User login
Placental allograft, cytology processor, cell-free RNA testing, and male infertility
Human placental allograft
For case reports involving Revita and for more information, visit https://www.stimlabs.com/revita.
FDA approval for cytology processor

For more information, visit: https://www.hologic.com/.
Cell-free RNA testing for pregnancy complications

Currently, Mirvie is recruiting for their Miracle of Life study, which requests that single gestation pregnant mothers who are not scheduled for cesarean delivery provide a blood sample during their second trimester. Women can see if they are eligible for study participation by visiting https://www.curebase.com/study/miracle/home.
For more information, visit: https://mirvie.com/.
Male fertility platform
![]()
For more information, visit: https://posterityhealth.com/.
Human placental allograft
For case reports involving Revita and for more information, visit https://www.stimlabs.com/revita.
FDA approval for cytology processor
For more information, visit: https://www.hologic.com/.
Cell-free RNA testing for pregnancy complications
Currently, Mirvie is recruiting for their Miracle of Life study, which requests that single gestation pregnant mothers who are not scheduled for cesarean delivery provide a blood sample during their second trimester. Women can see if they are eligible for study participation by visiting https://www.curebase.com/study/miracle/home.
For more information, visit: https://mirvie.com/.
Male fertility platform
![]()
For more information, visit: https://posterityhealth.com/.
Human placental allograft
For case reports involving Revita and for more information, visit https://www.stimlabs.com/revita.
FDA approval for cytology processor
For more information, visit: https://www.hologic.com/.
Cell-free RNA testing for pregnancy complications
Currently, Mirvie is recruiting for their Miracle of Life study, which requests that single gestation pregnant mothers who are not scheduled for cesarean delivery provide a blood sample during their second trimester. Women can see if they are eligible for study participation by visiting https://www.curebase.com/study/miracle/home.
For more information, visit: https://mirvie.com/.
Male fertility platform
![]()
For more information, visit: https://posterityhealth.com/.
Watchdog group demands removal of FDA leaders after aducanumab approval
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
OSHA issues new rules on COVID-19 safety for health care workers
The U.S. Occupational Safety and Health Administration issued its long-awaited Emergency Temporary Standard (ETS) for COVID-19 June 10, surprising many by including only health care workers in the new emergency workplace safety rules.
“The ETS is an overdue step toward protecting health care workers, especially those working in long-term care facilities and home health care who are at greatly increased risk of infection,” said George Washington University, Washington, professor and former Obama administration Assistant Secretary of Labor David Michaels, PhD, MPH. “OSHA’s failure to issue a COVID-specific standard in other high-risk industries, like meat and poultry processing, corrections, homeless shelters, and retail establishments is disappointing. If exposure is not controlled in these workplaces, they will continue to be important drivers of infections.”
With the new regulations in place, about 10.3 million health care workers at hospitals, nursing homes, and assisted living facilities, as well as emergency responders and home health care workers, should be guaranteed protection standards that replace former guidance.
The new protections include supplying personal protective equipment and ensuring proper usage (for example, mandatory seal checks on respirators); screening everyone who enters the facility for COVID-19; ensuring proper ventilation; and establishing physical distancing requirements (6 feet) for unvaccinated workers. It also requires employers to give workers time off for vaccination. An antiretaliation clause could shield workers who complain about unsafe conditions.
“The science tells us that health care workers, particularly those who come into regular contact with the virus, are most at risk at this point in the pandemic,” Labor Secretary Marty Walsh said on a press call. “So following an extensive review of the science and data, OSHA determined that a health care–specific safety requirement will make the biggest impact.”
But questions remain, said James Brudney, JD, a professor at Fordham Law School in New York and former chief counsel of the U.S. Senate Subcommittee on Labor. The standard doesn’t amplify or address existing rules regarding a right to refuse unsafe work, for example, so employees may still feel they are risking their jobs to complain, despite the antiretaliation clause.
And although vaccinated employees don’t have to adhere to the same distancing and masking standards in many instances, the standard doesn’t spell out how employers should determine their workers’ vaccination status – instead leaving that determination to employers through their own policies and procedures. (California’s state OSHA office rules specify the mechanism for documentation of vaccination.)
The Trump administration did not issue an ETS, saying OSHA’s general duty clause sufficed. President Joe Biden took the opposite approach, calling for an investigation into an ETS on his first day in office. But the process took months longer than promised.
“I know it’s been a long time coming,” Mr. Walsh acknowledged. “Our health care workers from the very beginning have been put at risk.
While health care unions had asked for mandated safety standards sooner, National Nurses United, the country’s largest labor union for registered nurses, still welcomed the rules.
“An ETS is a major step toward requiring accountability for hospitals who consistently put their budget goals and profits over our health and safety,” Zenei Triunfo-Cortez, RN, one of NNU’s three presidents, said in a statement June 9 anticipating the publication of the rules.
The rules do not apply to retail pharmacies, ambulatory care settings that screen nonemployees for COVID-19, or certain other settings in which all employees are vaccinated and people with suspected or confirmed COVID-19 cannot enter.
The agency said it will work with states that have already issued local regulations, including two states that issued temporary standards of their own, Virginia and California.
Employers will have 2 weeks to comply with most of the regulations after they’re published in the Federal Register. The standards will expire in 6 months but could then become permanent, as Virginia’s did in January.
A version of this article first appeared on Medscape.com.
The U.S. Occupational Safety and Health Administration issued its long-awaited Emergency Temporary Standard (ETS) for COVID-19 June 10, surprising many by including only health care workers in the new emergency workplace safety rules.
“The ETS is an overdue step toward protecting health care workers, especially those working in long-term care facilities and home health care who are at greatly increased risk of infection,” said George Washington University, Washington, professor and former Obama administration Assistant Secretary of Labor David Michaels, PhD, MPH. “OSHA’s failure to issue a COVID-specific standard in other high-risk industries, like meat and poultry processing, corrections, homeless shelters, and retail establishments is disappointing. If exposure is not controlled in these workplaces, they will continue to be important drivers of infections.”
With the new regulations in place, about 10.3 million health care workers at hospitals, nursing homes, and assisted living facilities, as well as emergency responders and home health care workers, should be guaranteed protection standards that replace former guidance.
The new protections include supplying personal protective equipment and ensuring proper usage (for example, mandatory seal checks on respirators); screening everyone who enters the facility for COVID-19; ensuring proper ventilation; and establishing physical distancing requirements (6 feet) for unvaccinated workers. It also requires employers to give workers time off for vaccination. An antiretaliation clause could shield workers who complain about unsafe conditions.
“The science tells us that health care workers, particularly those who come into regular contact with the virus, are most at risk at this point in the pandemic,” Labor Secretary Marty Walsh said on a press call. “So following an extensive review of the science and data, OSHA determined that a health care–specific safety requirement will make the biggest impact.”
But questions remain, said James Brudney, JD, a professor at Fordham Law School in New York and former chief counsel of the U.S. Senate Subcommittee on Labor. The standard doesn’t amplify or address existing rules regarding a right to refuse unsafe work, for example, so employees may still feel they are risking their jobs to complain, despite the antiretaliation clause.
And although vaccinated employees don’t have to adhere to the same distancing and masking standards in many instances, the standard doesn’t spell out how employers should determine their workers’ vaccination status – instead leaving that determination to employers through their own policies and procedures. (California’s state OSHA office rules specify the mechanism for documentation of vaccination.)
The Trump administration did not issue an ETS, saying OSHA’s general duty clause sufficed. President Joe Biden took the opposite approach, calling for an investigation into an ETS on his first day in office. But the process took months longer than promised.
“I know it’s been a long time coming,” Mr. Walsh acknowledged. “Our health care workers from the very beginning have been put at risk.
While health care unions had asked for mandated safety standards sooner, National Nurses United, the country’s largest labor union for registered nurses, still welcomed the rules.
“An ETS is a major step toward requiring accountability for hospitals who consistently put their budget goals and profits over our health and safety,” Zenei Triunfo-Cortez, RN, one of NNU’s three presidents, said in a statement June 9 anticipating the publication of the rules.
The rules do not apply to retail pharmacies, ambulatory care settings that screen nonemployees for COVID-19, or certain other settings in which all employees are vaccinated and people with suspected or confirmed COVID-19 cannot enter.
The agency said it will work with states that have already issued local regulations, including two states that issued temporary standards of their own, Virginia and California.
Employers will have 2 weeks to comply with most of the regulations after they’re published in the Federal Register. The standards will expire in 6 months but could then become permanent, as Virginia’s did in January.
A version of this article first appeared on Medscape.com.
The U.S. Occupational Safety and Health Administration issued its long-awaited Emergency Temporary Standard (ETS) for COVID-19 June 10, surprising many by including only health care workers in the new emergency workplace safety rules.
“The ETS is an overdue step toward protecting health care workers, especially those working in long-term care facilities and home health care who are at greatly increased risk of infection,” said George Washington University, Washington, professor and former Obama administration Assistant Secretary of Labor David Michaels, PhD, MPH. “OSHA’s failure to issue a COVID-specific standard in other high-risk industries, like meat and poultry processing, corrections, homeless shelters, and retail establishments is disappointing. If exposure is not controlled in these workplaces, they will continue to be important drivers of infections.”
With the new regulations in place, about 10.3 million health care workers at hospitals, nursing homes, and assisted living facilities, as well as emergency responders and home health care workers, should be guaranteed protection standards that replace former guidance.
The new protections include supplying personal protective equipment and ensuring proper usage (for example, mandatory seal checks on respirators); screening everyone who enters the facility for COVID-19; ensuring proper ventilation; and establishing physical distancing requirements (6 feet) for unvaccinated workers. It also requires employers to give workers time off for vaccination. An antiretaliation clause could shield workers who complain about unsafe conditions.
“The science tells us that health care workers, particularly those who come into regular contact with the virus, are most at risk at this point in the pandemic,” Labor Secretary Marty Walsh said on a press call. “So following an extensive review of the science and data, OSHA determined that a health care–specific safety requirement will make the biggest impact.”
But questions remain, said James Brudney, JD, a professor at Fordham Law School in New York and former chief counsel of the U.S. Senate Subcommittee on Labor. The standard doesn’t amplify or address existing rules regarding a right to refuse unsafe work, for example, so employees may still feel they are risking their jobs to complain, despite the antiretaliation clause.
And although vaccinated employees don’t have to adhere to the same distancing and masking standards in many instances, the standard doesn’t spell out how employers should determine their workers’ vaccination status – instead leaving that determination to employers through their own policies and procedures. (California’s state OSHA office rules specify the mechanism for documentation of vaccination.)
The Trump administration did not issue an ETS, saying OSHA’s general duty clause sufficed. President Joe Biden took the opposite approach, calling for an investigation into an ETS on his first day in office. But the process took months longer than promised.
“I know it’s been a long time coming,” Mr. Walsh acknowledged. “Our health care workers from the very beginning have been put at risk.
While health care unions had asked for mandated safety standards sooner, National Nurses United, the country’s largest labor union for registered nurses, still welcomed the rules.
“An ETS is a major step toward requiring accountability for hospitals who consistently put their budget goals and profits over our health and safety,” Zenei Triunfo-Cortez, RN, one of NNU’s three presidents, said in a statement June 9 anticipating the publication of the rules.
The rules do not apply to retail pharmacies, ambulatory care settings that screen nonemployees for COVID-19, or certain other settings in which all employees are vaccinated and people with suspected or confirmed COVID-19 cannot enter.
The agency said it will work with states that have already issued local regulations, including two states that issued temporary standards of their own, Virginia and California.
Employers will have 2 weeks to comply with most of the regulations after they’re published in the Federal Register. The standards will expire in 6 months but could then become permanent, as Virginia’s did in January.
A version of this article first appeared on Medscape.com.
Mistrust and Mandates: COVID-19 Vaccination in the Military
It is June and most of us are looking forward to a more normal summer than the one we had in 2020. Many Americans have been vaccinated and states are rolling back some (or all) masking requirements and restrictions on gatherings. In many sectors, including the US Department of Defense (DoD) and the US Department of Veterans Affairs (VA), worries from public health officials about vaccine supply and how to ethically allocate demand have given way to a new set of concerns: We have the shots, but for widespread protection we have to get them into arms.
The reluctance to roll up the sleeve is known as vaccine hesitancy. The National Academies of Science comments on vaccine hesitancy in its report on COVID-19 vaccination allocation. “Potential consequences of vaccine hesitancy—which the committee views as an attitude, preference, or motivational state—are the behaviors of vaccine refusal or delay.”2
On that count, there was encouraging albeit unexpected news in waning days of May. Media reported a sharp increase in the COVID-vaccination of military personnel. Unnamed DoD officials indicated, they had seen a 55% increase in the vaccination of active-duty service members over the previous month. This news represents a dramatic turnaround in a trend of vaccine hesitancy among military members that has persisted since the vaccine became available.3 Even last month, this would have been a very different column. The DoD has not disclosed the exact number of service members who have declined COVID-19 vaccination but multiple news outlets have documented that there was widespread and significant vaccine hesitancy among military personnel. In February, Military News reported that one-third of troops who were offered the vaccine declined it; and in April, USA Today stated that 40% of Marines had refused vaccination.4,5
Still, it is worth examining the data on vaccination among active duty service members. From December 2020 through March 2021, the military conducted the first study to evaluate rates of vaccine initiation and completion in the military in general and for service members from racial/ethnic minorities in particular. Black military personnel were 28% less likely than non-Hispanic White service members to initiate vaccination against coronavirus even after adjusting for other possible confounders. Just 29% of White, 25.5% of Hispanic, and 18.7% of Black service members had initiated the vaccine process in the survey.6
The authors suggest that in part, vaccine hesitancy explains the findings.4 Vaccine hesitancy among racial and ethnic minorities is even more tragic because these same already disadvantaged cohorts have disproportionately suffered from COVID-19 throughout the pandemic with higher rates of infection, serious illness requiring hospitalization, and infection-related morbidity.7
Vaccine hesitancy, delay, or refusal in Black Americans whether military or civilian often is attributed to the historical abuses like the Tuskegee syphilis experiments or the more recent example of cancer cell lines taken from Henrietta Lacks without consent.8 Such government sponsored betrayals no doubt are the soil in which hesitancy grows but recent commentators have opined that focusing solely on these infamous examples may ignore current systemic racism that is pervasively feeding Black Americans reluctance to consider or accept COVID-19 vaccination.9 Blaming infamous research also provides a convenient excuse for confronting contemporary racial discrimination in health care and taking responsibility as health care practitioners for reversing it. “Framing the conversation about distrust in COVID vaccines in terms of everyday racism rather than historical atrocities may increase underserved communities’ willingness to be vaccinated,” Bajaj and Stanford wrote in a recent recent New England Journal of Medicine commentary. “When we hyperfocus on Sims, Lacks, and Tuskegee, we ascribe the current Black health experience to past racism, rooting our present in immovable historical occurrences and undermining efforts to combat mistrust. Everyday racism, by contrast, can be tackled in the present.”9
The study of racial/ethnic disparities in COVID-19 vaccination in active-duty service members was a work product of the Armed Forces Health Surveillance Division. The authors underscore several factors that support the connection between discrimination and vaccine hesitancy in the military. Lack of access to and ability to obtain COVID-19 vaccination continues to be a major barrier that disadvantaged populations must overcome.10 The COVID-19 vaccine is widely available, easily obtained, and free of charge for all military personnel. Yet the vaccine hesitancy in the military parallels that of the civilian sector. This led the study authors to opine that, “forces external to the U.S. Military, such as interpersonal and societal factors also contribute to vaccine hesitancy among military service members.”6
Obviously, any unvaccinated active-duty service member reduces the combat readiness of the fighting force a consideration that led some in Congress to call for mandating vaccination. The vaccine is currently being administered under an emergency use authorization (EUA), which prevents even the military from mandating it.11 Even if President Joseph Biden obtained a waiver to make the vaccine mandatory, the implications of forcing service members who have volunteered to serve their country is ethically problematic. Those problems are exponentially amplified when applied to members of ethnic and racial minorities who have a past and present of health disparities and discrimination. Respecting the decision of those in uniform to decline COVID-19 vaccination is the first and perhaps most important step to rebuilding the trust that is the most promising means of reducing vaccine hesitancy.
Part of the accountability we all bear for health care inequity and racism is to continue the work of this landmark study to better understand vaccine hesitancy among military and veteran cohorts, develop counseling and education that target those attitudes, beliefs, and motivations with education, counseling, and support. All of us can in some small measure follow the ethical mandate “to dispel rumors and provide facts to people” of Secretary Austin, a Black retired 4-star Army general.1
1. Garmone J. Secretary of Defense Addresses Vaccine Hesitancy in the Military. Published February 25, 2021. Accessed May 26, 2021. https://www.defense.gov/Explore/News/Article/Article/2516511/secretary-of-defense-addresses-vaccine-hesitancy-in-military/
2. National Academies of Sciences, Engineering, and Medicine. Framework for Equitable Allocation of COVID-19 Vaccine . The National Academies of Science; 2020:188. doi:10.17226/25917
3. Liebermann O. US military sees 55% jump in COVID-19 vaccinations over last month. Published May 20, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/05/20/politics/us-military-covid-vaccinations/index.html
4. Kime P. Almost one-third of us troops are refusing COVID-19 vaccines, officials Say. Published February 17, 2021. Accessed May 26, 2021. https://www.military.com/daily-news/2021/02/17/almost-one-third-of-us-troops-are-refusing-covid-vaccines-officials-say.html
5. Elbeshbishi S. Nearly 40% of Marines decline COVID-19 vaccine, prompting some Democrats to urge Biden to set mandate for the military. USA Today. April 10, 2021. Accessed May 26, 2021. https://www.usatoday.com/story/news/politics/2021/04/10/covid-vaccine-nearly-forty-percent-us-marines-decline/7173918002/
6. Lang MA, Stahlman S, Wells NY, et al. Disparities in COVID-19 vaccine initiation and completion among active component service members and health care personnel, 11 December 2020-12 March 2021. MSMR. 2021;28(4):2-9.
7. Webb Hooper M, Nápoles AM, Pérez-Stable EJ. COVID-19 and racial/ethnic disparities. JAMA . 2020;323(24):2466-2467. doi:10.1001/jama.2020.8598
8. Kum D. Fueled by a history of mistreatment, Black Americans distrust the new COVID-19 vaccines. TIME. December 8, 2020. Accessed May 26, 2021.https://time.com/5925074/black-americans-covid-19-vaccine-distrust/
9. Bajaj SS, Stanford FC. Beyond Tuskegee - Vaccine Distrust and Everyday Racism. N Engl J Med. 2021;384(5):e12. doi:10.1056/NEJMpv2035827
10. Feldman N. Why Black and Latino people still lag on COVID-19 vaccines-and how to fix it. NPR. April 26, 2021. Accessed May 26, 2021. https://www.npr.org/sections/health-shots/2021/04/26/989962041/why-black-and-latino-people-still-lag-on-covid-vaccines-and-how-to-fix-it
11. Kaufman E. Lawmakers ask Biden to issue waiver to make COVID-19 vaccination mandatory of members of the military. Updated March 24, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/03/24/politics/congress-letter-military-vaccine/index.html
It is June and most of us are looking forward to a more normal summer than the one we had in 2020. Many Americans have been vaccinated and states are rolling back some (or all) masking requirements and restrictions on gatherings. In many sectors, including the US Department of Defense (DoD) and the US Department of Veterans Affairs (VA), worries from public health officials about vaccine supply and how to ethically allocate demand have given way to a new set of concerns: We have the shots, but for widespread protection we have to get them into arms.
The reluctance to roll up the sleeve is known as vaccine hesitancy. The National Academies of Science comments on vaccine hesitancy in its report on COVID-19 vaccination allocation. “Potential consequences of vaccine hesitancy—which the committee views as an attitude, preference, or motivational state—are the behaviors of vaccine refusal or delay.”2
On that count, there was encouraging albeit unexpected news in waning days of May. Media reported a sharp increase in the COVID-vaccination of military personnel. Unnamed DoD officials indicated, they had seen a 55% increase in the vaccination of active-duty service members over the previous month. This news represents a dramatic turnaround in a trend of vaccine hesitancy among military members that has persisted since the vaccine became available.3 Even last month, this would have been a very different column. The DoD has not disclosed the exact number of service members who have declined COVID-19 vaccination but multiple news outlets have documented that there was widespread and significant vaccine hesitancy among military personnel. In February, Military News reported that one-third of troops who were offered the vaccine declined it; and in April, USA Today stated that 40% of Marines had refused vaccination.4,5
Still, it is worth examining the data on vaccination among active duty service members. From December 2020 through March 2021, the military conducted the first study to evaluate rates of vaccine initiation and completion in the military in general and for service members from racial/ethnic minorities in particular. Black military personnel were 28% less likely than non-Hispanic White service members to initiate vaccination against coronavirus even after adjusting for other possible confounders. Just 29% of White, 25.5% of Hispanic, and 18.7% of Black service members had initiated the vaccine process in the survey.6
The authors suggest that in part, vaccine hesitancy explains the findings.4 Vaccine hesitancy among racial and ethnic minorities is even more tragic because these same already disadvantaged cohorts have disproportionately suffered from COVID-19 throughout the pandemic with higher rates of infection, serious illness requiring hospitalization, and infection-related morbidity.7
Vaccine hesitancy, delay, or refusal in Black Americans whether military or civilian often is attributed to the historical abuses like the Tuskegee syphilis experiments or the more recent example of cancer cell lines taken from Henrietta Lacks without consent.8 Such government sponsored betrayals no doubt are the soil in which hesitancy grows but recent commentators have opined that focusing solely on these infamous examples may ignore current systemic racism that is pervasively feeding Black Americans reluctance to consider or accept COVID-19 vaccination.9 Blaming infamous research also provides a convenient excuse for confronting contemporary racial discrimination in health care and taking responsibility as health care practitioners for reversing it. “Framing the conversation about distrust in COVID vaccines in terms of everyday racism rather than historical atrocities may increase underserved communities’ willingness to be vaccinated,” Bajaj and Stanford wrote in a recent recent New England Journal of Medicine commentary. “When we hyperfocus on Sims, Lacks, and Tuskegee, we ascribe the current Black health experience to past racism, rooting our present in immovable historical occurrences and undermining efforts to combat mistrust. Everyday racism, by contrast, can be tackled in the present.”9
The study of racial/ethnic disparities in COVID-19 vaccination in active-duty service members was a work product of the Armed Forces Health Surveillance Division. The authors underscore several factors that support the connection between discrimination and vaccine hesitancy in the military. Lack of access to and ability to obtain COVID-19 vaccination continues to be a major barrier that disadvantaged populations must overcome.10 The COVID-19 vaccine is widely available, easily obtained, and free of charge for all military personnel. Yet the vaccine hesitancy in the military parallels that of the civilian sector. This led the study authors to opine that, “forces external to the U.S. Military, such as interpersonal and societal factors also contribute to vaccine hesitancy among military service members.”6
Obviously, any unvaccinated active-duty service member reduces the combat readiness of the fighting force a consideration that led some in Congress to call for mandating vaccination. The vaccine is currently being administered under an emergency use authorization (EUA), which prevents even the military from mandating it.11 Even if President Joseph Biden obtained a waiver to make the vaccine mandatory, the implications of forcing service members who have volunteered to serve their country is ethically problematic. Those problems are exponentially amplified when applied to members of ethnic and racial minorities who have a past and present of health disparities and discrimination. Respecting the decision of those in uniform to decline COVID-19 vaccination is the first and perhaps most important step to rebuilding the trust that is the most promising means of reducing vaccine hesitancy.
Part of the accountability we all bear for health care inequity and racism is to continue the work of this landmark study to better understand vaccine hesitancy among military and veteran cohorts, develop counseling and education that target those attitudes, beliefs, and motivations with education, counseling, and support. All of us can in some small measure follow the ethical mandate “to dispel rumors and provide facts to people” of Secretary Austin, a Black retired 4-star Army general.1
It is June and most of us are looking forward to a more normal summer than the one we had in 2020. Many Americans have been vaccinated and states are rolling back some (or all) masking requirements and restrictions on gatherings. In many sectors, including the US Department of Defense (DoD) and the US Department of Veterans Affairs (VA), worries from public health officials about vaccine supply and how to ethically allocate demand have given way to a new set of concerns: We have the shots, but for widespread protection we have to get them into arms.
The reluctance to roll up the sleeve is known as vaccine hesitancy. The National Academies of Science comments on vaccine hesitancy in its report on COVID-19 vaccination allocation. “Potential consequences of vaccine hesitancy—which the committee views as an attitude, preference, or motivational state—are the behaviors of vaccine refusal or delay.”2
On that count, there was encouraging albeit unexpected news in waning days of May. Media reported a sharp increase in the COVID-vaccination of military personnel. Unnamed DoD officials indicated, they had seen a 55% increase in the vaccination of active-duty service members over the previous month. This news represents a dramatic turnaround in a trend of vaccine hesitancy among military members that has persisted since the vaccine became available.3 Even last month, this would have been a very different column. The DoD has not disclosed the exact number of service members who have declined COVID-19 vaccination but multiple news outlets have documented that there was widespread and significant vaccine hesitancy among military personnel. In February, Military News reported that one-third of troops who were offered the vaccine declined it; and in April, USA Today stated that 40% of Marines had refused vaccination.4,5
Still, it is worth examining the data on vaccination among active duty service members. From December 2020 through March 2021, the military conducted the first study to evaluate rates of vaccine initiation and completion in the military in general and for service members from racial/ethnic minorities in particular. Black military personnel were 28% less likely than non-Hispanic White service members to initiate vaccination against coronavirus even after adjusting for other possible confounders. Just 29% of White, 25.5% of Hispanic, and 18.7% of Black service members had initiated the vaccine process in the survey.6
The authors suggest that in part, vaccine hesitancy explains the findings.4 Vaccine hesitancy among racial and ethnic minorities is even more tragic because these same already disadvantaged cohorts have disproportionately suffered from COVID-19 throughout the pandemic with higher rates of infection, serious illness requiring hospitalization, and infection-related morbidity.7
Vaccine hesitancy, delay, or refusal in Black Americans whether military or civilian often is attributed to the historical abuses like the Tuskegee syphilis experiments or the more recent example of cancer cell lines taken from Henrietta Lacks without consent.8 Such government sponsored betrayals no doubt are the soil in which hesitancy grows but recent commentators have opined that focusing solely on these infamous examples may ignore current systemic racism that is pervasively feeding Black Americans reluctance to consider or accept COVID-19 vaccination.9 Blaming infamous research also provides a convenient excuse for confronting contemporary racial discrimination in health care and taking responsibility as health care practitioners for reversing it. “Framing the conversation about distrust in COVID vaccines in terms of everyday racism rather than historical atrocities may increase underserved communities’ willingness to be vaccinated,” Bajaj and Stanford wrote in a recent recent New England Journal of Medicine commentary. “When we hyperfocus on Sims, Lacks, and Tuskegee, we ascribe the current Black health experience to past racism, rooting our present in immovable historical occurrences and undermining efforts to combat mistrust. Everyday racism, by contrast, can be tackled in the present.”9
The study of racial/ethnic disparities in COVID-19 vaccination in active-duty service members was a work product of the Armed Forces Health Surveillance Division. The authors underscore several factors that support the connection between discrimination and vaccine hesitancy in the military. Lack of access to and ability to obtain COVID-19 vaccination continues to be a major barrier that disadvantaged populations must overcome.10 The COVID-19 vaccine is widely available, easily obtained, and free of charge for all military personnel. Yet the vaccine hesitancy in the military parallels that of the civilian sector. This led the study authors to opine that, “forces external to the U.S. Military, such as interpersonal and societal factors also contribute to vaccine hesitancy among military service members.”6
Obviously, any unvaccinated active-duty service member reduces the combat readiness of the fighting force a consideration that led some in Congress to call for mandating vaccination. The vaccine is currently being administered under an emergency use authorization (EUA), which prevents even the military from mandating it.11 Even if President Joseph Biden obtained a waiver to make the vaccine mandatory, the implications of forcing service members who have volunteered to serve their country is ethically problematic. Those problems are exponentially amplified when applied to members of ethnic and racial minorities who have a past and present of health disparities and discrimination. Respecting the decision of those in uniform to decline COVID-19 vaccination is the first and perhaps most important step to rebuilding the trust that is the most promising means of reducing vaccine hesitancy.
Part of the accountability we all bear for health care inequity and racism is to continue the work of this landmark study to better understand vaccine hesitancy among military and veteran cohorts, develop counseling and education that target those attitudes, beliefs, and motivations with education, counseling, and support. All of us can in some small measure follow the ethical mandate “to dispel rumors and provide facts to people” of Secretary Austin, a Black retired 4-star Army general.1
1. Garmone J. Secretary of Defense Addresses Vaccine Hesitancy in the Military. Published February 25, 2021. Accessed May 26, 2021. https://www.defense.gov/Explore/News/Article/Article/2516511/secretary-of-defense-addresses-vaccine-hesitancy-in-military/
2. National Academies of Sciences, Engineering, and Medicine. Framework for Equitable Allocation of COVID-19 Vaccine . The National Academies of Science; 2020:188. doi:10.17226/25917
3. Liebermann O. US military sees 55% jump in COVID-19 vaccinations over last month. Published May 20, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/05/20/politics/us-military-covid-vaccinations/index.html
4. Kime P. Almost one-third of us troops are refusing COVID-19 vaccines, officials Say. Published February 17, 2021. Accessed May 26, 2021. https://www.military.com/daily-news/2021/02/17/almost-one-third-of-us-troops-are-refusing-covid-vaccines-officials-say.html
5. Elbeshbishi S. Nearly 40% of Marines decline COVID-19 vaccine, prompting some Democrats to urge Biden to set mandate for the military. USA Today. April 10, 2021. Accessed May 26, 2021. https://www.usatoday.com/story/news/politics/2021/04/10/covid-vaccine-nearly-forty-percent-us-marines-decline/7173918002/
6. Lang MA, Stahlman S, Wells NY, et al. Disparities in COVID-19 vaccine initiation and completion among active component service members and health care personnel, 11 December 2020-12 March 2021. MSMR. 2021;28(4):2-9.
7. Webb Hooper M, Nápoles AM, Pérez-Stable EJ. COVID-19 and racial/ethnic disparities. JAMA . 2020;323(24):2466-2467. doi:10.1001/jama.2020.8598
8. Kum D. Fueled by a history of mistreatment, Black Americans distrust the new COVID-19 vaccines. TIME. December 8, 2020. Accessed May 26, 2021.https://time.com/5925074/black-americans-covid-19-vaccine-distrust/
9. Bajaj SS, Stanford FC. Beyond Tuskegee - Vaccine Distrust and Everyday Racism. N Engl J Med. 2021;384(5):e12. doi:10.1056/NEJMpv2035827
10. Feldman N. Why Black and Latino people still lag on COVID-19 vaccines-and how to fix it. NPR. April 26, 2021. Accessed May 26, 2021. https://www.npr.org/sections/health-shots/2021/04/26/989962041/why-black-and-latino-people-still-lag-on-covid-vaccines-and-how-to-fix-it
11. Kaufman E. Lawmakers ask Biden to issue waiver to make COVID-19 vaccination mandatory of members of the military. Updated March 24, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/03/24/politics/congress-letter-military-vaccine/index.html
1. Garmone J. Secretary of Defense Addresses Vaccine Hesitancy in the Military. Published February 25, 2021. Accessed May 26, 2021. https://www.defense.gov/Explore/News/Article/Article/2516511/secretary-of-defense-addresses-vaccine-hesitancy-in-military/
2. National Academies of Sciences, Engineering, and Medicine. Framework for Equitable Allocation of COVID-19 Vaccine . The National Academies of Science; 2020:188. doi:10.17226/25917
3. Liebermann O. US military sees 55% jump in COVID-19 vaccinations over last month. Published May 20, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/05/20/politics/us-military-covid-vaccinations/index.html
4. Kime P. Almost one-third of us troops are refusing COVID-19 vaccines, officials Say. Published February 17, 2021. Accessed May 26, 2021. https://www.military.com/daily-news/2021/02/17/almost-one-third-of-us-troops-are-refusing-covid-vaccines-officials-say.html
5. Elbeshbishi S. Nearly 40% of Marines decline COVID-19 vaccine, prompting some Democrats to urge Biden to set mandate for the military. USA Today. April 10, 2021. Accessed May 26, 2021. https://www.usatoday.com/story/news/politics/2021/04/10/covid-vaccine-nearly-forty-percent-us-marines-decline/7173918002/
6. Lang MA, Stahlman S, Wells NY, et al. Disparities in COVID-19 vaccine initiation and completion among active component service members and health care personnel, 11 December 2020-12 March 2021. MSMR. 2021;28(4):2-9.
