; and identification and treatment of comorbidities. Family physicians are well positioned to monitor patients with NF1 for age-specific disease manifestations and potential complications.9 All patients require:
- an annual physical examination by a physician who is familiar with the individual and with the disease
- annual ophthalmologic examination in early childhood; less frequent examination in older children and adults
- regular blood pressure monitoring
- other studies (eg, MRI) only as indicated on the basis of clinically apparent signs or symptoms
- monitoring by an appropriate specialist if there are abnormalities of the central nervous, skeletal, or cardiovascular systems
- referral to a neurologist for any unexplained neurological signs and symptoms. Referral should be urgent if there are acute symptoms of progressive sensory disturbance, motor deficit and incoordination, or sphincter disturbances since these might indicate an intracranial lesion or spinal cord compression. Headaches on waking, morning vomiting, and altered consciousness are suggestive of raised intracranial pressure.
Children with NF1 benefit from coordinated care between the FP and a pediatrician or other specialist familiar with the disease. In addition to providing usual well care, perform regular assessment of development and school performance. Pay careful attention to the cardiovascular system (particularly blood pressure) and evaluate for scoliosis.
Young adults should be continually monitored for all complications, especially hypertension. This population requires continued education about NF1 and its possible complications and may benefit from counseling about disease inheritance. Screen for anxiety and depression; offer psychological support.
Adults require monitoring based on patient preference and disease severity. For this population, blood pressure should be measured annually, or more frequently if the patient’s values indicate borderline hypertension. Provide education about complications, especially MPNSTs and spinal cord compression. Patients who have abnormalities of the central nervous, skeletal, or cardiovascular systems should be monitored by an appropriate specialist. If desired, the patient may be referred to a geneticist, especially if he or she expresses concern about inheritance. Cutaneous neurofibromas can be removed if they cause discomfort, although removal occasionally results in neurological deficit.
CORRESPONDENCE
T. Grant Phillips, MD, Associate Director, UPMC Altoona Family Physicians Residency, 501 Howard Avenue, Altoona, PA 16601-4899; [email protected]
Neurofibromatosis type 1 (NF1) is an autosomal dominant inherited disorder that is estimated to occur in 1:2500 births and to have a prevalence of 1:2000 to 1:4000.1,2 It was first described in 1882 by Friedrich Daniel Von Recklinghausen, who identified patients and their relatives with signs of neuroectodermal abnormalities (café-au-lait macules [CALMs], axillary and inguinal freckling, and neurofibromas).
NF1 may begin insidiously in childhood and evolves as the patient ages. It is associated with intracranial, intraspinal, and intraorbital neoplasms, although other organs and tissues can also be involved.
The family physician might be the first one to recognize the signs of this condition during a well-child exam and is in a unique position to coordinate a multidisciplinary approach to care.
A mutated allele and early manifestations on the skin
NF1 has been attributed to genetic mosaicism and is classified as segmental, generalized, or (less frequently) gonadal. The disorder results from germline mutations in the NF1 tumor-suppressor gene on chromosome 17, known to codify the cytoplasmic protein called neurofibromin.3 The penetrance of NF1 is complete, which means that 100% of patients with the mutated allele will develop the disease.
Patients typically have symptoms by the third decade of life, although many will show signs of the disease in early childhood. CALMs are the earliest expression of NF1. They manifest in the first 2 years of life and are found in almost all affected patients. The lesions are well defined and measure 10 to 40 mm. They are typically light brown, although they may darken with sun exposure.
Histologically, the lesions will show macromelanosomes and high concentrations of melanin but do not represent an increased risk for malignancy.4 Not all isolated CALMs are a sign of NF1. While children younger than 29 months with 6 or more CALMs have a high risk for NF1 (80.4%; 95% confidence interval [CI], 74.6% to 86.2%), those who are older than 29 months with at least 1 atypical CALM or fewer than 6 CALMs have just a 0.9% (95% CI, 0% to 2.6%) risk for constitutional NF1.5
Freckles are also observed in 90% of patients with NF1; these tend to develop after the third year of life. The breast and trunk are the most commonly affected areas in adults. The pathophysiology is unknown, but this freckling is believed to be related to skin friction, high humidity, and ambient temperature.6
Continue to: Neurofibromas are benign...
Neurofibromas are benign subcutaneous palpable lesions that grow within peripheral nerve tissue, including spinal, subcutaneous, plexiform, or dermal encapsulated nerves. Originating in Schwann cells, they are composed of fibroblasts, mast cells, macrophages, endothelial cells, and other perineural cells. Some patients show disfiguration when hundreds of these masses are present (FIGURE). These tumors increase in number as the patient ages or during pregnancy, which is thought to be secondary to hormonal changes.7 They are sometimes painful and can be pruritic. Their appearance can also cause patient distress.
The diagnosis is a clinical one
Suspicion for NF1 should be high in patients presenting with the dermatologic findings described, although CALMs and freckling are not exclusive to NF1. Diagnostic criteria for NF1, which distinguish it from other conditions, were first outlined in a National Institutes of Health Consensus Development Conference Statement in 1987.8 The list of criteria has subsequently been expanded.
While the presence of at least 2 criteria is required for diagnosis,2 NF1 should be suspected in individuals who have any of the following findings8,9:
- the presence of at least 6 CALMs that are > 5 mm in prepubertal children and > 15 mm in adults
- 2 or more neurofibromas of any type, or at least one plexiform neurofibroma
- axillary or groin freckling
- optic pathway glioma
- 2 or more Lisch nodules (iris hamartomas seen on slit-lamp examination)
- bony dysplasia (sphenoid wing dysplasia, bowing of long bone ± pseudarthrosis)
- first-degree relative with NF1.
What you’ll see as the disease progresses
NF1 can affect a variety of systems, and potential complications of the disease are numerous and varied (see TABLE9). Here is some of what you may see as the patient’s disease progresses to various organ systems:
Learning disabilities and other cognitive and behavioral problems, such as attention-deficit/hyperactivity disorder, may affect up to 70% of children with NF1. Additionally, children with NF1 have visual/spatial problems, impaired visual motor integration, and language deficits.10 The etiology of cognitive impairment in NF1 is unknown.11
Continue to: Hypertension
Hypertension is common and may contribute to premature death in patients with NF1. Up to 27% of patients will have significant cardiovascular anomalies, including pulmonary valve stenosis, hypertrophic cardiomyopathy in patients with complete deletions of the NF1 gene, intracardiac neurofibromas, renal artery stenosis, coarctation of the aorta, and cerebral infarctions.12 Renal artery stenosis occurs in approximately 2% of the NF1 population, and the diagnosis should be considered in hypertensive children, young adults, pregnant women, older individuals with refractory hypertension, and those with an abdominal bruit.13
Psychological issues. The disfigurement caused by neurofibromas and the uncertainty of an unpredictable disease course can cause psychological manifestations for patients with NF1. Anxiety and depression are common. Not surprisingly, patients with more severe disease report more adverse psychological effects.
Orthopedic deformities. Spinal deformities are the most common skeletal manifestation of NF1, with an incidence estimated from 10% to 25% in various studies. Bone mineral density, as measured by age- and gender-adjusted Z-scores, is significantly lower in NF1 patients than in the general population.14 Children may develop bowing of the long bones, particularly the tibia, and pseudarthrosis, a false joint in a long bone. Children with NF1 need yearly assessment of the spine. Patients with clinical evidence of scoliosis should be referred to Orthopedics for further evaluation.
Eye issues. A majority of adult patients develop neurofibroma-like nodules in the iris known as Lisch nodules. The nodules are not thought to cause any ophthalmologic complications. Patients may also develop palpebral neurofibroma, which may become large and sporadically show malignant transformation. Optic nerve glioma may cause strabismus and proptosis, and a large number of patients will also develop glaucoma and globe enlargement.15
Gastrointestinal lesions and cancer. Neurofibromas can grow in the stomach, liver, mesentery, retroperitoneum, and bowel. Adenocarcinoma developed in 23% of patients.16 Gastrointestinal tract bleeding, pseudo-obstruction, and protein-losing enteropathy also may occur.17
Continue to: Central nervous system manifestations
Central nervous system manifestations. Neurological manifestations have been observed in 55% of patients with NF1.18 These include headache, hydrocephalus, epilepsy, lacunar stroke, white matter disease, intraspinal neurofibroma, facial palsy, radiculopathy, and polyneuropathy. Tumors include optic pathway tumors, meningioma, and cerebral glioma. Glioma is the predominant tumor type in NF1 and occurs in all parts of the nervous system, with a predilection for the optic pathways, brainstem, and cerebellum.18
Malignant peripheral nerve sheath tumors. There is an 8% to 13% lifetime risk for malignant peripheral nerve sheath tumors (MPNST), predominantly in individuals between the ages of 20 and 35.19,20 Any change in neurofibroma from soft to hard, or a rapid increase in the size, is suspicious for MPNST. Other symptoms include persistent pain lasting for longer than a month, pain that disturbs sleep, and new neurological deficits. These cancers can be hard to detect, leading to poor prognosis secondary to metastasis.19,20 The greatest risk factors for MPNST are pain associated with a mass and the presence of cutaneous and subcutaneous neurofibromas.21
Treatment is symptom based, but there is a new option
Treatment is individualized to the patient’s symptoms. Neurofibromas that are disfiguring, disruptive, or malignant may be surgically removed.
