User login
Eruptive Syringoma Manifesting as a Widespread Rash in 3 Patients
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
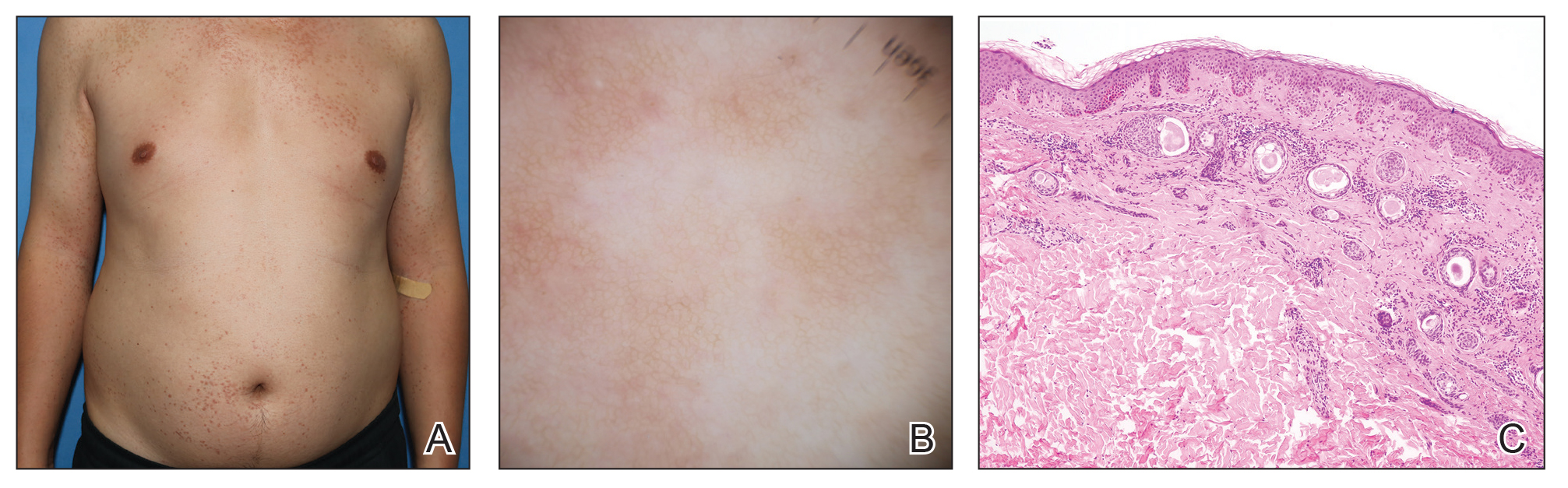
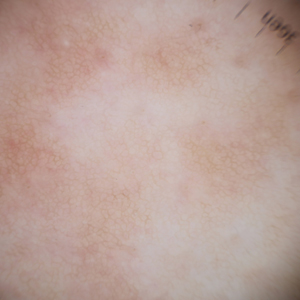
A 27-year-old woman presented with widespread asymptomatic papules

A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
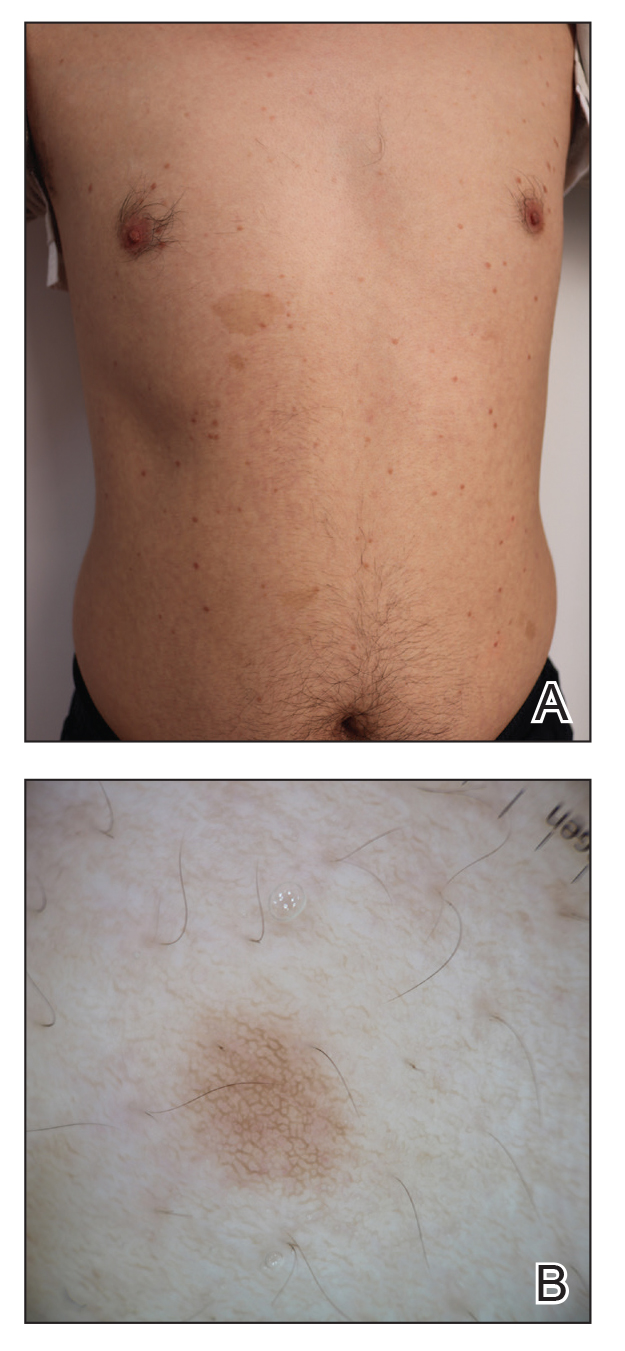
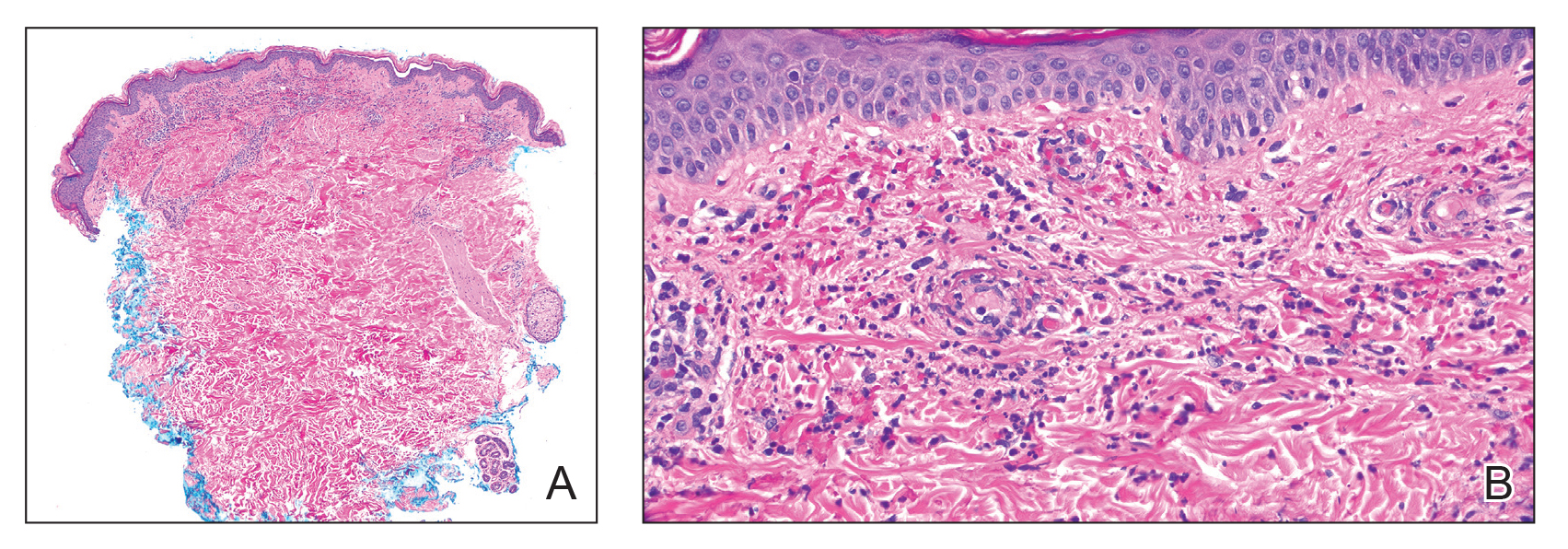
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
Practice Points
- Eruptive syringoma (ES) is a benign cutaneous adnexal neoplasm that typically does not require treatment.
- Dermoscopy and biopsy are helpful for the diagnosis of ES, which often is missed or misdiagnosed clinically.
Anti-Smith and Anti–Double-Stranded DNA Antibodies in a Patient With Henoch-Schönlein Purpura Following COVID-19 Vaccination
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
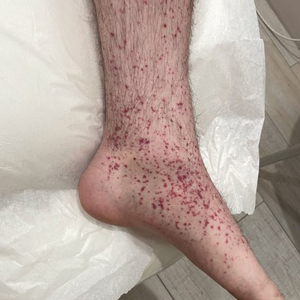
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
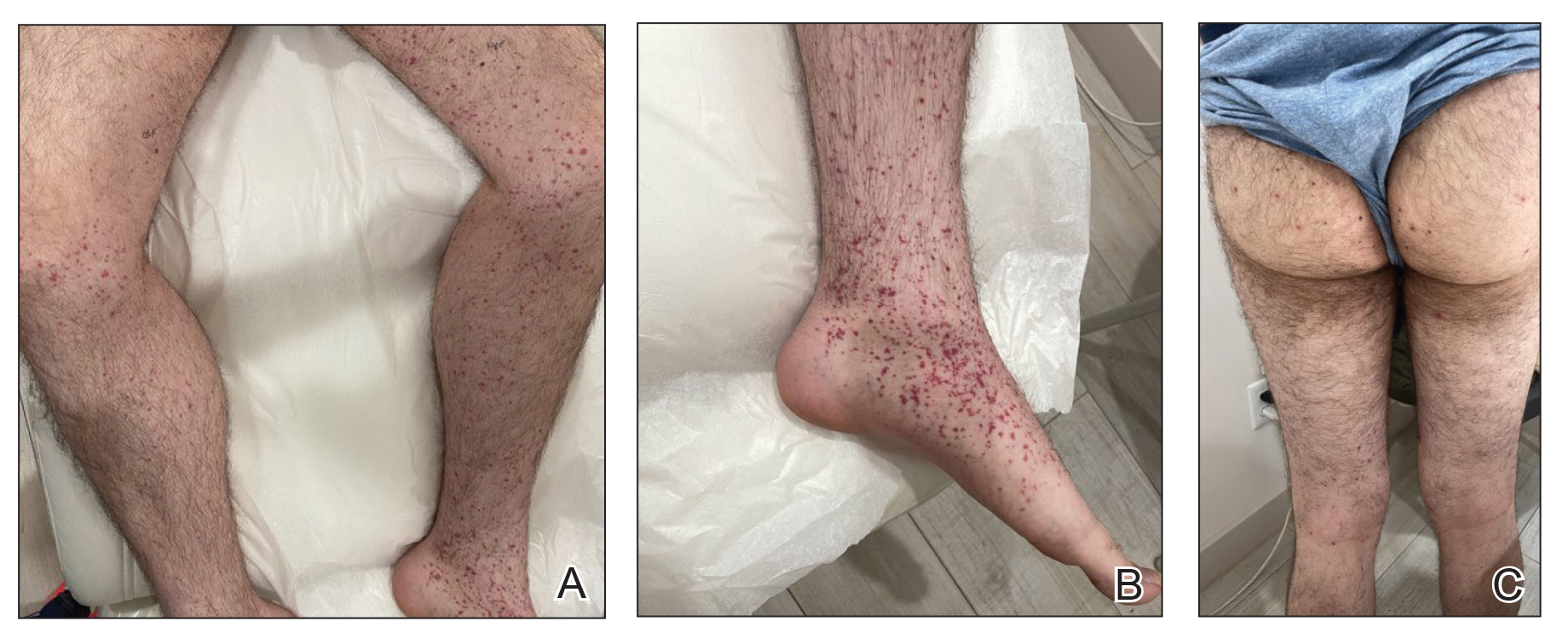
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
Practice Points
- Dermatologists should be vigilant for Henoch-Schönlein purpura (HSP) despite negative direct immunofluorescence of IgA deposition and unusual antibodies.
- Messenger RNA–based COVID-19 vaccines are associated with various cutaneous reactions, including HSP.
- Anti-Smith and anti–double-stranded DNA antibodies typically are not associated with HSP but may be seen in patients with coexisting systemic lupus erythematosus.
The Shield Sign of Cutaneous Metastases Is Associated With Carcinoma Hemorrhagiectoides
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
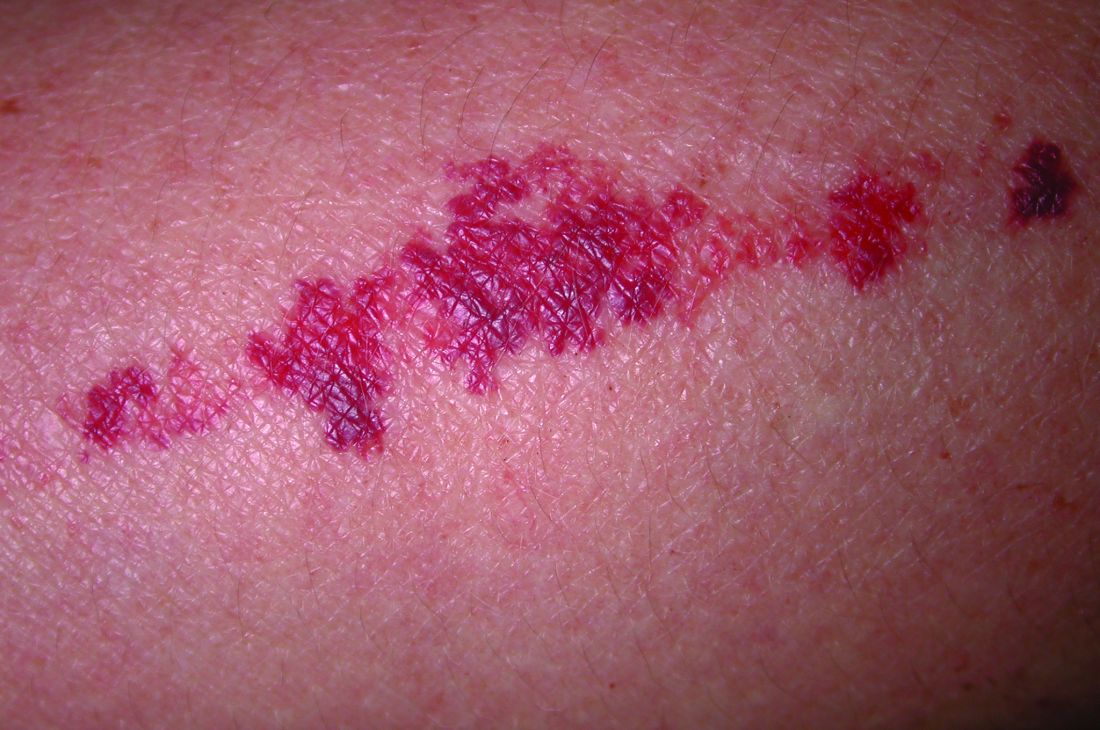
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
Dermatoporosis in Older Adults: A Condition That Requires Holistic, Creative Management
WASHINGTON — and conveys the skin’s vulnerability to serious medical complications, said Adam Friedman, MD, at the ElderDerm conference on dermatology in the older patient.