7. Webb Hooper M, Nápoles AM, Pérez-Stable EJ. COVID-19 and racial/ethnic disparities. JAMA . 2020;323(24):2466-2467. doi:10.1001/jama.2020.8598
8. Kum D. Fueled by a history of mistreatment, Black Americans distrust the new COVID-19 vaccines. TIME. December 8, 2020. Accessed May 26, 2021.https://time.com/5925074/black-americans-covid-19-vaccine-distrust/
9. Bajaj SS, Stanford FC. Beyond Tuskegee - Vaccine Distrust and Everyday Racism. N Engl J Med. 2021;384(5):e12. doi:10.1056/NEJMpv2035827
10. Feldman N. Why Black and Latino people still lag on COVID-19 vaccines-and how to fix it. NPR. April 26, 2021. Accessed May 26, 2021. https://www.npr.org/sections/health-shots/2021/04/26/989962041/why-black-and-latino-people-still-lag-on-covid-vaccines-and-how-to-fix-it
11. Kaufman E. Lawmakers ask Biden to issue waiver to make COVID-19 vaccination mandatory of members of the military. Updated March 24, 2021. Accessed May 26, 2021. https://www.cnn.com/2021/03/24/politics/congress-letter-military-vaccine/index.html
CDC director cites rise in hospitalizations in urging teen vaccinations
“I am deeply concerned by the numbers of hospitalized adolescents and saddened to see the number of adolescents who required treatment in intensive care units or mechanical ventilation,” CDC Director Rochelle Walensky, MD, said in a statement.
While urging teenagers to wear masks and take precautions around others, she asked “parents, relatives, and close friends to join me and talk with teens about the importance of these prevention strategies and to encourage them to get vaccinated.”
Dr. Walensky referred to the CDC’s Morbidity and Mortality Weekly Report that showed adolescent hospitalizations peaked at 2.1 per 100,000 in early January 2021, then dropped to 0.6 per 100,000 in mid-March.
Alarmingly, hospitalizations rose to 1.3 per 100,000 in April, and a number of teens required serious interventions.
“Among hospitalized adolescents, nearly one-third required intensive care unit admission, and 5% required invasive mechanical ventilation,” the report said. No deaths occurred.
The study looked at 376 adolescents aged 12-17 who were hospitalized and tested positive for coronavirus. Of that group, 204 were hospitalized for COVID-19 and the other 172 were hospitalized for reasons not directly related to COVID-19.
Of the 204 hospitalized for COVID-19, 70.6% had an underlying medical condition such as obesity or chronic lung disease.
The study noted that children and teenagers have lower hospitalization rates and generally show less severe symptoms than do older people.
Possible causes for the rise in adolescent COVID-19 hospitalizations include the arrival of variants, the growing number of children returning to in-person education, and the changes in mask-wearing and other safety precautions, the study said.
The American Academy of Pediatrics said that as of May 27, 4 million children have tested positive for COVID-19 since the pandemic began, with about 34,500 new child cases reported for the week ending May 27.
The AAP said children have represented 14.1% of total cases since the pandemic began, but for the week ending May 27, children represented 24.3% of new reported weekly COVID-19 cases.
On May 10, the FDA granted emergency use authorization for the Pfizer coronavirus vaccine to be given to children aged 12-15 years. Previously, the FDA had authorized the Pfizer vaccine for people aged 16 years and up, whereas the Moderna and Johnson & Johnson vaccines are authorized for people aged 18 years and up.
“Vaccination is our way out of this pandemic,” Dr. Walensky said in her statement. “I continue to see promising signs in CDC data that we are nearing the end of this pandemic in this country; however, we all have to do our part and get vaccinated to cross the finish line.”
A version of this article was first published on WebMD.com.
“I am deeply concerned by the numbers of hospitalized adolescents and saddened to see the number of adolescents who required treatment in intensive care units or mechanical ventilation,” CDC Director Rochelle Walensky, MD, said in a statement.
While urging teenagers to wear masks and take precautions around others, she asked “parents, relatives, and close friends to join me and talk with teens about the importance of these prevention strategies and to encourage them to get vaccinated.”
Dr. Walensky referred to the CDC’s Morbidity and Mortality Weekly Report that showed adolescent hospitalizations peaked at 2.1 per 100,000 in early January 2021, then dropped to 0.6 per 100,000 in mid-March.
Alarmingly, hospitalizations rose to 1.3 per 100,000 in April, and a number of teens required serious interventions.
“Among hospitalized adolescents, nearly one-third required intensive care unit admission, and 5% required invasive mechanical ventilation,” the report said. No deaths occurred.
The study looked at 376 adolescents aged 12-17 who were hospitalized and tested positive for coronavirus. Of that group, 204 were hospitalized for COVID-19 and the other 172 were hospitalized for reasons not directly related to COVID-19.
Of the 204 hospitalized for COVID-19, 70.6% had an underlying medical condition such as obesity or chronic lung disease.
The study noted that children and teenagers have lower hospitalization rates and generally show less severe symptoms than do older people.
Possible causes for the rise in adolescent COVID-19 hospitalizations include the arrival of variants, the growing number of children returning to in-person education, and the changes in mask-wearing and other safety precautions, the study said.
The American Academy of Pediatrics said that as of May 27, 4 million children have tested positive for COVID-19 since the pandemic began, with about 34,500 new child cases reported for the week ending May 27.
The AAP said children have represented 14.1% of total cases since the pandemic began, but for the week ending May 27, children represented 24.3% of new reported weekly COVID-19 cases.
On May 10, the FDA granted emergency use authorization for the Pfizer coronavirus vaccine to be given to children aged 12-15 years. Previously, the FDA had authorized the Pfizer vaccine for people aged 16 years and up, whereas the Moderna and Johnson & Johnson vaccines are authorized for people aged 18 years and up.
“Vaccination is our way out of this pandemic,” Dr. Walensky said in her statement. “I continue to see promising signs in CDC data that we are nearing the end of this pandemic in this country; however, we all have to do our part and get vaccinated to cross the finish line.”
A version of this article was first published on WebMD.com.
“I am deeply concerned by the numbers of hospitalized adolescents and saddened to see the number of adolescents who required treatment in intensive care units or mechanical ventilation,” CDC Director Rochelle Walensky, MD, said in a statement.
While urging teenagers to wear masks and take precautions around others, she asked “parents, relatives, and close friends to join me and talk with teens about the importance of these prevention strategies and to encourage them to get vaccinated.”
Dr. Walensky referred to the CDC’s Morbidity and Mortality Weekly Report that showed adolescent hospitalizations peaked at 2.1 per 100,000 in early January 2021, then dropped to 0.6 per 100,000 in mid-March.
Alarmingly, hospitalizations rose to 1.3 per 100,000 in April, and a number of teens required serious interventions.
“Among hospitalized adolescents, nearly one-third required intensive care unit admission, and 5% required invasive mechanical ventilation,” the report said. No deaths occurred.
The study looked at 376 adolescents aged 12-17 who were hospitalized and tested positive for coronavirus. Of that group, 204 were hospitalized for COVID-19 and the other 172 were hospitalized for reasons not directly related to COVID-19.
Of the 204 hospitalized for COVID-19, 70.6% had an underlying medical condition such as obesity or chronic lung disease.
The study noted that children and teenagers have lower hospitalization rates and generally show less severe symptoms than do older people.
Possible causes for the rise in adolescent COVID-19 hospitalizations include the arrival of variants, the growing number of children returning to in-person education, and the changes in mask-wearing and other safety precautions, the study said.
The American Academy of Pediatrics said that as of May 27, 4 million children have tested positive for COVID-19 since the pandemic began, with about 34,500 new child cases reported for the week ending May 27.
The AAP said children have represented 14.1% of total cases since the pandemic began, but for the week ending May 27, children represented 24.3% of new reported weekly COVID-19 cases.
On May 10, the FDA granted emergency use authorization for the Pfizer coronavirus vaccine to be given to children aged 12-15 years. Previously, the FDA had authorized the Pfizer vaccine for people aged 16 years and up, whereas the Moderna and Johnson & Johnson vaccines are authorized for people aged 18 years and up.
“Vaccination is our way out of this pandemic,” Dr. Walensky said in her statement. “I continue to see promising signs in CDC data that we are nearing the end of this pandemic in this country; however, we all have to do our part and get vaccinated to cross the finish line.”
A version of this article was first published on WebMD.com.
CDC: New botulism guidelines focus on mass casualty events
Botulinum toxin is said to be the most lethal substance known. Inhaling just 1-3 nanograms of toxin per kilogram of body mass constitutes a lethal dose.
The CDC has been working on these guidelines since 2015, initially establishing a technical development group and steering committee to prioritize topics for review and make recommendations. Since then, the agency published 15 systematic reviews in Clinical Infectious Diseases early in 2018. The reviews addressed the recognition of botulism clinically, treatment with botulinum antitoxin, and complications from that treatment. They also looked at the epidemiology of botulism outbreaks and botulism in the special populations of vulnerable pediatric and pregnant patients.
In 2016, the CDC held two extended forums and convened a workshop with 72 experts. In addition to the more standard topics of diagnosis and treatment, attention was given to crisis standards of care, caring for multiple patients at once, and ethical considerations in management.
Amesh Adalja, MD, senior scholar, Johns Hopkins Center for Health Security, Baltimore, said in an interview that the new guidance “was really specific [and] was meant to address the gap in guidance for mass casualty settings.”
While clinicians are used to focusing on an individual patient, in times of crises, with multiple patients from a food-borne outbreak or a bioterrorism attack, the focus must shift to the population rather than the individual. The workshop explored issues of triaging, adding beds, and caring for patients when a hospital is overwhelmed with an acute influx of severely ill patients.
Such a mass casualty event is similar to the stress encountered this past year with COVID-19 patients swamping the hospitals, which had too little oxygen, too few ventilators, and too few staff members to care for the sudden influx of critically ill patients.
Diagnosis
Leslie Edwards, MHS, BSN, a CDC epidemiologist and botulism expert, said that “botulism is rare and [so] could be difficult to diagnose.” The CDC “wanted to highlight some of those key clinical factors” to speed recognition.
Hospitals and health officials are being urged to develop crisis protocols as part of emergency preparedness plans. And clinicians should be able to recognize four major syndromes: botulism from food, wounds, and inhalation, as well as iatrogenic botulism (from exposure via injection of the neurotoxin).
Botulism has a characteristic and unusual pattern of symptoms, which begin with cranial nerve palsies. Then there is typically a descending, symmetric flaccid paralysis. Symptoms might progress to respiratory failure and death. Other critical clues that implicate botulism include a lack of sensory deficits and the absence of pain.
Symptoms are most likely to be mistaken for myasthenia gravis or Guillain-Barré syndrome, but the latter has an ascending paralysis. Cranial nerve involvement can present as blurred vision, ptosis (drooping lid), diplopia (double vision), ophthalmoplegia (weak eye muscles), or difficulty with speech and swallowing. Shortness of breath and abdominal discomfort can also occur. Respiratory failure may occur from weakness or paralysis of cranial nerves. Cranial nerve signs and symptoms in the absence of fever, along with a descending paralysis, should strongly suggest the diagnosis.
With food-borne botulism, vomiting occurs in half the patients. Improperly sterilized home-canned food is the major risk factor. While the toxin is rapidly destroyed by heat, the bacterial spores are not. Wound botulism is most commonly associated with the injection of drugs, particularly black tar heroin.
Dr. Edwards stressed that “time is of the essence when it comes to botulism diagnostics and treating. Timely administration of the botulism antitoxin early in the course of illness can arrest the progression of paralysis and possibly avert the need for intubation or ventilation.”
It’s essential to note that botulism is an urgent diagnosis that has to be made on clinical grounds. Lab assays for botulinum neurotoxins take too long and are only conducted in public health laboratories. The decision to use antitoxin must not be delayed to wait for confirmation.
Clinicians should immediately contact the local or state health department’s emergency on-call team if botulism is suspected. They will arrange for expert consultation.
Treatment
Botulinum antitoxin is the only specific therapy for this infection. If given early – preferably within 24-48 hours of symptom onset – it can stop the progression of paralysis. But antitoxin will not reverse existing paralysis. If paralysis is still progressing outside of that 24- to 48-hour window, the antitoxin should still provide benefit. The antitoxin is available only through state health departments and a request to the CDC.
Botulism antitoxin is made from horse serum and therefore may cause a variety of allergic reactions. The risk for anaphylaxis is less than 2%, far lower than the mortality from untreated botulism.
While these guidelines have an important focus on triaging and treating mass casualties from botulism, it’s important to note that food-borne outbreaks and prevention issues are covered elsewhere on the CDC site.
Dr. Edwards has disclosed no relevant financial relationships. Dr. Adalja is a consultant for Emergent BioSolutions, which makes the heptavalent botulism antitoxin.
Dr. Stone is an infectious disease specialist and author of “Resilience: One Family’s Story of Hope and Triumph Over Evil” and of “Conducting Clinical Research,” the essential guide to the topic. You can find her at drjudystone.com or on Twitter @drjudystone.
A version of this article first appeared on Medscape.com.
Botulinum toxin is said to be the most lethal substance known. Inhaling just 1-3 nanograms of toxin per kilogram of body mass constitutes a lethal dose.
The CDC has been working on these guidelines since 2015, initially establishing a technical development group and steering committee to prioritize topics for review and make recommendations. Since then, the agency published 15 systematic reviews in Clinical Infectious Diseases early in 2018. The reviews addressed the recognition of botulism clinically, treatment with botulinum antitoxin, and complications from that treatment. They also looked at the epidemiology of botulism outbreaks and botulism in the special populations of vulnerable pediatric and pregnant patients.
In 2016, the CDC held two extended forums and convened a workshop with 72 experts. In addition to the more standard topics of diagnosis and treatment, attention was given to crisis standards of care, caring for multiple patients at once, and ethical considerations in management.
Amesh Adalja, MD, senior scholar, Johns Hopkins Center for Health Security, Baltimore, said in an interview that the new guidance “was really specific [and] was meant to address the gap in guidance for mass casualty settings.”
While clinicians are used to focusing on an individual patient, in times of crises, with multiple patients from a food-borne outbreak or a bioterrorism attack, the focus must shift to the population rather than the individual. The workshop explored issues of triaging, adding beds, and caring for patients when a hospital is overwhelmed with an acute influx of severely ill patients.
Such a mass casualty event is similar to the stress encountered this past year with COVID-19 patients swamping the hospitals, which had too little oxygen, too few ventilators, and too few staff members to care for the sudden influx of critically ill patients.
Diagnosis
Leslie Edwards, MHS, BSN, a CDC epidemiologist and botulism expert, said that “botulism is rare and [so] could be difficult to diagnose.” The CDC “wanted to highlight some of those key clinical factors” to speed recognition.
Hospitals and health officials are being urged to develop crisis protocols as part of emergency preparedness plans. And clinicians should be able to recognize four major syndromes: botulism from food, wounds, and inhalation, as well as iatrogenic botulism (from exposure via injection of the neurotoxin).
Botulism has a characteristic and unusual pattern of symptoms, which begin with cranial nerve palsies. Then there is typically a descending, symmetric flaccid paralysis. Symptoms might progress to respiratory failure and death. Other critical clues that implicate botulism include a lack of sensory deficits and the absence of pain.
Symptoms are most likely to be mistaken for myasthenia gravis or Guillain-Barré syndrome, but the latter has an ascending paralysis. Cranial nerve involvement can present as blurred vision, ptosis (drooping lid), diplopia (double vision), ophthalmoplegia (weak eye muscles), or difficulty with speech and swallowing. Shortness of breath and abdominal discomfort can also occur. Respiratory failure may occur from weakness or paralysis of cranial nerves. Cranial nerve signs and symptoms in the absence of fever, along with a descending paralysis, should strongly suggest the diagnosis.
With food-borne botulism, vomiting occurs in half the patients. Improperly sterilized home-canned food is the major risk factor. While the toxin is rapidly destroyed by heat, the bacterial spores are not. Wound botulism is most commonly associated with the injection of drugs, particularly black tar heroin.
Dr. Edwards stressed that “time is of the essence when it comes to botulism diagnostics and treating. Timely administration of the botulism antitoxin early in the course of illness can arrest the progression of paralysis and possibly avert the need for intubation or ventilation.”
It’s essential to note that botulism is an urgent diagnosis that has to be made on clinical grounds. Lab assays for botulinum neurotoxins take too long and are only conducted in public health laboratories. The decision to use antitoxin must not be delayed to wait for confirmation.
Clinicians should immediately contact the local or state health department’s emergency on-call team if botulism is suspected. They will arrange for expert consultation.
Treatment
Botulinum antitoxin is the only specific therapy for this infection. If given early – preferably within 24-48 hours of symptom onset – it can stop the progression of paralysis. But antitoxin will not reverse existing paralysis. If paralysis is still progressing outside of that 24- to 48-hour window, the antitoxin should still provide benefit. The antitoxin is available only through state health departments and a request to the CDC.
Botulism antitoxin is made from horse serum and therefore may cause a variety of allergic reactions. The risk for anaphylaxis is less than 2%, far lower than the mortality from untreated botulism.
While these guidelines have an important focus on triaging and treating mass casualties from botulism, it’s important to note that food-borne outbreaks and prevention issues are covered elsewhere on the CDC site.
Dr. Edwards has disclosed no relevant financial relationships. Dr. Adalja is a consultant for Emergent BioSolutions, which makes the heptavalent botulism antitoxin.
Dr. Stone is an infectious disease specialist and author of “Resilience: One Family’s Story of Hope and Triumph Over Evil” and of “Conducting Clinical Research,” the essential guide to the topic. You can find her at drjudystone.com or on Twitter @drjudystone.
A version of this article first appeared on Medscape.com.
Botulinum toxin is said to be the most lethal substance known. Inhaling just 1-3 nanograms of toxin per kilogram of body mass constitutes a lethal dose.
The CDC has been working on these guidelines since 2015, initially establishing a technical development group and steering committee to prioritize topics for review and make recommendations. Since then, the agency published 15 systematic reviews in Clinical Infectious Diseases early in 2018. The reviews addressed the recognition of botulism clinically, treatment with botulinum antitoxin, and complications from that treatment. They also looked at the epidemiology of botulism outbreaks and botulism in the special populations of vulnerable pediatric and pregnant patients.
In 2016, the CDC held two extended forums and convened a workshop with 72 experts. In addition to the more standard topics of diagnosis and treatment, attention was given to crisis standards of care, caring for multiple patients at once, and ethical considerations in management.
Amesh Adalja, MD, senior scholar, Johns Hopkins Center for Health Security, Baltimore, said in an interview that the new guidance “was really specific [and] was meant to address the gap in guidance for mass casualty settings.”
While clinicians are used to focusing on an individual patient, in times of crises, with multiple patients from a food-borne outbreak or a bioterrorism attack, the focus must shift to the population rather than the individual. The workshop explored issues of triaging, adding beds, and caring for patients when a hospital is overwhelmed with an acute influx of severely ill patients.
Such a mass casualty event is similar to the stress encountered this past year with COVID-19 patients swamping the hospitals, which had too little oxygen, too few ventilators, and too few staff members to care for the sudden influx of critically ill patients.
Diagnosis
Leslie Edwards, MHS, BSN, a CDC epidemiologist and botulism expert, said that “botulism is rare and [so] could be difficult to diagnose.” The CDC “wanted to highlight some of those key clinical factors” to speed recognition.
Hospitals and health officials are being urged to develop crisis protocols as part of emergency preparedness plans. And clinicians should be able to recognize four major syndromes: botulism from food, wounds, and inhalation, as well as iatrogenic botulism (from exposure via injection of the neurotoxin).
Botulism has a characteristic and unusual pattern of symptoms, which begin with cranial nerve palsies. Then there is typically a descending, symmetric flaccid paralysis. Symptoms might progress to respiratory failure and death. Other critical clues that implicate botulism include a lack of sensory deficits and the absence of pain.
Symptoms are most likely to be mistaken for myasthenia gravis or Guillain-Barré syndrome, but the latter has an ascending paralysis. Cranial nerve involvement can present as blurred vision, ptosis (drooping lid), diplopia (double vision), ophthalmoplegia (weak eye muscles), or difficulty with speech and swallowing. Shortness of breath and abdominal discomfort can also occur. Respiratory failure may occur from weakness or paralysis of cranial nerves. Cranial nerve signs and symptoms in the absence of fever, along with a descending paralysis, should strongly suggest the diagnosis.
With food-borne botulism, vomiting occurs in half the patients. Improperly sterilized home-canned food is the major risk factor. While the toxin is rapidly destroyed by heat, the bacterial spores are not. Wound botulism is most commonly associated with the injection of drugs, particularly black tar heroin.
Dr. Edwards stressed that “time is of the essence when it comes to botulism diagnostics and treating. Timely administration of the botulism antitoxin early in the course of illness can arrest the progression of paralysis and possibly avert the need for intubation or ventilation.”
It’s essential to note that botulism is an urgent diagnosis that has to be made on clinical grounds. Lab assays for botulinum neurotoxins take too long and are only conducted in public health laboratories. The decision to use antitoxin must not be delayed to wait for confirmation.
Clinicians should immediately contact the local or state health department’s emergency on-call team if botulism is suspected. They will arrange for expert consultation.
Treatment
Botulinum antitoxin is the only specific therapy for this infection. If given early – preferably within 24-48 hours of symptom onset – it can stop the progression of paralysis. But antitoxin will not reverse existing paralysis. If paralysis is still progressing outside of that 24- to 48-hour window, the antitoxin should still provide benefit. The antitoxin is available only through state health departments and a request to the CDC.
Botulism antitoxin is made from horse serum and therefore may cause a variety of allergic reactions. The risk for anaphylaxis is less than 2%, far lower than the mortality from untreated botulism.
While these guidelines have an important focus on triaging and treating mass casualties from botulism, it’s important to note that food-borne outbreaks and prevention issues are covered elsewhere on the CDC site.
Dr. Edwards has disclosed no relevant financial relationships. Dr. Adalja is a consultant for Emergent BioSolutions, which makes the heptavalent botulism antitoxin.
Dr. Stone is an infectious disease specialist and author of “Resilience: One Family’s Story of Hope and Triumph Over Evil” and of “Conducting Clinical Research,” the essential guide to the topic. You can find her at drjudystone.com or on Twitter @drjudystone.
A version of this article first appeared on Medscape.com.
HbA1c Change in Patients With and Without Gaps in Pharmacist Visits at a Safety-Net Resident Physician Primary Care Clinic
From Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA (Drs. Chu and Ma and Mimi Lou), and Department of Family Medicine, Keck Medicine, University of Southern California, Los Angeles, CA (Dr. Suh).
Objective: The objective of this study is to describe HbA1c changes in patients who maintained continuous pharmacist care vs patients who had a gap in pharmacist care of 3 months or longer.
Methods: This retrospective study was conducted from October 1, 2018, to September 30, 2019. Electronic health record data from an academic-affiliated, safety-net resident physician primary care clinic were collected to observe HbA1c changes between patients with continuous pharmacist care and patients who had a gap of 3 months or longer in pharmacist care. A total of 189 patients met the inclusion criteria and were divided into 2 groups: those with continuous care and those with gaps in care. Data were analyzed using the Mann-Whitney test for continuous variables and the χ2 (or Fisher exact) test for categorical variables. The differences-in-differences model was used to compare the changes in HbA1c between the 2 groups.
Results: There was no significant difference in changes in HbA1c between the continuous care group and the gaps in care group, although the mean magnitude of HbA1c changes was numerically greater in the continuous care group (-1.48% vs -0.97%). Overall, both groups showed improvement in their HbA1c levels and had similar numbers of primary care physician visits and acute care utilizations, while the gaps in care group had longer duration with pharmacists and between the adjacent pharmacist visits.
Conclusion: Maintaining continuous, regular visits with a pharmacist at a safety-net resident physician primary care clinic did not show a significant difference in HbA1c changes compared to having gaps in pharmacist care. Future studies on socioeconomic and behavioral burden on HbA1c improvement and on pharmacist visits in these populations should be explored.
Keywords: clinical pharmacist; diabetes management; continuous visit; primary care clinic.
Pharmacists have unique skills in identifying and resolving problems related to the safety and efficacy of drug therapy while addressing medication adherence and access for patients. Their expertise is especially important to meet the care needs of a growing population with chronic conditions amidst a primary care physician shortage.1 As health care systems move toward value-based care, emphasis on improvement in quality and health measures have become central in care delivery. Pharmacists have been integrated into team-based care in primary care settings, but the value-based shift has opened more opportunities for pharmacists to address unmet quality standards.2-5
Many studies have reported that the integration of pharmacists into team-based care improves health outcomes and reduces overall health care costs.6-9 Specifically, when pharmacists were added to primary care teams to provide diabetes management, hemoglobin HbA1c levels were reduced compared to teams without pharmacists.10-13 Offering pharmacist visits as often as every 2 weeks to 3 months, with each patient having an average of 4.7 visits, resulted in improved therapeutic outcomes.3,7 During visits, pharmacists address the need for additional drug therapy, deprescribe unnecessary therapy, correct insufficient doses or durations, and switch patients to more cost-efficient drug therapy.9 Likewise, patients who visit pharmacists in addition to seeing their primary care physician can have medication-related concerns resolved and improve their therapeutic outcomes.10,11
Not much is known about the magnitude of HbA1c change based on the regularity of pharmacist visits. Although pharmacists offer follow-up appointments in reasonable time intervals, patients do not keep every appointment for a variety of reasons, including forgetfulness, personal issues, and a lack of transportation.14 Such missed appointments can negatively impact health outcomes.14-16 The purpose of this study is to describe HbA1c changes in patients who maintained continuous, regular pharmacist visits without a 3-month gap and in patients who had history of inconsistent pharmacist visits with a gap of 3 months or longer. Furthermore, this study describes the frequency of health care utilization for these 2 groups.
Methods
Setting
The Internal Medicine resident physician primary care clinic is 1 of 2 adult primary care clinics at an academic, urban, public medical center. It is in the heart of East Los Angeles, where predominantly Spanish-speaking and minority populations reside. The clinic has approximately 19000 empaneled patients and is the largest resident primary care clinic in the public health system. The clinical pharmacy service addresses unmet quality standards, specifically HbA1c. The clinical pharmacists are co-located and collaborate with resident physicians, attending physicians, care managers, nurses, social workers, and community health workers at the clinic. They operate under collaborative practice agreements with prescriptive authority, except for controlled substances, specialty drugs, and antipsychotic medications.
Pharmacist visit
Patients are primarily referred by resident physicians to clinical pharmacists when their HbA1c level is above 8% for an extended period, when poor adherence and low health literacy are evident regardless of HbA1c level, or when a complex medication regimen requires comprehensive medication review and reconciliation. The referral occurs through warm handoff by resident physicians as well as clinic nurses, and it is embedded in the clinic flow. Patients continue their visits with resident physicians for issues other than their referral to clinical pharmacists. The visits with pharmacists are appointment-based, occur independently from resident physician visits, and continue until the patient’s HbA1c level or adherence is optimized. Clinical pharmacists continue to follow up with patients who may have reached their target HbA1c level but still are deemed unstable due to inconsistency in their self-management and medication adherence.
After the desirable HbA1c target is achieved along with full adherence to medications and self-management, clinical pharmacists will hand off patients back to resident physicians. At each visit, pharmacists perform a comprehensive medication assessment and reconciliation that includes adjusting medication therapy, placing orders for necessary laboratory tests and prescriptions, and assessing medication adherence. They also evaluate patients’ signs and symptoms for hyperglycemic complications, hypoglycemia, and other potential treatment-related adverse events. These are all within the pharmacist’s scope of practice in comprehensive medication management. Patient education is provided with the teach-back method and includes lifestyle modifications and medication counseling (Table 1). Pharmacists offer face-to-face visits as frequently as every 1 to 2 weeks to every 4 to 6 weeks, depending on the level of complexity and the severity of a patient’s conditions and medications. For patients whose HbA1c has reached the target range but have not been deemed stable, pharmacists continue to check in with them every 2 months. Phone visits are also utilized as an additional care delivery method for patients having difficulty showing up for face-to-face visits or needing quick assessment of medication adherence and responses to changes in drug treatment in between the face-to-face visits. The maximal interval between pharmacist visits is offered no longer than every 8 weeks. Patients are contacted via phone or mail by the nursing staff to reschedule if they miss their appointments with pharmacists. Every pharmacy visit is documented in the patient’s electronic medical record.

Study design
This is a retrospective study describing the HbA1c changes in a patient group that maintained pharmacist visits, with each interval less than 3 months, and in another group, who had a history of a 3-month or longer gap between pharmacist visits. The data were obtained from patients’ electronic medical records during the study period of October 1, 2018, and September 30, 2019, and collected using a HIPAA-compliant, electronic data storage website, REDCap. The institutional review board approval was obtained under HS-19-00929. Patients 18 years and older who were referred by primary care resident physicians for diabetes management, and had 2 or more visits with a pharmacist within the study period, were included. Patients were excluded if they had only 1 HbA1c drawn during the study period, were referred to a pharmacist for reasons other than diabetes management, were concurrently managed by an endocrinologist, had only 1 visit with a pharmacist, or had no visits with their primary care resident physician for over a year. The patients were then divided into 2 groups: continuous care cohort (CCC) and gap in care cohort (GCC). Both face-to-face and phone visits were counted as pharmacist visits for each group.
Outcomes
The primary outcome was the change in HbA1c from baseline between the 2 groups. Baseline HbA1c was considered as the HbA1c value obtained within 3 months prior to, or within 1 month, of the first visit with the pharmacist during the study period. The final HbA1c was considered the value measured within 1 month of, or 3 months after, the patient’s last visit with the pharmacist during the study period.
Several subgroup analyses were conducted to examine the relationship between HbA1c and each group. Among patients whose baseline HbA1c was ≥ 8%, we looked at the percentage of patients reaching HbA1c < 8%, the percentage of patients showing any level of improvement in HbA1c, and the change in HbA1c for each group. We also looked at the percentage of patients with baseline HbA1c < 8% maintaining the level throughout the study period and the change in HbA1c for each group. Additionally, we looked at health care utilization, which included pharmacist visits, primary care physician visits, emergency room and urgent care visits, and hospitalizations for each group. The latter 3 types of utilization were grouped as acute care utilization and further analyzed for visit reasons, which were subsequently categorized as diabetes related and non-diabetes related. The diabetes related reasons linking to acute care utilization were defined as any episodes related to hypoglycemia, diabetic ketoacidosis (DKA), hyperosmolar hyperglycemic state (HHS), foot ulcers, retinopathy, and osteomyelitis infection. All other reasons leading to acute care utilization were categorized as non-diabetes related.
Statistical analysis
Descriptive analyses were conducted using the Mann-Whitney test for continuous data and χ2 (or Fisher exact) test for categorical data. A basic difference-in-differences (D-I-D) method was used to compare the changes of HbA1c between the CCC and GCC over 2 time points: baseline and final measurements. The repeated measures ANOVA was used for analyzing D-I-D. P < .05 was considered significant. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC).

Results
Baseline data
A total of 1272 patients were identified within the study period, and 189 met the study inclusion criteria. The CCC included 132 patients, the GCC 57. The mean age of patients in both groups was similar at 57 years old (P = .39). Most patients had Medicaid as their primary insurance. About one-third of patients in each group experienced clinical atherosclerotic cardiovascular disease, and about 12% overall had chronic kidney disease stage 3 and higher. The average number of days that patients were under pharmacist care during the study period was longer in the GCC compared to the CCC, and it was statistically significant (P < .001) (Table 2). The mean ± SD baseline HbA1c for the CCC and GCC was 10.0% ± 2.0% and 9.9% ± 1.7%, respectively, and the difference was not statistically significant (P = .93). About 86% of patients in the CCC and 90% in the GCC had a baseline HbA1c of ≥ 8%.

HbA1c
The mean change in HbA1c between the 2 groups was not statistically significant (-1.5% ± 2.0% in the CCC vs -1.0% ± 2.1% in the GCC, P = .36) (Table 3). However, an absolute mean HbA1c reduction of 1.3% was observed in both groups combined at the end of the study. Figure 1 shows a D-I-D model of the 2 groups. Based on the output, the P value of .11 on the interaction term (time*group) indicates that the D-I-D in HbA1c change from baseline to final between the CCC and GCC is not statistically different. However, the magnitude of the difference calculated from the LSMEANS results showed a trend. The HbA1c from baseline to final measurement of patients in the GCC declined by 0.97 percentage points (from 9.94% to 8.97%), while those in the CCC saw their HbA1c decline by 1.48 percentage points (from 9.96% to 8.48%), for a D-I-D of 0.51. In other words, those in the GCC had an HbA1c that decreased by 0.51% less than that of patients in the CCC, suggesting that the CCC shows a steeper line declining from baseline to final HbA1c compared to the GCC, whose line declines less sharply.

In the subgroup analysis of patients whose baseline HbA1c was ≥ 8%, about 42% in the CCC and 37% in the GCC achieved an HbA1c < 8% (P = .56) (Table 4). Approximately 83% of patients in the CCC had some degree of HbA1c improvement—the final HbA1c was lower than their baseline HbA1c—whereas this was observed in about 75% of patients in the GCC (P = .19). Of patients whose baseline HbA1c was < 8%, there was no significant difference in proportion of patients maintaining an HbA1c < 8% between the groups (P = .57), although some increases in HbA1c and HbA1c changes were observed in the GCC (Table 5).

