User login
Urinary Metals Linked to Increased Dementia Risk
TOPLINE:
METHODOLOGY:
- This multicenter prospective cohort study included 6303 participants from six US study centers from 2000 to 2002, with follow-up through 2018.
- Participants were aged 45-84 years (median age at baseline, 60 years; 52% women) and were free of diagnosed cardiovascular disease.
- Researchers measured urinary levels of arsenic, cadmium, cobalt, copper, lead, manganese, tungsten, uranium, and zinc.
- Neuropsychological assessments included the Digit Symbol Coding, Cognitive Abilities Screening Instrument, and Digit Span tests.
- The median follow-up duration was 11.7 years for participants with dementia and 16.8 years for those without; 559 cases of dementia were identified during the study.
TAKEAWAY:
- Lower Digit Symbol Coding scores were associated with higher urinary concentrations of arsenic (mean difference [MD] in score per interquartile range [IQR] increase, –0.03), cobalt (MD per IQR increase, –0.05), copper (MD per IQR increase, –0.05), uranium (MD per IQR increase, –0.04), and zinc (MD per IQR increase, –0.03).
- Effects for cobalt, uranium, and zinc were stronger in apolipoprotein epsilon 4 allele (APOE4) carriers vs noncarriers.
- Higher urinary levels of copper were associated with lower Digit Span scores (MD, –0.043) and elevated levels of copper (MD, –0.028) and zinc (MD, –0.024) were associated with lower global cognitive scores.
- Individuals with urinary levels of the nine-metal mixture at the 95th percentile had a 71% higher risk for dementia compared to those with levels at the 25th percentile, with the risk more pronounced in APOE4 carriers than in noncarriers (MD, –0.30 vs –0.10, respectively).
IN PRACTICE:
“We found an inverse association of essential and nonessential metals in urine, both individually and as a mixture, with the speed of mental operations, as well as a positive association of urinary metal levels with dementia risk. As metal exposure and levels in the body are modifiable, these findings could inform early screening and precision interventions for dementia prevention based on individuals’ metal exposure and genetic profiles,” the investigators wrote.
SOURCE:
The study was led by Arce Domingo-Relloso, PhD, Columbia University Mailman School of Public Health, New York City. It was published online in JAMA Network Open.
LIMITATIONS:
Data may have been missed for patients with dementia who were never hospitalized, died, or were lost to follow-up. The dementia diagnosis included nonspecific International Classification of Diseases codes, potentially leading to false-positive reports. In addition, the sample size was not sufficient to evaluate the associations between metal exposure and cognitive test scores for carriers of two APOE4 alleles.
DISCLOSURES:
The study was supported by the National Heart, Lung, and Blood Institute. Several authors reported receiving grants from the National Institutes of Health and consulting fees, editorial stipends, teaching fees, or unrelated grant funding from various sources, which are fully listed in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- This multicenter prospective cohort study included 6303 participants from six US study centers from 2000 to 2002, with follow-up through 2018.
- Participants were aged 45-84 years (median age at baseline, 60 years; 52% women) and were free of diagnosed cardiovascular disease.
- Researchers measured urinary levels of arsenic, cadmium, cobalt, copper, lead, manganese, tungsten, uranium, and zinc.
- Neuropsychological assessments included the Digit Symbol Coding, Cognitive Abilities Screening Instrument, and Digit Span tests.
- The median follow-up duration was 11.7 years for participants with dementia and 16.8 years for those without; 559 cases of dementia were identified during the study.
TAKEAWAY:
- Lower Digit Symbol Coding scores were associated with higher urinary concentrations of arsenic (mean difference [MD] in score per interquartile range [IQR] increase, –0.03), cobalt (MD per IQR increase, –0.05), copper (MD per IQR increase, –0.05), uranium (MD per IQR increase, –0.04), and zinc (MD per IQR increase, –0.03).
- Effects for cobalt, uranium, and zinc were stronger in apolipoprotein epsilon 4 allele (APOE4) carriers vs noncarriers.
- Higher urinary levels of copper were associated with lower Digit Span scores (MD, –0.043) and elevated levels of copper (MD, –0.028) and zinc (MD, –0.024) were associated with lower global cognitive scores.
- Individuals with urinary levels of the nine-metal mixture at the 95th percentile had a 71% higher risk for dementia compared to those with levels at the 25th percentile, with the risk more pronounced in APOE4 carriers than in noncarriers (MD, –0.30 vs –0.10, respectively).
IN PRACTICE:
“We found an inverse association of essential and nonessential metals in urine, both individually and as a mixture, with the speed of mental operations, as well as a positive association of urinary metal levels with dementia risk. As metal exposure and levels in the body are modifiable, these findings could inform early screening and precision interventions for dementia prevention based on individuals’ metal exposure and genetic profiles,” the investigators wrote.
SOURCE:
The study was led by Arce Domingo-Relloso, PhD, Columbia University Mailman School of Public Health, New York City. It was published online in JAMA Network Open.
