User login
Psoriasiform Dermatitis Associated With the Moderna COVID-19 Messenger RNA Vaccine
To the Editor:
The Moderna COVID-19 messenger RNA (mRNA) vaccine was authorized for use on December 18, 2020, with the second dose beginning on January 15, 2021.1-3 Some individuals who received the Moderna vaccine experienced an intense rash known as “COVID arm,” a harmless but bothersome adverse effect that typically appears within a week and is a localized and transient immunogenic response.4 COVID arm differs from most vaccine adverse effects. The rash emerges not immediately but 5 to 9 days after the initial dose—on average, 1 week later. Apart from being itchy, the rash does not appear to be harmful and is not a reason to hesitate getting vaccinated.
Dermatologists and allergists have been studying this adverse effect, which has been formally termed delayed cutaneous hypersensitivity. Of potential clinical consequence is that the efficacy of the mRNA COVID-19 vaccine may be harmed if postvaccination dermal reactions necessitate systemic corticosteroid therapy. Because this vaccine stimulates an immune response as viral RNA integrates in cells secondary to production of the spike protein of the virus, the skin may be affected secondarily and manifestations of any underlying disease may be aggravated.5 We report a patient who developed a psoriasiform dermatitis after the first dose of the Moderna vaccine.
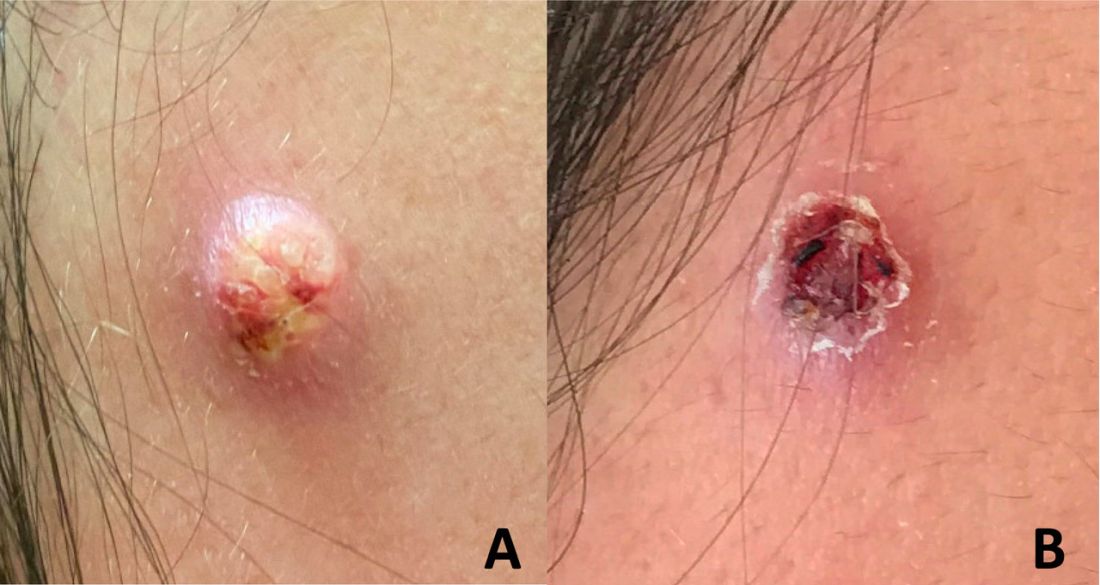
. B, Scattered scaly papules with mild macular erythema were present on the left upper chest and clavicular region, with pink to deep red–erythematous")
A 65-year-old woman presented to her primary care physician because of the severity of psoriasiform dermatitis that developed 5 days after she received the first dose of the Moderna COVID-19 mRNA vaccine. The patient had a medical history of Sjögren syndrome. Her medication history was negative, and her family history was negative for autoimmune disease. Physical examination by primary care revealed an erythematous scaly rash with plaques and papules on the neck and back (Figure 1). The patient presented again to primary care 2 days later with swollen, painful, discolored digits (Figure 2) and a stiff, sore neck.

Laboratory results were positive for anti–Sjögren syndrome–related antigens A and B. A complete blood cell count; comprehensive metabolic panel; erythrocyte sedimentation rate; and assays of rheumatoid factor, C-reactive protein, and anti–cyclic citrullinated peptide were within reference range. A biopsy of a lesion on the back showed psoriasiform dermatitis with confluent parakeratosis and scattered necrotic keratinocytes. There was superficial perivascular inflammation with rare eosinophils (Figure 3).
. B, Psoriasiform dermatitis with confluent parakeratosis and scattered necrotic keratinocytes also were noted")
The patient was treated with a course of systemic corticosteroids. The rash resolved in 1 week. She did not receive the second dose due to the rash.
Two mRNA COVID-19 vaccines—Pfizer BioNTech and Moderna—have been granted emergency use authorization by the US Food and Drug Administration.6 The safety profile of the mRNA-1273 vaccine for the median 2-month follow-up showed no safety concerns.3 Minor localized adverse effects (eg, pain, redness, swelling) have been observed more frequently with the vaccines than with placebo. Systemic symptoms, such as fever, fatigue, headache, and muscle and joint pain, also were seen somewhat more often with the vaccines than with placebo; most such effects occurred 24 to 48 hours after vaccination.3,6,7 The frequency of unsolicited adverse events and serious adverse events reported during the 28-day period after vaccination generally was similar among participants in the vaccine and placebo groups.3
There are 2 types of reactions to COVID-19 vaccination: immediate and delayed. Immediate reactions usually are due to anaphylaxis, requiring prompt recognition and treatment with epinephrine to stop rapid progression of life-threatening symptoms. Delayed reactions include localized reactions, such as urticaria and benign exanthema; serum sickness and serum sickness–like reactions; fever; and rare skin, organ, and neurologic sequelae.1,6-8
Cutaneous manifestations, present in 16% to 50% of patients with Sjögren syndrome, are considered one of the most common extraglandular presentations of the syndrome. They are classified as nonvascular (eg, xerosis, angular cheilitis, eyelid dermatitis, annular erythema) and vascular (eg, Raynaud phenomenon, vasculitis).9-11 Our patient did not have any of those findings. She had not taken any medications before the rash appeared, thereby ruling out a drug reaction.
The differential for our patient included post–urinary tract infection immune-reactive arthritis and rash, which is not typical with Escherichia coli infection but is described with infection with Chlamydia species and Salmonella species. Moreover, post–urinary tract infection immune-reactive arthritis and rash appear mostly on the palms and soles. Systemic lupus erythematosus–like rashes have a different histology and appear on sun-exposed areas; our patient’s rash was found mainly on unexposed areas.12
Because our patient received the Moderna vaccine 5 days before the rash appeared and later developed swelling of the digits with morning stiffness, a delayed serum sickness–like reaction secondary to COVID-19 vaccination was possible.3,6
COVID-19 mRNA vaccines developed by Pfizer-BioNTech and Moderna incorporate a lipid-based nanoparticle carrier system that prevents rapid enzymatic degradation of mRNA and facilitates in vivo delivery of mRNA. This lipid-based nanoparticle carrier system is further stabilized by a polyethylene glycol 2000 lipid conjugate that provides a hydrophilic layer, thus prolonging half-life. The presence of lipid polyethylene glycol 2000 in mRNA vaccines has led to concern that this component could be implicated in anaphylaxis.6
COVID-19 antigens can give rise to varying clinical manifestations that are directly related to viral tissue damage or are indirectly induced by the antiviral immune response.13,14 Hyperactivation of the immune system to eradicate COVID-19 may trigger autoimmunity; several immune-mediated disorders have been described in individuals infected with SARS-CoV-2. Dermal manifestations include cutaneous rash and vasculitis.13-16 Crucial immunologic steps occur during SARS-CoV-2 infection that may link autoimmunity to COVID-19.13,14 In preliminary published data on the efficacy of the Moderna vaccine on 45 trial enrollees, 3 did not receive the second dose of vaccination, including 1 who developed urticaria on both legs 5 days after the first dose.1
Introduction of viral RNA can induce autoimmunity that can be explained by various phenomena, including epitope spreading, molecular mimicry, cryptic antigen, and bystander activation. Remarkably, more than one-third of immunogenic proteins in SARS-CoV-2 have potentially problematic homology to proteins that are key to the human adaptive immune system.5
Moreover, SARS-CoV-2 seems to induce organ injury through alternative mechanisms beyond direct viral infection, including immunologic injury. In some situations, hyperactivation of the immune response to SARS-CoV-2 RNA can result in autoimmune disease. COVID-19 has been associated with immune-mediated systemic or organ-selective manifestations, some of which fulfill the diagnostic or classification criteria of specific autoimmune diseases. It is unclear whether those medical disorders are the result of transitory postinfectious epiphenomena.5
A few studies have shown that patients with rheumatic disease have an incidence and prevalence of COVID-19 that is similar to the general population. A similar pattern has been detected in COVID-19 morbidity and mortality rates, even among patients with an autoimmune disease, such as rheumatoid arthritis and Sjögren syndrome.5,17 Furthermore, exacerbation of preexisting rheumatic symptoms may be due to hyperactivation of antiviral pathways in a person with an autoimmune disease.17-19 The findings in our patient suggested a direct role for the vaccine in skin manifestations, rather than for reactivation or development of new systemic autoimmune processes, such as systemic lupus erythematosus.
Exacerbation of psoriasis following COVID-19 vaccination has been described20; however, the case patient did not have a history of psoriasis. The mechanism(s) of such exacerbation remain unclear; COVID-19 vaccine–induced helper T cells (TH17) may play a role.21 Other skin manifestations encountered following COVID-19 vaccination include lichen planus, leukocytoclastic vasculitic rash, erythema multiforme–like rash, and pityriasis rosea–like rash.22-25 The immune mechanisms of these manifestations remain unclear.
The clinical presentation of delayed vaccination reactions can be attributed to the timing of symptoms and, in this case, the immune-mediated background of a psoriasiform reaction. Although adverse reactions to the SARS-CoV-2 mRNA vaccine are rare, more individuals should be studied after vaccination to confirm and better understand this phenomenon.
- Jackson LA, Anderson EJ, Rouphael NG, et al; . An mRNA vaccine against SARS-CoV-2—preliminary report. N Engl J Med. 2020;383:1920-1931. doi:10.1056/NEJMoa2022483
- Anderson EJ, Rouphael NG, Widge AT, et al; . Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N Engl J Med. 2020;383:2427-2438. doi:10.1056/NEJMoa2028436
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403-416. doi:10.1056/NEJMoa2035389
- Weise E. ‘COVID arm’ rash seen after Moderna vaccine annoying but harmless, doctors say. USA Today. January 27, 2021. Accessed September 4, 2022. https://www.usatoday.com/story/news/health/2021/01/27/covid-arm-moderna-vaccine-rash-harmless-side-effect-doctors-say/4277725001/
- Talotta R, Robertson E. Autoimmunity as the comet tail of COVID-19 pandemic. World J Clin Cases. 2020;8:3621-3644. doi:10.12998/wjcc.v8.i17.3621
- Castells MC, Phillips EJ. Maintaining safety with SARS-CoV-2 vaccines. N Engl J Med. 2021;384:643-649. doi:10.1056/NEJMra2035343
- Polack FP, Thomas SJ, Kitchin N, et al; . Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603-2615. doi:10.1056/NEJMoa2034577
- Dooling K, McClung N, Chamberland M, et al. The Advisory Committee on Immunization Practices’ interim recommendation for allocating initial supplies of COVID-19 vaccine—United States, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1857-1859. doi:10.15585/mmwr.mm6949e1
- Roguedas AM, Misery L, Sassolas B, et al. Cutaneous manifestations of primary Sjögren’s syndrome are underestimated. Clin Exp Rheumatol. 2004;22:632-636.
- Katayama I. Dry skin manifestations in Sjögren syndrome and atopic dermatitis related to aberrant sudomotor function in inflammatory allergic skin diseases. Allergol Int. 2018;67:448-454. doi:10.1016/j.alit.2018.07.001
- Generali E, Costanzo A, Mainetti C, et al. Cutaneous and mucosal manifestations of Sjögren’s syndrome. Clin Rev Allergy Immunol. 2017;53:357-370. doi:10.1007/s12016-017-8639-y
- Chanprapaph K, Tankunakorn J, Suchonwanit P, et al. Dermatologic manifestations, histologic features and disease progression among cutaneous lupus erythematosus subtypes: a prospective observational study in Asians. Dermatol Ther (Heidelb). 2021;11:131-147. doi:10.1007/s13555-020-00471-y
- Ortega-Quijano D, Jimenez-Cauhe J, Selda-Enriquez G, et al. Algorithm for the classification of COVID-19 rashes. J Am Acad Dermatol. 2020;83:e103-e104. doi:10.1016/j.jaad.2020.05.034
- Rahimi H, Tehranchinia Z. A comprehensive review of cutaneous manifestations associated with COVID-19. Biomed Res Int. 2020;2020:1236520. doi:10.1155/2020/1236520
- Sachdeva M, Gianotti R, Shah M, et al. Cutaneous manifestations of COVID-19: report of three cases and a review of literature. J Dermatol Sci. 2020;98:75-81. doi:10.1016/j.jdermsci.2020.04.011
- Landa N, Mendieta-Eckert M, Fonda-Pascual P, et al. Chilblain-like lesions on feet and hands during the COVID-19 pandemic. Int J Dermatol. 2020;59:739-743. doi:10.1111/ijd.14937
- Dellavance A, Coelho Andrade LE. Immunologic derangement preceding clinical autoimmunity. Lupus. 2014;23:1305-1308. doi:10.1177/0961203314531346
- Parodi A, Gasparini G, Cozzani E. Could antiphospholipid antibodies contribute to coagulopathy in COVID-19? J Am Acad Dermatol. 2020;83:e249. doi:10.1016/j.jaad.2020.06.003
- Zhou Y, Han T, Chen J, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci. 2020;13:1077-1086. doi:10.1111/cts.12805
- Huang YW, Tsai TF. Exacerbation of psoriasis following COVID-19 vaccination: report from a single center. Front Med (Lausanne). 2021;8:812010. doi:10.3389/fmed.2021.812010
- Rouai M, Slimane MB, Sassi W, et al. Pustular rash triggered by Pfizer-BioNTech COVID-19 vaccination: a case report. Dermatol Ther. 2022:e15465. doi:10.1111/dth.15465
- Altun E, Kuzucular E. Leukocytoclastic vasculitis after COVID-19 vaccination. Dermatol Ther. 2022;35:e15279. doi:10.1111/dth.15279
- Buckley JE, Landis LN, Rapini RP. Pityriasis rosea-like rash after mRNA COVID-19 vaccination: a case report and review of the literature. JAAD Int. 2022;7:164-168. doi:10.1016/j.jdin.2022.01.009
- Gökçek GE, Öksüm Solak E, Çölgeçen E. Pityriasis rosea like eruption: a dermatological manifestation of Coronavac-COVID-19 vaccine. Dermatol Ther. 2022;35:e15256. doi:10.1111/dth.15256
- Kim MJ, Kim JW, Kim MS, et al. Generalized erythema multiforme-like skin rash following the first dose of COVID-19 vaccine (Pfizer-BioNTech). J Eur Acad Dermatol Venereol. 2022;36:e98-e100. doi:10.1111/jdv.17757
To the Editor:
The Moderna COVID-19 messenger RNA (mRNA) vaccine was authorized for use on December 18, 2020, with the second dose beginning on January 15, 2021.1-3 Some individuals who received the Moderna vaccine experienced an intense rash known as “COVID arm,” a harmless but bothersome adverse effect that typically appears within a week and is a localized and transient immunogenic response.4 COVID arm differs from most vaccine adverse effects. The rash emerges not immediately but 5 to 9 days after the initial dose—on average, 1 week later. Apart from being itchy, the rash does not appear to be harmful and is not a reason to hesitate getting vaccinated.
