User login
Would your patient benefit from a monoclonal antibody?
Small-molecule drugs such as aspirin, albuterol, atorvastatin, and lisinopril are the backbone of disease management in family medicine.1 However, large-molecule biological drugs such as monoclonal antibodies (MAbs) are increasingly prescribed to treat common conditions. In the past decade, MAbs comprised 20% of all drug approvals by the US Food and Drug Administration (FDA), and today they represent more than half of drugs currently in development.2 Fifteen MAbs have been approved by the FDA over the past decade for asthma, atopic dermatitis (AD), hyperlipidemia, osteoporosis, and migraine prevention.3 This review details what makes MAbs unique and what you should know about them.
The uniqueness of monoclonal antibodies
MAbs are biologics, but not all biologics are MAbs—eg, adalimumab (Humira) is a MAb, but etanercept (Enbrel) is not. MAbs are therapeutic proteins made possible by hybridoma technology used to create an antibody with single specificity.4-6 Monoclonal antibodies differ from small-molecule drugs in structure, dosing, route of administration, manufacturing, metabolism, drug interactions, and elimination (TABLE 17-9).

MAbs can be classified as naked, “without any drug or radioactive material attached to them,” or conjugated, “joined to a chemotherapy drug, radioactive isotope, or toxin.”10 MAbs work in several ways, including competitively inhibiting ligand-receptor binding, receptor blockade, or cell elimination from indirect immune system activities such as antibody-dependent cell-mediated cytotoxicity.11,12
Monoclonal antibody uses in family medicine
Asthma
Several MAbs have been approved for use in severe asthma, including but not limited to: omalizumab (Xolair),13 mepolizumab (Nucala),9,14 and dupilumab (Dupixent).15
Omalizumab is a humanized MAb that prevents IgE antibodies from binding to mast cells and basophils, thereby reducing inflammatory mediators.13 A systematic review found that, compared with placebo, omalizumab used in patients with inadequately controlled moderate-to-severe asthma led to significantly fewer asthma exacerbations (absolute risk reduction [ARR], 16% vs 26%; odds ratio [OR] = 0.55; 95% CI, 0.42-0.60; number needed to treat [NNT] = 10) and fewer hospitalizations (ARR, 0.5% vs 3%; OR = 0.16; 95% CI, 0.06-0.42; NNT = 40).13
Significantly more patients in the omalizumab group were able to withdraw from, or reduce, the dose of ICS. GINA recommends omalizumab for patients with positive skin sensitization, total serum IgE ≥ 30 IU/mL, weight within 30 kg to 150 kg, history of childhood asthma and recent exacerbations, and blood eosinophils ≥ 260/mcL.16 Omalizumab is also approved for use in chronic spontaneous urticaria and nasal polyps.
Mepolizumab
Continue to: Another trial found that...
Another trial found that mepolizumab reduced total OCS doses in patients with severe asthma by 50% without increasing exacerbations or worsening asthma control.18 All 3 anti-IL-5 drugs—including not only mepolizumab, but also benralizumab (Fasenra) and reslizumab (Cinqair)—appear to yield similar improvements. A 2017 systematic review found all anti-IL-5 treatments reduced rates of clinically significant asthma exacerbations (treatment with OCS for ≥ 3 days) by roughly 50% in patients with severe eosinophilic asthma and a history of ≥ 2 exacerbations in the past year.
Dupilumab is a humanized MAb that inhibits IL-4 and IL-13, which influence multiple cell types involved in inflammation (eg, mast cells, eosinophils) and inflammatory mediators (histamine, leukotrienes, cytokines).15 In a recent study of patients with uncontrolled asthma, dupilumab 200 mg every 2 weeks compared with placebo showed a modest reduction in the annualized rate of severe asthma exacerbations (0.46 exacerbations vs 0.87, respectively). Dupilumab was effective in patients with blood eosinophil counts ≥ 150/μL but was ineffective in patients with eosinophil counts < 150/μL.15
For patients ≥ 12 years old with severe eosinophilic asthma, GINA recommends using dupilumab as add-on therapy for an initial trial of 4 months at doses of 200 or 300 mg SC every 2 weeks, with preference for 300 mg SC every 2 weeks for OCS-dependent asthma. Dupilumab is approved for use in AD and chronic rhinosinusitis with nasal polyposis. If a biologic agent is not successful after a 4-month trial, consider a 6- to 12-month trial. If efficacy is still minimal, consider switching to an alternative biologic therapy approved for asthma.16
❯ Asthma: Test your skills
Subjective findings: A 19-year-old man presents to your clinic. He has a history of nasal polyps and allergic asthma. At age 18, he was given a diagnosis of severe persistent asthma. He has shortness of breath during waking hours 4 times per week, and treats each of these episodes with albuterol. He also wakes up about twice a week with shortness of breath and has some limitations in normal activities. He reports missing his prescribed fluticasone/salmeterol 500/50 μg, 1 inhalation bid, only once each month. In the last year, he has had 2 exacerbations requiring oral steroids.
Medications: Albuterol 90 μg, 1-2 inhalations, q6h prn; fluticasone/salmeterol 500/50 μg, 1 inhalation bid; tiotropium 1.25 μg, 2 puffs/d; montelukast 10 mg every morning; prednisone 10 mg/d.
Continue to: Objective data
Objective data: Patient is in no apparent distress and afebrile, and oxygen saturation on room air is 97%. Ht, 70 inches; wt, 75 kg. Labs: IgE, 15 IU/mL; serum eosinophils, 315/μL.
Which MAb would be appropriate for this patient? Given that the patient has a blood eosinophil level ≥ 300/μL and severe exacerbations, adult-onset asthma, nasal polyposis, and maintenance OCS at baseline, it would be reasonable to initiate mepolizumab 100 mg SC every 4 weeks, or dupilumab 600 mg once, then 300 mg SC every 2 weeks. Both agents can be self-administered.
Atopic dermatitis
Two MAbs—dupilumab and tralokinumab (Adbry; inhibits IL-13)—are approved for treatment of AD in adults that is uncontrolled with conventional therapy.15,19 Dupilumab is also approved for children ≥ 6 months old.20 Both MAbs are dosed at 600 mg SC, followed by 300 mg every 2 weeks. Dupilumab was compared with placebo in adult patients who had moderate-to-severe AD inadequately controlled on topical corticosteroids (TCSs), to determine the proportion of patients in each group achieving improvement of either 0 or 1 points or ≥ 2 points in the 5-point Investigator Global Assessment (IGA) score from baseline to 16 weeks.21 Thirty-seven percent of patients receiving dupilumab 300 mg SC weekly and 38% of patients receiving dupilumab 300 mg SC every 2 weeks achieved the primary outcome, compared with 10% of those receiving placebo (P < .001).21 Similar IGA scores were reported when dupilumab was combined with TCS, compared with placebo.22
It would be reasonable to consider dupilumab or tralokinumab in patients with: cutaneous atrophy or hypothalamic-pituitary-adrenal axis suppression with TCS, concerns of malignancy with topical calcineurin inhibitors, or problems with the alternative systemic therapies (cyclosporine-induced hypertension, nephrotoxicity, or immunosuppression; azathioprine-induced malignancy; or methotrexate-induced bone marrow suppression, renal impairment, hepatotoxicity, pneumonitis, or gastrointestinal toxicity).23
A distinct advantage of MAbs over other systemic agents in the management of AD is that MAbs do not require frequent monitoring of blood pressure, renal or liver function, complete blood count with differential, electrolytes, or uric acid. Additionally, MAbs have fewer black box warnings and adverse reactions when compared with other systemic agents.
Continue to: Hyperlipidemia
Hyperlipidemia
Three MAbs are approved for use in hyperlipidemia: the angiopoietin-like protein 3 (ANGPTL3) inhibitor evinacumab (Evkeeza)24 and 2 proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, evolocumab (Repatha)25 and alirocumab (Praluent).26
ANGPTL3 inhibitors block ANGPTL3 and reduce endothelial lipase and lipoprotein lipase activity, which in turn decreases low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglyceride formation. PCSK9 inhibitors prevent PCSK9 from binding to LDL receptors, thereby maintaining the number of active LDL receptors and increasing LDL-C removal.
Evinacumab is indicated for homozygous familial hypercholesterolemia and is administered intravenously every 4 weeks. Evinacumab has not been evaluated for effects on cardiovascular morbidity and mortality.
Evolocumab 140 mg SC every 2 weeks or 420 mg SC monthly has been studied in patients on statin therapy with LDL-C ≥ 70 mg/dL. Patients on evolocumab experienced significantly less of the composite endpoint of cardiovascular death, myocardial infarction (MI), stroke, hospitalization for unstable angina, or coronary revascularization compared with placebo (9.8% vs 11.3%; hazard ratio [HR] = 0.85; 95% CI, 0.79-0.92; NNT = 67.27
Alirocumab 75 mg SC every 2 weeks has also been studied in patients receiving statin therapy with LDL-C ≥ 70 mg/dL. Patients taking alirocumab experienced significantly less of the composite endpoint of death from coronary heart disease, nonfatal MI, ischemic stroke, or hospitalization for unstable angina compared with placebo (9.5% vs 11.1%; HR = 0.85; 95% CI, 0.78-0.93; NNT = 63).
Continue to: According to the 2018...
According to the 2018 AHA Cholesterol Guidelines, PCSK9 inhibitors are indicated for patients receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe) with LDL-C ≥ 70 mg/dL, if they have had multiple atherosclerotic cardiovascular disease (ASCVD) events or 1 major ASCVD event with multiple high-risk conditions (eg, heterozygous familial hypercholesterolemia, history of coronary artery bypass grafting or percutaneous coronary intervention, hypertension, estimated glomerular filtration rate of 15 to 59 mL/min/1.73m2).29 For patients without prior ASCVD events or high-risk conditions who are receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe), PCSK9 inhibitors are indicated if the LDL-C remains ≥ 100 mg/dL.
Osteoporosis
The 2 MAbs approved for use in osteoporosis are the receptor activator of nuclear factor kB ligand (RANKL) inhibitor denosumab (Prolia)30 and the sclerostin inhibitor romosozumab (Evenity).31
Denosumab prevents RANKL from binding to the RANK receptor, thereby inhibiting osteoclast formation and decreasing bone resorption. Denosumab is approved for use in women and men who are at high risk of osteoporotic fracture, including those taking OCSs, men receiving androgen deprivation therapy for prostate cancer, and women receiving adjuvant aromatase inhibitor therapy for breast cancer.
In a 3-year randomized trial, denosumab 60 mg SC every 6 months was compared with placebo in postmenopausal women with T-scores < –2.5, but not < –4.0 at the lumbar spine or total hip. Denosumab significantly reduced new radiographic vertebral fractures (2.3% vs 7.2%; risk ratio [RR] = 0.32; 95% CI, 0.26-0.41; NNT = 21), hip fracture (0.7% vs 1.2%), and nonvertebral fracture (6.5% vs 8.0%).32 Denosumab carries an increased risk of multiple vertebral fractures following discontinuation, skin infections, dermatologic reactions, and severe bone, joint, and muscle pain.
Romosozumab inhibits sclerostin, thereby increasing bone formation and, to a lesser degree, decreasing bone resorption. Romosozumab is approved for use in postmenopausal women at high risk for fracture (ie, those with a history of osteoporotic fracture or multiple risk factors for fracture) or in patients who have not benefited from or are intolerant of other therapies. In one study, postmenopausal women with a T-score of –2.5 to –3.5 at the total hip or femoral neck were randomly assigned to receive either romosozumab 210 mg SC or placebo for 12 months, then each group was switched to denosumab 60 mg SC for 12 months. After the first year, prior to initiating denosumab, patients taking romosozumab experienced significantly fewer new vertebral fractures than patients taking placebo (0.5% vs 1.8%; RR = 0.27; 95% CI, 0.16-0.47; NNT = 77); however, there was no significant difference between the 2 groups with nonvertebral fractures (HR = 0.75; 95% CI, 0.53-1.05).33
Continue to: In another study...
In another study, romosozumab 210 mg SC was compared with alendronate 70 mg weekly, followed by alendronate 70 mg weekly in both groups. Over the first 12 months, patients treated with romosozumab saw a significant reduction in the incidence of new vertebral fractures (4% vs 6.3%; RR = 0.63, P < .003; NNT = 44). Patients treated with romosozumab with alendronate added for another 12 months also saw a significant reduction in new incidence of vertebral fractures (6.2% vs 11.9%; RR = 0.52; P < .001; NNT = 18).34 There was a higher risk of cardiovascular events among patients receiving romosozumab compared with those treated with alendronate, so romosozumab should not be used in individuals who have had an MI or stroke within the previous year.34 Denosumab and romosozumab offer an advantage over some bisphosphonates in that they require less frequent dosing and can be used in patients with renal impairment (creatinine clearance < 35 mL/min, in which zoledronic acid is contraindicated and alendronate is not recommended; < 30 mL/min, in which risedronate and ibandronate are not recommended).
Migraine prevention
Four
Erenumab, fremanezumab, and galcanezumab are all available in subcutaneous autoinjectors (or syringe with fremanezumab). Eptinezumab is an intravenous (IV) infusion given every 3 months.
Erenumab is available in both 70-mg and 140-mg dosing options. Fremanezumab can be given as 225 mg monthly or 675 mg quarterly. Galcanezumab has an initial loading dose of 240 mg followed by 120 mg given monthly. Erenumab targets the CGRP receptor; the others target the CGRP ligand. Eptinezumab has 100% bioavailability and reaches maximum serum concentration sooner than the other antagonists (due to its route of administration), but it must be given in an infusion center. Few insurers approve the use of eptinezumab unless a trial of least 1 of the monthly injectables has failed.
There are no head-to-head studies of the medications in this class. Additionally, differing study designs, definitions, statistical analyses, endpoints, and responder-rate calculations make it challenging to compare them directly against one another. At the very least, all of the CGRP MAbs have efficacy comparable to conventional preventive migraine medications such as propranolol, amitriptyline, and topiramate.40
Continue to: The most commonly reported adverse...
The most commonly reported adverse effect for all 4 CGRPs is injection site reaction, which was highest with the quarterly fremanezumab dose (45%).37 Constipation was most notable with the 140-mg dose of erenumab (3%)35; with the other CGRP MAbs it is comparable to that seen with placebo (< 1%).
Erenumab-induced hypertension has been identified in 61 cases reported through the FDA Adverse Event Reporting System (FAERS) as of 2021.41 This was not reported during MAb development programs, nor was it noted during clinical trials. Blood pressure elevation was seen within 1 week of injection in nearly 50% of the cases, and nearly one-third had pre-existing hypertension.41 Due to these findings, the erenumab prescribing information was updated to include hypertension in its warnings and precautions. It is possible that hypertension could be a class effect, although trial data and posthoc studies have yet to bear that out. Since erenumab was the first CGRP antagonist brought to market (May 2018 vs September 2018 for fremanezumab and galcanezumab), it may have accumulated more FAERS reports. Nearly all studies exclude patients with older age, uncontrolled hypertension, and unstable cardiovascular disease, which could impact data.41
Overall, this class of medications is very well tolerated, easy to use (again, excluding eptinezumab), and maintains a low adverse effect profile, giving added value compared with conventional preventive migraine medications.
The American Headache Society recommends a preventive oral therapy for at least 3 months before trying an alternative medication. After treatment failure with at least 2 oral agents, CGRP MAbs are recommended.42 CGRP antagonists offer convenient dosing, bypass gastrointestinal metabolism (which is useful in patients with nausea/vomiting), and have fewer adverse effects than traditional oral medications.
Worth noting. Several newer oral agents have been recently approved for migraine prevention, including atogepant (Qulipta) and rimegepant (Nurtec), which are also CGRP antagonists. Rimegepant is approved for both acute migraine treatment and prevention.
Continue to: Migraine
❯ Migraine: Test your skills
Subjective findings: A 25-year-old woman presents to your clinic for management of episodic migraines with aura. Her baseline average migraine frequency is 9 headache days/month. Her migraines are becoming more frequent despite treatment. She fears IV medication use and avoids hospitals.
History: Hypertension, irritable bowel syndrome with constipation (IBS-C), and depression. The patient is not pregnant or trying to get pregnant.
Medications: Current medications (for previous 4 months) include propranolol 40 mg at bedtime, linaclotide 145 μg/d, citalopram 20 mg/d, and sumatriptan 50 mg prn. Past medications include venlafaxine 150 mg po bid for 5 months.
What would be appropriate for this patient? This patient meets the criteria for using a CGRP antagonist because she has tried 2 preventive treatments for more than 60 to 90 days. Erenumab is not the best option, given the patient’s history of hypertension and IBS-C. The patient fears hospitals and IV medications, making eptinezumab a less-than-ideal choice. Depending on her insurance, fremanezumab or galcanezumab would be good options at this time.
CGRP antagonists have not been studied or evaluated in pregnancy, but if this patient becomes pregnant, a first-line agent for prevention would be propranolol, and a second-line agent would be a tricyclic antidepressant, memantine, or verapamil. Avoid ergotamines and antiepileptics (topiramate or valproate) in pregnancy.43,44
Continue to: The challenges associated with MAbs
The challenges associated with MAbs
MAbs can be expensive (TABLE 2),45 some prohibitively so. On a population scale, biologics account for around 40% of prescription drug spending and may cost 22 times more than small-molecule drugs.46 Estimates in 2016 showed that MAbs comprise $90.2 billion (43%) of the biologic market.46

MAbs also require prior authorization forms to be submitted. Prior authorization criteria vary by state and by insurance plan. In my (ES) experience, submitting letters of medical necessity justifying the need for therapy or expertise in the disease states for which the MAb is being prescribed help your patient get the medication they need.
Expect to see additional MAbs approved in the future. If the costs come down, adoption of these agents into practice will likely increase.
