User login
Concurrent ‘never event’ prescribing may worsen dementia in Parkinson’s
Nearly half of Medicare beneficiaries with Parkinson’s disease were concurrently prescribed a high-potency anticholinergic medication and an acetylcholinesterase inhibitor, with higher rates of potential prescribing errors seen among women and Hispanic patients, according to a cross-sectional analysis of Centers for Medicare & Medicaid Services data published in JAMA Neurology.

“Coadministration of a drug with high anticholinergic activity and an [acetylcholinesterase inhibitor] represents a frank prescribing error because these drugs have opposing pharmacologic effects,” wrote Sneha Mantri, MD, of the Parkinson’s Disease Research, Education, and Clinical Center at the Philadelphia VA Medical Center, and her colleagues. “In patients with Parkinson disease, who bear additional risks of cognitive impairment and vulnerability to anticholinergic activity, coprescribing of an [acetylcholinesterase inhibitor] and a high-potency anticholinergic medication can be considered a never event because it is a medication error likely to contribute to disability.”
Dr. Mantri and her colleagues analyzed the inpatient, outpatient, and prescription data of 268,407 Medicare beneficiaries with Parkinson’s, of whom 73,093 patients (27.2%) were prescribed a minimum of one antidementia medication fill. Patients were mean 78.9 years old, and the demographics of the Medicare beneficiaries were 50.1% male, 86.7% white, 5.5% black, 2.7% Hispanic, 2.7% Asian, and 0.7% Native American. The most common antidementia prescriptions were donepezil hydrochloride (63.0%), memantine hydrochloride (41.8%), and rivastigmine tartrate (26.4%). The researchers measured medications in cases of coprescription with potential anticholinergic (ACH) activity using the Anticholinergic Cognitive Burden Scale.
They found antidementia medication use was associated with patients who were black (adjusted odds ratio, 1.33; 95% confidence interval, 1.28-1.38) and Hispanic (aOR, 1.28; 95% CI, 1.22-1.35). Meanwhile, a negative association was found between Native American patients and antidementia medication use (aOR, 0.62; 95% CI, 0.51-0.74) compared with white patients and women (aOR, 0.85; 95% CI, 0.84-0.87) compared with men. The researchers noted that 28,495 patients (44.5%) were prescribed concurrently one high-potency anticholinergic and acetylcholinesterase inhibitors, with higher rates of prescribing seen for Hispanic (aOR, 1.11; 95% CI, 1.00-1.23) and women (aOR, 1.30; 95% CI, 1.25-1.35). High prevalence clusters of this type of prescribing were statistically high in the Southern and Midwestern states, they added.
Limitations included the study of a single year of data and the absence of conclusive data of dementia prevalence among Parkinson’s patients based on antidementia medication use alone and potential off-label use of antidementia medication analyzed in the study, the researchers said.
“In determining whether anticholinergic drug exposure has a causal role in clinical dementia in Parkinson disease, future studies may take a clinical trial approach, in which high-potency anticholinergic medications are replaced with lower-potency alternatives, and the change in cognitive testing and cognitive trajectory are measured,” Dr. Mantri and her colleagues wrote. “Such an approach will allow the calculation of anticholinergic drug safety in terms that are easily understood, such as number needed to harm.”
This study was funded by a grant from the National Institute of Neurological Diseases and Stroke of the National Institutes of Health. The authors report no relevant conflicts of interest.
SOURCE: Mantri S et al. JAMA Neurol. 2018 Oct 1. doi: 10.1001/jamaneurol.2018.2820.
Describing the prescribing patterns of antidementia medication for patients with Parkinson’s disease is useful, particularly with regard to black and Hispanic patients, but the analysis by Sneha Mantri, MD, and her colleagues has methodological issues that limit its impact, wrote Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, wrote in an editorial.
While the researchers used the Anticholinergic Cognitive Burden (ACB) Scale and graded acetylcholinesterase inhibitor and high anticholinergic (ACH) activity medication combinations with the highest ACB score (3 on a scale of 0 to 3) and labeled them as a prescribing error, the ACB scale score alone should not be the sole determination of whether a prescription combination is deemed an error, the authors said.
“A problem, however, arises in defining the coadministration of an antidementia drug and a medication with high ACH activity as a prescribing error or a never event in all clinical circumstances,” the authors wrote. “Across the literature in this area, the most important principle repeatedly emphasized was that these resources are intended to identify potentially inappropriate medication (not inappropriate medications), and the recommendations and ratings provided cannot replace patient-specific clinical judgment.”
Further, medications such as clozapine and quetiapine fumarate are often prescribed for patients with Parkinson’s disease–related psychosis as a standard of care; those medications also have an ACB score of 3, which would label them as prescribing errors and never events under the study by Mantri et al., the authors continued.
“Variability in ACH activity scoring methods is an additional concern, as no criterion standard exists for ranking relative ACH activity and the scores for medications to treat Parkinson disease (such as quetiapine for Parkinson disease psychosis) can markedly differ between the scales commonly used (such as the ACB Scale),” they wrote.
Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, are affiliated with the Fixel Center for Neurological Diseases in the department of neurology at the University of Florida in Gainesville. These comments summarize their editorial (JAMA Neurol. 2018 Oct 1 doi: 10.1001/jamaneurol.2018.2826).They reported no relevant conflicts of interest.
Describing the prescribing patterns of antidementia medication for patients with Parkinson’s disease is useful, particularly with regard to black and Hispanic patients, but the analysis by Sneha Mantri, MD, and her colleagues has methodological issues that limit its impact, wrote Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, wrote in an editorial.
While the researchers used the Anticholinergic Cognitive Burden (ACB) Scale and graded acetylcholinesterase inhibitor and high anticholinergic (ACH) activity medication combinations with the highest ACB score (3 on a scale of 0 to 3) and labeled them as a prescribing error, the ACB scale score alone should not be the sole determination of whether a prescription combination is deemed an error, the authors said.
“A problem, however, arises in defining the coadministration of an antidementia drug and a medication with high ACH activity as a prescribing error or a never event in all clinical circumstances,” the authors wrote. “Across the literature in this area, the most important principle repeatedly emphasized was that these resources are intended to identify potentially inappropriate medication (not inappropriate medications), and the recommendations and ratings provided cannot replace patient-specific clinical judgment.”
Further, medications such as clozapine and quetiapine fumarate are often prescribed for patients with Parkinson’s disease–related psychosis as a standard of care; those medications also have an ACB score of 3, which would label them as prescribing errors and never events under the study by Mantri et al., the authors continued.
“Variability in ACH activity scoring methods is an additional concern, as no criterion standard exists for ranking relative ACH activity and the scores for medications to treat Parkinson disease (such as quetiapine for Parkinson disease psychosis) can markedly differ between the scales commonly used (such as the ACB Scale),” they wrote.
Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, are affiliated with the Fixel Center for Neurological Diseases in the department of neurology at the University of Florida in Gainesville. These comments summarize their editorial (JAMA Neurol. 2018 Oct 1 doi: 10.1001/jamaneurol.2018.2826).They reported no relevant conflicts of interest.
Describing the prescribing patterns of antidementia medication for patients with Parkinson’s disease is useful, particularly with regard to black and Hispanic patients, but the analysis by Sneha Mantri, MD, and her colleagues has methodological issues that limit its impact, wrote Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, wrote in an editorial.
While the researchers used the Anticholinergic Cognitive Burden (ACB) Scale and graded acetylcholinesterase inhibitor and high anticholinergic (ACH) activity medication combinations with the highest ACB score (3 on a scale of 0 to 3) and labeled them as a prescribing error, the ACB scale score alone should not be the sole determination of whether a prescription combination is deemed an error, the authors said.
“A problem, however, arises in defining the coadministration of an antidementia drug and a medication with high ACH activity as a prescribing error or a never event in all clinical circumstances,” the authors wrote. “Across the literature in this area, the most important principle repeatedly emphasized was that these resources are intended to identify potentially inappropriate medication (not inappropriate medications), and the recommendations and ratings provided cannot replace patient-specific clinical judgment.”
Further, medications such as clozapine and quetiapine fumarate are often prescribed for patients with Parkinson’s disease–related psychosis as a standard of care; those medications also have an ACB score of 3, which would label them as prescribing errors and never events under the study by Mantri et al., the authors continued.
“Variability in ACH activity scoring methods is an additional concern, as no criterion standard exists for ranking relative ACH activity and the scores for medications to treat Parkinson disease (such as quetiapine for Parkinson disease psychosis) can markedly differ between the scales commonly used (such as the ACB Scale),” they wrote.
Christopher W. Hess, MD, Michael S. Okun, MD, and Adolfo Ramirez-Zamora, MD, are affiliated with the Fixel Center for Neurological Diseases in the department of neurology at the University of Florida in Gainesville. These comments summarize their editorial (JAMA Neurol. 2018 Oct 1 doi: 10.1001/jamaneurol.2018.2826).They reported no relevant conflicts of interest.
Nearly half of Medicare beneficiaries with Parkinson’s disease were concurrently prescribed a high-potency anticholinergic medication and an acetylcholinesterase inhibitor, with higher rates of potential prescribing errors seen among women and Hispanic patients, according to a cross-sectional analysis of Centers for Medicare & Medicaid Services data published in JAMA Neurology.
“Coadministration of a drug with high anticholinergic activity and an [acetylcholinesterase inhibitor] represents a frank prescribing error because these drugs have opposing pharmacologic effects,” wrote Sneha Mantri, MD, of the Parkinson’s Disease Research, Education, and Clinical Center at the Philadelphia VA Medical Center, and her colleagues. “In patients with Parkinson disease, who bear additional risks of cognitive impairment and vulnerability to anticholinergic activity, coprescribing of an [acetylcholinesterase inhibitor] and a high-potency anticholinergic medication can be considered a never event because it is a medication error likely to contribute to disability.”
Dr. Mantri and her colleagues analyzed the inpatient, outpatient, and prescription data of 268,407 Medicare beneficiaries with Parkinson’s, of whom 73,093 patients (27.2%) were prescribed a minimum of one antidementia medication fill. Patients were mean 78.9 years old, and the demographics of the Medicare beneficiaries were 50.1% male, 86.7% white, 5.5% black, 2.7% Hispanic, 2.7% Asian, and 0.7% Native American. The most common antidementia prescriptions were donepezil hydrochloride (63.0%), memantine hydrochloride (41.8%), and rivastigmine tartrate (26.4%). The researchers measured medications in cases of coprescription with potential anticholinergic (ACH) activity using the Anticholinergic Cognitive Burden Scale.
They found antidementia medication use was associated with patients who were black (adjusted odds ratio, 1.33; 95% confidence interval, 1.28-1.38) and Hispanic (aOR, 1.28; 95% CI, 1.22-1.35). Meanwhile, a negative association was found between Native American patients and antidementia medication use (aOR, 0.62; 95% CI, 0.51-0.74) compared with white patients and women (aOR, 0.85; 95% CI, 0.84-0.87) compared with men. The researchers noted that 28,495 patients (44.5%) were prescribed concurrently one high-potency anticholinergic and acetylcholinesterase inhibitors, with higher rates of prescribing seen for Hispanic (aOR, 1.11; 95% CI, 1.00-1.23) and women (aOR, 1.30; 95% CI, 1.25-1.35). High prevalence clusters of this type of prescribing were statistically high in the Southern and Midwestern states, they added.
Limitations included the study of a single year of data and the absence of conclusive data of dementia prevalence among Parkinson’s patients based on antidementia medication use alone and potential off-label use of antidementia medication analyzed in the study, the researchers said.
“In determining whether anticholinergic drug exposure has a causal role in clinical dementia in Parkinson disease, future studies may take a clinical trial approach, in which high-potency anticholinergic medications are replaced with lower-potency alternatives, and the change in cognitive testing and cognitive trajectory are measured,” Dr. Mantri and her colleagues wrote. “Such an approach will allow the calculation of anticholinergic drug safety in terms that are easily understood, such as number needed to harm.”
This study was funded by a grant from the National Institute of Neurological Diseases and Stroke of the National Institutes of Health. The authors report no relevant conflicts of interest.
SOURCE: Mantri S et al. JAMA Neurol. 2018 Oct 1. doi: 10.1001/jamaneurol.2018.2820.
Nearly half of Medicare beneficiaries with Parkinson’s disease were concurrently prescribed a high-potency anticholinergic medication and an acetylcholinesterase inhibitor, with higher rates of potential prescribing errors seen among women and Hispanic patients, according to a cross-sectional analysis of Centers for Medicare & Medicaid Services data published in JAMA Neurology.
“Coadministration of a drug with high anticholinergic activity and an [acetylcholinesterase inhibitor] represents a frank prescribing error because these drugs have opposing pharmacologic effects,” wrote Sneha Mantri, MD, of the Parkinson’s Disease Research, Education, and Clinical Center at the Philadelphia VA Medical Center, and her colleagues. “In patients with Parkinson disease, who bear additional risks of cognitive impairment and vulnerability to anticholinergic activity, coprescribing of an [acetylcholinesterase inhibitor] and a high-potency anticholinergic medication can be considered a never event because it is a medication error likely to contribute to disability.”
Dr. Mantri and her colleagues analyzed the inpatient, outpatient, and prescription data of 268,407 Medicare beneficiaries with Parkinson’s, of whom 73,093 patients (27.2%) were prescribed a minimum of one antidementia medication fill. Patients were mean 78.9 years old, and the demographics of the Medicare beneficiaries were 50.1% male, 86.7% white, 5.5% black, 2.7% Hispanic, 2.7% Asian, and 0.7% Native American. The most common antidementia prescriptions were donepezil hydrochloride (63.0%), memantine hydrochloride (41.8%), and rivastigmine tartrate (26.4%). The researchers measured medications in cases of coprescription with potential anticholinergic (ACH) activity using the Anticholinergic Cognitive Burden Scale.
They found antidementia medication use was associated with patients who were black (adjusted odds ratio, 1.33; 95% confidence interval, 1.28-1.38) and Hispanic (aOR, 1.28; 95% CI, 1.22-1.35). Meanwhile, a negative association was found between Native American patients and antidementia medication use (aOR, 0.62; 95% CI, 0.51-0.74) compared with white patients and women (aOR, 0.85; 95% CI, 0.84-0.87) compared with men. The researchers noted that 28,495 patients (44.5%) were prescribed concurrently one high-potency anticholinergic and acetylcholinesterase inhibitors, with higher rates of prescribing seen for Hispanic (aOR, 1.11; 95% CI, 1.00-1.23) and women (aOR, 1.30; 95% CI, 1.25-1.35). High prevalence clusters of this type of prescribing were statistically high in the Southern and Midwestern states, they added.
Limitations included the study of a single year of data and the absence of conclusive data of dementia prevalence among Parkinson’s patients based on antidementia medication use alone and potential off-label use of antidementia medication analyzed in the study, the researchers said.
“In determining whether anticholinergic drug exposure has a causal role in clinical dementia in Parkinson disease, future studies may take a clinical trial approach, in which high-potency anticholinergic medications are replaced with lower-potency alternatives, and the change in cognitive testing and cognitive trajectory are measured,” Dr. Mantri and her colleagues wrote. “Such an approach will allow the calculation of anticholinergic drug safety in terms that are easily understood, such as number needed to harm.”
This study was funded by a grant from the National Institute of Neurological Diseases and Stroke of the National Institutes of Health. The authors report no relevant conflicts of interest.
SOURCE: Mantri S et al. JAMA Neurol. 2018 Oct 1. doi: 10.1001/jamaneurol.2018.2820.
FROM JAMA NEUROLOGY
Key clinical point: Medicare beneficiaries with Parkinson’s disease often are concurrently prescribed a high-potency anticholinergic medication and an acetylcholinesterase inhibitor.
Major finding: More than 44% of patients experienced at least one never event, with higher rates of potential prescribing errors seen in Hispanic patients (adjusted OR, 1.11) and women (adjusted OR, 1.30).
Study details: An analysis of inpatient, outpatient, and prescription data for 268,407 Medicare beneficiaries with Parkinson’s disease over 12 months.
Disclosures: This study was funded by a grant from the National Institute of Neurological Diseases and Stroke of the National Institutes of Health. The authors reported no relevant conflicts of interest.
Source: Mantri S et al. JAMA Neurol. 2018 Oct 1. doi: 10.1001/jamaneurol.2018.2820.
Low-dose ketamine controls pain from severe chest injury, while sparing opioid consumption
SAN DIEGO – while reducing opioid consumption.