Dermatologists and allergists have been studying this adverse effect, which has been formally termed delayed cutaneous hypersensitivity. Of potential clinical consequence is that the efficacy of the mRNA COVID-19 vaccine may be harmed if postvaccination dermal reactions necessitate systemic corticosteroid therapy. Because this vaccine stimulates an immune response as viral RNA integrates in cells secondary to production of the spike protein of the virus, the skin may be affected secondarily and manifestations of any underlying disease may be aggravated.5 We report a patient who developed a psoriasiform dermatitis after the first dose of the Moderna vaccine.
A 65-year-old woman presented to her primary care physician because of the severity of psoriasiform dermatitis that developed 5 days after she received the first dose of the Moderna COVID-19 mRNA vaccine. The patient had a medical history of Sjögren syndrome. Her medication history was negative, and her family history was negative for autoimmune disease. Physical examination by primary care revealed an erythematous scaly rash with plaques and papules on the neck and back (Figure 1). The patient presented again to primary care 2 days later with swollen, painful, discolored digits (Figure 2) and a stiff, sore neck.
Laboratory results were positive for anti–Sjögren syndrome–related antigens A and B. A complete blood cell count; comprehensive metabolic panel; erythrocyte sedimentation rate; and assays of rheumatoid factor, C-reactive protein, and anti–cyclic citrullinated peptide were within reference range. A biopsy of a lesion on the back showed psoriasiform dermatitis with confluent parakeratosis and scattered necrotic keratinocytes. There was superficial perivascular inflammation with rare eosinophils (Figure 3).
The patient was treated with a course of systemic corticosteroids. The rash resolved in 1 week. She did not receive the second dose due to the rash.
Two mRNA COVID-19 vaccines—Pfizer BioNTech and Moderna—have been granted emergency use authorization by the US Food and Drug Administration.6 The safety profile of the mRNA-1273 vaccine for the median 2-month follow-up showed no safety concerns.3 Minor localized adverse effects (eg, pain, redness, swelling) have been observed more frequently with the vaccines than with placebo. Systemic symptoms, such as fever, fatigue, headache, and muscle and joint pain, also were seen somewhat more often with the vaccines than with placebo; most such effects occurred 24 to 48 hours after vaccination.3,6,7 The frequency of unsolicited adverse events and serious adverse events reported during the 28-day period after vaccination generally was similar among participants in the vaccine and placebo groups.3
There are 2 types of reactions to COVID-19 vaccination: immediate and delayed. Immediate reactions usually are due to anaphylaxis, requiring prompt recognition and treatment with epinephrine to stop rapid progression of life-threatening symptoms. Delayed reactions include localized reactions, such as urticaria and benign exanthema; serum sickness and serum sickness–like reactions; fever; and rare skin, organ, and neurologic sequelae.1,6-8
Cutaneous manifestations, present in 16% to 50% of patients with Sjögren syndrome, are considered one of the most common extraglandular presentations of the syndrome. They are classified as nonvascular (eg, xerosis, angular cheilitis, eyelid dermatitis, annular erythema) and vascular (eg, Raynaud phenomenon, vasculitis).9-11 Our patient did not have any of those findings. She had not taken any medications before the rash appeared, thereby ruling out a drug reaction.
The differential for our patient included post–urinary tract infection immune-reactive arthritis and rash, which is not typical with Escherichia coli infection but is described with infection with Chlamydia species and Salmonella species. Moreover, post–urinary tract infection immune-reactive arthritis and rash appear mostly on the palms and soles. Systemic lupus erythematosus–like rashes have a different histology and appear on sun-exposed areas; our patient’s rash was found mainly on unexposed areas.12
Because our patient received the Moderna vaccine 5 days before the rash appeared and later developed swelling of the digits with morning stiffness, a delayed serum sickness–like reaction secondary to COVID-19 vaccination was possible.3,6
COVID-19 mRNA vaccines developed by Pfizer-BioNTech and Moderna incorporate a lipid-based nanoparticle carrier system that prevents rapid enzymatic degradation of mRNA and facilitates in vivo delivery of mRNA. This lipid-based nanoparticle carrier system is further stabilized by a polyethylene glycol 2000 lipid conjugate that provides a hydrophilic layer, thus prolonging half-life. The presence of lipid polyethylene glycol 2000 in mRNA vaccines has led to concern that this component could be implicated in anaphylaxis.6
COVID-19 antigens can give rise to varying clinical manifestations that are directly related to viral tissue damage or are indirectly induced by the antiviral immune response.13,14 Hyperactivation of the immune system to eradicate COVID-19 may trigger autoimmunity; several immune-mediated disorders have been described in individuals infected with SARS-CoV-2. Dermal manifestations include cutaneous rash and vasculitis.13-16 Crucial immunologic steps occur during SARS-CoV-2 infection that may link autoimmunity to COVID-19.13,14 In preliminary published data on the efficacy of the Moderna vaccine on 45 trial enrollees, 3 did not receive the second dose of vaccination, including 1 who developed urticaria on both legs 5 days after the first dose.1
Introduction of viral RNA can induce autoimmunity that can be explained by various phenomena, including epitope spreading, molecular mimicry, cryptic antigen, and bystander activation. Remarkably, more than one-third of immunogenic proteins in SARS-CoV-2 have potentially problematic homology to proteins that are key to the human adaptive immune system.5
Moreover, SARS-CoV-2 seems to induce organ injury through alternative mechanisms beyond direct viral infection, including immunologic injury. In some situations, hyperactivation of the immune response to SARS-CoV-2 RNA can result in autoimmune disease. COVID-19 has been associated with immune-mediated systemic or organ-selective manifestations, some of which fulfill the diagnostic or classification criteria of specific autoimmune diseases. It is unclear whether those medical disorders are the result of transitory postinfectious epiphenomena.5
A few studies have shown that patients with rheumatic disease have an incidence and prevalence of COVID-19 that is similar to the general population. A similar pattern has been detected in COVID-19 morbidity and mortality rates, even among patients with an autoimmune disease, such as rheumatoid arthritis and Sjögren syndrome.5,17 Furthermore, exacerbation of preexisting rheumatic symptoms may be due to hyperactivation of antiviral pathways in a person with an autoimmune disease.17-19 The findings in our patient suggested a direct role for the vaccine in skin manifestations, rather than for reactivation or development of new systemic autoimmune processes, such as systemic lupus erythematosus.
Exacerbation of psoriasis following COVID-19 vaccination has been described20; however, the case patient did not have a history of psoriasis. The mechanism(s) of such exacerbation remain unclear; COVID-19 vaccine–induced helper T cells (TH17) may play a role.21 Other skin manifestations encountered following COVID-19 vaccination include lichen planus, leukocytoclastic vasculitic rash, erythema multiforme–like rash, and pityriasis rosea–like rash.22-25 The immune mechanisms of these manifestations remain unclear.
The clinical presentation of delayed vaccination reactions can be attributed to the timing of symptoms and, in this case, the immune-mediated background of a psoriasiform reaction. Although adverse reactions to the SARS-CoV-2 mRNA vaccine are rare, more individuals should be studied after vaccination to confirm and better understand this phenomenon.
To the Editor:
The Moderna COVID-19 messenger RNA (mRNA) vaccine was authorized for use on December 18, 2020, with the second dose beginning on January 15, 2021.1-3 Some individuals who received the Moderna vaccine experienced an intense rash known as “COVID arm,” a harmless but bothersome adverse effect that typically appears within a week and is a localized and transient immunogenic response.4 COVID arm differs from most vaccine adverse effects. The rash emerges not immediately but 5 to 9 days after the initial dose—on average, 1 week later. Apart from being itchy, the rash does not appear to be harmful and is not a reason to hesitate getting vaccinated.
Dermatologists and allergists have been studying this adverse effect, which has been formally termed delayed cutaneous hypersensitivity. Of potential clinical consequence is that the efficacy of the mRNA COVID-19 vaccine may be harmed if postvaccination dermal reactions necessitate systemic corticosteroid therapy. Because this vaccine stimulates an immune response as viral RNA integrates in cells secondary to production of the spike protein of the virus, the skin may be affected secondarily and manifestations of any underlying disease may be aggravated.5 We report a patient who developed a psoriasiform dermatitis after the first dose of the Moderna vaccine.
A 65-year-old woman presented to her primary care physician because of the severity of psoriasiform dermatitis that developed 5 days after she received the first dose of the Moderna COVID-19 mRNA vaccine. The patient had a medical history of Sjögren syndrome. Her medication history was negative, and her family history was negative for autoimmune disease. Physical examination by primary care revealed an erythematous scaly rash with plaques and papules on the neck and back (Figure 1). The patient presented again to primary care 2 days later with swollen, painful, discolored digits (Figure 2) and a stiff, sore neck.
Laboratory results were positive for anti–Sjögren syndrome–related antigens A and B. A complete blood cell count; comprehensive metabolic panel; erythrocyte sedimentation rate; and assays of rheumatoid factor, C-reactive protein, and anti–cyclic citrullinated peptide were within reference range. A biopsy of a lesion on the back showed psoriasiform dermatitis with confluent parakeratosis and scattered necrotic keratinocytes. There was superficial perivascular inflammation with rare eosinophils (Figure 3).
The patient was treated with a course of systemic corticosteroids. The rash resolved in 1 week. She did not receive the second dose due to the rash.
Two mRNA COVID-19 vaccines—Pfizer BioNTech and Moderna—have been granted emergency use authorization by the US Food and Drug Administration.6 The safety profile of the mRNA-1273 vaccine for the median 2-month follow-up showed no safety concerns.3 Minor localized adverse effects (eg, pain, redness, swelling) have been observed more frequently with the vaccines than with placebo. Systemic symptoms, such as fever, fatigue, headache, and muscle and joint pain, also were seen somewhat more often with the vaccines than with placebo; most such effects occurred 24 to 48 hours after vaccination.3,6,7 The frequency of unsolicited adverse events and serious adverse events reported during the 28-day period after vaccination generally was similar among participants in the vaccine and placebo groups.3
There are 2 types of reactions to COVID-19 vaccination: immediate and delayed. Immediate reactions usually are due to anaphylaxis, requiring prompt recognition and treatment with epinephrine to stop rapid progression of life-threatening symptoms. Delayed reactions include localized reactions, such as urticaria and benign exanthema; serum sickness and serum sickness–like reactions; fever; and rare skin, organ, and neurologic sequelae.1,6-8
Cutaneous manifestations, present in 16% to 50% of patients with Sjögren syndrome, are considered one of the most common extraglandular presentations of the syndrome. They are classified as nonvascular (eg, xerosis, angular cheilitis, eyelid dermatitis, annular erythema) and vascular (eg, Raynaud phenomenon, vasculitis).9-11 Our patient did not have any of those findings. She had not taken any medications before the rash appeared, thereby ruling out a drug reaction.
The differential for our patient included post–urinary tract infection immune-reactive arthritis and rash, which is not typical with Escherichia coli infection but is described with infection with Chlamydia species and Salmonella species. Moreover, post–urinary tract infection immune-reactive arthritis and rash appear mostly on the palms and soles. Systemic lupus erythematosus–like rashes have a different histology and appear on sun-exposed areas; our patient’s rash was found mainly on unexposed areas.12
Because our patient received the Moderna vaccine 5 days before the rash appeared and later developed swelling of the digits with morning stiffness, a delayed serum sickness–like reaction secondary to COVID-19 vaccination was possible.3,6
COVID-19 mRNA vaccines developed by Pfizer-BioNTech and Moderna incorporate a lipid-based nanoparticle carrier system that prevents rapid enzymatic degradation of mRNA and facilitates in vivo delivery of mRNA. This lipid-based nanoparticle carrier system is further stabilized by a polyethylene glycol 2000 lipid conjugate that provides a hydrophilic layer, thus prolonging half-life. The presence of lipid polyethylene glycol 2000 in mRNA vaccines has led to concern that this component could be implicated in anaphylaxis.6
COVID-19 antigens can give rise to varying clinical manifestations that are directly related to viral tissue damage or are indirectly induced by the antiviral immune response.13,14 Hyperactivation of the immune system to eradicate COVID-19 may trigger autoimmunity; several immune-mediated disorders have been described in individuals infected with SARS-CoV-2. Dermal manifestations include cutaneous rash and vasculitis.13-16 Crucial immunologic steps occur during SARS-CoV-2 infection that may link autoimmunity to COVID-19.13,14 In preliminary published data on the efficacy of the Moderna vaccine on 45 trial enrollees, 3 did not receive the second dose of vaccination, including 1 who developed urticaria on both legs 5 days after the first dose.1
Introduction of viral RNA can induce autoimmunity that can be explained by various phenomena, including epitope spreading, molecular mimicry, cryptic antigen, and bystander activation. Remarkably, more than one-third of immunogenic proteins in SARS-CoV-2 have potentially problematic homology to proteins that are key to the human adaptive immune system.5
Moreover, SARS-CoV-2 seems to induce organ injury through alternative mechanisms beyond direct viral infection, including immunologic injury. In some situations, hyperactivation of the immune response to SARS-CoV-2 RNA can result in autoimmune disease. COVID-19 has been associated with immune-mediated systemic or organ-selective manifestations, some of which fulfill the diagnostic or classification criteria of specific autoimmune diseases. It is unclear whether those medical disorders are the result of transitory postinfectious epiphenomena.5
A few studies have shown that patients with rheumatic disease have an incidence and prevalence of COVID-19 that is similar to the general population. A similar pattern has been detected in COVID-19 morbidity and mortality rates, even among patients with an autoimmune disease, such as rheumatoid arthritis and Sjögren syndrome.5,17 Furthermore, exacerbation of preexisting rheumatic symptoms may be due to hyperactivation of antiviral pathways in a person with an autoimmune disease.17-19 The findings in our patient suggested a direct role for the vaccine in skin manifestations, rather than for reactivation or development of new systemic autoimmune processes, such as systemic lupus erythematosus.