Key features of dermatoporosis include atrophic skin, solar purpura, white pseudoscars, easily acquired skin lacerations and tears, bruises, and delayed healing. “We’re going to see more of this, and it will more and more be a chief complaint of patients,” said Dr. Friedman, professor and chair of dermatology at George Washington University (GWU) in Washington, and co-chair of the meeting. GWU hosted the conference, describing it as a first-of-its-kind meeting dedicated to improving dermatologic care for older adults.

Dermatoporosis was described in the literature in 2007 by dermatologists at the University of Geneva in Switzerland. “It is not only a cosmetic problem,” Dr. Friedman said. “This is a medical problem ... which can absolutely lead to comorbidities [such as deep dissecting hematomas] that are a huge strain on the healthcare system.”
Dermatologists can meet the moment with holistic, creative combination treatment and counseling approaches aimed at improving the mechanical strength of skin and preventing potential complications in older patients, Dr. Friedman said at the meeting.
He described the case of a 76-year-old woman who presented with dermatoporosis on her arms involving pronounced skin atrophy, solar purpura, and a small covered laceration. “This was a patient who was both devastated by the appearance” and impacted by the pain and burden of dressing frequent wounds, said Dr. Friedman, who is also the director of the Residency Program, of Translational Research, and of Supportive Oncodermatology, all within the Department of Dermatology at GWU.
With 11 months of topical treatment that included daily application of calcipotriene 0.05% ointment and nightly application of tazarotene 0.045% lotion and oral supplementation with 1000-mg vitamin C twice daily and 1000-mg citrus bioflavonoid complex daily, as well as no changes to the medications she took for various comorbidities, the solar purpura improved significantly and “we made a huge difference in the integrity of her skin,” he said.
Dr. Friedman also described this case in a recently published article in the Journal of Drugs in Dermatology titled “What’s Old Is New: An Emerging Focus on Dermatoporosis”.
Likely Pathophysiology
Advancing age and chronic ultraviolet (UV) radiation exposure are the chief drivers of dermatoporosis. In addition to UVA and UVB light, other secondary drivers include genetic susceptibility, topical and systematic corticosteroid use, and anticoagulant treatment.
Its pathogenesis is not well described in the literature but is easy to envision, Dr. Friedman said. For one, both advancing age and exposure to UV light lead to a reduction in hygroscopic glycosaminoglycans, including hyaluronate (HA), and the impact of this diminishment is believed to go “beyond [the loss of] buoyancy,” he noted. Researchers have “been showing these are not just water-loving molecules, they also have some biologic properties” relating to keratinocyte production and epidermal turnover that appear to be intricately linked to the pathogenesis of dermatoporosis.
HAs have been shown to interact with the cell surface receptor CD44 to stimulate keratinocyte proliferation, and low levels of CD44 have been reported in skin with dermatoporosis compared with a younger control population. (A newly characterized organelle, the hyaluronosome, serves as an HA factory and contains CD44 and heparin-binding epidermal growth factor, Dr. Friedman noted. Inadequate functioning may be involved in skin atrophy.)
Advancing age also brings an increase in matrix metalloproteinases (MMPs)–1, –2, and –3, which are “the demolition workers of the skin,” and downregulation of a tissue inhibitor of MMPs, he said.
Adding insult to injury, dermis-penetrating UVA also activates MMPs, “obliterating collagen and elastin.” UVB generates DNA photoproducts, including oxidative stress and damaging skin cell DNA. “That UV light induces breakdown [of the skin] through different mechanisms and inhibits buildup is a simple concept I think our patients can understand,” Dr. Friedman said.
Multifaceted Treatment
For an older adult, “there is never a wrong time to start sun-protective measures” to prevent or try to halt the progression of dermatoporosis, Dr. Friedman said, noting that “UV radiation is an immunosuppressant, so there are many good reasons to start” if the adult is not already taking measures on a regular basis.
Potential treatments for the syndrome of dermatoporosis are backed by few clinical studies, but dermatologists are skilled at translating the use of products from one disease state to another based on understandings of pathophysiology and mechanistic pathways, Dr. Friedman commented in an interview after the meeting.
For instance, “from decades of research, we know what retinoids will do to the skin,” he said in the interview. “We know they will turn on collagen-1 and -3 genes in the skin, and that they will increase the production of glycosaminoglycans ... By understanding the biology, we can translate this to dermatoporosis.” These changes were demonstrated, for instance, in a small study of topical retinol in older adults.
Studies of topical alpha hydroxy acid (AHA), moreover, have demonstrated epidermal thickening and firmness, and “some studies show they can limit steroid-induced atrophy,” Dr. Friedman said at the meeting. “And things like lactic acid and urea are super accessible.”
Topical dehydroepiandrosterone is backed by even less data than retinoids or AHAs are, “but it’s still something to consider” as part of a multimechanistic approach to dermatoporosis, Dr. Friedman shared, noting that a small study demonstrated beneficial effects on epidermal atrophy in aging skin.
The use of vitamin D analogues such as calcipotriene, which is approved for the treatment of psoriasis, may also be promising. “One concept is that [vitamin D analogues] increase calcium concentrations in the epidermis, and calcium is so central to keratinocyte differentiation” and epidermal function that calcipotriene in combination with topical steroid therapy has been shown to limit skin atrophy, he noted.
Nutritionally, low protein intake is a known problem in the older population and is associated with increased skin fragility and poorer healing. From a prevention and treatment standpoint, therefore, patients can be counseled to be attentive to their diets, Dr. Friedman said. Experts have recommended a higher protein intake for older adults than for younger adults; in 2013, an international group recommended a protein intake of 1-1.5 g/kg/d for healthy older adults and more for those with acute or chronic illness.
“Patients love talking about diet and skin disease ... and they love over-the-counter nutraceuticals as well because they want something natural,” Dr. Friedman said. “I like using bioflavonoids in combination with vitamin C, which can be effective especially for solar purpura.”
A 6-week randomized, placebo-controlled, double-blind trial involving 67 patients with purpura associated with aging found a 50% reduction in purpura lesions among those took a particular citrus bioflavonoid blend twice daily. “I thought this was a pretty well-done study,” he said, noting that patient self-assessment and investigator global assessment were utilized.
Skin Injury and Wound Prevention
In addition to recommending gentle skin cleansers and daily moisturizing, dermatologists should talk to their older patients with dermatoporosis about their home environments. “What is it like? Is there furniture with sharp edges?” Dr. Friedman advised. If so, could they use sleeves or protectors on their arms or legs “to protect against injury?”
In a later meeting session about lower-extremity wounds on geriatric patients, Michael Stempel, DPM, assistant professor of medicine and surgery and chief of podiatry at GWU, said that he was happy to hear the term dermatoporosis being used because like diabetes, it’s a risk factor for developing lower-extremity wounds and poor wound healing.
He shared the case of an older woman with dermatoporosis who “tripped and skinned her knee against a step and then self-treated it for over a month by pouring hydrogen peroxide over it and letting air get to it.” The wound developed into “full-thickness tissue loss,” said Dr. Stempel, also medical director of the Wound Healing and Limb Preservation Center at GWU Hospital.
Misperceptions are common among older patients about how a simple wound should be managed; for instance, the adage “just let it get air” is not uncommon. This makes anticipatory guidance about basic wound care — such as the importance of a moist and occlusive environment and the safe use of hydrogen peroxide — especially important for patients with dermatoporosis, Dr. Friedman commented after the meeting.
Dermatoporosis is quantifiable, Dr. Friedman said during the meeting, with a scoring system having been developed by the researchers in Switzerland who originally coined the term. Its use in practice is unnecessary, but its existence is “nice to share with patients who feel bothered because oftentimes, patients feel it’s been dismissed by other providers,” he said. “Telling your patients there’s an actual name for their problem, and that there are ways to quantify and measure changes over time, is validating.”
Its recognition as a medical condition, Dr. Friedman added, also enables the dermatologist to bring it up and counsel appropriately — without a patient feeling shame — when it is identified in the context of a skin excision, treatment of a primary inflammatory skin disease, or management of another dermatologic problem.
Dr. Friedman disclosed that he is a consultant/advisory board member for L’Oréal, La Roche-Posay, Galderma, and other companies; a speaker for Regeneron/Sanofi, Incyte, BMD, and Janssen; and has grants from Pfizer, Lilly, Incyte, and other companies. Dr. Stempel reported no disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON — and conveys the skin’s vulnerability to serious medical complications, said Adam Friedman, MD, at the ElderDerm conference on dermatology in the older patient.
Key features of dermatoporosis include atrophic skin, solar purpura, white pseudoscars, easily acquired skin lacerations and tears, bruises, and delayed healing. “We’re going to see more of this, and it will more and more be a chief complaint of patients,” said Dr. Friedman, professor and chair of dermatology at George Washington University (GWU) in Washington, and co-chair of the meeting. GWU hosted the conference, describing it as a first-of-its-kind meeting dedicated to improving dermatologic care for older adults.
Dermatoporosis was described in the literature in 2007 by dermatologists at the University of Geneva in Switzerland. “It is not only a cosmetic problem,” Dr. Friedman said. “This is a medical problem ... which can absolutely lead to comorbidities [such as deep dissecting hematomas] that are a huge strain on the healthcare system.”
Dermatologists can meet the moment with holistic, creative combination treatment and counseling approaches aimed at improving the mechanical strength of skin and preventing potential complications in older patients, Dr. Friedman said at the meeting.
He described the case of a 76-year-old woman who presented with dermatoporosis on her arms involving pronounced skin atrophy, solar purpura, and a small covered laceration. “This was a patient who was both devastated by the appearance” and impacted by the pain and burden of dressing frequent wounds, said Dr. Friedman, who is also the director of the Residency Program, of Translational Research, and of Supportive Oncodermatology, all within the Department of Dermatology at GWU.
With 11 months of topical treatment that included daily application of calcipotriene 0.05% ointment and nightly application of tazarotene 0.045% lotion and oral supplementation with 1000-mg vitamin C twice daily and 1000-mg citrus bioflavonoid complex daily, as well as no changes to the medications she took for various comorbidities, the solar purpura improved significantly and “we made a huge difference in the integrity of her skin,” he said.
Dr. Friedman also described this case in a recently published article in the Journal of Drugs in Dermatology titled “What’s Old Is New: An Emerging Focus on Dermatoporosis”.
Likely Pathophysiology
Advancing age and chronic ultraviolet (UV) radiation exposure are the chief drivers of dermatoporosis. In addition to UVA and UVB light, other secondary drivers include genetic susceptibility, topical and systematic corticosteroid use, and anticoagulant treatment.
Its pathogenesis is not well described in the literature but is easy to envision, Dr. Friedman said. For one, both advancing age and exposure to UV light lead to a reduction in hygroscopic glycosaminoglycans, including hyaluronate (HA), and the impact of this diminishment is believed to go “beyond [the loss of] buoyancy,” he noted. Researchers have “been showing these are not just water-loving molecules, they also have some biologic properties” relating to keratinocyte production and epidermal turnover that appear to be intricately linked to the pathogenesis of dermatoporosis.
HAs have been shown to interact with the cell surface receptor CD44 to stimulate keratinocyte proliferation, and low levels of CD44 have been reported in skin with dermatoporosis compared with a younger control population. (A newly characterized organelle, the hyaluronosome, serves as an HA factory and contains CD44 and heparin-binding epidermal growth factor, Dr. Friedman noted. Inadequate functioning may be involved in skin atrophy.)
Advancing age also brings an increase in matrix metalloproteinases (MMPs)–1, –2, and –3, which are “the demolition workers of the skin,” and downregulation of a tissue inhibitor of MMPs, he said.
Adding insult to injury, dermis-penetrating UVA also activates MMPs, “obliterating collagen and elastin.” UVB generates DNA photoproducts, including oxidative stress and damaging skin cell DNA. “That UV light induces breakdown [of the skin] through different mechanisms and inhibits buildup is a simple concept I think our patients can understand,” Dr. Friedman said.
Multifaceted Treatment
For an older adult, “there is never a wrong time to start sun-protective measures” to prevent or try to halt the progression of dermatoporosis, Dr. Friedman said, noting that “UV radiation is an immunosuppressant, so there are many good reasons to start” if the adult is not already taking measures on a regular basis.
Potential treatments for the syndrome of dermatoporosis are backed by few clinical studies, but dermatologists are skilled at translating the use of products from one disease state to another based on understandings of pathophysiology and mechanistic pathways, Dr. Friedman commented in an interview after the meeting.
For instance, “from decades of research, we know what retinoids will do to the skin,” he said in the interview. “We know they will turn on collagen-1 and -3 genes in the skin, and that they will increase the production of glycosaminoglycans ... By understanding the biology, we can translate this to dermatoporosis.” These changes were demonstrated, for instance, in a small study of topical retinol in older adults.
Studies of topical alpha hydroxy acid (AHA), moreover, have demonstrated epidermal thickening and firmness, and “some studies show they can limit steroid-induced atrophy,” Dr. Friedman said at the meeting. “And things like lactic acid and urea are super accessible.”
Topical dehydroepiandrosterone is backed by even less data than retinoids or AHAs are, “but it’s still something to consider” as part of a multimechanistic approach to dermatoporosis, Dr. Friedman shared, noting that a small study demonstrated beneficial effects on epidermal atrophy in aging skin.
The use of vitamin D analogues such as calcipotriene, which is approved for the treatment of psoriasis, may also be promising. “One concept is that [vitamin D analogues] increase calcium concentrations in the epidermis, and calcium is so central to keratinocyte differentiation” and epidermal function that calcipotriene in combination with topical steroid therapy has been shown to limit skin atrophy, he noted.
Nutritionally, low protein intake is a known problem in the older population and is associated with increased skin fragility and poorer healing. From a prevention and treatment standpoint, therefore, patients can be counseled to be attentive to their diets, Dr. Friedman said. Experts have recommended a higher protein intake for older adults than for younger adults; in 2013, an international group recommended a protein intake of 1-1.5 g/kg/d for healthy older adults and more for those with acute or chronic illness.
“Patients love talking about diet and skin disease ... and they love over-the-counter nutraceuticals as well because they want something natural,” Dr. Friedman said. “I like using bioflavonoids in combination with vitamin C, which can be effective especially for solar purpura.”
A 6-week randomized, placebo-controlled, double-blind trial involving 67 patients with purpura associated with aging found a 50% reduction in purpura lesions among those took a particular citrus bioflavonoid blend twice daily. “I thought this was a pretty well-done study,” he said, noting that patient self-assessment and investigator global assessment were utilized.
Skin Injury and Wound Prevention
In addition to recommending gentle skin cleansers and daily moisturizing, dermatologists should talk to their older patients with dermatoporosis about their home environments. “What is it like? Is there furniture with sharp edges?” Dr. Friedman advised. If so, could they use sleeves or protectors on their arms or legs “to protect against injury?”
In a later meeting session about lower-extremity wounds on geriatric patients, Michael Stempel, DPM, assistant professor of medicine and surgery and chief of podiatry at GWU, said that he was happy to hear the term dermatoporosis being used because like diabetes, it’s a risk factor for developing lower-extremity wounds and poor wound healing.
He shared the case of an older woman with dermatoporosis who “tripped and skinned her knee against a step and then self-treated it for over a month by pouring hydrogen peroxide over it and letting air get to it.” The wound developed into “full-thickness tissue loss,” said Dr. Stempel, also medical director of the Wound Healing and Limb Preservation Center at GWU Hospital.
