User login
Democratic Lawmakers Press Pfizer on Chemotherapy Drug Shortages
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
Unleashing Our Immune Response to Quash Cancer
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
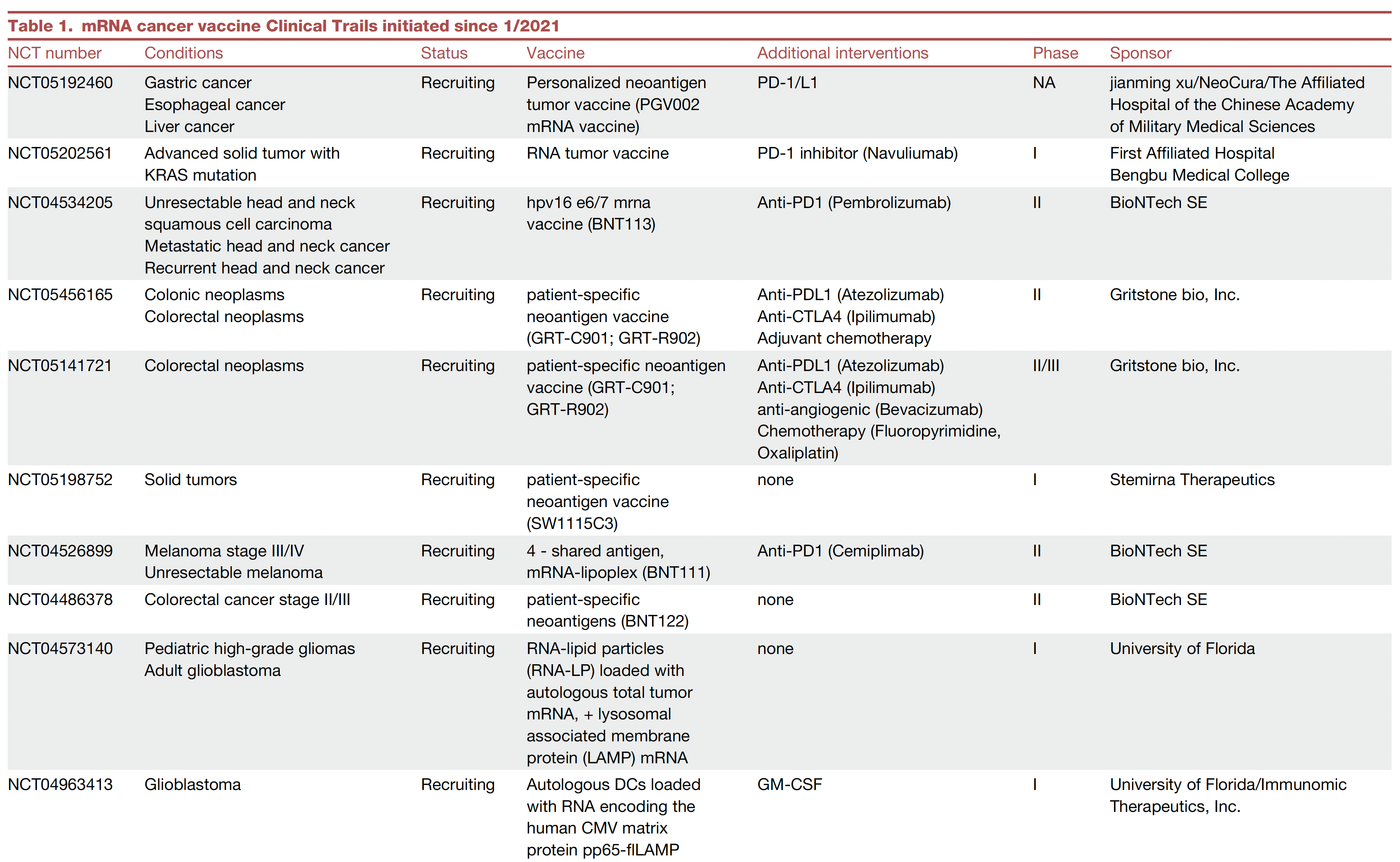
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
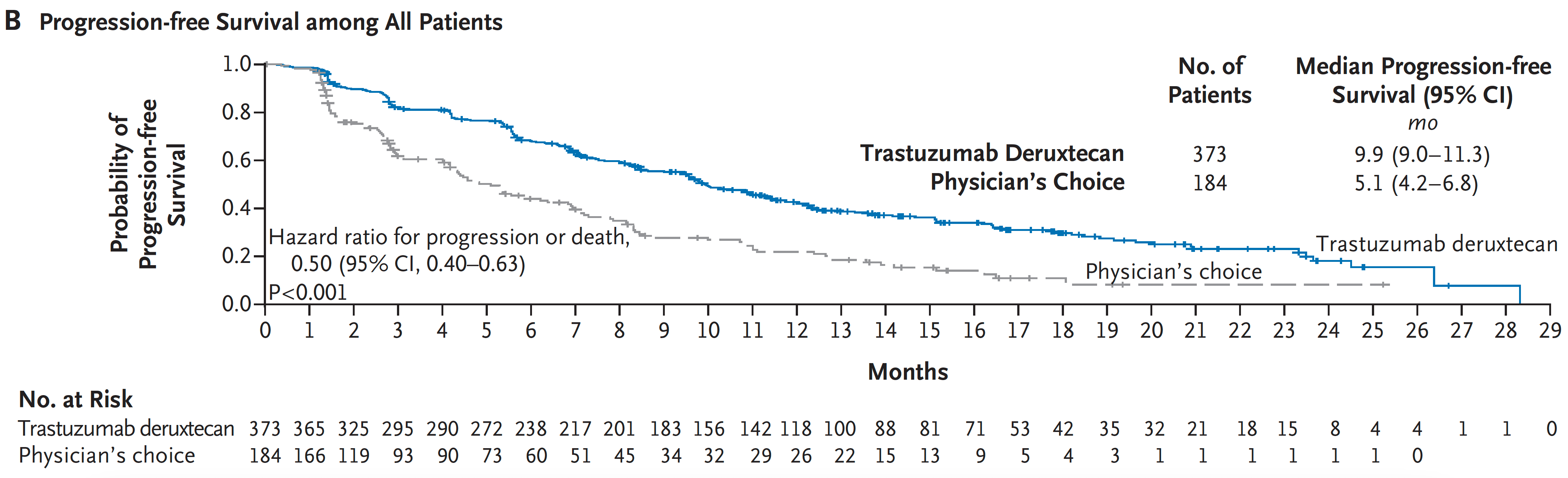
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
FDA OKs new agent to block chemotherapy-induced neutropenia
Efbemalenograstim joins other agents already on the U.S. market, including pegfilgrastim (Neulasta), that aim to reduce the incidence of chemotherapy-induced febrile neutropenia.
The approval of efbemalenograstim was based on two randomized trials. The first included 122 women with either metastatic or nonmetastatic breast cancer who were receiving doxorubicin and docetaxel. These patients were randomly assigned to receive either one subcutaneous injection of efbemalenograstim or placebo on the second day of their first chemotherapy cycle. All patients received efbemalenograstim on the second day of cycles two through four.
The mean duration of grade 4 neutropenia in the first cycle was 1.4 days with efbemalenograstim versus 4.3 days with placebo. Only 4.8% of patients who received efbemalenograstim experienced chemotherapy-induced febrile neutropenia, compared with 25.6% who received the placebo.
The new agent went up against pegfilgrastim in the second trial, which included 393 women who received docetaxel and cyclophosphamide as treatment for nonmetastatic breast cancer. These patients were randomly assigned to receive either a single subcutaneous injection of efbemalenograstim or pegfilgrastim on the second day of each cycle.
During the first cycle, patients in both arms of the trial experienced a mean of 0.2 days of grade 4 neutropenia.
The most common side effects associated with efbemalenograstim were nausea, anemia, and thrombocytopenia. Similar to pegfilgrastim’s label, efbemalenograstim’s label warns of possible splenic rupture, respiratory distress syndrome, sickle cell crisis, and other serious adverse events.
The FDA recommends a dose of 20 mg subcutaneous once per chemotherapy cycle.
A version of this article first appeared on Medscape.com.
Efbemalenograstim joins other agents already on the U.S. market, including pegfilgrastim (Neulasta), that aim to reduce the incidence of chemotherapy-induced febrile neutropenia.
The approval of efbemalenograstim was based on two randomized trials. The first included 122 women with either metastatic or nonmetastatic breast cancer who were receiving doxorubicin and docetaxel. These patients were randomly assigned to receive either one subcutaneous injection of efbemalenograstim or placebo on the second day of their first chemotherapy cycle. All patients received efbemalenograstim on the second day of cycles two through four.
The mean duration of grade 4 neutropenia in the first cycle was 1.4 days with efbemalenograstim versus 4.3 days with placebo. Only 4.8% of patients who received efbemalenograstim experienced chemotherapy-induced febrile neutropenia, compared with 25.6% who received the placebo.
The new agent went up against pegfilgrastim in the second trial, which included 393 women who received docetaxel and cyclophosphamide as treatment for nonmetastatic breast cancer. These patients were randomly assigned to receive either a single subcutaneous injection of efbemalenograstim or pegfilgrastim on the second day of each cycle.
During the first cycle, patients in both arms of the trial experienced a mean of 0.2 days of grade 4 neutropenia.
The most common side effects associated with efbemalenograstim were nausea, anemia, and thrombocytopenia. Similar to pegfilgrastim’s label, efbemalenograstim’s label warns of possible splenic rupture, respiratory distress syndrome, sickle cell crisis, and other serious adverse events.
The FDA recommends a dose of 20 mg subcutaneous once per chemotherapy cycle.
A version of this article first appeared on Medscape.com.
Efbemalenograstim joins other agents already on the U.S. market, including pegfilgrastim (Neulasta), that aim to reduce the incidence of chemotherapy-induced febrile neutropenia.
The approval of efbemalenograstim was based on two randomized trials. The first included 122 women with either metastatic or nonmetastatic breast cancer who were receiving doxorubicin and docetaxel. These patients were randomly assigned to receive either one subcutaneous injection of efbemalenograstim or placebo on the second day of their first chemotherapy cycle. All patients received efbemalenograstim on the second day of cycles two through four.
The mean duration of grade 4 neutropenia in the first cycle was 1.4 days with efbemalenograstim versus 4.3 days with placebo. Only 4.8% of patients who received efbemalenograstim experienced chemotherapy-induced febrile neutropenia, compared with 25.6% who received the placebo.
The new agent went up against pegfilgrastim in the second trial, which included 393 women who received docetaxel and cyclophosphamide as treatment for nonmetastatic breast cancer. These patients were randomly assigned to receive either a single subcutaneous injection of efbemalenograstim or pegfilgrastim on the second day of each cycle.
During the first cycle, patients in both arms of the trial experienced a mean of 0.2 days of grade 4 neutropenia.
The most common side effects associated with efbemalenograstim were nausea, anemia, and thrombocytopenia. Similar to pegfilgrastim’s label, efbemalenograstim’s label warns of possible splenic rupture, respiratory distress syndrome, sickle cell crisis, and other serious adverse events.
The FDA recommends a dose of 20 mg subcutaneous once per chemotherapy cycle.
A version of this article first appeared on Medscape.com.