In April 2020, the US Food and Drug Administration (FDA) approved selumetinib (Koselugo) for the treatment of pediatric patients (ages ≥ 2 years) with NF1 who have symptomatic, inoperable plexiform neurofibromas (PNs).22 In a clinical trial, patients received selumetinib 25 mg/m2 orally twice a day until they demonstrated disease progression or experienced “unacceptable” adverse events.22,23 The overall response rate was 66%, defined as “the percentage of patients with a complete response and those who experienced more than a 20% reduction in PN volume on MRI that was confirmed on a subsequent MRI within 3 to 6 months.”22
Of note, all patients had a partial, not complete, response. Common adverse effects included vomiting, rash, abdominal pain, diarrhea, and nausea.23 Selumetinib may also cause more serious adverse effects, including cardiomyopathy and ocular toxicity. Prior to treatment initiation and at regular intervals during treatment, patients should undergo cardiac and ophthalmic evaluation.22,23 Selumetinib was granted priority review and orphan drug status by the FDA.22
Continue to: You play a key role in ongoing monitoring
You play a key role in ongoing monitoring
In light of the condition’s heterogeneity, the goals of care include early recognition and treatment of complications, especially neoplasms; optimization of quality of life; and identification and treatment of comorbidities. Family physicians are well positioned to monitor patients with NF1 for age-specific disease manifestations and potential complications.9 All patients require:
- an annual physical examination by a physician who is familiar with the individual and with the disease
- annual ophthalmologic examination in early childhood; less frequent examination in older children and adults
- regular blood pressure monitoring
- other studies (eg, MRI) only as indicated on the basis of clinically apparent signs or symptoms
- monitoring by an appropriate specialist if there are abnormalities of the central nervous, skeletal, or cardiovascular systems
- referral to a neurologist for any unexplained neurological signs and symptoms. Referral should be urgent if there are acute symptoms of progressive sensory disturbance, motor deficit and incoordination, or sphincter disturbances since these might indicate an intracranial lesion or spinal cord compression. Headaches on waking, morning vomiting, and altered consciousness are suggestive of raised intracranial pressure.
Children with NF1 benefit from coordinated care between the FP and a pediatrician or other specialist familiar with the disease. In addition to providing usual well care, perform regular assessment of development and school performance. Pay careful attention to the cardiovascular system (particularly blood pressure) and evaluate for scoliosis.
Young adults should be continually monitored for all complications, especially hypertension. This population requires continued education about NF1 and its possible complications and may benefit from counseling about disease inheritance. Screen for anxiety and depression; offer psychological support.
Adults require monitoring based on patient preference and disease severity. For this population, blood pressure should be measured annually, or more frequently if the patient’s values indicate borderline hypertension. Provide education about complications, especially MPNSTs and spinal cord compression. Patients who have abnormalities of the central nervous, skeletal, or cardiovascular systems should be monitored by an appropriate specialist. If desired, the patient may be referred to a geneticist, especially if he or she expresses concern about inheritance. Cutaneous neurofibromas can be removed if they cause discomfort, although removal occasionally results in neurological deficit.
CORRESPONDENCE
T. Grant Phillips, MD, Associate Director, UPMC Altoona Family Physicians Residency, 501 Howard Avenue, Altoona, PA 16601-4899; [email protected]
1. Ly KI, Blakeley JO. The diagnosis and management of neurofibromatosis type 1. Med Clin North Am. 2019;103:1035-1054.
2. Miller DT, Freedenberg D, Schorry E, et al; Council on Genetics, American College of Medical Genetics and Genomics. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019;143:e20190660.
3. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 200l;61:1-14.
4. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834-844.
5. Ben-Shachar S, Dubov T, Toledano-Alhadef H, et al. Predicting neurofibromatosis type 1 risk among children with isolated café-au-lait macules. J Am Acad Dermatol. 2017;76:1077-1083.e3.
6. Friedman JM. Neurofibromatosis 1. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1109. Accessed Septemeber 28, 2020.
7. Roth TM, Petty EM, Barald KF. The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A. 2008;146A:1624-1633.
8. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, MD, July 13-15, 1987. Neurofibromatosis. 1988;1:172-178. https://consensus.nih.gov/1987/1987Neurofibramatosis064html.htm. Accessed Septemeber 28, 2020.
9. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81-88.
10. Koth CW, Cutting LE, Denckla MB. The association of neurofibromatosis type 1 and attention deficit hyperactivity disorder. Child Neuropsychol. 2000;6:185-194.
11. North KN, Riccardi VM, Samango‐Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121-1127.
12. Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
13. Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: a report of the NF1 Cardiovascular Task Force. Genet Med. 2003;4:105-111.
14. Lammert M, Kappler M, Mautner VF, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int. 2005;16:1161-1166.
15. Abdolrahimzadeh B, Piraino DC, Albanese G, et al. Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography. Clin Ophthalmol. 2016;10:851-860.
16. Bakker JR, Haber MM, Garcia FU. Gastrointestinal neurofibromatosis: an unusual cause of gastric outlet obstruction. Am Surg. 2005;71:100-105.
17. Rastogi R. Intra-abdominal manifestations of von Recklinghausen’s neurofibromatosis. Saudi J Gastroenterol. 2008;14:80-82.
18. Créange A, Zeller J, Rostaing-Rigattieri S, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122(pt 3):473-481.
19. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumours in neurofibromatosis 1. Cancer Res. 2002;62:1573-1577.
20. Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Med Genet. 2002;39:311-314.
21. King AA, Debaun MR, Riccardi VM, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am J Med Genet. 2000;93:388-392.
22. US Food and Drug Administration. FDA approves first therapy for children with debilitating and disfiguring rare disease [news release]. April 10, 2020. www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-children-debilitating-and-disfiguring-rare-disease. Accessed September 28, 2020.
23. Koselugo (selumetinib) [product information]. Wilmington, DC: AstraZeneca Pharmaceuticals LP; April 2020. www.accessdata.fda.gov/drugsatfda_docs/label/2020/213756s000lbl.pdf. Accessed September 24, 2020.
1. Ly KI, Blakeley JO. The diagnosis and management of neurofibromatosis type 1. Med Clin North Am. 2019;103:1035-1054.
2. Miller DT, Freedenberg D, Schorry E, et al; Council on Genetics, American College of Medical Genetics and Genomics. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019;143:e20190660.
3. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 200l;61:1-14.
4. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834-844.
5. Ben-Shachar S, Dubov T, Toledano-Alhadef H, et al. Predicting neurofibromatosis type 1 risk among children with isolated café-au-lait macules. J Am Acad Dermatol. 2017;76:1077-1083.e3.
6. Friedman JM. Neurofibromatosis 1. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1109. Accessed Septemeber 28, 2020.
7. Roth TM, Petty EM, Barald KF. The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A. 2008;146A:1624-1633.
8. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, MD, July 13-15, 1987. Neurofibromatosis. 1988;1:172-178. https://consensus.nih.gov/1987/1987Neurofibramatosis064html.htm. Accessed Septemeber 28, 2020.
9. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81-88.
10. Koth CW, Cutting LE, Denckla MB. The association of neurofibromatosis type 1 and attention deficit hyperactivity disorder. Child Neuropsychol. 2000;6:185-194.
11. North KN, Riccardi VM, Samango‐Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121-1127.
12. Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
13. Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: a report of the NF1 Cardiovascular Task Force. Genet Med. 2003;4:105-111.
14. Lammert M, Kappler M, Mautner VF, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int. 2005;16:1161-1166.
15. Abdolrahimzadeh B, Piraino DC, Albanese G, et al. Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography. Clin Ophthalmol. 2016;10:851-860.
16. Bakker JR, Haber MM, Garcia FU. Gastrointestinal neurofibromatosis: an unusual cause of gastric outlet obstruction. Am Surg. 2005;71:100-105.
17. Rastogi R. Intra-abdominal manifestations of von Recklinghausen’s neurofibromatosis. Saudi J Gastroenterol. 2008;14:80-82.
18. Créange A, Zeller J, Rostaing-Rigattieri S, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122(pt 3):473-481.
19. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumours in neurofibromatosis 1. Cancer Res. 2002;62:1573-1577.
20. Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Med Genet. 2002;39:311-314.
21. King AA, Debaun MR, Riccardi VM, et al. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. Am J Med Genet. 2000;93:388-392.
22. US Food and Drug Administration. FDA approves first therapy for children with debilitating and disfiguring rare disease [news release]. April 10, 2020. www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-children-debilitating-and-disfiguring-rare-disease. Accessed September 28, 2020.
23. Koselugo (selumetinib) [product information]. Wilmington, DC: AstraZeneca Pharmaceuticals LP; April 2020. www.accessdata.fda.gov/drugsatfda_docs/label/2020/213756s000lbl.pdf. Accessed September 24, 2020.