LIMITATIONS:
Data may have been missed for patients with dementia who were never hospitalized, died, or were lost to follow-up. The dementia diagnosis included nonspecific International Classification of Diseases codes, potentially leading to false-positive reports. In addition, the sample size was not sufficient to evaluate the associations between metal exposure and cognitive test scores for carriers of two APOE4 alleles.
DISCLOSURES:
The study was supported by the National Heart, Lung, and Blood Institute. Several authors reported receiving grants from the National Institutes of Health and consulting fees, editorial stipends, teaching fees, or unrelated grant funding from various sources, which are fully listed in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- This multicenter prospective cohort study included 6303 participants from six US study centers from 2000 to 2002, with follow-up through 2018.
- Participants were aged 45-84 years (median age at baseline, 60 years; 52% women) and were free of diagnosed cardiovascular disease.
- Researchers measured urinary levels of arsenic, cadmium, cobalt, copper, lead, manganese, tungsten, uranium, and zinc.
- Neuropsychological assessments included the Digit Symbol Coding, Cognitive Abilities Screening Instrument, and Digit Span tests.
- The median follow-up duration was 11.7 years for participants with dementia and 16.8 years for those without; 559 cases of dementia were identified during the study.
TAKEAWAY:
- Lower Digit Symbol Coding scores were associated with higher urinary concentrations of arsenic (mean difference [MD] in score per interquartile range [IQR] increase, –0.03), cobalt (MD per IQR increase, –0.05), copper (MD per IQR increase, –0.05), uranium (MD per IQR increase, –0.04), and zinc (MD per IQR increase, –0.03).
- Effects for cobalt, uranium, and zinc were stronger in apolipoprotein epsilon 4 allele (APOE4) carriers vs noncarriers.
- Higher urinary levels of copper were associated with lower Digit Span scores (MD, –0.043) and elevated levels of copper (MD, –0.028) and zinc (MD, –0.024) were associated with lower global cognitive scores.
- Individuals with urinary levels of the nine-metal mixture at the 95th percentile had a 71% higher risk for dementia compared to those with levels at the 25th percentile, with the risk more pronounced in APOE4 carriers than in noncarriers (MD, –0.30 vs –0.10, respectively).
IN PRACTICE:
“We found an inverse association of essential and nonessential metals in urine, both individually and as a mixture, with the speed of mental operations, as well as a positive association of urinary metal levels with dementia risk. As metal exposure and levels in the body are modifiable, these findings could inform early screening and precision interventions for dementia prevention based on individuals’ metal exposure and genetic profiles,” the investigators wrote.
SOURCE:
The study was led by Arce Domingo-Relloso, PhD, Columbia University Mailman School of Public Health, New York City. It was published online in JAMA Network Open.
LIMITATIONS:
Data may have been missed for patients with dementia who were never hospitalized, died, or were lost to follow-up. The dementia diagnosis included nonspecific International Classification of Diseases codes, potentially leading to false-positive reports. In addition, the sample size was not sufficient to evaluate the associations between metal exposure and cognitive test scores for carriers of two APOE4 alleles.
DISCLOSURES:
The study was supported by the National Heart, Lung, and Blood Institute. Several authors reported receiving grants from the National Institutes of Health and consulting fees, editorial stipends, teaching fees, or unrelated grant funding from various sources, which are fully listed in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Common Gut Infection Tied to Alzheimer’s Disease
Researchers are gaining new insight into the relationship between the human cytomegalovirus (HCMV), a common herpes virus found in the gut, and the immune response associated with CD83 antibody in some individuals with Alzheimer’s disease (AD).
Using tissue samples from deceased donors with AD, the study showed CD83-positive (CD83+) microglia in the superior frontal gyrus (SFG) are significantly associated with elevated immunoglobulin gamma 4 (IgG4) and HCMV in the transverse colon (TC), increased anti-HCMV IgG4 in the cerebrospinal fluid (CSF), and both HCMV and IgG4 in the SFG and vagus nerve.
“Our results indicate a complex, cross-tissue interaction between HCMV and the host adaptive immune response associated with CD83+ microglia in persons with AD,” noted the investigators, including Benjamin P. Readhead, MBBS, research associate professor, ASU-Banner Neurodegenerative Disease Research Center, Arizona State University, Tempe.
The results suggest antiviral therapy in patients with biomarker evidence of HCMV, IgG4, or CD83+ microglia might ward off dementia.
“We’re preparing to conduct a clinical trial to evaluate whether careful use of existing antivirals might be clinically helpful in preventing or slowing progression of CD83+ associated Alzheimer’s disease,” Readhead said in an interview.
The study was published on December 19, 2024, in Alzheimer’s & Dementia.
Vagus Nerve a Potential Pathway?
CMV is a common virus. In the United States, nearly one in three children are already infected with CMV by age 5 years. Over half the adults have been infected with CMV by age 40 years, the Centers for Disease Control and Prevention reported.
It is typically passed through bodily fluids and spread only when the virus is active. It’s not considered a sexually transmitted disease.
