User login
Severe skin reactions with enfortumab vedotin
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Accelerated approval now full for pembro in bladder cancer
But one such accelerated approval has now been converted to a full approval, with a small label change, by the Food and Drug Administration.
The new full approval is for pembrolizumab (Keytruda) for first-line use for patients with locally advanced or metastatic urothelial carcinoma (mUC) who are not eligible for any platinum-containing chemotherapy. (The small label change is that mention of PD-L1 status and testing for this have been removed.)
The move is in accordance with recommendations by experts at a recent meeting of the Oncologic Drugs Advisory Committee. They voted 5-3 in favor of this accelerated approval staying. They also voted 10-1 in favor of another immunotherapy, atezolizumab (Tecentriq), for the same indication.
One of the arguments put forward to support these accelerated approvals staying in place is that there is an unmet need in this population of patients who are ineligible for platinum chemotherapy.
But this argument doesn’t hold water – the mere existence of one of these negates the “unmet need” argument for the other, wrote Bishal Gyawali, MD, PhD, from Queen’s University, Kingston, Ont., in a commentary on why the FDA’s accelerated approval pathway is broken.
“Even if there is a genuine ‘unmet need’ in a particular setting, these drugs did not meet the standard of improving survival. An unmet need doesn’t imply that the treatment void should be filled with a drug that provides nothing of value to patients,” Dr. Gyawali wrote.
“When we talk about an unmet need, we are speaking of drugs that provide a clinical benefit; any true unmet needs will continue to exist despite maintaining these approvals,” he argued.
After obtaining the accelerated approval for pembrolizumab for patients with bladder cancer who are not eligible for cisplatin-containing chemotherapy, the manufacturer (Merck) carried out a subsequent clinical trial but conducted it in patients who were eligible for platinum-containing chemotherapy (KEYNOTE-361). However, this trial did not meet its prespecified dual primary endpoints of overall survival or progression-free survival in comparison with standard of care chemotherapy, the company noted in a press release.
“We are working with urgency to advance studies to help more patients living with bladder and other types of cancer,” commented Scot Ebbinghaus, MD, vice president of clinical research, Merck Research Laboratories. The company said it has “an extensive clinical development in bladder cancer” and is exploring pembrolizumab use in many settings.
In addition to the new full approval for the first-line indication for patients who are ineligible for platinum chemotherapy, pembrolizumab has two other approved indications in this therapeutic area: the treatment of patients with locally advanced urothelial carcinoma or mUC who experience disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy; and the treatment of patients with bacillus Calmette-Guérin–unresponsive, high-risk, non–muscle-invasive bladder cancer with carcinoma in situ, with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
A version of this article first appeared on Medscape.com.
But one such accelerated approval has now been converted to a full approval, with a small label change, by the Food and Drug Administration.
The new full approval is for pembrolizumab (Keytruda) for first-line use for patients with locally advanced or metastatic urothelial carcinoma (mUC) who are not eligible for any platinum-containing chemotherapy. (The small label change is that mention of PD-L1 status and testing for this have been removed.)
The move is in accordance with recommendations by experts at a recent meeting of the Oncologic Drugs Advisory Committee. They voted 5-3 in favor of this accelerated approval staying. They also voted 10-1 in favor of another immunotherapy, atezolizumab (Tecentriq), for the same indication.
One of the arguments put forward to support these accelerated approvals staying in place is that there is an unmet need in this population of patients who are ineligible for platinum chemotherapy.
But this argument doesn’t hold water – the mere existence of one of these negates the “unmet need” argument for the other, wrote Bishal Gyawali, MD, PhD, from Queen’s University, Kingston, Ont., in a commentary on why the FDA’s accelerated approval pathway is broken.
“Even if there is a genuine ‘unmet need’ in a particular setting, these drugs did not meet the standard of improving survival. An unmet need doesn’t imply that the treatment void should be filled with a drug that provides nothing of value to patients,” Dr. Gyawali wrote.
“When we talk about an unmet need, we are speaking of drugs that provide a clinical benefit; any true unmet needs will continue to exist despite maintaining these approvals,” he argued.
After obtaining the accelerated approval for pembrolizumab for patients with bladder cancer who are not eligible for cisplatin-containing chemotherapy, the manufacturer (Merck) carried out a subsequent clinical trial but conducted it in patients who were eligible for platinum-containing chemotherapy (KEYNOTE-361). However, this trial did not meet its prespecified dual primary endpoints of overall survival or progression-free survival in comparison with standard of care chemotherapy, the company noted in a press release.
“We are working with urgency to advance studies to help more patients living with bladder and other types of cancer,” commented Scot Ebbinghaus, MD, vice president of clinical research, Merck Research Laboratories. The company said it has “an extensive clinical development in bladder cancer” and is exploring pembrolizumab use in many settings.
In addition to the new full approval for the first-line indication for patients who are ineligible for platinum chemotherapy, pembrolizumab has two other approved indications in this therapeutic area: the treatment of patients with locally advanced urothelial carcinoma or mUC who experience disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy; and the treatment of patients with bacillus Calmette-Guérin–unresponsive, high-risk, non–muscle-invasive bladder cancer with carcinoma in situ, with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
A version of this article first appeared on Medscape.com.
But one such accelerated approval has now been converted to a full approval, with a small label change, by the Food and Drug Administration.
The new full approval is for pembrolizumab (Keytruda) for first-line use for patients with locally advanced or metastatic urothelial carcinoma (mUC) who are not eligible for any platinum-containing chemotherapy. (The small label change is that mention of PD-L1 status and testing for this have been removed.)
The move is in accordance with recommendations by experts at a recent meeting of the Oncologic Drugs Advisory Committee. They voted 5-3 in favor of this accelerated approval staying. They also voted 10-1 in favor of another immunotherapy, atezolizumab (Tecentriq), for the same indication.
One of the arguments put forward to support these accelerated approvals staying in place is that there is an unmet need in this population of patients who are ineligible for platinum chemotherapy.
But this argument doesn’t hold water – the mere existence of one of these negates the “unmet need” argument for the other, wrote Bishal Gyawali, MD, PhD, from Queen’s University, Kingston, Ont., in a commentary on why the FDA’s accelerated approval pathway is broken.
“Even if there is a genuine ‘unmet need’ in a particular setting, these drugs did not meet the standard of improving survival. An unmet need doesn’t imply that the treatment void should be filled with a drug that provides nothing of value to patients,” Dr. Gyawali wrote.
“When we talk about an unmet need, we are speaking of drugs that provide a clinical benefit; any true unmet needs will continue to exist despite maintaining these approvals,” he argued.
After obtaining the accelerated approval for pembrolizumab for patients with bladder cancer who are not eligible for cisplatin-containing chemotherapy, the manufacturer (Merck) carried out a subsequent clinical trial but conducted it in patients who were eligible for platinum-containing chemotherapy (KEYNOTE-361). However, this trial did not meet its prespecified dual primary endpoints of overall survival or progression-free survival in comparison with standard of care chemotherapy, the company noted in a press release.
“We are working with urgency to advance studies to help more patients living with bladder and other types of cancer,” commented Scot Ebbinghaus, MD, vice president of clinical research, Merck Research Laboratories. The company said it has “an extensive clinical development in bladder cancer” and is exploring pembrolizumab use in many settings.
In addition to the new full approval for the first-line indication for patients who are ineligible for platinum chemotherapy, pembrolizumab has two other approved indications in this therapeutic area: the treatment of patients with locally advanced urothelial carcinoma or mUC who experience disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy; and the treatment of patients with bacillus Calmette-Guérin–unresponsive, high-risk, non–muscle-invasive bladder cancer with carcinoma in situ, with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
A version of this article first appeared on Medscape.com.
Old saying about prostate cancer not true when it’s metastatic
.
The findings fill an information gap because, remarkably, “data are lacking” on causes of death among men whose prostate cancer has spread to other sites, say lead author Ahmed Elmehrath, MD, of Cairo University, Egypt, and colleagues.
“It was an important realization by our team that prostate cancer was the cause of death in 78% of patients,” said senior author Omar Alhalabi, MD, of University of Texas MD Anderson Cancer Center, Houston, in an email.
“Most patients with metastatic prostate cancer die from it, rather than other possible causes of death,” confirm Samuel Merriel, MSc, Tanimola Martins, PhD, and Sarah Bailey, PhD, University of Exeter, United Kingdom, in an accompanying editorial. The study was published last month in JAMA Network Open.
The findings represent the near opposite of a commonly held – and comforting – belief about early-stage disease: “You die with prostate cancer, not from it.”
That old saying is articulated in various ways, such as this from the Prostate Cancer Foundation: “We can confirm that there are those prostate cancers a man may die with and not of, while others are very aggressive.” The American Cancer Society says this: “Prostate cancer can be a serious disease, but most men diagnosed with prostate cancer do not die from it.”
However, these commonplace comments do not cover metastatic disease, which is what the authors of the new study decided to focus on.
The team used data from the Surveillance, Epidemiology, and End Results Program (SEER) database to gather a sample of 26,168 U.S. men who received a diagnosis of metastatic prostate cancer from January 2000 to December 2016. They then analyzed the data in 2020 and found that 16,732 men (64%) had died during the follow-up period.
The majority of these deaths (77.8%) were from prostate cancer, 5.5% were from other cancers, and 16.7% were from noncancer causes, including cardiovascular diseases, chronic obstructive pulmonary disease, and cerebrovascular diseases.
Senior author Dr. Alhalabi acknowledged a limitation in these findings – that the SEER database relies on causes of death extracted from death certificates. “Death certificates have limited granularity in terms of the details they can contain about the cause of death and also have reporting bias,” he said.
Most of the prostate cancer deaths (59%) occurred within 2 years. The 5-year overall survival rate in the study group was 26%.
The deadliness of metastatic disease “reinforces the need for innovations to promote early-stage diagnosis,” comment the editorialists. Striking a hopeful note, they also say that “new tests for prostate cancer detection may reduce the proportion of patients who receive a diagnosis at a late stage.”
Death from other causes
The mean age at metastatic prostate cancer diagnosis in the study was roughly 71 years. Most of the cohort was White (74.5%) and had a diagnosis of stage M1b metastatic prostate cancer (72.7%), which means the cancer had spread to the bones.
Among men in the cohort, the rates of death from septicemia, suicide, accidents, COPD, and cerebrovascular diseases were significantly increased compared with the general U.S. male population, the team observes.
Thus, the study authors were concerned with not only with death from metastatic prostate cancer but death from other causes.
That concern is rooted in the established fact that there is now improved survival among patients with prostate cancer in the U.S., including among men with advanced disease. “Patients tend to live long enough after a prostate cancer diagnosis for non–cancer-related comorbidities to be associated with their overall survival,” they write.
The editorialists agree: Prostate cancer “has a high long-term survival rate compared with almost all other cancer types and signals the need for greater holistic care for patients.”
As noted above, cardiovascular diseases were the most common cause of nonprostate cancer–related deaths in the new study.
As in the management of other cancers, there is concern among clinicians and researchers about the cardiotoxic effects of prostate cancer treatments.
The study authors point to a 2017 analysis that showed that men with prostate cancer and no prior cardiac disease had greater risk of heart failure after taking androgen deprivation therapy (ADT), a common treatment used when the disease recurs after definitive treatment. Another study suggested an association between cardiotoxic effects of ADT and myocardial infarction regardless of medical history in general.
The authors of the current study say that such findings highlight “the importance of multidisciplinary care for such patients and the role of primary care physicians in optimizing cardiovascular risk prevention and providing early referrals to cardiologists.”
Further, the team says that tailoring “ADT to each patient’s needs may be associated with improved survival, especially for patients with factors associated with cardiovascular disease.”
Who should lead the way in multidisciplinary care? “The answer probably is case-by-case,” said Dr. Alhalabi, adding that it might depend on the presence of underlying morbidities such as cardiovascular disease and COPD.
“It is also important for the oncologist (‘the gatekeeper’) to try to mitigate the potential metabolic effects of hormonal deprivation therapy such as weight gain, decreased muscle mass, hyperlipidemia, etc.,” he added.
The study had no specific funding. The study authors and editorialists have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
The findings fill an information gap because, remarkably, “data are lacking” on causes of death among men whose prostate cancer has spread to other sites, say lead author Ahmed Elmehrath, MD, of Cairo University, Egypt, and colleagues.
“It was an important realization by our team that prostate cancer was the cause of death in 78% of patients,” said senior author Omar Alhalabi, MD, of University of Texas MD Anderson Cancer Center, Houston, in an email.
“Most patients with metastatic prostate cancer die from it, rather than other possible causes of death,” confirm Samuel Merriel, MSc, Tanimola Martins, PhD, and Sarah Bailey, PhD, University of Exeter, United Kingdom, in an accompanying editorial. The study was published last month in JAMA Network Open.
The findings represent the near opposite of a commonly held – and comforting – belief about early-stage disease: “You die with prostate cancer, not from it.”
That old saying is articulated in various ways, such as this from the Prostate Cancer Foundation: “We can confirm that there are those prostate cancers a man may die with and not of, while others are very aggressive.” The American Cancer Society says this: “Prostate cancer can be a serious disease, but most men diagnosed with prostate cancer do not die from it.”
However, these commonplace comments do not cover metastatic disease, which is what the authors of the new study decided to focus on.
The team used data from the Surveillance, Epidemiology, and End Results Program (SEER) database to gather a sample of 26,168 U.S. men who received a diagnosis of metastatic prostate cancer from January 2000 to December 2016. They then analyzed the data in 2020 and found that 16,732 men (64%) had died during the follow-up period.