CORRESPONDENCE
Evelyn Sbar, MD, Texas Tech University Health Sciences Center, 1400 South Coulter Street, Suite 5100, Amarillo, TX 79106; [email protected]
1. Rui P, Okeyode T. National Ambulatory Medical Care Survey: 2016 national summary tables. National Center for Health Statistics. Accessed June 15, 2022. www.cdc.gov/nchs/data/ahcd/namcs_summary/2016_namcs_web_tables.pdf
2. IDBS. The future of biologics drug development is today. June 27, 2018. Accessed June 15, 2022. www.idbs.com/blog/2018/06/the-future-of-biologics-drug-development-is-today/
3. Antibody therapeutics approved or in regulatory review in the EU or US. Antibody Society. Accessed June 15, 2022. www.antibodysociety.org/resources/approved-antibodies/
4. FDA. Code of Federal Regulations, Title 21, Chapter I, Subchapter F biologics. March 29, 2022. Accessed June 15, 2022. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=600.3
5. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495-497. doi: 10.1038/256495a0
6. Raejewsky K. The advent and rise of monoclonal antibodies. Nature. November 4, 2019. Accessed June 15, 2022. www.nature.com/articles/d41586-019-02840-w
7. Flovent. Prescribing information. GlaxoSmithKline; 2010. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021433s015lbl.pdf
8. NLM. National Center for Biotechnology Information. PubChem. Method for the preparation of fluticasone and related 17beta-carbothioic esters using a novel carbothioic acid synthesis and novel purification methods. Accessed June 15, 2022. pubchem.ncbi.nlm.nih.gov/patent/WO-0162722-A2
9. Nucala. Prescribing information. GlaxoSmithKline; 2019. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761122s000lbl.pdf
10. Argyriou AA, Kalofonos HP. Recent advances relating to the clinical application of naked monoclonal antibodies in solid tumors. Mol Med. 2009;15:183-191. doi: 10.2119/molmed.2009.00007
11. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548-558. doi: 10.1038/clpt.2008.170
12. Zahavi D, AlDeghaither D, O’Connell A, et al. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1:7-12. doi: 10.1093/abt/tby002
13. Normansell R, Walker S, Milan SJ, et al. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev. 2014:CD003559. doi: 10.1002/14651858.CD003559.pub4
14. Farne HA, Wilson A, Powell C, et al. Anti-IL5 therapies for asthma. Cochrane Database Syst Rev. 2017;9:CD010834. doi: 10.1002/14651858.CD010834.pub3
15. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. 2018;378:2486-2496. doi: 10.1056/NEJMoa1804092
16. GINA. Global strategy for asthma management and prevention. 2022 Difficult-to-treat and severe asthma guide—slide set. Accessed June 23, 2022. https://ginasthma.org/severeasthma/
17. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198-1207. doi: 10.1056/NEJMoa1403290
18. Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189-1197. doi: 10.1056/NEJMoa1403291
19. Adbry. Prescribing information. Leo Pharma Inc; 2021. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761180Orig1s000lbl.pdf
20. Dupixent. Prescribing information. Regeneron Pharmaceuticals; 2022. Accessed October 5, 2022. https://www.regeneron.com/downloads/dupixent_fpi.pdf
21. Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348. doi: 10.1056/NEJMoa1610020
22. Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389:2287-2303. doi: 10.1016/s0140-6736(17)31191-1
23. Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349. doi: 10.1016/j.jaad.2014.03.030
24. Evkeeza. Prescribing information. Regeneron Pharmaceuticals; 2021. Accessed June 24, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
25. Repatha. Prescribing information. Amgen; 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125522s014lbl.pdf
26. Praluent. Prescribing information. Sanofi Aventis and Regeneron Pharmaceuticals. 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125559s002lbl.pdf
27. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713-1722. doi: 10.1056/NEJMoa1615664
28. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097-2107. doi:10.1056/NEJMoa1801174
29. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
30. Prolia. Prescribing information. Amgen; 2010. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2013/125320s094lbl.pdf
31. Evenity. Prescribing information. Amgen; 2019. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761062s000lbl.pdf
32. Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756-765. doi: 10.1056/NEJMoa0809493
33. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375:1532-1543. doi: 10.1056/NEJMoa1607948
34. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377:1417-1427. doi: 10.1056/NEJMoa1708322
35. Aimovig. Prescribing information. Amgen; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761077s000lbl.pdf
36. Vyepti. Prescribing information. Lundbeck Seattle BioPharmaceuticals; 2020. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2020/761119s000lbl.pdf
37. Ajovy. Prescribing information. Teva Pharmaceuticals; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761089s000lbl.pdf
38. Emgality. Prescribing information. Eli Lilly and Co.; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761063s000lbl.pdf
39. Edvinsson L, Haanes KA, Warfvinge K, et al. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338-350. doi: 10.1038/s41582-018-0003-1
40. Vandervorst F. Van Deun L, Van Dycke A, et al. CGRP monoclonal antibodies in migraine: an efficacy and tolerability comparison with standard prophylactic drugs. J Headache Pain. 2021;22:128. doi: 10.1186/s10194-021-01335-2
41. Saely S, Croteau D, Jawidzik L, et al. Hypertension: a new safety risk for patients treated with erenumab. Headache. 2021;61:202-208. doi: 10.1111/head.14051
42. American Headache Society. The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1-18. doi: 10.1111/head.13456
43. Burch R. Headache in pregnancy and the puerperium. Neurol Clin. 2019;37:31-51. doi: 10.1016/j.ncl.2018.09.004
44. Burch R. Epidemiology and treatment of menstrual migraine and migraine during pregnancy and lactation: a narrative review. Headache. 2020;60:200-216. doi: 10.1111/head.13665
45. Lexi-Comp. Lexi-drug database. Accessed April 4, 2022. https://online.lexi.com/lco/action/login
46. Walker N. Biologics: driving force in pharma. Pharma’s Almanac. June 5, 2017. Accessed June 15, 2020. www.pharmasalmanac.com/articles/biologics-driving-force-in-pharma
Small-molecule drugs such as aspirin, albuterol, atorvastatin, and lisinopril are the backbone of disease management in family medicine.1 However, large-molecule biological drugs such as monoclonal antibodies (MAbs) are increasingly prescribed to treat common conditions. In the past decade, MAbs comprised 20% of all drug approvals by the US Food and Drug Administration (FDA), and today they represent more than half of drugs currently in development.2 Fifteen MAbs have been approved by the FDA over the past decade for asthma, atopic dermatitis (AD), hyperlipidemia, osteoporosis, and migraine prevention.3 This review details what makes MAbs unique and what you should know about them.
The uniqueness of monoclonal antibodies
MAbs are biologics, but not all biologics are MAbs—eg, adalimumab (Humira) is a MAb, but etanercept (Enbrel) is not. MAbs are therapeutic proteins made possible by hybridoma technology used to create an antibody with single specificity.4-6 Monoclonal antibodies differ from small-molecule drugs in structure, dosing, route of administration, manufacturing, metabolism, drug interactions, and elimination (TABLE 17-9).
MAbs can be classified as naked, “without any drug or radioactive material attached to them,” or conjugated, “joined to a chemotherapy drug, radioactive isotope, or toxin.”10 MAbs work in several ways, including competitively inhibiting ligand-receptor binding, receptor blockade, or cell elimination from indirect immune system activities such as antibody-dependent cell-mediated cytotoxicity.11,12
Monoclonal antibody uses in family medicine
Asthma
Several MAbs have been approved for use in severe asthma, including but not limited to: omalizumab (Xolair),13 mepolizumab (Nucala),9,14 and dupilumab (Dupixent).15
Omalizumab is a humanized MAb that prevents IgE antibodies from binding to mast cells and basophils, thereby reducing inflammatory mediators.13 A systematic review found that, compared with placebo, omalizumab used in patients with inadequately controlled moderate-to-severe asthma led to significantly fewer asthma exacerbations (absolute risk reduction [ARR], 16% vs 26%; odds ratio [OR] = 0.55; 95% CI, 0.42-0.60; number needed to treat [NNT] = 10) and fewer hospitalizations (ARR, 0.5% vs 3%; OR = 0.16; 95% CI, 0.06-0.42; NNT = 40).13
Significantly more patients in the omalizumab group were able to withdraw from, or reduce, the dose of ICS. GINA recommends omalizumab for patients with positive skin sensitization, total serum IgE ≥ 30 IU/mL, weight within 30 kg to 150 kg, history of childhood asthma and recent exacerbations, and blood eosinophils ≥ 260/mcL.16 Omalizumab is also approved for use in chronic spontaneous urticaria and nasal polyps.
Mepolizumab
Continue to: Another trial found that...
Another trial found that mepolizumab reduced total OCS doses in patients with severe asthma by 50% without increasing exacerbations or worsening asthma control.18 All 3 anti-IL-5 drugs—including not only mepolizumab, but also benralizumab (Fasenra) and reslizumab (Cinqair)—appear to yield similar improvements. A 2017 systematic review found all anti-IL-5 treatments reduced rates of clinically significant asthma exacerbations (treatment with OCS for ≥ 3 days) by roughly 50% in patients with severe eosinophilic asthma and a history of ≥ 2 exacerbations in the past year.
Dupilumab is a humanized MAb that inhibits IL-4 and IL-13, which influence multiple cell types involved in inflammation (eg, mast cells, eosinophils) and inflammatory mediators (histamine, leukotrienes, cytokines).15 In a recent study of patients with uncontrolled asthma, dupilumab 200 mg every 2 weeks compared with placebo showed a modest reduction in the annualized rate of severe asthma exacerbations (0.46 exacerbations vs 0.87, respectively). Dupilumab was effective in patients with blood eosinophil counts ≥ 150/μL but was ineffective in patients with eosinophil counts < 150/μL.15
For patients ≥ 12 years old with severe eosinophilic asthma, GINA recommends using dupilumab as add-on therapy for an initial trial of 4 months at doses of 200 or 300 mg SC every 2 weeks, with preference for 300 mg SC every 2 weeks for OCS-dependent asthma. Dupilumab is approved for use in AD and chronic rhinosinusitis with nasal polyposis. If a biologic agent is not successful after a 4-month trial, consider a 6- to 12-month trial. If efficacy is still minimal, consider switching to an alternative biologic therapy approved for asthma.16
❯ Asthma: Test your skills
Subjective findings: A 19-year-old man presents to your clinic. He has a history of nasal polyps and allergic asthma. At age 18, he was given a diagnosis of severe persistent asthma. He has shortness of breath during waking hours 4 times per week, and treats each of these episodes with albuterol. He also wakes up about twice a week with shortness of breath and has some limitations in normal activities. He reports missing his prescribed fluticasone/salmeterol 500/50 μg, 1 inhalation bid, only once each month. In the last year, he has had 2 exacerbations requiring oral steroids.
Medications: Albuterol 90 μg, 1-2 inhalations, q6h prn; fluticasone/salmeterol 500/50 μg, 1 inhalation bid; tiotropium 1.25 μg, 2 puffs/d; montelukast 10 mg every morning; prednisone 10 mg/d.
Continue to: Objective data
Objective data: Patient is in no apparent distress and afebrile, and oxygen saturation on room air is 97%. Ht, 70 inches; wt, 75 kg. Labs: IgE, 15 IU/mL; serum eosinophils, 315/μL.
Which MAb would be appropriate for this patient? Given that the patient has a blood eosinophil level ≥ 300/μL and severe exacerbations, adult-onset asthma, nasal polyposis, and maintenance OCS at baseline, it would be reasonable to initiate mepolizumab 100 mg SC every 4 weeks, or dupilumab 600 mg once, then 300 mg SC every 2 weeks. Both agents can be self-administered.
Atopic dermatitis
Two MAbs—dupilumab and tralokinumab (Adbry; inhibits IL-13)—are approved for treatment of AD in adults that is uncontrolled with conventional therapy.15,19 Dupilumab is also approved for children ≥ 6 months old.20 Both MAbs are dosed at 600 mg SC, followed by 300 mg every 2 weeks. Dupilumab was compared with placebo in adult patients who had moderate-to-severe AD inadequately controlled on topical corticosteroids (TCSs), to determine the proportion of patients in each group achieving improvement of either 0 or 1 points or ≥ 2 points in the 5-point Investigator Global Assessment (IGA) score from baseline to 16 weeks.21 Thirty-seven percent of patients receiving dupilumab 300 mg SC weekly and 38% of patients receiving dupilumab 300 mg SC every 2 weeks achieved the primary outcome, compared with 10% of those receiving placebo (P < .001).21 Similar IGA scores were reported when dupilumab was combined with TCS, compared with placebo.22
It would be reasonable to consider dupilumab or tralokinumab in patients with: cutaneous atrophy or hypothalamic-pituitary-adrenal axis suppression with TCS, concerns of malignancy with topical calcineurin inhibitors, or problems with the alternative systemic therapies (cyclosporine-induced hypertension, nephrotoxicity, or immunosuppression; azathioprine-induced malignancy; or methotrexate-induced bone marrow suppression, renal impairment, hepatotoxicity, pneumonitis, or gastrointestinal toxicity).23
A distinct advantage of MAbs over other systemic agents in the management of AD is that MAbs do not require frequent monitoring of blood pressure, renal or liver function, complete blood count with differential, electrolytes, or uric acid. Additionally, MAbs have fewer black box warnings and adverse reactions when compared with other systemic agents.
Continue to: Hyperlipidemia
Hyperlipidemia
Three MAbs are approved for use in hyperlipidemia: the angiopoietin-like protein 3 (ANGPTL3) inhibitor evinacumab (Evkeeza)24 and 2 proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, evolocumab (Repatha)25 and alirocumab (Praluent).26
ANGPTL3 inhibitors block ANGPTL3 and reduce endothelial lipase and lipoprotein lipase activity, which in turn decreases low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglyceride formation. PCSK9 inhibitors prevent PCSK9 from binding to LDL receptors, thereby maintaining the number of active LDL receptors and increasing LDL-C removal.
Evinacumab is indicated for homozygous familial hypercholesterolemia and is administered intravenously every 4 weeks. Evinacumab has not been evaluated for effects on cardiovascular morbidity and mortality.
Evolocumab 140 mg SC every 2 weeks or 420 mg SC monthly has been studied in patients on statin therapy with LDL-C ≥ 70 mg/dL. Patients on evolocumab experienced significantly less of the composite endpoint of cardiovascular death, myocardial infarction (MI), stroke, hospitalization for unstable angina, or coronary revascularization compared with placebo (9.8% vs 11.3%; hazard ratio [HR] = 0.85; 95% CI, 0.79-0.92; NNT = 67.27
Alirocumab 75 mg SC every 2 weeks has also been studied in patients receiving statin therapy with LDL-C ≥ 70 mg/dL. Patients taking alirocumab experienced significantly less of the composite endpoint of death from coronary heart disease, nonfatal MI, ischemic stroke, or hospitalization for unstable angina compared with placebo (9.5% vs 11.1%; HR = 0.85; 95% CI, 0.78-0.93; NNT = 63).
Continue to: According to the 2018...
According to the 2018 AHA Cholesterol Guidelines, PCSK9 inhibitors are indicated for patients receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe) with LDL-C ≥ 70 mg/dL, if they have had multiple atherosclerotic cardiovascular disease (ASCVD) events or 1 major ASCVD event with multiple high-risk conditions (eg, heterozygous familial hypercholesterolemia, history of coronary artery bypass grafting or percutaneous coronary intervention, hypertension, estimated glomerular filtration rate of 15 to 59 mL/min/1.73m2).29 For patients without prior ASCVD events or high-risk conditions who are receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe), PCSK9 inhibitors are indicated if the LDL-C remains ≥ 100 mg/dL.
Osteoporosis
The 2 MAbs approved for use in osteoporosis are the receptor activator of nuclear factor kB ligand (RANKL) inhibitor denosumab (Prolia)30 and the sclerostin inhibitor romosozumab (Evenity).31
Denosumab prevents RANKL from binding to the RANK receptor, thereby inhibiting osteoclast formation and decreasing bone resorption. Denosumab is approved for use in women and men who are at high risk of osteoporotic fracture, including those taking OCSs, men receiving androgen deprivation therapy for prostate cancer, and women receiving adjuvant aromatase inhibitor therapy for breast cancer.
In a 3-year randomized trial, denosumab 60 mg SC every 6 months was compared with placebo in postmenopausal women with T-scores < –2.5, but not < –4.0 at the lumbar spine or total hip. Denosumab significantly reduced new radiographic vertebral fractures (2.3% vs 7.2%; risk ratio [RR] = 0.32; 95% CI, 0.26-0.41; NNT = 21), hip fracture (0.7% vs 1.2%), and nonvertebral fracture (6.5% vs 8.0%).32 Denosumab carries an increased risk of multiple vertebral fractures following discontinuation, skin infections, dermatologic reactions, and severe bone, joint, and muscle pain.
Romosozumab inhibits sclerostin, thereby increasing bone formation and, to a lesser degree, decreasing bone resorption. Romosozumab is approved for use in postmenopausal women at high risk for fracture (ie, those with a history of osteoporotic fracture or multiple risk factors for fracture) or in patients who have not benefited from or are intolerant of other therapies. In one study, postmenopausal women with a T-score of –2.5 to –3.5 at the total hip or femoral neck were randomly assigned to receive either romosozumab 210 mg SC or placebo for 12 months, then each group was switched to denosumab 60 mg SC for 12 months. After the first year, prior to initiating denosumab, patients taking romosozumab experienced significantly fewer new vertebral fractures than patients taking placebo (0.5% vs 1.8%; RR = 0.27; 95% CI, 0.16-0.47; NNT = 77); however, there was no significant difference between the 2 groups with nonvertebral fractures (HR = 0.75; 95% CI, 0.53-1.05).33
Continue to: In another study...
In another study, romosozumab 210 mg SC was compared with alendronate 70 mg weekly, followed by alendronate 70 mg weekly in both groups. Over the first 12 months, patients treated with romosozumab saw a significant reduction in the incidence of new vertebral fractures (4% vs 6.3%; RR = 0.63, P < .003; NNT = 44). Patients treated with romosozumab with alendronate added for another 12 months also saw a significant reduction in new incidence of vertebral fractures (6.2% vs 11.9%; RR = 0.52; P < .001; NNT = 18).34 There was a higher risk of cardiovascular events among patients receiving romosozumab compared with those treated with alendronate, so romosozumab should not be used in individuals who have had an MI or stroke within the previous year.34 Denosumab and romosozumab offer an advantage over some bisphosphonates in that they require less frequent dosing and can be used in patients with renal impairment (creatinine clearance < 35 mL/min, in which zoledronic acid is contraindicated and alendronate is not recommended; < 30 mL/min, in which risedronate and ibandronate are not recommended).
Migraine prevention
Four
Erenumab, fremanezumab, and galcanezumab are all available in subcutaneous autoinjectors (or syringe with fremanezumab). Eptinezumab is an intravenous (IV) infusion given every 3 months.
Erenumab is available in both 70-mg and 140-mg dosing options. Fremanezumab can be given as 225 mg monthly or 675 mg quarterly. Galcanezumab has an initial loading dose of 240 mg followed by 120 mg given monthly. Erenumab targets the CGRP receptor; the others target the CGRP ligand. Eptinezumab has 100% bioavailability and reaches maximum serum concentration sooner than the other antagonists (due to its route of administration), but it must be given in an infusion center. Few insurers approve the use of eptinezumab unless a trial of least 1 of the monthly injectables has failed.
There are no head-to-head studies of the medications in this class. Additionally, differing study designs, definitions, statistical analyses, endpoints, and responder-rate calculations make it challenging to compare them directly against one another. At the very least, all of the CGRP MAbs have efficacy comparable to conventional preventive migraine medications such as propranolol, amitriptyline, and topiramate.40
Continue to: The most commonly reported adverse...
The most commonly reported adverse effect for all 4 CGRPs is injection site reaction, which was highest with the quarterly fremanezumab dose (45%).37 Constipation was most notable with the 140-mg dose of erenumab (3%)35; with the other CGRP MAbs it is comparable to that seen with placebo (< 1%).
