User login
Mechanism for dust mite–triggered atopic dermatitis identified
Researchers have identified a possible mechanism by which house dust mites could trigger the development of atopic dermatitis in individuals with a genetic predisposition.
The study, published online Feb. 10 in Science Translational Medicine, took skin and blood samples from individuals with atopic dermatitis and healthy controls, then exposed the samples to house dust mite allergen.
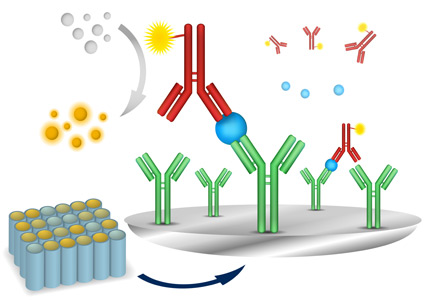
They found that in individuals with atopic dermatitis, this exposure modified phospholipids in the skin to release lipid antigens that then drove T-cell reactivity and inflammation (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad6833).
Furthermore, the study suggested that the skin barrier protein filaggrin can inhibit the modified phospholipid activity and decrease the skin inflammation caused by allergen exposure in atopic dermatitis; however, individuals with atopic dermatitis are more likely to have defective filaggrin.
“The data would support therapeutic approaches to inhibit allergen-derived PLA2 [phospholipase A2] activity, together with treatments that target the downstream immunological effector pathways,” wrote Dr. Rachael Jarrett of the University of Oxford (England) and coauthors.
The study was funded by the U.K. Medical Research Council and National Institute for Health Research Biomedical Research Centre, the National Institutes of Health, and the Burroughs Wellcome Fund in Translational Medicine. Several of the researchers acknowledged grants and pharmaceutical company support. No conflicts of interest were declared.
Researchers have identified a possible mechanism by which house dust mites could trigger the development of atopic dermatitis in individuals with a genetic predisposition.
The study, published online Feb. 10 in Science Translational Medicine, took skin and blood samples from individuals with atopic dermatitis and healthy controls, then exposed the samples to house dust mite allergen.
They found that in individuals with atopic dermatitis, this exposure modified phospholipids in the skin to release lipid antigens that then drove T-cell reactivity and inflammation (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad6833).
Furthermore, the study suggested that the skin barrier protein filaggrin can inhibit the modified phospholipid activity and decrease the skin inflammation caused by allergen exposure in atopic dermatitis; however, individuals with atopic dermatitis are more likely to have defective filaggrin.
“The data would support therapeutic approaches to inhibit allergen-derived PLA2 [phospholipase A2] activity, together with treatments that target the downstream immunological effector pathways,” wrote Dr. Rachael Jarrett of the University of Oxford (England) and coauthors.
The study was funded by the U.K. Medical Research Council and National Institute for Health Research Biomedical Research Centre, the National Institutes of Health, and the Burroughs Wellcome Fund in Translational Medicine. Several of the researchers acknowledged grants and pharmaceutical company support. No conflicts of interest were declared.
Researchers have identified a possible mechanism by which house dust mites could trigger the development of atopic dermatitis in individuals with a genetic predisposition.
The study, published online Feb. 10 in Science Translational Medicine, took skin and blood samples from individuals with atopic dermatitis and healthy controls, then exposed the samples to house dust mite allergen.
They found that in individuals with atopic dermatitis, this exposure modified phospholipids in the skin to release lipid antigens that then drove T-cell reactivity and inflammation (Sci Transl Med. 2016 Feb 10. doi: 10.1126/scitranslmed.aad6833).
Furthermore, the study suggested that the skin barrier protein filaggrin can inhibit the modified phospholipid activity and decrease the skin inflammation caused by allergen exposure in atopic dermatitis; however, individuals with atopic dermatitis are more likely to have defective filaggrin.
“The data would support therapeutic approaches to inhibit allergen-derived PLA2 [phospholipase A2] activity, together with treatments that target the downstream immunological effector pathways,” wrote Dr. Rachael Jarrett of the University of Oxford (England) and coauthors.
The study was funded by the U.K. Medical Research Council and National Institute for Health Research Biomedical Research Centre, the National Institutes of Health, and the Burroughs Wellcome Fund in Translational Medicine. Several of the researchers acknowledged grants and pharmaceutical company support. No conflicts of interest were declared.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: Dust mite allergen modifies skin phospholipids and triggers inflammation in atopic dermatitis.
Major finding: Individuals with atopic dermatitis have a defective skin protein that increases their reactivity to house dust mite allergen.
Data source: Laboratory study.
Disclosures: The study was funded by the U.K. Medical Research Council and National Institute for Health Research Biomedical Research Centre, the National Institutes of Health, and the Burroughs Wellcome Fund in Translational Medicine. Several of the researchers acknowledged grants and pharmaceutical company support. No conflicts of interest were declared.
Early-wheezing Patterns Prefigure Adolescent Respiratory Outcomes and Asthma
Wheezing patterns in early childhood can predict pulmonary function and the development of asthma in adolescence in a high-risk population, a study finds.
The study validates four clinically distinct early-life wheezing phenotypes identified by the landmark Tucson Children’s Respiratory Study in a high-risk population:
• Never.
• Transient early – wheezing before age 3 years but not at age 6 years.
• Late onset – wheezing at age 6 years but not before age 3 years.
• Persistent – wheezing before age 3 years and at age 6 years.

Previously, these phenotypes were shown to be associated with respiratory outcomes in adolescence but not with diagnosed asthma or in genetically predisposed children. The present study extends the associations with these early phenotypes to a high-risk adolescent population, said Meghan B. Azad, Ph.D., of the University of Manitoba, Winnipeg, and her associates.
The study findings were based on findings in 459 children previously enrolled in the Canadian Asthma Primary Prevention Study cohort; this was a prenatally randomized prevention trial in children at high genetic risk for asthma. The distribution of early-wheeze phenotypes was 51% never, 28% transient early, 9% late onset, and 13% persistent (JAMA Pediatrics. 2016 Feb 8. doi: 10.1001/jamapediatrics.2015.4127).
Across all four phenotypes, the authors found a strong gradient of decreasing lung function and increasing asthma risk by age 15 years. Asthma, assessed at 15 years in 320 adolescents, was associated with early-wheeze phenotypes: the prevalence of asthma was 5% among never, 19% among transient early, 27% among late onset, and 42% among persistent phenotypes.
At age 15, early-wheezing phenotypes were not associated with atopic dermatitis or allergic rhinitis. Atopy before 2 years of age was associated with persistent wheeze, which was in turn associated with a 12-fold increased risk of diagnosed asthma by age 15.
“Our results are consistent with other cohorts” and the study “extends these findings through adolescence in a high-risk cohort and demonstrates that asthma-associated deficits are already present at a young age. Collectively, these data show that early wheezing patterns provide clinically meaningful information and suggest that strategies to reduce early-life wheezing and atopic sensitization could have long-term health benefits,” Dr. Azad and her colleagues concluded.
Proven strategies to prevent wheezing include avoiding dust, pets, and tobacco smoke; encouragement of breastfeeding; and delayed introduction of solid foods.
The research was supported by the Canadian Institutes of Health Research. The investigators reported no relevant financial disclosures.
Wheezing patterns in early childhood can predict pulmonary function and the development of asthma in adolescence in a high-risk population, a study finds.
The study validates four clinically distinct early-life wheezing phenotypes identified by the landmark Tucson Children’s Respiratory Study in a high-risk population:
• Never.
• Transient early – wheezing before age 3 years but not at age 6 years.
• Late onset – wheezing at age 6 years but not before age 3 years.
• Persistent – wheezing before age 3 years and at age 6 years.
Previously, these phenotypes were shown to be associated with respiratory outcomes in adolescence but not with diagnosed asthma or in genetically predisposed children. The present study extends the associations with these early phenotypes to a high-risk adolescent population, said Meghan B. Azad, Ph.D., of the University of Manitoba, Winnipeg, and her associates.
The study findings were based on findings in 459 children previously enrolled in the Canadian Asthma Primary Prevention Study cohort; this was a prenatally randomized prevention trial in children at high genetic risk for asthma. The distribution of early-wheeze phenotypes was 51% never, 28% transient early, 9% late onset, and 13% persistent (JAMA Pediatrics. 2016 Feb 8. doi: 10.1001/jamapediatrics.2015.4127).
Across all four phenotypes, the authors found a strong gradient of decreasing lung function and increasing asthma risk by age 15 years. Asthma, assessed at 15 years in 320 adolescents, was associated with early-wheeze phenotypes: the prevalence of asthma was 5% among never, 19% among transient early, 27% among late onset, and 42% among persistent phenotypes.
At age 15, early-wheezing phenotypes were not associated with atopic dermatitis or allergic rhinitis. Atopy before 2 years of age was associated with persistent wheeze, which was in turn associated with a 12-fold increased risk of diagnosed asthma by age 15.
“Our results are consistent with other cohorts” and the study “extends these findings through adolescence in a high-risk cohort and demonstrates that asthma-associated deficits are already present at a young age. Collectively, these data show that early wheezing patterns provide clinically meaningful information and suggest that strategies to reduce early-life wheezing and atopic sensitization could have long-term health benefits,” Dr. Azad and her colleagues concluded.
Proven strategies to prevent wheezing include avoiding dust, pets, and tobacco smoke; encouragement of breastfeeding; and delayed introduction of solid foods.
The research was supported by the Canadian Institutes of Health Research. The investigators reported no relevant financial disclosures.
Wheezing patterns in early childhood can predict pulmonary function and the development of asthma in adolescence in a high-risk population, a study finds.
The study validates four clinically distinct early-life wheezing phenotypes identified by the landmark Tucson Children’s Respiratory Study in a high-risk population:
• Never.
• Transient early – wheezing before age 3 years but not at age 6 years.
• Late onset – wheezing at age 6 years but not before age 3 years.
• Persistent – wheezing before age 3 years and at age 6 years.
Previously, these phenotypes were shown to be associated with respiratory outcomes in adolescence but not with diagnosed asthma or in genetically predisposed children. The present study extends the associations with these early phenotypes to a high-risk adolescent population, said Meghan B. Azad, Ph.D., of the University of Manitoba, Winnipeg, and her associates.
The study findings were based on findings in 459 children previously enrolled in the Canadian Asthma Primary Prevention Study cohort; this was a prenatally randomized prevention trial in children at high genetic risk for asthma. The distribution of early-wheeze phenotypes was 51% never, 28% transient early, 9% late onset, and 13% persistent (JAMA Pediatrics. 2016 Feb 8. doi: 10.1001/jamapediatrics.2015.4127).
Across all four phenotypes, the authors found a strong gradient of decreasing lung function and increasing asthma risk by age 15 years. Asthma, assessed at 15 years in 320 adolescents, was associated with early-wheeze phenotypes: the prevalence of asthma was 5% among never, 19% among transient early, 27% among late onset, and 42% among persistent phenotypes.
At age 15, early-wheezing phenotypes were not associated with atopic dermatitis or allergic rhinitis. Atopy before 2 years of age was associated with persistent wheeze, which was in turn associated with a 12-fold increased risk of diagnosed asthma by age 15.
“Our results are consistent with other cohorts” and the study “extends these findings through adolescence in a high-risk cohort and demonstrates that asthma-associated deficits are already present at a young age. Collectively, these data show that early wheezing patterns provide clinically meaningful information and suggest that strategies to reduce early-life wheezing and atopic sensitization could have long-term health benefits,” Dr. Azad and her colleagues concluded.
Proven strategies to prevent wheezing include avoiding dust, pets, and tobacco smoke; encouragement of breastfeeding; and delayed introduction of solid foods.
The research was supported by the Canadian Institutes of Health Research. The investigators reported no relevant financial disclosures.
FROM JAMA PEDIATRICS

The intersection of obstructive lung disease and sleep apnea
Many patients who have obstructive lung disease, ie, chronic obstructive pulmonary disease (COPD) or asthma, also have obstructive sleep apnea (OSA), and vice versa.
The combination of COPD and OSA was first described almost 30 years ago by Flenley, who called it “overlap syndrome.”1 At that time, he recommended that a sleep study be considered in all obese patients with COPD who snore and in those who have frequent headaches after starting oxygen therapy. In the latter group, he doubted that nocturnal oxygen was the correct treatment. He also believed that the outcomes in patients with overlap syndrome were worse than those in patients with COPD or OSA alone. These opinions remain largely valid today.
We now also recognize the combination of asthma and OSA (alternative overlap syndrome) and collectively call both combinations obstructive lung disease-obstructive sleep apnea (OLDOSA) syndrome.2 Interestingly, these relationships are likely bidirectional, with one condition aggravating or predisposing to the other.
Knowing that a patient has one of these overlap syndromes, one can initiate continuous positive airway pressure (CPAP) therapy, which can improve clinical outcomes.3–6 Therefore, when evaluating a patient with asthma or COPD, one should consider OSA using a validated questionnaire and, if the findings suggest the diagnosis, polysomnography. Conversely, it is prudent to look for comorbid obstructive lung disease in patients with OSA, as interactions between upper and lower airway dysfunction may lead to distinctly different treatment and outcomes.
Here, we briefly review asthma and COPD, explore shared risk factors for sleep-disordered breathing and obstructive lung diseases, describe potential pathophysiologic mechanisms explaining these associations, and highlight the importance of recognizing and individually treating the overlaps of OSA and COPD or asthma.
COPD AND ASTHMA ARE VERY COMMON
About 10% of the US population have COPD,7 a preventable and treatable disease mainly caused by smoking, and a leading cause of sickness and death worldwide.8,9
About 8% of Americans have asthma,7 which has become one of the most common chronic conditions in the Western world, affecting about 1 in 7 children and about 1 in 12 adults. The World Health Organization estimates that 235 million people suffer from asthma worldwide, and by 2025 this number is projected to rise to 400 million.10,11
The prevalence of these conditions in a particular population depends on the frequency of risk factors and associated morbidities, including OSA. These factors may allow asthma or COPD to arise earlier or have more severe manifestations.8,12
Asthma and COPD: Similarities and differences
Asthma and COPD share several features. Both are inflammatory airway conditions triggered or perpetuated by allergens, viral infection, tobacco smoke, products of biomass or fossil fuel combustion, and other substances. In both diseases, airflow is “obstructed” or limited, with a low ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC). Symptoms can also be similar, with dyspnea, cough, wheezing, and chest tightness being the most frequent complaints. The similarities support the theory proposed by Orie et al13 (the “Dutch hypothesis”) that asthma and COPD may actually be manifestations of the same disease.
But there are also differences. COPD is strongly linked to cigarette smoking and has at least three phenotypes:
- Chronic bronchitis, defined clinically by cough and sputum production for more than 3 months per year for 2 consecutive years
- Emphysema, characterized anatomically by loss of lung parenchyma, as seen on tomographic imaging or examination of pathologic specimens
- A mixed form with bronchitic and emphysematous features, which is likely the most common.
Particularly in emphysematous COPD, smoking predisposes patients to gas-exchange abnormalities and low diffusing capacity for carbon monoxide.
In asthma, symptoms may be more episodic, the age of onset is often younger, and atopy is common, especially in allergic asthma. These episodic symptoms may correlate temporally with measurable airflow reversibility (≥ 12% and ≥ 200 mL improvement in FVC or in FEV1 after bronchodilator challenge).
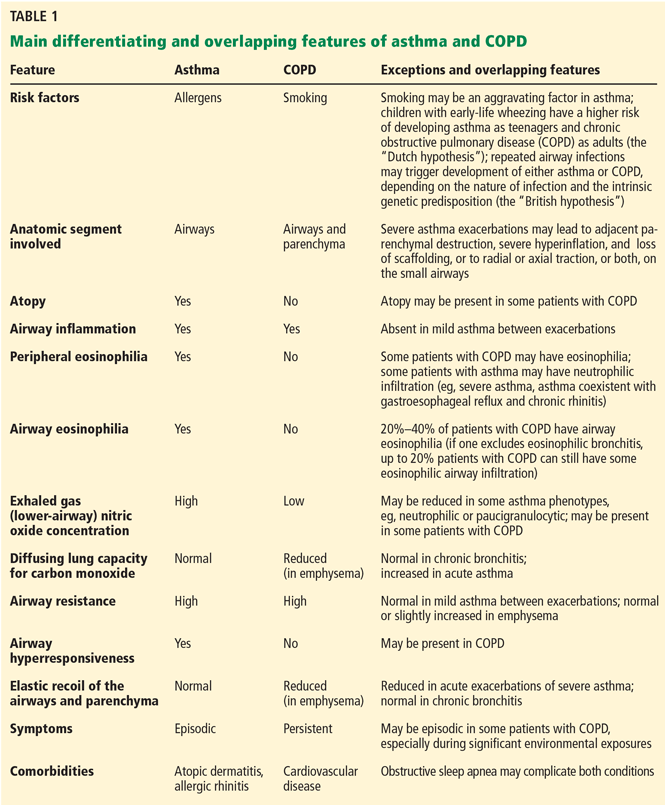
However, the current taxonomy does not unequivocally divide obstructive lung diseases into asthma and COPD, and major features such as airway hyperresponsiveness, airflow reversibility, neutrophilic or CD8 lymphocytic airway inflammation, and lower concentration of nitric oxide in the exhaled air may be present in different phenotypes of both conditions (Table 1).
AIRFLOW IN OBSTRUCTIVE LUNG DISEASES AND DURING SLEEP
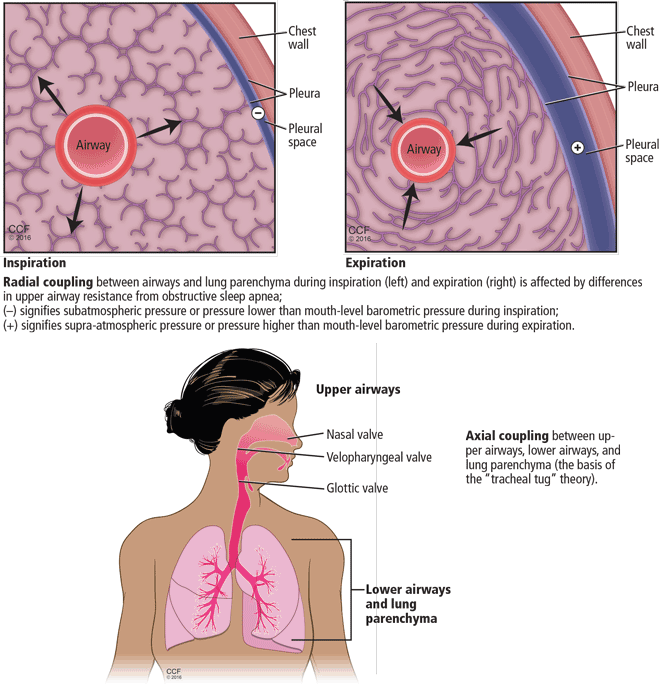
Normal airflow involves a complex interplay between airway resistance and elastic recoil of the entire respiratory system, including the airways, the lung parenchyma, and the chest wall (Figure 1).
In asthma and COPD, resistance to airflow is increased, predominantly in the upper airways (nasal passages, pharynx, and larynx) and in the first three or four subdivisions of the tracheobronchial tree. The problem is worse during exhalation, when elastic recoil of the lung parenchyma and chest wall also increases airway resistance, reduces airway caliber, and possibly even constricts the bronchi. This last effect may occur either due to mass loading of the bronchial smooth muscles or to large intrathoracic transmural pressure shifts that may increase extravasation of fluid in the bronchial walls, especially with higher vascular permeability in inflammatory conditions.
Furthermore, interactions between the airway and parenchyma and between the upper and lower airways, as well as radial and axial coupling of these anatomic and functional components, contribute to complex interplay between airway resistance and parenchymal-chest wall elastic energy—stretch or recoil.
The muscles of the upper and lower airway may not work together due to the loss of normal lung parenchyma (as in emphysema) or to the acute inflammation in the small airways and adjacent parenchyma (as in severe asthma exacerbations). This loss of coordination makes the upper airway more collapsible, a feature of OSA.
Additionally, obesity, gastroesophageal reflux, disease chronic rhinitis, nasal polyposis, and acute exacerbations of chronic systemic inflammation all contribute to more complex interactions between obstructive lung diseases and OSA.6
Sleep affects breathing, particularly in patients with respiratory comorbidities, and sleep-disordered breathing causes daytime symptoms and worsens quality of life.1,13–15 During sleep, respiratory centers become less sensitive to oxygen and carbon dioxide; breathing becomes more irregular, especially during rapid eye movement (REM) sleep; the chest wall moves less, so that the tidal volume and functional residual capacity are lower; sighs, yawns, and deep breaths become limited; and serum carbon dioxide concentration may rise.
OBSTRUCTIVE SLEEP APNEA
The prevalence of OSA, a form of sleep-disordered breathing characterized by limitation of inspiratory and (to a lesser degree) expiratory flow, has increased significantly in recent years, in parallel with the prevalence of its major risk factor, obesity.
OSA is generally defined as an apnea-hypopnea index of 5 or higher, ie, five or more episodes of apnea or hypopnea per hour.
OSA syndrome, ie, an apnea-hypopnea index of 5 or higher and excessive daytime sleepiness (defined by an Epworth Sleepiness Scale score > 10) was found in the initial analysis of the Wisconsin Sleep cohort in 1993 to be present in about 2% of women and 4% of men.16 A more recent longitudinal analysis showed a significant increase—for example, in people 50 to 70 years old the prevalence was up to 17.6% in men and 7.5% in women.17
Upper airway resistance syndrome, a milder form of sleep-disordered breathing, is now included under the diagnosis of OSA, as its pathophysiology is not significantly different.18
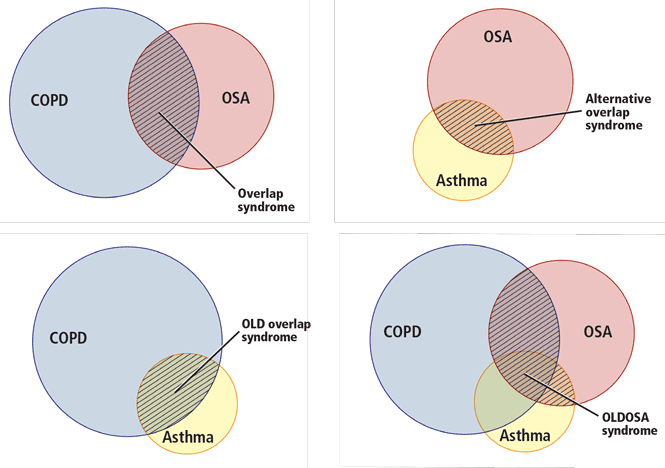
In the next section, we discuss what happens when OSA overlaps with COPD (overlap syndrome) and with asthma (“alternative overlap syndrome”)2,8 (Figure 2).
OSA AND COPD (OVERLAP SYNDROME)
Flenley1 hypothesized that patients with COPD in whom supplemental oxygen worsened hypercapnia may also have OSA and called this association overlap syndrome.
How common is overlap syndrome?
Since both COPD and OSA are prevalent conditions, overlap syndrome may also be common.
The reported prevalence of overlap syndrome varies widely, depending on the population studied and the methods used. In various studies, COPD was present in 9% to 56% of patients with OSA,19–23 and OSA was found in 5% to 85% of patients with COPD.24–27
Based on the prevalence of COPD in the general population (about 10%12) and that of sleep-disordered breathing (about 5% to 10%17), the expected prevalence of overlap syndrome in people over age 40 may be 0.5% to 1%.28 In a more inclusive estimate with “subclinical” forms of overlap syndrome—ie, OSA defined as an apnea-hypopnea index of 5 or more (about 25% of the population17) and COPD Global initiative for Chronic Obstructive Lung Disease (GOLD) stage 1 (16.8% in the National Health and Nutrition Education Survey12)—the expected prevalence of overlap is around 4%. Some studies found a higher prevalence of COPD in OSA patients than in the general population,21,29 while others did not.22,28,30 The studies differed in how they defined sleep-disordered breathing.
Larger studies are needed to better assess the true prevalence of sleep-disordered breathing in COPD. They should use more sensitive measures of airflow and standardized definitions of sleep-disordered breathing and should include patients with more severe COPD.
Fatigue and insomnia are common in COPD
Fatigue is strongly correlated with declining lung function, low exercise tolerance, and impaired quality of life in COPD.31 Factors that contribute to fatigue include dyspnea, depression, and impaired sleep.32 Some suggest that at least half of COPD patients have sleep complaints such as insomnia, sleep disruption, or sleep fragmentation.33 Insomnia, difficulty falling asleep, and early morning awakenings are the most common complaints (30%–70% of patients) and are associated with daytime fatigue.34 Conversely, comorbid OSA can contribute to fatigue and maintenance-type insomnia (ie, difficulty staying asleep and returning to sleep).
Multiple mechanisms of hypoxemia in overlap syndrome
Oxygenation abnormalities and increased work of breathing contribute to the pathophysiology of overlap syndrome. In patients with COPD, oxygenation during wakefulness is a strong predictor of gas exchange during sleep.35 Further, patients with overlap syndrome tend to have more severe hypoxia during sleep than patients with isolated COPD or OSA at rest or during exercise.36
In overlap syndrome, hypoxemia is the result of several mechanisms:
- Loss of upper airway muscle tone from intermittent episodes of obstructive apnea and hypopnea leads to upper airway collapse during sleep, particularly during REM sleep, increasing the severity of OSA.37
- Reductions in functional residual capacity from lying in the recumbent position and during REM sleep render patients with COPD more vulnerable, as compensatory use of accessory muscles to maintain near-normal ventilation in a hyperinflated state becomes impaired.37
- Alterations in pulmonary ventilation-perfusion matching may lead to altered carbon dioxide homeostasis and impaired oxygenation in patients with emphysema.
- Circadian variation in lower airway caliber may also be observed, in parallel with the bronchoconstriction caused by increased nocturnal vagotonia.
- Hypercapnia (Paco2 ≥ 45 mm Hg) may lead to overall reduced responsiveness of respiratory muscles and to a blunted response of respiratory centers to low oxygen and high carbon dioxide levels.38 Thus, hypercapnia is a better predictor of the severity of nocturnal hypoxemia than hypoxemia developing during exercise.39
In a person who is at near-maximal ventilatory capacity, even a mild increase in upper airway resistance (as seen with snoring, upper airway resistance syndrome, or OSA) increases the work of breathing. This phenomenon can lead to early arousals even before significant oxyhemoglobin desaturation occurs.