The majority of these deaths (77.8%) were from prostate cancer, 5.5% were from other cancers, and 16.7% were from noncancer causes, including cardiovascular diseases, chronic obstructive pulmonary disease, and cerebrovascular diseases.
Senior author Dr. Alhalabi acknowledged a limitation in these findings – that the SEER database relies on causes of death extracted from death certificates. “Death certificates have limited granularity in terms of the details they can contain about the cause of death and also have reporting bias,” he said.
Most of the prostate cancer deaths (59%) occurred within 2 years. The 5-year overall survival rate in the study group was 26%.
The deadliness of metastatic disease “reinforces the need for innovations to promote early-stage diagnosis,” comment the editorialists. Striking a hopeful note, they also say that “new tests for prostate cancer detection may reduce the proportion of patients who receive a diagnosis at a late stage.”
Death from other causes
The mean age at metastatic prostate cancer diagnosis in the study was roughly 71 years. Most of the cohort was White (74.5%) and had a diagnosis of stage M1b metastatic prostate cancer (72.7%), which means the cancer had spread to the bones.
Among men in the cohort, the rates of death from septicemia, suicide, accidents, COPD, and cerebrovascular diseases were significantly increased compared with the general U.S. male population, the team observes.
Thus, the study authors were concerned with not only with death from metastatic prostate cancer but death from other causes.
That concern is rooted in the established fact that there is now improved survival among patients with prostate cancer in the U.S., including among men with advanced disease. “Patients tend to live long enough after a prostate cancer diagnosis for non–cancer-related comorbidities to be associated with their overall survival,” they write.
The editorialists agree: Prostate cancer “has a high long-term survival rate compared with almost all other cancer types and signals the need for greater holistic care for patients.”
As noted above, cardiovascular diseases were the most common cause of nonprostate cancer–related deaths in the new study.
As in the management of other cancers, there is concern among clinicians and researchers about the cardiotoxic effects of prostate cancer treatments.
The study authors point to a 2017 analysis that showed that men with prostate cancer and no prior cardiac disease had greater risk of heart failure after taking androgen deprivation therapy (ADT), a common treatment used when the disease recurs after definitive treatment. Another study suggested an association between cardiotoxic effects of ADT and myocardial infarction regardless of medical history in general.
The authors of the current study say that such findings highlight “the importance of multidisciplinary care for such patients and the role of primary care physicians in optimizing cardiovascular risk prevention and providing early referrals to cardiologists.”
Further, the team says that tailoring “ADT to each patient’s needs may be associated with improved survival, especially for patients with factors associated with cardiovascular disease.”
Who should lead the way in multidisciplinary care? “The answer probably is case-by-case,” said Dr. Alhalabi, adding that it might depend on the presence of underlying morbidities such as cardiovascular disease and COPD.
“It is also important for the oncologist (‘the gatekeeper’) to try to mitigate the potential metabolic effects of hormonal deprivation therapy such as weight gain, decreased muscle mass, hyperlipidemia, etc.,” he added.
The study had no specific funding. The study authors and editorialists have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
The findings fill an information gap because, remarkably, “data are lacking” on causes of death among men whose prostate cancer has spread to other sites, say lead author Ahmed Elmehrath, MD, of Cairo University, Egypt, and colleagues.
“It was an important realization by our team that prostate cancer was the cause of death in 78% of patients,” said senior author Omar Alhalabi, MD, of University of Texas MD Anderson Cancer Center, Houston, in an email.
“Most patients with metastatic prostate cancer die from it, rather than other possible causes of death,” confirm Samuel Merriel, MSc, Tanimola Martins, PhD, and Sarah Bailey, PhD, University of Exeter, United Kingdom, in an accompanying editorial. The study was published last month in JAMA Network Open.
The findings represent the near opposite of a commonly held – and comforting – belief about early-stage disease: “You die with prostate cancer, not from it.”
That old saying is articulated in various ways, such as this from the Prostate Cancer Foundation: “We can confirm that there are those prostate cancers a man may die with and not of, while others are very aggressive.” The American Cancer Society says this: “Prostate cancer can be a serious disease, but most men diagnosed with prostate cancer do not die from it.”
However, these commonplace comments do not cover metastatic disease, which is what the authors of the new study decided to focus on.
The team used data from the Surveillance, Epidemiology, and End Results Program (SEER) database to gather a sample of 26,168 U.S. men who received a diagnosis of metastatic prostate cancer from January 2000 to December 2016. They then analyzed the data in 2020 and found that 16,732 men (64%) had died during the follow-up period.
The majority of these deaths (77.8%) were from prostate cancer, 5.5% were from other cancers, and 16.7% were from noncancer causes, including cardiovascular diseases, chronic obstructive pulmonary disease, and cerebrovascular diseases.
Senior author Dr. Alhalabi acknowledged a limitation in these findings – that the SEER database relies on causes of death extracted from death certificates. “Death certificates have limited granularity in terms of the details they can contain about the cause of death and also have reporting bias,” he said.
Most of the prostate cancer deaths (59%) occurred within 2 years. The 5-year overall survival rate in the study group was 26%.
The deadliness of metastatic disease “reinforces the need for innovations to promote early-stage diagnosis,” comment the editorialists. Striking a hopeful note, they also say that “new tests for prostate cancer detection may reduce the proportion of patients who receive a diagnosis at a late stage.”
Death from other causes
The mean age at metastatic prostate cancer diagnosis in the study was roughly 71 years. Most of the cohort was White (74.5%) and had a diagnosis of stage M1b metastatic prostate cancer (72.7%), which means the cancer had spread to the bones.
Among men in the cohort, the rates of death from septicemia, suicide, accidents, COPD, and cerebrovascular diseases were significantly increased compared with the general U.S. male population, the team observes.
Thus, the study authors were concerned with not only with death from metastatic prostate cancer but death from other causes.
That concern is rooted in the established fact that there is now improved survival among patients with prostate cancer in the U.S., including among men with advanced disease. “Patients tend to live long enough after a prostate cancer diagnosis for non–cancer-related comorbidities to be associated with their overall survival,” they write.
The editorialists agree: Prostate cancer “has a high long-term survival rate compared with almost all other cancer types and signals the need for greater holistic care for patients.”
As noted above, cardiovascular diseases were the most common cause of nonprostate cancer–related deaths in the new study.
As in the management of other cancers, there is concern among clinicians and researchers about the cardiotoxic effects of prostate cancer treatments.
The study authors point to a 2017 analysis that showed that men with prostate cancer and no prior cardiac disease had greater risk of heart failure after taking androgen deprivation therapy (ADT), a common treatment used when the disease recurs after definitive treatment. Another study suggested an association between cardiotoxic effects of ADT and myocardial infarction regardless of medical history in general.
The authors of the current study say that such findings highlight “the importance of multidisciplinary care for such patients and the role of primary care physicians in optimizing cardiovascular risk prevention and providing early referrals to cardiologists.”
Further, the team says that tailoring “ADT to each patient’s needs may be associated with improved survival, especially for patients with factors associated with cardiovascular disease.”
Who should lead the way in multidisciplinary care? “The answer probably is case-by-case,” said Dr. Alhalabi, adding that it might depend on the presence of underlying morbidities such as cardiovascular disease and COPD.
“It is also important for the oncologist (‘the gatekeeper’) to try to mitigate the potential metabolic effects of hormonal deprivation therapy such as weight gain, decreased muscle mass, hyperlipidemia, etc.,” he added.
The study had no specific funding. The study authors and editorialists have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
‘Dawn of a new era’ in the treatment of renal cell carcinoma
according to expert opinion.
The high hopes have been generated by results from the randomized, phase 3 KEYNOTE-564 trial, showing that monotherapy with pembrolizumab (Keytruda, Merck) was associated with significantly longer disease-free survival (DFS) after nephrectomy than placebo (77.3% vs. 68.1%, respectively). Median follow-up was 24 months.
The results come from the trial’s first interim analysis of data from 994 patients with clear-cell renal cell carcinoma (RCC) at high risk of recurrence.
For the pembrolizumab group, the estimated percentage alive at 24 months was 96.6%, compared with 93.5% in the placebo group (hazard ratio for death, 0.54), said Toni Choueiri, MD, of the Dana-Farber Cancer Institute, Boston, and colleagues.
However, grade 3 or higher adverse events (any cause) occurred at almost twice the rate in the pembrolizumab versus the placebo group (32.4% vs. 17.7%). The new study was published online Aug. 18, 2021, in the New England Journal of Medicine.
The study results were first presented at the 2021 American Society of Clinical Oncology annual meeting and described as likely to be practice changing in this setting, as reported by this news organization.
Currently, this patient population has “no options for adjuvant therapy to reduce the risk of recurrence that have high levels of supporting evidence,” observed the authors.
That’s about to change, as the trial results “herald the dawn of a new era in the treatment of renal cell carcinoma,” Rana McKay, MD, University of California San Diego Health, wrote in an accompanying editorial.
Multiple studies have investigated potential adjuvant therapies in RCC since the 1980s, she observed.
“For the first time, we now have an effective adjuvant immunotherapy option for patients with resected renal cell carcinoma at high risk of recurrence,” Dr. McKay said in an interview.
To date, the lack of clinically beneficial adjuvant therapy options in RCC has been “humbling,” Dr. Choueiri said in an interview. “We hope we can push the envelope further and get more patients with RCC some good options that make them live longer and better.”
Although the standard of care for patients diagnosed with locoregional RCC is partial or total nephrectomy, nearly half of patients eventually experience disease recurrence following surgery, Dr. Choueiri noted.
“No standard, globally approved adjuvant therapy options are currently available for this population,” he said. Clinical guidelines recommend patients at high risk of disease recurrence after surgery be entered into a clinical trial or undergo active surveillance.
Researchers will continue to follow the results for overall survival, a secondary endpoint. “The very early look suggests encouraging results [in overall survival] with an HR of 0.54,” Dr. Choueiri noted.
In the meantime, the prolongation of DFS represents a clear clinical benefit, said Dr. McKay, “given the magnitude of the increase” and “the limited incidence of toxic effects.”
KEYNOTE-564 will alter the adjuvant treatment landscape for RCC as a positive phase 3 trial of adjuvant immunotherapy for the disease, she added.
A number of earlier studies have investigated the use of adjuvant vascular endothelial growth factor–targeting agents in RCC. Only the 2016 Sunitinib Treatment of Renal Adjuvant Cancer (S-TRAC) trial showed improved DFS with sunitinib, compared with placebo (6.8 vs. 5.6 years). Subsequently, sunitinib was approved for adjuvant use in the United States. However, the S-TRAC trial also showed that sunitinib therapy was associated with an increased incidence of toxic effects and lower quality of life scores, and researchers did not observe any benefit in overall survival.
“Despite regulatory approval in the U.S., sunitinib is not approved for adjuvant use by the European Medicines Agency and has limited utilization in clinical practice given the low benefit-risk ratio,” Dr. McKay pointed out.
Study details
KEYNOTE-564 involved 996 patients with clear-cell RCC at high risk for recurrence after nephrectomy, with or without metastasectomy. They were randomly assigned in a 1:1 ratio to receive a 200-mg dose of adjuvant pembrolizumab or placebo given intravenously once every 3 weeks for up to 17 cycles for approximately 1 year.
The vast majority of patients enrolled in the study had localized disease with no evidence of metastases (M0) and intermediate to high or high risk of disease recurrence after partial or complete nephrectomy. However, 5.8% of patients in both the pembrolizumab and placebo groups had M1 NED (metastatic stage 1, no evidence of disease) status after nephrectomy and resection of metastatic lesions. These patients were also at intermediate to high or high risk of recurrence.
The benefit of pembrolizumab, compared with placebo, was maintained in this subgroup, said the investigators. “At this point, we continue to look at the data, but we know that there was a benefit for DFS in the population we included,” said Dr. Choueiri. “When we looked at several subgroups such as PD-L1 status, geography, gender, performance status, M0/M1, all HRs were less than 1 suggesting benefit from pembrolizumab over placebo.”
“Subset analyses by stage are going to be important to determine which group of patients will derive the most benefit,” asserted Dr. McKay. “While those with M1 NED appear to derive benefit with HR for DFS of 0.29, those with M1 NED comprise a small percentage of patient enrolled in the trial.”
Studies exploring tissue- and blood-based biomarkers, including circulating tumor DNA, will be key to identify patients at highest risk for recurrence or adjuvant treatment, Dr. McKay emphasized. “The adoption of adjuvant immune checkpoint inhibitors brings along new questions regarding patient selection, therapeutic use in patients with non–clear-cell renal cell carcinoma, and systemic treatment after recurrence during or after the receipt of adjuvant therapy.”
KEYNOTE-564 was funded by Merck. Multiple study authors including Dr. Choueiri have financial ties to the pharmaceutical industry, including Merck.
A version of this article first appeared on Medscape.com.
according to expert opinion.
The high hopes have been generated by results from the randomized, phase 3 KEYNOTE-564 trial, showing that monotherapy with pembrolizumab (Keytruda, Merck) was associated with significantly longer disease-free survival (DFS) after nephrectomy than placebo (77.3% vs. 68.1%, respectively). Median follow-up was 24 months.
The results come from the trial’s first interim analysis of data from 994 patients with clear-cell renal cell carcinoma (RCC) at high risk of recurrence.
For the pembrolizumab group, the estimated percentage alive at 24 months was 96.6%, compared with 93.5% in the placebo group (hazard ratio for death, 0.54), said Toni Choueiri, MD, of the Dana-Farber Cancer Institute, Boston, and colleagues.