Erenumab-induced hypertension has been identified in 61 cases reported through the FDA Adverse Event Reporting System (FAERS) as of 2021.41 This was not reported during MAb development programs, nor was it noted during clinical trials. Blood pressure elevation was seen within 1 week of injection in nearly 50% of the cases, and nearly one-third had pre-existing hypertension.41 Due to these findings, the erenumab prescribing information was updated to include hypertension in its warnings and precautions. It is possible that hypertension could be a class effect, although trial data and posthoc studies have yet to bear that out. Since erenumab was the first CGRP antagonist brought to market (May 2018 vs September 2018 for fremanezumab and galcanezumab), it may have accumulated more FAERS reports. Nearly all studies exclude patients with older age, uncontrolled hypertension, and unstable cardiovascular disease, which could impact data.41
Overall, this class of medications is very well tolerated, easy to use (again, excluding eptinezumab), and maintains a low adverse effect profile, giving added value compared with conventional preventive migraine medications.
The American Headache Society recommends a preventive oral therapy for at least 3 months before trying an alternative medication. After treatment failure with at least 2 oral agents, CGRP MAbs are recommended.42 CGRP antagonists offer convenient dosing, bypass gastrointestinal metabolism (which is useful in patients with nausea/vomiting), and have fewer adverse effects than traditional oral medications.
Worth noting. Several newer oral agents have been recently approved for migraine prevention, including atogepant (Qulipta) and rimegepant (Nurtec), which are also CGRP antagonists. Rimegepant is approved for both acute migraine treatment and prevention.
Continue to: Migraine
❯ Migraine: Test your skills
Subjective findings: A 25-year-old woman presents to your clinic for management of episodic migraines with aura. Her baseline average migraine frequency is 9 headache days/month. Her migraines are becoming more frequent despite treatment. She fears IV medication use and avoids hospitals.
History: Hypertension, irritable bowel syndrome with constipation (IBS-C), and depression. The patient is not pregnant or trying to get pregnant.
Medications: Current medications (for previous 4 months) include propranolol 40 mg at bedtime, linaclotide 145 μg/d, citalopram 20 mg/d, and sumatriptan 50 mg prn. Past medications include venlafaxine 150 mg po bid for 5 months.
What would be appropriate for this patient? This patient meets the criteria for using a CGRP antagonist because she has tried 2 preventive treatments for more than 60 to 90 days. Erenumab is not the best option, given the patient’s history of hypertension and IBS-C. The patient fears hospitals and IV medications, making eptinezumab a less-than-ideal choice. Depending on her insurance, fremanezumab or galcanezumab would be good options at this time.
CGRP antagonists have not been studied or evaluated in pregnancy, but if this patient becomes pregnant, a first-line agent for prevention would be propranolol, and a second-line agent would be a tricyclic antidepressant, memantine, or verapamil. Avoid ergotamines and antiepileptics (topiramate or valproate) in pregnancy.43,44
Continue to: The challenges associated with MAbs
The challenges associated with MAbs
MAbs can be expensive (TABLE 2),45 some prohibitively so. On a population scale, biologics account for around 40% of prescription drug spending and may cost 22 times more than small-molecule drugs.46 Estimates in 2016 showed that MAbs comprise $90.2 billion (43%) of the biologic market.46
MAbs also require prior authorization forms to be submitted. Prior authorization criteria vary by state and by insurance plan. In my (ES) experience, submitting letters of medical necessity justifying the need for therapy or expertise in the disease states for which the MAb is being prescribed help your patient get the medication they need.
Expect to see additional MAbs approved in the future. If the costs come down, adoption of these agents into practice will likely increase.
CORRESPONDENCE
Evelyn Sbar, MD, Texas Tech University Health Sciences Center, 1400 South Coulter Street, Suite 5100, Amarillo, TX 79106; [email protected]
Small-molecule drugs such as aspirin, albuterol, atorvastatin, and lisinopril are the backbone of disease management in family medicine.1 However, large-molecule biological drugs such as monoclonal antibodies (MAbs) are increasingly prescribed to treat common conditions. In the past decade, MAbs comprised 20% of all drug approvals by the US Food and Drug Administration (FDA), and today they represent more than half of drugs currently in development.2 Fifteen MAbs have been approved by the FDA over the past decade for asthma, atopic dermatitis (AD), hyperlipidemia, osteoporosis, and migraine prevention.3 This review details what makes MAbs unique and what you should know about them.
The uniqueness of monoclonal antibodies
MAbs are biologics, but not all biologics are MAbs—eg, adalimumab (Humira) is a MAb, but etanercept (Enbrel) is not. MAbs are therapeutic proteins made possible by hybridoma technology used to create an antibody with single specificity.4-6 Monoclonal antibodies differ from small-molecule drugs in structure, dosing, route of administration, manufacturing, metabolism, drug interactions, and elimination (TABLE 17-9).
MAbs can be classified as naked, “without any drug or radioactive material attached to them,” or conjugated, “joined to a chemotherapy drug, radioactive isotope, or toxin.”10 MAbs work in several ways, including competitively inhibiting ligand-receptor binding, receptor blockade, or cell elimination from indirect immune system activities such as antibody-dependent cell-mediated cytotoxicity.11,12
Monoclonal antibody uses in family medicine
Asthma
Several MAbs have been approved for use in severe asthma, including but not limited to: omalizumab (Xolair),13 mepolizumab (Nucala),9,14 and dupilumab (Dupixent).15
Omalizumab is a humanized MAb that prevents IgE antibodies from binding to mast cells and basophils, thereby reducing inflammatory mediators.13 A systematic review found that, compared with placebo, omalizumab used in patients with inadequately controlled moderate-to-severe asthma led to significantly fewer asthma exacerbations (absolute risk reduction [ARR], 16% vs 26%; odds ratio [OR] = 0.55; 95% CI, 0.42-0.60; number needed to treat [NNT] = 10) and fewer hospitalizations (ARR, 0.5% vs 3%; OR = 0.16; 95% CI, 0.06-0.42; NNT = 40).13
Significantly more patients in the omalizumab group were able to withdraw from, or reduce, the dose of ICS. GINA recommends omalizumab for patients with positive skin sensitization, total serum IgE ≥ 30 IU/mL, weight within 30 kg to 150 kg, history of childhood asthma and recent exacerbations, and blood eosinophils ≥ 260/mcL.16 Omalizumab is also approved for use in chronic spontaneous urticaria and nasal polyps.
Mepolizumab
Continue to: Another trial found that...
Another trial found that mepolizumab reduced total OCS doses in patients with severe asthma by 50% without increasing exacerbations or worsening asthma control.18 All 3 anti-IL-5 drugs—including not only mepolizumab, but also benralizumab (Fasenra) and reslizumab (Cinqair)—appear to yield similar improvements. A 2017 systematic review found all anti-IL-5 treatments reduced rates of clinically significant asthma exacerbations (treatment with OCS for ≥ 3 days) by roughly 50% in patients with severe eosinophilic asthma and a history of ≥ 2 exacerbations in the past year.
Dupilumab is a humanized MAb that inhibits IL-4 and IL-13, which influence multiple cell types involved in inflammation (eg, mast cells, eosinophils) and inflammatory mediators (histamine, leukotrienes, cytokines).15 In a recent study of patients with uncontrolled asthma, dupilumab 200 mg every 2 weeks compared with placebo showed a modest reduction in the annualized rate of severe asthma exacerbations (0.46 exacerbations vs 0.87, respectively). Dupilumab was effective in patients with blood eosinophil counts ≥ 150/μL but was ineffective in patients with eosinophil counts < 150/μL.15
For patients ≥ 12 years old with severe eosinophilic asthma, GINA recommends using dupilumab as add-on therapy for an initial trial of 4 months at doses of 200 or 300 mg SC every 2 weeks, with preference for 300 mg SC every 2 weeks for OCS-dependent asthma. Dupilumab is approved for use in AD and chronic rhinosinusitis with nasal polyposis. If a biologic agent is not successful after a 4-month trial, consider a 6- to 12-month trial. If efficacy is still minimal, consider switching to an alternative biologic therapy approved for asthma.16
❯ Asthma: Test your skills
Subjective findings: A 19-year-old man presents to your clinic. He has a history of nasal polyps and allergic asthma. At age 18, he was given a diagnosis of severe persistent asthma. He has shortness of breath during waking hours 4 times per week, and treats each of these episodes with albuterol. He also wakes up about twice a week with shortness of breath and has some limitations in normal activities. He reports missing his prescribed fluticasone/salmeterol 500/50 μg, 1 inhalation bid, only once each month. In the last year, he has had 2 exacerbations requiring oral steroids.
Medications: Albuterol 90 μg, 1-2 inhalations, q6h prn; fluticasone/salmeterol 500/50 μg, 1 inhalation bid; tiotropium 1.25 μg, 2 puffs/d; montelukast 10 mg every morning; prednisone 10 mg/d.
Continue to: Objective data
Objective data: Patient is in no apparent distress and afebrile, and oxygen saturation on room air is 97%. Ht, 70 inches; wt, 75 kg. Labs: IgE, 15 IU/mL; serum eosinophils, 315/μL.
Which MAb would be appropriate for this patient? Given that the patient has a blood eosinophil level ≥ 300/μL and severe exacerbations, adult-onset asthma, nasal polyposis, and maintenance OCS at baseline, it would be reasonable to initiate mepolizumab 100 mg SC every 4 weeks, or dupilumab 600 mg once, then 300 mg SC every 2 weeks. Both agents can be self-administered.
Atopic dermatitis
Two MAbs—dupilumab and tralokinumab (Adbry; inhibits IL-13)—are approved for treatment of AD in adults that is uncontrolled with conventional therapy.15,19 Dupilumab is also approved for children ≥ 6 months old.20 Both MAbs are dosed at 600 mg SC, followed by 300 mg every 2 weeks. Dupilumab was compared with placebo in adult patients who had moderate-to-severe AD inadequately controlled on topical corticosteroids (TCSs), to determine the proportion of patients in each group achieving improvement of either 0 or 1 points or ≥ 2 points in the 5-point Investigator Global Assessment (IGA) score from baseline to 16 weeks.21 Thirty-seven percent of patients receiving dupilumab 300 mg SC weekly and 38% of patients receiving dupilumab 300 mg SC every 2 weeks achieved the primary outcome, compared with 10% of those receiving placebo (P < .001).21 Similar IGA scores were reported when dupilumab was combined with TCS, compared with placebo.22
It would be reasonable to consider dupilumab or tralokinumab in patients with: cutaneous atrophy or hypothalamic-pituitary-adrenal axis suppression with TCS, concerns of malignancy with topical calcineurin inhibitors, or problems with the alternative systemic therapies (cyclosporine-induced hypertension, nephrotoxicity, or immunosuppression; azathioprine-induced malignancy; or methotrexate-induced bone marrow suppression, renal impairment, hepatotoxicity, pneumonitis, or gastrointestinal toxicity).23
A distinct advantage of MAbs over other systemic agents in the management of AD is that MAbs do not require frequent monitoring of blood pressure, renal or liver function, complete blood count with differential, electrolytes, or uric acid. Additionally, MAbs have fewer black box warnings and adverse reactions when compared with other systemic agents.
Continue to: Hyperlipidemia
Hyperlipidemia
Three MAbs are approved for use in hyperlipidemia: the angiopoietin-like protein 3 (ANGPTL3) inhibitor evinacumab (Evkeeza)24 and 2 proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, evolocumab (Repatha)25 and alirocumab (Praluent).26
ANGPTL3 inhibitors block ANGPTL3 and reduce endothelial lipase and lipoprotein lipase activity, which in turn decreases low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglyceride formation. PCSK9 inhibitors prevent PCSK9 from binding to LDL receptors, thereby maintaining the number of active LDL receptors and increasing LDL-C removal.
Evinacumab is indicated for homozygous familial hypercholesterolemia and is administered intravenously every 4 weeks. Evinacumab has not been evaluated for effects on cardiovascular morbidity and mortality.
Evolocumab 140 mg SC every 2 weeks or 420 mg SC monthly has been studied in patients on statin therapy with LDL-C ≥ 70 mg/dL. Patients on evolocumab experienced significantly less of the composite endpoint of cardiovascular death, myocardial infarction (MI), stroke, hospitalization for unstable angina, or coronary revascularization compared with placebo (9.8% vs 11.3%; hazard ratio [HR] = 0.85; 95% CI, 0.79-0.92; NNT = 67.27
Alirocumab 75 mg SC every 2 weeks has also been studied in patients receiving statin therapy with LDL-C ≥ 70 mg/dL. Patients taking alirocumab experienced significantly less of the composite endpoint of death from coronary heart disease, nonfatal MI, ischemic stroke, or hospitalization for unstable angina compared with placebo (9.5% vs 11.1%; HR = 0.85; 95% CI, 0.78-0.93; NNT = 63).
Continue to: According to the 2018...
According to the 2018 AHA Cholesterol Guidelines, PCSK9 inhibitors are indicated for patients receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe) with LDL-C ≥ 70 mg/dL, if they have had multiple atherosclerotic cardiovascular disease (ASCVD) events or 1 major ASCVD event with multiple high-risk conditions (eg, heterozygous familial hypercholesterolemia, history of coronary artery bypass grafting or percutaneous coronary intervention, hypertension, estimated glomerular filtration rate of 15 to 59 mL/min/1.73m2).29 For patients without prior ASCVD events or high-risk conditions who are receiving maximally tolerated LDL-C-lowering therapy (statin and ezetimibe), PCSK9 inhibitors are indicated if the LDL-C remains ≥ 100 mg/dL.
Osteoporosis
The 2 MAbs approved for use in osteoporosis are the receptor activator of nuclear factor kB ligand (RANKL) inhibitor denosumab (Prolia)30 and the sclerostin inhibitor romosozumab (Evenity).31
Denosumab prevents RANKL from binding to the RANK receptor, thereby inhibiting osteoclast formation and decreasing bone resorption. Denosumab is approved for use in women and men who are at high risk of osteoporotic fracture, including those taking OCSs, men receiving androgen deprivation therapy for prostate cancer, and women receiving adjuvant aromatase inhibitor therapy for breast cancer.
In a 3-year randomized trial, denosumab 60 mg SC every 6 months was compared with placebo in postmenopausal women with T-scores < –2.5, but not < –4.0 at the lumbar spine or total hip. Denosumab significantly reduced new radiographic vertebral fractures (2.3% vs 7.2%; risk ratio [RR] = 0.32; 95% CI, 0.26-0.41; NNT = 21), hip fracture (0.7% vs 1.2%), and nonvertebral fracture (6.5% vs 8.0%).32 Denosumab carries an increased risk of multiple vertebral fractures following discontinuation, skin infections, dermatologic reactions, and severe bone, joint, and muscle pain.
Romosozumab inhibits sclerostin, thereby increasing bone formation and, to a lesser degree, decreasing bone resorption. Romosozumab is approved for use in postmenopausal women at high risk for fracture (ie, those with a history of osteoporotic fracture or multiple risk factors for fracture) or in patients who have not benefited from or are intolerant of other therapies. In one study, postmenopausal women with a T-score of –2.5 to –3.5 at the total hip or femoral neck were randomly assigned to receive either romosozumab 210 mg SC or placebo for 12 months, then each group was switched to denosumab 60 mg SC for 12 months. After the first year, prior to initiating denosumab, patients taking romosozumab experienced significantly fewer new vertebral fractures than patients taking placebo (0.5% vs 1.8%; RR = 0.27; 95% CI, 0.16-0.47; NNT = 77); however, there was no significant difference between the 2 groups with nonvertebral fractures (HR = 0.75; 95% CI, 0.53-1.05).33
Continue to: In another study...
In another study, romosozumab 210 mg SC was compared with alendronate 70 mg weekly, followed by alendronate 70 mg weekly in both groups. Over the first 12 months, patients treated with romosozumab saw a significant reduction in the incidence of new vertebral fractures (4% vs 6.3%; RR = 0.63, P < .003; NNT = 44). Patients treated with romosozumab with alendronate added for another 12 months also saw a significant reduction in new incidence of vertebral fractures (6.2% vs 11.9%; RR = 0.52; P < .001; NNT = 18).34 There was a higher risk of cardiovascular events among patients receiving romosozumab compared with those treated with alendronate, so romosozumab should not be used in individuals who have had an MI or stroke within the previous year.34 Denosumab and romosozumab offer an advantage over some bisphosphonates in that they require less frequent dosing and can be used in patients with renal impairment (creatinine clearance < 35 mL/min, in which zoledronic acid is contraindicated and alendronate is not recommended; < 30 mL/min, in which risedronate and ibandronate are not recommended).
Migraine prevention
Four
Erenumab, fremanezumab, and galcanezumab are all available in subcutaneous autoinjectors (or syringe with fremanezumab). Eptinezumab is an intravenous (IV) infusion given every 3 months.
Erenumab is available in both 70-mg and 140-mg dosing options. Fremanezumab can be given as 225 mg monthly or 675 mg quarterly. Galcanezumab has an initial loading dose of 240 mg followed by 120 mg given monthly. Erenumab targets the CGRP receptor; the others target the CGRP ligand. Eptinezumab has 100% bioavailability and reaches maximum serum concentration sooner than the other antagonists (due to its route of administration), but it must be given in an infusion center. Few insurers approve the use of eptinezumab unless a trial of least 1 of the monthly injectables has failed.
There are no head-to-head studies of the medications in this class. Additionally, differing study designs, definitions, statistical analyses, endpoints, and responder-rate calculations make it challenging to compare them directly against one another. At the very least, all of the CGRP MAbs have efficacy comparable to conventional preventive migraine medications such as propranolol, amitriptyline, and topiramate.40
Continue to: The most commonly reported adverse...
The most commonly reported adverse effect for all 4 CGRPs is injection site reaction, which was highest with the quarterly fremanezumab dose (45%).37 Constipation was most notable with the 140-mg dose of erenumab (3%)35; with the other CGRP MAbs it is comparable to that seen with placebo (< 1%).
Erenumab-induced hypertension has been identified in 61 cases reported through the FDA Adverse Event Reporting System (FAERS) as of 2021.41 This was not reported during MAb development programs, nor was it noted during clinical trials. Blood pressure elevation was seen within 1 week of injection in nearly 50% of the cases, and nearly one-third had pre-existing hypertension.41 Due to these findings, the erenumab prescribing information was updated to include hypertension in its warnings and precautions. It is possible that hypertension could be a class effect, although trial data and posthoc studies have yet to bear that out. Since erenumab was the first CGRP antagonist brought to market (May 2018 vs September 2018 for fremanezumab and galcanezumab), it may have accumulated more FAERS reports. Nearly all studies exclude patients with older age, uncontrolled hypertension, and unstable cardiovascular disease, which could impact data.41
Overall, this class of medications is very well tolerated, easy to use (again, excluding eptinezumab), and maintains a low adverse effect profile, giving added value compared with conventional preventive migraine medications.