The anesthetic didn’t make much difference in pain control or opioid use overall in a randomized study of 93 patients with thoracic injury Nathan Kugler, MD, said at the annual meeting of the American Association for the Surgery of Trauma. But among severely injured patients, it cut the opioid mean equivalency dose by about 164 mg over the 48-hour infusion and by 328 mg over a mean hospital stay while maintaining pain control, said Dr. Kugler, a surgical resident at the Medical College of Wisconsin, Milwaukee.
“With increasing focus on multimodal pain strategies, opioid-based regimens continue to be the backbone of pain control,” he said. “We have used ketamine effectively for failure of maximum therapy and demonstrated an opioid-sparing effect.” This new research shows that the drug can be an effective adjunct for acute pain control for severely injured patients in the emergency setting.
The study recruited 93 patients with thoracic injury; they had a mean of six broken ribs, mostly caused by motor vehicle accidents. Most of the patients were male (75%), and their mean age was 46 years. The mean Injury Severity Score was about 15; about 30% had flail chest.
All patients received a standardized acute pain medication regime comprising acetaminophen, nonsteroidal anti-inflammatories, methocarbamol (Robaxin), and intravenous opioids. Regional therapies included rib block with an epidural catheter. In addition, they were randomized to placebo infusions or to 48 hours of IV ketamine at 2.5 mcg/kg per minute. “To put this in perspective, for a 70-kg patient, that is a mean of 10.5 mg/hour,” Dr. Kugler said.
The primary endpoint was a reduction of at least 2 points on an 11-point pain scale. Secondary endpoints included opioid use in oral morphine equivalents (OME); respiratory complications; and psychoactive events. The primary outcome was assessed with an area under the curve model.
In the overall group, there was no significant between-group difference in pain score. Nor were there differences in the total OME at 12-24 hours (184 mg ketamine vs. 230 mg placebo), or at 48 hours (86 vs. 113 mg).
Dr. Kugler also looked at these outcomes in patients who had only rib fractures independent of other chest injury. He saw no significant differences in pain scores or OME at 24 or 48 hours.
However, significant differences did emerge in the group of severely injured patients with an Injury Severity Score of more than 15. There were no differences in pain scores at either time point. However, ketamine allowed patients to achieve the same level of pain control with significantly less opioid medication. The OME at 12-24 hours was 50.5 mg vs 94 mg. At 24-48 hours, it was 87 mg vs. 64 mg.
This worked out to a mean OME savings of 148 mg over a patient’s entire hospitalization.
“We saw a very nice separation of opioid consumption that began early and continued to separate over the 48-hour infusion and even after it was discontinued,” Dr. Kugler said.
This benefit was achieved without any additional adverse events, he added. There were no significant differences in confusion; epidural placement; length of stay; respiratory event, sedation, hallucinations, delusions or disturbing dreams; or unplanned transfers to the ICU.
Dr. Kugler disclosed that he and primary investigator Thomas Carver, MD, also of the Medical College of Wisconsin, Milwaukee, are both paid consultants for InnoVital Systems.
SAN DIEGO – while reducing opioid consumption.
The anesthetic didn’t make much difference in pain control or opioid use overall in a randomized study of 93 patients with thoracic injury Nathan Kugler, MD, said at the annual meeting of the American Association for the Surgery of Trauma. But among severely injured patients, it cut the opioid mean equivalency dose by about 164 mg over the 48-hour infusion and by 328 mg over a mean hospital stay while maintaining pain control, said Dr. Kugler, a surgical resident at the Medical College of Wisconsin, Milwaukee.
“With increasing focus on multimodal pain strategies, opioid-based regimens continue to be the backbone of pain control,” he said. “We have used ketamine effectively for failure of maximum therapy and demonstrated an opioid-sparing effect.” This new research shows that the drug can be an effective adjunct for acute pain control for severely injured patients in the emergency setting.
The study recruited 93 patients with thoracic injury; they had a mean of six broken ribs, mostly caused by motor vehicle accidents. Most of the patients were male (75%), and their mean age was 46 years. The mean Injury Severity Score was about 15; about 30% had flail chest.
All patients received a standardized acute pain medication regime comprising acetaminophen, nonsteroidal anti-inflammatories, methocarbamol (Robaxin), and intravenous opioids. Regional therapies included rib block with an epidural catheter. In addition, they were randomized to placebo infusions or to 48 hours of IV ketamine at 2.5 mcg/kg per minute. “To put this in perspective, for a 70-kg patient, that is a mean of 10.5 mg/hour,” Dr. Kugler said.
The primary endpoint was a reduction of at least 2 points on an 11-point pain scale. Secondary endpoints included opioid use in oral morphine equivalents (OME); respiratory complications; and psychoactive events. The primary outcome was assessed with an area under the curve model.
In the overall group, there was no significant between-group difference in pain score. Nor were there differences in the total OME at 12-24 hours (184 mg ketamine vs. 230 mg placebo), or at 48 hours (86 vs. 113 mg).
Dr. Kugler also looked at these outcomes in patients who had only rib fractures independent of other chest injury. He saw no significant differences in pain scores or OME at 24 or 48 hours.
However, significant differences did emerge in the group of severely injured patients with an Injury Severity Score of more than 15. There were no differences in pain scores at either time point. However, ketamine allowed patients to achieve the same level of pain control with significantly less opioid medication. The OME at 12-24 hours was 50.5 mg vs 94 mg. At 24-48 hours, it was 87 mg vs. 64 mg.
This worked out to a mean OME savings of 148 mg over a patient’s entire hospitalization.
“We saw a very nice separation of opioid consumption that began early and continued to separate over the 48-hour infusion and even after it was discontinued,” Dr. Kugler said.
This benefit was achieved without any additional adverse events, he added. There were no significant differences in confusion; epidural placement; length of stay; respiratory event, sedation, hallucinations, delusions or disturbing dreams; or unplanned transfers to the ICU.
Dr. Kugler disclosed that he and primary investigator Thomas Carver, MD, also of the Medical College of Wisconsin, Milwaukee, are both paid consultants for InnoVital Systems.
SAN DIEGO – while reducing opioid consumption.
The anesthetic didn’t make much difference in pain control or opioid use overall in a randomized study of 93 patients with thoracic injury Nathan Kugler, MD, said at the annual meeting of the American Association for the Surgery of Trauma. But among severely injured patients, it cut the opioid mean equivalency dose by about 164 mg over the 48-hour infusion and by 328 mg over a mean hospital stay while maintaining pain control, said Dr. Kugler, a surgical resident at the Medical College of Wisconsin, Milwaukee.
“With increasing focus on multimodal pain strategies, opioid-based regimens continue to be the backbone of pain control,” he said. “We have used ketamine effectively for failure of maximum therapy and demonstrated an opioid-sparing effect.” This new research shows that the drug can be an effective adjunct for acute pain control for severely injured patients in the emergency setting.
The study recruited 93 patients with thoracic injury; they had a mean of six broken ribs, mostly caused by motor vehicle accidents. Most of the patients were male (75%), and their mean age was 46 years. The mean Injury Severity Score was about 15; about 30% had flail chest.
All patients received a standardized acute pain medication regime comprising acetaminophen, nonsteroidal anti-inflammatories, methocarbamol (Robaxin), and intravenous opioids. Regional therapies included rib block with an epidural catheter. In addition, they were randomized to placebo infusions or to 48 hours of IV ketamine at 2.5 mcg/kg per minute. “To put this in perspective, for a 70-kg patient, that is a mean of 10.5 mg/hour,” Dr. Kugler said.
The primary endpoint was a reduction of at least 2 points on an 11-point pain scale. Secondary endpoints included opioid use in oral morphine equivalents (OME); respiratory complications; and psychoactive events. The primary outcome was assessed with an area under the curve model.
In the overall group, there was no significant between-group difference in pain score. Nor were there differences in the total OME at 12-24 hours (184 mg ketamine vs. 230 mg placebo), or at 48 hours (86 vs. 113 mg).
Dr. Kugler also looked at these outcomes in patients who had only rib fractures independent of other chest injury. He saw no significant differences in pain scores or OME at 24 or 48 hours.
However, significant differences did emerge in the group of severely injured patients with an Injury Severity Score of more than 15. There were no differences in pain scores at either time point. However, ketamine allowed patients to achieve the same level of pain control with significantly less opioid medication. The OME at 12-24 hours was 50.5 mg vs 94 mg. At 24-48 hours, it was 87 mg vs. 64 mg.
This worked out to a mean OME savings of 148 mg over a patient’s entire hospitalization.
“We saw a very nice separation of opioid consumption that began early and continued to separate over the 48-hour infusion and even after it was discontinued,” Dr. Kugler said.
This benefit was achieved without any additional adverse events, he added. There were no significant differences in confusion; epidural placement; length of stay; respiratory event, sedation, hallucinations, delusions or disturbing dreams; or unplanned transfers to the ICU.
Dr. Kugler disclosed that he and primary investigator Thomas Carver, MD, also of the Medical College of Wisconsin, Milwaukee, are both paid consultants for InnoVital Systems.
REPORTING FROM THE AAST ANNUAL MEETING
Key clinical point: Low-dose ketamine controlled pain while reducing opioid use among patients with severe thoracic injury.
Major finding: Compared with placebo, ketamine reduced opioids conferred OME savings of 148 mg over a patient’s entire hospitalization.
Study details: The randomized study comprised 93 patients with thoracic injury.
Disclosures: Dr. Kugler disclosed that he and primary investigator Thomas Carver, MD, are both paid consultants for InnoVital Systems.
Source: Carver T et al. AAST 2018, Oral abstract 2
How effectively do ACE inhibitors and ARBs prevent migraines?
EVIDENCE SUMMARY
A network meta-analysis of 179 placebo-controlled trials of medications to treat migraine1 headache identified 3 trials involving ACE inhibitors and 3 involving ARBs (TABLE1). The authors of the meta-analysis gave 2 trials (one of lisinopril and one of candesartan) relatively high scores for methodologic quality.

Lisinopril reduces hours, days with headache and days with migraine
The first, a placebo-controlled lisinopril crossover trial, included 60 patients, 19 to 59 years of age, who experienced migraines with or without auras 2 to 6 times per month.2 Thirty patients received lisinopril 10 mg once daily for 1 week followed by 20 mg once daily (using 10-mg tablets) for 11 weeks. The other 30 patients received a similarly titrated placebo for 12 weeks. After a 2-week washout period, the groups were given the other therapy. Patients took triptan medications and analgesics as needed. Primary outcomes, extracted from headache diaries, included the number of hours and days with headache (of any type) and number of days with migraine specifically.
Out of the initial 60 participants, 47 completed the study. Using intention-to-treat analysis, lisinopril therapy resulted in fewer hours with headache (162 vs 138, a 15% difference; 95% confidence interval [CI], 0-30), fewer days with headache (25 vs 21, a 16% difference; 95% CI, 5-27), and fewer days with migraine (19 vs 15, a 22% change; 95% CI, 11-33), compared with placebo. Three patients discontinued lisinopril because of adverse events. Mean blood pressure reduction with lisinopril was 7 mm Hg systolic and 5 mm Hg diastolic more than placebo (P<.0001 for both comparisons).
Candesartan also decreases headaches and migraine
The other study given a high methodologic quality score by the network-meta-analysis authors was a placebo-controlled candesartan crossover trial.3 It enrolled 60 patients, 18 to 65 years of age, who experienced migraines with or without auras 2 to 6 times per month.
Thirty patients received 16 mg candesartan daily for 12 weeks, followed by a 4-week washout period before taking a placebo tablet daily for 12 weeks. The other 30 received placebo followed by candesartan. Patients took triptan medications and analgesics as needed. The primary outcome measure was days with headache, recorded by patients using daily diaries. Three patients didn’t complete the study.
Using intention-to-treat analysis, the mean number of days with headache was 18.5 with placebo and 13.6 with candesartan (P=.001). Secondary end points that also favored candesartan were hours with migraine (92 vs 59; P<.001), hours with headache (139 vs 95; P<.001), days with migraine (13 vs 9; P<.001), and days of sick leave (3.9 vs 1.4; P=.01). Adverse events, including dizziness, were similar with candesartan and placebo. Mean blood pressure reduction with candesartan was 11 mm Hg systolic and 7 mm Hg diastolic over placebo (P<.001 for both comparisons).
Continue to: Overall both drugs have a significant effect on number of headaches
Overall both drugs have a significant effect on number of headaches
Among all ACE inhibitor and ARB trials in the review, a network meta-analysis (designed to compare interventions never studied head-to-head) could be performed only on candesartan, which had a small effect size on headache frequency relative to placebo (2 trials, 118 patients; standardized mean difference [SMD]= −0.33; 95% CI, −0.59 to −0.7).1 (An SMD of 0.2 is considered small, 0.6 moderate, and 1.2 large). Combining data from all ACE inhibitor and ARB trials together in a standard meta-analysis yielded a large effect size on number of headaches per month compared with placebo (6 trials, 351 patients; SMD= −1.12; 95% CI, −1.97 to −0.27).1
RECOMMENDATIONS
In 2012, the American Academy of Neurology and the American Headache Society published guidelines on pharmacologic treatment for episodic migraine prevention in adults.4 The guidelines stated that lisinopril and candesartan were “possibly effective” for migraine prevention (level C recommendation based on a single lower-quality randomized clinical trial). They further advised clinicians to be “mindful of comorbid and coexistent conditions in patients with migraine to maximize potential treatment efficacy.”
1. Jackson JL, Cogbil E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PloS One. 2015;10:e0130733.
2. Schrader H, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with angiotensin converting enzyme inhibitor (lisinopril): randomized, placebo controlled, crossover study. BMJ. 2001;322:19-22.
3. Tronvik E, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA. 2003;289:65-69.
4. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
EVIDENCE SUMMARY
A network meta-analysis of 179 placebo-controlled trials of medications to treat migraine1 headache identified 3 trials involving ACE inhibitors and 3 involving ARBs (TABLE1). The authors of the meta-analysis gave 2 trials (one of lisinopril and one of candesartan) relatively high scores for methodologic quality.
Lisinopril reduces hours, days with headache and days with migraine
The first, a placebo-controlled lisinopril crossover trial, included 60 patients, 19 to 59 years of age, who experienced migraines with or without auras 2 to 6 times per month.2 Thirty patients received lisinopril 10 mg once daily for 1 week followed by 20 mg once daily (using 10-mg tablets) for 11 weeks. The other 30 patients received a similarly titrated placebo for 12 weeks. After a 2-week washout period, the groups were given the other therapy. Patients took triptan medications and analgesics as needed. Primary outcomes, extracted from headache diaries, included the number of hours and days with headache (of any type) and number of days with migraine specifically.
Out of the initial 60 participants, 47 completed the study. Using intention-to-treat analysis, lisinopril therapy resulted in fewer hours with headache (162 vs 138, a 15% difference; 95% confidence interval [CI], 0-30), fewer days with headache (25 vs 21, a 16% difference; 95% CI, 5-27), and fewer days with migraine (19 vs 15, a 22% change; 95% CI, 11-33), compared with placebo. Three patients discontinued lisinopril because of adverse events. Mean blood pressure reduction with lisinopril was 7 mm Hg systolic and 5 mm Hg diastolic more than placebo (P<.0001 for both comparisons).
Candesartan also decreases headaches and migraine
The other study given a high methodologic quality score by the network-meta-analysis authors was a placebo-controlled candesartan crossover trial.3 It enrolled 60 patients, 18 to 65 years of age, who experienced migraines with or without auras 2 to 6 times per month.
Thirty patients received 16 mg candesartan daily for 12 weeks, followed by a 4-week washout period before taking a placebo tablet daily for 12 weeks. The other 30 received placebo followed by candesartan. Patients took triptan medications and analgesics as needed. The primary outcome measure was days with headache, recorded by patients using daily diaries. Three patients didn’t complete the study.
Using intention-to-treat analysis, the mean number of days with headache was 18.5 with placebo and 13.6 with candesartan (P=.001). Secondary end points that also favored candesartan were hours with migraine (92 vs 59; P<.001), hours with headache (139 vs 95; P<.001), days with migraine (13 vs 9; P<.001), and days of sick leave (3.9 vs 1.4; P=.01). Adverse events, including dizziness, were similar with candesartan and placebo. Mean blood pressure reduction with candesartan was 11 mm Hg systolic and 7 mm Hg diastolic over placebo (P<.001 for both comparisons).
Continue to: Overall both drugs have a significant effect on number of headaches
Overall both drugs have a significant effect on number of headaches
Among all ACE inhibitor and ARB trials in the review, a network meta-analysis (designed to compare interventions never studied head-to-head) could be performed only on candesartan, which had a small effect size on headache frequency relative to placebo (2 trials, 118 patients; standardized mean difference [SMD]= −0.33; 95% CI, −0.59 to −0.7).1 (An SMD of 0.2 is considered small, 0.6 moderate, and 1.2 large). Combining data from all ACE inhibitor and ARB trials together in a standard meta-analysis yielded a large effect size on number of headaches per month compared with placebo (6 trials, 351 patients; SMD= −1.12; 95% CI, −1.97 to −0.27).1
RECOMMENDATIONS
In 2012, the American Academy of Neurology and the American Headache Society published guidelines on pharmacologic treatment for episodic migraine prevention in adults.4 The guidelines stated that lisinopril and candesartan were “possibly effective” for migraine prevention (level C recommendation based on a single lower-quality randomized clinical trial). They further advised clinicians to be “mindful of comorbid and coexistent conditions in patients with migraine to maximize potential treatment efficacy.”
EVIDENCE SUMMARY
A network meta-analysis of 179 placebo-controlled trials of medications to treat migraine1 headache identified 3 trials involving ACE inhibitors and 3 involving ARBs (TABLE1). The authors of the meta-analysis gave 2 trials (one of lisinopril and one of candesartan) relatively high scores for methodologic quality.
Lisinopril reduces hours, days with headache and days with migraine
The first, a placebo-controlled lisinopril crossover trial, included 60 patients, 19 to 59 years of age, who experienced migraines with or without auras 2 to 6 times per month.2 Thirty patients received lisinopril 10 mg once daily for 1 week followed by 20 mg once daily (using 10-mg tablets) for 11 weeks. The other 30 patients received a similarly titrated placebo for 12 weeks. After a 2-week washout period, the groups were given the other therapy. Patients took triptan medications and analgesics as needed. Primary outcomes, extracted from headache diaries, included the number of hours and days with headache (of any type) and number of days with migraine specifically.
Out of the initial 60 participants, 47 completed the study. Using intention-to-treat analysis, lisinopril therapy resulted in fewer hours with headache (162 vs 138, a 15% difference; 95% confidence interval [CI], 0-30), fewer days with headache (25 vs 21, a 16% difference; 95% CI, 5-27), and fewer days with migraine (19 vs 15, a 22% change; 95% CI, 11-33), compared with placebo. Three patients discontinued lisinopril because of adverse events. Mean blood pressure reduction with lisinopril was 7 mm Hg systolic and 5 mm Hg diastolic more than placebo (P<.0001 for both comparisons).
Candesartan also decreases headaches and migraine
The other study given a high methodologic quality score by the network-meta-analysis authors was a placebo-controlled candesartan crossover trial.3 It enrolled 60 patients, 18 to 65 years of age, who experienced migraines with or without auras 2 to 6 times per month.
Thirty patients received 16 mg candesartan daily for 12 weeks, followed by a 4-week washout period before taking a placebo tablet daily for 12 weeks. The other 30 received placebo followed by candesartan. Patients took triptan medications and analgesics as needed. The primary outcome measure was days with headache, recorded by patients using daily diaries. Three patients didn’t complete the study.
Using intention-to-treat analysis, the mean number of days with headache was 18.5 with placebo and 13.6 with candesartan (P=.001). Secondary end points that also favored candesartan were hours with migraine (92 vs 59; P<.001), hours with headache (139 vs 95; P<.001), days with migraine (13 vs 9; P<.001), and days of sick leave (3.9 vs 1.4; P=.01). Adverse events, including dizziness, were similar with candesartan and placebo. Mean blood pressure reduction with candesartan was 11 mm Hg systolic and 7 mm Hg diastolic over placebo (P<.001 for both comparisons).
Continue to: Overall both drugs have a significant effect on number of headaches
Overall both drugs have a significant effect on number of headaches
Among all ACE inhibitor and ARB trials in the review, a network meta-analysis (designed to compare interventions never studied head-to-head) could be performed only on candesartan, which had a small effect size on headache frequency relative to placebo (2 trials, 118 patients; standardized mean difference [SMD]= −0.33; 95% CI, −0.59 to −0.7).1 (An SMD of 0.2 is considered small, 0.6 moderate, and 1.2 large). Combining data from all ACE inhibitor and ARB trials together in a standard meta-analysis yielded a large effect size on number of headaches per month compared with placebo (6 trials, 351 patients; SMD= −1.12; 95% CI, −1.97 to −0.27).1
RECOMMENDATIONS
In 2012, the American Academy of Neurology and the American Headache Society published guidelines on pharmacologic treatment for episodic migraine prevention in adults.4 The guidelines stated that lisinopril and candesartan were “possibly effective” for migraine prevention (level C recommendation based on a single lower-quality randomized clinical trial). They further advised clinicians to be “mindful of comorbid and coexistent conditions in patients with migraine to maximize potential treatment efficacy.”
1. Jackson JL, Cogbil E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PloS One. 2015;10:e0130733.
2. Schrader H, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with angiotensin converting enzyme inhibitor (lisinopril): randomized, placebo controlled, crossover study. BMJ. 2001;322:19-22.
3. Tronvik E, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA. 2003;289:65-69.
4. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
1. Jackson JL, Cogbil E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PloS One. 2015;10:e0130733.
2. Schrader H, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with angiotensin converting enzyme inhibitor (lisinopril): randomized, placebo controlled, crossover study. BMJ. 2001;322:19-22.
3. Tronvik E, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA. 2003;289:65-69.
4. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
EVIDENCE-BASED ANSWER:
The angiotensin-converting enzyme (ACE) inhibitor lisinopril reduces the number of migraines by about 1.5 per month in patients experiencing 2 to 6 migraines monthly (strength of recommendation [SOR]: B, small crossover trial); the angiotensin II receptor blocker (ARB) candesartan may produce a similar reduction (SOR: C, conflicting crossover trials).
Considered as a group, ACE inhibitors and ARBs have a moderate to large effect on the frequency of migraine headaches (SOR: B, meta-analysis of small clinical trials), although only lisinopril and candesartan show fair to good evidence of efficacy.
Providers may consider lisinopril or candesartan for migraine prevention, taking into account their effect on other medical conditions (SOR: C, expert opinion).