However, grade 3 or higher adverse events (any cause) occurred at almost twice the rate in the pembrolizumab versus the placebo group (32.4% vs. 17.7%). The new study was published online Aug. 18, 2021, in the New England Journal of Medicine.
The study results were first presented at the 2021 American Society of Clinical Oncology annual meeting and described as likely to be practice changing in this setting, as reported by this news organization.
Currently, this patient population has “no options for adjuvant therapy to reduce the risk of recurrence that have high levels of supporting evidence,” observed the authors.
That’s about to change, as the trial results “herald the dawn of a new era in the treatment of renal cell carcinoma,” Rana McKay, MD, University of California San Diego Health, wrote in an accompanying editorial.
Multiple studies have investigated potential adjuvant therapies in RCC since the 1980s, she observed.
“For the first time, we now have an effective adjuvant immunotherapy option for patients with resected renal cell carcinoma at high risk of recurrence,” Dr. McKay said in an interview.
To date, the lack of clinically beneficial adjuvant therapy options in RCC has been “humbling,” Dr. Choueiri said in an interview. “We hope we can push the envelope further and get more patients with RCC some good options that make them live longer and better.”
Although the standard of care for patients diagnosed with locoregional RCC is partial or total nephrectomy, nearly half of patients eventually experience disease recurrence following surgery, Dr. Choueiri noted.
“No standard, globally approved adjuvant therapy options are currently available for this population,” he said. Clinical guidelines recommend patients at high risk of disease recurrence after surgery be entered into a clinical trial or undergo active surveillance.
Researchers will continue to follow the results for overall survival, a secondary endpoint. “The very early look suggests encouraging results [in overall survival] with an HR of 0.54,” Dr. Choueiri noted.
In the meantime, the prolongation of DFS represents a clear clinical benefit, said Dr. McKay, “given the magnitude of the increase” and “the limited incidence of toxic effects.”
KEYNOTE-564 will alter the adjuvant treatment landscape for RCC as a positive phase 3 trial of adjuvant immunotherapy for the disease, she added.
A number of earlier studies have investigated the use of adjuvant vascular endothelial growth factor–targeting agents in RCC. Only the 2016 Sunitinib Treatment of Renal Adjuvant Cancer (S-TRAC) trial showed improved DFS with sunitinib, compared with placebo (6.8 vs. 5.6 years). Subsequently, sunitinib was approved for adjuvant use in the United States. However, the S-TRAC trial also showed that sunitinib therapy was associated with an increased incidence of toxic effects and lower quality of life scores, and researchers did not observe any benefit in overall survival.
“Despite regulatory approval in the U.S., sunitinib is not approved for adjuvant use by the European Medicines Agency and has limited utilization in clinical practice given the low benefit-risk ratio,” Dr. McKay pointed out.
Study details
KEYNOTE-564 involved 996 patients with clear-cell RCC at high risk for recurrence after nephrectomy, with or without metastasectomy. They were randomly assigned in a 1:1 ratio to receive a 200-mg dose of adjuvant pembrolizumab or placebo given intravenously once every 3 weeks for up to 17 cycles for approximately 1 year.
The vast majority of patients enrolled in the study had localized disease with no evidence of metastases (M0) and intermediate to high or high risk of disease recurrence after partial or complete nephrectomy. However, 5.8% of patients in both the pembrolizumab and placebo groups had M1 NED (metastatic stage 1, no evidence of disease) status after nephrectomy and resection of metastatic lesions. These patients were also at intermediate to high or high risk of recurrence.
The benefit of pembrolizumab, compared with placebo, was maintained in this subgroup, said the investigators. “At this point, we continue to look at the data, but we know that there was a benefit for DFS in the population we included,” said Dr. Choueiri. “When we looked at several subgroups such as PD-L1 status, geography, gender, performance status, M0/M1, all HRs were less than 1 suggesting benefit from pembrolizumab over placebo.”
“Subset analyses by stage are going to be important to determine which group of patients will derive the most benefit,” asserted Dr. McKay. “While those with M1 NED appear to derive benefit with HR for DFS of 0.29, those with M1 NED comprise a small percentage of patient enrolled in the trial.”
Studies exploring tissue- and blood-based biomarkers, including circulating tumor DNA, will be key to identify patients at highest risk for recurrence or adjuvant treatment, Dr. McKay emphasized. “The adoption of adjuvant immune checkpoint inhibitors brings along new questions regarding patient selection, therapeutic use in patients with non–clear-cell renal cell carcinoma, and systemic treatment after recurrence during or after the receipt of adjuvant therapy.”
KEYNOTE-564 was funded by Merck. Multiple study authors including Dr. Choueiri have financial ties to the pharmaceutical industry, including Merck.
A version of this article first appeared on Medscape.com.
according to expert opinion.
The high hopes have been generated by results from the randomized, phase 3 KEYNOTE-564 trial, showing that monotherapy with pembrolizumab (Keytruda, Merck) was associated with significantly longer disease-free survival (DFS) after nephrectomy than placebo (77.3% vs. 68.1%, respectively). Median follow-up was 24 months.
The results come from the trial’s first interim analysis of data from 994 patients with clear-cell renal cell carcinoma (RCC) at high risk of recurrence.
For the pembrolizumab group, the estimated percentage alive at 24 months was 96.6%, compared with 93.5% in the placebo group (hazard ratio for death, 0.54), said Toni Choueiri, MD, of the Dana-Farber Cancer Institute, Boston, and colleagues.
However, grade 3 or higher adverse events (any cause) occurred at almost twice the rate in the pembrolizumab versus the placebo group (32.4% vs. 17.7%). The new study was published online Aug. 18, 2021, in the New England Journal of Medicine.
The study results were first presented at the 2021 American Society of Clinical Oncology annual meeting and described as likely to be practice changing in this setting, as reported by this news organization.
Currently, this patient population has “no options for adjuvant therapy to reduce the risk of recurrence that have high levels of supporting evidence,” observed the authors.
That’s about to change, as the trial results “herald the dawn of a new era in the treatment of renal cell carcinoma,” Rana McKay, MD, University of California San Diego Health, wrote in an accompanying editorial.
Multiple studies have investigated potential adjuvant therapies in RCC since the 1980s, she observed.
“For the first time, we now have an effective adjuvant immunotherapy option for patients with resected renal cell carcinoma at high risk of recurrence,” Dr. McKay said in an interview.
To date, the lack of clinically beneficial adjuvant therapy options in RCC has been “humbling,” Dr. Choueiri said in an interview. “We hope we can push the envelope further and get more patients with RCC some good options that make them live longer and better.”
Although the standard of care for patients diagnosed with locoregional RCC is partial or total nephrectomy, nearly half of patients eventually experience disease recurrence following surgery, Dr. Choueiri noted.
“No standard, globally approved adjuvant therapy options are currently available for this population,” he said. Clinical guidelines recommend patients at high risk of disease recurrence after surgery be entered into a clinical trial or undergo active surveillance.
Researchers will continue to follow the results for overall survival, a secondary endpoint. “The very early look suggests encouraging results [in overall survival] with an HR of 0.54,” Dr. Choueiri noted.
In the meantime, the prolongation of DFS represents a clear clinical benefit, said Dr. McKay, “given the magnitude of the increase” and “the limited incidence of toxic effects.”
KEYNOTE-564 will alter the adjuvant treatment landscape for RCC as a positive phase 3 trial of adjuvant immunotherapy for the disease, she added.
A number of earlier studies have investigated the use of adjuvant vascular endothelial growth factor–targeting agents in RCC. Only the 2016 Sunitinib Treatment of Renal Adjuvant Cancer (S-TRAC) trial showed improved DFS with sunitinib, compared with placebo (6.8 vs. 5.6 years). Subsequently, sunitinib was approved for adjuvant use in the United States. However, the S-TRAC trial also showed that sunitinib therapy was associated with an increased incidence of toxic effects and lower quality of life scores, and researchers did not observe any benefit in overall survival.
“Despite regulatory approval in the U.S., sunitinib is not approved for adjuvant use by the European Medicines Agency and has limited utilization in clinical practice given the low benefit-risk ratio,” Dr. McKay pointed out.
Study details
KEYNOTE-564 involved 996 patients with clear-cell RCC at high risk for recurrence after nephrectomy, with or without metastasectomy. They were randomly assigned in a 1:1 ratio to receive a 200-mg dose of adjuvant pembrolizumab or placebo given intravenously once every 3 weeks for up to 17 cycles for approximately 1 year.
The vast majority of patients enrolled in the study had localized disease with no evidence of metastases (M0) and intermediate to high or high risk of disease recurrence after partial or complete nephrectomy. However, 5.8% of patients in both the pembrolizumab and placebo groups had M1 NED (metastatic stage 1, no evidence of disease) status after nephrectomy and resection of metastatic lesions. These patients were also at intermediate to high or high risk of recurrence.
The benefit of pembrolizumab, compared with placebo, was maintained in this subgroup, said the investigators. “At this point, we continue to look at the data, but we know that there was a benefit for DFS in the population we included,” said Dr. Choueiri. “When we looked at several subgroups such as PD-L1 status, geography, gender, performance status, M0/M1, all HRs were less than 1 suggesting benefit from pembrolizumab over placebo.”
“Subset analyses by stage are going to be important to determine which group of patients will derive the most benefit,” asserted Dr. McKay. “While those with M1 NED appear to derive benefit with HR for DFS of 0.29, those with M1 NED comprise a small percentage of patient enrolled in the trial.”
Studies exploring tissue- and blood-based biomarkers, including circulating tumor DNA, will be key to identify patients at highest risk for recurrence or adjuvant treatment, Dr. McKay emphasized. “The adoption of adjuvant immune checkpoint inhibitors brings along new questions regarding patient selection, therapeutic use in patients with non–clear-cell renal cell carcinoma, and systemic treatment after recurrence during or after the receipt of adjuvant therapy.”
KEYNOTE-564 was funded by Merck. Multiple study authors including Dr. Choueiri have financial ties to the pharmaceutical industry, including Merck.
A version of this article first appeared on Medscape.com.
Although inconclusive, CV safety study of cancer therapy attracts attention
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.

“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).

In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.
“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).
In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.
“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).
In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
FROM ESC 2021
Three Primary Cancers in a Veteran With Agent Orange and Agent Blue Exposures
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
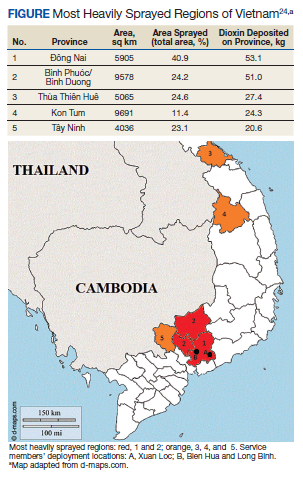
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
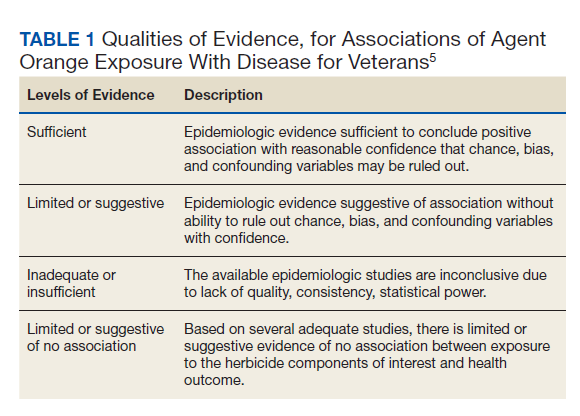
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
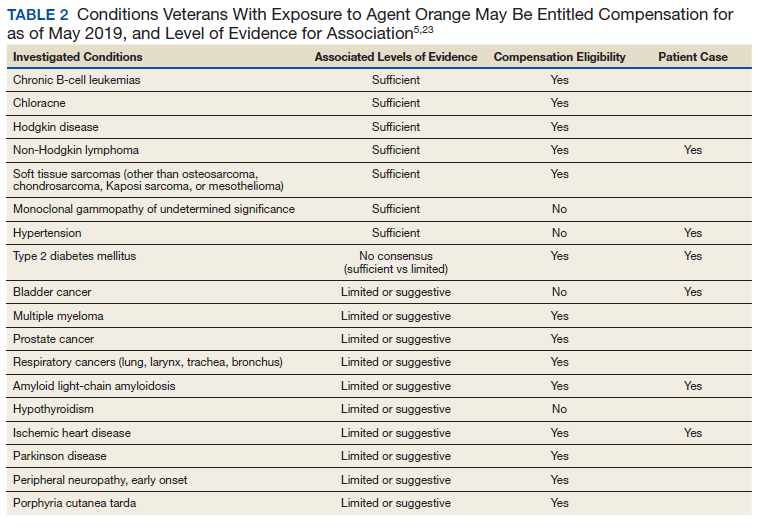
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420

31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
A Vietnam War veteran’s exposures likely contributed to his cancer diagnoses, but these associations are confounded by his substance use, particularly cigarette smoking.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
Known as the “6 rainbow herbicides,” based on their identifying color on storage containers, the United States widely deployed the herbicides agents orange, green, pink, purple, white, and blue during the Vietnam War to deny the enemy cover and destroy crops.1 Unfortunately, all these herbicides were found to have contained some form of carcinogen. Agent Blue’s active ingredient consisted of sodium cacodylate trihydrate (C2H6AsNaO2), a compound that is metabolized into the organic form of the carcinogen arsenic before eventually converting into its relatively less toxic inorganic form.2 Agent Orange’s defoliating agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). All rainbow herbicides except Agent Blue were unintentionally contaminated with carcinogenic dioxins. Agent Blue contained the carcinogen cacodylic acid, an organoarsenic acid. Today, herbicides no longer contain polychlorinated dibenzo-p-dioxins such as TCDD or arsenic due to strict manufacturing restrictions.2,3 In the treatment of veteran populations, knowledge of the 6 rainbow herbicides’ carcinogenic potential is important.