Health care utilization
Patients in the CCC visited pharmacists 5 times on average over 12 months, whereas patients in the GCC had an average of 6 visits (5 ± 2.6 in the CCC vs 6 ± 2.6 in the GCC, P = .01) (Table 6). The mean length between any 2 adjacent visits was significantly different, averaging about 33 days in the CCC compared to 64 days in the GCC (33.2 ± 10 in the CCC vs 63.7 ± 39.4 in the GCC, P < .001). As shown in Figure 2, the GCC shows wider ranges between any adjacent pharmacy visits throughout until the 10th visit. Both groups had a similar number of visits with primary care physicians during the same time period (4.6 ± 1.86 in the CCC vs 4.3 ± 2.51 in the GCC, P = .44). About 30% of patients in the CCC and 47% in the GCC had at least 1 visit to the emergency room or urgent care or had at least 1 hospital admission, for a total of 124 acute care utilizations between the 2 groups combined. Only a small fraction of acute care visits with or without hospitalizations were related to diabetes and its complications (23.1% in the CCC vs 22.0% in the GCC).

Discussion
This is a real-world study that describes HbA1c changes in patients who maintained pharmacy visits regularly and in those who had a history of a 3-month or longer gap in pharmacy visits. Although the study did not show statistically significant differences in HbA1c reduction between the 2 groups, pharmacists’ care, overall, provided mean HbA1c reductions of 1.3%. This result is consistent with those from multiple previous studies.10-13 It is worth noting that the final HbA1c was numerically lower in patients who followed up with pharmacists regularly than in patients with gaps in visits, with a difference of about 0.5 percentage points. This difference is considered clinically significant,17 and potentially could be even greater if the study duration was longer, as depicted by the slope of HbA1c reductions in the D-I-D model (Figure 1).

Previous studies have shown that pharmacist visits are conducted in shorter intervals than primary care physician visits to provide closer follow-up and to resolve any medication-related problems that may hinder therapeutic outcome improvements.3-4,7-9 Increasing access via pharmacists is particularly important in this clinic, where resident physician continuity and access is challenging. The pharmacist-driven program described in this study does not deviate from the norm, and this study confirms that pharmacist care, regardless of gaps in pharmacist visits, may still be beneficial.
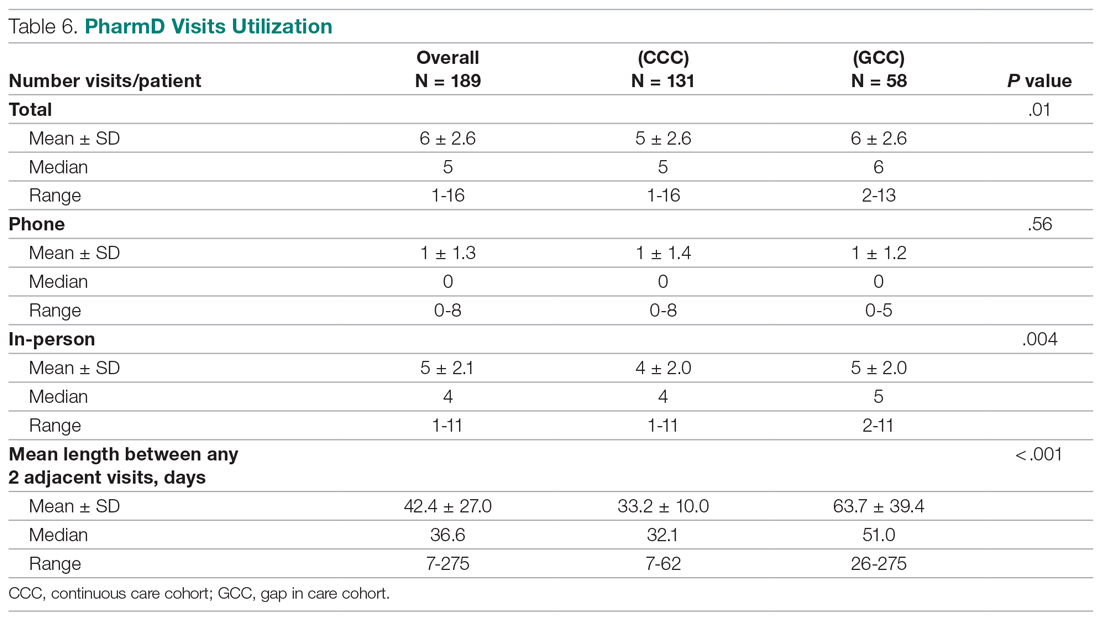
Another notable finding from this study was that although the average number of pharmacist visits per patient was significantly different, this difference of 1 visit did not result in a statistically significant improvement in HbA1c. In fact, the average number of pharmacist visits per patient seemed to be within the reported range by Choe et al in a similar setting.7 Conversely, patients with a history of a gap in pharmacist visits spent longer durations under pharmacist care compared to those who had continuous follow-up. This could mean that it may take longer times or 1 additional visit to achieve similar HbA1c results with continuous pharmacist care. Higher number of visits with pharmacists in the group with the history of gaps between pharmacist visits could have been facilitated by resident physicians, as both groups had a similar number of visits with them. Although this is not conclusive, identifying the optimal number of visits with pharmacists in this underserved population could be beneficial in strategizing pharmacist visits. Acute care utilization was not different between the 2 groups, and most cases that led to acute care utilization were not directly related to diabetes or its complications.
The average HbA1c at the end of the study did not measure < 8%, a target that was reached by less than half of patients from each group; however, this study is a snapshot of a series of ongoing clinical pharmacy services. About 25% of our patients started their first visit with a pharmacist less than 6 months from the study end date, and these patients may not have had enough time with pharmacists for their HbA1c to reach below the target goal. In addition, most patients in this clinic were enrolled in public health plans and may carry a significant burden of social and behavioral factors that can affect diabetes management.18,19 These patients may need longer care by pharmacists along with other integrated services, such as behavioral health and social work, to achieve optimal HbA1c levels.20
There are several limitations to this study, including the lack of a propensity matched control group of patients who only had resident physician visits; thus, it is hard to test the true impact of continuous or intermittent pharmacist visits on the therapeutic outcomes. The study also does not address potential social, economic, and physical environment factors that might have contributed to pharmacist visits and to overall diabetes care. These factors can negatively impact diabetes control and addressing them could help with an individualized diabetes management approach.17,18 Additionally, by nature of being a descriptive study, the results may be subject to undetermined confounding factors.
Conclusion
Patients maintaining continuous pharmacist visits do not have statistically significant differences in change in HbA1c compared to patients who had a history of 3-month or longer gaps in pharmacist visits at a resident physician primary care safety-net clinic. However, patients with diabetes will likely derive a benefit in HbA1c reduction regardless of regularity of pharmacist care. This finding still holds true in collaboration with resident physicians who also regularly meet with patients.
The study highlights that it is important to integrate clinical pharmacists into primary care teams for improved therapeutic outcomes. It is our hope that regular visits to pharmacists can be a gateway for behavioral health and social work referrals, thereby addressing pharmacist-identified social barriers. Furthermore, exploration of socioeconomic and behavioral barriers to pharmacist visits is necessary to address and improve the patient experience, health care delivery, and health outcomes.
Acknowledgments: The authors thank Roxanna Perez, PharmD, Amy Li, and Julie Dopheide, PharmD, BCPP, FASHP for their contributions to this project.
Corresponding author: Michelle Koun Lee Chu, PharmD, BCACP, APh, Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, 1985 Zonal Ave, Los Angeles, CA 90089-9121; [email protected].
Financial disclosures: None.
1. Manolakis PG, Skelton JB. Pharmacists’ contributions to primary care in the United States collaborating to address unmet patient care needs: the emerging role for pharmacists to address the shortage of primary care providers. Am J Pharm Educ. 2010;74(10):S7.
2. Scott MA, Hitch B, Ray L, Colvin G. Integration of pharmacists into a patient-centered medical home. J Am Pharm Assoc (2003). 2011;51(2):161‐166.
3. Wong SL, Barner JC, Sucic K, et al. Integration of pharmacists into patient-centered medical homes in federally qualified health centers in Texas. J Am Pharm Assoc (2003). 2017;57(3):375‐381.
4. Sapp ECH, Francis SM, Hincapie AL. Implementation of pharmacist-driven comprehensive medication management as part of an interdisciplinary team in primary care physicians’ offices. Am J Accountable Care. 2020;8(1):8-11.
5. Cowart K, Olson K. Impact of pharmacist care provision in value-based care settings: How are we measuring value-added services? J Am Pharm Assoc (2003). 2019;59(1):125-128.
6. Centers for Disease Control and Prevention. Pharmacy: Collaborative Practice Agreements to Enable Drug Therapy Management. January 16, 2018. Accessed April 17, 2021. https://www.cdc.gov/dhdsp/pubs/guides/best-practices/pharmacist-cdtm.htm
7. Choe HM, Farris KB, Stevenson JG, et al. Patient-centered medical home: developing, expanding, and sustaining a role for pharmacists. Am J Health Syst Pharm. 2012;69(12):1063-1071.
8. Coe AB, Choe HM. Pharmacists supporting population health in patient-centered medical homes. Am J Health Syst Pharm. 2017;74(18):1461-1466.
9. Luder HR, Shannon P, Kirby J, Frede SM. Community pharmacist collaboration with a patient-centered medical home: establishment of a patient-centered medical neighborhood and payment model. J Am Pharm Assoc (2003). 2018;58(1):44-50.
10. Matzke GR, Moczygemba LR, Williams KJ, et al. Impact of a pharmacist–physician collaborative care model on patient outcomes and health services utilization. 10.05Am J Health Syst Pharm. 2018;75(14):1039-1047.
11. Aneese NJ, Halalau A, Muench S, et al. Impact of a pharmacist-managed diabetes clinic on quality measures. Am J Manag Care. 2018;24(4 Spec No.):SP116-SP119.
12. Prudencio J, Cutler T, Roberts S, et al. The effect of clinical 10.05pharmacist-led comprehensive medication management on chronic disease state goal attainment in a patient-centered medical home. J Manag Care Spec Pharm. 2018;24(5):423-429.
13. Edwards HD, Webb RD, Scheid DC, et al. A pharmacist visit improves diabetes standards in a patient-centered medical home (PCMH). Am J Med Qual. 2012;27(6) 529-534.
14. Ullah S, Rajan S, Liu T, et al. Why do patients miss their appointments at primary care clinics? J Fam Med Dis Prev. 2018;4:090.
15. Moore CG, Wilson-Witherspoon P, Probst JC. Time and money: effects of no-shows at a family practice residency clinic. Fam Med. 2001;33(7):522-527.
16. Kheirkhah P, Feng Q, Travis LM, et al. Prevalence, predictors and economic consequences of no-shows. BMC Health Serv Res. 2016;16:13.
17. Little RR, Rohlfing C. The long and winding road to optimal HbA10.051c10.05 measurement. Clin Chim Acta. 2013;418:63-71.
18. Hill J, Nielsen M, Fox MH. Understanding the social factors that contribute to diabetes: a means to informing health care and social policies for the chronically ill. Perm J. 2013;17(2):67-72.
19. Gonzalez-Zacarias AA, Mavarez-Martinez A, Arias-Morales CE, et al. Impact of demographic, socioeconomic, and psychological factors on glycemic self-management in adults with type 2 diabetes mellitus. Front Public Health. 2016;4:195.
20. Pantalone KM, Misra-Hebert AD, Hobbs TD, et al. The probability of A1c goal attainment in patients with uncontrolled type 2 diabetes in a large integrated delivery system: a prediction model. Diabetes Care. 2020;43:1910-1919.
From Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA (Drs. Chu and Ma and Mimi Lou), and Department of Family Medicine, Keck Medicine, University of Southern California, Los Angeles, CA (Dr. Suh).
Objective: The objective of this study is to describe HbA1c changes in patients who maintained continuous pharmacist care vs patients who had a gap in pharmacist care of 3 months or longer.
Methods: This retrospective study was conducted from October 1, 2018, to September 30, 2019. Electronic health record data from an academic-affiliated, safety-net resident physician primary care clinic were collected to observe HbA1c changes between patients with continuous pharmacist care and patients who had a gap of 3 months or longer in pharmacist care. A total of 189 patients met the inclusion criteria and were divided into 2 groups: those with continuous care and those with gaps in care. Data were analyzed using the Mann-Whitney test for continuous variables and the χ2 (or Fisher exact) test for categorical variables. The differences-in-differences model was used to compare the changes in HbA1c between the 2 groups.
Results: There was no significant difference in changes in HbA1c between the continuous care group and the gaps in care group, although the mean magnitude of HbA1c changes was numerically greater in the continuous care group (-1.48% vs -0.97%). Overall, both groups showed improvement in their HbA1c levels and had similar numbers of primary care physician visits and acute care utilizations, while the gaps in care group had longer duration with pharmacists and between the adjacent pharmacist visits.
Conclusion: Maintaining continuous, regular visits with a pharmacist at a safety-net resident physician primary care clinic did not show a significant difference in HbA1c changes compared to having gaps in pharmacist care. Future studies on socioeconomic and behavioral burden on HbA1c improvement and on pharmacist visits in these populations should be explored.
Keywords: clinical pharmacist; diabetes management; continuous visit; primary care clinic.
Pharmacists have unique skills in identifying and resolving problems related to the safety and efficacy of drug therapy while addressing medication adherence and access for patients. Their expertise is especially important to meet the care needs of a growing population with chronic conditions amidst a primary care physician shortage.1 As health care systems move toward value-based care, emphasis on improvement in quality and health measures have become central in care delivery. Pharmacists have been integrated into team-based care in primary care settings, but the value-based shift has opened more opportunities for pharmacists to address unmet quality standards.2-5
Many studies have reported that the integration of pharmacists into team-based care improves health outcomes and reduces overall health care costs.6-9 Specifically, when pharmacists were added to primary care teams to provide diabetes management, hemoglobin HbA1c levels were reduced compared to teams without pharmacists.10-13 Offering pharmacist visits as often as every 2 weeks to 3 months, with each patient having an average of 4.7 visits, resulted in improved therapeutic outcomes.3,7 During visits, pharmacists address the need for additional drug therapy, deprescribe unnecessary therapy, correct insufficient doses or durations, and switch patients to more cost-efficient drug therapy.9 Likewise, patients who visit pharmacists in addition to seeing their primary care physician can have medication-related concerns resolved and improve their therapeutic outcomes.10,11
Not much is known about the magnitude of HbA1c change based on the regularity of pharmacist visits. Although pharmacists offer follow-up appointments in reasonable time intervals, patients do not keep every appointment for a variety of reasons, including forgetfulness, personal issues, and a lack of transportation.14 Such missed appointments can negatively impact health outcomes.14-16 The purpose of this study is to describe HbA1c changes in patients who maintained continuous, regular pharmacist visits without a 3-month gap and in patients who had history of inconsistent pharmacist visits with a gap of 3 months or longer. Furthermore, this study describes the frequency of health care utilization for these 2 groups.
Methods
Setting
The Internal Medicine resident physician primary care clinic is 1 of 2 adult primary care clinics at an academic, urban, public medical center. It is in the heart of East Los Angeles, where predominantly Spanish-speaking and minority populations reside. The clinic has approximately 19000 empaneled patients and is the largest resident primary care clinic in the public health system. The clinical pharmacy service addresses unmet quality standards, specifically HbA1c. The clinical pharmacists are co-located and collaborate with resident physicians, attending physicians, care managers, nurses, social workers, and community health workers at the clinic. They operate under collaborative practice agreements with prescriptive authority, except for controlled substances, specialty drugs, and antipsychotic medications.
Pharmacist visit
Patients are primarily referred by resident physicians to clinical pharmacists when their HbA1c level is above 8% for an extended period, when poor adherence and low health literacy are evident regardless of HbA1c level, or when a complex medication regimen requires comprehensive medication review and reconciliation. The referral occurs through warm handoff by resident physicians as well as clinic nurses, and it is embedded in the clinic flow. Patients continue their visits with resident physicians for issues other than their referral to clinical pharmacists. The visits with pharmacists are appointment-based, occur independently from resident physician visits, and continue until the patient’s HbA1c level or adherence is optimized. Clinical pharmacists continue to follow up with patients who may have reached their target HbA1c level but still are deemed unstable due to inconsistency in their self-management and medication adherence.
After the desirable HbA1c target is achieved along with full adherence to medications and self-management, clinical pharmacists will hand off patients back to resident physicians. At each visit, pharmacists perform a comprehensive medication assessment and reconciliation that includes adjusting medication therapy, placing orders for necessary laboratory tests and prescriptions, and assessing medication adherence. They also evaluate patients’ signs and symptoms for hyperglycemic complications, hypoglycemia, and other potential treatment-related adverse events. These are all within the pharmacist’s scope of practice in comprehensive medication management. Patient education is provided with the teach-back method and includes lifestyle modifications and medication counseling (Table 1). Pharmacists offer face-to-face visits as frequently as every 1 to 2 weeks to every 4 to 6 weeks, depending on the level of complexity and the severity of a patient’s conditions and medications. For patients whose HbA1c has reached the target range but have not been deemed stable, pharmacists continue to check in with them every 2 months. Phone visits are also utilized as an additional care delivery method for patients having difficulty showing up for face-to-face visits or needing quick assessment of medication adherence and responses to changes in drug treatment in between the face-to-face visits. The maximal interval between pharmacist visits is offered no longer than every 8 weeks. Patients are contacted via phone or mail by the nursing staff to reschedule if they miss their appointments with pharmacists. Every pharmacy visit is documented in the patient’s electronic medical record.
Study design
This is a retrospective study describing the HbA1c changes in a patient group that maintained pharmacist visits, with each interval less than 3 months, and in another group, who had a history of a 3-month or longer gap between pharmacist visits. The data were obtained from patients’ electronic medical records during the study period of October 1, 2018, and September 30, 2019, and collected using a HIPAA-compliant, electronic data storage website, REDCap. The institutional review board approval was obtained under HS-19-00929. Patients 18 years and older who were referred by primary care resident physicians for diabetes management, and had 2 or more visits with a pharmacist within the study period, were included. Patients were excluded if they had only 1 HbA1c drawn during the study period, were referred to a pharmacist for reasons other than diabetes management, were concurrently managed by an endocrinologist, had only 1 visit with a pharmacist, or had no visits with their primary care resident physician for over a year. The patients were then divided into 2 groups: continuous care cohort (CCC) and gap in care cohort (GCC). Both face-to-face and phone visits were counted as pharmacist visits for each group.
Outcomes
The primary outcome was the change in HbA1c from baseline between the 2 groups. Baseline HbA1c was considered as the HbA1c value obtained within 3 months prior to, or within 1 month, of the first visit with the pharmacist during the study period. The final HbA1c was considered the value measured within 1 month of, or 3 months after, the patient’s last visit with the pharmacist during the study period.
Several subgroup analyses were conducted to examine the relationship between HbA1c and each group. Among patients whose baseline HbA1c was ≥ 8%, we looked at the percentage of patients reaching HbA1c < 8%, the percentage of patients showing any level of improvement in HbA1c, and the change in HbA1c for each group. We also looked at the percentage of patients with baseline HbA1c < 8% maintaining the level throughout the study period and the change in HbA1c for each group. Additionally, we looked at health care utilization, which included pharmacist visits, primary care physician visits, emergency room and urgent care visits, and hospitalizations for each group. The latter 3 types of utilization were grouped as acute care utilization and further analyzed for visit reasons, which were subsequently categorized as diabetes related and non-diabetes related. The diabetes related reasons linking to acute care utilization were defined as any episodes related to hypoglycemia, diabetic ketoacidosis (DKA), hyperosmolar hyperglycemic state (HHS), foot ulcers, retinopathy, and osteomyelitis infection. All other reasons leading to acute care utilization were categorized as non-diabetes related.
Statistical analysis
Descriptive analyses were conducted using the Mann-Whitney test for continuous data and χ2 (or Fisher exact) test for categorical data. A basic difference-in-differences (D-I-D) method was used to compare the changes of HbA1c between the CCC and GCC over 2 time points: baseline and final measurements. The repeated measures ANOVA was used for analyzing D-I-D. P < .05 was considered significant. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC).
Results
Baseline data
A total of 1272 patients were identified within the study period, and 189 met the study inclusion criteria. The CCC included 132 patients, the GCC 57. The mean age of patients in both groups was similar at 57 years old (P = .39). Most patients had Medicaid as their primary insurance. About one-third of patients in each group experienced clinical atherosclerotic cardiovascular disease, and about 12% overall had chronic kidney disease stage 3 and higher. The average number of days that patients were under pharmacist care during the study period was longer in the GCC compared to the CCC, and it was statistically significant (P < .001) (Table 2). The mean ± SD baseline HbA1c for the CCC and GCC was 10.0% ± 2.0% and 9.9% ± 1.7%, respectively, and the difference was not statistically significant (P = .93). About 86% of patients in the CCC and 90% in the GCC had a baseline HbA1c of ≥ 8%.
HbA1c
The mean change in HbA1c between the 2 groups was not statistically significant (-1.5% ± 2.0% in the CCC vs -1.0% ± 2.1% in the GCC, P = .36) (Table 3). However, an absolute mean HbA1c reduction of 1.3% was observed in both groups combined at the end of the study. Figure 1 shows a D-I-D model of the 2 groups. Based on the output, the P value of .11 on the interaction term (time*group) indicates that the D-I-D in HbA1c change from baseline to final between the CCC and GCC is not statistically different. However, the magnitude of the difference calculated from the LSMEANS results showed a trend. The HbA1c from baseline to final measurement of patients in the GCC declined by 0.97 percentage points (from 9.94% to 8.97%), while those in the CCC saw their HbA1c decline by 1.48 percentage points (from 9.96% to 8.48%), for a D-I-D of 0.51. In other words, those in the GCC had an HbA1c that decreased by 0.51% less than that of patients in the CCC, suggesting that the CCC shows a steeper line declining from baseline to final HbA1c compared to the GCC, whose line declines less sharply.
In the subgroup analysis of patients whose baseline HbA1c was ≥ 8%, about 42% in the CCC and 37% in the GCC achieved an HbA1c < 8% (P = .56) (Table 4). Approximately 83% of patients in the CCC had some degree of HbA1c improvement—the final HbA1c was lower than their baseline HbA1c—whereas this was observed in about 75% of patients in the GCC (P = .19). Of patients whose baseline HbA1c was < 8%, there was no significant difference in proportion of patients maintaining an HbA1c < 8% between the groups (P = .57), although some increases in HbA1c and HbA1c changes were observed in the GCC (Table 5).
Health care utilization
Patients in the CCC visited pharmacists 5 times on average over 12 months, whereas patients in the GCC had an average of 6 visits (5 ± 2.6 in the CCC vs 6 ± 2.6 in the GCC, P = .01) (Table 6). The mean length between any 2 adjacent visits was significantly different, averaging about 33 days in the CCC compared to 64 days in the GCC (33.2 ± 10 in the CCC vs 63.7 ± 39.4 in the GCC, P < .001). As shown in Figure 2, the GCC shows wider ranges between any adjacent pharmacy visits throughout until the 10th visit. Both groups had a similar number of visits with primary care physicians during the same time period (4.6 ± 1.86 in the CCC vs 4.3 ± 2.51 in the GCC, P = .44). About 30% of patients in the CCC and 47% in the GCC had at least 1 visit to the emergency room or urgent care or had at least 1 hospital admission, for a total of 124 acute care utilizations between the 2 groups combined. Only a small fraction of acute care visits with or without hospitalizations were related to diabetes and its complications (23.1% in the CCC vs 22.0% in the GCC).
Discussion
This is a real-world study that describes HbA1c changes in patients who maintained pharmacy visits regularly and in those who had a history of a 3-month or longer gap in pharmacy visits. Although the study did not show statistically significant differences in HbA1c reduction between the 2 groups, pharmacists’ care, overall, provided mean HbA1c reductions of 1.3%. This result is consistent with those from multiple previous studies.10-13 It is worth noting that the final HbA1c was numerically lower in patients who followed up with pharmacists regularly than in patients with gaps in visits, with a difference of about 0.5 percentage points. This difference is considered clinically significant,17 and potentially could be even greater if the study duration was longer, as depicted by the slope of HbA1c reductions in the D-I-D model (Figure 1).
Previous studies have shown that pharmacist visits are conducted in shorter intervals than primary care physician visits to provide closer follow-up and to resolve any medication-related problems that may hinder therapeutic outcome improvements.3-4,7-9 Increasing access via pharmacists is particularly important in this clinic, where resident physician continuity and access is challenging. The pharmacist-driven program described in this study does not deviate from the norm, and this study confirms that pharmacist care, regardless of gaps in pharmacist visits, may still be beneficial.
Another notable finding from this study was that although the average number of pharmacist visits per patient was significantly different, this difference of 1 visit did not result in a statistically significant improvement in HbA1c. In fact, the average number of pharmacist visits per patient seemed to be within the reported range by Choe et al in a similar setting.7 Conversely, patients with a history of a gap in pharmacist visits spent longer durations under pharmacist care compared to those who had continuous follow-up. This could mean that it may take longer times or 1 additional visit to achieve similar HbA1c results with continuous pharmacist care. Higher number of visits with pharmacists in the group with the history of gaps between pharmacist visits could have been facilitated by resident physicians, as both groups had a similar number of visits with them. Although this is not conclusive, identifying the optimal number of visits with pharmacists in this underserved population could be beneficial in strategizing pharmacist visits. Acute care utilization was not different between the 2 groups, and most cases that led to acute care utilization were not directly related to diabetes or its complications.
The average HbA1c at the end of the study did not measure < 8%, a target that was reached by less than half of patients from each group; however, this study is a snapshot of a series of ongoing clinical pharmacy services. About 25% of our patients started their first visit with a pharmacist less than 6 months from the study end date, and these patients may not have had enough time with pharmacists for their HbA1c to reach below the target goal. In addition, most patients in this clinic were enrolled in public health plans and may carry a significant burden of social and behavioral factors that can affect diabetes management.18,19 These patients may need longer care by pharmacists along with other integrated services, such as behavioral health and social work, to achieve optimal HbA1c levels.20
There are several limitations to this study, including the lack of a propensity matched control group of patients who only had resident physician visits; thus, it is hard to test the true impact of continuous or intermittent pharmacist visits on the therapeutic outcomes. The study also does not address potential social, economic, and physical environment factors that might have contributed to pharmacist visits and to overall diabetes care. These factors can negatively impact diabetes control and addressing them could help with an individualized diabetes management approach.17,18 Additionally, by nature of being a descriptive study, the results may be subject to undetermined confounding factors.
Conclusion
Patients maintaining continuous pharmacist visits do not have statistically significant differences in change in HbA1c compared to patients who had a history of 3-month or longer gaps in pharmacist visits at a resident physician primary care safety-net clinic. However, patients with diabetes will likely derive a benefit in HbA1c reduction regardless of regularity of pharmacist care. This finding still holds true in collaboration with resident physicians who also regularly meet with patients.
The study highlights that it is important to integrate clinical pharmacists into primary care teams for improved therapeutic outcomes. It is our hope that regular visits to pharmacists can be a gateway for behavioral health and social work referrals, thereby addressing pharmacist-identified social barriers. Furthermore, exploration of socioeconomic and behavioral barriers to pharmacist visits is necessary to address and improve the patient experience, health care delivery, and health outcomes.
Acknowledgments: The authors thank Roxanna Perez, PharmD, Amy Li, and Julie Dopheide, PharmD, BCPP, FASHP for their contributions to this project.
Corresponding author: Michelle Koun Lee Chu, PharmD, BCACP, APh, Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, 1985 Zonal Ave, Los Angeles, CA 90089-9121; [email protected].
Financial disclosures: None.
From Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA (Drs. Chu and Ma and Mimi Lou), and Department of Family Medicine, Keck Medicine, University of Southern California, Los Angeles, CA (Dr. Suh).
Objective: The objective of this study is to describe HbA1c changes in patients who maintained continuous pharmacist care vs patients who had a gap in pharmacist care of 3 months or longer.
Methods: This retrospective study was conducted from October 1, 2018, to September 30, 2019. Electronic health record data from an academic-affiliated, safety-net resident physician primary care clinic were collected to observe HbA1c changes between patients with continuous pharmacist care and patients who had a gap of 3 months or longer in pharmacist care. A total of 189 patients met the inclusion criteria and were divided into 2 groups: those with continuous care and those with gaps in care. Data were analyzed using the Mann-Whitney test for continuous variables and the χ2 (or Fisher exact) test for categorical variables. The differences-in-differences model was used to compare the changes in HbA1c between the 2 groups.
Results: There was no significant difference in changes in HbA1c between the continuous care group and the gaps in care group, although the mean magnitude of HbA1c changes was numerically greater in the continuous care group (-1.48% vs -0.97%). Overall, both groups showed improvement in their HbA1c levels and had similar numbers of primary care physician visits and acute care utilizations, while the gaps in care group had longer duration with pharmacists and between the adjacent pharmacist visits.
Conclusion: Maintaining continuous, regular visits with a pharmacist at a safety-net resident physician primary care clinic did not show a significant difference in HbA1c changes compared to having gaps in pharmacist care. Future studies on socioeconomic and behavioral burden on HbA1c improvement and on pharmacist visits in these populations should be explored.
Keywords: clinical pharmacist; diabetes management; continuous visit; primary care clinic.
Pharmacists have unique skills in identifying and resolving problems related to the safety and efficacy of drug therapy while addressing medication adherence and access for patients. Their expertise is especially important to meet the care needs of a growing population with chronic conditions amidst a primary care physician shortage.1 As health care systems move toward value-based care, emphasis on improvement in quality and health measures have become central in care delivery. Pharmacists have been integrated into team-based care in primary care settings, but the value-based shift has opened more opportunities for pharmacists to address unmet quality standards.2-5
Many studies have reported that the integration of pharmacists into team-based care improves health outcomes and reduces overall health care costs.6-9 Specifically, when pharmacists were added to primary care teams to provide diabetes management, hemoglobin HbA1c levels were reduced compared to teams without pharmacists.10-13 Offering pharmacist visits as often as every 2 weeks to 3 months, with each patient having an average of 4.7 visits, resulted in improved therapeutic outcomes.3,7 During visits, pharmacists address the need for additional drug therapy, deprescribe unnecessary therapy, correct insufficient doses or durations, and switch patients to more cost-efficient drug therapy.9 Likewise, patients who visit pharmacists in addition to seeing their primary care physician can have medication-related concerns resolved and improve their therapeutic outcomes.10,11
Not much is known about the magnitude of HbA1c change based on the regularity of pharmacist visits. Although pharmacists offer follow-up appointments in reasonable time intervals, patients do not keep every appointment for a variety of reasons, including forgetfulness, personal issues, and a lack of transportation.14 Such missed appointments can negatively impact health outcomes.14-16 The purpose of this study is to describe HbA1c changes in patients who maintained continuous, regular pharmacist visits without a 3-month gap and in patients who had history of inconsistent pharmacist visits with a gap of 3 months or longer. Furthermore, this study describes the frequency of health care utilization for these 2 groups.
Methods
Setting
The Internal Medicine resident physician primary care clinic is 1 of 2 adult primary care clinics at an academic, urban, public medical center. It is in the heart of East Los Angeles, where predominantly Spanish-speaking and minority populations reside. The clinic has approximately 19000 empaneled patients and is the largest resident primary care clinic in the public health system. The clinical pharmacy service addresses unmet quality standards, specifically HbA1c. The clinical pharmacists are co-located and collaborate with resident physicians, attending physicians, care managers, nurses, social workers, and community health workers at the clinic. They operate under collaborative practice agreements with prescriptive authority, except for controlled substances, specialty drugs, and antipsychotic medications.
Pharmacist visit
Patients are primarily referred by resident physicians to clinical pharmacists when their HbA1c level is above 8% for an extended period, when poor adherence and low health literacy are evident regardless of HbA1c level, or when a complex medication regimen requires comprehensive medication review and reconciliation. The referral occurs through warm handoff by resident physicians as well as clinic nurses, and it is embedded in the clinic flow. Patients continue their visits with resident physicians for issues other than their referral to clinical pharmacists. The visits with pharmacists are appointment-based, occur independently from resident physician visits, and continue until the patient’s HbA1c level or adherence is optimized. Clinical pharmacists continue to follow up with patients who may have reached their target HbA1c level but still are deemed unstable due to inconsistency in their self-management and medication adherence.