Exacerbation of psoriasis following COVID-19 vaccination has been described20; however, the case patient did not have a history of psoriasis. The mechanism(s) of such exacerbation remain unclear; COVID-19 vaccine–induced helper T cells (TH17) may play a role.21 Other skin manifestations encountered following COVID-19 vaccination include lichen planus, leukocytoclastic vasculitic rash, erythema multiforme–like rash, and pityriasis rosea–like rash.22-25 The immune mechanisms of these manifestations remain unclear.
The clinical presentation of delayed vaccination reactions can be attributed to the timing of symptoms and, in this case, the immune-mediated background of a psoriasiform reaction. Although adverse reactions to the SARS-CoV-2 mRNA vaccine are rare, more individuals should be studied after vaccination to confirm and better understand this phenomenon.
- Jackson LA, Anderson EJ, Rouphael NG, et al; . An mRNA vaccine against SARS-CoV-2—preliminary report. N Engl J Med. 2020;383:1920-1931. doi:10.1056/NEJMoa2022483
- Anderson EJ, Rouphael NG, Widge AT, et al; . Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N Engl J Med. 2020;383:2427-2438. doi:10.1056/NEJMoa2028436
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403-416. doi:10.1056/NEJMoa2035389
- Weise E. ‘COVID arm’ rash seen after Moderna vaccine annoying but harmless, doctors say. USA Today. January 27, 2021. Accessed September 4, 2022. https://www.usatoday.com/story/news/health/2021/01/27/covid-arm-moderna-vaccine-rash-harmless-side-effect-doctors-say/4277725001/
- Talotta R, Robertson E. Autoimmunity as the comet tail of COVID-19 pandemic. World J Clin Cases. 2020;8:3621-3644. doi:10.12998/wjcc.v8.i17.3621
- Castells MC, Phillips EJ. Maintaining safety with SARS-CoV-2 vaccines. N Engl J Med. 2021;384:643-649. doi:10.1056/NEJMra2035343
- Polack FP, Thomas SJ, Kitchin N, et al; . Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603-2615. doi:10.1056/NEJMoa2034577
- Dooling K, McClung N, Chamberland M, et al. The Advisory Committee on Immunization Practices’ interim recommendation for allocating initial supplies of COVID-19 vaccine—United States, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1857-1859. doi:10.15585/mmwr.mm6949e1
- Roguedas AM, Misery L, Sassolas B, et al. Cutaneous manifestations of primary Sjögren’s syndrome are underestimated. Clin Exp Rheumatol. 2004;22:632-636.
- Katayama I. Dry skin manifestations in Sjögren syndrome and atopic dermatitis related to aberrant sudomotor function in inflammatory allergic skin diseases. Allergol Int. 2018;67:448-454. doi:10.1016/j.alit.2018.07.001
- Generali E, Costanzo A, Mainetti C, et al. Cutaneous and mucosal manifestations of Sjögren’s syndrome. Clin Rev Allergy Immunol. 2017;53:357-370. doi:10.1007/s12016-017-8639-y
- Chanprapaph K, Tankunakorn J, Suchonwanit P, et al. Dermatologic manifestations, histologic features and disease progression among cutaneous lupus erythematosus subtypes: a prospective observational study in Asians. Dermatol Ther (Heidelb). 2021;11:131-147. doi:10.1007/s13555-020-00471-y
- Ortega-Quijano D, Jimenez-Cauhe J, Selda-Enriquez G, et al. Algorithm for the classification of COVID-19 rashes. J Am Acad Dermatol. 2020;83:e103-e104. doi:10.1016/j.jaad.2020.05.034
- Rahimi H, Tehranchinia Z. A comprehensive review of cutaneous manifestations associated with COVID-19. Biomed Res Int. 2020;2020:1236520. doi:10.1155/2020/1236520
- Sachdeva M, Gianotti R, Shah M, et al. Cutaneous manifestations of COVID-19: report of three cases and a review of literature. J Dermatol Sci. 2020;98:75-81. doi:10.1016/j.jdermsci.2020.04.011
- Landa N, Mendieta-Eckert M, Fonda-Pascual P, et al. Chilblain-like lesions on feet and hands during the COVID-19 pandemic. Int J Dermatol. 2020;59:739-743. doi:10.1111/ijd.14937
- Dellavance A, Coelho Andrade LE. Immunologic derangement preceding clinical autoimmunity. Lupus. 2014;23:1305-1308. doi:10.1177/0961203314531346
- Parodi A, Gasparini G, Cozzani E. Could antiphospholipid antibodies contribute to coagulopathy in COVID-19? J Am Acad Dermatol. 2020;83:e249. doi:10.1016/j.jaad.2020.06.003
- Zhou Y, Han T, Chen J, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci. 2020;13:1077-1086. doi:10.1111/cts.12805
- Huang YW, Tsai TF. Exacerbation of psoriasis following COVID-19 vaccination: report from a single center. Front Med (Lausanne). 2021;8:812010. doi:10.3389/fmed.2021.812010
- Rouai M, Slimane MB, Sassi W, et al. Pustular rash triggered by Pfizer-BioNTech COVID-19 vaccination: a case report. Dermatol Ther. 2022:e15465. doi:10.1111/dth.15465
- Altun E, Kuzucular E. Leukocytoclastic vasculitis after COVID-19 vaccination. Dermatol Ther. 2022;35:e15279. doi:10.1111/dth.15279
- Buckley JE, Landis LN, Rapini RP. Pityriasis rosea-like rash after mRNA COVID-19 vaccination: a case report and review of the literature. JAAD Int. 2022;7:164-168. doi:10.1016/j.jdin.2022.01.009
- Gökçek GE, Öksüm Solak E, Çölgeçen E. Pityriasis rosea like eruption: a dermatological manifestation of Coronavac-COVID-19 vaccine. Dermatol Ther. 2022;35:e15256. doi:10.1111/dth.15256
- Kim MJ, Kim JW, Kim MS, et al. Generalized erythema multiforme-like skin rash following the first dose of COVID-19 vaccine (Pfizer-BioNTech). J Eur Acad Dermatol Venereol. 2022;36:e98-e100. doi:10.1111/jdv.17757
- Jackson LA, Anderson EJ, Rouphael NG, et al; . An mRNA vaccine against SARS-CoV-2—preliminary report. N Engl J Med. 2020;383:1920-1931. doi:10.1056/NEJMoa2022483
- Anderson EJ, Rouphael NG, Widge AT, et al; . Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N Engl J Med. 2020;383:2427-2438. doi:10.1056/NEJMoa2028436
- Baden LR, El Sahly HM, Essink B, et al; COVE Study Group. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403-416. doi:10.1056/NEJMoa2035389
- Weise E. ‘COVID arm’ rash seen after Moderna vaccine annoying but harmless, doctors say. USA Today. January 27, 2021. Accessed September 4, 2022. https://www.usatoday.com/story/news/health/2021/01/27/covid-arm-moderna-vaccine-rash-harmless-side-effect-doctors-say/4277725001/
- Talotta R, Robertson E. Autoimmunity as the comet tail of COVID-19 pandemic. World J Clin Cases. 2020;8:3621-3644. doi:10.12998/wjcc.v8.i17.3621
- Castells MC, Phillips EJ. Maintaining safety with SARS-CoV-2 vaccines. N Engl J Med. 2021;384:643-649. doi:10.1056/NEJMra2035343
- Polack FP, Thomas SJ, Kitchin N, et al; . Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603-2615. doi:10.1056/NEJMoa2034577
- Dooling K, McClung N, Chamberland M, et al. The Advisory Committee on Immunization Practices’ interim recommendation for allocating initial supplies of COVID-19 vaccine—United States, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1857-1859. doi:10.15585/mmwr.mm6949e1
- Roguedas AM, Misery L, Sassolas B, et al. Cutaneous manifestations of primary Sjögren’s syndrome are underestimated. Clin Exp Rheumatol. 2004;22:632-636.
- Katayama I. Dry skin manifestations in Sjögren syndrome and atopic dermatitis related to aberrant sudomotor function in inflammatory allergic skin diseases. Allergol Int. 2018;67:448-454. doi:10.1016/j.alit.2018.07.001
- Generali E, Costanzo A, Mainetti C, et al. Cutaneous and mucosal manifestations of Sjögren’s syndrome. Clin Rev Allergy Immunol. 2017;53:357-370. doi:10.1007/s12016-017-8639-y
- Chanprapaph K, Tankunakorn J, Suchonwanit P, et al. Dermatologic manifestations, histologic features and disease progression among cutaneous lupus erythematosus subtypes: a prospective observational study in Asians. Dermatol Ther (Heidelb). 2021;11:131-147. doi:10.1007/s13555-020-00471-y
- Ortega-Quijano D, Jimenez-Cauhe J, Selda-Enriquez G, et al. Algorithm for the classification of COVID-19 rashes. J Am Acad Dermatol. 2020;83:e103-e104. doi:10.1016/j.jaad.2020.05.034
- Rahimi H, Tehranchinia Z. A comprehensive review of cutaneous manifestations associated with COVID-19. Biomed Res Int. 2020;2020:1236520. doi:10.1155/2020/1236520
- Sachdeva M, Gianotti R, Shah M, et al. Cutaneous manifestations of COVID-19: report of three cases and a review of literature. J Dermatol Sci. 2020;98:75-81. doi:10.1016/j.jdermsci.2020.04.011
- Landa N, Mendieta-Eckert M, Fonda-Pascual P, et al. Chilblain-like lesions on feet and hands during the COVID-19 pandemic. Int J Dermatol. 2020;59:739-743. doi:10.1111/ijd.14937
- Dellavance A, Coelho Andrade LE. Immunologic derangement preceding clinical autoimmunity. Lupus. 2014;23:1305-1308. doi:10.1177/0961203314531346
- Parodi A, Gasparini G, Cozzani E. Could antiphospholipid antibodies contribute to coagulopathy in COVID-19? J Am Acad Dermatol. 2020;83:e249. doi:10.1016/j.jaad.2020.06.003
- Zhou Y, Han T, Chen J, et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci. 2020;13:1077-1086. doi:10.1111/cts.12805
- Huang YW, Tsai TF. Exacerbation of psoriasis following COVID-19 vaccination: report from a single center. Front Med (Lausanne). 2021;8:812010. doi:10.3389/fmed.2021.812010
- Rouai M, Slimane MB, Sassi W, et al. Pustular rash triggered by Pfizer-BioNTech COVID-19 vaccination: a case report. Dermatol Ther. 2022:e15465. doi:10.1111/dth.15465
- Altun E, Kuzucular E. Leukocytoclastic vasculitis after COVID-19 vaccination. Dermatol Ther. 2022;35:e15279. doi:10.1111/dth.15279
- Buckley JE, Landis LN, Rapini RP. Pityriasis rosea-like rash after mRNA COVID-19 vaccination: a case report and review of the literature. JAAD Int. 2022;7:164-168. doi:10.1016/j.jdin.2022.01.009
- Gökçek GE, Öksüm Solak E, Çölgeçen E. Pityriasis rosea like eruption: a dermatological manifestation of Coronavac-COVID-19 vaccine. Dermatol Ther. 2022;35:e15256. doi:10.1111/dth.15256
- Kim MJ, Kim JW, Kim MS, et al. Generalized erythema multiforme-like skin rash following the first dose of COVID-19 vaccine (Pfizer-BioNTech). J Eur Acad Dermatol Venereol. 2022;36:e98-e100. doi:10.1111/jdv.17757
PRACTICE POINTS
- The differential diagnosis for a new-onset psoriasiform rash in an elderly patient should include a vaccine-related rash.
- A rash following vaccination that necessitates systemic corticosteroid therapy can decrease vaccine efficacy.
.")
Yellow Papules and Plaques on a Child
The Diagnosis: Tuberous Xanthoma
The skin biopsy revealed a nodular collection of foam cells (quiz image [bottom]). Tuberous xanthoma was the most likely diagnosis based on the patient’s history as well as the clinical and histologic findings. Tuberous xanthomas are flat or elevated nodules in the dermis and subcutaneous tissue, commonly occurring on the skin over the joints.1 Smaller nodules and papules often are referred to as tuberoeruptive xanthomas and exist on a continuum with the larger tuberous xanthomas. All xanthomas appear histologically similar, with collections of foam cells present within the dermis.2 Foam cells form when serum lipoproteins diffuse through capillary walls, deposit in the skin or tendons, and are scavenged by monocytes.3 Tuberous xanthomas, along with tendinous, eruptive, and planar xanthomas, are the most likely to be associated with hyperlipidemia.4 They may indicate an underlying disorder of lipid metabolism, such as familial hypercholesterolemia.1,3 This is the most common cause of inheritable cardiovascular disease, with a prevalence of approximately 1:250.2 Premature cardiovascular disease risk increases 2 to 4 times in patients with familial hypercholesterolemia and tendinous xanthomas,1 illustrating that recognition of cutaneous lesions can lead to earlier diagnosis and prevention of patient morbidity and mortality.
