User login
Multiple basal cell carcinomas

These skin findings were the latest manifestation of a condition that the patient had been diagnosed with at age 32: basal cell nevus syndrome (BCNS), also called Gorlin syndrome. This syndrome is characterized by multiple biopsy-proven BCCs, palmar pitting, frontal bossing, scoliosis, and gum cysts. This patient had had gum cysts since she was 8 years old; her sister and mother had similar gum cysts and her mother had at least 1 BCC. BCNS is caused by an inheritable defect in the Patched 1 (PTCH1) gene, leading to various findings—including numerous BCCs at a young age.1
The Oncology team started the patient on the oral small-molecule chemotherapy agent vismodegib, 150 mg/d. The patient was also referred to Medical Genetics and Wound Care. Although her diagnosis had been made clinically years earlier, genetic testing was performed and confirmed a defect in the PTCH1 gene. This helped with surveillance plans. A computed tomography scan of the head revealed a nasal dermoid cyst of the ethmoid sinus that the Ear, Nose, & Throat and Neurology teams felt safe to observe.
After 3 months of therapy with vismodegib, the patient had significant improvement of facial lesions and significant re-epithelialization of the crown.
Patients on vismodegib often deal with adverse effects, but adjusted dosing regimens have proved to improve tolerability. This patient had substantial adverse effects including fatigue, hair loss, loss of taste, and weight loss (26 lbs). Because of these adverse effects, her regimen was adjusted to 1 month of every other day active treatment and 2 months off treatment, cycled continuously. With this regimen, her weight returned to normal and her sense of taste returned for most days in the treatment cycle.
She has been tolerating this regimen for 3 years with continued control of BCCs.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224. doi:10.1001/jamadermatol.2015.3210
These skin findings were the latest manifestation of a condition that the patient had been diagnosed with at age 32: basal cell nevus syndrome (BCNS), also called Gorlin syndrome. This syndrome is characterized by multiple biopsy-proven BCCs, palmar pitting, frontal bossing, scoliosis, and gum cysts. This patient had had gum cysts since she was 8 years old; her sister and mother had similar gum cysts and her mother had at least 1 BCC. BCNS is caused by an inheritable defect in the Patched 1 (PTCH1) gene, leading to various findings—including numerous BCCs at a young age.1
The Oncology team started the patient on the oral small-molecule chemotherapy agent vismodegib, 150 mg/d. The patient was also referred to Medical Genetics and Wound Care. Although her diagnosis had been made clinically years earlier, genetic testing was performed and confirmed a defect in the PTCH1 gene. This helped with surveillance plans. A computed tomography scan of the head revealed a nasal dermoid cyst of the ethmoid sinus that the Ear, Nose, & Throat and Neurology teams felt safe to observe.
After 3 months of therapy with vismodegib, the patient had significant improvement of facial lesions and significant re-epithelialization of the crown.
Patients on vismodegib often deal with adverse effects, but adjusted dosing regimens have proved to improve tolerability. This patient had substantial adverse effects including fatigue, hair loss, loss of taste, and weight loss (26 lbs). Because of these adverse effects, her regimen was adjusted to 1 month of every other day active treatment and 2 months off treatment, cycled continuously. With this regimen, her weight returned to normal and her sense of taste returned for most days in the treatment cycle.
She has been tolerating this regimen for 3 years with continued control of BCCs.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
These skin findings were the latest manifestation of a condition that the patient had been diagnosed with at age 32: basal cell nevus syndrome (BCNS), also called Gorlin syndrome. This syndrome is characterized by multiple biopsy-proven BCCs, palmar pitting, frontal bossing, scoliosis, and gum cysts. This patient had had gum cysts since she was 8 years old; her sister and mother had similar gum cysts and her mother had at least 1 BCC. BCNS is caused by an inheritable defect in the Patched 1 (PTCH1) gene, leading to various findings—including numerous BCCs at a young age.1
The Oncology team started the patient on the oral small-molecule chemotherapy agent vismodegib, 150 mg/d. The patient was also referred to Medical Genetics and Wound Care. Although her diagnosis had been made clinically years earlier, genetic testing was performed and confirmed a defect in the PTCH1 gene. This helped with surveillance plans. A computed tomography scan of the head revealed a nasal dermoid cyst of the ethmoid sinus that the Ear, Nose, & Throat and Neurology teams felt safe to observe.
After 3 months of therapy with vismodegib, the patient had significant improvement of facial lesions and significant re-epithelialization of the crown.
Patients on vismodegib often deal with adverse effects, but adjusted dosing regimens have proved to improve tolerability. This patient had substantial adverse effects including fatigue, hair loss, loss of taste, and weight loss (26 lbs). Because of these adverse effects, her regimen was adjusted to 1 month of every other day active treatment and 2 months off treatment, cycled continuously. With this regimen, her weight returned to normal and her sense of taste returned for most days in the treatment cycle.
She has been tolerating this regimen for 3 years with continued control of BCCs.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224. doi:10.1001/jamadermatol.2015.3210
1. Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224. doi:10.1001/jamadermatol.2015.3210

AAD updates guidelines for managing AD with phototherapy and systemic therapies
.
The guidelines cover approved and off-label uses of systemic therapies and phototherapy, including new treatments that have become available since the last guidelines were published almost a decade ago. These include biologics and oral Janus kinase (JAK) inhibitors, as well as older oral or injectable immunomodulators and antimetabolites, oral antibiotics, antihistamines, and phosphodiesterase-4 inhibitors. The guidelines rate the existing evidence as “strong” for dupilumab, tralokinumab, abrocitinib, baricitinib, and upadacitinib. They also conditionally recommend phototherapy, as well as cyclosporine, methotrexate, azathioprine, and mycophenolate, but recommend against the use of systemic corticosteroids.
The guidelines update the AAD’s 2014 recommendations for managing AD in adults with phototherapy and systemic therapies. “At that time, prednisone – universally agreed to be the least appropriate chronic therapy for AD – was the only Food and Drug Administration–approved agent,” Robert Sidbury, MD, MPH, who cochaired a 14-member multidisciplinary work group that assembled the guidelines, told this news organization. “This was the driver.”

The latest guidelines were published online in the Journal of the American Academy of Dermatology.
Broad evidence review
Dr. Sidbury, chief of the division of dermatology at Seattle Children’s Hospital, guidelines cochair Dawn M. R. Davis, MD, a dermatologist at the Mayo Clinic, Rochester, Minn., and colleagues conducted a systematic evidence review of phototherapy such as narrowband and broadband UVB and systemic therapies, including biologics such as dupilumab and tralokinumab, JAK inhibitors such as upadacitinib and abrocitinib, and immunosuppressants such as methotrexate and azathioprine.

Next, the work group applied the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) approach for assessing the certainty of the evidence and formulating and grading clinical recommendations based on relevant randomized trials in the medical literature.
Recommendations, future studies
Of the 11 evidence-based recommendations of therapies for adults with AD refractory to topical medications, the work group ranks 5 as “strong” based on the evidence and the rest as “conditional.” “Strong” implies the benefits clearly outweigh risks and burdens, they apply to most patients in most circumstances, and they fall under good clinical practice. “Conditional” means the benefits and risks are closely balanced for most patients, “but the appropriate action may different depending on the patient or other stakeholder values,” the authors wrote.
In their remarks about phototherapy, the work group noted that most published literature on the topic “reports on the efficacy and safety of narrow band UVB. Wherever possible, use a light source that minimizes the potential for harm under the supervision of a qualified clinician.”
In their remarks about cyclosporine, they noted that evidence suggests an initial dose of 3 mg/kg per day to 5 mg/kg per day is effective, but that the Food and Drug Administration has not approved cyclosporine for use in AD. “The FDA has approved limited-term use (up to 1 year) in psoriasis,” they wrote. “Comorbidities or drug interactions that may exacerbate toxicity make this intervention inappropriate for select patients.” The work group noted that significant research gaps remain in phototherapy, especially trials that compare different phototherapy modalities and those that compare phototherapy with other AD treatment strategies.
“Larger clinical trials would also be helpful for cyclosporine, methotrexate, azathioprine, and mycophenolate to improve the certainty of evidence for those medications,” they added. “Furthermore, formal cost-effectiveness analyses comparing older to newer treatments are needed.”
They recommended the inclusion of active comparator arms in randomized, controlled trials as new systemic therapies continue to be developed and tested.
The work group ranked the level of evidence they reviewed for the therapies from very low to moderate. No therapy was judged to have high evidence. They also cited the short duration of most randomized controlled trials of phototherapy.
Using the guidelines in clinical care
According to Dr. Davis, the topic of which agent if any should be considered “first line” generated robust discussion among the work group members.
“When there are not robust head-to-head trials – and there are not – it is often opinion that governs this decision, and opinion should not, when possible, govern a guideline,” Dr. Davis said. “Accordingly, we determined based upon the evidence agents – plural – that deserve to be considered ‘first line’ but not a single agent.”
In her opinion, the top three considerations regarding use of systemic therapy for AD relate to patient selection and shared decision making. One, standard therapy has failed. Two, diagnosis is assured. And three, “steroid phobia should be considered,” and patients should be “fully informed of risks and benefits of both treating and not treating,” she said.
Dr. Sidbury reported that he serves as an advisory board member for Pfizer, a principal investigator for Regeneron, an investigator for Brickell Biotech and Galderma USA, and a consultant for Galderma Global and Micreos. Dr. Davis reported having no relevant disclosures. Other work group members reported having financial disclosures with many pharmaceutical companies. The study was supported by internal funds from the American Academy of Dermatology.
.
The guidelines cover approved and off-label uses of systemic therapies and phototherapy, including new treatments that have become available since the last guidelines were published almost a decade ago. These include biologics and oral Janus kinase (JAK) inhibitors, as well as older oral or injectable immunomodulators and antimetabolites, oral antibiotics, antihistamines, and phosphodiesterase-4 inhibitors. The guidelines rate the existing evidence as “strong” for dupilumab, tralokinumab, abrocitinib, baricitinib, and upadacitinib. They also conditionally recommend phototherapy, as well as cyclosporine, methotrexate, azathioprine, and mycophenolate, but recommend against the use of systemic corticosteroids.
The guidelines update the AAD’s 2014 recommendations for managing AD in adults with phototherapy and systemic therapies. “At that time, prednisone – universally agreed to be the least appropriate chronic therapy for AD – was the only Food and Drug Administration–approved agent,” Robert Sidbury, MD, MPH, who cochaired a 14-member multidisciplinary work group that assembled the guidelines, told this news organization. “This was the driver.”
The latest guidelines were published online in the Journal of the American Academy of Dermatology.
Broad evidence review
Dr. Sidbury, chief of the division of dermatology at Seattle Children’s Hospital, guidelines cochair Dawn M. R. Davis, MD, a dermatologist at the Mayo Clinic, Rochester, Minn., and colleagues conducted a systematic evidence review of phototherapy such as narrowband and broadband UVB and systemic therapies, including biologics such as dupilumab and tralokinumab, JAK inhibitors such as upadacitinib and abrocitinib, and immunosuppressants such as methotrexate and azathioprine.
Next, the work group applied the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) approach for assessing the certainty of the evidence and formulating and grading clinical recommendations based on relevant randomized trials in the medical literature.
Recommendations, future studies
Of the 11 evidence-based recommendations of therapies for adults with AD refractory to topical medications, the work group ranks 5 as “strong” based on the evidence and the rest as “conditional.” “Strong” implies the benefits clearly outweigh risks and burdens, they apply to most patients in most circumstances, and they fall under good clinical practice. “Conditional” means the benefits and risks are closely balanced for most patients, “but the appropriate action may different depending on the patient or other stakeholder values,” the authors wrote.
In their remarks about phototherapy, the work group noted that most published literature on the topic “reports on the efficacy and safety of narrow band UVB. Wherever possible, use a light source that minimizes the potential for harm under the supervision of a qualified clinician.”