Misperceptions are common among older patients about how a simple wound should be managed; for instance, the adage “just let it get air” is not uncommon. This makes anticipatory guidance about basic wound care — such as the importance of a moist and occlusive environment and the safe use of hydrogen peroxide — especially important for patients with dermatoporosis, Dr. Friedman commented after the meeting.
Dermatoporosis is quantifiable, Dr. Friedman said during the meeting, with a scoring system having been developed by the researchers in Switzerland who originally coined the term. Its use in practice is unnecessary, but its existence is “nice to share with patients who feel bothered because oftentimes, patients feel it’s been dismissed by other providers,” he said. “Telling your patients there’s an actual name for their problem, and that there are ways to quantify and measure changes over time, is validating.”
Its recognition as a medical condition, Dr. Friedman added, also enables the dermatologist to bring it up and counsel appropriately — without a patient feeling shame — when it is identified in the context of a skin excision, treatment of a primary inflammatory skin disease, or management of another dermatologic problem.
Dr. Friedman disclosed that he is a consultant/advisory board member for L’Oréal, La Roche-Posay, Galderma, and other companies; a speaker for Regeneron/Sanofi, Incyte, BMD, and Janssen; and has grants from Pfizer, Lilly, Incyte, and other companies. Dr. Stempel reported no disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON — and conveys the skin’s vulnerability to serious medical complications, said Adam Friedman, MD, at the ElderDerm conference on dermatology in the older patient.
Key features of dermatoporosis include atrophic skin, solar purpura, white pseudoscars, easily acquired skin lacerations and tears, bruises, and delayed healing. “We’re going to see more of this, and it will more and more be a chief complaint of patients,” said Dr. Friedman, professor and chair of dermatology at George Washington University (GWU) in Washington, and co-chair of the meeting. GWU hosted the conference, describing it as a first-of-its-kind meeting dedicated to improving dermatologic care for older adults.
Dermatoporosis was described in the literature in 2007 by dermatologists at the University of Geneva in Switzerland. “It is not only a cosmetic problem,” Dr. Friedman said. “This is a medical problem ... which can absolutely lead to comorbidities [such as deep dissecting hematomas] that are a huge strain on the healthcare system.”
Dermatologists can meet the moment with holistic, creative combination treatment and counseling approaches aimed at improving the mechanical strength of skin and preventing potential complications in older patients, Dr. Friedman said at the meeting.
He described the case of a 76-year-old woman who presented with dermatoporosis on her arms involving pronounced skin atrophy, solar purpura, and a small covered laceration. “This was a patient who was both devastated by the appearance” and impacted by the pain and burden of dressing frequent wounds, said Dr. Friedman, who is also the director of the Residency Program, of Translational Research, and of Supportive Oncodermatology, all within the Department of Dermatology at GWU.
With 11 months of topical treatment that included daily application of calcipotriene 0.05% ointment and nightly application of tazarotene 0.045% lotion and oral supplementation with 1000-mg vitamin C twice daily and 1000-mg citrus bioflavonoid complex daily, as well as no changes to the medications she took for various comorbidities, the solar purpura improved significantly and “we made a huge difference in the integrity of her skin,” he said.
Dr. Friedman also described this case in a recently published article in the Journal of Drugs in Dermatology titled “What’s Old Is New: An Emerging Focus on Dermatoporosis”.
Likely Pathophysiology
Advancing age and chronic ultraviolet (UV) radiation exposure are the chief drivers of dermatoporosis. In addition to UVA and UVB light, other secondary drivers include genetic susceptibility, topical and systematic corticosteroid use, and anticoagulant treatment.
Its pathogenesis is not well described in the literature but is easy to envision, Dr. Friedman said. For one, both advancing age and exposure to UV light lead to a reduction in hygroscopic glycosaminoglycans, including hyaluronate (HA), and the impact of this diminishment is believed to go “beyond [the loss of] buoyancy,” he noted. Researchers have “been showing these are not just water-loving molecules, they also have some biologic properties” relating to keratinocyte production and epidermal turnover that appear to be intricately linked to the pathogenesis of dermatoporosis.
HAs have been shown to interact with the cell surface receptor CD44 to stimulate keratinocyte proliferation, and low levels of CD44 have been reported in skin with dermatoporosis compared with a younger control population. (A newly characterized organelle, the hyaluronosome, serves as an HA factory and contains CD44 and heparin-binding epidermal growth factor, Dr. Friedman noted. Inadequate functioning may be involved in skin atrophy.)
Advancing age also brings an increase in matrix metalloproteinases (MMPs)–1, –2, and –3, which are “the demolition workers of the skin,” and downregulation of a tissue inhibitor of MMPs, he said.
Adding insult to injury, dermis-penetrating UVA also activates MMPs, “obliterating collagen and elastin.” UVB generates DNA photoproducts, including oxidative stress and damaging skin cell DNA. “That UV light induces breakdown [of the skin] through different mechanisms and inhibits buildup is a simple concept I think our patients can understand,” Dr. Friedman said.
Multifaceted Treatment
For an older adult, “there is never a wrong time to start sun-protective measures” to prevent or try to halt the progression of dermatoporosis, Dr. Friedman said, noting that “UV radiation is an immunosuppressant, so there are many good reasons to start” if the adult is not already taking measures on a regular basis.
Potential treatments for the syndrome of dermatoporosis are backed by few clinical studies, but dermatologists are skilled at translating the use of products from one disease state to another based on understandings of pathophysiology and mechanistic pathways, Dr. Friedman commented in an interview after the meeting.
For instance, “from decades of research, we know what retinoids will do to the skin,” he said in the interview. “We know they will turn on collagen-1 and -3 genes in the skin, and that they will increase the production of glycosaminoglycans ... By understanding the biology, we can translate this to dermatoporosis.” These changes were demonstrated, for instance, in a small study of topical retinol in older adults.
Studies of topical alpha hydroxy acid (AHA), moreover, have demonstrated epidermal thickening and firmness, and “some studies show they can limit steroid-induced atrophy,” Dr. Friedman said at the meeting. “And things like lactic acid and urea are super accessible.”
Topical dehydroepiandrosterone is backed by even less data than retinoids or AHAs are, “but it’s still something to consider” as part of a multimechanistic approach to dermatoporosis, Dr. Friedman shared, noting that a small study demonstrated beneficial effects on epidermal atrophy in aging skin.
The use of vitamin D analogues such as calcipotriene, which is approved for the treatment of psoriasis, may also be promising. “One concept is that [vitamin D analogues] increase calcium concentrations in the epidermis, and calcium is so central to keratinocyte differentiation” and epidermal function that calcipotriene in combination with topical steroid therapy has been shown to limit skin atrophy, he noted.
Nutritionally, low protein intake is a known problem in the older population and is associated with increased skin fragility and poorer healing. From a prevention and treatment standpoint, therefore, patients can be counseled to be attentive to their diets, Dr. Friedman said. Experts have recommended a higher protein intake for older adults than for younger adults; in 2013, an international group recommended a protein intake of 1-1.5 g/kg/d for healthy older adults and more for those with acute or chronic illness.
“Patients love talking about diet and skin disease ... and they love over-the-counter nutraceuticals as well because they want something natural,” Dr. Friedman said. “I like using bioflavonoids in combination with vitamin C, which can be effective especially for solar purpura.”
A 6-week randomized, placebo-controlled, double-blind trial involving 67 patients with purpura associated with aging found a 50% reduction in purpura lesions among those took a particular citrus bioflavonoid blend twice daily. “I thought this was a pretty well-done study,” he said, noting that patient self-assessment and investigator global assessment were utilized.
Skin Injury and Wound Prevention
In addition to recommending gentle skin cleansers and daily moisturizing, dermatologists should talk to their older patients with dermatoporosis about their home environments. “What is it like? Is there furniture with sharp edges?” Dr. Friedman advised. If so, could they use sleeves or protectors on their arms or legs “to protect against injury?”
In a later meeting session about lower-extremity wounds on geriatric patients, Michael Stempel, DPM, assistant professor of medicine and surgery and chief of podiatry at GWU, said that he was happy to hear the term dermatoporosis being used because like diabetes, it’s a risk factor for developing lower-extremity wounds and poor wound healing.
He shared the case of an older woman with dermatoporosis who “tripped and skinned her knee against a step and then self-treated it for over a month by pouring hydrogen peroxide over it and letting air get to it.” The wound developed into “full-thickness tissue loss,” said Dr. Stempel, also medical director of the Wound Healing and Limb Preservation Center at GWU Hospital.
Misperceptions are common among older patients about how a simple wound should be managed; for instance, the adage “just let it get air” is not uncommon. This makes anticipatory guidance about basic wound care — such as the importance of a moist and occlusive environment and the safe use of hydrogen peroxide — especially important for patients with dermatoporosis, Dr. Friedman commented after the meeting.
Dermatoporosis is quantifiable, Dr. Friedman said during the meeting, with a scoring system having been developed by the researchers in Switzerland who originally coined the term. Its use in practice is unnecessary, but its existence is “nice to share with patients who feel bothered because oftentimes, patients feel it’s been dismissed by other providers,” he said. “Telling your patients there’s an actual name for their problem, and that there are ways to quantify and measure changes over time, is validating.”
Its recognition as a medical condition, Dr. Friedman added, also enables the dermatologist to bring it up and counsel appropriately — without a patient feeling shame — when it is identified in the context of a skin excision, treatment of a primary inflammatory skin disease, or management of another dermatologic problem.
Dr. Friedman disclosed that he is a consultant/advisory board member for L’Oréal, La Roche-Posay, Galderma, and other companies; a speaker for Regeneron/Sanofi, Incyte, BMD, and Janssen; and has grants from Pfizer, Lilly, Incyte, and other companies. Dr. Stempel reported no disclosures.
A version of this article first appeared on Medscape.com.
FROM ELDERDERM 2024
‘Emerging Threat’ Xylazine Use Continues to Spread Across the United States
Illicit use of the veterinary tranquilizer xylazine continues to spread across the United States. The drug, which is increasingly mixed with fentanyl, often fails to respond to the opioid overdose reversal medication naloxone and can cause severe necrotic lesions.
A report released by Millennium Health, a specialty lab that provides medication monitoring for pain management, drug treatment, and behavioral and substance use disorder treatment centers across the country, showed the number of urine specimens collected and tested at the US drug treatment centers were positive for xylazine in the most recent 6 months.
As previously reported by this news organization, in late 2022, the US Food and Drug Administration (FDA) issued a communication alerting clinicians about the special management required for opioid overdoses tainted with xylazine, which is also known as “tranq” or “tranq dope.”
Subsequently, in early 2023, The White House Office of National Drug Control Policy designated xylazine combined with fentanyl as an emerging threat to the United States.
Both the FDA and the Drug Enforcement Administration have taken steps to try to stop trafficking of the combination. However, despite these efforts, xylazine use has continued to spread.
The Millennium Health Signals report showed that the greatest increase in xylazine use was largely in the western United States. In the first 6 months of 2023, 3% of urine drug tests (UDTs) in Washington, Oregon, California, Hawaii, and Alaska were positive for xylazine. From November 2023 to April 2024, this rose to 8%, a 147% increase. In the Mountain West, xylazine-positive UDTs increased from 2% in 2023 to 4% in 2024, an increase of 94%. In addition to growth in the West, the report showed that xylazine use increased by more than 100% in New England — from 14% in 2023 to 28% in 2024.
Nationally, 16% of all urine specimens were positive for xylazine from late 2023 to April 2024, up slightly from 14% from April to October 2023.
Xylazine use was highest in the East and in the mid-Atlantic United States. Still, positivity rates in the mid-Atlantic dropped from 44% to 33%. The states included in that group were New York, Pennsylvania, Delaware, and New Jersey. East North Central states (Ohio, Michigan, Wisconsin, Indiana, and Illinois) also experienced a decline in positive tests from 32% to 30%.
The South Atlantic states, which include Maryland, Virginia, West Virginia, North and South Carolina, Georgia, and Florida, had a 17% increase in positivity — from 22% to 26%.
From April 2023 to April 2024 state-level UDT positivity rates were 40% in Pennsylvania, 37% in New York, and 35% in Ohio. But rates vary by locality. In Clermont and Hamilton counties in Ohio — both in the Cincinnati area — about 70% of specimens were positive for xylazine.
About one third of specimens in Maryland and South Carolina contained xylazine.
“Because xylazine exposure remains a significant challenge in the East and is a growing concern in the West, clinicians across the US need to be prepared to recognize and address the consequences of xylazine use — like diminished responses to naloxone and severe skin wounds that may lead to amputation — among people who use fentanyl,” Millennium Health Chief Clinical Officer Angela Huskey, PharmD, said in a press release.
The Health Signals Alert analyzed more than 50,000 fentanyl-positive UDT specimens collected between April 12, 2023, and April 11, 2024. Millennium Health researchers analyzed xylazine positivity rates in fentanyl-positive UDT specimens by the US Census Division and state.
A version of this article first appeared on Medscape.com.
Illicit use of the veterinary tranquilizer xylazine continues to spread across the United States. The drug, which is increasingly mixed with fentanyl, often fails to respond to the opioid overdose reversal medication naloxone and can cause severe necrotic lesions.
A report released by Millennium Health, a specialty lab that provides medication monitoring for pain management, drug treatment, and behavioral and substance use disorder treatment centers across the country, showed the number of urine specimens collected and tested at the US drug treatment centers were positive for xylazine in the most recent 6 months.
As previously reported by this news organization, in late 2022, the US Food and Drug Administration (FDA) issued a communication alerting clinicians about the special management required for opioid overdoses tainted with xylazine, which is also known as “tranq” or “tranq dope.”
Subsequently, in early 2023, The White House Office of National Drug Control Policy designated xylazine combined with fentanyl as an emerging threat to the United States.
Both the FDA and the Drug Enforcement Administration have taken steps to try to stop trafficking of the combination. However, despite these efforts, xylazine use has continued to spread.
The Millennium Health Signals report showed that the greatest increase in xylazine use was largely in the western United States. In the first 6 months of 2023, 3% of urine drug tests (UDTs) in Washington, Oregon, California, Hawaii, and Alaska were positive for xylazine. From November 2023 to April 2024, this rose to 8%, a 147% increase. In the Mountain West, xylazine-positive UDTs increased from 2% in 2023 to 4% in 2024, an increase of 94%. In addition to growth in the West, the report showed that xylazine use increased by more than 100% in New England — from 14% in 2023 to 28% in 2024.
Nationally, 16% of all urine specimens were positive for xylazine from late 2023 to April 2024, up slightly from 14% from April to October 2023.
Xylazine use was highest in the East and in the mid-Atlantic United States. Still, positivity rates in the mid-Atlantic dropped from 44% to 33%. The states included in that group were New York, Pennsylvania, Delaware, and New Jersey. East North Central states (Ohio, Michigan, Wisconsin, Indiana, and Illinois) also experienced a decline in positive tests from 32% to 30%.
The South Atlantic states, which include Maryland, Virginia, West Virginia, North and South Carolina, Georgia, and Florida, had a 17% increase in positivity — from 22% to 26%.
From April 2023 to April 2024 state-level UDT positivity rates were 40% in Pennsylvania, 37% in New York, and 35% in Ohio. But rates vary by locality. In Clermont and Hamilton counties in Ohio — both in the Cincinnati area — about 70% of specimens were positive for xylazine.
About one third of specimens in Maryland and South Carolina contained xylazine.
“Because xylazine exposure remains a significant challenge in the East and is a growing concern in the West, clinicians across the US need to be prepared to recognize and address the consequences of xylazine use — like diminished responses to naloxone and severe skin wounds that may lead to amputation — among people who use fentanyl,” Millennium Health Chief Clinical Officer Angela Huskey, PharmD, said in a press release.