Normally, inspiratory flow limitation is counteracted by increasing inspiratory time to maintain ventilation. Patients with COPD may not be able to do this, however, as they need more time to breathe out due to narrowing of their lower airways.40 The inability to compensate for upper airway resistance, similar to the increased work of breathing seen with exercise, may lead to early arousals and increased sleep fragmentation.
Consequences of overlap syndrome
Patients with overlap syndrome appear to have higher morbidity and mortality rates than those with COPD or sleep-disordered breathing alone.
Cor pulmonale. Nighttime hypoxia is more severe and persistent in overlap syndrome than with COPD or OSA alone. This may contribute to more significant pulmonary hypertension and to the development of cor pulmonale, in which the right ventricle is altered in structure (eg, hypertrophied, dilated) or reduced in function, or both, from severe pulmonary hypertension.
In contrast to right ventricular failure due to disorders of the left heart, cor pulmonale is a result of diseases of the vasculature (eg, idiopathic pulmonary arterial hypertension), lung parenchyma (eg, COPD), upper airway (eg, OSA), or chest wall (eg, severe kyphoscoliosis). COPD is the most common cause of cor pulmonale in the United States, accounting for up to 30% of cases of cor pulmonale.41–45 In OSA, cor pulmonale is seen in up to 20% of cases,43 while in overlap syndrome cor pulmonale is encountered even more often (ie, in up to 80%); these patients have a dismal 5-year survival rate of about 30%.46
Obesity hypoventilation syndrome is characterized by obesity (body mass index ≥ 30 kg/m2) and daytime hypercapnia (Paco2 ≥ 45 mm Hg) that cannot be fully attributed to an underlying cardiopulmonary or neurologic condition.18 Hypercapnia worsens during sleep (especially during REM sleep) and is often associated with severe arterial oxygen desaturation. Up to 90% of patients with obesity hypoventilation syndrome have comorbid OSA, and the rest generally have sleep-related hypoventilation, particularly during REM sleep.
In patients with obesity hypoventilation syndrome, daytime hypercapnia may improve or even normalize with adequate positive airway pressure treatment and sustained adherence to treatment.18 Many patients with obesity hypoventilation syndrome respond to CPAP or bilevel positive airway pressure (BPAP), with improvement in daytime Paco2. However, normalization of daytime Paco2 occurs only in a subgroup of patients. In contrast, treatment with oxygen therapy alone may worsen hypercapnia.
Oxygen therapy for pure COPD, but maybe not for overlap syndrome
Continuous oxygen therapy reduces mortality in COPD,47,48 but the duration and severity of hypoxemia that warrant oxygen therapy are less clear. Oxygen therapy in hypoxemic patients has been shown to improve sleep quality and reduce arousals.49
Indications for oxygen treatment of nocturnal hypoxemia are generally based on Medicare guidelines:
- At least 5 minutes of sleep with peripheral oxygen saturation ≤ 88% or Pao2 ≤ 55 mm Hg, or
- A decrease in Pao2 of more than 10 mm Hg or in peripheral oxygen saturation of more than 5% for at least 5 minutes of sleep and associated with signs or symptoms reasonably attributable to hypoxemia (group I criteria), or
- At least 5 minutes of sleep with peripheral oxygen saturation ≥ 89% or Pao2 56 to 59 mm Hg and pedal edema, pulmonary hypertension, cor pulmonale, or erythrocytosis (group II criteria).50
Approximately 47% of COPD patients who are hypoxemic during the day spend about 30% of sleep time with an oxygen saturation less than 90%, even while on continuous oxygen therapy.51 Current recommendations for nocturnal oxygen therapy are to increase the oxygen concentration by 1 L/minute above the baseline oxygen flow rate needed to maintain an oxygen saturation higher than 90% during resting wakefulness, using a nasal cannula or face mask.52
Caveat. In overlap syndrome, supplemental oxygen may prolong the duration of apnea episodes and worsen hypercapnia.
Positive airway pressure for OSA
Positive airway pressure therapy improves cardiovascular outcomes in OSA.53 Several studies54–58 compared the effectiveness of CPAP vs BPAP as initial therapy for OSA but did not provide enough evidence to favor one over the other in this setting. Similarly, the results are mixed for the use of fixed or auto-adjusting BPAP as salvage therapy in patients who cannot tolerate CPAP.59–61
In overlap syndrome, CPAP or BPAP with or without supplemental oxygen has been investigated in several studies.26,62–65 In general, the mortality rate of COPD patients who require oxygen therapy is quite high.47,66 In hypoxemic COPD patients with moderate to severe sleep-disordered breathing, the 5-year survival rate was 71% in those treated with CPAP plus oxygen, vs 26% in those on oxygen alone, independent of baseline postbronchodilator FEV1.67
There is no specific FEV1 cutoff for prescribing CPAP. In general, daytime hypercapnia and nocturnal hypoxemia despite supplemental oxygen therapy are indications for BPAP therapy, regardless of the presence of OSA. Whether noninvasive nocturnal ventilation for COPD patients who do not have OSA improves long-term COPD outcomes is not entirely clear.65,68,69
Adding nocturnal BPAP in spontaneous timed mode to pulmonary rehabilitation for severe hypercapnic COPD was found to improve quality of life, mood, dyspnea, gas exchange, and decline in lung function.70 Other studies noted that COPD patients hospitalized with respiratory failure who were randomized to noninvasive nocturnal ventilation plus oxygen therapy as opposed to oxygen alone experienced improvement in health-related quality of life and reduction in intensive-care-unit length of stay but no difference in mortality or subsequent hospitalizations.69 In stable hypercapnic COPD patients without OSA, there is no clear evidence that nocturnal noninvasive ventilation lessens the risk of death despite improved daytime gas exchange,71,72 but additional long-term studies are needed.
Lung volume reduction surgery, a procedure indicated for highly selected patients with severe COPD, has been shown to reduce hyperinflation, improve nocturnal hypoxemia, and improve total sleep time and sleep efficiency in patients without sleep-disordered breathing.73 More studies are needed to determine if reduction in lung hyperinflation has an impact on the occurrence of OSA and on morbidity related to sleep-disordered breathing.
Benefit of CPAP in overlap syndrome
In a nonrandomized study, Marin et al62 found that overlap syndrome is associated with an increased risk of death and hospitalization due to COPD exacerbations. CPAP therapy was associated with improved survival rates and decreased hospitalization rates in these patients.
Stanchina et al,74 in a post hoc analysis of an observational cohort, assessed the outcomes of 227 patients with overlap syndrome. Greater use of CPAP was found to be associated with lower mortality rates.
Jaoude et al75 found that hypercapnic patients with overlap syndrome who were adherent to CPAP therapy had a lower mortality rate than nonadherent hypercapnic patients (P = .04). In a multivariate analysis, the comorbidity index was the only independent predictor of mortality in normocapnic patients with overlap syndrome, while CPAP adherence was associated with improved survival.
Lastly, patients with overlap syndrome tend to need more healthcare and accrue higher medical costs than patients with COPD alone. An analysis of a state Medicaid database that included COPD patients showed that beneficiaries with overlap syndrome spent at least $4,000 more in medical expenditures than beneficiaries with “lone” COPD.24
In conclusion, CPAP is the first line of therapy for overlap syndrome, while daytime hypercapnia or nocturnal hypoxemia despite supplemental oxygen therapy are indications for nocturnal BPAP therapy, regardless of whether patients have OSA.
OSA AND ASTHMA (ALTERNATIVE OVERLAP SYNDROME)
Epidemiology and clinical features
The coexistence of asthma and OSA can begin in childhood and continue throughout adult life. A higher prevalence of lifetime asthma and OSA has been noted in children of racial and ethnic minorities, children of lower socioeconomic status, and those with atopy.76
In a pediatric asthma clinic, it was noted that 12 months into structured asthma management and optimization, children with sleep-disordered breathing were nearly four times more likely to have severe asthma at follow-up, even after adjusting for obesity, race, and gender.77
In adult patients with OSA, the prevalence of asthma is about 35%.78 Conversely, people with asthma are at higher risk of OSA. High risk of OSA was more prevalent in a group of patients with asthma than in a general medical clinic population (39.5% vs 27.2%, P < .05).79
Analysis of a large prospective cohort found that asthma was a risk factor for new-onset OSA. The incidence of OSA over 4 years in patients with self-reported asthma was 27%, compared with 16% without asthma. The relative risk adjusted for risk factors such as body mass index, age, and gender was 1.39 (95% confidence interval [CI] 15%–19%).80
Patients with asthma who are at high risk of OSA are more likely to have worse daytime and nighttime asthma symptoms. Interestingly, patients who are diagnosed with OSA and treated with CPAP seem to have better asthma control.
Patients with asthma who are more likely to have OSA are women (odds ratio [OR] 2.1), have greater asthma severity (OR 1.6), have gastroesophageal reflux disease (OR 2.7), and use inhaled corticosteroids (OR 4.0).81 These associations are different than the traditional, population-wide risk factors for OSA, such as male sex, excess body weight, and nocturnal nasal congestion.82
OSA also worsens asthma control. Teodorescu et al15 found that severe asthma was more frequent in older asthma patients (ages 60–75, prevalence 49%) than in younger patients (ages 18–59, 39%). Older adults with OSA were seven times as likely to have severe asthma (OR 6.6), whereas young adults with sleep apnea were only three times as likely (OR 2.6).
In a group of patients with difficult-to-treat asthma, OSA was significantly associated with frequent exacerbations (OR 3.4), an association similar in magnitude to that of psychological conditions (OR 10.8), severe sinus disease (OR 3.7), recurrent respiratory tract infections (OR 6.9), and gastroesophageal reflux disease (OR 4.9).83 More than half of the patients had at least three of these comorbid conditions.
Sleep quality can greatly affect asthma control, and its importance is often underestimated. Patients with severe asthma have worse sleep quality than patients with milder asthma or nonasthmatic patients, even after excluding patients with a high risk of OSA, patients on CPAP therapy, and patients with a history of gastroesophageal reflux disease. Furthermore, regardless of asthma severity, sleep quality is a significant predictor of asthma-related quality of life, even after accounting for body mass index, daytime sleepiness, and gastroesophageal reflux disease.84
Pathophysiology of alternative overlap syndrome
Sleep significantly affects respiratory pathophysiology in asthma. The underlying mechanisms include physical and mechanical stressors, neurohormonal changes, hypoxia, confounding medical conditions, and local and systemic inflammatory changes.
Patients with nocturnal asthma experience more pronounced obstruction when sleep-deprived, suggesting that sleep loss may contribute to worsening airflow limitation.14 Although changes in pulmonary mechanics and lung volumes may also have a role, volume-dependent airway narrowing does not appear to account for all observed nocturnal increases in airway resistance. Intrathoracic blood pooling may also contribute to nocturnal bronchoconstriction through stimulation of pulmonary C fibers and increased bronchial wall edema, a mechanism that may be similar to the “cardiac asthma” seen in left ventricular dysfunction.
Early studies of sleep-disordered breathing demonstrated that patients with asthma were breathing more irregularly (with hypopnea, apnea, and hyperpnea) in REM sleep than those without asthma.85 Interestingly, REM-related hypoxia has also been noted in children with asthma.86 This may be related to the increased cholinergic outflow that occurs during REM sleep, which in turn modulates the caliber and reactivity of the lower airways.
Physical changes such as upper airway collapse and reduced pharyngeal cross-sectional area may cause further mechanical strain.87 This can further propagate airway inflammation, alter airway mucosal muscle fibers, and stimulate neural reflexes, thereby increasing cholinergic tone and bronchoconstriction. Furthermore, heightened negative intrathoracic pressure during obstructive episodes can increase nocturnal pulmonary blood pooling.14 Hypoxia itself can augment airway hyperresponsiveness via vagal pathways or carotid body receptors, increasing reactive oxygen species and inflammatory mediators. Local inflammation can “spill over” into systemic inflammatory changes, while alterations in airway inflammatory markers in asthma seem to follow a circadian rhythm, in parallel with the nocturnal worsening of the asthma symptoms.88 Finally, altered sleep may be related to other comorbid conditions, such as gastroesophageal reflux disease, insomnia, and restless leg syndrome.
Management and outcomes of alternative overlap syndrome
Despite optimization of asthma management, OSA can still significantly affect asthma control and symptoms.84
Interestingly, medications that reduce airway inflammation (eg, corticosteroids) may promote OSA. This occurrence cannot be fully explained by an increase in body mass, as more respiratory disturbances occur during sleep with continuous corticosteroid treatment even without increases in body mass index.87 Therefore, these associations may be related to upper airway myopathy caused by the treatment, a small pharynx, facial dysmorphisms, or fat deposition.89
Does CPAP improve asthma?
OSA is often unrecognized in patients with asthma, and treating it can have an impact on asthma symptoms.
CPAP therapy has not been shown to significantly change airway responsiveness or lung function, but it has been noted to significantly improve both OSA-related and asthma-related quality of life and reduce the use of rescue bronchodilators.3,90 CPAP has demonstrated improvement of quality of life that positively correlated with body weight and apnea-hypopnea index at baseline, suggesting that asthmatic patients with greater obesity or worse OSA may benefit most from aggressive management.90
However, CPAP should be used only if the patient has confirmed OSA. Empiric use of CPAP without a diagnosis of OSA was poorly tolerated and failed to improve asthma symptoms or lung function.91 More importantly, using CPAP in a patient who does not have OSA may contribute to further sleep disruption.91
Second-line treatments such as mandibular advancing devices and airway or bariatric surgery have not yet been studied in alternative overlap syndrome.
A multidimensional assessment of asthma
The Western world is experiencing an epidemic of obesity and of asthma. Obesity contributes to the pathogenesis of OSA by altering the anatomy and collapsibility of the upper airway, affecting ventilatory control and increasing respiratory workload. Another paradigm, supported by some evidence, is that OSA itself may contribute to the development of obesity. Both OSA and obesity lead to activation of inflammatory biologic cascades, which are likely the pathogenic mechanisms for their cardiovascular and metabolic consequences. As such, early recognition of OSA is important, as effective treatments are available.
In some patients, obesity may cause asthma, as obesity precedes the onset of asthma in a significant proportion of patients, and bariatric surgery for morbid obesity may resolve asthma. The obese asthma phenotype seems to include chronic rhinosinusitis, gastroesophageal reflux disease, poorer asthma control, limited responsiveness to corticosteroids, and even different sets of biomarkers (eg, neutrophilic airway inflammation). A cohort of obese patients with poor asthma control demonstrated significant improvement in asthma symptoms, quality of life, and airway reactivity after weight loss from bariatric surgery.92
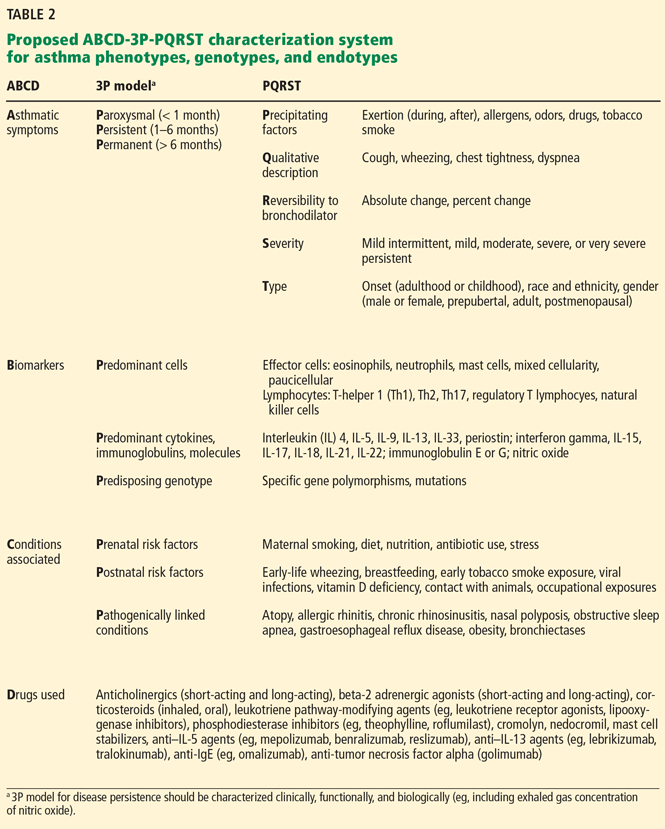
To improve our knowledge about airway disease phenotypes and endotypes and their response to therapy, we propose taking a multidimensional, structured assessment of all patients with asthma, using a schema we call “ABCD-3P-PQRST” (Table 2).
The purpose of using this type of system in clinics and research is to capture the multidimensionality of the disease and better develop future individualized therapeutic strategies by employing the latest advances in systems biology and computational methods such as cluster and principal component analysis.
Multidimensional assessments addressing airway problems such as asthma, COPD, OSA, other comorbidities and risk factors, and personalized management plans will need to be the basis of future therapeutic interventions. Increased attention to the complications of asthma and obstructive airway and lung diseases in our patients is imperative, specifically to develop effective systems of care, appropriate clinical guidelines, and research studies that lead to improved health outcomes.
- Flenley DC. Sleep in chronic obstructive lung disease. Clin Chest Med 1985; 6:651–661.
- Ioachimescu OC, Teodorescu M. Integrating the overlap of obstructive lung disease and obstructive sleep apnoea: OLDOSA syndrome. Respirology 2013; 18:421–431.
- Ciftci TU, Ciftci B, Guven SF, Kokturk O, Turktas H. Effect of nasal continuous positive airway pressure in uncontrolled nocturnal asthmatic patients with obstructive sleep apnea syndrome. Respiratory Med 2005; 99:529–534.
- Kim MY, Jo EJ, Kang SY, et al. Obstructive sleep apnea is associated with reduced quality of life in adult patients with asthma. Ann Allergy Asthma Immunol 2013; 110:253–257.
- Teodorescu M, Polomis DA, Teodorescu MC, et al. Association of obstructive sleep apnea risk or diagnosis with daytime asthma in adults. J Asthma 2012; 49:620–628.
- Puthalapattu S, Ioachimescu OC. Asthma and obstructive sleep apnea: clinical and pathogenic interactions. J Investig Med 2014; 62:665–675.
- National Institutes of Health. National Heart, Lung, and Blood Institute. NHLBI Factbook, Fiscal Year 2007. Chapter 4. Disease Statistics. www.nhlbi.nih.gov/about/factbook-07/chapter4.htm. Accessed November 11, 2015.
- Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- WHO. World Health Organization: Asthma. Fact sheet No 307. www.who.int/mediacentre/factsheets/fs307/en/. Accessed November 11, 2015.
- Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 2004; 59:469–478.
- WHO. Chronic obstructive pulmonary disease (COPD). Fact sheet No 315. www.who.int/mediacentre/factsheets/fs315/en/index.html. 2011. Accessed November 11, 2015.
- Ford ES, Mannino DM, Wheaton AG, Giles WH, Presley-Cantrell L, Croft JB. Trends in the prevalence of obstructive and restrictive lung function among adults in the United States: findings from the National Health and Nutrition Examination surveys from 1988–1994 to 2007–2010. Chest 2013; 143:1395–1406.
- Orie N, Sluiter H, de Vries K, Tammeling G, Witkop J. The host factor in bronchitis. Paper presented at: Bronchitis—an international symposium 1961; Assen, Netherlands.
- Ballard RD. Sleep, respiratory physiology, and nocturnal asthma. Chronobiol Int 1999; 16:565–580.
- Teodorescu M, Polomis DA, Gangnon RE, et al. Asthma control and its relationship with obstructive sleep apnea (OSA) in older adults. Sleep Disord 2013; 2013:251567.
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- International Classification of Sleep Disorders, 3rd ed.: Diagnostic and coding manual. Darien, Illinois: American Academy of Sleep Medicine. 2014.
- Lopez-Acevedo MN, Torres-Palacios A, Elena Ocasio-Tascon M, Campos-Santiago Z, Rodriguez-Cintron W. Overlap syndrome: an indication for sleep studies? A pilot study. Sleep Breath 2009; 13:409–413.
- Scharf C, Li P, Muntwyler J, et al. Rate-dependent AV delay optimization in cardiac resynchronization therapy. PACE 2005; 28:279–284.
- Chaouat A, Weitzenblum E, Krieger J, Ifoundza T, Oswald M, Kessler R. Association of chronic obstructive pulmonary disease and sleep apnea syndrome. Am J Respir Crit Care Med 1995;151:82–86.
- Bednarek M, Plywaczewski R, Jonczak L, Zielinski J. There is no relationship between chronic obstructive pulmonary disease and obstructive sleep apnea syndrome: a population study. Respiration 2005; 72:142–149.
- Fletcher EC. Chronic lung disease in the sleep apnea syndrome. Lung 1990; 168(suppl):751–761.
- Shaya FT, Lin PJ, Aljawadi MH, Scharf SM. Elevated economic burden in obstructive lung disease patients with concomitant sleep apnea syndrome. Sleep Breath 2009; 13:317–323.
- Larsson LG, Lindberg A, Franklin KA, Lundbäck B; Obstructive Lung Disease in Northern Sweden Studies. Obstructive sleep apnoea syndrome is common in subjects with chronic bronchitis. Report from the Obstructive Lung Disease in Northern Sweden studies. Respiration 2001; 68:250–255.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Resp J 2010; 35:132–137.
- Guilleminault C, Cummiskey J, Motta J. Chronic obstructive airflow disease and sleep studies. Am Rev Respir Dis 1980; 122:397–406.
- Weitzenblum E, Chaouat A, Kessler R, Canuet M. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc 2008; 5:237–241.
- Bradley TD, Rutherford R, Lue F, et al. Role of diffuse airway obstruction in the hypercapnia of obstructive sleep apnea. Am Rev Respir Dis 1986; 134:920–924.
- Sanders MH, Newman AB, Haggerty CL, et al. Sleep and sleep-disordered breathing in adults with predominantly mild obstructive airway disease. Am J Respir Crit Care Med 2003; 167:7–14.
- Breslin E, van der Schans C, Breukink S, et al. Perception of fatigue and quality of life in patients with COPD. Chest 1998; 114:958–964.
- Kapella MC, Larson JL, Patel MK, Covey MK, Berry JK. Subjective fatigue, influencing variables, and consequences in chronic obstructive pulmonary disease. Nurs Res 2006; 55:10–17.
- Klink M, Quan SF. Prevalence of reported sleep disturbances in a general adult population and their relationship to obstructive airways diseases. Chest 1987; 91:540–546.
- Bellia V, Catalano F, Scichilone N, et al. Sleep disorders in the elderly with and without chronic airflow obstruction: the SARA study. Sleep 2003; 26:318–323.
- Connaughton JJ, Catterall JR, Elton RA, Stradling JR, Douglas NJ. Do sleep studies contribute to the management of patients with severe chronic obstructive pulmonary disease? Am Rev Respir Dis 1988; 138:341–344.
- Mulloy E, McNicholas WT. Ventilation and gas exchange during sleep and exercise in severe COPD. Chest 1996; 109:387–394.
- Johnson MW, Remmers JE. Accessory muscle activity during sleep in chronic obstructive pulmonary disease. J Appl Physiol 1984; 57:1011–1017.
- Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax 1982; 37:840–844.
- Mulloy E, Fitzpatrick M, Bourke S, O’Regan A, McNicholas WT. Oxygen desaturation during sleep and exercise in patients with severe chronic obstructive pulmonary disease. Respir Med 1995; 89:193–198.
- Herpel LB, Brown CD, Goring KL, et al. COPD cannot compensate for upper airway obstruction during sleep (abstract). Am J Respir Crit Care Med 2007; 175:A71.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 2. Am J Respir Crit Care Med 1994; 150:1158–1168.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 1. Am J Respir Crit Care Med 1994; 150:833–852.
- Budev MM, Arroliga AC, Wiedemann HP, Matthay RA. Cor pulmonale: an overview. Semin Respir Crit Care Med 2003; 24:233–244.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(25 suppl):D34–D41.
- Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:20–22.
- Rasche K, Orth M, Kutscha A, Duchna HW. [Pulmonary diseases and heart function]. In German. Internist (Berl) 2007; 48:276–282.
- Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Nocturnal Oxygen Therapy Trial Group. Ann Intern Med 1980; 93:391–398.
- Newman AB, Foster G, Givelber R, Nieto FJ, Redline S, Young T. Progression and regression of sleep-disordered breathing with changes in weight: the Sleep Heart Health Study. Arch Intern Med 2005; 165:2408–2413.
- Calverley PM, Brezinova V, Douglas NJ, Catterall JR, Flenley DC. The effect of oxygenation on sleep quality in chronic bronchitis and emphysema. Am Rev Respir Dis 1982; 126:206–210.
- Centers for Medicare and Medicaid Services. National coverage determination (NCD) for home use of oxygen (240.2). www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=169&ncdver=1&NCAId=169&NcaName=Home+Use+of+Oxygen&IsPopup=y&bc=AAAAAAAAIAAA&. Accessed November 11, 2015.
- Plywaczewski R, Sliwinski P, Nowinski A, Kaminski D, Zielinski J. Incidence of nocturnal desaturation while breathing oxygen in COPD patients undergoing long-term oxygen therapy. Chest 2000; 117:679–683.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breathing 2007; 11:117–124.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Marin JM, DeAndres R, Alonso J, Sanchez A, Carrizo S. Long term mortality in the overlap syndrome. Eur Resp J 2008; 32(suppl 52):P865.
- Reeves-Hoche MK, Hudgel DW, Meck R, Witteman R, Ross A, Zwillich CW. Continuous versus bilevel positive airway pressure for obstructive sleep apnea. Am J Respir Crit Care Med 1995; 151:443–449.
- Gay PC, Herold DL, Olson EJ. A randomized, double-blind clinical trial comparing continuous positive airway pressure with a novel bilevel pressure system for treatment of obstructive sleep apnea syndrome. Sleep 2003; 26:864–869.
- Blau A, Minx M, Peter JG, et al. Auto bi-level pressure relief-PAP is as effective as CPAP in OSA patients—a pilot study. Sleep Breath 2012; 16:773–779.