Compared with other IgG subclasses, IgG4 is believed to be a less inflammatory, and therefore less damaging, immune response. But this response may be less effective at clearing infections and allow invasion of HCMV into the brain.
The researchers previously found a CD83+ microglial subtype in the SFG of 47% of brain donors with AD vs 25% of unaffected control individuals. They reported this subtype is associated with increased IgG4 in the TC.
The current analysis extends investigations of the potential etiology and clinicopathologic relevance of CD83+ microglia in the context of AD.
Researchers conducted experiments using donated tissue samples from deceased patients with AD and control individuals. Sources for these samples included the Banner cohort, for whom classifications for the presence of CD83+ microglia were available, as were tissue samples from the SFG, TC, and vagus nerve, and the Religious Orders Study and Rush Memory and Aging Project (ROSMAP), in which participants without known dementia are evaluated annually.
From the Banner cohort, researchers completed immunohistochemistry (IHC) studies on 34 SFG samples (21 AD and 13 control individuals) and included 25 TC samples (13 AD and 12 control individuals) and 8 vagal nerve samples (6 AD and 2 control individuals) in the study. From the ROSMAP cohort, they completed IHC studies on 27 prefrontal cortex samples from individuals with AD.
They carefully selected these samples to ensure matching for critical factors such as postmortem interval, age, and sex, as well as other relevant covariates, said the authors.
The study verified that CD83+ microglia are associated with IgG4 and HCMV in the TC and showed a significant association between CD83+ microglia and IgG4 immunoreactivity in the TC.
Investigators confirmed HCMV positivity in all nine CD83+ TC samples evaluated and in one CD83– TC sample, indicating a strong positive association between HCMV within the TC and CD83+ microglia within the SFG.
HCMV IgG seroprevalence is common, varies by age and comorbidity, and is present in 79% of 85-year-olds, the investigators noted. “Despite this, we note that HCMV presence in the TC was not ubiquitous and was significantly associated with CD83+ microglia and HCMV in the SFG,” they wrote.
This observation, they added, “may help reconcile how a common pathogen might contribute to a disease that most individuals do not develop.”
The experiments also uncovered increased anti-HCMV IgG4 in the CSF and evidence of HCMV and IgG4 in the vagus nerve.
“Overall, the histochemical staining patterns observed in TC, SFG, and vagus nerve of CD83+ subjects are consistent with active HCMV infection,” the investigators wrote. “Taken together, these results indicate a multiorgan presence of IgG4 and HCMV in subjects with CD83+ microglia within the SFG,” they added.
Accelerated AD Pathology
The team showed HCMV infection accelerates production of two pathologic features of AD — amyloid beta (Abeta) and tau — and causes neuronal death. “We observed high, positive correlations between the abundance of HCMV, and both Abeta42 and pTau-212,” they wrote.
As HCMV histochemistry is consistent with an active HCMV infection, the findings “may indicate an opportunity for the administration of antiviral therapy in subjects with AD and biomarker evidence of HCMV, IgG4, or CD83+ microglia,” they added.
In addition to planning a clinical trial of existing antivirals, the research team is developing a blood test that can help identify patients with an active HCMV infection who might benefit from such an intervention, said Readhead.
But he emphasized that the research is still in its infancy. “Our study is best understood as a series of interesting scientific findings that warrant further exploration, replication, and validation in additional study populations.”
Although it’s too early for the study to impact practice, “we’re motivated to understand whether these findings have implications for clinical care,” he added.
Tipping the Balance
A number of experts have weighed in on the research via the Science Media Center, an independent forum featuring the voices and views on science news from experts in the field.
Andrew Doig, PhD, professor, Division of Neuroscience, University of Manchester in England, said the new work supports the hypothesis that HCMV might be a trigger that tips the balance from a healthy brain to one with dementia. “If so, antiviral drugs against HCMV might be beneficial in reducing the risk of AD.”
Doig noted newly approved drugs for AD are expensive, provide only a small benefit, and have significant risks, such as causing brain hemorrhages. “Antiviral drugs are an attractive alternative that are well worth exploring.”
Richard Oakley, PhD, associate director of research and innovation, Alzheimer’s Society, cautioned the study only established a connection and didn’t directly show the virus leads to AD. “Also, the virus is not found in the brain of everyone with Alzheimer’s disease, the most common form of dementia.”
The significance of the new findings is “far from clear,” commented William McEwan, PhD, group leader at the UK Dementia Research Institute at Cambridge, England. “The study does not address how common this infection is in people without Alzheimer’s and therefore cannot by itself suggest that HCMV infection, or the associated immune response, is a driver of disease.”
The experts agreed follow-up research is needed to confirm these new findings and understand what they mean.
The study received support from the National Institute on Aging, National Institutes of Health, Global Lyme Alliance, National Institute of Neurological Disorders and Stroke, Arizona Alzheimer’s Consortium, The Benter Foundation, and NOMIS Stiftung. Readhead is a coinventor on a patent application for an IgG4-based peripheral biomarker for the detection of CD83+ microglia. Doig is a founder, director, and consultant for PharmaKure, which works on AD drugs and diagnostics, although not on viruses. He has cowritten a review on Viral Involvement in Alzheimer’s Disease. McEwan reported receiving research funding from Takeda Pharmaceuticals and is a founder and consultant to Trimtech Therapeutics.