The Health Signals Alert analyzed more than 50,000 fentanyl-positive UDT specimens collected between April 12, 2023, and April 11, 2024. Millennium Health researchers analyzed xylazine positivity rates in fentanyl-positive UDT specimens by the US Census Division and state.
A version of this article first appeared on Medscape.com.
Illicit use of the veterinary tranquilizer xylazine continues to spread across the United States. The drug, which is increasingly mixed with fentanyl, often fails to respond to the opioid overdose reversal medication naloxone and can cause severe necrotic lesions.
A report released by Millennium Health, a specialty lab that provides medication monitoring for pain management, drug treatment, and behavioral and substance use disorder treatment centers across the country, showed the number of urine specimens collected and tested at the US drug treatment centers were positive for xylazine in the most recent 6 months.
As previously reported by this news organization, in late 2022, the US Food and Drug Administration (FDA) issued a communication alerting clinicians about the special management required for opioid overdoses tainted with xylazine, which is also known as “tranq” or “tranq dope.”
Subsequently, in early 2023, The White House Office of National Drug Control Policy designated xylazine combined with fentanyl as an emerging threat to the United States.
Both the FDA and the Drug Enforcement Administration have taken steps to try to stop trafficking of the combination. However, despite these efforts, xylazine use has continued to spread.
The Millennium Health Signals report showed that the greatest increase in xylazine use was largely in the western United States. In the first 6 months of 2023, 3% of urine drug tests (UDTs) in Washington, Oregon, California, Hawaii, and Alaska were positive for xylazine. From November 2023 to April 2024, this rose to 8%, a 147% increase. In the Mountain West, xylazine-positive UDTs increased from 2% in 2023 to 4% in 2024, an increase of 94%. In addition to growth in the West, the report showed that xylazine use increased by more than 100% in New England — from 14% in 2023 to 28% in 2024.
Nationally, 16% of all urine specimens were positive for xylazine from late 2023 to April 2024, up slightly from 14% from April to October 2023.
Xylazine use was highest in the East and in the mid-Atlantic United States. Still, positivity rates in the mid-Atlantic dropped from 44% to 33%. The states included in that group were New York, Pennsylvania, Delaware, and New Jersey. East North Central states (Ohio, Michigan, Wisconsin, Indiana, and Illinois) also experienced a decline in positive tests from 32% to 30%.
The South Atlantic states, which include Maryland, Virginia, West Virginia, North and South Carolina, Georgia, and Florida, had a 17% increase in positivity — from 22% to 26%.
From April 2023 to April 2024 state-level UDT positivity rates were 40% in Pennsylvania, 37% in New York, and 35% in Ohio. But rates vary by locality. In Clermont and Hamilton counties in Ohio — both in the Cincinnati area — about 70% of specimens were positive for xylazine.
About one third of specimens in Maryland and South Carolina contained xylazine.
“Because xylazine exposure remains a significant challenge in the East and is a growing concern in the West, clinicians across the US need to be prepared to recognize and address the consequences of xylazine use — like diminished responses to naloxone and severe skin wounds that may lead to amputation — among people who use fentanyl,” Millennium Health Chief Clinical Officer Angela Huskey, PharmD, said in a press release.
The Health Signals Alert analyzed more than 50,000 fentanyl-positive UDT specimens collected between April 12, 2023, and April 11, 2024. Millennium Health researchers analyzed xylazine positivity rates in fentanyl-positive UDT specimens by the US Census Division and state.
A version of this article first appeared on Medscape.com.
Progressive Eyelash Loss and Scale of the Right Eyelid
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
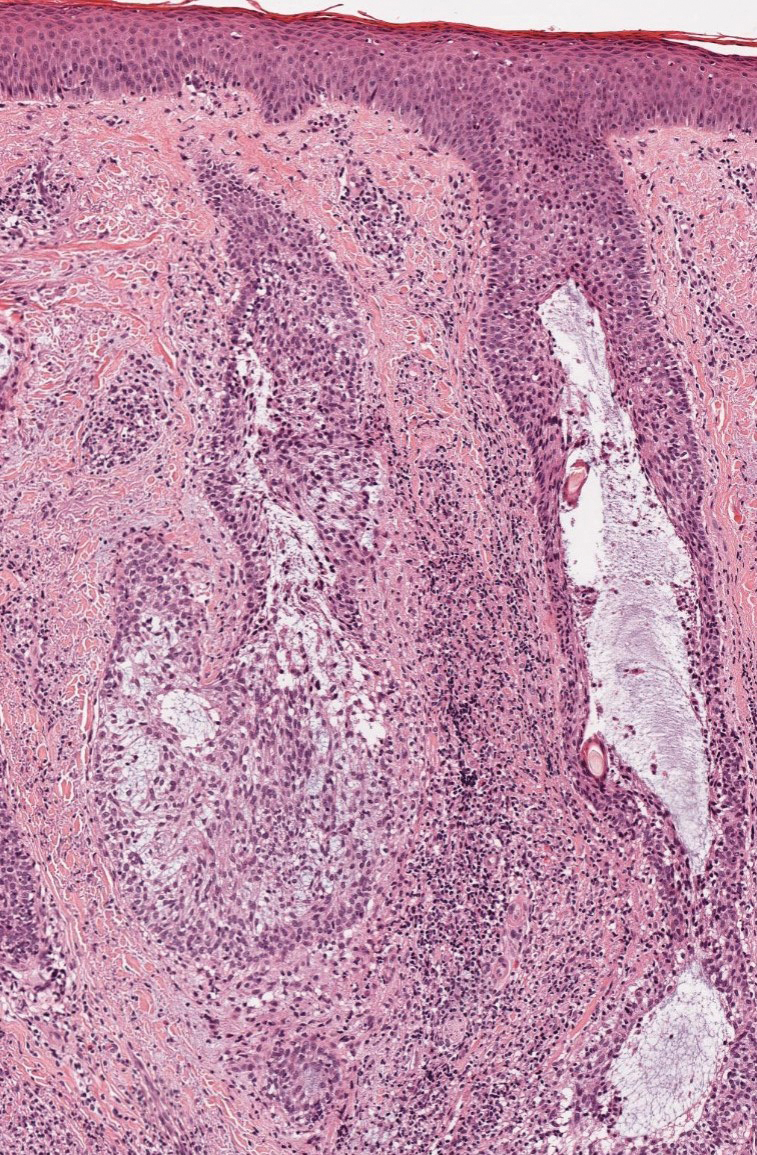
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
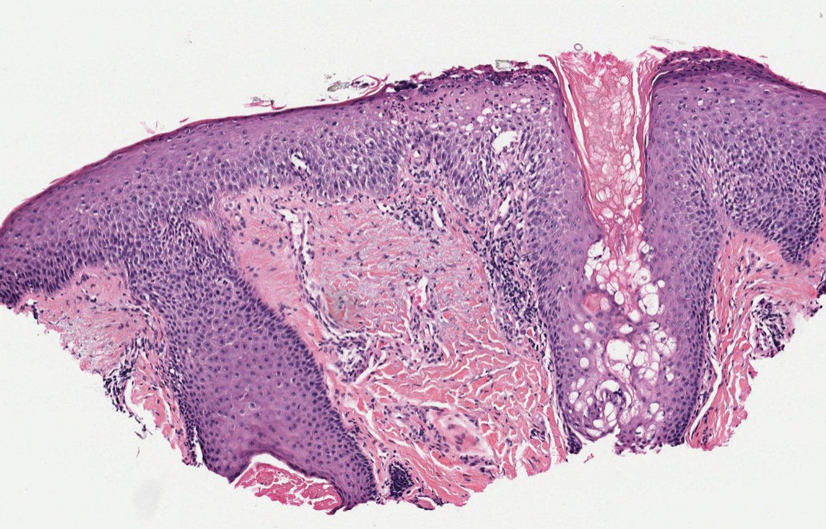
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
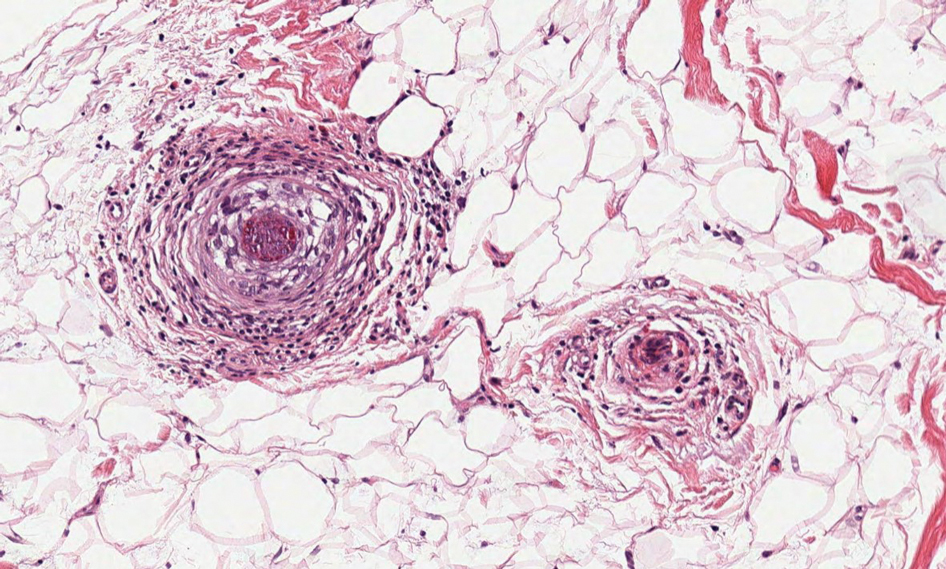
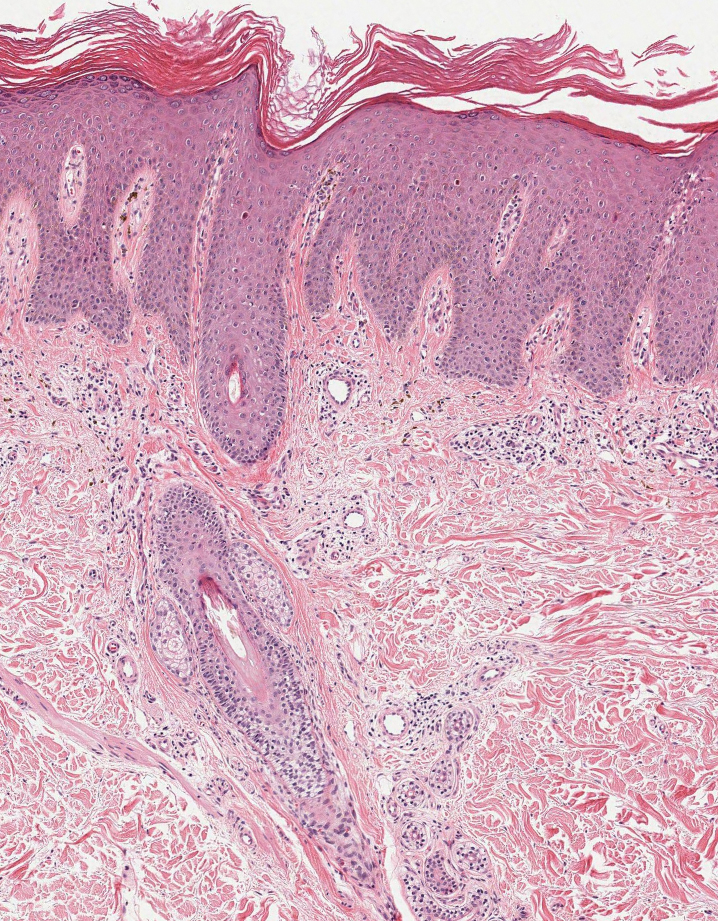
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
An 88-year-old man presented with progressive eyelash loss and scale involving the right eyelids (top). Dermatopathologic examination was performed (bottom).
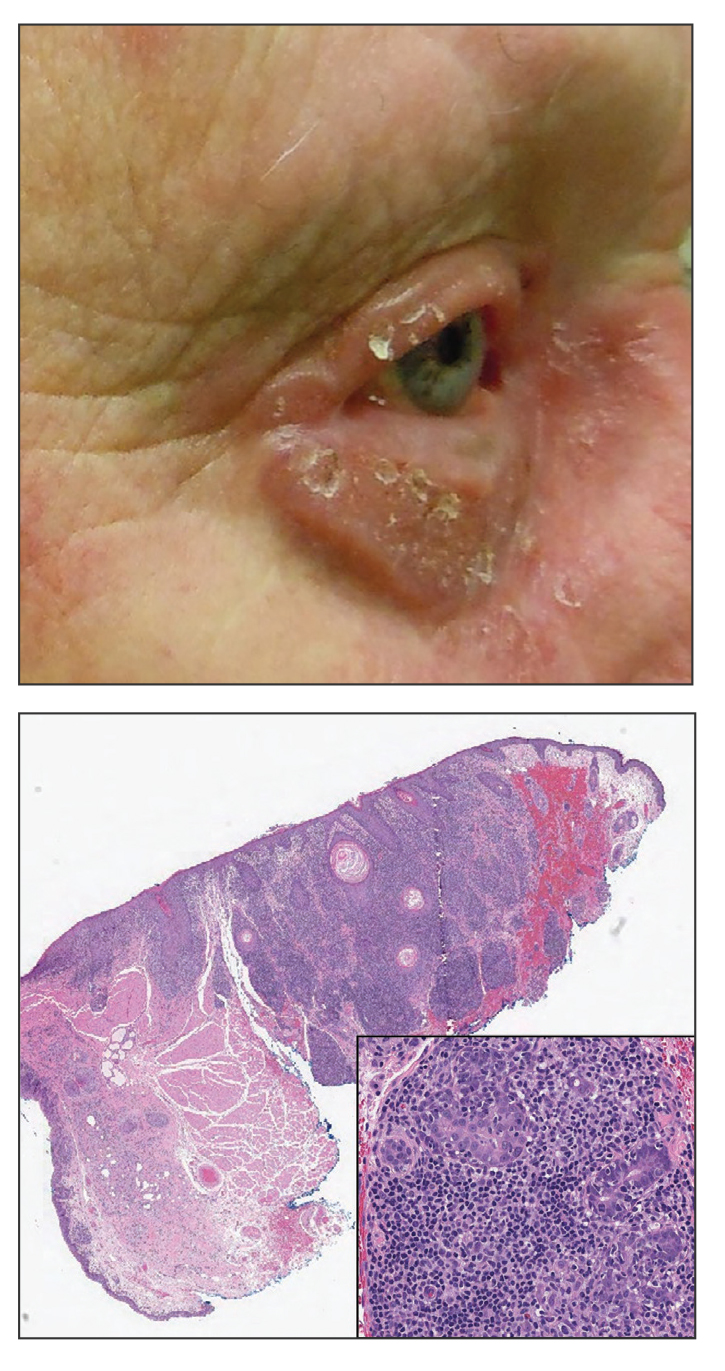
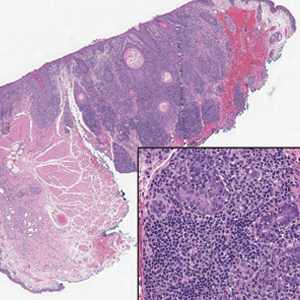
Pigmented Lesion on the Left Shoulder in an Older Woman
The Diagnosis: Pigmented Nodular Basal Cell Carcinoma
Dermoscopy of our patient’s irregular dark brown papule revealed large blue clustered clods and radial lines converging to a central dot (middle quiz image). Histopathology revealed nests of basaloid cells with peripheral palisading, small horn pseudocysts, and deposits of melanin extending into the dermis (Figure). These findings were consistent with a diagnosis of pigmented nodular basal cell carcinoma (BCC).