{kind=link}
{kind=link}
FDA panel voices concerns over 2 lymphoma accelerated approvals
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
FDA’s Project Optimus aims to transform early cancer research
SAN DIEGO –
The goal is “to better identify and characterize optimized doses” in early stages of research and move away from the default of the traditional maximum tolerated dose strategy, hematologist-oncologist Marc R. Theoret, MD, deputy director of the FDA’s Oncology Center of Excellence, said in a presentation at the 2023 Society for Immunotherapy of Cancer annual meeting.
Earlier this year, the FDA released a draft guidance regarding the changes it hopes to see. The agency supported randomized, parallel dose-response trials when feasible, and “strong rationale for choice of dosage should be provided before initiating a registration trial(s) to support a subsequent indication and usage.”
The goal of controlling toxicity is “very highly important” in hematology research since blood cancer drugs can cause significant adverse effects in areas such as the lungs and heart, said Cecilia Yeung, MD, who led the SITC session about Project Optimus. Dr. Yeung is a clinical pathologist who works on investigational trials at Fred Hutchinson Cancer Research Center in Seattle.
In an interview, Dr. Yeung, who has a subspecialty in hematopathology, explained why the foundations of cancer research are changing and what hematologist-oncologists can expect to see on the horizon.
Q: Project Optimus aims to move beyond the traditional dose-escalation approach to the development of cancer drugs. How does that strategy work?
Dr. Yeung: Prior to Project Optimus, they’d use a 3+3 strategy in phase 1 trials: They’d give a dose to three fairly healthy patients, then they’d go up by escalating doses in more patients. They’d keep going up until two-thirds of patients at a specific dose suffered from bad side effects, then they’d back off to the last dose.
Q: This approach, which aims to identify the “maximum tolerated dose,” seemed to work well over decades of research into chemotherapy drugs. But worries arose as targeted therapies appeared in oncology areas such as blood cancer. Why did things change?
Dr. Yeung: With 3+3, you could tell pretty quickly how toxic chemotherapy was. But in targeted therapy, we were finding that these studies are not representative of actual toxicity. You’re not treating these patients for a very long time in phase 1, while patients on targeted therapy may be on these drugs for years. Concerns actually started with the first targeted drugs to treat leukemias and lymphomas. They were shown to have unexpected toxicity. A 2016 study found that drug developers had to reduce the original phase 1 dose in 45% of phase 3 trials [of small molecule and monoclonal antibody targeted agents] approved by the FDA over 12 years because of toxicity.
Q: What is FDA’s goal for Project Optimus?
Dr. Yeung: They want to have a second piece, to balance that maximum tolerated dose with a safe and tolerable dose for most people.
Q: What kind of resistance is the FDA getting from drug companies?
Dr. Yeung: The FDA makes a good argument that the system wasn’t working. But drug companies say this will drive up the cost of clinical trials and won’t allow them to treat patients with the maximal doses they could give them. I see arguments from both sides. There has to be a balance between the two.
Q: How will all this affect drug development?
Dr. Yeung: Drugs may become more expensive because much more testing will happen during clinical trials.
Q: Could this reduce the number of investigational drugs?
Dr. Yeung: Hopefully not, but this is huge endeavor for smaller companies that are strapped for funding.
Q: What do you think the future holds?
Dr. Yeung: Ultimately, this is a good thing because if everything works out, we’ll have fewer toxic side effects. But we’re going to have to go through a period of growing pains.
SAN DIEGO –
The goal is “to better identify and characterize optimized doses” in early stages of research and move away from the default of the traditional maximum tolerated dose strategy, hematologist-oncologist Marc R. Theoret, MD, deputy director of the FDA’s Oncology Center of Excellence, said in a presentation at the 2023 Society for Immunotherapy of Cancer annual meeting.
Earlier this year, the FDA released a draft guidance regarding the changes it hopes to see. The agency supported randomized, parallel dose-response trials when feasible, and “strong rationale for choice of dosage should be provided before initiating a registration trial(s) to support a subsequent indication and usage.”
The goal of controlling toxicity is “very highly important” in hematology research since blood cancer drugs can cause significant adverse effects in areas such as the lungs and heart, said Cecilia Yeung, MD, who led the SITC session about Project Optimus. Dr. Yeung is a clinical pathologist who works on investigational trials at Fred Hutchinson Cancer Research Center in Seattle.
In an interview, Dr. Yeung, who has a subspecialty in hematopathology, explained why the foundations of cancer research are changing and what hematologist-oncologists can expect to see on the horizon.
Q: Project Optimus aims to move beyond the traditional dose-escalation approach to the development of cancer drugs. How does that strategy work?
Dr. Yeung: Prior to Project Optimus, they’d use a 3+3 strategy in phase 1 trials: They’d give a dose to three fairly healthy patients, then they’d go up by escalating doses in more patients. They’d keep going up until two-thirds of patients at a specific dose suffered from bad side effects, then they’d back off to the last dose.
Q: This approach, which aims to identify the “maximum tolerated dose,” seemed to work well over decades of research into chemotherapy drugs. But worries arose as targeted therapies appeared in oncology areas such as blood cancer. Why did things change?
Dr. Yeung: With 3+3, you could tell pretty quickly how toxic chemotherapy was. But in targeted therapy, we were finding that these studies are not representative of actual toxicity. You’re not treating these patients for a very long time in phase 1, while patients on targeted therapy may be on these drugs for years. Concerns actually started with the first targeted drugs to treat leukemias and lymphomas. They were shown to have unexpected toxicity. A 2016 study found that drug developers had to reduce the original phase 1 dose in 45% of phase 3 trials [of small molecule and monoclonal antibody targeted agents] approved by the FDA over 12 years because of toxicity.
Q: What is FDA’s goal for Project Optimus?
Dr. Yeung: They want to have a second piece, to balance that maximum tolerated dose with a safe and tolerable dose for most people.
Q: What kind of resistance is the FDA getting from drug companies?
Dr. Yeung: The FDA makes a good argument that the system wasn’t working. But drug companies say this will drive up the cost of clinical trials and won’t allow them to treat patients with the maximal doses they could give them. I see arguments from both sides. There has to be a balance between the two.
Q: How will all this affect drug development?
Dr. Yeung: Drugs may become more expensive because much more testing will happen during clinical trials.
Q: Could this reduce the number of investigational drugs?
Dr. Yeung: Hopefully not, but this is huge endeavor for smaller companies that are strapped for funding.
Q: What do you think the future holds?
Dr. Yeung: Ultimately, this is a good thing because if everything works out, we’ll have fewer toxic side effects. But we’re going to have to go through a period of growing pains.
SAN DIEGO –
The goal is “to better identify and characterize optimized doses” in early stages of research and move away from the default of the traditional maximum tolerated dose strategy, hematologist-oncologist Marc R. Theoret, MD, deputy director of the FDA’s Oncology Center of Excellence, said in a presentation at the 2023 Society for Immunotherapy of Cancer annual meeting.
Earlier this year, the FDA released a draft guidance regarding the changes it hopes to see. The agency supported randomized, parallel dose-response trials when feasible, and “strong rationale for choice of dosage should be provided before initiating a registration trial(s) to support a subsequent indication and usage.”
The goal of controlling toxicity is “very highly important” in hematology research since blood cancer drugs can cause significant adverse effects in areas such as the lungs and heart, said Cecilia Yeung, MD, who led the SITC session about Project Optimus. Dr. Yeung is a clinical pathologist who works on investigational trials at Fred Hutchinson Cancer Research Center in Seattle.
In an interview, Dr. Yeung, who has a subspecialty in hematopathology, explained why the foundations of cancer research are changing and what hematologist-oncologists can expect to see on the horizon.
Q: Project Optimus aims to move beyond the traditional dose-escalation approach to the development of cancer drugs. How does that strategy work?
Dr. Yeung: Prior to Project Optimus, they’d use a 3+3 strategy in phase 1 trials: They’d give a dose to three fairly healthy patients, then they’d go up by escalating doses in more patients. They’d keep going up until two-thirds of patients at a specific dose suffered from bad side effects, then they’d back off to the last dose.
Q: This approach, which aims to identify the “maximum tolerated dose,” seemed to work well over decades of research into chemotherapy drugs. But worries arose as targeted therapies appeared in oncology areas such as blood cancer. Why did things change?
Dr. Yeung: With 3+3, you could tell pretty quickly how toxic chemotherapy was. But in targeted therapy, we were finding that these studies are not representative of actual toxicity. You’re not treating these patients for a very long time in phase 1, while patients on targeted therapy may be on these drugs for years. Concerns actually started with the first targeted drugs to treat leukemias and lymphomas. They were shown to have unexpected toxicity. A 2016 study found that drug developers had to reduce the original phase 1 dose in 45% of phase 3 trials [of small molecule and monoclonal antibody targeted agents] approved by the FDA over 12 years because of toxicity.
Q: What is FDA’s goal for Project Optimus?
Dr. Yeung: They want to have a second piece, to balance that maximum tolerated dose with a safe and tolerable dose for most people.
Q: What kind of resistance is the FDA getting from drug companies?
Dr. Yeung: The FDA makes a good argument that the system wasn’t working. But drug companies say this will drive up the cost of clinical trials and won’t allow them to treat patients with the maximal doses they could give them. I see arguments from both sides. There has to be a balance between the two.
Q: How will all this affect drug development?
Dr. Yeung: Drugs may become more expensive because much more testing will happen during clinical trials.
Q: Could this reduce the number of investigational drugs?
Dr. Yeung: Hopefully not, but this is huge endeavor for smaller companies that are strapped for funding.
Q: What do you think the future holds?
Dr. Yeung: Ultimately, this is a good thing because if everything works out, we’ll have fewer toxic side effects. But we’re going to have to go through a period of growing pains.
AT SITC 2023
Antibody shows promise in preventing GVHD
Early, intriguing research suggests that preventing acute graft-versus-host disease (GVHD) in the gut – a potentially life-threatening complication of allogeneic hematopoietic cell transplantation (allo-HCT) – could be accomplished by the administration of a single antibody that targets the anti-DLL4 Notch signaling pathway, without compromising the stem cell transplant.
“The major surprise was that none of the anti–DLL4-treated animals developed acute gastrointestinal GVHD for the entire duration of the study. This was a remarkable finding, given that intestinal GVHD is otherwise seen in the vast majority of nonhuman primate transplant recipients that receive either no prophylaxis, or prophylaxis with agents other than anti-DLL4 antibodies,” co–senior author Ivan Maillard, MD, PhD, a professor of medicine and vice chief for research in hematology-oncology at the University of Pennsylvania, Philadelphia, said in an interview.
“The timing was critical,” the authors noted in the study, recently published in Science Translational Medicine. “Intervening before any symptoms of GvHD appear made the long-term protection possible.”
While GVHD may be mild to moderate in chronic forms, acute cases can be serious, if not fatal, and nearly all severe acute GVHD prominently involves the gastrointestinal tract, which can drive activation of pathogenic T cells and potentially lead to tissue damage following allo-HCT.
Systemic corticosteroids are standard first-line treatment for acute GVHD. However, response rates generally range only from 40% to 60%, and there are concerns of side effects. Meanwhile, second-line treatments are of inconsistent benefit.
With previous studies on mice showing benefits of targeting Notch pathway inhibition, particularly DLL4, Dr. Maillard and colleagues further investigated the effects in nonhuman primates that were allo-HCT recipients, using the anti-DLL4 antibody REGN421, which has pharmacokinetic and toxicity information available from previous studies.
The nonhuman primates were treated with one of two dosing regimens: a single dose of REGN421 3 mg/kg at baseline, post HCT, (n = 7) or three weekly doses at days 0, 7 and 14, post transplant (n = 4). Those primates were compared with 11 primates receiving allo-HCT transplants that received supportive care only.