The American Headache Society recommends a preventive oral therapy for at least 3 months before trying an alternative medication. After treatment failure with at least 2 oral agents, CGRP MAbs are recommended.42 CGRP antagonists offer convenient dosing, bypass gastrointestinal metabolism (which is useful in patients with nausea/vomiting), and have fewer adverse effects than traditional oral medications.
Worth noting. Several newer oral agents have been recently approved for migraine prevention, including atogepant (Qulipta) and rimegepant (Nurtec), which are also CGRP antagonists. Rimegepant is approved for both acute migraine treatment and prevention.
Continue to: Migraine
❯ Migraine: Test your skills
Subjective findings: A 25-year-old woman presents to your clinic for management of episodic migraines with aura. Her baseline average migraine frequency is 9 headache days/month. Her migraines are becoming more frequent despite treatment. She fears IV medication use and avoids hospitals.
History: Hypertension, irritable bowel syndrome with constipation (IBS-C), and depression. The patient is not pregnant or trying to get pregnant.
Medications: Current medications (for previous 4 months) include propranolol 40 mg at bedtime, linaclotide 145 μg/d, citalopram 20 mg/d, and sumatriptan 50 mg prn. Past medications include venlafaxine 150 mg po bid for 5 months.
What would be appropriate for this patient? This patient meets the criteria for using a CGRP antagonist because she has tried 2 preventive treatments for more than 60 to 90 days. Erenumab is not the best option, given the patient’s history of hypertension and IBS-C. The patient fears hospitals and IV medications, making eptinezumab a less-than-ideal choice. Depending on her insurance, fremanezumab or galcanezumab would be good options at this time.
CGRP antagonists have not been studied or evaluated in pregnancy, but if this patient becomes pregnant, a first-line agent for prevention would be propranolol, and a second-line agent would be a tricyclic antidepressant, memantine, or verapamil. Avoid ergotamines and antiepileptics (topiramate or valproate) in pregnancy.43,44
Continue to: The challenges associated with MAbs
The challenges associated with MAbs
MAbs can be expensive (TABLE 2),45 some prohibitively so. On a population scale, biologics account for around 40% of prescription drug spending and may cost 22 times more than small-molecule drugs.46 Estimates in 2016 showed that MAbs comprise $90.2 billion (43%) of the biologic market.46
MAbs also require prior authorization forms to be submitted. Prior authorization criteria vary by state and by insurance plan. In my (ES) experience, submitting letters of medical necessity justifying the need for therapy or expertise in the disease states for which the MAb is being prescribed help your patient get the medication they need.
Expect to see additional MAbs approved in the future. If the costs come down, adoption of these agents into practice will likely increase.
CORRESPONDENCE
Evelyn Sbar, MD, Texas Tech University Health Sciences Center, 1400 South Coulter Street, Suite 5100, Amarillo, TX 79106; [email protected]
1. Rui P, Okeyode T. National Ambulatory Medical Care Survey: 2016 national summary tables. National Center for Health Statistics. Accessed June 15, 2022. www.cdc.gov/nchs/data/ahcd/namcs_summary/2016_namcs_web_tables.pdf
2. IDBS. The future of biologics drug development is today. June 27, 2018. Accessed June 15, 2022. www.idbs.com/blog/2018/06/the-future-of-biologics-drug-development-is-today/
3. Antibody therapeutics approved or in regulatory review in the EU or US. Antibody Society. Accessed June 15, 2022. www.antibodysociety.org/resources/approved-antibodies/
4. FDA. Code of Federal Regulations, Title 21, Chapter I, Subchapter F biologics. March 29, 2022. Accessed June 15, 2022. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=600.3
5. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495-497. doi: 10.1038/256495a0
6. Raejewsky K. The advent and rise of monoclonal antibodies. Nature. November 4, 2019. Accessed June 15, 2022. www.nature.com/articles/d41586-019-02840-w
7. Flovent. Prescribing information. GlaxoSmithKline; 2010. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021433s015lbl.pdf
8. NLM. National Center for Biotechnology Information. PubChem. Method for the preparation of fluticasone and related 17beta-carbothioic esters using a novel carbothioic acid synthesis and novel purification methods. Accessed June 15, 2022. pubchem.ncbi.nlm.nih.gov/patent/WO-0162722-A2
9. Nucala. Prescribing information. GlaxoSmithKline; 2019. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761122s000lbl.pdf
10. Argyriou AA, Kalofonos HP. Recent advances relating to the clinical application of naked monoclonal antibodies in solid tumors. Mol Med. 2009;15:183-191. doi: 10.2119/molmed.2009.00007
11. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548-558. doi: 10.1038/clpt.2008.170
12. Zahavi D, AlDeghaither D, O’Connell A, et al. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1:7-12. doi: 10.1093/abt/tby002
13. Normansell R, Walker S, Milan SJ, et al. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev. 2014:CD003559. doi: 10.1002/14651858.CD003559.pub4
14. Farne HA, Wilson A, Powell C, et al. Anti-IL5 therapies for asthma. Cochrane Database Syst Rev. 2017;9:CD010834. doi: 10.1002/14651858.CD010834.pub3
15. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. 2018;378:2486-2496. doi: 10.1056/NEJMoa1804092
16. GINA. Global strategy for asthma management and prevention. 2022 Difficult-to-treat and severe asthma guide—slide set. Accessed June 23, 2022. https://ginasthma.org/severeasthma/
17. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198-1207. doi: 10.1056/NEJMoa1403290
18. Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189-1197. doi: 10.1056/NEJMoa1403291
19. Adbry. Prescribing information. Leo Pharma Inc; 2021. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761180Orig1s000lbl.pdf
20. Dupixent. Prescribing information. Regeneron Pharmaceuticals; 2022. Accessed October 5, 2022. https://www.regeneron.com/downloads/dupixent_fpi.pdf
21. Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348. doi: 10.1056/NEJMoa1610020
22. Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389:2287-2303. doi: 10.1016/s0140-6736(17)31191-1
23. Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349. doi: 10.1016/j.jaad.2014.03.030
24. Evkeeza. Prescribing information. Regeneron Pharmaceuticals; 2021. Accessed June 24, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
25. Repatha. Prescribing information. Amgen; 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125522s014lbl.pdf
26. Praluent. Prescribing information. Sanofi Aventis and Regeneron Pharmaceuticals. 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125559s002lbl.pdf
27. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713-1722. doi: 10.1056/NEJMoa1615664
28. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097-2107. doi:10.1056/NEJMoa1801174
29. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
30. Prolia. Prescribing information. Amgen; 2010. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2013/125320s094lbl.pdf
31. Evenity. Prescribing information. Amgen; 2019. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761062s000lbl.pdf
32. Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756-765. doi: 10.1056/NEJMoa0809493
33. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375:1532-1543. doi: 10.1056/NEJMoa1607948
34. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377:1417-1427. doi: 10.1056/NEJMoa1708322
35. Aimovig. Prescribing information. Amgen; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761077s000lbl.pdf
36. Vyepti. Prescribing information. Lundbeck Seattle BioPharmaceuticals; 2020. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2020/761119s000lbl.pdf
37. Ajovy. Prescribing information. Teva Pharmaceuticals; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761089s000lbl.pdf
38. Emgality. Prescribing information. Eli Lilly and Co.; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761063s000lbl.pdf
39. Edvinsson L, Haanes KA, Warfvinge K, et al. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338-350. doi: 10.1038/s41582-018-0003-1
40. Vandervorst F. Van Deun L, Van Dycke A, et al. CGRP monoclonal antibodies in migraine: an efficacy and tolerability comparison with standard prophylactic drugs. J Headache Pain. 2021;22:128. doi: 10.1186/s10194-021-01335-2
41. Saely S, Croteau D, Jawidzik L, et al. Hypertension: a new safety risk for patients treated with erenumab. Headache. 2021;61:202-208. doi: 10.1111/head.14051
42. American Headache Society. The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1-18. doi: 10.1111/head.13456
43. Burch R. Headache in pregnancy and the puerperium. Neurol Clin. 2019;37:31-51. doi: 10.1016/j.ncl.2018.09.004
44. Burch R. Epidemiology and treatment of menstrual migraine and migraine during pregnancy and lactation: a narrative review. Headache. 2020;60:200-216. doi: 10.1111/head.13665
45. Lexi-Comp. Lexi-drug database. Accessed April 4, 2022. https://online.lexi.com/lco/action/login
46. Walker N. Biologics: driving force in pharma. Pharma’s Almanac. June 5, 2017. Accessed June 15, 2020. www.pharmasalmanac.com/articles/biologics-driving-force-in-pharma
1. Rui P, Okeyode T. National Ambulatory Medical Care Survey: 2016 national summary tables. National Center for Health Statistics. Accessed June 15, 2022. www.cdc.gov/nchs/data/ahcd/namcs_summary/2016_namcs_web_tables.pdf
2. IDBS. The future of biologics drug development is today. June 27, 2018. Accessed June 15, 2022. www.idbs.com/blog/2018/06/the-future-of-biologics-drug-development-is-today/
3. Antibody therapeutics approved or in regulatory review in the EU or US. Antibody Society. Accessed June 15, 2022. www.antibodysociety.org/resources/approved-antibodies/
4. FDA. Code of Federal Regulations, Title 21, Chapter I, Subchapter F biologics. March 29, 2022. Accessed June 15, 2022. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=600.3
5. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495-497. doi: 10.1038/256495a0
6. Raejewsky K. The advent and rise of monoclonal antibodies. Nature. November 4, 2019. Accessed June 15, 2022. www.nature.com/articles/d41586-019-02840-w
7. Flovent. Prescribing information. GlaxoSmithKline; 2010. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021433s015lbl.pdf
8. NLM. National Center for Biotechnology Information. PubChem. Method for the preparation of fluticasone and related 17beta-carbothioic esters using a novel carbothioic acid synthesis and novel purification methods. Accessed June 15, 2022. pubchem.ncbi.nlm.nih.gov/patent/WO-0162722-A2
9. Nucala. Prescribing information. GlaxoSmithKline; 2019. Accessed June 15, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761122s000lbl.pdf
10. Argyriou AA, Kalofonos HP. Recent advances relating to the clinical application of naked monoclonal antibodies in solid tumors. Mol Med. 2009;15:183-191. doi: 10.2119/molmed.2009.00007
11. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548-558. doi: 10.1038/clpt.2008.170
12. Zahavi D, AlDeghaither D, O’Connell A, et al. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1:7-12. doi: 10.1093/abt/tby002
13. Normansell R, Walker S, Milan SJ, et al. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev. 2014:CD003559. doi: 10.1002/14651858.CD003559.pub4
14. Farne HA, Wilson A, Powell C, et al. Anti-IL5 therapies for asthma. Cochrane Database Syst Rev. 2017;9:CD010834. doi: 10.1002/14651858.CD010834.pub3
15. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. 2018;378:2486-2496. doi: 10.1056/NEJMoa1804092
16. GINA. Global strategy for asthma management and prevention. 2022 Difficult-to-treat and severe asthma guide—slide set. Accessed June 23, 2022. https://ginasthma.org/severeasthma/
17. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198-1207. doi: 10.1056/NEJMoa1403290
18. Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189-1197. doi: 10.1056/NEJMoa1403291
19. Adbry. Prescribing information. Leo Pharma Inc; 2021. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761180Orig1s000lbl.pdf
20. Dupixent. Prescribing information. Regeneron Pharmaceuticals; 2022. Accessed October 5, 2022. https://www.regeneron.com/downloads/dupixent_fpi.pdf
21. Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348. doi: 10.1056/NEJMoa1610020
22. Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389:2287-2303. doi: 10.1016/s0140-6736(17)31191-1
23. Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349. doi: 10.1016/j.jaad.2014.03.030
24. Evkeeza. Prescribing information. Regeneron Pharmaceuticals; 2021. Accessed June 24, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
25. Repatha. Prescribing information. Amgen; 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125522s014lbl.pdf
26. Praluent. Prescribing information. Sanofi Aventis and Regeneron Pharmaceuticals. 2015. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2017/125559s002lbl.pdf
27. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713-1722. doi: 10.1056/NEJMoa1615664
28. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097-2107. doi:10.1056/NEJMoa1801174
29. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:e285-e350. doi: 10.1016/j.jacc.2018.11.003
30. Prolia. Prescribing information. Amgen; 2010. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2013/125320s094lbl.pdf
31. Evenity. Prescribing information. Amgen; 2019. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761062s000lbl.pdf
32. Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756-765. doi: 10.1056/NEJMoa0809493
33. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375:1532-1543. doi: 10.1056/NEJMoa1607948
34. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377:1417-1427. doi: 10.1056/NEJMoa1708322
35. Aimovig. Prescribing information. Amgen; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761077s000lbl.pdf
36. Vyepti. Prescribing information. Lundbeck Seattle BioPharmaceuticals; 2020. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2020/761119s000lbl.pdf
37. Ajovy. Prescribing information. Teva Pharmaceuticals; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761089s000lbl.pdf
38. Emgality. Prescribing information. Eli Lilly and Co.; 2018. Accessed June 24, 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761063s000lbl.pdf
39. Edvinsson L, Haanes KA, Warfvinge K, et al. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338-350. doi: 10.1038/s41582-018-0003-1
40. Vandervorst F. Van Deun L, Van Dycke A, et al. CGRP monoclonal antibodies in migraine: an efficacy and tolerability comparison with standard prophylactic drugs. J Headache Pain. 2021;22:128. doi: 10.1186/s10194-021-01335-2
41. Saely S, Croteau D, Jawidzik L, et al. Hypertension: a new safety risk for patients treated with erenumab. Headache. 2021;61:202-208. doi: 10.1111/head.14051
42. American Headache Society. The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1-18. doi: 10.1111/head.13456
43. Burch R. Headache in pregnancy and the puerperium. Neurol Clin. 2019;37:31-51. doi: 10.1016/j.ncl.2018.09.004
44. Burch R. Epidemiology and treatment of menstrual migraine and migraine during pregnancy and lactation: a narrative review. Headache. 2020;60:200-216. doi: 10.1111/head.13665
45. Lexi-Comp. Lexi-drug database. Accessed April 4, 2022. https://online.lexi.com/lco/action/login
46. Walker N. Biologics: driving force in pharma. Pharma’s Almanac. June 5, 2017. Accessed June 15, 2020. www.pharmasalmanac.com/articles/biologics-driving-force-in-pharma
PRACTICE RECOMMENDATIONS
› Consider anti-immunoglobulin E, anti-interleukin 5, or anti-interleukin 4/interleukin 13 for patients with moderate-to-severe asthma and type 2 airway inflammation. B
› Consider dupilumab for patients with moderate-to-severe atopic dermatitis (with or without topical corticosteroids), or when traditional oral therapies are inadequate or contraindicated. B
› Consider proprotein convertase subtilisin/kexin type 9 inhibitors for patients with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease when maximally tolerated statins or ezetimibe have not lowered low-density lipoprotein cholesterol levels far enough. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Tinea capitis
THE COMPARISON
A Areas of alopecia with erythema and scale in a young Black boy with tinea capitis. He also had an enlarged posterior cervical lymph node (arrow) from this fungal infection.
B White patches of scale from tinea capitis in a young Black boy with no obvious hair loss; however, a potassium hydroxide preparation from the scale was positive for fungus.
C A subtle area of tinea capitis on the scalp of a Latina girl showed comma hairs.

Tinea capitis is a common dermatophyte infection of the scalp in school-aged children. The infection is spread by close contact with infected people or with their personal items, including combs, brushes, pillowcases, and hats, as well as animals. It is uncommon in adults.
Epidemiology
Tinea capitis is the most common fungal infection among school-aged children worldwide.1 In a US-based study of more than 10,000 school-aged children, the prevalence of tinea capitis ranged from 0% to 19.4%, with Black children having the highest rates of infection at 12.9%.2 However, people of all races and ages may develop tinea capitis.3
Tinea capitis most commonly is caused by Trichophyton tonsurans and Microsporum canis. Dermatophyte scalp infections caused by T tonsurans produce fungal spores that may occur within the hair shaft (endothrix) or with fungal elements external to the hair shaft (exothrix) caused by M canis. M canis usually fluoresces an apple green color on Wood lamp examination because of the location of the spores.
Key clinical features
Tinea capitis has a variety of clinical presentations:
- broken hairs that appear as black dots on the scalp
- diffuse scale mimicking seborrheic dermatitis
- well-demarcated annular plaques
- exudate and tenderness caused by inflammation
- scalp pruritus
- occipital scalp lymphadenopathy.
Worth noting
Tinea capitis impacts all patient groups, not just Black patients. In the United States, Black and Hispanic children are most commonly affected.4 Due to a tendency to have dry hair and hair breakage, those with more tightly coiled, textured hair may routinely apply oil and/or grease to the scalp. However, the application of heavy emollients, oils, and grease to camouflage scale contributes to false-negative fungal cultures of the scalp if applied within 1 week of the fungal culture, which may delay diagnosis. If tinea capitis is suspected, occipital lymphadenopathy on physical examination should prompt treatment for tinea capitis, even without a fungal culture.5
Health disparity highlight
A risk factor for tinea capitis is crowded living environments. Some families may live in crowded environments due to economic and housing disparities. This close contact increases the risk for conditions such as tinea capitis.6 Treatment delays may occur due to some cultural practices of applying oils and grease to the hair and scalp, camouflaging the clinical signs of tinea capitis.
1. Gupta AK, Mays RR, Versteeg SG, et al. Tinea capitis in children: a systematic review of management. J Eur Acad Dermatol Venereol. 2018;32:2264-2274. doi: 10.1111/jdv.15088
2. Abdel-Rahman SM, Farrand N, Schuenemann E, et al. The prevalence of infections with Trichophyton tonsurans in schoolchildren: the CAPITIS study. Pediatrics. 2010;125:966-973. doi: 10.1542/peds.2009-2522
3. Silverberg NB, Weinberg JM, DeLeo VA. Tinea capitis: focus on African American women. J Am Acad Dermatol. 2002;46(2 suppl understanding):S120-S124. doi: 10.1067/mjd.2002.120793
4. Alvarez MS, Silverberg NB. Tinea capitis. In: Kelly AP, Taylor SC, eds. Dermatology for Skin of Color. McGraw Hill Medical; 2009:246-255.
5. Nguyen CV, Collier S, Merten AH, et al. Tinea capitis: a singleinstitution retrospective review from 2010 to 2015. Pediatr Dermatol. 2020;37:305-310. doi: 10.1111/pde.14092
6. Emele FE, Oyeka CA. Tinea capitis among primary school children in Anambra state of Nigeria. Mycoses. 2008;51:536-541. doi: 10.1111/j.1439-0507.2008.01507.x
THE COMPARISON
A Areas of alopecia with erythema and scale in a young Black boy with tinea capitis. He also had an enlarged posterior cervical lymph node (arrow) from this fungal infection.