Nodular BCC represents 60% to 80% of all BCC cases; pigmented BCC represents 6% of BCC cases.1 Basal cell carcinomas frequently manifest as pearly papules with areas of pigment, surface telangiectases, and foci of ulceration. Dermoscopic features include fine arborizing vessels, blue-gray ovoid nests, spoke wheel–like structures, leaflike structures, and focal ulceration.1 Histopathology shows well-defined dermal nodules comprising basaloid epithelial cells with peripheral palisading, mucinous stroma, focal melanin deposits, and surrounding clefting.2 Arborizing vessels correspond to dilated vessels in the dermis.3 Blue-gray ovoid nests are wellcircumscribed ovoid or elongated structures that correspond histologically to well-defined large tumor nests with melanin aggregates invading the dermis. Spoke wheel–like structures are well-circumscribed radial projections connected to a pigmented central axis that correspond histologically to tumor nests near the epidermis and that appear as fingerlike projections with centrally located melanin deposits.3
The differential diagnosis of our patient’s lesion included nodular melanoma, lentigo maligna melanoma, deep penetrating nevus, and cellular blue nevus. Nodular melanoma is an invasive melanoma that lacks a radial growth phase. Dermoscopically, the more common features are a bluewhite veil, atypical vascular pattern, asymmetric pigmentation, atypical pigment network, and peripheral black globules.4 Histopathology reveals atypical melanocytes and architectural disorder.2 Pigmented nodular BCC also can display dark globules on dermoscopy but typically has smaller and more arborizing blood vessels and does not have a pigmented network. Furthermore, BCC would not have atypical melanocytes on histopathology.4,5
Dermoscopy of lentigo maligna melanoma displays hyperpigmented follicular openings, an annular-granular pattern, pigmented rhomboidal structures, and obliterated hair follicles.6 Histopathology demonstrates epidermal atrophy, increased pigmentation in basal keratinocytes, prominent solar elastosis, and an increased number of melanocytes that extend beyond the epidermis. 7 Pigmented nodular BCC can be distinguished from lentigo maligna melanoma dermoscopically by the presence of arborizing vessels, blue-gray ovoid nests, and lack of a pigment network.
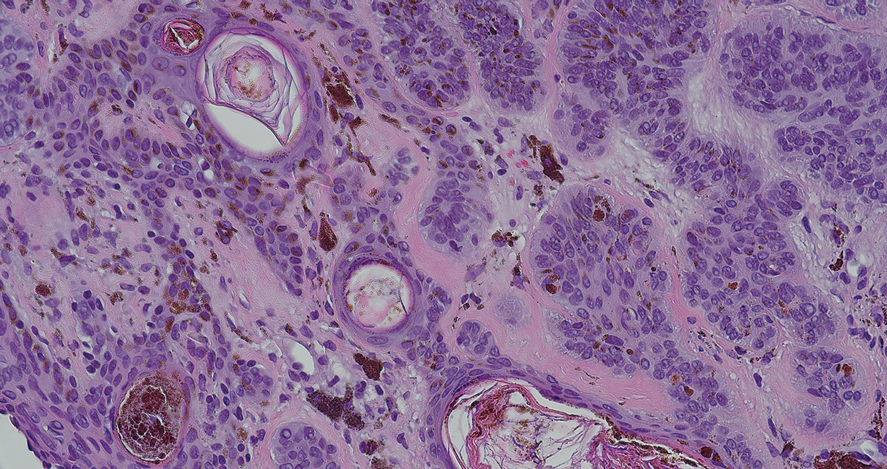
Deep penetrating nevus is a darkly pigmented melanocytic lesion that infiltrates deeply into the reticular dermis.8 Specific dermoscopic features have not been well established; however, a uniformly dark blue or black pattern is common. Histologically, this type of nevus is symmetric and wedge shaped with a broad base extending to the deep dermis and subcutaneous fat.8 Melanocytes do not exhibit atypia or bizarre mitoses. Although pigmented nodular BCC can appear similar to deep penetrating nevus, histologically there will be atypical basaloid epithelial cells in BCC.
Blue nevi clinically appear as a smooth blue-gray lesion with a steel blue ground-glass pattern on dermoscopy. Histopathology shows spindle-shaped melanocytes in the dermis, which distinguishes this lesion from BCC.9
Consider pigmented BCC when a patient presents with a pigmented lesion. Dermoscopy can help appreciate a pigmented BCC by looking for features such as a spoke wheel– like pattern, blue ovoid nests, arborizing blood vessels, and lack of a pigment network. Because pigmented BCC constitutes a small fraction of all BCCs, it is important to be familiar with its presentation and dermoscopic features.
- Heath MS, Bar A. Basal cell carcinoma. Dermatol Clin. 2023;41:13-21. doi:10.1016/j.det.2022.07.005
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014; 28:1005-1012.
- Wozniak-Rito A, Zalaudek I, Rudnicka L. Dermoscopy of basal cell carcinoma. Clin Exp Dermatol. 2018;43:241-247. doi:10.1111/ced.13387
- Menzies SW, Moloney FJ, Byth K, et al. Dermoscopic valuation of nodular melanoma. JAMA Dermatol. 2013;149:699-709. doi:10.1001 /jamadermatol.2013.2466
- Pizzichetta MA, Kittler H, Stanganelli I, et al; Italian Melanoma Intergroup. Pigmented nodular melanoma: the predictive value of dermoscopic features using multivariate analysis. Br J Dermatol. 2015;173:106-114. doi:10.1111/bjd.13861
- Pralong P, Bathelier E, Dalle S, et al. Dermoscopy of lentigo maligna melanoma: report of 125 cases. Br J Dermatol. 2012;167:280-287. doi:10.1111/j.1365-2133.2012.10932.x
- Reed JA, Shea CR. Lentigo maligna: melanoma in situ on chronically sun-damaged skin. Arch Pathol Lab Med. 2011;135:838-841. doi:10.5858/2011-0051-RAIR.1
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240. doi:10.1016/j .jaad.2014.07.026
- Ferrera G, Argenziano G. Blue nevus. In: Soyer HP, Argenziano G, Hofmann-Wellenhof R, et al, eds. Color Atlas of Melanocytic Lesions of the Skin. Springer; 2007:78-86.
The Diagnosis: Pigmented Nodular Basal Cell Carcinoma
Dermoscopy of our patient’s irregular dark brown papule revealed large blue clustered clods and radial lines converging to a central dot (middle quiz image). Histopathology revealed nests of basaloid cells with peripheral palisading, small horn pseudocysts, and deposits of melanin extending into the dermis (Figure). These findings were consistent with a diagnosis of pigmented nodular basal cell carcinoma (BCC).
Nodular BCC represents 60% to 80% of all BCC cases; pigmented BCC represents 6% of BCC cases.1 Basal cell carcinomas frequently manifest as pearly papules with areas of pigment, surface telangiectases, and foci of ulceration. Dermoscopic features include fine arborizing vessels, blue-gray ovoid nests, spoke wheel–like structures, leaflike structures, and focal ulceration.1 Histopathology shows well-defined dermal nodules comprising basaloid epithelial cells with peripheral palisading, mucinous stroma, focal melanin deposits, and surrounding clefting.2 Arborizing vessels correspond to dilated vessels in the dermis.3 Blue-gray ovoid nests are wellcircumscribed ovoid or elongated structures that correspond histologically to well-defined large tumor nests with melanin aggregates invading the dermis. Spoke wheel–like structures are well-circumscribed radial projections connected to a pigmented central axis that correspond histologically to tumor nests near the epidermis and that appear as fingerlike projections with centrally located melanin deposits.3
The differential diagnosis of our patient’s lesion included nodular melanoma, lentigo maligna melanoma, deep penetrating nevus, and cellular blue nevus. Nodular melanoma is an invasive melanoma that lacks a radial growth phase. Dermoscopically, the more common features are a bluewhite veil, atypical vascular pattern, asymmetric pigmentation, atypical pigment network, and peripheral black globules.4 Histopathology reveals atypical melanocytes and architectural disorder.2 Pigmented nodular BCC also can display dark globules on dermoscopy but typically has smaller and more arborizing blood vessels and does not have a pigmented network. Furthermore, BCC would not have atypical melanocytes on histopathology.4,5
Dermoscopy of lentigo maligna melanoma displays hyperpigmented follicular openings, an annular-granular pattern, pigmented rhomboidal structures, and obliterated hair follicles.6 Histopathology demonstrates epidermal atrophy, increased pigmentation in basal keratinocytes, prominent solar elastosis, and an increased number of melanocytes that extend beyond the epidermis. 7 Pigmented nodular BCC can be distinguished from lentigo maligna melanoma dermoscopically by the presence of arborizing vessels, blue-gray ovoid nests, and lack of a pigment network.
Deep penetrating nevus is a darkly pigmented melanocytic lesion that infiltrates deeply into the reticular dermis.8 Specific dermoscopic features have not been well established; however, a uniformly dark blue or black pattern is common. Histologically, this type of nevus is symmetric and wedge shaped with a broad base extending to the deep dermis and subcutaneous fat.8 Melanocytes do not exhibit atypia or bizarre mitoses. Although pigmented nodular BCC can appear similar to deep penetrating nevus, histologically there will be atypical basaloid epithelial cells in BCC.
Blue nevi clinically appear as a smooth blue-gray lesion with a steel blue ground-glass pattern on dermoscopy. Histopathology shows spindle-shaped melanocytes in the dermis, which distinguishes this lesion from BCC.9
Consider pigmented BCC when a patient presents with a pigmented lesion. Dermoscopy can help appreciate a pigmented BCC by looking for features such as a spoke wheel– like pattern, blue ovoid nests, arborizing blood vessels, and lack of a pigment network. Because pigmented BCC constitutes a small fraction of all BCCs, it is important to be familiar with its presentation and dermoscopic features.
The Diagnosis: Pigmented Nodular Basal Cell Carcinoma
Dermoscopy of our patient’s irregular dark brown papule revealed large blue clustered clods and radial lines converging to a central dot (middle quiz image). Histopathology revealed nests of basaloid cells with peripheral palisading, small horn pseudocysts, and deposits of melanin extending into the dermis (Figure). These findings were consistent with a diagnosis of pigmented nodular basal cell carcinoma (BCC).
Nodular BCC represents 60% to 80% of all BCC cases; pigmented BCC represents 6% of BCC cases.1 Basal cell carcinomas frequently manifest as pearly papules with areas of pigment, surface telangiectases, and foci of ulceration. Dermoscopic features include fine arborizing vessels, blue-gray ovoid nests, spoke wheel–like structures, leaflike structures, and focal ulceration.1 Histopathology shows well-defined dermal nodules comprising basaloid epithelial cells with peripheral palisading, mucinous stroma, focal melanin deposits, and surrounding clefting.2 Arborizing vessels correspond to dilated vessels in the dermis.3 Blue-gray ovoid nests are wellcircumscribed ovoid or elongated structures that correspond histologically to well-defined large tumor nests with melanin aggregates invading the dermis. Spoke wheel–like structures are well-circumscribed radial projections connected to a pigmented central axis that correspond histologically to tumor nests near the epidermis and that appear as fingerlike projections with centrally located melanin deposits.3
The differential diagnosis of our patient’s lesion included nodular melanoma, lentigo maligna melanoma, deep penetrating nevus, and cellular blue nevus. Nodular melanoma is an invasive melanoma that lacks a radial growth phase. Dermoscopically, the more common features are a bluewhite veil, atypical vascular pattern, asymmetric pigmentation, atypical pigment network, and peripheral black globules.4 Histopathology reveals atypical melanocytes and architectural disorder.2 Pigmented nodular BCC also can display dark globules on dermoscopy but typically has smaller and more arborizing blood vessels and does not have a pigmented network. Furthermore, BCC would not have atypical melanocytes on histopathology.4,5
Dermoscopy of lentigo maligna melanoma displays hyperpigmented follicular openings, an annular-granular pattern, pigmented rhomboidal structures, and obliterated hair follicles.6 Histopathology demonstrates epidermal atrophy, increased pigmentation in basal keratinocytes, prominent solar elastosis, and an increased number of melanocytes that extend beyond the epidermis. 7 Pigmented nodular BCC can be distinguished from lentigo maligna melanoma dermoscopically by the presence of arborizing vessels, blue-gray ovoid nests, and lack of a pigment network.
Deep penetrating nevus is a darkly pigmented melanocytic lesion that infiltrates deeply into the reticular dermis.8 Specific dermoscopic features have not been well established; however, a uniformly dark blue or black pattern is common. Histologically, this type of nevus is symmetric and wedge shaped with a broad base extending to the deep dermis and subcutaneous fat.8 Melanocytes do not exhibit atypia or bizarre mitoses. Although pigmented nodular BCC can appear similar to deep penetrating nevus, histologically there will be atypical basaloid epithelial cells in BCC.
Blue nevi clinically appear as a smooth blue-gray lesion with a steel blue ground-glass pattern on dermoscopy. Histopathology shows spindle-shaped melanocytes in the dermis, which distinguishes this lesion from BCC.9
Consider pigmented BCC when a patient presents with a pigmented lesion. Dermoscopy can help appreciate a pigmented BCC by looking for features such as a spoke wheel– like pattern, blue ovoid nests, arborizing blood vessels, and lack of a pigment network. Because pigmented BCC constitutes a small fraction of all BCCs, it is important to be familiar with its presentation and dermoscopic features.
- Heath MS, Bar A. Basal cell carcinoma. Dermatol Clin. 2023;41:13-21. doi:10.1016/j.det.2022.07.005
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014; 28:1005-1012.
- Wozniak-Rito A, Zalaudek I, Rudnicka L. Dermoscopy of basal cell carcinoma. Clin Exp Dermatol. 2018;43:241-247. doi:10.1111/ced.13387
- Menzies SW, Moloney FJ, Byth K, et al. Dermoscopic valuation of nodular melanoma. JAMA Dermatol. 2013;149:699-709. doi:10.1001 /jamadermatol.2013.2466
- Pizzichetta MA, Kittler H, Stanganelli I, et al; Italian Melanoma Intergroup. Pigmented nodular melanoma: the predictive value of dermoscopic features using multivariate analysis. Br J Dermatol. 2015;173:106-114. doi:10.1111/bjd.13861
- Pralong P, Bathelier E, Dalle S, et al. Dermoscopy of lentigo maligna melanoma: report of 125 cases. Br J Dermatol. 2012;167:280-287. doi:10.1111/j.1365-2133.2012.10932.x
- Reed JA, Shea CR. Lentigo maligna: melanoma in situ on chronically sun-damaged skin. Arch Pathol Lab Med. 2011;135:838-841. doi:10.5858/2011-0051-RAIR.1
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240. doi:10.1016/j .jaad.2014.07.026
- Ferrera G, Argenziano G. Blue nevus. In: Soyer HP, Argenziano G, Hofmann-Wellenhof R, et al, eds. Color Atlas of Melanocytic Lesions of the Skin. Springer; 2007:78-86.
- Heath MS, Bar A. Basal cell carcinoma. Dermatol Clin. 2023;41:13-21. doi:10.1016/j.det.2022.07.005
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014; 28:1005-1012.