A 31-year-old with a 3-week history of a waxing and waning, mildly pruritic eruption on his neck, chest, and back
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1

The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.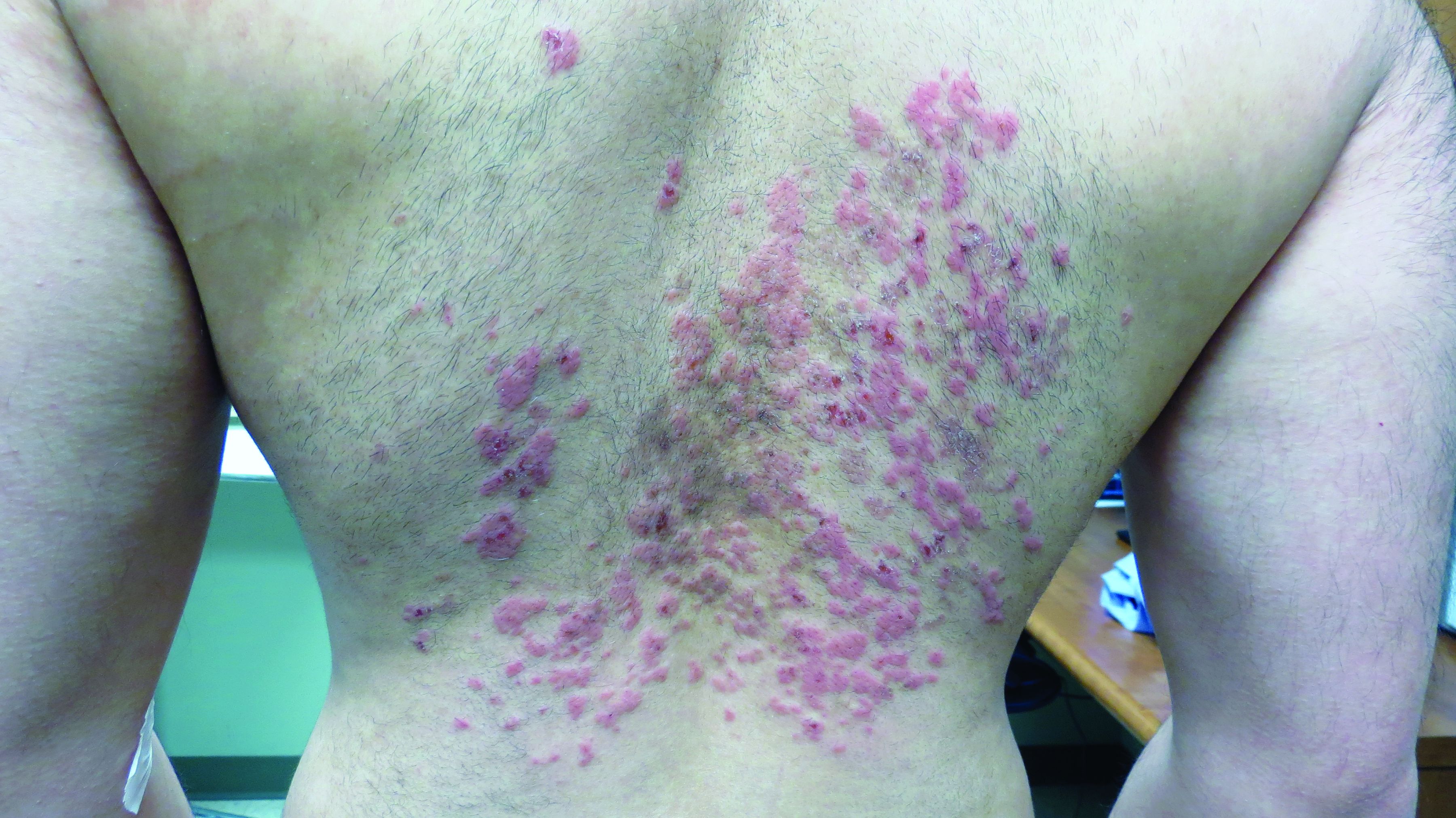
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1
The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1
The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
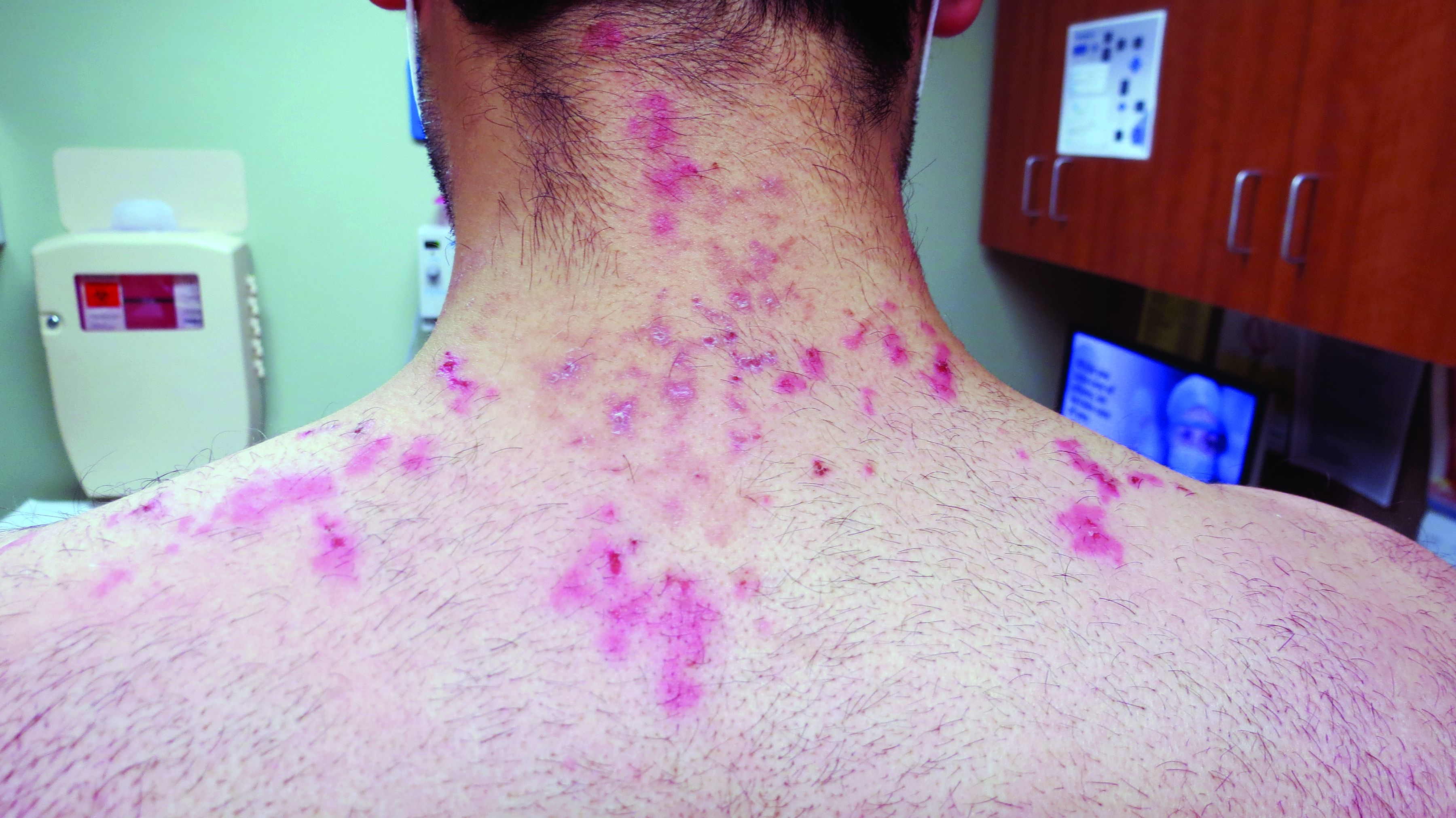
Chronic, nonhealing leg ulcer
An 80-year-old woman with a history of hypertension, hyperlipidemia, psoriasis vulgaris with associated pruritus, and well-controlled type 2 diabetes mellitus presented with a slowly enlarging ulceration on her left leg of 1 year’s duration. She noted that this lesion healed less rapidly than previous stasis leg ulcerations, despite using the same treatment approach that included dressings, elevation, and diuretics to decrease pedal edema.
Physical examination revealed plaques with white micaceous scaling over her extensor surfaces and scalp, as well as guttate lesions on the trunk, typical of psoriasis vulgaris. A 5.8 × 7.2-cm malodorous ulceration was superimposed on a large psoriatic plaque on her left anterior lower leg (FIGURE 1). A 4-mm punch biopsy was obtained from the peripheral margin.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Basal cell carcinoma
Histopathological examination revealed elongated strands of closely packed basaloid cells embedded in a dense fibrous stroma with overlying ulceration and crusting (FIGURE 2). Immunohistochemical staining with cytokeratin (CK) 5/6 decorated the cytoplasm of the tumor cells, which confirmed that the tumor was a keratinocyte cancer. CK 20 was negative, excluding the possibility of a Merkel cell carcinoma. Scout biopsies from 3 additional areas of ulceration confirmed that the entire ulceration was infiltrated by basal cell carcinoma (BCC).

A surprise hidden in a chronic ulcer
More than 6 million Americans have chronic ulcers and most occur on the legs.1 The majority of these chronic ulcerations are etiologically related to venous stasis, arterial insufficiency, or neuropathy.2
Bacterial pyoderma, chronic infection caused by atypical acid-fast bacilli or deep fungal infection, pyoderma gangrenosum, cutaneous vasculitis, calciphylaxis, and venous ulceration were all considered to explain this patient’s nonhealing wound. A biopsy was required to fully assess these possibilities.