B White patches of scale from tinea capitis in a young Black boy with no obvious hair loss; however, a potassium hydroxide preparation from the scale was positive for fungus.
C A subtle area of tinea capitis on the scalp of a Latina girl showed comma hairs.
Tinea capitis is a common dermatophyte infection of the scalp in school-aged children. The infection is spread by close contact with infected people or with their personal items, including combs, brushes, pillowcases, and hats, as well as animals. It is uncommon in adults.
Epidemiology
Tinea capitis is the most common fungal infection among school-aged children worldwide.1 In a US-based study of more than 10,000 school-aged children, the prevalence of tinea capitis ranged from 0% to 19.4%, with Black children having the highest rates of infection at 12.9%.2 However, people of all races and ages may develop tinea capitis.3
Tinea capitis most commonly is caused by Trichophyton tonsurans and Microsporum canis. Dermatophyte scalp infections caused by T tonsurans produce fungal spores that may occur within the hair shaft (endothrix) or with fungal elements external to the hair shaft (exothrix) caused by M canis. M canis usually fluoresces an apple green color on Wood lamp examination because of the location of the spores.
Key clinical features
Tinea capitis has a variety of clinical presentations:
- broken hairs that appear as black dots on the scalp
- diffuse scale mimicking seborrheic dermatitis
- well-demarcated annular plaques
- exudate and tenderness caused by inflammation
- scalp pruritus
- occipital scalp lymphadenopathy.
Worth noting
Tinea capitis impacts all patient groups, not just Black patients. In the United States, Black and Hispanic children are most commonly affected.4 Due to a tendency to have dry hair and hair breakage, those with more tightly coiled, textured hair may routinely apply oil and/or grease to the scalp. However, the application of heavy emollients, oils, and grease to camouflage scale contributes to false-negative fungal cultures of the scalp if applied within 1 week of the fungal culture, which may delay diagnosis. If tinea capitis is suspected, occipital lymphadenopathy on physical examination should prompt treatment for tinea capitis, even without a fungal culture.5
Health disparity highlight
A risk factor for tinea capitis is crowded living environments. Some families may live in crowded environments due to economic and housing disparities. This close contact increases the risk for conditions such as tinea capitis.6 Treatment delays may occur due to some cultural practices of applying oils and grease to the hair and scalp, camouflaging the clinical signs of tinea capitis.
THE COMPARISON
A Areas of alopecia with erythema and scale in a young Black boy with tinea capitis. He also had an enlarged posterior cervical lymph node (arrow) from this fungal infection.
B White patches of scale from tinea capitis in a young Black boy with no obvious hair loss; however, a potassium hydroxide preparation from the scale was positive for fungus.
C A subtle area of tinea capitis on the scalp of a Latina girl showed comma hairs.
Tinea capitis is a common dermatophyte infection of the scalp in school-aged children. The infection is spread by close contact with infected people or with their personal items, including combs, brushes, pillowcases, and hats, as well as animals. It is uncommon in adults.
Epidemiology
Tinea capitis is the most common fungal infection among school-aged children worldwide.1 In a US-based study of more than 10,000 school-aged children, the prevalence of tinea capitis ranged from 0% to 19.4%, with Black children having the highest rates of infection at 12.9%.2 However, people of all races and ages may develop tinea capitis.3
Tinea capitis most commonly is caused by Trichophyton tonsurans and Microsporum canis. Dermatophyte scalp infections caused by T tonsurans produce fungal spores that may occur within the hair shaft (endothrix) or with fungal elements external to the hair shaft (exothrix) caused by M canis. M canis usually fluoresces an apple green color on Wood lamp examination because of the location of the spores.
Key clinical features
Tinea capitis has a variety of clinical presentations:
- broken hairs that appear as black dots on the scalp
- diffuse scale mimicking seborrheic dermatitis
- well-demarcated annular plaques
- exudate and tenderness caused by inflammation
- scalp pruritus
- occipital scalp lymphadenopathy.
Worth noting
Tinea capitis impacts all patient groups, not just Black patients. In the United States, Black and Hispanic children are most commonly affected.4 Due to a tendency to have dry hair and hair breakage, those with more tightly coiled, textured hair may routinely apply oil and/or grease to the scalp. However, the application of heavy emollients, oils, and grease to camouflage scale contributes to false-negative fungal cultures of the scalp if applied within 1 week of the fungal culture, which may delay diagnosis. If tinea capitis is suspected, occipital lymphadenopathy on physical examination should prompt treatment for tinea capitis, even without a fungal culture.5
Health disparity highlight
A risk factor for tinea capitis is crowded living environments. Some families may live in crowded environments due to economic and housing disparities. This close contact increases the risk for conditions such as tinea capitis.6 Treatment delays may occur due to some cultural practices of applying oils and grease to the hair and scalp, camouflaging the clinical signs of tinea capitis.
1. Gupta AK, Mays RR, Versteeg SG, et al. Tinea capitis in children: a systematic review of management. J Eur Acad Dermatol Venereol. 2018;32:2264-2274. doi: 10.1111/jdv.15088
2. Abdel-Rahman SM, Farrand N, Schuenemann E, et al. The prevalence of infections with Trichophyton tonsurans in schoolchildren: the CAPITIS study. Pediatrics. 2010;125:966-973. doi: 10.1542/peds.2009-2522
3. Silverberg NB, Weinberg JM, DeLeo VA. Tinea capitis: focus on African American women. J Am Acad Dermatol. 2002;46(2 suppl understanding):S120-S124. doi: 10.1067/mjd.2002.120793
4. Alvarez MS, Silverberg NB. Tinea capitis. In: Kelly AP, Taylor SC, eds. Dermatology for Skin of Color. McGraw Hill Medical; 2009:246-255.
5. Nguyen CV, Collier S, Merten AH, et al. Tinea capitis: a singleinstitution retrospective review from 2010 to 2015. Pediatr Dermatol. 2020;37:305-310. doi: 10.1111/pde.14092
6. Emele FE, Oyeka CA. Tinea capitis among primary school children in Anambra state of Nigeria. Mycoses. 2008;51:536-541. doi: 10.1111/j.1439-0507.2008.01507.x
1. Gupta AK, Mays RR, Versteeg SG, et al. Tinea capitis in children: a systematic review of management. J Eur Acad Dermatol Venereol. 2018;32:2264-2274. doi: 10.1111/jdv.15088
2. Abdel-Rahman SM, Farrand N, Schuenemann E, et al. The prevalence of infections with Trichophyton tonsurans in schoolchildren: the CAPITIS study. Pediatrics. 2010;125:966-973. doi: 10.1542/peds.2009-2522
3. Silverberg NB, Weinberg JM, DeLeo VA. Tinea capitis: focus on African American women. J Am Acad Dermatol. 2002;46(2 suppl understanding):S120-S124. doi: 10.1067/mjd.2002.120793
4. Alvarez MS, Silverberg NB. Tinea capitis. In: Kelly AP, Taylor SC, eds. Dermatology for Skin of Color. McGraw Hill Medical; 2009:246-255.
5. Nguyen CV, Collier S, Merten AH, et al. Tinea capitis: a singleinstitution retrospective review from 2010 to 2015. Pediatr Dermatol. 2020;37:305-310. doi: 10.1111/pde.14092
6. Emele FE, Oyeka CA. Tinea capitis among primary school children in Anambra state of Nigeria. Mycoses. 2008;51:536-541. doi: 10.1111/j.1439-0507.2008.01507.x

IVIG proves effective for dermatomyositis in phase 3 trial
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.

“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
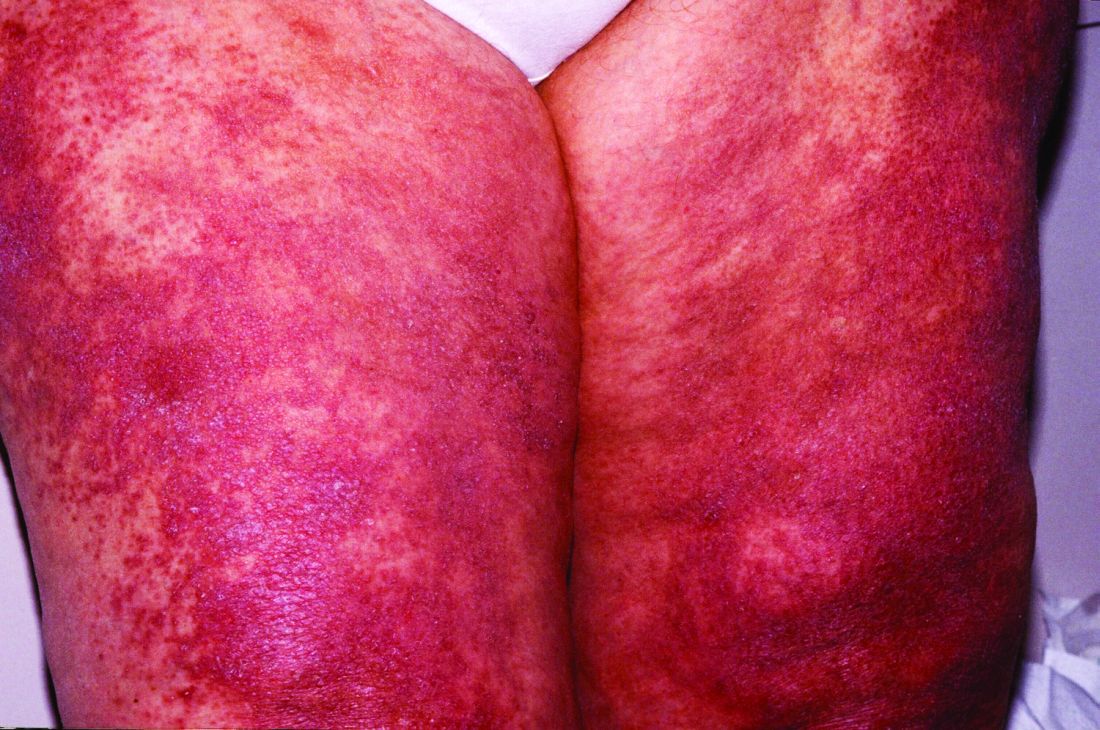
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.
“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
With use of intravenous immunoglobulin for the treatment of adults with dermatomyositis, a significantly higher percentage of patients experienced at least minimal improvement in disease activity in comparison with placebo in the first-ever phase 3 trial of the blood-product therapy for the condition.
Until this trial, published in the New England Journal of Medicine, there had not been an extensive evaluation of IVIG for the treatment of dermatomyositis, the study’s authors noted.
Glucocorticoids are typically offered as first-line therapy, followed by various immunosuppressants. IVIG is composed of purified liquid IgG concentrates from human plasma. It has been prescribed off label as second- or third-line therapy for dermatomyositis, usually along with immunosuppressive drugs. In European guidelines, it has been recommended as a glucocorticoid-sparing agent for patients with this condition.
“The study provides support that IVIG is effective in treating the signs and symptoms of patients with dermatomyositis, at least in the short term,” said David Fiorentino, MD, PhD, professor of dermatology and associate residency program director at Stanford Health Care, Stanford, California, who was not involved in the study.
“IVIG appears to be effective for patients with any severity level and works relatively quickly [within 1 month of therapy],” he added. “IVIG is effective in treating both the muscle symptoms as well as the rash of dermatomyositis, which is important, as both organ systems can cause significant patient morbidity in this disease.”
Time to improvement was shorter with IVIG than with placebo (a median of 35 days vs. 115 days), said Kathryn H. Dao, MD, associate professor in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, who was not involved in the study.
The study’s greatest strengths are its international, multicenter, randomized, placebo-controlled design, Dr. Dao said. In addition, “these patients were permitted to be on background medicines that we typically use in real-world situations.”
Study methodology
Researchers led by Rohit Aggarwal, MD, of the division of rheumatology and clinical immunology at the University of Pittsburgh, recruited patients aged 18-80 years with active dermatomyositis. Individuals were randomly assigned in a 1:1 ratio to receive either IVIG at a dose of 2.0 g/kg of body weight or placebo (0.9% sodium chloride) every 4 weeks for 16 weeks.
Those who were administered placebo and those who did not experience confirmed clinical deterioration while receiving IVIG could participate in an open-label extension phase for another 24 weeks.
The primary endpoint was a response, defined as a Total Improvement Score (TIS) of at least 20 (indicating at least minimal improvement) at week 16 and no confirmed deterioration up to week 16. The TIS is a weighted composite score that reflects the change in a core set of six measures of myositis activity over time. Scores span from 0 to 100, with higher scores indicating more significant improvement.
Secondary endpoints
Key secondary endpoints included moderate improvement (TIS ≥ 40) and major improvement (TIS ≥ 60) and change in score on the Cutaneous Dermatomyositis Disease Area and Severity Index.
A total of 95 patients underwent randomization; 47 patients received IVIG and 48 received placebo. At 16 weeks, a TIS of at least 20 occurred in 37 of 47 (79%) patients who received IVIG and in 21 of 48 (44%) patients with placebo (difference, 35%; 95% confidence interval, 17%-53%; P < .001).
The results with respect to the secondary endpoints, including at least moderate improvement and major improvement, were generally in the same direction as the results of the primary endpoint analysis, except for change in creatine kinase (CK) level (an individual core measure of the TIS), which did not differ meaningfully between the two groups.
Adverse events
Over the course of 40 weeks, 282 treatment-related adverse events were documented among patients who received IVIG. Headache was experienced by 42%, pyrexia by 19%, and nausea by 16%. Nine serious adverse events occurred and were believed to be associated with IVIG, including six thromboembolic events.
Despite the favorable outcome observed with IVIG, in an editorial that accompanied the study, Anthony A. Amato, MD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, noted that “most of the core components of the TIS are subjective. Because of the high percentage of patients who had a response with placebo, large numbers of patients will be needed in future trials to show a significant difference between trial groups, or the primary endpoint would need to be set higher (e.g., a TIS of ≥40).”
Dr. Dao thought it was significant that the study proactively assessed patients for venous thrombotic events (VTEs) after each infusion. There were eight events in six patients who received IVIG. “Of interest and possibly practice changing is the finding that slowing the IVIG infusion rate from 0.12 to 0.04 mL/kg per minute reduced the incidence of VTEs from 1.54/100 patient-months to 0.54/100 patient-months,” she said. “This is important, as it informs clinicians that IVIG infusion rates should be slower for patients with active dermatomyositis to reduce the risk for blood clots.”
Study weaknesses
A considerable proportion of patients with dermatomyositis do not have clinical muscle involvement but do have rash and do not substantially differ in any other ways from those with classic dermatomyositis, Dr. Fiorentino said.
“These patients were not eligible to enter the trial, and so we have no data on the efficacy of IVIG in this population,” he said. “Unfortunately, these patients might now be denied insurance reimbursement for IVIG therapy, given that they are not part of the indicated patient population in the label.”
In addition, there is limited information about Black, Asian, or Hispanic patients because few of those patients participated in the study. That is also the case for patients younger than 18, which for this disease is relevant because incidence peaks in younger patients (juvenile dermatomyositis), Dr. Fiorentino noted.
Among the study’s weaknesses, Dr. Dao noted that more than 70% of participants were women. The study was short in duration, fewer than half of patients underwent muscle biopsy to confirm myositis, and only two thirds of patients underwent electromyography/nerve conduction studies to show evidence of myositis. There was a high placebo response (44%), the CK values were not high at the start of the trial, and they did not change with treatment.
No analysis was performed to evaluate the efficacy of IVIG across dermatomyositis subgroups – defined by autoantibodies – but the study likely was not powered to do so. These subgroups might respond differently to IVIG, yielding important information, Fiorentino said.
The study provided efficacy data for only one formulation of IVIG, Octagam 10%, which was approved for dermatomyositis by the Food and Drug Administration in 2021 on the basis of this trial. However, in the United States, patients with dermatomyositis are treated with multiple brands of IVIG. “The decision around IVIG brand is largely determined by third-party payers, and for the most part, the different brands are used interchangeably from the standpoint of the treating provider,” Dr. Fiorentino said. “This will likely continue to be the case, as the results of this study are generally being extrapolated to all brands of IVIG.”
Multiple IVIG brands that have been used for immune-mediated diseases differ in concentration, content of IgA, sugar concentration, additives, and preparations (for example, the need for reconstitution vs. being ready to use), Dr. Dao said. Octagam 10% is the only brand approved by the FDA for adult dermatomyositis; hence, cost can be an issue for patients if other brands are used off label. The typical cost of IVIG is $100-$400 per gram; a typical course of treatment is estimated to be $30,000-$40,000 per month. “However, if Octagam is not available or a patient has a reaction to it, clinicians may use other IVIG brands as deemed medically necessary to treat their patients,” she said.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Some of the coauthors were employees of Octapharma or had financial relationships with the company. Dr. Dao disclosed no relevant financial relationships. Dr. Fiorentino has conducted sponsored research for Pfizer and Argenyx, has received research funding from Serono, and is a paid adviser to Bristol-Myers Squibb, Janssen, Acelyrin, and Corbus.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
How do patients with chronic urticaria fare during pregnancy?
In addition, the rates of preterm births and medical problems of newborns in patients with CU are similar to those of the normal population and not linked to treatment used during pregnancy.
Those are the key findings from an analysis of new data from PREG-CU, an international, multicenter study of the Urticaria Centers of Reference and Excellence (UCARE) network. Results from the first PREG-CU analysis published in 2021 found that CU improved in about half of patients with CU during pregnancy. “However, two in five patients reported acute exacerbations of CU especially at the beginning and end of pregnancy,” investigators led by Emek Kocatürk, MD, of the department of dermatology and UCARE at Koç University School of Medicine, Istanbul, wrote in the new study, recently published in the Journal of the European Academy of Dermatology and Venereology.

“In addition, 1 in 10 pregnant CU patients required urticaria emergency care and 1 of 6 had angioedema during pregnancy,” they said. Risk factors for worsening CU during pregnancy, they added, were “mild disease and no angioedema before pregnancy, not taking treatment before pregnancy, chronic inducible urticaria, CU worsening during a previous pregnancy, stress as a driver of exacerbations, and treatment during pregnancy.”
Analysis involved 288 pregnant women
To optimize treatment of CU during pregnancy and to better understand how treatment affects pregnancy outcomes, the researchers analyzed 288 pregnancies in 288 women with CU from 13 countries and 21 centers worldwide. Their mean age at pregnancy was 32.1 years, and their mean duration of CU was 84.9 months. Prior to pregnancy, 35.7% of patients rated the severity of their CU symptoms as mild, 34.2% rated it as moderate, and 29.7% rated it as severe.
The researchers found that during pregnancy, 60% of patients used urticaria medication, including standard-dose second-generation H1-antihistamines (35.1%), first-generation H1-antihistamines (7.6%), high-dose second-generation H1-antihistamines (5.6%), and omalizumab (5.6%). The preterm birth rate was 10.2%, which was similar between patients who did and did not receive treatment during pregnancy (11.6% vs. 8.7%, respectively; P = .59).