After the desirable HbA1c target is achieved along with full adherence to medications and self-management, clinical pharmacists will hand off patients back to resident physicians. At each visit, pharmacists perform a comprehensive medication assessment and reconciliation that includes adjusting medication therapy, placing orders for necessary laboratory tests and prescriptions, and assessing medication adherence. They also evaluate patients’ signs and symptoms for hyperglycemic complications, hypoglycemia, and other potential treatment-related adverse events. These are all within the pharmacist’s scope of practice in comprehensive medication management. Patient education is provided with the teach-back method and includes lifestyle modifications and medication counseling (Table 1). Pharmacists offer face-to-face visits as frequently as every 1 to 2 weeks to every 4 to 6 weeks, depending on the level of complexity and the severity of a patient’s conditions and medications. For patients whose HbA1c has reached the target range but have not been deemed stable, pharmacists continue to check in with them every 2 months. Phone visits are also utilized as an additional care delivery method for patients having difficulty showing up for face-to-face visits or needing quick assessment of medication adherence and responses to changes in drug treatment in between the face-to-face visits. The maximal interval between pharmacist visits is offered no longer than every 8 weeks. Patients are contacted via phone or mail by the nursing staff to reschedule if they miss their appointments with pharmacists. Every pharmacy visit is documented in the patient’s electronic medical record.
Study design
This is a retrospective study describing the HbA1c changes in a patient group that maintained pharmacist visits, with each interval less than 3 months, and in another group, who had a history of a 3-month or longer gap between pharmacist visits. The data were obtained from patients’ electronic medical records during the study period of October 1, 2018, and September 30, 2019, and collected using a HIPAA-compliant, electronic data storage website, REDCap. The institutional review board approval was obtained under HS-19-00929. Patients 18 years and older who were referred by primary care resident physicians for diabetes management, and had 2 or more visits with a pharmacist within the study period, were included. Patients were excluded if they had only 1 HbA1c drawn during the study period, were referred to a pharmacist for reasons other than diabetes management, were concurrently managed by an endocrinologist, had only 1 visit with a pharmacist, or had no visits with their primary care resident physician for over a year. The patients were then divided into 2 groups: continuous care cohort (CCC) and gap in care cohort (GCC). Both face-to-face and phone visits were counted as pharmacist visits for each group.
Outcomes
The primary outcome was the change in HbA1c from baseline between the 2 groups. Baseline HbA1c was considered as the HbA1c value obtained within 3 months prior to, or within 1 month, of the first visit with the pharmacist during the study period. The final HbA1c was considered the value measured within 1 month of, or 3 months after, the patient’s last visit with the pharmacist during the study period.
Several subgroup analyses were conducted to examine the relationship between HbA1c and each group. Among patients whose baseline HbA1c was ≥ 8%, we looked at the percentage of patients reaching HbA1c < 8%, the percentage of patients showing any level of improvement in HbA1c, and the change in HbA1c for each group. We also looked at the percentage of patients with baseline HbA1c < 8% maintaining the level throughout the study period and the change in HbA1c for each group. Additionally, we looked at health care utilization, which included pharmacist visits, primary care physician visits, emergency room and urgent care visits, and hospitalizations for each group. The latter 3 types of utilization were grouped as acute care utilization and further analyzed for visit reasons, which were subsequently categorized as diabetes related and non-diabetes related. The diabetes related reasons linking to acute care utilization were defined as any episodes related to hypoglycemia, diabetic ketoacidosis (DKA), hyperosmolar hyperglycemic state (HHS), foot ulcers, retinopathy, and osteomyelitis infection. All other reasons leading to acute care utilization were categorized as non-diabetes related.
Statistical analysis
Descriptive analyses were conducted using the Mann-Whitney test for continuous data and χ2 (or Fisher exact) test for categorical data. A basic difference-in-differences (D-I-D) method was used to compare the changes of HbA1c between the CCC and GCC over 2 time points: baseline and final measurements. The repeated measures ANOVA was used for analyzing D-I-D. P < .05 was considered significant. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC).
Results
Baseline data
A total of 1272 patients were identified within the study period, and 189 met the study inclusion criteria. The CCC included 132 patients, the GCC 57. The mean age of patients in both groups was similar at 57 years old (P = .39). Most patients had Medicaid as their primary insurance. About one-third of patients in each group experienced clinical atherosclerotic cardiovascular disease, and about 12% overall had chronic kidney disease stage 3 and higher. The average number of days that patients were under pharmacist care during the study period was longer in the GCC compared to the CCC, and it was statistically significant (P < .001) (Table 2). The mean ± SD baseline HbA1c for the CCC and GCC was 10.0% ± 2.0% and 9.9% ± 1.7%, respectively, and the difference was not statistically significant (P = .93). About 86% of patients in the CCC and 90% in the GCC had a baseline HbA1c of ≥ 8%.
HbA1c
The mean change in HbA1c between the 2 groups was not statistically significant (-1.5% ± 2.0% in the CCC vs -1.0% ± 2.1% in the GCC, P = .36) (Table 3). However, an absolute mean HbA1c reduction of 1.3% was observed in both groups combined at the end of the study. Figure 1 shows a D-I-D model of the 2 groups. Based on the output, the P value of .11 on the interaction term (time*group) indicates that the D-I-D in HbA1c change from baseline to final between the CCC and GCC is not statistically different. However, the magnitude of the difference calculated from the LSMEANS results showed a trend. The HbA1c from baseline to final measurement of patients in the GCC declined by 0.97 percentage points (from 9.94% to 8.97%), while those in the CCC saw their HbA1c decline by 1.48 percentage points (from 9.96% to 8.48%), for a D-I-D of 0.51. In other words, those in the GCC had an HbA1c that decreased by 0.51% less than that of patients in the CCC, suggesting that the CCC shows a steeper line declining from baseline to final HbA1c compared to the GCC, whose line declines less sharply.
In the subgroup analysis of patients whose baseline HbA1c was ≥ 8%, about 42% in the CCC and 37% in the GCC achieved an HbA1c < 8% (P = .56) (Table 4). Approximately 83% of patients in the CCC had some degree of HbA1c improvement—the final HbA1c was lower than their baseline HbA1c—whereas this was observed in about 75% of patients in the GCC (P = .19). Of patients whose baseline HbA1c was < 8%, there was no significant difference in proportion of patients maintaining an HbA1c < 8% between the groups (P = .57), although some increases in HbA1c and HbA1c changes were observed in the GCC (Table 5).
Health care utilization
Patients in the CCC visited pharmacists 5 times on average over 12 months, whereas patients in the GCC had an average of 6 visits (5 ± 2.6 in the CCC vs 6 ± 2.6 in the GCC, P = .01) (Table 6). The mean length between any 2 adjacent visits was significantly different, averaging about 33 days in the CCC compared to 64 days in the GCC (33.2 ± 10 in the CCC vs 63.7 ± 39.4 in the GCC, P < .001). As shown in Figure 2, the GCC shows wider ranges between any adjacent pharmacy visits throughout until the 10th visit. Both groups had a similar number of visits with primary care physicians during the same time period (4.6 ± 1.86 in the CCC vs 4.3 ± 2.51 in the GCC, P = .44). About 30% of patients in the CCC and 47% in the GCC had at least 1 visit to the emergency room or urgent care or had at least 1 hospital admission, for a total of 124 acute care utilizations between the 2 groups combined. Only a small fraction of acute care visits with or without hospitalizations were related to diabetes and its complications (23.1% in the CCC vs 22.0% in the GCC).
Discussion
This is a real-world study that describes HbA1c changes in patients who maintained pharmacy visits regularly and in those who had a history of a 3-month or longer gap in pharmacy visits. Although the study did not show statistically significant differences in HbA1c reduction between the 2 groups, pharmacists’ care, overall, provided mean HbA1c reductions of 1.3%. This result is consistent with those from multiple previous studies.10-13 It is worth noting that the final HbA1c was numerically lower in patients who followed up with pharmacists regularly than in patients with gaps in visits, with a difference of about 0.5 percentage points. This difference is considered clinically significant,17 and potentially could be even greater if the study duration was longer, as depicted by the slope of HbA1c reductions in the D-I-D model (Figure 1).
Previous studies have shown that pharmacist visits are conducted in shorter intervals than primary care physician visits to provide closer follow-up and to resolve any medication-related problems that may hinder therapeutic outcome improvements.3-4,7-9 Increasing access via pharmacists is particularly important in this clinic, where resident physician continuity and access is challenging. The pharmacist-driven program described in this study does not deviate from the norm, and this study confirms that pharmacist care, regardless of gaps in pharmacist visits, may still be beneficial.
Another notable finding from this study was that although the average number of pharmacist visits per patient was significantly different, this difference of 1 visit did not result in a statistically significant improvement in HbA1c. In fact, the average number of pharmacist visits per patient seemed to be within the reported range by Choe et al in a similar setting.7 Conversely, patients with a history of a gap in pharmacist visits spent longer durations under pharmacist care compared to those who had continuous follow-up. This could mean that it may take longer times or 1 additional visit to achieve similar HbA1c results with continuous pharmacist care. Higher number of visits with pharmacists in the group with the history of gaps between pharmacist visits could have been facilitated by resident physicians, as both groups had a similar number of visits with them. Although this is not conclusive, identifying the optimal number of visits with pharmacists in this underserved population could be beneficial in strategizing pharmacist visits. Acute care utilization was not different between the 2 groups, and most cases that led to acute care utilization were not directly related to diabetes or its complications.
The average HbA1c at the end of the study did not measure < 8%, a target that was reached by less than half of patients from each group; however, this study is a snapshot of a series of ongoing clinical pharmacy services. About 25% of our patients started their first visit with a pharmacist less than 6 months from the study end date, and these patients may not have had enough time with pharmacists for their HbA1c to reach below the target goal. In addition, most patients in this clinic were enrolled in public health plans and may carry a significant burden of social and behavioral factors that can affect diabetes management.18,19 These patients may need longer care by pharmacists along with other integrated services, such as behavioral health and social work, to achieve optimal HbA1c levels.20
There are several limitations to this study, including the lack of a propensity matched control group of patients who only had resident physician visits; thus, it is hard to test the true impact of continuous or intermittent pharmacist visits on the therapeutic outcomes. The study also does not address potential social, economic, and physical environment factors that might have contributed to pharmacist visits and to overall diabetes care. These factors can negatively impact diabetes control and addressing them could help with an individualized diabetes management approach.17,18 Additionally, by nature of being a descriptive study, the results may be subject to undetermined confounding factors.
Conclusion
Patients maintaining continuous pharmacist visits do not have statistically significant differences in change in HbA1c compared to patients who had a history of 3-month or longer gaps in pharmacist visits at a resident physician primary care safety-net clinic. However, patients with diabetes will likely derive a benefit in HbA1c reduction regardless of regularity of pharmacist care. This finding still holds true in collaboration with resident physicians who also regularly meet with patients.
The study highlights that it is important to integrate clinical pharmacists into primary care teams for improved therapeutic outcomes. It is our hope that regular visits to pharmacists can be a gateway for behavioral health and social work referrals, thereby addressing pharmacist-identified social barriers. Furthermore, exploration of socioeconomic and behavioral barriers to pharmacist visits is necessary to address and improve the patient experience, health care delivery, and health outcomes.
Acknowledgments: The authors thank Roxanna Perez, PharmD, Amy Li, and Julie Dopheide, PharmD, BCPP, FASHP for their contributions to this project.
Corresponding author: Michelle Koun Lee Chu, PharmD, BCACP, APh, Titus Family Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, 1985 Zonal Ave, Los Angeles, CA 90089-9121; [email protected].
Financial disclosures: None.
1. Manolakis PG, Skelton JB. Pharmacists’ contributions to primary care in the United States collaborating to address unmet patient care needs: the emerging role for pharmacists to address the shortage of primary care providers. Am J Pharm Educ. 2010;74(10):S7.
2. Scott MA, Hitch B, Ray L, Colvin G. Integration of pharmacists into a patient-centered medical home. J Am Pharm Assoc (2003). 2011;51(2):161‐166.
3. Wong SL, Barner JC, Sucic K, et al. Integration of pharmacists into patient-centered medical homes in federally qualified health centers in Texas. J Am Pharm Assoc (2003). 2017;57(3):375‐381.
4. Sapp ECH, Francis SM, Hincapie AL. Implementation of pharmacist-driven comprehensive medication management as part of an interdisciplinary team in primary care physicians’ offices. Am J Accountable Care. 2020;8(1):8-11.
5. Cowart K, Olson K. Impact of pharmacist care provision in value-based care settings: How are we measuring value-added services? J Am Pharm Assoc (2003). 2019;59(1):125-128.
6. Centers for Disease Control and Prevention. Pharmacy: Collaborative Practice Agreements to Enable Drug Therapy Management. January 16, 2018. Accessed April 17, 2021. https://www.cdc.gov/dhdsp/pubs/guides/best-practices/pharmacist-cdtm.htm
7. Choe HM, Farris KB, Stevenson JG, et al. Patient-centered medical home: developing, expanding, and sustaining a role for pharmacists. Am J Health Syst Pharm. 2012;69(12):1063-1071.
8. Coe AB, Choe HM. Pharmacists supporting population health in patient-centered medical homes. Am J Health Syst Pharm. 2017;74(18):1461-1466.
9. Luder HR, Shannon P, Kirby J, Frede SM. Community pharmacist collaboration with a patient-centered medical home: establishment of a patient-centered medical neighborhood and payment model. J Am Pharm Assoc (2003). 2018;58(1):44-50.
10. Matzke GR, Moczygemba LR, Williams KJ, et al. Impact of a pharmacist–physician collaborative care model on patient outcomes and health services utilization. 10.05Am J Health Syst Pharm. 2018;75(14):1039-1047.
11. Aneese NJ, Halalau A, Muench S, et al. Impact of a pharmacist-managed diabetes clinic on quality measures. Am J Manag Care. 2018;24(4 Spec No.):SP116-SP119.
12. Prudencio J, Cutler T, Roberts S, et al. The effect of clinical 10.05pharmacist-led comprehensive medication management on chronic disease state goal attainment in a patient-centered medical home. J Manag Care Spec Pharm. 2018;24(5):423-429.
13. Edwards HD, Webb RD, Scheid DC, et al. A pharmacist visit improves diabetes standards in a patient-centered medical home (PCMH). Am J Med Qual. 2012;27(6) 529-534.
14. Ullah S, Rajan S, Liu T, et al. Why do patients miss their appointments at primary care clinics? J Fam Med Dis Prev. 2018;4:090.
15. Moore CG, Wilson-Witherspoon P, Probst JC. Time and money: effects of no-shows at a family practice residency clinic. Fam Med. 2001;33(7):522-527.
16. Kheirkhah P, Feng Q, Travis LM, et al. Prevalence, predictors and economic consequences of no-shows. BMC Health Serv Res. 2016;16:13.
17. Little RR, Rohlfing C. The long and winding road to optimal HbA10.051c10.05 measurement. Clin Chim Acta. 2013;418:63-71.
18. Hill J, Nielsen M, Fox MH. Understanding the social factors that contribute to diabetes: a means to informing health care and social policies for the chronically ill. Perm J. 2013;17(2):67-72.
19. Gonzalez-Zacarias AA, Mavarez-Martinez A, Arias-Morales CE, et al. Impact of demographic, socioeconomic, and psychological factors on glycemic self-management in adults with type 2 diabetes mellitus. Front Public Health. 2016;4:195.
20. Pantalone KM, Misra-Hebert AD, Hobbs TD, et al. The probability of A1c goal attainment in patients with uncontrolled type 2 diabetes in a large integrated delivery system: a prediction model. Diabetes Care. 2020;43:1910-1919.
1. Manolakis PG, Skelton JB. Pharmacists’ contributions to primary care in the United States collaborating to address unmet patient care needs: the emerging role for pharmacists to address the shortage of primary care providers. Am J Pharm Educ. 2010;74(10):S7.
2. Scott MA, Hitch B, Ray L, Colvin G. Integration of pharmacists into a patient-centered medical home. J Am Pharm Assoc (2003). 2011;51(2):161‐166.
3. Wong SL, Barner JC, Sucic K, et al. Integration of pharmacists into patient-centered medical homes in federally qualified health centers in Texas. J Am Pharm Assoc (2003). 2017;57(3):375‐381.
4. Sapp ECH, Francis SM, Hincapie AL. Implementation of pharmacist-driven comprehensive medication management as part of an interdisciplinary team in primary care physicians’ offices. Am J Accountable Care. 2020;8(1):8-11.
5. Cowart K, Olson K. Impact of pharmacist care provision in value-based care settings: How are we measuring value-added services? J Am Pharm Assoc (2003). 2019;59(1):125-128.
6. Centers for Disease Control and Prevention. Pharmacy: Collaborative Practice Agreements to Enable Drug Therapy Management. January 16, 2018. Accessed April 17, 2021. https://www.cdc.gov/dhdsp/pubs/guides/best-practices/pharmacist-cdtm.htm
7. Choe HM, Farris KB, Stevenson JG, et al. Patient-centered medical home: developing, expanding, and sustaining a role for pharmacists. Am J Health Syst Pharm. 2012;69(12):1063-1071.
8. Coe AB, Choe HM. Pharmacists supporting population health in patient-centered medical homes. Am J Health Syst Pharm. 2017;74(18):1461-1466.
9. Luder HR, Shannon P, Kirby J, Frede SM. Community pharmacist collaboration with a patient-centered medical home: establishment of a patient-centered medical neighborhood and payment model. J Am Pharm Assoc (2003). 2018;58(1):44-50.
10. Matzke GR, Moczygemba LR, Williams KJ, et al. Impact of a pharmacist–physician collaborative care model on patient outcomes and health services utilization. 10.05Am J Health Syst Pharm. 2018;75(14):1039-1047.
11. Aneese NJ, Halalau A, Muench S, et al. Impact of a pharmacist-managed diabetes clinic on quality measures. Am J Manag Care. 2018;24(4 Spec No.):SP116-SP119.
12. Prudencio J, Cutler T, Roberts S, et al. The effect of clinical 10.05pharmacist-led comprehensive medication management on chronic disease state goal attainment in a patient-centered medical home. J Manag Care Spec Pharm. 2018;24(5):423-429.
13. Edwards HD, Webb RD, Scheid DC, et al. A pharmacist visit improves diabetes standards in a patient-centered medical home (PCMH). Am J Med Qual. 2012;27(6) 529-534.
14. Ullah S, Rajan S, Liu T, et al. Why do patients miss their appointments at primary care clinics? J Fam Med Dis Prev. 2018;4:090.
15. Moore CG, Wilson-Witherspoon P, Probst JC. Time and money: effects of no-shows at a family practice residency clinic. Fam Med. 2001;33(7):522-527.
16. Kheirkhah P, Feng Q, Travis LM, et al. Prevalence, predictors and economic consequences of no-shows. BMC Health Serv Res. 2016;16:13.
17. Little RR, Rohlfing C. The long and winding road to optimal HbA10.051c10.05 measurement. Clin Chim Acta. 2013;418:63-71.
18. Hill J, Nielsen M, Fox MH. Understanding the social factors that contribute to diabetes: a means to informing health care and social policies for the chronically ill. Perm J. 2013;17(2):67-72.
19. Gonzalez-Zacarias AA, Mavarez-Martinez A, Arias-Morales CE, et al. Impact of demographic, socioeconomic, and psychological factors on glycemic self-management in adults with type 2 diabetes mellitus. Front Public Health. 2016;4:195.
20. Pantalone KM, Misra-Hebert AD, Hobbs TD, et al. The probability of A1c goal attainment in patients with uncontrolled type 2 diabetes in a large integrated delivery system: a prediction model. Diabetes Care. 2020;43:1910-1919.
Impact of Hospitalist Programs on Perceived Care Quality, Interprofessional Collaboration, and Communication: Lessons from Implementation of 3 Hospital Medicine Programs in Canada
From the Fraser Health Authority, Surrey, BC, Canada (Drs. Yousefi and Paletta), and Catalyst Consulting Inc., Vancouver, BC, Canada (Elayne McIvor).
Objective: Despite the ongoing growth in the number of hospitalist programs in Canada, their impact on the quality of interprofessional communication, teamwork, and staff satisfaction is not well known. This study aimed to evaluate perceptions of frontline care providers and hospital managers about the impact of the implementation of 3 new hospitalist services on care quality, teamwork, and interprofessional communication.
Design: We used an online survey and semistructured interviews to evaluate respondents’ views on quality of interprofessional communication and collaboration, impact of the new services on quality of care, and overall staff satisfaction with the new inpatient care model.
Setting: Integrated Regional Health Authority in British Columbia, Canada.
Participants: Participants included hospital administrators, frontline care providers (across a range of professions), and hospital and community-based physicians.
Results: The majority of respondents reported high levels of satisfaction with their new hospital medicine services. They identified improvements in interprofessional collaboration and communication between hospitalists and other professionals, which were attributed to enhanced onsite presence of physicians. They also perceived improvements in quality of care and efficiency. On the other hand, they identified a number of challenges with the change process, and raised concerns about the impact of patient handoffs on care quality and efficiency.
Conclusion: Across 3 very different acute care settings, the implementation of a hospitalist service was widely perceived to have resulted in improved teamwork, quality of care, and interprofessional communication.
Keywords: hospital medicine; hospitalist; teamwork; interprofessional collaboration.
Over the past 2 decades, the hospitalist model has become prevalent in Canada and internationally.1 Hospitalist care has been associated with improvements in efficiency and quality of care.2-6 However, less is known about its impact on the quality of interprofessional communication, teamwork, and staff satisfaction. In a 2012 study of a specialized orthopedic facility in the Greater Toronto Area (GTA), Ontario, Webster et al found a pervasive perception among interviewees that the addition of a hospitalist resulted in improved patient safety, expedited transfers, enhanced communication with Primary Care Providers (PCPs), and better continuity of care.7 They also identified enhanced collaboration among providers since the addition of the hospitalist to the care team. In another study of 5 community hospitals in the GTA, Conn et al8 found that staff on General Internal Medicine wards where hospitalists worked described superior interprofessional collaboration, deeper interpersonal relationships between physicians and other care team members, and a higher sense of “team-based care.”
Fraser Health Authority (FH) is an integrated regional health system with one of the largest regional Hospital Medicine (HM) networks in Canada.9 Over the past 2 decades, FH has implemented a number of HM services in its acute care facilities across a range of small and large community and academic hospitals. More recently, 3 hospitalist services were implemented over a 2-year period: new HM services in a tertiary referral center (Site A, July 2016) and a small community hospital (Site B, December 2016), and reintroduction of a hospitalist service in a medium-sized community hospital (Site C, January 2017). This provided a unique opportunity to assess the impact of the implementation of the hospitalist model across a range of facilities. The main objectives of this evaluation were to understand the level of physician, nursing, allied staff, and hospital administration satisfaction with the new hospitalist model, as well as the perceived impact of the service on efficiency and quality of care. As such, FH engaged an external consultant (EM) to conduct a comprehensive evaluation of the introduction of its latest HM services.
Methods
Setting
Hospital medicine services are currently available in 10 of 12 acute care facilities within the FH system. The 3 sites described in this evaluation constitute the most recent sites where a hospitalist service was implemented.
Site A is a 272-bed tertiary referral center situated in a rapidly growing community. At the time of our evaluation, 21 Full Time Equivalent (FTE) hospitalists cared for an average of 126 patients, which constituted the majority of adult medical patients. Each day, 8 individuals rounded on admitted patients (average individual census: 16) with another person providing in-house, evening, and overnight coverage. An additional flexible shift during the early afternoon helped with Emergency Department (ED) admissions.
Site B is small, 45-bed community hospital in a semi-rural community. The hospitalist service began in December 2016, with 4 FTE hospitalists caring for an average of 28 patients daily. This constituted 2 hospitalists rounding daily on admitted patients, with on-call coverage provided from home.
Site C is a 188-bed community hospital with a hospitalist service initially introduced in 2005. In 2016, the program was disbanded and the site moved back to a primarily community-based model, in which family physicians in the community were invited to assume the care of hospitalized patients. However, the hospitalist program had to be reintroduced in January 2017 due to poor uptake among PCPs in the community. At the time of evaluation, 19 FTE hospitalists (with 7 hospitalists working daily) provided most responsible physician care to a daily census of 116 patients (average individual census: 16). The program also covered ED admissions in-house until midnight, with overnight call provided from home.
Approach
We adopted a utilization-focused evaluation approach to guide our investigation. In this approach, the assessment is deliberately planned and conducted in a way that it maximizes the likelihood that findings would be used by the organization to inform learning, adaptations, and decision-making.11 To enable this, the evaluator identified the primary intended recipients and engaged them at the start of the evaluation process to understand the main intended uses of the project. Moreover, the evaluator ensured that these intended uses of the evaluation guided all other decisions made throughout the process.
We collected data using an online survey of the staff at the 3 facilities, complemented by a series of semistructured qualitative interviews with FH administrators and frontline providers.
Online survey
We conducted an open online survey of a broad range of stakeholders who worked in the 3 facilities. To develop the questionnaire, we searched our department’s archives for previous surveys conducted from 2001 to 2005. We also interviewed the regional HM program management team to identify priority areas and reached out to the local leadership of the 3 acute care facilities for their input and support of the project. We refined the survey through several iterations, seeking input from experts in the FH Department of Evaluation and Research. The final questionnaire contained 10 items, including a mix of closed- and open-ended questions (Appendix A).
To reach the target audience, we collaborated with each hospital’s local leadership as well as the Divisions of Family Practice (DFP) that support local community PCPs in each hospital community.10 Existing email lists were compiled to create a master electronic survey distribution list. The initial invitation and 3 subsequent reminders were disseminated to the following target groups: hospital physicians (both hospitalists and nonhospitalists), PCPs, nursing and other allied professionals, administrators, and DFP leadership.
The survey consent form, background information, questions, and online platform (SimpleSurvey, Montreal, QC) were approved by FH’s Privacy Department. All respondents were required to provide their consent and able to withdraw at any time. Survey responses were kept anonymous and confidential, with results captured automatically into a spreadsheet by the survey platform. As an incentive for participation, respondents had the opportunity to win 1 of 3 $100 Visa gift cards. Personal contact information provided for the prize draw was collected in a separate survey that could not link back to respondents’ answers. The survey was trialed several times by the evaluation team to address any technical challenges before dissemination to the targeted participants.
Qualitative interviews
We conducted semistructured interviews with a purposive sample of FH administrators and frontline providers (Appendix B). The interview questions broadly mirrored the survey but allowed for more in-depth exploration of constructs. Interviewees were recruited through email invitations to selected senior and mid-level local and regional administrators, asking interviewees to refer our team to other contacts, and inviting survey respondents to voluntarily participate in a follow-up interview. One of the authors (EM), a Credentialed Evaluator, conducted all the one-time interviews either in-person at the individual participant’s workplace or by telephone. She did not have pre-existing relationships with any of the interviewees. Interviews were recorded and transcribed for analysis. Interviewees were required to consent to participate and understood that they could withdraw at any point. They were not offered incentives to participate. Interviews were carried out until thematic saturation was reached.
Analysis
A content analysis approach was employed for all qualitative data, which included open-ended responses from the online survey and interview transcripts. One of the authors (EM) conducted the analysis. The following steps were followed in the inductive content analysis process: repeated reading of the raw data, generation of initial thematic codes, organizing and sorting codes into categories (ie, main vs subcategories), coding of all data, quantifying codes, and interpreting themes. When responding to open-ended questions, respondents often provided multiple answers per question. Each of the respondents’ answers were coded. In alignment with the inductive nature of the analysis process, themes emerged organically from the data rather than the researchers using preconceived theories and categories to code the text. This was achieved by postponing the review of relevant literature on the topic until after the analysis was complete and using an external evaluation consultant (with no prior relationship to FH and limited theoretical knowledge of the topic matter) to analyze the data. Descriptive statistics were run on quantitative data in SPSS (v.24, IBM, Armonk, NY). For survey responses to be included in the analysis, the respondents needed to indicate which site they worked at and were required to answer at least 1 other survey question. One interviewee was excluded from the analysis since they were not familiar with the hospitalist model at their site.
Ethics approval
The evaluation protocol was reviewed by FH Department of Evaluation and Research and was deemed exempt from formal research ethics review.
Results
A total of 377 individuals responded to the online survey between January 8 and February 28, 2018 (response rate 14%). The distribution of respondents generally reflected the size of the respective acute care facilities. Compared to the overall sampled population, fewer nurses participated in the survey (45% vs 64%) while the rate of participation for Unit Clerks (14% vs 16%) and allied professionals (12% vs 16%) were similar.
")
Out of the 45 people approached for an interview, a total of 38 were conducted from January 3 to March 5, 2018 (response rate 84%). The interviews lasted an average of 42 minutes. Interviewees represented a range of administrative and health professional roles (Figure 1). Some interviewees held multiple positions.

Satisfaction with HM service
Across all sites, survey respondents reported high levels of satisfaction with their respective HM services and identified positive impacts on their job satisfaction (Figure 2). Almost all interviewees similarly expressed high satisfaction levels with their HM services (95%; n = 36).
")
Perceptions of HM service performance
Survey respondents rated the strength of hospitalists’ interprofessional communication and collaboration with other physicians and with care teams. Roughly two-thirds reported that overall hospitalist communication was “good” or “very good.” We also asked participants to rate the frequency at which hospitalists met best practice expectations related to interprofessional teamwork. Across all sites, similar proportions of respondents (23% to 39%) reported that these best practices were met “most of the time” or “always” (Figure 3). Survey questions also assessed perceptions of respondents about the quality and safety of care provided by hospitalists (Figure 4).
")
Perceptions of the impact of the HM service postimplementation
The majority of survey respondents reported improvements in the quality of communication, professional relationships, and coordination of inpatient care at transition points after the implementation of the HM service (Figure 5). This was also reflected in interviews, where some indicated that it was easier to communicate with hospitalists due to their on-site presence, accessibility, and 24/7 availability (n = 21). They also described improved collaboration within the care teams (n = 7), and easier communication with hospitalists because they were approachable, willing, and receptive (n = 4).
")
We also asked the survey respondents to assess the impact of the new hospitalist model on different dimensions of care quality, including patient satisfaction, patient experience, efficiency, and overall quality of care (Figure 6). Findings were comparable across these dimensions, with roughly 50-60% of respondents noting positive changes compared to before the implementation of the programs. However, most interviewees identified both positive and negative effects in these areas. Positive impacts included hospitalist on-site presence leading to better accessibility and timeliness of care (n = 5), hospitalists providing continuity to patients/families by working for weeklong rotations (n = 6), hospitalists being particularly skilled at managing complex clinical presentations (n = 2), and hospitalists being able to spend more time with patients (n = 2). On the other hand, some interviewees noted that patients and families did not like seeing multiple doctors due to frequent handoffs between hospitalists (n = 12). They also raised concerns that hospitalists did not know patients’ histories or had relationships with them, potentially leading to longer length of stay and unnecessary investigations (n = 8).
")
Site-to-site ratings of satisfaction and performance
Survey respondents’ satisfaction and performance ratings varied substantially site-to-site. Across all areas assessed, ratings were consistently highest at Site B (the smallest institution in our evaluation and the most recent addition to the HM network in the health authority). These differences were statistically significant across all survey questions asked.
Discussion
Findings from this study provide insight into the experiences of frontline health care professionals and administrators with the implementation of new HM services across a range of small to large acute care facilities. They indicate that the majority of respondents reported high levels of satisfaction with their hospitalist services. Most also indicated that the service had resulted in improvements compared to prior inpatient care models.
Over half of the survey respondents, and the majority of interviewees, reported a positive impact on interprofessional communication and collaboration. This was largely attributed to enhanced accessibility and availability of hospitalists:
- "Being on-site lends itself to better communication because they’re accessible. Hospitalists always answer the phone, but the general practitioners (GP) don’t always since they may be with other patients." (Dietician, Site A)
- "A big strength is that we have physician presence on the unit all day during scheduled hours, which makes us more accessible to nurses and more able to follow up on patients that we have concerns about." (Physician Leader, Site B)
However, the ratings dropped substantially when they were asked to assess adherence to specific best practices of such communication and collaboration, such as participation in daily check-ins or attendance at team care rounds (Figure 3). Interdisciplinary clinical rounds have been identified as a tool to improve the effectiveness of care teams.12 A number of elements have been identified as key components of effective rounds.13 Bedside rounds have also been found to enhance communication and teamwork.14,15 In our study, the discrepancy between overall high levels of satisfaction with hospitalists’ communication/collaboration despite low scores on participation in more concrete activities may illustrate the importance of informal and ad hoc opportunities for interactions between hospitalists and other care providers that result from the enhanced presence of hospitalists on care units.8 Outside of formal rounds, hospitalists have the ability to interact with other care providers throughout their shifts. Prior studies have shown that hospitalists spend a significant portion of their time communicating with other care team members throughout their workdays.16 At the same time, the amount of time spent on communication should be balanced against the need for provision of direct care at the bedside. Future research should aim to identify the right balance between these competing priorities, and to understand the nature and quality of the communication between various care providers.