Nausea and vomiting • sensitivity to smell • history of hypertension and alcohol abuse • Dx?
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m

DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; [email protected].
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m
DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; [email protected].
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m
DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; [email protected].
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.

Office approach to small fiber neuropathy
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES

Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.

Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.

INITIAL TESTING FOR AN UNDERLYING CAUSE

Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control

Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies

PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES
Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.
Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.
INITIAL TESTING FOR AN UNDERLYING CAUSE
Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control
Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies
PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES
Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.
Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.
INITIAL TESTING FOR AN UNDERLYING CAUSE
Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control
Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies
PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
KEY POINTS
- Patients typically develop a symmetric “stocking-glove” pattern of sensory loss in the feet and hands.
- The diagnosis may be confirmed with skin biopsy for nerve fiber density, which can easily be done in a clinic setting with commercially available kits.
- Diabetes is the most common identifiable cause of small fiber neuropathy.
- Serologic testing can help uncover a vitamin deficiency or other potentially treatable condition.
- Antiepileptics, antidepressants, and topical agents are first-line drugs for managing pain.
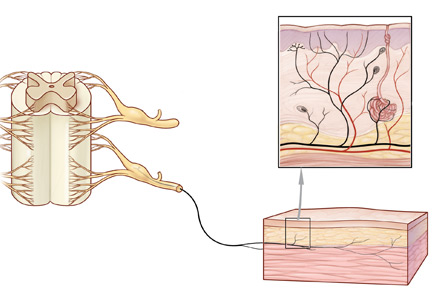
Small fibers, large impact