A version of this article first appeared on Medscape.com.
Researchers are gaining new insight into the relationship between the human cytomegalovirus (HCMV), a common herpes virus found in the gut, and the immune response associated with CD83 antibody in some individuals with Alzheimer’s disease (AD).
Using tissue samples from deceased donors with AD, the study showed CD83-positive (CD83+) microglia in the superior frontal gyrus (SFG) are significantly associated with elevated immunoglobulin gamma 4 (IgG4) and HCMV in the transverse colon (TC), increased anti-HCMV IgG4 in the cerebrospinal fluid (CSF), and both HCMV and IgG4 in the SFG and vagus nerve.
“Our results indicate a complex, cross-tissue interaction between HCMV and the host adaptive immune response associated with CD83+ microglia in persons with AD,” noted the investigators, including Benjamin P. Readhead, MBBS, research associate professor, ASU-Banner Neurodegenerative Disease Research Center, Arizona State University, Tempe.
The results suggest antiviral therapy in patients with biomarker evidence of HCMV, IgG4, or CD83+ microglia might ward off dementia.
“We’re preparing to conduct a clinical trial to evaluate whether careful use of existing antivirals might be clinically helpful in preventing or slowing progression of CD83+ associated Alzheimer’s disease,” Readhead said in an interview.
The study was published on December 19, 2024, in Alzheimer’s & Dementia.
Vagus Nerve a Potential Pathway?
CMV is a common virus. In the United States, nearly one in three children are already infected with CMV by age 5 years. Over half the adults have been infected with CMV by age 40 years, the Centers for Disease Control and Prevention reported.
It is typically passed through bodily fluids and spread only when the virus is active. It’s not considered a sexually transmitted disease.
Compared with other IgG subclasses, IgG4 is believed to be a less inflammatory, and therefore less damaging, immune response. But this response may be less effective at clearing infections and allow invasion of HCMV into the brain.
The researchers previously found a CD83+ microglial subtype in the SFG of 47% of brain donors with AD vs 25% of unaffected control individuals. They reported this subtype is associated with increased IgG4 in the TC.
The current analysis extends investigations of the potential etiology and clinicopathologic relevance of CD83+ microglia in the context of AD.
Researchers conducted experiments using donated tissue samples from deceased patients with AD and control individuals. Sources for these samples included the Banner cohort, for whom classifications for the presence of CD83+ microglia were available, as were tissue samples from the SFG, TC, and vagus nerve, and the Religious Orders Study and Rush Memory and Aging Project (ROSMAP), in which participants without known dementia are evaluated annually.
From the Banner cohort, researchers completed immunohistochemistry (IHC) studies on 34 SFG samples (21 AD and 13 control individuals) and included 25 TC samples (13 AD and 12 control individuals) and 8 vagal nerve samples (6 AD and 2 control individuals) in the study. From the ROSMAP cohort, they completed IHC studies on 27 prefrontal cortex samples from individuals with AD.
They carefully selected these samples to ensure matching for critical factors such as postmortem interval, age, and sex, as well as other relevant covariates, said the authors.
The study verified that CD83+ microglia are associated with IgG4 and HCMV in the TC and showed a significant association between CD83+ microglia and IgG4 immunoreactivity in the TC.
Investigators confirmed HCMV positivity in all nine CD83+ TC samples evaluated and in one CD83– TC sample, indicating a strong positive association between HCMV within the TC and CD83+ microglia within the SFG.
HCMV IgG seroprevalence is common, varies by age and comorbidity, and is present in 79% of 85-year-olds, the investigators noted. “Despite this, we note that HCMV presence in the TC was not ubiquitous and was significantly associated with CD83+ microglia and HCMV in the SFG,” they wrote.
This observation, they added, “may help reconcile how a common pathogen might contribute to a disease that most individuals do not develop.”
The experiments also uncovered increased anti-HCMV IgG4 in the CSF and evidence of HCMV and IgG4 in the vagus nerve.
“Overall, the histochemical staining patterns observed in TC, SFG, and vagus nerve of CD83+ subjects are consistent with active HCMV infection,” the investigators wrote. “Taken together, these results indicate a multiorgan presence of IgG4 and HCMV in subjects with CD83+ microglia within the SFG,” they added.
Accelerated AD Pathology
The team showed HCMV infection accelerates production of two pathologic features of AD — amyloid beta (Abeta) and tau — and causes neuronal death. “We observed high, positive correlations between the abundance of HCMV, and both Abeta42 and pTau-212,” they wrote.
As HCMV histochemistry is consistent with an active HCMV infection, the findings “may indicate an opportunity for the administration of antiviral therapy in subjects with AD and biomarker evidence of HCMV, IgG4, or CD83+ microglia,” they added.