We also aimed to understand the perceptions of study participants about the impact of the HM service on quality of care. Survey participants not only expressed reasonable satisfaction with various aspects of hospitalists’ performance, but also described a positive impact on care quality after the implementation of their new services. This was also reflected in the interviews:
- "The clinical knowledge of the new hospitalists is far better. Some are internal medicine trained, so they bring better knowledge and skills. I feel comfortable that they can take patients and manage them. I wasn’t always comfortable with doing that in the past." (Emergency Physician, Site C)
- "Hospitalists are really familiar with acute care and how it works. They’ve become more familiar with the discharge planning system and thus know more about the resources available. And even something as simple as knowing which forms to use." (Dietician, Site A)
It must be noted that these observations should ideally be corroborated through a robust before-after analysis of various quality measures. While such an analysis was beyond the scope of our current project, we have previously demonstrated that across our network (including the 3 sites included in our evaluation) hospitalist care is associated with lower mortality and readmission rates.4 Our findings appear to confirm previous suggestions that hospitalists’ dedicated focus on inpatient care may allow them to develop enhanced skills in the management of common conditions in the acute care setting17 which can be perceived to be of value to other hospital-based care providers.
The issue of frequent handover among hospitalists was the most commonly identified challenge by both survey respondents and interviewees:
- "They’re very reluctant to discharge patients if it’s their first day with the patient. Even if the previous hospitalist said they were ready for discharge, the new doc wants to run all of their own tests before they feel comfortable. Maybe it’s a trust issue between hospitalists when they hand patients over. It’s also being personally liable for patients if you discharge them." (Patient Care Coordinator, Site A)
- "Communication is an issue. There’s lots of turnover in hospitalists. Relationships were closer with GPs because we had so much more interaction with particular individuals." (Hospitalist Physician Leader, Site A)
It must be noted that we conducted our evaluation in a relatively short time span (within 2 years) after the 3 services were implemented. Developing trust among a large number of hospitalists newly recruited to these programs can take time and may be a factor that can explain the reluctance of some to discharge patients after handoffs. However, concerns about discontinuity of care inherent in the hospitalist model are not new.18,19 Better continuity has been associated with higher probability of patient discharges20 and improved outcomes.21 To address this challenge, the hospitalist community has focused on defining the core competencies associated with high quality handovers,22 and deliberate efforts to improve the quality of handoffs through quality improvement methodologies.23 Our study participants similarly identified these measures as potential solutions. Despite this, addressing hospitalist continuity of care remains a pressing challenge for the broader hospitalist community.24
Our evaluation has a number of methodological limitations. First, the survey response rate was only 14%, which raises questions about nonresponse bias and the representativeness of the findings to the larger population of interest. While the distribution of respondents was largely similar to the overall sampled population, a number of factors may have impacted our response rate. For example, we were only able to distribute our survey to health care providers’ institutional email addresses. Moreover, while we provided incentives for participation and sent out a number of reminders, we solely relied on one communication modality (ie, electronic communication) and did not utilize other methods (such as posters, reminder at meetings, in-person invitations). Second, while the survey included a number of open-ended questions, many of these responses were at times brief and difficult to interpret and were not included in the analysis. Third, all data collected were self-reported. For example, we could not corroborate comments about participation in interdisciplinary rounds by objective measures such as attendance records or direct observation. Self-report data is subjective in nature and is vulnerable to a range of biases, such as social desirability bias.25 Finally, patient satisfaction and experience with hospitalist care were not assessed by patients themselves. Ideally, standardized cross-site indicators should validate our patient-related results.
As mentioned above, hospitalist performance ratings varied substantially from site-to-site and were consistently higher at Site B (a small community hospital in a semi-rural area), followed by Site C (a medium-sized community hospital) and Site A (a tertiary referral center). The variability in program ratings and perceived hospitalist impacts between sites could be due to a variety of factors, such as the degree of change between the past and current models at each site, differences in hospitalist hiring processes, hospital size and culture, and differences in service design and operations. It may also be related to the timing of the introduction of the HM service, as Site B was the most recent site where the service was established. As such, there may be an element of recall bias behind the observed discrepancies. This highlights the importance of local context on respondent perceptions and suggests that our results may not be generalizable to other institutions with different attributes and characteristics.
Conclusion
Findings from this study have demonstrated that the recent hospitalist services in our health system have improved overall levels of interprofessional communication and teamwork, as well as perceptions of care quality among the majority of participants who reported high levels of satisfaction with their programs. Our findings further highlight the issue of frequent handovers among hospitalists as a pressing and ongoing challenge.
Corresponding Author: Vandad Yousefi, MD, CCFP, Past Regional Department Head – Hospital Medicine, Fraser Health Authority, Central City Tower, Suite 400, 13450 – 102nd Ave, Surrey, BC V3T 0H1; [email protected].
Financial disclosures: This project was funded by the Fraser Health Authority, which provided the funding for hiring of the external consultant to design, implement, and analyze the results of the evaluation program in collaboration with the Regional Hospitalist Program at Fraser Health.
1. Yousefi V, Wilton D. Re-designing Hospital Care: Learning from the Experience of Hospital Medicine in Canada. Journal of Global Health Care Systems. 2011;1(3).
2. White HL. Assessing the Prevalence, Penetration and Performance of Hospital Physicians in Ontario: Implications for the Quality and Efficiency of Inpatient Care. Doctoral Thesis; 2016.
3. Yousefi V, Chong CA. Does implementation of a hospitalist program in a Canadian community hospital improve measures of quality of care and utilization? An observational comparative analysis of hospitalists vs. traditional care providers. BMC Health Serv Res. 2013;13:204.
4. Yousefi V, Hejazi S, Lam A. Impact of Hospitalists on Care Outcomes in a Large Integrated Health System in British Columbia. Journal of Clinical Outcomes Management. 2020;27(2):59-72.
5. Salim SA, Elmaraezy A, Pamarthy A, et al. Impact of hospitalists on the efficiency of inpatient care and patient satisfaction: a systematic review and meta-analysis. J Community Hosp Intern Med Perspect. 2019;9(2):121-134.
6. Peterson MC. A systematic review of outcomes and quality measures in adult patients cared for by hospitalists vs nonhospitalists. Mayo Clinic Proc. 2009;84(3):248-254.
7. Webster F, Bremner S, Jackson M, et al. The impact of a hospitalist on role boundaries in an orthopedic environment. J Multidiscip Healthc. 2012;5:249-256.
8. Gotlib Conn L, Reeves S, Dainty K, et al. Interprofessional communication with hospitalist and consultant physicians in general internal medicine: a qualitative study. BMC Health Serv Res. 2012; 12:437.
9. About Fraser Health. Fraser Health Authority. Updated 2018. Accessed January 30, 2019. https://www.fraserhealth.ca/about-us/about-fraser-health#.XFJrl9JKiUk
10. Divisions of Family Practice. Accessed May 2, 2020. https://www.divisionsbc.ca/provincial/about-us
11. Patton MQ. Essentials of Utilization-Focused Evaluation. 2012. Sage Publications, Inc; 2011.
12. Buljac-Samardzic M, Doekhie KD, van Wijngaarden JDH. Interventions to improve team effectiveness within health care: a systematic review of the past decade. Hum Resour Health. 2020;18(1):2.
13. Verhaegh KJ, Seller-Boersma A, Simons R, et al. An exploratory study of healthcare professionals’ perceptions of interprofessional communication and collaboration. J Interprof Care. 2017;31(3):397-400.
14. O’Leary KJ, Johnson JK, Manojlovich M, et al. Redesigning systems to improve teamwork and quality for hospitalized patients (RESET): study protocol evaluating the effect of mentored implementation to redesign clinical microsystems. BMC Health Serv Res. 2019;19(1):293.
15. Stein J, Payne C, Methvin A, et al. Reorganizing a hospital ward as an accountable care unit. J Hosp Med. 2015;10(1):36-40.
16. Yousefi V. How Canadian hospitalists spend their time - A work-sampling study within a hospital medicine program in Ontario. Journal of Clinical Outcomes Management. 2011;18(4):159.
17. Marinella MA: Hospitalists-Where They Came from, Who They Are, and What They Do. Hosp Physician. 2002;38(5):32-36.
18. Wachter RM. An introduction to the hospitalist model. Ann Intern Med. 1999;130(4 Pt 2):338-342.
19. Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287(4):487-494.
20. van Walraven C. The Influence of Inpatient Physician Continuity on Hospital Discharge. J Gen Intern Med. 2019;34(9):1709-1714.
21. Goodwin JS, Li S, Kuo YF. Association of the Work Schedules of Hospitalists With Patient Outcomes of Hospitalization. JAMA Intern Med. 2020;180(2):215-222.
22. Nichani S, Fitterman N, Lukela M, Crocker J, the Society of Hospital Medicine, Patient Handoff. 2017 Hospital Medicine Revised Core Competencies. J Hosp Med. 2017;4:S74.
23. Lo HY, Mullan PC, Lye C, et al. A QI initiative: implementing a patient handoff checklist for pediatric hospitalist attendings. BMJ Qual Improv Rep. 2016;5(1):u212920.w5661.
24. Wachter RM, Goldman L. Zero to 50,000 - The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375(11):1009-1011.
25. Grimm, P. Social Desirability Bias. In: Sheth J, Malhotra N, eds. Wiley International Encyclopedia of Marketing. John Wiley & Sons, Ltd; 2010.
From the Fraser Health Authority, Surrey, BC, Canada (Drs. Yousefi and Paletta), and Catalyst Consulting Inc., Vancouver, BC, Canada (Elayne McIvor).
Objective: Despite the ongoing growth in the number of hospitalist programs in Canada, their impact on the quality of interprofessional communication, teamwork, and staff satisfaction is not well known. This study aimed to evaluate perceptions of frontline care providers and hospital managers about the impact of the implementation of 3 new hospitalist services on care quality, teamwork, and interprofessional communication.
Design: We used an online survey and semistructured interviews to evaluate respondents’ views on quality of interprofessional communication and collaboration, impact of the new services on quality of care, and overall staff satisfaction with the new inpatient care model.
Setting: Integrated Regional Health Authority in British Columbia, Canada.
Participants: Participants included hospital administrators, frontline care providers (across a range of professions), and hospital and community-based physicians.
Results: The majority of respondents reported high levels of satisfaction with their new hospital medicine services. They identified improvements in interprofessional collaboration and communication between hospitalists and other professionals, which were attributed to enhanced onsite presence of physicians. They also perceived improvements in quality of care and efficiency. On the other hand, they identified a number of challenges with the change process, and raised concerns about the impact of patient handoffs on care quality and efficiency.
Conclusion: Across 3 very different acute care settings, the implementation of a hospitalist service was widely perceived to have resulted in improved teamwork, quality of care, and interprofessional communication.
Keywords: hospital medicine; hospitalist; teamwork; interprofessional collaboration.
Over the past 2 decades, the hospitalist model has become prevalent in Canada and internationally.1 Hospitalist care has been associated with improvements in efficiency and quality of care.2-6 However, less is known about its impact on the quality of interprofessional communication, teamwork, and staff satisfaction. In a 2012 study of a specialized orthopedic facility in the Greater Toronto Area (GTA), Ontario, Webster et al found a pervasive perception among interviewees that the addition of a hospitalist resulted in improved patient safety, expedited transfers, enhanced communication with Primary Care Providers (PCPs), and better continuity of care.7 They also identified enhanced collaboration among providers since the addition of the hospitalist to the care team. In another study of 5 community hospitals in the GTA, Conn et al8 found that staff on General Internal Medicine wards where hospitalists worked described superior interprofessional collaboration, deeper interpersonal relationships between physicians and other care team members, and a higher sense of “team-based care.”
Fraser Health Authority (FH) is an integrated regional health system with one of the largest regional Hospital Medicine (HM) networks in Canada.9 Over the past 2 decades, FH has implemented a number of HM services in its acute care facilities across a range of small and large community and academic hospitals. More recently, 3 hospitalist services were implemented over a 2-year period: new HM services in a tertiary referral center (Site A, July 2016) and a small community hospital (Site B, December 2016), and reintroduction of a hospitalist service in a medium-sized community hospital (Site C, January 2017). This provided a unique opportunity to assess the impact of the implementation of the hospitalist model across a range of facilities. The main objectives of this evaluation were to understand the level of physician, nursing, allied staff, and hospital administration satisfaction with the new hospitalist model, as well as the perceived impact of the service on efficiency and quality of care. As such, FH engaged an external consultant (EM) to conduct a comprehensive evaluation of the introduction of its latest HM services.
Methods
Setting
Hospital medicine services are currently available in 10 of 12 acute care facilities within the FH system. The 3 sites described in this evaluation constitute the most recent sites where a hospitalist service was implemented.
Site A is a 272-bed tertiary referral center situated in a rapidly growing community. At the time of our evaluation, 21 Full Time Equivalent (FTE) hospitalists cared for an average of 126 patients, which constituted the majority of adult medical patients. Each day, 8 individuals rounded on admitted patients (average individual census: 16) with another person providing in-house, evening, and overnight coverage. An additional flexible shift during the early afternoon helped with Emergency Department (ED) admissions.
Site B is small, 45-bed community hospital in a semi-rural community. The hospitalist service began in December 2016, with 4 FTE hospitalists caring for an average of 28 patients daily. This constituted 2 hospitalists rounding daily on admitted patients, with on-call coverage provided from home.
Site C is a 188-bed community hospital with a hospitalist service initially introduced in 2005. In 2016, the program was disbanded and the site moved back to a primarily community-based model, in which family physicians in the community were invited to assume the care of hospitalized patients. However, the hospitalist program had to be reintroduced in January 2017 due to poor uptake among PCPs in the community. At the time of evaluation, 19 FTE hospitalists (with 7 hospitalists working daily) provided most responsible physician care to a daily census of 116 patients (average individual census: 16). The program also covered ED admissions in-house until midnight, with overnight call provided from home.
Approach
We adopted a utilization-focused evaluation approach to guide our investigation. In this approach, the assessment is deliberately planned and conducted in a way that it maximizes the likelihood that findings would be used by the organization to inform learning, adaptations, and decision-making.11 To enable this, the evaluator identified the primary intended recipients and engaged them at the start of the evaluation process to understand the main intended uses of the project. Moreover, the evaluator ensured that these intended uses of the evaluation guided all other decisions made throughout the process.
We collected data using an online survey of the staff at the 3 facilities, complemented by a series of semistructured qualitative interviews with FH administrators and frontline providers.
Online survey
We conducted an open online survey of a broad range of stakeholders who worked in the 3 facilities. To develop the questionnaire, we searched our department’s archives for previous surveys conducted from 2001 to 2005. We also interviewed the regional HM program management team to identify priority areas and reached out to the local leadership of the 3 acute care facilities for their input and support of the project. We refined the survey through several iterations, seeking input from experts in the FH Department of Evaluation and Research. The final questionnaire contained 10 items, including a mix of closed- and open-ended questions (Appendix A).
To reach the target audience, we collaborated with each hospital’s local leadership as well as the Divisions of Family Practice (DFP) that support local community PCPs in each hospital community.10 Existing email lists were compiled to create a master electronic survey distribution list. The initial invitation and 3 subsequent reminders were disseminated to the following target groups: hospital physicians (both hospitalists and nonhospitalists), PCPs, nursing and other allied professionals, administrators, and DFP leadership.
The survey consent form, background information, questions, and online platform (SimpleSurvey, Montreal, QC) were approved by FH’s Privacy Department. All respondents were required to provide their consent and able to withdraw at any time. Survey responses were kept anonymous and confidential, with results captured automatically into a spreadsheet by the survey platform. As an incentive for participation, respondents had the opportunity to win 1 of 3 $100 Visa gift cards. Personal contact information provided for the prize draw was collected in a separate survey that could not link back to respondents’ answers. The survey was trialed several times by the evaluation team to address any technical challenges before dissemination to the targeted participants.
Qualitative interviews
We conducted semistructured interviews with a purposive sample of FH administrators and frontline providers (Appendix B). The interview questions broadly mirrored the survey but allowed for more in-depth exploration of constructs. Interviewees were recruited through email invitations to selected senior and mid-level local and regional administrators, asking interviewees to refer our team to other contacts, and inviting survey respondents to voluntarily participate in a follow-up interview. One of the authors (EM), a Credentialed Evaluator, conducted all the one-time interviews either in-person at the individual participant’s workplace or by telephone. She did not have pre-existing relationships with any of the interviewees. Interviews were recorded and transcribed for analysis. Interviewees were required to consent to participate and understood that they could withdraw at any point. They were not offered incentives to participate. Interviews were carried out until thematic saturation was reached.
Analysis
A content analysis approach was employed for all qualitative data, which included open-ended responses from the online survey and interview transcripts. One of the authors (EM) conducted the analysis. The following steps were followed in the inductive content analysis process: repeated reading of the raw data, generation of initial thematic codes, organizing and sorting codes into categories (ie, main vs subcategories), coding of all data, quantifying codes, and interpreting themes. When responding to open-ended questions, respondents often provided multiple answers per question. Each of the respondents’ answers were coded. In alignment with the inductive nature of the analysis process, themes emerged organically from the data rather than the researchers using preconceived theories and categories to code the text. This was achieved by postponing the review of relevant literature on the topic until after the analysis was complete and using an external evaluation consultant (with no prior relationship to FH and limited theoretical knowledge of the topic matter) to analyze the data. Descriptive statistics were run on quantitative data in SPSS (v.24, IBM, Armonk, NY). For survey responses to be included in the analysis, the respondents needed to indicate which site they worked at and were required to answer at least 1 other survey question. One interviewee was excluded from the analysis since they were not familiar with the hospitalist model at their site.
Ethics approval
The evaluation protocol was reviewed by FH Department of Evaluation and Research and was deemed exempt from formal research ethics review.
Results
A total of 377 individuals responded to the online survey between January 8 and February 28, 2018 (response rate 14%). The distribution of respondents generally reflected the size of the respective acute care facilities. Compared to the overall sampled population, fewer nurses participated in the survey (45% vs 64%) while the rate of participation for Unit Clerks (14% vs 16%) and allied professionals (12% vs 16%) were similar.
Out of the 45 people approached for an interview, a total of 38 were conducted from January 3 to March 5, 2018 (response rate 84%). The interviews lasted an average of 42 minutes. Interviewees represented a range of administrative and health professional roles (Figure 1). Some interviewees held multiple positions.
Satisfaction with HM service
Across all sites, survey respondents reported high levels of satisfaction with their respective HM services and identified positive impacts on their job satisfaction (Figure 2). Almost all interviewees similarly expressed high satisfaction levels with their HM services (95%; n = 36).
Perceptions of HM service performance
Survey respondents rated the strength of hospitalists’ interprofessional communication and collaboration with other physicians and with care teams. Roughly two-thirds reported that overall hospitalist communication was “good” or “very good.” We also asked participants to rate the frequency at which hospitalists met best practice expectations related to interprofessional teamwork. Across all sites, similar proportions of respondents (23% to 39%) reported that these best practices were met “most of the time” or “always” (Figure 3). Survey questions also assessed perceptions of respondents about the quality and safety of care provided by hospitalists (Figure 4).
Perceptions of the impact of the HM service postimplementation
The majority of survey respondents reported improvements in the quality of communication, professional relationships, and coordination of inpatient care at transition points after the implementation of the HM service (Figure 5). This was also reflected in interviews, where some indicated that it was easier to communicate with hospitalists due to their on-site presence, accessibility, and 24/7 availability (n = 21). They also described improved collaboration within the care teams (n = 7), and easier communication with hospitalists because they were approachable, willing, and receptive (n = 4).
We also asked the survey respondents to assess the impact of the new hospitalist model on different dimensions of care quality, including patient satisfaction, patient experience, efficiency, and overall quality of care (Figure 6). Findings were comparable across these dimensions, with roughly 50-60% of respondents noting positive changes compared to before the implementation of the programs. However, most interviewees identified both positive and negative effects in these areas. Positive impacts included hospitalist on-site presence leading to better accessibility and timeliness of care (n = 5), hospitalists providing continuity to patients/families by working for weeklong rotations (n = 6), hospitalists being particularly skilled at managing complex clinical presentations (n = 2), and hospitalists being able to spend more time with patients (n = 2). On the other hand, some interviewees noted that patients and families did not like seeing multiple doctors due to frequent handoffs between hospitalists (n = 12). They also raised concerns that hospitalists did not know patients’ histories or had relationships with them, potentially leading to longer length of stay and unnecessary investigations (n = 8).
Site-to-site ratings of satisfaction and performance
Survey respondents’ satisfaction and performance ratings varied substantially site-to-site. Across all areas assessed, ratings were consistently highest at Site B (the smallest institution in our evaluation and the most recent addition to the HM network in the health authority). These differences were statistically significant across all survey questions asked.
Discussion
Findings from this study provide insight into the experiences of frontline health care professionals and administrators with the implementation of new HM services across a range of small to large acute care facilities. They indicate that the majority of respondents reported high levels of satisfaction with their hospitalist services. Most also indicated that the service had resulted in improvements compared to prior inpatient care models.
Over half of the survey respondents, and the majority of interviewees, reported a positive impact on interprofessional communication and collaboration. This was largely attributed to enhanced accessibility and availability of hospitalists:
- "Being on-site lends itself to better communication because they’re accessible. Hospitalists always answer the phone, but the general practitioners (GP) don’t always since they may be with other patients." (Dietician, Site A)
- "A big strength is that we have physician presence on the unit all day during scheduled hours, which makes us more accessible to nurses and more able to follow up on patients that we have concerns about." (Physician Leader, Site B)
However, the ratings dropped substantially when they were asked to assess adherence to specific best practices of such communication and collaboration, such as participation in daily check-ins or attendance at team care rounds (Figure 3). Interdisciplinary clinical rounds have been identified as a tool to improve the effectiveness of care teams.12 A number of elements have been identified as key components of effective rounds.13 Bedside rounds have also been found to enhance communication and teamwork.14,15 In our study, the discrepancy between overall high levels of satisfaction with hospitalists’ communication/collaboration despite low scores on participation in more concrete activities may illustrate the importance of informal and ad hoc opportunities for interactions between hospitalists and other care providers that result from the enhanced presence of hospitalists on care units.8 Outside of formal rounds, hospitalists have the ability to interact with other care providers throughout their shifts. Prior studies have shown that hospitalists spend a significant portion of their time communicating with other care team members throughout their workdays.16 At the same time, the amount of time spent on communication should be balanced against the need for provision of direct care at the bedside. Future research should aim to identify the right balance between these competing priorities, and to understand the nature and quality of the communication between various care providers.
We also aimed to understand the perceptions of study participants about the impact of the HM service on quality of care. Survey participants not only expressed reasonable satisfaction with various aspects of hospitalists’ performance, but also described a positive impact on care quality after the implementation of their new services. This was also reflected in the interviews:
- "The clinical knowledge of the new hospitalists is far better. Some are internal medicine trained, so they bring better knowledge and skills. I feel comfortable that they can take patients and manage them. I wasn’t always comfortable with doing that in the past." (Emergency Physician, Site C)
- "Hospitalists are really familiar with acute care and how it works. They’ve become more familiar with the discharge planning system and thus know more about the resources available. And even something as simple as knowing which forms to use." (Dietician, Site A)
It must be noted that these observations should ideally be corroborated through a robust before-after analysis of various quality measures. While such an analysis was beyond the scope of our current project, we have previously demonstrated that across our network (including the 3 sites included in our evaluation) hospitalist care is associated with lower mortality and readmission rates.4 Our findings appear to confirm previous suggestions that hospitalists’ dedicated focus on inpatient care may allow them to develop enhanced skills in the management of common conditions in the acute care setting17 which can be perceived to be of value to other hospital-based care providers.
The issue of frequent handover among hospitalists was the most commonly identified challenge by both survey respondents and interviewees:
- "They’re very reluctant to discharge patients if it’s their first day with the patient. Even if the previous hospitalist said they were ready for discharge, the new doc wants to run all of their own tests before they feel comfortable. Maybe it’s a trust issue between hospitalists when they hand patients over. It’s also being personally liable for patients if you discharge them." (Patient Care Coordinator, Site A)
- "Communication is an issue. There’s lots of turnover in hospitalists. Relationships were closer with GPs because we had so much more interaction with particular individuals." (Hospitalist Physician Leader, Site A)
It must be noted that we conducted our evaluation in a relatively short time span (within 2 years) after the 3 services were implemented. Developing trust among a large number of hospitalists newly recruited to these programs can take time and may be a factor that can explain the reluctance of some to discharge patients after handoffs. However, concerns about discontinuity of care inherent in the hospitalist model are not new.18,19 Better continuity has been associated with higher probability of patient discharges20 and improved outcomes.21 To address this challenge, the hospitalist community has focused on defining the core competencies associated with high quality handovers,22 and deliberate efforts to improve the quality of handoffs through quality improvement methodologies.23 Our study participants similarly identified these measures as potential solutions. Despite this, addressing hospitalist continuity of care remains a pressing challenge for the broader hospitalist community.24
Our evaluation has a number of methodological limitations. First, the survey response rate was only 14%, which raises questions about nonresponse bias and the representativeness of the findings to the larger population of interest. While the distribution of respondents was largely similar to the overall sampled population, a number of factors may have impacted our response rate. For example, we were only able to distribute our survey to health care providers’ institutional email addresses. Moreover, while we provided incentives for participation and sent out a number of reminders, we solely relied on one communication modality (ie, electronic communication) and did not utilize other methods (such as posters, reminder at meetings, in-person invitations). Second, while the survey included a number of open-ended questions, many of these responses were at times brief and difficult to interpret and were not included in the analysis. Third, all data collected were self-reported. For example, we could not corroborate comments about participation in interdisciplinary rounds by objective measures such as attendance records or direct observation. Self-report data is subjective in nature and is vulnerable to a range of biases, such as social desirability bias.25 Finally, patient satisfaction and experience with hospitalist care were not assessed by patients themselves. Ideally, standardized cross-site indicators should validate our patient-related results.
As mentioned above, hospitalist performance ratings varied substantially from site-to-site and were consistently higher at Site B (a small community hospital in a semi-rural area), followed by Site C (a medium-sized community hospital) and Site A (a tertiary referral center). The variability in program ratings and perceived hospitalist impacts between sites could be due to a variety of factors, such as the degree of change between the past and current models at each site, differences in hospitalist hiring processes, hospital size and culture, and differences in service design and operations. It may also be related to the timing of the introduction of the HM service, as Site B was the most recent site where the service was established. As such, there may be an element of recall bias behind the observed discrepancies. This highlights the importance of local context on respondent perceptions and suggests that our results may not be generalizable to other institutions with different attributes and characteristics.
Conclusion
Findings from this study have demonstrated that the recent hospitalist services in our health system have improved overall levels of interprofessional communication and teamwork, as well as perceptions of care quality among the majority of participants who reported high levels of satisfaction with their programs. Our findings further highlight the issue of frequent handovers among hospitalists as a pressing and ongoing challenge.
Corresponding Author: Vandad Yousefi, MD, CCFP, Past Regional Department Head – Hospital Medicine, Fraser Health Authority, Central City Tower, Suite 400, 13450 – 102nd Ave, Surrey, BC V3T 0H1; [email protected].
Financial disclosures: This project was funded by the Fraser Health Authority, which provided the funding for hiring of the external consultant to design, implement, and analyze the results of the evaluation program in collaboration with the Regional Hospitalist Program at Fraser Health.
From the Fraser Health Authority, Surrey, BC, Canada (Drs. Yousefi and Paletta), and Catalyst Consulting Inc., Vancouver, BC, Canada (Elayne McIvor).
Objective: Despite the ongoing growth in the number of hospitalist programs in Canada, their impact on the quality of interprofessional communication, teamwork, and staff satisfaction is not well known. This study aimed to evaluate perceptions of frontline care providers and hospital managers about the impact of the implementation of 3 new hospitalist services on care quality, teamwork, and interprofessional communication.
Design: We used an online survey and semistructured interviews to evaluate respondents’ views on quality of interprofessional communication and collaboration, impact of the new services on quality of care, and overall staff satisfaction with the new inpatient care model.
Setting: Integrated Regional Health Authority in British Columbia, Canada.
Participants: Participants included hospital administrators, frontline care providers (across a range of professions), and hospital and community-based physicians.
Results: The majority of respondents reported high levels of satisfaction with their new hospital medicine services. They identified improvements in interprofessional collaboration and communication between hospitalists and other professionals, which were attributed to enhanced onsite presence of physicians. They also perceived improvements in quality of care and efficiency. On the other hand, they identified a number of challenges with the change process, and raised concerns about the impact of patient handoffs on care quality and efficiency.
Conclusion: Across 3 very different acute care settings, the implementation of a hospitalist service was widely perceived to have resulted in improved teamwork, quality of care, and interprofessional communication.
Keywords: hospital medicine; hospitalist; teamwork; interprofessional collaboration.
Over the past 2 decades, the hospitalist model has become prevalent in Canada and internationally.1 Hospitalist care has been associated with improvements in efficiency and quality of care.2-6 However, less is known about its impact on the quality of interprofessional communication, teamwork, and staff satisfaction. In a 2012 study of a specialized orthopedic facility in the Greater Toronto Area (GTA), Ontario, Webster et al found a pervasive perception among interviewees that the addition of a hospitalist resulted in improved patient safety, expedited transfers, enhanced communication with Primary Care Providers (PCPs), and better continuity of care.7 They also identified enhanced collaboration among providers since the addition of the hospitalist to the care team. In another study of 5 community hospitals in the GTA, Conn et al8 found that staff on General Internal Medicine wards where hospitalists worked described superior interprofessional collaboration, deeper interpersonal relationships between physicians and other care team members, and a higher sense of “team-based care.”
Fraser Health Authority (FH) is an integrated regional health system with one of the largest regional Hospital Medicine (HM) networks in Canada.9 Over the past 2 decades, FH has implemented a number of HM services in its acute care facilities across a range of small and large community and academic hospitals. More recently, 3 hospitalist services were implemented over a 2-year period: new HM services in a tertiary referral center (Site A, July 2016) and a small community hospital (Site B, December 2016), and reintroduction of a hospitalist service in a medium-sized community hospital (Site C, January 2017). This provided a unique opportunity to assess the impact of the implementation of the hospitalist model across a range of facilities. The main objectives of this evaluation were to understand the level of physician, nursing, allied staff, and hospital administration satisfaction with the new hospitalist model, as well as the perceived impact of the service on efficiency and quality of care. As such, FH engaged an external consultant (EM) to conduct a comprehensive evaluation of the introduction of its latest HM services.
Methods
Setting
Hospital medicine services are currently available in 10 of 12 acute care facilities within the FH system. The 3 sites described in this evaluation constitute the most recent sites where a hospitalist service was implemented.
Site A is a 272-bed tertiary referral center situated in a rapidly growing community. At the time of our evaluation, 21 Full Time Equivalent (FTE) hospitalists cared for an average of 126 patients, which constituted the majority of adult medical patients. Each day, 8 individuals rounded on admitted patients (average individual census: 16) with another person providing in-house, evening, and overnight coverage. An additional flexible shift during the early afternoon helped with Emergency Department (ED) admissions.
Site B is small, 45-bed community hospital in a semi-rural community. The hospitalist service began in December 2016, with 4 FTE hospitalists caring for an average of 28 patients daily. This constituted 2 hospitalists rounding daily on admitted patients, with on-call coverage provided from home.
Site C is a 188-bed community hospital with a hospitalist service initially introduced in 2005. In 2016, the program was disbanded and the site moved back to a primarily community-based model, in which family physicians in the community were invited to assume the care of hospitalized patients. However, the hospitalist program had to be reintroduced in January 2017 due to poor uptake among PCPs in the community. At the time of evaluation, 19 FTE hospitalists (with 7 hospitalists working daily) provided most responsible physician care to a daily census of 116 patients (average individual census: 16). The program also covered ED admissions in-house until midnight, with overnight call provided from home.
Approach
We adopted a utilization-focused evaluation approach to guide our investigation. In this approach, the assessment is deliberately planned and conducted in a way that it maximizes the likelihood that findings would be used by the organization to inform learning, adaptations, and decision-making.11 To enable this, the evaluator identified the primary intended recipients and engaged them at the start of the evaluation process to understand the main intended uses of the project. Moreover, the evaluator ensured that these intended uses of the evaluation guided all other decisions made throughout the process.
We collected data using an online survey of the staff at the 3 facilities, complemented by a series of semistructured qualitative interviews with FH administrators and frontline providers.
Online survey
We conducted an open online survey of a broad range of stakeholders who worked in the 3 facilities. To develop the questionnaire, we searched our department’s archives for previous surveys conducted from 2001 to 2005. We also interviewed the regional HM program management team to identify priority areas and reached out to the local leadership of the 3 acute care facilities for their input and support of the project. We refined the survey through several iterations, seeking input from experts in the FH Department of Evaluation and Research. The final questionnaire contained 10 items, including a mix of closed- and open-ended questions (Appendix A).