The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
When stroke runs in the family
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
.")
.")
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
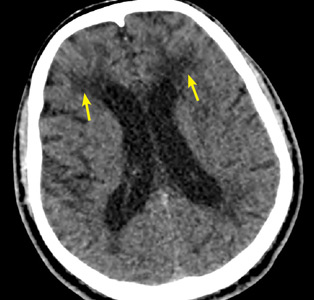
Postsurgical hypoparathyroidism is not primary hypoparathyroidism
To the Editor: I read with interest the case of a 67-year-old woman with bilateral hand numbness, published in the March 2018 issue of the Journal, and I would like to suggest 2 important corrections to this article.1
The authors present a case of hypocalcemia secondary to postsurgical hypoparathyroidism but describe it as due to primary hypoparathyroidism. The patient had undergone thyroidectomy 10 years earlier and since then had hypocalcemia, secondary to postsurgical hypoparathyroidism, that was treated with calcium and vitamin D, until she stopped taking these agents. Postsurgical hypothyroidism is the most common cause of acquired or secondary hypoparathyroidism and is not primary hypoparathyroidism. I strongly feel that this requires an update or correction to the article. This patient may have associated malabsorption, as the authors alluded to, as the cause of her “normal” serum parathyroid hormone level.
The patient also had hypomagnesemia, which the authors state could have been due to furosemide use and “uncontrolled” diabetes mellitus. Diabetes doesn’t need to be uncontrolled to cause hypomagnesemia. Hypomagnesemia is common in patients with type 2 diabetes mellitus, with a prevalence of 14% to 48% in patients with diabetes compared with 2.5% to 15% in the general population.2 It is often multifactorial and may be secondary to one or more of the following factors: poor dietary intake, autonomic dysfunction, altered insulin resistance, glomerular hyperfiltration, osmotic diuresis (uncontrolled diabetes), recurrent metabolic acidosis, hypophosphatemia, hypokalemia, and therapy with drugs such as metformin and sulfonylureas.
Patients with type 2 diabetes and hypomagnesemia often enter a vicious cycle in which hypomagnesemia worsens insulin resistance and insulin resistance, by reducing the activity of renal magnesium channel transient receptor potential melastatin (TRPM) type 6, perpetuates hypomagnesemia.3
- Radwan SS, Hamo KN, Zayed AA. A 67-year-old woman with bilateral hand numbness. Cleve Clin J Med 2018; 85(3):200–208. doi:10.3949/ccjm.85a.17026
- Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol 2007; 2(2):366–373. doi:10.2215/CJN.02960906
- Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes 2016; 65(1):3–13. doi:10.2337/db15-1028
To the Editor: I read with interest the case of a 67-year-old woman with bilateral hand numbness, published in the March 2018 issue of the Journal, and I would like to suggest 2 important corrections to this article.1
The authors present a case of hypocalcemia secondary to postsurgical hypoparathyroidism but describe it as due to primary hypoparathyroidism. The patient had undergone thyroidectomy 10 years earlier and since then had hypocalcemia, secondary to postsurgical hypoparathyroidism, that was treated with calcium and vitamin D, until she stopped taking these agents. Postsurgical hypothyroidism is the most common cause of acquired or secondary hypoparathyroidism and is not primary hypoparathyroidism. I strongly feel that this requires an update or correction to the article. This patient may have associated malabsorption, as the authors alluded to, as the cause of her “normal” serum parathyroid hormone level.
The patient also had hypomagnesemia, which the authors state could have been due to furosemide use and “uncontrolled” diabetes mellitus. Diabetes doesn’t need to be uncontrolled to cause hypomagnesemia. Hypomagnesemia is common in patients with type 2 diabetes mellitus, with a prevalence of 14% to 48% in patients with diabetes compared with 2.5% to 15% in the general population.2 It is often multifactorial and may be secondary to one or more of the following factors: poor dietary intake, autonomic dysfunction, altered insulin resistance, glomerular hyperfiltration, osmotic diuresis (uncontrolled diabetes), recurrent metabolic acidosis, hypophosphatemia, hypokalemia, and therapy with drugs such as metformin and sulfonylureas.
Patients with type 2 diabetes and hypomagnesemia often enter a vicious cycle in which hypomagnesemia worsens insulin resistance and insulin resistance, by reducing the activity of renal magnesium channel transient receptor potential melastatin (TRPM) type 6, perpetuates hypomagnesemia.3
To the Editor: I read with interest the case of a 67-year-old woman with bilateral hand numbness, published in the March 2018 issue of the Journal, and I would like to suggest 2 important corrections to this article.1
The authors present a case of hypocalcemia secondary to postsurgical hypoparathyroidism but describe it as due to primary hypoparathyroidism. The patient had undergone thyroidectomy 10 years earlier and since then had hypocalcemia, secondary to postsurgical hypoparathyroidism, that was treated with calcium and vitamin D, until she stopped taking these agents. Postsurgical hypothyroidism is the most common cause of acquired or secondary hypoparathyroidism and is not primary hypoparathyroidism. I strongly feel that this requires an update or correction to the article. This patient may have associated malabsorption, as the authors alluded to, as the cause of her “normal” serum parathyroid hormone level.
The patient also had hypomagnesemia, which the authors state could have been due to furosemide use and “uncontrolled” diabetes mellitus. Diabetes doesn’t need to be uncontrolled to cause hypomagnesemia. Hypomagnesemia is common in patients with type 2 diabetes mellitus, with a prevalence of 14% to 48% in patients with diabetes compared with 2.5% to 15% in the general population.2 It is often multifactorial and may be secondary to one or more of the following factors: poor dietary intake, autonomic dysfunction, altered insulin resistance, glomerular hyperfiltration, osmotic diuresis (uncontrolled diabetes), recurrent metabolic acidosis, hypophosphatemia, hypokalemia, and therapy with drugs such as metformin and sulfonylureas.
Patients with type 2 diabetes and hypomagnesemia often enter a vicious cycle in which hypomagnesemia worsens insulin resistance and insulin resistance, by reducing the activity of renal magnesium channel transient receptor potential melastatin (TRPM) type 6, perpetuates hypomagnesemia.3
- Radwan SS, Hamo KN, Zayed AA. A 67-year-old woman with bilateral hand numbness. Cleve Clin J Med 2018; 85(3):200–208. doi:10.3949/ccjm.85a.17026
- Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol 2007; 2(2):366–373. doi:10.2215/CJN.02960906
- Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes 2016; 65(1):3–13. doi:10.2337/db15-1028
- Radwan SS, Hamo KN, Zayed AA. A 67-year-old woman with bilateral hand numbness. Cleve Clin J Med 2018; 85(3):200–208. doi:10.3949/ccjm.85a.17026
- Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol 2007; 2(2):366–373. doi:10.2215/CJN.02960906
- Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes 2016; 65(1):3–13. doi:10.2337/db15-1028
In reply: Postsurgical hypoparathyroidism is not primary hypoparathyroidism
In Reply: We thank Dr. Parmar and appreciate his important comments.
Regarding the difference between primary and secondary hypoparathyroidism, the definition varies among investigators. Some define primary hypoparathyroidism as a condition characterized by primary absence or deficiency of parathyroid hormone (PTH), which results in hypocalcemia and which can be congenital or acquired, including postsurgical hypoparathyroidism.1–4 In principle, this is similar to the classification of disorders affecting other endocrine glands as primary and secondary. For example, primary hypothyroidism refers to a state of low thyroid hormones resulting from impairment or loss of function of the thyroid gland itself, such as in Hashimoto thyroiditis, radioactive iodine therapy, or thyroidectomy, among others.5 We adopted this definition in our article. In contrast, secondary hypoparathyroidism is characterized by low PTH secretion in response to certain conditions that cause hypercalcemia. Non-PTH-mediated hypercalcemia is a more common term used to describe this state of secondary hypoparathyroidism.
Other investigators restrict the term “primary hypoparathyroidism” to nonacquired (congenital or hereditary) etiologies, while applying the term “secondary hypoparathyroidism” to acquired etiologies.6
Concerning the association between diabetes mellitus and hypomagnesemia, we agree that diabetes does not need to be uncontrolled to cause hypomagnesemia. However, the patient described in our article presented with severe hypomagnesemia (serum level 0.6 mg/dL), which is not commonly associated with diabetes. Most cases of hypomagnesemia in patients with type 2 diabetes mellitus are mild and asymptomatic, whereas severe manifestations including seizures, cardiac arrhythmias, and acute tetany are rarely encountered in clinical practice.7 Furthermore, numerous studies have shown a negative correlation between serum magnesium level and glycemic control.7–11 A recent study reported that plasma triglyceride and glucose levels are the main determinants of the plasma magnesium concentration in patients with type 2 diabetes.12
Our patient’s diabetes was uncontrolled, as evidenced by her hemoglobin A1c level of 9.7% and her random serum glucose level of 224 mg/dL. Therefore, it is more likely that “uncontrolled diabetes mellitus” (in addition to diuretic use) was the cause of her symptomatic severe hypomagnesemia rather than controlled diabetes mellitus.
- Mendes EM, Meireles-Brandão L, Meira C, Morais N, Ribeiro C, Guerra D. Primary hypoparathyroidism presenting as basal ganglia calcification secondary to extreme hypocalcemia. Clin Pract 2018; 8(1):1007. doi:10.4081/cp.2018.1007
- Vadiveloo T, Donnan PT, Leese GP. A population-based study of the epidemiology of chronic hypoparathyroidism. J Bone Miner Res 2018; 33(3):478-485. doi:10.1002/jbmr.3329
- Hendy GN, Cole DEC, Bastepe M. Hypoparathyroidism and pseudohypoparathyroidism. In: De Groot LJ, Chrousos G, Dungan K, et al, eds. Endotext [Internet], South Dartmouth (MA): MDText.com, Inc.; 2017. www.ncbi.nlm.nih.gov/books/NBK279165. Accessed August 20, 2018.
- Rosa RG, Barros AJ, de Lima AR, et al. Mood disorder as a manifestation of primary hypoparathyroidism: a case report. J Med Case Rep 2014; 8:326. doi:10.1186/1752-1947-8-326
- Almandoz JP, Gharib H. Hypothyroidism: etiology, diagnosis, and management. Med Clin North Am 2012; 96(2):203–221. doi:10.1016/j.mcna.2012.01.005
- Fouda UM, Fouda RM, Ammar HM, Salem M, Darouti ME. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J 2009; 2:9338. doi:10.1186/1757-1626-2-9338
- Tosiello L. Hypomagnesemia and diabetes mellitus. A review of clinical implications. Arch Intern Med 1996; 156(11):1143–1148. pmid: 8639008
- Pham PC, Pham PM, Pham PA, et al. Lower serum magnesium levels are associated with more rapid decline of renal function in patients with diabetes mellitus type 2. Clin Nephrol 2005; 63(6):429–436. pmid:15960144
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20(1):3–17. doi:10.1177/0885066604271539
- Resnick LM, Altura BT, Gupta RK, Laragh JH, Alderman MH, Altura BM. Intracellular and extracellular magnesium depletion in type 2 (non-insulin-independent) diabetes mellitus. Diabetologia 1993; 36(8):767–770. pmid:8405745
- Pun KK, Ho PW. Subclinical hyponatremia, hyperkalemia and hypomagnesemia in patients with poorly controlled diabetes mellitus. Diabetes Res Clin Pract 1989; 7(3)163–167. pmid: 2605984
- Kurstjens S, de Baaij JH, Bouras H, Bindels RJ, Tack CJ, Hoenderop JG. Determinants of hypomagnesemia in patients with type 2 diabetes mellitus. Eur J Endocrinol 2017; 176(1):11–19. doi:10.1530/EJE-16-0517
In Reply: We thank Dr. Parmar and appreciate his important comments.
Regarding the difference between primary and secondary hypoparathyroidism, the definition varies among investigators. Some define primary hypoparathyroidism as a condition characterized by primary absence or deficiency of parathyroid hormone (PTH), which results in hypocalcemia and which can be congenital or acquired, including postsurgical hypoparathyroidism.1–4 In principle, this is similar to the classification of disorders affecting other endocrine glands as primary and secondary. For example, primary hypothyroidism refers to a state of low thyroid hormones resulting from impairment or loss of function of the thyroid gland itself, such as in Hashimoto thyroiditis, radioactive iodine therapy, or thyroidectomy, among others.5 We adopted this definition in our article. In contrast, secondary hypoparathyroidism is characterized by low PTH secretion in response to certain conditions that cause hypercalcemia. Non-PTH-mediated hypercalcemia is a more common term used to describe this state of secondary hypoparathyroidism.
Other investigators restrict the term “primary hypoparathyroidism” to nonacquired (congenital or hereditary) etiologies, while applying the term “secondary hypoparathyroidism” to acquired etiologies.6
Concerning the association between diabetes mellitus and hypomagnesemia, we agree that diabetes does not need to be uncontrolled to cause hypomagnesemia. However, the patient described in our article presented with severe hypomagnesemia (serum level 0.6 mg/dL), which is not commonly associated with diabetes. Most cases of hypomagnesemia in patients with type 2 diabetes mellitus are mild and asymptomatic, whereas severe manifestations including seizures, cardiac arrhythmias, and acute tetany are rarely encountered in clinical practice.7 Furthermore, numerous studies have shown a negative correlation between serum magnesium level and glycemic control.7–11 A recent study reported that plasma triglyceride and glucose levels are the main determinants of the plasma magnesium concentration in patients with type 2 diabetes.12
Our patient’s diabetes was uncontrolled, as evidenced by her hemoglobin A1c level of 9.7% and her random serum glucose level of 224 mg/dL. Therefore, it is more likely that “uncontrolled diabetes mellitus” (in addition to diuretic use) was the cause of her symptomatic severe hypomagnesemia rather than controlled diabetes mellitus.
In Reply: We thank Dr. Parmar and appreciate his important comments.
Regarding the difference between primary and secondary hypoparathyroidism, the definition varies among investigators. Some define primary hypoparathyroidism as a condition characterized by primary absence or deficiency of parathyroid hormone (PTH), which results in hypocalcemia and which can be congenital or acquired, including postsurgical hypoparathyroidism.1–4 In principle, this is similar to the classification of disorders affecting other endocrine glands as primary and secondary. For example, primary hypothyroidism refers to a state of low thyroid hormones resulting from impairment or loss of function of the thyroid gland itself, such as in Hashimoto thyroiditis, radioactive iodine therapy, or thyroidectomy, among others.5 We adopted this definition in our article. In contrast, secondary hypoparathyroidism is characterized by low PTH secretion in response to certain conditions that cause hypercalcemia. Non-PTH-mediated hypercalcemia is a more common term used to describe this state of secondary hypoparathyroidism.
Other investigators restrict the term “primary hypoparathyroidism” to nonacquired (congenital or hereditary) etiologies, while applying the term “secondary hypoparathyroidism” to acquired etiologies.6
Concerning the association between diabetes mellitus and hypomagnesemia, we agree that diabetes does not need to be uncontrolled to cause hypomagnesemia. However, the patient described in our article presented with severe hypomagnesemia (serum level 0.6 mg/dL), which is not commonly associated with diabetes. Most cases of hypomagnesemia in patients with type 2 diabetes mellitus are mild and asymptomatic, whereas severe manifestations including seizures, cardiac arrhythmias, and acute tetany are rarely encountered in clinical practice.7 Furthermore, numerous studies have shown a negative correlation between serum magnesium level and glycemic control.7–11 A recent study reported that plasma triglyceride and glucose levels are the main determinants of the plasma magnesium concentration in patients with type 2 diabetes.12
Our patient’s diabetes was uncontrolled, as evidenced by her hemoglobin A1c level of 9.7% and her random serum glucose level of 224 mg/dL. Therefore, it is more likely that “uncontrolled diabetes mellitus” (in addition to diuretic use) was the cause of her symptomatic severe hypomagnesemia rather than controlled diabetes mellitus.
- Mendes EM, Meireles-Brandão L, Meira C, Morais N, Ribeiro C, Guerra D. Primary hypoparathyroidism presenting as basal ganglia calcification secondary to extreme hypocalcemia. Clin Pract 2018; 8(1):1007. doi:10.4081/cp.2018.1007
- Vadiveloo T, Donnan PT, Leese GP. A population-based study of the epidemiology of chronic hypoparathyroidism. J Bone Miner Res 2018; 33(3):478-485. doi:10.1002/jbmr.3329
- Hendy GN, Cole DEC, Bastepe M. Hypoparathyroidism and pseudohypoparathyroidism. In: De Groot LJ, Chrousos G, Dungan K, et al, eds. Endotext [Internet], South Dartmouth (MA): MDText.com, Inc.; 2017. www.ncbi.nlm.nih.gov/books/NBK279165. Accessed August 20, 2018.
- Rosa RG, Barros AJ, de Lima AR, et al. Mood disorder as a manifestation of primary hypoparathyroidism: a case report. J Med Case Rep 2014; 8:326. doi:10.1186/1752-1947-8-326
- Almandoz JP, Gharib H. Hypothyroidism: etiology, diagnosis, and management. Med Clin North Am 2012; 96(2):203–221. doi:10.1016/j.mcna.2012.01.005
- Fouda UM, Fouda RM, Ammar HM, Salem M, Darouti ME. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J 2009; 2:9338. doi:10.1186/1757-1626-2-9338
- Tosiello L. Hypomagnesemia and diabetes mellitus. A review of clinical implications. Arch Intern Med 1996; 156(11):1143–1148. pmid: 8639008
- Pham PC, Pham PM, Pham PA, et al. Lower serum magnesium levels are associated with more rapid decline of renal function in patients with diabetes mellitus type 2. Clin Nephrol 2005; 63(6):429–436. pmid:15960144
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20(1):3–17. doi:10.1177/0885066604271539
- Resnick LM, Altura BT, Gupta RK, Laragh JH, Alderman MH, Altura BM. Intracellular and extracellular magnesium depletion in type 2 (non-insulin-independent) diabetes mellitus. Diabetologia 1993; 36(8):767–770. pmid:8405745
- Pun KK, Ho PW. Subclinical hyponatremia, hyperkalemia and hypomagnesemia in patients with poorly controlled diabetes mellitus. Diabetes Res Clin Pract 1989; 7(3)163–167. pmid: 2605984
- Kurstjens S, de Baaij JH, Bouras H, Bindels RJ, Tack CJ, Hoenderop JG. Determinants of hypomagnesemia in patients with type 2 diabetes mellitus. Eur J Endocrinol 2017; 176(1):11–19. doi:10.1530/EJE-16-0517
- Mendes EM, Meireles-Brandão L, Meira C, Morais N, Ribeiro C, Guerra D. Primary hypoparathyroidism presenting as basal ganglia calcification secondary to extreme hypocalcemia. Clin Pract 2018; 8(1):1007. doi:10.4081/cp.2018.1007
- Vadiveloo T, Donnan PT, Leese GP. A population-based study of the epidemiology of chronic hypoparathyroidism. J Bone Miner Res 2018; 33(3):478-485. doi:10.1002/jbmr.3329
- Hendy GN, Cole DEC, Bastepe M. Hypoparathyroidism and pseudohypoparathyroidism. In: De Groot LJ, Chrousos G, Dungan K, et al, eds. Endotext [Internet], South Dartmouth (MA): MDText.com, Inc.; 2017. www.ncbi.nlm.nih.gov/books/NBK279165. Accessed August 20, 2018.
- Rosa RG, Barros AJ, de Lima AR, et al. Mood disorder as a manifestation of primary hypoparathyroidism: a case report. J Med Case Rep 2014; 8:326. doi:10.1186/1752-1947-8-326
- Almandoz JP, Gharib H. Hypothyroidism: etiology, diagnosis, and management. Med Clin North Am 2012; 96(2):203–221. doi:10.1016/j.mcna.2012.01.005
- Fouda UM, Fouda RM, Ammar HM, Salem M, Darouti ME. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J 2009; 2:9338. doi:10.1186/1757-1626-2-9338
- Tosiello L. Hypomagnesemia and diabetes mellitus. A review of clinical implications. Arch Intern Med 1996; 156(11):1143–1148. pmid: 8639008
- Pham PC, Pham PM, Pham PA, et al. Lower serum magnesium levels are associated with more rapid decline of renal function in patients with diabetes mellitus type 2. Clin Nephrol 2005; 63(6):429–436. pmid:15960144
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20(1):3–17. doi:10.1177/0885066604271539
- Resnick LM, Altura BT, Gupta RK, Laragh JH, Alderman MH, Altura BM. Intracellular and extracellular magnesium depletion in type 2 (non-insulin-independent) diabetes mellitus. Diabetologia 1993; 36(8):767–770. pmid:8405745
- Pun KK, Ho PW. Subclinical hyponatremia, hyperkalemia and hypomagnesemia in patients with poorly controlled diabetes mellitus. Diabetes Res Clin Pract 1989; 7(3)163–167. pmid: 2605984
- Kurstjens S, de Baaij JH, Bouras H, Bindels RJ, Tack CJ, Hoenderop JG. Determinants of hypomagnesemia in patients with type 2 diabetes mellitus. Eur J Endocrinol 2017; 176(1):11–19. doi:10.1530/EJE-16-0517

What’s the Buzz? Treatment Strategies in Chronic Subjective Tinnitus
CE/CME No: CR-1810
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Distinguish primary tinnitus from secondary tinnitus.
• Understand and implement a full clinical evaluation of tinnitus, including imaging studies when appropriate.
• Discuss expectations regarding treatment options and realistic outcomes of currently recommended therapy.
• Direct patients to specialist care for cognitive behavioral therapy or tinnitus retraining therapy.
• Know when pharmacotherapeutic intervention is indicated.
FACULTY
Wendy Gillian Ross practices urgent care medicine in Lake Grove, New York, and primary care in Patchogue, New York. Randy Danielsen is Professor and Dean, Arizona School of Health Sciences, and Director, Center for the Future of the Health Professions, both at A.T. Still University, in Mesa, Arizona. He is Physician Assistant Editor-in-Chief of Clinician Reviews.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through September 30, 2019.
Article begins on next page >>
Tinnitus can be a debilitating condition that affects quality of life and is often not treated according to guidelines. Cognitive behavioral therapy and tinnitus retraining therapy have been successful in reducing tinnitus bother; pharmacotherapy is not widely accepted as successful, and can, in fact, be deleterious. This article describes pathophysiologic disturbances of hearing and how they relate to chronic subjective tinnitus, discusses the clinical evaluation of tinnitus as a presenting symptom, and reviews current treatments.
Primary chronic subjective tinnitus, often thought of more as a symptom than a diagnosis, affects millions of people worldwide. This troublesome condition has been chronicled as far back as the first century
It is estimated that only 20% of people who experience tinnitus actively seek treatment.2 In the United States, 2 to 3 million of the 12 million patients who do request treatment report lasting symptoms that they describe as debilitating.3 For patients who seek help, the treatment recommended by physicians is typically pharmacotherapeutic—which does not follow guidelines.4
The aim of this article is to reinforce a greater understanding of the mechanisms of tinnitus and integrate that knowledge into treatment guidelines. The article does not discuss surgical treatment of tinnitus.
DEFINITION AND CLASSIFICATION
A universal standard definition of chronic tinnitus does not exist; Trevis et al define it as a phantom sound that persists for more than three months.5 The quality and loudness of tinnitus is variable but is often described as a buzz, hiss, or ringing. Prevalence increases with age, smoking, male gender, and ethnicity, with the non-Latino white population statistically at greater risk.3 Comorbid conditions (eg, diabetes and other autoimmune diseases) are risk factors for tinnitus. A history of exposure to loud sound—occupational, environmental, or recreational—also can predispose a person to tinnitus.3
The American Academy of Otolaryngology–Head and Neck Surgery (AAO–HNS) classifies tinnitus as primary (subjective) or secondary (objective). Primary tinnitus—representing the majority of cases—has no identifiable cause; there may be accompanying sensorineural hearing loss or hyperacusis. Secondary tinnitus can also be associated with sensorineural hearing loss but has an identifiable underlying cause.6 The differential diagnosis of tinnitus is listed in the Table.7

Tinnitus is further defined by its persistence. Persistent tinnitus is defined as tinnitus lasting more than six months, slightly longer than the duration offered by Trevis et al, who also define tinnitus as bothersome or non-bothersome, depending on its impact on quality of life.5,6 Causes of reduced quality of life include depression, anxiety, insomnia, and neurocognitive decline—all of which have been associated with chronic subjective tinnitus.8
Continue to: Researchers have discovered that...
Researchers have discovered that tinnitus is not simply a cochlear phenomenon. The pathology extends well beyond the auditory complex, having a deleterious effect on both the somatosensory and central nervous systems, providing some explanation for the prevalence of anxiety and depression associated with the disorder (see "Pathophysiology of tinnitus").9-17