- Randerath WJ, Galetke W, Ruhle KH. Auto-adjusting CPAP based on impedance versus bilevel pressure in difficult-to-treat sleep apnea syndrome: a prospective randomized crossover study. Med Sci Monit 2003; 9:CR353–CR358.
- Schwartz SW, Rosas J, Iannacone MR, Foulis PR, Anderson WM. Correlates of a prescription for bilevel positive airway pressure for treatment of obstructive sleep apnea among veterans. J Clin Sleep Med 2013; 9:327–335.
- Gentina T, Fortin F, Douay B, et al. Auto bi-level with pressure relief during exhalation as a rescue therapy for optimally treated obstructive sleep apnoea patients with poor compliance to continuous positive airways pressure therapy--a pilot study. Sleep Breathing 2011; 15:21–27.
- Ballard RD, Gay PC, Strollo PJ. Interventions to improve compliance in sleep apnea patients previously non-compliant with continuous positive airway pressure. J Clinical Sleep Med 2007; 3:706–712.
- Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med 2010; 182:325–331.
- de Miguel J, Cabello J, Sanchez-Alarcos JM, Alvarez-Sala R, Espinos D, Alvarez-Sala JL. Long-term effects of treatment with nasal continuous positive airway pressure on lung function in patients with overlap syndrome. Sleep Breath 2002; 6:3–10.
- Mansfield D, Naughton MT. Effects of continuous positive airway pressure on lung function in patients with chronic obstructive pulmonary disease and sleep disordered breathing. Respirology 1999; 4:365–370.
- McEvoy RD, Pierce RJ, Hillman D, et al. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet 1981; 1:681–686.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Respir J 2010; 35:132–137.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Clini E, Sturani C, Rossi A, et al. The Italian multicentre study on noninvasive ventilation in chronic obstructive pulmonary disease patients. Eur Respir J 2002; 20:529–538.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Gay PC, Hubmayr RD, Stroetz RW. Efficacy of nocturnal nasal ventilation in stable, severe chronic obstructive pulmonary disease during a 3-month controlled trial. Mayo Clin Proc 1996; 71:533–542.
- Meecham Jones DJ, Paul EA, Jones PW, Wedzicha JA. Nasal pressure support ventilation plus oxygen compared with oxygen therapy alone in hypercapnic COPD. Am J Respir Crit Care Med 1995; 152:538–544.
- Krachman SL, Chatila W, Martin UJ, et al. Effects of lung volume reduction surgery on sleep quality and nocturnal gas exchange in patients with severe emphysema. Chest 2005; 128:3221–3228.
- Stanchina ML, Welicky LM, Donat W, Lee D, Corrao W, Malhotra A. Impact of CPAP use and age on mortality in patients with combined COPD and obstructive sleep apnea: the overlap syndrome. J Clin Sleep Med 2013; 9:767–772.
- Jaoude P, Kufel T, El-Solh AA. Survival benefit of CPAP favors hypercapnic patients with the overlap syndrome. Lung 2014; 192:251–258.
- Ramagopal M, Mehta A, Roberts DW, et al. Asthma as a predictor of obstructive sleep apnea in urban African-American children. J Asthma 2009; 46:895–899.
- Ross KR, Storfer-Isser A, Hart MA, et al. Sleep-disordered breathing is associated with asthma severity in children. J Ped 2012; 160:736–742.
- Alharbi M, Almutairi A, Alotaibi D, Alotaibi A, Shaikh S, Bahammam AS. The prevalence of asthma in patients with obstructive sleep apnoea. Prim Care Respir J 2009; 18:328–330.
- Auckley D, Moallem M, Shaman Z, Mustafa M. Findings of a Berlin Questionnaire survey: comparison between patients seen in an asthma clinic versus internal medicine clinic. Sleep Med 2008; 9:494–499.
- Teodorescu M, Barnet JH, Hagen EW, Palta M, Young TB, Peppard PE. Association between asthma and risk of developing obstructive sleep apnea. JAMA 2015; 313:156–164.
- Teodorescu M, Polomis DA, Hall SV, et al. Association of obstructive sleep apnea risk with asthma control in adults. Chest 2010; 138:543–550.
- Larsson LG, Lindberg A, Franklin KA, Lundback B. Gender differences in symptoms related to sleep apnea in a general population and in relation to referral to sleep clinic. Chest 2003; 124:204–211.
- ten Brinke A, Sterk PJ, Masclee AA, et al. Risk factors of frequent exacerbations in difficult-to-treat asthma. Eur Respir J 2005; 26:812–818.
- Luyster FS, Teodorescu M, Bleecker E, et al. Sleep quality and asthma control and quality of life in non-severe and severe asthma. Sleep Breath 2012; 16:1129–1137.
- Catterall JR, Douglas NJ, Calverley PM, et al. Irregular breathing and hypoxaemia during sleep in chronic stable asthma. Lancet 1982; 1:301–304.
- Perez GF, Gutierrez MJ, Huseni S, et al. Oximetry signal processing identifies REM sleep-related vulnerability trait in asthmatic children. Sleep Disord 2013; 2013:406157.
- Yigla M, Tov N, Solomonov A, Rubin AH, Harlev D. Difficult-to-control asthma and obstructive sleep apnea. J Asthma 2003; 40:865–871.
- Kelly EA, Houtman JJ, Jarjour NN. Inflammatory changes associated with circadian variation in pulmonary function in subjects with mild asthma. Clin Exper Allergy 2004; 34:227–233.
- Bohadana AB, Hannhart B, Teculescu DB. Nocturnal worsening of asthma and sleep-disordered breathing. J Asthma 2002; 39:85–100.
- Lafond C, Series F, Lemiere C. Impact of CPAP on asthmatic patients with obstructive sleep apnoea. Eur Respir J 2007; 29:307–311.
- Martin RJ, Pak J. Nasal CPAP in nonapneic nocturnal asthma. Chest 1991; 100:1024–1027.
- Dixon AE, Pratley RE, Forgione PM, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol 2011; 128:508–515 e501–502.
Many patients who have obstructive lung disease, ie, chronic obstructive pulmonary disease (COPD) or asthma, also have obstructive sleep apnea (OSA), and vice versa.
The combination of COPD and OSA was first described almost 30 years ago by Flenley, who called it “overlap syndrome.”1 At that time, he recommended that a sleep study be considered in all obese patients with COPD who snore and in those who have frequent headaches after starting oxygen therapy. In the latter group, he doubted that nocturnal oxygen was the correct treatment. He also believed that the outcomes in patients with overlap syndrome were worse than those in patients with COPD or OSA alone. These opinions remain largely valid today.
We now also recognize the combination of asthma and OSA (alternative overlap syndrome) and collectively call both combinations obstructive lung disease-obstructive sleep apnea (OLDOSA) syndrome.2 Interestingly, these relationships are likely bidirectional, with one condition aggravating or predisposing to the other.
Knowing that a patient has one of these overlap syndromes, one can initiate continuous positive airway pressure (CPAP) therapy, which can improve clinical outcomes.3–6 Therefore, when evaluating a patient with asthma or COPD, one should consider OSA using a validated questionnaire and, if the findings suggest the diagnosis, polysomnography. Conversely, it is prudent to look for comorbid obstructive lung disease in patients with OSA, as interactions between upper and lower airway dysfunction may lead to distinctly different treatment and outcomes.
Here, we briefly review asthma and COPD, explore shared risk factors for sleep-disordered breathing and obstructive lung diseases, describe potential pathophysiologic mechanisms explaining these associations, and highlight the importance of recognizing and individually treating the overlaps of OSA and COPD or asthma.
COPD AND ASTHMA ARE VERY COMMON
About 10% of the US population have COPD,7 a preventable and treatable disease mainly caused by smoking, and a leading cause of sickness and death worldwide.8,9
About 8% of Americans have asthma,7 which has become one of the most common chronic conditions in the Western world, affecting about 1 in 7 children and about 1 in 12 adults. The World Health Organization estimates that 235 million people suffer from asthma worldwide, and by 2025 this number is projected to rise to 400 million.10,11
The prevalence of these conditions in a particular population depends on the frequency of risk factors and associated morbidities, including OSA. These factors may allow asthma or COPD to arise earlier or have more severe manifestations.8,12
Asthma and COPD: Similarities and differences
Asthma and COPD share several features. Both are inflammatory airway conditions triggered or perpetuated by allergens, viral infection, tobacco smoke, products of biomass or fossil fuel combustion, and other substances. In both diseases, airflow is “obstructed” or limited, with a low ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC). Symptoms can also be similar, with dyspnea, cough, wheezing, and chest tightness being the most frequent complaints. The similarities support the theory proposed by Orie et al13 (the “Dutch hypothesis”) that asthma and COPD may actually be manifestations of the same disease.
But there are also differences. COPD is strongly linked to cigarette smoking and has at least three phenotypes:
- Chronic bronchitis, defined clinically by cough and sputum production for more than 3 months per year for 2 consecutive years
- Emphysema, characterized anatomically by loss of lung parenchyma, as seen on tomographic imaging or examination of pathologic specimens
- A mixed form with bronchitic and emphysematous features, which is likely the most common.
Particularly in emphysematous COPD, smoking predisposes patients to gas-exchange abnormalities and low diffusing capacity for carbon monoxide.
In asthma, symptoms may be more episodic, the age of onset is often younger, and atopy is common, especially in allergic asthma. These episodic symptoms may correlate temporally with measurable airflow reversibility (≥ 12% and ≥ 200 mL improvement in FVC or in FEV1 after bronchodilator challenge).
However, the current taxonomy does not unequivocally divide obstructive lung diseases into asthma and COPD, and major features such as airway hyperresponsiveness, airflow reversibility, neutrophilic or CD8 lymphocytic airway inflammation, and lower concentration of nitric oxide in the exhaled air may be present in different phenotypes of both conditions (Table 1).
AIRFLOW IN OBSTRUCTIVE LUNG DISEASES AND DURING SLEEP
Normal airflow involves a complex interplay between airway resistance and elastic recoil of the entire respiratory system, including the airways, the lung parenchyma, and the chest wall (Figure 1).
In asthma and COPD, resistance to airflow is increased, predominantly in the upper airways (nasal passages, pharynx, and larynx) and in the first three or four subdivisions of the tracheobronchial tree. The problem is worse during exhalation, when elastic recoil of the lung parenchyma and chest wall also increases airway resistance, reduces airway caliber, and possibly even constricts the bronchi. This last effect may occur either due to mass loading of the bronchial smooth muscles or to large intrathoracic transmural pressure shifts that may increase extravasation of fluid in the bronchial walls, especially with higher vascular permeability in inflammatory conditions.
Furthermore, interactions between the airway and parenchyma and between the upper and lower airways, as well as radial and axial coupling of these anatomic and functional components, contribute to complex interplay between airway resistance and parenchymal-chest wall elastic energy—stretch or recoil.
The muscles of the upper and lower airway may not work together due to the loss of normal lung parenchyma (as in emphysema) or to the acute inflammation in the small airways and adjacent parenchyma (as in severe asthma exacerbations). This loss of coordination makes the upper airway more collapsible, a feature of OSA.
Additionally, obesity, gastroesophageal reflux, disease chronic rhinitis, nasal polyposis, and acute exacerbations of chronic systemic inflammation all contribute to more complex interactions between obstructive lung diseases and OSA.6
Sleep affects breathing, particularly in patients with respiratory comorbidities, and sleep-disordered breathing causes daytime symptoms and worsens quality of life.1,13–15 During sleep, respiratory centers become less sensitive to oxygen and carbon dioxide; breathing becomes more irregular, especially during rapid eye movement (REM) sleep; the chest wall moves less, so that the tidal volume and functional residual capacity are lower; sighs, yawns, and deep breaths become limited; and serum carbon dioxide concentration may rise.
OBSTRUCTIVE SLEEP APNEA
The prevalence of OSA, a form of sleep-disordered breathing characterized by limitation of inspiratory and (to a lesser degree) expiratory flow, has increased significantly in recent years, in parallel with the prevalence of its major risk factor, obesity.
OSA is generally defined as an apnea-hypopnea index of 5 or higher, ie, five or more episodes of apnea or hypopnea per hour.
OSA syndrome, ie, an apnea-hypopnea index of 5 or higher and excessive daytime sleepiness (defined by an Epworth Sleepiness Scale score > 10) was found in the initial analysis of the Wisconsin Sleep cohort in 1993 to be present in about 2% of women and 4% of men.16 A more recent longitudinal analysis showed a significant increase—for example, in people 50 to 70 years old the prevalence was up to 17.6% in men and 7.5% in women.17
Upper airway resistance syndrome, a milder form of sleep-disordered breathing, is now included under the diagnosis of OSA, as its pathophysiology is not significantly different.18
In the next section, we discuss what happens when OSA overlaps with COPD (overlap syndrome) and with asthma (“alternative overlap syndrome”)2,8 (Figure 2).
OSA AND COPD (OVERLAP SYNDROME)
Flenley1 hypothesized that patients with COPD in whom supplemental oxygen worsened hypercapnia may also have OSA and called this association overlap syndrome.
How common is overlap syndrome?
Since both COPD and OSA are prevalent conditions, overlap syndrome may also be common.
The reported prevalence of overlap syndrome varies widely, depending on the population studied and the methods used. In various studies, COPD was present in 9% to 56% of patients with OSA,19–23 and OSA was found in 5% to 85% of patients with COPD.24–27
Based on the prevalence of COPD in the general population (about 10%12) and that of sleep-disordered breathing (about 5% to 10%17), the expected prevalence of overlap syndrome in people over age 40 may be 0.5% to 1%.28 In a more inclusive estimate with “subclinical” forms of overlap syndrome—ie, OSA defined as an apnea-hypopnea index of 5 or more (about 25% of the population17) and COPD Global initiative for Chronic Obstructive Lung Disease (GOLD) stage 1 (16.8% in the National Health and Nutrition Education Survey12)—the expected prevalence of overlap is around 4%. Some studies found a higher prevalence of COPD in OSA patients than in the general population,21,29 while others did not.22,28,30 The studies differed in how they defined sleep-disordered breathing.
Larger studies are needed to better assess the true prevalence of sleep-disordered breathing in COPD. They should use more sensitive measures of airflow and standardized definitions of sleep-disordered breathing and should include patients with more severe COPD.
Fatigue and insomnia are common in COPD
Fatigue is strongly correlated with declining lung function, low exercise tolerance, and impaired quality of life in COPD.31 Factors that contribute to fatigue include dyspnea, depression, and impaired sleep.32 Some suggest that at least half of COPD patients have sleep complaints such as insomnia, sleep disruption, or sleep fragmentation.33 Insomnia, difficulty falling asleep, and early morning awakenings are the most common complaints (30%–70% of patients) and are associated with daytime fatigue.34 Conversely, comorbid OSA can contribute to fatigue and maintenance-type insomnia (ie, difficulty staying asleep and returning to sleep).
Multiple mechanisms of hypoxemia in overlap syndrome
Oxygenation abnormalities and increased work of breathing contribute to the pathophysiology of overlap syndrome. In patients with COPD, oxygenation during wakefulness is a strong predictor of gas exchange during sleep.35 Further, patients with overlap syndrome tend to have more severe hypoxia during sleep than patients with isolated COPD or OSA at rest or during exercise.36
In overlap syndrome, hypoxemia is the result of several mechanisms:
- Loss of upper airway muscle tone from intermittent episodes of obstructive apnea and hypopnea leads to upper airway collapse during sleep, particularly during REM sleep, increasing the severity of OSA.37
- Reductions in functional residual capacity from lying in the recumbent position and during REM sleep render patients with COPD more vulnerable, as compensatory use of accessory muscles to maintain near-normal ventilation in a hyperinflated state becomes impaired.37
- Alterations in pulmonary ventilation-perfusion matching may lead to altered carbon dioxide homeostasis and impaired oxygenation in patients with emphysema.
- Circadian variation in lower airway caliber may also be observed, in parallel with the bronchoconstriction caused by increased nocturnal vagotonia.
- Hypercapnia (Paco2 ≥ 45 mm Hg) may lead to overall reduced responsiveness of respiratory muscles and to a blunted response of respiratory centers to low oxygen and high carbon dioxide levels.38 Thus, hypercapnia is a better predictor of the severity of nocturnal hypoxemia than hypoxemia developing during exercise.39
In a person who is at near-maximal ventilatory capacity, even a mild increase in upper airway resistance (as seen with snoring, upper airway resistance syndrome, or OSA) increases the work of breathing. This phenomenon can lead to early arousals even before significant oxyhemoglobin desaturation occurs.
Normally, inspiratory flow limitation is counteracted by increasing inspiratory time to maintain ventilation. Patients with COPD may not be able to do this, however, as they need more time to breathe out due to narrowing of their lower airways.40 The inability to compensate for upper airway resistance, similar to the increased work of breathing seen with exercise, may lead to early arousals and increased sleep fragmentation.
Consequences of overlap syndrome
Patients with overlap syndrome appear to have higher morbidity and mortality rates than those with COPD or sleep-disordered breathing alone.
Cor pulmonale. Nighttime hypoxia is more severe and persistent in overlap syndrome than with COPD or OSA alone. This may contribute to more significant pulmonary hypertension and to the development of cor pulmonale, in which the right ventricle is altered in structure (eg, hypertrophied, dilated) or reduced in function, or both, from severe pulmonary hypertension.
In contrast to right ventricular failure due to disorders of the left heart, cor pulmonale is a result of diseases of the vasculature (eg, idiopathic pulmonary arterial hypertension), lung parenchyma (eg, COPD), upper airway (eg, OSA), or chest wall (eg, severe kyphoscoliosis). COPD is the most common cause of cor pulmonale in the United States, accounting for up to 30% of cases of cor pulmonale.41–45 In OSA, cor pulmonale is seen in up to 20% of cases,43 while in overlap syndrome cor pulmonale is encountered even more often (ie, in up to 80%); these patients have a dismal 5-year survival rate of about 30%.46
Obesity hypoventilation syndrome is characterized by obesity (body mass index ≥ 30 kg/m2) and daytime hypercapnia (Paco2 ≥ 45 mm Hg) that cannot be fully attributed to an underlying cardiopulmonary or neurologic condition.18 Hypercapnia worsens during sleep (especially during REM sleep) and is often associated with severe arterial oxygen desaturation. Up to 90% of patients with obesity hypoventilation syndrome have comorbid OSA, and the rest generally have sleep-related hypoventilation, particularly during REM sleep.
In patients with obesity hypoventilation syndrome, daytime hypercapnia may improve or even normalize with adequate positive airway pressure treatment and sustained adherence to treatment.18 Many patients with obesity hypoventilation syndrome respond to CPAP or bilevel positive airway pressure (BPAP), with improvement in daytime Paco2. However, normalization of daytime Paco2 occurs only in a subgroup of patients. In contrast, treatment with oxygen therapy alone may worsen hypercapnia.
Oxygen therapy for pure COPD, but maybe not for overlap syndrome
Continuous oxygen therapy reduces mortality in COPD,47,48 but the duration and severity of hypoxemia that warrant oxygen therapy are less clear. Oxygen therapy in hypoxemic patients has been shown to improve sleep quality and reduce arousals.49
Indications for oxygen treatment of nocturnal hypoxemia are generally based on Medicare guidelines:
- At least 5 minutes of sleep with peripheral oxygen saturation ≤ 88% or Pao2 ≤ 55 mm Hg, or
- A decrease in Pao2 of more than 10 mm Hg or in peripheral oxygen saturation of more than 5% for at least 5 minutes of sleep and associated with signs or symptoms reasonably attributable to hypoxemia (group I criteria), or
- At least 5 minutes of sleep with peripheral oxygen saturation ≥ 89% or Pao2 56 to 59 mm Hg and pedal edema, pulmonary hypertension, cor pulmonale, or erythrocytosis (group II criteria).50
Approximately 47% of COPD patients who are hypoxemic during the day spend about 30% of sleep time with an oxygen saturation less than 90%, even while on continuous oxygen therapy.51 Current recommendations for nocturnal oxygen therapy are to increase the oxygen concentration by 1 L/minute above the baseline oxygen flow rate needed to maintain an oxygen saturation higher than 90% during resting wakefulness, using a nasal cannula or face mask.52
Caveat. In overlap syndrome, supplemental oxygen may prolong the duration of apnea episodes and worsen hypercapnia.
Positive airway pressure for OSA
Positive airway pressure therapy improves cardiovascular outcomes in OSA.53 Several studies54–58 compared the effectiveness of CPAP vs BPAP as initial therapy for OSA but did not provide enough evidence to favor one over the other in this setting. Similarly, the results are mixed for the use of fixed or auto-adjusting BPAP as salvage therapy in patients who cannot tolerate CPAP.59–61
In overlap syndrome, CPAP or BPAP with or without supplemental oxygen has been investigated in several studies.26,62–65 In general, the mortality rate of COPD patients who require oxygen therapy is quite high.47,66 In hypoxemic COPD patients with moderate to severe sleep-disordered breathing, the 5-year survival rate was 71% in those treated with CPAP plus oxygen, vs 26% in those on oxygen alone, independent of baseline postbronchodilator FEV1.67
There is no specific FEV1 cutoff for prescribing CPAP. In general, daytime hypercapnia and nocturnal hypoxemia despite supplemental oxygen therapy are indications for BPAP therapy, regardless of the presence of OSA. Whether noninvasive nocturnal ventilation for COPD patients who do not have OSA improves long-term COPD outcomes is not entirely clear.65,68,69
Adding nocturnal BPAP in spontaneous timed mode to pulmonary rehabilitation for severe hypercapnic COPD was found to improve quality of life, mood, dyspnea, gas exchange, and decline in lung function.70 Other studies noted that COPD patients hospitalized with respiratory failure who were randomized to noninvasive nocturnal ventilation plus oxygen therapy as opposed to oxygen alone experienced improvement in health-related quality of life and reduction in intensive-care-unit length of stay but no difference in mortality or subsequent hospitalizations.69 In stable hypercapnic COPD patients without OSA, there is no clear evidence that nocturnal noninvasive ventilation lessens the risk of death despite improved daytime gas exchange,71,72 but additional long-term studies are needed.
Lung volume reduction surgery, a procedure indicated for highly selected patients with severe COPD, has been shown to reduce hyperinflation, improve nocturnal hypoxemia, and improve total sleep time and sleep efficiency in patients without sleep-disordered breathing.73 More studies are needed to determine if reduction in lung hyperinflation has an impact on the occurrence of OSA and on morbidity related to sleep-disordered breathing.
Benefit of CPAP in overlap syndrome
In a nonrandomized study, Marin et al62 found that overlap syndrome is associated with an increased risk of death and hospitalization due to COPD exacerbations. CPAP therapy was associated with improved survival rates and decreased hospitalization rates in these patients.
Stanchina et al,74 in a post hoc analysis of an observational cohort, assessed the outcomes of 227 patients with overlap syndrome. Greater use of CPAP was found to be associated with lower mortality rates.
Jaoude et al75 found that hypercapnic patients with overlap syndrome who were adherent to CPAP therapy had a lower mortality rate than nonadherent hypercapnic patients (P = .04). In a multivariate analysis, the comorbidity index was the only independent predictor of mortality in normocapnic patients with overlap syndrome, while CPAP adherence was associated with improved survival.
Lastly, patients with overlap syndrome tend to need more healthcare and accrue higher medical costs than patients with COPD alone. An analysis of a state Medicaid database that included COPD patients showed that beneficiaries with overlap syndrome spent at least $4,000 more in medical expenditures than beneficiaries with “lone” COPD.24
In conclusion, CPAP is the first line of therapy for overlap syndrome, while daytime hypercapnia or nocturnal hypoxemia despite supplemental oxygen therapy are indications for nocturnal BPAP therapy, regardless of whether patients have OSA.
OSA AND ASTHMA (ALTERNATIVE OVERLAP SYNDROME)
Epidemiology and clinical features
The coexistence of asthma and OSA can begin in childhood and continue throughout adult life. A higher prevalence of lifetime asthma and OSA has been noted in children of racial and ethnic minorities, children of lower socioeconomic status, and those with atopy.76
In a pediatric asthma clinic, it was noted that 12 months into structured asthma management and optimization, children with sleep-disordered breathing were nearly four times more likely to have severe asthma at follow-up, even after adjusting for obesity, race, and gender.77
In adult patients with OSA, the prevalence of asthma is about 35%.78 Conversely, people with asthma are at higher risk of OSA. High risk of OSA was more prevalent in a group of patients with asthma than in a general medical clinic population (39.5% vs 27.2%, P < .05).79
Analysis of a large prospective cohort found that asthma was a risk factor for new-onset OSA. The incidence of OSA over 4 years in patients with self-reported asthma was 27%, compared with 16% without asthma. The relative risk adjusted for risk factors such as body mass index, age, and gender was 1.39 (95% confidence interval [CI] 15%–19%).80
Patients with asthma who are at high risk of OSA are more likely to have worse daytime and nighttime asthma symptoms. Interestingly, patients who are diagnosed with OSA and treated with CPAP seem to have better asthma control.
Patients with asthma who are more likely to have OSA are women (odds ratio [OR] 2.1), have greater asthma severity (OR 1.6), have gastroesophageal reflux disease (OR 2.7), and use inhaled corticosteroids (OR 4.0).81 These associations are different than the traditional, population-wide risk factors for OSA, such as male sex, excess body weight, and nocturnal nasal congestion.82
OSA also worsens asthma control. Teodorescu et al15 found that severe asthma was more frequent in older asthma patients (ages 60–75, prevalence 49%) than in younger patients (ages 18–59, 39%). Older adults with OSA were seven times as likely to have severe asthma (OR 6.6), whereas young adults with sleep apnea were only three times as likely (OR 2.6).
In a group of patients with difficult-to-treat asthma, OSA was significantly associated with frequent exacerbations (OR 3.4), an association similar in magnitude to that of psychological conditions (OR 10.8), severe sinus disease (OR 3.7), recurrent respiratory tract infections (OR 6.9), and gastroesophageal reflux disease (OR 4.9).83 More than half of the patients had at least three of these comorbid conditions.