To reach the target audience, we collaborated with each hospital’s local leadership as well as the Divisions of Family Practice (DFP) that support local community PCPs in each hospital community.10 Existing email lists were compiled to create a master electronic survey distribution list. The initial invitation and 3 subsequent reminders were disseminated to the following target groups: hospital physicians (both hospitalists and nonhospitalists), PCPs, nursing and other allied professionals, administrators, and DFP leadership.
The survey consent form, background information, questions, and online platform (SimpleSurvey, Montreal, QC) were approved by FH’s Privacy Department. All respondents were required to provide their consent and able to withdraw at any time. Survey responses were kept anonymous and confidential, with results captured automatically into a spreadsheet by the survey platform. As an incentive for participation, respondents had the opportunity to win 1 of 3 $100 Visa gift cards. Personal contact information provided for the prize draw was collected in a separate survey that could not link back to respondents’ answers. The survey was trialed several times by the evaluation team to address any technical challenges before dissemination to the targeted participants.
Qualitative interviews
We conducted semistructured interviews with a purposive sample of FH administrators and frontline providers (Appendix B). The interview questions broadly mirrored the survey but allowed for more in-depth exploration of constructs. Interviewees were recruited through email invitations to selected senior and mid-level local and regional administrators, asking interviewees to refer our team to other contacts, and inviting survey respondents to voluntarily participate in a follow-up interview. One of the authors (EM), a Credentialed Evaluator, conducted all the one-time interviews either in-person at the individual participant’s workplace or by telephone. She did not have pre-existing relationships with any of the interviewees. Interviews were recorded and transcribed for analysis. Interviewees were required to consent to participate and understood that they could withdraw at any point. They were not offered incentives to participate. Interviews were carried out until thematic saturation was reached.
Analysis
A content analysis approach was employed for all qualitative data, which included open-ended responses from the online survey and interview transcripts. One of the authors (EM) conducted the analysis. The following steps were followed in the inductive content analysis process: repeated reading of the raw data, generation of initial thematic codes, organizing and sorting codes into categories (ie, main vs subcategories), coding of all data, quantifying codes, and interpreting themes. When responding to open-ended questions, respondents often provided multiple answers per question. Each of the respondents’ answers were coded. In alignment with the inductive nature of the analysis process, themes emerged organically from the data rather than the researchers using preconceived theories and categories to code the text. This was achieved by postponing the review of relevant literature on the topic until after the analysis was complete and using an external evaluation consultant (with no prior relationship to FH and limited theoretical knowledge of the topic matter) to analyze the data. Descriptive statistics were run on quantitative data in SPSS (v.24, IBM, Armonk, NY). For survey responses to be included in the analysis, the respondents needed to indicate which site they worked at and were required to answer at least 1 other survey question. One interviewee was excluded from the analysis since they were not familiar with the hospitalist model at their site.
Ethics approval
The evaluation protocol was reviewed by FH Department of Evaluation and Research and was deemed exempt from formal research ethics review.
Results
A total of 377 individuals responded to the online survey between January 8 and February 28, 2018 (response rate 14%). The distribution of respondents generally reflected the size of the respective acute care facilities. Compared to the overall sampled population, fewer nurses participated in the survey (45% vs 64%) while the rate of participation for Unit Clerks (14% vs 16%) and allied professionals (12% vs 16%) were similar.
Out of the 45 people approached for an interview, a total of 38 were conducted from January 3 to March 5, 2018 (response rate 84%). The interviews lasted an average of 42 minutes. Interviewees represented a range of administrative and health professional roles (Figure 1). Some interviewees held multiple positions.
Satisfaction with HM service
Across all sites, survey respondents reported high levels of satisfaction with their respective HM services and identified positive impacts on their job satisfaction (Figure 2). Almost all interviewees similarly expressed high satisfaction levels with their HM services (95%; n = 36).
Perceptions of HM service performance
Survey respondents rated the strength of hospitalists’ interprofessional communication and collaboration with other physicians and with care teams. Roughly two-thirds reported that overall hospitalist communication was “good” or “very good.” We also asked participants to rate the frequency at which hospitalists met best practice expectations related to interprofessional teamwork. Across all sites, similar proportions of respondents (23% to 39%) reported that these best practices were met “most of the time” or “always” (Figure 3). Survey questions also assessed perceptions of respondents about the quality and safety of care provided by hospitalists (Figure 4).
Perceptions of the impact of the HM service postimplementation
The majority of survey respondents reported improvements in the quality of communication, professional relationships, and coordination of inpatient care at transition points after the implementation of the HM service (Figure 5). This was also reflected in interviews, where some indicated that it was easier to communicate with hospitalists due to their on-site presence, accessibility, and 24/7 availability (n = 21). They also described improved collaboration within the care teams (n = 7), and easier communication with hospitalists because they were approachable, willing, and receptive (n = 4).
We also asked the survey respondents to assess the impact of the new hospitalist model on different dimensions of care quality, including patient satisfaction, patient experience, efficiency, and overall quality of care (Figure 6). Findings were comparable across these dimensions, with roughly 50-60% of respondents noting positive changes compared to before the implementation of the programs. However, most interviewees identified both positive and negative effects in these areas. Positive impacts included hospitalist on-site presence leading to better accessibility and timeliness of care (n = 5), hospitalists providing continuity to patients/families by working for weeklong rotations (n = 6), hospitalists being particularly skilled at managing complex clinical presentations (n = 2), and hospitalists being able to spend more time with patients (n = 2). On the other hand, some interviewees noted that patients and families did not like seeing multiple doctors due to frequent handoffs between hospitalists (n = 12). They also raised concerns that hospitalists did not know patients’ histories or had relationships with them, potentially leading to longer length of stay and unnecessary investigations (n = 8).
Site-to-site ratings of satisfaction and performance
Survey respondents’ satisfaction and performance ratings varied substantially site-to-site. Across all areas assessed, ratings were consistently highest at Site B (the smallest institution in our evaluation and the most recent addition to the HM network in the health authority). These differences were statistically significant across all survey questions asked.
Discussion
Findings from this study provide insight into the experiences of frontline health care professionals and administrators with the implementation of new HM services across a range of small to large acute care facilities. They indicate that the majority of respondents reported high levels of satisfaction with their hospitalist services. Most also indicated that the service had resulted in improvements compared to prior inpatient care models.
Over half of the survey respondents, and the majority of interviewees, reported a positive impact on interprofessional communication and collaboration. This was largely attributed to enhanced accessibility and availability of hospitalists:
- "Being on-site lends itself to better communication because they’re accessible. Hospitalists always answer the phone, but the general practitioners (GP) don’t always since they may be with other patients." (Dietician, Site A)
- "A big strength is that we have physician presence on the unit all day during scheduled hours, which makes us more accessible to nurses and more able to follow up on patients that we have concerns about." (Physician Leader, Site B)
However, the ratings dropped substantially when they were asked to assess adherence to specific best practices of such communication and collaboration, such as participation in daily check-ins or attendance at team care rounds (Figure 3). Interdisciplinary clinical rounds have been identified as a tool to improve the effectiveness of care teams.12 A number of elements have been identified as key components of effective rounds.13 Bedside rounds have also been found to enhance communication and teamwork.14,15 In our study, the discrepancy between overall high levels of satisfaction with hospitalists’ communication/collaboration despite low scores on participation in more concrete activities may illustrate the importance of informal and ad hoc opportunities for interactions between hospitalists and other care providers that result from the enhanced presence of hospitalists on care units.8 Outside of formal rounds, hospitalists have the ability to interact with other care providers throughout their shifts. Prior studies have shown that hospitalists spend a significant portion of their time communicating with other care team members throughout their workdays.16 At the same time, the amount of time spent on communication should be balanced against the need for provision of direct care at the bedside. Future research should aim to identify the right balance between these competing priorities, and to understand the nature and quality of the communication between various care providers.
We also aimed to understand the perceptions of study participants about the impact of the HM service on quality of care. Survey participants not only expressed reasonable satisfaction with various aspects of hospitalists’ performance, but also described a positive impact on care quality after the implementation of their new services. This was also reflected in the interviews:
- "The clinical knowledge of the new hospitalists is far better. Some are internal medicine trained, so they bring better knowledge and skills. I feel comfortable that they can take patients and manage them. I wasn’t always comfortable with doing that in the past." (Emergency Physician, Site C)
- "Hospitalists are really familiar with acute care and how it works. They’ve become more familiar with the discharge planning system and thus know more about the resources available. And even something as simple as knowing which forms to use." (Dietician, Site A)
It must be noted that these observations should ideally be corroborated through a robust before-after analysis of various quality measures. While such an analysis was beyond the scope of our current project, we have previously demonstrated that across our network (including the 3 sites included in our evaluation) hospitalist care is associated with lower mortality and readmission rates.4 Our findings appear to confirm previous suggestions that hospitalists’ dedicated focus on inpatient care may allow them to develop enhanced skills in the management of common conditions in the acute care setting17 which can be perceived to be of value to other hospital-based care providers.
The issue of frequent handover among hospitalists was the most commonly identified challenge by both survey respondents and interviewees:
- "They’re very reluctant to discharge patients if it’s their first day with the patient. Even if the previous hospitalist said they were ready for discharge, the new doc wants to run all of their own tests before they feel comfortable. Maybe it’s a trust issue between hospitalists when they hand patients over. It’s also being personally liable for patients if you discharge them." (Patient Care Coordinator, Site A)
- "Communication is an issue. There’s lots of turnover in hospitalists. Relationships were closer with GPs because we had so much more interaction with particular individuals." (Hospitalist Physician Leader, Site A)
It must be noted that we conducted our evaluation in a relatively short time span (within 2 years) after the 3 services were implemented. Developing trust among a large number of hospitalists newly recruited to these programs can take time and may be a factor that can explain the reluctance of some to discharge patients after handoffs. However, concerns about discontinuity of care inherent in the hospitalist model are not new.18,19 Better continuity has been associated with higher probability of patient discharges20 and improved outcomes.21 To address this challenge, the hospitalist community has focused on defining the core competencies associated with high quality handovers,22 and deliberate efforts to improve the quality of handoffs through quality improvement methodologies.23 Our study participants similarly identified these measures as potential solutions. Despite this, addressing hospitalist continuity of care remains a pressing challenge for the broader hospitalist community.24
Our evaluation has a number of methodological limitations. First, the survey response rate was only 14%, which raises questions about nonresponse bias and the representativeness of the findings to the larger population of interest. While the distribution of respondents was largely similar to the overall sampled population, a number of factors may have impacted our response rate. For example, we were only able to distribute our survey to health care providers’ institutional email addresses. Moreover, while we provided incentives for participation and sent out a number of reminders, we solely relied on one communication modality (ie, electronic communication) and did not utilize other methods (such as posters, reminder at meetings, in-person invitations). Second, while the survey included a number of open-ended questions, many of these responses were at times brief and difficult to interpret and were not included in the analysis. Third, all data collected were self-reported. For example, we could not corroborate comments about participation in interdisciplinary rounds by objective measures such as attendance records or direct observation. Self-report data is subjective in nature and is vulnerable to a range of biases, such as social desirability bias.25 Finally, patient satisfaction and experience with hospitalist care were not assessed by patients themselves. Ideally, standardized cross-site indicators should validate our patient-related results.
As mentioned above, hospitalist performance ratings varied substantially from site-to-site and were consistently higher at Site B (a small community hospital in a semi-rural area), followed by Site C (a medium-sized community hospital) and Site A (a tertiary referral center). The variability in program ratings and perceived hospitalist impacts between sites could be due to a variety of factors, such as the degree of change between the past and current models at each site, differences in hospitalist hiring processes, hospital size and culture, and differences in service design and operations. It may also be related to the timing of the introduction of the HM service, as Site B was the most recent site where the service was established. As such, there may be an element of recall bias behind the observed discrepancies. This highlights the importance of local context on respondent perceptions and suggests that our results may not be generalizable to other institutions with different attributes and characteristics.
Conclusion
Findings from this study have demonstrated that the recent hospitalist services in our health system have improved overall levels of interprofessional communication and teamwork, as well as perceptions of care quality among the majority of participants who reported high levels of satisfaction with their programs. Our findings further highlight the issue of frequent handovers among hospitalists as a pressing and ongoing challenge.
Corresponding Author: Vandad Yousefi, MD, CCFP, Past Regional Department Head – Hospital Medicine, Fraser Health Authority, Central City Tower, Suite 400, 13450 – 102nd Ave, Surrey, BC V3T 0H1; [email protected].
Financial disclosures: This project was funded by the Fraser Health Authority, which provided the funding for hiring of the external consultant to design, implement, and analyze the results of the evaluation program in collaboration with the Regional Hospitalist Program at Fraser Health.
1. Yousefi V, Wilton D. Re-designing Hospital Care: Learning from the Experience of Hospital Medicine in Canada. Journal of Global Health Care Systems. 2011;1(3).
2. White HL. Assessing the Prevalence, Penetration and Performance of Hospital Physicians in Ontario: Implications for the Quality and Efficiency of Inpatient Care. Doctoral Thesis; 2016.
3. Yousefi V, Chong CA. Does implementation of a hospitalist program in a Canadian community hospital improve measures of quality of care and utilization? An observational comparative analysis of hospitalists vs. traditional care providers. BMC Health Serv Res. 2013;13:204.
4. Yousefi V, Hejazi S, Lam A. Impact of Hospitalists on Care Outcomes in a Large Integrated Health System in British Columbia. Journal of Clinical Outcomes Management. 2020;27(2):59-72.
5. Salim SA, Elmaraezy A, Pamarthy A, et al. Impact of hospitalists on the efficiency of inpatient care and patient satisfaction: a systematic review and meta-analysis. J Community Hosp Intern Med Perspect. 2019;9(2):121-134.
6. Peterson MC. A systematic review of outcomes and quality measures in adult patients cared for by hospitalists vs nonhospitalists. Mayo Clinic Proc. 2009;84(3):248-254.
7. Webster F, Bremner S, Jackson M, et al. The impact of a hospitalist on role boundaries in an orthopedic environment. J Multidiscip Healthc. 2012;5:249-256.
8. Gotlib Conn L, Reeves S, Dainty K, et al. Interprofessional communication with hospitalist and consultant physicians in general internal medicine: a qualitative study. BMC Health Serv Res. 2012; 12:437.
9. About Fraser Health. Fraser Health Authority. Updated 2018. Accessed January 30, 2019. https://www.fraserhealth.ca/about-us/about-fraser-health#.XFJrl9JKiUk
10. Divisions of Family Practice. Accessed May 2, 2020. https://www.divisionsbc.ca/provincial/about-us
11. Patton MQ. Essentials of Utilization-Focused Evaluation. 2012. Sage Publications, Inc; 2011.
12. Buljac-Samardzic M, Doekhie KD, van Wijngaarden JDH. Interventions to improve team effectiveness within health care: a systematic review of the past decade. Hum Resour Health. 2020;18(1):2.
13. Verhaegh KJ, Seller-Boersma A, Simons R, et al. An exploratory study of healthcare professionals’ perceptions of interprofessional communication and collaboration. J Interprof Care. 2017;31(3):397-400.
14. O’Leary KJ, Johnson JK, Manojlovich M, et al. Redesigning systems to improve teamwork and quality for hospitalized patients (RESET): study protocol evaluating the effect of mentored implementation to redesign clinical microsystems. BMC Health Serv Res. 2019;19(1):293.
15. Stein J, Payne C, Methvin A, et al. Reorganizing a hospital ward as an accountable care unit. J Hosp Med. 2015;10(1):36-40.
16. Yousefi V. How Canadian hospitalists spend their time - A work-sampling study within a hospital medicine program in Ontario. Journal of Clinical Outcomes Management. 2011;18(4):159.
17. Marinella MA: Hospitalists-Where They Came from, Who They Are, and What They Do. Hosp Physician. 2002;38(5):32-36.
18. Wachter RM. An introduction to the hospitalist model. Ann Intern Med. 1999;130(4 Pt 2):338-342.
19. Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287(4):487-494.
20. van Walraven C. The Influence of Inpatient Physician Continuity on Hospital Discharge. J Gen Intern Med. 2019;34(9):1709-1714.
21. Goodwin JS, Li S, Kuo YF. Association of the Work Schedules of Hospitalists With Patient Outcomes of Hospitalization. JAMA Intern Med. 2020;180(2):215-222.
22. Nichani S, Fitterman N, Lukela M, Crocker J, the Society of Hospital Medicine, Patient Handoff. 2017 Hospital Medicine Revised Core Competencies. J Hosp Med. 2017;4:S74.
23. Lo HY, Mullan PC, Lye C, et al. A QI initiative: implementing a patient handoff checklist for pediatric hospitalist attendings. BMJ Qual Improv Rep. 2016;5(1):u212920.w5661.
24. Wachter RM, Goldman L. Zero to 50,000 - The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375(11):1009-1011.
25. Grimm, P. Social Desirability Bias. In: Sheth J, Malhotra N, eds. Wiley International Encyclopedia of Marketing. John Wiley & Sons, Ltd; 2010.
1. Yousefi V, Wilton D. Re-designing Hospital Care: Learning from the Experience of Hospital Medicine in Canada. Journal of Global Health Care Systems. 2011;1(3).
2. White HL. Assessing the Prevalence, Penetration and Performance of Hospital Physicians in Ontario: Implications for the Quality and Efficiency of Inpatient Care. Doctoral Thesis; 2016.
3. Yousefi V, Chong CA. Does implementation of a hospitalist program in a Canadian community hospital improve measures of quality of care and utilization? An observational comparative analysis of hospitalists vs. traditional care providers. BMC Health Serv Res. 2013;13:204.
4. Yousefi V, Hejazi S, Lam A. Impact of Hospitalists on Care Outcomes in a Large Integrated Health System in British Columbia. Journal of Clinical Outcomes Management. 2020;27(2):59-72.
5. Salim SA, Elmaraezy A, Pamarthy A, et al. Impact of hospitalists on the efficiency of inpatient care and patient satisfaction: a systematic review and meta-analysis. J Community Hosp Intern Med Perspect. 2019;9(2):121-134.
6. Peterson MC. A systematic review of outcomes and quality measures in adult patients cared for by hospitalists vs nonhospitalists. Mayo Clinic Proc. 2009;84(3):248-254.
7. Webster F, Bremner S, Jackson M, et al. The impact of a hospitalist on role boundaries in an orthopedic environment. J Multidiscip Healthc. 2012;5:249-256.
8. Gotlib Conn L, Reeves S, Dainty K, et al. Interprofessional communication with hospitalist and consultant physicians in general internal medicine: a qualitative study. BMC Health Serv Res. 2012; 12:437.
9. About Fraser Health. Fraser Health Authority. Updated 2018. Accessed January 30, 2019. https://www.fraserhealth.ca/about-us/about-fraser-health#.XFJrl9JKiUk
10. Divisions of Family Practice. Accessed May 2, 2020. https://www.divisionsbc.ca/provincial/about-us
11. Patton MQ. Essentials of Utilization-Focused Evaluation. 2012. Sage Publications, Inc; 2011.
12. Buljac-Samardzic M, Doekhie KD, van Wijngaarden JDH. Interventions to improve team effectiveness within health care: a systematic review of the past decade. Hum Resour Health. 2020;18(1):2.
13. Verhaegh KJ, Seller-Boersma A, Simons R, et al. An exploratory study of healthcare professionals’ perceptions of interprofessional communication and collaboration. J Interprof Care. 2017;31(3):397-400.
14. O’Leary KJ, Johnson JK, Manojlovich M, et al. Redesigning systems to improve teamwork and quality for hospitalized patients (RESET): study protocol evaluating the effect of mentored implementation to redesign clinical microsystems. BMC Health Serv Res. 2019;19(1):293.
15. Stein J, Payne C, Methvin A, et al. Reorganizing a hospital ward as an accountable care unit. J Hosp Med. 2015;10(1):36-40.
16. Yousefi V. How Canadian hospitalists spend their time - A work-sampling study within a hospital medicine program in Ontario. Journal of Clinical Outcomes Management. 2011;18(4):159.
17. Marinella MA: Hospitalists-Where They Came from, Who They Are, and What They Do. Hosp Physician. 2002;38(5):32-36.
18. Wachter RM. An introduction to the hospitalist model. Ann Intern Med. 1999;130(4 Pt 2):338-342.
19. Wachter RM, Goldman L. The hospitalist movement 5 years later. JAMA. 2002;287(4):487-494.
20. van Walraven C. The Influence of Inpatient Physician Continuity on Hospital Discharge. J Gen Intern Med. 2019;34(9):1709-1714.
21. Goodwin JS, Li S, Kuo YF. Association of the Work Schedules of Hospitalists With Patient Outcomes of Hospitalization. JAMA Intern Med. 2020;180(2):215-222.
22. Nichani S, Fitterman N, Lukela M, Crocker J, the Society of Hospital Medicine, Patient Handoff. 2017 Hospital Medicine Revised Core Competencies. J Hosp Med. 2017;4:S74.
23. Lo HY, Mullan PC, Lye C, et al. A QI initiative: implementing a patient handoff checklist for pediatric hospitalist attendings. BMJ Qual Improv Rep. 2016;5(1):u212920.w5661.
24. Wachter RM, Goldman L. Zero to 50,000 - The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375(11):1009-1011.
25. Grimm, P. Social Desirability Bias. In: Sheth J, Malhotra N, eds. Wiley International Encyclopedia of Marketing. John Wiley & Sons, Ltd; 2010.
Implementation of a Symptom–Triggered Protocol for Severe Alcohol Withdrawal Treatment in a Medical Step-down Unit
From Stamford Hospital, Stamford, CT.
Objective: This single-center, quasi-experimental study of adult patients admitted or transferred to a medical step-down unit with alcohol withdrawal diagnoses sought to determine if symptom–triggered therapy (STT) is more effective than combined fixed-scheduled (FS) and STT in severe alcohol withdrawal.
Methods: In the preintervention group (72 episodes), patients were treated with FS and STT based on physician preference. In the postintervention group (69 episodes), providers were required to utilize only the STT protocol.
Results: Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001) and a decrease in average length of stay from 8.0 days to 5.1 days (P < .001). Secondary safety measures included a reduction in the proportion of patients who experienced delirium tremens from 47.5% to 22.5% (P < .001), and a reduction in intubation rates from 13.8% to 1.3% (P = .003).
Conclusion: The STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients requires frequent monitoring to assess withdrawal severity combined with appropriate and timely dosing of benzodiazepines.
Keywords: alcohol withdrawal delirium; alcohol withdrawal syndrome; treatment protocol; benzodiazepine; lorazepam.
Management of severe alcohol withdrawal and delirium tremens (DT) is challenging and requires significant resources, including close monitoring and intensive treatment, frequently in an intensive care unit (ICU).1 Early diagnosis and therapeutic intervention are important to limit potential complications associated with DT.2 Benzodiazepines are first-line therapeutic agents, but the definition of optimal use and dosing regimens has been limited, due to a lack of randomized controlled trials. In lower acuity patients admitted to a detoxification unit, systematic symptom–triggered benzodiazepine therapy (STT) has been established to be more effective than fixed-schedule (FS) dosing.3-5 Patients treated using STT require lower total benzodiazepine dosing and achieve shorter treatment durations. However, in higher-acuity patients admitted to general medical services, analyses have not shown an advantage of STT over combined FS and STT.6
Methods
The purpose of this study was to determine whether implementation of STT is more effective than FS dosing combined with episodic STT in the management of hospitalized high-acuity alcohol withdrawal patients. We conducted a preintervention and postintervention quasi-experimental study in the step-down unit (SDU) of a 305-bed community teaching hospital. The study population consisted of adult inpatients 18 years or older admitted or transferred to the 12-bed SDU with alcohol withdrawal, as defined by primary or secondary International Classification of Diseases, Tenth Revision diagnoses. SDU admission criteria included patients with prior DT or those who had received multiple doses of benzodiazepines in the emergency department. In-hospital transfer to the SDU was at the physician’s discretion, if the patient required escalating doses of benzodiazepines or the use of increasing resources, such as those for behavioral emergencies. The majority of patients admitted or transferred to the SDU were assigned to medical house staff teams under hospitalist supervision, and, on occasion, under community physicians. The nurse-to-patient ratio in the SDU was 1:3.
Study groups
The preintervention group consisted of 80 successive treatment episodes involving patients admitted or transferred to the SDU from
In the preintervention group, fixed, scheduled doses of lorazepam or chlordiazepoxide and as-needed lorazepam were prescribed and adjusted based upon physician judgment. Monitoring of symptom severity was scored using the revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar). Benzodiazepine dosing occurred if the CIWA-Ar score had increased 2 or more points from the last score.
In the postintervention group, the STT protocol included the creation of a standardized physician order set for benzodiazepine “sliding scale” administration. The STT protocol allowed for escalating doses for higher withdrawal scores. Symptom severity was scored using MINDS (Minnesota Detoxification Scale) criteria.1 Lorazepam as-needed dosing was based upon MINDS scores. A MINDS score less than 10 resulted in no medication, MINDS 10-12 required 2 mg, MINDS 13-16 required 4 mg, MINDS 17-19 required 6 mg, and MINDS 20 required 8 mg and a call to the physician. Transfer to the ICU was recommended if the MINDS score was ≥ 20 for 3 consecutive hours. Monitoring intervals occurred more frequently at 30 minutes unless the MINDS score was less than 10. After 7 days, the MINDS protocol was recommended to be discontinued, as the patient might have had iatrogenic delirium.
The STT protocol was introduced during a didactic session for the hospitalists and a separate session for internal medicine and family residents. Each registered nurse working in the SDU was trained in the use of the STT protocol and MINDS during nursing huddles.
Patients were excluded from evaluation if they were transferred to the SDU after 7 or more days in the hospital, if they had stayed in the hospital more than 30 days, were chronically on benzodiazepine therapy (to avoid confounding withdrawal symptoms), or if they left the hospital against medical advice (AMA). To avoid bias in the results, the patients with early discontinuation of treatment were included in analyses of secondary outcomes, thus resulting in all 80 episodes analyzed.
Measures and data
The primary outcome measure was benzodiazepine dose intensity, expressed in total lorazepam-equivalents. Secondary measures included average length of stay (including general medical, surgical, and ICU days), seizure incidence, DT incidence, sitter use, behavioral emergency responses, rates of leaving AMA, intubation, transfer to the ICU, and death.
Benzodiazepine dosing and length of stay were obtained from the data warehouse of the hospital’s electronic health record (EHR; Meditech). Benzodiazepine dosing was expressed in total lorazepam-equivalents, with conversion as follows: lorazepam orally and intravenously 1 mg = chlordiazepoxide 25 mg = diazepam 5 mg. All other measures were obtained from chart review of the patients’ EMR entries. The Stamford Hospital Institutional Review Board approved this study.
Analysis
Data analyses for this study were performed using SPSS version 25.0 (IBM). Categorical data were reported as frequency (count) and percent within category. Continuous data were reported as mean (SD). Categorical data were analyzed using χ2 analysis; continuous data were analyzed using t-tests. A P value of .05 was considered significant for each analysis.
Results
During the preintervention period, 72 episodes (58 patients) met inclusion criteria, and 69 episodes (55 patients) met inclusion criteria during the postintervention period. Ten patients were represented in both groups. Eight preintervention episodes were excluded from the primary analysis because the patient left AMA. Eleven postintervention episodes were excluded: 9 due to patients leaving AMA, 1 due to chronic benzodiazepine usage, and 1 due to transfer to the SDU unit after 7 days. Baseline characteristics and medication use profiles of the preintervention and postintervention groups are summarized in Table 1.

Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001), as shown in Table 2. Average length of stay decreased from 8.0 days to 5.1 days (P < .001). Secondary safety measures were notable for a reduction in DT incidence, from 47.5% to 22.5% (P < .001), and lower rates of intubation, from 13.8% to 1.3% (P = .003). Seven-day readmission rates were 0% preintervention and 1.4% postintervention.

Discussion
We found that hospitalized patients with severe alcohol withdrawal treated with STT required fewer benzodiazepines and had a lower length of stay than patients treated with a conventional combined STT and FS regimen. Implementation of the change from the STT and FS approach to the STT approach in the SDU resulted in concerns that waiting for symptoms to appear could result in more severe withdrawal and prolonged treatment.3 To address this, the intervention included monitoring and dosing every 30 minutes, as compared to monitoring and dosing every 1 hour preintervention. In addition, a sliding-scale approach to match alcohol withdrawal score with dosage was employed in postintervention patients.
Employment of the STT protocol also resulted in decreased complications, including lower rates of DT and transfer to the ICU. This new intervention resulted in significantly decreased time required to control severe symptoms. In the preintervention phase, if a patient’s symptoms escalated despite administration of the as-needed dose of benzodiazepine, there was often a delay in administration of additional doses due to the time needed for nurses to reach a physician and subsequent placement of a new order. In the postintervention phase, the STT protocol allowed nursing staff to give benzodiazepines without delay when needed. We believe this reduced the number of calls by nursing staff to physicians requesting additional medications, and that this improved teamwork when managing these patients.
As part of the intervention, a decision was made to use the MINDS scale rather than the CIWA-Ar scale to assess withdrawal severity. This was because the CIWA-Ar has only been validated in patients with uncomplicated alcohol withdrawal syndrome and has not been researched extensively in patients requiring ICU-level care.1 MINDS assessment has proven to be reliable and reflects severity of withdrawal. Furthermore, MINDS requires less time to administer—3 to 5 minutes vs 5 to 15 minutes for the CIWA-Ar scale. CIWA-Ar, unlike MINDS, requires subjective input from the patient, which is less reliable for higher acuity patients. Our study is unique in that it focused on high-acuity patients and it showed both a significant reduction in quantity of benzodiazepines prescribed and length of stay. Previous studies on lower acuity patients in detoxification units have confirmed that STT is more effective than a FS approach.3-5 In patients of higher acuity, STT has not proven to be superior.
A key lesson learned was the need for proper education of nursing staff. Concurrent nursing audits were necessary to ensure that scoring was performed in an accurate and timely manner. In addition, it was challenging to predict which patients might develop DTs versus those requiring a brief inpatient stay. While there was initial concern that an STT protocol could result in underdosing, we found that patients had fewer DT episodes and fewer ICU transfers.
This study had several limitations. These include a relatively small sample size and the data being less recent. As there has been no intervening change to the therapeutic paradigm of DT treatment, the findings remain pertinent to the present time. The study employed a simple pre/post design and was conducted in a single setting. We are not aware of any temporal or local trends likely to influence these results. Admissions and transfers to the SDU for severe alcohol withdrawal were based on physician discretion. However, patient characteristics in both groups were similar (Table 1). We note that the postintervention STT protocol allowed for more frequent benzodiazepine dosing, though benzodiazepine use did decrease. Different alcohol withdrawal scores (MINDS vs. CIWA-Ar) were used for postintervention and preintervention, although previous research has shown that MINDS and CIWA-Ar scores correlate well.7 Finally, some patients of higher acuity and complexity were excluded, potentially limiting the generalizability of our results.
Conclusion
Our STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients also requires frequent monitoring using the MINDS scale, integrated with benzodiazepine sliding-scale dosing to match symptom severity. This bundled approach resulted in a significant reduction of benzodiazepine usage and reduced length of stay. Timely treatment of these patients also reduced the percent of patients developing DTs, and reduced intubation rates and transfers to the ICU. Further studies may be warranted at other sites to confirm the effectiveness of this STT protocol.
Corresponding author: Paul W. Huang, MD, Stamford Hospital, One Hospital Plaza, PO Box 9317, Stamford, CT 06904; [email protected].
Financial disclosures: None.
1. DeCarolis DD, Rice KL, Ho L, et al. Symptom-driven lorazepam protocol for treatment of severe alcohol withdrawal delirium in the intensive care unit. Pharmacotherapy. 2007;27(4):510-518.
2. DeBellis R, Smith BS, Choi S, Malloy M. Management of delirium tremens. J Intensive Care Med. 2005;20(3):164-173.
3. Saitz R, Mayo-Smith MF, Roberts MS, et al. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA. 1994;272(7):519-523.
4. Sachdeva A, Chandra M, Deshpande SN. A comparative study of fixed tapering dose regimen versus symptom-triggered regimen of lorazepam for alcohol detoxification. Alcohol Alcohol. 2014;49(3):287-291.
5. Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med. 2002;162(10):1117-1121.
6. Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc. 2001;76(7):695-701.
7. Littlefield AJ, Heavner MS, Eng CC, et al. Correlation Between mMINDS and CIWA-Ar Scoring Tools in Patients With Alcohol Withdrawal Syndrome. Am J Crit Care. 2018;27(4):280-286.
From Stamford Hospital, Stamford, CT.
Objective: This single-center, quasi-experimental study of adult patients admitted or transferred to a medical step-down unit with alcohol withdrawal diagnoses sought to determine if symptom–triggered therapy (STT) is more effective than combined fixed-scheduled (FS) and STT in severe alcohol withdrawal.
Methods: In the preintervention group (72 episodes), patients were treated with FS and STT based on physician preference. In the postintervention group (69 episodes), providers were required to utilize only the STT protocol.
