User login
Cross-sectional study finds chronic skin conditions have highest opioid prescribing rates
at dermatology visits.
“Overall, opioid prescribing rates among dermatologists were low. However, dermatologists should remain aware of risk factors for long-term opioid use and consider using nonnarcotic or nonpharmacologic interventions when possible,” Sarah P. Pourali, a medical student at Vanderbilt University, Nashville, said at the annual meeting of the Society for Investigative Dermatology, where she presented the results.
Ms. Pourali said that although Mohs surgery and dermatologic procedures are the focus of “much of the literature” concerning opioid use in dermatology, there are limited data on medication prescribing patterns for other skin conditions treated by dermatologists.
She and her colleagues performed a cross-sectional study using data from the National Ambulatory Medical Care Survey (NAMCS) from 2009 to 2016 on 288,462,610 weighted dermatology visits. The researchers used International Classification of Diseases, Ninth Revision (ICD-9) and ICD-10 codes to identify dermatologic diseases. They also identified and grouped oral pain medication into the following categories: opiate analgesics, nonsteroidal anti-inflammatory drugs, acetaminophen, and gabapentin. A linear regression analysis was used to evaluate pain medicine prescribing each year, and the researchers used a logistic regression analysis to explore how opiate prescriptions were connected to patient clinical characteristics. The analysis was adjusted for age, gender, race, ethnicity, and region.
Overall, most dermatology visits were for patients older than 65 years (36.2%) and 45-64 years old (32.1%). Over half of the dermatologist visits were for women (56.4%) and most (92.2%) visits were for patients who were White (5.1 % were for patients who were Black); most were non-Hispanic or Latino (93.5%). Most dermatology visits were in the South (35.4%) and West (25.2%), followed by the Northeast (21.9%) and Midwest (17.5%).
Opioids were prescribed in 1.3% of the visits, Ms. Pourali said. In addition, 4.7% of visits included an NSAID prescription, 0.7% an acetaminophen prescription, and 0.6% a gabapentin prescription.
Dermatologic procedure visits accounted for 43.1% of opioid prescriptions, she noted. The most common skin conditions for which opioids were prescribed included vitiligo (10.3%), hemangioma (3.8%), pemphigus (3.6%), atopic dermatitis (3.4%), and psoriasis (2.5%).
Although patients older than 65 years accounted for 36.2% of visits to dermatologists, 58.5% of opioids prescribed by dermatologists were for patients in this age group. “We hypothesize that this may be due to a higher proportion of older patients requiring skin cancer surgeries where a lot of opioids are prescribed within dermatology,” Ms. Pourali said.
The highest population-adjusted prescription rates for opiates were in the Northeast and Western regions of the United States, which “partially corroborates” previous studies that have found “higher rates of opioid prescribing in the southern and western U.S.,” she noted.
When evaluating risk-factors for long-term opiate use, Ms. Pourali and colleagues found opioids were also prescribed in 13.2% of visits where a benzodiazepine was prescribed (adjusted odds ratio, 8.17; 95% confidence interval, 5.3-12.7), 8.4% of visits where the patient had a substance abuse disorder (adjusted OR, 9.40; 95% CI, 2.0-44.4), 5.2% of visits with a patient who had depression (adjusted OR, 3.28; 95% CI, 2.0-5.4), and 2.4% of visits with a patient who used tobacco (adjusted OR, 1.09; 95% CI, 1.0-1.1).
Consider nonopioid postoperative pain management options
In an interview, Sailesh Konda, MD, associate clinical professor of dermatology and director of Mohs surgery and surgical dermatology at the University of Florida, Gainesville, who was not involved with the research, noted the finding in the study that vitiligo, hemangioma, pemphigus, AD, and psoriasis were diagnoses with the highest rates of opioid prescription was surprising. “In general, these are conditions that are not routinely managed with opioids,” he said.
NAMCS contains a primary diagnosis field and space for four additional diagnoses such as chronic conditions, as well as thirty fields for medications. “If an opioid was prescribed at a visit, it could have been prescribed for any of the diagnoses related to the visit,” Dr. Konda said. “Additionally, for those opioid prescriptions associated with dermatologic procedures, it would have been helpful to have a breakdown of the specific procedures.”
Dr. Konda compared these results to a recent study of opioid prescribing patterns in the dermatology Medicare population, which found that 93.9% of the top 1% of opioid prescribers were dermatologists working in a surgical practice.
He said that recommendations for opioid prescribing should be developed for general dermatology as they have been for Mohs surgery and dermatologic surgery. For dermatologists currently prescribing opioids, he recommended monitoring prescribing patterns and to “consider nonopioid interventions, such as acetaminophen plus ibuprofen, which has been found to effectively control postoperative pain with fewer complications.”
Ms. Pourali reports no relevant financial disclosures. Her coauthors included the principal investigator, April Armstrong, MD, MPH, professor of dermatology, University of Southern California, Los Angeles. Dr. Konda reports no relevant financial disclosures.
at dermatology visits.
“Overall, opioid prescribing rates among dermatologists were low. However, dermatologists should remain aware of risk factors for long-term opioid use and consider using nonnarcotic or nonpharmacologic interventions when possible,” Sarah P. Pourali, a medical student at Vanderbilt University, Nashville, said at the annual meeting of the Society for Investigative Dermatology, where she presented the results.
Ms. Pourali said that although Mohs surgery and dermatologic procedures are the focus of “much of the literature” concerning opioid use in dermatology, there are limited data on medication prescribing patterns for other skin conditions treated by dermatologists.
She and her colleagues performed a cross-sectional study using data from the National Ambulatory Medical Care Survey (NAMCS) from 2009 to 2016 on 288,462,610 weighted dermatology visits. The researchers used International Classification of Diseases, Ninth Revision (ICD-9) and ICD-10 codes to identify dermatologic diseases. They also identified and grouped oral pain medication into the following categories: opiate analgesics, nonsteroidal anti-inflammatory drugs, acetaminophen, and gabapentin. A linear regression analysis was used to evaluate pain medicine prescribing each year, and the researchers used a logistic regression analysis to explore how opiate prescriptions were connected to patient clinical characteristics. The analysis was adjusted for age, gender, race, ethnicity, and region.
Overall, most dermatology visits were for patients older than 65 years (36.2%) and 45-64 years old (32.1%). Over half of the dermatologist visits were for women (56.4%) and most (92.2%) visits were for patients who were White (5.1 % were for patients who were Black); most were non-Hispanic or Latino (93.5%). Most dermatology visits were in the South (35.4%) and West (25.2%), followed by the Northeast (21.9%) and Midwest (17.5%).
Opioids were prescribed in 1.3% of the visits, Ms. Pourali said. In addition, 4.7% of visits included an NSAID prescription, 0.7% an acetaminophen prescription, and 0.6% a gabapentin prescription.
Dermatologic procedure visits accounted for 43.1% of opioid prescriptions, she noted. The most common skin conditions for which opioids were prescribed included vitiligo (10.3%), hemangioma (3.8%), pemphigus (3.6%), atopic dermatitis (3.4%), and psoriasis (2.5%).
Although patients older than 65 years accounted for 36.2% of visits to dermatologists, 58.5% of opioids prescribed by dermatologists were for patients in this age group. “We hypothesize that this may be due to a higher proportion of older patients requiring skin cancer surgeries where a lot of opioids are prescribed within dermatology,” Ms. Pourali said.
The highest population-adjusted prescription rates for opiates were in the Northeast and Western regions of the United States, which “partially corroborates” previous studies that have found “higher rates of opioid prescribing in the southern and western U.S.,” she noted.
When evaluating risk-factors for long-term opiate use, Ms. Pourali and colleagues found opioids were also prescribed in 13.2% of visits where a benzodiazepine was prescribed (adjusted odds ratio, 8.17; 95% confidence interval, 5.3-12.7), 8.4% of visits where the patient had a substance abuse disorder (adjusted OR, 9.40; 95% CI, 2.0-44.4), 5.2% of visits with a patient who had depression (adjusted OR, 3.28; 95% CI, 2.0-5.4), and 2.4% of visits with a patient who used tobacco (adjusted OR, 1.09; 95% CI, 1.0-1.1).
Consider nonopioid postoperative pain management options
In an interview, Sailesh Konda, MD, associate clinical professor of dermatology and director of Mohs surgery and surgical dermatology at the University of Florida, Gainesville, who was not involved with the research, noted the finding in the study that vitiligo, hemangioma, pemphigus, AD, and psoriasis were diagnoses with the highest rates of opioid prescription was surprising. “In general, these are conditions that are not routinely managed with opioids,” he said.
NAMCS contains a primary diagnosis field and space for four additional diagnoses such as chronic conditions, as well as thirty fields for medications. “If an opioid was prescribed at a visit, it could have been prescribed for any of the diagnoses related to the visit,” Dr. Konda said. “Additionally, for those opioid prescriptions associated with dermatologic procedures, it would have been helpful to have a breakdown of the specific procedures.”
Dr. Konda compared these results to a recent study of opioid prescribing patterns in the dermatology Medicare population, which found that 93.9% of the top 1% of opioid prescribers were dermatologists working in a surgical practice.
He said that recommendations for opioid prescribing should be developed for general dermatology as they have been for Mohs surgery and dermatologic surgery. For dermatologists currently prescribing opioids, he recommended monitoring prescribing patterns and to “consider nonopioid interventions, such as acetaminophen plus ibuprofen, which has been found to effectively control postoperative pain with fewer complications.”
Ms. Pourali reports no relevant financial disclosures. Her coauthors included the principal investigator, April Armstrong, MD, MPH, professor of dermatology, University of Southern California, Los Angeles. Dr. Konda reports no relevant financial disclosures.
at dermatology visits.
“Overall, opioid prescribing rates among dermatologists were low. However, dermatologists should remain aware of risk factors for long-term opioid use and consider using nonnarcotic or nonpharmacologic interventions when possible,” Sarah P. Pourali, a medical student at Vanderbilt University, Nashville, said at the annual meeting of the Society for Investigative Dermatology, where she presented the results.
Ms. Pourali said that although Mohs surgery and dermatologic procedures are the focus of “much of the literature” concerning opioid use in dermatology, there are limited data on medication prescribing patterns for other skin conditions treated by dermatologists.
She and her colleagues performed a cross-sectional study using data from the National Ambulatory Medical Care Survey (NAMCS) from 2009 to 2016 on 288,462,610 weighted dermatology visits. The researchers used International Classification of Diseases, Ninth Revision (ICD-9) and ICD-10 codes to identify dermatologic diseases. They also identified and grouped oral pain medication into the following categories: opiate analgesics, nonsteroidal anti-inflammatory drugs, acetaminophen, and gabapentin. A linear regression analysis was used to evaluate pain medicine prescribing each year, and the researchers used a logistic regression analysis to explore how opiate prescriptions were connected to patient clinical characteristics. The analysis was adjusted for age, gender, race, ethnicity, and region.
Overall, most dermatology visits were for patients older than 65 years (36.2%) and 45-64 years old (32.1%). Over half of the dermatologist visits were for women (56.4%) and most (92.2%) visits were for patients who were White (5.1 % were for patients who were Black); most were non-Hispanic or Latino (93.5%). Most dermatology visits were in the South (35.4%) and West (25.2%), followed by the Northeast (21.9%) and Midwest (17.5%).
Opioids were prescribed in 1.3% of the visits, Ms. Pourali said. In addition, 4.7% of visits included an NSAID prescription, 0.7% an acetaminophen prescription, and 0.6% a gabapentin prescription.
Dermatologic procedure visits accounted for 43.1% of opioid prescriptions, she noted. The most common skin conditions for which opioids were prescribed included vitiligo (10.3%), hemangioma (3.8%), pemphigus (3.6%), atopic dermatitis (3.4%), and psoriasis (2.5%).
Although patients older than 65 years accounted for 36.2% of visits to dermatologists, 58.5% of opioids prescribed by dermatologists were for patients in this age group. “We hypothesize that this may be due to a higher proportion of older patients requiring skin cancer surgeries where a lot of opioids are prescribed within dermatology,” Ms. Pourali said.
The highest population-adjusted prescription rates for opiates were in the Northeast and Western regions of the United States, which “partially corroborates” previous studies that have found “higher rates of opioid prescribing in the southern and western U.S.,” she noted.
When evaluating risk-factors for long-term opiate use, Ms. Pourali and colleagues found opioids were also prescribed in 13.2% of visits where a benzodiazepine was prescribed (adjusted odds ratio, 8.17; 95% confidence interval, 5.3-12.7), 8.4% of visits where the patient had a substance abuse disorder (adjusted OR, 9.40; 95% CI, 2.0-44.4), 5.2% of visits with a patient who had depression (adjusted OR, 3.28; 95% CI, 2.0-5.4), and 2.4% of visits with a patient who used tobacco (adjusted OR, 1.09; 95% CI, 1.0-1.1).
Consider nonopioid postoperative pain management options
In an interview, Sailesh Konda, MD, associate clinical professor of dermatology and director of Mohs surgery and surgical dermatology at the University of Florida, Gainesville, who was not involved with the research, noted the finding in the study that vitiligo, hemangioma, pemphigus, AD, and psoriasis were diagnoses with the highest rates of opioid prescription was surprising. “In general, these are conditions that are not routinely managed with opioids,” he said.
NAMCS contains a primary diagnosis field and space for four additional diagnoses such as chronic conditions, as well as thirty fields for medications. “If an opioid was prescribed at a visit, it could have been prescribed for any of the diagnoses related to the visit,” Dr. Konda said. “Additionally, for those opioid prescriptions associated with dermatologic procedures, it would have been helpful to have a breakdown of the specific procedures.”
Dr. Konda compared these results to a recent study of opioid prescribing patterns in the dermatology Medicare population, which found that 93.9% of the top 1% of opioid prescribers were dermatologists working in a surgical practice.