In addition to planning a clinical trial of existing antivirals, the research team is developing a blood test that can help identify patients with an active HCMV infection who might benefit from such an intervention, said Readhead.
But he emphasized that the research is still in its infancy. “Our study is best understood as a series of interesting scientific findings that warrant further exploration, replication, and validation in additional study populations.”
Although it’s too early for the study to impact practice, “we’re motivated to understand whether these findings have implications for clinical care,” he added.
Tipping the Balance
A number of experts have weighed in on the research via the Science Media Center, an independent forum featuring the voices and views on science news from experts in the field.
Andrew Doig, PhD, professor, Division of Neuroscience, University of Manchester in England, said the new work supports the hypothesis that HCMV might be a trigger that tips the balance from a healthy brain to one with dementia. “If so, antiviral drugs against HCMV might be beneficial in reducing the risk of AD.”
Doig noted newly approved drugs for AD are expensive, provide only a small benefit, and have significant risks, such as causing brain hemorrhages. “Antiviral drugs are an attractive alternative that are well worth exploring.”
Richard Oakley, PhD, associate director of research and innovation, Alzheimer’s Society, cautioned the study only established a connection and didn’t directly show the virus leads to AD. “Also, the virus is not found in the brain of everyone with Alzheimer’s disease, the most common form of dementia.”
The significance of the new findings is “far from clear,” commented William McEwan, PhD, group leader at the UK Dementia Research Institute at Cambridge, England. “The study does not address how common this infection is in people without Alzheimer’s and therefore cannot by itself suggest that HCMV infection, or the associated immune response, is a driver of disease.”
The experts agreed follow-up research is needed to confirm these new findings and understand what they mean.
The study received support from the National Institute on Aging, National Institutes of Health, Global Lyme Alliance, National Institute of Neurological Disorders and Stroke, Arizona Alzheimer’s Consortium, The Benter Foundation, and NOMIS Stiftung. Readhead is a coinventor on a patent application for an IgG4-based peripheral biomarker for the detection of CD83+ microglia. Doig is a founder, director, and consultant for PharmaKure, which works on AD drugs and diagnostics, although not on viruses. He has cowritten a review on Viral Involvement in Alzheimer’s Disease. McEwan reported receiving research funding from Takeda Pharmaceuticals and is a founder and consultant to Trimtech Therapeutics.
A version of this article first appeared on Medscape.com.
Researchers are gaining new insight into the relationship between the human cytomegalovirus (HCMV), a common herpes virus found in the gut, and the immune response associated with CD83 antibody in some individuals with Alzheimer’s disease (AD).
Using tissue samples from deceased donors with AD, the study showed CD83-positive (CD83+) microglia in the superior frontal gyrus (SFG) are significantly associated with elevated immunoglobulin gamma 4 (IgG4) and HCMV in the transverse colon (TC), increased anti-HCMV IgG4 in the cerebrospinal fluid (CSF), and both HCMV and IgG4 in the SFG and vagus nerve.
“Our results indicate a complex, cross-tissue interaction between HCMV and the host adaptive immune response associated with CD83+ microglia in persons with AD,” noted the investigators, including Benjamin P. Readhead, MBBS, research associate professor, ASU-Banner Neurodegenerative Disease Research Center, Arizona State University, Tempe.
The results suggest antiviral therapy in patients with biomarker evidence of HCMV, IgG4, or CD83+ microglia might ward off dementia.
“We’re preparing to conduct a clinical trial to evaluate whether careful use of existing antivirals might be clinically helpful in preventing or slowing progression of CD83+ associated Alzheimer’s disease,” Readhead said in an interview.
The study was published on December 19, 2024, in Alzheimer’s & Dementia.
Vagus Nerve a Potential Pathway?
CMV is a common virus. In the United States, nearly one in three children are already infected with CMV by age 5 years. Over half the adults have been infected with CMV by age 40 years, the Centers for Disease Control and Prevention reported.
It is typically passed through bodily fluids and spread only when the virus is active. It’s not considered a sexually transmitted disease.
Compared with other IgG subclasses, IgG4 is believed to be a less inflammatory, and therefore less damaging, immune response. But this response may be less effective at clearing infections and allow invasion of HCMV into the brain.
The researchers previously found a CD83+ microglial subtype in the SFG of 47% of brain donors with AD vs 25% of unaffected control individuals. They reported this subtype is associated with increased IgG4 in the TC.
The current analysis extends investigations of the potential etiology and clinicopathologic relevance of CD83+ microglia in the context of AD.
Researchers conducted experiments using donated tissue samples from deceased patients with AD and control individuals. Sources for these samples included the Banner cohort, for whom classifications for the presence of CD83+ microglia were available, as were tissue samples from the SFG, TC, and vagus nerve, and the Religious Orders Study and Rush Memory and Aging Project (ROSMAP), in which participants without known dementia are evaluated annually.