- Wozniak-Rito A, Zalaudek I, Rudnicka L. Dermoscopy of basal cell carcinoma. Clin Exp Dermatol. 2018;43:241-247. doi:10.1111/ced.13387
- Menzies SW, Moloney FJ, Byth K, et al. Dermoscopic valuation of nodular melanoma. JAMA Dermatol. 2013;149:699-709. doi:10.1001 /jamadermatol.2013.2466
- Pizzichetta MA, Kittler H, Stanganelli I, et al; Italian Melanoma Intergroup. Pigmented nodular melanoma: the predictive value of dermoscopic features using multivariate analysis. Br J Dermatol. 2015;173:106-114. doi:10.1111/bjd.13861
- Pralong P, Bathelier E, Dalle S, et al. Dermoscopy of lentigo maligna melanoma: report of 125 cases. Br J Dermatol. 2012;167:280-287. doi:10.1111/j.1365-2133.2012.10932.x
- Reed JA, Shea CR. Lentigo maligna: melanoma in situ on chronically sun-damaged skin. Arch Pathol Lab Med. 2011;135:838-841. doi:10.5858/2011-0051-RAIR.1
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240. doi:10.1016/j .jaad.2014.07.026
- Ferrera G, Argenziano G. Blue nevus. In: Soyer HP, Argenziano G, Hofmann-Wellenhof R, et al, eds. Color Atlas of Melanocytic Lesions of the Skin. Springer; 2007:78-86.
A 92-year-old woman presented to dermatology as a new patient for a full-body skin examination. She had a history of sarcoidosis and a liposarcoma that had been excised more than 20 years prior. She had no history of skin cancer; however, her granddaughter recently was diagnosed with melanoma. Physical examination revealed a 5-mm, irregular, dark brown papule on the left shoulder (top) that was evaluated by dermoscopy (middle). A tangential biopsy was performed for histopathologic analysis (bottom).
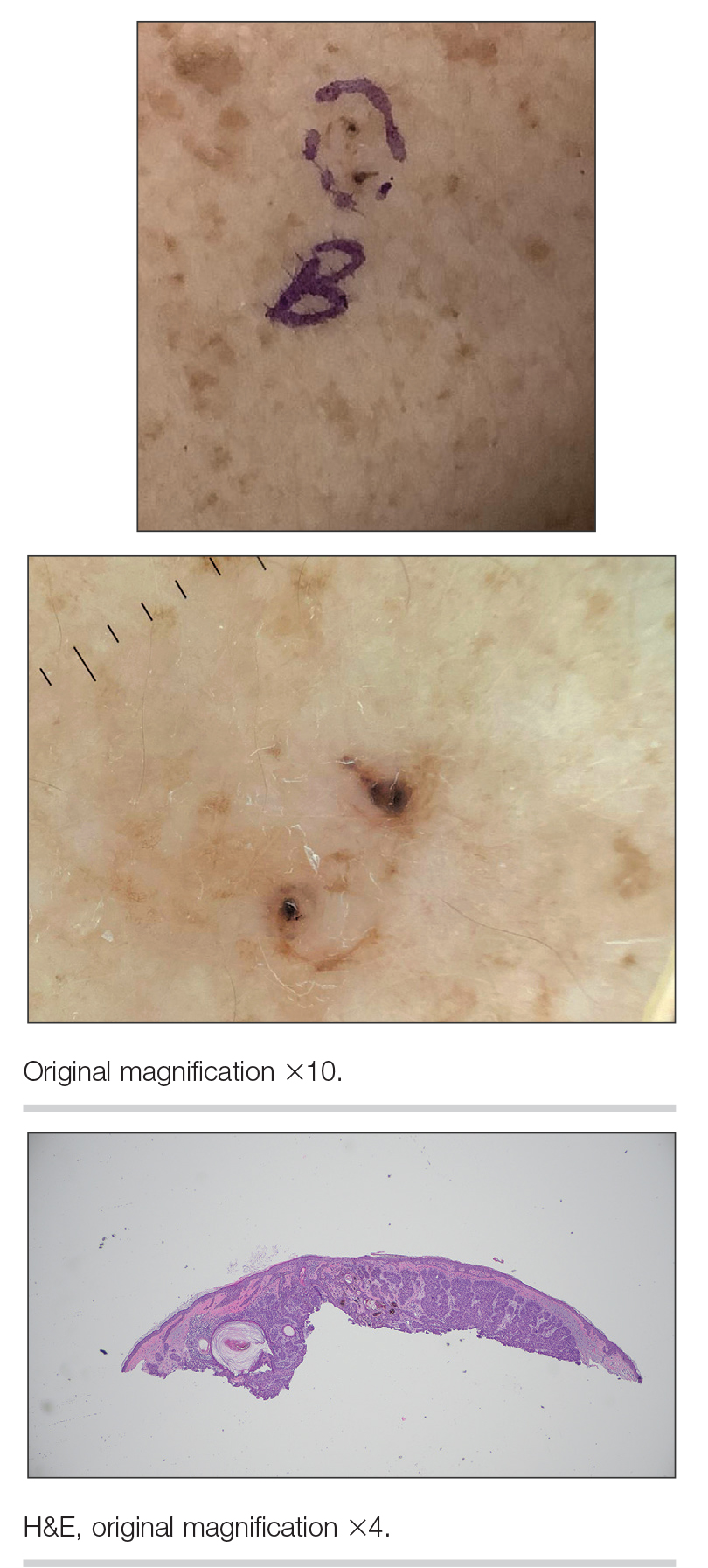
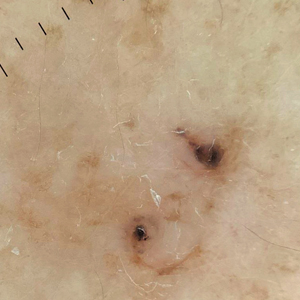
Erythematous Flaky Rash on the Toe
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
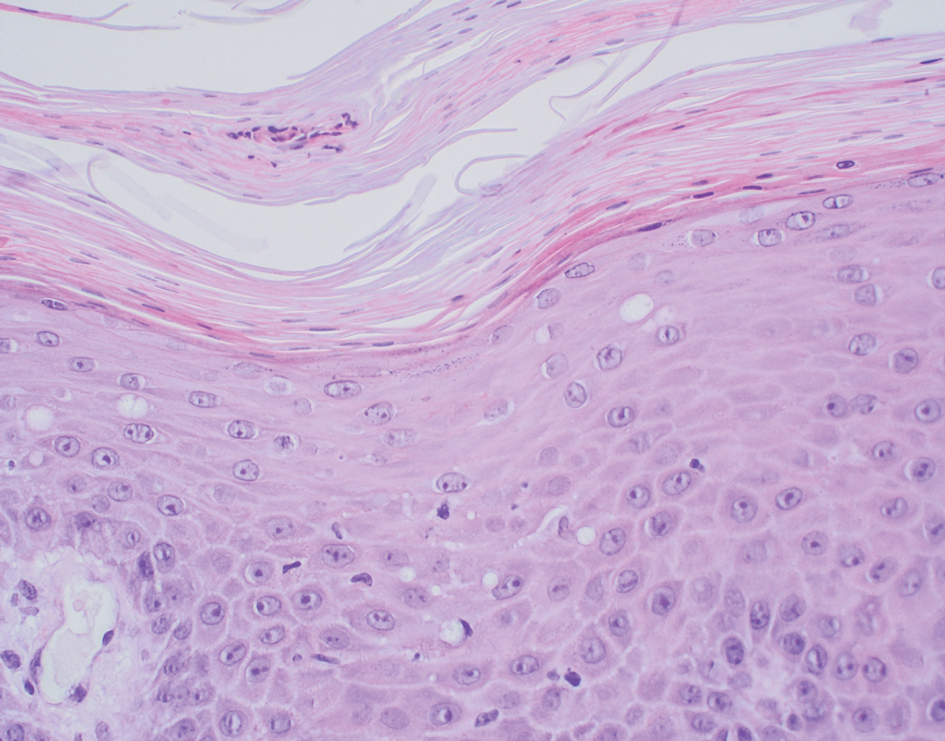
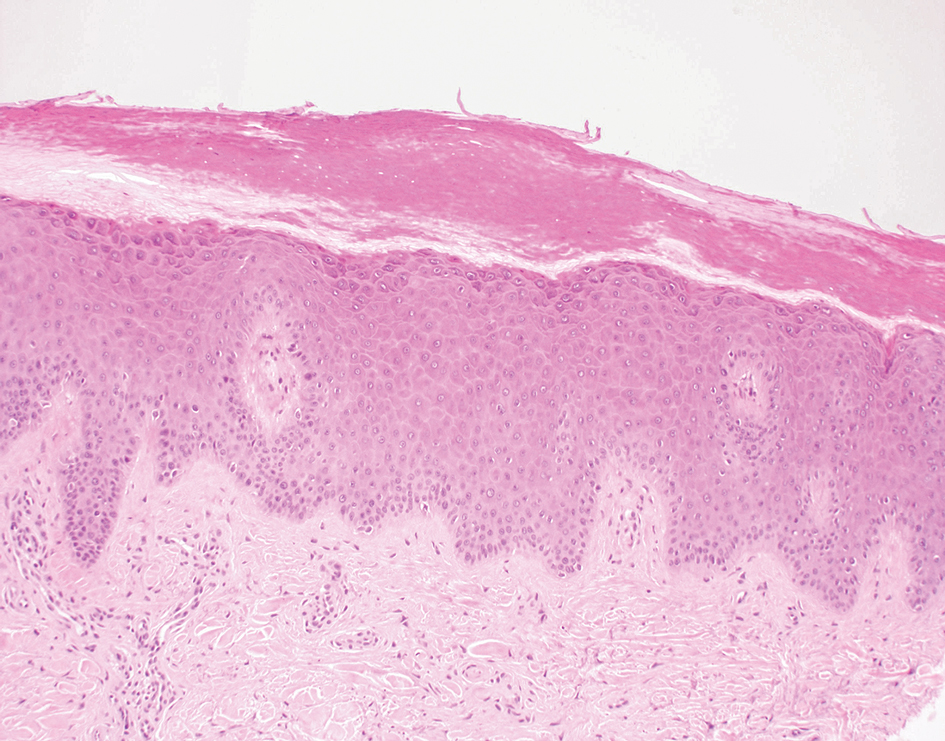
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
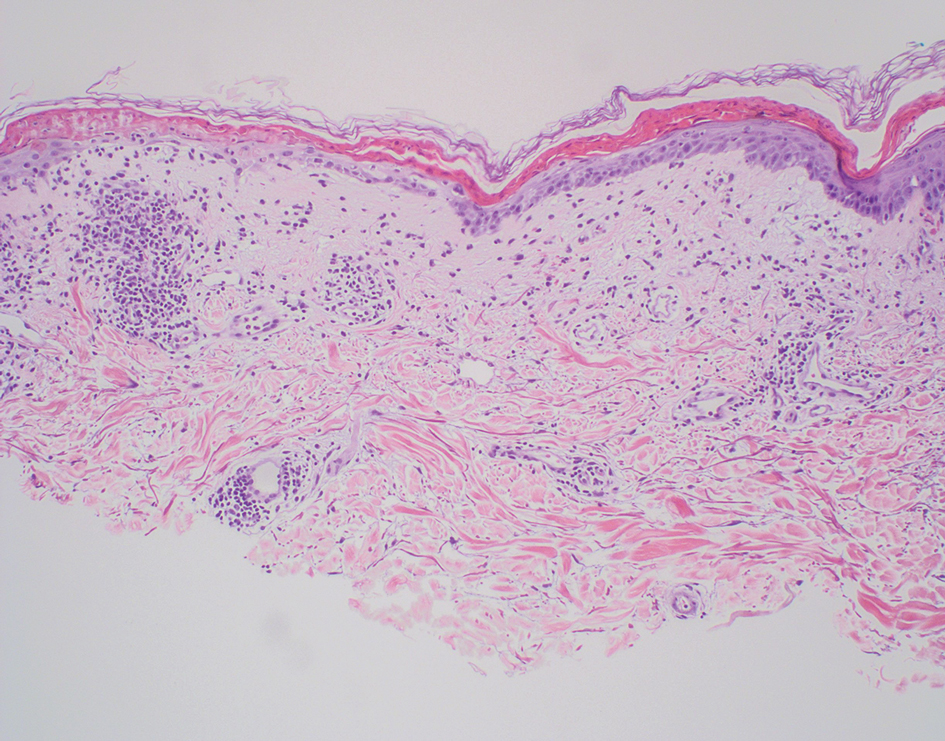
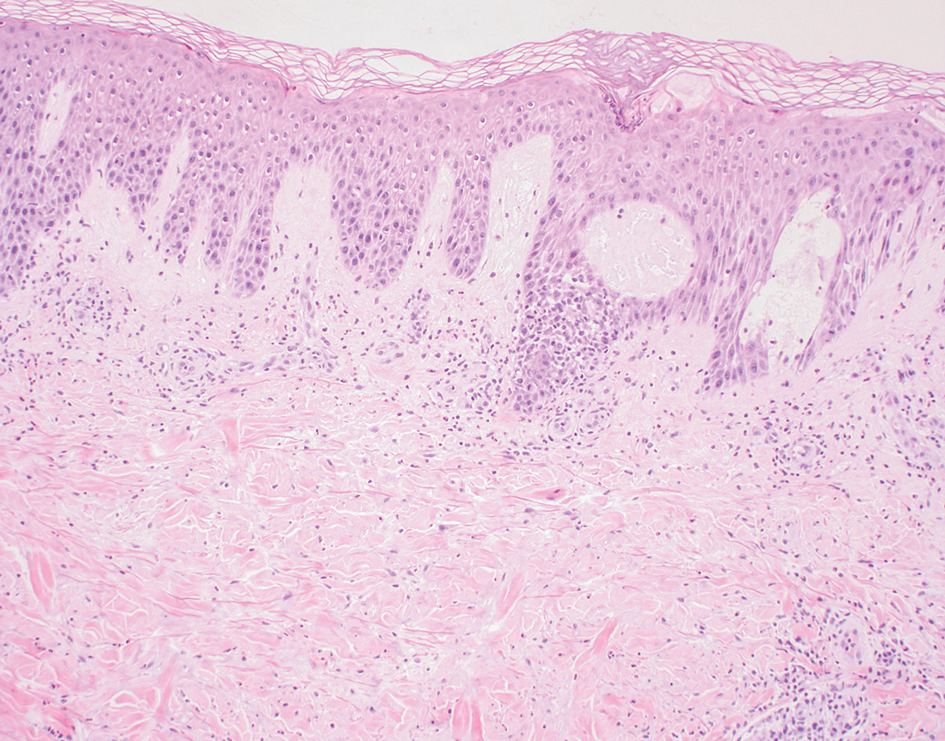
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
A 62-year-old man presented with an erythematous flaky rash associated with burning pain on the right medial second toe that persisted for several months. Prior treatment with econazole, ciclopirox, and oral amoxicillin had failed. A shave biopsy was performed.