He said that recommendations for opioid prescribing should be developed for general dermatology as they have been for Mohs surgery and dermatologic surgery. For dermatologists currently prescribing opioids, he recommended monitoring prescribing patterns and to “consider nonopioid interventions, such as acetaminophen plus ibuprofen, which has been found to effectively control postoperative pain with fewer complications.”
Ms. Pourali reports no relevant financial disclosures. Her coauthors included the principal investigator, April Armstrong, MD, MPH, professor of dermatology, University of Southern California, Los Angeles. Dr. Konda reports no relevant financial disclosures.
FROM SID 2021
Lenabasum missed mark for systemic sclerosis but may show promise for adjunctive therapy
Although a phase 3 trial of lenabasum did not meet its primary endpoint for treatment of diffuse cutaneous systemic sclerosis (dcSSc), the drug led to more improvement in participants who were not receiving background immunosuppressant therapy during the trial than that seen in participants who received the placebo. Lenabasum also had a favorable safety profile, according to findings presented at the annual European Congress of Rheumatology.
The double-blind, randomized, placebo-controlled trial involved 363 adults who had had dcSSc for up to 6 years. One third of the participants received 5 mg of oral lenabasum, one third received 20 mg, and one third received a placebo. Patients already receiving immunosuppressant therapy could continue to receive it during the trial if the dose had been stable for at least 8 weeks before screening and corticosteroid therapy did not exceed 10 mg prednisone per day or the equivalent.
“The decision to allow background immunosuppressant therapies was made to reflect real-world clinical practice,” coprincipal investigator Robert Dr. Spiera, MD, director of the Vasculitis and Scleroderma Program at the Hospital for Special Surgery, New York, told attendees.
“It is surprising that we do not see any added efficacy of lenabasum in this trial, given the fact that the previous phase 2 trial in 42 patients did show a clear benefit of lenabasum over placebo in the same population,” Jeska K. de Vries-Bouwstra, MD, PhD, a rheumatologist at Leiden (the Netherlands) University Medical Center told this news organization. “Even more, the clinical response in the phase 2 study was supported by a greater change in gene expression in skin tissue of pathways involved in inflammation and fibrosis with lenabasum as compared to placebo.”
Background immunosuppressants contribute to unprecedented placebo responses
The researchers compared the ACR CRISS (Combined Response Index in Diffuse Cutaneous Systemic Sclerosis) score and several secondary endpoints at 52 weeks between the 123 participants who received the placebo and the 120 participants who received 20 mg of lenabasum. A total of 60% of the lenabasum group and 66% of the placebo group had a disease duration of 3 or fewer years, and the modified Rodnan skin score (mRSS) was 22 in the lenabasum group and 23.3 in the placebo group at baseline.
A large majority of participants in both groups – 89% in the lenabasum group and 84% in the placebo group – were receiving background immunosuppressant therapy during the trial. Specifically, 53% of each group was taking mycophenolate, and 23% of the lenabasum group and 32% of the placebo group were taking corticosteroids. In addition, 22% of the lenabasum group and 12% of the placebo group were on methotrexate, and 27% of the lenabasum group and 22% of the placebo group were on another immunosuppressant therapy.
Half of the placebo group and 58% of the lenabasum group were taking only one immunosuppressive therapy. About one-third of the lenabasum (32%) and placebo (34%) groups were taking two or more immunosuppressive therapies.
The primary endpoint at 52 weeks was not significantly different between the two groups: a CRISS score of 0.888 in the lenabasum group and 0.887 in the placebo group. A CRISS score of 0.6 or higher indicates likelihood that a patient improved on treatment. Patients with significant worsening of renal or cardiopulmonary involvement are classified as not improved (score of 0), regardless of improvements in other core items.
“We had very high CRISS scores in all three groups, and they were comparable in all three groups,” Dr. Spiera reported. Because improvement in placebo group far exceeded expectations, the researchers were unable to discern the treatment effect of lenabasum on top of the placebo effect.
The placebo group had better outcomes than expected because of the background immunosuppressant therapy, particularly the use of mycophenolate. When the researchers looked only at placebo participants, the CRISS score was 0.936 in the 97 patients receiving background immunosuppressant therapy of any kind and 0.935 in the 29 patients taking only mycophenolate with no other immunosuppressant therapy, compared with 0.417 in the 16 patients not receiving any background therapy.
In a prespecified analysis, the researchers investigated background immunosuppressive therapy as a mediator. The CRISS score for the 10 lenabasum participants not receiving background therapy was 0.811, compared with 0.417 seen in the placebo group patients not on background therapy.
Among the 173 participants taking mycophenolate in particular, the mycophenolate “had a statistically significant improvement on CRISS score that increased with each visit,” Dr. Spiera reported. The duration of mycophenolate therapy also affected efficacy results.
Patients who had been taking mycophenolate longer saw less improvement in their CRISS score over time. Those taking it more than 2 years at baseline had a CRISS score of 0.86, compared with 0.96 for those taking it for 1-2 years at baseline and 0.98 for those taking it from 6 months to 1 year at baseline. Those who had only been taking mycophenolate for up to 6 months at baseline had a CRISS score of 0.99. Meanwhile, patients not taking any background immunosuppressant therapies only had a CRISS score of about 0.35.
Changes in secondary endpoints followed same pattern as CRISS
The secondary endpoints similarly showed no statistically significant difference when comparing the lenabasum and placebo groups overall. These endpoints included change in mRSS score, change in forced vital capacity (FVC) percentage and volume, and change in the Health Assessment Questionnaire Disability Index (HAQ-DI) score.
However, the researchers again found that duration of background therapy affected FVC.
“You were more likely to have declined [in FVC] if you were on placebo and more likely to have improved or stayed stable if you were on lenabasum if you were a patient on more than 2 years of immunomodulatory therapy at baseline,” Dr. Spiera reported. “There was evidence for an effect of lenabasum on FVC suggested by post-hoc analyses that considered the effect of background immunosuppressive therapies on outcomes, but those results would require confirmation in additional studies to determine the potential of lenabasum for treating patients with diffuse cutaneous systemic sclerosis,” Dr. Spiera noted in his conclusions.
Serious adverse events occurred in 9.2% of the lenabasum group and 5.8% of the placebo group. Rates of severe adverse events were similar between the lenabasum (14.6%) and placebo (13%) groups.
Is there a subgroup for whom lenabasum would be efficacious?
Although De Vries-Bouwstra of Leiden University Medical Center acknowledged the role of mycophenolate in the trial, she does not think background therapy can totally explain the observation and speculated on other possibilities.
“For example, there were fewer males in the placebo group as compared to the phase 2 study. From previous cohort studies we know that males have higher risk of worsening of skin disease,” she said. “In addition, it could be worthwhile to evaluate antibody profiles of the population under study; some subpopulations defined by autoantibody have higher risk for skin progression, while others can show spontaneous improvement.”
Dr. De Vries-Bouwstra said that, although it’s not currently appropriate to advocate for lenabasum to treat dcSSc, it may eventually become an additional treatment in those who still show active skin or lung disease after 2 years of mycophenolate treatment if future research identifies a benefit from that application. She would also like to see an evaluation of lenabasum’s possible benefits in patients with very early and active inflammatory disease. “Ideally, one could stratify patients based on biomarkers reflecting activation in relevant pathways, for example by using gene expression analysis from skin tissue to stratify,” she said.
Jacob M. van Laar, MD, PhD, professor of rheumatology at University Medical Center Utrecht (the Netherlands), also commented on the potential differences in using the drug in early versus later disease.
“Based on ex vivo analyses of skin samples from systemic sclerosis patients, one would expect such a mechanism of action to be particularly relevant in very early disease, so the observation that it might also be effective at a later disease stage is interesting,” Dr. van Laar told this news organization. “We still have a lot to learn about this complex disease.”
Given that safety does not appear to be a major concern and that there may be a benefit in a subgroup of patients, Dr. van Laar also said he hoped “the company is not deterred by the seemingly negative result of the primary endpoint.”
Dr. Spiera expressed optimism about what this trial’s findings have revealed about management of dcSSc.
“Independent of what lenabasum did or didn’t do in this trial, I think there’s going to be a lot that we’re going to learn from this trial and that we’re already learning and analyzing right now about treating scleroderma,” he said in an interview.
He reiterated the value of allowing background therapy in the trial to ensure it better replicated real-world clinical practice.
“You’re not withholding therapies that we think are probably active from patients with active disease that, once you incur organ damage, is probably not going to be reversible,” Dr. Spiera said. “The downside is that it makes it harder to see an effect of a drug on top of the background therapy if that background therapy is effective. So what we saw in terms of this absence of benefit from lenabasum really may have been a ceiling effect.”
Nevertheless, Dr. Spiera said the findings still strongly suggest that lenabasum is an active compound.
“It’s not an enormously powerful effect, but it probably has a role as an adjunctive therapy in people on stable background therapy who have either plateaued or are getting worse,” he said. “The thing we have to keep in mind also is this was an incredibly safe therapy. It’s not immunosuppressive.”
The trial was funded by Corbus. Dr. Spiera has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, Boehringer Ingelheim, Chemocentryx, Corbus, Formation Biologics, Inflarx, Kadmon, AstraZeneca, AbbVie, CSL Behring, Sanofi, and Janssen. Dr. De Vries-Bouwstra has received consulting fees from AbbVie and Boehringer Ingelheim and research grants from Galapagos and Janssen. Dr. Van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Although a phase 3 trial of lenabasum did not meet its primary endpoint for treatment of diffuse cutaneous systemic sclerosis (dcSSc), the drug led to more improvement in participants who were not receiving background immunosuppressant therapy during the trial than that seen in participants who received the placebo. Lenabasum also had a favorable safety profile, according to findings presented at the annual European Congress of Rheumatology.
The double-blind, randomized, placebo-controlled trial involved 363 adults who had had dcSSc for up to 6 years. One third of the participants received 5 mg of oral lenabasum, one third received 20 mg, and one third received a placebo. Patients already receiving immunosuppressant therapy could continue to receive it during the trial if the dose had been stable for at least 8 weeks before screening and corticosteroid therapy did not exceed 10 mg prednisone per day or the equivalent.
“The decision to allow background immunosuppressant therapies was made to reflect real-world clinical practice,” coprincipal investigator Robert Dr. Spiera, MD, director of the Vasculitis and Scleroderma Program at the Hospital for Special Surgery, New York, told attendees.
“It is surprising that we do not see any added efficacy of lenabasum in this trial, given the fact that the previous phase 2 trial in 42 patients did show a clear benefit of lenabasum over placebo in the same population,” Jeska K. de Vries-Bouwstra, MD, PhD, a rheumatologist at Leiden (the Netherlands) University Medical Center told this news organization. “Even more, the clinical response in the phase 2 study was supported by a greater change in gene expression in skin tissue of pathways involved in inflammation and fibrosis with lenabasum as compared to placebo.”
Background immunosuppressants contribute to unprecedented placebo responses
The researchers compared the ACR CRISS (Combined Response Index in Diffuse Cutaneous Systemic Sclerosis) score and several secondary endpoints at 52 weeks between the 123 participants who received the placebo and the 120 participants who received 20 mg of lenabasum. A total of 60% of the lenabasum group and 66% of the placebo group had a disease duration of 3 or fewer years, and the modified Rodnan skin score (mRSS) was 22 in the lenabasum group and 23.3 in the placebo group at baseline.
A large majority of participants in both groups – 89% in the lenabasum group and 84% in the placebo group – were receiving background immunosuppressant therapy during the trial. Specifically, 53% of each group was taking mycophenolate, and 23% of the lenabasum group and 32% of the placebo group were taking corticosteroids. In addition, 22% of the lenabasum group and 12% of the placebo group were on methotrexate, and 27% of the lenabasum group and 22% of the placebo group were on another immunosuppressant therapy.
Half of the placebo group and 58% of the lenabasum group were taking only one immunosuppressive therapy. About one-third of the lenabasum (32%) and placebo (34%) groups were taking two or more immunosuppressive therapies.
The primary endpoint at 52 weeks was not significantly different between the two groups: a CRISS score of 0.888 in the lenabasum group and 0.887 in the placebo group. A CRISS score of 0.6 or higher indicates likelihood that a patient improved on treatment. Patients with significant worsening of renal or cardiopulmonary involvement are classified as not improved (score of 0), regardless of improvements in other core items.
“We had very high CRISS scores in all three groups, and they were comparable in all three groups,” Dr. Spiera reported. Because improvement in placebo group far exceeded expectations, the researchers were unable to discern the treatment effect of lenabasum on top of the placebo effect.
The placebo group had better outcomes than expected because of the background immunosuppressant therapy, particularly the use of mycophenolate. When the researchers looked only at placebo participants, the CRISS score was 0.936 in the 97 patients receiving background immunosuppressant therapy of any kind and 0.935 in the 29 patients taking only mycophenolate with no other immunosuppressant therapy, compared with 0.417 in the 16 patients not receiving any background therapy.
In a prespecified analysis, the researchers investigated background immunosuppressive therapy as a mediator. The CRISS score for the 10 lenabasum participants not receiving background therapy was 0.811, compared with 0.417 seen in the placebo group patients not on background therapy.
Among the 173 participants taking mycophenolate in particular, the mycophenolate “had a statistically significant improvement on CRISS score that increased with each visit,” Dr. Spiera reported. The duration of mycophenolate therapy also affected efficacy results.
Patients who had been taking mycophenolate longer saw less improvement in their CRISS score over time. Those taking it more than 2 years at baseline had a CRISS score of 0.86, compared with 0.96 for those taking it for 1-2 years at baseline and 0.98 for those taking it from 6 months to 1 year at baseline. Those who had only been taking mycophenolate for up to 6 months at baseline had a CRISS score of 0.99. Meanwhile, patients not taking any background immunosuppressant therapies only had a CRISS score of about 0.35.