Don’t overlook the possibility of malignancy. In a cross-sectional, multicenter study by Senet et al,3 144 patients with 154 total chronic leg ulcers were evaluated in tertiary care centers for malignancy, which was found to occur at a rate of 10.4%. Similarly, Ghasemi et al4 demonstrated a malignancy rate of 16.1% in 124 patients who underwent biopsy; the anterior shin was determined to be the most frequent location for malignancy. The most common skin cancer identified within the setting of chronic ulcers is squamous cell carcinoma.3 Although rare, there are reports of BCC identified in chronic wounds.3-7
Morphological signs suggestive of malignancy in chronic ulcerations include hyperkeratosis, granulation tissue surrounded by a raised border, unusual pain or bleeding, and increased tissue friability. Our patient had none of these signs and symptoms. However, it is possible that she had a tumor that ulcerated and would not heal.
Continue to: Which came first?
Which came first? It’s difficult to know in this case whether a persistent BCC ulcerated, forming this lesion, or if scarring associated with a chronic ulceration led to the development of the BCC.6 Based on biopsies taken at an earlier date, Schnirring-Judge and Belpedio7 concluded that a chronic leg ulcer could, indeed, transform into a BCC; however, pre-existing BCC more commonly ulcerates and then does not heal.
Treatment options
While smaller, superficial BCCs can be treated with topical imiquimod, photodynamic therapy, or electrodesiccation and curettage, larger lesions should be treated with Mohs micrographic surgery and excisional surgery with grafting. Inoperable tumors may be treated with radiation therapy and vismodegib.
Our patient. Once the diagnosis of BCC was established, treatment options were discussed, including excision, local radiation therapy, and oral hedgehog inhibitor drug therapy.8 Our patient opted to undergo a wide local excision of the lesion followed by negative-pressure wound therapy, which led to complete healing.
CORRESPONDENCE
David Crasto, DO, William Carey University College of Osteopathic Medicine, 498 Tuscan Avenue, Hattiesburg, MS 39401; [email protected]
1. Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
2. Fox JD, Baquerizo Nole KL, Berriman SJ, et al. Chronic wounds: the need for greater emphasis in medical schools, post-graduate training and public health discussions. Ann Surg. 2016;264:241-243.
3. Senet P, Combemale P, Debure C, et al. Malignancy and chronic leg ulcers. Arch Dermatol. 2012;148:704-708.
4. Ghasemi F, Anooshirvani N, Sibbald RG, et al. The point prevalence of malignancy in a wound clinic. Int J Low Extrem Wounds. 2016;15:58-62.
5. Labropoulos N, Manalo D, Patel N, et al. Uncommon leg ulcers in the lower extremity. J Vasc Surg. 2007;45:568-573.
6. Tchanque-Fossuo CN, Millsop J, Johnson MA, et al. Ulcerated basal cell carcinomas masquerading as venous leg ulcers. Adv Skin Wound Care. 2018;31:130-134.
7. Schnirring-Judge M, Belpedio D. Malignant transformation of a chronic venous stasis ulcer to basal cell carcinoma in a diabetic patient: case and review of the pathophysiology. J Foot Ankle Surg. 2010;49:75-79.
8. Puig S, Berrocal A. Management of high-risk and advanced basal cell carcinoma. Clin Transl Oncol. 2015;17:497-503.
An 80-year-old woman with a history of hypertension, hyperlipidemia, psoriasis vulgaris with associated pruritus, and well-controlled type 2 diabetes mellitus presented with a slowly enlarging ulceration on her left leg of 1 year’s duration. She noted that this lesion healed less rapidly than previous stasis leg ulcerations, despite using the same treatment approach that included dressings, elevation, and diuretics to decrease pedal edema.
Physical examination revealed plaques with white micaceous scaling over her extensor surfaces and scalp, as well as guttate lesions on the trunk, typical of psoriasis vulgaris. A 5.8 × 7.2-cm malodorous ulceration was superimposed on a large psoriatic plaque on her left anterior lower leg (FIGURE 1). A 4-mm punch biopsy was obtained from the peripheral margin.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Basal cell carcinoma
Histopathological examination revealed elongated strands of closely packed basaloid cells embedded in a dense fibrous stroma with overlying ulceration and crusting (FIGURE 2). Immunohistochemical staining with cytokeratin (CK) 5/6 decorated the cytoplasm of the tumor cells, which confirmed that the tumor was a keratinocyte cancer. CK 20 was negative, excluding the possibility of a Merkel cell carcinoma. Scout biopsies from 3 additional areas of ulceration confirmed that the entire ulceration was infiltrated by basal cell carcinoma (BCC).
A surprise hidden in a chronic ulcer
More than 6 million Americans have chronic ulcers and most occur on the legs.1 The majority of these chronic ulcerations are etiologically related to venous stasis, arterial insufficiency, or neuropathy.2
Bacterial pyoderma, chronic infection caused by atypical acid-fast bacilli or deep fungal infection, pyoderma gangrenosum, cutaneous vasculitis, calciphylaxis, and venous ulceration were all considered to explain this patient’s nonhealing wound. A biopsy was required to fully assess these possibilities.
Don’t overlook the possibility of malignancy. In a cross-sectional, multicenter study by Senet et al,3 144 patients with 154 total chronic leg ulcers were evaluated in tertiary care centers for malignancy, which was found to occur at a rate of 10.4%. Similarly, Ghasemi et al4 demonstrated a malignancy rate of 16.1% in 124 patients who underwent biopsy; the anterior shin was determined to be the most frequent location for malignancy. The most common skin cancer identified within the setting of chronic ulcers is squamous cell carcinoma.3 Although rare, there are reports of BCC identified in chronic wounds.3-7
Morphological signs suggestive of malignancy in chronic ulcerations include hyperkeratosis, granulation tissue surrounded by a raised border, unusual pain or bleeding, and increased tissue friability. Our patient had none of these signs and symptoms. However, it is possible that she had a tumor that ulcerated and would not heal.
Continue to: Which came first?
Which came first? It’s difficult to know in this case whether a persistent BCC ulcerated, forming this lesion, or if scarring associated with a chronic ulceration led to the development of the BCC.6 Based on biopsies taken at an earlier date, Schnirring-Judge and Belpedio7 concluded that a chronic leg ulcer could, indeed, transform into a BCC; however, pre-existing BCC more commonly ulcerates and then does not heal.
Treatment options
While smaller, superficial BCCs can be treated with topical imiquimod, photodynamic therapy, or electrodesiccation and curettage, larger lesions should be treated with Mohs micrographic surgery and excisional surgery with grafting. Inoperable tumors may be treated with radiation therapy and vismodegib.
Our patient. Once the diagnosis of BCC was established, treatment options were discussed, including excision, local radiation therapy, and oral hedgehog inhibitor drug therapy.8 Our patient opted to undergo a wide local excision of the lesion followed by negative-pressure wound therapy, which led to complete healing.
CORRESPONDENCE
David Crasto, DO, William Carey University College of Osteopathic Medicine, 498 Tuscan Avenue, Hattiesburg, MS 39401; [email protected]
An 80-year-old woman with a history of hypertension, hyperlipidemia, psoriasis vulgaris with associated pruritus, and well-controlled type 2 diabetes mellitus presented with a slowly enlarging ulceration on her left leg of 1 year’s duration. She noted that this lesion healed less rapidly than previous stasis leg ulcerations, despite using the same treatment approach that included dressings, elevation, and diuretics to decrease pedal edema.
Physical examination revealed plaques with white micaceous scaling over her extensor surfaces and scalp, as well as guttate lesions on the trunk, typical of psoriasis vulgaris. A 5.8 × 7.2-cm malodorous ulceration was superimposed on a large psoriatic plaque on her left anterior lower leg (FIGURE 1). A 4-mm punch biopsy was obtained from the peripheral margin.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Basal cell carcinoma
Histopathological examination revealed elongated strands of closely packed basaloid cells embedded in a dense fibrous stroma with overlying ulceration and crusting (FIGURE 2). Immunohistochemical staining with cytokeratin (CK) 5/6 decorated the cytoplasm of the tumor cells, which confirmed that the tumor was a keratinocyte cancer. CK 20 was negative, excluding the possibility of a Merkel cell carcinoma. Scout biopsies from 3 additional areas of ulceration confirmed that the entire ulceration was infiltrated by basal cell carcinoma (BCC).
A surprise hidden in a chronic ulcer
More than 6 million Americans have chronic ulcers and most occur on the legs.1 The majority of these chronic ulcerations are etiologically related to venous stasis, arterial insufficiency, or neuropathy.2
Bacterial pyoderma, chronic infection caused by atypical acid-fast bacilli or deep fungal infection, pyoderma gangrenosum, cutaneous vasculitis, calciphylaxis, and venous ulceration were all considered to explain this patient’s nonhealing wound. A biopsy was required to fully assess these possibilities.
Don’t overlook the possibility of malignancy. In a cross-sectional, multicenter study by Senet et al,3 144 patients with 154 total chronic leg ulcers were evaluated in tertiary care centers for malignancy, which was found to occur at a rate of 10.4%. Similarly, Ghasemi et al4 demonstrated a malignancy rate of 16.1% in 124 patients who underwent biopsy; the anterior shin was determined to be the most frequent location for malignancy. The most common skin cancer identified within the setting of chronic ulcers is squamous cell carcinoma.3 Although rare, there are reports of BCC identified in chronic wounds.3-7
Morphological signs suggestive of malignancy in chronic ulcerations include hyperkeratosis, granulation tissue surrounded by a raised border, unusual pain or bleeding, and increased tissue friability. Our patient had none of these signs and symptoms. However, it is possible that she had a tumor that ulcerated and would not heal.