From the Banner cohort, researchers completed immunohistochemistry (IHC) studies on 34 SFG samples (21 AD and 13 control individuals) and included 25 TC samples (13 AD and 12 control individuals) and 8 vagal nerve samples (6 AD and 2 control individuals) in the study. From the ROSMAP cohort, they completed IHC studies on 27 prefrontal cortex samples from individuals with AD.
They carefully selected these samples to ensure matching for critical factors such as postmortem interval, age, and sex, as well as other relevant covariates, said the authors.
The study verified that CD83+ microglia are associated with IgG4 and HCMV in the TC and showed a significant association between CD83+ microglia and IgG4 immunoreactivity in the TC.
Investigators confirmed HCMV positivity in all nine CD83+ TC samples evaluated and in one CD83– TC sample, indicating a strong positive association between HCMV within the TC and CD83+ microglia within the SFG.
HCMV IgG seroprevalence is common, varies by age and comorbidity, and is present in 79% of 85-year-olds, the investigators noted. “Despite this, we note that HCMV presence in the TC was not ubiquitous and was significantly associated with CD83+ microglia and HCMV in the SFG,” they wrote.
This observation, they added, “may help reconcile how a common pathogen might contribute to a disease that most individuals do not develop.”
The experiments also uncovered increased anti-HCMV IgG4 in the CSF and evidence of HCMV and IgG4 in the vagus nerve.
“Overall, the histochemical staining patterns observed in TC, SFG, and vagus nerve of CD83+ subjects are consistent with active HCMV infection,” the investigators wrote. “Taken together, these results indicate a multiorgan presence of IgG4 and HCMV in subjects with CD83+ microglia within the SFG,” they added.
Accelerated AD Pathology
The team showed HCMV infection accelerates production of two pathologic features of AD — amyloid beta (Abeta) and tau — and causes neuronal death. “We observed high, positive correlations between the abundance of HCMV, and both Abeta42 and pTau-212,” they wrote.
As HCMV histochemistry is consistent with an active HCMV infection, the findings “may indicate an opportunity for the administration of antiviral therapy in subjects with AD and biomarker evidence of HCMV, IgG4, or CD83+ microglia,” they added.
In addition to planning a clinical trial of existing antivirals, the research team is developing a blood test that can help identify patients with an active HCMV infection who might benefit from such an intervention, said Readhead.
But he emphasized that the research is still in its infancy. “Our study is best understood as a series of interesting scientific findings that warrant further exploration, replication, and validation in additional study populations.”
Although it’s too early for the study to impact practice, “we’re motivated to understand whether these findings have implications for clinical care,” he added.
Tipping the Balance
A number of experts have weighed in on the research via the Science Media Center, an independent forum featuring the voices and views on science news from experts in the field.
Andrew Doig, PhD, professor, Division of Neuroscience, University of Manchester in England, said the new work supports the hypothesis that HCMV might be a trigger that tips the balance from a healthy brain to one with dementia. “If so, antiviral drugs against HCMV might be beneficial in reducing the risk of AD.”
Doig noted newly approved drugs for AD are expensive, provide only a small benefit, and have significant risks, such as causing brain hemorrhages. “Antiviral drugs are an attractive alternative that are well worth exploring.”
Richard Oakley, PhD, associate director of research and innovation, Alzheimer’s Society, cautioned the study only established a connection and didn’t directly show the virus leads to AD. “Also, the virus is not found in the brain of everyone with Alzheimer’s disease, the most common form of dementia.”
The significance of the new findings is “far from clear,” commented William McEwan, PhD, group leader at the UK Dementia Research Institute at Cambridge, England. “The study does not address how common this infection is in people without Alzheimer’s and therefore cannot by itself suggest that HCMV infection, or the associated immune response, is a driver of disease.”
The experts agreed follow-up research is needed to confirm these new findings and understand what they mean.
The study received support from the National Institute on Aging, National Institutes of Health, Global Lyme Alliance, National Institute of Neurological Disorders and Stroke, Arizona Alzheimer’s Consortium, The Benter Foundation, and NOMIS Stiftung. Readhead is a coinventor on a patent application for an IgG4-based peripheral biomarker for the detection of CD83+ microglia. Doig is a founder, director, and consultant for PharmaKure, which works on AD drugs and diagnostics, although not on viruses. He has cowritten a review on Viral Involvement in Alzheimer’s Disease. McEwan reported receiving research funding from Takeda Pharmaceuticals and is a founder and consultant to Trimtech Therapeutics.
A version of this article first appeared on Medscape.com.
FROM ALZHEIMER’S & DEMENTIA
Study Supports Pediatric Concussion Management Approach
“With that result, it means we don’t need to change management protocols” depending on the cause of the concussion, study author Andrée-Anne Ledoux, PhD, a scientist at Children’s Hospital of Eastern Ontario Research Institute in Ottawa, Ontario, Canada, said in an interview. “That’s kind of good news. We’re applying the right management protocols with them.”
The data were published on December 4 in JAMA Network Open.