Changes in secondary endpoints followed same pattern as CRISS
The secondary endpoints similarly showed no statistically significant difference when comparing the lenabasum and placebo groups overall. These endpoints included change in mRSS score, change in forced vital capacity (FVC) percentage and volume, and change in the Health Assessment Questionnaire Disability Index (HAQ-DI) score.
However, the researchers again found that duration of background therapy affected FVC.
“You were more likely to have declined [in FVC] if you were on placebo and more likely to have improved or stayed stable if you were on lenabasum if you were a patient on more than 2 years of immunomodulatory therapy at baseline,” Dr. Spiera reported. “There was evidence for an effect of lenabasum on FVC suggested by post-hoc analyses that considered the effect of background immunosuppressive therapies on outcomes, but those results would require confirmation in additional studies to determine the potential of lenabasum for treating patients with diffuse cutaneous systemic sclerosis,” Dr. Spiera noted in his conclusions.
Serious adverse events occurred in 9.2% of the lenabasum group and 5.8% of the placebo group. Rates of severe adverse events were similar between the lenabasum (14.6%) and placebo (13%) groups.
Is there a subgroup for whom lenabasum would be efficacious?
Although De Vries-Bouwstra of Leiden University Medical Center acknowledged the role of mycophenolate in the trial, she does not think background therapy can totally explain the observation and speculated on other possibilities.
“For example, there were fewer males in the placebo group as compared to the phase 2 study. From previous cohort studies we know that males have higher risk of worsening of skin disease,” she said. “In addition, it could be worthwhile to evaluate antibody profiles of the population under study; some subpopulations defined by autoantibody have higher risk for skin progression, while others can show spontaneous improvement.”
Dr. De Vries-Bouwstra said that, although it’s not currently appropriate to advocate for lenabasum to treat dcSSc, it may eventually become an additional treatment in those who still show active skin or lung disease after 2 years of mycophenolate treatment if future research identifies a benefit from that application. She would also like to see an evaluation of lenabasum’s possible benefits in patients with very early and active inflammatory disease. “Ideally, one could stratify patients based on biomarkers reflecting activation in relevant pathways, for example by using gene expression analysis from skin tissue to stratify,” she said.
Jacob M. van Laar, MD, PhD, professor of rheumatology at University Medical Center Utrecht (the Netherlands), also commented on the potential differences in using the drug in early versus later disease.
“Based on ex vivo analyses of skin samples from systemic sclerosis patients, one would expect such a mechanism of action to be particularly relevant in very early disease, so the observation that it might also be effective at a later disease stage is interesting,” Dr. van Laar told this news organization. “We still have a lot to learn about this complex disease.”
Given that safety does not appear to be a major concern and that there may be a benefit in a subgroup of patients, Dr. van Laar also said he hoped “the company is not deterred by the seemingly negative result of the primary endpoint.”
Dr. Spiera expressed optimism about what this trial’s findings have revealed about management of dcSSc.
“Independent of what lenabasum did or didn’t do in this trial, I think there’s going to be a lot that we’re going to learn from this trial and that we’re already learning and analyzing right now about treating scleroderma,” he said in an interview.
He reiterated the value of allowing background therapy in the trial to ensure it better replicated real-world clinical practice.
“You’re not withholding therapies that we think are probably active from patients with active disease that, once you incur organ damage, is probably not going to be reversible,” Dr. Spiera said. “The downside is that it makes it harder to see an effect of a drug on top of the background therapy if that background therapy is effective. So what we saw in terms of this absence of benefit from lenabasum really may have been a ceiling effect.”
Nevertheless, Dr. Spiera said the findings still strongly suggest that lenabasum is an active compound.
“It’s not an enormously powerful effect, but it probably has a role as an adjunctive therapy in people on stable background therapy who have either plateaued or are getting worse,” he said. “The thing we have to keep in mind also is this was an incredibly safe therapy. It’s not immunosuppressive.”
The trial was funded by Corbus. Dr. Spiera has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, Boehringer Ingelheim, Chemocentryx, Corbus, Formation Biologics, Inflarx, Kadmon, AstraZeneca, AbbVie, CSL Behring, Sanofi, and Janssen. Dr. De Vries-Bouwstra has received consulting fees from AbbVie and Boehringer Ingelheim and research grants from Galapagos and Janssen. Dr. Van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Although a phase 3 trial of lenabasum did not meet its primary endpoint for treatment of diffuse cutaneous systemic sclerosis (dcSSc), the drug led to more improvement in participants who were not receiving background immunosuppressant therapy during the trial than that seen in participants who received the placebo. Lenabasum also had a favorable safety profile, according to findings presented at the annual European Congress of Rheumatology.
The double-blind, randomized, placebo-controlled trial involved 363 adults who had had dcSSc for up to 6 years. One third of the participants received 5 mg of oral lenabasum, one third received 20 mg, and one third received a placebo. Patients already receiving immunosuppressant therapy could continue to receive it during the trial if the dose had been stable for at least 8 weeks before screening and corticosteroid therapy did not exceed 10 mg prednisone per day or the equivalent.
“The decision to allow background immunosuppressant therapies was made to reflect real-world clinical practice,” coprincipal investigator Robert Dr. Spiera, MD, director of the Vasculitis and Scleroderma Program at the Hospital for Special Surgery, New York, told attendees.
“It is surprising that we do not see any added efficacy of lenabasum in this trial, given the fact that the previous phase 2 trial in 42 patients did show a clear benefit of lenabasum over placebo in the same population,” Jeska K. de Vries-Bouwstra, MD, PhD, a rheumatologist at Leiden (the Netherlands) University Medical Center told this news organization. “Even more, the clinical response in the phase 2 study was supported by a greater change in gene expression in skin tissue of pathways involved in inflammation and fibrosis with lenabasum as compared to placebo.”
Background immunosuppressants contribute to unprecedented placebo responses
The researchers compared the ACR CRISS (Combined Response Index in Diffuse Cutaneous Systemic Sclerosis) score and several secondary endpoints at 52 weeks between the 123 participants who received the placebo and the 120 participants who received 20 mg of lenabasum. A total of 60% of the lenabasum group and 66% of the placebo group had a disease duration of 3 or fewer years, and the modified Rodnan skin score (mRSS) was 22 in the lenabasum group and 23.3 in the placebo group at baseline.
A large majority of participants in both groups – 89% in the lenabasum group and 84% in the placebo group – were receiving background immunosuppressant therapy during the trial. Specifically, 53% of each group was taking mycophenolate, and 23% of the lenabasum group and 32% of the placebo group were taking corticosteroids. In addition, 22% of the lenabasum group and 12% of the placebo group were on methotrexate, and 27% of the lenabasum group and 22% of the placebo group were on another immunosuppressant therapy.
Half of the placebo group and 58% of the lenabasum group were taking only one immunosuppressive therapy. About one-third of the lenabasum (32%) and placebo (34%) groups were taking two or more immunosuppressive therapies.
The primary endpoint at 52 weeks was not significantly different between the two groups: a CRISS score of 0.888 in the lenabasum group and 0.887 in the placebo group. A CRISS score of 0.6 or higher indicates likelihood that a patient improved on treatment. Patients with significant worsening of renal or cardiopulmonary involvement are classified as not improved (score of 0), regardless of improvements in other core items.
“We had very high CRISS scores in all three groups, and they were comparable in all three groups,” Dr. Spiera reported. Because improvement in placebo group far exceeded expectations, the researchers were unable to discern the treatment effect of lenabasum on top of the placebo effect.
The placebo group had better outcomes than expected because of the background immunosuppressant therapy, particularly the use of mycophenolate. When the researchers looked only at placebo participants, the CRISS score was 0.936 in the 97 patients receiving background immunosuppressant therapy of any kind and 0.935 in the 29 patients taking only mycophenolate with no other immunosuppressant therapy, compared with 0.417 in the 16 patients not receiving any background therapy.
In a prespecified analysis, the researchers investigated background immunosuppressive therapy as a mediator. The CRISS score for the 10 lenabasum participants not receiving background therapy was 0.811, compared with 0.417 seen in the placebo group patients not on background therapy.
Among the 173 participants taking mycophenolate in particular, the mycophenolate “had a statistically significant improvement on CRISS score that increased with each visit,” Dr. Spiera reported. The duration of mycophenolate therapy also affected efficacy results.
Patients who had been taking mycophenolate longer saw less improvement in their CRISS score over time. Those taking it more than 2 years at baseline had a CRISS score of 0.86, compared with 0.96 for those taking it for 1-2 years at baseline and 0.98 for those taking it from 6 months to 1 year at baseline. Those who had only been taking mycophenolate for up to 6 months at baseline had a CRISS score of 0.99. Meanwhile, patients not taking any background immunosuppressant therapies only had a CRISS score of about 0.35.
Changes in secondary endpoints followed same pattern as CRISS
The secondary endpoints similarly showed no statistically significant difference when comparing the lenabasum and placebo groups overall. These endpoints included change in mRSS score, change in forced vital capacity (FVC) percentage and volume, and change in the Health Assessment Questionnaire Disability Index (HAQ-DI) score.
However, the researchers again found that duration of background therapy affected FVC.
“You were more likely to have declined [in FVC] if you were on placebo and more likely to have improved or stayed stable if you were on lenabasum if you were a patient on more than 2 years of immunomodulatory therapy at baseline,” Dr. Spiera reported. “There was evidence for an effect of lenabasum on FVC suggested by post-hoc analyses that considered the effect of background immunosuppressive therapies on outcomes, but those results would require confirmation in additional studies to determine the potential of lenabasum for treating patients with diffuse cutaneous systemic sclerosis,” Dr. Spiera noted in his conclusions.
Serious adverse events occurred in 9.2% of the lenabasum group and 5.8% of the placebo group. Rates of severe adverse events were similar between the lenabasum (14.6%) and placebo (13%) groups.
Is there a subgroup for whom lenabasum would be efficacious?
Although De Vries-Bouwstra of Leiden University Medical Center acknowledged the role of mycophenolate in the trial, she does not think background therapy can totally explain the observation and speculated on other possibilities.
“For example, there were fewer males in the placebo group as compared to the phase 2 study. From previous cohort studies we know that males have higher risk of worsening of skin disease,” she said. “In addition, it could be worthwhile to evaluate antibody profiles of the population under study; some subpopulations defined by autoantibody have higher risk for skin progression, while others can show spontaneous improvement.”
Dr. De Vries-Bouwstra said that, although it’s not currently appropriate to advocate for lenabasum to treat dcSSc, it may eventually become an additional treatment in those who still show active skin or lung disease after 2 years of mycophenolate treatment if future research identifies a benefit from that application. She would also like to see an evaluation of lenabasum’s possible benefits in patients with very early and active inflammatory disease. “Ideally, one could stratify patients based on biomarkers reflecting activation in relevant pathways, for example by using gene expression analysis from skin tissue to stratify,” she said.
Jacob M. van Laar, MD, PhD, professor of rheumatology at University Medical Center Utrecht (the Netherlands), also commented on the potential differences in using the drug in early versus later disease.
“Based on ex vivo analyses of skin samples from systemic sclerosis patients, one would expect such a mechanism of action to be particularly relevant in very early disease, so the observation that it might also be effective at a later disease stage is interesting,” Dr. van Laar told this news organization. “We still have a lot to learn about this complex disease.”
Given that safety does not appear to be a major concern and that there may be a benefit in a subgroup of patients, Dr. van Laar also said he hoped “the company is not deterred by the seemingly negative result of the primary endpoint.”
Dr. Spiera expressed optimism about what this trial’s findings have revealed about management of dcSSc.
“Independent of what lenabasum did or didn’t do in this trial, I think there’s going to be a lot that we’re going to learn from this trial and that we’re already learning and analyzing right now about treating scleroderma,” he said in an interview.
He reiterated the value of allowing background therapy in the trial to ensure it better replicated real-world clinical practice.
“You’re not withholding therapies that we think are probably active from patients with active disease that, once you incur organ damage, is probably not going to be reversible,” Dr. Spiera said. “The downside is that it makes it harder to see an effect of a drug on top of the background therapy if that background therapy is effective. So what we saw in terms of this absence of benefit from lenabasum really may have been a ceiling effect.”
Nevertheless, Dr. Spiera said the findings still strongly suggest that lenabasum is an active compound.
“It’s not an enormously powerful effect, but it probably has a role as an adjunctive therapy in people on stable background therapy who have either plateaued or are getting worse,” he said. “The thing we have to keep in mind also is this was an incredibly safe therapy. It’s not immunosuppressive.”
The trial was funded by Corbus. Dr. Spiera has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, Boehringer Ingelheim, Chemocentryx, Corbus, Formation Biologics, Inflarx, Kadmon, AstraZeneca, AbbVie, CSL Behring, Sanofi, and Janssen. Dr. De Vries-Bouwstra has received consulting fees from AbbVie and Boehringer Ingelheim and research grants from Galapagos and Janssen. Dr. Van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Intravenous immunoglobulin controls dermatomyositis in phase 3 trial
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
Nearly 50% achieve moderate improvement or better
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Rituximab superior to mycophenolate mofetil in pemphigus vulgaris study
Mycophenolate mofetil, commonly used as a first-line corticosteroid-sparing agent for moderate to severe cases of the autoimmune blistering skin condition pemphigus vulgaris, has been found to be inferior to the biologic agent rituximab.

Mycophenolate mofetil is widely accepted as a first-in-line corticosteroid-sparing agent for pemphigus vulgaris, but few studies have compared the effectiveness of the two treatments for pemphigus vulgaris. The European Academy of Dermatology and Venereology recommends rituximab (Rituxan), a CD20 inhibitor, as first-line treatment for patients with new-onset cases of moderate to severe intensity or for patients who fail to achieve clinical remission with systemic corticosteroids with or without other immunosuppressive treatments.