In their remarks about cyclosporine, they noted that evidence suggests an initial dose of 3 mg/kg per day to 5 mg/kg per day is effective, but that the Food and Drug Administration has not approved cyclosporine for use in AD. “The FDA has approved limited-term use (up to 1 year) in psoriasis,” they wrote. “Comorbidities or drug interactions that may exacerbate toxicity make this intervention inappropriate for select patients.” The work group noted that significant research gaps remain in phototherapy, especially trials that compare different phototherapy modalities and those that compare phototherapy with other AD treatment strategies.
“Larger clinical trials would also be helpful for cyclosporine, methotrexate, azathioprine, and mycophenolate to improve the certainty of evidence for those medications,” they added. “Furthermore, formal cost-effectiveness analyses comparing older to newer treatments are needed.”
They recommended the inclusion of active comparator arms in randomized, controlled trials as new systemic therapies continue to be developed and tested.
The work group ranked the level of evidence they reviewed for the therapies from very low to moderate. No therapy was judged to have high evidence. They also cited the short duration of most randomized controlled trials of phototherapy.
Using the guidelines in clinical care
According to Dr. Davis, the topic of which agent if any should be considered “first line” generated robust discussion among the work group members.
“When there are not robust head-to-head trials – and there are not – it is often opinion that governs this decision, and opinion should not, when possible, govern a guideline,” Dr. Davis said. “Accordingly, we determined based upon the evidence agents – plural – that deserve to be considered ‘first line’ but not a single agent.”
In her opinion, the top three considerations regarding use of systemic therapy for AD relate to patient selection and shared decision making. One, standard therapy has failed. Two, diagnosis is assured. And three, “steroid phobia should be considered,” and patients should be “fully informed of risks and benefits of both treating and not treating,” she said.
Dr. Sidbury reported that he serves as an advisory board member for Pfizer, a principal investigator for Regeneron, an investigator for Brickell Biotech and Galderma USA, and a consultant for Galderma Global and Micreos. Dr. Davis reported having no relevant disclosures. Other work group members reported having financial disclosures with many pharmaceutical companies. The study was supported by internal funds from the American Academy of Dermatology.
.
The guidelines cover approved and off-label uses of systemic therapies and phototherapy, including new treatments that have become available since the last guidelines were published almost a decade ago. These include biologics and oral Janus kinase (JAK) inhibitors, as well as older oral or injectable immunomodulators and antimetabolites, oral antibiotics, antihistamines, and phosphodiesterase-4 inhibitors. The guidelines rate the existing evidence as “strong” for dupilumab, tralokinumab, abrocitinib, baricitinib, and upadacitinib. They also conditionally recommend phototherapy, as well as cyclosporine, methotrexate, azathioprine, and mycophenolate, but recommend against the use of systemic corticosteroids.
The guidelines update the AAD’s 2014 recommendations for managing AD in adults with phototherapy and systemic therapies. “At that time, prednisone – universally agreed to be the least appropriate chronic therapy for AD – was the only Food and Drug Administration–approved agent,” Robert Sidbury, MD, MPH, who cochaired a 14-member multidisciplinary work group that assembled the guidelines, told this news organization. “This was the driver.”
The latest guidelines were published online in the Journal of the American Academy of Dermatology.
Broad evidence review
Dr. Sidbury, chief of the division of dermatology at Seattle Children’s Hospital, guidelines cochair Dawn M. R. Davis, MD, a dermatologist at the Mayo Clinic, Rochester, Minn., and colleagues conducted a systematic evidence review of phototherapy such as narrowband and broadband UVB and systemic therapies, including biologics such as dupilumab and tralokinumab, JAK inhibitors such as upadacitinib and abrocitinib, and immunosuppressants such as methotrexate and azathioprine.
Next, the work group applied the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) approach for assessing the certainty of the evidence and formulating and grading clinical recommendations based on relevant randomized trials in the medical literature.
Recommendations, future studies
Of the 11 evidence-based recommendations of therapies for adults with AD refractory to topical medications, the work group ranks 5 as “strong” based on the evidence and the rest as “conditional.” “Strong” implies the benefits clearly outweigh risks and burdens, they apply to most patients in most circumstances, and they fall under good clinical practice. “Conditional” means the benefits and risks are closely balanced for most patients, “but the appropriate action may different depending on the patient or other stakeholder values,” the authors wrote.
In their remarks about phototherapy, the work group noted that most published literature on the topic “reports on the efficacy and safety of narrow band UVB. Wherever possible, use a light source that minimizes the potential for harm under the supervision of a qualified clinician.”
In their remarks about cyclosporine, they noted that evidence suggests an initial dose of 3 mg/kg per day to 5 mg/kg per day is effective, but that the Food and Drug Administration has not approved cyclosporine for use in AD. “The FDA has approved limited-term use (up to 1 year) in psoriasis,” they wrote. “Comorbidities or drug interactions that may exacerbate toxicity make this intervention inappropriate for select patients.” The work group noted that significant research gaps remain in phototherapy, especially trials that compare different phototherapy modalities and those that compare phototherapy with other AD treatment strategies.
“Larger clinical trials would also be helpful for cyclosporine, methotrexate, azathioprine, and mycophenolate to improve the certainty of evidence for those medications,” they added. “Furthermore, formal cost-effectiveness analyses comparing older to newer treatments are needed.”
They recommended the inclusion of active comparator arms in randomized, controlled trials as new systemic therapies continue to be developed and tested.
The work group ranked the level of evidence they reviewed for the therapies from very low to moderate. No therapy was judged to have high evidence. They also cited the short duration of most randomized controlled trials of phototherapy.
Using the guidelines in clinical care
According to Dr. Davis, the topic of which agent if any should be considered “first line” generated robust discussion among the work group members.
“When there are not robust head-to-head trials – and there are not – it is often opinion that governs this decision, and opinion should not, when possible, govern a guideline,” Dr. Davis said. “Accordingly, we determined based upon the evidence agents – plural – that deserve to be considered ‘first line’ but not a single agent.”
In her opinion, the top three considerations regarding use of systemic therapy for AD relate to patient selection and shared decision making. One, standard therapy has failed. Two, diagnosis is assured. And three, “steroid phobia should be considered,” and patients should be “fully informed of risks and benefits of both treating and not treating,” she said.
Dr. Sidbury reported that he serves as an advisory board member for Pfizer, a principal investigator for Regeneron, an investigator for Brickell Biotech and Galderma USA, and a consultant for Galderma Global and Micreos. Dr. Davis reported having no relevant disclosures. Other work group members reported having financial disclosures with many pharmaceutical companies. The study was supported by internal funds from the American Academy of Dermatology.
FROM JAMA DERMATOLOGY
Specialty-trained pathologists more likely to make higher-grade diagnoses for melanocytic lesions
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
, results from an exploratory study showed.
The findings “could in part play a role in the rising incidence of early-stage melanoma with low risk of progression or patient morbidity, thereby contributing to increasing rates of overdiagnosis,” researchers led by co–senior authors Joann G. Elmore, MD, MPH, of the University of California, Los Angeles, and Raymond L. Barnhill, MD, MBA, of the Institut Curie, Paris, wrote in their study, published online in JAMA Dermatology.
To investigate the characteristics associated with rendering higher-grade diagnoses, including invasive melanoma, the researchers drew from two national data sets: the Melanoma Pathology (M-Path) study, conducted from July 2013 to May 2016, and the Reducing Errors in Melanocytic Interpretations (REMI) study, conducted from August 2018 to March 2021. In both studies, pathologists who interpreted melanocytic lesions in their clinical practices interpreted study cases in glass slide format. For the current study, researchers used logistic regression to examine the association of pathologist characteristics with diagnosis of a study case as higher grade (including severely dysplastic and melanoma in situ) vs. lower grade (including mild to moderately dysplastic nevi) and diagnosis of invasive melanoma vs. any less severe diagnosis.
A total of 338 pathologists were included in the analysis. Of these, 113 were general pathologists and 225 were dermatopathologists (those who were board certified and/or fellowship trained in dermatopathology).
The researchers found that, compared with general pathologists, dermatopathologists were 2.63 times more likely to render higher-grade diagnoses and 1.95 times more likely to diagnose invasive melanoma (P < .001 for both associations). Diagnoses of stage pT1a melanomas with no mitotic activity completely accounted for the difference between dermatopathologists and general pathologists in diagnosing invasive melanoma.
For the analysis limited to the 225 dermatopathologists, those with a higher practice caseload of melanocytic lesions were more likely to assign higher-grade diagnoses (odds ratio for trend, 1.27; P = .02), while those affiliated with an academic center had lower odds of diagnosing invasive melanoma (OR, 0.61; P = .049).
The researchers acknowledged limitations of their analysis, including the lack of data on patient outcomes, “so we could not make conclusions about the clinical outcome of any particular diagnosis by a study participant,” they wrote. “While our analyses revealed pathologist characteristics associated with assigning more vs. less severe diagnoses of melanocytic lesions, we could not conclude that any particular diagnosis by a study participant was overcalling or undercalling. However, the epidemiologic evidence that melanoma is overdiagnosed suggests that overcalling by some pathologists may be contributing to increasing rates of low-risk melanoma diagnoses.”
In an accompanying editorial, authors Klaus J. Busam, MD, of the department of pathology and laboratory medicine at Memorial Sloan Kettering Cancer Center, New York, Pedram Gerami, MD, of the department of dermatology at Northwestern University, Chicago, and Richard A. Scolyer, MD, of the Melanoma Institute, Wollstonecraft, Australia, wrote that the study findings “raise the question of whether subspecialization in dermatopathology may be a factor contributing to the epidemiologic phenomenon of overdiagnosis – that is, the discordance in the rise of melanoma incidence and relatively constant annual mortality rates over many decades. The findings also invite a discussion about strategies to minimize harm from overdiagnosis for both patients and the health care system.”
To minimize misdiagnoses, they continued, efforts to facilitate diagnostic accuracy should be encouraged. “Excisional (rather than partial) biopsies and provision of relevant clinical information would facilitate rendering of the correct histopathologic diagnosis,” they wrote. “When the diagnosis is uncertain, this is best acknowledged. If felt necessary, a reexcision of a lesion with an uncertain diagnosis can be recommended without upgrading the diagnosis.”
In addition, “improvements in prognosis are needed beyond American Joint Committee on Cancer staging,” they noted. “This will likely require a multimodal approach with novel methods, including artificial intelligence and biomarkers that help distinguish low-risk melanomas, for which a conservative approach may be appropriate, from those that require surgical intervention.”
The study was supported by the National Center for Advancing Translational Sciences and by the National Institutes of Health. One author disclosed receiving grants from the National Cancer Institute during the conduct of the study, and another disclosed serving as editor in chief of Primary Care topics at UpToDate; other authors had no disclosures. Dr. Busam reported receiving nonfinancial support from the American Society of Dermatopathology. Dr. Gerami reported receiving consulting fees from Castle Biosciences. Dr. Scolyer reported receiving an investigator grant from the National Health and Medical Research Council of Australia during the conduct of the study and personal fees from several pharmaceutical companies outside the submitted work.
FROM JAMA DERMATOLOGY
Prurigo nodularis diagnosis delay in skin of color gains added significance
NEW YORK – according to an expert evaluating current approaches at the Skin of Color Update 2023.
“As dermatologists, prurigo nodularis is one of the most severe diseases we treat, said Shawn G. Kwatra, MD, director of the Johns Hopkins Itch Center, Baltimore. Now with one approved therapy and more coming, “it offers one of the most important opportunities we have to dramatically improve someone’s entire life.”