Continue to: Which came first?
Which came first? It’s difficult to know in this case whether a persistent BCC ulcerated, forming this lesion, or if scarring associated with a chronic ulceration led to the development of the BCC.6 Based on biopsies taken at an earlier date, Schnirring-Judge and Belpedio7 concluded that a chronic leg ulcer could, indeed, transform into a BCC; however, pre-existing BCC more commonly ulcerates and then does not heal.
Treatment options
While smaller, superficial BCCs can be treated with topical imiquimod, photodynamic therapy, or electrodesiccation and curettage, larger lesions should be treated with Mohs micrographic surgery and excisional surgery with grafting. Inoperable tumors may be treated with radiation therapy and vismodegib.
Our patient. Once the diagnosis of BCC was established, treatment options were discussed, including excision, local radiation therapy, and oral hedgehog inhibitor drug therapy.8 Our patient opted to undergo a wide local excision of the lesion followed by negative-pressure wound therapy, which led to complete healing.
CORRESPONDENCE
David Crasto, DO, William Carey University College of Osteopathic Medicine, 498 Tuscan Avenue, Hattiesburg, MS 39401; [email protected]
1. Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
2. Fox JD, Baquerizo Nole KL, Berriman SJ, et al. Chronic wounds: the need for greater emphasis in medical schools, post-graduate training and public health discussions. Ann Surg. 2016;264:241-243.
3. Senet P, Combemale P, Debure C, et al. Malignancy and chronic leg ulcers. Arch Dermatol. 2012;148:704-708.
4. Ghasemi F, Anooshirvani N, Sibbald RG, et al. The point prevalence of malignancy in a wound clinic. Int J Low Extrem Wounds. 2016;15:58-62.
5. Labropoulos N, Manalo D, Patel N, et al. Uncommon leg ulcers in the lower extremity. J Vasc Surg. 2007;45:568-573.
6. Tchanque-Fossuo CN, Millsop J, Johnson MA, et al. Ulcerated basal cell carcinomas masquerading as venous leg ulcers. Adv Skin Wound Care. 2018;31:130-134.
7. Schnirring-Judge M, Belpedio D. Malignant transformation of a chronic venous stasis ulcer to basal cell carcinoma in a diabetic patient: case and review of the pathophysiology. J Foot Ankle Surg. 2010;49:75-79.
8. Puig S, Berrocal A. Management of high-risk and advanced basal cell carcinoma. Clin Transl Oncol. 2015;17:497-503.
1. Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
2. Fox JD, Baquerizo Nole KL, Berriman SJ, et al. Chronic wounds: the need for greater emphasis in medical schools, post-graduate training and public health discussions. Ann Surg. 2016;264:241-243.
3. Senet P, Combemale P, Debure C, et al. Malignancy and chronic leg ulcers. Arch Dermatol. 2012;148:704-708.
4. Ghasemi F, Anooshirvani N, Sibbald RG, et al. The point prevalence of malignancy in a wound clinic. Int J Low Extrem Wounds. 2016;15:58-62.
5. Labropoulos N, Manalo D, Patel N, et al. Uncommon leg ulcers in the lower extremity. J Vasc Surg. 2007;45:568-573.
6. Tchanque-Fossuo CN, Millsop J, Johnson MA, et al. Ulcerated basal cell carcinomas masquerading as venous leg ulcers. Adv Skin Wound Care. 2018;31:130-134.
7. Schnirring-Judge M, Belpedio D. Malignant transformation of a chronic venous stasis ulcer to basal cell carcinoma in a diabetic patient: case and review of the pathophysiology. J Foot Ankle Surg. 2010;49:75-79.
8. Puig S, Berrocal A. Management of high-risk and advanced basal cell carcinoma. Clin Transl Oncol. 2015;17:497-503.

Don’t discount ultrapotent topical corticosteroids for vulvar lichen sclerosus
according to an expert speaking at the virtual conference on diseases of the vulva and vagina.

If needed, intralesional steroid injections or calcineurin inhibitors can be added to a topical corticosteroid regimen, Libby Edwards, MD, suggested at the meeting, hosted by the International Society for the Study of Vulvovaginal Disease. In addition, early reports indicate that newer interventions such as fractional CO2 laser treatments may help patients with refractory disease.
Still, “there is no question, there is no argument: First-, second- and third-line treatment for lichen sclerosus is an ultrapotent or superpotent topical corticosteroid,” she said. Steroids include halobetasol, clobetasol, or betamethasone dipropionate in augmented vehicle ointment once or twice per day. Patients should continue this regimen until the skin texture is normal or the disease is controlled as well as possible, which usually takes several months, said Dr. Edwards, of Southeast Vulvar Clinic in Charlotte, N.C.
Patients then should continue treatment, but less frequently or with a lower potency steroid.
Although corticosteroids are not Food and Drug Administration–approved for the treatment of lichen sclerosus, double-blind, placebo-controlled trials support their use, Dr. Edwards said.
Getting patients to use topical corticosteroids as directed can be a challenge, however, and patient education is crucial.
After about 10 days, many patients start to feel better and stop the medication prematurely, which may lead to recurrence.
“That is such an important counseling point,” Aruna Venkatesan, MD, chief of dermatology and director of the genital dermatology clinic at Santa Clara Valley Medical Center in San Jose, Calif., said during a panel discussion. “Tell them, listen, I may not see you back for a couple months, and you may start feeling better sooner. But I want you to keep using this at this frequency so that when you come back we can make a good decision about whether you’re ready” for a lower potency regimen.
To encourage daily use, Hope K. Haefner, MD, asks patients whether they brush their teeth every night. “When they say yes, I tell them to put the steroid ointment by their toothpaste and use it after brushing,” Dr. Haefner, the Harold A. Furlong Professor of Women’s Health at Michigan Medicine in Ann Arbor, said during the discussion. “But don’t mix up the tubes.”
Once lichen sclerosus is controlled, options include decreasing the superpotent steroid to once, three times per week or changing to a midpotency steroid such as triamcinolone ointment every day, Dr. Edwards said.
Evidence suggests that controlling lichen sclerosus may prevent squamous cell carcinoma and scarring. In a study of more than 500 patients, about 70% complied with treatment instructions, whereas about 30% were considered partially compliant (JAMA Dermatol. 2015 Oct;151[10]:1061-7.). Patients who adhered to their therapy were less likely to have cancer or ongoing scarring during an average of 4.7 years of follow-up.
Beyond topical steroids
“Almost always, topical steroids are all you need,” said Dr. Edwards. “Before I go beyond that, I think of other issues that may be causing symptoms,” such as atrophic vagina, steroid dermatitis, or vulvodynia.
For patients with refractory lichen sclerosus, other treatments “can add more oomph to your topical steroid, but they are not better,” she said.
Intralesional corticosteroid injections are one option.
Another option is adding a calcineurin inhibitor such as tacrolimus or pimecrolimus, although these medications can burn with application and irritate. In addition, they carry warnings about rare cases of cancer associated with their use.
Dr. Edwards also uses methotrexate, which is supported by case reports and an open-label study. In a recently published study that included 21 patients with vulvar lichen sclerosus and 24 patients with extragenital lichen sclerosus, about half improved after receiving methotrexate (Dermatol Ther. 2020 Apr 29;e13473.).
What about lasers?
Fractional CO2 laser treatments, which are pulsed to minimize damage from heat, have “a lot of providers very excited,” Dr. Edwards said. In one open-label study of 40 patients, most reported a decrease in symptoms. (J Low Genit Tract Dis. 2020 Apr;24[2]:225-8.)
“We’re awaiting blinded, controlled studies,” Dr. Edwards said. “We don’t have those available yet although they are in progress.”
Ten of Dr. Edwards’ patients who did not improve enough with medication have received laser treatments. One patient stopped laser therapy after one treatment. One did not improve. Two were completely cleared, and six had significant improvement.
If patients who improved stopped steroids against recommendations, lichen sclerosus recurred, Dr. Edwards said.
The ISSVD does not recommend laser for the routine treatment of lichen sclerosus because of a lack of adequate studies and long-term safety data and biologic implausibility, Dr. Edwards noted (J Low Genit Tract Dis. 2019 Apr;23[2]:151-60.) Laser treatments for lichen sclerosus should not be used outside of clinical trials or without special arrangements for clinical governance, consent, and audit, according to a consensus document from the society.
“I mostly agree with that,” Dr. Edwards said. “But I now think that this is a reasonable thing to use when other treatments have been exhausted.”
Dr. Edwards and Dr. Venkatesan had no conflicts of interest. Dr. Haefner is an author for UpToDate.
according to an expert speaking at the virtual conference on diseases of the vulva and vagina.
If needed, intralesional steroid injections or calcineurin inhibitors can be added to a topical corticosteroid regimen, Libby Edwards, MD, suggested at the meeting, hosted by the International Society for the Study of Vulvovaginal Disease. In addition, early reports indicate that newer interventions such as fractional CO2 laser treatments may help patients with refractory disease.