In the current study, published online on May 19, 2021, in the New England Journal of Medicine, researchers led by Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, conducted a randomized, controlled trial of 135 patients (mean age, 48 years; 53% women) with moderate to severe pemphigus vulgaris with 67 receiving rituximab and 68 receiving mycophenolate mofetil (99% of patients in the rituximab group and 85% of patients in the mycophenolate mofetil group completed the trial).
Patients in the rituximab group received 1,000 mg of IV rituximab on days 1, 15, 168, and 182 of the study, plus twice-daily oral placebo. Intravenous methylprednisolone at 100 mg was administered before each rituximab infusion to reduce infusion-related reactions. Patients in the second group were given mycophenolate mofetil orally twice daily, starting at 1 g/day in divided doses and adjusted to 2 g/day in divided doses by week 2. They also received placebo infusions on days 1, 15, 168, and 182 of the study.
Patients in both groups received oral glucocorticoids throughout the course of the trial: an average of 3,545 mg for the rituximab treatment group and a cumulative dose of 5,140 mg for the group treated with mycophenolate mofetil, a statistically significant difference (P < .001). Outcomes based on 62 patients treated with rituximab and 63 on MMF, a modified intention-to-treat group.
By week 52, 25 patients (40%) who were treated with rituximab experienced complete sustained remission (the primary endpoint), compared with 6 patients (10%) in the mycophenolate mofetil group (95% confidence interval, 15-45, P < .001).
Only six patients in the rituximab group experienced a disease flare as compared with 44 patients in the mycophenolate mofetil group (adjusted rate ratio, 0.12; 95% CI, 0.05-0.29; P < .001). Serious adverse events occurred in 15 of 67 patients (22%) in the rituximab group and in 10 of 68 (15%) in the mycophenolate mofetil group with 3 patients in the rituximab group and 26 in the mycophenolate mofetil receiving rescue therapy.
Second to remission, the goal of treatment for pemphigus vulgaris is to reduce the use of glucocorticoids, Dr. Werth and colleagues wrote, adding: “The results of this trial showed that rituximab was superior to mycophenolate mofetil in producing sustained complete remission over 52 weeks among patients with moderate to severe pemphigus vulgaris. Rituximab had a greater glucocorticoid-sparing effect than mycophenolate mofetil, but more patients in this group had serious adverse events.”
Most adverse events in the rituximab group were limited to infusion-related reactions, but serious adverse events occurred in 15 patients (including pneumonia and upper respiratory tract infection, cellulitis and acute pyelonephritis, viral pneumonia, and skin infection). Ten patients in the mycophenolate mofetil group experienced serious adverse events (pneumonia and influenza, cellulitis and sepsis, herpes zoster, and pyelonephritis).
The current study had several limitations, primarily its small size. Plus, the authors noted a short follow-up period after glucocorticoids were stopped.
Mycophenolate mofetil, along with immunosuppressants, is approved in the United States as a treatment for organ rejection in patients who have received kidney, heart or liver transplants. But it is also used off label for pemphigus vulgaris and in rheumatology as a treatment for lupus, rheumatoid arthritis, vasculitis, inflammatory bowel disease (Crohn’s disease), inflammatory eye disease (uveitis) as well as kidney and skin disorders.
In the 2018 treatment guidelines for pemphigus by the European Dermatology Forum and the EADV, mycophenolate mofetil is recommended as a first-line corticosteroid sparing agent for pemphigus vulgaris.
Rituximab was approved in 2018 as the first biologic therapy for patients with pemphigus vulgaris and is currently recommended as a treatment for patients with pemphigus. But how well it works in comparison with the long-established mycophenolate mofetil hasn’t been extensively studied.
Other smaller studies show that mycophenolate mofetil has a treatment effect, but those studies were small. The Ritux 3 trial, published in The Lancet showed that rituximab plus glucocorticoids as opposed to glucocorticoids alone was beneficial in treating pemphigus.
“Rituximab has moved toward first-line therapy for moderate to severe pemphigus as recommended by an international panel of experts,” Dr. Werth said in an interview.
In her practice, Dr. Werth said that she has observed similar outcomes in clinical practice for patients prescribed oral mycophenolate mofetil. “Patients take a long time to get to remission and frequently end up staying on prednisone and long-term mycophenolate mofetil,” she said. She uses mycophenolate mofetil less often since rituximab has been shown to be effective for many patients, but mycophenolate mofetil “still has a place for patients who don’t want, or can’t tolerate, rituximab, or for cases in which rituximab doesn’t work.”
This study was supported by a grant from Hoffmann–La Roche. Dr. Werth disclosed having served as a consultant to Genentech on pemphigus, and that the University of Pennsylvania has received a grant/contract to perform a rituximab–mycophenolate mofetil trial for pemphigus vulgaris.
Mycophenolate mofetil, commonly used as a first-line corticosteroid-sparing agent for moderate to severe cases of the autoimmune blistering skin condition pemphigus vulgaris, has been found to be inferior to the biologic agent rituximab.
Mycophenolate mofetil is widely accepted as a first-in-line corticosteroid-sparing agent for pemphigus vulgaris, but few studies have compared the effectiveness of the two treatments for pemphigus vulgaris. The European Academy of Dermatology and Venereology recommends rituximab (Rituxan), a CD20 inhibitor, as first-line treatment for patients with new-onset cases of moderate to severe intensity or for patients who fail to achieve clinical remission with systemic corticosteroids with or without other immunosuppressive treatments.
In the current study, published online on May 19, 2021, in the New England Journal of Medicine, researchers led by Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, conducted a randomized, controlled trial of 135 patients (mean age, 48 years; 53% women) with moderate to severe pemphigus vulgaris with 67 receiving rituximab and 68 receiving mycophenolate mofetil (99% of patients in the rituximab group and 85% of patients in the mycophenolate mofetil group completed the trial).
Patients in the rituximab group received 1,000 mg of IV rituximab on days 1, 15, 168, and 182 of the study, plus twice-daily oral placebo. Intravenous methylprednisolone at 100 mg was administered before each rituximab infusion to reduce infusion-related reactions. Patients in the second group were given mycophenolate mofetil orally twice daily, starting at 1 g/day in divided doses and adjusted to 2 g/day in divided doses by week 2. They also received placebo infusions on days 1, 15, 168, and 182 of the study.
Patients in both groups received oral glucocorticoids throughout the course of the trial: an average of 3,545 mg for the rituximab treatment group and a cumulative dose of 5,140 mg for the group treated with mycophenolate mofetil, a statistically significant difference (P < .001). Outcomes based on 62 patients treated with rituximab and 63 on MMF, a modified intention-to-treat group.
By week 52, 25 patients (40%) who were treated with rituximab experienced complete sustained remission (the primary endpoint), compared with 6 patients (10%) in the mycophenolate mofetil group (95% confidence interval, 15-45, P < .001).
Only six patients in the rituximab group experienced a disease flare as compared with 44 patients in the mycophenolate mofetil group (adjusted rate ratio, 0.12; 95% CI, 0.05-0.29; P < .001). Serious adverse events occurred in 15 of 67 patients (22%) in the rituximab group and in 10 of 68 (15%) in the mycophenolate mofetil group with 3 patients in the rituximab group and 26 in the mycophenolate mofetil receiving rescue therapy.
Second to remission, the goal of treatment for pemphigus vulgaris is to reduce the use of glucocorticoids, Dr. Werth and colleagues wrote, adding: “The results of this trial showed that rituximab was superior to mycophenolate mofetil in producing sustained complete remission over 52 weeks among patients with moderate to severe pemphigus vulgaris. Rituximab had a greater glucocorticoid-sparing effect than mycophenolate mofetil, but more patients in this group had serious adverse events.”
Most adverse events in the rituximab group were limited to infusion-related reactions, but serious adverse events occurred in 15 patients (including pneumonia and upper respiratory tract infection, cellulitis and acute pyelonephritis, viral pneumonia, and skin infection). Ten patients in the mycophenolate mofetil group experienced serious adverse events (pneumonia and influenza, cellulitis and sepsis, herpes zoster, and pyelonephritis).
The current study had several limitations, primarily its small size. Plus, the authors noted a short follow-up period after glucocorticoids were stopped.
Mycophenolate mofetil, along with immunosuppressants, is approved in the United States as a treatment for organ rejection in patients who have received kidney, heart or liver transplants. But it is also used off label for pemphigus vulgaris and in rheumatology as a treatment for lupus, rheumatoid arthritis, vasculitis, inflammatory bowel disease (Crohn’s disease), inflammatory eye disease (uveitis) as well as kidney and skin disorders.
In the 2018 treatment guidelines for pemphigus by the European Dermatology Forum and the EADV, mycophenolate mofetil is recommended as a first-line corticosteroid sparing agent for pemphigus vulgaris.
Rituximab was approved in 2018 as the first biologic therapy for patients with pemphigus vulgaris and is currently recommended as a treatment for patients with pemphigus. But how well it works in comparison with the long-established mycophenolate mofetil hasn’t been extensively studied.
Other smaller studies show that mycophenolate mofetil has a treatment effect, but those studies were small. The Ritux 3 trial, published in The Lancet showed that rituximab plus glucocorticoids as opposed to glucocorticoids alone was beneficial in treating pemphigus.
“Rituximab has moved toward first-line therapy for moderate to severe pemphigus as recommended by an international panel of experts,” Dr. Werth said in an interview.
In her practice, Dr. Werth said that she has observed similar outcomes in clinical practice for patients prescribed oral mycophenolate mofetil. “Patients take a long time to get to remission and frequently end up staying on prednisone and long-term mycophenolate mofetil,” she said. She uses mycophenolate mofetil less often since rituximab has been shown to be effective for many patients, but mycophenolate mofetil “still has a place for patients who don’t want, or can’t tolerate, rituximab, or for cases in which rituximab doesn’t work.”
This study was supported by a grant from Hoffmann–La Roche. Dr. Werth disclosed having served as a consultant to Genentech on pemphigus, and that the University of Pennsylvania has received a grant/contract to perform a rituximab–mycophenolate mofetil trial for pemphigus vulgaris.
Mycophenolate mofetil, commonly used as a first-line corticosteroid-sparing agent for moderate to severe cases of the autoimmune blistering skin condition pemphigus vulgaris, has been found to be inferior to the biologic agent rituximab.
Mycophenolate mofetil is widely accepted as a first-in-line corticosteroid-sparing agent for pemphigus vulgaris, but few studies have compared the effectiveness of the two treatments for pemphigus vulgaris. The European Academy of Dermatology and Venereology recommends rituximab (Rituxan), a CD20 inhibitor, as first-line treatment for patients with new-onset cases of moderate to severe intensity or for patients who fail to achieve clinical remission with systemic corticosteroids with or without other immunosuppressive treatments.
In the current study, published online on May 19, 2021, in the New England Journal of Medicine, researchers led by Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, conducted a randomized, controlled trial of 135 patients (mean age, 48 years; 53% women) with moderate to severe pemphigus vulgaris with 67 receiving rituximab and 68 receiving mycophenolate mofetil (99% of patients in the rituximab group and 85% of patients in the mycophenolate mofetil group completed the trial).
Patients in the rituximab group received 1,000 mg of IV rituximab on days 1, 15, 168, and 182 of the study, plus twice-daily oral placebo. Intravenous methylprednisolone at 100 mg was administered before each rituximab infusion to reduce infusion-related reactions. Patients in the second group were given mycophenolate mofetil orally twice daily, starting at 1 g/day in divided doses and adjusted to 2 g/day in divided doses by week 2. They also received placebo infusions on days 1, 15, 168, and 182 of the study.
Patients in both groups received oral glucocorticoids throughout the course of the trial: an average of 3,545 mg for the rituximab treatment group and a cumulative dose of 5,140 mg for the group treated with mycophenolate mofetil, a statistically significant difference (P < .001). Outcomes based on 62 patients treated with rituximab and 63 on MMF, a modified intention-to-treat group.
By week 52, 25 patients (40%) who were treated with rituximab experienced complete sustained remission (the primary endpoint), compared with 6 patients (10%) in the mycophenolate mofetil group (95% confidence interval, 15-45, P < .001).
Only six patients in the rituximab group experienced a disease flare as compared with 44 patients in the mycophenolate mofetil group (adjusted rate ratio, 0.12; 95% CI, 0.05-0.29; P < .001). Serious adverse events occurred in 15 of 67 patients (22%) in the rituximab group and in 10 of 68 (15%) in the mycophenolate mofetil group with 3 patients in the rituximab group and 26 in the mycophenolate mofetil receiving rescue therapy.
Second to remission, the goal of treatment for pemphigus vulgaris is to reduce the use of glucocorticoids, Dr. Werth and colleagues wrote, adding: “The results of this trial showed that rituximab was superior to mycophenolate mofetil in producing sustained complete remission over 52 weeks among patients with moderate to severe pemphigus vulgaris. Rituximab had a greater glucocorticoid-sparing effect than mycophenolate mofetil, but more patients in this group had serious adverse events.”
Most adverse events in the rituximab group were limited to infusion-related reactions, but serious adverse events occurred in 15 patients (including pneumonia and upper respiratory tract infection, cellulitis and acute pyelonephritis, viral pneumonia, and skin infection). Ten patients in the mycophenolate mofetil group experienced serious adverse events (pneumonia and influenza, cellulitis and sepsis, herpes zoster, and pyelonephritis).
The current study had several limitations, primarily its small size. Plus, the authors noted a short follow-up period after glucocorticoids were stopped.
Mycophenolate mofetil, along with immunosuppressants, is approved in the United States as a treatment for organ rejection in patients who have received kidney, heart or liver transplants. But it is also used off label for pemphigus vulgaris and in rheumatology as a treatment for lupus, rheumatoid arthritis, vasculitis, inflammatory bowel disease (Crohn’s disease), inflammatory eye disease (uveitis) as well as kidney and skin disorders.