Sleep quality can greatly affect asthma control, and its importance is often underestimated. Patients with severe asthma have worse sleep quality than patients with milder asthma or nonasthmatic patients, even after excluding patients with a high risk of OSA, patients on CPAP therapy, and patients with a history of gastroesophageal reflux disease. Furthermore, regardless of asthma severity, sleep quality is a significant predictor of asthma-related quality of life, even after accounting for body mass index, daytime sleepiness, and gastroesophageal reflux disease.84
Pathophysiology of alternative overlap syndrome
Sleep significantly affects respiratory pathophysiology in asthma. The underlying mechanisms include physical and mechanical stressors, neurohormonal changes, hypoxia, confounding medical conditions, and local and systemic inflammatory changes.
Patients with nocturnal asthma experience more pronounced obstruction when sleep-deprived, suggesting that sleep loss may contribute to worsening airflow limitation.14 Although changes in pulmonary mechanics and lung volumes may also have a role, volume-dependent airway narrowing does not appear to account for all observed nocturnal increases in airway resistance. Intrathoracic blood pooling may also contribute to nocturnal bronchoconstriction through stimulation of pulmonary C fibers and increased bronchial wall edema, a mechanism that may be similar to the “cardiac asthma” seen in left ventricular dysfunction.
Early studies of sleep-disordered breathing demonstrated that patients with asthma were breathing more irregularly (with hypopnea, apnea, and hyperpnea) in REM sleep than those without asthma.85 Interestingly, REM-related hypoxia has also been noted in children with asthma.86 This may be related to the increased cholinergic outflow that occurs during REM sleep, which in turn modulates the caliber and reactivity of the lower airways.
Physical changes such as upper airway collapse and reduced pharyngeal cross-sectional area may cause further mechanical strain.87 This can further propagate airway inflammation, alter airway mucosal muscle fibers, and stimulate neural reflexes, thereby increasing cholinergic tone and bronchoconstriction. Furthermore, heightened negative intrathoracic pressure during obstructive episodes can increase nocturnal pulmonary blood pooling.14 Hypoxia itself can augment airway hyperresponsiveness via vagal pathways or carotid body receptors, increasing reactive oxygen species and inflammatory mediators. Local inflammation can “spill over” into systemic inflammatory changes, while alterations in airway inflammatory markers in asthma seem to follow a circadian rhythm, in parallel with the nocturnal worsening of the asthma symptoms.88 Finally, altered sleep may be related to other comorbid conditions, such as gastroesophageal reflux disease, insomnia, and restless leg syndrome.
Management and outcomes of alternative overlap syndrome
Despite optimization of asthma management, OSA can still significantly affect asthma control and symptoms.84
Interestingly, medications that reduce airway inflammation (eg, corticosteroids) may promote OSA. This occurrence cannot be fully explained by an increase in body mass, as more respiratory disturbances occur during sleep with continuous corticosteroid treatment even without increases in body mass index.87 Therefore, these associations may be related to upper airway myopathy caused by the treatment, a small pharynx, facial dysmorphisms, or fat deposition.89
Does CPAP improve asthma?
OSA is often unrecognized in patients with asthma, and treating it can have an impact on asthma symptoms.
CPAP therapy has not been shown to significantly change airway responsiveness or lung function, but it has been noted to significantly improve both OSA-related and asthma-related quality of life and reduce the use of rescue bronchodilators.3,90 CPAP has demonstrated improvement of quality of life that positively correlated with body weight and apnea-hypopnea index at baseline, suggesting that asthmatic patients with greater obesity or worse OSA may benefit most from aggressive management.90
However, CPAP should be used only if the patient has confirmed OSA. Empiric use of CPAP without a diagnosis of OSA was poorly tolerated and failed to improve asthma symptoms or lung function.91 More importantly, using CPAP in a patient who does not have OSA may contribute to further sleep disruption.91
Second-line treatments such as mandibular advancing devices and airway or bariatric surgery have not yet been studied in alternative overlap syndrome.
A multidimensional assessment of asthma
The Western world is experiencing an epidemic of obesity and of asthma. Obesity contributes to the pathogenesis of OSA by altering the anatomy and collapsibility of the upper airway, affecting ventilatory control and increasing respiratory workload. Another paradigm, supported by some evidence, is that OSA itself may contribute to the development of obesity. Both OSA and obesity lead to activation of inflammatory biologic cascades, which are likely the pathogenic mechanisms for their cardiovascular and metabolic consequences. As such, early recognition of OSA is important, as effective treatments are available.
In some patients, obesity may cause asthma, as obesity precedes the onset of asthma in a significant proportion of patients, and bariatric surgery for morbid obesity may resolve asthma. The obese asthma phenotype seems to include chronic rhinosinusitis, gastroesophageal reflux disease, poorer asthma control, limited responsiveness to corticosteroids, and even different sets of biomarkers (eg, neutrophilic airway inflammation). A cohort of obese patients with poor asthma control demonstrated significant improvement in asthma symptoms, quality of life, and airway reactivity after weight loss from bariatric surgery.92
To improve our knowledge about airway disease phenotypes and endotypes and their response to therapy, we propose taking a multidimensional, structured assessment of all patients with asthma, using a schema we call “ABCD-3P-PQRST” (Table 2).
The purpose of using this type of system in clinics and research is to capture the multidimensionality of the disease and better develop future individualized therapeutic strategies by employing the latest advances in systems biology and computational methods such as cluster and principal component analysis.
Multidimensional assessments addressing airway problems such as asthma, COPD, OSA, other comorbidities and risk factors, and personalized management plans will need to be the basis of future therapeutic interventions. Increased attention to the complications of asthma and obstructive airway and lung diseases in our patients is imperative, specifically to develop effective systems of care, appropriate clinical guidelines, and research studies that lead to improved health outcomes.
Many patients who have obstructive lung disease, ie, chronic obstructive pulmonary disease (COPD) or asthma, also have obstructive sleep apnea (OSA), and vice versa.
The combination of COPD and OSA was first described almost 30 years ago by Flenley, who called it “overlap syndrome.”1 At that time, he recommended that a sleep study be considered in all obese patients with COPD who snore and in those who have frequent headaches after starting oxygen therapy. In the latter group, he doubted that nocturnal oxygen was the correct treatment. He also believed that the outcomes in patients with overlap syndrome were worse than those in patients with COPD or OSA alone. These opinions remain largely valid today.
We now also recognize the combination of asthma and OSA (alternative overlap syndrome) and collectively call both combinations obstructive lung disease-obstructive sleep apnea (OLDOSA) syndrome.2 Interestingly, these relationships are likely bidirectional, with one condition aggravating or predisposing to the other.
Knowing that a patient has one of these overlap syndromes, one can initiate continuous positive airway pressure (CPAP) therapy, which can improve clinical outcomes.3–6 Therefore, when evaluating a patient with asthma or COPD, one should consider OSA using a validated questionnaire and, if the findings suggest the diagnosis, polysomnography. Conversely, it is prudent to look for comorbid obstructive lung disease in patients with OSA, as interactions between upper and lower airway dysfunction may lead to distinctly different treatment and outcomes.
Here, we briefly review asthma and COPD, explore shared risk factors for sleep-disordered breathing and obstructive lung diseases, describe potential pathophysiologic mechanisms explaining these associations, and highlight the importance of recognizing and individually treating the overlaps of OSA and COPD or asthma.
COPD AND ASTHMA ARE VERY COMMON
About 10% of the US population have COPD,7 a preventable and treatable disease mainly caused by smoking, and a leading cause of sickness and death worldwide.8,9
About 8% of Americans have asthma,7 which has become one of the most common chronic conditions in the Western world, affecting about 1 in 7 children and about 1 in 12 adults. The World Health Organization estimates that 235 million people suffer from asthma worldwide, and by 2025 this number is projected to rise to 400 million.10,11
The prevalence of these conditions in a particular population depends on the frequency of risk factors and associated morbidities, including OSA. These factors may allow asthma or COPD to arise earlier or have more severe manifestations.8,12
Asthma and COPD: Similarities and differences
Asthma and COPD share several features. Both are inflammatory airway conditions triggered or perpetuated by allergens, viral infection, tobacco smoke, products of biomass or fossil fuel combustion, and other substances. In both diseases, airflow is “obstructed” or limited, with a low ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC). Symptoms can also be similar, with dyspnea, cough, wheezing, and chest tightness being the most frequent complaints. The similarities support the theory proposed by Orie et al13 (the “Dutch hypothesis”) that asthma and COPD may actually be manifestations of the same disease.
But there are also differences. COPD is strongly linked to cigarette smoking and has at least three phenotypes:
- Chronic bronchitis, defined clinically by cough and sputum production for more than 3 months per year for 2 consecutive years
- Emphysema, characterized anatomically by loss of lung parenchyma, as seen on tomographic imaging or examination of pathologic specimens
- A mixed form with bronchitic and emphysematous features, which is likely the most common.
Particularly in emphysematous COPD, smoking predisposes patients to gas-exchange abnormalities and low diffusing capacity for carbon monoxide.
In asthma, symptoms may be more episodic, the age of onset is often younger, and atopy is common, especially in allergic asthma. These episodic symptoms may correlate temporally with measurable airflow reversibility (≥ 12% and ≥ 200 mL improvement in FVC or in FEV1 after bronchodilator challenge).
However, the current taxonomy does not unequivocally divide obstructive lung diseases into asthma and COPD, and major features such as airway hyperresponsiveness, airflow reversibility, neutrophilic or CD8 lymphocytic airway inflammation, and lower concentration of nitric oxide in the exhaled air may be present in different phenotypes of both conditions (Table 1).
AIRFLOW IN OBSTRUCTIVE LUNG DISEASES AND DURING SLEEP
Normal airflow involves a complex interplay between airway resistance and elastic recoil of the entire respiratory system, including the airways, the lung parenchyma, and the chest wall (Figure 1).
In asthma and COPD, resistance to airflow is increased, predominantly in the upper airways (nasal passages, pharynx, and larynx) and in the first three or four subdivisions of the tracheobronchial tree. The problem is worse during exhalation, when elastic recoil of the lung parenchyma and chest wall also increases airway resistance, reduces airway caliber, and possibly even constricts the bronchi. This last effect may occur either due to mass loading of the bronchial smooth muscles or to large intrathoracic transmural pressure shifts that may increase extravasation of fluid in the bronchial walls, especially with higher vascular permeability in inflammatory conditions.
Furthermore, interactions between the airway and parenchyma and between the upper and lower airways, as well as radial and axial coupling of these anatomic and functional components, contribute to complex interplay between airway resistance and parenchymal-chest wall elastic energy—stretch or recoil.
The muscles of the upper and lower airway may not work together due to the loss of normal lung parenchyma (as in emphysema) or to the acute inflammation in the small airways and adjacent parenchyma (as in severe asthma exacerbations). This loss of coordination makes the upper airway more collapsible, a feature of OSA.
Additionally, obesity, gastroesophageal reflux, disease chronic rhinitis, nasal polyposis, and acute exacerbations of chronic systemic inflammation all contribute to more complex interactions between obstructive lung diseases and OSA.6
Sleep affects breathing, particularly in patients with respiratory comorbidities, and sleep-disordered breathing causes daytime symptoms and worsens quality of life.1,13–15 During sleep, respiratory centers become less sensitive to oxygen and carbon dioxide; breathing becomes more irregular, especially during rapid eye movement (REM) sleep; the chest wall moves less, so that the tidal volume and functional residual capacity are lower; sighs, yawns, and deep breaths become limited; and serum carbon dioxide concentration may rise.
OBSTRUCTIVE SLEEP APNEA
The prevalence of OSA, a form of sleep-disordered breathing characterized by limitation of inspiratory and (to a lesser degree) expiratory flow, has increased significantly in recent years, in parallel with the prevalence of its major risk factor, obesity.
OSA is generally defined as an apnea-hypopnea index of 5 or higher, ie, five or more episodes of apnea or hypopnea per hour.
OSA syndrome, ie, an apnea-hypopnea index of 5 or higher and excessive daytime sleepiness (defined by an Epworth Sleepiness Scale score > 10) was found in the initial analysis of the Wisconsin Sleep cohort in 1993 to be present in about 2% of women and 4% of men.16 A more recent longitudinal analysis showed a significant increase—for example, in people 50 to 70 years old the prevalence was up to 17.6% in men and 7.5% in women.17
Upper airway resistance syndrome, a milder form of sleep-disordered breathing, is now included under the diagnosis of OSA, as its pathophysiology is not significantly different.18
In the next section, we discuss what happens when OSA overlaps with COPD (overlap syndrome) and with asthma (“alternative overlap syndrome”)2,8 (Figure 2).
OSA AND COPD (OVERLAP SYNDROME)
Flenley1 hypothesized that patients with COPD in whom supplemental oxygen worsened hypercapnia may also have OSA and called this association overlap syndrome.
How common is overlap syndrome?
Since both COPD and OSA are prevalent conditions, overlap syndrome may also be common.
The reported prevalence of overlap syndrome varies widely, depending on the population studied and the methods used. In various studies, COPD was present in 9% to 56% of patients with OSA,19–23 and OSA was found in 5% to 85% of patients with COPD.24–27
Based on the prevalence of COPD in the general population (about 10%12) and that of sleep-disordered breathing (about 5% to 10%17), the expected prevalence of overlap syndrome in people over age 40 may be 0.5% to 1%.28 In a more inclusive estimate with “subclinical” forms of overlap syndrome—ie, OSA defined as an apnea-hypopnea index of 5 or more (about 25% of the population17) and COPD Global initiative for Chronic Obstructive Lung Disease (GOLD) stage 1 (16.8% in the National Health and Nutrition Education Survey12)—the expected prevalence of overlap is around 4%. Some studies found a higher prevalence of COPD in OSA patients than in the general population,21,29 while others did not.22,28,30 The studies differed in how they defined sleep-disordered breathing.
Larger studies are needed to better assess the true prevalence of sleep-disordered breathing in COPD. They should use more sensitive measures of airflow and standardized definitions of sleep-disordered breathing and should include patients with more severe COPD.
Fatigue and insomnia are common in COPD
Fatigue is strongly correlated with declining lung function, low exercise tolerance, and impaired quality of life in COPD.31 Factors that contribute to fatigue include dyspnea, depression, and impaired sleep.32 Some suggest that at least half of COPD patients have sleep complaints such as insomnia, sleep disruption, or sleep fragmentation.33 Insomnia, difficulty falling asleep, and early morning awakenings are the most common complaints (30%–70% of patients) and are associated with daytime fatigue.34 Conversely, comorbid OSA can contribute to fatigue and maintenance-type insomnia (ie, difficulty staying asleep and returning to sleep).
Multiple mechanisms of hypoxemia in overlap syndrome
Oxygenation abnormalities and increased work of breathing contribute to the pathophysiology of overlap syndrome. In patients with COPD, oxygenation during wakefulness is a strong predictor of gas exchange during sleep.35 Further, patients with overlap syndrome tend to have more severe hypoxia during sleep than patients with isolated COPD or OSA at rest or during exercise.36
In overlap syndrome, hypoxemia is the result of several mechanisms:
- Loss of upper airway muscle tone from intermittent episodes of obstructive apnea and hypopnea leads to upper airway collapse during sleep, particularly during REM sleep, increasing the severity of OSA.37
- Reductions in functional residual capacity from lying in the recumbent position and during REM sleep render patients with COPD more vulnerable, as compensatory use of accessory muscles to maintain near-normal ventilation in a hyperinflated state becomes impaired.37
- Alterations in pulmonary ventilation-perfusion matching may lead to altered carbon dioxide homeostasis and impaired oxygenation in patients with emphysema.
- Circadian variation in lower airway caliber may also be observed, in parallel with the bronchoconstriction caused by increased nocturnal vagotonia.
- Hypercapnia (Paco2 ≥ 45 mm Hg) may lead to overall reduced responsiveness of respiratory muscles and to a blunted response of respiratory centers to low oxygen and high carbon dioxide levels.38 Thus, hypercapnia is a better predictor of the severity of nocturnal hypoxemia than hypoxemia developing during exercise.39
In a person who is at near-maximal ventilatory capacity, even a mild increase in upper airway resistance (as seen with snoring, upper airway resistance syndrome, or OSA) increases the work of breathing. This phenomenon can lead to early arousals even before significant oxyhemoglobin desaturation occurs.
Normally, inspiratory flow limitation is counteracted by increasing inspiratory time to maintain ventilation. Patients with COPD may not be able to do this, however, as they need more time to breathe out due to narrowing of their lower airways.40 The inability to compensate for upper airway resistance, similar to the increased work of breathing seen with exercise, may lead to early arousals and increased sleep fragmentation.
Consequences of overlap syndrome
Patients with overlap syndrome appear to have higher morbidity and mortality rates than those with COPD or sleep-disordered breathing alone.
Cor pulmonale. Nighttime hypoxia is more severe and persistent in overlap syndrome than with COPD or OSA alone. This may contribute to more significant pulmonary hypertension and to the development of cor pulmonale, in which the right ventricle is altered in structure (eg, hypertrophied, dilated) or reduced in function, or both, from severe pulmonary hypertension.
In contrast to right ventricular failure due to disorders of the left heart, cor pulmonale is a result of diseases of the vasculature (eg, idiopathic pulmonary arterial hypertension), lung parenchyma (eg, COPD), upper airway (eg, OSA), or chest wall (eg, severe kyphoscoliosis). COPD is the most common cause of cor pulmonale in the United States, accounting for up to 30% of cases of cor pulmonale.41–45 In OSA, cor pulmonale is seen in up to 20% of cases,43 while in overlap syndrome cor pulmonale is encountered even more often (ie, in up to 80%); these patients have a dismal 5-year survival rate of about 30%.46
Obesity hypoventilation syndrome is characterized by obesity (body mass index ≥ 30 kg/m2) and daytime hypercapnia (Paco2 ≥ 45 mm Hg) that cannot be fully attributed to an underlying cardiopulmonary or neurologic condition.18 Hypercapnia worsens during sleep (especially during REM sleep) and is often associated with severe arterial oxygen desaturation. Up to 90% of patients with obesity hypoventilation syndrome have comorbid OSA, and the rest generally have sleep-related hypoventilation, particularly during REM sleep.
In patients with obesity hypoventilation syndrome, daytime hypercapnia may improve or even normalize with adequate positive airway pressure treatment and sustained adherence to treatment.18 Many patients with obesity hypoventilation syndrome respond to CPAP or bilevel positive airway pressure (BPAP), with improvement in daytime Paco2. However, normalization of daytime Paco2 occurs only in a subgroup of patients. In contrast, treatment with oxygen therapy alone may worsen hypercapnia.
Oxygen therapy for pure COPD, but maybe not for overlap syndrome
Continuous oxygen therapy reduces mortality in COPD,47,48 but the duration and severity of hypoxemia that warrant oxygen therapy are less clear. Oxygen therapy in hypoxemic patients has been shown to improve sleep quality and reduce arousals.49
Indications for oxygen treatment of nocturnal hypoxemia are generally based on Medicare guidelines:
- At least 5 minutes of sleep with peripheral oxygen saturation ≤ 88% or Pao2 ≤ 55 mm Hg, or
- A decrease in Pao2 of more than 10 mm Hg or in peripheral oxygen saturation of more than 5% for at least 5 minutes of sleep and associated with signs or symptoms reasonably attributable to hypoxemia (group I criteria), or
- At least 5 minutes of sleep with peripheral oxygen saturation ≥ 89% or Pao2 56 to 59 mm Hg and pedal edema, pulmonary hypertension, cor pulmonale, or erythrocytosis (group II criteria).50
Approximately 47% of COPD patients who are hypoxemic during the day spend about 30% of sleep time with an oxygen saturation less than 90%, even while on continuous oxygen therapy.51 Current recommendations for nocturnal oxygen therapy are to increase the oxygen concentration by 1 L/minute above the baseline oxygen flow rate needed to maintain an oxygen saturation higher than 90% during resting wakefulness, using a nasal cannula or face mask.52
Caveat. In overlap syndrome, supplemental oxygen may prolong the duration of apnea episodes and worsen hypercapnia.
Positive airway pressure for OSA
Positive airway pressure therapy improves cardiovascular outcomes in OSA.53 Several studies54–58 compared the effectiveness of CPAP vs BPAP as initial therapy for OSA but did not provide enough evidence to favor one over the other in this setting. Similarly, the results are mixed for the use of fixed or auto-adjusting BPAP as salvage therapy in patients who cannot tolerate CPAP.59–61
In overlap syndrome, CPAP or BPAP with or without supplemental oxygen has been investigated in several studies.26,62–65 In general, the mortality rate of COPD patients who require oxygen therapy is quite high.47,66 In hypoxemic COPD patients with moderate to severe sleep-disordered breathing, the 5-year survival rate was 71% in those treated with CPAP plus oxygen, vs 26% in those on oxygen alone, independent of baseline postbronchodilator FEV1.67
There is no specific FEV1 cutoff for prescribing CPAP. In general, daytime hypercapnia and nocturnal hypoxemia despite supplemental oxygen therapy are indications for BPAP therapy, regardless of the presence of OSA. Whether noninvasive nocturnal ventilation for COPD patients who do not have OSA improves long-term COPD outcomes is not entirely clear.65,68,69
Adding nocturnal BPAP in spontaneous timed mode to pulmonary rehabilitation for severe hypercapnic COPD was found to improve quality of life, mood, dyspnea, gas exchange, and decline in lung function.70 Other studies noted that COPD patients hospitalized with respiratory failure who were randomized to noninvasive nocturnal ventilation plus oxygen therapy as opposed to oxygen alone experienced improvement in health-related quality of life and reduction in intensive-care-unit length of stay but no difference in mortality or subsequent hospitalizations.69 In stable hypercapnic COPD patients without OSA, there is no clear evidence that nocturnal noninvasive ventilation lessens the risk of death despite improved daytime gas exchange,71,72 but additional long-term studies are needed.
Lung volume reduction surgery, a procedure indicated for highly selected patients with severe COPD, has been shown to reduce hyperinflation, improve nocturnal hypoxemia, and improve total sleep time and sleep efficiency in patients without sleep-disordered breathing.73 More studies are needed to determine if reduction in lung hyperinflation has an impact on the occurrence of OSA and on morbidity related to sleep-disordered breathing.
Benefit of CPAP in overlap syndrome
In a nonrandomized study, Marin et al62 found that overlap syndrome is associated with an increased risk of death and hospitalization due to COPD exacerbations. CPAP therapy was associated with improved survival rates and decreased hospitalization rates in these patients.
Stanchina et al,74 in a post hoc analysis of an observational cohort, assessed the outcomes of 227 patients with overlap syndrome. Greater use of CPAP was found to be associated with lower mortality rates.
Jaoude et al75 found that hypercapnic patients with overlap syndrome who were adherent to CPAP therapy had a lower mortality rate than nonadherent hypercapnic patients (P = .04). In a multivariate analysis, the comorbidity index was the only independent predictor of mortality in normocapnic patients with overlap syndrome, while CPAP adherence was associated with improved survival.
Lastly, patients with overlap syndrome tend to need more healthcare and accrue higher medical costs than patients with COPD alone. An analysis of a state Medicaid database that included COPD patients showed that beneficiaries with overlap syndrome spent at least $4,000 more in medical expenditures than beneficiaries with “lone” COPD.24
In conclusion, CPAP is the first line of therapy for overlap syndrome, while daytime hypercapnia or nocturnal hypoxemia despite supplemental oxygen therapy are indications for nocturnal BPAP therapy, regardless of whether patients have OSA.
OSA AND ASTHMA (ALTERNATIVE OVERLAP SYNDROME)
Epidemiology and clinical features
The coexistence of asthma and OSA can begin in childhood and continue throughout adult life. A higher prevalence of lifetime asthma and OSA has been noted in children of racial and ethnic minorities, children of lower socioeconomic status, and those with atopy.76
In a pediatric asthma clinic, it was noted that 12 months into structured asthma management and optimization, children with sleep-disordered breathing were nearly four times more likely to have severe asthma at follow-up, even after adjusting for obesity, race, and gender.77
In adult patients with OSA, the prevalence of asthma is about 35%.78 Conversely, people with asthma are at higher risk of OSA. High risk of OSA was more prevalent in a group of patients with asthma than in a general medical clinic population (39.5% vs 27.2%, P < .05).79
Analysis of a large prospective cohort found that asthma was a risk factor for new-onset OSA. The incidence of OSA over 4 years in patients with self-reported asthma was 27%, compared with 16% without asthma. The relative risk adjusted for risk factors such as body mass index, age, and gender was 1.39 (95% confidence interval [CI] 15%–19%).80
Patients with asthma who are at high risk of OSA are more likely to have worse daytime and nighttime asthma symptoms. Interestingly, patients who are diagnosed with OSA and treated with CPAP seem to have better asthma control.
Patients with asthma who are more likely to have OSA are women (odds ratio [OR] 2.1), have greater asthma severity (OR 1.6), have gastroesophageal reflux disease (OR 2.7), and use inhaled corticosteroids (OR 4.0).81 These associations are different than the traditional, population-wide risk factors for OSA, such as male sex, excess body weight, and nocturnal nasal congestion.82
OSA also worsens asthma control. Teodorescu et al15 found that severe asthma was more frequent in older asthma patients (ages 60–75, prevalence 49%) than in younger patients (ages 18–59, 39%). Older adults with OSA were seven times as likely to have severe asthma (OR 6.6), whereas young adults with sleep apnea were only three times as likely (OR 2.6).
In a group of patients with difficult-to-treat asthma, OSA was significantly associated with frequent exacerbations (OR 3.4), an association similar in magnitude to that of psychological conditions (OR 10.8), severe sinus disease (OR 3.7), recurrent respiratory tract infections (OR 6.9), and gastroesophageal reflux disease (OR 4.9).83 More than half of the patients had at least three of these comorbid conditions.
Sleep quality can greatly affect asthma control, and its importance is often underestimated. Patients with severe asthma have worse sleep quality than patients with milder asthma or nonasthmatic patients, even after excluding patients with a high risk of OSA, patients on CPAP therapy, and patients with a history of gastroesophageal reflux disease. Furthermore, regardless of asthma severity, sleep quality is a significant predictor of asthma-related quality of life, even after accounting for body mass index, daytime sleepiness, and gastroesophageal reflux disease.84
Pathophysiology of alternative overlap syndrome
Sleep significantly affects respiratory pathophysiology in asthma. The underlying mechanisms include physical and mechanical stressors, neurohormonal changes, hypoxia, confounding medical conditions, and local and systemic inflammatory changes.