Because of the insidious nature of tinnitus and lack of standard measures of severity, true prevalence is difficult to calculate.18
CLINICAL EVALUATION
Tinnitus can be a presenting complaint or elicited during history-taking. Symptomatic patients should receive full evaluation, including a complete physical exam, medication history, and laboratory workup.
Adverse effect of drugs
Medications that commonly cause tinnitus symptoms are NSAIDs, chemotherapeutic agents, and antibiotics (eg, macrolides and fluoroquinolones). Amiodarone, ACE inhibitors, proton-pump inhibitors, and calcium-channel blockers have also been implicated. Paradoxically, anxiolytics and tricyclic antidepressants, which are sometimes used to treat tinnitus, have been linked to causing the condition.7
Laboratory tests and imaging
Testing should include investigation for infectious disease, autoimmune disorders, and vitamin deficiency.7 According to the American College of Radiology, imaging is unnecessary in the workup of primary tinnitus. Any suspicion of a vascular cause noted on the physical exam (eg, an associated bruit or venous hum), however, should be explored with imaging. Furthermore, any case of tinnitus that lateralizes also requires additional investigation. Modalities of choice are MRI, CT, and CT angiography.19
Continue to: Referral for audiology evaluation
Referral for audiology evaluation
When no underlying pathology can be identified for tinnitus, the patient should be sent for a full audiology evaluation to screen for associated hearing loss. Discussion of audiology screening tests is beyond the scope of this article; however, testing includes otoscopy, audiography, tympanography, otoacoustic emission testing, auditory brainstem-response testing, and vestibular evoked myogenic potential testing.7
Probing nonphysical impacts
Quality of life and overall emotional wellness, including cognitive function, should be investigated in patients with tinnitus. Two questionnaires commonly used in the assessment of tinnitus bother are the Tinnitus Handicap Inventory and the Tinnitus Reaction Questionnaire.7 In a large, systematic review, Trevis et al report that “64% of studies investigating depression found an increase in depressive symptoms in people with chronic tinnitus compared to hearing control groups, and 62% of studies investigating anxiety reported significantly increased anxiety symptoms.”5
MANAGEMENT
Tinnitus management should be viewed two ways: treatment of perceived loudness and treatment of comorbid symptoms relating to tinnitus bother.6 In the same meta-analysis, Trevis and colleagues found that patients with tinnitus had higher rates of anxiety, depression, and overall decline in cognitive function, including processing speed, concentration, and sleep disorders.5 It is useful to keep this observation in mind when reviewing treatment options for tinnitus.
Five classic pharmacotherapeutic approaches to tinnitus management are
- Anticonvulsants
- Antidepressants
- Anesthetics
- Anxiolytics
- Lidocaine.
Newer medications that show some promise are N-methyl-D-aspartate (NMDA) receptor antagonists, notably neramexane. Alternative pharmaceuticals include vitamin-based treatments, cannabinoids, and herbal compounds.
Continue to: The AAOS-HNS supports...
The AAO–HNS supports nonpharmacotherapeutic treatment of tinnitus; its guidelines include a recommendation for cognitive behavioral therapy (CBT) as primary therapy.6 In addition, tinnitus-retraining therapy, tinnitus-masking therapy/sound therapy, meditation/mindfulness, and yoga all have been studied for their ability to alleviate tinnitus bother.
Pharmacotherapeutic management
Anticonvulsants have failed to provide strong evidence of usefulness in the treatment of tinnitus and are not supported by the AAO–HNS as such.6 This conclusion notwithstanding, the anticonvulsants carbamazepine and gabapentin have historically been two of the more common medications used to treat tinnitus.
Carbamazepine is a glutamate receptor antagonist that suppresses seizure activity. Based on prior research suggesting that spontaneous firing within the auditory complex is similar to seizure activity, Iranian researchers explored the hypothesis that carbamazepine might lessen tinnitus severity. Their study revealed, however, that carbamazepine did not statistically significantly reduce the severity of tinnitus, compared to placebo.20 While carbamazepine may be of limited use in the treatment of subjective tinnitus, recent literature confirms that it is not only useful, but also diagnostic, in typewriter tinnitus (ie, having a staccato quality, like the sound of typewriter keys being depressed). Typewriter tinnitus is a secondary cause of tinnitus related to disruption of the stapes in the middle ear.21
Gabapentin works by promoting gamma-aminobutyric acid (GABA) production in the brain. GABA is an inhibitory neurotransmitter, thus slowing down signals between neurons. Following on preliminary research that detected low levels of GABA in the inferior colliculus of rodents with salicylate-induced tinnitus, Aazh and colleagues conducted a double-blind study of gabapentin—and concluded that it yielded no improvement in symptoms, compared to placebo.22
Valproic acid has not been formally investigated but is commonly incorporated in the treatment of tinnitus.23 Lamotrigine has provided similarly disappointing results in the treatment of tinnitus.24
Continue to: Antidepressants and anxiolytics
Antidepressants and anxiolytics. Based on the results of their early clinical trials, Sullivan and colleagues concluded that tricyclic antidepressants produced significant improvement in tinnitus symptoms, due to the analgesic effects of these drugs. The researchers studied nortriptyline specifically; in severely depressed patients, the drug reduced the loudness of tinnitus and depressive symptoms. In non-depressed subjects, however, nortriptyline was not as efficacious.25
Selective serotonin reuptake inhibitors have not had the same success as nortriptyline. In a study of paroxetine conducted by Oishi and colleagues, there was little evidence that the drug reduced the loudness of tinnitus, although overall, it did reduce tinnitus bother and anxiety.26
Included in the category of anxiolytics, benzodiazepines have long been used to treat severe tinnitus-induced anxiety, with some success. However, as Elgoyhen and Langguth point out, studies of benzodiazepines for tinnitus have been limited in size.23
The AAO–HNS does not support routine use of antidepressants and anxiolytics for tinnitus bother.7
NMDA receptor antagonists. In a recent clinical trial, neramexane was studied for its efficacy in tinnitus. Neramexane acts at the cholinergic nicotinic and NMDA receptors in the efferent auditory system. Its complex reaction is thought to prevent transmission of unwanted sound not only to structures within the auditory system but beyond, to the medial geniculate body and lateral nucleus of the amygdala. The trial has proved some benefit concerning overall perception of tinnitus loudness; a phase 2 trial is being conducted.27
Continue to: Intra-tympanic anesthetics
Intra-tympanic anesthetics. Anesthetics, such as lidocaine, have had limited success and results have not been found to be sustained.
Alternative medical managements
Traditional Chinese herbal medications have been used for centuries and are increasingly popular in Western culture. Hilton and colleagues studied Ginkgo biloba, or maidenhair tree, a traditional Chinese herbal supplement available as an extract and as dried leaves. The main action of the extract is vasoregulatory; antiplatelet effects are also seen. Adverse effects include gastrointestinal upset and headache. In a systematic review, Hilton and colleagues concluded that Ginkgo did not reduce overall tinnitus loudness or severity; the review was limited, however, by the fact that only two studies met criteria for inclusion.28
Vitamins, lipoflavinoids, zinc, manganese, and melatonin are all supplements marketed to improve tinnitus symptoms. However, a cross-sectional study confirmed prior research that did not show any benefit from the use of these supplements.29
Cannabinoids are being studied for their proposed antiepileptic effects. There is a popular misconception of Cannabis as a singular chemical when in fact, it is a plant that contains hundreds of chemicals that each act differently on the brain. In a review, Smith and Zheng30 explain that two cannabinoid receptors, CB1 and CB2, are represented, and exert their effects, in different areas of the brain. CB1 receptors block calcium influx in presynaptic terminals, resulting in an inhibitory effect on neurotransmitter release.
CB1 receptors have been found in the dorsal cochlear nuclei, prompting research interest in how cannabinoids affect neurotransmission of unwanted sounds of tinnitus. To date, however, there are conflicting data concerning the benefit of cannabinoids and tinnitus. In fact, Smith and Zheng state that some data suggest that cannabinoids might make tinnitus worse.30
Continue to: Nonpharmacotherapeutic management
Nonpharmacotherapeutic management
Cognitive behavioral therapy. Conceptualized by Aaron T. Beck in the 1960s, cognitive behavioral therapy (CBT) is the leading recommendation made by the AAO–HNS in its tinnitus treatment guidelines.6 Beck’s work centered on the idea that behaviors are modifiable thoughts, through analysis of past experiences and assumptions based on those experiences. By understanding the core belief that a patient attaches to a feeling, Beck hypothesized that behaviors or responses to those feelings could be changed; this is accomplished through discussion to dispel unwarranted fears and by teaching coping mechanisms, such as relaxation. The idea behind CBT in the management of tinnitus is clear: The sound cannot be eliminated, but the patient’s response to the sound can be modified. Ultimately, through this modified response or habituation, the patient can relax and live with the sound.31
Since anxiety, depression, and insomnia are common comorbidities of tinnitus, a psychologic approach remains in the forefront of treatment recommendations. Hoare and colleagues reported that in “a meta-analysis of 10 randomized trials evaluating different forms of CBT (by the therapist and over the Internet), CBT improved tinnitus symptoms compared to non-CBT controls.”7
Tinnitus retraining therapy (TRT) is another form of habituation therapy, introduced by Jastreboff in the 1990s. His work furthered the idea that tinnitus could be reframed, as it is in CBT. Simply, he proposed that systems outside the auditory complex—namely the autonomic nervous system and the limbic system—respond to the signal produced by damaged hair cells in the cochlear nuclei. TRT retrains connections to block or ignore these signals.13 Unlike CBT, the aim of TRT is to eliminate the perception of sound.
By educating patients about the physiologic mechanisms of tinnitus, TRT reduces patient anxiety related to the sound. The process of habituation follows counseling. To accomplish this, the patient wears a sound generator, similar in appearance to hearing aids, using broadband noise. The sound does not mask the tinnitus but closes the gap between silence and the perception of tinnitus. The sound generator is worn for six hours daily for approximately 12 months.
Multiple studies have employed Jastreboff’s original technique, including a clinical trial by Bauer and colleagues. The published outcome of this study confirmed that patients experienced a positive and lasting effect with TRT.32 In addition, a small study of TRT conducted by Barozzi and colleagues, using different colors of sound (ie, how the frequency of a given sound corresponds to the light-wave frequency of a particular color), found statistically significant improvement. Allowing patients to pick a sound that they found more pleasant increased the effectiveness of the treatment.33 (Patients can learn more about TRT by visiting www.tinnitus-pjj.com, hosted by tinnitus researcher Pawel J. Jastreboff.)
Continue to: Alternative nonmedical therapies...
Alternative nonmedical therapies have become popular; they include meditation, yoga, physical therapy, mindfulness, and tinnitus-masking treatment with sound.
Results of a study of yoga and meditation showed that patients felt more relaxed, but that these interventions had no effect on the severity of tinnitus. The principle behind yoga practice, according to Köksoy and colleagues, is that the discipline is thought to affect the limbic system by deactivating the sympathetic response to stimulation from surrounding sounds. In addition, Köksoy states, other researchers have provided evidence that yoga increases circulating levels of antioxidants, which in turn reduce oxidative stress.34
Particularly among members of the millennial generation, mindfulness has become a buzzword. The practice refers to a “method for facing, exploring, and alleviating suffering by relating to present experiences.”35 Roland and colleagues conducted a clinical trial of mindfulness practiced by a cohort of patients with bothersome tinnitus; results were based on scores gleaned from standard rating scales (eg, Global Bothersome Scale, Cognitive and Affective Mindfulness Scale-Revised, Cognitive Failures Questionnaire, Tinnitus Handicap Inventory, and Tinnitus Functional Index). Evaluated before and four weeks after cessation of therapy, subjects reported that tinnitus bother was reduced, but none showed statistically significant improvement in depression, anxiety, or cognitive ability.35
Used for more than 40 years, sound-based therapy has been discussed in conjunction with TRT.36 It is recognized as an approved but optional treatment by the AAO–HNS. In response to a 2010 study by Hobson that used sound-based therapy alone for tinnitus, Tunkel and colleagues cautioned that the modality showed little benefit. The major downside to acoustic therapy, according to the AAO–HNS clinical guidelines, is cost and patients’ excessive expectation of effectiveness.6
According to the AAO–HNS, repetitive-transcranial magnetic stimulation is not supported as a valid treatment for tinnitus because it can lead to seizures in patients who are taking medication that lowers the seizure threshold or who have a secondary cause of tinnitus, such as a tumor—therefore creating risk that outweighs any benefit.6
Continue to: CONCLUSION
CONCLUSION
For a large percentage of the population, chronic subjective tinnitus is a significant variable in the evaluation of quality of life. The condition is not completely understood and often displays features unique to the individual. Much of the initial response to research linking tinnitus with shared pathways typical for chronic pain, anxiety, and depression has resulted in pharmacotherapeutic management that is not always warranted—or successful.
Clinical research into the pathophysiology of tinnitus is providing a better understanding of the neurophysiologic mechanisms that underpin the science of chronic tinnitus. With this information, researchers can one day design medical management that targets specific receptors, resulting in greater management success.
The psychologic impact of tinnitus cannot be underestimated. When almost one-third of patients complain of debilitating symptoms that can also result in neurocognitive decline, tinnitus becomes a condition that cannot be ignored. Guidelines set forth by the AAO–HNS state that CBT and TRT offer some reprieve from symptoms and teach patients habituation without further damage to hearing. The use of broad-based sound generators has been well established as a useful management tool, although it is not curative.
The limitations of some studies that reviewed alternative medicines include small sample size and difficulty comparing research analysis because of disparities in tinnitus rating scales. Also, age bias, comorbid conditions, and study drop-out rates affected overall statistical significance of some studies. Additional, high-quality research is warranted in this area.
Continue to: Prevention of tinnitus...
Prevention of tinnitus through education on hearing loss and its causes should be regarded as implicit; occupational noise and recreational use of music devices put people at heightened risk for hearing loss and tinnitus. Information and open discussion that include the discovery of tinnitus symptoms during routine physical examination are recommended.
Last, providers who adhere to recognized guidelines will aid patients in coping with the challenges that tinnitus presents. As research continues to unravel the complex interaction between neurons, medical science is hopeful that curative treatments will become available.
1. Maltby MT. Ancient voices on tinnitus: the pathology and treatment of tinnitus in Celsus and the Hippocratic Corpus compared and contrasted. Int Tinnitus J. 2012;17(2):140-145.
2. Wolever RQ, Price R, Hazelton GA, et al. Complementary therapies for significant dysfunction from tinnitus: treatment review and potential for integrative medicine. Evid Based Complement Alternat Med. 2015;15:931418.
3. Shargorodsky J, Curhan GC, Farwell WR. Prevalence and characteristics of tinnitus among US adults. Am J Med. 2010;123(8):711-718.
4. Bhatt JM, Lin HW, Bhattacharyya N. Prevalence, severity, exposures, and treatment patterns of tinnitus in the United States. JAMA Otolaryngol Head Neck Surg. 2016;142(10):959-965.
5. Trevis KJ, McLachlan NM, Wilson SJ. A systematic review and meta-analysis of psychological functioning in chronic tinnitus. Clin Psychol Rev. 2018;60:62-86.
6. Tunkel DE, Bauer CA, Sun GH, et al. Clinical practice guideline: tinnitus. Otolaryngol Head Neck Surg. 2014;151(suppl 2):S1-S40.
7. Dinces EA. Treatment of tinnitus. UpToDate. April 12, 2018. www.uptodate.com/contents/treatment-of-tinnitus. Accessed September 17, 2018.
8. Gudwani S, Munjal SK, Panda NK, Kohli A. Association of chronic subjective tinnitus with neuro-cognitive performance. Int Tinnitus J. 2017;21:90-97.
9. Jastreboff PJ. 25 years of tinnitus retraining therapy. HNO. 2015;63:307-311.
10. Pujol R. Journey into the world of hearing. 2016. www.cochlea.eu/en. Accessed September 17, 2018.
11. Adjamian P, Hall DA, Palmer AR, et al. Neuroanatomical abnormalities in chronic tinnitus in the human brain.Neurosci Biobehav Rev. 2014;45:119-133.
12. Shore SE, Roberts LE, Langguth B. Maladaptive plasticity in tinnitus—triggers, mechanisms and treatment. Nat Rev Neurol. 2016;12(3):150-160.
13. Jastreboff PJ, Gray WC, Gold SL. Neurophysiological approach to tinnitus patients. Am J Otol. 1996;17(2):236-240.
14. Kaltenbach JA. Tinnitus: models and mechanisms. Hear Res. 2011;276:52-60.
15. Rauschecker JP, Leaver AM, Mühlau M. Tuning out the noise: limbic-auditory interactions in tinnitus. Neuron. 2010;66(6):819-826.
16. Møller AR. Sensorineural tinnitus: its pathology and probable therapies. Int J Otolaryngol. 2016;2016:2830157.