Primates receiving three weekly doses of REGN421 showed antibody concentrations of greater than 2 mcg/mL for more than 30 days post HCT. A single dose of REGN421 was associated with protection from acute GVHD at day 0, while three weekly doses showed protection at day 0, 7, and 14, consistent with an impact of REGN421 during the early phases of T-cell activation.
Compared with animals receiving only supportive care, prophylaxis with REGN421 was associated with delayed acute GVHD onset and lengthened survival.
Of the 11 primates treated with REGN421, none developed clinical signs of gastrointestinal acute GVHD, whereas the majority of those receiving standard care or other preventive interventions did.
“Detailed analysis of acute GVHD clinical presentations in REGN421-treated animals in comparison to no treatment controls revealed near complete protection from GI-acute GvHD with REGN421,” the authors reported.
Furthermore, pathology scores in the gastrointestinal tract were lower with REGN421 treatment, compared with the no-treatment cohort, and the scores matched those of healthy nontransplanted nonhuman primates.
The primates treated with REGN421 did ultimately develop other clinical and pathologic signs of skin, hepatic or pulmonary acute GVHD, but without gastrointestinal disease.
The treatment was not associated with any adverse effects on the allo-HCT, with primates receiving either a single dose or three weekly doses of REGN421 showing rapid donor engraftment after allo-HCT, including high bone marrow, whole blood, and T-cell donor chimerism.
“Reassuringly, short-term systemic DLL4 blockade with REGN421 did not trigger unexpected side effects in our nonhuman primate model, while preserving rapid engraftment as well hematopoietic and immune reconstitution.”
The mechanism preserving the engraftment, described as a “major surprise,” specifically involved DLL4 inhibition blocking the homing of pathogenic T cells to the gut while preserving homing of regulatory T cells that dampen the immune response, Dr. Maillard explained.
“This effect turned out to be at least in part through a posttranslational effect of DLL4/Notch blockade on integrin pairing at the T-cell surface,” he explained. “This was a novel and quite unexpected mechanism of action conserved from mice to nonhuman primates.”
The results are encouraging in terms of translating to humans because of their closer similarities in various physiological factors, Dr. Maillard said.
“The nonhuman primate model of transplantation [offers] a transplantation model very close to what is being performed in humans, as well as the opportunity to study an immune system very similar to that of humans in nonhuman primates,” he said.
Dr. Maillard noted that, while trials in humans are not underway yet, “we are in active discussions about it,” and the team is indeed interested in testing REGN421 itself, with the effects likely to be as a prophylactic strategy.
There are currently no approved anti-DLL4 antibody drugs for use in humans.
“Our approach is mostly promising as a preventive treatment, rather than as a secondary treatment for GVHD, because DLL4/Notch blockade seems most active when applied early after transplantation during the time of initial seeding of the gut by T cells (in mice, we had observed the critical time window for a successful intervention to be within 48 hours of transplantation),” Dr. Maillard said.“There remain questions about which other prophylactic treatments we should ideally combine anti-DLL4 antibodies with.”
Dr. Maillard has received research funding from Regeneron and Genentech and is a member of Garuda Therapeutics’s scientific advisory board.
Early, intriguing research suggests that preventing acute graft-versus-host disease (GVHD) in the gut – a potentially life-threatening complication of allogeneic hematopoietic cell transplantation (allo-HCT) – could be accomplished by the administration of a single antibody that targets the anti-DLL4 Notch signaling pathway, without compromising the stem cell transplant.
“The major surprise was that none of the anti–DLL4-treated animals developed acute gastrointestinal GVHD for the entire duration of the study. This was a remarkable finding, given that intestinal GVHD is otherwise seen in the vast majority of nonhuman primate transplant recipients that receive either no prophylaxis, or prophylaxis with agents other than anti-DLL4 antibodies,” co–senior author Ivan Maillard, MD, PhD, a professor of medicine and vice chief for research in hematology-oncology at the University of Pennsylvania, Philadelphia, said in an interview.
“The timing was critical,” the authors noted in the study, recently published in Science Translational Medicine. “Intervening before any symptoms of GvHD appear made the long-term protection possible.”
While GVHD may be mild to moderate in chronic forms, acute cases can be serious, if not fatal, and nearly all severe acute GVHD prominently involves the gastrointestinal tract, which can drive activation of pathogenic T cells and potentially lead to tissue damage following allo-HCT.
Systemic corticosteroids are standard first-line treatment for acute GVHD. However, response rates generally range only from 40% to 60%, and there are concerns of side effects. Meanwhile, second-line treatments are of inconsistent benefit.
With previous studies on mice showing benefits of targeting Notch pathway inhibition, particularly DLL4, Dr. Maillard and colleagues further investigated the effects in nonhuman primates that were allo-HCT recipients, using the anti-DLL4 antibody REGN421, which has pharmacokinetic and toxicity information available from previous studies.
The nonhuman primates were treated with one of two dosing regimens: a single dose of REGN421 3 mg/kg at baseline, post HCT, (n = 7) or three weekly doses at days 0, 7 and 14, post transplant (n = 4). Those primates were compared with 11 primates receiving allo-HCT transplants that received supportive care only.
Primates receiving three weekly doses of REGN421 showed antibody concentrations of greater than 2 mcg/mL for more than 30 days post HCT. A single dose of REGN421 was associated with protection from acute GVHD at day 0, while three weekly doses showed protection at day 0, 7, and 14, consistent with an impact of REGN421 during the early phases of T-cell activation.
Compared with animals receiving only supportive care, prophylaxis with REGN421 was associated with delayed acute GVHD onset and lengthened survival.
Of the 11 primates treated with REGN421, none developed clinical signs of gastrointestinal acute GVHD, whereas the majority of those receiving standard care or other preventive interventions did.
“Detailed analysis of acute GVHD clinical presentations in REGN421-treated animals in comparison to no treatment controls revealed near complete protection from GI-acute GvHD with REGN421,” the authors reported.
Furthermore, pathology scores in the gastrointestinal tract were lower with REGN421 treatment, compared with the no-treatment cohort, and the scores matched those of healthy nontransplanted nonhuman primates.
The primates treated with REGN421 did ultimately develop other clinical and pathologic signs of skin, hepatic or pulmonary acute GVHD, but without gastrointestinal disease.
The treatment was not associated with any adverse effects on the allo-HCT, with primates receiving either a single dose or three weekly doses of REGN421 showing rapid donor engraftment after allo-HCT, including high bone marrow, whole blood, and T-cell donor chimerism.
“Reassuringly, short-term systemic DLL4 blockade with REGN421 did not trigger unexpected side effects in our nonhuman primate model, while preserving rapid engraftment as well hematopoietic and immune reconstitution.”
The mechanism preserving the engraftment, described as a “major surprise,” specifically involved DLL4 inhibition blocking the homing of pathogenic T cells to the gut while preserving homing of regulatory T cells that dampen the immune response, Dr. Maillard explained.
“This effect turned out to be at least in part through a posttranslational effect of DLL4/Notch blockade on integrin pairing at the T-cell surface,” he explained. “This was a novel and quite unexpected mechanism of action conserved from mice to nonhuman primates.”
The results are encouraging in terms of translating to humans because of their closer similarities in various physiological factors, Dr. Maillard said.
“The nonhuman primate model of transplantation [offers] a transplantation model very close to what is being performed in humans, as well as the opportunity to study an immune system very similar to that of humans in nonhuman primates,” he said.
Dr. Maillard noted that, while trials in humans are not underway yet, “we are in active discussions about it,” and the team is indeed interested in testing REGN421 itself, with the effects likely to be as a prophylactic strategy.
There are currently no approved anti-DLL4 antibody drugs for use in humans.
“Our approach is mostly promising as a preventive treatment, rather than as a secondary treatment for GVHD, because DLL4/Notch blockade seems most active when applied early after transplantation during the time of initial seeding of the gut by T cells (in mice, we had observed the critical time window for a successful intervention to be within 48 hours of transplantation),” Dr. Maillard said.“There remain questions about which other prophylactic treatments we should ideally combine anti-DLL4 antibodies with.”
Dr. Maillard has received research funding from Regeneron and Genentech and is a member of Garuda Therapeutics’s scientific advisory board.
Early, intriguing research suggests that preventing acute graft-versus-host disease (GVHD) in the gut – a potentially life-threatening complication of allogeneic hematopoietic cell transplantation (allo-HCT) – could be accomplished by the administration of a single antibody that targets the anti-DLL4 Notch signaling pathway, without compromising the stem cell transplant.
“The major surprise was that none of the anti–DLL4-treated animals developed acute gastrointestinal GVHD for the entire duration of the study. This was a remarkable finding, given that intestinal GVHD is otherwise seen in the vast majority of nonhuman primate transplant recipients that receive either no prophylaxis, or prophylaxis with agents other than anti-DLL4 antibodies,” co–senior author Ivan Maillard, MD, PhD, a professor of medicine and vice chief for research in hematology-oncology at the University of Pennsylvania, Philadelphia, said in an interview.
“The timing was critical,” the authors noted in the study, recently published in Science Translational Medicine. “Intervening before any symptoms of GvHD appear made the long-term protection possible.”
While GVHD may be mild to moderate in chronic forms, acute cases can be serious, if not fatal, and nearly all severe acute GVHD prominently involves the gastrointestinal tract, which can drive activation of pathogenic T cells and potentially lead to tissue damage following allo-HCT.
Systemic corticosteroids are standard first-line treatment for acute GVHD. However, response rates generally range only from 40% to 60%, and there are concerns of side effects. Meanwhile, second-line treatments are of inconsistent benefit.
With previous studies on mice showing benefits of targeting Notch pathway inhibition, particularly DLL4, Dr. Maillard and colleagues further investigated the effects in nonhuman primates that were allo-HCT recipients, using the anti-DLL4 antibody REGN421, which has pharmacokinetic and toxicity information available from previous studies.
The nonhuman primates were treated with one of two dosing regimens: a single dose of REGN421 3 mg/kg at baseline, post HCT, (n = 7) or three weekly doses at days 0, 7 and 14, post transplant (n = 4). Those primates were compared with 11 primates receiving allo-HCT transplants that received supportive care only.
Primates receiving three weekly doses of REGN421 showed antibody concentrations of greater than 2 mcg/mL for more than 30 days post HCT. A single dose of REGN421 was associated with protection from acute GVHD at day 0, while three weekly doses showed protection at day 0, 7, and 14, consistent with an impact of REGN421 during the early phases of T-cell activation.
Compared with animals receiving only supportive care, prophylaxis with REGN421 was associated with delayed acute GVHD onset and lengthened survival.
Of the 11 primates treated with REGN421, none developed clinical signs of gastrointestinal acute GVHD, whereas the majority of those receiving standard care or other preventive interventions did.
“Detailed analysis of acute GVHD clinical presentations in REGN421-treated animals in comparison to no treatment controls revealed near complete protection from GI-acute GvHD with REGN421,” the authors reported.
Furthermore, pathology scores in the gastrointestinal tract were lower with REGN421 treatment, compared with the no-treatment cohort, and the scores matched those of healthy nontransplanted nonhuman primates.
The primates treated with REGN421 did ultimately develop other clinical and pathologic signs of skin, hepatic or pulmonary acute GVHD, but without gastrointestinal disease.
The treatment was not associated with any adverse effects on the allo-HCT, with primates receiving either a single dose or three weekly doses of REGN421 showing rapid donor engraftment after allo-HCT, including high bone marrow, whole blood, and T-cell donor chimerism.