Still, “there is no question, there is no argument: First-, second- and third-line treatment for lichen sclerosus is an ultrapotent or superpotent topical corticosteroid,” she said. Steroids include halobetasol, clobetasol, or betamethasone dipropionate in augmented vehicle ointment once or twice per day. Patients should continue this regimen until the skin texture is normal or the disease is controlled as well as possible, which usually takes several months, said Dr. Edwards, of Southeast Vulvar Clinic in Charlotte, N.C.
Patients then should continue treatment, but less frequently or with a lower potency steroid.
Although corticosteroids are not Food and Drug Administration–approved for the treatment of lichen sclerosus, double-blind, placebo-controlled trials support their use, Dr. Edwards said.
Getting patients to use topical corticosteroids as directed can be a challenge, however, and patient education is crucial.
After about 10 days, many patients start to feel better and stop the medication prematurely, which may lead to recurrence.
“That is such an important counseling point,” Aruna Venkatesan, MD, chief of dermatology and director of the genital dermatology clinic at Santa Clara Valley Medical Center in San Jose, Calif., said during a panel discussion. “Tell them, listen, I may not see you back for a couple months, and you may start feeling better sooner. But I want you to keep using this at this frequency so that when you come back we can make a good decision about whether you’re ready” for a lower potency regimen.
To encourage daily use, Hope K. Haefner, MD, asks patients whether they brush their teeth every night. “When they say yes, I tell them to put the steroid ointment by their toothpaste and use it after brushing,” Dr. Haefner, the Harold A. Furlong Professor of Women’s Health at Michigan Medicine in Ann Arbor, said during the discussion. “But don’t mix up the tubes.”
Once lichen sclerosus is controlled, options include decreasing the superpotent steroid to once, three times per week or changing to a midpotency steroid such as triamcinolone ointment every day, Dr. Edwards said.
Evidence suggests that controlling lichen sclerosus may prevent squamous cell carcinoma and scarring. In a study of more than 500 patients, about 70% complied with treatment instructions, whereas about 30% were considered partially compliant (JAMA Dermatol. 2015 Oct;151[10]:1061-7.). Patients who adhered to their therapy were less likely to have cancer or ongoing scarring during an average of 4.7 years of follow-up.
Beyond topical steroids
“Almost always, topical steroids are all you need,” said Dr. Edwards. “Before I go beyond that, I think of other issues that may be causing symptoms,” such as atrophic vagina, steroid dermatitis, or vulvodynia.
For patients with refractory lichen sclerosus, other treatments “can add more oomph to your topical steroid, but they are not better,” she said.
Intralesional corticosteroid injections are one option.
Another option is adding a calcineurin inhibitor such as tacrolimus or pimecrolimus, although these medications can burn with application and irritate. In addition, they carry warnings about rare cases of cancer associated with their use.
Dr. Edwards also uses methotrexate, which is supported by case reports and an open-label study. In a recently published study that included 21 patients with vulvar lichen sclerosus and 24 patients with extragenital lichen sclerosus, about half improved after receiving methotrexate (Dermatol Ther. 2020 Apr 29;e13473.).
What about lasers?
Fractional CO2 laser treatments, which are pulsed to minimize damage from heat, have “a lot of providers very excited,” Dr. Edwards said. In one open-label study of 40 patients, most reported a decrease in symptoms. (J Low Genit Tract Dis. 2020 Apr;24[2]:225-8.)
“We’re awaiting blinded, controlled studies,” Dr. Edwards said. “We don’t have those available yet although they are in progress.”
Ten of Dr. Edwards’ patients who did not improve enough with medication have received laser treatments. One patient stopped laser therapy after one treatment. One did not improve. Two were completely cleared, and six had significant improvement.
If patients who improved stopped steroids against recommendations, lichen sclerosus recurred, Dr. Edwards said.
The ISSVD does not recommend laser for the routine treatment of lichen sclerosus because of a lack of adequate studies and long-term safety data and biologic implausibility, Dr. Edwards noted (J Low Genit Tract Dis. 2019 Apr;23[2]:151-60.) Laser treatments for lichen sclerosus should not be used outside of clinical trials or without special arrangements for clinical governance, consent, and audit, according to a consensus document from the society.
“I mostly agree with that,” Dr. Edwards said. “But I now think that this is a reasonable thing to use when other treatments have been exhausted.”
Dr. Edwards and Dr. Venkatesan had no conflicts of interest. Dr. Haefner is an author for UpToDate.
according to an expert speaking at the virtual conference on diseases of the vulva and vagina.
If needed, intralesional steroid injections or calcineurin inhibitors can be added to a topical corticosteroid regimen, Libby Edwards, MD, suggested at the meeting, hosted by the International Society for the Study of Vulvovaginal Disease. In addition, early reports indicate that newer interventions such as fractional CO2 laser treatments may help patients with refractory disease.
Still, “there is no question, there is no argument: First-, second- and third-line treatment for lichen sclerosus is an ultrapotent or superpotent topical corticosteroid,” she said. Steroids include halobetasol, clobetasol, or betamethasone dipropionate in augmented vehicle ointment once or twice per day. Patients should continue this regimen until the skin texture is normal or the disease is controlled as well as possible, which usually takes several months, said Dr. Edwards, of Southeast Vulvar Clinic in Charlotte, N.C.
Patients then should continue treatment, but less frequently or with a lower potency steroid.
Although corticosteroids are not Food and Drug Administration–approved for the treatment of lichen sclerosus, double-blind, placebo-controlled trials support their use, Dr. Edwards said.
Getting patients to use topical corticosteroids as directed can be a challenge, however, and patient education is crucial.
After about 10 days, many patients start to feel better and stop the medication prematurely, which may lead to recurrence.
“That is such an important counseling point,” Aruna Venkatesan, MD, chief of dermatology and director of the genital dermatology clinic at Santa Clara Valley Medical Center in San Jose, Calif., said during a panel discussion. “Tell them, listen, I may not see you back for a couple months, and you may start feeling better sooner. But I want you to keep using this at this frequency so that when you come back we can make a good decision about whether you’re ready” for a lower potency regimen.
To encourage daily use, Hope K. Haefner, MD, asks patients whether they brush their teeth every night. “When they say yes, I tell them to put the steroid ointment by their toothpaste and use it after brushing,” Dr. Haefner, the Harold A. Furlong Professor of Women’s Health at Michigan Medicine in Ann Arbor, said during the discussion. “But don’t mix up the tubes.”
Once lichen sclerosus is controlled, options include decreasing the superpotent steroid to once, three times per week or changing to a midpotency steroid such as triamcinolone ointment every day, Dr. Edwards said.
Evidence suggests that controlling lichen sclerosus may prevent squamous cell carcinoma and scarring. In a study of more than 500 patients, about 70% complied with treatment instructions, whereas about 30% were considered partially compliant (JAMA Dermatol. 2015 Oct;151[10]:1061-7.). Patients who adhered to their therapy were less likely to have cancer or ongoing scarring during an average of 4.7 years of follow-up.
Beyond topical steroids
“Almost always, topical steroids are all you need,” said Dr. Edwards. “Before I go beyond that, I think of other issues that may be causing symptoms,” such as atrophic vagina, steroid dermatitis, or vulvodynia.
For patients with refractory lichen sclerosus, other treatments “can add more oomph to your topical steroid, but they are not better,” she said.
Intralesional corticosteroid injections are one option.
Another option is adding a calcineurin inhibitor such as tacrolimus or pimecrolimus, although these medications can burn with application and irritate. In addition, they carry warnings about rare cases of cancer associated with their use.
Dr. Edwards also uses methotrexate, which is supported by case reports and an open-label study. In a recently published study that included 21 patients with vulvar lichen sclerosus and 24 patients with extragenital lichen sclerosus, about half improved after receiving methotrexate (Dermatol Ther. 2020 Apr 29;e13473.).
What about lasers?
Fractional CO2 laser treatments, which are pulsed to minimize damage from heat, have “a lot of providers very excited,” Dr. Edwards said. In one open-label study of 40 patients, most reported a decrease in symptoms. (J Low Genit Tract Dis. 2020 Apr;24[2]:225-8.)
“We’re awaiting blinded, controlled studies,” Dr. Edwards said. “We don’t have those available yet although they are in progress.”
Ten of Dr. Edwards’ patients who did not improve enough with medication have received laser treatments. One patient stopped laser therapy after one treatment. One did not improve. Two were completely cleared, and six had significant improvement.
If patients who improved stopped steroids against recommendations, lichen sclerosus recurred, Dr. Edwards said.
The ISSVD does not recommend laser for the routine treatment of lichen sclerosus because of a lack of adequate studies and long-term safety data and biologic implausibility, Dr. Edwards noted (J Low Genit Tract Dis. 2019 Apr;23[2]:151-60.) Laser treatments for lichen sclerosus should not be used outside of clinical trials or without special arrangements for clinical governance, consent, and audit, according to a consensus document from the society.
“I mostly agree with that,” Dr. Edwards said. “But I now think that this is a reasonable thing to use when other treatments have been exhausted.”
Dr. Edwards and Dr. Venkatesan had no conflicts of interest. Dr. Haefner is an author for UpToDate.