Prior to the September 2022 approval of dupilumab for the treatment of prurigo nodularis (the first treatment approved for this indication), Dr. Kwatra said that the limited options for control of pruritus made him anxious. Prurigo nodularis is characterized by highly itchy nodules that can produce symptoms patients describe as unbearable.
Itch typically severe
On a scale for which 10 represents the worst itch imaginable, scores of 8 or greater are not unusual, according to Dr. Kwatra. Nodules on the trunk and the extensor surfaces of the arms and legs are characteristic, but the persistent itch is the immediate target of treatment once the diagnosis is made. For that reason, he urged clinicians to be familiar with the presentation in patients with darker skin types to reduce time to treatment.
In addition to the difficulty of seeing the characteristic red that is typical of erythema in lighter skin, patients with darker skin types tend to have larger nodules that might vary in shape relative to lighter skin types, Dr. Kwatra said. Given that the presentation of prurigo nodularis is highly heterogeneous even among the same skin types, the nuances in patients with darker skin can be that much more confusing for those without prior experience.
Among Blacks in particular, the nodules in some cases “can be huge,” he added. “They can almost look like keloids due to their thickened and fibrotic appearance.”
Phenotypes appear to be racially linked
In Black patients, the appearance can vary enough relative to lighter skin individuals, that “there seems to be something a little bit different going on,” he said, and this is, in fact, supported by a cluster analysis of circulating biomarkers reported by Dr. Kwatra and colleagues in 2022, in the Journal of Investigative Dermatology.
In that study, the biomarker profile distinguished two distinct groups. Whites were more common in a cluster with relatively low expression of inflammatory markers (cluster 1), while Blacks were more common in a cluster with an inflammatory plasma profile (cluster 2), with higher relative expression of multiple cytokines, C-reactive protein, eosinophils, and other markers of up-regulated inflammation.
In addition to a lower rate of myelopathy in cluster 2 than cluster 1 (18% vs. 67%; P = .028), patients in cluster 2 had a significantly worse itch than those in cluster 1 on the Numeric Rating Scale for itch and a significantly lower quality of life based on the Dermatology Life Quality Index score.
Other work at Dr. Kwatra’s center that is based on genetic sequencing has provided evidence that Blacks – and Asians to a lesser extent – are predisposed genetically to develop nodules, perhaps explaining why the nodules tend to be larger than those seen in Whites.
The significance of the evidence that prurigo nodularis is associated with a more up-regulated inflammatory profile in Blacks than in Whites is that they might be particularly likely to respond to dupilumab or other targeted immunomodulating therapies that are in development, according to Dr. Kwatra. Although he did not provide data on response by race, he did provide several case examples of complete itch control following dupilumab therapy in Black patients.
In his experience, high levels of blood eosinophils and other inflammatory markers are predictors of response to dupilumab regardless of skin type, but he expressed concern that time to diagnosis is sometimes longer in Black patients if the nuances of disease expression are not appreciated.
For treating prurigo nodularis in Blacks as well as Whites, Dr. Kwatra suggested that clinicians stay current with what he predicted will be a growing array of treatment options. He did not discuss nemolizumab, an interleukin-31 receptor alpha antagonist. Soon after the meeting, results of a phase 3 trial of nemolizumab in patients with moderate to severe prurigo nodularis were published in the New England Journal of Medicine. (Dr. Kwatra is the lead author of the study but did not specifically discuss this treatment at the meeting.)
In the international placebo-controlled trial, called OLYMPIA 2, treatment was associated with a significant reduction in the signs and symptoms of prurigo nodularis, including reductions in itch, at 16 weeks, although only 4% of patients in the study were Black.
Given the expanding array of therapies, the message of considering prurigo nodularis in Black patients in order to accelerate the time to diagnosis is timely, Andrew F. Alexis, MD, MPH, professor of clinical dermatology and vice-chair for diversity and inclusion for the department of dermatology, Weill Cornell Medicine, New York.
“Current studies suggest a higher prevalence and greater severity of prurigo nodularis among Black patients compared to White patients,” said Dr. Alexis, agreeing with Dr. Kwatra. Referring to evidence that Blacks might mount a greater inflammatory response to prurigo nodularis than Whites, Dr. Alexis called for “a better understanding of the pathomechanisms” of this disease in order “to address unmet needs and reduce disparities for our diverse population of patients who suffer from prurigo nodularis.’
Dr. Kwatra reported financial relationships with AbbVie, Amgen, Arcutis, ASLAN, Cara, Castle Biosciences, Celldex, Galderma, Incyte, Johnson & Johnson, LEO pharma, Novartis, Pfizer, Regeneron, and Sanofi.
NEW YORK – according to an expert evaluating current approaches at the Skin of Color Update 2023.
“As dermatologists, prurigo nodularis is one of the most severe diseases we treat, said Shawn G. Kwatra, MD, director of the Johns Hopkins Itch Center, Baltimore. Now with one approved therapy and more coming, “it offers one of the most important opportunities we have to dramatically improve someone’s entire life.”
Prior to the September 2022 approval of dupilumab for the treatment of prurigo nodularis (the first treatment approved for this indication), Dr. Kwatra said that the limited options for control of pruritus made him anxious. Prurigo nodularis is characterized by highly itchy nodules that can produce symptoms patients describe as unbearable.
Itch typically severe
On a scale for which 10 represents the worst itch imaginable, scores of 8 or greater are not unusual, according to Dr. Kwatra. Nodules on the trunk and the extensor surfaces of the arms and legs are characteristic, but the persistent itch is the immediate target of treatment once the diagnosis is made. For that reason, he urged clinicians to be familiar with the presentation in patients with darker skin types to reduce time to treatment.
In addition to the difficulty of seeing the characteristic red that is typical of erythema in lighter skin, patients with darker skin types tend to have larger nodules that might vary in shape relative to lighter skin types, Dr. Kwatra said. Given that the presentation of prurigo nodularis is highly heterogeneous even among the same skin types, the nuances in patients with darker skin can be that much more confusing for those without prior experience.
Among Blacks in particular, the nodules in some cases “can be huge,” he added. “They can almost look like keloids due to their thickened and fibrotic appearance.”
Phenotypes appear to be racially linked
In Black patients, the appearance can vary enough relative to lighter skin individuals, that “there seems to be something a little bit different going on,” he said, and this is, in fact, supported by a cluster analysis of circulating biomarkers reported by Dr. Kwatra and colleagues in 2022, in the Journal of Investigative Dermatology.
In that study, the biomarker profile distinguished two distinct groups. Whites were more common in a cluster with relatively low expression of inflammatory markers (cluster 1), while Blacks were more common in a cluster with an inflammatory plasma profile (cluster 2), with higher relative expression of multiple cytokines, C-reactive protein, eosinophils, and other markers of up-regulated inflammation.
In addition to a lower rate of myelopathy in cluster 2 than cluster 1 (18% vs. 67%; P = .028), patients in cluster 2 had a significantly worse itch than those in cluster 1 on the Numeric Rating Scale for itch and a significantly lower quality of life based on the Dermatology Life Quality Index score.
Other work at Dr. Kwatra’s center that is based on genetic sequencing has provided evidence that Blacks – and Asians to a lesser extent – are predisposed genetically to develop nodules, perhaps explaining why the nodules tend to be larger than those seen in Whites.
The significance of the evidence that prurigo nodularis is associated with a more up-regulated inflammatory profile in Blacks than in Whites is that they might be particularly likely to respond to dupilumab or other targeted immunomodulating therapies that are in development, according to Dr. Kwatra. Although he did not provide data on response by race, he did provide several case examples of complete itch control following dupilumab therapy in Black patients.
In his experience, high levels of blood eosinophils and other inflammatory markers are predictors of response to dupilumab regardless of skin type, but he expressed concern that time to diagnosis is sometimes longer in Black patients if the nuances of disease expression are not appreciated.
For treating prurigo nodularis in Blacks as well as Whites, Dr. Kwatra suggested that clinicians stay current with what he predicted will be a growing array of treatment options. He did not discuss nemolizumab, an interleukin-31 receptor alpha antagonist. Soon after the meeting, results of a phase 3 trial of nemolizumab in patients with moderate to severe prurigo nodularis were published in the New England Journal of Medicine. (Dr. Kwatra is the lead author of the study but did not specifically discuss this treatment at the meeting.)
In the international placebo-controlled trial, called OLYMPIA 2, treatment was associated with a significant reduction in the signs and symptoms of prurigo nodularis, including reductions in itch, at 16 weeks, although only 4% of patients in the study were Black.
Given the expanding array of therapies, the message of considering prurigo nodularis in Black patients in order to accelerate the time to diagnosis is timely, Andrew F. Alexis, MD, MPH, professor of clinical dermatology and vice-chair for diversity and inclusion for the department of dermatology, Weill Cornell Medicine, New York.
“Current studies suggest a higher prevalence and greater severity of prurigo nodularis among Black patients compared to White patients,” said Dr. Alexis, agreeing with Dr. Kwatra. Referring to evidence that Blacks might mount a greater inflammatory response to prurigo nodularis than Whites, Dr. Alexis called for “a better understanding of the pathomechanisms” of this disease in order “to address unmet needs and reduce disparities for our diverse population of patients who suffer from prurigo nodularis.’
Dr. Kwatra reported financial relationships with AbbVie, Amgen, Arcutis, ASLAN, Cara, Castle Biosciences, Celldex, Galderma, Incyte, Johnson & Johnson, LEO pharma, Novartis, Pfizer, Regeneron, and Sanofi.
NEW YORK – according to an expert evaluating current approaches at the Skin of Color Update 2023.
“As dermatologists, prurigo nodularis is one of the most severe diseases we treat, said Shawn G. Kwatra, MD, director of the Johns Hopkins Itch Center, Baltimore. Now with one approved therapy and more coming, “it offers one of the most important opportunities we have to dramatically improve someone’s entire life.”
Prior to the September 2022 approval of dupilumab for the treatment of prurigo nodularis (the first treatment approved for this indication), Dr. Kwatra said that the limited options for control of pruritus made him anxious. Prurigo nodularis is characterized by highly itchy nodules that can produce symptoms patients describe as unbearable.
Itch typically severe
On a scale for which 10 represents the worst itch imaginable, scores of 8 or greater are not unusual, according to Dr. Kwatra. Nodules on the trunk and the extensor surfaces of the arms and legs are characteristic, but the persistent itch is the immediate target of treatment once the diagnosis is made. For that reason, he urged clinicians to be familiar with the presentation in patients with darker skin types to reduce time to treatment.
In addition to the difficulty of seeing the characteristic red that is typical of erythema in lighter skin, patients with darker skin types tend to have larger nodules that might vary in shape relative to lighter skin types, Dr. Kwatra said. Given that the presentation of prurigo nodularis is highly heterogeneous even among the same skin types, the nuances in patients with darker skin can be that much more confusing for those without prior experience.
Among Blacks in particular, the nodules in some cases “can be huge,” he added. “They can almost look like keloids due to their thickened and fibrotic appearance.”
Phenotypes appear to be racially linked
In Black patients, the appearance can vary enough relative to lighter skin individuals, that “there seems to be something a little bit different going on,” he said, and this is, in fact, supported by a cluster analysis of circulating biomarkers reported by Dr. Kwatra and colleagues in 2022, in the Journal of Investigative Dermatology.
In that study, the biomarker profile distinguished two distinct groups. Whites were more common in a cluster with relatively low expression of inflammatory markers (cluster 1), while Blacks were more common in a cluster with an inflammatory plasma profile (cluster 2), with higher relative expression of multiple cytokines, C-reactive protein, eosinophils, and other markers of up-regulated inflammation.
In addition to a lower rate of myelopathy in cluster 2 than cluster 1 (18% vs. 67%; P = .028), patients in cluster 2 had a significantly worse itch than those in cluster 1 on the Numeric Rating Scale for itch and a significantly lower quality of life based on the Dermatology Life Quality Index score.