“Reassuringly, short-term systemic DLL4 blockade with REGN421 did not trigger unexpected side effects in our nonhuman primate model, while preserving rapid engraftment as well hematopoietic and immune reconstitution.”
The mechanism preserving the engraftment, described as a “major surprise,” specifically involved DLL4 inhibition blocking the homing of pathogenic T cells to the gut while preserving homing of regulatory T cells that dampen the immune response, Dr. Maillard explained.
“This effect turned out to be at least in part through a posttranslational effect of DLL4/Notch blockade on integrin pairing at the T-cell surface,” he explained. “This was a novel and quite unexpected mechanism of action conserved from mice to nonhuman primates.”
The results are encouraging in terms of translating to humans because of their closer similarities in various physiological factors, Dr. Maillard said.
“The nonhuman primate model of transplantation [offers] a transplantation model very close to what is being performed in humans, as well as the opportunity to study an immune system very similar to that of humans in nonhuman primates,” he said.
Dr. Maillard noted that, while trials in humans are not underway yet, “we are in active discussions about it,” and the team is indeed interested in testing REGN421 itself, with the effects likely to be as a prophylactic strategy.
There are currently no approved anti-DLL4 antibody drugs for use in humans.
“Our approach is mostly promising as a preventive treatment, rather than as a secondary treatment for GVHD, because DLL4/Notch blockade seems most active when applied early after transplantation during the time of initial seeding of the gut by T cells (in mice, we had observed the critical time window for a successful intervention to be within 48 hours of transplantation),” Dr. Maillard said.“There remain questions about which other prophylactic treatments we should ideally combine anti-DLL4 antibodies with.”
Dr. Maillard has received research funding from Regeneron and Genentech and is a member of Garuda Therapeutics’s scientific advisory board.
FROM SCIENCE TRANSLATIONAL MEDICINE
ESMO helps hematologists assess new cancer drugs
It consists of 11 2- to 3-page forms with checklists to grade treatment trials on the extent to which they meet efficacy and safety thresholds. Each of the 11 forms covers a specific trial scenario, such as a randomized controlled trial with curative intent or a trial of a therapy that is not likely to be curative with a primary endpoint of overall survival.
Treatments with curative intent are graded A, B, or C, while treatments in the noncurative setting are graded on a descending scale from 5 to 1. Scores of A and B in the curative setting and 5 and 4 in the noncurative setting represent substantial benefit.
On the form for RCTs with curative intent, for instance, a survival improvement of 5% or more garners an A but an improvement of less than 3% gets a C. Scores are also annotated for serious acute and/or persistent toxicity if present.
The tool, dubbed the ESMO-MCBS:H (European Society for Medical Oncology Magnitude of Clinical Benefit Scale: Hematology), is explained in an article published in Annals of Oncology. The evaluation forms are available online.
The idea behind the work is to help health care professionals and others to more “accurately assess the value of and prioritise therapies for patients with blood cancers. For clinicians, ESMO-MCBS:H will aid in their clinical decision-making and in the development of evidence-based practice and guidelines,” ESMO said in a press release.
To develop ESMO-MCBS:H, the group tailored its tool for evaluating solid tumor therapies, the ESMO-MCBS, to account for the sometimes different endpoints used in hematologic malignancy trials and the very indolent nature of some blood cancers, such as follicular lymphoma, which hampers development of mature data.
Specific changes include adding a new evaluation form to grade single-arm trials with curative intent, such as those used for CAR-T-cell therapies; incorporating molecular surrogate endpoints used in CML trials; and adding a way to grade outcomes for indolent cancers, among others.
The development process included applying the solid tumor tool to 80 blood cancer studies to identify shortcomings and improve its applicability. The final tool was field tested with 51 international experts from EHA and ESMO who largely agreed on the reasonableness of the trial scores.
ESMO said it expects ESMO-MCBS:H will be useful. The solid tumor tool, first published in 2015, is used by the World Health Organization to screen medications for its essential medicines list as well as by ESMO to generate guidelines and oncology centers across Europe to help with resource allocation decisions.
It consists of 11 2- to 3-page forms with checklists to grade treatment trials on the extent to which they meet efficacy and safety thresholds. Each of the 11 forms covers a specific trial scenario, such as a randomized controlled trial with curative intent or a trial of a therapy that is not likely to be curative with a primary endpoint of overall survival.
Treatments with curative intent are graded A, B, or C, while treatments in the noncurative setting are graded on a descending scale from 5 to 1. Scores of A and B in the curative setting and 5 and 4 in the noncurative setting represent substantial benefit.
On the form for RCTs with curative intent, for instance, a survival improvement of 5% or more garners an A but an improvement of less than 3% gets a C. Scores are also annotated for serious acute and/or persistent toxicity if present.
The tool, dubbed the ESMO-MCBS:H (European Society for Medical Oncology Magnitude of Clinical Benefit Scale: Hematology), is explained in an article published in Annals of Oncology. The evaluation forms are available online.
The idea behind the work is to help health care professionals and others to more “accurately assess the value of and prioritise therapies for patients with blood cancers. For clinicians, ESMO-MCBS:H will aid in their clinical decision-making and in the development of evidence-based practice and guidelines,” ESMO said in a press release.
To develop ESMO-MCBS:H, the group tailored its tool for evaluating solid tumor therapies, the ESMO-MCBS, to account for the sometimes different endpoints used in hematologic malignancy trials and the very indolent nature of some blood cancers, such as follicular lymphoma, which hampers development of mature data.
Specific changes include adding a new evaluation form to grade single-arm trials with curative intent, such as those used for CAR-T-cell therapies; incorporating molecular surrogate endpoints used in CML trials; and adding a way to grade outcomes for indolent cancers, among others.
The development process included applying the solid tumor tool to 80 blood cancer studies to identify shortcomings and improve its applicability. The final tool was field tested with 51 international experts from EHA and ESMO who largely agreed on the reasonableness of the trial scores.
ESMO said it expects ESMO-MCBS:H will be useful. The solid tumor tool, first published in 2015, is used by the World Health Organization to screen medications for its essential medicines list as well as by ESMO to generate guidelines and oncology centers across Europe to help with resource allocation decisions.
It consists of 11 2- to 3-page forms with checklists to grade treatment trials on the extent to which they meet efficacy and safety thresholds. Each of the 11 forms covers a specific trial scenario, such as a randomized controlled trial with curative intent or a trial of a therapy that is not likely to be curative with a primary endpoint of overall survival.
Treatments with curative intent are graded A, B, or C, while treatments in the noncurative setting are graded on a descending scale from 5 to 1. Scores of A and B in the curative setting and 5 and 4 in the noncurative setting represent substantial benefit.
On the form for RCTs with curative intent, for instance, a survival improvement of 5% or more garners an A but an improvement of less than 3% gets a C. Scores are also annotated for serious acute and/or persistent toxicity if present.
The tool, dubbed the ESMO-MCBS:H (European Society for Medical Oncology Magnitude of Clinical Benefit Scale: Hematology), is explained in an article published in Annals of Oncology. The evaluation forms are available online.
The idea behind the work is to help health care professionals and others to more “accurately assess the value of and prioritise therapies for patients with blood cancers. For clinicians, ESMO-MCBS:H will aid in their clinical decision-making and in the development of evidence-based practice and guidelines,” ESMO said in a press release.
To develop ESMO-MCBS:H, the group tailored its tool for evaluating solid tumor therapies, the ESMO-MCBS, to account for the sometimes different endpoints used in hematologic malignancy trials and the very indolent nature of some blood cancers, such as follicular lymphoma, which hampers development of mature data.
Specific changes include adding a new evaluation form to grade single-arm trials with curative intent, such as those used for CAR-T-cell therapies; incorporating molecular surrogate endpoints used in CML trials; and adding a way to grade outcomes for indolent cancers, among others.
The development process included applying the solid tumor tool to 80 blood cancer studies to identify shortcomings and improve its applicability. The final tool was field tested with 51 international experts from EHA and ESMO who largely agreed on the reasonableness of the trial scores.
ESMO said it expects ESMO-MCBS:H will be useful. The solid tumor tool, first published in 2015, is used by the World Health Organization to screen medications for its essential medicines list as well as by ESMO to generate guidelines and oncology centers across Europe to help with resource allocation decisions.
FROM ANNALS OF ONCOLOGY
NORD: Making Progress Through Collaboration

While people living with rare cancers continue to face daunting obstacles, progress is being made, and there are reasons to hope for a better future. Advances in genomic testing and precision medicine provide increasing evidence that rare cancers can be more efficiently and effectively diagnosed and treated. Genomic tests examine tumor DNA to identify mutations that are unique to an individual’s cancer. This genetic information enables a more precise diagnosis and targeted treatment approach. Jim Palma, Co-Lead of the NORD Rare Cancer Coalition, said “There is promise for rare cancer patients due to increased legislative efforts to cover the costs of genomic testing coupled by an increase in FDA approvals for targeted and tissue agnostic therapies.”
In 2019, the National Cancer Institute established MyPART, a vast pediatric and adult rare tumor network that aims to bolster patient involvement in research and develop effective therapies through tumor sample collection, shared data, shared samples, new methods to test treatments, and new trial designs. In 2022, MyPART welcomed NORD’s Rare Cancer Coalition as an advocacy partner.
Meanwhile, advocacy organizations are giving rare cancer a rising voice. NORD’s Rare Cancer Coalition unites rare cancer patient advocacy organizations and helps them drive progress together. The coalition promotes research and awareness through its annual Rare Cancer Day (September 30) campaign. Additionally, NORD has produced over 22 continuing medical education modules on rare cancers in collaboration with PlatformQ Health, providing updates on new therapies and treatment approaches. NORD also offers rare disease reports and educational videos on rare cancers, sessions inclusive of rare cancer topics at the annual NORD Summit, and a quarterly e-newsletter, “Caring for Rare” for healthcare professionals. Please visit us at rarediseases.org to access these resources.
Much work on rare cancers remains to be done, but the progress over recent years points to better outcomes moving forward. We are grateful for the work you do and your dedication to your patients, including those with rare cancers and other rare conditions. We hope you will find the information in this special issue useful for your clinical practice.
– Katie Kowalski, MPH
Associate Director of Education
National Organization for Rare Disorders
- About Rare Cancers. National Cancer Institute. Posted February 27, 2019. Accessed April 28, 2023. http://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/about-rare-cancers
- Gatta G, Capocaccia R, Botta L, et al. Burden and centralized treatment in Europe of rare tumours: Results of RARECAREnet-a population-based study. Lancet Oncol. 2017,18(8):1022–1039. doi:10.1016/S1470-2045(17)30445-X
While people living with rare cancers continue to face daunting obstacles, progress is being made, and there are reasons to hope for a better future. Advances in genomic testing and precision medicine provide increasing evidence that rare cancers can be more efficiently and effectively diagnosed and treated. Genomic tests examine tumor DNA to identify mutations that are unique to an individual’s cancer. This genetic information enables a more precise diagnosis and targeted treatment approach. Jim Palma, Co-Lead of the NORD Rare Cancer Coalition, said “There is promise for rare cancer patients due to increased legislative efforts to cover the costs of genomic testing coupled by an increase in FDA approvals for targeted and tissue agnostic therapies.”
In 2019, the National Cancer Institute established MyPART, a vast pediatric and adult rare tumor network that aims to bolster patient involvement in research and develop effective therapies through tumor sample collection, shared data, shared samples, new methods to test treatments, and new trial designs. In 2022, MyPART welcomed NORD’s Rare Cancer Coalition as an advocacy partner.