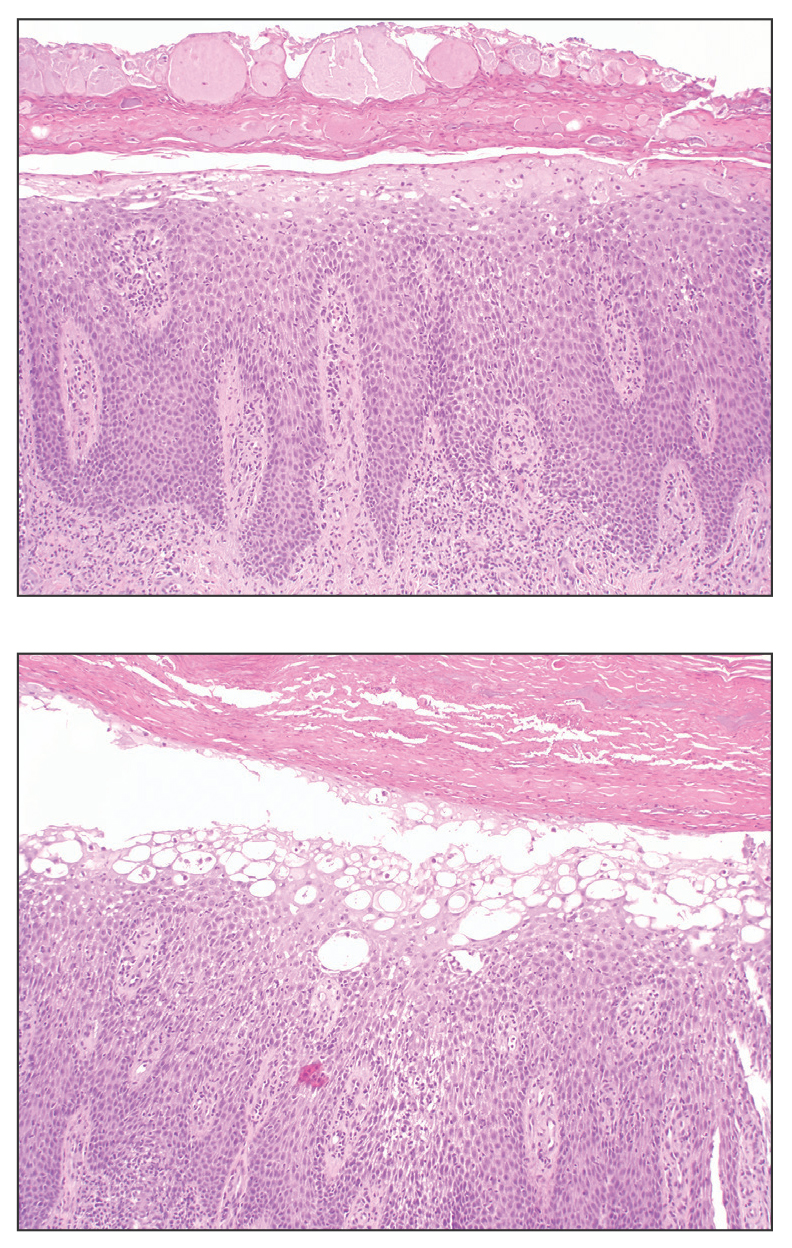
Need a Wood Lamp Alternative? Grab Your Smartphone
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
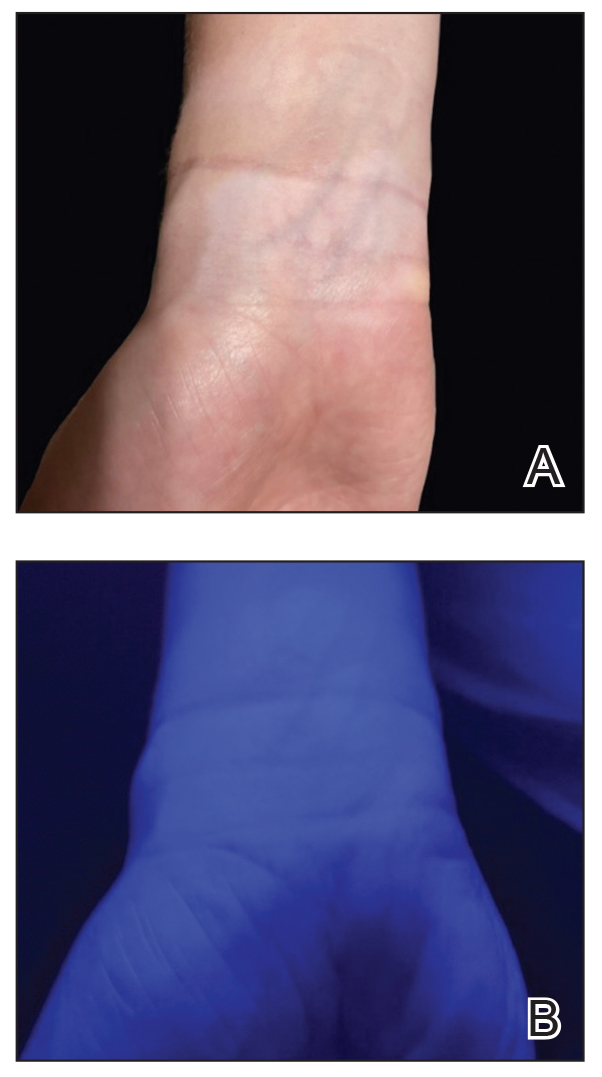
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
Practice Gap
The Wood lamp commonly is used as a diagnostic tool for pigmentary skin conditions (eg, vitiligo) or skin conditions that exhibit fluorescence (eg, erythrasma).1 Recently, its diagnostic efficacy has extended to scabies, in which it unveils a distinctive wavy, bluish-white, linear fluorescence upon illumination.2
Functionally, the Wood lamp operates by subjecting phosphors to UV light within the wavelength range of 320 to 400 nm, inducing fluorescence in substances such as collagen and elastin. In the context of vitiligo, this process manifests as a preferential chalk white fluorescence in areas lacking melanin.1
Despite its demonstrated effectiveness, the Wood lamp is not without limitations. It comes with a notable financial investment ranging from $70 to $500, requires periodic maintenance such as light bulb replacements, and can be unwieldy.3 Furthermore, its reliance on a power source poses a challenge in settings where immediate access to convenient power outlets is limited, such as inpatient and rural dermatology clinics. These limitations underscore the need for alternative solutions and innovations to address challenges and ensure accessibility in diverse health care environments.
The Tools
Free smartphone applications (apps), such as Ultraviolet Light-UV Lamp by AppBrain or Blacklight UV Light Simulator by That Smile, can simulate UV light and functionally serve as a Wood lamp.
The Technique
UV light apps use LED or organic LED screen pixels to emit a blue light equivalent at 467 nm.4 Although these apps are not designed specifically for dermatologic uses, they are mostly free, widely available for Android and iPhone users, and portable. Importantly, they can demonstrate good performance in visualizing vitiligo, as shown in Figure 1—albeit perhaps not reaching the same level as the Wood lamp (Figure 2).
Because these UV light apps are not regulated and their efficacy for medical use has not been firmly established, the Wood lamp remains the gold standard. Therefore, we propose the use of UV light apps in situations when a Wood lamp is not available or convenient, such as in rural, inpatient, or international health care settings.
Practice Implications
Exploring and adopting these free alternatives can contribute to improved accessibility and diagnostic capabilities in diverse health care environments, particularly for communities facing financial constraints. Continued research and validation of these apps in clinical settings will be essential to establish their reliability and effectiveness in enhancing diagnostic practices.
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
- Dyer JM, Foy VM. Revealing the unseen: a review of Wood’s lamp in dermatology. J Clin Aesthet Dermatol. 2022;15:25-30.
- Scanni G. Facilitations in the clinical diagnosis of human scabies through the use of ultraviolet light (UV-scab scanning): a case-series study. Trop Med Infect Dis. 2022;7:422. doi:10.3390/tropicalmed7120422
- USA Medical and Surgical Supplies. Top 9 medical diagnostic applications for a Woods lamp. February 26, 2019. Accessed May 20, 2024.
- Huang Y, Hsiang E-L, Deng M-Y, et al. Mini-led, micro-led and OLED displays: present status and future perspectives. Light Sci Appl. 2020;9:105. doi:10.1038/s41377-020-0341-9
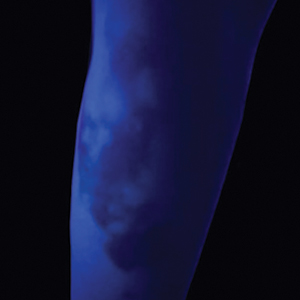
Celiac Disease: Five Things to Know
Celiac disease is a chronic, immune-mediated, systemic disorder caused by intolerance to gluten — a protein present in rye, barley, and wheat grains — that affects genetically predisposed individuals.
Due to its wide spectrum of clinical manifestations, celiac disease resembles a multisystemic disorder. Its most common gastrointestinal (GI) symptoms include chronic diarrhea, weight loss, and abdominal distention. However, celiac disease can also manifest in myriad extraintestinal symptoms, ranging from headache and fatigue to delayed puberty and psychiatric disorders, with differing presentations in children and adults.
To date, the only treatment is adopting a gluten-free diet (GFD). Although key to preventing persistent villous atrophy, the main cause of complications in celiac disease, lifelong adherence to GFD is challenging and may not resolve all clinical issues. These shortcomings have driven recent efforts to develop novel therapeutic options for patients with this disease.
Here are five things to know about celiac disease.
1. Rising Prevalence of Celiac Disease and Other Autoimmune Disorders Suggests Environmental Factors May Be at Play
Gluten was first identified as the cause of celiac disease in the 1950s. At that time, the condition was thought to be a relatively rare GI disease of childhood that primarily affected people of European descent, but it is now known to be a common disease affecting those of various ages, races, and ethnicities.
A 2018 meta-analysis found the pooled global prevalence of celiac disease was 1.4%. Incidence has increased by as much as 7.5% annually over the past several decades.
Increased awareness among clinicians and improved detection likely play a role in the trend. However, the growth in celiac disease is consistent with that seen for other autoimmune disorders, according to a 2024 update of evidence surrounding celiac disease. Shared environmental factors have been proposed as triggers for celiac disease and other autoimmune diseases and appear to be influencing their rise, the authors noted. These factors include migration and population growth, changing dietary patterns and food processing practices, and altered wheat consumption.
2. No-Biopsy Diagnosis Is Accepted for Children and Shows Promise for Adults
It is estimated that almost 60 million people worldwide have celiac disease, but most remain undiagnosed or misdiagnosed, or they experience significant diagnostic delays.
Prospective data indicate that children with first-degree relatives with celiac disease are at a significantly higher risk of developing the condition, which should prompt screening efforts in this population.
The 2023 updated guidelines from the American College of Gastroenterology (ACG) state that serology testing plays a central role in screening. This commonly involves serological testing for positive serological markers of the disease, including immunoglobulin A (IgA), anti-tissue transglutaminase IgA (tTG-IgA), anti-deamidated gliadin peptide, or endomysial antibodies.
To confirm diagnosis, clinicians have relied on intestinal biopsy since the late 1950s. The ACG still recommends esophagogastroduodenoscopy with multiple duodenal biopsies for confirmation of diagnosis in both children and adults with suspicion of celiac disease. However, recent years have seen a shift toward a no-biopsy approach.
For more than a decade in Europe, a no-biopsy approach has been established practice in pediatric patients, for whom the burden of obtaining a histological confirmation is understandably greater. Most guidelines now permit children to be diagnosed with celiac disease in the absence of a biopsy under specific circumstances (eg, characteristic symptoms of celiac disease and tTG-IgA levels > 10 times the upper limit of normal). The ACG guidelines state that “this approach is a reasonable alternative to the standard approach to a [celiac disease] diagnosis in selected children.”
The ACG does not recommend a no-biopsy approach in adults, noting that, in comparison with children, there is a relative lack of data indicating that serology is predictive in this population. However, it does recognize that physicians may encounter patients for whom a biopsy diagnosis may not be safe or practical. In such cases, an “after-the-fact” diagnosis of likely celiac disease can be given to symptomatic adult patients with a ≥ 10-fold elevation of tTG-IgA and a positive endomysial antibody in a second blood sample.
A 2024 meta-analysis of 18 studies involving over 12,103 adult patients from 15 countries concluded that a no-biopsy approach using tTG-IgA antibody levels ≥ 10 times the upper limit of normal was highly specific and predictive of celiac disease.
3. Celiac Disease Is Associated With Several Life-Threatening Conditions
Emerging data indicate that gastroenterologists should be vigilant in screening patients with celiac disease for several other GI conditions.
Inflammatory bowel disease and celiac disease have a strong bidirectional association, suggesting a possible genetic link between the conditions and indicating that physicians should consider the alternate diagnosis when symptoms persist after treatment.
Given the hypervigilance around food and diet inherent to celiac disease, patients are at an increased risk of developing avoidant/restrictive food intake disorder, according to a 2022 retrospective study.
In 2023, Italian investigators showed that children with celiac disease have an elevated prevalence of functional GI disorders even after adopting a GFD for a year, regardless of whether they consumed processed or natural foods. It was unclear whether this was due to a chronic inflammatory process or to nutritional factors.
Complications resulting from celiac disease are not limited to GI disorders. For a variety of underlying pathophysiological reasons, including intestinal permeability, hyposplenism, and malabsorption of nutrients, patients with celiac disease may be at a higher risk for non-GI conditions, such as osteopenia, women’s health disorders (eg, ovarian failure, endometriosis, or pregnancy loss), juvenile idiopathic arthritis in children and rheumatoid arthritis in adults, certain forms of cancer, infectious diseases, and cardiomyopathy.
4. GFD Is the Only Treatment, but It’s Imperfect and Frustrating for Patients
GFD is the only treatment for celiac disease and must be adhered to without deviation throughout a patient’s life.
Maintaining unwavering adherence reaps considerable benefits: Improved clinical symptoms, robust mucosal healing, and normalization of serological markers. Yet it also takes a considerable toll on patients. Patients with celiac disease struggle with a host of negative physical, psychological, and social impacts. They also report a higher treatment burden than those with gastroesophageal reflux disease or hypertension, and comparable with end-stage renal disease.
GFD also poses financial challenges. Although the price of gluten-free products has decreased in recent years, they still cost significantly more than items with gluten.
Adherence to GFD does not always equate to complete mucosal recovery. While mucosal recovery is achieved in 95% of children within 2 years of the diet’s adoption, only 34% and 66% of adults obtain it within 2 and 5 years, respectively.
GFD may lead to nutrient imbalances because gluten-free foods are typically low in alimentary fiber, micronutrients (eg, vitamin D, vitamin B12, or folate), and minerals (eg, iron, zinc, magnesium, or calcium). With higher sugar and fat content, GFD may leave patients susceptible to unwanted weight gain.
The pervasiveness of gluten in the food production system makes the risk for cross-contamination high. Gluten is often found in both naturally gluten-free foods and products labeled as such. Gluten-sensing technologies, some of which can be used via smartphone apps, have been developed to help patients identify possible cross-contamination. However, the ACG guidelines recommend against the use of these technologies until there is sufficient evidence supporting their ability to improve adherence and clinical outcomes.
5. Novel Therapies for Celiac Disease Are in the Pipeline
The limitations of GFD as the standard treatment for celiac disease have led to an increased focus on developing novel therapeutic interventions. They can be sorted into five key categories: Modulation of the immunostimulatory effects of toxic gluten peptides, elimination of toxic gluten peptides before they reach the intestine, induction of gluten tolerance, modulation of intestinal permeability, and restoration of gut microbiota balance.
Three therapies designed to block antigen presentation by HLA-DQ2/8, the gene alleles that predispose people to celiac disease, show promise: TPM502, an agent that contains three gluten-specific antigenic peptides with overlapping T-cell epitopes for the HLA-DQ2.5 gene; KAN-101, designed to induce gluten tolerance by targeting receptors on the liver; and DONQ52, a multi-specific antibody that targets HLA-DQ2. The KAN-101 therapy received Fast Track designation by the US Food and Drug Administration in 2022.
These and several other agents in clinical and preclinical development are discussed in detail in a 2024 review article. Although no therapies have reached phase 3 testing, when they do, it will undoubtedly be welcomed by those with celiac disease.