Other work at Dr. Kwatra’s center that is based on genetic sequencing has provided evidence that Blacks – and Asians to a lesser extent – are predisposed genetically to develop nodules, perhaps explaining why the nodules tend to be larger than those seen in Whites.
The significance of the evidence that prurigo nodularis is associated with a more up-regulated inflammatory profile in Blacks than in Whites is that they might be particularly likely to respond to dupilumab or other targeted immunomodulating therapies that are in development, according to Dr. Kwatra. Although he did not provide data on response by race, he did provide several case examples of complete itch control following dupilumab therapy in Black patients.
In his experience, high levels of blood eosinophils and other inflammatory markers are predictors of response to dupilumab regardless of skin type, but he expressed concern that time to diagnosis is sometimes longer in Black patients if the nuances of disease expression are not appreciated.
For treating prurigo nodularis in Blacks as well as Whites, Dr. Kwatra suggested that clinicians stay current with what he predicted will be a growing array of treatment options. He did not discuss nemolizumab, an interleukin-31 receptor alpha antagonist. Soon after the meeting, results of a phase 3 trial of nemolizumab in patients with moderate to severe prurigo nodularis were published in the New England Journal of Medicine. (Dr. Kwatra is the lead author of the study but did not specifically discuss this treatment at the meeting.)
In the international placebo-controlled trial, called OLYMPIA 2, treatment was associated with a significant reduction in the signs and symptoms of prurigo nodularis, including reductions in itch, at 16 weeks, although only 4% of patients in the study were Black.
Given the expanding array of therapies, the message of considering prurigo nodularis in Black patients in order to accelerate the time to diagnosis is timely, Andrew F. Alexis, MD, MPH, professor of clinical dermatology and vice-chair for diversity and inclusion for the department of dermatology, Weill Cornell Medicine, New York.
“Current studies suggest a higher prevalence and greater severity of prurigo nodularis among Black patients compared to White patients,” said Dr. Alexis, agreeing with Dr. Kwatra. Referring to evidence that Blacks might mount a greater inflammatory response to prurigo nodularis than Whites, Dr. Alexis called for “a better understanding of the pathomechanisms” of this disease in order “to address unmet needs and reduce disparities for our diverse population of patients who suffer from prurigo nodularis.’
Dr. Kwatra reported financial relationships with AbbVie, Amgen, Arcutis, ASLAN, Cara, Castle Biosciences, Celldex, Galderma, Incyte, Johnson & Johnson, LEO pharma, Novartis, Pfizer, Regeneron, and Sanofi.
AT SOC 2023
The challenges of palmoplantar pustulosis and other acral psoriatic disease
WASHINGTON – The approval last year of the interleukin (IL)-36 receptor antagonist spesolimab for treating generalized pustular psoriasis flares brightened the treatment landscape for this rare condition, and a recently published phase 2 study suggests a potential role of spesolimab for flare prevention. , according to speakers at the annual research symposium of the National Psoriasis Foundation.
“The IL-36 receptor antagonists don’t seem to be quite the answer for [palmoplantar pustulosis] that they are for generalized pustular psoriasis [GPP],” Megan H. Noe, MD, MPH, assistant professor of dermatology at Harvard Medical School and a dermatologist at Brigham and Women’s Hospital, Boston, said at the meeting.

Psoriasis affecting the hands and feet – both pustular and nonpustular – has a higher impact on quality of life and higher functional disability than does non-acral psoriasis, is less responsive to treatment, and has a “very confusing nomenclature” that complicates research and thus management, said Jason Ezra Hawkes, MD, a dermatologist in Rocklin, Calif., and former faculty member of several departments of dermatology. Both he and Dr. Noe spoke during a tough-to-treat session at the NPF meeting.
IL-17 and IL-23 blockade, as well as tumor necrosis factor (TNF) inhibition, are effective overall for palmoplantar psoriasis (nonpustular), but in general, responses are lower than for plaque psoriasis. Apremilast (Otezla), a phosphodiesterase-4 inhibitor, has some efficacy for pustular variants, but for hyperkeratotic variants it “does not perform as well as more selective inhibition of IL-17 and IL-23 blockade,” he said.

In general, ”what’s happening in the acral sites is different from an immune perspective than what’s happening in the non-acral sites,” and more research utilizing a clearer, descriptive nomenclature is needed to tease out differing immunophenotypes, explained Dr. Hawkes, who has led multiple clinical trials of treatments for psoriasis and other inflammatory skin conditions.
Palmoplantar pustulosis, and a word on generalized disease
Dermatologists are using a variety of treatments for palmoplantar pustulosis, with no clear first-line choices, Dr. Noe said. In a case series of almost 200 patients with palmoplantar pustulosis across 20 dermatology practices, published in JAMA Dermatology, 35% of patients received a systemic therapy prescription at their initial encounter – most commonly acitretin, followed by methotrexate and phototherapy. “Biologics were used, but use was varied and not as often as with oral agents,” said Dr. Noe, a coauthor of the study.
TNF blockers led to improvements ranging from 57% to 84%, depending on the agent, in a 2020 retrospective study of patients with palmoplantar pustulosis or acrodermatitis continua of Hallopeau, Dr. Noe noted. However, rates of complete clearance were only 20%-29%.
Apremilast showed modest efficacy after 5 months of treatment, with 62% of patients achieving at least a 50% improvement in the Palmoplantar Pustulosis Psoriasis Area and Severity Index (PPPASI) in a 2021 open-label, phase 2 study involving 21 patients. “This may represent a potential treatment option,” Dr. Noe said. “It’s something, but not what we’re used to seeing in our plaque psoriasis patients.”
A 2021 phase 2a, double-blind, randomized, placebo-controlled study of spesolimab in patients with palmoplantar pustulosis, meanwhile, failed to meet its primary endpoint, with only 32% of patients achieving a 50% improvement at 16 weeks, compared with 24% of patients in the placebo arm. And a recently published network meta-analysis found that none of the five drugs studied in seven randomized controlled trials – biologic or oral – was more effective than placebo for clearance or improvement of palmoplantar pustulosis.
The spesolimab (Spevigo) results have been disappointing considering the biologic’s newfound efficacy and role as the first Food and Drug Administration–approved therapy for generalized pustular disease, according to Dr. Noe. The ability of a single 900-mg intravenous dose of the IL-36 receptor antagonist to completely clear pustules at 1 week in 54% of patients with generalized disease, compared with 6% of the placebo group, was “groundbreaking,” she said, referring to results of the pivotal trial published in the New England Journal of Medicine.
And given that “preventing GPP flares is ultimately what we want,” she said, more good news was reported this year in The Lancet: The finding from an international, randomized, placebo-controlled study that high-dose subcutaneous spesolimab significantly reduced the risk of a flare over 48 weeks. “There are lots of ongoing studies right now to understand the best way to dose spesolimab,” she said.
Moreover, another IL-36 receptor antagonist, imsidolimab, is being investigated in a phase 3 trial for generalized pustular disease, she noted. A phase 2, open-label study of patients with GPP found that “more than half of patients were very much improved at 4 weeks, and some patients started showing improvement at day 3,” Dr. Noe said.
An area of research she is interested in is the potential for Janus kinase (JAK) inhibitors as a treatment for palmoplantar pustulosis. For pustulosis on the hands and feet, recent case reports describing the efficacy of JAK inhibitors have caught her eye. “Right now, all we have is this case report data, mostly with tofacitinib, but I think it’s exciting,” she said, noting a recently published report in the British Journal of Dermatology.
Palmoplantar psoriasis
Pustular psoriatic disease can be localized to the hand and/or feet only, or can co-occur with generalized pustular disease, just as palmoplantar psoriasis can be localized to the hands and/or feet or, more commonly, can co-occur with widespread plaque psoriasis. Research has shown, Dr. Hawkes said, that with both types of acral disease, many patients have or have had plaque psoriasis outside of acral sites.
The nomenclature and acronyms for palmoplantar psoriatic disease have complicated patient education, communication, and research, Dr. Hawkes said. Does PPP refer to palmoplantar psoriasis, or palmoplantar pustulosis, for instance? What is the difference between palmoplantar pustulosis (coined PPP) and palmoplantar pustular psoriasis (referred to as PPPP)?
What if disease is only on the hands, only on the feet, or only on the backs of the hands? And at what point is disease not classified as palmoplantar psoriasis, but plaque psoriasis with involvement of the hands and feet? Inconsistencies and lack of clarification lead to “confusing” literature, he said.
Heterogeneity in populations across trials resulting from “inconsistent categorization and phenotype inclusion” may partly account for the recalcitrance to treatment reported in the literature, he said. Misdiagnosis as psoriasis in cases of localized disease (confusion with eczema, for instance), and the fact that hands and feet are subject to increased trauma and injury, compared with non-acral sites, are also at play.
Trials may also allow insufficient time for improvement, compared with non-acral sites. “What we’ve learned about the hands and feet is that it takes a much longer time for disease to improve,” Dr. Hawkes said, so primary endpoints must take this into account.
There is unique immunologic signaling in palmoplantar disease that differs from the predominant signaling in traditional plaque psoriasis, he emphasized, and “mixed immunophenotypes” that need to be unraveled.
Dr. Hawkes disclosed ties with AbbVie, Arcutis, Bristol-Myers Squibb, Boehringer Ingelheim, Janssen, LEO, Lilly, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma, and UCB. Dr. Noe disclosed ties to Bristol-Myers Squibb and Boehringer Ingelheim.
WASHINGTON – The approval last year of the interleukin (IL)-36 receptor antagonist spesolimab for treating generalized pustular psoriasis flares brightened the treatment landscape for this rare condition, and a recently published phase 2 study suggests a potential role of spesolimab for flare prevention. , according to speakers at the annual research symposium of the National Psoriasis Foundation.
“The IL-36 receptor antagonists don’t seem to be quite the answer for [palmoplantar pustulosis] that they are for generalized pustular psoriasis [GPP],” Megan H. Noe, MD, MPH, assistant professor of dermatology at Harvard Medical School and a dermatologist at Brigham and Women’s Hospital, Boston, said at the meeting.
Psoriasis affecting the hands and feet – both pustular and nonpustular – has a higher impact on quality of life and higher functional disability than does non-acral psoriasis, is less responsive to treatment, and has a “very confusing nomenclature” that complicates research and thus management, said Jason Ezra Hawkes, MD, a dermatologist in Rocklin, Calif., and former faculty member of several departments of dermatology. Both he and Dr. Noe spoke during a tough-to-treat session at the NPF meeting.
IL-17 and IL-23 blockade, as well as tumor necrosis factor (TNF) inhibition, are effective overall for palmoplantar psoriasis (nonpustular), but in general, responses are lower than for plaque psoriasis. Apremilast (Otezla), a phosphodiesterase-4 inhibitor, has some efficacy for pustular variants, but for hyperkeratotic variants it “does not perform as well as more selective inhibition of IL-17 and IL-23 blockade,” he said.
In general, ”what’s happening in the acral sites is different from an immune perspective than what’s happening in the non-acral sites,” and more research utilizing a clearer, descriptive nomenclature is needed to tease out differing immunophenotypes, explained Dr. Hawkes, who has led multiple clinical trials of treatments for psoriasis and other inflammatory skin conditions.
Palmoplantar pustulosis, and a word on generalized disease
Dermatologists are using a variety of treatments for palmoplantar pustulosis, with no clear first-line choices, Dr. Noe said. In a case series of almost 200 patients with palmoplantar pustulosis across 20 dermatology practices, published in JAMA Dermatology, 35% of patients received a systemic therapy prescription at their initial encounter – most commonly acitretin, followed by methotrexate and phototherapy. “Biologics were used, but use was varied and not as often as with oral agents,” said Dr. Noe, a coauthor of the study.