Meanwhile, advocacy organizations are giving rare cancer a rising voice. NORD’s Rare Cancer Coalition unites rare cancer patient advocacy organizations and helps them drive progress together. The coalition promotes research and awareness through its annual Rare Cancer Day (September 30) campaign. Additionally, NORD has produced over 22 continuing medical education modules on rare cancers in collaboration with PlatformQ Health, providing updates on new therapies and treatment approaches. NORD also offers rare disease reports and educational videos on rare cancers, sessions inclusive of rare cancer topics at the annual NORD Summit, and a quarterly e-newsletter, “Caring for Rare” for healthcare professionals. Please visit us at rarediseases.org to access these resources.
Much work on rare cancers remains to be done, but the progress over recent years points to better outcomes moving forward. We are grateful for the work you do and your dedication to your patients, including those with rare cancers and other rare conditions. We hope you will find the information in this special issue useful for your clinical practice.
– Katie Kowalski, MPH
Associate Director of Education
National Organization for Rare Disorders
While people living with rare cancers continue to face daunting obstacles, progress is being made, and there are reasons to hope for a better future. Advances in genomic testing and precision medicine provide increasing evidence that rare cancers can be more efficiently and effectively diagnosed and treated. Genomic tests examine tumor DNA to identify mutations that are unique to an individual’s cancer. This genetic information enables a more precise diagnosis and targeted treatment approach. Jim Palma, Co-Lead of the NORD Rare Cancer Coalition, said “There is promise for rare cancer patients due to increased legislative efforts to cover the costs of genomic testing coupled by an increase in FDA approvals for targeted and tissue agnostic therapies.”
In 2019, the National Cancer Institute established MyPART, a vast pediatric and adult rare tumor network that aims to bolster patient involvement in research and develop effective therapies through tumor sample collection, shared data, shared samples, new methods to test treatments, and new trial designs. In 2022, MyPART welcomed NORD’s Rare Cancer Coalition as an advocacy partner.
Meanwhile, advocacy organizations are giving rare cancer a rising voice. NORD’s Rare Cancer Coalition unites rare cancer patient advocacy organizations and helps them drive progress together. The coalition promotes research and awareness through its annual Rare Cancer Day (September 30) campaign. Additionally, NORD has produced over 22 continuing medical education modules on rare cancers in collaboration with PlatformQ Health, providing updates on new therapies and treatment approaches. NORD also offers rare disease reports and educational videos on rare cancers, sessions inclusive of rare cancer topics at the annual NORD Summit, and a quarterly e-newsletter, “Caring for Rare” for healthcare professionals. Please visit us at rarediseases.org to access these resources.
Much work on rare cancers remains to be done, but the progress over recent years points to better outcomes moving forward. We are grateful for the work you do and your dedication to your patients, including those with rare cancers and other rare conditions. We hope you will find the information in this special issue useful for your clinical practice.
– Katie Kowalski, MPH
Associate Director of Education
National Organization for Rare Disorders
- About Rare Cancers. National Cancer Institute. Posted February 27, 2019. Accessed April 28, 2023. http://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/about-rare-cancers
- Gatta G, Capocaccia R, Botta L, et al. Burden and centralized treatment in Europe of rare tumours: Results of RARECAREnet-a population-based study. Lancet Oncol. 2017,18(8):1022–1039. doi:10.1016/S1470-2045(17)30445-X
- About Rare Cancers. National Cancer Institute. Posted February 27, 2019. Accessed April 28, 2023. http://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/about-rare-cancers
- Gatta G, Capocaccia R, Botta L, et al. Burden and centralized treatment in Europe of rare tumours: Results of RARECAREnet-a population-based study. Lancet Oncol. 2017,18(8):1022–1039. doi:10.1016/S1470-2045(17)30445-X
2023 Rare Diseases Report: Cancers
This edition of Rare Diseases Report: Cancers highlights the latest breakthroughs and remaining unmet needs in the management of rare cancers. In addition to celebrating the great progress that has been made in recent years, we also discuss new challenges, such as how the healthcare system can prepare to manage the growing number of rare cancer survivors who are living longer due to improvements in disease management.
INTRODUCTION
NORD: Making Progress Through Collaboration
By Katie Kowalski, MPH
IN THIS ISSUE
The Complex Challenge of Survival After HPV-Associated Oropharyngeal Cancer
By Vlad C. Sandulache, MD, PhD
Progress in Ovarian Cancer: Discovery of Fallopian Tube Involvement
By Ronny Drapkin, MD, PhD
An Evolving Understanding of Adenosquamous Carcinoma of the Lung
By Rajwanth Veluswamy, MD, MSCR
Gastrointestinal Stromal Tumor: Reflecting on 2 Decades of Clinical Advancements
By Jason K. Sicklick, MD, FACS
Progress in Treating Testicular Cancer
By Liang Cheng, MD
Strategies to Improve Long-Term Outcomes in Younger Patients with Hodgkin Lymphoma
By Ann LaCasce, MD, MMSc
Targeted Therapies in Younger and Older Patients with Mantle Cell Lymphoma
By Reem Karmali, MD, MS
Advances in Management of Relapsed/Refractory Hairy Cell Leukemia
By Robert J. Kreitman, MD
Treatment Needs of Older Adults With Newly Diagnosed Acute Myeloid Leukemia
By Harry Erba, MD, PhD
Progress in Management of Advanced Acute Lymphocytic Leukemia in Children
By Susan Colace, MD, MSCI
This edition of Rare Diseases Report: Cancers highlights the latest breakthroughs and remaining unmet needs in the management of rare cancers. In addition to celebrating the great progress that has been made in recent years, we also discuss new challenges, such as how the healthcare system can prepare to manage the growing number of rare cancer survivors who are living longer due to improvements in disease management.
INTRODUCTION
NORD: Making Progress Through Collaboration
By Katie Kowalski, MPH
IN THIS ISSUE
The Complex Challenge of Survival After HPV-Associated Oropharyngeal Cancer
By Vlad C. Sandulache, MD, PhD
Progress in Ovarian Cancer: Discovery of Fallopian Tube Involvement
By Ronny Drapkin, MD, PhD
An Evolving Understanding of Adenosquamous Carcinoma of the Lung
By Rajwanth Veluswamy, MD, MSCR
Gastrointestinal Stromal Tumor: Reflecting on 2 Decades of Clinical Advancements
By Jason K. Sicklick, MD, FACS
Progress in Treating Testicular Cancer
By Liang Cheng, MD
Strategies to Improve Long-Term Outcomes in Younger Patients with Hodgkin Lymphoma
By Ann LaCasce, MD, MMSc
Targeted Therapies in Younger and Older Patients with Mantle Cell Lymphoma
By Reem Karmali, MD, MS
Advances in Management of Relapsed/Refractory Hairy Cell Leukemia
By Robert J. Kreitman, MD
Treatment Needs of Older Adults With Newly Diagnosed Acute Myeloid Leukemia
By Harry Erba, MD, PhD
Progress in Management of Advanced Acute Lymphocytic Leukemia in Children
By Susan Colace, MD, MSCI
This edition of Rare Diseases Report: Cancers highlights the latest breakthroughs and remaining unmet needs in the management of rare cancers. In addition to celebrating the great progress that has been made in recent years, we also discuss new challenges, such as how the healthcare system can prepare to manage the growing number of rare cancer survivors who are living longer due to improvements in disease management.
INTRODUCTION
NORD: Making Progress Through Collaboration
By Katie Kowalski, MPH
IN THIS ISSUE
The Complex Challenge of Survival After HPV-Associated Oropharyngeal Cancer
By Vlad C. Sandulache, MD, PhD
Progress in Ovarian Cancer: Discovery of Fallopian Tube Involvement
By Ronny Drapkin, MD, PhD
An Evolving Understanding of Adenosquamous Carcinoma of the Lung
By Rajwanth Veluswamy, MD, MSCR
Gastrointestinal Stromal Tumor: Reflecting on 2 Decades of Clinical Advancements
By Jason K. Sicklick, MD, FACS
Progress in Treating Testicular Cancer
By Liang Cheng, MD
Strategies to Improve Long-Term Outcomes in Younger Patients with Hodgkin Lymphoma
By Ann LaCasce, MD, MMSc
Targeted Therapies in Younger and Older Patients with Mantle Cell Lymphoma
By Reem Karmali, MD, MS
Advances in Management of Relapsed/Refractory Hairy Cell Leukemia
By Robert J. Kreitman, MD
Treatment Needs of Older Adults With Newly Diagnosed Acute Myeloid Leukemia
By Harry Erba, MD, PhD
Progress in Management of Advanced Acute Lymphocytic Leukemia in Children
By Susan Colace, MD, MSCI
Strategies to Improve Long-Term Outcomes in Younger Patients With Hodgkin Lymphoma

Background
Hodgkin lymphoma occurs in fewer than 9,000 individuals in the United States each year,1 but it is one of the most common types of cancer in AYAs.2 For the purposes of cHL, AYA is typically defined as an age range of 18 to 39 years, which covers the first of 2 bimodal peaks in incidence but stops short of the second.3,4 The first of these peaks occurs between the ages of 15 and 34 years, while the second begins at about age 55.5 Children younger than 15 years of age can also develop Hodgkin lymphoma, but it is less common.6
In AYAs and in adults, more than 90% of patients with Hodgkin lymphoma have cHL.7 Most AYAs present with the nodular sclerosis subtype, but cHL is managed differently in pediatric patients versus in adult centers.8,9 Evidence suggests that the specific risks of common treatment protocols, although similar, are not the same in AYAs as in adults.10,11 Even though the literature evaluating the presentation and management of AYA cHL has been growing since 2005, when the AYA Oncology Progress Review Group called for AYAs to be recognized
as a distinct group, clinical trials specific to AYA cHL remain limited.9
Major Hodgkin lymphoma guidelines only partially address AYAs as a distinct group. In guidelines issued by the National Cancer Institute, the differences in clinical presentation of AYAs are described for young children, AYAs, and older adults, but there are no treatment recommendations specific to AYAs.12 Guidelines from the EuroNet Paediatric Hodgkin Lymphoma Group offer recommendations for relapsed and refractory Hodgkin lymphoma, but do not differentiate between children and adolescents.13 The National Comprehensive Cancer Network (NCCN) provides separate treatment recommendations for patients 18 years or younger and those who are older than 18.14,15 For Hodgkin lymphoma, AYA is not addressed as a separate category even though the NCCN has provided general guidelines for treatment of malignancies in AYA.16
First-line therapies are effective in children, AYAs, and adults. Survival rates at 5 years have increased steadily, approaching or exceeding 90% across age groups even for patients with unfavorable risk characteristics.17 This success has permitted greater focus on developing strategies that preserve efficacy with lower acute and long-term risks.
Risk-Adapted Therapies
While the potential for new and novel therapies to reduce the risk of long-term toxicities continues to be explored, adjusting existing regimens to reduce these risks has proven to be a viable strategy. This adjustment is a standard of care in the pediatric setting based on results from such studies as German GPOH-HD 95, which suggested that doses of radiotherapy, a major contributor to late toxicities,18 can be omitted in patients with a complete response after chemotherapy.11 This pediatric trial contained both younger children and adolescents, but subsequent secondary analyses looking specifically at AYAs in this and other trials have suggested that efficacy is similarly preserved with risk-adapted strategies.9
However, due to AYA patients with cHL being treated using both pediatric and adult approaches, the persistent debate about optimal therapies in this age group complicates the effort to define a well-accepted strategy for risk adjustment. While risk-adapted strategies that rely on interim positron emission tomography (PET) to calibrate treatment intensity are now being used routinely across age stratifications, other initiatives are creating additional opportunities to gauge the impact on late effects in AYAs. These include strategies to improve collaboration across groups of trialists and data generated by observational cohorts, which can evaluate late effects not captured in time-limited clinical trials.