In the 2018 treatment guidelines for pemphigus by the European Dermatology Forum and the EADV, mycophenolate mofetil is recommended as a first-line corticosteroid sparing agent for pemphigus vulgaris.
Rituximab was approved in 2018 as the first biologic therapy for patients with pemphigus vulgaris and is currently recommended as a treatment for patients with pemphigus. But how well it works in comparison with the long-established mycophenolate mofetil hasn’t been extensively studied.
Other smaller studies show that mycophenolate mofetil has a treatment effect, but those studies were small. The Ritux 3 trial, published in The Lancet showed that rituximab plus glucocorticoids as opposed to glucocorticoids alone was beneficial in treating pemphigus.
“Rituximab has moved toward first-line therapy for moderate to severe pemphigus as recommended by an international panel of experts,” Dr. Werth said in an interview.
In her practice, Dr. Werth said that she has observed similar outcomes in clinical practice for patients prescribed oral mycophenolate mofetil. “Patients take a long time to get to remission and frequently end up staying on prednisone and long-term mycophenolate mofetil,” she said. She uses mycophenolate mofetil less often since rituximab has been shown to be effective for many patients, but mycophenolate mofetil “still has a place for patients who don’t want, or can’t tolerate, rituximab, or for cases in which rituximab doesn’t work.”
This study was supported by a grant from Hoffmann–La Roche. Dr. Werth disclosed having served as a consultant to Genentech on pemphigus, and that the University of Pennsylvania has received a grant/contract to perform a rituximab–mycophenolate mofetil trial for pemphigus vulgaris.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
An infant girl presents with a growing pink-red leg nodule
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.

A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.

Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
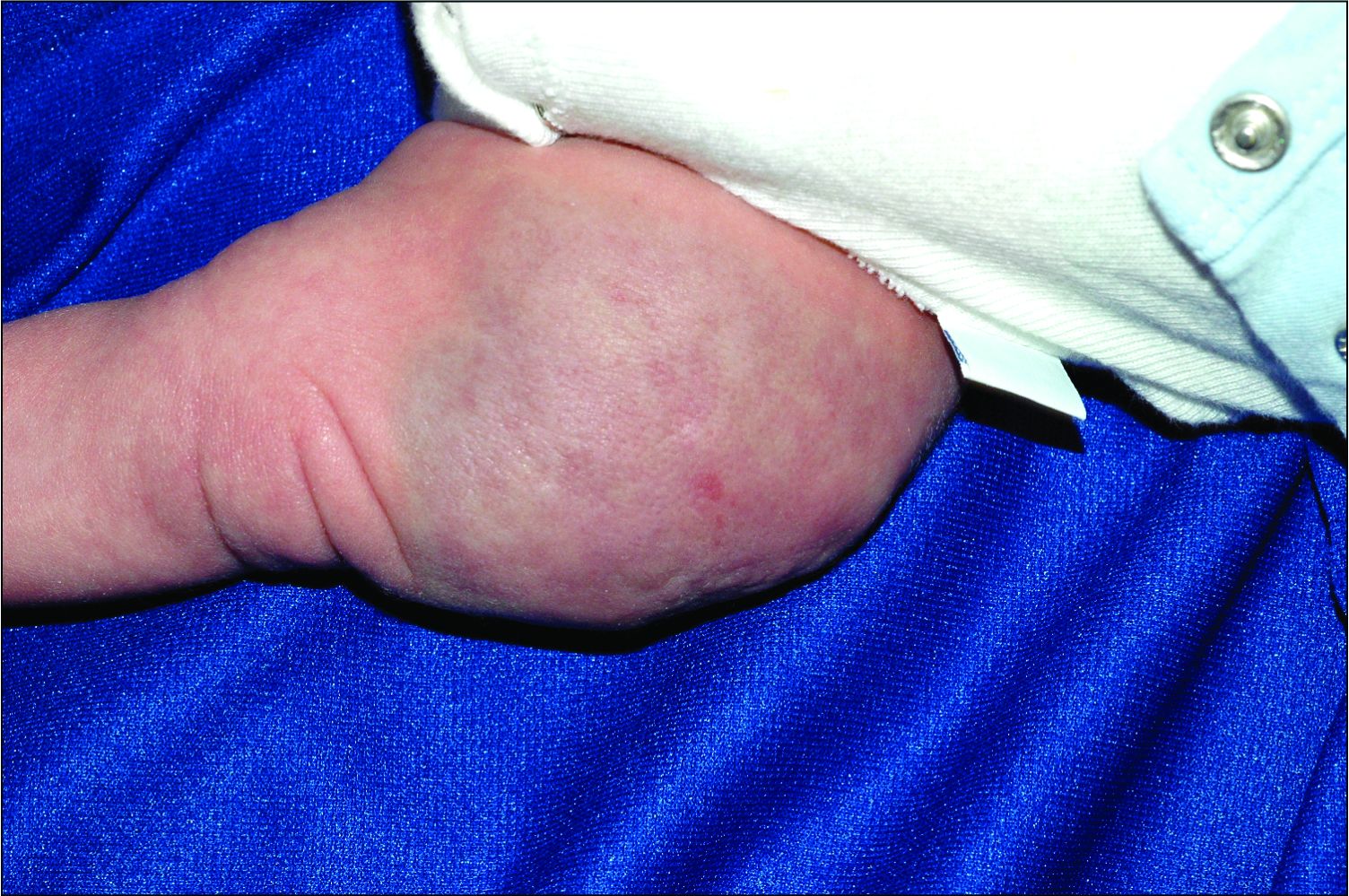
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
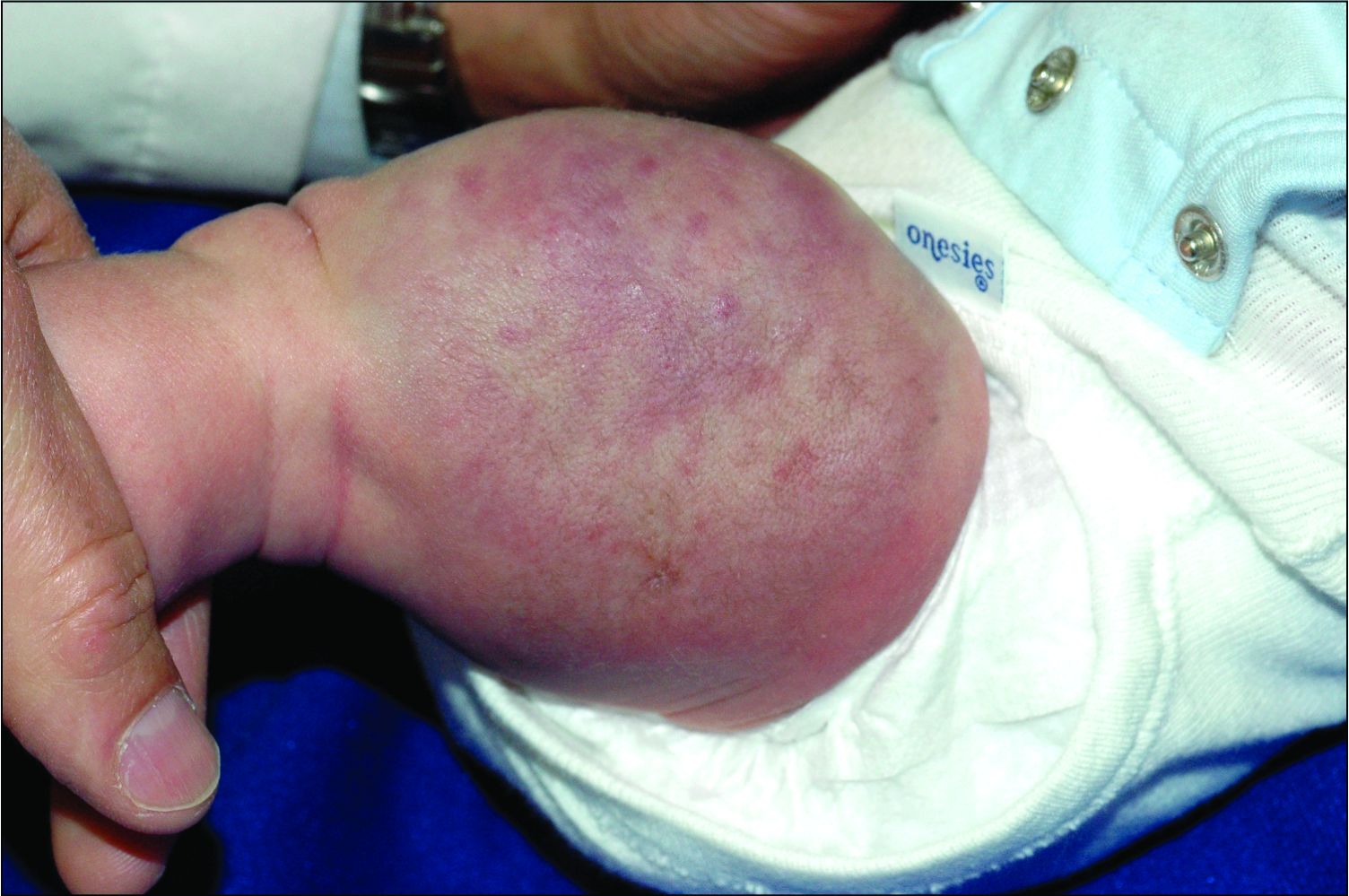
On physical exam, you see an infant with a mass of the left lower extremity. Close examination reveals an approximately 7 cm x 8 cm poorly defined mass with overlying central erythematous to violaceous color of the left anterior upper leg with a lumpy texture. The lesion is moderately firm and mildly tender on palpation.
FDA panel narrowly backs avacopan approval
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.

At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.

“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”

Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.

During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
In pemphigus, phase 2 results with BTK inhibitor raise hopes for phase 3 trial
In patients with newly diagnosed or relapsing pemphigus vulgaris, an update of the phase 2 BELIEVE study with the Bruton tyrosine kinase (BTK) inhibitor rilzabrutinib has raised hopes that the ongoing phase 3 trial will confirm that this drug is a breakthrough therapy, according to an investigator who presented the data at the American Academy of Dermatology Virtual Meeting Experience.

Among the highlights of the phase 2 data presented during a late-breaking research session was that a substantial minority of patients achieved a complete response within 12 weeks of starting treatment with rilzabrutinib. Treatment was associated with mostly mild and transient adverse events, according to Dedee F. Murrell, MD, director of dermatology, St. George Hospital, University of New South Wales, Sydney.
Many of the phase 2 results have been presented previously and the phase 3 trial, called PEGASUS, has now completed enrollment.
Focusing on part A of the BELIEVE study, Dr. Murrell reported that about one-third of the 27 patients enrolled had newly diagnosed pemphigus. The remaining patients had relapsing disease after a mean 8.9 years after diagnosis. The disease was judged moderate to severe in 59%. The daily oral dose of rilzabrutinib ranged from 400 mg to 600 mg twice daily.
For the primary endpoint of control of disease activity (CDA), meaning no formation of new lesions with diminishing activity of existing lesions, 52% had responded by week 4 and 70% had responded by week 12, which was the end of active treatment. Responses at both time points were comparable among patients with newly diagnosed disease (56% at week 4 and 67% at week 12) relapsing disease (50% and 72%, respectively), moderate disease severity at baseline (55% and 64%, respectively) and more severe disease (50% and 75%, respectively), Dr. Murrell noted.
“A complete response was achieved by 22% of patients at week 4 and nearly 30% by the end of the study,” she said.
These response rates were reflected in the Pemphigus Disease Area Index (PDAI) and the Autoimmune Bullous Disease Quality of Life (ABQOL) Score. From a baseline score of 20, the PDAI fell to 10 at 4 weeks and then to 6 at 12 weeks in the newly diagnosed cohort. In the relapsing cohort, the score fell from a baseline of 18 to 13 at week 4 and then to 7 at week 12.
“The improvement corresponded to a reduction in steroid doses,” Dr. Murrell reported. By the end of the study, the mean daily dose of corticosteroids fell to 10 mg from a baseline of 20 mg. In a 12-week follow-up, corticosteroid doses rose slowly and did not reach baseline levels until about eight weeks after rilzabrutinib was discontinued.
The ABQOL scores fell most rapidly in the newly diagnosed cohort. By week 12, there was about a 6.6-point reduction. In the relapsing group, the score fell by 3.7 points from a similar baseline level. Both reductions are considered highly clinically meaningful, according to Dr. Murrell. At the end of the 12 weeks of follow-up after the drug was discontinued, ABQOL scores had increased but remained below the baseline.
Nausea was reported by 15% of patients, making it the most commonly reported adverse event. All cases were grade 1 severity. Three patients had grade 2 abdominal pain. The only grade 3 event in this series was a case of cellulitis in a patient who had developed steroid-induced diabetes mellitus. With treatment, the cellulitis resolved, and the patient completed the study.
Data from part B of the BELIEVE study, which was similarly designed and enrolled 15 patients with newly diagnosed or relapsing pemphigus vulgaris, was not updated by Dr. Murrell at the meeting, but these data have been presented before and showed similar results, including achievement of CDA in the majority of patients accompanied by a reduction in corticosteroid doses.
“In summary, rilzabrutinib produced a rapid clinical effect with an overall favorable benefit-to-risk profile,” said Dr. Murrell, who reiterated that the improvement in quality of life underscored meaningful activity.
Three BTK inhibitors, ibrutinib, acalabrutinib, and zanubrutinib, have been approved for the treatment of hematologic malignancies. These have also been well tolerated. The shared mechanism of action of these drugs is a reduction in B-cell activity achieved by blocking BTK enzyme signaling. The autoimmune activity of pemphigus vulgaris is at least partially mediated by B cells.