Secondary Analysis
The results stem from a planned secondary analysis of the prospective Predicting and Preventing Postconcussive Problems in Pediatrics study. Conducted from August 2013 to June 2015 at nine pediatric emergency departments in Canada, it included children of different ages (5 to < 18 years), genders, demographic characteristics, and comorbidities. All participants had a concussion.
The secondary analysis focused on study participants who were aged 5-12 years and had presented within 48 hours of injury. The primary outcome was symptom change, which was defined as current ratings minus preinjury ratings, across time (1, 2, 4, 8, and 12 weeks), measured using the Post-Concussion Symptom Inventory.
No significant differences in postinjury recovery curves were found between participants with sport-related concussions (SRC) and those with non-SRC. The latter injuries resulted from causes such as falls and objects dropped on heads. SRC and non-SRC showed a nonlinear association with time, with symptoms decreasing over time.
Perhaps surprisingly, the researchers also reported a higher rate of persisting symptoms after concussion (PSAC) following limited contact sports than following contact sports such as hockey, soccer, rugby, lacrosse, and football. Limited contact sports include activities such as bicycling, horseback riding, tobogganing, gymnastics, and cheerleading.
This finding suggests that the management of SRC may not require distinct strategies based on sports classification, the researchers wrote. “Instead, it may be more appropriate for clinicians to consider the specific dynamics of the activity, such as velocity and risk of falls from heights. This nuanced perspective can aid in assessing the likelihood of persisting symptoms.” The researchers urged more investigation of this question. “A larger sample with more information on injury height and velocity would be required to confirm whether an association exists.”
In addition, the researchers cited guidelines that include a recommendation for a gradual return to low to moderate physical and cognitive activity starting 24-48 hours after a concussion at a level that does not result in recurrence or exacerbation of symptoms.
“Children do need to return to their lives. They need to return to school,” said Ledoux. “They can have accommodations while they return to school, but just returning to school has huge benefits because you’re reintegrating the child into their typical lifestyle and socialization as well.”
A potential limitation of the study was its reliance on participants who had been seen in emergency departments and thus may have been experiencing more intense symptoms than those seen elsewhere.
The researchers also excluded cases of concussion resulting from assaults and motor vehicle crashes. This decision may explain why they didn’t reproduce the previous observation that patients with SRC tended to recover faster than those with concussions from other causes.
Injuries resulting from assaults and motor vehicle crashes can involve damage beyond concussions, Ledoux said. Including these cases would not allow for an apples-to-apples comparison of SRC and non-SRC.
‘Don’t Cocoon Kids’
The authors of an accompanying editorial wrote that the researchers had done “a beautiful job highlighting this important nuance.” Noncontact sports with seemingly little risk “actually carry substantial risks when one imagines the high-impact forces that can occur with a fall from height, albeit rare,” Scott Zuckerman, MD, MPH, assistant professor of neurological surgery at Vanderbilt University Medical Center, Nashville, Tennessee, and colleagues wrote.
The new analysis suggests a need to rethink a “somewhat archaic way of classifying sport risk, which may oversimplify how we categorize risk of brain and spine injuries.”
The commentary also noted how the researchers used the term PSAC to describe lingering symptoms instead of more widely used terms like “persistent postconcussive symptoms” or “postconcussive syndrome.”
“These traditional terms often connote a permanent syndrome or assumption that the concussion itself is solely responsible for 100% of symptoms, which can be harmful to a patient’s recovery,” the editorialists wrote. “Conversely, PSAC offers room for the clinician to discuss how other causes may be maintaining, magnifying, or mimicking concussion symptoms.”
Commenting on the findings, Richard Figler, MD, an orthopedic surgeon at the Cleveland Clinic, Cleveland, praised the researchers for addressing concussion in younger children, a field in which little research has been conducted. The research supports the current approaches to treatment. The approach has shifted toward easing children quickly and safely back into normal routines. “We don’t cocoon kids. We don’t send them to dark rooms,” Figler added.
He also commended the researchers’ decision to examine data about concussions linked to limited contact sports. In contact sports, participants may be more likely to anticipate and prepare for a hit. That’s not the case with injuries sustained in limited contact sports.
“Dodgeball is basically a sucker punch. That’s why these kids have so many concussions,” said Figler. “They typically don’t see the ball coming, or they can’t get out of the way, and they can’t tense themselves to take that blow.”
The Predicting and Preventing Postconcussive Problems in Pediatrics study was funded by the Canadian Institutes of Health Research and the Canadian Institutes of Health Research-Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team. Ledoux reported receiving grants from the Children’s Hospital of Eastern Ontario Foundation, Ontario Brain Institute, and University of Ottawa Brain and Mind Research Institute. She received nonfinancial support from Mobio Interactive outside the submitted work. Zuckerman reported receiving personal fees from the National Football League and Medtronic outside the submitted work. Figler had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
“With that result, it means we don’t need to change management protocols” depending on the cause of the concussion, study author Andrée-Anne Ledoux, PhD, a scientist at Children’s Hospital of Eastern Ontario Research Institute in Ottawa, Ontario, Canada, said in an interview. “That’s kind of good news. We’re applying the right management protocols with them.”