On multivariate logistic regression, two predictors for preterm birth emerged: giving birth to twins (a 13.3-fold increased risk; P = .016) and emergency referrals for CU (a 4.3-fold increased risk; P =.016). The cesarean delivery rate was 51.3%, and more than 90% of newborns were healthy at birth. There was no link between any patient or disease characteristics or treatments and medical problems at birth.
In other findings, 78.8% of women with CU breastfed their babies. Of the 58 patients who did not breastfeed, 20.7% indicated severe urticaria/angioedema and/or taking medications as the main reason for not breastfeeding.
“Most CU patients use treatment during pregnancy and such treatments, especially second generation H1 antihistamines, seem to be safe during pregnancy regardless of the trimester,” the researchers concluded. “Outcomes of pregnancy in patients with CU were similar compared to the general population and not linked to treatment used during pregnancy. Notably, emergency referral for CU was an independent risk factor for preterm birth,” and the high cesarean delivery rate was “probably linked to comorbidities associated with the disease,” they added. “Overall, these findings suggest that patients should continue their treatments using an individualized dose to provide optimal symptom control.”
International guidelines
The authors noted that international guidelines for the management of urticaria published in 2022 suggest that modern second-generation H1-antihistamines should be used for pregnant patients, preferably loratadine with a possible extrapolation to desloratadine, cetirizine, or levocetirizine.
“Similarly, in this population, we found that cetirizine and loratadine were the most commonly used antihistamines, followed by levocetirizine and fexofenadine,” Dr. Kocatürk and colleagues wrote.
“Guidelines also suggest that the use of first-generation H1-antihistamines should be avoided given their sedative effects; but if these are to be given, it would be wise to know that use of first-generation H1-antihistamines immediately before parturition could cause respiratory depression and other adverse effects in the neonate,” they added, noting that chlorpheniramine and diphenhydramine are the first-generation H1-antihistamines with the greatest evidence of safety in pregnancy.
They acknowledged certain limitations of the analysis, including its retrospective design and the fact that there were no data on low birth weight, small for gestational age, or miscarriage rates. In addition, disease activity or severity during pregnancy and after birth were not monitored.
Asked to comment on these results, Raj Chovatiya, MD, PhD, who directs the center for eczema and itch in the department of dermatology at Northwestern University, Chicago, noted that despite a higher prevalence of CU among females compared with males, very little is known about how the condition is managed during pregnancy. “This retrospective study shows that most patients continue to utilize CU treatment during pregnancy (primarily second-generation antihistamines), with similar birth outcomes as the general population,” he said. “Interestingly, cesarean rates were higher among mothers with CU, and emergency CU referral was a risk factor for preterm birth. While additional prospective studies are needed, these results suggest that CU patients should be carefully managed, particularly during pregnancy, when treatment should be optimized.”
Dr. Kocatürk reported having received personal fees from Novartis, Ibrahim Etem-Menarini, and Sanofi, outside the submitted work. Many coauthors reported having numerous financial disclosures. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arcutis, Arena, Incyte, Pfizer, Regeneron, and Sanofi Genzyme.
In addition, the rates of preterm births and medical problems of newborns in patients with CU are similar to those of the normal population and not linked to treatment used during pregnancy.
Those are the key findings from an analysis of new data from PREG-CU, an international, multicenter study of the Urticaria Centers of Reference and Excellence (UCARE) network. Results from the first PREG-CU analysis published in 2021 found that CU improved in about half of patients with CU during pregnancy. “However, two in five patients reported acute exacerbations of CU especially at the beginning and end of pregnancy,” investigators led by Emek Kocatürk, MD, of the department of dermatology and UCARE at Koç University School of Medicine, Istanbul, wrote in the new study, recently published in the Journal of the European Academy of Dermatology and Venereology.
“In addition, 1 in 10 pregnant CU patients required urticaria emergency care and 1 of 6 had angioedema during pregnancy,” they said. Risk factors for worsening CU during pregnancy, they added, were “mild disease and no angioedema before pregnancy, not taking treatment before pregnancy, chronic inducible urticaria, CU worsening during a previous pregnancy, stress as a driver of exacerbations, and treatment during pregnancy.”
Analysis involved 288 pregnant women
To optimize treatment of CU during pregnancy and to better understand how treatment affects pregnancy outcomes, the researchers analyzed 288 pregnancies in 288 women with CU from 13 countries and 21 centers worldwide. Their mean age at pregnancy was 32.1 years, and their mean duration of CU was 84.9 months. Prior to pregnancy, 35.7% of patients rated the severity of their CU symptoms as mild, 34.2% rated it as moderate, and 29.7% rated it as severe.
The researchers found that during pregnancy, 60% of patients used urticaria medication, including standard-dose second-generation H1-antihistamines (35.1%), first-generation H1-antihistamines (7.6%), high-dose second-generation H1-antihistamines (5.6%), and omalizumab (5.6%). The preterm birth rate was 10.2%, which was similar between patients who did and did not receive treatment during pregnancy (11.6% vs. 8.7%, respectively; P = .59).
On multivariate logistic regression, two predictors for preterm birth emerged: giving birth to twins (a 13.3-fold increased risk; P = .016) and emergency referrals for CU (a 4.3-fold increased risk; P =.016). The cesarean delivery rate was 51.3%, and more than 90% of newborns were healthy at birth. There was no link between any patient or disease characteristics or treatments and medical problems at birth.
In other findings, 78.8% of women with CU breastfed their babies. Of the 58 patients who did not breastfeed, 20.7% indicated severe urticaria/angioedema and/or taking medications as the main reason for not breastfeeding.
“Most CU patients use treatment during pregnancy and such treatments, especially second generation H1 antihistamines, seem to be safe during pregnancy regardless of the trimester,” the researchers concluded. “Outcomes of pregnancy in patients with CU were similar compared to the general population and not linked to treatment used during pregnancy. Notably, emergency referral for CU was an independent risk factor for preterm birth,” and the high cesarean delivery rate was “probably linked to comorbidities associated with the disease,” they added. “Overall, these findings suggest that patients should continue their treatments using an individualized dose to provide optimal symptom control.”
International guidelines
The authors noted that international guidelines for the management of urticaria published in 2022 suggest that modern second-generation H1-antihistamines should be used for pregnant patients, preferably loratadine with a possible extrapolation to desloratadine, cetirizine, or levocetirizine.
“Similarly, in this population, we found that cetirizine and loratadine were the most commonly used antihistamines, followed by levocetirizine and fexofenadine,” Dr. Kocatürk and colleagues wrote.
“Guidelines also suggest that the use of first-generation H1-antihistamines should be avoided given their sedative effects; but if these are to be given, it would be wise to know that use of first-generation H1-antihistamines immediately before parturition could cause respiratory depression and other adverse effects in the neonate,” they added, noting that chlorpheniramine and diphenhydramine are the first-generation H1-antihistamines with the greatest evidence of safety in pregnancy.
They acknowledged certain limitations of the analysis, including its retrospective design and the fact that there were no data on low birth weight, small for gestational age, or miscarriage rates. In addition, disease activity or severity during pregnancy and after birth were not monitored.
Asked to comment on these results, Raj Chovatiya, MD, PhD, who directs the center for eczema and itch in the department of dermatology at Northwestern University, Chicago, noted that despite a higher prevalence of CU among females compared with males, very little is known about how the condition is managed during pregnancy. “This retrospective study shows that most patients continue to utilize CU treatment during pregnancy (primarily second-generation antihistamines), with similar birth outcomes as the general population,” he said. “Interestingly, cesarean rates were higher among mothers with CU, and emergency CU referral was a risk factor for preterm birth. While additional prospective studies are needed, these results suggest that CU patients should be carefully managed, particularly during pregnancy, when treatment should be optimized.”
Dr. Kocatürk reported having received personal fees from Novartis, Ibrahim Etem-Menarini, and Sanofi, outside the submitted work. Many coauthors reported having numerous financial disclosures. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arcutis, Arena, Incyte, Pfizer, Regeneron, and Sanofi Genzyme.
In addition, the rates of preterm births and medical problems of newborns in patients with CU are similar to those of the normal population and not linked to treatment used during pregnancy.
Those are the key findings from an analysis of new data from PREG-CU, an international, multicenter study of the Urticaria Centers of Reference and Excellence (UCARE) network. Results from the first PREG-CU analysis published in 2021 found that CU improved in about half of patients with CU during pregnancy. “However, two in five patients reported acute exacerbations of CU especially at the beginning and end of pregnancy,” investigators led by Emek Kocatürk, MD, of the department of dermatology and UCARE at Koç University School of Medicine, Istanbul, wrote in the new study, recently published in the Journal of the European Academy of Dermatology and Venereology.
“In addition, 1 in 10 pregnant CU patients required urticaria emergency care and 1 of 6 had angioedema during pregnancy,” they said. Risk factors for worsening CU during pregnancy, they added, were “mild disease and no angioedema before pregnancy, not taking treatment before pregnancy, chronic inducible urticaria, CU worsening during a previous pregnancy, stress as a driver of exacerbations, and treatment during pregnancy.”
Analysis involved 288 pregnant women
To optimize treatment of CU during pregnancy and to better understand how treatment affects pregnancy outcomes, the researchers analyzed 288 pregnancies in 288 women with CU from 13 countries and 21 centers worldwide. Their mean age at pregnancy was 32.1 years, and their mean duration of CU was 84.9 months. Prior to pregnancy, 35.7% of patients rated the severity of their CU symptoms as mild, 34.2% rated it as moderate, and 29.7% rated it as severe.
The researchers found that during pregnancy, 60% of patients used urticaria medication, including standard-dose second-generation H1-antihistamines (35.1%), first-generation H1-antihistamines (7.6%), high-dose second-generation H1-antihistamines (5.6%), and omalizumab (5.6%). The preterm birth rate was 10.2%, which was similar between patients who did and did not receive treatment during pregnancy (11.6% vs. 8.7%, respectively; P = .59).
On multivariate logistic regression, two predictors for preterm birth emerged: giving birth to twins (a 13.3-fold increased risk; P = .016) and emergency referrals for CU (a 4.3-fold increased risk; P =.016). The cesarean delivery rate was 51.3%, and more than 90% of newborns were healthy at birth. There was no link between any patient or disease characteristics or treatments and medical problems at birth.
In other findings, 78.8% of women with CU breastfed their babies. Of the 58 patients who did not breastfeed, 20.7% indicated severe urticaria/angioedema and/or taking medications as the main reason for not breastfeeding.
“Most CU patients use treatment during pregnancy and such treatments, especially second generation H1 antihistamines, seem to be safe during pregnancy regardless of the trimester,” the researchers concluded. “Outcomes of pregnancy in patients with CU were similar compared to the general population and not linked to treatment used during pregnancy. Notably, emergency referral for CU was an independent risk factor for preterm birth,” and the high cesarean delivery rate was “probably linked to comorbidities associated with the disease,” they added. “Overall, these findings suggest that patients should continue their treatments using an individualized dose to provide optimal symptom control.”
International guidelines
The authors noted that international guidelines for the management of urticaria published in 2022 suggest that modern second-generation H1-antihistamines should be used for pregnant patients, preferably loratadine with a possible extrapolation to desloratadine, cetirizine, or levocetirizine.
“Similarly, in this population, we found that cetirizine and loratadine were the most commonly used antihistamines, followed by levocetirizine and fexofenadine,” Dr. Kocatürk and colleagues wrote.
“Guidelines also suggest that the use of first-generation H1-antihistamines should be avoided given their sedative effects; but if these are to be given, it would be wise to know that use of first-generation H1-antihistamines immediately before parturition could cause respiratory depression and other adverse effects in the neonate,” they added, noting that chlorpheniramine and diphenhydramine are the first-generation H1-antihistamines with the greatest evidence of safety in pregnancy.
They acknowledged certain limitations of the analysis, including its retrospective design and the fact that there were no data on low birth weight, small for gestational age, or miscarriage rates. In addition, disease activity or severity during pregnancy and after birth were not monitored.
Asked to comment on these results, Raj Chovatiya, MD, PhD, who directs the center for eczema and itch in the department of dermatology at Northwestern University, Chicago, noted that despite a higher prevalence of CU among females compared with males, very little is known about how the condition is managed during pregnancy. “This retrospective study shows that most patients continue to utilize CU treatment during pregnancy (primarily second-generation antihistamines), with similar birth outcomes as the general population,” he said. “Interestingly, cesarean rates were higher among mothers with CU, and emergency CU referral was a risk factor for preterm birth. While additional prospective studies are needed, these results suggest that CU patients should be carefully managed, particularly during pregnancy, when treatment should be optimized.”
Dr. Kocatürk reported having received personal fees from Novartis, Ibrahim Etem-Menarini, and Sanofi, outside the submitted work. Many coauthors reported having numerous financial disclosures. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arcutis, Arena, Incyte, Pfizer, Regeneron, and Sanofi Genzyme.
FROM JEADV

Velvety brown lesion

Dermoscopy revealed a uniform, sharply demarcated, slightly scaly lesion on a background of occasional scale and solar-damaged skin. This appearance, paired with the absence of abnormal blood vessels or suspicious, irregular pigmentation, pointed to a diagnosis of benign lichenoid keratosis also known as lichenoid keratosis (LK) or lichen planus-like keratosis. (It’s worth noting that in some cases, a dermoscopic evaluation will reveal blue-grey dots rather than the uniform, velvety brown pigmentation that was seen here.)
LK is a benign reactive inflammatory lesion that usually manifests as a solitary lesion in middle age. LKs can be found on the trunk or lower extremities. As the alternative name “lichen planus-like keratosis” implies, the lesions can be purple, polygonal, raised, and have stria. The etiology is unknown but thought to be a reaction to a lentigo or another lesion, resulting in an inflammatory infiltrate.1
If dermoscopic evaluation of the lesion is unclear, biopsy is warranted. Maor et al1 reported the pathology results of 263 consecutive patients with a histologic diagnosis of LK. Of those cases, 47% were clinically thought to be basal cell carcinoma (BCC) and 18% were submitted with a diagnosis of seborrheic keratosis.1 The high rate of concern for BCC and not listing a diagnosis of LK may have been the result of clinicians doing biopsies on the atypical lesions and clinically following the typical banal lesions.
At the patient’s request, he was given a written list of the diagnoses of his various skin lesions and advised that his LK was benign and did not require treatment. He was advised to continue coming in for serial skin examinations and report any concerning lesions in the interim.
Image and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
1. Maor D, Ondhia C, Yu LL, et al. Lichenoid keratosis is frequently misdiagnosed as basal cell carcinoma. Clin Exp Dermatol. 2017;42:663-666. doi: 10.1111/ced.13178
Dermoscopy revealed a uniform, sharply demarcated, slightly scaly lesion on a background of occasional scale and solar-damaged skin. This appearance, paired with the absence of abnormal blood vessels or suspicious, irregular pigmentation, pointed to a diagnosis of benign lichenoid keratosis also known as lichenoid keratosis (LK) or lichen planus-like keratosis. (It’s worth noting that in some cases, a dermoscopic evaluation will reveal blue-grey dots rather than the uniform, velvety brown pigmentation that was seen here.)
LK is a benign reactive inflammatory lesion that usually manifests as a solitary lesion in middle age. LKs can be found on the trunk or lower extremities. As the alternative name “lichen planus-like keratosis” implies, the lesions can be purple, polygonal, raised, and have stria. The etiology is unknown but thought to be a reaction to a lentigo or another lesion, resulting in an inflammatory infiltrate.1
If dermoscopic evaluation of the lesion is unclear, biopsy is warranted. Maor et al1 reported the pathology results of 263 consecutive patients with a histologic diagnosis of LK. Of those cases, 47% were clinically thought to be basal cell carcinoma (BCC) and 18% were submitted with a diagnosis of seborrheic keratosis.1 The high rate of concern for BCC and not listing a diagnosis of LK may have been the result of clinicians doing biopsies on the atypical lesions and clinically following the typical banal lesions.
At the patient’s request, he was given a written list of the diagnoses of his various skin lesions and advised that his LK was benign and did not require treatment. He was advised to continue coming in for serial skin examinations and report any concerning lesions in the interim.
Image and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
Dermoscopy revealed a uniform, sharply demarcated, slightly scaly lesion on a background of occasional scale and solar-damaged skin. This appearance, paired with the absence of abnormal blood vessels or suspicious, irregular pigmentation, pointed to a diagnosis of benign lichenoid keratosis also known as lichenoid keratosis (LK) or lichen planus-like keratosis. (It’s worth noting that in some cases, a dermoscopic evaluation will reveal blue-grey dots rather than the uniform, velvety brown pigmentation that was seen here.)
LK is a benign reactive inflammatory lesion that usually manifests as a solitary lesion in middle age. LKs can be found on the trunk or lower extremities. As the alternative name “lichen planus-like keratosis” implies, the lesions can be purple, polygonal, raised, and have stria. The etiology is unknown but thought to be a reaction to a lentigo or another lesion, resulting in an inflammatory infiltrate.1
If dermoscopic evaluation of the lesion is unclear, biopsy is warranted. Maor et al1 reported the pathology results of 263 consecutive patients with a histologic diagnosis of LK. Of those cases, 47% were clinically thought to be basal cell carcinoma (BCC) and 18% were submitted with a diagnosis of seborrheic keratosis.1 The high rate of concern for BCC and not listing a diagnosis of LK may have been the result of clinicians doing biopsies on the atypical lesions and clinically following the typical banal lesions.
At the patient’s request, he was given a written list of the diagnoses of his various skin lesions and advised that his LK was benign and did not require treatment. He was advised to continue coming in for serial skin examinations and report any concerning lesions in the interim.
Image and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
1. Maor D, Ondhia C, Yu LL, et al. Lichenoid keratosis is frequently misdiagnosed as basal cell carcinoma. Clin Exp Dermatol. 2017;42:663-666. doi: 10.1111/ced.13178
1. Maor D, Ondhia C, Yu LL, et al. Lichenoid keratosis is frequently misdiagnosed as basal cell carcinoma. Clin Exp Dermatol. 2017;42:663-666. doi: 10.1111/ced.13178

Dupilumab study outlines benefits, safety profile in infants, preschoolers with atopic dermatitis
at 31 treatment centers in North America and Europe.
Children younger than 6 years with moderate to severe AD have few options if their symptoms are uncontrolled with topical therapies, and persistent itchiness has a negative impact on quality of life for patients and families, Amy S. Paller, MD, professor and chair of dermatology, and professor of pediatrics at Northwestern University, Chicago, and colleagues wrote in the study, published in the Lancet.

The study was the basis of the Food and Drug Administration expanded approval of dupilumab in June 2022, to include children aged 6 months to 5 years with moderate to severe AD, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Regulatory submission for this age group is under review by the European Medicines Agency, and by regulatory authorities in other countries, according to the manufacturers.