TNF blockers led to improvements ranging from 57% to 84%, depending on the agent, in a 2020 retrospective study of patients with palmoplantar pustulosis or acrodermatitis continua of Hallopeau, Dr. Noe noted. However, rates of complete clearance were only 20%-29%.
Apremilast showed modest efficacy after 5 months of treatment, with 62% of patients achieving at least a 50% improvement in the Palmoplantar Pustulosis Psoriasis Area and Severity Index (PPPASI) in a 2021 open-label, phase 2 study involving 21 patients. “This may represent a potential treatment option,” Dr. Noe said. “It’s something, but not what we’re used to seeing in our plaque psoriasis patients.”
A 2021 phase 2a, double-blind, randomized, placebo-controlled study of spesolimab in patients with palmoplantar pustulosis, meanwhile, failed to meet its primary endpoint, with only 32% of patients achieving a 50% improvement at 16 weeks, compared with 24% of patients in the placebo arm. And a recently published network meta-analysis found that none of the five drugs studied in seven randomized controlled trials – biologic or oral – was more effective than placebo for clearance or improvement of palmoplantar pustulosis.
The spesolimab (Spevigo) results have been disappointing considering the biologic’s newfound efficacy and role as the first Food and Drug Administration–approved therapy for generalized pustular disease, according to Dr. Noe. The ability of a single 900-mg intravenous dose of the IL-36 receptor antagonist to completely clear pustules at 1 week in 54% of patients with generalized disease, compared with 6% of the placebo group, was “groundbreaking,” she said, referring to results of the pivotal trial published in the New England Journal of Medicine.
And given that “preventing GPP flares is ultimately what we want,” she said, more good news was reported this year in The Lancet: The finding from an international, randomized, placebo-controlled study that high-dose subcutaneous spesolimab significantly reduced the risk of a flare over 48 weeks. “There are lots of ongoing studies right now to understand the best way to dose spesolimab,” she said.
Moreover, another IL-36 receptor antagonist, imsidolimab, is being investigated in a phase 3 trial for generalized pustular disease, she noted. A phase 2, open-label study of patients with GPP found that “more than half of patients were very much improved at 4 weeks, and some patients started showing improvement at day 3,” Dr. Noe said.
An area of research she is interested in is the potential for Janus kinase (JAK) inhibitors as a treatment for palmoplantar pustulosis. For pustulosis on the hands and feet, recent case reports describing the efficacy of JAK inhibitors have caught her eye. “Right now, all we have is this case report data, mostly with tofacitinib, but I think it’s exciting,” she said, noting a recently published report in the British Journal of Dermatology.
Palmoplantar psoriasis
Pustular psoriatic disease can be localized to the hand and/or feet only, or can co-occur with generalized pustular disease, just as palmoplantar psoriasis can be localized to the hands and/or feet or, more commonly, can co-occur with widespread plaque psoriasis. Research has shown, Dr. Hawkes said, that with both types of acral disease, many patients have or have had plaque psoriasis outside of acral sites.
The nomenclature and acronyms for palmoplantar psoriatic disease have complicated patient education, communication, and research, Dr. Hawkes said. Does PPP refer to palmoplantar psoriasis, or palmoplantar pustulosis, for instance? What is the difference between palmoplantar pustulosis (coined PPP) and palmoplantar pustular psoriasis (referred to as PPPP)?
What if disease is only on the hands, only on the feet, or only on the backs of the hands? And at what point is disease not classified as palmoplantar psoriasis, but plaque psoriasis with involvement of the hands and feet? Inconsistencies and lack of clarification lead to “confusing” literature, he said.
Heterogeneity in populations across trials resulting from “inconsistent categorization and phenotype inclusion” may partly account for the recalcitrance to treatment reported in the literature, he said. Misdiagnosis as psoriasis in cases of localized disease (confusion with eczema, for instance), and the fact that hands and feet are subject to increased trauma and injury, compared with non-acral sites, are also at play.
Trials may also allow insufficient time for improvement, compared with non-acral sites. “What we’ve learned about the hands and feet is that it takes a much longer time for disease to improve,” Dr. Hawkes said, so primary endpoints must take this into account.
There is unique immunologic signaling in palmoplantar disease that differs from the predominant signaling in traditional plaque psoriasis, he emphasized, and “mixed immunophenotypes” that need to be unraveled.
Dr. Hawkes disclosed ties with AbbVie, Arcutis, Bristol-Myers Squibb, Boehringer Ingelheim, Janssen, LEO, Lilly, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma, and UCB. Dr. Noe disclosed ties to Bristol-Myers Squibb and Boehringer Ingelheim.
WASHINGTON – The approval last year of the interleukin (IL)-36 receptor antagonist spesolimab for treating generalized pustular psoriasis flares brightened the treatment landscape for this rare condition, and a recently published phase 2 study suggests a potential role of spesolimab for flare prevention. , according to speakers at the annual research symposium of the National Psoriasis Foundation.
“The IL-36 receptor antagonists don’t seem to be quite the answer for [palmoplantar pustulosis] that they are for generalized pustular psoriasis [GPP],” Megan H. Noe, MD, MPH, assistant professor of dermatology at Harvard Medical School and a dermatologist at Brigham and Women’s Hospital, Boston, said at the meeting.
Psoriasis affecting the hands and feet – both pustular and nonpustular – has a higher impact on quality of life and higher functional disability than does non-acral psoriasis, is less responsive to treatment, and has a “very confusing nomenclature” that complicates research and thus management, said Jason Ezra Hawkes, MD, a dermatologist in Rocklin, Calif., and former faculty member of several departments of dermatology. Both he and Dr. Noe spoke during a tough-to-treat session at the NPF meeting.
IL-17 and IL-23 blockade, as well as tumor necrosis factor (TNF) inhibition, are effective overall for palmoplantar psoriasis (nonpustular), but in general, responses are lower than for plaque psoriasis. Apremilast (Otezla), a phosphodiesterase-4 inhibitor, has some efficacy for pustular variants, but for hyperkeratotic variants it “does not perform as well as more selective inhibition of IL-17 and IL-23 blockade,” he said.
In general, ”what’s happening in the acral sites is different from an immune perspective than what’s happening in the non-acral sites,” and more research utilizing a clearer, descriptive nomenclature is needed to tease out differing immunophenotypes, explained Dr. Hawkes, who has led multiple clinical trials of treatments for psoriasis and other inflammatory skin conditions.
Palmoplantar pustulosis, and a word on generalized disease
Dermatologists are using a variety of treatments for palmoplantar pustulosis, with no clear first-line choices, Dr. Noe said. In a case series of almost 200 patients with palmoplantar pustulosis across 20 dermatology practices, published in JAMA Dermatology, 35% of patients received a systemic therapy prescription at their initial encounter – most commonly acitretin, followed by methotrexate and phototherapy. “Biologics were used, but use was varied and not as often as with oral agents,” said Dr. Noe, a coauthor of the study.
TNF blockers led to improvements ranging from 57% to 84%, depending on the agent, in a 2020 retrospective study of patients with palmoplantar pustulosis or acrodermatitis continua of Hallopeau, Dr. Noe noted. However, rates of complete clearance were only 20%-29%.
Apremilast showed modest efficacy after 5 months of treatment, with 62% of patients achieving at least a 50% improvement in the Palmoplantar Pustulosis Psoriasis Area and Severity Index (PPPASI) in a 2021 open-label, phase 2 study involving 21 patients. “This may represent a potential treatment option,” Dr. Noe said. “It’s something, but not what we’re used to seeing in our plaque psoriasis patients.”
A 2021 phase 2a, double-blind, randomized, placebo-controlled study of spesolimab in patients with palmoplantar pustulosis, meanwhile, failed to meet its primary endpoint, with only 32% of patients achieving a 50% improvement at 16 weeks, compared with 24% of patients in the placebo arm. And a recently published network meta-analysis found that none of the five drugs studied in seven randomized controlled trials – biologic or oral – was more effective than placebo for clearance or improvement of palmoplantar pustulosis.
The spesolimab (Spevigo) results have been disappointing considering the biologic’s newfound efficacy and role as the first Food and Drug Administration–approved therapy for generalized pustular disease, according to Dr. Noe. The ability of a single 900-mg intravenous dose of the IL-36 receptor antagonist to completely clear pustules at 1 week in 54% of patients with generalized disease, compared with 6% of the placebo group, was “groundbreaking,” she said, referring to results of the pivotal trial published in the New England Journal of Medicine.
And given that “preventing GPP flares is ultimately what we want,” she said, more good news was reported this year in The Lancet: The finding from an international, randomized, placebo-controlled study that high-dose subcutaneous spesolimab significantly reduced the risk of a flare over 48 weeks. “There are lots of ongoing studies right now to understand the best way to dose spesolimab,” she said.
Moreover, another IL-36 receptor antagonist, imsidolimab, is being investigated in a phase 3 trial for generalized pustular disease, she noted. A phase 2, open-label study of patients with GPP found that “more than half of patients were very much improved at 4 weeks, and some patients started showing improvement at day 3,” Dr. Noe said.
An area of research she is interested in is the potential for Janus kinase (JAK) inhibitors as a treatment for palmoplantar pustulosis. For pustulosis on the hands and feet, recent case reports describing the efficacy of JAK inhibitors have caught her eye. “Right now, all we have is this case report data, mostly with tofacitinib, but I think it’s exciting,” she said, noting a recently published report in the British Journal of Dermatology.
Palmoplantar psoriasis
Pustular psoriatic disease can be localized to the hand and/or feet only, or can co-occur with generalized pustular disease, just as palmoplantar psoriasis can be localized to the hands and/or feet or, more commonly, can co-occur with widespread plaque psoriasis. Research has shown, Dr. Hawkes said, that with both types of acral disease, many patients have or have had plaque psoriasis outside of acral sites.
The nomenclature and acronyms for palmoplantar psoriatic disease have complicated patient education, communication, and research, Dr. Hawkes said. Does PPP refer to palmoplantar psoriasis, or palmoplantar pustulosis, for instance? What is the difference between palmoplantar pustulosis (coined PPP) and palmoplantar pustular psoriasis (referred to as PPPP)?
What if disease is only on the hands, only on the feet, or only on the backs of the hands? And at what point is disease not classified as palmoplantar psoriasis, but plaque psoriasis with involvement of the hands and feet? Inconsistencies and lack of clarification lead to “confusing” literature, he said.
Heterogeneity in populations across trials resulting from “inconsistent categorization and phenotype inclusion” may partly account for the recalcitrance to treatment reported in the literature, he said. Misdiagnosis as psoriasis in cases of localized disease (confusion with eczema, for instance), and the fact that hands and feet are subject to increased trauma and injury, compared with non-acral sites, are also at play.
Trials may also allow insufficient time for improvement, compared with non-acral sites. “What we’ve learned about the hands and feet is that it takes a much longer time for disease to improve,” Dr. Hawkes said, so primary endpoints must take this into account.
There is unique immunologic signaling in palmoplantar disease that differs from the predominant signaling in traditional plaque psoriasis, he emphasized, and “mixed immunophenotypes” that need to be unraveled.
Dr. Hawkes disclosed ties with AbbVie, Arcutis, Bristol-Myers Squibb, Boehringer Ingelheim, Janssen, LEO, Lilly, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma, and UCB. Dr. Noe disclosed ties to Bristol-Myers Squibb and Boehringer Ingelheim.
AT THE NPF RESEARCH SYMPOSIUM 2023
An 88-year-old Black woman presented with 3 months duration of asymptomatic, violaceous patches on the left breast
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.

Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.
Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
Angiosarcomas are uncommon, high-grade malignant tumors of endothelial cell origin that can arise via the lymphatics or vasculature. They typically occur spontaneously; however, there have been cases reported of benign vascular transformation. These tumors are more commonly found in elderly men on the head and neck in sun-damaged skin. . This is a late complication, typically occurring about 5-10 years after radiation. Stewart-Treves syndrome, chronic lymphedema occurring after breast cancer treatment with axillary node dissection, increases the risk of angiosarcoma. As a vascular tumor, angiosarcoma spreads hematogenously and carries a poor prognosis if not caught early. Differential diagnoses include other vascular tumors such as retiform hemangioendothelioma. In this specific patient, the differential diagnosis includes Paget’s disease, chronic radiation skin changes, and eczema.
Histopathologically, angiosarcomas exhibit abnormal, pleomorphic, malignant endothelial cells. As the tumor progresses, the cell architecture becomes more distorted and cells form layers with papillary projections into the vascular lumen. Malignant cells may stain positive for CD31, CD34, the oncogene ERG and the proto-oncogene FLI-1. Histology in this patient revealed radiation changes in the dermis, as well as few vascular channels lined by large endothelial cells with marked nuclear atypia, in the form of large nucleoli and variably coarse chromatin. The cells were positive for MYC.
Treatment of angiosarcoma involves a multidisciplinary approach. Resection with wide margins is generally the treatment of choice. However, recurrence is relatively common, which may be a result of microsatellite deposits of the tumor. Perioperative radiation is recommended, and adjuvant chemotherapy often is recommended for metastatic disease. Specifically, paclitaxel has been found to promote survival in some cases of cutaneous angiosarcoma. Metastatic disease may be treated with cytotoxic drugs such as anthracyclines and taxanes. Additionally, targeted therapy including anti-VEGF drugs and tyrosine kinase inhibitors have been tested.
The case and photo were submitted by Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Dr. Bilu Martin. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cohen-Hallaleh RB et al. Clin Sarcoma Res. 2017 Aug 7:7:15.
Cozzi S et al. Rep Pract Oncol Radiother. 2021 Sep 30;26(5):827-32.
Spiker AM, Mangla A, Ramsey ML. Angiosarcoma. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing; 2023 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK441983/
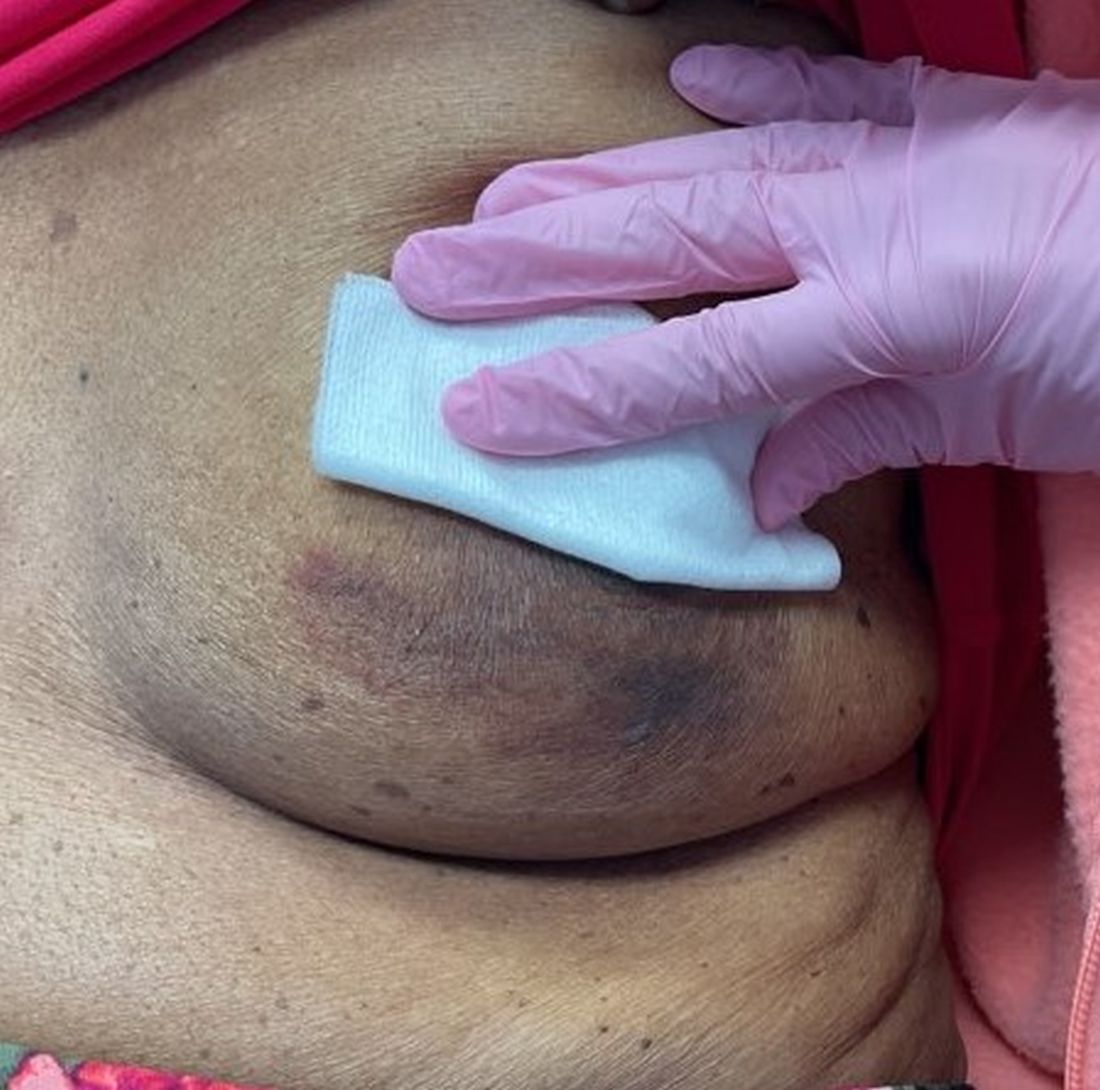
Review estimates acne risk with JAK inhibitor therapy
TOPLINE:
, according to an analysis of 25 JAK inhibitor studies.
METHODOLOGY:
- Acne has been reported to be an adverse effect of JAK inhibitors, but not much is known about how common acne is overall and how incidence differs between different JAK inhibitors and the disease being treated.
- For the systematic review and meta-analysis, researchers identified 25 phase 2 or 3 randomized, controlled trials that reported acne as an adverse event associated with the use of JAK inhibitors.
- The study population included 10,839 participants (54% male, 46% female).
- The primary outcome was the incidence of acne following a period of JAK inhibitor use.
TAKEAWAY:
- Overall, the risk of acne was significantly higher among those treated with JAK inhibitors in comparison with patients given placebo in a pooled analysis (odds ratio [OR], 3.83).
- The risk of acne was highest with abrocitinib (OR, 13.47), followed by baricitinib (OR, 4.96), upadacitinib (OR, 4.79), deuruxolitinib (OR, 3.30), and deucravacitinib (OR, 2.64). By JAK inhibitor class, results were as follows: JAK1-specific inhibitors (OR, 4.69), combined JAK1 and JAK2 inhibitors (OR, 3.43), and tyrosine kinase 2 inhibitors (OR, 2.64).
- In a subgroup analysis, risk of acne was higher among patients using JAK inhibitors for dermatologic conditions in comparison with those using JAK inhibitors for nondermatologic conditions (OR, 4.67 vs 1.18).
- Age and gender had no apparent impact on the effect of JAK inhibitor use on acne risk.
IN PRACTICE:
“The occurrence of acne following treatment with certain classes of JAK inhibitors is of potential concern, as this adverse effect may jeopardize treatment adherence among some patients,” the researchers wrote. More studies are needed “to characterize the underlying mechanism of acne with JAK inhibitor use and to identify best practices for treatment,” they added.
SOURCE:
The lead author was Jeremy Martinez, MPH, of Harvard Medical School, Boston. The study was published online in JAMA Dermatology.
LIMITATIONS:
The review was limited by the variable classification and reporting of acne across studies, the potential exclusion of relevant studies, and the small number of studies for certain drugs.
DISCLOSURES:
The studies were mainly funded by the pharmaceutical industry. Mr. Martinez disclosed no relevant financial relationships. Several coauthors have ties with Dexcel Pharma Technologies, AbbVie, Concert, Pfizer, 3Derm Systems, Incyte, Aclaris, Eli Lilly, Concert, Equillium, ASLAN, ACOM, and Boehringer Ingelheim.
A version of this article appeared on Medscape.com.
TOPLINE:
, according to an analysis of 25 JAK inhibitor studies.
METHODOLOGY:
- Acne has been reported to be an adverse effect of JAK inhibitors, but not much is known about how common acne is overall and how incidence differs between different JAK inhibitors and the disease being treated.
- For the systematic review and meta-analysis, researchers identified 25 phase 2 or 3 randomized, controlled trials that reported acne as an adverse event associated with the use of JAK inhibitors.
- The study population included 10,839 participants (54% male, 46% female).
- The primary outcome was the incidence of acne following a period of JAK inhibitor use.
TAKEAWAY:
- Overall, the risk of acne was significantly higher among those treated with JAK inhibitors in comparison with patients given placebo in a pooled analysis (odds ratio [OR], 3.83).
- The risk of acne was highest with abrocitinib (OR, 13.47), followed by baricitinib (OR, 4.96), upadacitinib (OR, 4.79), deuruxolitinib (OR, 3.30), and deucravacitinib (OR, 2.64). By JAK inhibitor class, results were as follows: JAK1-specific inhibitors (OR, 4.69), combined JAK1 and JAK2 inhibitors (OR, 3.43), and tyrosine kinase 2 inhibitors (OR, 2.64).
- In a subgroup analysis, risk of acne was higher among patients using JAK inhibitors for dermatologic conditions in comparison with those using JAK inhibitors for nondermatologic conditions (OR, 4.67 vs 1.18).
- Age and gender had no apparent impact on the effect of JAK inhibitor use on acne risk.
IN PRACTICE:
“The occurrence of acne following treatment with certain classes of JAK inhibitors is of potential concern, as this adverse effect may jeopardize treatment adherence among some patients,” the researchers wrote. More studies are needed “to characterize the underlying mechanism of acne with JAK inhibitor use and to identify best practices for treatment,” they added.
SOURCE:
The lead author was Jeremy Martinez, MPH, of Harvard Medical School, Boston. The study was published online in JAMA Dermatology.
LIMITATIONS:
The review was limited by the variable classification and reporting of acne across studies, the potential exclusion of relevant studies, and the small number of studies for certain drugs.
DISCLOSURES:
The studies were mainly funded by the pharmaceutical industry. Mr. Martinez disclosed no relevant financial relationships. Several coauthors have ties with Dexcel Pharma Technologies, AbbVie, Concert, Pfizer, 3Derm Systems, Incyte, Aclaris, Eli Lilly, Concert, Equillium, ASLAN, ACOM, and Boehringer Ingelheim.
A version of this article appeared on Medscape.com.
TOPLINE:
, according to an analysis of 25 JAK inhibitor studies.
METHODOLOGY:
- Acne has been reported to be an adverse effect of JAK inhibitors, but not much is known about how common acne is overall and how incidence differs between different JAK inhibitors and the disease being treated.
- For the systematic review and meta-analysis, researchers identified 25 phase 2 or 3 randomized, controlled trials that reported acne as an adverse event associated with the use of JAK inhibitors.
- The study population included 10,839 participants (54% male, 46% female).
- The primary outcome was the incidence of acne following a period of JAK inhibitor use.
TAKEAWAY:
- Overall, the risk of acne was significantly higher among those treated with JAK inhibitors in comparison with patients given placebo in a pooled analysis (odds ratio [OR], 3.83).
- The risk of acne was highest with abrocitinib (OR, 13.47), followed by baricitinib (OR, 4.96), upadacitinib (OR, 4.79), deuruxolitinib (OR, 3.30), and deucravacitinib (OR, 2.64). By JAK inhibitor class, results were as follows: JAK1-specific inhibitors (OR, 4.69), combined JAK1 and JAK2 inhibitors (OR, 3.43), and tyrosine kinase 2 inhibitors (OR, 2.64).