“Rilzabrutinib is the first BTK inhibitor tried in pemphigus,” said Dr. Murrell, who cited evidence that pemphigus is at least partially mediated by B-cell activity. The proof-of-concept phase 2 study has increased expectations for the phase 3 PEGASUS trial, which is scheduled for completion in about 1 year, she said.
Dr. Murrell reports financial relationship with multiple pharmaceutical companies, including Principia Biopharma, a Sanofi subsidiary that is developing rilzabrutinib and sponsored the BELIEVE trial.
In patients with newly diagnosed or relapsing pemphigus vulgaris, an update of the phase 2 BELIEVE study with the Bruton tyrosine kinase (BTK) inhibitor rilzabrutinib has raised hopes that the ongoing phase 3 trial will confirm that this drug is a breakthrough therapy, according to an investigator who presented the data at the American Academy of Dermatology Virtual Meeting Experience.
Among the highlights of the phase 2 data presented during a late-breaking research session was that a substantial minority of patients achieved a complete response within 12 weeks of starting treatment with rilzabrutinib. Treatment was associated with mostly mild and transient adverse events, according to Dedee F. Murrell, MD, director of dermatology, St. George Hospital, University of New South Wales, Sydney.
Many of the phase 2 results have been presented previously and the phase 3 trial, called PEGASUS, has now completed enrollment.
Focusing on part A of the BELIEVE study, Dr. Murrell reported that about one-third of the 27 patients enrolled had newly diagnosed pemphigus. The remaining patients had relapsing disease after a mean 8.9 years after diagnosis. The disease was judged moderate to severe in 59%. The daily oral dose of rilzabrutinib ranged from 400 mg to 600 mg twice daily.
For the primary endpoint of control of disease activity (CDA), meaning no formation of new lesions with diminishing activity of existing lesions, 52% had responded by week 4 and 70% had responded by week 12, which was the end of active treatment. Responses at both time points were comparable among patients with newly diagnosed disease (56% at week 4 and 67% at week 12) relapsing disease (50% and 72%, respectively), moderate disease severity at baseline (55% and 64%, respectively) and more severe disease (50% and 75%, respectively), Dr. Murrell noted.
“A complete response was achieved by 22% of patients at week 4 and nearly 30% by the end of the study,” she said.
These response rates were reflected in the Pemphigus Disease Area Index (PDAI) and the Autoimmune Bullous Disease Quality of Life (ABQOL) Score. From a baseline score of 20, the PDAI fell to 10 at 4 weeks and then to 6 at 12 weeks in the newly diagnosed cohort. In the relapsing cohort, the score fell from a baseline of 18 to 13 at week 4 and then to 7 at week 12.
“The improvement corresponded to a reduction in steroid doses,” Dr. Murrell reported. By the end of the study, the mean daily dose of corticosteroids fell to 10 mg from a baseline of 20 mg. In a 12-week follow-up, corticosteroid doses rose slowly and did not reach baseline levels until about eight weeks after rilzabrutinib was discontinued.
The ABQOL scores fell most rapidly in the newly diagnosed cohort. By week 12, there was about a 6.6-point reduction. In the relapsing group, the score fell by 3.7 points from a similar baseline level. Both reductions are considered highly clinically meaningful, according to Dr. Murrell. At the end of the 12 weeks of follow-up after the drug was discontinued, ABQOL scores had increased but remained below the baseline.
Nausea was reported by 15% of patients, making it the most commonly reported adverse event. All cases were grade 1 severity. Three patients had grade 2 abdominal pain. The only grade 3 event in this series was a case of cellulitis in a patient who had developed steroid-induced diabetes mellitus. With treatment, the cellulitis resolved, and the patient completed the study.
Data from part B of the BELIEVE study, which was similarly designed and enrolled 15 patients with newly diagnosed or relapsing pemphigus vulgaris, was not updated by Dr. Murrell at the meeting, but these data have been presented before and showed similar results, including achievement of CDA in the majority of patients accompanied by a reduction in corticosteroid doses.
“In summary, rilzabrutinib produced a rapid clinical effect with an overall favorable benefit-to-risk profile,” said Dr. Murrell, who reiterated that the improvement in quality of life underscored meaningful activity.
Three BTK inhibitors, ibrutinib, acalabrutinib, and zanubrutinib, have been approved for the treatment of hematologic malignancies. These have also been well tolerated. The shared mechanism of action of these drugs is a reduction in B-cell activity achieved by blocking BTK enzyme signaling. The autoimmune activity of pemphigus vulgaris is at least partially mediated by B cells.
“Rilzabrutinib is the first BTK inhibitor tried in pemphigus,” said Dr. Murrell, who cited evidence that pemphigus is at least partially mediated by B-cell activity. The proof-of-concept phase 2 study has increased expectations for the phase 3 PEGASUS trial, which is scheduled for completion in about 1 year, she said.
Dr. Murrell reports financial relationship with multiple pharmaceutical companies, including Principia Biopharma, a Sanofi subsidiary that is developing rilzabrutinib and sponsored the BELIEVE trial.
In patients with newly diagnosed or relapsing pemphigus vulgaris, an update of the phase 2 BELIEVE study with the Bruton tyrosine kinase (BTK) inhibitor rilzabrutinib has raised hopes that the ongoing phase 3 trial will confirm that this drug is a breakthrough therapy, according to an investigator who presented the data at the American Academy of Dermatology Virtual Meeting Experience.
Among the highlights of the phase 2 data presented during a late-breaking research session was that a substantial minority of patients achieved a complete response within 12 weeks of starting treatment with rilzabrutinib. Treatment was associated with mostly mild and transient adverse events, according to Dedee F. Murrell, MD, director of dermatology, St. George Hospital, University of New South Wales, Sydney.
Many of the phase 2 results have been presented previously and the phase 3 trial, called PEGASUS, has now completed enrollment.
Focusing on part A of the BELIEVE study, Dr. Murrell reported that about one-third of the 27 patients enrolled had newly diagnosed pemphigus. The remaining patients had relapsing disease after a mean 8.9 years after diagnosis. The disease was judged moderate to severe in 59%. The daily oral dose of rilzabrutinib ranged from 400 mg to 600 mg twice daily.
For the primary endpoint of control of disease activity (CDA), meaning no formation of new lesions with diminishing activity of existing lesions, 52% had responded by week 4 and 70% had responded by week 12, which was the end of active treatment. Responses at both time points were comparable among patients with newly diagnosed disease (56% at week 4 and 67% at week 12) relapsing disease (50% and 72%, respectively), moderate disease severity at baseline (55% and 64%, respectively) and more severe disease (50% and 75%, respectively), Dr. Murrell noted.
“A complete response was achieved by 22% of patients at week 4 and nearly 30% by the end of the study,” she said.
These response rates were reflected in the Pemphigus Disease Area Index (PDAI) and the Autoimmune Bullous Disease Quality of Life (ABQOL) Score. From a baseline score of 20, the PDAI fell to 10 at 4 weeks and then to 6 at 12 weeks in the newly diagnosed cohort. In the relapsing cohort, the score fell from a baseline of 18 to 13 at week 4 and then to 7 at week 12.
“The improvement corresponded to a reduction in steroid doses,” Dr. Murrell reported. By the end of the study, the mean daily dose of corticosteroids fell to 10 mg from a baseline of 20 mg. In a 12-week follow-up, corticosteroid doses rose slowly and did not reach baseline levels until about eight weeks after rilzabrutinib was discontinued.
The ABQOL scores fell most rapidly in the newly diagnosed cohort. By week 12, there was about a 6.6-point reduction. In the relapsing group, the score fell by 3.7 points from a similar baseline level. Both reductions are considered highly clinically meaningful, according to Dr. Murrell. At the end of the 12 weeks of follow-up after the drug was discontinued, ABQOL scores had increased but remained below the baseline.
Nausea was reported by 15% of patients, making it the most commonly reported adverse event. All cases were grade 1 severity. Three patients had grade 2 abdominal pain. The only grade 3 event in this series was a case of cellulitis in a patient who had developed steroid-induced diabetes mellitus. With treatment, the cellulitis resolved, and the patient completed the study.
Data from part B of the BELIEVE study, which was similarly designed and enrolled 15 patients with newly diagnosed or relapsing pemphigus vulgaris, was not updated by Dr. Murrell at the meeting, but these data have been presented before and showed similar results, including achievement of CDA in the majority of patients accompanied by a reduction in corticosteroid doses.
“In summary, rilzabrutinib produced a rapid clinical effect with an overall favorable benefit-to-risk profile,” said Dr. Murrell, who reiterated that the improvement in quality of life underscored meaningful activity.
Three BTK inhibitors, ibrutinib, acalabrutinib, and zanubrutinib, have been approved for the treatment of hematologic malignancies. These have also been well tolerated. The shared mechanism of action of these drugs is a reduction in B-cell activity achieved by blocking BTK enzyme signaling. The autoimmune activity of pemphigus vulgaris is at least partially mediated by B cells.
“Rilzabrutinib is the first BTK inhibitor tried in pemphigus,” said Dr. Murrell, who cited evidence that pemphigus is at least partially mediated by B-cell activity. The proof-of-concept phase 2 study has increased expectations for the phase 3 PEGASUS trial, which is scheduled for completion in about 1 year, she said.
Dr. Murrell reports financial relationship with multiple pharmaceutical companies, including Principia Biopharma, a Sanofi subsidiary that is developing rilzabrutinib and sponsored the BELIEVE trial.
FROM AAD VMX 2021
For diagnosing skin lesions, AI risks failing in skin of color
In the analysis of images for detecting potential pathology, if training does not specifically address these skin types, according to Adewole S. Adamson, MD, who outlined this issue at the American Academy of Dermatology Virtual Meeting Experience.

“Machine learning algorithms are only as good as the inputs through which they learn. Without representation from individuals with skin of color, we are at risk of creating a new source of racial disparity in patient care,” Dr. Adamson, assistant professor in the division of dermatology, department of internal medicine, University of Texas at Austin, said at the meeting.
Diagnostic algorithms using AI are typically based on deep learning, a subset of machine learning that depends on artificial neural networks. In the case of image processing, neural networks can “learn” to recognize objects, faces, or, in the realm of health care, disease, from exposure to multiple images.
There are many other variables that affect the accuracy of deep learning for diagnostic algorithms, including the depth of the layering through which the process distills multiple inputs of information, but the number of inputs is critical. In the case of skin lesions, machines cannot learn to recognize features of different skin types without exposure.
“There are studies demonstrating that dermatologists can be outperformed for detection of skin cancers by AI, so this is going to be an increasingly powerful tool,” Dr. Adamson said. The problem is that “there has been very little representation in darker skin types” in the algorithms developed so far.
The risk is that AI will exacerbate an existing problem. Skin cancer in darker skin is less common but already underdiagnosed, independent of AI. Per 100,000 males in the United States, the rate of melanoma is about 30-fold greater in White men than in Black men (33.0 vs. 1.0). Among females, the racial difference is smaller but still enormous (20.2 vs. 1.2 per 100,000 females), according to U.S. data.
For the low representation of darker skin in studies so far with AI, “one of the arguments is that skin cancer is not a big deal in darker skin types,” Dr. Adamson said.
It might be the other way around. The relative infrequency with which skin cancer occurs in the Black population in the United States might explain a low level of suspicion and ultimately delays in diagnosis, which, in turn, leads to worse outcomes. According to one analysis drawn from the Surveillance, Epidemiology and End-Result (SEER) database (1998-2011), the proportion of patients with regionally advanced or distant disease was nearly twice as great (11.6% vs. 6.0%; P < .05) in Black patients, relative to White patients.
Not surprisingly, given the importance of early diagnosis of cancers overall and skin cancer specifically, the mean survival for malignant melanoma in Black patients was almost 4 years lower than in White patients (10.8 vs. 14.6 years; P < .001) for nodular melanoma, the same study found.
In humans, bias is reasonably attributed in many cases to judgments made on a small sample size. The problem in AI is analogous. Dr. Adamson, who has published research on the potential for machine learning to contribute to health care disparities in dermatology, cited work done by Joy Buolamwini, a graduate researcher in the media lab at the Massachusetts Institute of Technology. In one study she conducted, the rate of AI facial recognition failure was 1% in White males, 7% in White females, 12% in skin-of-color males, and 35% in skin-of-color females. Fewer inputs of skin of color is the likely explanation, Dr. Adamson said.
The potential for racial bias from AI in the diagnosis of disease increases and becomes more complex when inputs beyond imaging, such as past medical history, are included. Dr. Adamson warned of the potential for “bias to creep in” when there is failure to account for societal, cultural, or other differences that distinguish one patient group from another. However, for skin cancer or other diseases based on images alone, he said there are solutions.
“We are in the early days, and there is time to change this,” Dr. Adamson said, referring to the low representation of skin of color in AI training sets. In addition to including more skin types to train recognition, creating AI algorithms specifically for dark skin is another potential approach.
However, his key point was the importance of recognizing the need for solutions.
“AI is the future, but we must apply the same rigor to AI as to other medical interventions to ensure that the technology is not applied in a biased fashion,” he said.
Susan M. Swetter, MD, professor of dermatology and director of the pigmented lesion and melanoma program at Stanford (Calif.) University Medical Center and Cancer Institute, agreed. As someone who has been following the progress of AI in the diagnosis of skin cancer, Dr. Swetter recognizes the potential for this technology to increase diagnostic efficiency and accuracy, but she also called for studies specific to skin of color.

The algorithms “have not yet been adequately evaluated in people of color, particularly Black patients in whom dermoscopic criteria for benign versus malignant melanocytic neoplasms differ from those with lighter skin types,” Dr. Swetter said in an interview.
She sees the same fix as that proposed by Dr. Adamson.