17. Chen YC, Xia W, Chen H, et al. Tinnitus distress is linked to enhanced resting‐state functional connectivity from the limbic system to the auditory cortex. Hum Brain Mapp. 2017;38(5):2384-2397.
18. McCormack A, Edmonson-Jones M, Somerset S, Hall D. A systematic review of the reporting of tinnitus prevalence and severity. Hear Res. 2016;337:70-79.
19. Kessler MM, Moussa M, Bykowski J, et al; Expert Panel on Neurologic Imaging. ACR Appropriateness Criteria® Tinnitus. J Am Coll Radiol. 2017;14:S584-S591.
20. Gerami H, Saberi A, Nemati, S, et al. Effects of oxcarbazepine versus carbamazepine on tinnitus: a randomized double-blind placebo-controlled clinical trial. Iran J Neurol. 2012;11(3):106-110.
21. Sunwoo W, Jeon YJ, Bae YJ, et al. Typewriter tinnitus revisited: the typical symptoms and the initial response to carbamazepine are the most reliable diagnostic clues. Sci Rep. 2017;7:10615.
22. Aazh H, El Refaie A, Humphriss R. Gabapentin for tinnitus: a systematic review. Am J Audiol. 2011;20:151-158.
23. Elgoyhen AB, Langguth B. Pharmacological approaches to the treatment of tinnitus. Drug Discov Today. 2010;15:300-305.
24. Langguth B, Kreuzer PM, Kleinjung T, De Ridder D. Tinnitus: causes and clinical management. Lancet Neurol. 2013;12(9):920-930.
25. Sullivan M, Katon W, Russo J, et al. A randomized trial of nortriptyline for severe chronic tinnitus. Effects on depression, disability, and tinnitus symptoms. Arch Intern Med. 1993;153(19):2251-2259.
26. Oishi N, Kanzaki S, Shinden S, et al. Effects of selective serotonin reuptake inhibitor on treating tinnitus in patients stratified for presence of depression or anxiety. Audiol Neurootol. 2010;15(3):187-193.
27. Suckfüll M, Althaus M, Ellers-Lenz B, et al. A randomized, double-blind, placebo-controlled clinical trial to evaluate the efficacy and safety of neramexane in patients with moderate to severe subjective tinnitus. BMC Ear Nose Throat Disord. 2011;11:1.
28. Hilton MP, Zimmermann EF, Hunt WT. Ginkgo biloba for tinnitus. Cochrane Database Syst Rev. 2013;CD003852. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD003852.pub3/full. Accessed September 17, 2018.
29. Coelho C, Tyler R, Ji H, et al. Survey on the effectiveness of dietary supplements to treat tinnitus. Am J Audiol. 2016;25:184-205.
30. Smith PF, Zheng Y. Cannabinoids, cannabinoid receptors and tinnitus. Hear Res. 2015;332:210-216.
31. Martinez-Devesa P, Perera R, Theodoulou M, Waddell A. Cognitive behavioural therapy for tinnitus. Cochrane Database Syst Rev. 2010:CD005233. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD005233.pub3/full. Accessed September 17, 2018.
32. Bauer CA, Berry JL, Brozoski TJ. The effect of tinnitus retraining therapy on chronic tinnitus: a controlled trial. Laryngoscope Investig Otolaryngol. 2017;2(4):166-177.
33. Barozzi S, Ambrosetti U, Callaway SL, et al. Effects of tinnitus retraining therapy with different colours of sound. Int Tinnitus J. 2017;21:139-143.
34. Köksoy S, Eti CM, Karatas¸ M, Vayisoglu Y. The effects of yoga in patients suffering from subjective tinnitus. Int Arch Otorhinolaryngol. 2018;22(1):9-13.
35. Roland LT, Lenze EJ, Hardin FM, et al. Effects of mindfulness based stress reduction therapy on subjective bother and neural connectivity in chronic tinnitus. Otolaryngol Head Neck Surg. 2015;152(5):919-926.
36. Ibarra D, Tavira-Sanchez F, Recuero-Lopez M, Anthony BW. In-ear medical devices for acoustic therapies in tinnitus treatments, state of the art. Auris Nasus Larynx. 2018;45:6-12.
CE/CME No: CR-1810
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Distinguish primary tinnitus from secondary tinnitus.
• Understand and implement a full clinical evaluation of tinnitus, including imaging studies when appropriate.
• Discuss expectations regarding treatment options and realistic outcomes of currently recommended therapy.
• Direct patients to specialist care for cognitive behavioral therapy or tinnitus retraining therapy.
• Know when pharmacotherapeutic intervention is indicated.
FACULTY
Wendy Gillian Ross practices urgent care medicine in Lake Grove, New York, and primary care in Patchogue, New York. Randy Danielsen is Professor and Dean, Arizona School of Health Sciences, and Director, Center for the Future of the Health Professions, both at A.T. Still University, in Mesa, Arizona. He is Physician Assistant Editor-in-Chief of Clinician Reviews.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through September 30, 2019.
Article begins on next page >>
Tinnitus can be a debilitating condition that affects quality of life and is often not treated according to guidelines. Cognitive behavioral therapy and tinnitus retraining therapy have been successful in reducing tinnitus bother; pharmacotherapy is not widely accepted as successful, and can, in fact, be deleterious. This article describes pathophysiologic disturbances of hearing and how they relate to chronic subjective tinnitus, discusses the clinical evaluation of tinnitus as a presenting symptom, and reviews current treatments.
Primary chronic subjective tinnitus, often thought of more as a symptom than a diagnosis, affects millions of people worldwide. This troublesome condition has been chronicled as far back as the first century
It is estimated that only 20% of people who experience tinnitus actively seek treatment.2 In the United States, 2 to 3 million of the 12 million patients who do request treatment report lasting symptoms that they describe as debilitating.3 For patients who seek help, the treatment recommended by physicians is typically pharmacotherapeutic—which does not follow guidelines.4
The aim of this article is to reinforce a greater understanding of the mechanisms of tinnitus and integrate that knowledge into treatment guidelines. The article does not discuss surgical treatment of tinnitus.
DEFINITION AND CLASSIFICATION
A universal standard definition of chronic tinnitus does not exist; Trevis et al define it as a phantom sound that persists for more than three months.5 The quality and loudness of tinnitus is variable but is often described as a buzz, hiss, or ringing. Prevalence increases with age, smoking, male gender, and ethnicity, with the non-Latino white population statistically at greater risk.3 Comorbid conditions (eg, diabetes and other autoimmune diseases) are risk factors for tinnitus. A history of exposure to loud sound—occupational, environmental, or recreational—also can predispose a person to tinnitus.3
The American Academy of Otolaryngology–Head and Neck Surgery (AAO–HNS) classifies tinnitus as primary (subjective) or secondary (objective). Primary tinnitus—representing the majority of cases—has no identifiable cause; there may be accompanying sensorineural hearing loss or hyperacusis. Secondary tinnitus can also be associated with sensorineural hearing loss but has an identifiable underlying cause.6 The differential diagnosis of tinnitus is listed in the Table.7
Tinnitus is further defined by its persistence. Persistent tinnitus is defined as tinnitus lasting more than six months, slightly longer than the duration offered by Trevis et al, who also define tinnitus as bothersome or non-bothersome, depending on its impact on quality of life.5,6 Causes of reduced quality of life include depression, anxiety, insomnia, and neurocognitive decline—all of which have been associated with chronic subjective tinnitus.8
Continue to: Researchers have discovered that...
Researchers have discovered that tinnitus is not simply a cochlear phenomenon. The pathology extends well beyond the auditory complex, having a deleterious effect on both the somatosensory and central nervous systems, providing some explanation for the prevalence of anxiety and depression associated with the disorder (see "Pathophysiology of tinnitus").9-17
Because of the insidious nature of tinnitus and lack of standard measures of severity, true prevalence is difficult to calculate.18
CLINICAL EVALUATION
Tinnitus can be a presenting complaint or elicited during history-taking. Symptomatic patients should receive full evaluation, including a complete physical exam, medication history, and laboratory workup.
Adverse effect of drugs
Medications that commonly cause tinnitus symptoms are NSAIDs, chemotherapeutic agents, and antibiotics (eg, macrolides and fluoroquinolones). Amiodarone, ACE inhibitors, proton-pump inhibitors, and calcium-channel blockers have also been implicated. Paradoxically, anxiolytics and tricyclic antidepressants, which are sometimes used to treat tinnitus, have been linked to causing the condition.7
Laboratory tests and imaging
Testing should include investigation for infectious disease, autoimmune disorders, and vitamin deficiency.7 According to the American College of Radiology, imaging is unnecessary in the workup of primary tinnitus. Any suspicion of a vascular cause noted on the physical exam (eg, an associated bruit or venous hum), however, should be explored with imaging. Furthermore, any case of tinnitus that lateralizes also requires additional investigation. Modalities of choice are MRI, CT, and CT angiography.19
Continue to: Referral for audiology evaluation
Referral for audiology evaluation
When no underlying pathology can be identified for tinnitus, the patient should be sent for a full audiology evaluation to screen for associated hearing loss. Discussion of audiology screening tests is beyond the scope of this article; however, testing includes otoscopy, audiography, tympanography, otoacoustic emission testing, auditory brainstem-response testing, and vestibular evoked myogenic potential testing.7
Probing nonphysical impacts
Quality of life and overall emotional wellness, including cognitive function, should be investigated in patients with tinnitus. Two questionnaires commonly used in the assessment of tinnitus bother are the Tinnitus Handicap Inventory and the Tinnitus Reaction Questionnaire.7 In a large, systematic review, Trevis et al report that “64% of studies investigating depression found an increase in depressive symptoms in people with chronic tinnitus compared to hearing control groups, and 62% of studies investigating anxiety reported significantly increased anxiety symptoms.”5
MANAGEMENT
Tinnitus management should be viewed two ways: treatment of perceived loudness and treatment of comorbid symptoms relating to tinnitus bother.6 In the same meta-analysis, Trevis and colleagues found that patients with tinnitus had higher rates of anxiety, depression, and overall decline in cognitive function, including processing speed, concentration, and sleep disorders.5 It is useful to keep this observation in mind when reviewing treatment options for tinnitus.
Five classic pharmacotherapeutic approaches to tinnitus management are
- Anticonvulsants
- Antidepressants
- Anesthetics
- Anxiolytics
- Lidocaine.
Newer medications that show some promise are N-methyl-D-aspartate (NMDA) receptor antagonists, notably neramexane. Alternative pharmaceuticals include vitamin-based treatments, cannabinoids, and herbal compounds.
Continue to: The AAOS-HNS supports...
The AAO–HNS supports nonpharmacotherapeutic treatment of tinnitus; its guidelines include a recommendation for cognitive behavioral therapy (CBT) as primary therapy.6 In addition, tinnitus-retraining therapy, tinnitus-masking therapy/sound therapy, meditation/mindfulness, and yoga all have been studied for their ability to alleviate tinnitus bother.
Pharmacotherapeutic management
Anticonvulsants have failed to provide strong evidence of usefulness in the treatment of tinnitus and are not supported by the AAO–HNS as such.6 This conclusion notwithstanding, the anticonvulsants carbamazepine and gabapentin have historically been two of the more common medications used to treat tinnitus.
Carbamazepine is a glutamate receptor antagonist that suppresses seizure activity. Based on prior research suggesting that spontaneous firing within the auditory complex is similar to seizure activity, Iranian researchers explored the hypothesis that carbamazepine might lessen tinnitus severity. Their study revealed, however, that carbamazepine did not statistically significantly reduce the severity of tinnitus, compared to placebo.20 While carbamazepine may be of limited use in the treatment of subjective tinnitus, recent literature confirms that it is not only useful, but also diagnostic, in typewriter tinnitus (ie, having a staccato quality, like the sound of typewriter keys being depressed). Typewriter tinnitus is a secondary cause of tinnitus related to disruption of the stapes in the middle ear.21
Gabapentin works by promoting gamma-aminobutyric acid (GABA) production in the brain. GABA is an inhibitory neurotransmitter, thus slowing down signals between neurons. Following on preliminary research that detected low levels of GABA in the inferior colliculus of rodents with salicylate-induced tinnitus, Aazh and colleagues conducted a double-blind study of gabapentin—and concluded that it yielded no improvement in symptoms, compared to placebo.22
Valproic acid has not been formally investigated but is commonly incorporated in the treatment of tinnitus.23 Lamotrigine has provided similarly disappointing results in the treatment of tinnitus.24
Continue to: Antidepressants and anxiolytics
Antidepressants and anxiolytics. Based on the results of their early clinical trials, Sullivan and colleagues concluded that tricyclic antidepressants produced significant improvement in tinnitus symptoms, due to the analgesic effects of these drugs. The researchers studied nortriptyline specifically; in severely depressed patients, the drug reduced the loudness of tinnitus and depressive symptoms. In non-depressed subjects, however, nortriptyline was not as efficacious.25
Selective serotonin reuptake inhibitors have not had the same success as nortriptyline. In a study of paroxetine conducted by Oishi and colleagues, there was little evidence that the drug reduced the loudness of tinnitus, although overall, it did reduce tinnitus bother and anxiety.26
Included in the category of anxiolytics, benzodiazepines have long been used to treat severe tinnitus-induced anxiety, with some success. However, as Elgoyhen and Langguth point out, studies of benzodiazepines for tinnitus have been limited in size.23
The AAO–HNS does not support routine use of antidepressants and anxiolytics for tinnitus bother.7
NMDA receptor antagonists. In a recent clinical trial, neramexane was studied for its efficacy in tinnitus. Neramexane acts at the cholinergic nicotinic and NMDA receptors in the efferent auditory system. Its complex reaction is thought to prevent transmission of unwanted sound not only to structures within the auditory system but beyond, to the medial geniculate body and lateral nucleus of the amygdala. The trial has proved some benefit concerning overall perception of tinnitus loudness; a phase 2 trial is being conducted.27
Continue to: Intra-tympanic anesthetics
Intra-tympanic anesthetics. Anesthetics, such as lidocaine, have had limited success and results have not been found to be sustained.
Alternative medical managements
Traditional Chinese herbal medications have been used for centuries and are increasingly popular in Western culture. Hilton and colleagues studied Ginkgo biloba, or maidenhair tree, a traditional Chinese herbal supplement available as an extract and as dried leaves. The main action of the extract is vasoregulatory; antiplatelet effects are also seen. Adverse effects include gastrointestinal upset and headache. In a systematic review, Hilton and colleagues concluded that Ginkgo did not reduce overall tinnitus loudness or severity; the review was limited, however, by the fact that only two studies met criteria for inclusion.28
Vitamins, lipoflavinoids, zinc, manganese, and melatonin are all supplements marketed to improve tinnitus symptoms. However, a cross-sectional study confirmed prior research that did not show any benefit from the use of these supplements.29
Cannabinoids are being studied for their proposed antiepileptic effects. There is a popular misconception of Cannabis as a singular chemical when in fact, it is a plant that contains hundreds of chemicals that each act differently on the brain. In a review, Smith and Zheng30 explain that two cannabinoid receptors, CB1 and CB2, are represented, and exert their effects, in different areas of the brain. CB1 receptors block calcium influx in presynaptic terminals, resulting in an inhibitory effect on neurotransmitter release.
CB1 receptors have been found in the dorsal cochlear nuclei, prompting research interest in how cannabinoids affect neurotransmission of unwanted sounds of tinnitus. To date, however, there are conflicting data concerning the benefit of cannabinoids and tinnitus. In fact, Smith and Zheng state that some data suggest that cannabinoids might make tinnitus worse.30
Continue to: Nonpharmacotherapeutic management
Nonpharmacotherapeutic management
Cognitive behavioral therapy. Conceptualized by Aaron T. Beck in the 1960s, cognitive behavioral therapy (CBT) is the leading recommendation made by the AAO–HNS in its tinnitus treatment guidelines.6 Beck’s work centered on the idea that behaviors are modifiable thoughts, through analysis of past experiences and assumptions based on those experiences. By understanding the core belief that a patient attaches to a feeling, Beck hypothesized that behaviors or responses to those feelings could be changed; this is accomplished through discussion to dispel unwarranted fears and by teaching coping mechanisms, such as relaxation. The idea behind CBT in the management of tinnitus is clear: The sound cannot be eliminated, but the patient’s response to the sound can be modified. Ultimately, through this modified response or habituation, the patient can relax and live with the sound.31
Since anxiety, depression, and insomnia are common comorbidities of tinnitus, a psychologic approach remains in the forefront of treatment recommendations. Hoare and colleagues reported that in “a meta-analysis of 10 randomized trials evaluating different forms of CBT (by the therapist and over the Internet), CBT improved tinnitus symptoms compared to non-CBT controls.”7
Tinnitus retraining therapy (TRT) is another form of habituation therapy, introduced by Jastreboff in the 1990s. His work furthered the idea that tinnitus could be reframed, as it is in CBT. Simply, he proposed that systems outside the auditory complex—namely the autonomic nervous system and the limbic system—respond to the signal produced by damaged hair cells in the cochlear nuclei. TRT retrains connections to block or ignore these signals.13 Unlike CBT, the aim of TRT is to eliminate the perception of sound.
By educating patients about the physiologic mechanisms of tinnitus, TRT reduces patient anxiety related to the sound. The process of habituation follows counseling. To accomplish this, the patient wears a sound generator, similar in appearance to hearing aids, using broadband noise. The sound does not mask the tinnitus but closes the gap between silence and the perception of tinnitus. The sound generator is worn for six hours daily for approximately 12 months.