- In a subgroup analysis, risk of acne was higher among patients using JAK inhibitors for dermatologic conditions in comparison with those using JAK inhibitors for nondermatologic conditions (OR, 4.67 vs 1.18).
- Age and gender had no apparent impact on the effect of JAK inhibitor use on acne risk.
IN PRACTICE:
“The occurrence of acne following treatment with certain classes of JAK inhibitors is of potential concern, as this adverse effect may jeopardize treatment adherence among some patients,” the researchers wrote. More studies are needed “to characterize the underlying mechanism of acne with JAK inhibitor use and to identify best practices for treatment,” they added.
SOURCE:
The lead author was Jeremy Martinez, MPH, of Harvard Medical School, Boston. The study was published online in JAMA Dermatology.
LIMITATIONS:
The review was limited by the variable classification and reporting of acne across studies, the potential exclusion of relevant studies, and the small number of studies for certain drugs.
DISCLOSURES:
The studies were mainly funded by the pharmaceutical industry. Mr. Martinez disclosed no relevant financial relationships. Several coauthors have ties with Dexcel Pharma Technologies, AbbVie, Concert, Pfizer, 3Derm Systems, Incyte, Aclaris, Eli Lilly, Concert, Equillium, ASLAN, ACOM, and Boehringer Ingelheim.
A version of this article appeared on Medscape.com.
Topical ivermectin study sheds light on dysbiosis in rosacea
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
FROM EADV 2023
Papules on lip

Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120
Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120

Researchers tease apart multiple biologic failure in psoriasis, PsA
WASHINGTON – Multiple biologic failure in a minority of patients with psoriasis may have several causes, from genetic endotypes and immunologic factors to lower serum drug levels, the presence of anti-drug antibody levels, female sex, and certain comorbidities, Wilson Liao, MD, said at the annual research symposium of the National Psoriasis Foundation.
“Tough-to-treat psoriasis remains a challenge despite newer therapies ... Why do we still have this sub-population of patients who seem to be refractory?” said Dr. Liao, professor and associate vice chair of research in the department of dermatology at the University of California, San Francisco, who coauthored a 2015-2022 prospective cohort analysis that documented about 6% of patients failing two or more biologic agents of different mechanistic classes.
“These patients are really suffering,” he said. “We need to have better guidelines and treatment algorithms for these patients.”
A significant number of patients with psoriatic arthritis (PsA), meanwhile, are inadequate responders to tumor necrosis factor (TNF) inhibition, Christopher T. Ritchlin, MD, PhD, professor of medicine in the division of allergy/immunology and rheumatology and the Center of Musculoskeletal Research at the University of Rochester (N.Y.), said during another session at the meeting.
The long-term “persistence,” or usage, of first-line biologics in patients with PsA – and of second-line biologics in patients who failed one TNF-inhibitor – is low, but the literature offers little information on the reasons for TNF-inhibitor discontinuation, said Dr. Ritchlin, who coauthored a perspective piece in Arthritis & Rheumatology on managing the patient with PsA who fails one TNF inhibitor.
Dr. Ritchlin and his coauthors were asked to provide evidence-informed advice and algorithms, but the task was difficult. “It’s hard to know what to recommend for the next step if we don’t know why patients failed the first,” he said. “The point is, we need more data. [Clinical trials] are not recording the kind of information we need.”
Anti-drug antibodies, genetics, other factors in psoriasis
Research shows that in large cohorts, “all the biologics do seem to lose efficacy over time,” said Dr. Liao, who directs the UCSF Psoriasis and Skin Treatment Center. “Some are better than others, but we do see a loss of effectiveness over time.”
A cohort study published in 2022 in JAMA Dermatology, for instance, documented declining “drug survival” associated with ineffectiveness during 2 years of treatment for each of five biologics studied (adalimumab [Humira], ustekinumab [Stelara], secukinumab [Cosentyx], guselkumab [Tremfya], and ixekizumab [Taltz]).
“There have been a number of theories put forward” as to why that’s the case, including lower serum drug levels, “which of course can be related to anti-drug antibody production,” he said.
He pointed to two studies of ustekinumab: One prospective observational cohort study that reported an association of lower early drug levels of the IL-12/23 receptor antagonist with lower Psoriasis Area and Severity Index (PASI) scores, and another observational study that documented an association between anti-drug antibody positivity with lower ustekinumab levels and impaired clinical response.
“We also now know ... that there are genetic endotypes in psoriasis, and that patients who are [HLA-C*06:02]-positive tend to respond a little better to drugs like ustekinumab, and those who are [HLA-C*06:02]-negative tend to do a little better with the TNF inhibitors,” Dr. Liao said. The human leukocyte antigen (HLA) allele HLA-C*06:02 is associated with susceptibility to psoriasis.
In a study using a national psoriasis registry, HLA-C*06:02-negative patients were 3 times more likely to achieve PASI90 status in response to adalimumab, a TNF-alpha inhibitor, than with ustekinumab treatment. And in a meta-analysis covering eight studies with more than 1,000 patients with psoriasis, the median PASI75 response rate after 6 months of ustekinumab therapy was 92% in the HLA-C*06:02-positive group and 67% in HLA-C*06:02-negative patients.
The recently published cohort study showing a 6% rate of multiple biologic failure evaluated patients in the multicenter CorEvitas Psoriasis Registry who initiated their first biologic between 2015 and 2020 and were followed for 2 or more years. Investigators looked for sociodemographic and clinical differences between the patients who continued use of their first biologic for at least 2 years (“good response”), and those who discontinued two or more biologics of different classes, each used for at least 90 days, because of inadequate efficacy.
Of 1,039 evaluated patients, 490 (47.2%) had good clinical response to their first biologic and 65 (6.3%) had multiple biologic failure. All biologic classes were represented among those who failed multiple biologics. The first and second biologic classes used were attempted for a mean duration of 10 months – “an adequate trial” of each, Dr. Liao said.
In multivariable regression analysis, six variables were significantly associated with multiple biologic failure: female sex at birth, shorter disease duration, earlier year of biologic initiation, prior nonbiologic systemic therapy, having Medicaid insurance, and a history of hyperlipidemia. The latter is “interesting because other studies have shown that metabolic syndrome, of which hyperlipidemia is a component, can also relate to poor response to biologics,” Dr. Liao said.
The most common sequences of first-to-second biologics among those with multiple biologic failure were TNF inhibitor to IL-17 inhibitor (30.8%); IL-12/23 inhibitor to IL-17 inhibitor (21.5%); TNF inhibitor to IL-12/23 inhibitor (12.3%); and IL-17 inhibitor to IL-23 inhibitor (10.8%).
The vast majority of patients failed more than two biologics, however, and “more than 20% had five or more biologics tried over a relatively short period,” Dr. Liao said.
Comorbidities and biologic failure in psoriasis, PsA
In practice, it was said during a discussion period, biologic failures in psoriasis can be of two types: a primary inadequate response or initial failure, or a secondary failure with initial improvement followed by declining or no response. “I agree 100% that these probably represent two different endotypes,” Dr. Liao said. “There’s research emerging that psoriasis isn’t necessarily a clean phenotype.”
The option of focusing on comorbidities in the face of biologic failure was another point of discussion. “Maybe the next biologic is not the answer,” a meeting participant said. “Maybe we should focus on metabolic syndrome.”
“I agree,” Dr. Liao said. “In clinic, there are people who may not respond to therapies but have other comorbidities and factors that make it difficult to manage [their psoriasis] ... that may be causative for psoriasis. Maybe if we treat the comorbidities, it will make it easier to treat the psoriasis.”
Addressing comorbidities and “extra-articular traits” such as poorly controlled diabetes, centralized pain, anxiety and depression, and obesity is something Dr. Ritchlin advocates for PsA. “Centralized pain, I believe, is a major driver of nonresponse,” he said at the meeting. “We have to be careful about blaming nonresponse and lack of efficacy of biologics when it could be a wholly different mechanism the biologic won’t treat ... for example, centralized pain.”
As with psoriasis, the emergence of antidrug antibodies may be one reason for the secondary failure of biologic agents for PsA, Dr. Ritchlin and his coauthors wrote in their paper on management of PsA after failure of one TNF inhibitor. Other areas to consider in evaluating failure, they wrote, are compliance and time of dosing, and financial barriers.
Low long-term persistence of second-line biologics for patients with PsA was demonstrated in a national cohort study utilizing the French health insurance database, Dr. Ritchlin noted at the research meeting.
The French study covered almost 3,000 patients who started a second biologic after discontinuing a TNF inhibitor during 2015-2020. Overall, 1-year and 3-year persistence rates were 42% and 17%, respectively.
Dr. Liao disclosed research grant funding from AbbVie, Amgen, Janssen, Leo, Novartis, Pfizer, Regeneron, and Trex Bio. Dr. Ritchlin reported no disclosures.
WASHINGTON – Multiple biologic failure in a minority of patients with psoriasis may have several causes, from genetic endotypes and immunologic factors to lower serum drug levels, the presence of anti-drug antibody levels, female sex, and certain comorbidities, Wilson Liao, MD, said at the annual research symposium of the National Psoriasis Foundation.
“Tough-to-treat psoriasis remains a challenge despite newer therapies ... Why do we still have this sub-population of patients who seem to be refractory?” said Dr. Liao, professor and associate vice chair of research in the department of dermatology at the University of California, San Francisco, who coauthored a 2015-2022 prospective cohort analysis that documented about 6% of patients failing two or more biologic agents of different mechanistic classes.
“These patients are really suffering,” he said. “We need to have better guidelines and treatment algorithms for these patients.”
A significant number of patients with psoriatic arthritis (PsA), meanwhile, are inadequate responders to tumor necrosis factor (TNF) inhibition, Christopher T. Ritchlin, MD, PhD, professor of medicine in the division of allergy/immunology and rheumatology and the Center of Musculoskeletal Research at the University of Rochester (N.Y.), said during another session at the meeting.
The long-term “persistence,” or usage, of first-line biologics in patients with PsA – and of second-line biologics in patients who failed one TNF-inhibitor – is low, but the literature offers little information on the reasons for TNF-inhibitor discontinuation, said Dr. Ritchlin, who coauthored a perspective piece in Arthritis & Rheumatology on managing the patient with PsA who fails one TNF inhibitor.
Dr. Ritchlin and his coauthors were asked to provide evidence-informed advice and algorithms, but the task was difficult. “It’s hard to know what to recommend for the next step if we don’t know why patients failed the first,” he said. “The point is, we need more data. [Clinical trials] are not recording the kind of information we need.”
Anti-drug antibodies, genetics, other factors in psoriasis
Research shows that in large cohorts, “all the biologics do seem to lose efficacy over time,” said Dr. Liao, who directs the UCSF Psoriasis and Skin Treatment Center. “Some are better than others, but we do see a loss of effectiveness over time.”
A cohort study published in 2022 in JAMA Dermatology, for instance, documented declining “drug survival” associated with ineffectiveness during 2 years of treatment for each of five biologics studied (adalimumab [Humira], ustekinumab [Stelara], secukinumab [Cosentyx], guselkumab [Tremfya], and ixekizumab [Taltz]).
“There have been a number of theories put forward” as to why that’s the case, including lower serum drug levels, “which of course can be related to anti-drug antibody production,” he said.
He pointed to two studies of ustekinumab: One prospective observational cohort study that reported an association of lower early drug levels of the IL-12/23 receptor antagonist with lower Psoriasis Area and Severity Index (PASI) scores, and another observational study that documented an association between anti-drug antibody positivity with lower ustekinumab levels and impaired clinical response.