“Efforts to include skin of color in AI algorithms for validation and further training are needed to prevent potential harms of over- or underdiagnosis in darker skin patients,” she pointed out.
Dr. Adamson reports no potential conflicts of interest relevant to this topic. Dr. Swetter had no relevant disclosures.
In the analysis of images for detecting potential pathology, if training does not specifically address these skin types, according to Adewole S. Adamson, MD, who outlined this issue at the American Academy of Dermatology Virtual Meeting Experience.
“Machine learning algorithms are only as good as the inputs through which they learn. Without representation from individuals with skin of color, we are at risk of creating a new source of racial disparity in patient care,” Dr. Adamson, assistant professor in the division of dermatology, department of internal medicine, University of Texas at Austin, said at the meeting.
Diagnostic algorithms using AI are typically based on deep learning, a subset of machine learning that depends on artificial neural networks. In the case of image processing, neural networks can “learn” to recognize objects, faces, or, in the realm of health care, disease, from exposure to multiple images.
There are many other variables that affect the accuracy of deep learning for diagnostic algorithms, including the depth of the layering through which the process distills multiple inputs of information, but the number of inputs is critical. In the case of skin lesions, machines cannot learn to recognize features of different skin types without exposure.
“There are studies demonstrating that dermatologists can be outperformed for detection of skin cancers by AI, so this is going to be an increasingly powerful tool,” Dr. Adamson said. The problem is that “there has been very little representation in darker skin types” in the algorithms developed so far.
The risk is that AI will exacerbate an existing problem. Skin cancer in darker skin is less common but already underdiagnosed, independent of AI. Per 100,000 males in the United States, the rate of melanoma is about 30-fold greater in White men than in Black men (33.0 vs. 1.0). Among females, the racial difference is smaller but still enormous (20.2 vs. 1.2 per 100,000 females), according to U.S. data.
For the low representation of darker skin in studies so far with AI, “one of the arguments is that skin cancer is not a big deal in darker skin types,” Dr. Adamson said.
It might be the other way around. The relative infrequency with which skin cancer occurs in the Black population in the United States might explain a low level of suspicion and ultimately delays in diagnosis, which, in turn, leads to worse outcomes. According to one analysis drawn from the Surveillance, Epidemiology and End-Result (SEER) database (1998-2011), the proportion of patients with regionally advanced or distant disease was nearly twice as great (11.6% vs. 6.0%; P < .05) in Black patients, relative to White patients.
Not surprisingly, given the importance of early diagnosis of cancers overall and skin cancer specifically, the mean survival for malignant melanoma in Black patients was almost 4 years lower than in White patients (10.8 vs. 14.6 years; P < .001) for nodular melanoma, the same study found.
In humans, bias is reasonably attributed in many cases to judgments made on a small sample size. The problem in AI is analogous. Dr. Adamson, who has published research on the potential for machine learning to contribute to health care disparities in dermatology, cited work done by Joy Buolamwini, a graduate researcher in the media lab at the Massachusetts Institute of Technology. In one study she conducted, the rate of AI facial recognition failure was 1% in White males, 7% in White females, 12% in skin-of-color males, and 35% in skin-of-color females. Fewer inputs of skin of color is the likely explanation, Dr. Adamson said.
The potential for racial bias from AI in the diagnosis of disease increases and becomes more complex when inputs beyond imaging, such as past medical history, are included. Dr. Adamson warned of the potential for “bias to creep in” when there is failure to account for societal, cultural, or other differences that distinguish one patient group from another. However, for skin cancer or other diseases based on images alone, he said there are solutions.
“We are in the early days, and there is time to change this,” Dr. Adamson said, referring to the low representation of skin of color in AI training sets. In addition to including more skin types to train recognition, creating AI algorithms specifically for dark skin is another potential approach.
However, his key point was the importance of recognizing the need for solutions.
“AI is the future, but we must apply the same rigor to AI as to other medical interventions to ensure that the technology is not applied in a biased fashion,” he said.
Susan M. Swetter, MD, professor of dermatology and director of the pigmented lesion and melanoma program at Stanford (Calif.) University Medical Center and Cancer Institute, agreed. As someone who has been following the progress of AI in the diagnosis of skin cancer, Dr. Swetter recognizes the potential for this technology to increase diagnostic efficiency and accuracy, but she also called for studies specific to skin of color.
The algorithms “have not yet been adequately evaluated in people of color, particularly Black patients in whom dermoscopic criteria for benign versus malignant melanocytic neoplasms differ from those with lighter skin types,” Dr. Swetter said in an interview.
She sees the same fix as that proposed by Dr. Adamson.
“Efforts to include skin of color in AI algorithms for validation and further training are needed to prevent potential harms of over- or underdiagnosis in darker skin patients,” she pointed out.
Dr. Adamson reports no potential conflicts of interest relevant to this topic. Dr. Swetter had no relevant disclosures.
In the analysis of images for detecting potential pathology, if training does not specifically address these skin types, according to Adewole S. Adamson, MD, who outlined this issue at the American Academy of Dermatology Virtual Meeting Experience.
“Machine learning algorithms are only as good as the inputs through which they learn. Without representation from individuals with skin of color, we are at risk of creating a new source of racial disparity in patient care,” Dr. Adamson, assistant professor in the division of dermatology, department of internal medicine, University of Texas at Austin, said at the meeting.
Diagnostic algorithms using AI are typically based on deep learning, a subset of machine learning that depends on artificial neural networks. In the case of image processing, neural networks can “learn” to recognize objects, faces, or, in the realm of health care, disease, from exposure to multiple images.
There are many other variables that affect the accuracy of deep learning for diagnostic algorithms, including the depth of the layering through which the process distills multiple inputs of information, but the number of inputs is critical. In the case of skin lesions, machines cannot learn to recognize features of different skin types without exposure.
“There are studies demonstrating that dermatologists can be outperformed for detection of skin cancers by AI, so this is going to be an increasingly powerful tool,” Dr. Adamson said. The problem is that “there has been very little representation in darker skin types” in the algorithms developed so far.
The risk is that AI will exacerbate an existing problem. Skin cancer in darker skin is less common but already underdiagnosed, independent of AI. Per 100,000 males in the United States, the rate of melanoma is about 30-fold greater in White men than in Black men (33.0 vs. 1.0). Among females, the racial difference is smaller but still enormous (20.2 vs. 1.2 per 100,000 females), according to U.S. data.
For the low representation of darker skin in studies so far with AI, “one of the arguments is that skin cancer is not a big deal in darker skin types,” Dr. Adamson said.
It might be the other way around. The relative infrequency with which skin cancer occurs in the Black population in the United States might explain a low level of suspicion and ultimately delays in diagnosis, which, in turn, leads to worse outcomes. According to one analysis drawn from the Surveillance, Epidemiology and End-Result (SEER) database (1998-2011), the proportion of patients with regionally advanced or distant disease was nearly twice as great (11.6% vs. 6.0%; P < .05) in Black patients, relative to White patients.
Not surprisingly, given the importance of early diagnosis of cancers overall and skin cancer specifically, the mean survival for malignant melanoma in Black patients was almost 4 years lower than in White patients (10.8 vs. 14.6 years; P < .001) for nodular melanoma, the same study found.
In humans, bias is reasonably attributed in many cases to judgments made on a small sample size. The problem in AI is analogous. Dr. Adamson, who has published research on the potential for machine learning to contribute to health care disparities in dermatology, cited work done by Joy Buolamwini, a graduate researcher in the media lab at the Massachusetts Institute of Technology. In one study she conducted, the rate of AI facial recognition failure was 1% in White males, 7% in White females, 12% in skin-of-color males, and 35% in skin-of-color females. Fewer inputs of skin of color is the likely explanation, Dr. Adamson said.
The potential for racial bias from AI in the diagnosis of disease increases and becomes more complex when inputs beyond imaging, such as past medical history, are included. Dr. Adamson warned of the potential for “bias to creep in” when there is failure to account for societal, cultural, or other differences that distinguish one patient group from another. However, for skin cancer or other diseases based on images alone, he said there are solutions.
“We are in the early days, and there is time to change this,” Dr. Adamson said, referring to the low representation of skin of color in AI training sets. In addition to including more skin types to train recognition, creating AI algorithms specifically for dark skin is another potential approach.
However, his key point was the importance of recognizing the need for solutions.
“AI is the future, but we must apply the same rigor to AI as to other medical interventions to ensure that the technology is not applied in a biased fashion,” he said.
Susan M. Swetter, MD, professor of dermatology and director of the pigmented lesion and melanoma program at Stanford (Calif.) University Medical Center and Cancer Institute, agreed. As someone who has been following the progress of AI in the diagnosis of skin cancer, Dr. Swetter recognizes the potential for this technology to increase diagnostic efficiency and accuracy, but she also called for studies specific to skin of color.
The algorithms “have not yet been adequately evaluated in people of color, particularly Black patients in whom dermoscopic criteria for benign versus malignant melanocytic neoplasms differ from those with lighter skin types,” Dr. Swetter said in an interview.
She sees the same fix as that proposed by Dr. Adamson.
“Efforts to include skin of color in AI algorithms for validation and further training are needed to prevent potential harms of over- or underdiagnosis in darker skin patients,” she pointed out.
Dr. Adamson reports no potential conflicts of interest relevant to this topic. Dr. Swetter had no relevant disclosures.
FROM AAD VMX 2021
Novel BRAF-inhibitor cream ameliorates rash from EGFR inhibitors
The results come from a first-in-human, phase 1 clinical trial conducted in 10 patients with metastatic colorectal cancer who were receiving treatment with either cetuximab or panitumumab and who developed a grade 1 or grade 2 rash while on treatment.
All were treated with the novel topical cream, dubbed LUTO14 (under development by Lutris Pharma).
For 6 of the 10 patients, the acneiform rash improved, according to investigator Mario Lacouture, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
The study was published online in Cancer Discovery.
“Based on preclinical modeling and early clinical trial testing, we conclude that improving a topmost adverse event of EGFR inhibitor therapy with topical LUT014 could allow [maintenance of] quality of life and dose intensity, thereby maximizing the antitumor effects [from EGFR inhibitor therapy] while locally inhibiting dose-limiting skin toxicities,” the investigators wrote.
The cream was well tolerated, and no dose-limiting toxicity or maximum tolerated dose was observed, although the cream did appear to be more effective at lower doses.
Rash is a common side effect of EGFR inhibitors. Previous studies have reported that 75%-90% of patients experience “some form of papulopustular, acneiform rash, which frequently leads to ... suboptimal anticancer treatment due to treatment interruptions, dose reductions, or permanent discontinuation of EGFR inhibitor therapy,” the investigators noted.
Paradoxical mechanism of action
How the novel cream containing a BRAF inhibitor helps ameliorate EGFR inhibitor–induced skin toxicity is complicated, but at a cellular level, the mechanism seems somewhat paradoxical, the team commented.
Skin toxicity experienced in the setting of EGFR inhibitor therapy is induced by inhibition of the mitogen-activated protein kinase (MAPK) pathway. Downstream inhibition of the MAPK pathway results in, among other effects, inflammatory changes in epithelial cells that mediate the acneiform rash on the skin.
In contrast, “BRAF inhibitors given systemically have an opposite effect on epithelial cells, resulting in paradoxical activation of the MAPK pathway,” the authors explained. They hypothesized that topical administration of BRAF inhibitors similarly activates the MAPK pathway in epithelial cells, although it was important to develop a specific BRAF inhibitor that would optimally induce paradoxical MAPK activation. That they managed to do so was shown when they evaluated LUT014 in cell culture systems.
The next phase of the study is designed to include approximately 120 patients recruited from centers in the United States and Israel. Interim results are expected by the end of 2021.
The study was funded by Lutris Pharma, the company developing LUT014.
A version of this article first appeared on Medscape.com.
The results come from a first-in-human, phase 1 clinical trial conducted in 10 patients with metastatic colorectal cancer who were receiving treatment with either cetuximab or panitumumab and who developed a grade 1 or grade 2 rash while on treatment.
All were treated with the novel topical cream, dubbed LUTO14 (under development by Lutris Pharma).
For 6 of the 10 patients, the acneiform rash improved, according to investigator Mario Lacouture, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
The study was published online in Cancer Discovery.
“Based on preclinical modeling and early clinical trial testing, we conclude that improving a topmost adverse event of EGFR inhibitor therapy with topical LUT014 could allow [maintenance of] quality of life and dose intensity, thereby maximizing the antitumor effects [from EGFR inhibitor therapy] while locally inhibiting dose-limiting skin toxicities,” the investigators wrote.
The cream was well tolerated, and no dose-limiting toxicity or maximum tolerated dose was observed, although the cream did appear to be more effective at lower doses.
Rash is a common side effect of EGFR inhibitors. Previous studies have reported that 75%-90% of patients experience “some form of papulopustular, acneiform rash, which frequently leads to ... suboptimal anticancer treatment due to treatment interruptions, dose reductions, or permanent discontinuation of EGFR inhibitor therapy,” the investigators noted.
Paradoxical mechanism of action
How the novel cream containing a BRAF inhibitor helps ameliorate EGFR inhibitor–induced skin toxicity is complicated, but at a cellular level, the mechanism seems somewhat paradoxical, the team commented.
Skin toxicity experienced in the setting of EGFR inhibitor therapy is induced by inhibition of the mitogen-activated protein kinase (MAPK) pathway. Downstream inhibition of the MAPK pathway results in, among other effects, inflammatory changes in epithelial cells that mediate the acneiform rash on the skin.
In contrast, “BRAF inhibitors given systemically have an opposite effect on epithelial cells, resulting in paradoxical activation of the MAPK pathway,” the authors explained. They hypothesized that topical administration of BRAF inhibitors similarly activates the MAPK pathway in epithelial cells, although it was important to develop a specific BRAF inhibitor that would optimally induce paradoxical MAPK activation. That they managed to do so was shown when they evaluated LUT014 in cell culture systems.