Patients with nocturnal asthma experience more pronounced obstruction when sleep-deprived, suggesting that sleep loss may contribute to worsening airflow limitation.14 Although changes in pulmonary mechanics and lung volumes may also have a role, volume-dependent airway narrowing does not appear to account for all observed nocturnal increases in airway resistance. Intrathoracic blood pooling may also contribute to nocturnal bronchoconstriction through stimulation of pulmonary C fibers and increased bronchial wall edema, a mechanism that may be similar to the “cardiac asthma” seen in left ventricular dysfunction.
Early studies of sleep-disordered breathing demonstrated that patients with asthma were breathing more irregularly (with hypopnea, apnea, and hyperpnea) in REM sleep than those without asthma.85 Interestingly, REM-related hypoxia has also been noted in children with asthma.86 This may be related to the increased cholinergic outflow that occurs during REM sleep, which in turn modulates the caliber and reactivity of the lower airways.
Physical changes such as upper airway collapse and reduced pharyngeal cross-sectional area may cause further mechanical strain.87 This can further propagate airway inflammation, alter airway mucosal muscle fibers, and stimulate neural reflexes, thereby increasing cholinergic tone and bronchoconstriction. Furthermore, heightened negative intrathoracic pressure during obstructive episodes can increase nocturnal pulmonary blood pooling.14 Hypoxia itself can augment airway hyperresponsiveness via vagal pathways or carotid body receptors, increasing reactive oxygen species and inflammatory mediators. Local inflammation can “spill over” into systemic inflammatory changes, while alterations in airway inflammatory markers in asthma seem to follow a circadian rhythm, in parallel with the nocturnal worsening of the asthma symptoms.88 Finally, altered sleep may be related to other comorbid conditions, such as gastroesophageal reflux disease, insomnia, and restless leg syndrome.
Management and outcomes of alternative overlap syndrome
Despite optimization of asthma management, OSA can still significantly affect asthma control and symptoms.84
Interestingly, medications that reduce airway inflammation (eg, corticosteroids) may promote OSA. This occurrence cannot be fully explained by an increase in body mass, as more respiratory disturbances occur during sleep with continuous corticosteroid treatment even without increases in body mass index.87 Therefore, these associations may be related to upper airway myopathy caused by the treatment, a small pharynx, facial dysmorphisms, or fat deposition.89
Does CPAP improve asthma?
OSA is often unrecognized in patients with asthma, and treating it can have an impact on asthma symptoms.
CPAP therapy has not been shown to significantly change airway responsiveness or lung function, but it has been noted to significantly improve both OSA-related and asthma-related quality of life and reduce the use of rescue bronchodilators.3,90 CPAP has demonstrated improvement of quality of life that positively correlated with body weight and apnea-hypopnea index at baseline, suggesting that asthmatic patients with greater obesity or worse OSA may benefit most from aggressive management.90
However, CPAP should be used only if the patient has confirmed OSA. Empiric use of CPAP without a diagnosis of OSA was poorly tolerated and failed to improve asthma symptoms or lung function.91 More importantly, using CPAP in a patient who does not have OSA may contribute to further sleep disruption.91
Second-line treatments such as mandibular advancing devices and airway or bariatric surgery have not yet been studied in alternative overlap syndrome.
A multidimensional assessment of asthma
The Western world is experiencing an epidemic of obesity and of asthma. Obesity contributes to the pathogenesis of OSA by altering the anatomy and collapsibility of the upper airway, affecting ventilatory control and increasing respiratory workload. Another paradigm, supported by some evidence, is that OSA itself may contribute to the development of obesity. Both OSA and obesity lead to activation of inflammatory biologic cascades, which are likely the pathogenic mechanisms for their cardiovascular and metabolic consequences. As such, early recognition of OSA is important, as effective treatments are available.
In some patients, obesity may cause asthma, as obesity precedes the onset of asthma in a significant proportion of patients, and bariatric surgery for morbid obesity may resolve asthma. The obese asthma phenotype seems to include chronic rhinosinusitis, gastroesophageal reflux disease, poorer asthma control, limited responsiveness to corticosteroids, and even different sets of biomarkers (eg, neutrophilic airway inflammation). A cohort of obese patients with poor asthma control demonstrated significant improvement in asthma symptoms, quality of life, and airway reactivity after weight loss from bariatric surgery.92
To improve our knowledge about airway disease phenotypes and endotypes and their response to therapy, we propose taking a multidimensional, structured assessment of all patients with asthma, using a schema we call “ABCD-3P-PQRST” (Table 2).
The purpose of using this type of system in clinics and research is to capture the multidimensionality of the disease and better develop future individualized therapeutic strategies by employing the latest advances in systems biology and computational methods such as cluster and principal component analysis.
Multidimensional assessments addressing airway problems such as asthma, COPD, OSA, other comorbidities and risk factors, and personalized management plans will need to be the basis of future therapeutic interventions. Increased attention to the complications of asthma and obstructive airway and lung diseases in our patients is imperative, specifically to develop effective systems of care, appropriate clinical guidelines, and research studies that lead to improved health outcomes.
- Flenley DC. Sleep in chronic obstructive lung disease. Clin Chest Med 1985; 6:651–661.
- Ioachimescu OC, Teodorescu M. Integrating the overlap of obstructive lung disease and obstructive sleep apnoea: OLDOSA syndrome. Respirology 2013; 18:421–431.
- Ciftci TU, Ciftci B, Guven SF, Kokturk O, Turktas H. Effect of nasal continuous positive airway pressure in uncontrolled nocturnal asthmatic patients with obstructive sleep apnea syndrome. Respiratory Med 2005; 99:529–534.
- Kim MY, Jo EJ, Kang SY, et al. Obstructive sleep apnea is associated with reduced quality of life in adult patients with asthma. Ann Allergy Asthma Immunol 2013; 110:253–257.
- Teodorescu M, Polomis DA, Teodorescu MC, et al. Association of obstructive sleep apnea risk or diagnosis with daytime asthma in adults. J Asthma 2012; 49:620–628.
- Puthalapattu S, Ioachimescu OC. Asthma and obstructive sleep apnea: clinical and pathogenic interactions. J Investig Med 2014; 62:665–675.
- National Institutes of Health. National Heart, Lung, and Blood Institute. NHLBI Factbook, Fiscal Year 2007. Chapter 4. Disease Statistics. www.nhlbi.nih.gov/about/factbook-07/chapter4.htm. Accessed November 11, 2015.
- Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- WHO. World Health Organization: Asthma. Fact sheet No 307. www.who.int/mediacentre/factsheets/fs307/en/. Accessed November 11, 2015.
- Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 2004; 59:469–478.
- WHO. Chronic obstructive pulmonary disease (COPD). Fact sheet No 315. www.who.int/mediacentre/factsheets/fs315/en/index.html. 2011. Accessed November 11, 2015.
- Ford ES, Mannino DM, Wheaton AG, Giles WH, Presley-Cantrell L, Croft JB. Trends in the prevalence of obstructive and restrictive lung function among adults in the United States: findings from the National Health and Nutrition Examination surveys from 1988–1994 to 2007–2010. Chest 2013; 143:1395–1406.
- Orie N, Sluiter H, de Vries K, Tammeling G, Witkop J. The host factor in bronchitis. Paper presented at: Bronchitis—an international symposium 1961; Assen, Netherlands.
- Ballard RD. Sleep, respiratory physiology, and nocturnal asthma. Chronobiol Int 1999; 16:565–580.
- Teodorescu M, Polomis DA, Gangnon RE, et al. Asthma control and its relationship with obstructive sleep apnea (OSA) in older adults. Sleep Disord 2013; 2013:251567.
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- International Classification of Sleep Disorders, 3rd ed.: Diagnostic and coding manual. Darien, Illinois: American Academy of Sleep Medicine. 2014.
- Lopez-Acevedo MN, Torres-Palacios A, Elena Ocasio-Tascon M, Campos-Santiago Z, Rodriguez-Cintron W. Overlap syndrome: an indication for sleep studies? A pilot study. Sleep Breath 2009; 13:409–413.
- Scharf C, Li P, Muntwyler J, et al. Rate-dependent AV delay optimization in cardiac resynchronization therapy. PACE 2005; 28:279–284.
- Chaouat A, Weitzenblum E, Krieger J, Ifoundza T, Oswald M, Kessler R. Association of chronic obstructive pulmonary disease and sleep apnea syndrome. Am J Respir Crit Care Med 1995;151:82–86.
- Bednarek M, Plywaczewski R, Jonczak L, Zielinski J. There is no relationship between chronic obstructive pulmonary disease and obstructive sleep apnea syndrome: a population study. Respiration 2005; 72:142–149.
- Fletcher EC. Chronic lung disease in the sleep apnea syndrome. Lung 1990; 168(suppl):751–761.
- Shaya FT, Lin PJ, Aljawadi MH, Scharf SM. Elevated economic burden in obstructive lung disease patients with concomitant sleep apnea syndrome. Sleep Breath 2009; 13:317–323.
- Larsson LG, Lindberg A, Franklin KA, Lundbäck B; Obstructive Lung Disease in Northern Sweden Studies. Obstructive sleep apnoea syndrome is common in subjects with chronic bronchitis. Report from the Obstructive Lung Disease in Northern Sweden studies. Respiration 2001; 68:250–255.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Resp J 2010; 35:132–137.
- Guilleminault C, Cummiskey J, Motta J. Chronic obstructive airflow disease and sleep studies. Am Rev Respir Dis 1980; 122:397–406.
- Weitzenblum E, Chaouat A, Kessler R, Canuet M. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc 2008; 5:237–241.
- Bradley TD, Rutherford R, Lue F, et al. Role of diffuse airway obstruction in the hypercapnia of obstructive sleep apnea. Am Rev Respir Dis 1986; 134:920–924.
- Sanders MH, Newman AB, Haggerty CL, et al. Sleep and sleep-disordered breathing in adults with predominantly mild obstructive airway disease. Am J Respir Crit Care Med 2003; 167:7–14.
- Breslin E, van der Schans C, Breukink S, et al. Perception of fatigue and quality of life in patients with COPD. Chest 1998; 114:958–964.
- Kapella MC, Larson JL, Patel MK, Covey MK, Berry JK. Subjective fatigue, influencing variables, and consequences in chronic obstructive pulmonary disease. Nurs Res 2006; 55:10–17.
- Klink M, Quan SF. Prevalence of reported sleep disturbances in a general adult population and their relationship to obstructive airways diseases. Chest 1987; 91:540–546.
- Bellia V, Catalano F, Scichilone N, et al. Sleep disorders in the elderly with and without chronic airflow obstruction: the SARA study. Sleep 2003; 26:318–323.
- Connaughton JJ, Catterall JR, Elton RA, Stradling JR, Douglas NJ. Do sleep studies contribute to the management of patients with severe chronic obstructive pulmonary disease? Am Rev Respir Dis 1988; 138:341–344.
- Mulloy E, McNicholas WT. Ventilation and gas exchange during sleep and exercise in severe COPD. Chest 1996; 109:387–394.
- Johnson MW, Remmers JE. Accessory muscle activity during sleep in chronic obstructive pulmonary disease. J Appl Physiol 1984; 57:1011–1017.
- Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax 1982; 37:840–844.
- Mulloy E, Fitzpatrick M, Bourke S, O’Regan A, McNicholas WT. Oxygen desaturation during sleep and exercise in patients with severe chronic obstructive pulmonary disease. Respir Med 1995; 89:193–198.
- Herpel LB, Brown CD, Goring KL, et al. COPD cannot compensate for upper airway obstruction during sleep (abstract). Am J Respir Crit Care Med 2007; 175:A71.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 2. Am J Respir Crit Care Med 1994; 150:1158–1168.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 1. Am J Respir Crit Care Med 1994; 150:833–852.
- Budev MM, Arroliga AC, Wiedemann HP, Matthay RA. Cor pulmonale: an overview. Semin Respir Crit Care Med 2003; 24:233–244.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(25 suppl):D34–D41.
- Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:20–22.
- Rasche K, Orth M, Kutscha A, Duchna HW. [Pulmonary diseases and heart function]. In German. Internist (Berl) 2007; 48:276–282.
- Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Nocturnal Oxygen Therapy Trial Group. Ann Intern Med 1980; 93:391–398.
- Newman AB, Foster G, Givelber R, Nieto FJ, Redline S, Young T. Progression and regression of sleep-disordered breathing with changes in weight: the Sleep Heart Health Study. Arch Intern Med 2005; 165:2408–2413.
- Calverley PM, Brezinova V, Douglas NJ, Catterall JR, Flenley DC. The effect of oxygenation on sleep quality in chronic bronchitis and emphysema. Am Rev Respir Dis 1982; 126:206–210.
- Centers for Medicare and Medicaid Services. National coverage determination (NCD) for home use of oxygen (240.2). www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=169&ncdver=1&NCAId=169&NcaName=Home+Use+of+Oxygen&IsPopup=y&bc=AAAAAAAAIAAA&. Accessed November 11, 2015.
- Plywaczewski R, Sliwinski P, Nowinski A, Kaminski D, Zielinski J. Incidence of nocturnal desaturation while breathing oxygen in COPD patients undergoing long-term oxygen therapy. Chest 2000; 117:679–683.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breathing 2007; 11:117–124.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Marin JM, DeAndres R, Alonso J, Sanchez A, Carrizo S. Long term mortality in the overlap syndrome. Eur Resp J 2008; 32(suppl 52):P865.
- Reeves-Hoche MK, Hudgel DW, Meck R, Witteman R, Ross A, Zwillich CW. Continuous versus bilevel positive airway pressure for obstructive sleep apnea. Am J Respir Crit Care Med 1995; 151:443–449.
- Gay PC, Herold DL, Olson EJ. A randomized, double-blind clinical trial comparing continuous positive airway pressure with a novel bilevel pressure system for treatment of obstructive sleep apnea syndrome. Sleep 2003; 26:864–869.
- Blau A, Minx M, Peter JG, et al. Auto bi-level pressure relief-PAP is as effective as CPAP in OSA patients—a pilot study. Sleep Breath 2012; 16:773–779.
- Randerath WJ, Galetke W, Ruhle KH. Auto-adjusting CPAP based on impedance versus bilevel pressure in difficult-to-treat sleep apnea syndrome: a prospective randomized crossover study. Med Sci Monit 2003; 9:CR353–CR358.
- Schwartz SW, Rosas J, Iannacone MR, Foulis PR, Anderson WM. Correlates of a prescription for bilevel positive airway pressure for treatment of obstructive sleep apnea among veterans. J Clin Sleep Med 2013; 9:327–335.
- Gentina T, Fortin F, Douay B, et al. Auto bi-level with pressure relief during exhalation as a rescue therapy for optimally treated obstructive sleep apnoea patients with poor compliance to continuous positive airways pressure therapy--a pilot study. Sleep Breathing 2011; 15:21–27.
- Ballard RD, Gay PC, Strollo PJ. Interventions to improve compliance in sleep apnea patients previously non-compliant with continuous positive airway pressure. J Clinical Sleep Med 2007; 3:706–712.
- Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med 2010; 182:325–331.
- de Miguel J, Cabello J, Sanchez-Alarcos JM, Alvarez-Sala R, Espinos D, Alvarez-Sala JL. Long-term effects of treatment with nasal continuous positive airway pressure on lung function in patients with overlap syndrome. Sleep Breath 2002; 6:3–10.
- Mansfield D, Naughton MT. Effects of continuous positive airway pressure on lung function in patients with chronic obstructive pulmonary disease and sleep disordered breathing. Respirology 1999; 4:365–370.
- McEvoy RD, Pierce RJ, Hillman D, et al. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet 1981; 1:681–686.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Respir J 2010; 35:132–137.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Clini E, Sturani C, Rossi A, et al. The Italian multicentre study on noninvasive ventilation in chronic obstructive pulmonary disease patients. Eur Respir J 2002; 20:529–538.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Gay PC, Hubmayr RD, Stroetz RW. Efficacy of nocturnal nasal ventilation in stable, severe chronic obstructive pulmonary disease during a 3-month controlled trial. Mayo Clin Proc 1996; 71:533–542.
- Meecham Jones DJ, Paul EA, Jones PW, Wedzicha JA. Nasal pressure support ventilation plus oxygen compared with oxygen therapy alone in hypercapnic COPD. Am J Respir Crit Care Med 1995; 152:538–544.
- Krachman SL, Chatila W, Martin UJ, et al. Effects of lung volume reduction surgery on sleep quality and nocturnal gas exchange in patients with severe emphysema. Chest 2005; 128:3221–3228.
- Stanchina ML, Welicky LM, Donat W, Lee D, Corrao W, Malhotra A. Impact of CPAP use and age on mortality in patients with combined COPD and obstructive sleep apnea: the overlap syndrome. J Clin Sleep Med 2013; 9:767–772.
- Jaoude P, Kufel T, El-Solh AA. Survival benefit of CPAP favors hypercapnic patients with the overlap syndrome. Lung 2014; 192:251–258.
- Ramagopal M, Mehta A, Roberts DW, et al. Asthma as a predictor of obstructive sleep apnea in urban African-American children. J Asthma 2009; 46:895–899.
- Ross KR, Storfer-Isser A, Hart MA, et al. Sleep-disordered breathing is associated with asthma severity in children. J Ped 2012; 160:736–742.
- Alharbi M, Almutairi A, Alotaibi D, Alotaibi A, Shaikh S, Bahammam AS. The prevalence of asthma in patients with obstructive sleep apnoea. Prim Care Respir J 2009; 18:328–330.
- Auckley D, Moallem M, Shaman Z, Mustafa M. Findings of a Berlin Questionnaire survey: comparison between patients seen in an asthma clinic versus internal medicine clinic. Sleep Med 2008; 9:494–499.
- Teodorescu M, Barnet JH, Hagen EW, Palta M, Young TB, Peppard PE. Association between asthma and risk of developing obstructive sleep apnea. JAMA 2015; 313:156–164.
- Teodorescu M, Polomis DA, Hall SV, et al. Association of obstructive sleep apnea risk with asthma control in adults. Chest 2010; 138:543–550.
- Larsson LG, Lindberg A, Franklin KA, Lundback B. Gender differences in symptoms related to sleep apnea in a general population and in relation to referral to sleep clinic. Chest 2003; 124:204–211.
- ten Brinke A, Sterk PJ, Masclee AA, et al. Risk factors of frequent exacerbations in difficult-to-treat asthma. Eur Respir J 2005; 26:812–818.
- Luyster FS, Teodorescu M, Bleecker E, et al. Sleep quality and asthma control and quality of life in non-severe and severe asthma. Sleep Breath 2012; 16:1129–1137.
- Catterall JR, Douglas NJ, Calverley PM, et al. Irregular breathing and hypoxaemia during sleep in chronic stable asthma. Lancet 1982; 1:301–304.
- Perez GF, Gutierrez MJ, Huseni S, et al. Oximetry signal processing identifies REM sleep-related vulnerability trait in asthmatic children. Sleep Disord 2013; 2013:406157.
- Yigla M, Tov N, Solomonov A, Rubin AH, Harlev D. Difficult-to-control asthma and obstructive sleep apnea. J Asthma 2003; 40:865–871.
- Kelly EA, Houtman JJ, Jarjour NN. Inflammatory changes associated with circadian variation in pulmonary function in subjects with mild asthma. Clin Exper Allergy 2004; 34:227–233.
- Bohadana AB, Hannhart B, Teculescu DB. Nocturnal worsening of asthma and sleep-disordered breathing. J Asthma 2002; 39:85–100.
- Lafond C, Series F, Lemiere C. Impact of CPAP on asthmatic patients with obstructive sleep apnoea. Eur Respir J 2007; 29:307–311.
- Martin RJ, Pak J. Nasal CPAP in nonapneic nocturnal asthma. Chest 1991; 100:1024–1027.
- Dixon AE, Pratley RE, Forgione PM, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol 2011; 128:508–515 e501–502.
- Flenley DC. Sleep in chronic obstructive lung disease. Clin Chest Med 1985; 6:651–661.
- Ioachimescu OC, Teodorescu M. Integrating the overlap of obstructive lung disease and obstructive sleep apnoea: OLDOSA syndrome. Respirology 2013; 18:421–431.
- Ciftci TU, Ciftci B, Guven SF, Kokturk O, Turktas H. Effect of nasal continuous positive airway pressure in uncontrolled nocturnal asthmatic patients with obstructive sleep apnea syndrome. Respiratory Med 2005; 99:529–534.
- Kim MY, Jo EJ, Kang SY, et al. Obstructive sleep apnea is associated with reduced quality of life in adult patients with asthma. Ann Allergy Asthma Immunol 2013; 110:253–257.
- Teodorescu M, Polomis DA, Teodorescu MC, et al. Association of obstructive sleep apnea risk or diagnosis with daytime asthma in adults. J Asthma 2012; 49:620–628.
- Puthalapattu S, Ioachimescu OC. Asthma and obstructive sleep apnea: clinical and pathogenic interactions. J Investig Med 2014; 62:665–675.
- National Institutes of Health. National Heart, Lung, and Blood Institute. NHLBI Factbook, Fiscal Year 2007. Chapter 4. Disease Statistics. www.nhlbi.nih.gov/about/factbook-07/chapter4.htm. Accessed November 11, 2015.
- Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- WHO. World Health Organization: Asthma. Fact sheet No 307. www.who.int/mediacentre/factsheets/fs307/en/. Accessed November 11, 2015.
- Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 2004; 59:469–478.
- WHO. Chronic obstructive pulmonary disease (COPD). Fact sheet No 315. www.who.int/mediacentre/factsheets/fs315/en/index.html. 2011. Accessed November 11, 2015.
- Ford ES, Mannino DM, Wheaton AG, Giles WH, Presley-Cantrell L, Croft JB. Trends in the prevalence of obstructive and restrictive lung function among adults in the United States: findings from the National Health and Nutrition Examination surveys from 1988–1994 to 2007–2010. Chest 2013; 143:1395–1406.
- Orie N, Sluiter H, de Vries K, Tammeling G, Witkop J. The host factor in bronchitis. Paper presented at: Bronchitis—an international symposium 1961; Assen, Netherlands.
- Ballard RD. Sleep, respiratory physiology, and nocturnal asthma. Chronobiol Int 1999; 16:565–580.
- Teodorescu M, Polomis DA, Gangnon RE, et al. Asthma control and its relationship with obstructive sleep apnea (OSA) in older adults. Sleep Disord 2013; 2013:251567.
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- International Classification of Sleep Disorders, 3rd ed.: Diagnostic and coding manual. Darien, Illinois: American Academy of Sleep Medicine. 2014.
- Lopez-Acevedo MN, Torres-Palacios A, Elena Ocasio-Tascon M, Campos-Santiago Z, Rodriguez-Cintron W. Overlap syndrome: an indication for sleep studies? A pilot study. Sleep Breath 2009; 13:409–413.
- Scharf C, Li P, Muntwyler J, et al. Rate-dependent AV delay optimization in cardiac resynchronization therapy. PACE 2005; 28:279–284.
- Chaouat A, Weitzenblum E, Krieger J, Ifoundza T, Oswald M, Kessler R. Association of chronic obstructive pulmonary disease and sleep apnea syndrome. Am J Respir Crit Care Med 1995;151:82–86.
- Bednarek M, Plywaczewski R, Jonczak L, Zielinski J. There is no relationship between chronic obstructive pulmonary disease and obstructive sleep apnea syndrome: a population study. Respiration 2005; 72:142–149.
- Fletcher EC. Chronic lung disease in the sleep apnea syndrome. Lung 1990; 168(suppl):751–761.
- Shaya FT, Lin PJ, Aljawadi MH, Scharf SM. Elevated economic burden in obstructive lung disease patients with concomitant sleep apnea syndrome. Sleep Breath 2009; 13:317–323.
- Larsson LG, Lindberg A, Franklin KA, Lundbäck B; Obstructive Lung Disease in Northern Sweden Studies. Obstructive sleep apnoea syndrome is common in subjects with chronic bronchitis. Report from the Obstructive Lung Disease in Northern Sweden studies. Respiration 2001; 68:250–255.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Resp J 2010; 35:132–137.
- Guilleminault C, Cummiskey J, Motta J. Chronic obstructive airflow disease and sleep studies. Am Rev Respir Dis 1980; 122:397–406.
- Weitzenblum E, Chaouat A, Kessler R, Canuet M. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc 2008; 5:237–241.
- Bradley TD, Rutherford R, Lue F, et al. Role of diffuse airway obstruction in the hypercapnia of obstructive sleep apnea. Am Rev Respir Dis 1986; 134:920–924.
- Sanders MH, Newman AB, Haggerty CL, et al. Sleep and sleep-disordered breathing in adults with predominantly mild obstructive airway disease. Am J Respir Crit Care Med 2003; 167:7–14.
- Breslin E, van der Schans C, Breukink S, et al. Perception of fatigue and quality of life in patients with COPD. Chest 1998; 114:958–964.
- Kapella MC, Larson JL, Patel MK, Covey MK, Berry JK. Subjective fatigue, influencing variables, and consequences in chronic obstructive pulmonary disease. Nurs Res 2006; 55:10–17.
- Klink M, Quan SF. Prevalence of reported sleep disturbances in a general adult population and their relationship to obstructive airways diseases. Chest 1987; 91:540–546.
- Bellia V, Catalano F, Scichilone N, et al. Sleep disorders in the elderly with and without chronic airflow obstruction: the SARA study. Sleep 2003; 26:318–323.
- Connaughton JJ, Catterall JR, Elton RA, Stradling JR, Douglas NJ. Do sleep studies contribute to the management of patients with severe chronic obstructive pulmonary disease? Am Rev Respir Dis 1988; 138:341–344.
- Mulloy E, McNicholas WT. Ventilation and gas exchange during sleep and exercise in severe COPD. Chest 1996; 109:387–394.
- Johnson MW, Remmers JE. Accessory muscle activity during sleep in chronic obstructive pulmonary disease. J Appl Physiol 1984; 57:1011–1017.
- Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax 1982; 37:840–844.
- Mulloy E, Fitzpatrick M, Bourke S, O’Regan A, McNicholas WT. Oxygen desaturation during sleep and exercise in patients with severe chronic obstructive pulmonary disease. Respir Med 1995; 89:193–198.