Among recent data supporting risk-adjusted therapy, the toxicity outcomes from a multicenter trial of PET-guided intensive treatment in patients with newly diagnosed advanced cHL were presented at the 2022 annual meeting of the American Society of Hematology.19 This phase 3 trial enrolled patients younger than 60 years, 79% of whom were younger than 45 years. Building on previous evidence that PET guidance improves the safety of eBEACOPP (escalated doses of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), nearly 1,500 patients were randomized to this strategy or to PET-guided BrECADD, a modified eBEACOPP in which the antibody conjugate brentuximab vedotin (BV) was substituted for bleomycin. For an adjudicated endpoint of treatment-related morbidity, the experimental BrECAAD regimen reduced the risk by nearly 30% (hazard ratio [HR] 0.72). It is unclear whether this strategy will be used in the United States, where trials have been built on ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) rather than BEACOPP.
Efficacy data from this trial are not yet available, and these data will be important. There is concern that PET-directed therapy might result in lower toxicity at a cost of reduced rates of disease control. It is possible that the serious consequences of late toxicities—including infertility, compromised cardiovascular function, secondary cancers, and other organ damage—might need to be balanced against some loss of efficacy.
Novel Targeted Therapies
The goal of reducing late toxicities of cHL therapy in AYAs is also likely to be advanced by novel therapies. Research endeavors include a multicenter collaboration between US and Canadian investigators that is exploring the combination of nivolumab (a checkpoint inhibitor) plus BV.20 The trial recently completed accrual and includes both adult and pediatric patients. If novel agents prove effective for improving efficacy while reducing the risk of late complications in AYAs, they are expected to have a profound effect on clinical practice.
Arguably, the era of targeted and novel therapies in cHL was initiated more than 10 years ago with the introduction of BV for the treatment of advanced disease in older adults.21 BV was moved into the front line for patients 18 years of age or older with advanced cHL in a trial that compared the standard of ABVD to the same drugs with BV substituted for bleomycin.22 In this study, the BV-containing regimen was associated with a significantly improved progression-free survival (PFS) (P = .04) and a lower rate of adverse events, including pulmonary toxicity (1% vs 3%) after 2 years of follow-up.
A similar study recently associated a BV-containing regimen with even greater efficacy in pediatric high-risk cHL.23 In this multicenter study with 600 treatment-naïve patients ranging in age from 2 to 21 years, the standard pediatric regimen of doxorubicin, bleomycin, vincristine, etoposide, prednisone, and cyclophosphamide was compared to the same regimen with BV substituted for bleomycin. With event-free survival as the primary endpoint, the experimental regimen was associated with a nearly 60% reduction in the risk of an adverse event or death (HR 0.41). However, no substantial differences were noted in toxicity after a follow-up of 42 months. It not yet clear whether the elimination of bleomycin will translate into less late toxicity, such as pulmonary or cardiovascular morbidity.
In the era of targeted therapies, the experience with BV has been a step toward more effective treatments using novel mechanisms of action to improve outcomes when used in the first-line treatment of patients with high-risk disease. Historically, many regimens and treatments that have demonstrated efficacy in relapsed and refractory cHL have found their way into the first-line setting. This trend might also be true of the checkpoint inhibitors, which have been tested extensively in relapsed/refractory cHL. In AYA patients with cHL, the rationale for these treatments might not only include a poor predicted response to current regimens, but a reduced risk of late toxicities if long-term follow-up demonstrates these treatments reduce late complications, such as secondary malignancies, which are associated with standard strategies, particularly those that include radiotherapy.
If targeted therapies do preserve efficacy and reduce risk of late complications, strategies to individualize therapy will remain relevant. Many of the emerging targeted therapies involve challenging and costly treatment protocols that demand selective application. Efforts to develop simpler and more precise biomarkers might streamline this task. Of promising developments in this area, cell-free DNA (cfDNA) appears to be near routine clinical application. A small study of cfDNA conducted in 121 patients found that minimal residual disease assessment by repeat cfDNA sequencing predicted response and PFS when performed as early as a week after treatment initiation.24 If larger studies confirm accuracy, this biomarker strategy might prove simpler and more convenient than PET imaging.
Summary
In the treatment of hematologic malignancies, cHL is widely regarded as a success story with high rates of extended survival among children, AYAs, and older adults. This level of success does not obviate the need for even more effective treatments, and also permits more attention to be directed to reducing the risk of late toxicities. For the AYA population, which represents a large group with cHL, the current directions of clinical research offer the promise of imminent changes in how the disease is controlled and a reduction in treatment-related late morbidity and mortality.
- Hodgkin Lymphoma. American Cancer Society. Accessed March 20, 2023. https://www.cancer.org/cancer/hodgkin-lymphoma.html
- Aben KK, van Gaal C, van Gils NA, van der Graaf WT, Zielhuis GA. Cancer in adolescents and young adults (15-29 years): a population-based study in the Netherlands 1989-2009. Acta Oncol. 2012;51(7):922-933. doi:10.3109/0284186X.2012.705891
- Ansell SM. Hodgkin lymphoma: 2016 update on diagnosis, risk-stratification, and
management. Am J Hematol. 2016;91(4):434-442. doi:10.1002/ajh.24272 - Cartwright RA, Watkins G. Epidemiology of Hodgkin’s disease: a review. Hematol Oncol. 2004;22(1):11-26. doi:10.1002/hon.723
- Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506-1514. doi:10.1056/NEJM199811193392104
- Bleyer A, Barr R, Hayes-Lattin B, et al. The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer. 2008;8(4):288-298. doi:10.1038/nrc2349
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116-132. doi:10.3322/caac.21438
- Bigenwald C, Galimard JE, Quero L, et al. Hodgkin lymphoma in adolescent and young adults: insights from an adult tertiary single-center cohort of 349 patients. Oncotarget. 2017;8(45):80073-80082. doi:10.18632/oncotarget.20684
- Kahn JM, Kelly KM. Adolescent and young adult Hodgkin lymphoma: raising the bar through collaborative science and multidisciplinary care. Pediatr Blood Cancer. 2018;65(7):e27033. doi:10.1002/pbc.27033
- Yung L, Smith P, Hancock BW, et al. Long term outcome in adolescents with Hodgkin’s lymphoma: poor results using regimens designed for adults. Leuk Lymphoma. 2004;45(8):1579-1585. doi:10.1080/1042819042000209404
- Dorffel W, Ruhl U, Luders H, et al. Treatment of children and adolescents with Hodgkin lymphoma without radiotherapy for patients in complete remission after chemotherapy: final results of the multinational trial GPOH-HD95. J Clin Oncol. 2013;31(12):1562-1568. doi:10.1200/JCO.2012.45.3266
- National Cancer Institute. Childhood Hodgkin lymphoma treatment (PDQ®)–Health Professional Version. National Institutes of Health. Updated February 14, 2023. Accessed March 20, 2023. https://www.cancer.gov/types/lymphoma/hp/child-hodgkin-treatment-pdq
- Daw S, Hasenclever D, Mascarin M, et al. Risk and response adapted treatment guidelines for managing first relapsed and refractory classical Hodgkin lymphoma in children and young people. Recommendations from the EuroNet Pediatric Hodgkin Lymphoma Group. Hemasphere. 2020;4(1):e329. doi:10.1097/HS9.0000000000000329
- Flerlage JE, Hiniker SM, Armenian S, et al. Pediatric Hodgkin lymphoma, version 3.2021. J Natl Compr Canc Netw. 2021;19(6):733-754. doi:10.6004/jnccn.2021.0027
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Hodgkin lymphoma. Version 2.2023. November 8, 2022. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hodgkins.pdf
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Adolescent and young adult (AYA) oncology. Version 3.2023. January 9, 2023. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/aya.pdf
- Mohty R, Dulery R, Bazarbachi AH, et al. Latest advances in the management of classical Hodgkin lymphoma: the era of novel therapies. Blood Cancer J. 2021;11(7):126. doi:10.1038/s41408-021-00518-z
- Witkowska M, Majchrzak A, Smolewski P. The role of radiotherapy in Hodgkin’s lymphoma: what has been achieved during the last 50 years? Biomed Res Int. 2015;2015:485071. doi:10.1155/2015/485071
- Borchmann P, Moccia A, Greil R, et al. Treatment-related morbidity in patients with classical Hodgkin lymphoma: results of the ongoing, randomized phase II HD21 trial by the German Hodgkin Study Group. Hemasphere. 2022;6(suppl ):1-2. doi:10.1097/01.HS9.0000890576.23258.1c
- Immunotherapy (nivolumab or brentuximab vedotin) plus combination chemotherapy in treating patients with newly diagnosed stage III-IV classic Hodgkin lymphoma. ClinicalTrials.gov. Updated March 8, 2023. Accessed March 20, 2023. https://clinicaltrials.gov/ct2/show/NCT03907488
- Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183-2189. doi:10.1200/JCO.2011.38.0410
- Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378(4):331-344. doi:10.1056/NEJMoa1708984
- Castellino SM, Pei Q, Parsons SK, et al. Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma. N Engl J Med. 2022;387(18):1649-1660. doi:10.1056/NEJMoa2206660
- Sobesky S, Mammadova L, Cirillo M, et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med (N Y). 2021;2(10):1171-1193.e11. doi:10.1016/j.medj.2021.09.002
Background
Hodgkin lymphoma occurs in fewer than 9,000 individuals in the United States each year,1 but it is one of the most common types of cancer in AYAs.2 For the purposes of cHL, AYA is typically defined as an age range of 18 to 39 years, which covers the first of 2 bimodal peaks in incidence but stops short of the second.3,4 The first of these peaks occurs between the ages of 15 and 34 years, while the second begins at about age 55.5 Children younger than 15 years of age can also develop Hodgkin lymphoma, but it is less common.6
In AYAs and in adults, more than 90% of patients with Hodgkin lymphoma have cHL.7 Most AYAs present with the nodular sclerosis subtype, but cHL is managed differently in pediatric patients versus in adult centers.8,9 Evidence suggests that the specific risks of common treatment protocols, although similar, are not the same in AYAs as in adults.10,11 Even though the literature evaluating the presentation and management of AYA cHL has been growing since 2005, when the AYA Oncology Progress Review Group called for AYAs to be recognized
as a distinct group, clinical trials specific to AYA cHL remain limited.9
Major Hodgkin lymphoma guidelines only partially address AYAs as a distinct group. In guidelines issued by the National Cancer Institute, the differences in clinical presentation of AYAs are described for young children, AYAs, and older adults, but there are no treatment recommendations specific to AYAs.12 Guidelines from the EuroNet Paediatric Hodgkin Lymphoma Group offer recommendations for relapsed and refractory Hodgkin lymphoma, but do not differentiate between children and adolescents.13 The National Comprehensive Cancer Network (NCCN) provides separate treatment recommendations for patients 18 years or younger and those who are older than 18.14,15 For Hodgkin lymphoma, AYA is not addressed as a separate category even though the NCCN has provided general guidelines for treatment of malignancies in AYA.16
First-line therapies are effective in children, AYAs, and adults. Survival rates at 5 years have increased steadily, approaching or exceeding 90% across age groups even for patients with unfavorable risk characteristics.17 This success has permitted greater focus on developing strategies that preserve efficacy with lower acute and long-term risks.