Results: Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001) and a decrease in average length of stay from 8.0 days to 5.1 days (P < .001). Secondary safety measures included a reduction in the proportion of patients who experienced delirium tremens from 47.5% to 22.5% (P < .001), and a reduction in intubation rates from 13.8% to 1.3% (P = .003).
Conclusion: The STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients requires frequent monitoring to assess withdrawal severity combined with appropriate and timely dosing of benzodiazepines.
Keywords: alcohol withdrawal delirium; alcohol withdrawal syndrome; treatment protocol; benzodiazepine; lorazepam.
Management of severe alcohol withdrawal and delirium tremens (DT) is challenging and requires significant resources, including close monitoring and intensive treatment, frequently in an intensive care unit (ICU).1 Early diagnosis and therapeutic intervention are important to limit potential complications associated with DT.2 Benzodiazepines are first-line therapeutic agents, but the definition of optimal use and dosing regimens has been limited, due to a lack of randomized controlled trials. In lower acuity patients admitted to a detoxification unit, systematic symptom–triggered benzodiazepine therapy (STT) has been established to be more effective than fixed-schedule (FS) dosing.3-5 Patients treated using STT require lower total benzodiazepine dosing and achieve shorter treatment durations. However, in higher-acuity patients admitted to general medical services, analyses have not shown an advantage of STT over combined FS and STT.6
Methods
The purpose of this study was to determine whether implementation of STT is more effective than FS dosing combined with episodic STT in the management of hospitalized high-acuity alcohol withdrawal patients. We conducted a preintervention and postintervention quasi-experimental study in the step-down unit (SDU) of a 305-bed community teaching hospital. The study population consisted of adult inpatients 18 years or older admitted or transferred to the 12-bed SDU with alcohol withdrawal, as defined by primary or secondary International Classification of Diseases, Tenth Revision diagnoses. SDU admission criteria included patients with prior DT or those who had received multiple doses of benzodiazepines in the emergency department. In-hospital transfer to the SDU was at the physician’s discretion, if the patient required escalating doses of benzodiazepines or the use of increasing resources, such as those for behavioral emergencies. The majority of patients admitted or transferred to the SDU were assigned to medical house staff teams under hospitalist supervision, and, on occasion, under community physicians. The nurse-to-patient ratio in the SDU was 1:3.
Study groups
The preintervention group consisted of 80 successive treatment episodes involving patients admitted or transferred to the SDU from
In the preintervention group, fixed, scheduled doses of lorazepam or chlordiazepoxide and as-needed lorazepam were prescribed and adjusted based upon physician judgment. Monitoring of symptom severity was scored using the revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar). Benzodiazepine dosing occurred if the CIWA-Ar score had increased 2 or more points from the last score.
In the postintervention group, the STT protocol included the creation of a standardized physician order set for benzodiazepine “sliding scale” administration. The STT protocol allowed for escalating doses for higher withdrawal scores. Symptom severity was scored using MINDS (Minnesota Detoxification Scale) criteria.1 Lorazepam as-needed dosing was based upon MINDS scores. A MINDS score less than 10 resulted in no medication, MINDS 10-12 required 2 mg, MINDS 13-16 required 4 mg, MINDS 17-19 required 6 mg, and MINDS 20 required 8 mg and a call to the physician. Transfer to the ICU was recommended if the MINDS score was ≥ 20 for 3 consecutive hours. Monitoring intervals occurred more frequently at 30 minutes unless the MINDS score was less than 10. After 7 days, the MINDS protocol was recommended to be discontinued, as the patient might have had iatrogenic delirium.
The STT protocol was introduced during a didactic session for the hospitalists and a separate session for internal medicine and family residents. Each registered nurse working in the SDU was trained in the use of the STT protocol and MINDS during nursing huddles.
Patients were excluded from evaluation if they were transferred to the SDU after 7 or more days in the hospital, if they had stayed in the hospital more than 30 days, were chronically on benzodiazepine therapy (to avoid confounding withdrawal symptoms), or if they left the hospital against medical advice (AMA). To avoid bias in the results, the patients with early discontinuation of treatment were included in analyses of secondary outcomes, thus resulting in all 80 episodes analyzed.
Measures and data
The primary outcome measure was benzodiazepine dose intensity, expressed in total lorazepam-equivalents. Secondary measures included average length of stay (including general medical, surgical, and ICU days), seizure incidence, DT incidence, sitter use, behavioral emergency responses, rates of leaving AMA, intubation, transfer to the ICU, and death.
Benzodiazepine dosing and length of stay were obtained from the data warehouse of the hospital’s electronic health record (EHR; Meditech). Benzodiazepine dosing was expressed in total lorazepam-equivalents, with conversion as follows: lorazepam orally and intravenously 1 mg = chlordiazepoxide 25 mg = diazepam 5 mg. All other measures were obtained from chart review of the patients’ EMR entries. The Stamford Hospital Institutional Review Board approved this study.
Analysis
Data analyses for this study were performed using SPSS version 25.0 (IBM). Categorical data were reported as frequency (count) and percent within category. Continuous data were reported as mean (SD). Categorical data were analyzed using χ2 analysis; continuous data were analyzed using t-tests. A P value of .05 was considered significant for each analysis.
Results
During the preintervention period, 72 episodes (58 patients) met inclusion criteria, and 69 episodes (55 patients) met inclusion criteria during the postintervention period. Ten patients were represented in both groups. Eight preintervention episodes were excluded from the primary analysis because the patient left AMA. Eleven postintervention episodes were excluded: 9 due to patients leaving AMA, 1 due to chronic benzodiazepine usage, and 1 due to transfer to the SDU unit after 7 days. Baseline characteristics and medication use profiles of the preintervention and postintervention groups are summarized in Table 1.
Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001), as shown in Table 2. Average length of stay decreased from 8.0 days to 5.1 days (P < .001). Secondary safety measures were notable for a reduction in DT incidence, from 47.5% to 22.5% (P < .001), and lower rates of intubation, from 13.8% to 1.3% (P = .003). Seven-day readmission rates were 0% preintervention and 1.4% postintervention.
Discussion
We found that hospitalized patients with severe alcohol withdrawal treated with STT required fewer benzodiazepines and had a lower length of stay than patients treated with a conventional combined STT and FS regimen. Implementation of the change from the STT and FS approach to the STT approach in the SDU resulted in concerns that waiting for symptoms to appear could result in more severe withdrawal and prolonged treatment.3 To address this, the intervention included monitoring and dosing every 30 minutes, as compared to monitoring and dosing every 1 hour preintervention. In addition, a sliding-scale approach to match alcohol withdrawal score with dosage was employed in postintervention patients.
Employment of the STT protocol also resulted in decreased complications, including lower rates of DT and transfer to the ICU. This new intervention resulted in significantly decreased time required to control severe symptoms. In the preintervention phase, if a patient’s symptoms escalated despite administration of the as-needed dose of benzodiazepine, there was often a delay in administration of additional doses due to the time needed for nurses to reach a physician and subsequent placement of a new order. In the postintervention phase, the STT protocol allowed nursing staff to give benzodiazepines without delay when needed. We believe this reduced the number of calls by nursing staff to physicians requesting additional medications, and that this improved teamwork when managing these patients.
As part of the intervention, a decision was made to use the MINDS scale rather than the CIWA-Ar scale to assess withdrawal severity. This was because the CIWA-Ar has only been validated in patients with uncomplicated alcohol withdrawal syndrome and has not been researched extensively in patients requiring ICU-level care.1 MINDS assessment has proven to be reliable and reflects severity of withdrawal. Furthermore, MINDS requires less time to administer—3 to 5 minutes vs 5 to 15 minutes for the CIWA-Ar scale. CIWA-Ar, unlike MINDS, requires subjective input from the patient, which is less reliable for higher acuity patients. Our study is unique in that it focused on high-acuity patients and it showed both a significant reduction in quantity of benzodiazepines prescribed and length of stay. Previous studies on lower acuity patients in detoxification units have confirmed that STT is more effective than a FS approach.3-5 In patients of higher acuity, STT has not proven to be superior.
A key lesson learned was the need for proper education of nursing staff. Concurrent nursing audits were necessary to ensure that scoring was performed in an accurate and timely manner. In addition, it was challenging to predict which patients might develop DTs versus those requiring a brief inpatient stay. While there was initial concern that an STT protocol could result in underdosing, we found that patients had fewer DT episodes and fewer ICU transfers.
This study had several limitations. These include a relatively small sample size and the data being less recent. As there has been no intervening change to the therapeutic paradigm of DT treatment, the findings remain pertinent to the present time. The study employed a simple pre/post design and was conducted in a single setting. We are not aware of any temporal or local trends likely to influence these results. Admissions and transfers to the SDU for severe alcohol withdrawal were based on physician discretion. However, patient characteristics in both groups were similar (Table 1). We note that the postintervention STT protocol allowed for more frequent benzodiazepine dosing, though benzodiazepine use did decrease. Different alcohol withdrawal scores (MINDS vs. CIWA-Ar) were used for postintervention and preintervention, although previous research has shown that MINDS and CIWA-Ar scores correlate well.7 Finally, some patients of higher acuity and complexity were excluded, potentially limiting the generalizability of our results.
Conclusion
Our STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients also requires frequent monitoring using the MINDS scale, integrated with benzodiazepine sliding-scale dosing to match symptom severity. This bundled approach resulted in a significant reduction of benzodiazepine usage and reduced length of stay. Timely treatment of these patients also reduced the percent of patients developing DTs, and reduced intubation rates and transfers to the ICU. Further studies may be warranted at other sites to confirm the effectiveness of this STT protocol.
Corresponding author: Paul W. Huang, MD, Stamford Hospital, One Hospital Plaza, PO Box 9317, Stamford, CT 06904; [email protected].
Financial disclosures: None.
From Stamford Hospital, Stamford, CT.
Objective: This single-center, quasi-experimental study of adult patients admitted or transferred to a medical step-down unit with alcohol withdrawal diagnoses sought to determine if symptom–triggered therapy (STT) is more effective than combined fixed-scheduled (FS) and STT in severe alcohol withdrawal.
Methods: In the preintervention group (72 episodes), patients were treated with FS and STT based on physician preference. In the postintervention group (69 episodes), providers were required to utilize only the STT protocol.
Results: Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001) and a decrease in average length of stay from 8.0 days to 5.1 days (P < .001). Secondary safety measures included a reduction in the proportion of patients who experienced delirium tremens from 47.5% to 22.5% (P < .001), and a reduction in intubation rates from 13.8% to 1.3% (P = .003).
Conclusion: The STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients requires frequent monitoring to assess withdrawal severity combined with appropriate and timely dosing of benzodiazepines.
Keywords: alcohol withdrawal delirium; alcohol withdrawal syndrome; treatment protocol; benzodiazepine; lorazepam.
Management of severe alcohol withdrawal and delirium tremens (DT) is challenging and requires significant resources, including close monitoring and intensive treatment, frequently in an intensive care unit (ICU).1 Early diagnosis and therapeutic intervention are important to limit potential complications associated with DT.2 Benzodiazepines are first-line therapeutic agents, but the definition of optimal use and dosing regimens has been limited, due to a lack of randomized controlled trials. In lower acuity patients admitted to a detoxification unit, systematic symptom–triggered benzodiazepine therapy (STT) has been established to be more effective than fixed-schedule (FS) dosing.3-5 Patients treated using STT require lower total benzodiazepine dosing and achieve shorter treatment durations. However, in higher-acuity patients admitted to general medical services, analyses have not shown an advantage of STT over combined FS and STT.6
Methods
The purpose of this study was to determine whether implementation of STT is more effective than FS dosing combined with episodic STT in the management of hospitalized high-acuity alcohol withdrawal patients. We conducted a preintervention and postintervention quasi-experimental study in the step-down unit (SDU) of a 305-bed community teaching hospital. The study population consisted of adult inpatients 18 years or older admitted or transferred to the 12-bed SDU with alcohol withdrawal, as defined by primary or secondary International Classification of Diseases, Tenth Revision diagnoses. SDU admission criteria included patients with prior DT or those who had received multiple doses of benzodiazepines in the emergency department. In-hospital transfer to the SDU was at the physician’s discretion, if the patient required escalating doses of benzodiazepines or the use of increasing resources, such as those for behavioral emergencies. The majority of patients admitted or transferred to the SDU were assigned to medical house staff teams under hospitalist supervision, and, on occasion, under community physicians. The nurse-to-patient ratio in the SDU was 1:3.
Study groups
The preintervention group consisted of 80 successive treatment episodes involving patients admitted or transferred to the SDU from
In the preintervention group, fixed, scheduled doses of lorazepam or chlordiazepoxide and as-needed lorazepam were prescribed and adjusted based upon physician judgment. Monitoring of symptom severity was scored using the revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar). Benzodiazepine dosing occurred if the CIWA-Ar score had increased 2 or more points from the last score.
In the postintervention group, the STT protocol included the creation of a standardized physician order set for benzodiazepine “sliding scale” administration. The STT protocol allowed for escalating doses for higher withdrawal scores. Symptom severity was scored using MINDS (Minnesota Detoxification Scale) criteria.1 Lorazepam as-needed dosing was based upon MINDS scores. A MINDS score less than 10 resulted in no medication, MINDS 10-12 required 2 mg, MINDS 13-16 required 4 mg, MINDS 17-19 required 6 mg, and MINDS 20 required 8 mg and a call to the physician. Transfer to the ICU was recommended if the MINDS score was ≥ 20 for 3 consecutive hours. Monitoring intervals occurred more frequently at 30 minutes unless the MINDS score was less than 10. After 7 days, the MINDS protocol was recommended to be discontinued, as the patient might have had iatrogenic delirium.
The STT protocol was introduced during a didactic session for the hospitalists and a separate session for internal medicine and family residents. Each registered nurse working in the SDU was trained in the use of the STT protocol and MINDS during nursing huddles.
Patients were excluded from evaluation if they were transferred to the SDU after 7 or more days in the hospital, if they had stayed in the hospital more than 30 days, were chronically on benzodiazepine therapy (to avoid confounding withdrawal symptoms), or if they left the hospital against medical advice (AMA). To avoid bias in the results, the patients with early discontinuation of treatment were included in analyses of secondary outcomes, thus resulting in all 80 episodes analyzed.
Measures and data
The primary outcome measure was benzodiazepine dose intensity, expressed in total lorazepam-equivalents. Secondary measures included average length of stay (including general medical, surgical, and ICU days), seizure incidence, DT incidence, sitter use, behavioral emergency responses, rates of leaving AMA, intubation, transfer to the ICU, and death.
Benzodiazepine dosing and length of stay were obtained from the data warehouse of the hospital’s electronic health record (EHR; Meditech). Benzodiazepine dosing was expressed in total lorazepam-equivalents, with conversion as follows: lorazepam orally and intravenously 1 mg = chlordiazepoxide 25 mg = diazepam 5 mg. All other measures were obtained from chart review of the patients’ EMR entries. The Stamford Hospital Institutional Review Board approved this study.
Analysis
Data analyses for this study were performed using SPSS version 25.0 (IBM). Categorical data were reported as frequency (count) and percent within category. Continuous data were reported as mean (SD). Categorical data were analyzed using χ2 analysis; continuous data were analyzed using t-tests. A P value of .05 was considered significant for each analysis.
Results
During the preintervention period, 72 episodes (58 patients) met inclusion criteria, and 69 episodes (55 patients) met inclusion criteria during the postintervention period. Ten patients were represented in both groups. Eight preintervention episodes were excluded from the primary analysis because the patient left AMA. Eleven postintervention episodes were excluded: 9 due to patients leaving AMA, 1 due to chronic benzodiazepine usage, and 1 due to transfer to the SDU unit after 7 days. Baseline characteristics and medication use profiles of the preintervention and postintervention groups are summarized in Table 1.
Implementation of the intervention was associated with a significant reduction in average (per patient) cumulative benzodiazepine dose, from 250 mg to 96 mg (P < .001), as shown in Table 2. Average length of stay decreased from 8.0 days to 5.1 days (P < .001). Secondary safety measures were notable for a reduction in DT incidence, from 47.5% to 22.5% (P < .001), and lower rates of intubation, from 13.8% to 1.3% (P = .003). Seven-day readmission rates were 0% preintervention and 1.4% postintervention.
Discussion
We found that hospitalized patients with severe alcohol withdrawal treated with STT required fewer benzodiazepines and had a lower length of stay than patients treated with a conventional combined STT and FS regimen. Implementation of the change from the STT and FS approach to the STT approach in the SDU resulted in concerns that waiting for symptoms to appear could result in more severe withdrawal and prolonged treatment.3 To address this, the intervention included monitoring and dosing every 30 minutes, as compared to monitoring and dosing every 1 hour preintervention. In addition, a sliding-scale approach to match alcohol withdrawal score with dosage was employed in postintervention patients.
Employment of the STT protocol also resulted in decreased complications, including lower rates of DT and transfer to the ICU. This new intervention resulted in significantly decreased time required to control severe symptoms. In the preintervention phase, if a patient’s symptoms escalated despite administration of the as-needed dose of benzodiazepine, there was often a delay in administration of additional doses due to the time needed for nurses to reach a physician and subsequent placement of a new order. In the postintervention phase, the STT protocol allowed nursing staff to give benzodiazepines without delay when needed. We believe this reduced the number of calls by nursing staff to physicians requesting additional medications, and that this improved teamwork when managing these patients.
As part of the intervention, a decision was made to use the MINDS scale rather than the CIWA-Ar scale to assess withdrawal severity. This was because the CIWA-Ar has only been validated in patients with uncomplicated alcohol withdrawal syndrome and has not been researched extensively in patients requiring ICU-level care.1 MINDS assessment has proven to be reliable and reflects severity of withdrawal. Furthermore, MINDS requires less time to administer—3 to 5 minutes vs 5 to 15 minutes for the CIWA-Ar scale. CIWA-Ar, unlike MINDS, requires subjective input from the patient, which is less reliable for higher acuity patients. Our study is unique in that it focused on high-acuity patients and it showed both a significant reduction in quantity of benzodiazepines prescribed and length of stay. Previous studies on lower acuity patients in detoxification units have confirmed that STT is more effective than a FS approach.3-5 In patients of higher acuity, STT has not proven to be superior.
A key lesson learned was the need for proper education of nursing staff. Concurrent nursing audits were necessary to ensure that scoring was performed in an accurate and timely manner. In addition, it was challenging to predict which patients might develop DTs versus those requiring a brief inpatient stay. While there was initial concern that an STT protocol could result in underdosing, we found that patients had fewer DT episodes and fewer ICU transfers.
This study had several limitations. These include a relatively small sample size and the data being less recent. As there has been no intervening change to the therapeutic paradigm of DT treatment, the findings remain pertinent to the present time. The study employed a simple pre/post design and was conducted in a single setting. We are not aware of any temporal or local trends likely to influence these results. Admissions and transfers to the SDU for severe alcohol withdrawal were based on physician discretion. However, patient characteristics in both groups were similar (Table 1). We note that the postintervention STT protocol allowed for more frequent benzodiazepine dosing, though benzodiazepine use did decrease. Different alcohol withdrawal scores (MINDS vs. CIWA-Ar) were used for postintervention and preintervention, although previous research has shown that MINDS and CIWA-Ar scores correlate well.7 Finally, some patients of higher acuity and complexity were excluded, potentially limiting the generalizability of our results.
Conclusion
Our STT protocol proved to be more effective and safer in treating severe alcohol withdrawal patients than usual care employing STT with FS. We believe the successful implementation of a STT protocol in high-acuity patients also requires frequent monitoring using the MINDS scale, integrated with benzodiazepine sliding-scale dosing to match symptom severity. This bundled approach resulted in a significant reduction of benzodiazepine usage and reduced length of stay. Timely treatment of these patients also reduced the percent of patients developing DTs, and reduced intubation rates and transfers to the ICU. Further studies may be warranted at other sites to confirm the effectiveness of this STT protocol.
Corresponding author: Paul W. Huang, MD, Stamford Hospital, One Hospital Plaza, PO Box 9317, Stamford, CT 06904; [email protected].
Financial disclosures: None.
1. DeCarolis DD, Rice KL, Ho L, et al. Symptom-driven lorazepam protocol for treatment of severe alcohol withdrawal delirium in the intensive care unit. Pharmacotherapy. 2007;27(4):510-518.
2. DeBellis R, Smith BS, Choi S, Malloy M. Management of delirium tremens. J Intensive Care Med. 2005;20(3):164-173.
3. Saitz R, Mayo-Smith MF, Roberts MS, et al. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA. 1994;272(7):519-523.
4. Sachdeva A, Chandra M, Deshpande SN. A comparative study of fixed tapering dose regimen versus symptom-triggered regimen of lorazepam for alcohol detoxification. Alcohol Alcohol. 2014;49(3):287-291.
5. Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med. 2002;162(10):1117-1121.
6. Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc. 2001;76(7):695-701.
7. Littlefield AJ, Heavner MS, Eng CC, et al. Correlation Between mMINDS and CIWA-Ar Scoring Tools in Patients With Alcohol Withdrawal Syndrome. Am J Crit Care. 2018;27(4):280-286.
1. DeCarolis DD, Rice KL, Ho L, et al. Symptom-driven lorazepam protocol for treatment of severe alcohol withdrawal delirium in the intensive care unit. Pharmacotherapy. 2007;27(4):510-518.
2. DeBellis R, Smith BS, Choi S, Malloy M. Management of delirium tremens. J Intensive Care Med. 2005;20(3):164-173.
3. Saitz R, Mayo-Smith MF, Roberts MS, et al. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA. 1994;272(7):519-523.
4. Sachdeva A, Chandra M, Deshpande SN. A comparative study of fixed tapering dose regimen versus symptom-triggered regimen of lorazepam for alcohol detoxification. Alcohol Alcohol. 2014;49(3):287-291.
5. Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med. 2002;162(10):1117-1121.
6. Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc. 2001;76(7):695-701.
7. Littlefield AJ, Heavner MS, Eng CC, et al. Correlation Between mMINDS and CIWA-Ar Scoring Tools in Patients With Alcohol Withdrawal Syndrome. Am J Crit Care. 2018;27(4):280-286.
A Service Evaluation of Acute Neurological Patients Managed on Clinically Inappropriate Wards
From Western Sussex Hospitals NHS Foundation Trust, Physiotherapy Department, Chichester, UK (Richard J. Holmes), and Western Sussex Hospitals NHS Foundation Trust, Department of Occupational Therapy, Chichester, UK (Sophie Stratford).
Objective: Despite the benefits of early and frequent input from a neurologist, there is wide variation in the availability of this service, especially in district general hospitals, with many patients managed on clinically inappropriate wards. The purpose of this service evaluation was to explore the impact this had on patient care.
Methods: A retrospective service evaluation was undertaken at a National Health Service hospital by reviewing patient records over a 6-month period. Data related to demographics, processes within the patient’s care, and secondary complications were recorded. Findings were compared with those of stroke patients managed on a specialist stroke ward.
Results: A total of 63 patients were identified, with a mean age of 72 years. The mean length of stay was 25.9 days, with a readmission rate of 16.7%. Only 15.9% of patients were reviewed by a neurologist. There was a high rate of secondary complications, with a number of patients experiencing falls (11.1%), pressure ulcers (14.3%), and health care–acquired infections (33.3%) during their admission.
Conclusions: The lack of specialist input from a neurologist and the management of patients on clinically inappropriate wards may have negatively impacted length of stay, readmission rates, and the frequency of secondary complications.
Keywords: evaluation; clinical safety; neurology; patient-centered care; clinical outcomes; length of stay.
It is estimated that 10% of acute admissions to district general hospitals (DGHs) of the National Health Service (NHS) in the United Kingdom are due to a neurological problem other than stroke.1 In 2011, a joint report from the Royal College of Physicians and the Association of British Neurologists (ABN) recommended that all of these patients should be admitted under the care of a neurologist and be regularly reviewed by a neurologist during their admission.2 The rationale for this recommendation is clear. The involvement of a neurologist has been shown to improve accuracy of the diagnosis3 and significantly reduce length of stay.4,5 Studies have also shown that the involvement of a neurologist has led to a change in the management plan in as high as 79%6 to 89%3 of cases, suggesting that a high proportion of neurological patients not seen by a neurologist are being managed suboptimally.
Despite this, a recent ABN survey of acute neurology services found ongoing wide variations in the availability of this specialist care, with a large proportion of DGHs having limited or no access to a neurologist and very few having dedicated neurology beds.7 While it is recognized that services have been structured in response to the reduced numbers of neurologists within the United Kingdom,8 it is prudent to assess the impact that such services have on patient care.
With this in mind, we planned to evaluate the current provision of care provided to neurological patients in a real-world setting. This was conducted in the context of a neurology liaison service at a DGH with no dedicated neurology beds.
Methods
A retrospective service evaluation was undertaken at a DGH in the southeast of England. The NHS hospital has neurologists on site who provide diagnostic and therapeutic consultations on the wards, but there are no dedicated beds for patients with neurological conditions. Patients requiring neurosurgical input are referred to a tertiary neurosciences center.
Patients were selected from the neurotherapy database if they were referred into the service between August 1, 2019, and January 31, 2020. The neurotherapy database was used as this was the only source that held thorough data on this patient group and allowed for the identification of patients who were not referred into the neurologist’s service. Patients were included if they had a new neurological condition as their primary diagnosis or if they had an exacerbation of an already established neurological condition. If a patient was admitted with more than 1 neurological diagnosis then the primary diagnosis for the admission was to be used in the analysis, though this did not occur during this evaluation. Patients with a primary diagnosis of a stroke were included if they were not managed on the acute stroke ward. Those managed on the stroke ward were excluded so that an analysis of patients managed on wards that were deemed clinically inappropriate could be undertaken. Patients were not included if they had a pre-existing neurological condition (ie, dementia, multiple sclerosis) but were admitted due to a non-neurological cause such as a fall or infection. All patients who met the criteria were included.
A team member independently reviewed each set of patient notes. Demographic data extracted from the medical notes included the patient’s age (on admission), gender, and diagnosis. Medical, nursing, and therapy notes were reviewed to identify secondary complications that arose during the patient’s admission. The secondary complications reviewed were falls (defined as the patient unexpectedly coming to the ground or other lower level), health care–acquired infections (HAIs) (defined as any infection acquired during the hospital admission), and pressure ulcers (defined as injuries to the skin or underlying tissue during the hospital admission). Other details, obtained from the patient administration system, included the length of stay (days), the number of ward moves the patient experienced, the speciality of the consultant responsible for the patient’s care, the discharge destination, and whether the patient was readmitted for any cause within 30 days. All data collected were stored on a password-protected computer and no patient-identifiable data were included.
The results were collated using descriptive statistics. The χ2 test was used to compare categorical data between those patients who were and were not reviewed by a neurologist, and the Mann-Whitney U test was used to compare differences in the length of stay between these 2 groups.
No national data relating to this specific patient group were available within the literature. Therefore, to provide a comparator of neurological patients within the same hospital, data were collected on stroke patients managed on the stroke ward. This group was deemed most appropriate for comparison as they present with similar neurological symptoms but are cared for on a specialist ward. During the evaluation period, 284 stroke patients were admitted to the stroke ward. A sample of 75 patients was randomly selected using a random number generator, and the procedure for data collection was repeated. It was not appropriate to make direct comparative analysis on these 2 groups due to the inherent differences, but it was felt important to provide context with regards to what usual care was like on a specialist ward within the same hospital.
Ethical approval was not required as this was a service evaluation of routinely collected data within a single hospital site.
Results
In total, 63 patients were identified: 26 females and 37 males. The median age of patients was 74 years (range, 39-92 years). These demographic details and comparisons to stroke patients managed on a specialist ward can be seen in Table 1. To quantify the range of diagnoses, the condition groups defined by GIRFT Neurology Methodology9 were used. The most common diagnoses were tumors of the nervous system (25.4%) and traumatic brain and spine injury (23.8%). The other conditions included in the analysis can be seen in Table 2.

Despite having a neurological condition as their primary diagnosis, only 15.9% of patients were reviewed by a neurologist during their hospital admission. Patients were most commonly under the care of a geriatrician (60.3%), but they were also managed by orthopedics (12.6%), acute medicine (7.9%), respiratory (6.3%), cardiology (4.8%), gastroenterology (3.2%), and surgery (3.2%). One patient (1.6%) was managed by intensivists.

The average length of stay was 25.9 days (range, 2-78 days). This was more than double the average length of stay on the stroke ward (11.4 days) (Table 1) and the national average for patients with neurological conditions (9.78 days).10 During their stay, 33% had 2 or more ward moves, with 1 patient moving wards a total of 6 times. Just over half (52.4%) of the patients returned to their usual residence on discharge. The remainder were discharged to rehabilitation units (15.9%), nursing homes (14.3%), residential homes (6.3%), tertiary centers (4.8%), and hospice (1.6%). Unfortunately, 3 patients (4.8%) passed away. Of those still alive (n = 60), 16.7% were readmitted to the hospital within 30 days, compared to a readmission rate of 11% on the stroke ward. None of the patients who were readmitted were seen by a neurologist during their initial admission.
The frequency of secondary complications was reviewed as a measure of the multidisciplinary management of this patient group. It was noted that 11.1% had a fall on the ward, which was similar to a rate of 10.7% on the stroke ward. More striking was the fact that 14.3% of patients developed a pressure ulcer and 33.3% developed an HAI during their admission, compared with rates of 1.3% and 10.7%, respectively, on the stroke ward (Table 1).
There were no significant differences found in length of stay between those who were and were not reviewed by a neurologist (P = .73). This was also true for categorical data, whereby readmission rate (P = .13), frequency of falls (P = .22), frequency of pressure ulcers (P = .67), and HAIs (P = .81) all failed to show a significant difference between groups.
Discussion
The findings of this service evaluation show markedly poorer outcomes for neurological patients compared to stroke patients managed on a specialist stroke ward. It is suggested that these results are in part due to the lack of specialist input from a neurologist in the majority of cases and the fact that all were managed on clinically inappropriate wards. Only 15.9% of neurological patients were seen by a neurologist. This is a slight improvement compared to previous studies in DGHs that showed rates of 10%1 and 11%,11 but it is still a far cry from the goal of 100% set out in recommendations.2 In addition, the increased readmission rate may be suggestive of suboptimal management, especially given that none of those readmitted had been reviewed by a neurologist. There are undoubtedly other factors that may influence readmissions, such as comorbidities, the severity/complexity of the condition, and the strength of community services. However, the impact of a lack of input from a specialist should not be underestimated, and further evaluation of this factor (with confounding factors controlled) would be beneficial.
The result of an extended length of stay was also a predictable outcome based on previous evidence.4,5 With the potential for suboptimal management plans and inaccurate diagnoses, it is inevitable that the patient’s movement through the hospital system will be impeded. In our example, it is possible that the extended length of stay was influenced by the fact that patients included in the evaluation were managed on nonspecialist wards and a large proportion had multiple ward changes.
Given that the evidence clearly shows that stroke patients are most effectively managed by a multidisciplinary team (MDT) with specialist skills,12 it is likely that other neurological patients, who have similar multifactorial needs, would also benefit. The patients in our evaluation were cared for by nursing staff who lacked specific skills and experience in neurology. The allied health professionals involved were specialists in neurotherapy but were not based on the ward and not directly linked to the ward MDT. A review by Epstein found that the benefits of having a MDT, in any speciality, working together on a ward included improved communication, reduced adverse events, and a reduced length of stay.13 This lack of an effective MDT approach may provide some explanation as to why the average length of stay and the rates of some secondary complications were at such elevated levels.
A systematic review exploring the impact of patients admitted to clinically inappropriate wards in a range of specialities found that these patients were associated with worse outcomes.14 This is supported by our findings, in which a higher rate of pressure ulcers and HAIs were observed when compared to rates in the specialist stroke ward. Again, a potential explanation for this is the impact of patients being managed by clinicians who lack the specialist knowledge of the patient group and the risks they face. Another explanation could be due to the high number of ward moves the patients experienced. Blay et al found that ward moves increased length of stay and carried an associated clinical risk, with the odds of falls and HAIs increasing with each move.15 A case example of this is apparent within our analysis in that the patient who experienced 6 ward moves not only had the longest length of stay (78 days), but also developed a pressure ulcer and 2 HAIs during their admission.
This service evaluation had a number of limitations that should be considered when interpreting the results. First, despite including all patients who met the criteria within the stipulated time frame, the sample size was relatively small, making it difficult to identify consistent patterns of behavior within the data.
Furthermore, caution should be applied when interpreting the comparators used, as the patient groups are not equivalent. The use of comparison against a standard is not a prerequisite in a service evaluation of this nature, but comparators were included to help frame the context for the reader. As such, they should only be used in this way rather than to make any firm conclusions.
Finally, as the evaluation was limited to the use of routinely collected data, there are several variables, other than those reported, which may have influenced the results. For example, it was not possible to ascertain certain demographic details, such as body mass index and socioeconomic factors, nor lifestyle factors such as smoking status, alcohol consumption, and exercise levels, all of which could impact negatively on the outcomes of interest. Furthermore, data were not collected on follow-up services after discharge to evaluate whether these had any impact on readmission rates.