Juvenile xanthogranuloma typically presents as smooth yellow papules or nodules on the head and neck, with a characteristic “setting-sun” appearance (ie, yellow center with an erythematous halo) on dermoscopy.5 Histologically, juvenile xanthogranulomas are composed of foam cells and a mixed lymphohistiocytic infiltrate with eosinophils within the dermis. Giant cells with a ring of nuclei surrounded by cytoplasm containing lipid vacuoles (called Touton giant cells) are characteristic (Figure 1). In contrast to tuberous xanthomas, juvenile xanthogranulomas often present within the first year of life.6
. Reference bar indicates 50 mm.")
Keloid scars are more prevalent in patients with skin of color. They are characterized by eosinophilic keloidal collagen with a whorled proliferation of fibroblasts on histology (Figure 2).7 They occur spontaneously or at sites of injury and present as bluish-red or flesh-colored firm papules or nodules.8 In our patient, keloid scars were an unlikely diagnosis due to the lack of trauma and the absence of keloidal collagen on histology.
.")
Necrobiosis lipoidica diabeticorum typically presents as an erythematous, yellow-brown, circular plaque on the anterior lower leg in patients with diabetes mellitus; it rarely occurs in children.9 Microscopy shows palisaded granulomas surrounding necrobiotic collagen arranged horizontally in a layer cake–like fashion (Figure 3).9,10 The etiology of necrobiosis lipoidica diabeticorum currently is unknown, though immune complex deposition may contribute to its pathology. It has been associated with type 1 diabetes mellitus, though severity of the lesions is not associated with extent of glycemic control.10
.")
Rosai-Dorfman disease is an uncommon disorder characterized by a proliferation of histiocytes that most often presents as bilateral cervical lymphadenopathy in children and young adults but rarely can present with cutaneous lesions when extranodal involvement is present.11,12 The cutaneous form most commonly presents as red papules or nodules. On histology, the lesions exhibit a nodular dermal proliferation of histiocytes and smaller lymphocytoid cells with a marbled or starry sky–like appearance on low power (Figure 4). On higher magnification, the characteristic finding of emperipolesis can be seen.11 On immunohistochemistry, the histiocytes stain positively for CD68 and S-100. Although the pathogenesis currently is unknown, evidence of clonality indicates the disease may be related to a neoplastic process.12
.")
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188. doi:10.5507/bp.2014.016
- Ison HE, Clarke SL, Knowles JW. Familial hypercholesterolemia. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews. University of Washington, Seattle; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK174884/
- Sathiyakumar V, Jones SR, Martin SS. Xanthomas and lipoprotein disorders. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw Hill; 2019.
- Massangale WT. Xanthomas. In: Bolognia JL, Schaffer JV, Cerroni L, et al, eds. Dermatology. Elsevier; 2018:1634-1643.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls. StatPearls Publishing; 2021. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Hernández-San Martín MJ, Vargas-Mora P, Aranibar L. Juvenile xanthogranuloma: an entity with a wide clinical spectrum. Actas Dermosifiliogr (Engl Ed). 2020;111:725-733. doi:10.1016/j.ad.2020.07.004
- Lee JY, Yang C, Chao S, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathology. 2004;26:379-384.
- Wolff K, Johnson R, Saavedra AP, et al. Benign neoplasms and hyperplasias. In: Wolff K, Johnson R, Saavedra AP, et al, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw Hill; 2017:141-188.
- Bonura C, Frontino G, Rigamonti A, et al. Necrobiosis lipoidica diabeticorum: a pediatric case report. Dermatoendocrinol. 2014;6:E27790. doi:10.4161/derm.27790
- Lepe K, Riley CA, Salazar FJ. Necrobiosis lipoidica. StatPearls. StatPearls Publishing; 2021. https://www-ncbi-nlm-nih-gov.proxy.kumc.edu/books/NBK459318/
- Parrent T, Clark T, Hall D. Cutaneous Rosai-Dorfman disease. Cutis. 2012;90:237-238.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697-705. doi:10.1136/jclinpath-2020-206733
The Diagnosis: Tuberous Xanthoma
The skin biopsy revealed a nodular collection of foam cells (quiz image [bottom]). Tuberous xanthoma was the most likely diagnosis based on the patient’s history as well as the clinical and histologic findings. Tuberous xanthomas are flat or elevated nodules in the dermis and subcutaneous tissue, commonly occurring on the skin over the joints.1 Smaller nodules and papules often are referred to as tuberoeruptive xanthomas and exist on a continuum with the larger tuberous xanthomas. All xanthomas appear histologically similar, with collections of foam cells present within the dermis.2 Foam cells form when serum lipoproteins diffuse through capillary walls, deposit in the skin or tendons, and are scavenged by monocytes.3 Tuberous xanthomas, along with tendinous, eruptive, and planar xanthomas, are the most likely to be associated with hyperlipidemia.4 They may indicate an underlying disorder of lipid metabolism, such as familial hypercholesterolemia.1,3 This is the most common cause of inheritable cardiovascular disease, with a prevalence of approximately 1:250.2 Premature cardiovascular disease risk increases 2 to 4 times in patients with familial hypercholesterolemia and tendinous xanthomas,1 illustrating that recognition of cutaneous lesions can lead to earlier diagnosis and prevention of patient morbidity and mortality.
Juvenile xanthogranuloma typically presents as smooth yellow papules or nodules on the head and neck, with a characteristic “setting-sun” appearance (ie, yellow center with an erythematous halo) on dermoscopy.5 Histologically, juvenile xanthogranulomas are composed of foam cells and a mixed lymphohistiocytic infiltrate with eosinophils within the dermis. Giant cells with a ring of nuclei surrounded by cytoplasm containing lipid vacuoles (called Touton giant cells) are characteristic (Figure 1). In contrast to tuberous xanthomas, juvenile xanthogranulomas often present within the first year of life.6
Keloid scars are more prevalent in patients with skin of color. They are characterized by eosinophilic keloidal collagen with a whorled proliferation of fibroblasts on histology (Figure 2).7 They occur spontaneously or at sites of injury and present as bluish-red or flesh-colored firm papules or nodules.8 In our patient, keloid scars were an unlikely diagnosis due to the lack of trauma and the absence of keloidal collagen on histology.
Necrobiosis lipoidica diabeticorum typically presents as an erythematous, yellow-brown, circular plaque on the anterior lower leg in patients with diabetes mellitus; it rarely occurs in children.9 Microscopy shows palisaded granulomas surrounding necrobiotic collagen arranged horizontally in a layer cake–like fashion (Figure 3).9,10 The etiology of necrobiosis lipoidica diabeticorum currently is unknown, though immune complex deposition may contribute to its pathology. It has been associated with type 1 diabetes mellitus, though severity of the lesions is not associated with extent of glycemic control.10
Rosai-Dorfman disease is an uncommon disorder characterized by a proliferation of histiocytes that most often presents as bilateral cervical lymphadenopathy in children and young adults but rarely can present with cutaneous lesions when extranodal involvement is present.11,12 The cutaneous form most commonly presents as red papules or nodules. On histology, the lesions exhibit a nodular dermal proliferation of histiocytes and smaller lymphocytoid cells with a marbled or starry sky–like appearance on low power (Figure 4). On higher magnification, the characteristic finding of emperipolesis can be seen.11 On immunohistochemistry, the histiocytes stain positively for CD68 and S-100. Although the pathogenesis currently is unknown, evidence of clonality indicates the disease may be related to a neoplastic process.12
The Diagnosis: Tuberous Xanthoma
The skin biopsy revealed a nodular collection of foam cells (quiz image [bottom]). Tuberous xanthoma was the most likely diagnosis based on the patient’s history as well as the clinical and histologic findings. Tuberous xanthomas are flat or elevated nodules in the dermis and subcutaneous tissue, commonly occurring on the skin over the joints.1 Smaller nodules and papules often are referred to as tuberoeruptive xanthomas and exist on a continuum with the larger tuberous xanthomas. All xanthomas appear histologically similar, with collections of foam cells present within the dermis.2 Foam cells form when serum lipoproteins diffuse through capillary walls, deposit in the skin or tendons, and are scavenged by monocytes.3 Tuberous xanthomas, along with tendinous, eruptive, and planar xanthomas, are the most likely to be associated with hyperlipidemia.4 They may indicate an underlying disorder of lipid metabolism, such as familial hypercholesterolemia.1,3 This is the most common cause of inheritable cardiovascular disease, with a prevalence of approximately 1:250.2 Premature cardiovascular disease risk increases 2 to 4 times in patients with familial hypercholesterolemia and tendinous xanthomas,1 illustrating that recognition of cutaneous lesions can lead to earlier diagnosis and prevention of patient morbidity and mortality.
Juvenile xanthogranuloma typically presents as smooth yellow papules or nodules on the head and neck, with a characteristic “setting-sun” appearance (ie, yellow center with an erythematous halo) on dermoscopy.5 Histologically, juvenile xanthogranulomas are composed of foam cells and a mixed lymphohistiocytic infiltrate with eosinophils within the dermis. Giant cells with a ring of nuclei surrounded by cytoplasm containing lipid vacuoles (called Touton giant cells) are characteristic (Figure 1). In contrast to tuberous xanthomas, juvenile xanthogranulomas often present within the first year of life.6
Keloid scars are more prevalent in patients with skin of color. They are characterized by eosinophilic keloidal collagen with a whorled proliferation of fibroblasts on histology (Figure 2).7 They occur spontaneously or at sites of injury and present as bluish-red or flesh-colored firm papules or nodules.8 In our patient, keloid scars were an unlikely diagnosis due to the lack of trauma and the absence of keloidal collagen on histology.
Necrobiosis lipoidica diabeticorum typically presents as an erythematous, yellow-brown, circular plaque on the anterior lower leg in patients with diabetes mellitus; it rarely occurs in children.9 Microscopy shows palisaded granulomas surrounding necrobiotic collagen arranged horizontally in a layer cake–like fashion (Figure 3).9,10 The etiology of necrobiosis lipoidica diabeticorum currently is unknown, though immune complex deposition may contribute to its pathology. It has been associated with type 1 diabetes mellitus, though severity of the lesions is not associated with extent of glycemic control.10
Rosai-Dorfman disease is an uncommon disorder characterized by a proliferation of histiocytes that most often presents as bilateral cervical lymphadenopathy in children and young adults but rarely can present with cutaneous lesions when extranodal involvement is present.11,12 The cutaneous form most commonly presents as red papules or nodules. On histology, the lesions exhibit a nodular dermal proliferation of histiocytes and smaller lymphocytoid cells with a marbled or starry sky–like appearance on low power (Figure 4). On higher magnification, the characteristic finding of emperipolesis can be seen.11 On immunohistochemistry, the histiocytes stain positively for CD68 and S-100. Although the pathogenesis currently is unknown, evidence of clonality indicates the disease may be related to a neoplastic process.12
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188. doi:10.5507/bp.2014.016
- Ison HE, Clarke SL, Knowles JW. Familial hypercholesterolemia. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews. University of Washington, Seattle; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK174884/
- Sathiyakumar V, Jones SR, Martin SS. Xanthomas and lipoprotein disorders. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw Hill; 2019.
- Massangale WT. Xanthomas. In: Bolognia JL, Schaffer JV, Cerroni L, et al, eds. Dermatology. Elsevier; 2018:1634-1643.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls. StatPearls Publishing; 2021. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Hernández-San Martín MJ, Vargas-Mora P, Aranibar L. Juvenile xanthogranuloma: an entity with a wide clinical spectrum. Actas Dermosifiliogr (Engl Ed). 2020;111:725-733. doi:10.1016/j.ad.2020.07.004
- Lee JY, Yang C, Chao S, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathology. 2004;26:379-384.
- Wolff K, Johnson R, Saavedra AP, et al. Benign neoplasms and hyperplasias. In: Wolff K, Johnson R, Saavedra AP, et al, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw Hill; 2017:141-188.
- Bonura C, Frontino G, Rigamonti A, et al. Necrobiosis lipoidica diabeticorum: a pediatric case report. Dermatoendocrinol. 2014;6:E27790. doi:10.4161/derm.27790
- Lepe K, Riley CA, Salazar FJ. Necrobiosis lipoidica. StatPearls. StatPearls Publishing; 2021. https://www-ncbi-nlm-nih-gov.proxy.kumc.edu/books/NBK459318/
- Parrent T, Clark T, Hall D. Cutaneous Rosai-Dorfman disease. Cutis. 2012;90:237-238.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697-705. doi:10.1136/jclinpath-2020-206733
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188. doi:10.5507/bp.2014.016
- Ison HE, Clarke SL, Knowles JW. Familial hypercholesterolemia. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews. University of Washington, Seattle; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK174884/
- Sathiyakumar V, Jones SR, Martin SS. Xanthomas and lipoprotein disorders. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw Hill; 2019.
- Massangale WT. Xanthomas. In: Bolognia JL, Schaffer JV, Cerroni L, et al, eds. Dermatology. Elsevier; 2018:1634-1643.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls. StatPearls Publishing; 2021. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Hernández-San Martín MJ, Vargas-Mora P, Aranibar L. Juvenile xanthogranuloma: an entity with a wide clinical spectrum. Actas Dermosifiliogr (Engl Ed). 2020;111:725-733. doi:10.1016/j.ad.2020.07.004
- Lee JY, Yang C, Chao S, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathology. 2004;26:379-384.
- Wolff K, Johnson R, Saavedra AP, et al. Benign neoplasms and hyperplasias. In: Wolff K, Johnson R, Saavedra AP, et al, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 8th ed. McGraw Hill; 2017:141-188.
- Bonura C, Frontino G, Rigamonti A, et al. Necrobiosis lipoidica diabeticorum: a pediatric case report. Dermatoendocrinol. 2014;6:E27790. doi:10.4161/derm.27790
- Lepe K, Riley CA, Salazar FJ. Necrobiosis lipoidica. StatPearls. StatPearls Publishing; 2021. https://www-ncbi-nlm-nih-gov.proxy.kumc.edu/books/NBK459318/
- Parrent T, Clark T, Hall D. Cutaneous Rosai-Dorfman disease. Cutis. 2012;90:237-238.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697-705. doi:10.1136/jclinpath-2020-206733
A 3-year-old girl presented with raised, firm, enlarging, asymptomatic, well-defined, subcutaneous papules, plaques, and nodules on the hands, knees, and posterior ankles of 1 year’s duration. The patient’s mother stated that the lesions began on the ankles (top), and she initially believed them to be due to friction from the child’s shoes until the more recent involvement of the knees and hands. The patient’s father, paternal grandfather, and paternal great-grandfather had a history of elevated cholesterol levels. A shave biopsy was performed (bottom).