“We also now know ... that there are genetic endotypes in psoriasis, and that patients who are [HLA-C*06:02]-positive tend to respond a little better to drugs like ustekinumab, and those who are [HLA-C*06:02]-negative tend to do a little better with the TNF inhibitors,” Dr. Liao said. The human leukocyte antigen (HLA) allele HLA-C*06:02 is associated with susceptibility to psoriasis.
In a study using a national psoriasis registry, HLA-C*06:02-negative patients were 3 times more likely to achieve PASI90 status in response to adalimumab, a TNF-alpha inhibitor, than with ustekinumab treatment. And in a meta-analysis covering eight studies with more than 1,000 patients with psoriasis, the median PASI75 response rate after 6 months of ustekinumab therapy was 92% in the HLA-C*06:02-positive group and 67% in HLA-C*06:02-negative patients.
The recently published cohort study showing a 6% rate of multiple biologic failure evaluated patients in the multicenter CorEvitas Psoriasis Registry who initiated their first biologic between 2015 and 2020 and were followed for 2 or more years. Investigators looked for sociodemographic and clinical differences between the patients who continued use of their first biologic for at least 2 years (“good response”), and those who discontinued two or more biologics of different classes, each used for at least 90 days, because of inadequate efficacy.
Of 1,039 evaluated patients, 490 (47.2%) had good clinical response to their first biologic and 65 (6.3%) had multiple biologic failure. All biologic classes were represented among those who failed multiple biologics. The first and second biologic classes used were attempted for a mean duration of 10 months – “an adequate trial” of each, Dr. Liao said.
In multivariable regression analysis, six variables were significantly associated with multiple biologic failure: female sex at birth, shorter disease duration, earlier year of biologic initiation, prior nonbiologic systemic therapy, having Medicaid insurance, and a history of hyperlipidemia. The latter is “interesting because other studies have shown that metabolic syndrome, of which hyperlipidemia is a component, can also relate to poor response to biologics,” Dr. Liao said.
The most common sequences of first-to-second biologics among those with multiple biologic failure were TNF inhibitor to IL-17 inhibitor (30.8%); IL-12/23 inhibitor to IL-17 inhibitor (21.5%); TNF inhibitor to IL-12/23 inhibitor (12.3%); and IL-17 inhibitor to IL-23 inhibitor (10.8%).
The vast majority of patients failed more than two biologics, however, and “more than 20% had five or more biologics tried over a relatively short period,” Dr. Liao said.
Comorbidities and biologic failure in psoriasis, PsA
In practice, it was said during a discussion period, biologic failures in psoriasis can be of two types: a primary inadequate response or initial failure, or a secondary failure with initial improvement followed by declining or no response. “I agree 100% that these probably represent two different endotypes,” Dr. Liao said. “There’s research emerging that psoriasis isn’t necessarily a clean phenotype.”
The option of focusing on comorbidities in the face of biologic failure was another point of discussion. “Maybe the next biologic is not the answer,” a meeting participant said. “Maybe we should focus on metabolic syndrome.”
“I agree,” Dr. Liao said. “In clinic, there are people who may not respond to therapies but have other comorbidities and factors that make it difficult to manage [their psoriasis] ... that may be causative for psoriasis. Maybe if we treat the comorbidities, it will make it easier to treat the psoriasis.”
Addressing comorbidities and “extra-articular traits” such as poorly controlled diabetes, centralized pain, anxiety and depression, and obesity is something Dr. Ritchlin advocates for PsA. “Centralized pain, I believe, is a major driver of nonresponse,” he said at the meeting. “We have to be careful about blaming nonresponse and lack of efficacy of biologics when it could be a wholly different mechanism the biologic won’t treat ... for example, centralized pain.”
As with psoriasis, the emergence of antidrug antibodies may be one reason for the secondary failure of biologic agents for PsA, Dr. Ritchlin and his coauthors wrote in their paper on management of PsA after failure of one TNF inhibitor. Other areas to consider in evaluating failure, they wrote, are compliance and time of dosing, and financial barriers.
Low long-term persistence of second-line biologics for patients with PsA was demonstrated in a national cohort study utilizing the French health insurance database, Dr. Ritchlin noted at the research meeting.
The French study covered almost 3,000 patients who started a second biologic after discontinuing a TNF inhibitor during 2015-2020. Overall, 1-year and 3-year persistence rates were 42% and 17%, respectively.
Dr. Liao disclosed research grant funding from AbbVie, Amgen, Janssen, Leo, Novartis, Pfizer, Regeneron, and Trex Bio. Dr. Ritchlin reported no disclosures.
WASHINGTON – Multiple biologic failure in a minority of patients with psoriasis may have several causes, from genetic endotypes and immunologic factors to lower serum drug levels, the presence of anti-drug antibody levels, female sex, and certain comorbidities, Wilson Liao, MD, said at the annual research symposium of the National Psoriasis Foundation.
“Tough-to-treat psoriasis remains a challenge despite newer therapies ... Why do we still have this sub-population of patients who seem to be refractory?” said Dr. Liao, professor and associate vice chair of research in the department of dermatology at the University of California, San Francisco, who coauthored a 2015-2022 prospective cohort analysis that documented about 6% of patients failing two or more biologic agents of different mechanistic classes.
“These patients are really suffering,” he said. “We need to have better guidelines and treatment algorithms for these patients.”
A significant number of patients with psoriatic arthritis (PsA), meanwhile, are inadequate responders to tumor necrosis factor (TNF) inhibition, Christopher T. Ritchlin, MD, PhD, professor of medicine in the division of allergy/immunology and rheumatology and the Center of Musculoskeletal Research at the University of Rochester (N.Y.), said during another session at the meeting.
The long-term “persistence,” or usage, of first-line biologics in patients with PsA – and of second-line biologics in patients who failed one TNF-inhibitor – is low, but the literature offers little information on the reasons for TNF-inhibitor discontinuation, said Dr. Ritchlin, who coauthored a perspective piece in Arthritis & Rheumatology on managing the patient with PsA who fails one TNF inhibitor.
Dr. Ritchlin and his coauthors were asked to provide evidence-informed advice and algorithms, but the task was difficult. “It’s hard to know what to recommend for the next step if we don’t know why patients failed the first,” he said. “The point is, we need more data. [Clinical trials] are not recording the kind of information we need.”
Anti-drug antibodies, genetics, other factors in psoriasis
Research shows that in large cohorts, “all the biologics do seem to lose efficacy over time,” said Dr. Liao, who directs the UCSF Psoriasis and Skin Treatment Center. “Some are better than others, but we do see a loss of effectiveness over time.”
A cohort study published in 2022 in JAMA Dermatology, for instance, documented declining “drug survival” associated with ineffectiveness during 2 years of treatment for each of five biologics studied (adalimumab [Humira], ustekinumab [Stelara], secukinumab [Cosentyx], guselkumab [Tremfya], and ixekizumab [Taltz]).
“There have been a number of theories put forward” as to why that’s the case, including lower serum drug levels, “which of course can be related to anti-drug antibody production,” he said.
He pointed to two studies of ustekinumab: One prospective observational cohort study that reported an association of lower early drug levels of the IL-12/23 receptor antagonist with lower Psoriasis Area and Severity Index (PASI) scores, and another observational study that documented an association between anti-drug antibody positivity with lower ustekinumab levels and impaired clinical response.
“We also now know ... that there are genetic endotypes in psoriasis, and that patients who are [HLA-C*06:02]-positive tend to respond a little better to drugs like ustekinumab, and those who are [HLA-C*06:02]-negative tend to do a little better with the TNF inhibitors,” Dr. Liao said. The human leukocyte antigen (HLA) allele HLA-C*06:02 is associated with susceptibility to psoriasis.
In a study using a national psoriasis registry, HLA-C*06:02-negative patients were 3 times more likely to achieve PASI90 status in response to adalimumab, a TNF-alpha inhibitor, than with ustekinumab treatment. And in a meta-analysis covering eight studies with more than 1,000 patients with psoriasis, the median PASI75 response rate after 6 months of ustekinumab therapy was 92% in the HLA-C*06:02-positive group and 67% in HLA-C*06:02-negative patients.
The recently published cohort study showing a 6% rate of multiple biologic failure evaluated patients in the multicenter CorEvitas Psoriasis Registry who initiated their first biologic between 2015 and 2020 and were followed for 2 or more years. Investigators looked for sociodemographic and clinical differences between the patients who continued use of their first biologic for at least 2 years (“good response”), and those who discontinued two or more biologics of different classes, each used for at least 90 days, because of inadequate efficacy.
Of 1,039 evaluated patients, 490 (47.2%) had good clinical response to their first biologic and 65 (6.3%) had multiple biologic failure. All biologic classes were represented among those who failed multiple biologics. The first and second biologic classes used were attempted for a mean duration of 10 months – “an adequate trial” of each, Dr. Liao said.
In multivariable regression analysis, six variables were significantly associated with multiple biologic failure: female sex at birth, shorter disease duration, earlier year of biologic initiation, prior nonbiologic systemic therapy, having Medicaid insurance, and a history of hyperlipidemia. The latter is “interesting because other studies have shown that metabolic syndrome, of which hyperlipidemia is a component, can also relate to poor response to biologics,” Dr. Liao said.
The most common sequences of first-to-second biologics among those with multiple biologic failure were TNF inhibitor to IL-17 inhibitor (30.8%); IL-12/23 inhibitor to IL-17 inhibitor (21.5%); TNF inhibitor to IL-12/23 inhibitor (12.3%); and IL-17 inhibitor to IL-23 inhibitor (10.8%).
The vast majority of patients failed more than two biologics, however, and “more than 20% had five or more biologics tried over a relatively short period,” Dr. Liao said.
Comorbidities and biologic failure in psoriasis, PsA
In practice, it was said during a discussion period, biologic failures in psoriasis can be of two types: a primary inadequate response or initial failure, or a secondary failure with initial improvement followed by declining or no response. “I agree 100% that these probably represent two different endotypes,” Dr. Liao said. “There’s research emerging that psoriasis isn’t necessarily a clean phenotype.”
The option of focusing on comorbidities in the face of biologic failure was another point of discussion. “Maybe the next biologic is not the answer,” a meeting participant said. “Maybe we should focus on metabolic syndrome.”
“I agree,” Dr. Liao said. “In clinic, there are people who may not respond to therapies but have other comorbidities and factors that make it difficult to manage [their psoriasis] ... that may be causative for psoriasis. Maybe if we treat the comorbidities, it will make it easier to treat the psoriasis.”
Addressing comorbidities and “extra-articular traits” such as poorly controlled diabetes, centralized pain, anxiety and depression, and obesity is something Dr. Ritchlin advocates for PsA. “Centralized pain, I believe, is a major driver of nonresponse,” he said at the meeting. “We have to be careful about blaming nonresponse and lack of efficacy of biologics when it could be a wholly different mechanism the biologic won’t treat ... for example, centralized pain.”
As with psoriasis, the emergence of antidrug antibodies may be one reason for the secondary failure of biologic agents for PsA, Dr. Ritchlin and his coauthors wrote in their paper on management of PsA after failure of one TNF inhibitor. Other areas to consider in evaluating failure, they wrote, are compliance and time of dosing, and financial barriers.
Low long-term persistence of second-line biologics for patients with PsA was demonstrated in a national cohort study utilizing the French health insurance database, Dr. Ritchlin noted at the research meeting.
The French study covered almost 3,000 patients who started a second biologic after discontinuing a TNF inhibitor during 2015-2020. Overall, 1-year and 3-year persistence rates were 42% and 17%, respectively.
Dr. Liao disclosed research grant funding from AbbVie, Amgen, Janssen, Leo, Novartis, Pfizer, Regeneron, and Trex Bio. Dr. Ritchlin reported no disclosures.
AT THE NPF RESEARCH SYMPOSIUM 2023