Risk-Adapted Therapies
While the potential for new and novel therapies to reduce the risk of long-term toxicities continues to be explored, adjusting existing regimens to reduce these risks has proven to be a viable strategy. This adjustment is a standard of care in the pediatric setting based on results from such studies as German GPOH-HD 95, which suggested that doses of radiotherapy, a major contributor to late toxicities,18 can be omitted in patients with a complete response after chemotherapy.11 This pediatric trial contained both younger children and adolescents, but subsequent secondary analyses looking specifically at AYAs in this and other trials have suggested that efficacy is similarly preserved with risk-adapted strategies.9
However, due to AYA patients with cHL being treated using both pediatric and adult approaches, the persistent debate about optimal therapies in this age group complicates the effort to define a well-accepted strategy for risk adjustment. While risk-adapted strategies that rely on interim positron emission tomography (PET) to calibrate treatment intensity are now being used routinely across age stratifications, other initiatives are creating additional opportunities to gauge the impact on late effects in AYAs. These include strategies to improve collaboration across groups of trialists and data generated by observational cohorts, which can evaluate late effects not captured in time-limited clinical trials.
Among recent data supporting risk-adjusted therapy, the toxicity outcomes from a multicenter trial of PET-guided intensive treatment in patients with newly diagnosed advanced cHL were presented at the 2022 annual meeting of the American Society of Hematology.19 This phase 3 trial enrolled patients younger than 60 years, 79% of whom were younger than 45 years. Building on previous evidence that PET guidance improves the safety of eBEACOPP (escalated doses of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), nearly 1,500 patients were randomized to this strategy or to PET-guided BrECADD, a modified eBEACOPP in which the antibody conjugate brentuximab vedotin (BV) was substituted for bleomycin. For an adjudicated endpoint of treatment-related morbidity, the experimental BrECAAD regimen reduced the risk by nearly 30% (hazard ratio [HR] 0.72). It is unclear whether this strategy will be used in the United States, where trials have been built on ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) rather than BEACOPP.
Efficacy data from this trial are not yet available, and these data will be important. There is concern that PET-directed therapy might result in lower toxicity at a cost of reduced rates of disease control. It is possible that the serious consequences of late toxicities—including infertility, compromised cardiovascular function, secondary cancers, and other organ damage—might need to be balanced against some loss of efficacy.
Novel Targeted Therapies
The goal of reducing late toxicities of cHL therapy in AYAs is also likely to be advanced by novel therapies. Research endeavors include a multicenter collaboration between US and Canadian investigators that is exploring the combination of nivolumab (a checkpoint inhibitor) plus BV.20 The trial recently completed accrual and includes both adult and pediatric patients. If novel agents prove effective for improving efficacy while reducing the risk of late complications in AYAs, they are expected to have a profound effect on clinical practice.
Arguably, the era of targeted and novel therapies in cHL was initiated more than 10 years ago with the introduction of BV for the treatment of advanced disease in older adults.21 BV was moved into the front line for patients 18 years of age or older with advanced cHL in a trial that compared the standard of ABVD to the same drugs with BV substituted for bleomycin.22 In this study, the BV-containing regimen was associated with a significantly improved progression-free survival (PFS) (P = .04) and a lower rate of adverse events, including pulmonary toxicity (1% vs 3%) after 2 years of follow-up.
A similar study recently associated a BV-containing regimen with even greater efficacy in pediatric high-risk cHL.23 In this multicenter study with 600 treatment-naïve patients ranging in age from 2 to 21 years, the standard pediatric regimen of doxorubicin, bleomycin, vincristine, etoposide, prednisone, and cyclophosphamide was compared to the same regimen with BV substituted for bleomycin. With event-free survival as the primary endpoint, the experimental regimen was associated with a nearly 60% reduction in the risk of an adverse event or death (HR 0.41). However, no substantial differences were noted in toxicity after a follow-up of 42 months. It not yet clear whether the elimination of bleomycin will translate into less late toxicity, such as pulmonary or cardiovascular morbidity.
In the era of targeted therapies, the experience with BV has been a step toward more effective treatments using novel mechanisms of action to improve outcomes when used in the first-line treatment of patients with high-risk disease. Historically, many regimens and treatments that have demonstrated efficacy in relapsed and refractory cHL have found their way into the first-line setting. This trend might also be true of the checkpoint inhibitors, which have been tested extensively in relapsed/refractory cHL. In AYA patients with cHL, the rationale for these treatments might not only include a poor predicted response to current regimens, but a reduced risk of late toxicities if long-term follow-up demonstrates these treatments reduce late complications, such as secondary malignancies, which are associated with standard strategies, particularly those that include radiotherapy.
If targeted therapies do preserve efficacy and reduce risk of late complications, strategies to individualize therapy will remain relevant. Many of the emerging targeted therapies involve challenging and costly treatment protocols that demand selective application. Efforts to develop simpler and more precise biomarkers might streamline this task. Of promising developments in this area, cell-free DNA (cfDNA) appears to be near routine clinical application. A small study of cfDNA conducted in 121 patients found that minimal residual disease assessment by repeat cfDNA sequencing predicted response and PFS when performed as early as a week after treatment initiation.24 If larger studies confirm accuracy, this biomarker strategy might prove simpler and more convenient than PET imaging.
Summary
In the treatment of hematologic malignancies, cHL is widely regarded as a success story with high rates of extended survival among children, AYAs, and older adults. This level of success does not obviate the need for even more effective treatments, and also permits more attention to be directed to reducing the risk of late toxicities. For the AYA population, which represents a large group with cHL, the current directions of clinical research offer the promise of imminent changes in how the disease is controlled and a reduction in treatment-related late morbidity and mortality.
Background
Hodgkin lymphoma occurs in fewer than 9,000 individuals in the United States each year,1 but it is one of the most common types of cancer in AYAs.2 For the purposes of cHL, AYA is typically defined as an age range of 18 to 39 years, which covers the first of 2 bimodal peaks in incidence but stops short of the second.3,4 The first of these peaks occurs between the ages of 15 and 34 years, while the second begins at about age 55.5 Children younger than 15 years of age can also develop Hodgkin lymphoma, but it is less common.6
In AYAs and in adults, more than 90% of patients with Hodgkin lymphoma have cHL.7 Most AYAs present with the nodular sclerosis subtype, but cHL is managed differently in pediatric patients versus in adult centers.8,9 Evidence suggests that the specific risks of common treatment protocols, although similar, are not the same in AYAs as in adults.10,11 Even though the literature evaluating the presentation and management of AYA cHL has been growing since 2005, when the AYA Oncology Progress Review Group called for AYAs to be recognized
as a distinct group, clinical trials specific to AYA cHL remain limited.9
Major Hodgkin lymphoma guidelines only partially address AYAs as a distinct group. In guidelines issued by the National Cancer Institute, the differences in clinical presentation of AYAs are described for young children, AYAs, and older adults, but there are no treatment recommendations specific to AYAs.12 Guidelines from the EuroNet Paediatric Hodgkin Lymphoma Group offer recommendations for relapsed and refractory Hodgkin lymphoma, but do not differentiate between children and adolescents.13 The National Comprehensive Cancer Network (NCCN) provides separate treatment recommendations for patients 18 years or younger and those who are older than 18.14,15 For Hodgkin lymphoma, AYA is not addressed as a separate category even though the NCCN has provided general guidelines for treatment of malignancies in AYA.16
First-line therapies are effective in children, AYAs, and adults. Survival rates at 5 years have increased steadily, approaching or exceeding 90% across age groups even for patients with unfavorable risk characteristics.17 This success has permitted greater focus on developing strategies that preserve efficacy with lower acute and long-term risks.
Risk-Adapted Therapies
While the potential for new and novel therapies to reduce the risk of long-term toxicities continues to be explored, adjusting existing regimens to reduce these risks has proven to be a viable strategy. This adjustment is a standard of care in the pediatric setting based on results from such studies as German GPOH-HD 95, which suggested that doses of radiotherapy, a major contributor to late toxicities,18 can be omitted in patients with a complete response after chemotherapy.11 This pediatric trial contained both younger children and adolescents, but subsequent secondary analyses looking specifically at AYAs in this and other trials have suggested that efficacy is similarly preserved with risk-adapted strategies.9
However, due to AYA patients with cHL being treated using both pediatric and adult approaches, the persistent debate about optimal therapies in this age group complicates the effort to define a well-accepted strategy for risk adjustment. While risk-adapted strategies that rely on interim positron emission tomography (PET) to calibrate treatment intensity are now being used routinely across age stratifications, other initiatives are creating additional opportunities to gauge the impact on late effects in AYAs. These include strategies to improve collaboration across groups of trialists and data generated by observational cohorts, which can evaluate late effects not captured in time-limited clinical trials.
Among recent data supporting risk-adjusted therapy, the toxicity outcomes from a multicenter trial of PET-guided intensive treatment in patients with newly diagnosed advanced cHL were presented at the 2022 annual meeting of the American Society of Hematology.19 This phase 3 trial enrolled patients younger than 60 years, 79% of whom were younger than 45 years. Building on previous evidence that PET guidance improves the safety of eBEACOPP (escalated doses of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), nearly 1,500 patients were randomized to this strategy or to PET-guided BrECADD, a modified eBEACOPP in which the antibody conjugate brentuximab vedotin (BV) was substituted for bleomycin. For an adjudicated endpoint of treatment-related morbidity, the experimental BrECAAD regimen reduced the risk by nearly 30% (hazard ratio [HR] 0.72). It is unclear whether this strategy will be used in the United States, where trials have been built on ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) rather than BEACOPP.
Efficacy data from this trial are not yet available, and these data will be important. There is concern that PET-directed therapy might result in lower toxicity at a cost of reduced rates of disease control. It is possible that the serious consequences of late toxicities—including infertility, compromised cardiovascular function, secondary cancers, and other organ damage—might need to be balanced against some loss of efficacy.
Novel Targeted Therapies
The goal of reducing late toxicities of cHL therapy in AYAs is also likely to be advanced by novel therapies. Research endeavors include a multicenter collaboration between US and Canadian investigators that is exploring the combination of nivolumab (a checkpoint inhibitor) plus BV.20 The trial recently completed accrual and includes both adult and pediatric patients. If novel agents prove effective for improving efficacy while reducing the risk of late complications in AYAs, they are expected to have a profound effect on clinical practice.
Arguably, the era of targeted and novel therapies in cHL was initiated more than 10 years ago with the introduction of BV for the treatment of advanced disease in older adults.21 BV was moved into the front line for patients 18 years of age or older with advanced cHL in a trial that compared the standard of ABVD to the same drugs with BV substituted for bleomycin.22 In this study, the BV-containing regimen was associated with a significantly improved progression-free survival (PFS) (P = .04) and a lower rate of adverse events, including pulmonary toxicity (1% vs 3%) after 2 years of follow-up.
A similar study recently associated a BV-containing regimen with even greater efficacy in pediatric high-risk cHL.23 In this multicenter study with 600 treatment-naïve patients ranging in age from 2 to 21 years, the standard pediatric regimen of doxorubicin, bleomycin, vincristine, etoposide, prednisone, and cyclophosphamide was compared to the same regimen with BV substituted for bleomycin. With event-free survival as the primary endpoint, the experimental regimen was associated with a nearly 60% reduction in the risk of an adverse event or death (HR 0.41). However, no substantial differences were noted in toxicity after a follow-up of 42 months. It not yet clear whether the elimination of bleomycin will translate into less late toxicity, such as pulmonary or cardiovascular morbidity.