. Reference bar indicates 50 mm.")
An adolescent male presents with an eroded bump on the temple
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.

Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.

Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.
Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.
Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.
Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.
Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
Painful and Pruritic Eruptions on the Entire Body
The Diagnosis: IgA Pemphigus
Histopathology revealed a neutrophilic pustule and vesicle formation underlying the corneal layer (Figure). Direct immunofluorescence (DIF) showed weak positive staining for IgA within the intercellular keratinocyte in the epithelial compartment and a negative pattern with IgG, IgM, C3, and fibrinogen. The patient received a 40-mg intralesional triamcinolone injection and was placed on an oral prednisone 50-mg taper within 5 days. The plaques, bullae, and pustules began to resolve, but the lesions returned 1 day later. Oral prednisone 10 mg daily was initiated for 1 month, which resulted in full resolution of the lesions.
.")
IgA pemphigus is a rare autoimmune disorder characterized by the occurrence of painful pruritic blisters caused by circulating IgA antibodies, which react against keratinocyte cellular components responsible for mediating cell-to-cell adherence.1 The etiology of IgA pemphigus presently remains elusive, though it has been reported to occur concomitantly with several chronic malignancies and inflammatory conditions. Although its etiology is unknown, IgA pemphigus most commonly is treated with oral dapsone and corticosteroids.2
IgA pemphigus can be divided into 2 primary subtypes: subcorneal pustular dermatosis and intraepidermal neutrophilic dermatosis.1,3 The former is characterized by intercellular deposition of IgA that reacts to the glycoprotein desmocollin-1 in the upper layer of the epidermis. Intraepidermal neutrophilic dermatosis is distinguished by the presence of autoantibodies against the desmoglein members of the cadherin superfamily of proteins. Additionally, unlike subcorneal pustular dermatosis, intraepidermal neutrophilic dermatosis autoantibody reactivity occurs in the lower epidermis.4
The differential includes dermatitis herpetiformis, which is commonly seen on the elbows, knees, and buttocks, with DIF showing IgA deposition at the dermal papillae. Pemphigus foliaceus is distributed on the scalp, face, and trunk, with DIF showing IgG intercellular deposition. Pustular psoriasis presents as erythematous sterile pustules in a more localized annular pattern. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) has similar clinical and histological findings to IgA pemphigus; however, DIF is negative.
- Kridin K, Patel PM, Jones VA, et al. IgA pemphigus: a systematic review. J Am Acad Dermatol. 2020;82:1386-1392.
- Moreno ACL, Santi CG, Gabbi TVB, et al. IgA pemphigus: case series with emphasis on therapeutic response. J Am Acad Dermatol. 2014;70:200-201.
- Niimi Y, Kawana S, Kusunoki T. IgA pemphigus: a case report and its characteristic clinical features compared with subcorneal pustular dermatosis. J Am Acad Dermatol. 2000;43:546-549.
- Aslanova M, Yarrarapu SNS, Zito PM. IgA pemphigus. StatPearls. StatPearls Publishing; 2021.
The Diagnosis: IgA Pemphigus
Histopathology revealed a neutrophilic pustule and vesicle formation underlying the corneal layer (Figure). Direct immunofluorescence (DIF) showed weak positive staining for IgA within the intercellular keratinocyte in the epithelial compartment and a negative pattern with IgG, IgM, C3, and fibrinogen. The patient received a 40-mg intralesional triamcinolone injection and was placed on an oral prednisone 50-mg taper within 5 days. The plaques, bullae, and pustules began to resolve, but the lesions returned 1 day later. Oral prednisone 10 mg daily was initiated for 1 month, which resulted in full resolution of the lesions.
IgA pemphigus is a rare autoimmune disorder characterized by the occurrence of painful pruritic blisters caused by circulating IgA antibodies, which react against keratinocyte cellular components responsible for mediating cell-to-cell adherence.1 The etiology of IgA pemphigus presently remains elusive, though it has been reported to occur concomitantly with several chronic malignancies and inflammatory conditions. Although its etiology is unknown, IgA pemphigus most commonly is treated with oral dapsone and corticosteroids.2
IgA pemphigus can be divided into 2 primary subtypes: subcorneal pustular dermatosis and intraepidermal neutrophilic dermatosis.1,3 The former is characterized by intercellular deposition of IgA that reacts to the glycoprotein desmocollin-1 in the upper layer of the epidermis. Intraepidermal neutrophilic dermatosis is distinguished by the presence of autoantibodies against the desmoglein members of the cadherin superfamily of proteins. Additionally, unlike subcorneal pustular dermatosis, intraepidermal neutrophilic dermatosis autoantibody reactivity occurs in the lower epidermis.4
The differential includes dermatitis herpetiformis, which is commonly seen on the elbows, knees, and buttocks, with DIF showing IgA deposition at the dermal papillae. Pemphigus foliaceus is distributed on the scalp, face, and trunk, with DIF showing IgG intercellular deposition. Pustular psoriasis presents as erythematous sterile pustules in a more localized annular pattern. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) has similar clinical and histological findings to IgA pemphigus; however, DIF is negative.
The Diagnosis: IgA Pemphigus
Histopathology revealed a neutrophilic pustule and vesicle formation underlying the corneal layer (Figure). Direct immunofluorescence (DIF) showed weak positive staining for IgA within the intercellular keratinocyte in the epithelial compartment and a negative pattern with IgG, IgM, C3, and fibrinogen. The patient received a 40-mg intralesional triamcinolone injection and was placed on an oral prednisone 50-mg taper within 5 days. The plaques, bullae, and pustules began to resolve, but the lesions returned 1 day later. Oral prednisone 10 mg daily was initiated for 1 month, which resulted in full resolution of the lesions.
IgA pemphigus is a rare autoimmune disorder characterized by the occurrence of painful pruritic blisters caused by circulating IgA antibodies, which react against keratinocyte cellular components responsible for mediating cell-to-cell adherence.1 The etiology of IgA pemphigus presently remains elusive, though it has been reported to occur concomitantly with several chronic malignancies and inflammatory conditions. Although its etiology is unknown, IgA pemphigus most commonly is treated with oral dapsone and corticosteroids.2
IgA pemphigus can be divided into 2 primary subtypes: subcorneal pustular dermatosis and intraepidermal neutrophilic dermatosis.1,3 The former is characterized by intercellular deposition of IgA that reacts to the glycoprotein desmocollin-1 in the upper layer of the epidermis. Intraepidermal neutrophilic dermatosis is distinguished by the presence of autoantibodies against the desmoglein members of the cadherin superfamily of proteins. Additionally, unlike subcorneal pustular dermatosis, intraepidermal neutrophilic dermatosis autoantibody reactivity occurs in the lower epidermis.4
The differential includes dermatitis herpetiformis, which is commonly seen on the elbows, knees, and buttocks, with DIF showing IgA deposition at the dermal papillae. Pemphigus foliaceus is distributed on the scalp, face, and trunk, with DIF showing IgG intercellular deposition. Pustular psoriasis presents as erythematous sterile pustules in a more localized annular pattern. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) has similar clinical and histological findings to IgA pemphigus; however, DIF is negative.
- Kridin K, Patel PM, Jones VA, et al. IgA pemphigus: a systematic review. J Am Acad Dermatol. 2020;82:1386-1392.
- Moreno ACL, Santi CG, Gabbi TVB, et al. IgA pemphigus: case series with emphasis on therapeutic response. J Am Acad Dermatol. 2014;70:200-201.
- Niimi Y, Kawana S, Kusunoki T. IgA pemphigus: a case report and its characteristic clinical features compared with subcorneal pustular dermatosis. J Am Acad Dermatol. 2000;43:546-549.
- Aslanova M, Yarrarapu SNS, Zito PM. IgA pemphigus. StatPearls. StatPearls Publishing; 2021.
- Kridin K, Patel PM, Jones VA, et al. IgA pemphigus: a systematic review. J Am Acad Dermatol. 2020;82:1386-1392.
- Moreno ACL, Santi CG, Gabbi TVB, et al. IgA pemphigus: case series with emphasis on therapeutic response. J Am Acad Dermatol. 2014;70:200-201.
- Niimi Y, Kawana S, Kusunoki T. IgA pemphigus: a case report and its characteristic clinical features compared with subcorneal pustular dermatosis. J Am Acad Dermatol. 2000;43:546-549.
- Aslanova M, Yarrarapu SNS, Zito PM. IgA pemphigus. StatPearls. StatPearls Publishing; 2021.
A 36-year-old man presented with painful tender blisters and rashes on the entire body, including the ears and tongue. The rash began as a few pinpointed red dots on the abdomen, which subsequently increased in size and spread over the last week. He initially felt red and flushed and noticed new lesions appearing throughout the day. He did not attempt any specific treatment for these lesions. The patient tested positive for COVID-19 four months prior to the skin eruption. He denied systemic symptoms, smoking, or recent travel. He had no history of skin cancer, skin disorders, HIV, or hepatitis. He had no known medication allergies. Physical examination revealed multiple disseminated pustules on the ears, superficial ulcerations on the tongue, and blisters on the right lip. Few lesions were tender to the touch and drained clear fluid. Bacterial, viral, HIV, herpes, and rapid plasma reagin culture and laboratory screenings were negative. He was started on valaciclovir and cephalexin; however, no improvement was noticed. Punch biopsies were taken from the blisters on the chest and perilesional area.


Cutaneous and Subcutaneous Perineuriomas in 2 Pediatric Patients
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
. Reference bar indicates 500 µm.")
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
(original magnification ×15,000).")
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
. Reference bar indicates 100 µm.")
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
. Reference bar indicates 100 µm.")
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
Perineuriomas are benign, slow-growing tumors derived from perineurial cells,1 which form the structurally supportive perineurium that surrounds individual nerve fascicles.2,3 Perineuriomas are classified into 2 main forms: intraneural or extraneural.4 Intraneural perineuriomas are found within the border of the peripheral nerve,5 while extraneural perineuriomas usually are found in soft tissue and skin. Extraneural perineuriomas can be further classified into variants based on their histologic appearance, including reticular, sclerosing, and plexiform subtypes. Extraneural perineuriomas usually present on the extremities or trunk of young to middle-aged adults as a well-circumscribed, painless, subcutaneous masses.1 These tumors are especially unusual in children.4 We present 2 extraneural perineurioma cases in children, and we review the pertinent diagnostic features of perineurioma as well as the presentation in the pediatric population.
Case Reports
Patient 1—A 10-year-old boy with a history of cerebral palsy and related comorbidities presented to the clinic for evaluation of a lesion on the thigh with no associated pain, irritation, erythema, or drainage. Physical examination revealed a soft, pedunculated, mobile nodule on the right medial thigh. An elliptical excision was performed. Gross examination demonstrated a 2.0×2.0×1.8-cm polypoid nodule. Histologic examination showed a dermal-based proliferation of bland spindle cells (Figure 1). The cytomorphology was characterized by elongated tapering nuclei and many areas with delicate bipolar cytoplasmic processes. The constituent cells were arranged in a whorled pattern in a variably myxoid to collagenous stroma. The tumor cells were multifocally positive for CD34; focally positive for smooth muscle actin (SMA); and negative for S-100, epithelial membrane antigen (EMA), GLUT1, claudin-1, STAT6, and desmin. Rb protein was intact. The CD34 immunostain highlighted the cytoplasmic processes. Electron microscopy was performed because the immunohistochemical results were nonspecific despite the favorable histologic features for perineurioma and showed pinocytic vesicles with delicate cytoplasmic processes, characteristic of perineurioma (Figure 2). Follow-up visits were related to the management of multiple comorbidities; no known recurrence of the lesion was documented.
Patient 2—A 15-year-old adolescent boy with no notable medical history presented to the pediatric clinic for a bump on the right upper arm of 4 to 5 months’ duration. He did not recall an injury to the area and denied change in size, redness, bruising, or pain of the lesion. Ultrasonography demonstrated a 2.6×2.3×1.3-cm hypoechoic and slightly heterogeneous, well-circumscribed, subcutaneous mass with internal vascularity. The patient was then referred to a pediatric surgeon. The clinical differential included a lipoma, lymphadenopathy, or sebaceous cyst. An excision was performed. Gross inspection demonstrated a 7-g, 2.8×2.6×1.8-cm, homogeneous, tan-pink, rubbery nodule with minimal surrounding soft tissue. Histologic examination showed a bland proliferation of spindle cells with storiform and whorled patterns (Figure 3). No notable nuclear atypia or necrosis was identified. The tumor cells were focally positive for EMA (Figure 4), claudin-1, and CD34 and negative for S-100, SOX10, GLUT1, desmin, STAT6, pankeratin AE1/AE3, and SMA. The diagnosis of perineurioma was rendered. No recurrence of the lesion was appreciated clinically on a 6-month follow-up examination.