- Herpel LB, Brown CD, Goring KL, et al. COPD cannot compensate for upper airway obstruction during sleep (abstract). Am J Respir Crit Care Med 2007; 175:A71.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 2. Am J Respir Crit Care Med 1994; 150:1158–1168.
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part 1. Am J Respir Crit Care Med 1994; 150:833–852.
- Budev MM, Arroliga AC, Wiedemann HP, Matthay RA. Cor pulmonale: an overview. Semin Respir Crit Care Med 2003; 24:233–244.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(25 suppl):D34–D41.
- Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:20–22.
- Rasche K, Orth M, Kutscha A, Duchna HW. [Pulmonary diseases and heart function]. In German. Internist (Berl) 2007; 48:276–282.
- Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Nocturnal Oxygen Therapy Trial Group. Ann Intern Med 1980; 93:391–398.
- Newman AB, Foster G, Givelber R, Nieto FJ, Redline S, Young T. Progression and regression of sleep-disordered breathing with changes in weight: the Sleep Heart Health Study. Arch Intern Med 2005; 165:2408–2413.
- Calverley PM, Brezinova V, Douglas NJ, Catterall JR, Flenley DC. The effect of oxygenation on sleep quality in chronic bronchitis and emphysema. Am Rev Respir Dis 1982; 126:206–210.
- Centers for Medicare and Medicaid Services. National coverage determination (NCD) for home use of oxygen (240.2). www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=169&ncdver=1&NCAId=169&NcaName=Home+Use+of+Oxygen&IsPopup=y&bc=AAAAAAAAIAAA&. Accessed November 11, 2015.
- Plywaczewski R, Sliwinski P, Nowinski A, Kaminski D, Zielinski J. Incidence of nocturnal desaturation while breathing oxygen in COPD patients undergoing long-term oxygen therapy. Chest 2000; 117:679–683.
- Mokhlesi B, Tulaimat A, Faibussowitsch I, Wang Y, Evans AT. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep Breathing 2007; 11:117–124.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Marin JM, DeAndres R, Alonso J, Sanchez A, Carrizo S. Long term mortality in the overlap syndrome. Eur Resp J 2008; 32(suppl 52):P865.
- Reeves-Hoche MK, Hudgel DW, Meck R, Witteman R, Ross A, Zwillich CW. Continuous versus bilevel positive airway pressure for obstructive sleep apnea. Am J Respir Crit Care Med 1995; 151:443–449.
- Gay PC, Herold DL, Olson EJ. A randomized, double-blind clinical trial comparing continuous positive airway pressure with a novel bilevel pressure system for treatment of obstructive sleep apnea syndrome. Sleep 2003; 26:864–869.
- Blau A, Minx M, Peter JG, et al. Auto bi-level pressure relief-PAP is as effective as CPAP in OSA patients—a pilot study. Sleep Breath 2012; 16:773–779.
- Randerath WJ, Galetke W, Ruhle KH. Auto-adjusting CPAP based on impedance versus bilevel pressure in difficult-to-treat sleep apnea syndrome: a prospective randomized crossover study. Med Sci Monit 2003; 9:CR353–CR358.
- Schwartz SW, Rosas J, Iannacone MR, Foulis PR, Anderson WM. Correlates of a prescription for bilevel positive airway pressure for treatment of obstructive sleep apnea among veterans. J Clin Sleep Med 2013; 9:327–335.
- Gentina T, Fortin F, Douay B, et al. Auto bi-level with pressure relief during exhalation as a rescue therapy for optimally treated obstructive sleep apnoea patients with poor compliance to continuous positive airways pressure therapy--a pilot study. Sleep Breathing 2011; 15:21–27.
- Ballard RD, Gay PC, Strollo PJ. Interventions to improve compliance in sleep apnea patients previously non-compliant with continuous positive airway pressure. J Clinical Sleep Med 2007; 3:706–712.
- Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med 2010; 182:325–331.
- de Miguel J, Cabello J, Sanchez-Alarcos JM, Alvarez-Sala R, Espinos D, Alvarez-Sala JL. Long-term effects of treatment with nasal continuous positive airway pressure on lung function in patients with overlap syndrome. Sleep Breath 2002; 6:3–10.
- Mansfield D, Naughton MT. Effects of continuous positive airway pressure on lung function in patients with chronic obstructive pulmonary disease and sleep disordered breathing. Respirology 1999; 4:365–370.
- McEvoy RD, Pierce RJ, Hillman D, et al. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet 1981; 1:681–686.
- Machado MC, Vollmer WM, Togeiro SM, et al. CPAP and survival in moderate-to-severe obstructive sleep apnoea syndrome and hypoxaemic COPD. Eur Respir J 2010; 35:132–137.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Clini E, Sturani C, Rossi A, et al. The Italian multicentre study on noninvasive ventilation in chronic obstructive pulmonary disease patients. Eur Respir J 2002; 20:529–538.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Gay PC, Hubmayr RD, Stroetz RW. Efficacy of nocturnal nasal ventilation in stable, severe chronic obstructive pulmonary disease during a 3-month controlled trial. Mayo Clin Proc 1996; 71:533–542.
- Meecham Jones DJ, Paul EA, Jones PW, Wedzicha JA. Nasal pressure support ventilation plus oxygen compared with oxygen therapy alone in hypercapnic COPD. Am J Respir Crit Care Med 1995; 152:538–544.
- Krachman SL, Chatila W, Martin UJ, et al. Effects of lung volume reduction surgery on sleep quality and nocturnal gas exchange in patients with severe emphysema. Chest 2005; 128:3221–3228.
- Stanchina ML, Welicky LM, Donat W, Lee D, Corrao W, Malhotra A. Impact of CPAP use and age on mortality in patients with combined COPD and obstructive sleep apnea: the overlap syndrome. J Clin Sleep Med 2013; 9:767–772.
- Jaoude P, Kufel T, El-Solh AA. Survival benefit of CPAP favors hypercapnic patients with the overlap syndrome. Lung 2014; 192:251–258.
- Ramagopal M, Mehta A, Roberts DW, et al. Asthma as a predictor of obstructive sleep apnea in urban African-American children. J Asthma 2009; 46:895–899.
- Ross KR, Storfer-Isser A, Hart MA, et al. Sleep-disordered breathing is associated with asthma severity in children. J Ped 2012; 160:736–742.
- Alharbi M, Almutairi A, Alotaibi D, Alotaibi A, Shaikh S, Bahammam AS. The prevalence of asthma in patients with obstructive sleep apnoea. Prim Care Respir J 2009; 18:328–330.
- Auckley D, Moallem M, Shaman Z, Mustafa M. Findings of a Berlin Questionnaire survey: comparison between patients seen in an asthma clinic versus internal medicine clinic. Sleep Med 2008; 9:494–499.
- Teodorescu M, Barnet JH, Hagen EW, Palta M, Young TB, Peppard PE. Association between asthma and risk of developing obstructive sleep apnea. JAMA 2015; 313:156–164.
- Teodorescu M, Polomis DA, Hall SV, et al. Association of obstructive sleep apnea risk with asthma control in adults. Chest 2010; 138:543–550.
- Larsson LG, Lindberg A, Franklin KA, Lundback B. Gender differences in symptoms related to sleep apnea in a general population and in relation to referral to sleep clinic. Chest 2003; 124:204–211.
- ten Brinke A, Sterk PJ, Masclee AA, et al. Risk factors of frequent exacerbations in difficult-to-treat asthma. Eur Respir J 2005; 26:812–818.
- Luyster FS, Teodorescu M, Bleecker E, et al. Sleep quality and asthma control and quality of life in non-severe and severe asthma. Sleep Breath 2012; 16:1129–1137.
- Catterall JR, Douglas NJ, Calverley PM, et al. Irregular breathing and hypoxaemia during sleep in chronic stable asthma. Lancet 1982; 1:301–304.
- Perez GF, Gutierrez MJ, Huseni S, et al. Oximetry signal processing identifies REM sleep-related vulnerability trait in asthmatic children. Sleep Disord 2013; 2013:406157.
- Yigla M, Tov N, Solomonov A, Rubin AH, Harlev D. Difficult-to-control asthma and obstructive sleep apnea. J Asthma 2003; 40:865–871.
- Kelly EA, Houtman JJ, Jarjour NN. Inflammatory changes associated with circadian variation in pulmonary function in subjects with mild asthma. Clin Exper Allergy 2004; 34:227–233.
- Bohadana AB, Hannhart B, Teculescu DB. Nocturnal worsening of asthma and sleep-disordered breathing. J Asthma 2002; 39:85–100.
- Lafond C, Series F, Lemiere C. Impact of CPAP on asthmatic patients with obstructive sleep apnoea. Eur Respir J 2007; 29:307–311.
- Martin RJ, Pak J. Nasal CPAP in nonapneic nocturnal asthma. Chest 1991; 100:1024–1027.
- Dixon AE, Pratley RE, Forgione PM, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol 2011; 128:508–515 e501–502.
KEY POINTS
- Obstructive lung diseases and OSA are both common and may exacerbate each other.
- When assessing a patient with COPD, it may be prudent to think about whether the patient also has OSA, and vice versa.
- Oxygen therapy lowers the risk of death in patients with COPD but may worsen hypercapnia and apneic episodes in those with OSA.
- Continuous positive airway pressure is the first line of therapy for overlap syndrome. Daytime hypercapnia and nocturnal hypoxemia despite supplemental oxygen therapy are indications for nocturnal bilevel positive airway pressure therapy, regardless of the presence of OSA.
Rash, Reaction, or Red Flag?
1. The patient had just recovered from a sore throat and noticed discrete red nodules, which eventually coalesced into a single large edematous plaque over the right anterior tibia. The deep intradermal and subdermal edema is exquisitely tender to touch, considerably warmer than the surrounding skin, and highly blanchable.
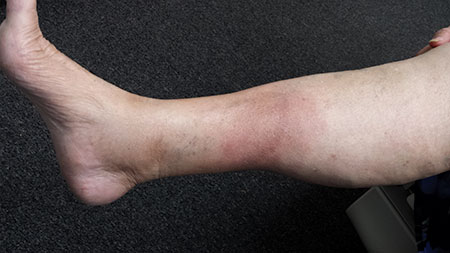
Diagnosis: Erythema nodosum is a reactive form of septal panniculitis with many potential triggers. Notable triggers include Crohn disease flares and use of drugs such as sulfa, gold salts, and oral contraceptives. Several infections have been identified as triggers, including strep, mycoplasma, and campylobacter, as well as deep fungal infections (histoplasmosis, blastomycosis, coccidioidomycosis, and sporotrichosis). More unusual causes include pregnancy and diseases such as sarcoidosis, tuberculosis, Behçet disease, and leukemia/lymphoma.
For more information, see “Painful Lesion Hasn’t Responded to Antibiotics.” Clin Rev. 2015;25(11):10,12.
For the next photograph, proceed to the next page >>
2. A 16-year-old high school student joins her friends in a 2K run one morning. The next day, her shins are so painful she can hardly walk. She applied ice packs to her legs, using elastic bandages to hold them in place until the ice cubes melt. As her legs rewarm, a rash appears where the ice packs contacted the skin.
Diagnosis: This condition is urticarial in nature—albeit an unusual form, triggered by cold. Though it appears counterintuitive, cold uriticaria typically appears only on rewarming of the affected area and is marked by the sudden appearance of “welts” or “hives” that usually clear (with or without treatment) within hours.
Uncomplicated urticaria resolves without leaving any signs (eg, purpura, ecchymosis) that might otherwise suggest the presence of a vasculitic component, such as that seen with lupus or other autoimmune diseases. Blanchability on digital pressure is one way to confirm benignancy, since blood tends to leak from vessels damaged by vasculitis, emptying into the surrounding interstitial spaces and presenting as nonblanchable petechiae, purpura, or ecchymosis. The relatively benign nature of this patient’s urticaria was also suggested by additional history taking, in which she denied having fever, malaise, or arthralgia. These are all symptoms we might have seen with more serious underlying causes.
Cold urticaria is one of the so-called physical urticarias, a group that includes urticaria caused by vibration, pressure, heat, sun, and even exposure to water. Thought to comprise up to 20% of all urticarias, the physical urticarias occur most frequently in persons ages 17 to 40. Dermatographism is the most common form, occurring in the linear track of a vigorous scratch as a wheal that manifests rapidly, lasts a few minutes, then disappears without a trace. Its presence is purposely sought by the examiner to confirm the diagnosis of urticaria (most often the chronic idiopathic variety).
For more information, see “Inexperienced runner develops leg rash.” Clin Rev. 2012;22(8):W3
For the next photograph, proceed to the next page >>
Source: PhotoStock-Israel / Science Source
3. Typically manifesting with edema, pruritus, warmth, and tenderness, this lesion is usually associated with a history of recent trauma or pharyngitis followed by malaise, chills, and high fever. The lesion is usually raised with a clear line of demarcation at the edge.
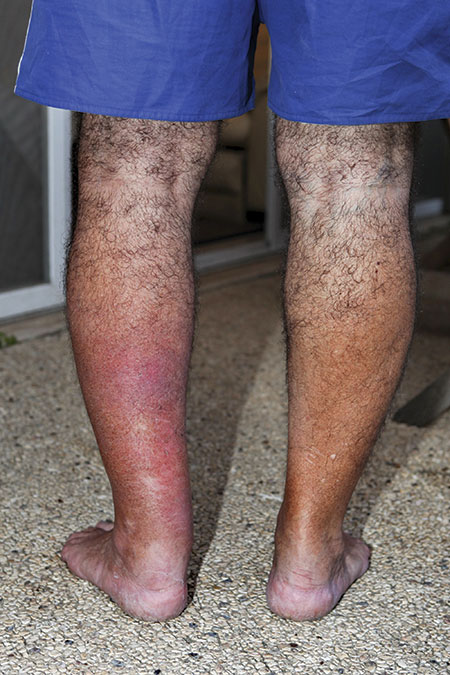
Diagnosis: Erysipelas, an acute infection of the skin and subcutaneous tissue, is caused by beta-hemolytic streptococci invading tissues via a disruption to the skin barrier. Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. Associated comorbidities in erysipelas include diabetes mellitus, as well as hypertension, chronic venous insufficiency, and other cardiovascular diseases.
For more information, see “Painful rash on face.” J Fam Pract. 2010;59(8):459-462.
For the next photograph, proceed to the next page >>
Source: CDC Public Health Image Library.
4. This patient presented with a red, expanding rash on the lateral aspect of the left thigh. Affecting any part of the body, this illness may present with fever, chills, sweats, muscle aches, fatigue, nausea and joint pain. Some patients have a rash or Bell’s palsy.
Diagnosis: Lyme disease, caused by B. burgdorferi bacteria, is transmitted to humans through the bite of infected Ixodes ticks. Typical symptoms include fever, headache, fatigue, and a characteristic skin rash called erythema migrans. If left untreated, infection can spread to joints, the heart, and the nervous system.
Because its symptoms mimic many other diseases, diagnosing Lyme disease can be difficult. The diagnosis is based on symptoms, physical findings, eg, rash, and the possibility of exposure to infected ticks; laboratory testing is helpful if used correctly and performed with validated methods.
Treatment choice depends on the whether the disease is early or late. Most cases of early Lyme disease can be treated successfully with a few weeks of antibiotics.
For more information, see “Lyme Disease Presents Differently in Men and Women.”
1. The patient had just recovered from a sore throat and noticed discrete red nodules, which eventually coalesced into a single large edematous plaque over the right anterior tibia. The deep intradermal and subdermal edema is exquisitely tender to touch, considerably warmer than the surrounding skin, and highly blanchable.
Diagnosis: Erythema nodosum is a reactive form of septal panniculitis with many potential triggers. Notable triggers include Crohn disease flares and use of drugs such as sulfa, gold salts, and oral contraceptives. Several infections have been identified as triggers, including strep, mycoplasma, and campylobacter, as well as deep fungal infections (histoplasmosis, blastomycosis, coccidioidomycosis, and sporotrichosis). More unusual causes include pregnancy and diseases such as sarcoidosis, tuberculosis, Behçet disease, and leukemia/lymphoma.
For more information, see “Painful Lesion Hasn’t Responded to Antibiotics.” Clin Rev. 2015;25(11):10,12.
For the next photograph, proceed to the next page >>
2. A 16-year-old high school student joins her friends in a 2K run one morning. The next day, her shins are so painful she can hardly walk. She applied ice packs to her legs, using elastic bandages to hold them in place until the ice cubes melt. As her legs rewarm, a rash appears where the ice packs contacted the skin.
Diagnosis: This condition is urticarial in nature—albeit an unusual form, triggered by cold. Though it appears counterintuitive, cold uriticaria typically appears only on rewarming of the affected area and is marked by the sudden appearance of “welts” or “hives” that usually clear (with or without treatment) within hours.
Uncomplicated urticaria resolves without leaving any signs (eg, purpura, ecchymosis) that might otherwise suggest the presence of a vasculitic component, such as that seen with lupus or other autoimmune diseases. Blanchability on digital pressure is one way to confirm benignancy, since blood tends to leak from vessels damaged by vasculitis, emptying into the surrounding interstitial spaces and presenting as nonblanchable petechiae, purpura, or ecchymosis. The relatively benign nature of this patient’s urticaria was also suggested by additional history taking, in which she denied having fever, malaise, or arthralgia. These are all symptoms we might have seen with more serious underlying causes.
Cold urticaria is one of the so-called physical urticarias, a group that includes urticaria caused by vibration, pressure, heat, sun, and even exposure to water. Thought to comprise up to 20% of all urticarias, the physical urticarias occur most frequently in persons ages 17 to 40. Dermatographism is the most common form, occurring in the linear track of a vigorous scratch as a wheal that manifests rapidly, lasts a few minutes, then disappears without a trace. Its presence is purposely sought by the examiner to confirm the diagnosis of urticaria (most often the chronic idiopathic variety).
For more information, see “Inexperienced runner develops leg rash.” Clin Rev. 2012;22(8):W3
For the next photograph, proceed to the next page >>
Source: PhotoStock-Israel / Science Source
3. Typically manifesting with edema, pruritus, warmth, and tenderness, this lesion is usually associated with a history of recent trauma or pharyngitis followed by malaise, chills, and high fever. The lesion is usually raised with a clear line of demarcation at the edge.
Diagnosis: Erysipelas, an acute infection of the skin and subcutaneous tissue, is caused by beta-hemolytic streptococci invading tissues via a disruption to the skin barrier. Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. Associated comorbidities in erysipelas include diabetes mellitus, as well as hypertension, chronic venous insufficiency, and other cardiovascular diseases.
For more information, see “Painful rash on face.” J Fam Pract. 2010;59(8):459-462.
For the next photograph, proceed to the next page >>
Source: CDC Public Health Image Library.
4. This patient presented with a red, expanding rash on the lateral aspect of the left thigh. Affecting any part of the body, this illness may present with fever, chills, sweats, muscle aches, fatigue, nausea and joint pain. Some patients have a rash or Bell’s palsy.
Diagnosis: Lyme disease, caused by B. burgdorferi bacteria, is transmitted to humans through the bite of infected Ixodes ticks. Typical symptoms include fever, headache, fatigue, and a characteristic skin rash called erythema migrans. If left untreated, infection can spread to joints, the heart, and the nervous system.
Because its symptoms mimic many other diseases, diagnosing Lyme disease can be difficult. The diagnosis is based on symptoms, physical findings, eg, rash, and the possibility of exposure to infected ticks; laboratory testing is helpful if used correctly and performed with validated methods.
Treatment choice depends on the whether the disease is early or late. Most cases of early Lyme disease can be treated successfully with a few weeks of antibiotics.
For more information, see “Lyme Disease Presents Differently in Men and Women.”
1. The patient had just recovered from a sore throat and noticed discrete red nodules, which eventually coalesced into a single large edematous plaque over the right anterior tibia. The deep intradermal and subdermal edema is exquisitely tender to touch, considerably warmer than the surrounding skin, and highly blanchable.
Diagnosis: Erythema nodosum is a reactive form of septal panniculitis with many potential triggers. Notable triggers include Crohn disease flares and use of drugs such as sulfa, gold salts, and oral contraceptives. Several infections have been identified as triggers, including strep, mycoplasma, and campylobacter, as well as deep fungal infections (histoplasmosis, blastomycosis, coccidioidomycosis, and sporotrichosis). More unusual causes include pregnancy and diseases such as sarcoidosis, tuberculosis, Behçet disease, and leukemia/lymphoma.
For more information, see “Painful Lesion Hasn’t Responded to Antibiotics.” Clin Rev. 2015;25(11):10,12.
For the next photograph, proceed to the next page >>
2. A 16-year-old high school student joins her friends in a 2K run one morning. The next day, her shins are so painful she can hardly walk. She applied ice packs to her legs, using elastic bandages to hold them in place until the ice cubes melt. As her legs rewarm, a rash appears where the ice packs contacted the skin.
Diagnosis: This condition is urticarial in nature—albeit an unusual form, triggered by cold. Though it appears counterintuitive, cold uriticaria typically appears only on rewarming of the affected area and is marked by the sudden appearance of “welts” or “hives” that usually clear (with or without treatment) within hours.
Uncomplicated urticaria resolves without leaving any signs (eg, purpura, ecchymosis) that might otherwise suggest the presence of a vasculitic component, such as that seen with lupus or other autoimmune diseases. Blanchability on digital pressure is one way to confirm benignancy, since blood tends to leak from vessels damaged by vasculitis, emptying into the surrounding interstitial spaces and presenting as nonblanchable petechiae, purpura, or ecchymosis. The relatively benign nature of this patient’s urticaria was also suggested by additional history taking, in which she denied having fever, malaise, or arthralgia. These are all symptoms we might have seen with more serious underlying causes.
Cold urticaria is one of the so-called physical urticarias, a group that includes urticaria caused by vibration, pressure, heat, sun, and even exposure to water. Thought to comprise up to 20% of all urticarias, the physical urticarias occur most frequently in persons ages 17 to 40. Dermatographism is the most common form, occurring in the linear track of a vigorous scratch as a wheal that manifests rapidly, lasts a few minutes, then disappears without a trace. Its presence is purposely sought by the examiner to confirm the diagnosis of urticaria (most often the chronic idiopathic variety).
For more information, see “Inexperienced runner develops leg rash.” Clin Rev. 2012;22(8):W3
For the next photograph, proceed to the next page >>
Source: PhotoStock-Israel / Science Source
3. Typically manifesting with edema, pruritus, warmth, and tenderness, this lesion is usually associated with a history of recent trauma or pharyngitis followed by malaise, chills, and high fever. The lesion is usually raised with a clear line of demarcation at the edge.
Diagnosis: Erysipelas, an acute infection of the skin and subcutaneous tissue, is caused by beta-hemolytic streptococci invading tissues via a disruption to the skin barrier. Streptococcus strains are susceptible to penicillin and 99.5% are susceptible to clindamycin. Associated comorbidities in erysipelas include diabetes mellitus, as well as hypertension, chronic venous insufficiency, and other cardiovascular diseases.
For more information, see “Painful rash on face.” J Fam Pract. 2010;59(8):459-462.
For the next photograph, proceed to the next page >>
Source: CDC Public Health Image Library.
4. This patient presented with a red, expanding rash on the lateral aspect of the left thigh. Affecting any part of the body, this illness may present with fever, chills, sweats, muscle aches, fatigue, nausea and joint pain. Some patients have a rash or Bell’s palsy.
Diagnosis: Lyme disease, caused by B. burgdorferi bacteria, is transmitted to humans through the bite of infected Ixodes ticks. Typical symptoms include fever, headache, fatigue, and a characteristic skin rash called erythema migrans. If left untreated, infection can spread to joints, the heart, and the nervous system.
Because its symptoms mimic many other diseases, diagnosing Lyme disease can be difficult. The diagnosis is based on symptoms, physical findings, eg, rash, and the possibility of exposure to infected ticks; laboratory testing is helpful if used correctly and performed with validated methods.
Treatment choice depends on the whether the disease is early or late. Most cases of early Lyme disease can be treated successfully with a few weeks of antibiotics.
For more information, see “Lyme Disease Presents Differently in Men and Women.”
High-dose vitamin D in pregnancy fails to prevent wheezing risk in children
Among pregnant women at high risk for having a child with asthma, high doses of vitamin D administered during the third trimester failed to prevent persistent wheezing illness in their children at age 3, according to two separate reports published online Jan. 26 in JAMA.
Both studies were conducted because vitamin D insufficiency during pregnancy is commonplace and is thought to affect fetal immune programming and to contribute to asthma pathogenesis. In addition, observational studies have found an association between low levels of vitamin D in cord blood and later asthma in the child.

The two randomized, double-blind placebo-controlled clinical trials found that neither 2,800 IU/day nor 4,400 IU/day of vitamin D significantly reduced the risk of persistent wheeze in the offspring through 3 years of age. However, both research groups noted that their studies may have been underpowered to detect a clinically important protective effect, and both recommended longer-term observation of their study participants, as well as further studies using larger sample sizes, higher doses of vitamin D, administration earlier in pregnancy, and postnatal supplementation to establish a definitive result.
In the first study – conducted as part of the Copenhagen Prospective Studies on Asthma in Childhood 2010 cohort – 623 Danish women already taking the standard 400 IU of vitamin D3 during pregnancy were randomly assigned to receive an additional 2,400 IU (315 women) or a matching placebo (308 women) from 22 to 26 weeks’ gestation until delivery. After exclusions, researchers analyzed data on 581 children.
Maternal serum vitamin D levels increased markedly in the active-treatment group. “Correspondingly, the percentage of women with sufficient levels of vitamin D (greater than 30 ng/mL) after the intervention was 81% in the vitamin D group, compared with 44% in the control group,” wrote Dr. Bo L. Chawes of Copenhagen Prospective Studies on Asthma in Childhood, University of Copenhagen, and his associates.
Persistent wheeze developed in 104 (18%) of the 581 children: 47 (16%) in the vitamin D group and 57 (20%) in the control group, a nonsignificant difference. Similarly, asthma was diagnosed in 79 children: 32 (12%) in the vitamin D group and 47 (14%) in the control group, another nonsignificant difference.