Multiple studies have employed Jastreboff’s original technique, including a clinical trial by Bauer and colleagues. The published outcome of this study confirmed that patients experienced a positive and lasting effect with TRT.32 In addition, a small study of TRT conducted by Barozzi and colleagues, using different colors of sound (ie, how the frequency of a given sound corresponds to the light-wave frequency of a particular color), found statistically significant improvement. Allowing patients to pick a sound that they found more pleasant increased the effectiveness of the treatment.33 (Patients can learn more about TRT by visiting www.tinnitus-pjj.com, hosted by tinnitus researcher Pawel J. Jastreboff.)
Continue to: Alternative nonmedical therapies...
Alternative nonmedical therapies have become popular; they include meditation, yoga, physical therapy, mindfulness, and tinnitus-masking treatment with sound.
Results of a study of yoga and meditation showed that patients felt more relaxed, but that these interventions had no effect on the severity of tinnitus. The principle behind yoga practice, according to Köksoy and colleagues, is that the discipline is thought to affect the limbic system by deactivating the sympathetic response to stimulation from surrounding sounds. In addition, Köksoy states, other researchers have provided evidence that yoga increases circulating levels of antioxidants, which in turn reduce oxidative stress.34
Particularly among members of the millennial generation, mindfulness has become a buzzword. The practice refers to a “method for facing, exploring, and alleviating suffering by relating to present experiences.”35 Roland and colleagues conducted a clinical trial of mindfulness practiced by a cohort of patients with bothersome tinnitus; results were based on scores gleaned from standard rating scales (eg, Global Bothersome Scale, Cognitive and Affective Mindfulness Scale-Revised, Cognitive Failures Questionnaire, Tinnitus Handicap Inventory, and Tinnitus Functional Index). Evaluated before and four weeks after cessation of therapy, subjects reported that tinnitus bother was reduced, but none showed statistically significant improvement in depression, anxiety, or cognitive ability.35
Used for more than 40 years, sound-based therapy has been discussed in conjunction with TRT.36 It is recognized as an approved but optional treatment by the AAO–HNS. In response to a 2010 study by Hobson that used sound-based therapy alone for tinnitus, Tunkel and colleagues cautioned that the modality showed little benefit. The major downside to acoustic therapy, according to the AAO–HNS clinical guidelines, is cost and patients’ excessive expectation of effectiveness.6
According to the AAO–HNS, repetitive-transcranial magnetic stimulation is not supported as a valid treatment for tinnitus because it can lead to seizures in patients who are taking medication that lowers the seizure threshold or who have a secondary cause of tinnitus, such as a tumor—therefore creating risk that outweighs any benefit.6
Continue to: CONCLUSION
CONCLUSION
For a large percentage of the population, chronic subjective tinnitus is a significant variable in the evaluation of quality of life. The condition is not completely understood and often displays features unique to the individual. Much of the initial response to research linking tinnitus with shared pathways typical for chronic pain, anxiety, and depression has resulted in pharmacotherapeutic management that is not always warranted—or successful.
Clinical research into the pathophysiology of tinnitus is providing a better understanding of the neurophysiologic mechanisms that underpin the science of chronic tinnitus. With this information, researchers can one day design medical management that targets specific receptors, resulting in greater management success.
The psychologic impact of tinnitus cannot be underestimated. When almost one-third of patients complain of debilitating symptoms that can also result in neurocognitive decline, tinnitus becomes a condition that cannot be ignored. Guidelines set forth by the AAO–HNS state that CBT and TRT offer some reprieve from symptoms and teach patients habituation without further damage to hearing. The use of broad-based sound generators has been well established as a useful management tool, although it is not curative.
The limitations of some studies that reviewed alternative medicines include small sample size and difficulty comparing research analysis because of disparities in tinnitus rating scales. Also, age bias, comorbid conditions, and study drop-out rates affected overall statistical significance of some studies. Additional, high-quality research is warranted in this area.
Continue to: Prevention of tinnitus...
Prevention of tinnitus through education on hearing loss and its causes should be regarded as implicit; occupational noise and recreational use of music devices put people at heightened risk for hearing loss and tinnitus. Information and open discussion that include the discovery of tinnitus symptoms during routine physical examination are recommended.
Last, providers who adhere to recognized guidelines will aid patients in coping with the challenges that tinnitus presents. As research continues to unravel the complex interaction between neurons, medical science is hopeful that curative treatments will become available.
CE/CME No: CR-1810
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Distinguish primary tinnitus from secondary tinnitus.
• Understand and implement a full clinical evaluation of tinnitus, including imaging studies when appropriate.
• Discuss expectations regarding treatment options and realistic outcomes of currently recommended therapy.
• Direct patients to specialist care for cognitive behavioral therapy or tinnitus retraining therapy.
• Know when pharmacotherapeutic intervention is indicated.
FACULTY
Wendy Gillian Ross practices urgent care medicine in Lake Grove, New York, and primary care in Patchogue, New York. Randy Danielsen is Professor and Dean, Arizona School of Health Sciences, and Director, Center for the Future of the Health Professions, both at A.T. Still University, in Mesa, Arizona. He is Physician Assistant Editor-in-Chief of Clinician Reviews.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through September 30, 2019.
Article begins on next page >>
Tinnitus can be a debilitating condition that affects quality of life and is often not treated according to guidelines. Cognitive behavioral therapy and tinnitus retraining therapy have been successful in reducing tinnitus bother; pharmacotherapy is not widely accepted as successful, and can, in fact, be deleterious. This article describes pathophysiologic disturbances of hearing and how they relate to chronic subjective tinnitus, discusses the clinical evaluation of tinnitus as a presenting symptom, and reviews current treatments.
Primary chronic subjective tinnitus, often thought of more as a symptom than a diagnosis, affects millions of people worldwide. This troublesome condition has been chronicled as far back as the first century
It is estimated that only 20% of people who experience tinnitus actively seek treatment.2 In the United States, 2 to 3 million of the 12 million patients who do request treatment report lasting symptoms that they describe as debilitating.3 For patients who seek help, the treatment recommended by physicians is typically pharmacotherapeutic—which does not follow guidelines.4
The aim of this article is to reinforce a greater understanding of the mechanisms of tinnitus and integrate that knowledge into treatment guidelines. The article does not discuss surgical treatment of tinnitus.
DEFINITION AND CLASSIFICATION
A universal standard definition of chronic tinnitus does not exist; Trevis et al define it as a phantom sound that persists for more than three months.5 The quality and loudness of tinnitus is variable but is often described as a buzz, hiss, or ringing. Prevalence increases with age, smoking, male gender, and ethnicity, with the non-Latino white population statistically at greater risk.3 Comorbid conditions (eg, diabetes and other autoimmune diseases) are risk factors for tinnitus. A history of exposure to loud sound—occupational, environmental, or recreational—also can predispose a person to tinnitus.3
The American Academy of Otolaryngology–Head and Neck Surgery (AAO–HNS) classifies tinnitus as primary (subjective) or secondary (objective). Primary tinnitus—representing the majority of cases—has no identifiable cause; there may be accompanying sensorineural hearing loss or hyperacusis. Secondary tinnitus can also be associated with sensorineural hearing loss but has an identifiable underlying cause.6 The differential diagnosis of tinnitus is listed in the Table.7
Tinnitus is further defined by its persistence. Persistent tinnitus is defined as tinnitus lasting more than six months, slightly longer than the duration offered by Trevis et al, who also define tinnitus as bothersome or non-bothersome, depending on its impact on quality of life.5,6 Causes of reduced quality of life include depression, anxiety, insomnia, and neurocognitive decline—all of which have been associated with chronic subjective tinnitus.8
Continue to: Researchers have discovered that...
Researchers have discovered that tinnitus is not simply a cochlear phenomenon. The pathology extends well beyond the auditory complex, having a deleterious effect on both the somatosensory and central nervous systems, providing some explanation for the prevalence of anxiety and depression associated with the disorder (see "Pathophysiology of tinnitus").9-17
Because of the insidious nature of tinnitus and lack of standard measures of severity, true prevalence is difficult to calculate.18
CLINICAL EVALUATION
Tinnitus can be a presenting complaint or elicited during history-taking. Symptomatic patients should receive full evaluation, including a complete physical exam, medication history, and laboratory workup.
Adverse effect of drugs
Medications that commonly cause tinnitus symptoms are NSAIDs, chemotherapeutic agents, and antibiotics (eg, macrolides and fluoroquinolones). Amiodarone, ACE inhibitors, proton-pump inhibitors, and calcium-channel blockers have also been implicated. Paradoxically, anxiolytics and tricyclic antidepressants, which are sometimes used to treat tinnitus, have been linked to causing the condition.7
Laboratory tests and imaging
Testing should include investigation for infectious disease, autoimmune disorders, and vitamin deficiency.7 According to the American College of Radiology, imaging is unnecessary in the workup of primary tinnitus. Any suspicion of a vascular cause noted on the physical exam (eg, an associated bruit or venous hum), however, should be explored with imaging. Furthermore, any case of tinnitus that lateralizes also requires additional investigation. Modalities of choice are MRI, CT, and CT angiography.19
Continue to: Referral for audiology evaluation
Referral for audiology evaluation
When no underlying pathology can be identified for tinnitus, the patient should be sent for a full audiology evaluation to screen for associated hearing loss. Discussion of audiology screening tests is beyond the scope of this article; however, testing includes otoscopy, audiography, tympanography, otoacoustic emission testing, auditory brainstem-response testing, and vestibular evoked myogenic potential testing.7
Probing nonphysical impacts
Quality of life and overall emotional wellness, including cognitive function, should be investigated in patients with tinnitus. Two questionnaires commonly used in the assessment of tinnitus bother are the Tinnitus Handicap Inventory and the Tinnitus Reaction Questionnaire.7 In a large, systematic review, Trevis et al report that “64% of studies investigating depression found an increase in depressive symptoms in people with chronic tinnitus compared to hearing control groups, and 62% of studies investigating anxiety reported significantly increased anxiety symptoms.”5
MANAGEMENT
Tinnitus management should be viewed two ways: treatment of perceived loudness and treatment of comorbid symptoms relating to tinnitus bother.6 In the same meta-analysis, Trevis and colleagues found that patients with tinnitus had higher rates of anxiety, depression, and overall decline in cognitive function, including processing speed, concentration, and sleep disorders.5 It is useful to keep this observation in mind when reviewing treatment options for tinnitus.
Five classic pharmacotherapeutic approaches to tinnitus management are
- Anticonvulsants
- Antidepressants
- Anesthetics
- Anxiolytics
- Lidocaine.
Newer medications that show some promise are N-methyl-D-aspartate (NMDA) receptor antagonists, notably neramexane. Alternative pharmaceuticals include vitamin-based treatments, cannabinoids, and herbal compounds.
Continue to: The AAOS-HNS supports...
The AAO–HNS supports nonpharmacotherapeutic treatment of tinnitus; its guidelines include a recommendation for cognitive behavioral therapy (CBT) as primary therapy.6 In addition, tinnitus-retraining therapy, tinnitus-masking therapy/sound therapy, meditation/mindfulness, and yoga all have been studied for their ability to alleviate tinnitus bother.
Pharmacotherapeutic management
Anticonvulsants have failed to provide strong evidence of usefulness in the treatment of tinnitus and are not supported by the AAO–HNS as such.6 This conclusion notwithstanding, the anticonvulsants carbamazepine and gabapentin have historically been two of the more common medications used to treat tinnitus.
Carbamazepine is a glutamate receptor antagonist that suppresses seizure activity. Based on prior research suggesting that spontaneous firing within the auditory complex is similar to seizure activity, Iranian researchers explored the hypothesis that carbamazepine might lessen tinnitus severity. Their study revealed, however, that carbamazepine did not statistically significantly reduce the severity of tinnitus, compared to placebo.20 While carbamazepine may be of limited use in the treatment of subjective tinnitus, recent literature confirms that it is not only useful, but also diagnostic, in typewriter tinnitus (ie, having a staccato quality, like the sound of typewriter keys being depressed). Typewriter tinnitus is a secondary cause of tinnitus related to disruption of the stapes in the middle ear.21
Gabapentin works by promoting gamma-aminobutyric acid (GABA) production in the brain. GABA is an inhibitory neurotransmitter, thus slowing down signals between neurons. Following on preliminary research that detected low levels of GABA in the inferior colliculus of rodents with salicylate-induced tinnitus, Aazh and colleagues conducted a double-blind study of gabapentin—and concluded that it yielded no improvement in symptoms, compared to placebo.22
Valproic acid has not been formally investigated but is commonly incorporated in the treatment of tinnitus.23 Lamotrigine has provided similarly disappointing results in the treatment of tinnitus.24
Continue to: Antidepressants and anxiolytics
Antidepressants and anxiolytics. Based on the results of their early clinical trials, Sullivan and colleagues concluded that tricyclic antidepressants produced significant improvement in tinnitus symptoms, due to the analgesic effects of these drugs. The researchers studied nortriptyline specifically; in severely depressed patients, the drug reduced the loudness of tinnitus and depressive symptoms. In non-depressed subjects, however, nortriptyline was not as efficacious.25
Selective serotonin reuptake inhibitors have not had the same success as nortriptyline. In a study of paroxetine conducted by Oishi and colleagues, there was little evidence that the drug reduced the loudness of tinnitus, although overall, it did reduce tinnitus bother and anxiety.26
Included in the category of anxiolytics, benzodiazepines have long been used to treat severe tinnitus-induced anxiety, with some success. However, as Elgoyhen and Langguth point out, studies of benzodiazepines for tinnitus have been limited in size.23
The AAO–HNS does not support routine use of antidepressants and anxiolytics for tinnitus bother.7
NMDA receptor antagonists. In a recent clinical trial, neramexane was studied for its efficacy in tinnitus. Neramexane acts at the cholinergic nicotinic and NMDA receptors in the efferent auditory system. Its complex reaction is thought to prevent transmission of unwanted sound not only to structures within the auditory system but beyond, to the medial geniculate body and lateral nucleus of the amygdala. The trial has proved some benefit concerning overall perception of tinnitus loudness; a phase 2 trial is being conducted.27
Continue to: Intra-tympanic anesthetics
Intra-tympanic anesthetics. Anesthetics, such as lidocaine, have had limited success and results have not been found to be sustained.
Alternative medical managements
Traditional Chinese herbal medications have been used for centuries and are increasingly popular in Western culture. Hilton and colleagues studied Ginkgo biloba, or maidenhair tree, a traditional Chinese herbal supplement available as an extract and as dried leaves. The main action of the extract is vasoregulatory; antiplatelet effects are also seen. Adverse effects include gastrointestinal upset and headache. In a systematic review, Hilton and colleagues concluded that Ginkgo did not reduce overall tinnitus loudness or severity; the review was limited, however, by the fact that only two studies met criteria for inclusion.28
Vitamins, lipoflavinoids, zinc, manganese, and melatonin are all supplements marketed to improve tinnitus symptoms. However, a cross-sectional study confirmed prior research that did not show any benefit from the use of these supplements.29
Cannabinoids are being studied for their proposed antiepileptic effects. There is a popular misconception of Cannabis as a singular chemical when in fact, it is a plant that contains hundreds of chemicals that each act differently on the brain. In a review, Smith and Zheng30 explain that two cannabinoid receptors, CB1 and CB2, are represented, and exert their effects, in different areas of the brain. CB1 receptors block calcium influx in presynaptic terminals, resulting in an inhibitory effect on neurotransmitter release.
CB1 receptors have been found in the dorsal cochlear nuclei, prompting research interest in how cannabinoids affect neurotransmission of unwanted sounds of tinnitus. To date, however, there are conflicting data concerning the benefit of cannabinoids and tinnitus. In fact, Smith and Zheng state that some data suggest that cannabinoids might make tinnitus worse.30
Continue to: Nonpharmacotherapeutic management
Nonpharmacotherapeutic management