Comment
Characteristics of Perineuriomas—On gross evaluation, perineuriomas are firm, gray-white, and well circumscribed but not encapsulated. Histologically, perineuriomas can have a storiform, whorled, or lamellar pattern of spindle cells. Perivascular whorls can be a histologic clue. The spindle cells are bland appearing and typically are elongated and slender but can appear slightly ovoid and plump. The background stroma can be myxoid, collagenous, or mixed. There usually is no atypia, and mitotic figures are rare.2,3,6,7 Intraneural perineuriomas vary architecturally in that they display a unique onion bulb–like appearance in which whorls of cytoplasmic material of variable sizes surround central axons.3
Diagnosis—The diagnosis of perineuriomas usually requires characteristic immunohistochemical and sometimes ultrastructural features. Perineuriomas are positive for EMA and GLUT1 and variable for CD34.6 Approximately 20% to 91% will be positive for claudin-1, a tight junction protein associated with perineuriomas.8 Of note, EMA and GLUT1 usually are positive in both neoplastic and nonneoplastic perineurial cells.9,10 Occasionally, these tumors can be focally positive for SMA and negative for S-100 and glial fibrillary acidic protein. The bipolar, thin, delicate, cytoplasmic processes with long-tapering nuclei may be easier to appreciate on electron microscopy than on conventional light microscopy. In addition, the cells contain pinocytotic vesicles and a discontinuous external lamina, which may be helpful for diagnosis.10
Genetics—Genetic alterations in perineurioma continue to be elucidated. Although many soft tissue perineuriomas possess deletion of chromosome 22q material, this is not a consistent finding and is not pathognomonic. Notably, the NF2 tumor suppressor gene is found on chromosome 22.11 For the sclerosing variant of perineurioma, rearrangements or deletions of chromosome 10q have been described. A study of 14 soft tissue/extraneural perineuriomas using whole-exome sequencing and single nucleotide polymorphism array showed 6 cases of recurrent chromosome 22q deletions containing the NF2 locus and 4 cases with a previously unreported finding of chromosome 17q deletions containing the NF1 locus that were mutually exclusive events in all but 1 case.12 Although perineuriomas can harbor NF1 or NF2 mutations, perineuriomas are not considered to be associated with neurofibromatosis type 1 or 2 (NF1 or NF2, respectively). Patients with NF1 or NF2 and perineurioma are exceedingly rare. One pediatric patient with both soft tissue perineurioma and NF1 has been reported in the literature.13
Differential Diagnosis—Perineuriomas should be distinguished from other benign neural neoplasms of the skin and soft tissue. Commonly considered in the differential diagnosis is schwannoma and neurofibroma. Schwannomas are encapsulated epineurial nerve sheath tumors comprised of a neoplastic proliferation of Schwann cells. Schwannomas morphologically differ from perineuriomas because of the presence of the hypercellular Antoni A with Verocay bodies and the hypocellular myxoid Antoni B patterns of spindle cells with elongated wavy nuclei and tapered ends. Other features include hyalinized vessels, hemosiderin deposition, cystic degeneration, and/or degenerative atypia.3,14 Importantly, the constituent cells of schwannomas are positive for S-100 and SOX10 and negative for EMA.3 Neurofibromas consist of fascicles and whorls of Schwann cells in a background myxoid stroma with scattered mast cells, lymphocytes, fibroblasts, and perineurial cells. Similar to schwannomas, neurofibromas also are positive for S-100 and negative for EMA.3,14 Neurofibromas can have either a somatic or germline mutation of the biallelic NF1 gene on chromosome 17q11.2 with subsequent loss of protein neurofibromin activity.15 Less common but still a consideration are the hybrid peripheral nerve sheath tumors that may present with a biphasic or intermingled morphology. Combinations include neurofibroma-schwannoma, schwannoma-perineurioma, and neurofibroma-perineurioma. The hybrid schwannoma-perineurioma has a mixture of thin and plump spindle cells with tapered nuclei as well as patchy S-100 positivity corresponding to schwannian areas. Similarly, S-100 will highlight the wavy Schwann cells in neurofibroma-perineurioma as well as CD34-highlighting fibroblasts.7,15 In both aforementioned hybrid tumors, EMA will be positive in the perineurial areas. Another potential diagnostic consideration that can occur in both pediatric and adult populations is dermatofibrosarcoma protuberans (DFSP), which is comprised of a dermal proliferation of monomorphic fusiform spindle cells. Although both perineuriomas and DFSP can have a storiform architecture, DFSP is more asymmetric and infiltrative. Dermatofibrosarcoma protuberans is recognized in areas of individual adipocyte trapping, referred to as honeycombing. Dermatofibrosarcoma protuberans typically does not express EMA, though the sclerosing variant of DFSP has been reported to sometimes demonstrate focal EMA reactivity.11,14,16 For morphologically challenging cases, cytogenetic studies will show t(17;22) translocation fusing the COL1A1 and PDGFRB genes.16 Finally, for subcutaneous or deep-seated tumors, one also may consider other mesenchymal neoplasms, including solitary fibrous tumor, low-grade fibromyxoid sarcoma, or low-grade malignant peripheral nerve sheath tumor (MPNST).11
Management—Perineuriomas are considered benign. The presence of mitotic figures, pleomorphism, and degenerative nuclear atypia akin to ancient change, as seen in ancient schwannoma, does not affect their benign clinical behavior. Treatment of a perineurioma typically is surgical excision with conservative margins and minimal chance of recurrence.1,11 So-called malignant perineuriomas are better classified as MPNSTs with perineural differentiation or perineurial MPNST. They also are positive for EMA and may be distinguished from perineurioma by the presence of major atypia and an infiltrative growth pattern.17,18
Considerations in the Pediatric Population—Few pediatric soft tissue perineuriomas have been reported. A clinicopathologic analysis by Hornick and Fletcher1 of patients with soft tissue perineurioma showed that only 6 of 81 patients were younger than 20 years. The youngest reported case of perineurioma occurred as an extraneural perineurioma on the scalp in an infant.19 Only 1 soft tissue perineural MPNST has been reported in the pediatric population, arising on the face of an 11-year-old boy. In a case series of 11 pediatric perineuriomas, including extraneural and intraneural, there was no evidence of recurrence or metastasis at follow-up.4
Conclusion
Perineuriomas are rare benign peripheral nerve sheath tumors with unique histologic and immunohistochemical features. Soft tissue perineuriomas in the pediatric population are an important diagnostic consideration, especially for the pediatrician or dermatologist when encountering a well-circumscribed nodular soft tissue lesion of the extremity or when encountering a neural-appearing tumor in the subcutaneous tissue.
Acknowledgment—We would like to thank Christopher Fletcher, MD (Boston, Massachusetts), for his expertise in outside consultation for patient 1.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
- Hornick J, Fletcher C. Soft tissue perineurioma. Am J Surg Pathol. 2005;29:845-858.
- Tsang WY, Chan JK, Chow LT, et al. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol. 1992;16:756-763.
- Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88:443-456.
- Balarezo FS, Muller RC, Weiss RG, et al. Soft tissue perineuriomas in children: report of three cases and review of the literature. Pediatr Dev Pathol. 2003;6:137-141. Published correction appears in Pediatr Dev Pathol. 2003;6:following 364.
- Macarenco R, Ellinger F, Oliveira A. Perineurioma: a distinctive and underrecognized peripheral nerve sheath neoplasm. Arch Pathol Lab Med. 2007;131:625-636.
- Agaimy A, Buslei R, Coras R, et al. Comparative study of soft tissue perineurioma and meningioma using a five-marker immunohistochemical panel. Histopathology. 2014;65:60-70.
- Greenson JK, Hornick JL, Longacre TA, et al. Sternberg’s Diagnostic Surgical Pathology. Wolters Kluwer; 2015.
- Folpe A, Billings S, McKenney J, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620-1626.
- Hirose T, Tani T, Shimada T, et al. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293-298.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. Perineurioma. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2013:176-178.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier Saunders; 2013.
- Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708-1714.
- Al-Adnani M. Soft tissue perineurioma in a child with neurofibromatosis type 1: a case report and review of the literature. Pediatr Dev Pathol. 2017;20:444-448.
- Reddy VB, David O, Spitz DJ, et al. Gattuso’s Differential Diagnosis in Surgical Pathology. Elsevier Saunders; 2022.
- Michal M, Kazakov DV, Michal M. Hybrid peripheral nerve sheath tumors: a review. Cesk Patol. 2017;53:81-88.
- Abdaljaleel MY, North JP. Sclerosing dermatofibrosarcoma protuberans shows significant overlap with sclerotic fibroma in both routine and immunohistochemical analysis: a potential diagnostic pitfall. Am J Dermatopathol. 2017;39:83-88.
- Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma.” J Cutan Pathol. 2002;29:362-367.
- Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation). Clin Neuropathol. 2012;31:424-429.
- Duhan A, Rana P, Beniwal K, et al. Perineurioma of scalp in an infant: a case report with short review of literature. Asian J Neurosurg. 2016;11:81-83.
Practice Points
- Perineuriomas are rare benign peripheral nerve sheath tumors that most commonly occur in young to middle-aged adults but rarely can present in children.
- Immunohistochemically, perineuriomas show positive staining with epithelial membrane antigen, GLUT1, claudin-1, and frequently with CD34; they are negative for S-100 and glial fibrillary acidic protein.
- Perineuriomas should be considered in the differential diagnosis in children who present with a well-circumscribed nodular lesion in the subcutaneous tissue.
(original magnification ×15,000).")
Acquired Acrodermatitis Enteropathica in an Infant
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.

Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.

The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.

Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Practice Points
- Although clinically characterized by the triad of acral and periorificial dermatitis, alopecia, and diarrhea, most cases of acrodermatitis enteropathica (AE) present with only partial features of this syndrome.
- Low levels of zinc-dependent enzymes such as alkaline phosphatase may support the diagnosis of AE.

Asymptomatic Umbilical Nodule
The Diagnosis: Sister Mary Joseph Nodule
Histopathologic analysis of the biopsy specimen revealed a dense infiltrate of large, hyperchromatic, mucin-producing cells exhibiting varying degrees of nuclear pleomorphism (Figure 1). Immunohistochemical (IHC) staining was negative for cytokeratin (CK) 20; however, CK7 was found positive (Figure 2), which confirmed the presence of a metastatic adenocarcinoma, consistent with the clinical diagnosis of a Sister Mary Joseph nodule (SMJN). Subsequent IHC workup to determine the site of origin revealed densely positive expression of both cancer antigen 125 and paired homeobox gene 8 (PAX-8)(Figure 3), consistent with primary ovarian disease. Furthermore, expression of estrogen receptor and p53 both were positive within the nuclei, illustrating an aberrant expression pattern. On the other hand, cancer antigen 19-9, caudal-type homeobox 2, gross cystic disease fluid protein 15, and mammaglobin were all determined negative, thus leading to the pathologic diagnosis of a metastatic ovarian adenocarcinoma. Additional workup with computed tomography of the abdomen and pelvis highlighted a large left ovarian mass with multiple omental nodules as well as enlarged retroperitoneal and pelvic lymph nodes.
.")
The SMJN is a rare presentation of internal malignancy that appears as a nodule that metastasizes to the umbilicus. It may be ulcerated or necrotic and is seen in up to 10% of patients with cutaneous metastases from internal malignancy.1 These nodules are named after Sister Mary Joseph, the surgical assistant of Dr. William Mayo who first described the relationship between umbilical nodules seen in patients with gastrointestinal and genitourinary cancer. The most common underlying malignancies include primary gastrointestinal and gynecologic adenocarcinomas. In a retrospective study of 34 patients by Chalya et al,2 the stomach was found to be the most common primary site (41.1%). The presence of an SMJN affords a poor prognosis, with a mean overall survival of 11 months from the time of diagnosis.3 The mechanism of disease dissemination remains unknown but is thought to occur through lymphovascular invasion of tumor cells and spread via the umbilical ligament.1,4
.")
Merkel cell carcinoma is a cutaneous neuroendocrine tumor that most commonly presents in elderly patients as red-violet nodules or plaques. Although Merkel cell carcinoma most frequently is encountered on sun-exposed skin, they also can arise on the trunk and abdomen. Positive IHC staining for CK20 would be expected; however, it was negative in our case.5
. B, Paired homeobox gene 8 (PAX-8) immunohistochemical staining displayed the uptake in the tumor cells")
Cutaneous endometriosis is a rare disease presentation and most commonly occurs as a secondary process due to surgical inoculation of the abdominal wall. Primary cutaneous endometriosis in which there is no history of abdominal surgery less frequently is encountered. Patients typically will report pain and cyclical bleeding with menses. Pathology demonstrates ectopic endometrial tissue with glands and uterine myxoid stroma.6
Amelanotic melanoma is an uncommon subtype of malignant melanoma that presents as nonpigmented nodules that have a propensity to ulcerate and bleed. Furthermore, the umbilicus is an exceedingly rare location for primary melanoma. However, one report does exist, and amelanotic melanoma should be considered in the differential for patients with umbilical nodules.7
Dermoid cysts are benign congenital lesions that typically present as a painless, slow-growing, and wellcircumscribed nodule, as similarly experienced by our patient. They most commonly are found on the testicles and ovaries but also are known to arise in embryologic fusion planes, and reports of umbilical lesions exist.8 Dermoid cysts are diagnosed based on histopathology, supporting the need for a biopsy to distinguish a malignant process from benign lesions.9
- Gabriele R, Conte M, Egidi F, et al. Umbilical metastases: current viewpoint. World J Surg Oncol. 2005;3:13.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph’s nodule at a university teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Leyrat B, Bernadach M, Ginzac A, et al. Sister Mary Joseph nodules: a case report about a rare location of skin metastasis. Case Rep Oncol. 2021;14:664-670.
- Yendluri V, Centeno B, Springett GM. Pancreatic cancer presenting as a Sister Mary Joseph’s nodule: case report and update of the literature. Pancreas. 2007;34:161-164.
- Uchi H. Merkel cell carcinoma: an update and immunotherapy. Front Oncol. 2018;8:48.
- Bittar PG, Hryneewycz KT, Bryant EA. Primary cutaneous endometriosis presenting as an umbilical nodule. JAMA Dermatol. 2021;157:1227.
- Kovitwanichkanont T, Joseph S, Yip L. Hidden in plain sight: umbilical melanoma [published online January 28, 2020]. Med J Aust. 2020;212:154-155.e1.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Akinci O, Turker C, Erturk MS, et al. Umbilical dermoid cyst: a rare case. Cerrahpasa Med J. 2020;44:51-53.
The Diagnosis: Sister Mary Joseph Nodule
Histopathologic analysis of the biopsy specimen revealed a dense infiltrate of large, hyperchromatic, mucin-producing cells exhibiting varying degrees of nuclear pleomorphism (Figure 1). Immunohistochemical (IHC) staining was negative for cytokeratin (CK) 20; however, CK7 was found positive (Figure 2), which confirmed the presence of a metastatic adenocarcinoma, consistent with the clinical diagnosis of a Sister Mary Joseph nodule (SMJN). Subsequent IHC workup to determine the site of origin revealed densely positive expression of both cancer antigen 125 and paired homeobox gene 8 (PAX-8)(Figure 3), consistent with primary ovarian disease. Furthermore, expression of estrogen receptor and p53 both were positive within the nuclei, illustrating an aberrant expression pattern. On the other hand, cancer antigen 19-9, caudal-type homeobox 2, gross cystic disease fluid protein 15, and mammaglobin were all determined negative, thus leading to the pathologic diagnosis of a metastatic ovarian adenocarcinoma. Additional workup with computed tomography of the abdomen and pelvis highlighted a large left ovarian mass with multiple omental nodules as well as enlarged retroperitoneal and pelvic lymph nodes.