EXPERT ANALYSIS FROM THE ISSVD BIENNIAL CONFERENCE
New lupus classification criteria perform well in children, young adults
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
FROM ARTHRITIS CARE & RESEARCH
Hidradenitis suppurativa therapy options should be patient guided
of their most challenging symptoms, according to an expert summary presented at the Skin of Color Update 2020.

“If your patient is only focused on the appearance of the lesions or the presence of sinus tracts, they might not think your treatment is working,” said Ginette A. Okoye, MD, professor and chair, department of dermatology, Howard University, Washington.
Instead, she advised working with patients to define priorities, allowing them to measure and appreciate improvement. The most difficult symptoms for one patient, such as pain or persistent abscess drainage, might not be the same for another.
There is a large array of treatment options for HS. These were once typically employed in stepwise manner, moving from steroids to hormonal therapies, antibiotics, and on to biologics and lasers, but Dr. Okoye reported that she layers on treatments, guided by patient priorities and responses. “Most of my patients are not on just one treatment at a time,” she said.
In addition to patient goals, her treatment choices are also influenced by the presence of comorbidities such as metabolic syndrome, polycystic ovarian syndrome (PCOS), or inflammatory bowel disease (IBD). For example, she reported she is more likely to include metformin among treatment options in patients with central obesity or insulin resistance, whereas she moves more quickly to a biologic for those with another systemic inflammatory disease such as IBD.
Although multiple factors appear to contribute to the symptoms of HS, the pathophysiology remains incompletely understood, but follicular occlusion is often “a primary inciting event,” Dr. Okoye said.
For this reason, laser hair removal can provide substantial benefit, she noted. Not only does it eliminate the occlusion, but the heat generated by the laser eliminates some of the pathogens, such as Porphyromonas gingivalis, associated with HS.
“Lasers work well for preventing new lesions from forming but also in making active lesions go away faster,” said Dr. Okoye, who relies on the Nd:YAG laser when treating this disease in darker skin. She has found lasers to be particularly effective in mild to moderate disease.
When using lasers, one challenge is third-party insurance, according to Dr. Okoye, who reported that she has tried repeatedly to convince payers that this treatment is medically indicated for HS, but claims have been routinely denied. As a result, she has had to significantly discount the cost of laser at her center in order to provide access to “a modality that actually works.”
Incision and drainage of inflamed painful lesions is a common intervention in HS, but Dr. Okoye discourages this approach. Because of the high recurrence rates, the benefits are temporary. Instead, she recommends an intralesional injection of triamcinolone acetonide diluted with equal amounts of lidocaine.
With this injection, “there is immediate pain relief followed by significant resolution of the inflammation,” she said. Because of the likelihood that patients seeking care in the emergency department for acutely inflamed lesions will receive surgical treatment, Dr. Okoye recommends offering patients urgent appointments for steroid injections when painful and inflamed lesions need immediate attention.
In contrast, marsupialization of abscesses or sinus tracts, often called deroofing, is associated with a relatively low risk of recurrence, can be done under local anesthesia in an office, and can lead to resolution of persistent nodules in patients with mild disease.
“This is an easy procedure that takes relatively little time,” advised Dr. Okoye, who provided CPT codes (10060 and 10061) that will provide reimbursement as long as procedural notes describe the rationale.
Metformin is an attractive adjunctive therapy for HS in patients with type 2 diabetes or features that suggest metabolic disturbances, such as central obesity, hypercholesterolemia, hypertension, or hypertriglyceridemia. It should also be considered in patients with PCOS because metformin decreases ovarian androgen production, she said.
When prescribing metformin in HS, which is an off-label indication, “I prefer the extended release formulation. It has a better profile in regard to gastrointestinal side effects and it can be taken once-daily,” Dr. Okoye said.
Citing a study that suggests patients with HS have even worse quality of life scores than do patients with diabetes, Dr. Okoye also emphasized the importance of psychosocial support and lifestyle modification as part of a holistic approach. With multiple manifestations of varying severity, individualizing therapy to control symptoms that the patient finds most bothersome is essential for optimizing patient well being.
Tien Viet Nguyen, MD, who practices dermatology and conducts clinical research in Bellevue, Wash., agrees that a comprehensive treatment program is needed. First author of a recent review article on HS, Dr. Nguyen agreed that common comorbidities like IBD, PCOS, and diabetes are accompanied frequently by a host of mental health and behavioral issues that contribute to impaired quality of life, such as depression, low self-esteem, sexual dysfunction, impaired sleep, and substance use disorders.
“Therefore, addressing these important comorbidities and quality of life issues with other health care professionals as a team is the best approach to improving health outcomes,” he said in an interview.
Dr. Nguyen also recently authored a chapter on quality of life issues associated with HS in the soon-to-be-published Comprehensive Guide to Hidradenitis Suppurativa (1st Edition, Dermatology Clinics). He agreed that optimal outcomes are achieved by an interdisciplinary team of health care providers who can address the sometimes independent but often interrelated comorbidities associated with this disorder.
Dr. Okoye has financial relationships with Pfizer and Unilver, but neither is relevant to this topic.
of their most challenging symptoms, according to an expert summary presented at the Skin of Color Update 2020.
“If your patient is only focused on the appearance of the lesions or the presence of sinus tracts, they might not think your treatment is working,” said Ginette A. Okoye, MD, professor and chair, department of dermatology, Howard University, Washington.
Instead, she advised working with patients to define priorities, allowing them to measure and appreciate improvement. The most difficult symptoms for one patient, such as pain or persistent abscess drainage, might not be the same for another.
There is a large array of treatment options for HS. These were once typically employed in stepwise manner, moving from steroids to hormonal therapies, antibiotics, and on to biologics and lasers, but Dr. Okoye reported that she layers on treatments, guided by patient priorities and responses. “Most of my patients are not on just one treatment at a time,” she said.
In addition to patient goals, her treatment choices are also influenced by the presence of comorbidities such as metabolic syndrome, polycystic ovarian syndrome (PCOS), or inflammatory bowel disease (IBD). For example, she reported she is more likely to include metformin among treatment options in patients with central obesity or insulin resistance, whereas she moves more quickly to a biologic for those with another systemic inflammatory disease such as IBD.
Although multiple factors appear to contribute to the symptoms of HS, the pathophysiology remains incompletely understood, but follicular occlusion is often “a primary inciting event,” Dr. Okoye said.
For this reason, laser hair removal can provide substantial benefit, she noted. Not only does it eliminate the occlusion, but the heat generated by the laser eliminates some of the pathogens, such as Porphyromonas gingivalis, associated with HS.
“Lasers work well for preventing new lesions from forming but also in making active lesions go away faster,” said Dr. Okoye, who relies on the Nd:YAG laser when treating this disease in darker skin. She has found lasers to be particularly effective in mild to moderate disease.
When using lasers, one challenge is third-party insurance, according to Dr. Okoye, who reported that she has tried repeatedly to convince payers that this treatment is medically indicated for HS, but claims have been routinely denied. As a result, she has had to significantly discount the cost of laser at her center in order to provide access to “a modality that actually works.”
Incision and drainage of inflamed painful lesions is a common intervention in HS, but Dr. Okoye discourages this approach. Because of the high recurrence rates, the benefits are temporary. Instead, she recommends an intralesional injection of triamcinolone acetonide diluted with equal amounts of lidocaine.
With this injection, “there is immediate pain relief followed by significant resolution of the inflammation,” she said. Because of the likelihood that patients seeking care in the emergency department for acutely inflamed lesions will receive surgical treatment, Dr. Okoye recommends offering patients urgent appointments for steroid injections when painful and inflamed lesions need immediate attention.
In contrast, marsupialization of abscesses or sinus tracts, often called deroofing, is associated with a relatively low risk of recurrence, can be done under local anesthesia in an office, and can lead to resolution of persistent nodules in patients with mild disease.
“This is an easy procedure that takes relatively little time,” advised Dr. Okoye, who provided CPT codes (10060 and 10061) that will provide reimbursement as long as procedural notes describe the rationale.
Metformin is an attractive adjunctive therapy for HS in patients with type 2 diabetes or features that suggest metabolic disturbances, such as central obesity, hypercholesterolemia, hypertension, or hypertriglyceridemia. It should also be considered in patients with PCOS because metformin decreases ovarian androgen production, she said.
When prescribing metformin in HS, which is an off-label indication, “I prefer the extended release formulation. It has a better profile in regard to gastrointestinal side effects and it can be taken once-daily,” Dr. Okoye said.
Citing a study that suggests patients with HS have even worse quality of life scores than do patients with diabetes, Dr. Okoye also emphasized the importance of psychosocial support and lifestyle modification as part of a holistic approach. With multiple manifestations of varying severity, individualizing therapy to control symptoms that the patient finds most bothersome is essential for optimizing patient well being.
Tien Viet Nguyen, MD, who practices dermatology and conducts clinical research in Bellevue, Wash., agrees that a comprehensive treatment program is needed. First author of a recent review article on HS, Dr. Nguyen agreed that common comorbidities like IBD, PCOS, and diabetes are accompanied frequently by a host of mental health and behavioral issues that contribute to impaired quality of life, such as depression, low self-esteem, sexual dysfunction, impaired sleep, and substance use disorders.
“Therefore, addressing these important comorbidities and quality of life issues with other health care professionals as a team is the best approach to improving health outcomes,” he said in an interview.