Between 1962 and 1971, the United States sprayed more than 45 million liters of Agent Orange on Vietnam and at least 366 kg of TCDD on South Vietnam.1,4 However, because Agent Orange was not a known carcinogen during the Vietnam War, records of exposure are poor. Additionally, individuals in Vietnam during this period were not the only ones exposed to this carcinogen as Agent Orange also was sprayed in Thailand and Korea.5 Even today there are still locations in Vietnam where Agent Orange concentrations exceed internationally acceptable levels. The Da Nang, Bien Hoa, and Phu Cat airports in Vietnam have been found to have dioxin levels exceeding 1000 ppt (parts of dioxin per trillion parts of lipid) toxicity equivalence in the soil. Although the Vietnam government is working toward decontaminating these and many other dioxin hotspots, residents in these locations are exposed to higher than internationally acceptable levels of dioxin.6
Despite receiving less media attention, Vietnam War veterans and Vietnamese soldiers and civilians were exposed to significant amounts of arsenic-based Agent Blue. Arsenic is a compound which has no environmental half-life and is carcinogenic humans if inhaled or ingested.2 Between 1962 and 1971, the United States distributed 7.8 million liters of Agent Blue containing 1,232,400 kg of arsenic across 300,000 hectares of rice paddies, 100,000 hectares of forest, and perimeters of all military bases during the Vietnam War.2,5 According to a review by Saha and colleagues, lower levels of arsenic exposure are associated with acute and chronic diseases, including cancers, of all organ systems.7
The following case presentation involves a Vietnam War veteran aged 70 years who was exposed to Agent Orange and developed 3 primary cancers, including cutaneous large B-cell non-Hodgkin lymphoma (NHL), high-grade urothelial carcinoma, and anal carcinoma in situ. Epidemiologically, this is an uncommon occurrence as only 8% of cancer survivors in the United States have been diagnosed with > 1 cancer.8
With no family history of cancer, the development of multiple malignancies raises concern for a history of toxin exposure. This report of a Vietnam War veteran with multiple conditions found to be associated with Agent Orange exposure provides an opportunity to discuss the role this exposure may have on the development of a comprehensive list of medical conditions as described by the literature. Additionally, the potential contributions of other confounding toxin exposures such as cigarette smoking, excessive alcohol use, and potential Agent Blue exposure on our patients’ health will be discussed.
Case Presentation
A male aged 70 years with Stage IV primary cutaneous large B-cell NHL, incompletely resected high-grade urothelial cancer, carcinoma in situ of the anal canal, and peripheral arterial disease (PAD) presented to the primary care clinic at the Washington DC Veterans Affairs Medical Center (DCVAMC) with concern for left leg ischemia. He also reported 2 large telangiectasias on his back for 6 months accompanied by lymphadenopathy and intermittent night sweats.
He was last seen at the DCVAMC 15 months prior after his twelfth dose of rituximab treatment for NHL. However, the patient failed to return for completion of his treatment due to frustration with the lengthy chemotherapy and follow-up process. Additionally, the patient's history included 3 failed arterial stents with complete nonadherence to the prescribed clopidogrel, resulting in the failure of 3 more subsequent graft placements. On presentation, the patient continued to report nonadherence with the clopidogrel.
The patient’s medical history included coronary artery disease (CAD) status after 2 stents in the left anterior descending artery and 1 stent in the proximal circumflex artery placed 4 years prior. He also had a history of hypertension, type 2 diabetes mellitus (T2DM), amyloid light-chain (AL) amyloidosis, aortic aneurysm, cataracts, obesity, treated hepatitis B and C, and posttraumatic stress disorder. He had no family history of cancer or AL amyloidosis; however, he noted that he was estranged from his family.
His social history was notable for active cigarette smoking up to 3 packs per day for 40 years and consuming large quantities of alcohol—at one point as many as 20 beers per day over a period of 4.5 years. He had a distant history of cocaine use but no current use, which was supported with negative urinary toxicology screens for illicit drugs over the past year.
Our patient also reported a history of Agent Orange exposure. As an artilleryman in the US Army III Corps, he was deployed for about 1 year in the most heavily sprayed regions of Vietnam, including Bien Hua, Long Binh, Xuan Loc, and Camp Zion for about 2 to 4 months at each location.
Hospital Course
The patient was treated on an inpatient basis for expedited workup and treatment for his urothelial carcinoma, NHL, and ischemic limb. His urothelial carcinoma was successfully resected, and the telangiectasias on his back were biopsied and found to be consistent with his known cutaneous large B-cell NHL, for which plans to resume outpatient chemotherapy were made. The patient’s 3 arterial grafts in his left leg were confirmed to have failed, and the patient was counseled that he would soon likely require an amputation of his ischemic leg.
Discussion
We must rely on our patient’s historical recall as there are no widely available laboratory tests or physical examination findings to confirm and/or determine the magnitude of TCDD or arsenic exposure.9-11
Exposures
The patient was stationed in Bien Hoa, the second highest dioxin-contaminated air base in Vietnam (Figure).6 Dioxin also is known to be a particularly persistent environmental pollutant, such that in January 2018, Bien Hoa was found to still have dioxin levels higher than what is considered internationally acceptable. In fact, these levels were deemed significant enough to lead the United States and Vietnamese government to sign a memorandum of intent to begin cleanup of this airport.6 TCDD is known to have a half-life of about 7.6 years, and its long half-life is mainly attributed to its slow elimination process from its stores within the liver and fat, consisting of passive excretion through the gut wall and slow metabolism by the liver.12,13 Thus, as an artilleryman mainly operating 105 howitzers within the foliage of Vietnam, our patient was exposed not only to high levels of this persistent environmental pollutant on a daily basis, but this toxin likely remained within his system for many years after his return from Vietnam.
Our patient also had a convincing history for potential Agent Blue exposure through both inhalation and ingestion of contaminated food and water. Additionally, his description of deforestations occurring within a matter of days increased the level of suspicion for Agent Blue exposure. This is because Agent Blue was the herbicide of choice for missions requiring rapid deforestation, achieving defoliation as quickly as 1 to 2 days.14 Additionally, our patient was stationed within cities in southern Vietnam near Agent Blue hot spots, such as Da Nang and Saigon, and Agent Blue was sprayed along the perimeter of all military bases.2
Levels of Evidence
Using the Veterans and Agent Orange Update in 2018 as our guide, we reviewed the quality of evidence suggesting an association between many of our patient’s comorbidities to Agent Orange exposure.5 This publication categorizes the level of evidence for association between health conditions and Agent Orange exposure in 4 main categories (Table 1).
In the Veterans and Agent Orange Update, NHL notably has a sufficient level of evidence of association with Agent Orange exposure.5 Although our patient’s extensive history of polysubstance use confounds the effect Agent Orange may have had on his health, cutaneous large B-cell NHL is an interesting exception as literature does not support even a correlative link between smoking and excessive alcohol use with primary cutaneous large B-cell NHL. Several case-control studies have found little to no association with cigarette smoking and the large B-cell subtype of NHL.15,16 Moreover, several studies have found that moderate- to-heavy alcohol use, especially beer, may have a protective effect against the development of NHL.17 Of note, our patient’s alcoholic beverage of choice was beer. Regarding our patient’s distant history of cocaine use, it has been reported that cocaine use, in the absence of an HIV infection, has not been found to increase the risk of developing NHL.18 Similarly, arsenic exposure has not been associated with NHL in the literature.19,20
The 2018 update also upgraded bladder carcinoma from having inadequate or insufficient to a limited or suggestive level of evidence for association.5 However, our patient’s most significant risk factor for bladder cancer was smoking, with a meta-analysis of 430,000 patients reporting a risk ratio (RR) of 3.14 for current cigarette smokers.21 The patient’s arsenic exposure from Agent Blue also increased his risk of developing bladder cancer. Several studies suggest a strong association between environmental arsenic exposure and bladder cancer.22-26 A 30-year meta-analysis of 40 studies by Saint-Jacques and colleagues reported that the incidence of bladder cancer was found to increase in a dose-dependent manner, with higher concentrations of arsenic contaminated wate, with incidence rising from 2.7 to 5.8 times as the amount of arsenic contamination water increased from 10 to 150 mg/L.
Our patient’s history is concerning for higher than average Agent Blue exposure compared with that of most Vietnam War veterans. Given the dose-dependent effect of arsenic on bladder cancer risk, both our patient’s history of smoking and Agent Blue exposure are risk factors in the development of his bladder cancer.22 These likely played a more significant role in his development of bladder cancer than did his Agent Orange exposure.
Finally, smoking is the most significant risk factor in our patient’s development of anal carcinoma in situ. The 2018 Agent Orange update does report limited/suggested evidence of no association between Agent Orange and anal carcinoma.5 It also is unknown whether Agent Blue exposure is a contributing cause to his development of anal carcinoma in situ.27 However, current smokers are at significant risk of developing anal cancer independent of age.28-30 Given our patient’s extensive smoking history, this is the most likely contributing factor.
Our patient also had several noncancer-related comorbidities with correlative associations with Agent Orange exposure of varying degrees (Table 2). Somewhat surprising, the development of our patient’s hypertension and T2DM may be associated in some way with his history of Agent Orange exposure. Hypertension had been recategorized from having limited or suggestive evidence to sufficient evidence in this committee’s most recent publication, and the committee is undecided on whether T2DM has a sufficient vs limited level of evidence for association with Agent Orange exposure.5 On the other hand, the committee continues to classify both ischemic heart disease and AL amyloidosis as having a limited or suggestive level of evidence that links Agent Orange exposure to these conditions.5
Arsenic may be another risk factor for our patient’s development of CAD and arterial insufficiency. Arsenic exposure is theorized to cause a direct toxic effect on coronary arteries, and arsenic exposure has been linked to PAD, CAD, and hypertension.31-34 Other significant and compelling risk factors for cardiovascular disease in our patient included his extensive history of heavy cigarette smoking, poorly controlled T2DM, obesity, and hypertension.35-37 AL amyloidosis is a rare disorder with an incidence of only 9 to 14 cases per million person-years.38,39 This disorder has not been linked to smoking or arsenic exposure in the literature. As our patient does not have a history of plasma dyscrasias or a family history of AL amyloidosis, the only known risk factors for AL amyloidosis that apply to our patient included NHL and Agent Orange exposure—NHL being a condition that is noted to be strongly correlated with Agent Orange exposure as discussed previously.5,36,40,41
Conclusions
This case describes a Vietnam War veteran with significant exposure to rainbow herbicides and considerable polysubstance who developed 3 primary cancers and several chronic medical conditions. His exposure to Agents Orange and Blue likely contributed to his medical problems, but these associations are confounded by his substance use, particularly cigarette smoking. Of all his comorbidities, our patient’s NHL is the condition most likely to be associated with his history of Agent Orange exposure. His Agent Blue exposure also increased his risk for developing bladder cancer, cardiovascular disease, and PAD.
This case also highlights the importance of evaluating Vietnam War veterans for rainbow herbicide exposure and the complexity associated with attributing diseases to these exposures. All veterans who served in the inland waterways of Vietnam between 1962 and 1975; in the Korean Demilitarized Zone between April 1, 1968 and August 31, 1971; or in Thailand between February 28, 1961 and May 7, 1975 were at risk of rainbow herbicide exposure. These veterans may not only be eligible for disability compensation but also should be screened for associated comorbidities as outlined by current research.42 We hope that this report will serve as an aid in achieving this mission.
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420
31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp
1. Stellman JM, Stellman SD, Christian R, Weber T, Tomasallo C. The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature. 2003;422(6933):681-687. doi:10.1038/nature01537
2. Olson K, Cihacek L. The fate of Agent Blue, the arsenic based herbicide, used in South Vietnam during the Vietnam War. Open J Soil Sci. 2020;10:518-577. doi:10.4236/ojss.2020.1011027
3. Lee Chang A, Dym AA, Venegas-Borsellino C, et al. Comparison between simulation-based training and lecture-based education in teaching situation awareness. a randomized controlled study. Ann Am Thorac Soc. 2017;14(4):529-535. doi:10.1513/AnnalsATS.201612-950OC
4. Stellman SD. Agent Orange during the Vietnam War: the lingering issue of its civilian and military health impact. Am J Public Health. 2018;108(6):726-728. doi:10.2105/AJPH.2018.304426
5. National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 11 (2018). The National Academies Press; 2018. doi:10.17226/25137
6. Martin MF. US Agent Orange/dioxin assistance to Vietnam. Updated January 15, 2021. Accessed June 17, 2021. https://fas.org/sgp/crs/row/R44268.pdf
7. Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC. A review of arsenic poisoning and its effects on human health. Crit Rev Environ Sci Technol. 1999;29(3):281-313. doi:10.1080/10643389991259227
8. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
9. American Cancer Society. Agent Orange and cancer risk. Updated June 9, 2020. Accessed June 17, 2021. https://www.cancer.org/cancer/cancer-causes/agent-orange-and-cancer.html
10. US Department of Veterans Affairs. Veterans’ diseases associated with Agent Orange. Updated June 16, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/conditions/index.asp
11. Katz SA. On the use of hair analysis for assessing arsenic intoxication. Int J Environ Res Public Health. 2019;16(6):977. Published 2019 Mar 18. doi:10.3390/ijerph16060977
12. Chang ET, Boffetta P, Adami HO, Mandel JS. A critical review of the epidemiology of Agent Orange or 2,3,7,8-tetrachlorodibenzo-p-dioxin and lymphoid malignancies. Ann Epidemiol. 2015;25(4):275-292.e30. doi:10.1016/j.annepidem.2015.01.002
13. Kramárová E, Kogevinas M, Anh CT, et al. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environ Health Perspect. 1998;106 Suppl 2(suppl 2):671-678. doi:10.1289/ehp.106-1533419
14. Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. National Academies Press US; 1994.
15. Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14(4):925-933. doi:10.1158/1055-9965.EPI-04-0693
16. Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin’s lymphoma--a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1791-1796. doi:10.1158/1055-9965.EPI-05-0077
17. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486-495. doi:10.1002/ijc.31330
18. Aujla AS, Lee SH. Association between cocaine use and hematological malignancies. J Clin Oncol. 2016;34(15_suppl):e19072-e19072. doi:10.1200/JCO.2016.34.15_suppl.e19072
19. Mao Y, Hu J, Ugnat AM, White K. Non-Hodgkin’s lymphoma and occupational exposure to chemicals in Canada. Canadian Cancer Registries Epidemiology Research Group. Ann Oncol. 2000;11 (suppl 1):69-73. doi:10.1093/annonc/11.suppl_1.S69
20. Kelekci KH, Bilgin I, Ermete M. Arsenical keratoses and non-Hodgkin’s lymphoma: arsenic-induced or coincidental conditions? J Pakistan Assoc Dermatol. 2012;22(4):366-369.
21. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96-108. doi:10.1016/j.eururo.2016.06.010
22. Saint-Jacques N, Parker L, Brown P, Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014;13:44. Published 2014 Jun 2. doi:10.1186/1476-069X-13-44
23. Radosavljevic′ V, Jakovljevic′ B. Arsenic and bladder cancer: observations and suggestions. J Environ Health. 2008;71(3):40-42.
24. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076(1):810-821. doi:10.1196/annals.1371.031
25. Mendez WM Jr, Eftim S, Cohen J, et al. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 2017;27(3):235-243. doi:10.1038/jes.2016.58
26. Pal DK, Agrawal A, Ghosh S, Ghosh A. Association of arsenic with recurrence of urinary bladder cancer. Trop Doct. 2020;50(4):325-330. doi:10.1177/0049475520930155
27. García-Esquinas E, Pollán M, Umans JG, et al. Arsenic exposure and cancer mortality in a US-based prospective cohort: the strong heart study [published correction appears in Cancer Epidemiol Biomarkers Prev. 2013;22(8):1479]. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1944-1953. doi:10.1158/1055-9965.EPI-13-0234-T
28. Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270-280. doi:10.1002/cncr.20365
29. Bertisch B, Franceschi S, Lise M, et al; Swiss HIV Cohort Study Investigators. Risk factors for anal cancer in persons infected with HIV: a nested case-control study in the Swiss HIV Cohort Study. Am J Epidemiol. 2013;178(6):877-884. doi:10.1093/aje/kwt153
30. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54-58. doi:10.1093/oxfordjournals.aje.a116420
31. Newman JD, Navas-Acien A, Kuo CC, et al. Peripheral arterial disease and its association with arsenic exposure and metabolism in the Strong Heart Study. Am J Epidemiol. 2016;184(11):806-817. doi:10.1093/aje/kww002
32. Moon KA, Guallar E, Umans JG, et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease. A prospective cohort study. Ann Intern Med. 2013;159(10):649-659. doi:10.7326/0003-4819-159-10-201311190-00719
33. Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542-555. doi:10.1007/s11883-012-0280-x
34. Stea F, Bianchi F, Cori L, Sicari R. Cardiovascular effects of arsenic: clinical and epidemiological findings. Environ Sci Pollut Res Int. 2014;21(1):244-251. doi:10.1007/s11356-013-2113-z
35. Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46(1):11-29. doi:10.1016/s0033-0620(03)00079-3
36. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. Published 2018 Oct 25. doi:10.1038/s41572-018-0034-3
37. Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008;21(3):160-165. doi:10.2337/diaspect.21.3.160
38. Vaxman I, Gertz M. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. 2019;141(2):93-106. doi:10.1159/000495455
39. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053. doi:10.1182/bloodadvances.2018016402
40. Basset M, Defrancesco I, Milani P, et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood. 2020;135(4):293-296. doi:10.1182/blood.2019002762
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569. doi:10.1056/NEJMoa01133202
42. US Department of Veterans Affairs. Agent Orange registry health exam for veterans. Updated May 28, 2021. Accessed June 17, 2021. https://www.publichealth.va.gov/exposures/agentorange/benefits/registry-exam.asp
‘Routine’ use of focal therapy for prostate cancer in next 5 years
They maintain that focal therapy (FT) offers a “middle ground” between two extremes: Treating the whole gland with radical prostatectomy or radiotherapy and not treating immediately via active surveillance or watchful waiting.
Focal therapy typically treats the primary lesion within the prostate, while leaving the rest of the gland intact. Most often performed with cryoablation or high-intensity focused ultrasound (HIFU), it can also be carried out with a variety of technologies, including transurethral ultrasound ablation and focal laser ablation.
The shift to focal therapy will coincide with maturing, long-term data from studies with various technologies, predict the authors, led by Amir Lebastchi, MD, a urologist at the University of Southern California.
“Standard adoption of focal therapy is ultimately dependent on the availability of robust level I evidence, which in turn will drive medical societies and payees,” the authors also write.
But payees are already making changes, even without such data, they add.
For example, in January the American Medical Association announced a new code for high-intensity focal ultrasound (HIFU): This approach now has a Current Procedural Terminology (CPT) code from the U.S. Centers for Medicare & Medicaid Services
This news organization reached out to Matthew Cooperberg, MD, MPH, a urologist at the University of California, San Francisco (UCSF), for comments about the essay’s optimism; he has questioned focal therapy in the past because of a lack of strong supporting evidence.
“While ‘routine’ is a bit of a vague term, now that HIFU has a CPT code, I do expect its use will in fact increase in the next 5 years,” Dr. Cooperberg wrote in an email. “The question is whether its use will increase appropriately.”
The challenge with focal therapy – regardless of energy modality – remains patient selection and accurate ablation zone definition, he added.
Notably, UCSF has launched a new HIFU program – and Dr. Cooperberg has referred selected patients. “I’m both enthusiastic and cautious about the future, and we need to track our outcomes very closely across various practice settings,” he said.
While waiting for CHRONOS, select wisely
The goal of focal therapy is to treat only the area with the most aggressive tumor, known as the index tumor, while leaving the remaining gland and its surrounding structures alone, according to Derek Lomas, MD, PharmD, a urologist at the Mayo Clinic in Rochester, Minn., in an explanatory article. “This approach is widely accepted in other types of cancer. For example, we commonly treat kidney cancers by removing or ablating only the tumor while leaving the rest of the kidney intact.”
However, some focal therapies also include approaches known as hemiablations, in which a full half of the prostate is destroyed, and approaches that leave very little of the gland behind.
Each of the modalities used for focal therapy has “unique indications, risks, and benefits and uses a different energy source for ablation,” Dr. Lebastchi and colleagues write in their essay.
They assert that focal therapy can provide oncological efficacy similar to radical prostatectomy or radiotherapy “while considerably reducing or even eliminating functional morbidities, such as incontinence and erectile dysfunction.”
Overall, they say focal therapy offers an opportunity for improved care because there is “an increasing body of emerging evidence demonstrating a favorable adverse effect profile with oncological control similar to whole-gland treatment options.”
What is that evidence?
In the essay, Dr. Lebastchi and colleagues point to a number of single-arm studies with encouraging efficacy and safety results. They also highlight a phase 3, randomized trial that they were involved in: This compared focal therapy (partial gland ablation with vascular-targeted photodynamic therapy) with active surveillance in early-stage disease and uniformly showed better post-treatment biopsy (disease/no disease) and conversion-to-prostatectomy results with the focal therapy out to 4 years (J Urol. 2018;200:786-793).
However, that study did not have an active treatment comparator. For that gold standard, there is now anticipation for results from the CHRONOS trial in the United Kingdom, especially part A of the trial, which compares radical therapy to focal therapy (HIFU or cryotherapy), with 5-year progression-free survival as the primary outcome. That trial is slated for completion in 2027.
Until then, the lack of prospective randomized clinical trials and long-term follow-up “hinders acceptance [of focal therapy] in the urology community,” the essay authors comment.
Meanwhile, careful patient selection is very important, they say.
The latest relevant guidelines state that appropriate candidates are men with a solitary, well-defined index lesion; patients with bilateral multifocal lesions; or very advanced tumors that are not appropriate for the focal approach.
A multidisciplinary international expert panel recently convened to establish guidance for clinicians offering focal therapies and then published a consensus statement to advise practitioners and researchers.
UCSF’s Dr. Cooperberg sees plenty of room for improvement among focal therapy practitioners and investigators. “From an outcomes standpoint, follow-up protocols and definitions of success remain inconsistent. I believe we’re making progress in all these areas, but we’re not there yet,” he says.
To date, some patients have been managed poorly, Dr. Cooperberg added. “We certainly see many patients who have been inadequately counseled as to HIFU’s advantages and disadvantages, with sometimes disastrous results.”
Some of those unfortunate results may have arisen from the U.S. Food and Drug Administration’s initial approval of HIFU in 2015, which was for use in ablating prostate tissue in general and not cancer specifically. This approval generated confusion, one expert commented at the time: “The FDA doesn’t specify whether it’s for benign or malignant disease; it’s a bit vague, like saying you can drive this car, but we’re not going to tell you how to drive it,” said Manoj Monga, MD, from the Cleveland Clinic.
Dr. Lebastchi has disclosed no relevant financial relationships; co-author Inderbir Gill, MD, is an unpaid consultant for Steba Biotech, and co-author Andre Luis Abreu, MD, is a consultant for Koelis and was a proctor in training for Steba Biotech. Dr. Cooperberg is a consultant for Alessa Therapeutics.
A version of this article first appeared on Medscape.com.
They maintain that focal therapy (FT) offers a “middle ground” between two extremes: Treating the whole gland with radical prostatectomy or radiotherapy and not treating immediately via active surveillance or watchful waiting.
Focal therapy typically treats the primary lesion within the prostate, while leaving the rest of the gland intact. Most often performed with cryoablation or high-intensity focused ultrasound (HIFU), it can also be carried out with a variety of technologies, including transurethral ultrasound ablation and focal laser ablation.
The shift to focal therapy will coincide with maturing, long-term data from studies with various technologies, predict the authors, led by Amir Lebastchi, MD, a urologist at the University of Southern California.
“Standard adoption of focal therapy is ultimately dependent on the availability of robust level I evidence, which in turn will drive medical societies and payees,” the authors also write.
But payees are already making changes, even without such data, they add.
For example, in January the American Medical Association announced a new code for high-intensity focal ultrasound (HIFU): This approach now has a Current Procedural Terminology (CPT) code from the U.S. Centers for Medicare & Medicaid Services
This news organization reached out to Matthew Cooperberg, MD, MPH, a urologist at the University of California, San Francisco (UCSF), for comments about the essay’s optimism; he has questioned focal therapy in the past because of a lack of strong supporting evidence.
“While ‘routine’ is a bit of a vague term, now that HIFU has a CPT code, I do expect its use will in fact increase in the next 5 years,” Dr. Cooperberg wrote in an email. “The question is whether its use will increase appropriately.”
The challenge with focal therapy – regardless of energy modality – remains patient selection and accurate ablation zone definition, he added.
Notably, UCSF has launched a new HIFU program – and Dr. Cooperberg has referred selected patients. “I’m both enthusiastic and cautious about the future, and we need to track our outcomes very closely across various practice settings,” he said.
While waiting for CHRONOS, select wisely
The goal of focal therapy is to treat only the area with the most aggressive tumor, known as the index tumor, while leaving the remaining gland and its surrounding structures alone, according to Derek Lomas, MD, PharmD, a urologist at the Mayo Clinic in Rochester, Minn., in an explanatory article. “This approach is widely accepted in other types of cancer. For example, we commonly treat kidney cancers by removing or ablating only the tumor while leaving the rest of the kidney intact.”
However, some focal therapies also include approaches known as hemiablations, in which a full half of the prostate is destroyed, and approaches that leave very little of the gland behind.
Each of the modalities used for focal therapy has “unique indications, risks, and benefits and uses a different energy source for ablation,” Dr. Lebastchi and colleagues write in their essay.
They assert that focal therapy can provide oncological efficacy similar to radical prostatectomy or radiotherapy “while considerably reducing or even eliminating functional morbidities, such as incontinence and erectile dysfunction.”
Overall, they say focal therapy offers an opportunity for improved care because there is “an increasing body of emerging evidence demonstrating a favorable adverse effect profile with oncological control similar to whole-gland treatment options.”
What is that evidence?
In the essay, Dr. Lebastchi and colleagues point to a number of single-arm studies with encouraging efficacy and safety results. They also highlight a phase 3, randomized trial that they were involved in: This compared focal therapy (partial gland ablation with vascular-targeted photodynamic therapy) with active surveillance in early-stage disease and uniformly showed better post-treatment biopsy (disease/no disease) and conversion-to-prostatectomy results with the focal therapy out to 4 years (J Urol. 2018;200:786-793).