The SMJN is a rare presentation of internal malignancy that appears as a nodule that metastasizes to the umbilicus. It may be ulcerated or necrotic and is seen in up to 10% of patients with cutaneous metastases from internal malignancy.1 These nodules are named after Sister Mary Joseph, the surgical assistant of Dr. William Mayo who first described the relationship between umbilical nodules seen in patients with gastrointestinal and genitourinary cancer. The most common underlying malignancies include primary gastrointestinal and gynecologic adenocarcinomas. In a retrospective study of 34 patients by Chalya et al,2 the stomach was found to be the most common primary site (41.1%). The presence of an SMJN affords a poor prognosis, with a mean overall survival of 11 months from the time of diagnosis.3 The mechanism of disease dissemination remains unknown but is thought to occur through lymphovascular invasion of tumor cells and spread via the umbilical ligament.1,4
Merkel cell carcinoma is a cutaneous neuroendocrine tumor that most commonly presents in elderly patients as red-violet nodules or plaques. Although Merkel cell carcinoma most frequently is encountered on sun-exposed skin, they also can arise on the trunk and abdomen. Positive IHC staining for CK20 would be expected; however, it was negative in our case.5
Cutaneous endometriosis is a rare disease presentation and most commonly occurs as a secondary process due to surgical inoculation of the abdominal wall. Primary cutaneous endometriosis in which there is no history of abdominal surgery less frequently is encountered. Patients typically will report pain and cyclical bleeding with menses. Pathology demonstrates ectopic endometrial tissue with glands and uterine myxoid stroma.6
Amelanotic melanoma is an uncommon subtype of malignant melanoma that presents as nonpigmented nodules that have a propensity to ulcerate and bleed. Furthermore, the umbilicus is an exceedingly rare location for primary melanoma. However, one report does exist, and amelanotic melanoma should be considered in the differential for patients with umbilical nodules.7
Dermoid cysts are benign congenital lesions that typically present as a painless, slow-growing, and wellcircumscribed nodule, as similarly experienced by our patient. They most commonly are found on the testicles and ovaries but also are known to arise in embryologic fusion planes, and reports of umbilical lesions exist.8 Dermoid cysts are diagnosed based on histopathology, supporting the need for a biopsy to distinguish a malignant process from benign lesions.9
The Diagnosis: Sister Mary Joseph Nodule
Histopathologic analysis of the biopsy specimen revealed a dense infiltrate of large, hyperchromatic, mucin-producing cells exhibiting varying degrees of nuclear pleomorphism (Figure 1). Immunohistochemical (IHC) staining was negative for cytokeratin (CK) 20; however, CK7 was found positive (Figure 2), which confirmed the presence of a metastatic adenocarcinoma, consistent with the clinical diagnosis of a Sister Mary Joseph nodule (SMJN). Subsequent IHC workup to determine the site of origin revealed densely positive expression of both cancer antigen 125 and paired homeobox gene 8 (PAX-8)(Figure 3), consistent with primary ovarian disease. Furthermore, expression of estrogen receptor and p53 both were positive within the nuclei, illustrating an aberrant expression pattern. On the other hand, cancer antigen 19-9, caudal-type homeobox 2, gross cystic disease fluid protein 15, and mammaglobin were all determined negative, thus leading to the pathologic diagnosis of a metastatic ovarian adenocarcinoma. Additional workup with computed tomography of the abdomen and pelvis highlighted a large left ovarian mass with multiple omental nodules as well as enlarged retroperitoneal and pelvic lymph nodes.
The SMJN is a rare presentation of internal malignancy that appears as a nodule that metastasizes to the umbilicus. It may be ulcerated or necrotic and is seen in up to 10% of patients with cutaneous metastases from internal malignancy.1 These nodules are named after Sister Mary Joseph, the surgical assistant of Dr. William Mayo who first described the relationship between umbilical nodules seen in patients with gastrointestinal and genitourinary cancer. The most common underlying malignancies include primary gastrointestinal and gynecologic adenocarcinomas. In a retrospective study of 34 patients by Chalya et al,2 the stomach was found to be the most common primary site (41.1%). The presence of an SMJN affords a poor prognosis, with a mean overall survival of 11 months from the time of diagnosis.3 The mechanism of disease dissemination remains unknown but is thought to occur through lymphovascular invasion of tumor cells and spread via the umbilical ligament.1,4
Merkel cell carcinoma is a cutaneous neuroendocrine tumor that most commonly presents in elderly patients as red-violet nodules or plaques. Although Merkel cell carcinoma most frequently is encountered on sun-exposed skin, they also can arise on the trunk and abdomen. Positive IHC staining for CK20 would be expected; however, it was negative in our case.5
Cutaneous endometriosis is a rare disease presentation and most commonly occurs as a secondary process due to surgical inoculation of the abdominal wall. Primary cutaneous endometriosis in which there is no history of abdominal surgery less frequently is encountered. Patients typically will report pain and cyclical bleeding with menses. Pathology demonstrates ectopic endometrial tissue with glands and uterine myxoid stroma.6
Amelanotic melanoma is an uncommon subtype of malignant melanoma that presents as nonpigmented nodules that have a propensity to ulcerate and bleed. Furthermore, the umbilicus is an exceedingly rare location for primary melanoma. However, one report does exist, and amelanotic melanoma should be considered in the differential for patients with umbilical nodules.7
Dermoid cysts are benign congenital lesions that typically present as a painless, slow-growing, and wellcircumscribed nodule, as similarly experienced by our patient. They most commonly are found on the testicles and ovaries but also are known to arise in embryologic fusion planes, and reports of umbilical lesions exist.8 Dermoid cysts are diagnosed based on histopathology, supporting the need for a biopsy to distinguish a malignant process from benign lesions.9
- Gabriele R, Conte M, Egidi F, et al. Umbilical metastases: current viewpoint. World J Surg Oncol. 2005;3:13.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph’s nodule at a university teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Leyrat B, Bernadach M, Ginzac A, et al. Sister Mary Joseph nodules: a case report about a rare location of skin metastasis. Case Rep Oncol. 2021;14:664-670.
- Yendluri V, Centeno B, Springett GM. Pancreatic cancer presenting as a Sister Mary Joseph’s nodule: case report and update of the literature. Pancreas. 2007;34:161-164.
- Uchi H. Merkel cell carcinoma: an update and immunotherapy. Front Oncol. 2018;8:48.
- Bittar PG, Hryneewycz KT, Bryant EA. Primary cutaneous endometriosis presenting as an umbilical nodule. JAMA Dermatol. 2021;157:1227.
- Kovitwanichkanont T, Joseph S, Yip L. Hidden in plain sight: umbilical melanoma [published online January 28, 2020]. Med J Aust. 2020;212:154-155.e1.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Akinci O, Turker C, Erturk MS, et al. Umbilical dermoid cyst: a rare case. Cerrahpasa Med J. 2020;44:51-53.
- Gabriele R, Conte M, Egidi F, et al. Umbilical metastases: current viewpoint. World J Surg Oncol. 2005;3:13.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph’s nodule at a university teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Leyrat B, Bernadach M, Ginzac A, et al. Sister Mary Joseph nodules: a case report about a rare location of skin metastasis. Case Rep Oncol. 2021;14:664-670.
- Yendluri V, Centeno B, Springett GM. Pancreatic cancer presenting as a Sister Mary Joseph’s nodule: case report and update of the literature. Pancreas. 2007;34:161-164.
- Uchi H. Merkel cell carcinoma: an update and immunotherapy. Front Oncol. 2018;8:48.
- Bittar PG, Hryneewycz KT, Bryant EA. Primary cutaneous endometriosis presenting as an umbilical nodule. JAMA Dermatol. 2021;157:1227.
- Kovitwanichkanont T, Joseph S, Yip L. Hidden in plain sight: umbilical melanoma [published online January 28, 2020]. Med J Aust. 2020;212:154-155.e1.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Akinci O, Turker C, Erturk MS, et al. Umbilical dermoid cyst: a rare case. Cerrahpasa Med J. 2020;44:51-53.
A 64-year-old woman with no notable medical history was referred to our dermatology clinic with an intermittent eczematous rash around the eyelids of 3 months’ duration. While performing a total-body skin examination, a firm pink nodule with a smooth surface incidentally was discovered on the umbilicus. The patient was uncertain when the lesion first appeared and denied any associated symptoms including pain and bleeding. Additionally, a lymph node examination revealed right inguinal lymphadenopathy. Upon further questioning, she reported worsening muscle weakness, fatigue, night sweats, and an unintentional weight loss of 10 pounds. A 6-mm punch biopsy of the umbilical lesion was obtained for routine histopathology.


An infant with a tender bump on her ear
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.

Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A 4-month-old female was referred to our clinic for evaluation of a bump on the right ear. The lesion was first noted at 2 months of age as a little pimple. She was evaluated by her pediatrician and was treated with topical and oral antibiotics without resolution of the lesion. The bump continued to grow and seemed tender to palpation, so she was referred to dermatology for evaluation.
She was born via normal vaginal delivery at 40 weeks. Her mother has no medical conditions and the pregnancy was uneventful. She has been growing and developing well. She takes vitamin D and is currently breast fed.
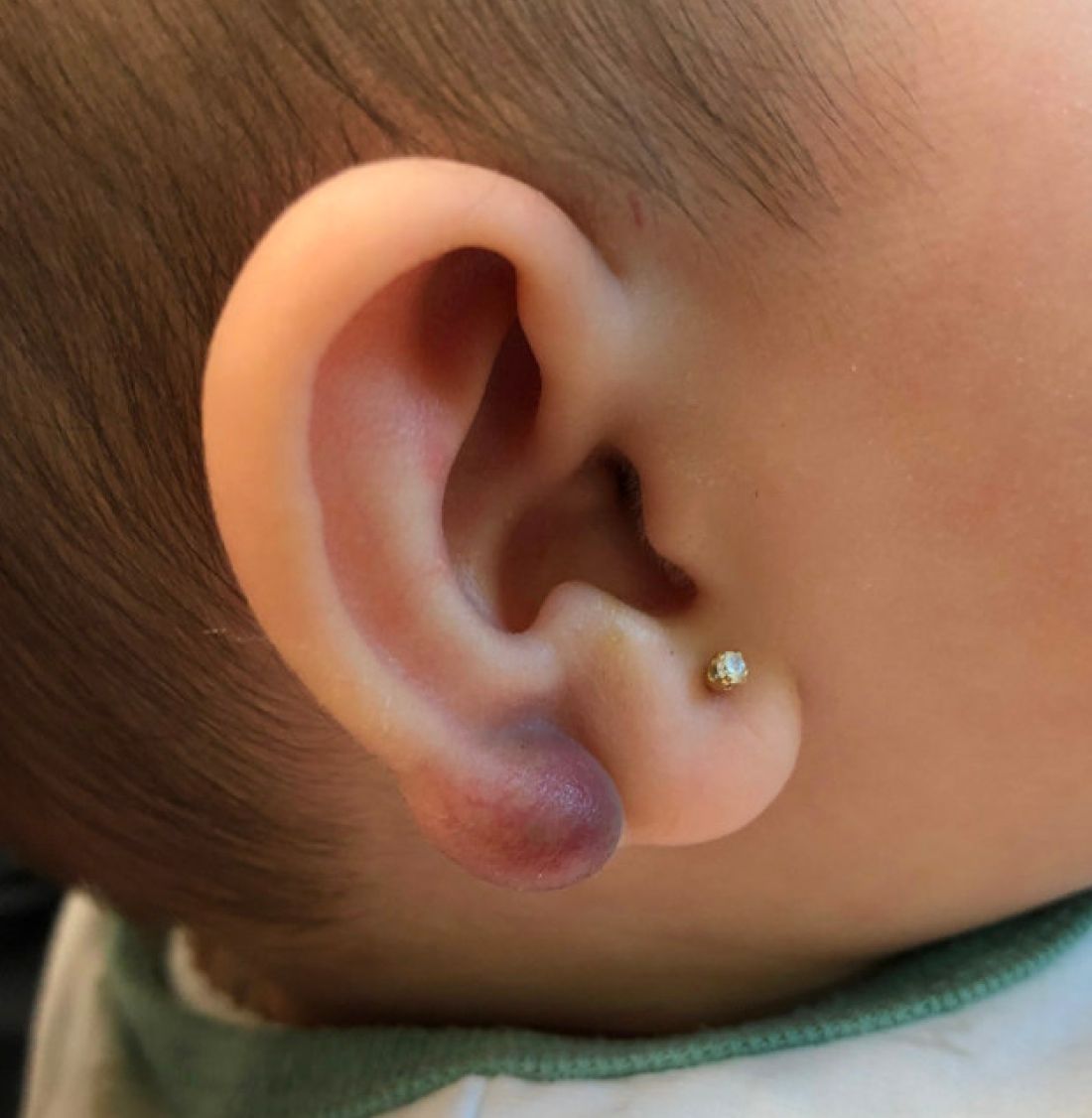
There have been no other family members with similar lesions. She had her ears pierced at a month of age without any complications.
On skin examination she has a firm red nodule on the right ear that appears slightly tender to touch. She has no other skin lesions of concern. She has normal muscle tone and there are no other abnormalities noted on the physical exam. She has no hepatomegaly, splenomegaly, or lymphadenopathy.
Nonblanching Rash on the Legs and Chest
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
.")
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
The Diagnosis: Leukemia Cutis
Hematoxylin and eosin staining revealed an infiltration of monomorphic atypical myeloid cells with cleaved nuclei within the dermis, with a relatively uninvolved epidermis (Figure, A). The cells formed aggregates in single-file lines along dermal collagen bundles. Occasional Auer rods, which are crystal aggregates of the enzyme myeloperoxidase, a marker unique to cells of the myeloid lineage (Figure, B) were appreciated.
Immunohistochemical staining for myeloperoxidase was weakly positive; however, flow cytometric evaluation of the bone marrow aspirate revealed that approximately 20% of all CD45+ cells were myeloid blasts. These findings confirmed the diagnosis of recurrent acute myeloid leukemia (AML). The diagnosis of AML can be confirmed with a bone marrow biopsy demonstrating more than 20% of the total cells in blast form as well as evidence that the cells are of myeloid origin, which can be inferred by the presence of Auer rods, positive myeloperoxidase staining, or immunophenotyping. In our patient, the Auer rods, myeloperoxidase staining, and atypical myeloid cells on skin biopsy, in conjunction with the bone marrow biopsy results, confirmed leukemia cutis.