Vitamin D supplementation also made no difference in infants’ levels of C-reactive protein, interleukin-6, tumor necrosis factor–alpha, or CXCL8, nor in the number of upper respiratory tract infections (5.2 per year vs 5.3 per year), the number of lower respiratory tract infections (32% vs 33%), the risk of allergic sensitization as measured by skin prick test or specific IgE level, or the development of eczema (23% vs 25%).
However, the risk of persistent wheeze was higher in children whose mothers’ vitamin D levels were lowest, compared with those whose mothers’ vitamin D levels were in the middle and upper tertiles. And high-dose vitamin D was protective with regard to some secondary endpoints, such as preventing more episodes of “troublesome lung symptoms” (5.9 vs. 7.2).
This finding, together with the study’s somewhat reduced statistical power, mean that a clinically important protective effect cannot be ruled out. In addition, the supplementation dose may have been too low or may have been given too late in the course of pregnancy to produce a significant effect, Dr. Chawes and his associates wrote (JAMA. 2016;315[4]:353-61. doi: 10.1001/jama.2015.18318).In the second study – the Vitamin D Antenatal Asthma Reduction Trial – 876 pregnant women in Boston, St. Louis, and San Diego who were already taking the standard 400 IU of vitamin D were randomly assigned to receive either an additional 4,000 IU/day (440 participants) or a matching placebo (436 participants). Maternal levels of vitamin D rose markedly in the active-treatment group (mean, 39.2 ng/mL), compared with the control group (mean, 26.8 ng/mL), and the proportion of women who achieved higher than “inadequate” levels was much greater (74.9% vs 34.0%), reported Dr. Augusto A. Litonjua of Brigham and Women’s Hospital, Boston, and his associates.
A total of 24.3% of the vitamin D group and 30.4% of the control group developed asthma or recurrent wheeze by age 3 years, a nonsignificant difference. However, the incidence of asthma was so much lower than anticipated in both study groups that the study may have lost statistical power to detect a clinically meaningful difference, according to the investigators (JAMA. 2016;315[4]:362-70. doi: 10.1001/jama.2015.18589).
It remains unclear whether vitamin D supplementation during pregnancy will reduce asthma and persistent wheezing in the offspring. “Larger studies and longer follow-up of the children in this study will be needed to answer the question,” the investigators wrote. “If additional studies identify a significant effect, given the high prevalence of low vitamin D levels in pregnant women, the effect of this inexpensive intervention on child health could be substantial.”
The first study was supported by the Copenhagen Prospective Study on Asthma in Childhood, which is funded by private and public research groups. One of the coauthors reported receiving consulting fees from Chiesi. The Vitamin D Antenatal Asthma Reduction Trial was supported by the U.S. National Heart, Lung, and Blood Institute and the National Centers for Advancing Translational Sciences. The lead author, Dr. Litonjua, reported receiving personal fees from UpToDate and Springer Humana Press; his associates reported ties to numerous industry sources.
These are sobering findings. Even if we assume that prenatal vitamin D supplementation will prove more protective as the children in these studies grow older, vitamin D insufficiency still would explain only a small portion of the current asthma epidemic.
But neither study showed any unwanted effects from supplementation, so it seems reasonable for clinicians to prescribe vitamin D to mothers at high risk of having children with asthma by virtue of their own asthma, eczema, or allergic rhinitis – especially if those mothers are deficient in vitamin D. However, the data in these two clinical trials do not support the use of very high-dose vitamin D, since any beneficial effects achieved with 4,400 IU/day were identical to those achieved with approximately half as high a dose.
Dr. Erika von Mutius is at Ludwig Maximilians University, Munich. Dr. Fernando D. Martinez is at the asthma and airway disease research center and the department of pediatrics at the University of Arizona, Tucson. Both reported having no relevant financial disclosures. Their remarks are adapted from an editorial accompanying the two reports (JAMA 2016;315[4]:347-8.).
These are sobering findings. Even if we assume that prenatal vitamin D supplementation will prove more protective as the children in these studies grow older, vitamin D insufficiency still would explain only a small portion of the current asthma epidemic.
But neither study showed any unwanted effects from supplementation, so it seems reasonable for clinicians to prescribe vitamin D to mothers at high risk of having children with asthma by virtue of their own asthma, eczema, or allergic rhinitis – especially if those mothers are deficient in vitamin D. However, the data in these two clinical trials do not support the use of very high-dose vitamin D, since any beneficial effects achieved with 4,400 IU/day were identical to those achieved with approximately half as high a dose.
Dr. Erika von Mutius is at Ludwig Maximilians University, Munich. Dr. Fernando D. Martinez is at the asthma and airway disease research center and the department of pediatrics at the University of Arizona, Tucson. Both reported having no relevant financial disclosures. Their remarks are adapted from an editorial accompanying the two reports (JAMA 2016;315[4]:347-8.).
These are sobering findings. Even if we assume that prenatal vitamin D supplementation will prove more protective as the children in these studies grow older, vitamin D insufficiency still would explain only a small portion of the current asthma epidemic.
But neither study showed any unwanted effects from supplementation, so it seems reasonable for clinicians to prescribe vitamin D to mothers at high risk of having children with asthma by virtue of their own asthma, eczema, or allergic rhinitis – especially if those mothers are deficient in vitamin D. However, the data in these two clinical trials do not support the use of very high-dose vitamin D, since any beneficial effects achieved with 4,400 IU/day were identical to those achieved with approximately half as high a dose.
Dr. Erika von Mutius is at Ludwig Maximilians University, Munich. Dr. Fernando D. Martinez is at the asthma and airway disease research center and the department of pediatrics at the University of Arizona, Tucson. Both reported having no relevant financial disclosures. Their remarks are adapted from an editorial accompanying the two reports (JAMA 2016;315[4]:347-8.).
Among pregnant women at high risk for having a child with asthma, high doses of vitamin D administered during the third trimester failed to prevent persistent wheezing illness in their children at age 3, according to two separate reports published online Jan. 26 in JAMA.
Both studies were conducted because vitamin D insufficiency during pregnancy is commonplace and is thought to affect fetal immune programming and to contribute to asthma pathogenesis. In addition, observational studies have found an association between low levels of vitamin D in cord blood and later asthma in the child.
The two randomized, double-blind placebo-controlled clinical trials found that neither 2,800 IU/day nor 4,400 IU/day of vitamin D significantly reduced the risk of persistent wheeze in the offspring through 3 years of age. However, both research groups noted that their studies may have been underpowered to detect a clinically important protective effect, and both recommended longer-term observation of their study participants, as well as further studies using larger sample sizes, higher doses of vitamin D, administration earlier in pregnancy, and postnatal supplementation to establish a definitive result.
In the first study – conducted as part of the Copenhagen Prospective Studies on Asthma in Childhood 2010 cohort – 623 Danish women already taking the standard 400 IU of vitamin D3 during pregnancy were randomly assigned to receive an additional 2,400 IU (315 women) or a matching placebo (308 women) from 22 to 26 weeks’ gestation until delivery. After exclusions, researchers analyzed data on 581 children.
Maternal serum vitamin D levels increased markedly in the active-treatment group. “Correspondingly, the percentage of women with sufficient levels of vitamin D (greater than 30 ng/mL) after the intervention was 81% in the vitamin D group, compared with 44% in the control group,” wrote Dr. Bo L. Chawes of Copenhagen Prospective Studies on Asthma in Childhood, University of Copenhagen, and his associates.
Persistent wheeze developed in 104 (18%) of the 581 children: 47 (16%) in the vitamin D group and 57 (20%) in the control group, a nonsignificant difference. Similarly, asthma was diagnosed in 79 children: 32 (12%) in the vitamin D group and 47 (14%) in the control group, another nonsignificant difference.
Vitamin D supplementation also made no difference in infants’ levels of C-reactive protein, interleukin-6, tumor necrosis factor–alpha, or CXCL8, nor in the number of upper respiratory tract infections (5.2 per year vs 5.3 per year), the number of lower respiratory tract infections (32% vs 33%), the risk of allergic sensitization as measured by skin prick test or specific IgE level, or the development of eczema (23% vs 25%).
However, the risk of persistent wheeze was higher in children whose mothers’ vitamin D levels were lowest, compared with those whose mothers’ vitamin D levels were in the middle and upper tertiles. And high-dose vitamin D was protective with regard to some secondary endpoints, such as preventing more episodes of “troublesome lung symptoms” (5.9 vs. 7.2).
This finding, together with the study’s somewhat reduced statistical power, mean that a clinically important protective effect cannot be ruled out. In addition, the supplementation dose may have been too low or may have been given too late in the course of pregnancy to produce a significant effect, Dr. Chawes and his associates wrote (JAMA. 2016;315[4]:353-61. doi: 10.1001/jama.2015.18318).In the second study – the Vitamin D Antenatal Asthma Reduction Trial – 876 pregnant women in Boston, St. Louis, and San Diego who were already taking the standard 400 IU of vitamin D were randomly assigned to receive either an additional 4,000 IU/day (440 participants) or a matching placebo (436 participants). Maternal levels of vitamin D rose markedly in the active-treatment group (mean, 39.2 ng/mL), compared with the control group (mean, 26.8 ng/mL), and the proportion of women who achieved higher than “inadequate” levels was much greater (74.9% vs 34.0%), reported Dr. Augusto A. Litonjua of Brigham and Women’s Hospital, Boston, and his associates.
A total of 24.3% of the vitamin D group and 30.4% of the control group developed asthma or recurrent wheeze by age 3 years, a nonsignificant difference. However, the incidence of asthma was so much lower than anticipated in both study groups that the study may have lost statistical power to detect a clinically meaningful difference, according to the investigators (JAMA. 2016;315[4]:362-70. doi: 10.1001/jama.2015.18589).
It remains unclear whether vitamin D supplementation during pregnancy will reduce asthma and persistent wheezing in the offspring. “Larger studies and longer follow-up of the children in this study will be needed to answer the question,” the investigators wrote. “If additional studies identify a significant effect, given the high prevalence of low vitamin D levels in pregnant women, the effect of this inexpensive intervention on child health could be substantial.”
The first study was supported by the Copenhagen Prospective Study on Asthma in Childhood, which is funded by private and public research groups. One of the coauthors reported receiving consulting fees from Chiesi. The Vitamin D Antenatal Asthma Reduction Trial was supported by the U.S. National Heart, Lung, and Blood Institute and the National Centers for Advancing Translational Sciences. The lead author, Dr. Litonjua, reported receiving personal fees from UpToDate and Springer Humana Press; his associates reported ties to numerous industry sources.
Among pregnant women at high risk for having a child with asthma, high doses of vitamin D administered during the third trimester failed to prevent persistent wheezing illness in their children at age 3, according to two separate reports published online Jan. 26 in JAMA.
Both studies were conducted because vitamin D insufficiency during pregnancy is commonplace and is thought to affect fetal immune programming and to contribute to asthma pathogenesis. In addition, observational studies have found an association between low levels of vitamin D in cord blood and later asthma in the child.
The two randomized, double-blind placebo-controlled clinical trials found that neither 2,800 IU/day nor 4,400 IU/day of vitamin D significantly reduced the risk of persistent wheeze in the offspring through 3 years of age. However, both research groups noted that their studies may have been underpowered to detect a clinically important protective effect, and both recommended longer-term observation of their study participants, as well as further studies using larger sample sizes, higher doses of vitamin D, administration earlier in pregnancy, and postnatal supplementation to establish a definitive result.
In the first study – conducted as part of the Copenhagen Prospective Studies on Asthma in Childhood 2010 cohort – 623 Danish women already taking the standard 400 IU of vitamin D3 during pregnancy were randomly assigned to receive an additional 2,400 IU (315 women) or a matching placebo (308 women) from 22 to 26 weeks’ gestation until delivery. After exclusions, researchers analyzed data on 581 children.
Maternal serum vitamin D levels increased markedly in the active-treatment group. “Correspondingly, the percentage of women with sufficient levels of vitamin D (greater than 30 ng/mL) after the intervention was 81% in the vitamin D group, compared with 44% in the control group,” wrote Dr. Bo L. Chawes of Copenhagen Prospective Studies on Asthma in Childhood, University of Copenhagen, and his associates.
Persistent wheeze developed in 104 (18%) of the 581 children: 47 (16%) in the vitamin D group and 57 (20%) in the control group, a nonsignificant difference. Similarly, asthma was diagnosed in 79 children: 32 (12%) in the vitamin D group and 47 (14%) in the control group, another nonsignificant difference.
Vitamin D supplementation also made no difference in infants’ levels of C-reactive protein, interleukin-6, tumor necrosis factor–alpha, or CXCL8, nor in the number of upper respiratory tract infections (5.2 per year vs 5.3 per year), the number of lower respiratory tract infections (32% vs 33%), the risk of allergic sensitization as measured by skin prick test or specific IgE level, or the development of eczema (23% vs 25%).
However, the risk of persistent wheeze was higher in children whose mothers’ vitamin D levels were lowest, compared with those whose mothers’ vitamin D levels were in the middle and upper tertiles. And high-dose vitamin D was protective with regard to some secondary endpoints, such as preventing more episodes of “troublesome lung symptoms” (5.9 vs. 7.2).
This finding, together with the study’s somewhat reduced statistical power, mean that a clinically important protective effect cannot be ruled out. In addition, the supplementation dose may have been too low or may have been given too late in the course of pregnancy to produce a significant effect, Dr. Chawes and his associates wrote (JAMA. 2016;315[4]:353-61. doi: 10.1001/jama.2015.18318).In the second study – the Vitamin D Antenatal Asthma Reduction Trial – 876 pregnant women in Boston, St. Louis, and San Diego who were already taking the standard 400 IU of vitamin D were randomly assigned to receive either an additional 4,000 IU/day (440 participants) or a matching placebo (436 participants). Maternal levels of vitamin D rose markedly in the active-treatment group (mean, 39.2 ng/mL), compared with the control group (mean, 26.8 ng/mL), and the proportion of women who achieved higher than “inadequate” levels was much greater (74.9% vs 34.0%), reported Dr. Augusto A. Litonjua of Brigham and Women’s Hospital, Boston, and his associates.
A total of 24.3% of the vitamin D group and 30.4% of the control group developed asthma or recurrent wheeze by age 3 years, a nonsignificant difference. However, the incidence of asthma was so much lower than anticipated in both study groups that the study may have lost statistical power to detect a clinically meaningful difference, according to the investigators (JAMA. 2016;315[4]:362-70. doi: 10.1001/jama.2015.18589).
It remains unclear whether vitamin D supplementation during pregnancy will reduce asthma and persistent wheezing in the offspring. “Larger studies and longer follow-up of the children in this study will be needed to answer the question,” the investigators wrote. “If additional studies identify a significant effect, given the high prevalence of low vitamin D levels in pregnant women, the effect of this inexpensive intervention on child health could be substantial.”
The first study was supported by the Copenhagen Prospective Study on Asthma in Childhood, which is funded by private and public research groups. One of the coauthors reported receiving consulting fees from Chiesi. The Vitamin D Antenatal Asthma Reduction Trial was supported by the U.S. National Heart, Lung, and Blood Institute and the National Centers for Advancing Translational Sciences. The lead author, Dr. Litonjua, reported receiving personal fees from UpToDate and Springer Humana Press; his associates reported ties to numerous industry sources.
FROM JAMA
Key clinical point: High doses of vitamin D administered during the third trimester didn’t prevent wheezing in children at age 3.
Major finding: In a study of 581 children, persistent wheeze developed in 16% in the vitamin D group and in 20% in the control group. In a separate trial of 806 infants, there was a 6.1% reduction in asthma or recurrent wheezing at age 3, which was not statistically significant.
Data source: Two separate randomized, double-blind placebo-controlled trials involving a total of nearly 1,500 pregnant women.
Disclosures: The first study was supported by the Copenhagen Prospective Study on Asthma in Childhood, which is funded by private and public research groups. One of the coauthors reported receiving consulting fees from Chiesi. The Vitamin D Antenatal Asthma Reduction Trial was supported by the U.S. National Heart, Lung, and Blood Institute and the National Centers for Advancing Translational Sciences. The lead author, Dr. Litonjua, reported receiving personal fees from UpToDate and Springer Humana Press; his associates reported ties to numerous industry sources.

VIDEO: Treating your atopic dermatitis patients more effectively
ORLANDO – A “hard and soft” approach to treating atopic dermatitis – treating more frequently while symptoms persist, then pulling back on treatment to keep symptoms at bay – is an effective way to keep the condition manageable for your patients.
“This is not an easy disease to manage, and that’s the key,” explained Dr. Adam Friedman of the George Washington University in Washington, at the Orlando Dermatology Aesthetic and Clinical annual meeting, adding that it’s important to educate patients that atopic dermatitis is “something that they will always have” and does not have one-shot cures.
In this video interview, Dr. Friedman, who is a Dermatology News board member, discusses the best way to talk to new patients about what atopic dermatitis treatment will entail, and offers methods to make management of the disease more effective and, consequently, improve patients’ quality of life.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – A “hard and soft” approach to treating atopic dermatitis – treating more frequently while symptoms persist, then pulling back on treatment to keep symptoms at bay – is an effective way to keep the condition manageable for your patients.
“This is not an easy disease to manage, and that’s the key,” explained Dr. Adam Friedman of the George Washington University in Washington, at the Orlando Dermatology Aesthetic and Clinical annual meeting, adding that it’s important to educate patients that atopic dermatitis is “something that they will always have” and does not have one-shot cures.
In this video interview, Dr. Friedman, who is a Dermatology News board member, discusses the best way to talk to new patients about what atopic dermatitis treatment will entail, and offers methods to make management of the disease more effective and, consequently, improve patients’ quality of life.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – A “hard and soft” approach to treating atopic dermatitis – treating more frequently while symptoms persist, then pulling back on treatment to keep symptoms at bay – is an effective way to keep the condition manageable for your patients.
“This is not an easy disease to manage, and that’s the key,” explained Dr. Adam Friedman of the George Washington University in Washington, at the Orlando Dermatology Aesthetic and Clinical annual meeting, adding that it’s important to educate patients that atopic dermatitis is “something that they will always have” and does not have one-shot cures.
In this video interview, Dr. Friedman, who is a Dermatology News board member, discusses the best way to talk to new patients about what atopic dermatitis treatment will entail, and offers methods to make management of the disease more effective and, consequently, improve patients’ quality of life.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ODAC 2016

Single and double dose of MenACWY-CRM series are immunogenic
Both single-dose and two-dose meningococcal CRM-conjugate vaccine series (MenACWY-CRM) were immunogenic, but antibody responses were greater in patients after two doses, especially for patients aged 2-5 years old, according to a new study.
The multicenter, randomized, observer-blind, placebo-controlled study, conducted in the United States between October 2012 and May 2014, included 713 subjects who were up to date with their routine childhood immunizations. The subjects were broken into two age groups: children aged 2-5 years and children aged 6-10 years. The two age groups were randomized 1:1 to receive two doses of MenACWY-CRM or one dose of placebo followed by one dose of MenACWY-CRM, 2 months apart. Immunogenicity was measured using serum bactericidal activity with human complement.

The researchers’ primary objective was to assess the noninferiority and superiority of antibody responses of the two doses, compared with the single dose against Neisseria meningitidis serogroups A,C, W, and Y, at 1-month after the last vaccination. Among the exclusion criteria for participation in the study were previous or suspected disease caused by N. meningitidis or immunization with any investigational or licensed vaccines containing meningococcal antigens.
Noninferiority of two doses of MenACWY-CRM, compared with a single dose, was demonstrated for all four serogroups. Additionally, at 1 month after the last vaccination, the superiority of receiving the two doses over the single dose was demonstrated for serogroups C and Y, for patients in the 2- to 5-year-old age cohort, while the superiority of two doses over one dose was demonstrated for the serogroup Y, for patients in the 6- to 10-year-old cohort.
The safety profile of the vaccine was similar in both groups. Medically attended adverse events that were considered to have been “possibly or probably related to the study vaccine” occurred in five subjects (as six events) in the 2- to 5-year-old age cohort and three subjects (as three events) in the 6- to 10-year-age cohort. One subject in the cohort aged 6-10 years withdrew from the study, because of vomiting and diarrhea, “which were considered to be unrelated to the study vaccine.” Eight serious adverse events were reported by five subjects, with one of the events “considered to be possibly related to the study vaccine.” That event was idiopathic thrombocytopenia, which occurred in a 6-year-old who received the single dose.
“[B]oth the single-dose and the two-dose primary series of MenACWY-CRM are immunogenic and have an acceptable safety profile in 2- to 10-year-old children. The two-dose primary series induced a higher immune response than [did] a single dose, although this difference in response decreases over time,” Dr. William Johnston Jr. of Alabama Clinical Therapeutics in Birmingham and his colleagues said.
Read the study in the Pediatric Infectious Disease Journal (doi: 10.1097/INF.0000000000000931).
Both single-dose and two-dose meningococcal CRM-conjugate vaccine series (MenACWY-CRM) were immunogenic, but antibody responses were greater in patients after two doses, especially for patients aged 2-5 years old, according to a new study.
The multicenter, randomized, observer-blind, placebo-controlled study, conducted in the United States between October 2012 and May 2014, included 713 subjects who were up to date with their routine childhood immunizations. The subjects were broken into two age groups: children aged 2-5 years and children aged 6-10 years. The two age groups were randomized 1:1 to receive two doses of MenACWY-CRM or one dose of placebo followed by one dose of MenACWY-CRM, 2 months apart. Immunogenicity was measured using serum bactericidal activity with human complement.
The researchers’ primary objective was to assess the noninferiority and superiority of antibody responses of the two doses, compared with the single dose against Neisseria meningitidis serogroups A,C, W, and Y, at 1-month after the last vaccination. Among the exclusion criteria for participation in the study were previous or suspected disease caused by N. meningitidis or immunization with any investigational or licensed vaccines containing meningococcal antigens.
Noninferiority of two doses of MenACWY-CRM, compared with a single dose, was demonstrated for all four serogroups. Additionally, at 1 month after the last vaccination, the superiority of receiving the two doses over the single dose was demonstrated for serogroups C and Y, for patients in the 2- to 5-year-old age cohort, while the superiority of two doses over one dose was demonstrated for the serogroup Y, for patients in the 6- to 10-year-old cohort.
The safety profile of the vaccine was similar in both groups. Medically attended adverse events that were considered to have been “possibly or probably related to the study vaccine” occurred in five subjects (as six events) in the 2- to 5-year-old age cohort and three subjects (as three events) in the 6- to 10-year-age cohort. One subject in the cohort aged 6-10 years withdrew from the study, because of vomiting and diarrhea, “which were considered to be unrelated to the study vaccine.” Eight serious adverse events were reported by five subjects, with one of the events “considered to be possibly related to the study vaccine.” That event was idiopathic thrombocytopenia, which occurred in a 6-year-old who received the single dose.
“[B]oth the single-dose and the two-dose primary series of MenACWY-CRM are immunogenic and have an acceptable safety profile in 2- to 10-year-old children. The two-dose primary series induced a higher immune response than [did] a single dose, although this difference in response decreases over time,” Dr. William Johnston Jr. of Alabama Clinical Therapeutics in Birmingham and his colleagues said.
Read the study in the Pediatric Infectious Disease Journal (doi: 10.1097/INF.0000000000000931).
Both single-dose and two-dose meningococcal CRM-conjugate vaccine series (MenACWY-CRM) were immunogenic, but antibody responses were greater in patients after two doses, especially for patients aged 2-5 years old, according to a new study.
The multicenter, randomized, observer-blind, placebo-controlled study, conducted in the United States between October 2012 and May 2014, included 713 subjects who were up to date with their routine childhood immunizations. The subjects were broken into two age groups: children aged 2-5 years and children aged 6-10 years. The two age groups were randomized 1:1 to receive two doses of MenACWY-CRM or one dose of placebo followed by one dose of MenACWY-CRM, 2 months apart. Immunogenicity was measured using serum bactericidal activity with human complement.
The researchers’ primary objective was to assess the noninferiority and superiority of antibody responses of the two doses, compared with the single dose against Neisseria meningitidis serogroups A,C, W, and Y, at 1-month after the last vaccination. Among the exclusion criteria for participation in the study were previous or suspected disease caused by N. meningitidis or immunization with any investigational or licensed vaccines containing meningococcal antigens.
Noninferiority of two doses of MenACWY-CRM, compared with a single dose, was demonstrated for all four serogroups. Additionally, at 1 month after the last vaccination, the superiority of receiving the two doses over the single dose was demonstrated for serogroups C and Y, for patients in the 2- to 5-year-old age cohort, while the superiority of two doses over one dose was demonstrated for the serogroup Y, for patients in the 6- to 10-year-old cohort.
The safety profile of the vaccine was similar in both groups. Medically attended adverse events that were considered to have been “possibly or probably related to the study vaccine” occurred in five subjects (as six events) in the 2- to 5-year-old age cohort and three subjects (as three events) in the 6- to 10-year-age cohort. One subject in the cohort aged 6-10 years withdrew from the study, because of vomiting and diarrhea, “which were considered to be unrelated to the study vaccine.” Eight serious adverse events were reported by five subjects, with one of the events “considered to be possibly related to the study vaccine.” That event was idiopathic thrombocytopenia, which occurred in a 6-year-old who received the single dose.
“[B]oth the single-dose and the two-dose primary series of MenACWY-CRM are immunogenic and have an acceptable safety profile in 2- to 10-year-old children. The two-dose primary series induced a higher immune response than [did] a single dose, although this difference in response decreases over time,” Dr. William Johnston Jr. of Alabama Clinical Therapeutics in Birmingham and his colleagues said.
Read the study in the Pediatric Infectious Disease Journal (doi: 10.1097/INF.0000000000000931).
FROM THE PEDIATRIC INFECTIOUS DISEASE JOURNAL
Many Providers Don’t Teach Epinephrine Use for Food Allergies
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.

The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.
The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.
The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).

Many physicians don’t teach epinephrine use for food allergies
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.

The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.
The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).
Many pediatricians and allergists failed to provide instructions for when and how to use epinephrine and a written emergency action plan to the parents of children with food allergies, a study found.