However, that study did not have an active treatment comparator. For that gold standard, there is now anticipation for results from the CHRONOS trial in the United Kingdom, especially part A of the trial, which compares radical therapy to focal therapy (HIFU or cryotherapy), with 5-year progression-free survival as the primary outcome. That trial is slated for completion in 2027.
Until then, the lack of prospective randomized clinical trials and long-term follow-up “hinders acceptance [of focal therapy] in the urology community,” the essay authors comment.
Meanwhile, careful patient selection is very important, they say.
The latest relevant guidelines state that appropriate candidates are men with a solitary, well-defined index lesion; patients with bilateral multifocal lesions; or very advanced tumors that are not appropriate for the focal approach.
A multidisciplinary international expert panel recently convened to establish guidance for clinicians offering focal therapies and then published a consensus statement to advise practitioners and researchers.
UCSF’s Dr. Cooperberg sees plenty of room for improvement among focal therapy practitioners and investigators. “From an outcomes standpoint, follow-up protocols and definitions of success remain inconsistent. I believe we’re making progress in all these areas, but we’re not there yet,” he says.
To date, some patients have been managed poorly, Dr. Cooperberg added. “We certainly see many patients who have been inadequately counseled as to HIFU’s advantages and disadvantages, with sometimes disastrous results.”
Some of those unfortunate results may have arisen from the U.S. Food and Drug Administration’s initial approval of HIFU in 2015, which was for use in ablating prostate tissue in general and not cancer specifically. This approval generated confusion, one expert commented at the time: “The FDA doesn’t specify whether it’s for benign or malignant disease; it’s a bit vague, like saying you can drive this car, but we’re not going to tell you how to drive it,” said Manoj Monga, MD, from the Cleveland Clinic.
Dr. Lebastchi has disclosed no relevant financial relationships; co-author Inderbir Gill, MD, is an unpaid consultant for Steba Biotech, and co-author Andre Luis Abreu, MD, is a consultant for Koelis and was a proctor in training for Steba Biotech. Dr. Cooperberg is a consultant for Alessa Therapeutics.
A version of this article first appeared on Medscape.com.
They maintain that focal therapy (FT) offers a “middle ground” between two extremes: Treating the whole gland with radical prostatectomy or radiotherapy and not treating immediately via active surveillance or watchful waiting.
Focal therapy typically treats the primary lesion within the prostate, while leaving the rest of the gland intact. Most often performed with cryoablation or high-intensity focused ultrasound (HIFU), it can also be carried out with a variety of technologies, including transurethral ultrasound ablation and focal laser ablation.
The shift to focal therapy will coincide with maturing, long-term data from studies with various technologies, predict the authors, led by Amir Lebastchi, MD, a urologist at the University of Southern California.
“Standard adoption of focal therapy is ultimately dependent on the availability of robust level I evidence, which in turn will drive medical societies and payees,” the authors also write.
But payees are already making changes, even without such data, they add.
For example, in January the American Medical Association announced a new code for high-intensity focal ultrasound (HIFU): This approach now has a Current Procedural Terminology (CPT) code from the U.S. Centers for Medicare & Medicaid Services
This news organization reached out to Matthew Cooperberg, MD, MPH, a urologist at the University of California, San Francisco (UCSF), for comments about the essay’s optimism; he has questioned focal therapy in the past because of a lack of strong supporting evidence.
“While ‘routine’ is a bit of a vague term, now that HIFU has a CPT code, I do expect its use will in fact increase in the next 5 years,” Dr. Cooperberg wrote in an email. “The question is whether its use will increase appropriately.”
The challenge with focal therapy – regardless of energy modality – remains patient selection and accurate ablation zone definition, he added.
Notably, UCSF has launched a new HIFU program – and Dr. Cooperberg has referred selected patients. “I’m both enthusiastic and cautious about the future, and we need to track our outcomes very closely across various practice settings,” he said.
While waiting for CHRONOS, select wisely
The goal of focal therapy is to treat only the area with the most aggressive tumor, known as the index tumor, while leaving the remaining gland and its surrounding structures alone, according to Derek Lomas, MD, PharmD, a urologist at the Mayo Clinic in Rochester, Minn., in an explanatory article. “This approach is widely accepted in other types of cancer. For example, we commonly treat kidney cancers by removing or ablating only the tumor while leaving the rest of the kidney intact.”
However, some focal therapies also include approaches known as hemiablations, in which a full half of the prostate is destroyed, and approaches that leave very little of the gland behind.
Each of the modalities used for focal therapy has “unique indications, risks, and benefits and uses a different energy source for ablation,” Dr. Lebastchi and colleagues write in their essay.
They assert that focal therapy can provide oncological efficacy similar to radical prostatectomy or radiotherapy “while considerably reducing or even eliminating functional morbidities, such as incontinence and erectile dysfunction.”
Overall, they say focal therapy offers an opportunity for improved care because there is “an increasing body of emerging evidence demonstrating a favorable adverse effect profile with oncological control similar to whole-gland treatment options.”
What is that evidence?
In the essay, Dr. Lebastchi and colleagues point to a number of single-arm studies with encouraging efficacy and safety results. They also highlight a phase 3, randomized trial that they were involved in: This compared focal therapy (partial gland ablation with vascular-targeted photodynamic therapy) with active surveillance in early-stage disease and uniformly showed better post-treatment biopsy (disease/no disease) and conversion-to-prostatectomy results with the focal therapy out to 4 years (J Urol. 2018;200:786-793).
However, that study did not have an active treatment comparator. For that gold standard, there is now anticipation for results from the CHRONOS trial in the United Kingdom, especially part A of the trial, which compares radical therapy to focal therapy (HIFU or cryotherapy), with 5-year progression-free survival as the primary outcome. That trial is slated for completion in 2027.
Until then, the lack of prospective randomized clinical trials and long-term follow-up “hinders acceptance [of focal therapy] in the urology community,” the essay authors comment.
Meanwhile, careful patient selection is very important, they say.
The latest relevant guidelines state that appropriate candidates are men with a solitary, well-defined index lesion; patients with bilateral multifocal lesions; or very advanced tumors that are not appropriate for the focal approach.
A multidisciplinary international expert panel recently convened to establish guidance for clinicians offering focal therapies and then published a consensus statement to advise practitioners and researchers.
UCSF’s Dr. Cooperberg sees plenty of room for improvement among focal therapy practitioners and investigators. “From an outcomes standpoint, follow-up protocols and definitions of success remain inconsistent. I believe we’re making progress in all these areas, but we’re not there yet,” he says.
To date, some patients have been managed poorly, Dr. Cooperberg added. “We certainly see many patients who have been inadequately counseled as to HIFU’s advantages and disadvantages, with sometimes disastrous results.”
Some of those unfortunate results may have arisen from the U.S. Food and Drug Administration’s initial approval of HIFU in 2015, which was for use in ablating prostate tissue in general and not cancer specifically. This approval generated confusion, one expert commented at the time: “The FDA doesn’t specify whether it’s for benign or malignant disease; it’s a bit vague, like saying you can drive this car, but we’re not going to tell you how to drive it,” said Manoj Monga, MD, from the Cleveland Clinic.
Dr. Lebastchi has disclosed no relevant financial relationships; co-author Inderbir Gill, MD, is an unpaid consultant for Steba Biotech, and co-author Andre Luis Abreu, MD, is a consultant for Koelis and was a proctor in training for Steba Biotech. Dr. Cooperberg is a consultant for Alessa Therapeutics.
A version of this article first appeared on Medscape.com.
One in three cancer articles on social media has wrong info
Of the 200 most popular articles (50 each for prostate, lung, breast, and colorectal cancer), about a third (32.5%, n = 65) contained misinformation.
Among these articles containing misinformation, 76.9% (50/65) contained harmful information.
“The Internet is a leading source of health misinformation,” the study authors wrote. This is “particularly true for social media, where false information spreads faster and more broadly than fact-checked information,” they said, citing other research.
“We need to address these issues head on,” said lead author Skyler Johnson, MD, of the University of Utah’s Huntsman Cancer Institute in Salt Lake City.
“As a medical community, we can’t ignore the problem of cancer misinformation on social media or ask our patients to ignore it. We must empathize with our patients and help them when they encounter this type of information,” he said in a statement. “My goal is to help answer their questions, and provide cancer patients with accurate information that will give them the best chance for the best outcome.”
The study was published online July 22 in the Journal of the National Cancer Institute.
The study period ran from 2018 to 2019, and looked at articles posted on social media platforms Facebook, Reddit, Twitter, or Pinterest. Popularity was measured by engagement with readers, such as upvotes, comments, reactions, and shares.
Some of the articles came from long-established news entities such as CBS News, The New York Times, and medical journals, while others came from fleeting crowdfunding web pages and fledging nontraditional news sites.
One example of popular and harmful misinformation highlighted by Dr. Johnson in an interview was titled, “44-Year-Old Mother Claims CBD Oil Cured Her of Breast Cancer within 5 Months.” Posted on truththeory.com in February 2018, the article is tagged as “opinion” by the publisher and in turn links to another news story about the same woman in the UK’s Daily Mail newspaper.
The ideas and claims in such articles can be very influential, Jennifer L. Lycette, MD, suggested in a recent blog post.
“After 18 years as a cancer doctor, it sadly doesn’t come as a surprise anymore when a patient declines treatment recommendations and instead opts for ‘alternative’ treatment,” she wrote.
Sometimes, misinformation is not sensational but is still effective via clever wording and presentation, observed Brian G. Southwell, PhD, of Duke University, Durham, N.C., who has studied patients and misinformation.
“It isn’t the falsehood that is somehow magically attractive, per se, but the way that misinformation is often framed that can make it attractive,” he said in an interview.
Dr. Southwell recommends that clinicians be proactive about medical misinformation.
“Rather than expect patients to raise concerns without prompting, health care providers should invite conversations about potential misinformation with their patients,” he wrote in a recent essay in the American Journal of Public Health.
In short, ask patients what they know about the treatment of their cancer, he suggests.
“Patients don’t typically know that the misinformation they are encountering is misinformation,” said Dr. Southwell. “Approaching patients with compassion and empathy is a good first step.”
Study details
For the study, reported by Johnson et al., two National Comprehensive Cancer Network panel members were selected as content experts for each of the four cancers and were tasked with reviewing the primary medical claims in each article. The experts then completed a set of ratings to arrive at the proportion of misinformation and potential for harm in each article.
Of the 200 articles, 41.5% were from nontraditional news (digital only), 37.5% were from traditional news sources (online versions of print and/or broadcast media), 17% were from medical journals, 3% were from a crowdfunding site, and 1% were from personal blogs.
This expert review concluded that nearly one-third of the articles contained misinformation, as noted above. The misinformation was described as misleading (title not supported by text or statistics/data do not support conclusion, 28.8%), strength of the evidence mischaracterized (weak evidence portrayed as strong or vice versa, 27.7%) and unproven therapies (not studied or insufficient evidence, 26.7%).
Notably, the median number of engagements, such as likes on Twitter, for articles with misinformation was greater than that of factual articles (median, 2,300 vs. 1,600; P = .05).
In total, 30.5% of all 200 articles contained harmful information. This was described as harmful inaction (could lead to delay or not seeking medical attention for treatable/curable condition, 31.0%), economic harm (out-of-pocket financial costs associated with treatment/travel, 27.7%), harmful action (potentially toxic effects of the suggested test/treatment, 17.0%), and harmful interactions (known/unknown medical interactions with curative therapies, 16.2%).
The median number of engagements for articles with harmful information was statistically significantly greater than that of articles with correct information (median, 2,300 vs. 1,500; P = .007).
A limitation of the study is that it included only the most popular English language cancer articles.
This study was funded in part by the Huntsman Cancer Institute. Dr. Johnson, Dr. Lycette, and Dr. Southwell have disclosed no relevant financial relationships. Some study authors have ties to the pharmaceutical industry.
A version of this article first appeared on Medscape.com.
Of the 200 most popular articles (50 each for prostate, lung, breast, and colorectal cancer), about a third (32.5%, n = 65) contained misinformation.
Among these articles containing misinformation, 76.9% (50/65) contained harmful information.
“The Internet is a leading source of health misinformation,” the study authors wrote. This is “particularly true for social media, where false information spreads faster and more broadly than fact-checked information,” they said, citing other research.
“We need to address these issues head on,” said lead author Skyler Johnson, MD, of the University of Utah’s Huntsman Cancer Institute in Salt Lake City.
“As a medical community, we can’t ignore the problem of cancer misinformation on social media or ask our patients to ignore it. We must empathize with our patients and help them when they encounter this type of information,” he said in a statement. “My goal is to help answer their questions, and provide cancer patients with accurate information that will give them the best chance for the best outcome.”
The study was published online July 22 in the Journal of the National Cancer Institute.
The study period ran from 2018 to 2019, and looked at articles posted on social media platforms Facebook, Reddit, Twitter, or Pinterest. Popularity was measured by engagement with readers, such as upvotes, comments, reactions, and shares.
Some of the articles came from long-established news entities such as CBS News, The New York Times, and medical journals, while others came from fleeting crowdfunding web pages and fledging nontraditional news sites.
One example of popular and harmful misinformation highlighted by Dr. Johnson in an interview was titled, “44-Year-Old Mother Claims CBD Oil Cured Her of Breast Cancer within 5 Months.” Posted on truththeory.com in February 2018, the article is tagged as “opinion” by the publisher and in turn links to another news story about the same woman in the UK’s Daily Mail newspaper.
The ideas and claims in such articles can be very influential, Jennifer L. Lycette, MD, suggested in a recent blog post.
“After 18 years as a cancer doctor, it sadly doesn’t come as a surprise anymore when a patient declines treatment recommendations and instead opts for ‘alternative’ treatment,” she wrote.