Dupilumab (Dupixent), which inhibits the signaling of the interleukin-4 and IL-13 pathways, was first approved in 2017 for treating adults with moderate to severe AD.
“There has not been a biologic approved before at such a young age, and for such a common disease,” Dr. Paller said in an interview. “This is the drug that has revolutionized care of the most common inflammatory skin disease in children, and this is the pivotal study that brought it to market for the youngest children who suffer from the severe forms.”
The study also sets a precedent for a lower threshold for starting systemic medication in young children for treating moderate to severe disease given the absence of severe side effects and no need for lab monitoring, Dr. Paller noted. However, dupilumab will also be closely watched “for both impact on the developing immune system and the possibility that it will alter the long-term course of the eczema and the development of allergic comorbidities, such as lowering the risk of developing asthma, GI, allergy, and possibly other conditions.”
In the study, the researchers randomized 83 children aged 6 months up to 6 years to treatment with dupilumab, administered subcutaneously, and 79 to placebo every 4 weeks for 16 weeks; both groups also received topical corticosteroids. Dosage of dupilumab was based on body weight; those with a body weight of 5-15 kg received 200 mg, while those with a body weight of 15-30 kg received 300 mg. The primary endpoint was the proportion of patients with clear or almost clear skin at 16 weeks, defined as scores of 0 or 1 on the Investigator’s Global Assessment.
After 16 weeks, 28% of dupilumab patients met the primary endpoint versus 4% of those on placebo (P < .0001). In addition, 53% of dupilumab patients met the key secondary endpoint of a 75% improvement from baseline in Eczema Area and Severity Index, compared with 11% of patients on placebo (P < .0001). Treatment with dupilumab also resulted in significantly greater improvements in pruritus and skin pain, and sleep quality, as well as improved quality of life for patients and their caregivers, the authors reported.
Overall, adverse event rates were slightly lower in the dupilumab-treated patients, compared with patients on placebo (64% vs. 74%); there were no adverse events related to dupilumab that were serious or resulted in treatment discontinuation. Treatment-emergent adverse effects that were reported in 3% or more of patients and affected more of those on dupilumab than those on placebo included molluscum contagiosum (5% vs. 3%), viral gastroenteritis (4% vs. 0), rhinorrhea (5% vs. 1%), dental caries (5% vs. 0), and conjunctivitis (4% vs 0).
The rate of skin infections among the children on dupilumab was 12% vs. 24% among those on placebo.
Severe and treatment-related adverse events also were similar in both subgroups of body weight.
The findings were limited by the small number of patients younger than 2 years and the lack of study sites outside of North America and Europe, the researchers noted. However, the results were strengthened by the randomized, double-blind design and use of background topical therapy to provide a real-world safety and efficacy assessment in a very young population.
Overcoming injection issues
The safety profile for dupilumab, which is of the highest importance, “did not surprise me at all,” Dr. Paller said in an interview. “My only surprise is that the placebo injections actually led to more injection site reactions than [with] dupilumab, but numbers were quite low in both groups.” (Rates were 2% among those on dupilumab and 3% among those on placebo.)
The major barrier to the use of dupilumab in clinical practice is the requirement for injection, which, she explained, can be “unbearable for some young children, and thus becomes impossible for parents because of lack of cooperation and their intensified concern about giving the injection,” because of their child’s response.
“We like to administer the first dose in the office, allowing us to teach parents a few tricks related to proper technique,” including audio and visual distraction, tactile stimulation before and during the injection, use of topical anesthetic if helpful, “and making sure that the medication is at room temperature before administration,” she said. Cost is another potential barrier; however, even public insurance has been covering the medication, often after optimized use of topical medications has been unsuccessful.
Future research questions
As for additional research, the current study had a relatively small number of patients younger than 2 years, and more data are needed for this age group, said Dr. Paller. “We also need better understanding of the safety of dupilumab administration when live vaccines are administered. Finally, we certainly want to know what additional effects dupilumab may have beyond just the efficacy for treating eczema.”
In particular, these questions include whether dupilumab modifies the long-term course of the disease, possibly reducing the risk of persistence of disease with advancing age, or even cures the disease if started at a young age, she said. In addition, research has yet to show whether dupilumab might reduce the risk of other atopic disorders, such as asthma, food allergy, and allergic rhinitis.
“Ongoing studies and real-life experiences in the next several years will help us to answer these questions,” Dr. Paller said.
Data support safety, efficacy, quality of life
AD is associated with immense quality of life impairment, Raj Chovatiya, MD, of Northwestern University, Chicago, said in an interview. Most AD is initially diagnosed in early childhood, but previous treatment options for those with moderate to severe disease have been limited by safety concerns, which adds to the burden on infants and young children, and their parents and caregivers, said Dr. Chovatiya, who was not involved in the study.

“This phase 3 study showed that dupilumab, a fully human monoclonal antibody that selectively inhibits IL-4 and IL-13 mediated type 2 inflammatory signaling, provided both meaningful and statistically significant improvement in AD severity, extent of disease, and itch in patients,” he said. Dupilumab also improved children’s sleep quality and the overall quality of life in both patients and caregivers.
“These findings were quite similar to those described in older children and adults, where dupilumab is already approved for the treatment of moderate-severe AD and has demonstrated real-world safety and efficacy,” said Dr. Chovatiya. However, “the current study was limited to only a short-term analysis of 16 weeks, an ongoing open-label study should further address long-term treatment responses.”
The study was supported by Sanofi and Regeneron Pharmaceuticals. In addition to being an investigator for Regeneron, and several other pharmaceutical companies, Dr. Paller has been a consultant with honorarium for Regeneron, Sanofi, and multiple other companies. Dr. Chovatiya disclosed serving as a consultant and speaker for Regeneron and Sanofi, but was not involved in the current study.
at 31 treatment centers in North America and Europe.
Children younger than 6 years with moderate to severe AD have few options if their symptoms are uncontrolled with topical therapies, and persistent itchiness has a negative impact on quality of life for patients and families, Amy S. Paller, MD, professor and chair of dermatology, and professor of pediatrics at Northwestern University, Chicago, and colleagues wrote in the study, published in the Lancet.
The study was the basis of the Food and Drug Administration expanded approval of dupilumab in June 2022, to include children aged 6 months to 5 years with moderate to severe AD, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Regulatory submission for this age group is under review by the European Medicines Agency, and by regulatory authorities in other countries, according to the manufacturers.
Dupilumab (Dupixent), which inhibits the signaling of the interleukin-4 and IL-13 pathways, was first approved in 2017 for treating adults with moderate to severe AD.
“There has not been a biologic approved before at such a young age, and for such a common disease,” Dr. Paller said in an interview. “This is the drug that has revolutionized care of the most common inflammatory skin disease in children, and this is the pivotal study that brought it to market for the youngest children who suffer from the severe forms.”
The study also sets a precedent for a lower threshold for starting systemic medication in young children for treating moderate to severe disease given the absence of severe side effects and no need for lab monitoring, Dr. Paller noted. However, dupilumab will also be closely watched “for both impact on the developing immune system and the possibility that it will alter the long-term course of the eczema and the development of allergic comorbidities, such as lowering the risk of developing asthma, GI, allergy, and possibly other conditions.”
In the study, the researchers randomized 83 children aged 6 months up to 6 years to treatment with dupilumab, administered subcutaneously, and 79 to placebo every 4 weeks for 16 weeks; both groups also received topical corticosteroids. Dosage of dupilumab was based on body weight; those with a body weight of 5-15 kg received 200 mg, while those with a body weight of 15-30 kg received 300 mg. The primary endpoint was the proportion of patients with clear or almost clear skin at 16 weeks, defined as scores of 0 or 1 on the Investigator’s Global Assessment.
After 16 weeks, 28% of dupilumab patients met the primary endpoint versus 4% of those on placebo (P < .0001). In addition, 53% of dupilumab patients met the key secondary endpoint of a 75% improvement from baseline in Eczema Area and Severity Index, compared with 11% of patients on placebo (P < .0001). Treatment with dupilumab also resulted in significantly greater improvements in pruritus and skin pain, and sleep quality, as well as improved quality of life for patients and their caregivers, the authors reported.
Overall, adverse event rates were slightly lower in the dupilumab-treated patients, compared with patients on placebo (64% vs. 74%); there were no adverse events related to dupilumab that were serious or resulted in treatment discontinuation. Treatment-emergent adverse effects that were reported in 3% or more of patients and affected more of those on dupilumab than those on placebo included molluscum contagiosum (5% vs. 3%), viral gastroenteritis (4% vs. 0), rhinorrhea (5% vs. 1%), dental caries (5% vs. 0), and conjunctivitis (4% vs 0).
The rate of skin infections among the children on dupilumab was 12% vs. 24% among those on placebo.
Severe and treatment-related adverse events also were similar in both subgroups of body weight.
The findings were limited by the small number of patients younger than 2 years and the lack of study sites outside of North America and Europe, the researchers noted. However, the results were strengthened by the randomized, double-blind design and use of background topical therapy to provide a real-world safety and efficacy assessment in a very young population.
Overcoming injection issues
The safety profile for dupilumab, which is of the highest importance, “did not surprise me at all,” Dr. Paller said in an interview. “My only surprise is that the placebo injections actually led to more injection site reactions than [with] dupilumab, but numbers were quite low in both groups.” (Rates were 2% among those on dupilumab and 3% among those on placebo.)
The major barrier to the use of dupilumab in clinical practice is the requirement for injection, which, she explained, can be “unbearable for some young children, and thus becomes impossible for parents because of lack of cooperation and their intensified concern about giving the injection,” because of their child’s response.
“We like to administer the first dose in the office, allowing us to teach parents a few tricks related to proper technique,” including audio and visual distraction, tactile stimulation before and during the injection, use of topical anesthetic if helpful, “and making sure that the medication is at room temperature before administration,” she said. Cost is another potential barrier; however, even public insurance has been covering the medication, often after optimized use of topical medications has been unsuccessful.
Future research questions
As for additional research, the current study had a relatively small number of patients younger than 2 years, and more data are needed for this age group, said Dr. Paller. “We also need better understanding of the safety of dupilumab administration when live vaccines are administered. Finally, we certainly want to know what additional effects dupilumab may have beyond just the efficacy for treating eczema.”
In particular, these questions include whether dupilumab modifies the long-term course of the disease, possibly reducing the risk of persistence of disease with advancing age, or even cures the disease if started at a young age, she said. In addition, research has yet to show whether dupilumab might reduce the risk of other atopic disorders, such as asthma, food allergy, and allergic rhinitis.
“Ongoing studies and real-life experiences in the next several years will help us to answer these questions,” Dr. Paller said.
Data support safety, efficacy, quality of life
AD is associated with immense quality of life impairment, Raj Chovatiya, MD, of Northwestern University, Chicago, said in an interview. Most AD is initially diagnosed in early childhood, but previous treatment options for those with moderate to severe disease have been limited by safety concerns, which adds to the burden on infants and young children, and their parents and caregivers, said Dr. Chovatiya, who was not involved in the study.
“This phase 3 study showed that dupilumab, a fully human monoclonal antibody that selectively inhibits IL-4 and IL-13 mediated type 2 inflammatory signaling, provided both meaningful and statistically significant improvement in AD severity, extent of disease, and itch in patients,” he said. Dupilumab also improved children’s sleep quality and the overall quality of life in both patients and caregivers.
“These findings were quite similar to those described in older children and adults, where dupilumab is already approved for the treatment of moderate-severe AD and has demonstrated real-world safety and efficacy,” said Dr. Chovatiya. However, “the current study was limited to only a short-term analysis of 16 weeks, an ongoing open-label study should further address long-term treatment responses.”
The study was supported by Sanofi and Regeneron Pharmaceuticals. In addition to being an investigator for Regeneron, and several other pharmaceutical companies, Dr. Paller has been a consultant with honorarium for Regeneron, Sanofi, and multiple other companies. Dr. Chovatiya disclosed serving as a consultant and speaker for Regeneron and Sanofi, but was not involved in the current study.
at 31 treatment centers in North America and Europe.
Children younger than 6 years with moderate to severe AD have few options if their symptoms are uncontrolled with topical therapies, and persistent itchiness has a negative impact on quality of life for patients and families, Amy S. Paller, MD, professor and chair of dermatology, and professor of pediatrics at Northwestern University, Chicago, and colleagues wrote in the study, published in the Lancet.
The study was the basis of the Food and Drug Administration expanded approval of dupilumab in June 2022, to include children aged 6 months to 5 years with moderate to severe AD, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Regulatory submission for this age group is under review by the European Medicines Agency, and by regulatory authorities in other countries, according to the manufacturers.
Dupilumab (Dupixent), which inhibits the signaling of the interleukin-4 and IL-13 pathways, was first approved in 2017 for treating adults with moderate to severe AD.
“There has not been a biologic approved before at such a young age, and for such a common disease,” Dr. Paller said in an interview. “This is the drug that has revolutionized care of the most common inflammatory skin disease in children, and this is the pivotal study that brought it to market for the youngest children who suffer from the severe forms.”
The study also sets a precedent for a lower threshold for starting systemic medication in young children for treating moderate to severe disease given the absence of severe side effects and no need for lab monitoring, Dr. Paller noted. However, dupilumab will also be closely watched “for both impact on the developing immune system and the possibility that it will alter the long-term course of the eczema and the development of allergic comorbidities, such as lowering the risk of developing asthma, GI, allergy, and possibly other conditions.”
In the study, the researchers randomized 83 children aged 6 months up to 6 years to treatment with dupilumab, administered subcutaneously, and 79 to placebo every 4 weeks for 16 weeks; both groups also received topical corticosteroids. Dosage of dupilumab was based on body weight; those with a body weight of 5-15 kg received 200 mg, while those with a body weight of 15-30 kg received 300 mg. The primary endpoint was the proportion of patients with clear or almost clear skin at 16 weeks, defined as scores of 0 or 1 on the Investigator’s Global Assessment.
After 16 weeks, 28% of dupilumab patients met the primary endpoint versus 4% of those on placebo (P < .0001). In addition, 53% of dupilumab patients met the key secondary endpoint of a 75% improvement from baseline in Eczema Area and Severity Index, compared with 11% of patients on placebo (P < .0001). Treatment with dupilumab also resulted in significantly greater improvements in pruritus and skin pain, and sleep quality, as well as improved quality of life for patients and their caregivers, the authors reported.
Overall, adverse event rates were slightly lower in the dupilumab-treated patients, compared with patients on placebo (64% vs. 74%); there were no adverse events related to dupilumab that were serious or resulted in treatment discontinuation. Treatment-emergent adverse effects that were reported in 3% or more of patients and affected more of those on dupilumab than those on placebo included molluscum contagiosum (5% vs. 3%), viral gastroenteritis (4% vs. 0), rhinorrhea (5% vs. 1%), dental caries (5% vs. 0), and conjunctivitis (4% vs 0).
The rate of skin infections among the children on dupilumab was 12% vs. 24% among those on placebo.
Severe and treatment-related adverse events also were similar in both subgroups of body weight.
The findings were limited by the small number of patients younger than 2 years and the lack of study sites outside of North America and Europe, the researchers noted. However, the results were strengthened by the randomized, double-blind design and use of background topical therapy to provide a real-world safety and efficacy assessment in a very young population.
Overcoming injection issues
The safety profile for dupilumab, which is of the highest importance, “did not surprise me at all,” Dr. Paller said in an interview. “My only surprise is that the placebo injections actually led to more injection site reactions than [with] dupilumab, but numbers were quite low in both groups.” (Rates were 2% among those on dupilumab and 3% among those on placebo.)
The major barrier to the use of dupilumab in clinical practice is the requirement for injection, which, she explained, can be “unbearable for some young children, and thus becomes impossible for parents because of lack of cooperation and their intensified concern about giving the injection,” because of their child’s response.
“We like to administer the first dose in the office, allowing us to teach parents a few tricks related to proper technique,” including audio and visual distraction, tactile stimulation before and during the injection, use of topical anesthetic if helpful, “and making sure that the medication is at room temperature before administration,” she said. Cost is another potential barrier; however, even public insurance has been covering the medication, often after optimized use of topical medications has been unsuccessful.
Future research questions
As for additional research, the current study had a relatively small number of patients younger than 2 years, and more data are needed for this age group, said Dr. Paller. “We also need better understanding of the safety of dupilumab administration when live vaccines are administered. Finally, we certainly want to know what additional effects dupilumab may have beyond just the efficacy for treating eczema.”
In particular, these questions include whether dupilumab modifies the long-term course of the disease, possibly reducing the risk of persistence of disease with advancing age, or even cures the disease if started at a young age, she said. In addition, research has yet to show whether dupilumab might reduce the risk of other atopic disorders, such as asthma, food allergy, and allergic rhinitis.
“Ongoing studies and real-life experiences in the next several years will help us to answer these questions,” Dr. Paller said.
Data support safety, efficacy, quality of life
AD is associated with immense quality of life impairment, Raj Chovatiya, MD, of Northwestern University, Chicago, said in an interview. Most AD is initially diagnosed in early childhood, but previous treatment options for those with moderate to severe disease have been limited by safety concerns, which adds to the burden on infants and young children, and their parents and caregivers, said Dr. Chovatiya, who was not involved in the study.
“This phase 3 study showed that dupilumab, a fully human monoclonal antibody that selectively inhibits IL-4 and IL-13 mediated type 2 inflammatory signaling, provided both meaningful and statistically significant improvement in AD severity, extent of disease, and itch in patients,” he said. Dupilumab also improved children’s sleep quality and the overall quality of life in both patients and caregivers.
“These findings were quite similar to those described in older children and adults, where dupilumab is already approved for the treatment of moderate-severe AD and has demonstrated real-world safety and efficacy,” said Dr. Chovatiya. However, “the current study was limited to only a short-term analysis of 16 weeks, an ongoing open-label study should further address long-term treatment responses.”
The study was supported by Sanofi and Regeneron Pharmaceuticals. In addition to being an investigator for Regeneron, and several other pharmaceutical companies, Dr. Paller has been a consultant with honorarium for Regeneron, Sanofi, and multiple other companies. Dr. Chovatiya disclosed serving as a consultant and speaker for Regeneron and Sanofi, but was not involved in the current study.
FROM THE LANCET
Liquid injectable silicone safe for acne scarring in dark-skinned patients, study finds
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”

Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”
Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”
Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
AT ASDS 2022
Expert makes the case for not subtyping patients with rosacea
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
FROM MEDSCAPE LIVE COASTAL DERM
Analysis of PsA guidelines reveals much room for improvement on conflicts of interest
, according to a retrospective analysis of all authors on the most recent guidelines issued by the American College of Rheumatology (ACR) and the Japanese Dermatological Association (JDA).