Leukemia cutis is the infiltration of neoplastic proliferating leukocytes in the epidermis, dermis, or subcutis from a primary or more commonly metastatic malignancy. Leukemic cutaneous involvement is seen in up to 13% of leukemia patients and most commonly is seen in monocytic or myelomonocytic forms of AML.1 It may present anywhere on the body but mostly is found on the back, trunk, and head. It also may have a predilection for areas with a history of trauma or inflammation. The lesions most often are firm, erythematous to violaceous papules and nodules, though leukemia cutis can present with hemorrhagic ulcers, purpura, or other cutaneous manifestations of concomitant thrombocytopenia such as petechiae and ecchymoses.2 Involvement of the lower extremities mimicking venous stasis dermatitis has been described.3,4
Treatment of leukemia cutis requires targeting the underlying leukemia2 under the guidance of hematology and oncology as well as the use of chemotherapeutic agents.5 The presence of leukemia cutis is a poor prognostic sign, and a discussion regarding goals of care often is appropriate. Our patient initially responded to FLAG (fludarabine, cytarabine, filgrastim) chemotherapy induction and consolidation, which was followed by midostaurin maintenance. However, she ultimately regressed, requiring decitabine and gilteritinib treatment, and died 9 months later from the course of the disease.
Although typically asymptomatic and presenting on the lower limbs, capillaritis (also known as the pigmented purpuric dermatoses) consists of a set of cutaneous conditions that often are chronic and relapsing in nature, as opposed to our patient’s subacute presentation. These benign conditions have several distinct morphologies; some are characterized by pigmented macules or pinpoint red-brown petechiae that most often are found on the legs but also are seen on the trunk and upper extremities.6 Of the various clinical presentations of capillaritis, our patient’s skin findings may be most consistent with pigmented purpuric lichenoid dermatitis of Gougerot and Blum, in which purpuric red-brown papules coalesce into plaques, though her lesions were not raised. The other pigmented purpuric dermatoses can present with cayenne pepper–colored petechiae, golden-brown macules, pruritic purpuric patches, or red-brown annular patches,6 which were not seen in our patient.
Venous stasis dermatitis also favors the lower extremities7; however, it classically includes the medial malleolus and often presents with scaling and hyperpigmentation from hemosiderin deposition.8 It often is associated with pruritus, as opposed to the nonpruritic nonpainful lesions in leukemia cutis. Other signs of venous insufficiency also may be appreciated, including edema or varicose veins,7 which were not evident in our patient.
Leukocytoclastic vasculitis, a small vessel vasculitis, also appears as palpable or macular purpura, which classically is asymptomatic and erupts on the shins approximately 1 week after an inciting exposure,9 such as medications, pathogens, or autoimmune diseases. One of the least distinctive vasculitides is polyarteritis nodosa, a form of medium vessel vasculitis, which presents most often with palpable purpura or painful nodules on the lower extremities and may be accompanied by livedo reticularis or digital necrosis.9 Acute leukemia may be accompanied by inflammatory paraneoplastic conditions including vasculitis, which is thought to be due to leukemic cells infiltrating and damaging blood vessels.10
Pretibial myxedema is closely associated with Graves disease and shares some features seen in the presentation of our patient’s leukemia cutis. It is asymptomatic, classically affects the pretibial regions, and most commonly affects older adults and women.11,12 Pretibial myxedema presents with thick indurated plaques rather than patches. Our patient did not demonstrate ophthalmopathy, which nearly always precedes pretibial myxedema.12 The most common form of pretibial myxedema is nonpitting, though nodular, plaquelike, polypoid, and elephantiasic forms also exist.11 Pretibial myxedema classically favors the shins; however, it also can affect the ankles, dorsal aspects of the feet, and toes. The characteristic induration of the skin is believed to be the result of excess fibroblast production of glycosaminoglycans in the dermis and subcutis likely triggered by stimulation of fibroblast thyroid stimulating hormone receptors.11
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
- Bakst RL, Tallman MS, Douer D, et al. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118:3785-3793.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:973-977.
- Papadavid E, Panayiotides I, Katoulis A, et al. Stasis dermatitis-like leukaemic infiltration in a patient with myelodysplastic syndrome. Clin Exp Dermatol. 2008;33:298-300.
- Chang HY, Wong KM, Bosenberg M, et al. Myelogenous leukemia cutis resembling stasis dermatitis. J Am Acad Dermatol. 2003;49:128-129.
- Aguilera SB, Zarraga M, Rosen L. Leukemia cutis in a patient with acute myelogenous leukemia: a case report and review of the literature. Cutis. 2010;85:31-36.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other eczematous eruptions. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. 2nd ed. Elsevier; 2014:103-108.
- Krooks JA, Weatherall AG. Leukemia cutis in acute myeloid leukemia signifies a poor prognosis. Cutis. 2018;102:266, 271-272.
- Wetter DA, Dutz JP, Shinkai K, et al. Cutaneous vasculitis. In: Bolognia JL, Schaffer JV, Lorenzo C, eds. Dermatology. 4th ed. Elsevier; 2018:409-439.
- Jones D, Dorfman DM, Barnhill RL, et al. Leukemic vasculitis: a feature of leukemia cutis in some patients. Am J Clin Pathol. 1997;107:637-642.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7.
A 67-year-old woman with history of atrial fibrillation and leukemia presented with a nonpruritic nonpainful rash of 10 days' duration that began on the distal lower extremities (top) and then spread superiorly. She reported having a sore throat and mouth, cough, night sweats, unintentional weight loss, and lymphadenopathy. Physical examination revealed pink-purple nonblanching macules and patches on the lower extremities extending from the ankles to the knees. She also had firm pink papules on the chest (bottom) and back. Punch biopsies of the skin on the chest and leg were obtained for histologic examination and immunohistochemical staining.


Firm Mobile Nodule on the Scalp
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12

Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
.")
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
.")
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19

Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12
Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19
Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
The Diagnosis: Metastatic Carcinoid Tumor
Carcinoid tumors are derived from neuroendocrine cell compartments and generally arise in the gastrointestinal tract, with a quarter of carcinoids arising in the small bowel.1 Carcinoid tumors have an incidence of approximately 2 to 5 per 100,000 patients.2 Metastasis of carcinoids is approximately 31.2% to 46.7%.1 Metastasis to the skin is uncommon; we present a rare case of a carcinoid tumor of the terminal ileum with metastasis to the scalp.
Unlike our patient, most patients with carcinoid tumors have an indolent clinical course. The most common cutaneous symptom is flushing, which occurs in 75% of patients.3 Secreted vasoactive peptides such as serotonin may cause other symptoms such as tachycardia, diarrhea, and bronchospasm; together, these symptoms comprise carcinoid syndrome. Carcinoid syndrome requires metastasis of the tumor to the liver or a site outside of the gastrointestinal tract because the liver will metabolize the secreted serotonin. However, even in patients with liver metastasis, carcinoid syndrome only occurs in approximately 10% of patients.4 Common skin findings of carcinoid syndrome include pellagralike dermatitis, flushing, and sclerodermalike changes.5 Our patient experienced several episodes of presyncope with symptoms of dyspnea, lightheadedness, and flushing but did not have bronchospasm or recurrent diarrhea. Intramuscular octreotide improved some symptoms.
The scalp accounts for approximately 15% of cutaneous metastases, the most common being from the lung, renal, and breast cancers.6 Cutaneous metastases of carcinoid tumors are rare. A PubMed search of articles indexed for MEDLINE using the terms metastatic AND [carcinoid OR neuroendocrine] tumors AND [skin OR cutaneous] revealed 47 cases.7-11 Similar to other skin metastases, cutaneous metastases of carcinoid tumors commonly present as firm erythematous nodules of varying sizes that may be asymptomatic, tender, or pruritic (Figure 1). Cases of carcinoid tumors with cutaneous metastasis as the initial and only presenting sign are exceedingly rare.12
Histology of carcinoid tumors reveals a dermal neoplasm composed of loosely cohesive, mildly atypical, polygonal cells with salt-and-pepper chromatin and eosinophilic cytoplasm, which are similar findings to the primary tumor. The cells may grow in the typical trabecular or organoid neuroendocrine pattern or exhibit a pseudoglandular growth pattern with prominent vessels (quiz image, top).12 Positive chromogranin and synaptophysin immunostaining are the most common and reliable markers used for the diagnosis of carcinoid tumors.
An important histopathologic differential diagnosis is the aggressive Merkel cell carcinoma, which also demonstrates homogenous salt-and-pepper chromatin but exhibits a higher mitotic rate and positive cytokeratin 20 staining (Figure 2).13 Basal cell carcinoma (BCC) also may display similar features, including a blue tumor at scanning magnification and nodular or infiltrative growth patterns. The cell morphology of BCC is characterized by islands of basaloid cells with minimal cytoplasm and frequent apoptosis, connecting to the epidermis with peripheral palisading, retraction artifact, and a myxoid stroma; BCC lacks the salt-and-pepper chromatin commonly seen in carcinoid tumors (Figure 3). Basal cell carcinoma is characterized by positive BerEP4 (epithelial cell adhesion molecule immunostain), cytokeratin 5/6, and cytokeratin 14 uptake. Cytokeratin 20, often used to diagnose Merkel cell carcinoma, is negative in BCC. Chromogranin and synaptophysin occasionally may be positive in BCC.14
The superficial Ewing sarcoma family of tumors also may be included in the differential diagnosis of small round cell tumors of the skin, but they are very rare. These tumors possess strong positive membranous staining of cytokeratin 99 and also can stain positively for synaptophysin and chromogranin.15 Epithelial membrane antigen, which is negative in Ewing sarcomas, is positive in carcinoid tumors.16 Neuroendocrine tumors of all sites share similar basic morphologic patterns, and multiple primary tumors should be considered, including small cell lung carcinoma (Figure 4).17,18 Red granulations and true glandular lumina typically are not seen in the lungs but are common in gastrointestinal carcinoids.18 Regarding immunohistochemistry, TTF-1 is negative and CDX2 is positive in gastroenteropancreatic carcinoids, suggesting that these 2 markers can help distinguish carcinoids of unknown primary origin.19
Metastases in carcinoid tumors are common, with one study noting that the highest frequency of small intestinal metastases was from the ileal subset.20 At the time of diagnosis, 58% to 64% of patients with small intestine carcinoid tumors already had nonlocalized disease, with frequent sites being the lymph nodes (89.8%), liver (44.1%), lungs (13.6%), and peritoneum (13.6%). Regional and distant metastases are associated with substantially worse prognoses, with survival rates of 71.7% and 38.5%, respectively.1 Treatment of symptomatic unresectable disease focuses on symptomatic management with somatostatin analogs that also control tumor growth.21
We present a rare case of scalp metastasis of a carcinoid tumor of the terminal ileum. Distant metastasis is associated with poorer prognosis and should be considered in patients with a known history of a carcinoid tumor.
Acknowledgment—We would like to acknowledge the Research Histology and Tissue Imaging Core at University of Illinois Chicago Research Resources Center for the immunohistochemistry studies.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1-18, vii.
- Sabir S, James WD, Schuchter LM. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999;11:139-144.
- Tomassetti P. Clinical aspects of carcinoid tumours. Italian J Gastroenterol Hepatol. 1999;31(suppl 2):S143-S146.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2 pt 1):228-236.
- Garcia A, Mays S, Silapunt S. Metastatic neuroendocrine carcinoma in the skin. Dermatol Online J. 2017;23:13030/qt9052w9x1.
- Ciliberti MP, Carbonara R, Grillo A, et al. Unexpected response to palliative radiotherapy for subcutaneous metastases of an advanced small cell pancreatic neuroendocrine carcinoma: a case report of two different radiation schedules. BMC Cancer. 2020;20:311.
- Devnani B, Kumar R, Pathy S, et al. Cutaneous metastases from neuroendocrine carcinoma of the cervix: an unusual metastatic lesion from an uncommon malignancy. Curr Probl Cancer. 2018; 42:527-533.
- Falto-Aizpurua L, Seyfer S, Krishnan B, et al. Cutaneous metastasis of a pulmonary carcinoid tumor. Cutis. 2017;99:E13-E15.
- Dhingra R, Tse JY, Saif MW. Cutaneous metastasis of gastroenteropancreatic neuroendocrine tumors (GEP-Nets)[published online September 8, 2018]. JOP. 2018;19.
- Jedrych J, Busam K, Klimstra DS, et al. Cutaneous metastases as an initial manifestation of visceral well-differentiated neuroendocrine tumor: a report of four cases and a review of literature. J Cutan Pathol. 2014;41:113-122.
- Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol. 2003;14:293-301.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502.
- Machado I, Llombart B, Calabuig-Fariñas S, et al. Superficial Ewing’s sarcoma family of tumors: a clinicopathological study with differential diagnoses. J Cutan Pathol. 2011;38:636-643.
- D’Cruze L, Dutta R, Rao S, et al. The role of immunohistochemistry in the analysis of the spectrum of small round cell tumours at a tertiary care centre. J Clin Diagn Res. 2013;7:1377-1382.
- Chirila DN, Turdeanu NA, Constantea NA, et al. Multiple malignant tumors. Chirurgia (Bucur). 2013;108:498-502.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-1638.
- Lin X, Saad RS, Luckasevic TM, et al. Diagnostic value of CDX-2 and TTF-1 expressions in separating metastatic neuroendocrine neoplasms of unknown origin. Appl Immunohistochem Mol Morphol. 2007;15:407-414.
- Olney JR, Urdaneta LF, Al-Jurf AS, et al. Carcinoid tumors of the gastrointestinal tract. Am Surg. 1985;51:37-41.
- Strosberg JR, Halfdanarson TR, Bellizzi AM, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. 2017;46:707-714.
A 47-year-old woman was admitted to the hospital with abdominal pain and flushing. She had a history of a midgut carcinoid that originated in the ileum with metastasis to the colon, liver, and pancreas. Dermatologic examination revealed a firm, nontender, mobile, 7-mm scalp nodule with a pink-purple overlying epidermis. The lesion was associated with a slight decrease in hair density. A 4-mm punch biopsy was performed.