Researchers asked the parents of 859 children with allergies to share their level of satisfaction with the care and delivery of care from the pediatricians and allergists treating their children over the course of the previous 6 months. While more than 75% of the doctors treated the parents courteously and respectfully and provided clear explanations of children’s allergies, a high percentage of the doctors neglected to provide other information essential for caring for children with food allergies, the parents reported.
The parents most often said they were missing explanations on when and how their children should be receiving epinephrine to treat allergic reactions and instructions on exactly what to do if their children had a medical emergency related to their specific allergies and circumstances. More pediatricians than allergists failed to share such information.
Among allergists, 68% explained when to use epinephrine, while 37% of pediatricians provided such information, the parents reported. Instructions on how to use epinephrine were provided to parents by 47% of allergists and 20% of pediatricians. Written emergency health care plans customized to each child were provided by 56% of the allergists and 24% of the pediatricians
“Although pediatricians might be relying on allergists to deliver [an emergency action plan and training in epinephrine autoinjectors use], with the rise in food allergies, guidelines recommend that pediatricians conduct these steps and not rely solely on the allergist,” said Jesse A. Blumenstock of Northwestern University, Chicago, and colleagues. “With our understanding of food allergy and anaphylaxis constantly evolving, guidelines and recommendations for how and when to give epinephrine and the need for an action plan need to be reinforced by physicians.”
Read the study in the Journal of Allergy and Clinical Immunology: In Practice (doi: 10.1016/j.jaip.2015.10.011).

Serum allergen-specific IgE testing: How much is too much?
A 25-year-old man is evaluated for angioedema (swelling of lips and tongue) after eating paella at a Spanish restaurant. He has no history of allergies, but he says he had never eaten such a large variety of seafood before, especially shellfish.
He suspects that he is allergic to shellfish and asks the attending physician to order blood tests for seafood allergies, as he heard from a friend that blood tests are superior to other types of tests for allergy. The physician requests a serum immunoglobulin E (IgE) food panel test for this patient.
SERUM ALLERGEN-SPECIFIC IgE TESTING
Many methods of testing for allergy are available, including the skin-prick test, double-blind and single-blind placebo-controlled food challenges, open food challenges, inhalant challenges, drug challenges, and serum IgE tests. In clinical practice, these tests are often used in combination because when used individually, few of them are both highly sensitive and specific (Table 1).1–6
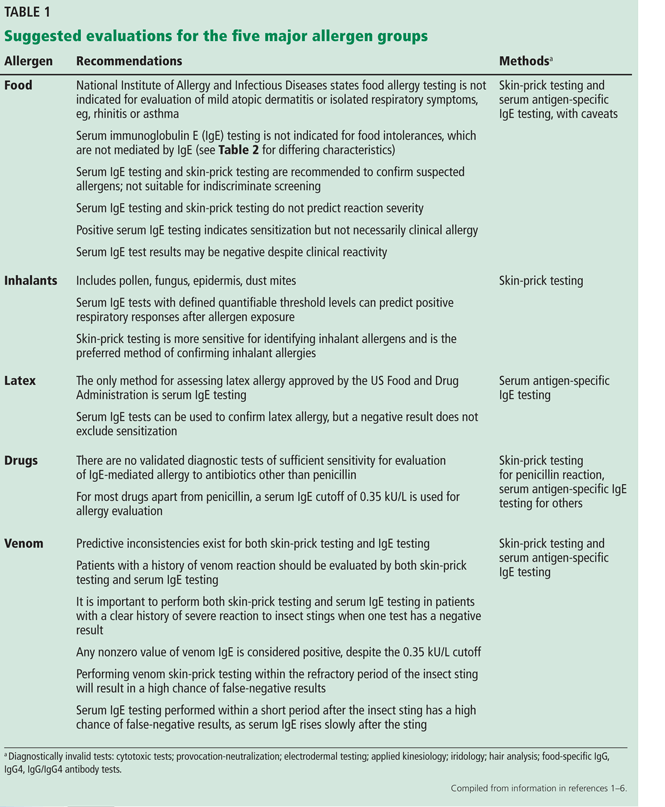
Skin-prick testing is generally the method of choice for the preliminary evaluation of IgE-mediated allergies because it is more sensitive and requires less time to get a result.1 But it is not the preferred test if the patient is at risk of a systemic reaction or has widespread dermatitis, nor is it useful if the patient is taking drugs that suppress the histamine response, such as antihistamines or tricyclic antidepressants.6 Moreover, skin-prick testing is more invasive and time-consuming than serum IgE testing.
Serum IgE testing is an attractive alternative, and it is more convenient because it requires only a single blood draw and poses a lower risk of adverse effects.
NOT A RELIABLE DIAGNOSTIC TOOL
As serum IgE testing has gained popularity, researchers have tried to improve its diagnostic power (ie, maximize its sensitivity and specificity) by determining the best cutoff values for IgE against specific antigens. Unfortunately, these values are difficult to determine because of confounding factors such as the lack of a reference standard, population diversity, patient atopy, and the overwhelming number of allergens that must be examined.
In addition, some researchers have used positive and negative predictive values to evaluate diagnostic cutoffs for serum antigen-specific IgE values. But these are not the most suitable performance measure to evaluate because they depend on disease prevalence and population characteristics.
Despite these efforts, results are still conflicting, and serum antigen-specific IgE testing is not a reliable diagnostic tool.
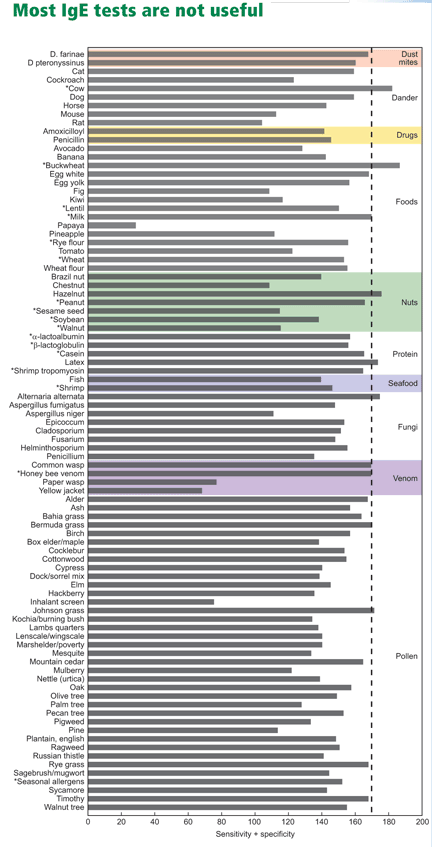
In an effort to gain insight from the available research data, we evaluated the clinical usefulness of 89 antigen-specific IgE tests, using an approach of summing their sensitivity and specificity. Previously, Wians7 proposed that a test is likely to be clinically useful if the sum of its sensitivity and specificity is equal to or greater than 170. Figure 1 shows the 89 tests, grouped into categories, and their summed sensitivities and specificities. The dashed line indicates a cutoff of 170; any bar that touches or crosses that line indicates that the test may be clinically useful, according to Wians.7
Only 7 of the 89 tests (cow, buckwheat, hazelnut, latex, Alternaria alternata, honey bee venom, and Johnson grass) satisfied this criterion. This suggests that a significant number of serum antigen-specific IgE tests perform suboptimally, and we are left with the question of why they are so commonly ordered.
Inappropriate use can lead to false-positive results, a situation in which patients may be subjected to unnecessary food avoidance that can result in nutritional deficiencies and decreased quality of life. It can also lead to false-negative results, when life-threatening diagnoses are missed and further excessive downstream testing is required—all leading; to negative outcomes for both patients and healthcare providers.
CHOOSING WISELY
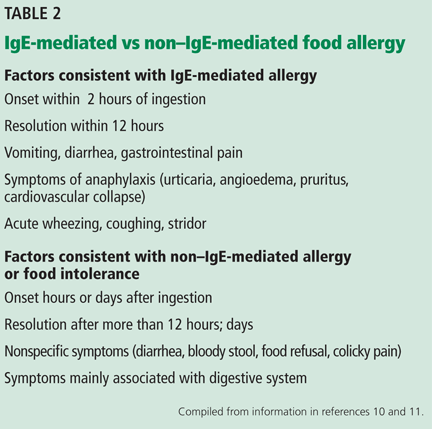
The Choosing Wisely campaign in the United States has partnered with the American Academy of Allergy, Asthma, and Immunology to advocate against indiscriminate IgE testing in evaluating allergy.8 Allergy diagnosis and evaluation should be based on a combination of clinical history and judicious ordering of specific IgE tests, whether through skin or blood testing. Ordering of serum allergen-specific IgE tests for food allergies should be consistent with a clinical history of potential IgE-mediated food allergy8 and not food intolerance (Table 2).4,5
Some jurisdictions in Canada have followed suit by restricting the number of serum IgE tests each physician is allowed to order per patient, to encourage more responsible ordering and to lower the number of potential false-positive results, which can lead to increased downstream costs as well as unnecessary patient worry and lifestyle modification.
CLINICAL BOTTOM LINE
Ordering diagnostic tests that have little clinical utility has long-term detrimental effects on both patient safety and healthcare sustainability.
In the case of the 25-year-old evaluated for shellfish allergy, the clinician should first explain that the swelling of the lips and tongue (angioedema) does suggest an IgE-mediated allergic reaction and not a non–IgE-mediated allergic reaction or a food intolerance. Non–IgE-mediated food allergies and food intolerances are marked by symptoms relating mainly to nonimmune aspects of the digestive system, whereas IgE-mediated food allergies affect the immune system and can involve a multitude of organs, including the skin and the respiratory and digestive systems (Table 2).
However, clinicians should avoid indiscriminately ordering food allergen IgE panels and instead should focus on foods likely to be the culprits based on the clinical history.9 Indiscriminate testing can lead to false-positive results and unnecessary food avoidance.
Since the patient developed symptoms of angioedema when he was exposed to his allergen, he may be apprehensive about a skin- prick test and the possibility of being subjected to the same discomfort. Therefore, in this situation, it may be best to perform serum IgE tests, but on a few targeted seafoods rather than the food panel the physician had ordered. A patient can be sensitized to an allergen (possess IgE antibodies) but not experience symptoms when exposed to it (ie, have tolerance).5 Also, false-negative results may occur, so a negative serum allergen-specific IgE test should likewise be interpreted in light of the pretest probability of allergy to a specific antigen.
If the history and the results of testing are not clear and congruent, the patient should be referred to an allergist for diagnosis or for management. The allergist can provide management techniques and periodic assessment as to the progression and resolution of the allergy. Table 2 highlights symptoms that differentiate an IgE-mediated from a non–IgE-mediated food allergy.10,11 Table 1 presents clinical indications and suggested diagnostic methods to the five most common allergen groups and the diagnostically invalid tests.1–6
The bottom line is that we must consider the poor performance of serum allergen-specific IgE tests when diagnosing and treating suspected type I allergies and avoid ordering food allergen IgE panels whenever possible.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Bird JA, Crain M, Varshney P. Food allergen panel testing often results in misdiagnosis of food allergy. J Pediatr 2015; 166:97–100.
- Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am 2015; 35:61–76.
- Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update-2014. J Allergy Clin Immunol 2014; 134:1016–1025.e43.
- Sicherer SH, Sampson HA. Food allergy: epidemiology, pathogenesis, diagnosis, and treatment. J Allergy Clin Immunol 2014; 133:291–308.
- Siles RI, Hsieh FH. Allergy blood testing: a practical guide for clinicians. Cleve Clin J Med 2011; 78:585–592.
- Wians FH Jr. Clinical laboratory tests: which, why, and what do the results mean? Lab Medicine 2009; 40:105–113.
- Choosing Wisely. American Academy of Allergy, Asthma & Immunology. Ten Things Physicians and Patients Should Question. www.choosingwisely.org/doctor-patient-lists/american-academy-of-allergy-asthma-immunology/. Accessed December 3, 2015.
- Fleischer DM, Burks AW. Pitfalls in food allergy diagnosis: serum IgE testing. J Pediatr 2015; 166: 8-10.
- Boyce JA, Assa'ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Stiefel G, Roberts G. How to use serum-specific IgE measurements in diagnosing and monitoring food allergy. Arch Dis Child Educ Pract Ed 2012; 97:29–36.
A 25-year-old man is evaluated for angioedema (swelling of lips and tongue) after eating paella at a Spanish restaurant. He has no history of allergies, but he says he had never eaten such a large variety of seafood before, especially shellfish.
He suspects that he is allergic to shellfish and asks the attending physician to order blood tests for seafood allergies, as he heard from a friend that blood tests are superior to other types of tests for allergy. The physician requests a serum immunoglobulin E (IgE) food panel test for this patient.
SERUM ALLERGEN-SPECIFIC IgE TESTING
Many methods of testing for allergy are available, including the skin-prick test, double-blind and single-blind placebo-controlled food challenges, open food challenges, inhalant challenges, drug challenges, and serum IgE tests. In clinical practice, these tests are often used in combination because when used individually, few of them are both highly sensitive and specific (Table 1).1–6
Skin-prick testing is generally the method of choice for the preliminary evaluation of IgE-mediated allergies because it is more sensitive and requires less time to get a result.1 But it is not the preferred test if the patient is at risk of a systemic reaction or has widespread dermatitis, nor is it useful if the patient is taking drugs that suppress the histamine response, such as antihistamines or tricyclic antidepressants.6 Moreover, skin-prick testing is more invasive and time-consuming than serum IgE testing.
Serum IgE testing is an attractive alternative, and it is more convenient because it requires only a single blood draw and poses a lower risk of adverse effects.
NOT A RELIABLE DIAGNOSTIC TOOL
As serum IgE testing has gained popularity, researchers have tried to improve its diagnostic power (ie, maximize its sensitivity and specificity) by determining the best cutoff values for IgE against specific antigens. Unfortunately, these values are difficult to determine because of confounding factors such as the lack of a reference standard, population diversity, patient atopy, and the overwhelming number of allergens that must be examined.
In addition, some researchers have used positive and negative predictive values to evaluate diagnostic cutoffs for serum antigen-specific IgE values. But these are not the most suitable performance measure to evaluate because they depend on disease prevalence and population characteristics.
Despite these efforts, results are still conflicting, and serum antigen-specific IgE testing is not a reliable diagnostic tool.
In an effort to gain insight from the available research data, we evaluated the clinical usefulness of 89 antigen-specific IgE tests, using an approach of summing their sensitivity and specificity. Previously, Wians7 proposed that a test is likely to be clinically useful if the sum of its sensitivity and specificity is equal to or greater than 170. Figure 1 shows the 89 tests, grouped into categories, and their summed sensitivities and specificities. The dashed line indicates a cutoff of 170; any bar that touches or crosses that line indicates that the test may be clinically useful, according to Wians.7
Only 7 of the 89 tests (cow, buckwheat, hazelnut, latex, Alternaria alternata, honey bee venom, and Johnson grass) satisfied this criterion. This suggests that a significant number of serum antigen-specific IgE tests perform suboptimally, and we are left with the question of why they are so commonly ordered.
Inappropriate use can lead to false-positive results, a situation in which patients may be subjected to unnecessary food avoidance that can result in nutritional deficiencies and decreased quality of life. It can also lead to false-negative results, when life-threatening diagnoses are missed and further excessive downstream testing is required—all leading; to negative outcomes for both patients and healthcare providers.
CHOOSING WISELY
The Choosing Wisely campaign in the United States has partnered with the American Academy of Allergy, Asthma, and Immunology to advocate against indiscriminate IgE testing in evaluating allergy.8 Allergy diagnosis and evaluation should be based on a combination of clinical history and judicious ordering of specific IgE tests, whether through skin or blood testing. Ordering of serum allergen-specific IgE tests for food allergies should be consistent with a clinical history of potential IgE-mediated food allergy8 and not food intolerance (Table 2).4,5
Some jurisdictions in Canada have followed suit by restricting the number of serum IgE tests each physician is allowed to order per patient, to encourage more responsible ordering and to lower the number of potential false-positive results, which can lead to increased downstream costs as well as unnecessary patient worry and lifestyle modification.
CLINICAL BOTTOM LINE
Ordering diagnostic tests that have little clinical utility has long-term detrimental effects on both patient safety and healthcare sustainability.
In the case of the 25-year-old evaluated for shellfish allergy, the clinician should first explain that the swelling of the lips and tongue (angioedema) does suggest an IgE-mediated allergic reaction and not a non–IgE-mediated allergic reaction or a food intolerance. Non–IgE-mediated food allergies and food intolerances are marked by symptoms relating mainly to nonimmune aspects of the digestive system, whereas IgE-mediated food allergies affect the immune system and can involve a multitude of organs, including the skin and the respiratory and digestive systems (Table 2).
However, clinicians should avoid indiscriminately ordering food allergen IgE panels and instead should focus on foods likely to be the culprits based on the clinical history.9 Indiscriminate testing can lead to false-positive results and unnecessary food avoidance.
Since the patient developed symptoms of angioedema when he was exposed to his allergen, he may be apprehensive about a skin- prick test and the possibility of being subjected to the same discomfort. Therefore, in this situation, it may be best to perform serum IgE tests, but on a few targeted seafoods rather than the food panel the physician had ordered. A patient can be sensitized to an allergen (possess IgE antibodies) but not experience symptoms when exposed to it (ie, have tolerance).5 Also, false-negative results may occur, so a negative serum allergen-specific IgE test should likewise be interpreted in light of the pretest probability of allergy to a specific antigen.
If the history and the results of testing are not clear and congruent, the patient should be referred to an allergist for diagnosis or for management. The allergist can provide management techniques and periodic assessment as to the progression and resolution of the allergy. Table 2 highlights symptoms that differentiate an IgE-mediated from a non–IgE-mediated food allergy.10,11 Table 1 presents clinical indications and suggested diagnostic methods to the five most common allergen groups and the diagnostically invalid tests.1–6
The bottom line is that we must consider the poor performance of serum allergen-specific IgE tests when diagnosing and treating suspected type I allergies and avoid ordering food allergen IgE panels whenever possible.
A 25-year-old man is evaluated for angioedema (swelling of lips and tongue) after eating paella at a Spanish restaurant. He has no history of allergies, but he says he had never eaten such a large variety of seafood before, especially shellfish.
He suspects that he is allergic to shellfish and asks the attending physician to order blood tests for seafood allergies, as he heard from a friend that blood tests are superior to other types of tests for allergy. The physician requests a serum immunoglobulin E (IgE) food panel test for this patient.
SERUM ALLERGEN-SPECIFIC IgE TESTING
Many methods of testing for allergy are available, including the skin-prick test, double-blind and single-blind placebo-controlled food challenges, open food challenges, inhalant challenges, drug challenges, and serum IgE tests. In clinical practice, these tests are often used in combination because when used individually, few of them are both highly sensitive and specific (Table 1).1–6
Skin-prick testing is generally the method of choice for the preliminary evaluation of IgE-mediated allergies because it is more sensitive and requires less time to get a result.1 But it is not the preferred test if the patient is at risk of a systemic reaction or has widespread dermatitis, nor is it useful if the patient is taking drugs that suppress the histamine response, such as antihistamines or tricyclic antidepressants.6 Moreover, skin-prick testing is more invasive and time-consuming than serum IgE testing.
Serum IgE testing is an attractive alternative, and it is more convenient because it requires only a single blood draw and poses a lower risk of adverse effects.
NOT A RELIABLE DIAGNOSTIC TOOL
As serum IgE testing has gained popularity, researchers have tried to improve its diagnostic power (ie, maximize its sensitivity and specificity) by determining the best cutoff values for IgE against specific antigens. Unfortunately, these values are difficult to determine because of confounding factors such as the lack of a reference standard, population diversity, patient atopy, and the overwhelming number of allergens that must be examined.
In addition, some researchers have used positive and negative predictive values to evaluate diagnostic cutoffs for serum antigen-specific IgE values. But these are not the most suitable performance measure to evaluate because they depend on disease prevalence and population characteristics.
Despite these efforts, results are still conflicting, and serum antigen-specific IgE testing is not a reliable diagnostic tool.
In an effort to gain insight from the available research data, we evaluated the clinical usefulness of 89 antigen-specific IgE tests, using an approach of summing their sensitivity and specificity. Previously, Wians7 proposed that a test is likely to be clinically useful if the sum of its sensitivity and specificity is equal to or greater than 170. Figure 1 shows the 89 tests, grouped into categories, and their summed sensitivities and specificities. The dashed line indicates a cutoff of 170; any bar that touches or crosses that line indicates that the test may be clinically useful, according to Wians.7
Only 7 of the 89 tests (cow, buckwheat, hazelnut, latex, Alternaria alternata, honey bee venom, and Johnson grass) satisfied this criterion. This suggests that a significant number of serum antigen-specific IgE tests perform suboptimally, and we are left with the question of why they are so commonly ordered.
Inappropriate use can lead to false-positive results, a situation in which patients may be subjected to unnecessary food avoidance that can result in nutritional deficiencies and decreased quality of life. It can also lead to false-negative results, when life-threatening diagnoses are missed and further excessive downstream testing is required—all leading; to negative outcomes for both patients and healthcare providers.
CHOOSING WISELY
The Choosing Wisely campaign in the United States has partnered with the American Academy of Allergy, Asthma, and Immunology to advocate against indiscriminate IgE testing in evaluating allergy.8 Allergy diagnosis and evaluation should be based on a combination of clinical history and judicious ordering of specific IgE tests, whether through skin or blood testing. Ordering of serum allergen-specific IgE tests for food allergies should be consistent with a clinical history of potential IgE-mediated food allergy8 and not food intolerance (Table 2).4,5
Some jurisdictions in Canada have followed suit by restricting the number of serum IgE tests each physician is allowed to order per patient, to encourage more responsible ordering and to lower the number of potential false-positive results, which can lead to increased downstream costs as well as unnecessary patient worry and lifestyle modification.
CLINICAL BOTTOM LINE
Ordering diagnostic tests that have little clinical utility has long-term detrimental effects on both patient safety and healthcare sustainability.
In the case of the 25-year-old evaluated for shellfish allergy, the clinician should first explain that the swelling of the lips and tongue (angioedema) does suggest an IgE-mediated allergic reaction and not a non–IgE-mediated allergic reaction or a food intolerance. Non–IgE-mediated food allergies and food intolerances are marked by symptoms relating mainly to nonimmune aspects of the digestive system, whereas IgE-mediated food allergies affect the immune system and can involve a multitude of organs, including the skin and the respiratory and digestive systems (Table 2).
However, clinicians should avoid indiscriminately ordering food allergen IgE panels and instead should focus on foods likely to be the culprits based on the clinical history.9 Indiscriminate testing can lead to false-positive results and unnecessary food avoidance.
Since the patient developed symptoms of angioedema when he was exposed to his allergen, he may be apprehensive about a skin- prick test and the possibility of being subjected to the same discomfort. Therefore, in this situation, it may be best to perform serum IgE tests, but on a few targeted seafoods rather than the food panel the physician had ordered. A patient can be sensitized to an allergen (possess IgE antibodies) but not experience symptoms when exposed to it (ie, have tolerance).5 Also, false-negative results may occur, so a negative serum allergen-specific IgE test should likewise be interpreted in light of the pretest probability of allergy to a specific antigen.
If the history and the results of testing are not clear and congruent, the patient should be referred to an allergist for diagnosis or for management. The allergist can provide management techniques and periodic assessment as to the progression and resolution of the allergy. Table 2 highlights symptoms that differentiate an IgE-mediated from a non–IgE-mediated food allergy.10,11 Table 1 presents clinical indications and suggested diagnostic methods to the five most common allergen groups and the diagnostically invalid tests.1–6
The bottom line is that we must consider the poor performance of serum allergen-specific IgE tests when diagnosing and treating suspected type I allergies and avoid ordering food allergen IgE panels whenever possible.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Bird JA, Crain M, Varshney P. Food allergen panel testing often results in misdiagnosis of food allergy. J Pediatr 2015; 166:97–100.
- Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am 2015; 35:61–76.
- Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update-2014. J Allergy Clin Immunol 2014; 134:1016–1025.e43.
- Sicherer SH, Sampson HA. Food allergy: epidemiology, pathogenesis, diagnosis, and treatment. J Allergy Clin Immunol 2014; 133:291–308.
- Siles RI, Hsieh FH. Allergy blood testing: a practical guide for clinicians. Cleve Clin J Med 2011; 78:585–592.
- Wians FH Jr. Clinical laboratory tests: which, why, and what do the results mean? Lab Medicine 2009; 40:105–113.
- Choosing Wisely. American Academy of Allergy, Asthma & Immunology. Ten Things Physicians and Patients Should Question. www.choosingwisely.org/doctor-patient-lists/american-academy-of-allergy-asthma-immunology/. Accessed December 3, 2015.
- Fleischer DM, Burks AW. Pitfalls in food allergy diagnosis: serum IgE testing. J Pediatr 2015; 166: 8-10.
- Boyce JA, Assa'ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Stiefel G, Roberts G. How to use serum-specific IgE measurements in diagnosing and monitoring food allergy. Arch Dis Child Educ Pract Ed 2012; 97:29–36.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Bird JA, Crain M, Varshney P. Food allergen panel testing often results in misdiagnosis of food allergy. J Pediatr 2015; 166:97–100.
- Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am 2015; 35:61–76.
- Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update-2014. J Allergy Clin Immunol 2014; 134:1016–1025.e43.
- Sicherer SH, Sampson HA. Food allergy: epidemiology, pathogenesis, diagnosis, and treatment. J Allergy Clin Immunol 2014; 133:291–308.
- Siles RI, Hsieh FH. Allergy blood testing: a practical guide for clinicians. Cleve Clin J Med 2011; 78:585–592.
- Wians FH Jr. Clinical laboratory tests: which, why, and what do the results mean? Lab Medicine 2009; 40:105–113.
- Choosing Wisely. American Academy of Allergy, Asthma & Immunology. Ten Things Physicians and Patients Should Question. www.choosingwisely.org/doctor-patient-lists/american-academy-of-allergy-asthma-immunology/. Accessed December 3, 2015.
- Fleischer DM, Burks AW. Pitfalls in food allergy diagnosis: serum IgE testing. J Pediatr 2015; 166: 8-10.
- Boyce JA, Assa'ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Stiefel G, Roberts G. How to use serum-specific IgE measurements in diagnosing and monitoring food allergy. Arch Dis Child Educ Pract Ed 2012; 97:29–36.