Sometimes, misinformation is not sensational but is still effective via clever wording and presentation, observed Brian G. Southwell, PhD, of Duke University, Durham, N.C., who has studied patients and misinformation.
“It isn’t the falsehood that is somehow magically attractive, per se, but the way that misinformation is often framed that can make it attractive,” he said in an interview.
Dr. Southwell recommends that clinicians be proactive about medical misinformation.
“Rather than expect patients to raise concerns without prompting, health care providers should invite conversations about potential misinformation with their patients,” he wrote in a recent essay in the American Journal of Public Health.
In short, ask patients what they know about the treatment of their cancer, he suggests.
“Patients don’t typically know that the misinformation they are encountering is misinformation,” said Dr. Southwell. “Approaching patients with compassion and empathy is a good first step.”
Study details
For the study, reported by Johnson et al., two National Comprehensive Cancer Network panel members were selected as content experts for each of the four cancers and were tasked with reviewing the primary medical claims in each article. The experts then completed a set of ratings to arrive at the proportion of misinformation and potential for harm in each article.
Of the 200 articles, 41.5% were from nontraditional news (digital only), 37.5% were from traditional news sources (online versions of print and/or broadcast media), 17% were from medical journals, 3% were from a crowdfunding site, and 1% were from personal blogs.
This expert review concluded that nearly one-third of the articles contained misinformation, as noted above. The misinformation was described as misleading (title not supported by text or statistics/data do not support conclusion, 28.8%), strength of the evidence mischaracterized (weak evidence portrayed as strong or vice versa, 27.7%) and unproven therapies (not studied or insufficient evidence, 26.7%).
Notably, the median number of engagements, such as likes on Twitter, for articles with misinformation was greater than that of factual articles (median, 2,300 vs. 1,600; P = .05).
In total, 30.5% of all 200 articles contained harmful information. This was described as harmful inaction (could lead to delay or not seeking medical attention for treatable/curable condition, 31.0%), economic harm (out-of-pocket financial costs associated with treatment/travel, 27.7%), harmful action (potentially toxic effects of the suggested test/treatment, 17.0%), and harmful interactions (known/unknown medical interactions with curative therapies, 16.2%).
The median number of engagements for articles with harmful information was statistically significantly greater than that of articles with correct information (median, 2,300 vs. 1,500; P = .007).
A limitation of the study is that it included only the most popular English language cancer articles.
This study was funded in part by the Huntsman Cancer Institute. Dr. Johnson, Dr. Lycette, and Dr. Southwell have disclosed no relevant financial relationships. Some study authors have ties to the pharmaceutical industry.
A version of this article first appeared on Medscape.com.
Of the 200 most popular articles (50 each for prostate, lung, breast, and colorectal cancer), about a third (32.5%, n = 65) contained misinformation.
Among these articles containing misinformation, 76.9% (50/65) contained harmful information.
“The Internet is a leading source of health misinformation,” the study authors wrote. This is “particularly true for social media, where false information spreads faster and more broadly than fact-checked information,” they said, citing other research.
“We need to address these issues head on,” said lead author Skyler Johnson, MD, of the University of Utah’s Huntsman Cancer Institute in Salt Lake City.
“As a medical community, we can’t ignore the problem of cancer misinformation on social media or ask our patients to ignore it. We must empathize with our patients and help them when they encounter this type of information,” he said in a statement. “My goal is to help answer their questions, and provide cancer patients with accurate information that will give them the best chance for the best outcome.”
The study was published online July 22 in the Journal of the National Cancer Institute.
The study period ran from 2018 to 2019, and looked at articles posted on social media platforms Facebook, Reddit, Twitter, or Pinterest. Popularity was measured by engagement with readers, such as upvotes, comments, reactions, and shares.
Some of the articles came from long-established news entities such as CBS News, The New York Times, and medical journals, while others came from fleeting crowdfunding web pages and fledging nontraditional news sites.
One example of popular and harmful misinformation highlighted by Dr. Johnson in an interview was titled, “44-Year-Old Mother Claims CBD Oil Cured Her of Breast Cancer within 5 Months.” Posted on truththeory.com in February 2018, the article is tagged as “opinion” by the publisher and in turn links to another news story about the same woman in the UK’s Daily Mail newspaper.
The ideas and claims in such articles can be very influential, Jennifer L. Lycette, MD, suggested in a recent blog post.
“After 18 years as a cancer doctor, it sadly doesn’t come as a surprise anymore when a patient declines treatment recommendations and instead opts for ‘alternative’ treatment,” she wrote.
Sometimes, misinformation is not sensational but is still effective via clever wording and presentation, observed Brian G. Southwell, PhD, of Duke University, Durham, N.C., who has studied patients and misinformation.
“It isn’t the falsehood that is somehow magically attractive, per se, but the way that misinformation is often framed that can make it attractive,” he said in an interview.
Dr. Southwell recommends that clinicians be proactive about medical misinformation.
“Rather than expect patients to raise concerns without prompting, health care providers should invite conversations about potential misinformation with their patients,” he wrote in a recent essay in the American Journal of Public Health.
In short, ask patients what they know about the treatment of their cancer, he suggests.
“Patients don’t typically know that the misinformation they are encountering is misinformation,” said Dr. Southwell. “Approaching patients with compassion and empathy is a good first step.”
Study details
For the study, reported by Johnson et al., two National Comprehensive Cancer Network panel members were selected as content experts for each of the four cancers and were tasked with reviewing the primary medical claims in each article. The experts then completed a set of ratings to arrive at the proportion of misinformation and potential for harm in each article.
Of the 200 articles, 41.5% were from nontraditional news (digital only), 37.5% were from traditional news sources (online versions of print and/or broadcast media), 17% were from medical journals, 3% were from a crowdfunding site, and 1% were from personal blogs.
This expert review concluded that nearly one-third of the articles contained misinformation, as noted above. The misinformation was described as misleading (title not supported by text or statistics/data do not support conclusion, 28.8%), strength of the evidence mischaracterized (weak evidence portrayed as strong or vice versa, 27.7%) and unproven therapies (not studied or insufficient evidence, 26.7%).
Notably, the median number of engagements, such as likes on Twitter, for articles with misinformation was greater than that of factual articles (median, 2,300 vs. 1,600; P = .05).
In total, 30.5% of all 200 articles contained harmful information. This was described as harmful inaction (could lead to delay or not seeking medical attention for treatable/curable condition, 31.0%), economic harm (out-of-pocket financial costs associated with treatment/travel, 27.7%), harmful action (potentially toxic effects of the suggested test/treatment, 17.0%), and harmful interactions (known/unknown medical interactions with curative therapies, 16.2%).
The median number of engagements for articles with harmful information was statistically significantly greater than that of articles with correct information (median, 2,300 vs. 1,500; P = .007).
A limitation of the study is that it included only the most popular English language cancer articles.
This study was funded in part by the Huntsman Cancer Institute. Dr. Johnson, Dr. Lycette, and Dr. Southwell have disclosed no relevant financial relationships. Some study authors have ties to the pharmaceutical industry.
A version of this article first appeared on Medscape.com.
Bone drugs for prostate cancer may result in survival benefit
Results from a retrospective study show that the addition of bone resorption inhibitors (BRIs), including zoledronic acid and denosumab (Xgeva), to abiraterone plus prednisolone was associated with significantly longer overall survival (OS). The median OS was increased by nearly 9 months among recipients, compared with men who didn’t receive these drugs in this setting.
The findings were published online July 22, 2021, in JAMA Network Open.
All men with prostate cancer should receive BRIs “as the disease reaches the castration resistance with bone metastases stage, as recommended by the international guidelines,” lead author Edoardo Francini, MD, PhD, of the University of Florence (Italy) said in a comment.
While there is no evidence that BRIs – when used alone – may improve survival in metastatic castration-resistant prostate cancer (mCRPC) with bone involvement, there has been a “suggestion” of a survival benefit with BRIs when combined with other anticancer therapies in this setting, say the authors.
So Dr. Francini and a team of international coinvestigators looked at the medical records of men with mCRPC and bone metastases treated at eight institutions in Canada, Europe, and the United States and focused on patients who received abiraterone acetate (with prednisone) because it is the most common first-line therapy in this setting.
Patients were classified by receipt versus nonreceipt of concomitant BRIs and subclassified by volume of disease (high or low volume).
There were two cohorts in the study population: 529 men (71.0%) who received abiraterone alone and 216 men (29.0%) who received abiraterone plus BRIs. The median follow-up was 23.5 months.
Patients in the BRI cohort experienced significantly longer OS compared with those in the abiraterone alone cohort (31.8 vs. 23.0 months; hazard ratio, 0.65; P < .001).
Notably, the OS benefit in the BRI cohort was greater for patients with high-volume versus low-volume disease (33.6 vs. 19.7 months; HR, 0.51; P < .001).
Dr. Francini hopes the new study results can effect change. “Hopefully, clinicians will be more inclined to use bone resorption inhibitors in combination with abiraterone acetate plus prednisone as soon as the disease reaches the castration-resistance with bone metastases stage, as recommended by the international guidelines.”
Importance of bone-targeted drugs
“This study highlights the importance of bone-targeted therapy in current practice for men with mCRPC and bone metastases,” Samuel Takvorian, MD, and Naomi Haas, MD, of the University of Pennsylvania, Philadelphia, wrote in an accompanying editorial.
But the study also reveals that work needs to be done to get clinicians to prescribe BRIs, they said, and that clinical pathways and behavioral “nudges” could help promote adoption.
Most (71%) of the men in this study did not get bone protective drug therapy, they pointed out, even though they were being treated at major hospital systems.
So, why aren’t more men receiving BRIs?
“I think this is less likely due to poor communication from professional societies (the guidelines are clear) and more likely due to bone health being low on the list of priorities for these patients and clinician uncertainty and/or lack of appreciation of the clinical benefit of these agents,” Dr. Takvorian said in an interview.
“When prostate cancer progresses to the castration-resistant phase, clinicians (and patients) rightfully are focused on the next cancer-directed therapy. However, this may be at the expense of supportive care, like bone agents, which often gets short shrift,” he added.
As would be expected, the men who were taking BRIs had a significantly shorter time to first skeletal-related events (SREs), compared with those who were not (32.4 vs. 42.7 months; HR, 1.27; P = .04), and the risk of a first SRE was more than double in the subgroup with low-volume disease (HR, 2.29; P < .001).
“These SREs collectively represent a clinically meaningful outcome that is often measured in clinical trials,” the editorialists observed. In the current study, SREs were comprised of pathological fractures, spinal cord compression, or the need for surgery or radiotherapy to bone.
“Up to one-half of men with mCRPC, the advanced and often fatal stage of disease, experience SREs, which are associated with considerable morbidity, decreased survival, and increased health care utilization and costs,” they wrote.
Costly vs. inexpensive BRI
The study found no difference in the OS benefit between the different BRIs used, that is, between that seen with zoledronic acid versus denosumab.
The editorialists suggested that this finding is important, even though it “must be considered preliminary given the limitations of a retrospective study.” These results add “to data suggesting that these agents are comparably beneficial; thus, decisions between them should focus on clinical factors, such as kidney function, patient preference, and cost.”
The two agents differ mechanistically, they added, with zoledronic acid preferentially inhibiting osteoclast proliferation and denosumab inhibiting an important factor in osteoclast maturation.
In terms of having differentiating characteristics, the editorialists say that zoledronic acid is “more often associated with acute phase reactions and required monitoring of kidney function” while “denosumab conferred a higher risk of hypocalcemia.” Rates of osteonecrosis of the jaw are comparable.
International guidelines endorse the use of either agent for the treatment of men with mCRPC. But “some argue that the marginal benefit of denosumab must be weighed against its dramatically higher cost (the annual cost of zoledronic acid is approximately $140 vs. $29,000 for denosumab),” the editorialists said.
The dramatically higher cost of denosumab versus zoledronic acid has also been noted by other oncologists treating patients with other cancers, including multiple myeloma.
In addition to drug costs, there is another issue at stake: the prescribing oncologist is reimbursed by Medicare Part D at 6% for whichever drug is chosen, which represents a “financial conflict” for oncologists, said Vincent Rajkumar, MD, professor of medicine and a hematologist/oncologist at the Mayo Clinic, Rochester, Minn.
There is also a difference in how the drugs are administered, which may influence patient preference, the myeloma experts noted. Zoledronic acid is given intravenously every 3 months and requires a 15-minute infusion at a center, while denosumab needs to be given more frequently (every month) but is administered by subcutaneous injection.
Dr. Francini reported receiving grants from Roche Italia and personal fees for research travel from Janssen-Cilag outside the submitted work. A number of other authors disclosed financial ties to Janssen or Amgen, makers of abiraterone and denosumab, respectively. The editorialists reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Results from a retrospective study show that the addition of bone resorption inhibitors (BRIs), including zoledronic acid and denosumab (Xgeva), to abiraterone plus prednisolone was associated with significantly longer overall survival (OS). The median OS was increased by nearly 9 months among recipients, compared with men who didn’t receive these drugs in this setting.
The findings were published online July 22, 2021, in JAMA Network Open.
All men with prostate cancer should receive BRIs “as the disease reaches the castration resistance with bone metastases stage, as recommended by the international guidelines,” lead author Edoardo Francini, MD, PhD, of the University of Florence (Italy) said in a comment.
While there is no evidence that BRIs – when used alone – may improve survival in metastatic castration-resistant prostate cancer (mCRPC) with bone involvement, there has been a “suggestion” of a survival benefit with BRIs when combined with other anticancer therapies in this setting, say the authors.