Dr. Nguyen also recently authored a chapter on quality of life issues associated with HS in the soon-to-be-published Comprehensive Guide to Hidradenitis Suppurativa (1st Edition, Dermatology Clinics). He agreed that optimal outcomes are achieved by an interdisciplinary team of health care providers who can address the sometimes independent but often interrelated comorbidities associated with this disorder.
Dr. Okoye has financial relationships with Pfizer and Unilver, but neither is relevant to this topic.
of their most challenging symptoms, according to an expert summary presented at the Skin of Color Update 2020.
“If your patient is only focused on the appearance of the lesions or the presence of sinus tracts, they might not think your treatment is working,” said Ginette A. Okoye, MD, professor and chair, department of dermatology, Howard University, Washington.
Instead, she advised working with patients to define priorities, allowing them to measure and appreciate improvement. The most difficult symptoms for one patient, such as pain or persistent abscess drainage, might not be the same for another.
There is a large array of treatment options for HS. These were once typically employed in stepwise manner, moving from steroids to hormonal therapies, antibiotics, and on to biologics and lasers, but Dr. Okoye reported that she layers on treatments, guided by patient priorities and responses. “Most of my patients are not on just one treatment at a time,” she said.
In addition to patient goals, her treatment choices are also influenced by the presence of comorbidities such as metabolic syndrome, polycystic ovarian syndrome (PCOS), or inflammatory bowel disease (IBD). For example, she reported she is more likely to include metformin among treatment options in patients with central obesity or insulin resistance, whereas she moves more quickly to a biologic for those with another systemic inflammatory disease such as IBD.
Although multiple factors appear to contribute to the symptoms of HS, the pathophysiology remains incompletely understood, but follicular occlusion is often “a primary inciting event,” Dr. Okoye said.
For this reason, laser hair removal can provide substantial benefit, she noted. Not only does it eliminate the occlusion, but the heat generated by the laser eliminates some of the pathogens, such as Porphyromonas gingivalis, associated with HS.
“Lasers work well for preventing new lesions from forming but also in making active lesions go away faster,” said Dr. Okoye, who relies on the Nd:YAG laser when treating this disease in darker skin. She has found lasers to be particularly effective in mild to moderate disease.
When using lasers, one challenge is third-party insurance, according to Dr. Okoye, who reported that she has tried repeatedly to convince payers that this treatment is medically indicated for HS, but claims have been routinely denied. As a result, she has had to significantly discount the cost of laser at her center in order to provide access to “a modality that actually works.”
Incision and drainage of inflamed painful lesions is a common intervention in HS, but Dr. Okoye discourages this approach. Because of the high recurrence rates, the benefits are temporary. Instead, she recommends an intralesional injection of triamcinolone acetonide diluted with equal amounts of lidocaine.
With this injection, “there is immediate pain relief followed by significant resolution of the inflammation,” she said. Because of the likelihood that patients seeking care in the emergency department for acutely inflamed lesions will receive surgical treatment, Dr. Okoye recommends offering patients urgent appointments for steroid injections when painful and inflamed lesions need immediate attention.
In contrast, marsupialization of abscesses or sinus tracts, often called deroofing, is associated with a relatively low risk of recurrence, can be done under local anesthesia in an office, and can lead to resolution of persistent nodules in patients with mild disease.
“This is an easy procedure that takes relatively little time,” advised Dr. Okoye, who provided CPT codes (10060 and 10061) that will provide reimbursement as long as procedural notes describe the rationale.
Metformin is an attractive adjunctive therapy for HS in patients with type 2 diabetes or features that suggest metabolic disturbances, such as central obesity, hypercholesterolemia, hypertension, or hypertriglyceridemia. It should also be considered in patients with PCOS because metformin decreases ovarian androgen production, she said.
When prescribing metformin in HS, which is an off-label indication, “I prefer the extended release formulation. It has a better profile in regard to gastrointestinal side effects and it can be taken once-daily,” Dr. Okoye said.
Citing a study that suggests patients with HS have even worse quality of life scores than do patients with diabetes, Dr. Okoye also emphasized the importance of psychosocial support and lifestyle modification as part of a holistic approach. With multiple manifestations of varying severity, individualizing therapy to control symptoms that the patient finds most bothersome is essential for optimizing patient well being.
Tien Viet Nguyen, MD, who practices dermatology and conducts clinical research in Bellevue, Wash., agrees that a comprehensive treatment program is needed. First author of a recent review article on HS, Dr. Nguyen agreed that common comorbidities like IBD, PCOS, and diabetes are accompanied frequently by a host of mental health and behavioral issues that contribute to impaired quality of life, such as depression, low self-esteem, sexual dysfunction, impaired sleep, and substance use disorders.
“Therefore, addressing these important comorbidities and quality of life issues with other health care professionals as a team is the best approach to improving health outcomes,” he said in an interview.
Dr. Nguyen also recently authored a chapter on quality of life issues associated with HS in the soon-to-be-published Comprehensive Guide to Hidradenitis Suppurativa (1st Edition, Dermatology Clinics). He agreed that optimal outcomes are achieved by an interdisciplinary team of health care providers who can address the sometimes independent but often interrelated comorbidities associated with this disorder.
Dr. Okoye has financial relationships with Pfizer and Unilver, but neither is relevant to this topic.
FROM SOC 2020
Hyperpigmented patch on the back

Large unilateral hyperpigmented patches on the trunk with onset around puberty are the hallmark of a Becker nevus, also more descriptively called pigmented hairy epidermal nevus.
Becker nevi are a form of epidermal nevus that usually occur on the upper back or chest. They most commonly develop during puberty when there are increasing circulating levels of androgens. (Becker nevus cells are androgen sensitive.) This is consistent with this patient’s history of the lesion developing in her teens when the lesions become hyperpigmented and noticeable. The localized androgen sensitivity also can lead to unilateral hypoplastic breast growth when it occurs on the chest in young women.
The lesions are more common in males than females and often have associated hypertrichosis. The etiology is not certain but is thought to be due to regional loss of heterozygosity during embryogenesis leading to the abnormally elevated levels of androgen receptors and increased androgen sensitivity in the basal keratinocytes and dermal fibroblasts.
Laser is the most effective therapy for the hyperpigmentation and for hypertrichosis when present. If a young woman with a Becker nevus has breast hypoplasia, spironolactone (an antiandrogen) has been helpful in restoring breast growth. For this patient, the hyperpigmented patch was asymptomatic and not troublesome, so she opted not to treat it.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Patel P, Malik K, Khachemoune A. Sebaceus and Becker’s nevus: overview of their presentation, pathogenesis, associations, and treatment. Am J Clin Dermatol. 2015;16:197-204.
Large unilateral hyperpigmented patches on the trunk with onset around puberty are the hallmark of a Becker nevus, also more descriptively called pigmented hairy epidermal nevus.
Becker nevi are a form of epidermal nevus that usually occur on the upper back or chest. They most commonly develop during puberty when there are increasing circulating levels of androgens. (Becker nevus cells are androgen sensitive.) This is consistent with this patient’s history of the lesion developing in her teens when the lesions become hyperpigmented and noticeable. The localized androgen sensitivity also can lead to unilateral hypoplastic breast growth when it occurs on the chest in young women.
The lesions are more common in males than females and often have associated hypertrichosis. The etiology is not certain but is thought to be due to regional loss of heterozygosity during embryogenesis leading to the abnormally elevated levels of androgen receptors and increased androgen sensitivity in the basal keratinocytes and dermal fibroblasts.
Laser is the most effective therapy for the hyperpigmentation and for hypertrichosis when present. If a young woman with a Becker nevus has breast hypoplasia, spironolactone (an antiandrogen) has been helpful in restoring breast growth. For this patient, the hyperpigmented patch was asymptomatic and not troublesome, so she opted not to treat it.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Large unilateral hyperpigmented patches on the trunk with onset around puberty are the hallmark of a Becker nevus, also more descriptively called pigmented hairy epidermal nevus.
Becker nevi are a form of epidermal nevus that usually occur on the upper back or chest. They most commonly develop during puberty when there are increasing circulating levels of androgens. (Becker nevus cells are androgen sensitive.) This is consistent with this patient’s history of the lesion developing in her teens when the lesions become hyperpigmented and noticeable. The localized androgen sensitivity also can lead to unilateral hypoplastic breast growth when it occurs on the chest in young women.
The lesions are more common in males than females and often have associated hypertrichosis. The etiology is not certain but is thought to be due to regional loss of heterozygosity during embryogenesis leading to the abnormally elevated levels of androgen receptors and increased androgen sensitivity in the basal keratinocytes and dermal fibroblasts.
Laser is the most effective therapy for the hyperpigmentation and for hypertrichosis when present. If a young woman with a Becker nevus has breast hypoplasia, spironolactone (an antiandrogen) has been helpful in restoring breast growth. For this patient, the hyperpigmented patch was asymptomatic and not troublesome, so she opted not to treat it.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Patel P, Malik K, Khachemoune A. Sebaceus and Becker’s nevus: overview of their presentation, pathogenesis, associations, and treatment. Am J Clin Dermatol. 2015;16:197-204.
Patel P, Malik K, Khachemoune A. Sebaceus and Becker’s nevus: overview of their presentation, pathogenesis, associations, and treatment. Am J Clin Dermatol. 2015;16:197-204.