Conclusion
This service evaluation highlights the potential impact of managing neurological patients on clinically inappropriate wards with limited input from a neurologist. There is the potential to ameliorate these impacts by cohorting these patients in neurologist-led beds with a specialist MDT. While there are limitations in the design of our study, including the lack of a controlled comparison, the small sample size, and the fact that this is an evaluation of a single service, the negative impacts to patients are concerning and warrant further investigation.
Corresponding author: Richard J. Holmes, MSc, Physiotherapy Department, St. Richard’s Hospital, Chichester, West Sussex, PO19 6SE; [email protected].
Financial disclosures: None.
1. Kanagaratnam M, Boodhoo A, MacDonald BK, Nitkunan A. Prevalence of acute neurology: a 2-week snapshot in a district general hospital. Clin Med (Lond). 2020;20(2):169-173.
2. Royal College of Physicians. Local adult neurology services for the next decade. Report of a working party. June 2011. Accessed October 29, 2020. https://www.mstrust.org.uk/sites/default/files/files/Local%20adult%20neurology%20services%20for%20the%20next%20decade.pdf
3. McColgan P, Carr AS, McCarron MO. The value of a liaison neurology service in a district general hospital. Postgrad Med J. 2011;87(1025):166-169.
4. Forbes R, Craig J, Callender M, Patterson V. Liaison neurology for acute medical admissions. Clin Med (Lond). 2004;4(3):290.
5. Craig J, Chua R, Russell C, et al. A cohort study of early neurological consultation by telemedicine on the care of neurological inpatients. J Neurol Neurosurg Psychiatry. 2004;75(7):1031-1035.
6. Ali E, Chaila E, Hutchinson M, Tubridy N. The ‘hidden work’ of a hospital neurologist: 1000 consults later. Eur J Neurol. 2010;17(4):e28-e32.
7. Association of British Neurologists. Acute Neurology services survey 2017. Accessed October 29, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/ABN_2017_Acute_Neurology_Survey.pdf
8. Nitkunan A, Lawrence J, Reilly MM. Neurology Workforce Survey. January 28, 2020. Accessed October 28, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/2020_ABN_Neurology_Workforce_Survey_2018-19_28_Jan_2020.pdf
9. Fuller G, Connolly M, Mummery C, Williams A. GIRT Neurology Methodology and Initial Summary of Regional Data. September 2019. Accessed October 26, 2020. https://gettingitrightfirsttime.co.uk/wp-content/uploads/2017/07/GIRFT-neurology-methodology-090919-FINAL.pdf
10. The Neurological Alliance. Neuro Numbers 2019. Accessed October 28, 2020. https://www.neural.org.uk/wp-content/uploads/2019/07/neuro-numbers-2019.pdf
11. Cai A, Brex P. A survey of acute neurology at a general hospital in the UK. Clin Med (Lond). 2010;10(6):642-643.
12. Langhorne P, Ramachandra S; Stroke Unit Trialists’ Collaboration. Organised inpatient (stroke unit) care for stroke: network meta-analysis. Cochrane Database Syst Rev. 2020;4(4):CD000197.
13. Epstein NE. Multidisciplinary in-hospital teams improve patient outcomes: A review. Surg Neurol Int. 2014;5(Suppl 7):S295-S303.
14. La Regina M, Guarneri F, Romano E, et al. What Quality and Safety of Care for Patients Admitted to Clinically Inappropriate Wards: a Systematic Review. J Gen Intern Med. 2019;34(7):1314-1321.
15. Blay N, Roche M, Duffield C, Xu X. Intrahospital transfers and adverse patient outcomes: An analysis of administrative health data. J Clin Nurs. 2017;26(23-24):4927-4935.
From Western Sussex Hospitals NHS Foundation Trust, Physiotherapy Department, Chichester, UK (Richard J. Holmes), and Western Sussex Hospitals NHS Foundation Trust, Department of Occupational Therapy, Chichester, UK (Sophie Stratford).
Objective: Despite the benefits of early and frequent input from a neurologist, there is wide variation in the availability of this service, especially in district general hospitals, with many patients managed on clinically inappropriate wards. The purpose of this service evaluation was to explore the impact this had on patient care.
Methods: A retrospective service evaluation was undertaken at a National Health Service hospital by reviewing patient records over a 6-month period. Data related to demographics, processes within the patient’s care, and secondary complications were recorded. Findings were compared with those of stroke patients managed on a specialist stroke ward.
Results: A total of 63 patients were identified, with a mean age of 72 years. The mean length of stay was 25.9 days, with a readmission rate of 16.7%. Only 15.9% of patients were reviewed by a neurologist. There was a high rate of secondary complications, with a number of patients experiencing falls (11.1%), pressure ulcers (14.3%), and health care–acquired infections (33.3%) during their admission.
Conclusions: The lack of specialist input from a neurologist and the management of patients on clinically inappropriate wards may have negatively impacted length of stay, readmission rates, and the frequency of secondary complications.
Keywords: evaluation; clinical safety; neurology; patient-centered care; clinical outcomes; length of stay.
It is estimated that 10% of acute admissions to district general hospitals (DGHs) of the National Health Service (NHS) in the United Kingdom are due to a neurological problem other than stroke.1 In 2011, a joint report from the Royal College of Physicians and the Association of British Neurologists (ABN) recommended that all of these patients should be admitted under the care of a neurologist and be regularly reviewed by a neurologist during their admission.2 The rationale for this recommendation is clear. The involvement of a neurologist has been shown to improve accuracy of the diagnosis3 and significantly reduce length of stay.4,5 Studies have also shown that the involvement of a neurologist has led to a change in the management plan in as high as 79%6 to 89%3 of cases, suggesting that a high proportion of neurological patients not seen by a neurologist are being managed suboptimally.
Despite this, a recent ABN survey of acute neurology services found ongoing wide variations in the availability of this specialist care, with a large proportion of DGHs having limited or no access to a neurologist and very few having dedicated neurology beds.7 While it is recognized that services have been structured in response to the reduced numbers of neurologists within the United Kingdom,8 it is prudent to assess the impact that such services have on patient care.
With this in mind, we planned to evaluate the current provision of care provided to neurological patients in a real-world setting. This was conducted in the context of a neurology liaison service at a DGH with no dedicated neurology beds.
Methods
A retrospective service evaluation was undertaken at a DGH in the southeast of England. The NHS hospital has neurologists on site who provide diagnostic and therapeutic consultations on the wards, but there are no dedicated beds for patients with neurological conditions. Patients requiring neurosurgical input are referred to a tertiary neurosciences center.
Patients were selected from the neurotherapy database if they were referred into the service between August 1, 2019, and January 31, 2020. The neurotherapy database was used as this was the only source that held thorough data on this patient group and allowed for the identification of patients who were not referred into the neurologist’s service. Patients were included if they had a new neurological condition as their primary diagnosis or if they had an exacerbation of an already established neurological condition. If a patient was admitted with more than 1 neurological diagnosis then the primary diagnosis for the admission was to be used in the analysis, though this did not occur during this evaluation. Patients with a primary diagnosis of a stroke were included if they were not managed on the acute stroke ward. Those managed on the stroke ward were excluded so that an analysis of patients managed on wards that were deemed clinically inappropriate could be undertaken. Patients were not included if they had a pre-existing neurological condition (ie, dementia, multiple sclerosis) but were admitted due to a non-neurological cause such as a fall or infection. All patients who met the criteria were included.
A team member independently reviewed each set of patient notes. Demographic data extracted from the medical notes included the patient’s age (on admission), gender, and diagnosis. Medical, nursing, and therapy notes were reviewed to identify secondary complications that arose during the patient’s admission. The secondary complications reviewed were falls (defined as the patient unexpectedly coming to the ground or other lower level), health care–acquired infections (HAIs) (defined as any infection acquired during the hospital admission), and pressure ulcers (defined as injuries to the skin or underlying tissue during the hospital admission). Other details, obtained from the patient administration system, included the length of stay (days), the number of ward moves the patient experienced, the speciality of the consultant responsible for the patient’s care, the discharge destination, and whether the patient was readmitted for any cause within 30 days. All data collected were stored on a password-protected computer and no patient-identifiable data were included.
The results were collated using descriptive statistics. The χ2 test was used to compare categorical data between those patients who were and were not reviewed by a neurologist, and the Mann-Whitney U test was used to compare differences in the length of stay between these 2 groups.
No national data relating to this specific patient group were available within the literature. Therefore, to provide a comparator of neurological patients within the same hospital, data were collected on stroke patients managed on the stroke ward. This group was deemed most appropriate for comparison as they present with similar neurological symptoms but are cared for on a specialist ward. During the evaluation period, 284 stroke patients were admitted to the stroke ward. A sample of 75 patients was randomly selected using a random number generator, and the procedure for data collection was repeated. It was not appropriate to make direct comparative analysis on these 2 groups due to the inherent differences, but it was felt important to provide context with regards to what usual care was like on a specialist ward within the same hospital.
Ethical approval was not required as this was a service evaluation of routinely collected data within a single hospital site.
Results
In total, 63 patients were identified: 26 females and 37 males. The median age of patients was 74 years (range, 39-92 years). These demographic details and comparisons to stroke patients managed on a specialist ward can be seen in Table 1. To quantify the range of diagnoses, the condition groups defined by GIRFT Neurology Methodology9 were used. The most common diagnoses were tumors of the nervous system (25.4%) and traumatic brain and spine injury (23.8%). The other conditions included in the analysis can be seen in Table 2.
Despite having a neurological condition as their primary diagnosis, only 15.9% of patients were reviewed by a neurologist during their hospital admission. Patients were most commonly under the care of a geriatrician (60.3%), but they were also managed by orthopedics (12.6%), acute medicine (7.9%), respiratory (6.3%), cardiology (4.8%), gastroenterology (3.2%), and surgery (3.2%). One patient (1.6%) was managed by intensivists.
The average length of stay was 25.9 days (range, 2-78 days). This was more than double the average length of stay on the stroke ward (11.4 days) (Table 1) and the national average for patients with neurological conditions (9.78 days).10 During their stay, 33% had 2 or more ward moves, with 1 patient moving wards a total of 6 times. Just over half (52.4%) of the patients returned to their usual residence on discharge. The remainder were discharged to rehabilitation units (15.9%), nursing homes (14.3%), residential homes (6.3%), tertiary centers (4.8%), and hospice (1.6%). Unfortunately, 3 patients (4.8%) passed away. Of those still alive (n = 60), 16.7% were readmitted to the hospital within 30 days, compared to a readmission rate of 11% on the stroke ward. None of the patients who were readmitted were seen by a neurologist during their initial admission.
The frequency of secondary complications was reviewed as a measure of the multidisciplinary management of this patient group. It was noted that 11.1% had a fall on the ward, which was similar to a rate of 10.7% on the stroke ward. More striking was the fact that 14.3% of patients developed a pressure ulcer and 33.3% developed an HAI during their admission, compared with rates of 1.3% and 10.7%, respectively, on the stroke ward (Table 1).
There were no significant differences found in length of stay between those who were and were not reviewed by a neurologist (P = .73). This was also true for categorical data, whereby readmission rate (P = .13), frequency of falls (P = .22), frequency of pressure ulcers (P = .67), and HAIs (P = .81) all failed to show a significant difference between groups.
Discussion
The findings of this service evaluation show markedly poorer outcomes for neurological patients compared to stroke patients managed on a specialist stroke ward. It is suggested that these results are in part due to the lack of specialist input from a neurologist in the majority of cases and the fact that all were managed on clinically inappropriate wards. Only 15.9% of neurological patients were seen by a neurologist. This is a slight improvement compared to previous studies in DGHs that showed rates of 10%1 and 11%,11 but it is still a far cry from the goal of 100% set out in recommendations.2 In addition, the increased readmission rate may be suggestive of suboptimal management, especially given that none of those readmitted had been reviewed by a neurologist. There are undoubtedly other factors that may influence readmissions, such as comorbidities, the severity/complexity of the condition, and the strength of community services. However, the impact of a lack of input from a specialist should not be underestimated, and further evaluation of this factor (with confounding factors controlled) would be beneficial.
The result of an extended length of stay was also a predictable outcome based on previous evidence.4,5 With the potential for suboptimal management plans and inaccurate diagnoses, it is inevitable that the patient’s movement through the hospital system will be impeded. In our example, it is possible that the extended length of stay was influenced by the fact that patients included in the evaluation were managed on nonspecialist wards and a large proportion had multiple ward changes.
Given that the evidence clearly shows that stroke patients are most effectively managed by a multidisciplinary team (MDT) with specialist skills,12 it is likely that other neurological patients, who have similar multifactorial needs, would also benefit. The patients in our evaluation were cared for by nursing staff who lacked specific skills and experience in neurology. The allied health professionals involved were specialists in neurotherapy but were not based on the ward and not directly linked to the ward MDT. A review by Epstein found that the benefits of having a MDT, in any speciality, working together on a ward included improved communication, reduced adverse events, and a reduced length of stay.13 This lack of an effective MDT approach may provide some explanation as to why the average length of stay and the rates of some secondary complications were at such elevated levels.
A systematic review exploring the impact of patients admitted to clinically inappropriate wards in a range of specialities found that these patients were associated with worse outcomes.14 This is supported by our findings, in which a higher rate of pressure ulcers and HAIs were observed when compared to rates in the specialist stroke ward. Again, a potential explanation for this is the impact of patients being managed by clinicians who lack the specialist knowledge of the patient group and the risks they face. Another explanation could be due to the high number of ward moves the patients experienced. Blay et al found that ward moves increased length of stay and carried an associated clinical risk, with the odds of falls and HAIs increasing with each move.15 A case example of this is apparent within our analysis in that the patient who experienced 6 ward moves not only had the longest length of stay (78 days), but also developed a pressure ulcer and 2 HAIs during their admission.
This service evaluation had a number of limitations that should be considered when interpreting the results. First, despite including all patients who met the criteria within the stipulated time frame, the sample size was relatively small, making it difficult to identify consistent patterns of behavior within the data.
Furthermore, caution should be applied when interpreting the comparators used, as the patient groups are not equivalent. The use of comparison against a standard is not a prerequisite in a service evaluation of this nature, but comparators were included to help frame the context for the reader. As such, they should only be used in this way rather than to make any firm conclusions.
Finally, as the evaluation was limited to the use of routinely collected data, there are several variables, other than those reported, which may have influenced the results. For example, it was not possible to ascertain certain demographic details, such as body mass index and socioeconomic factors, nor lifestyle factors such as smoking status, alcohol consumption, and exercise levels, all of which could impact negatively on the outcomes of interest. Furthermore, data were not collected on follow-up services after discharge to evaluate whether these had any impact on readmission rates.
Conclusion
This service evaluation highlights the potential impact of managing neurological patients on clinically inappropriate wards with limited input from a neurologist. There is the potential to ameliorate these impacts by cohorting these patients in neurologist-led beds with a specialist MDT. While there are limitations in the design of our study, including the lack of a controlled comparison, the small sample size, and the fact that this is an evaluation of a single service, the negative impacts to patients are concerning and warrant further investigation.
Corresponding author: Richard J. Holmes, MSc, Physiotherapy Department, St. Richard’s Hospital, Chichester, West Sussex, PO19 6SE; [email protected].
Financial disclosures: None.
From Western Sussex Hospitals NHS Foundation Trust, Physiotherapy Department, Chichester, UK (Richard J. Holmes), and Western Sussex Hospitals NHS Foundation Trust, Department of Occupational Therapy, Chichester, UK (Sophie Stratford).
Objective: Despite the benefits of early and frequent input from a neurologist, there is wide variation in the availability of this service, especially in district general hospitals, with many patients managed on clinically inappropriate wards. The purpose of this service evaluation was to explore the impact this had on patient care.
Methods: A retrospective service evaluation was undertaken at a National Health Service hospital by reviewing patient records over a 6-month period. Data related to demographics, processes within the patient’s care, and secondary complications were recorded. Findings were compared with those of stroke patients managed on a specialist stroke ward.
Results: A total of 63 patients were identified, with a mean age of 72 years. The mean length of stay was 25.9 days, with a readmission rate of 16.7%. Only 15.9% of patients were reviewed by a neurologist. There was a high rate of secondary complications, with a number of patients experiencing falls (11.1%), pressure ulcers (14.3%), and health care–acquired infections (33.3%) during their admission.
Conclusions: The lack of specialist input from a neurologist and the management of patients on clinically inappropriate wards may have negatively impacted length of stay, readmission rates, and the frequency of secondary complications.
Keywords: evaluation; clinical safety; neurology; patient-centered care; clinical outcomes; length of stay.
It is estimated that 10% of acute admissions to district general hospitals (DGHs) of the National Health Service (NHS) in the United Kingdom are due to a neurological problem other than stroke.1 In 2011, a joint report from the Royal College of Physicians and the Association of British Neurologists (ABN) recommended that all of these patients should be admitted under the care of a neurologist and be regularly reviewed by a neurologist during their admission.2 The rationale for this recommendation is clear. The involvement of a neurologist has been shown to improve accuracy of the diagnosis3 and significantly reduce length of stay.4,5 Studies have also shown that the involvement of a neurologist has led to a change in the management plan in as high as 79%6 to 89%3 of cases, suggesting that a high proportion of neurological patients not seen by a neurologist are being managed suboptimally.
Despite this, a recent ABN survey of acute neurology services found ongoing wide variations in the availability of this specialist care, with a large proportion of DGHs having limited or no access to a neurologist and very few having dedicated neurology beds.7 While it is recognized that services have been structured in response to the reduced numbers of neurologists within the United Kingdom,8 it is prudent to assess the impact that such services have on patient care.
With this in mind, we planned to evaluate the current provision of care provided to neurological patients in a real-world setting. This was conducted in the context of a neurology liaison service at a DGH with no dedicated neurology beds.
Methods
A retrospective service evaluation was undertaken at a DGH in the southeast of England. The NHS hospital has neurologists on site who provide diagnostic and therapeutic consultations on the wards, but there are no dedicated beds for patients with neurological conditions. Patients requiring neurosurgical input are referred to a tertiary neurosciences center.
Patients were selected from the neurotherapy database if they were referred into the service between August 1, 2019, and January 31, 2020. The neurotherapy database was used as this was the only source that held thorough data on this patient group and allowed for the identification of patients who were not referred into the neurologist’s service. Patients were included if they had a new neurological condition as their primary diagnosis or if they had an exacerbation of an already established neurological condition. If a patient was admitted with more than 1 neurological diagnosis then the primary diagnosis for the admission was to be used in the analysis, though this did not occur during this evaluation. Patients with a primary diagnosis of a stroke were included if they were not managed on the acute stroke ward. Those managed on the stroke ward were excluded so that an analysis of patients managed on wards that were deemed clinically inappropriate could be undertaken. Patients were not included if they had a pre-existing neurological condition (ie, dementia, multiple sclerosis) but were admitted due to a non-neurological cause such as a fall or infection. All patients who met the criteria were included.
A team member independently reviewed each set of patient notes. Demographic data extracted from the medical notes included the patient’s age (on admission), gender, and diagnosis. Medical, nursing, and therapy notes were reviewed to identify secondary complications that arose during the patient’s admission. The secondary complications reviewed were falls (defined as the patient unexpectedly coming to the ground or other lower level), health care–acquired infections (HAIs) (defined as any infection acquired during the hospital admission), and pressure ulcers (defined as injuries to the skin or underlying tissue during the hospital admission). Other details, obtained from the patient administration system, included the length of stay (days), the number of ward moves the patient experienced, the speciality of the consultant responsible for the patient’s care, the discharge destination, and whether the patient was readmitted for any cause within 30 days. All data collected were stored on a password-protected computer and no patient-identifiable data were included.
The results were collated using descriptive statistics. The χ2 test was used to compare categorical data between those patients who were and were not reviewed by a neurologist, and the Mann-Whitney U test was used to compare differences in the length of stay between these 2 groups.
No national data relating to this specific patient group were available within the literature. Therefore, to provide a comparator of neurological patients within the same hospital, data were collected on stroke patients managed on the stroke ward. This group was deemed most appropriate for comparison as they present with similar neurological symptoms but are cared for on a specialist ward. During the evaluation period, 284 stroke patients were admitted to the stroke ward. A sample of 75 patients was randomly selected using a random number generator, and the procedure for data collection was repeated. It was not appropriate to make direct comparative analysis on these 2 groups due to the inherent differences, but it was felt important to provide context with regards to what usual care was like on a specialist ward within the same hospital.
Ethical approval was not required as this was a service evaluation of routinely collected data within a single hospital site.
Results
In total, 63 patients were identified: 26 females and 37 males. The median age of patients was 74 years (range, 39-92 years). These demographic details and comparisons to stroke patients managed on a specialist ward can be seen in Table 1. To quantify the range of diagnoses, the condition groups defined by GIRFT Neurology Methodology9 were used. The most common diagnoses were tumors of the nervous system (25.4%) and traumatic brain and spine injury (23.8%). The other conditions included in the analysis can be seen in Table 2.
Despite having a neurological condition as their primary diagnosis, only 15.9% of patients were reviewed by a neurologist during their hospital admission. Patients were most commonly under the care of a geriatrician (60.3%), but they were also managed by orthopedics (12.6%), acute medicine (7.9%), respiratory (6.3%), cardiology (4.8%), gastroenterology (3.2%), and surgery (3.2%). One patient (1.6%) was managed by intensivists.
The average length of stay was 25.9 days (range, 2-78 days). This was more than double the average length of stay on the stroke ward (11.4 days) (Table 1) and the national average for patients with neurological conditions (9.78 days).10 During their stay, 33% had 2 or more ward moves, with 1 patient moving wards a total of 6 times. Just over half (52.4%) of the patients returned to their usual residence on discharge. The remainder were discharged to rehabilitation units (15.9%), nursing homes (14.3%), residential homes (6.3%), tertiary centers (4.8%), and hospice (1.6%). Unfortunately, 3 patients (4.8%) passed away. Of those still alive (n = 60), 16.7% were readmitted to the hospital within 30 days, compared to a readmission rate of 11% on the stroke ward. None of the patients who were readmitted were seen by a neurologist during their initial admission.
The frequency of secondary complications was reviewed as a measure of the multidisciplinary management of this patient group. It was noted that 11.1% had a fall on the ward, which was similar to a rate of 10.7% on the stroke ward. More striking was the fact that 14.3% of patients developed a pressure ulcer and 33.3% developed an HAI during their admission, compared with rates of 1.3% and 10.7%, respectively, on the stroke ward (Table 1).
There were no significant differences found in length of stay between those who were and were not reviewed by a neurologist (P = .73). This was also true for categorical data, whereby readmission rate (P = .13), frequency of falls (P = .22), frequency of pressure ulcers (P = .67), and HAIs (P = .81) all failed to show a significant difference between groups.
Discussion
The findings of this service evaluation show markedly poorer outcomes for neurological patients compared to stroke patients managed on a specialist stroke ward. It is suggested that these results are in part due to the lack of specialist input from a neurologist in the majority of cases and the fact that all were managed on clinically inappropriate wards. Only 15.9% of neurological patients were seen by a neurologist. This is a slight improvement compared to previous studies in DGHs that showed rates of 10%1 and 11%,11 but it is still a far cry from the goal of 100% set out in recommendations.2 In addition, the increased readmission rate may be suggestive of suboptimal management, especially given that none of those readmitted had been reviewed by a neurologist. There are undoubtedly other factors that may influence readmissions, such as comorbidities, the severity/complexity of the condition, and the strength of community services. However, the impact of a lack of input from a specialist should not be underestimated, and further evaluation of this factor (with confounding factors controlled) would be beneficial.
The result of an extended length of stay was also a predictable outcome based on previous evidence.4,5 With the potential for suboptimal management plans and inaccurate diagnoses, it is inevitable that the patient’s movement through the hospital system will be impeded. In our example, it is possible that the extended length of stay was influenced by the fact that patients included in the evaluation were managed on nonspecialist wards and a large proportion had multiple ward changes.
Given that the evidence clearly shows that stroke patients are most effectively managed by a multidisciplinary team (MDT) with specialist skills,12 it is likely that other neurological patients, who have similar multifactorial needs, would also benefit. The patients in our evaluation were cared for by nursing staff who lacked specific skills and experience in neurology. The allied health professionals involved were specialists in neurotherapy but were not based on the ward and not directly linked to the ward MDT. A review by Epstein found that the benefits of having a MDT, in any speciality, working together on a ward included improved communication, reduced adverse events, and a reduced length of stay.13 This lack of an effective MDT approach may provide some explanation as to why the average length of stay and the rates of some secondary complications were at such elevated levels.
A systematic review exploring the impact of patients admitted to clinically inappropriate wards in a range of specialities found that these patients were associated with worse outcomes.14 This is supported by our findings, in which a higher rate of pressure ulcers and HAIs were observed when compared to rates in the specialist stroke ward. Again, a potential explanation for this is the impact of patients being managed by clinicians who lack the specialist knowledge of the patient group and the risks they face. Another explanation could be due to the high number of ward moves the patients experienced. Blay et al found that ward moves increased length of stay and carried an associated clinical risk, with the odds of falls and HAIs increasing with each move.15 A case example of this is apparent within our analysis in that the patient who experienced 6 ward moves not only had the longest length of stay (78 days), but also developed a pressure ulcer and 2 HAIs during their admission.
This service evaluation had a number of limitations that should be considered when interpreting the results. First, despite including all patients who met the criteria within the stipulated time frame, the sample size was relatively small, making it difficult to identify consistent patterns of behavior within the data.
Furthermore, caution should be applied when interpreting the comparators used, as the patient groups are not equivalent. The use of comparison against a standard is not a prerequisite in a service evaluation of this nature, but comparators were included to help frame the context for the reader. As such, they should only be used in this way rather than to make any firm conclusions.
Finally, as the evaluation was limited to the use of routinely collected data, there are several variables, other than those reported, which may have influenced the results. For example, it was not possible to ascertain certain demographic details, such as body mass index and socioeconomic factors, nor lifestyle factors such as smoking status, alcohol consumption, and exercise levels, all of which could impact negatively on the outcomes of interest. Furthermore, data were not collected on follow-up services after discharge to evaluate whether these had any impact on readmission rates.
Conclusion
This service evaluation highlights the potential impact of managing neurological patients on clinically inappropriate wards with limited input from a neurologist. There is the potential to ameliorate these impacts by cohorting these patients in neurologist-led beds with a specialist MDT. While there are limitations in the design of our study, including the lack of a controlled comparison, the small sample size, and the fact that this is an evaluation of a single service, the negative impacts to patients are concerning and warrant further investigation.
Corresponding author: Richard J. Holmes, MSc, Physiotherapy Department, St. Richard’s Hospital, Chichester, West Sussex, PO19 6SE; [email protected].
Financial disclosures: None.
1. Kanagaratnam M, Boodhoo A, MacDonald BK, Nitkunan A. Prevalence of acute neurology: a 2-week snapshot in a district general hospital. Clin Med (Lond). 2020;20(2):169-173.
2. Royal College of Physicians. Local adult neurology services for the next decade. Report of a working party. June 2011. Accessed October 29, 2020. https://www.mstrust.org.uk/sites/default/files/files/Local%20adult%20neurology%20services%20for%20the%20next%20decade.pdf
3. McColgan P, Carr AS, McCarron MO. The value of a liaison neurology service in a district general hospital. Postgrad Med J. 2011;87(1025):166-169.
4. Forbes R, Craig J, Callender M, Patterson V. Liaison neurology for acute medical admissions. Clin Med (Lond). 2004;4(3):290.
5. Craig J, Chua R, Russell C, et al. A cohort study of early neurological consultation by telemedicine on the care of neurological inpatients. J Neurol Neurosurg Psychiatry. 2004;75(7):1031-1035.
6. Ali E, Chaila E, Hutchinson M, Tubridy N. The ‘hidden work’ of a hospital neurologist: 1000 consults later. Eur J Neurol. 2010;17(4):e28-e32.
7. Association of British Neurologists. Acute Neurology services survey 2017. Accessed October 29, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/ABN_2017_Acute_Neurology_Survey.pdf
8. Nitkunan A, Lawrence J, Reilly MM. Neurology Workforce Survey. January 28, 2020. Accessed October 28, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/2020_ABN_Neurology_Workforce_Survey_2018-19_28_Jan_2020.pdf
9. Fuller G, Connolly M, Mummery C, Williams A. GIRT Neurology Methodology and Initial Summary of Regional Data. September 2019. Accessed October 26, 2020. https://gettingitrightfirsttime.co.uk/wp-content/uploads/2017/07/GIRFT-neurology-methodology-090919-FINAL.pdf
10. The Neurological Alliance. Neuro Numbers 2019. Accessed October 28, 2020. https://www.neural.org.uk/wp-content/uploads/2019/07/neuro-numbers-2019.pdf
11. Cai A, Brex P. A survey of acute neurology at a general hospital in the UK. Clin Med (Lond). 2010;10(6):642-643.
12. Langhorne P, Ramachandra S; Stroke Unit Trialists’ Collaboration. Organised inpatient (stroke unit) care for stroke: network meta-analysis. Cochrane Database Syst Rev. 2020;4(4):CD000197.
13. Epstein NE. Multidisciplinary in-hospital teams improve patient outcomes: A review. Surg Neurol Int. 2014;5(Suppl 7):S295-S303.
14. La Regina M, Guarneri F, Romano E, et al. What Quality and Safety of Care for Patients Admitted to Clinically Inappropriate Wards: a Systematic Review. J Gen Intern Med. 2019;34(7):1314-1321.
15. Blay N, Roche M, Duffield C, Xu X. Intrahospital transfers and adverse patient outcomes: An analysis of administrative health data. J Clin Nurs. 2017;26(23-24):4927-4935.
1. Kanagaratnam M, Boodhoo A, MacDonald BK, Nitkunan A. Prevalence of acute neurology: a 2-week snapshot in a district general hospital. Clin Med (Lond). 2020;20(2):169-173.
2. Royal College of Physicians. Local adult neurology services for the next decade. Report of a working party. June 2011. Accessed October 29, 2020. https://www.mstrust.org.uk/sites/default/files/files/Local%20adult%20neurology%20services%20for%20the%20next%20decade.pdf
3. McColgan P, Carr AS, McCarron MO. The value of a liaison neurology service in a district general hospital. Postgrad Med J. 2011;87(1025):166-169.
4. Forbes R, Craig J, Callender M, Patterson V. Liaison neurology for acute medical admissions. Clin Med (Lond). 2004;4(3):290.
5. Craig J, Chua R, Russell C, et al. A cohort study of early neurological consultation by telemedicine on the care of neurological inpatients. J Neurol Neurosurg Psychiatry. 2004;75(7):1031-1035.
6. Ali E, Chaila E, Hutchinson M, Tubridy N. The ‘hidden work’ of a hospital neurologist: 1000 consults later. Eur J Neurol. 2010;17(4):e28-e32.
7. Association of British Neurologists. Acute Neurology services survey 2017. Accessed October 29, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/ABN_2017_Acute_Neurology_Survey.pdf
8. Nitkunan A, Lawrence J, Reilly MM. Neurology Workforce Survey. January 28, 2020. Accessed October 28, 2020. https://cdn.ymaws.com/www.theabn.org/resource/collection/219B4A48-4D25-4726-97AA-0EB6090769BE/2020_ABN_Neurology_Workforce_Survey_2018-19_28_Jan_2020.pdf
9. Fuller G, Connolly M, Mummery C, Williams A. GIRT Neurology Methodology and Initial Summary of Regional Data. September 2019. Accessed October 26, 2020. https://gettingitrightfirsttime.co.uk/wp-content/uploads/2017/07/GIRFT-neurology-methodology-090919-FINAL.pdf
10. The Neurological Alliance. Neuro Numbers 2019. Accessed October 28, 2020. https://www.neural.org.uk/wp-content/uploads/2019/07/neuro-numbers-2019.pdf
11. Cai A, Brex P. A survey of acute neurology at a general hospital in the UK. Clin Med (Lond). 2010;10(6):642-643.
12. Langhorne P, Ramachandra S; Stroke Unit Trialists’ Collaboration. Organised inpatient (stroke unit) care for stroke: network meta-analysis. Cochrane Database Syst Rev. 2020;4(4):CD000197.
13. Epstein NE. Multidisciplinary in-hospital teams improve patient outcomes: A review. Surg Neurol Int. 2014;5(Suppl 7):S295-S303.
14. La Regina M, Guarneri F, Romano E, et al. What Quality and Safety of Care for Patients Admitted to Clinically Inappropriate Wards: a Systematic Review. J Gen Intern Med. 2019;34(7):1314-1321.
15. Blay N, Roche M, Duffield C, Xu X. Intrahospital transfers and adverse patient outcomes: An analysis of administrative health data. J Clin Nurs. 2017;26(23-24):4927-4935.