So Dr. Francini and a team of international coinvestigators looked at the medical records of men with mCRPC and bone metastases treated at eight institutions in Canada, Europe, and the United States and focused on patients who received abiraterone acetate (with prednisone) because it is the most common first-line therapy in this setting.
Patients were classified by receipt versus nonreceipt of concomitant BRIs and subclassified by volume of disease (high or low volume).
There were two cohorts in the study population: 529 men (71.0%) who received abiraterone alone and 216 men (29.0%) who received abiraterone plus BRIs. The median follow-up was 23.5 months.
Patients in the BRI cohort experienced significantly longer OS compared with those in the abiraterone alone cohort (31.8 vs. 23.0 months; hazard ratio, 0.65; P < .001).
Notably, the OS benefit in the BRI cohort was greater for patients with high-volume versus low-volume disease (33.6 vs. 19.7 months; HR, 0.51; P < .001).
Dr. Francini hopes the new study results can effect change. “Hopefully, clinicians will be more inclined to use bone resorption inhibitors in combination with abiraterone acetate plus prednisone as soon as the disease reaches the castration-resistance with bone metastases stage, as recommended by the international guidelines.”
Importance of bone-targeted drugs
“This study highlights the importance of bone-targeted therapy in current practice for men with mCRPC and bone metastases,” Samuel Takvorian, MD, and Naomi Haas, MD, of the University of Pennsylvania, Philadelphia, wrote in an accompanying editorial.
But the study also reveals that work needs to be done to get clinicians to prescribe BRIs, they said, and that clinical pathways and behavioral “nudges” could help promote adoption.
Most (71%) of the men in this study did not get bone protective drug therapy, they pointed out, even though they were being treated at major hospital systems.
So, why aren’t more men receiving BRIs?
“I think this is less likely due to poor communication from professional societies (the guidelines are clear) and more likely due to bone health being low on the list of priorities for these patients and clinician uncertainty and/or lack of appreciation of the clinical benefit of these agents,” Dr. Takvorian said in an interview.
“When prostate cancer progresses to the castration-resistant phase, clinicians (and patients) rightfully are focused on the next cancer-directed therapy. However, this may be at the expense of supportive care, like bone agents, which often gets short shrift,” he added.
As would be expected, the men who were taking BRIs had a significantly shorter time to first skeletal-related events (SREs), compared with those who were not (32.4 vs. 42.7 months; HR, 1.27; P = .04), and the risk of a first SRE was more than double in the subgroup with low-volume disease (HR, 2.29; P < .001).
“These SREs collectively represent a clinically meaningful outcome that is often measured in clinical trials,” the editorialists observed. In the current study, SREs were comprised of pathological fractures, spinal cord compression, or the need for surgery or radiotherapy to bone.
“Up to one-half of men with mCRPC, the advanced and often fatal stage of disease, experience SREs, which are associated with considerable morbidity, decreased survival, and increased health care utilization and costs,” they wrote.
Costly vs. inexpensive BRI
The study found no difference in the OS benefit between the different BRIs used, that is, between that seen with zoledronic acid versus denosumab.
The editorialists suggested that this finding is important, even though it “must be considered preliminary given the limitations of a retrospective study.” These results add “to data suggesting that these agents are comparably beneficial; thus, decisions between them should focus on clinical factors, such as kidney function, patient preference, and cost.”
The two agents differ mechanistically, they added, with zoledronic acid preferentially inhibiting osteoclast proliferation and denosumab inhibiting an important factor in osteoclast maturation.
In terms of having differentiating characteristics, the editorialists say that zoledronic acid is “more often associated with acute phase reactions and required monitoring of kidney function” while “denosumab conferred a higher risk of hypocalcemia.” Rates of osteonecrosis of the jaw are comparable.
International guidelines endorse the use of either agent for the treatment of men with mCRPC. But “some argue that the marginal benefit of denosumab must be weighed against its dramatically higher cost (the annual cost of zoledronic acid is approximately $140 vs. $29,000 for denosumab),” the editorialists said.
The dramatically higher cost of denosumab versus zoledronic acid has also been noted by other oncologists treating patients with other cancers, including multiple myeloma.
In addition to drug costs, there is another issue at stake: the prescribing oncologist is reimbursed by Medicare Part D at 6% for whichever drug is chosen, which represents a “financial conflict” for oncologists, said Vincent Rajkumar, MD, professor of medicine and a hematologist/oncologist at the Mayo Clinic, Rochester, Minn.
There is also a difference in how the drugs are administered, which may influence patient preference, the myeloma experts noted. Zoledronic acid is given intravenously every 3 months and requires a 15-minute infusion at a center, while denosumab needs to be given more frequently (every month) but is administered by subcutaneous injection.
Dr. Francini reported receiving grants from Roche Italia and personal fees for research travel from Janssen-Cilag outside the submitted work. A number of other authors disclosed financial ties to Janssen or Amgen, makers of abiraterone and denosumab, respectively. The editorialists reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Results from a retrospective study show that the addition of bone resorption inhibitors (BRIs), including zoledronic acid and denosumab (Xgeva), to abiraterone plus prednisolone was associated with significantly longer overall survival (OS). The median OS was increased by nearly 9 months among recipients, compared with men who didn’t receive these drugs in this setting.
The findings were published online July 22, 2021, in JAMA Network Open.
All men with prostate cancer should receive BRIs “as the disease reaches the castration resistance with bone metastases stage, as recommended by the international guidelines,” lead author Edoardo Francini, MD, PhD, of the University of Florence (Italy) said in a comment.
While there is no evidence that BRIs – when used alone – may improve survival in metastatic castration-resistant prostate cancer (mCRPC) with bone involvement, there has been a “suggestion” of a survival benefit with BRIs when combined with other anticancer therapies in this setting, say the authors.
So Dr. Francini and a team of international coinvestigators looked at the medical records of men with mCRPC and bone metastases treated at eight institutions in Canada, Europe, and the United States and focused on patients who received abiraterone acetate (with prednisone) because it is the most common first-line therapy in this setting.
Patients were classified by receipt versus nonreceipt of concomitant BRIs and subclassified by volume of disease (high or low volume).
There were two cohorts in the study population: 529 men (71.0%) who received abiraterone alone and 216 men (29.0%) who received abiraterone plus BRIs. The median follow-up was 23.5 months.
Patients in the BRI cohort experienced significantly longer OS compared with those in the abiraterone alone cohort (31.8 vs. 23.0 months; hazard ratio, 0.65; P < .001).
Notably, the OS benefit in the BRI cohort was greater for patients with high-volume versus low-volume disease (33.6 vs. 19.7 months; HR, 0.51; P < .001).
Dr. Francini hopes the new study results can effect change. “Hopefully, clinicians will be more inclined to use bone resorption inhibitors in combination with abiraterone acetate plus prednisone as soon as the disease reaches the castration-resistance with bone metastases stage, as recommended by the international guidelines.”
Importance of bone-targeted drugs
“This study highlights the importance of bone-targeted therapy in current practice for men with mCRPC and bone metastases,” Samuel Takvorian, MD, and Naomi Haas, MD, of the University of Pennsylvania, Philadelphia, wrote in an accompanying editorial.
But the study also reveals that work needs to be done to get clinicians to prescribe BRIs, they said, and that clinical pathways and behavioral “nudges” could help promote adoption.
Most (71%) of the men in this study did not get bone protective drug therapy, they pointed out, even though they were being treated at major hospital systems.
So, why aren’t more men receiving BRIs?
“I think this is less likely due to poor communication from professional societies (the guidelines are clear) and more likely due to bone health being low on the list of priorities for these patients and clinician uncertainty and/or lack of appreciation of the clinical benefit of these agents,” Dr. Takvorian said in an interview.
“When prostate cancer progresses to the castration-resistant phase, clinicians (and patients) rightfully are focused on the next cancer-directed therapy. However, this may be at the expense of supportive care, like bone agents, which often gets short shrift,” he added.
As would be expected, the men who were taking BRIs had a significantly shorter time to first skeletal-related events (SREs), compared with those who were not (32.4 vs. 42.7 months; HR, 1.27; P = .04), and the risk of a first SRE was more than double in the subgroup with low-volume disease (HR, 2.29; P < .001).
“These SREs collectively represent a clinically meaningful outcome that is often measured in clinical trials,” the editorialists observed. In the current study, SREs were comprised of pathological fractures, spinal cord compression, or the need for surgery or radiotherapy to bone.
“Up to one-half of men with mCRPC, the advanced and often fatal stage of disease, experience SREs, which are associated with considerable morbidity, decreased survival, and increased health care utilization and costs,” they wrote.
Costly vs. inexpensive BRI
The study found no difference in the OS benefit between the different BRIs used, that is, between that seen with zoledronic acid versus denosumab.
The editorialists suggested that this finding is important, even though it “must be considered preliminary given the limitations of a retrospective study.” These results add “to data suggesting that these agents are comparably beneficial; thus, decisions between them should focus on clinical factors, such as kidney function, patient preference, and cost.”
The two agents differ mechanistically, they added, with zoledronic acid preferentially inhibiting osteoclast proliferation and denosumab inhibiting an important factor in osteoclast maturation.
In terms of having differentiating characteristics, the editorialists say that zoledronic acid is “more often associated with acute phase reactions and required monitoring of kidney function” while “denosumab conferred a higher risk of hypocalcemia.” Rates of osteonecrosis of the jaw are comparable.
International guidelines endorse the use of either agent for the treatment of men with mCRPC. But “some argue that the marginal benefit of denosumab must be weighed against its dramatically higher cost (the annual cost of zoledronic acid is approximately $140 vs. $29,000 for denosumab),” the editorialists said.
The dramatically higher cost of denosumab versus zoledronic acid has also been noted by other oncologists treating patients with other cancers, including multiple myeloma.
In addition to drug costs, there is another issue at stake: the prescribing oncologist is reimbursed by Medicare Part D at 6% for whichever drug is chosen, which represents a “financial conflict” for oncologists, said Vincent Rajkumar, MD, professor of medicine and a hematologist/oncologist at the Mayo Clinic, Rochester, Minn.
There is also a difference in how the drugs are administered, which may influence patient preference, the myeloma experts noted. Zoledronic acid is given intravenously every 3 months and requires a 15-minute infusion at a center, while denosumab needs to be given more frequently (every month) but is administered by subcutaneous injection.
Dr. Francini reported receiving grants from Roche Italia and personal fees for research travel from Janssen-Cilag outside the submitted work. A number of other authors disclosed financial ties to Janssen or Amgen, makers of abiraterone and denosumab, respectively. The editorialists reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Delayed Coronary Vasospasm in a Patient with Metastatic Gastric Cancer Receiving FOLFOX Therapy
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
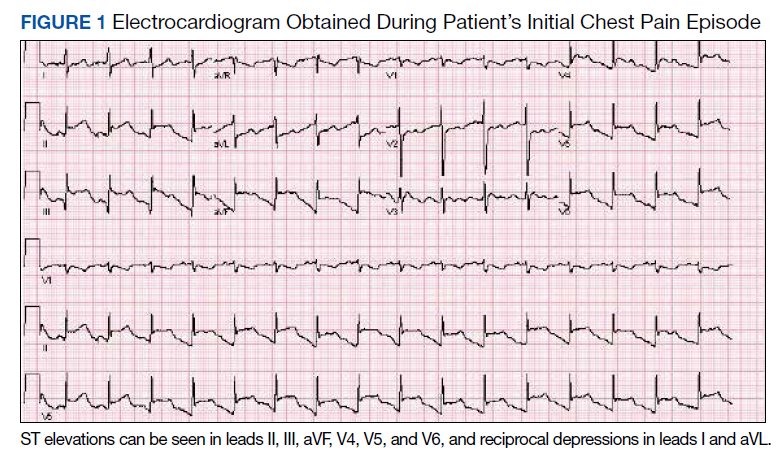
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
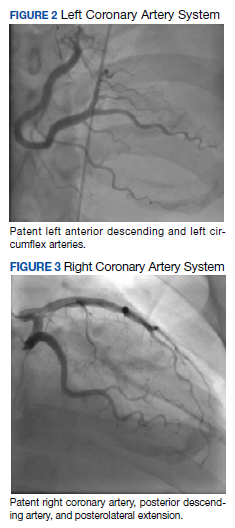
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
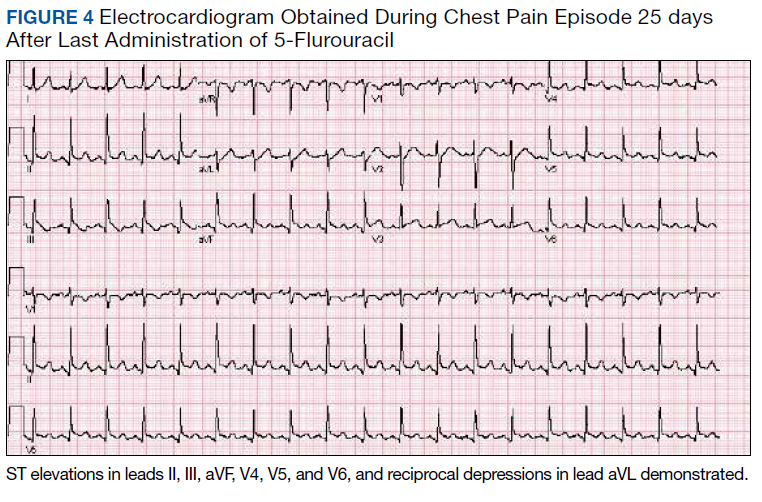
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877