A version of this article first appeared on Medscape.com.
Celiac disease is a chronic, immune-mediated, systemic disorder caused by intolerance to gluten — a protein present in rye, barley, and wheat grains — that affects genetically predisposed individuals.
Due to its wide spectrum of clinical manifestations, celiac disease resembles a multisystemic disorder. Its most common gastrointestinal (GI) symptoms include chronic diarrhea, weight loss, and abdominal distention. However, celiac disease can also manifest in myriad extraintestinal symptoms, ranging from headache and fatigue to delayed puberty and psychiatric disorders, with differing presentations in children and adults.
To date, the only treatment is adopting a gluten-free diet (GFD). Although key to preventing persistent villous atrophy, the main cause of complications in celiac disease, lifelong adherence to GFD is challenging and may not resolve all clinical issues. These shortcomings have driven recent efforts to develop novel therapeutic options for patients with this disease.
Here are five things to know about celiac disease.
1. Rising Prevalence of Celiac Disease and Other Autoimmune Disorders Suggests Environmental Factors May Be at Play
Gluten was first identified as the cause of celiac disease in the 1950s. At that time, the condition was thought to be a relatively rare GI disease of childhood that primarily affected people of European descent, but it is now known to be a common disease affecting those of various ages, races, and ethnicities.
A 2018 meta-analysis found the pooled global prevalence of celiac disease was 1.4%. Incidence has increased by as much as 7.5% annually over the past several decades.
Increased awareness among clinicians and improved detection likely play a role in the trend. However, the growth in celiac disease is consistent with that seen for other autoimmune disorders, according to a 2024 update of evidence surrounding celiac disease. Shared environmental factors have been proposed as triggers for celiac disease and other autoimmune diseases and appear to be influencing their rise, the authors noted. These factors include migration and population growth, changing dietary patterns and food processing practices, and altered wheat consumption.
2. No-Biopsy Diagnosis Is Accepted for Children and Shows Promise for Adults
It is estimated that almost 60 million people worldwide have celiac disease, but most remain undiagnosed or misdiagnosed, or they experience significant diagnostic delays.
Prospective data indicate that children with first-degree relatives with celiac disease are at a significantly higher risk of developing the condition, which should prompt screening efforts in this population.
The 2023 updated guidelines from the American College of Gastroenterology (ACG) state that serology testing plays a central role in screening. This commonly involves serological testing for positive serological markers of the disease, including immunoglobulin A (IgA), anti-tissue transglutaminase IgA (tTG-IgA), anti-deamidated gliadin peptide, or endomysial antibodies.
To confirm diagnosis, clinicians have relied on intestinal biopsy since the late 1950s. The ACG still recommends esophagogastroduodenoscopy with multiple duodenal biopsies for confirmation of diagnosis in both children and adults with suspicion of celiac disease. However, recent years have seen a shift toward a no-biopsy approach.
For more than a decade in Europe, a no-biopsy approach has been established practice in pediatric patients, for whom the burden of obtaining a histological confirmation is understandably greater. Most guidelines now permit children to be diagnosed with celiac disease in the absence of a biopsy under specific circumstances (eg, characteristic symptoms of celiac disease and tTG-IgA levels > 10 times the upper limit of normal). The ACG guidelines state that “this approach is a reasonable alternative to the standard approach to a [celiac disease] diagnosis in selected children.”
The ACG does not recommend a no-biopsy approach in adults, noting that, in comparison with children, there is a relative lack of data indicating that serology is predictive in this population. However, it does recognize that physicians may encounter patients for whom a biopsy diagnosis may not be safe or practical. In such cases, an “after-the-fact” diagnosis of likely celiac disease can be given to symptomatic adult patients with a ≥ 10-fold elevation of tTG-IgA and a positive endomysial antibody in a second blood sample.
A 2024 meta-analysis of 18 studies involving over 12,103 adult patients from 15 countries concluded that a no-biopsy approach using tTG-IgA antibody levels ≥ 10 times the upper limit of normal was highly specific and predictive of celiac disease.
3. Celiac Disease Is Associated With Several Life-Threatening Conditions
Emerging data indicate that gastroenterologists should be vigilant in screening patients with celiac disease for several other GI conditions.
Inflammatory bowel disease and celiac disease have a strong bidirectional association, suggesting a possible genetic link between the conditions and indicating that physicians should consider the alternate diagnosis when symptoms persist after treatment.
Given the hypervigilance around food and diet inherent to celiac disease, patients are at an increased risk of developing avoidant/restrictive food intake disorder, according to a 2022 retrospective study.
In 2023, Italian investigators showed that children with celiac disease have an elevated prevalence of functional GI disorders even after adopting a GFD for a year, regardless of whether they consumed processed or natural foods. It was unclear whether this was due to a chronic inflammatory process or to nutritional factors.
Complications resulting from celiac disease are not limited to GI disorders. For a variety of underlying pathophysiological reasons, including intestinal permeability, hyposplenism, and malabsorption of nutrients, patients with celiac disease may be at a higher risk for non-GI conditions, such as osteopenia, women’s health disorders (eg, ovarian failure, endometriosis, or pregnancy loss), juvenile idiopathic arthritis in children and rheumatoid arthritis in adults, certain forms of cancer, infectious diseases, and cardiomyopathy.
4. GFD Is the Only Treatment, but It’s Imperfect and Frustrating for Patients
GFD is the only treatment for celiac disease and must be adhered to without deviation throughout a patient’s life.
Maintaining unwavering adherence reaps considerable benefits: Improved clinical symptoms, robust mucosal healing, and normalization of serological markers. Yet it also takes a considerable toll on patients. Patients with celiac disease struggle with a host of negative physical, psychological, and social impacts. They also report a higher treatment burden than those with gastroesophageal reflux disease or hypertension, and comparable with end-stage renal disease.
GFD also poses financial challenges. Although the price of gluten-free products has decreased in recent years, they still cost significantly more than items with gluten.
Adherence to GFD does not always equate to complete mucosal recovery. While mucosal recovery is achieved in 95% of children within 2 years of the diet’s adoption, only 34% and 66% of adults obtain it within 2 and 5 years, respectively.
GFD may lead to nutrient imbalances because gluten-free foods are typically low in alimentary fiber, micronutrients (eg, vitamin D, vitamin B12, or folate), and minerals (eg, iron, zinc, magnesium, or calcium). With higher sugar and fat content, GFD may leave patients susceptible to unwanted weight gain.
The pervasiveness of gluten in the food production system makes the risk for cross-contamination high. Gluten is often found in both naturally gluten-free foods and products labeled as such. Gluten-sensing technologies, some of which can be used via smartphone apps, have been developed to help patients identify possible cross-contamination. However, the ACG guidelines recommend against the use of these technologies until there is sufficient evidence supporting their ability to improve adherence and clinical outcomes.
5. Novel Therapies for Celiac Disease Are in the Pipeline
The limitations of GFD as the standard treatment for celiac disease have led to an increased focus on developing novel therapeutic interventions. They can be sorted into five key categories: Modulation of the immunostimulatory effects of toxic gluten peptides, elimination of toxic gluten peptides before they reach the intestine, induction of gluten tolerance, modulation of intestinal permeability, and restoration of gut microbiota balance.
Three therapies designed to block antigen presentation by HLA-DQ2/8, the gene alleles that predispose people to celiac disease, show promise: TPM502, an agent that contains three gluten-specific antigenic peptides with overlapping T-cell epitopes for the HLA-DQ2.5 gene; KAN-101, designed to induce gluten tolerance by targeting receptors on the liver; and DONQ52, a multi-specific antibody that targets HLA-DQ2. The KAN-101 therapy received Fast Track designation by the US Food and Drug Administration in 2022.
These and several other agents in clinical and preclinical development are discussed in detail in a 2024 review article. Although no therapies have reached phase 3 testing, when they do, it will undoubtedly be welcomed by those with celiac disease.
A version of this article first appeared on Medscape.com.
Celiac disease is a chronic, immune-mediated, systemic disorder caused by intolerance to gluten — a protein present in rye, barley, and wheat grains — that affects genetically predisposed individuals.
Due to its wide spectrum of clinical manifestations, celiac disease resembles a multisystemic disorder. Its most common gastrointestinal (GI) symptoms include chronic diarrhea, weight loss, and abdominal distention. However, celiac disease can also manifest in myriad extraintestinal symptoms, ranging from headache and fatigue to delayed puberty and psychiatric disorders, with differing presentations in children and adults.
To date, the only treatment is adopting a gluten-free diet (GFD). Although key to preventing persistent villous atrophy, the main cause of complications in celiac disease, lifelong adherence to GFD is challenging and may not resolve all clinical issues. These shortcomings have driven recent efforts to develop novel therapeutic options for patients with this disease.
Here are five things to know about celiac disease.
1. Rising Prevalence of Celiac Disease and Other Autoimmune Disorders Suggests Environmental Factors May Be at Play
Gluten was first identified as the cause of celiac disease in the 1950s. At that time, the condition was thought to be a relatively rare GI disease of childhood that primarily affected people of European descent, but it is now known to be a common disease affecting those of various ages, races, and ethnicities.
A 2018 meta-analysis found the pooled global prevalence of celiac disease was 1.4%. Incidence has increased by as much as 7.5% annually over the past several decades.
Increased awareness among clinicians and improved detection likely play a role in the trend. However, the growth in celiac disease is consistent with that seen for other autoimmune disorders, according to a 2024 update of evidence surrounding celiac disease. Shared environmental factors have been proposed as triggers for celiac disease and other autoimmune diseases and appear to be influencing their rise, the authors noted. These factors include migration and population growth, changing dietary patterns and food processing practices, and altered wheat consumption.
2. No-Biopsy Diagnosis Is Accepted for Children and Shows Promise for Adults
It is estimated that almost 60 million people worldwide have celiac disease, but most remain undiagnosed or misdiagnosed, or they experience significant diagnostic delays.
Prospective data indicate that children with first-degree relatives with celiac disease are at a significantly higher risk of developing the condition, which should prompt screening efforts in this population.
The 2023 updated guidelines from the American College of Gastroenterology (ACG) state that serology testing plays a central role in screening. This commonly involves serological testing for positive serological markers of the disease, including immunoglobulin A (IgA), anti-tissue transglutaminase IgA (tTG-IgA), anti-deamidated gliadin peptide, or endomysial antibodies.
To confirm diagnosis, clinicians have relied on intestinal biopsy since the late 1950s. The ACG still recommends esophagogastroduodenoscopy with multiple duodenal biopsies for confirmation of diagnosis in both children and adults with suspicion of celiac disease. However, recent years have seen a shift toward a no-biopsy approach.
For more than a decade in Europe, a no-biopsy approach has been established practice in pediatric patients, for whom the burden of obtaining a histological confirmation is understandably greater. Most guidelines now permit children to be diagnosed with celiac disease in the absence of a biopsy under specific circumstances (eg, characteristic symptoms of celiac disease and tTG-IgA levels > 10 times the upper limit of normal). The ACG guidelines state that “this approach is a reasonable alternative to the standard approach to a [celiac disease] diagnosis in selected children.”
The ACG does not recommend a no-biopsy approach in adults, noting that, in comparison with children, there is a relative lack of data indicating that serology is predictive in this population. However, it does recognize that physicians may encounter patients for whom a biopsy diagnosis may not be safe or practical. In such cases, an “after-the-fact” diagnosis of likely celiac disease can be given to symptomatic adult patients with a ≥ 10-fold elevation of tTG-IgA and a positive endomysial antibody in a second blood sample.
A 2024 meta-analysis of 18 studies involving over 12,103 adult patients from 15 countries concluded that a no-biopsy approach using tTG-IgA antibody levels ≥ 10 times the upper limit of normal was highly specific and predictive of celiac disease.
3. Celiac Disease Is Associated With Several Life-Threatening Conditions
Emerging data indicate that gastroenterologists should be vigilant in screening patients with celiac disease for several other GI conditions.
Inflammatory bowel disease and celiac disease have a strong bidirectional association, suggesting a possible genetic link between the conditions and indicating that physicians should consider the alternate diagnosis when symptoms persist after treatment.
Given the hypervigilance around food and diet inherent to celiac disease, patients are at an increased risk of developing avoidant/restrictive food intake disorder, according to a 2022 retrospective study.
In 2023, Italian investigators showed that children with celiac disease have an elevated prevalence of functional GI disorders even after adopting a GFD for a year, regardless of whether they consumed processed or natural foods. It was unclear whether this was due to a chronic inflammatory process or to nutritional factors.
Complications resulting from celiac disease are not limited to GI disorders. For a variety of underlying pathophysiological reasons, including intestinal permeability, hyposplenism, and malabsorption of nutrients, patients with celiac disease may be at a higher risk for non-GI conditions, such as osteopenia, women’s health disorders (eg, ovarian failure, endometriosis, or pregnancy loss), juvenile idiopathic arthritis in children and rheumatoid arthritis in adults, certain forms of cancer, infectious diseases, and cardiomyopathy.
4. GFD Is the Only Treatment, but It’s Imperfect and Frustrating for Patients
GFD is the only treatment for celiac disease and must be adhered to without deviation throughout a patient’s life.
Maintaining unwavering adherence reaps considerable benefits: Improved clinical symptoms, robust mucosal healing, and normalization of serological markers. Yet it also takes a considerable toll on patients. Patients with celiac disease struggle with a host of negative physical, psychological, and social impacts. They also report a higher treatment burden than those with gastroesophageal reflux disease or hypertension, and comparable with end-stage renal disease.
GFD also poses financial challenges. Although the price of gluten-free products has decreased in recent years, they still cost significantly more than items with gluten.
Adherence to GFD does not always equate to complete mucosal recovery. While mucosal recovery is achieved in 95% of children within 2 years of the diet’s adoption, only 34% and 66% of adults obtain it within 2 and 5 years, respectively.
GFD may lead to nutrient imbalances because gluten-free foods are typically low in alimentary fiber, micronutrients (eg, vitamin D, vitamin B12, or folate), and minerals (eg, iron, zinc, magnesium, or calcium). With higher sugar and fat content, GFD may leave patients susceptible to unwanted weight gain.
The pervasiveness of gluten in the food production system makes the risk for cross-contamination high. Gluten is often found in both naturally gluten-free foods and products labeled as such. Gluten-sensing technologies, some of which can be used via smartphone apps, have been developed to help patients identify possible cross-contamination. However, the ACG guidelines recommend against the use of these technologies until there is sufficient evidence supporting their ability to improve adherence and clinical outcomes.
5. Novel Therapies for Celiac Disease Are in the Pipeline
The limitations of GFD as the standard treatment for celiac disease have led to an increased focus on developing novel therapeutic interventions. They can be sorted into five key categories: Modulation of the immunostimulatory effects of toxic gluten peptides, elimination of toxic gluten peptides before they reach the intestine, induction of gluten tolerance, modulation of intestinal permeability, and restoration of gut microbiota balance.
Three therapies designed to block antigen presentation by HLA-DQ2/8, the gene alleles that predispose people to celiac disease, show promise: TPM502, an agent that contains three gluten-specific antigenic peptides with overlapping T-cell epitopes for the HLA-DQ2.5 gene; KAN-101, designed to induce gluten tolerance by targeting receptors on the liver; and DONQ52, a multi-specific antibody that targets HLA-DQ2. The KAN-101 therapy received Fast Track designation by the US Food and Drug Administration in 2022.
These and several other agents in clinical and preclinical development are discussed in detail in a 2024 review article. Although no therapies have reached phase 3 testing, when they do, it will undoubtedly be welcomed by those with celiac disease.
A version of this article first appeared on Medscape.com.