In addition to finding that the majority of the authors of psoriatic arthritis (PsA) clinical practice guidelines (CPGs) issued by the JDA and ACR received substantial personal payments from pharmaceutical companies before and during CPG development, researchers led by Hanano Mamada and Anju Murayama of the Medical Governance Research Institute, Tokyo, wrote in Arthritis Care & Research that “several CPG authors self-cited their articles without the disclosure of NFCOI [nonfinancial conflicts of interest], and most of the recommendations were based on low or very low quality of evidence. Although the COI policies used by JDA and ACR are clearly inadequate, no significant revisions have been made for the last 3 years.”
Based on their findings, which were made using payment data from major Japanese pharmaceutical companies and the U.S. Open Payments Database from 2016 to 2018, the researchers suggested that the medical societies should:
- Adopt global standard COI policies from organizations such as the National Academy of Medicine and Guidelines International Network, including a 3-year lookback period for COI declaration.
- Consider a comprehensive definition and rigorous management with full disclosure of NFCOI.
- Publish a list of authors making each recommendation to grasp the implications of COI in clinical practice guidelines.
- Mention the detailed date of the COI disclosure, which should be close to the publication date as much as possible.
Financial conflicts of interest
The researchers used payment data published between 2016 and 2018 for all 83 companies belonging to the Japan Pharmaceutical Manufacturers Association, focusing on personal payments (for lecturing, writing, and consultancy) and excluding research payments, “since in Japan, the name, institution, and position of the author or researcher who received the research payment is not disclosed, which makes assessing research payments difficult.” To evaluate authors’ FCOI in the ACR’s CPG, the researchers analyzed the U.S. Open Payments Database “for all categories of general payments such as speaking, consulting, meals, and travel expenses 3 years from before the guideline’s first online publication on November 30, 2018.”
The 2018 ACR/National Psoriasis Foundation Guideline for the Treatment of Psoriatic Arthritis had 36 authors and the JDA’s Clinical Practice Guideline for the Treatment of Psoriatic Arthritis 2019 had 23. Overall, 61% of JDA authors and half of ACR authors voluntarily declared FCOI with pharmaceutical companies; 25 of the ACR authors were U.S. physicians and could be included in the Open Payments Database search.
A total of 21 (91.3%) JDA authors and 21 (84.0%) ACR authors received at least one payment, with the combined total of $3,335,413 and $4,081,629 payments, respectively, over the 3 years. The average and median personal payments were $145,018 and $123,876 for JDA authors and $162,825 and $58,826 for ACR authors. When the payments to ACR authors were limited to lecturing, writing, and consulting fees that are required under the ACR’s COI policy, the mean was $130,102 and median was $39,375. The corresponding payments for JDA authors were $123,876 and $8,170, respectively,
The researchers found undisclosed payments for more than three-quarters of physician authors of the Japanese guideline, and nearly half of the doctors authoring the American guideline had undisclosed payments. These added up to $474,000 for the JDA, which amounted to 38% of the total for personal payments that must be reported to the JDA based on its COI policy for clinical practice guidelines, and $218,000 for the ACR, amounting to 18% of the total for personal payments that must be reported to the society based on its COI policy.
Of the 11 ACR authors who were not eligible for the U.S. Open Payments Database search, 5 declared FCOI with pharmaceutical companies in the guideline, meaning that 26 (72%) of the 36 authors had FCOI with pharmaceutical companies.
The ACR only required authors to declare FCOI covering 1 year before and during guideline development, and although the JDA required authors to declare their FCOI for the past 3 years of guideline development, the study authors noted that the JDA guideline disclosed them for only 2 years (between Jan. 1, 2017, and Dec. 31, 2018).
“It is true that influential doctors such as clinical practice guideline authors tend to receive various types of payments from pharmaceutical companies and that it is difficult to conduct research without funding from pharmaceutical companies. However, our current research mainly focuses on personal payments from pharmaceutical companies such as lecture fees and consulting fees. These payments are recognized as pocket money and are not used for research. Thus, it is questionable that the observed relationships are something evitable,” the researchers wrote.
Nonfinancial conflicts of interest
Many authors of the ACR’s CPG and the JDA’s CPG also had NFCOI, defined objectively in this study as self-citation rate. NFCOI have been more broadly defined by the International Committee of Medical Journal Editors (ICMJE) as “conflicts, such as personal relationships or rivalries, academic competition, and intellectual beliefs”; the ICMJE recommends reporting NFCOI on its COI form.
The JDA guideline included self-citations by 78% of its authors, compared with 32% of the ACR guideline authors, but this weighed differently among the two guidelines in that only 12 of the 354 (3.4%) citations in the JDA guideline were self-cited, compared with 46 of 137 (34%) citations in the ACR guideline.
The researchers noted that while the self-citation rates between JDA and ACR authors “differed remarkably,” the impact of ACR authors on CPG recommendations was much more direct. Three-quarters of JDA authors’ self-cited articles were about observational studies, whereas 52% of the ACR authors’ self-cited articles were clinical trials, most of which were randomized, controlled studies, and these NFCOI were not disclosed in the guideline.
Half of the strong recommendations in the JDA guideline were based on low or very low quality of evidence, whereas the ACR guideline had no strong recommendations based on low or very low quality of evidence.
This study was supported by the nonprofit Medical Governance Research Institute, which receives donations from Ain Pharmacies Inc., other organizations, and private individuals. The study also received support from the Tansa (formerly known as the Waseda Chronicle), an independent nonprofit news organization dedicated to investigative journalism. Three authors reported receiving personal fees from several pharmaceutical companies for work outside of the scope of this study.
, according to a retrospective analysis of all authors on the most recent guidelines issued by the American College of Rheumatology (ACR) and the Japanese Dermatological Association (JDA).
In addition to finding that the majority of the authors of psoriatic arthritis (PsA) clinical practice guidelines (CPGs) issued by the JDA and ACR received substantial personal payments from pharmaceutical companies before and during CPG development, researchers led by Hanano Mamada and Anju Murayama of the Medical Governance Research Institute, Tokyo, wrote in Arthritis Care & Research that “several CPG authors self-cited their articles without the disclosure of NFCOI [nonfinancial conflicts of interest], and most of the recommendations were based on low or very low quality of evidence. Although the COI policies used by JDA and ACR are clearly inadequate, no significant revisions have been made for the last 3 years.”
Based on their findings, which were made using payment data from major Japanese pharmaceutical companies and the U.S. Open Payments Database from 2016 to 2018, the researchers suggested that the medical societies should:
- Adopt global standard COI policies from organizations such as the National Academy of Medicine and Guidelines International Network, including a 3-year lookback period for COI declaration.
- Consider a comprehensive definition and rigorous management with full disclosure of NFCOI.
- Publish a list of authors making each recommendation to grasp the implications of COI in clinical practice guidelines.
- Mention the detailed date of the COI disclosure, which should be close to the publication date as much as possible.
Financial conflicts of interest
The researchers used payment data published between 2016 and 2018 for all 83 companies belonging to the Japan Pharmaceutical Manufacturers Association, focusing on personal payments (for lecturing, writing, and consultancy) and excluding research payments, “since in Japan, the name, institution, and position of the author or researcher who received the research payment is not disclosed, which makes assessing research payments difficult.” To evaluate authors’ FCOI in the ACR’s CPG, the researchers analyzed the U.S. Open Payments Database “for all categories of general payments such as speaking, consulting, meals, and travel expenses 3 years from before the guideline’s first online publication on November 30, 2018.”
The 2018 ACR/National Psoriasis Foundation Guideline for the Treatment of Psoriatic Arthritis had 36 authors and the JDA’s Clinical Practice Guideline for the Treatment of Psoriatic Arthritis 2019 had 23. Overall, 61% of JDA authors and half of ACR authors voluntarily declared FCOI with pharmaceutical companies; 25 of the ACR authors were U.S. physicians and could be included in the Open Payments Database search.
A total of 21 (91.3%) JDA authors and 21 (84.0%) ACR authors received at least one payment, with the combined total of $3,335,413 and $4,081,629 payments, respectively, over the 3 years. The average and median personal payments were $145,018 and $123,876 for JDA authors and $162,825 and $58,826 for ACR authors. When the payments to ACR authors were limited to lecturing, writing, and consulting fees that are required under the ACR’s COI policy, the mean was $130,102 and median was $39,375. The corresponding payments for JDA authors were $123,876 and $8,170, respectively,
The researchers found undisclosed payments for more than three-quarters of physician authors of the Japanese guideline, and nearly half of the doctors authoring the American guideline had undisclosed payments. These added up to $474,000 for the JDA, which amounted to 38% of the total for personal payments that must be reported to the JDA based on its COI policy for clinical practice guidelines, and $218,000 for the ACR, amounting to 18% of the total for personal payments that must be reported to the society based on its COI policy.
Of the 11 ACR authors who were not eligible for the U.S. Open Payments Database search, 5 declared FCOI with pharmaceutical companies in the guideline, meaning that 26 (72%) of the 36 authors had FCOI with pharmaceutical companies.
The ACR only required authors to declare FCOI covering 1 year before and during guideline development, and although the JDA required authors to declare their FCOI for the past 3 years of guideline development, the study authors noted that the JDA guideline disclosed them for only 2 years (between Jan. 1, 2017, and Dec. 31, 2018).
“It is true that influential doctors such as clinical practice guideline authors tend to receive various types of payments from pharmaceutical companies and that it is difficult to conduct research without funding from pharmaceutical companies. However, our current research mainly focuses on personal payments from pharmaceutical companies such as lecture fees and consulting fees. These payments are recognized as pocket money and are not used for research. Thus, it is questionable that the observed relationships are something evitable,” the researchers wrote.
Nonfinancial conflicts of interest
Many authors of the ACR’s CPG and the JDA’s CPG also had NFCOI, defined objectively in this study as self-citation rate. NFCOI have been more broadly defined by the International Committee of Medical Journal Editors (ICMJE) as “conflicts, such as personal relationships or rivalries, academic competition, and intellectual beliefs”; the ICMJE recommends reporting NFCOI on its COI form.
The JDA guideline included self-citations by 78% of its authors, compared with 32% of the ACR guideline authors, but this weighed differently among the two guidelines in that only 12 of the 354 (3.4%) citations in the JDA guideline were self-cited, compared with 46 of 137 (34%) citations in the ACR guideline.
The researchers noted that while the self-citation rates between JDA and ACR authors “differed remarkably,” the impact of ACR authors on CPG recommendations was much more direct. Three-quarters of JDA authors’ self-cited articles were about observational studies, whereas 52% of the ACR authors’ self-cited articles were clinical trials, most of which were randomized, controlled studies, and these NFCOI were not disclosed in the guideline.
Half of the strong recommendations in the JDA guideline were based on low or very low quality of evidence, whereas the ACR guideline had no strong recommendations based on low or very low quality of evidence.
This study was supported by the nonprofit Medical Governance Research Institute, which receives donations from Ain Pharmacies Inc., other organizations, and private individuals. The study also received support from the Tansa (formerly known as the Waseda Chronicle), an independent nonprofit news organization dedicated to investigative journalism. Three authors reported receiving personal fees from several pharmaceutical companies for work outside of the scope of this study.
, according to a retrospective analysis of all authors on the most recent guidelines issued by the American College of Rheumatology (ACR) and the Japanese Dermatological Association (JDA).
In addition to finding that the majority of the authors of psoriatic arthritis (PsA) clinical practice guidelines (CPGs) issued by the JDA and ACR received substantial personal payments from pharmaceutical companies before and during CPG development, researchers led by Hanano Mamada and Anju Murayama of the Medical Governance Research Institute, Tokyo, wrote in Arthritis Care & Research that “several CPG authors self-cited their articles without the disclosure of NFCOI [nonfinancial conflicts of interest], and most of the recommendations were based on low or very low quality of evidence. Although the COI policies used by JDA and ACR are clearly inadequate, no significant revisions have been made for the last 3 years.”
Based on their findings, which were made using payment data from major Japanese pharmaceutical companies and the U.S. Open Payments Database from 2016 to 2018, the researchers suggested that the medical societies should:
- Adopt global standard COI policies from organizations such as the National Academy of Medicine and Guidelines International Network, including a 3-year lookback period for COI declaration.
- Consider a comprehensive definition and rigorous management with full disclosure of NFCOI.
- Publish a list of authors making each recommendation to grasp the implications of COI in clinical practice guidelines.
- Mention the detailed date of the COI disclosure, which should be close to the publication date as much as possible.
Financial conflicts of interest
The researchers used payment data published between 2016 and 2018 for all 83 companies belonging to the Japan Pharmaceutical Manufacturers Association, focusing on personal payments (for lecturing, writing, and consultancy) and excluding research payments, “since in Japan, the name, institution, and position of the author or researcher who received the research payment is not disclosed, which makes assessing research payments difficult.” To evaluate authors’ FCOI in the ACR’s CPG, the researchers analyzed the U.S. Open Payments Database “for all categories of general payments such as speaking, consulting, meals, and travel expenses 3 years from before the guideline’s first online publication on November 30, 2018.”
The 2018 ACR/National Psoriasis Foundation Guideline for the Treatment of Psoriatic Arthritis had 36 authors and the JDA’s Clinical Practice Guideline for the Treatment of Psoriatic Arthritis 2019 had 23. Overall, 61% of JDA authors and half of ACR authors voluntarily declared FCOI with pharmaceutical companies; 25 of the ACR authors were U.S. physicians and could be included in the Open Payments Database search.
A total of 21 (91.3%) JDA authors and 21 (84.0%) ACR authors received at least one payment, with the combined total of $3,335,413 and $4,081,629 payments, respectively, over the 3 years. The average and median personal payments were $145,018 and $123,876 for JDA authors and $162,825 and $58,826 for ACR authors. When the payments to ACR authors were limited to lecturing, writing, and consulting fees that are required under the ACR’s COI policy, the mean was $130,102 and median was $39,375. The corresponding payments for JDA authors were $123,876 and $8,170, respectively,
The researchers found undisclosed payments for more than three-quarters of physician authors of the Japanese guideline, and nearly half of the doctors authoring the American guideline had undisclosed payments. These added up to $474,000 for the JDA, which amounted to 38% of the total for personal payments that must be reported to the JDA based on its COI policy for clinical practice guidelines, and $218,000 for the ACR, amounting to 18% of the total for personal payments that must be reported to the society based on its COI policy.
Of the 11 ACR authors who were not eligible for the U.S. Open Payments Database search, 5 declared FCOI with pharmaceutical companies in the guideline, meaning that 26 (72%) of the 36 authors had FCOI with pharmaceutical companies.
The ACR only required authors to declare FCOI covering 1 year before and during guideline development, and although the JDA required authors to declare their FCOI for the past 3 years of guideline development, the study authors noted that the JDA guideline disclosed them for only 2 years (between Jan. 1, 2017, and Dec. 31, 2018).
“It is true that influential doctors such as clinical practice guideline authors tend to receive various types of payments from pharmaceutical companies and that it is difficult to conduct research without funding from pharmaceutical companies. However, our current research mainly focuses on personal payments from pharmaceutical companies such as lecture fees and consulting fees. These payments are recognized as pocket money and are not used for research. Thus, it is questionable that the observed relationships are something evitable,” the researchers wrote.
Nonfinancial conflicts of interest
Many authors of the ACR’s CPG and the JDA’s CPG also had NFCOI, defined objectively in this study as self-citation rate. NFCOI have been more broadly defined by the International Committee of Medical Journal Editors (ICMJE) as “conflicts, such as personal relationships or rivalries, academic competition, and intellectual beliefs”; the ICMJE recommends reporting NFCOI on its COI form.
The JDA guideline included self-citations by 78% of its authors, compared with 32% of the ACR guideline authors, but this weighed differently among the two guidelines in that only 12 of the 354 (3.4%) citations in the JDA guideline were self-cited, compared with 46 of 137 (34%) citations in the ACR guideline.
The researchers noted that while the self-citation rates between JDA and ACR authors “differed remarkably,” the impact of ACR authors on CPG recommendations was much more direct. Three-quarters of JDA authors’ self-cited articles were about observational studies, whereas 52% of the ACR authors’ self-cited articles were clinical trials, most of which were randomized, controlled studies, and these NFCOI were not disclosed in the guideline.
Half of the strong recommendations in the JDA guideline were based on low or very low quality of evidence, whereas the ACR guideline had no strong recommendations based on low or very low quality of evidence.
This study was supported by the nonprofit Medical Governance Research Institute, which receives donations from Ain Pharmacies Inc., other organizations, and private individuals. The study also received support from the Tansa (formerly known as the Waseda Chronicle), an independent nonprofit news organization dedicated to investigative journalism. Three authors reported receiving personal fees from several pharmaceutical companies for work outside of the scope of this study.
FROM ARTHRITIS CARE & RESEARCH
Recurrent drainage from an old gunshot wound

An x-ray revealed a metal density in the area of concern that was consistent with a bullet fragment or other metallic foreign body. Since there were no lucencies on x-ray or tracking from the area of concern to the metacarpal, the diagnosis was confirmed as an infected foreign body. The history was very concerning for osteomyelitis, given that the patient had sustained a GSW and had undergone surgical repair with hardware. (Shifting hardware can also lead to callus formation and skin breakdown.)
The patient was told that he’d retained a bullet fragment or foreign body that caused a chronic infection and the recurrent drainage. In addition, the hardware spanning the gap between the remnants of his proximal and distal metacarpal had broken as a result of fatigue. He was referred to a surgeon to remove the foreign body and treat the infection. The patient was advised that he might also need replacement hardware and a bone graft.
Images and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
An x-ray revealed a metal density in the area of concern that was consistent with a bullet fragment or other metallic foreign body. Since there were no lucencies on x-ray or tracking from the area of concern to the metacarpal, the diagnosis was confirmed as an infected foreign body. The history was very concerning for osteomyelitis, given that the patient had sustained a GSW and had undergone surgical repair with hardware. (Shifting hardware can also lead to callus formation and skin breakdown.)
The patient was told that he’d retained a bullet fragment or foreign body that caused a chronic infection and the recurrent drainage. In addition, the hardware spanning the gap between the remnants of his proximal and distal metacarpal had broken as a result of fatigue. He was referred to a surgeon to remove the foreign body and treat the infection. The patient was advised that he might also need replacement hardware and a bone graft.
Images and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
An x-ray revealed a metal density in the area of concern that was consistent with a bullet fragment or other metallic foreign body. Since there were no lucencies on x-ray or tracking from the area of concern to the metacarpal, the diagnosis was confirmed as an infected foreign body. The history was very concerning for osteomyelitis, given that the patient had sustained a GSW and had undergone surgical repair with hardware. (Shifting hardware can also lead to callus formation and skin breakdown.)
The patient was told that he’d retained a bullet fragment or foreign body that caused a chronic infection and the recurrent drainage. In addition, the hardware spanning the gap between the remnants of his proximal and distal metacarpal had broken as a result of fatigue. He was referred to a surgeon to remove the foreign body and treat the infection. The patient was advised that he might also need replacement hardware and a bone graft.
Images and text courtesy of Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo.