Cognitive behavioral therapy. Conceptualized by Aaron T. Beck in the 1960s, cognitive behavioral therapy (CBT) is the leading recommendation made by the AAO–HNS in its tinnitus treatment guidelines.6 Beck’s work centered on the idea that behaviors are modifiable thoughts, through analysis of past experiences and assumptions based on those experiences. By understanding the core belief that a patient attaches to a feeling, Beck hypothesized that behaviors or responses to those feelings could be changed; this is accomplished through discussion to dispel unwarranted fears and by teaching coping mechanisms, such as relaxation. The idea behind CBT in the management of tinnitus is clear: The sound cannot be eliminated, but the patient’s response to the sound can be modified. Ultimately, through this modified response or habituation, the patient can relax and live with the sound.31
Since anxiety, depression, and insomnia are common comorbidities of tinnitus, a psychologic approach remains in the forefront of treatment recommendations. Hoare and colleagues reported that in “a meta-analysis of 10 randomized trials evaluating different forms of CBT (by the therapist and over the Internet), CBT improved tinnitus symptoms compared to non-CBT controls.”7
Tinnitus retraining therapy (TRT) is another form of habituation therapy, introduced by Jastreboff in the 1990s. His work furthered the idea that tinnitus could be reframed, as it is in CBT. Simply, he proposed that systems outside the auditory complex—namely the autonomic nervous system and the limbic system—respond to the signal produced by damaged hair cells in the cochlear nuclei. TRT retrains connections to block or ignore these signals.13 Unlike CBT, the aim of TRT is to eliminate the perception of sound.
By educating patients about the physiologic mechanisms of tinnitus, TRT reduces patient anxiety related to the sound. The process of habituation follows counseling. To accomplish this, the patient wears a sound generator, similar in appearance to hearing aids, using broadband noise. The sound does not mask the tinnitus but closes the gap between silence and the perception of tinnitus. The sound generator is worn for six hours daily for approximately 12 months.
Multiple studies have employed Jastreboff’s original technique, including a clinical trial by Bauer and colleagues. The published outcome of this study confirmed that patients experienced a positive and lasting effect with TRT.32 In addition, a small study of TRT conducted by Barozzi and colleagues, using different colors of sound (ie, how the frequency of a given sound corresponds to the light-wave frequency of a particular color), found statistically significant improvement. Allowing patients to pick a sound that they found more pleasant increased the effectiveness of the treatment.33 (Patients can learn more about TRT by visiting www.tinnitus-pjj.com, hosted by tinnitus researcher Pawel J. Jastreboff.)
Continue to: Alternative nonmedical therapies...
Alternative nonmedical therapies have become popular; they include meditation, yoga, physical therapy, mindfulness, and tinnitus-masking treatment with sound.
Results of a study of yoga and meditation showed that patients felt more relaxed, but that these interventions had no effect on the severity of tinnitus. The principle behind yoga practice, according to Köksoy and colleagues, is that the discipline is thought to affect the limbic system by deactivating the sympathetic response to stimulation from surrounding sounds. In addition, Köksoy states, other researchers have provided evidence that yoga increases circulating levels of antioxidants, which in turn reduce oxidative stress.34
Particularly among members of the millennial generation, mindfulness has become a buzzword. The practice refers to a “method for facing, exploring, and alleviating suffering by relating to present experiences.”35 Roland and colleagues conducted a clinical trial of mindfulness practiced by a cohort of patients with bothersome tinnitus; results were based on scores gleaned from standard rating scales (eg, Global Bothersome Scale, Cognitive and Affective Mindfulness Scale-Revised, Cognitive Failures Questionnaire, Tinnitus Handicap Inventory, and Tinnitus Functional Index). Evaluated before and four weeks after cessation of therapy, subjects reported that tinnitus bother was reduced, but none showed statistically significant improvement in depression, anxiety, or cognitive ability.35
Used for more than 40 years, sound-based therapy has been discussed in conjunction with TRT.36 It is recognized as an approved but optional treatment by the AAO–HNS. In response to a 2010 study by Hobson that used sound-based therapy alone for tinnitus, Tunkel and colleagues cautioned that the modality showed little benefit. The major downside to acoustic therapy, according to the AAO–HNS clinical guidelines, is cost and patients’ excessive expectation of effectiveness.6
According to the AAO–HNS, repetitive-transcranial magnetic stimulation is not supported as a valid treatment for tinnitus because it can lead to seizures in patients who are taking medication that lowers the seizure threshold or who have a secondary cause of tinnitus, such as a tumor—therefore creating risk that outweighs any benefit.6
Continue to: CONCLUSION
CONCLUSION
For a large percentage of the population, chronic subjective tinnitus is a significant variable in the evaluation of quality of life. The condition is not completely understood and often displays features unique to the individual. Much of the initial response to research linking tinnitus with shared pathways typical for chronic pain, anxiety, and depression has resulted in pharmacotherapeutic management that is not always warranted—or successful.
Clinical research into the pathophysiology of tinnitus is providing a better understanding of the neurophysiologic mechanisms that underpin the science of chronic tinnitus. With this information, researchers can one day design medical management that targets specific receptors, resulting in greater management success.
The psychologic impact of tinnitus cannot be underestimated. When almost one-third of patients complain of debilitating symptoms that can also result in neurocognitive decline, tinnitus becomes a condition that cannot be ignored. Guidelines set forth by the AAO–HNS state that CBT and TRT offer some reprieve from symptoms and teach patients habituation without further damage to hearing. The use of broad-based sound generators has been well established as a useful management tool, although it is not curative.
The limitations of some studies that reviewed alternative medicines include small sample size and difficulty comparing research analysis because of disparities in tinnitus rating scales. Also, age bias, comorbid conditions, and study drop-out rates affected overall statistical significance of some studies. Additional, high-quality research is warranted in this area.
Continue to: Prevention of tinnitus...
Prevention of tinnitus through education on hearing loss and its causes should be regarded as implicit; occupational noise and recreational use of music devices put people at heightened risk for hearing loss and tinnitus. Information and open discussion that include the discovery of tinnitus symptoms during routine physical examination are recommended.
Last, providers who adhere to recognized guidelines will aid patients in coping with the challenges that tinnitus presents. As research continues to unravel the complex interaction between neurons, medical science is hopeful that curative treatments will become available.
1. Maltby MT. Ancient voices on tinnitus: the pathology and treatment of tinnitus in Celsus and the Hippocratic Corpus compared and contrasted. Int Tinnitus J. 2012;17(2):140-145.
2. Wolever RQ, Price R, Hazelton GA, et al. Complementary therapies for significant dysfunction from tinnitus: treatment review and potential for integrative medicine. Evid Based Complement Alternat Med. 2015;15:931418.
3. Shargorodsky J, Curhan GC, Farwell WR. Prevalence and characteristics of tinnitus among US adults. Am J Med. 2010;123(8):711-718.
4. Bhatt JM, Lin HW, Bhattacharyya N. Prevalence, severity, exposures, and treatment patterns of tinnitus in the United States. JAMA Otolaryngol Head Neck Surg. 2016;142(10):959-965.
5. Trevis KJ, McLachlan NM, Wilson SJ. A systematic review and meta-analysis of psychological functioning in chronic tinnitus. Clin Psychol Rev. 2018;60:62-86.
6. Tunkel DE, Bauer CA, Sun GH, et al. Clinical practice guideline: tinnitus. Otolaryngol Head Neck Surg. 2014;151(suppl 2):S1-S40.
7. Dinces EA. Treatment of tinnitus. UpToDate. April 12, 2018. www.uptodate.com/contents/treatment-of-tinnitus. Accessed September 17, 2018.
8. Gudwani S, Munjal SK, Panda NK, Kohli A. Association of chronic subjective tinnitus with neuro-cognitive performance. Int Tinnitus J. 2017;21:90-97.
9. Jastreboff PJ. 25 years of tinnitus retraining therapy. HNO. 2015;63:307-311.
10. Pujol R. Journey into the world of hearing. 2016. www.cochlea.eu/en. Accessed September 17, 2018.
11. Adjamian P, Hall DA, Palmer AR, et al. Neuroanatomical abnormalities in chronic tinnitus in the human brain.Neurosci Biobehav Rev. 2014;45:119-133.
12. Shore SE, Roberts LE, Langguth B. Maladaptive plasticity in tinnitus—triggers, mechanisms and treatment. Nat Rev Neurol. 2016;12(3):150-160.
13. Jastreboff PJ, Gray WC, Gold SL. Neurophysiological approach to tinnitus patients. Am J Otol. 1996;17(2):236-240.
14. Kaltenbach JA. Tinnitus: models and mechanisms. Hear Res. 2011;276:52-60.
15. Rauschecker JP, Leaver AM, Mühlau M. Tuning out the noise: limbic-auditory interactions in tinnitus. Neuron. 2010;66(6):819-826.
16. Møller AR. Sensorineural tinnitus: its pathology and probable therapies. Int J Otolaryngol. 2016;2016:2830157.
17. Chen YC, Xia W, Chen H, et al. Tinnitus distress is linked to enhanced resting‐state functional connectivity from the limbic system to the auditory cortex. Hum Brain Mapp. 2017;38(5):2384-2397.
18. McCormack A, Edmonson-Jones M, Somerset S, Hall D. A systematic review of the reporting of tinnitus prevalence and severity. Hear Res. 2016;337:70-79.
19. Kessler MM, Moussa M, Bykowski J, et al; Expert Panel on Neurologic Imaging. ACR Appropriateness Criteria® Tinnitus. J Am Coll Radiol. 2017;14:S584-S591.
20. Gerami H, Saberi A, Nemati, S, et al. Effects of oxcarbazepine versus carbamazepine on tinnitus: a randomized double-blind placebo-controlled clinical trial. Iran J Neurol. 2012;11(3):106-110.
21. Sunwoo W, Jeon YJ, Bae YJ, et al. Typewriter tinnitus revisited: the typical symptoms and the initial response to carbamazepine are the most reliable diagnostic clues. Sci Rep. 2017;7:10615.
22. Aazh H, El Refaie A, Humphriss R. Gabapentin for tinnitus: a systematic review. Am J Audiol. 2011;20:151-158.
23. Elgoyhen AB, Langguth B. Pharmacological approaches to the treatment of tinnitus. Drug Discov Today. 2010;15:300-305.
24. Langguth B, Kreuzer PM, Kleinjung T, De Ridder D. Tinnitus: causes and clinical management. Lancet Neurol. 2013;12(9):920-930.
25. Sullivan M, Katon W, Russo J, et al. A randomized trial of nortriptyline for severe chronic tinnitus. Effects on depression, disability, and tinnitus symptoms. Arch Intern Med. 1993;153(19):2251-2259.
26. Oishi N, Kanzaki S, Shinden S, et al. Effects of selective serotonin reuptake inhibitor on treating tinnitus in patients stratified for presence of depression or anxiety. Audiol Neurootol. 2010;15(3):187-193.
27. Suckfüll M, Althaus M, Ellers-Lenz B, et al. A randomized, double-blind, placebo-controlled clinical trial to evaluate the efficacy and safety of neramexane in patients with moderate to severe subjective tinnitus. BMC Ear Nose Throat Disord. 2011;11:1.
28. Hilton MP, Zimmermann EF, Hunt WT. Ginkgo biloba for tinnitus. Cochrane Database Syst Rev. 2013;CD003852. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD003852.pub3/full. Accessed September 17, 2018.
29. Coelho C, Tyler R, Ji H, et al. Survey on the effectiveness of dietary supplements to treat tinnitus. Am J Audiol. 2016;25:184-205.
30. Smith PF, Zheng Y. Cannabinoids, cannabinoid receptors and tinnitus. Hear Res. 2015;332:210-216.
31. Martinez-Devesa P, Perera R, Theodoulou M, Waddell A. Cognitive behavioural therapy for tinnitus. Cochrane Database Syst Rev. 2010:CD005233. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD005233.pub3/full. Accessed September 17, 2018.
32. Bauer CA, Berry JL, Brozoski TJ. The effect of tinnitus retraining therapy on chronic tinnitus: a controlled trial. Laryngoscope Investig Otolaryngol. 2017;2(4):166-177.
33. Barozzi S, Ambrosetti U, Callaway SL, et al. Effects of tinnitus retraining therapy with different colours of sound. Int Tinnitus J. 2017;21:139-143.
34. Köksoy S, Eti CM, Karatas¸ M, Vayisoglu Y. The effects of yoga in patients suffering from subjective tinnitus. Int Arch Otorhinolaryngol. 2018;22(1):9-13.
35. Roland LT, Lenze EJ, Hardin FM, et al. Effects of mindfulness based stress reduction therapy on subjective bother and neural connectivity in chronic tinnitus. Otolaryngol Head Neck Surg. 2015;152(5):919-926.
36. Ibarra D, Tavira-Sanchez F, Recuero-Lopez M, Anthony BW. In-ear medical devices for acoustic therapies in tinnitus treatments, state of the art. Auris Nasus Larynx. 2018;45:6-12.
1. Maltby MT. Ancient voices on tinnitus: the pathology and treatment of tinnitus in Celsus and the Hippocratic Corpus compared and contrasted. Int Tinnitus J. 2012;17(2):140-145.
2. Wolever RQ, Price R, Hazelton GA, et al. Complementary therapies for significant dysfunction from tinnitus: treatment review and potential for integrative medicine. Evid Based Complement Alternat Med. 2015;15:931418.
3. Shargorodsky J, Curhan GC, Farwell WR. Prevalence and characteristics of tinnitus among US adults. Am J Med. 2010;123(8):711-718.
4. Bhatt JM, Lin HW, Bhattacharyya N. Prevalence, severity, exposures, and treatment patterns of tinnitus in the United States. JAMA Otolaryngol Head Neck Surg. 2016;142(10):959-965.
5. Trevis KJ, McLachlan NM, Wilson SJ. A systematic review and meta-analysis of psychological functioning in chronic tinnitus. Clin Psychol Rev. 2018;60:62-86.
6. Tunkel DE, Bauer CA, Sun GH, et al. Clinical practice guideline: tinnitus. Otolaryngol Head Neck Surg. 2014;151(suppl 2):S1-S40.
7. Dinces EA. Treatment of tinnitus. UpToDate. April 12, 2018. www.uptodate.com/contents/treatment-of-tinnitus. Accessed September 17, 2018.
8. Gudwani S, Munjal SK, Panda NK, Kohli A. Association of chronic subjective tinnitus with neuro-cognitive performance. Int Tinnitus J. 2017;21:90-97.
9. Jastreboff PJ. 25 years of tinnitus retraining therapy. HNO. 2015;63:307-311.
10. Pujol R. Journey into the world of hearing. 2016. www.cochlea.eu/en. Accessed September 17, 2018.
11. Adjamian P, Hall DA, Palmer AR, et al. Neuroanatomical abnormalities in chronic tinnitus in the human brain.Neurosci Biobehav Rev. 2014;45:119-133.
12. Shore SE, Roberts LE, Langguth B. Maladaptive plasticity in tinnitus—triggers, mechanisms and treatment. Nat Rev Neurol. 2016;12(3):150-160.
13. Jastreboff PJ, Gray WC, Gold SL. Neurophysiological approach to tinnitus patients. Am J Otol. 1996;17(2):236-240.
14. Kaltenbach JA. Tinnitus: models and mechanisms. Hear Res. 2011;276:52-60.
15. Rauschecker JP, Leaver AM, Mühlau M. Tuning out the noise: limbic-auditory interactions in tinnitus. Neuron. 2010;66(6):819-826.
16. Møller AR. Sensorineural tinnitus: its pathology and probable therapies. Int J Otolaryngol. 2016;2016:2830157.
17. Chen YC, Xia W, Chen H, et al. Tinnitus distress is linked to enhanced resting‐state functional connectivity from the limbic system to the auditory cortex. Hum Brain Mapp. 2017;38(5):2384-2397.
18. McCormack A, Edmonson-Jones M, Somerset S, Hall D. A systematic review of the reporting of tinnitus prevalence and severity. Hear Res. 2016;337:70-79.
19. Kessler MM, Moussa M, Bykowski J, et al; Expert Panel on Neurologic Imaging. ACR Appropriateness Criteria® Tinnitus. J Am Coll Radiol. 2017;14:S584-S591.
20. Gerami H, Saberi A, Nemati, S, et al. Effects of oxcarbazepine versus carbamazepine on tinnitus: a randomized double-blind placebo-controlled clinical trial. Iran J Neurol. 2012;11(3):106-110.
21. Sunwoo W, Jeon YJ, Bae YJ, et al. Typewriter tinnitus revisited: the typical symptoms and the initial response to carbamazepine are the most reliable diagnostic clues. Sci Rep. 2017;7:10615.
22. Aazh H, El Refaie A, Humphriss R. Gabapentin for tinnitus: a systematic review. Am J Audiol. 2011;20:151-158.
23. Elgoyhen AB, Langguth B. Pharmacological approaches to the treatment of tinnitus. Drug Discov Today. 2010;15:300-305.
24. Langguth B, Kreuzer PM, Kleinjung T, De Ridder D. Tinnitus: causes and clinical management. Lancet Neurol. 2013;12(9):920-930.
25. Sullivan M, Katon W, Russo J, et al. A randomized trial of nortriptyline for severe chronic tinnitus. Effects on depression, disability, and tinnitus symptoms. Arch Intern Med. 1993;153(19):2251-2259.
26. Oishi N, Kanzaki S, Shinden S, et al. Effects of selective serotonin reuptake inhibitor on treating tinnitus in patients stratified for presence of depression or anxiety. Audiol Neurootol. 2010;15(3):187-193.
27. Suckfüll M, Althaus M, Ellers-Lenz B, et al. A randomized, double-blind, placebo-controlled clinical trial to evaluate the efficacy and safety of neramexane in patients with moderate to severe subjective tinnitus. BMC Ear Nose Throat Disord. 2011;11:1.
28. Hilton MP, Zimmermann EF, Hunt WT. Ginkgo biloba for tinnitus. Cochrane Database Syst Rev. 2013;CD003852. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD003852.pub3/full. Accessed September 17, 2018.
29. Coelho C, Tyler R, Ji H, et al. Survey on the effectiveness of dietary supplements to treat tinnitus. Am J Audiol. 2016;25:184-205.
30. Smith PF, Zheng Y. Cannabinoids, cannabinoid receptors and tinnitus. Hear Res. 2015;332:210-216.
31. Martinez-Devesa P, Perera R, Theodoulou M, Waddell A. Cognitive behavioural therapy for tinnitus. Cochrane Database Syst Rev. 2010:CD005233. http://cochranelibrary-wiley.com/doi/10.1002/14651858.CD005233.pub3/full. Accessed September 17, 2018.
32. Bauer CA, Berry JL, Brozoski TJ. The effect of tinnitus retraining therapy on chronic tinnitus: a controlled trial. Laryngoscope Investig Otolaryngol. 2017;2(4):166-177.
33. Barozzi S, Ambrosetti U, Callaway SL, et al. Effects of tinnitus retraining therapy with different colours of sound. Int Tinnitus J. 2017;21:139-143.
34. Köksoy S, Eti CM, Karatas¸ M, Vayisoglu Y. The effects of yoga in patients suffering from subjective tinnitus. Int Arch Otorhinolaryngol. 2018;22(1):9-13.
35. Roland LT, Lenze EJ, Hardin FM, et al. Effects of mindfulness based stress reduction therapy on subjective bother and neural connectivity in chronic tinnitus. Otolaryngol Head Neck Surg. 2015;152(5):919-926.
36. Ibarra D, Tavira-Sanchez F, Recuero-Lopez M, Anthony BW. In-ear medical devices for acoustic therapies in tinnitus treatments, state of the art. Auris Nasus Larynx. 2018;45:6-12.