The next phase of the study is designed to include approximately 120 patients recruited from centers in the United States and Israel. Interim results are expected by the end of 2021.
The study was funded by Lutris Pharma, the company developing LUT014.
A version of this article first appeared on Medscape.com.
The results come from a first-in-human, phase 1 clinical trial conducted in 10 patients with metastatic colorectal cancer who were receiving treatment with either cetuximab or panitumumab and who developed a grade 1 or grade 2 rash while on treatment.
All were treated with the novel topical cream, dubbed LUTO14 (under development by Lutris Pharma).
For 6 of the 10 patients, the acneiform rash improved, according to investigator Mario Lacouture, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues.
The study was published online in Cancer Discovery.
“Based on preclinical modeling and early clinical trial testing, we conclude that improving a topmost adverse event of EGFR inhibitor therapy with topical LUT014 could allow [maintenance of] quality of life and dose intensity, thereby maximizing the antitumor effects [from EGFR inhibitor therapy] while locally inhibiting dose-limiting skin toxicities,” the investigators wrote.
The cream was well tolerated, and no dose-limiting toxicity or maximum tolerated dose was observed, although the cream did appear to be more effective at lower doses.
Rash is a common side effect of EGFR inhibitors. Previous studies have reported that 75%-90% of patients experience “some form of papulopustular, acneiform rash, which frequently leads to ... suboptimal anticancer treatment due to treatment interruptions, dose reductions, or permanent discontinuation of EGFR inhibitor therapy,” the investigators noted.
Paradoxical mechanism of action
How the novel cream containing a BRAF inhibitor helps ameliorate EGFR inhibitor–induced skin toxicity is complicated, but at a cellular level, the mechanism seems somewhat paradoxical, the team commented.
Skin toxicity experienced in the setting of EGFR inhibitor therapy is induced by inhibition of the mitogen-activated protein kinase (MAPK) pathway. Downstream inhibition of the MAPK pathway results in, among other effects, inflammatory changes in epithelial cells that mediate the acneiform rash on the skin.
In contrast, “BRAF inhibitors given systemically have an opposite effect on epithelial cells, resulting in paradoxical activation of the MAPK pathway,” the authors explained. They hypothesized that topical administration of BRAF inhibitors similarly activates the MAPK pathway in epithelial cells, although it was important to develop a specific BRAF inhibitor that would optimally induce paradoxical MAPK activation. That they managed to do so was shown when they evaluated LUT014 in cell culture systems.
The next phase of the study is designed to include approximately 120 patients recruited from centers in the United States and Israel. Interim results are expected by the end of 2021.
The study was funded by Lutris Pharma, the company developing LUT014.
A version of this article first appeared on Medscape.com.
Tofacitinib: Small study shows big cutaneous sarcoidosis response
Researchers are reporting impressive results in a small, , and all patients improved by an average of 83% via a scoring system.

“Not only did patients get better, but they were in many cases able to come off their baseline immunosuppressive regimen, including prednisone and methotrexate. They’d get off prednisone entirely or, in some cases, decrease it substantially,” study investigator William Damsky, MD, PhD, reported at the American Academy of Dermatology Virtual Meeting Experience.
Sarcoidosis is a common disease that affects an estimated 1 in 25 Black women and is believed to contribute to the deaths of about 4,000 people in the United States each year, noted Dr. Damsky of the department of dermatology, Yale University, New Haven, Conn. One famous patient is comedian Bernie Mac, who died from the condition in 2008.
“Approximately one third of patients have cutaneous involvement,” Dr. Damsky said, and skin may be the only manifestation of the disease. There is no Food and Drug Administration-approved therapy for cutaneous sarcoidosis, he added. Prednisone, the first-line therapy in skin manifestations, is approved only for pulmonary sarcoidosis.
“Oftentimes, there’s an attempt to transition either partially or fully to other therapies, including methotrexate and TNF-alpha blockers. But there’s been mixed success in doing that,” he said. This is not always possible, “so a lot of patients end up on prednisone.”
Earlier, a team at Yale prescribed 5 mg tofacitinib (Xeljanz) for several patients with severe cutaneous sarcoidosis and saw impressive results, Dr. Damsky said, including a patient with pulmonary sarcoidosis that also improved. He noted that there are case reports in the medical literature with similar findings.
Those positive results inspired the new study. Researchers recruited 10 patients with cutaneous sarcoidosis (9 with internal organ involvement) with a Cutaneous Sarcoidosis Activity and Morphology Instrument ( CSAMI ) score of 10 or higher. Nine patients were in their 50s, one was aged 63 years, and five were men. Skin colors of the patients ranged from Fitzpatrick skin types I to VI, and all had been taking at least two medications, typically methotrexate and prednisone.
The patients received 5 mg of tofacitinib twice a day for 6 months. “Everyone got better during the study, and six patients had a complete response, which we defined as a CSAMI score of zero activity,” Dr. Damsky said. “It’s really quite remarkable to see that.” Overall, the patients saw an 83% improvement in CSAMI scores.
In regard to safety, “all patients completed the study,” he said. “Tofacitinib was well tolerated, and there were no serious adverse effects or events.”
Tofacitinib is approved for treating rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular course juvenile idiopathic arthritis.
A month’s supply of twice-daily 5 mg tofacitinib pills would cost $4,900-$5,100 with free coupons, according to information accessed on April 24, 2021, on GoodRx.com. Generics are not available.
In an interview, Sotonye Imadojemu, MD, of the department of dermatology, Brigham and Women’s Hospital, Boston, praised the study, and said “tofacitinib is a reasonable treatment for treatment-refractory or extensive cutaneous sarcoidosis,” although it will be helpful to get results from randomized-controlled trials.
She cautioned that the drug “is a powerful immunosuppressant, so the risk of infection must be discussed with patients before prescribing. Screening for chronic infections such as viral hepatitis, tuberculosis, and HIV should be completed prior to treatment initiation. Blood counts, liver function, and lipid panels should be regularly monitored. The vaccines necessary for those who are immunosuppressed should be administered as able, and age-appropriate cancer screening must be kept up to date.”
The study was funded by Pfizer, the Dermatology Foundation, and the Yale Department of Dermatology. Dr. Damsky disclosed research support (Pfizer), consulting fees (Eli Lilly, Pfizer, TWi Biotechnology), and licensing fees (EMD Millipore/MillporeSigma). Dr. Imadojemu has no disclosures.
This article was updated 5/5/21.
Researchers are reporting impressive results in a small, , and all patients improved by an average of 83% via a scoring system.
“Not only did patients get better, but they were in many cases able to come off their baseline immunosuppressive regimen, including prednisone and methotrexate. They’d get off prednisone entirely or, in some cases, decrease it substantially,” study investigator William Damsky, MD, PhD, reported at the American Academy of Dermatology Virtual Meeting Experience.
Sarcoidosis is a common disease that affects an estimated 1 in 25 Black women and is believed to contribute to the deaths of about 4,000 people in the United States each year, noted Dr. Damsky of the department of dermatology, Yale University, New Haven, Conn. One famous patient is comedian Bernie Mac, who died from the condition in 2008.
“Approximately one third of patients have cutaneous involvement,” Dr. Damsky said, and skin may be the only manifestation of the disease. There is no Food and Drug Administration-approved therapy for cutaneous sarcoidosis, he added. Prednisone, the first-line therapy in skin manifestations, is approved only for pulmonary sarcoidosis.
“Oftentimes, there’s an attempt to transition either partially or fully to other therapies, including methotrexate and TNF-alpha blockers. But there’s been mixed success in doing that,” he said. This is not always possible, “so a lot of patients end up on prednisone.”
Earlier, a team at Yale prescribed 5 mg tofacitinib (Xeljanz) for several patients with severe cutaneous sarcoidosis and saw impressive results, Dr. Damsky said, including a patient with pulmonary sarcoidosis that also improved. He noted that there are case reports in the medical literature with similar findings.
Those positive results inspired the new study. Researchers recruited 10 patients with cutaneous sarcoidosis (9 with internal organ involvement) with a Cutaneous Sarcoidosis Activity and Morphology Instrument ( CSAMI ) score of 10 or higher. Nine patients were in their 50s, one was aged 63 years, and five were men. Skin colors of the patients ranged from Fitzpatrick skin types I to VI, and all had been taking at least two medications, typically methotrexate and prednisone.
The patients received 5 mg of tofacitinib twice a day for 6 months. “Everyone got better during the study, and six patients had a complete response, which we defined as a CSAMI score of zero activity,” Dr. Damsky said. “It’s really quite remarkable to see that.” Overall, the patients saw an 83% improvement in CSAMI scores.
In regard to safety, “all patients completed the study,” he said. “Tofacitinib was well tolerated, and there were no serious adverse effects or events.”
Tofacitinib is approved for treating rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular course juvenile idiopathic arthritis.
A month’s supply of twice-daily 5 mg tofacitinib pills would cost $4,900-$5,100 with free coupons, according to information accessed on April 24, 2021, on GoodRx.com. Generics are not available.
In an interview, Sotonye Imadojemu, MD, of the department of dermatology, Brigham and Women’s Hospital, Boston, praised the study, and said “tofacitinib is a reasonable treatment for treatment-refractory or extensive cutaneous sarcoidosis,” although it will be helpful to get results from randomized-controlled trials.
She cautioned that the drug “is a powerful immunosuppressant, so the risk of infection must be discussed with patients before prescribing. Screening for chronic infections such as viral hepatitis, tuberculosis, and HIV should be completed prior to treatment initiation. Blood counts, liver function, and lipid panels should be regularly monitored. The vaccines necessary for those who are immunosuppressed should be administered as able, and age-appropriate cancer screening must be kept up to date.”
The study was funded by Pfizer, the Dermatology Foundation, and the Yale Department of Dermatology. Dr. Damsky disclosed research support (Pfizer), consulting fees (Eli Lilly, Pfizer, TWi Biotechnology), and licensing fees (EMD Millipore/MillporeSigma). Dr. Imadojemu has no disclosures.
This article was updated 5/5/21.
Researchers are reporting impressive results in a small, , and all patients improved by an average of 83% via a scoring system.
“Not only did patients get better, but they were in many cases able to come off their baseline immunosuppressive regimen, including prednisone and methotrexate. They’d get off prednisone entirely or, in some cases, decrease it substantially,” study investigator William Damsky, MD, PhD, reported at the American Academy of Dermatology Virtual Meeting Experience.
Sarcoidosis is a common disease that affects an estimated 1 in 25 Black women and is believed to contribute to the deaths of about 4,000 people in the United States each year, noted Dr. Damsky of the department of dermatology, Yale University, New Haven, Conn. One famous patient is comedian Bernie Mac, who died from the condition in 2008.
“Approximately one third of patients have cutaneous involvement,” Dr. Damsky said, and skin may be the only manifestation of the disease. There is no Food and Drug Administration-approved therapy for cutaneous sarcoidosis, he added. Prednisone, the first-line therapy in skin manifestations, is approved only for pulmonary sarcoidosis.
“Oftentimes, there’s an attempt to transition either partially or fully to other therapies, including methotrexate and TNF-alpha blockers. But there’s been mixed success in doing that,” he said. This is not always possible, “so a lot of patients end up on prednisone.”
Earlier, a team at Yale prescribed 5 mg tofacitinib (Xeljanz) for several patients with severe cutaneous sarcoidosis and saw impressive results, Dr. Damsky said, including a patient with pulmonary sarcoidosis that also improved. He noted that there are case reports in the medical literature with similar findings.
Those positive results inspired the new study. Researchers recruited 10 patients with cutaneous sarcoidosis (9 with internal organ involvement) with a Cutaneous Sarcoidosis Activity and Morphology Instrument ( CSAMI ) score of 10 or higher. Nine patients were in their 50s, one was aged 63 years, and five were men. Skin colors of the patients ranged from Fitzpatrick skin types I to VI, and all had been taking at least two medications, typically methotrexate and prednisone.
The patients received 5 mg of tofacitinib twice a day for 6 months. “Everyone got better during the study, and six patients had a complete response, which we defined as a CSAMI score of zero activity,” Dr. Damsky said. “It’s really quite remarkable to see that.” Overall, the patients saw an 83% improvement in CSAMI scores.
In regard to safety, “all patients completed the study,” he said. “Tofacitinib was well tolerated, and there were no serious adverse effects or events.”
Tofacitinib is approved for treating rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, and polyarticular course juvenile idiopathic arthritis.
A month’s supply of twice-daily 5 mg tofacitinib pills would cost $4,900-$5,100 with free coupons, according to information accessed on April 24, 2021, on GoodRx.com. Generics are not available.
In an interview, Sotonye Imadojemu, MD, of the department of dermatology, Brigham and Women’s Hospital, Boston, praised the study, and said “tofacitinib is a reasonable treatment for treatment-refractory or extensive cutaneous sarcoidosis,” although it will be helpful to get results from randomized-controlled trials.
She cautioned that the drug “is a powerful immunosuppressant, so the risk of infection must be discussed with patients before prescribing. Screening for chronic infections such as viral hepatitis, tuberculosis, and HIV should be completed prior to treatment initiation. Blood counts, liver function, and lipid panels should be regularly monitored. The vaccines necessary for those who are immunosuppressed should be administered as able, and age-appropriate cancer screening must be kept up to date.”
The study was funded by Pfizer, the Dermatology Foundation, and the Yale Department of Dermatology. Dr. Damsky disclosed research support (Pfizer), consulting fees (Eli Lilly, Pfizer, TWi Biotechnology), and licensing fees (EMD Millipore/MillporeSigma). Dr. Imadojemu has no disclosures.
This article was updated 5/5/21.
REPORTING FROM AAD VMX 2021