In the era of targeted therapies, the experience with BV has been a step toward more effective treatments using novel mechanisms of action to improve outcomes when used in the first-line treatment of patients with high-risk disease. Historically, many regimens and treatments that have demonstrated efficacy in relapsed and refractory cHL have found their way into the first-line setting. This trend might also be true of the checkpoint inhibitors, which have been tested extensively in relapsed/refractory cHL. In AYA patients with cHL, the rationale for these treatments might not only include a poor predicted response to current regimens, but a reduced risk of late toxicities if long-term follow-up demonstrates these treatments reduce late complications, such as secondary malignancies, which are associated with standard strategies, particularly those that include radiotherapy.
If targeted therapies do preserve efficacy and reduce risk of late complications, strategies to individualize therapy will remain relevant. Many of the emerging targeted therapies involve challenging and costly treatment protocols that demand selective application. Efforts to develop simpler and more precise biomarkers might streamline this task. Of promising developments in this area, cell-free DNA (cfDNA) appears to be near routine clinical application. A small study of cfDNA conducted in 121 patients found that minimal residual disease assessment by repeat cfDNA sequencing predicted response and PFS when performed as early as a week after treatment initiation.24 If larger studies confirm accuracy, this biomarker strategy might prove simpler and more convenient than PET imaging.
Summary
In the treatment of hematologic malignancies, cHL is widely regarded as a success story with high rates of extended survival among children, AYAs, and older adults. This level of success does not obviate the need for even more effective treatments, and also permits more attention to be directed to reducing the risk of late toxicities. For the AYA population, which represents a large group with cHL, the current directions of clinical research offer the promise of imminent changes in how the disease is controlled and a reduction in treatment-related late morbidity and mortality.
- Hodgkin Lymphoma. American Cancer Society. Accessed March 20, 2023. https://www.cancer.org/cancer/hodgkin-lymphoma.html
- Aben KK, van Gaal C, van Gils NA, van der Graaf WT, Zielhuis GA. Cancer in adolescents and young adults (15-29 years): a population-based study in the Netherlands 1989-2009. Acta Oncol. 2012;51(7):922-933. doi:10.3109/0284186X.2012.705891
- Ansell SM. Hodgkin lymphoma: 2016 update on diagnosis, risk-stratification, and
management. Am J Hematol. 2016;91(4):434-442. doi:10.1002/ajh.24272 - Cartwright RA, Watkins G. Epidemiology of Hodgkin’s disease: a review. Hematol Oncol. 2004;22(1):11-26. doi:10.1002/hon.723
- Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506-1514. doi:10.1056/NEJM199811193392104
- Bleyer A, Barr R, Hayes-Lattin B, et al. The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer. 2008;8(4):288-298. doi:10.1038/nrc2349
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116-132. doi:10.3322/caac.21438
- Bigenwald C, Galimard JE, Quero L, et al. Hodgkin lymphoma in adolescent and young adults: insights from an adult tertiary single-center cohort of 349 patients. Oncotarget. 2017;8(45):80073-80082. doi:10.18632/oncotarget.20684
- Kahn JM, Kelly KM. Adolescent and young adult Hodgkin lymphoma: raising the bar through collaborative science and multidisciplinary care. Pediatr Blood Cancer. 2018;65(7):e27033. doi:10.1002/pbc.27033
- Yung L, Smith P, Hancock BW, et al. Long term outcome in adolescents with Hodgkin’s lymphoma: poor results using regimens designed for adults. Leuk Lymphoma. 2004;45(8):1579-1585. doi:10.1080/1042819042000209404
- Dorffel W, Ruhl U, Luders H, et al. Treatment of children and adolescents with Hodgkin lymphoma without radiotherapy for patients in complete remission after chemotherapy: final results of the multinational trial GPOH-HD95. J Clin Oncol. 2013;31(12):1562-1568. doi:10.1200/JCO.2012.45.3266
- National Cancer Institute. Childhood Hodgkin lymphoma treatment (PDQ®)–Health Professional Version. National Institutes of Health. Updated February 14, 2023. Accessed March 20, 2023. https://www.cancer.gov/types/lymphoma/hp/child-hodgkin-treatment-pdq
- Daw S, Hasenclever D, Mascarin M, et al. Risk and response adapted treatment guidelines for managing first relapsed and refractory classical Hodgkin lymphoma in children and young people. Recommendations from the EuroNet Pediatric Hodgkin Lymphoma Group. Hemasphere. 2020;4(1):e329. doi:10.1097/HS9.0000000000000329
- Flerlage JE, Hiniker SM, Armenian S, et al. Pediatric Hodgkin lymphoma, version 3.2021. J Natl Compr Canc Netw. 2021;19(6):733-754. doi:10.6004/jnccn.2021.0027
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Hodgkin lymphoma. Version 2.2023. November 8, 2022. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hodgkins.pdf
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Adolescent and young adult (AYA) oncology. Version 3.2023. January 9, 2023. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/aya.pdf
- Mohty R, Dulery R, Bazarbachi AH, et al. Latest advances in the management of classical Hodgkin lymphoma: the era of novel therapies. Blood Cancer J. 2021;11(7):126. doi:10.1038/s41408-021-00518-z
- Witkowska M, Majchrzak A, Smolewski P. The role of radiotherapy in Hodgkin’s lymphoma: what has been achieved during the last 50 years? Biomed Res Int. 2015;2015:485071. doi:10.1155/2015/485071
- Borchmann P, Moccia A, Greil R, et al. Treatment-related morbidity in patients with classical Hodgkin lymphoma: results of the ongoing, randomized phase II HD21 trial by the German Hodgkin Study Group. Hemasphere. 2022;6(suppl ):1-2. doi:10.1097/01.HS9.0000890576.23258.1c
- Immunotherapy (nivolumab or brentuximab vedotin) plus combination chemotherapy in treating patients with newly diagnosed stage III-IV classic Hodgkin lymphoma. ClinicalTrials.gov. Updated March 8, 2023. Accessed March 20, 2023. https://clinicaltrials.gov/ct2/show/NCT03907488
- Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183-2189. doi:10.1200/JCO.2011.38.0410
- Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378(4):331-344. doi:10.1056/NEJMoa1708984
- Castellino SM, Pei Q, Parsons SK, et al. Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma. N Engl J Med. 2022;387(18):1649-1660. doi:10.1056/NEJMoa2206660
- Sobesky S, Mammadova L, Cirillo M, et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med (N Y). 2021;2(10):1171-1193.e11. doi:10.1016/j.medj.2021.09.002
- Hodgkin Lymphoma. American Cancer Society. Accessed March 20, 2023. https://www.cancer.org/cancer/hodgkin-lymphoma.html
- Aben KK, van Gaal C, van Gils NA, van der Graaf WT, Zielhuis GA. Cancer in adolescents and young adults (15-29 years): a population-based study in the Netherlands 1989-2009. Acta Oncol. 2012;51(7):922-933. doi:10.3109/0284186X.2012.705891
- Ansell SM. Hodgkin lymphoma: 2016 update on diagnosis, risk-stratification, and
management. Am J Hematol. 2016;91(4):434-442. doi:10.1002/ajh.24272 - Cartwright RA, Watkins G. Epidemiology of Hodgkin’s disease: a review. Hematol Oncol. 2004;22(1):11-26. doi:10.1002/hon.723
- Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506-1514. doi:10.1056/NEJM199811193392104
- Bleyer A, Barr R, Hayes-Lattin B, et al. The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer. 2008;8(4):288-298. doi:10.1038/nrc2349
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116-132. doi:10.3322/caac.21438
- Bigenwald C, Galimard JE, Quero L, et al. Hodgkin lymphoma in adolescent and young adults: insights from an adult tertiary single-center cohort of 349 patients. Oncotarget. 2017;8(45):80073-80082. doi:10.18632/oncotarget.20684
- Kahn JM, Kelly KM. Adolescent and young adult Hodgkin lymphoma: raising the bar through collaborative science and multidisciplinary care. Pediatr Blood Cancer. 2018;65(7):e27033. doi:10.1002/pbc.27033
- Yung L, Smith P, Hancock BW, et al. Long term outcome in adolescents with Hodgkin’s lymphoma: poor results using regimens designed for adults. Leuk Lymphoma. 2004;45(8):1579-1585. doi:10.1080/1042819042000209404
- Dorffel W, Ruhl U, Luders H, et al. Treatment of children and adolescents with Hodgkin lymphoma without radiotherapy for patients in complete remission after chemotherapy: final results of the multinational trial GPOH-HD95. J Clin Oncol. 2013;31(12):1562-1568. doi:10.1200/JCO.2012.45.3266
- National Cancer Institute. Childhood Hodgkin lymphoma treatment (PDQ®)–Health Professional Version. National Institutes of Health. Updated February 14, 2023. Accessed March 20, 2023. https://www.cancer.gov/types/lymphoma/hp/child-hodgkin-treatment-pdq
- Daw S, Hasenclever D, Mascarin M, et al. Risk and response adapted treatment guidelines for managing first relapsed and refractory classical Hodgkin lymphoma in children and young people. Recommendations from the EuroNet Pediatric Hodgkin Lymphoma Group. Hemasphere. 2020;4(1):e329. doi:10.1097/HS9.0000000000000329
- Flerlage JE, Hiniker SM, Armenian S, et al. Pediatric Hodgkin lymphoma, version 3.2021. J Natl Compr Canc Netw. 2021;19(6):733-754. doi:10.6004/jnccn.2021.0027
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Hodgkin lymphoma. Version 2.2023. November 8, 2022. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hodgkins.pdf
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Adolescent and young adult (AYA) oncology. Version 3.2023. January 9, 2023. Accessed March 20, 2023. https://www.nccn.org/professionals/physician_gls/pdf/aya.pdf
- Mohty R, Dulery R, Bazarbachi AH, et al. Latest advances in the management of classical Hodgkin lymphoma: the era of novel therapies. Blood Cancer J. 2021;11(7):126. doi:10.1038/s41408-021-00518-z
- Witkowska M, Majchrzak A, Smolewski P. The role of radiotherapy in Hodgkin’s lymphoma: what has been achieved during the last 50 years? Biomed Res Int. 2015;2015:485071. doi:10.1155/2015/485071
- Borchmann P, Moccia A, Greil R, et al. Treatment-related morbidity in patients with classical Hodgkin lymphoma: results of the ongoing, randomized phase II HD21 trial by the German Hodgkin Study Group. Hemasphere. 2022;6(suppl ):1-2. doi:10.1097/01.HS9.0000890576.23258.1c
- Immunotherapy (nivolumab or brentuximab vedotin) plus combination chemotherapy in treating patients with newly diagnosed stage III-IV classic Hodgkin lymphoma. ClinicalTrials.gov. Updated March 8, 2023. Accessed March 20, 2023. https://clinicaltrials.gov/ct2/show/NCT03907488
- Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183-2189. doi:10.1200/JCO.2011.38.0410
- Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378(4):331-344. doi:10.1056/NEJMoa1708984
- Castellino SM, Pei Q, Parsons SK, et al. Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma. N Engl J Med. 2022;387(18):1649-1660. doi:10.1056/NEJMoa2206660
- Sobesky S, Mammadova L, Cirillo M, et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med (N Y). 2021;2(10):1171-1193.e11. doi:10.1016/j.medj.2021.09.002