The data were published on December 4 in JAMA Network Open.
Secondary Analysis
The results stem from a planned secondary analysis of the prospective Predicting and Preventing Postconcussive Problems in Pediatrics study. Conducted from August 2013 to June 2015 at nine pediatric emergency departments in Canada, it included children of different ages (5 to < 18 years), genders, demographic characteristics, and comorbidities. All participants had a concussion.
The secondary analysis focused on study participants who were aged 5-12 years and had presented within 48 hours of injury. The primary outcome was symptom change, which was defined as current ratings minus preinjury ratings, across time (1, 2, 4, 8, and 12 weeks), measured using the Post-Concussion Symptom Inventory.
No significant differences in postinjury recovery curves were found between participants with sport-related concussions (SRC) and those with non-SRC. The latter injuries resulted from causes such as falls and objects dropped on heads. SRC and non-SRC showed a nonlinear association with time, with symptoms decreasing over time.
Perhaps surprisingly, the researchers also reported a higher rate of persisting symptoms after concussion (PSAC) following limited contact sports than following contact sports such as hockey, soccer, rugby, lacrosse, and football. Limited contact sports include activities such as bicycling, horseback riding, tobogganing, gymnastics, and cheerleading.
This finding suggests that the management of SRC may not require distinct strategies based on sports classification, the researchers wrote. “Instead, it may be more appropriate for clinicians to consider the specific dynamics of the activity, such as velocity and risk of falls from heights. This nuanced perspective can aid in assessing the likelihood of persisting symptoms.” The researchers urged more investigation of this question. “A larger sample with more information on injury height and velocity would be required to confirm whether an association exists.”
In addition, the researchers cited guidelines that include a recommendation for a gradual return to low to moderate physical and cognitive activity starting 24-48 hours after a concussion at a level that does not result in recurrence or exacerbation of symptoms.
“Children do need to return to their lives. They need to return to school,” said Ledoux. “They can have accommodations while they return to school, but just returning to school has huge benefits because you’re reintegrating the child into their typical lifestyle and socialization as well.”
A potential limitation of the study was its reliance on participants who had been seen in emergency departments and thus may have been experiencing more intense symptoms than those seen elsewhere.
The researchers also excluded cases of concussion resulting from assaults and motor vehicle crashes. This decision may explain why they didn’t reproduce the previous observation that patients with SRC tended to recover faster than those with concussions from other causes.
Injuries resulting from assaults and motor vehicle crashes can involve damage beyond concussions, Ledoux said. Including these cases would not allow for an apples-to-apples comparison of SRC and non-SRC.
‘Don’t Cocoon Kids’
The authors of an accompanying editorial wrote that the researchers had done “a beautiful job highlighting this important nuance.” Noncontact sports with seemingly little risk “actually carry substantial risks when one imagines the high-impact forces that can occur with a fall from height, albeit rare,” Scott Zuckerman, MD, MPH, assistant professor of neurological surgery at Vanderbilt University Medical Center, Nashville, Tennessee, and colleagues wrote.
The new analysis suggests a need to rethink a “somewhat archaic way of classifying sport risk, which may oversimplify how we categorize risk of brain and spine injuries.”
The commentary also noted how the researchers used the term PSAC to describe lingering symptoms instead of more widely used terms like “persistent postconcussive symptoms” or “postconcussive syndrome.”
“These traditional terms often connote a permanent syndrome or assumption that the concussion itself is solely responsible for 100% of symptoms, which can be harmful to a patient’s recovery,” the editorialists wrote. “Conversely, PSAC offers room for the clinician to discuss how other causes may be maintaining, magnifying, or mimicking concussion symptoms.”
Commenting on the findings, Richard Figler, MD, an orthopedic surgeon at the Cleveland Clinic, Cleveland, praised the researchers for addressing concussion in younger children, a field in which little research has been conducted. The research supports the current approaches to treatment. The approach has shifted toward easing children quickly and safely back into normal routines. “We don’t cocoon kids. We don’t send them to dark rooms,” Figler added.
He also commended the researchers’ decision to examine data about concussions linked to limited contact sports. In contact sports, participants may be more likely to anticipate and prepare for a hit. That’s not the case with injuries sustained in limited contact sports.
“Dodgeball is basically a sucker punch. That’s why these kids have so many concussions,” said Figler. “They typically don’t see the ball coming, or they can’t get out of the way, and they can’t tense themselves to take that blow.”
The Predicting and Preventing Postconcussive Problems in Pediatrics study was funded by the Canadian Institutes of Health Research and the Canadian Institutes of Health Research-Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team. Ledoux reported receiving grants from the Children’s Hospital of Eastern Ontario Foundation, Ontario Brain Institute, and University of Ottawa Brain and Mind Research Institute. She received nonfinancial support from Mobio Interactive outside the submitted work. Zuckerman reported receiving personal fees from the National Football League and Medtronic outside the submitted work. Figler had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
“With that result, it means we don’t need to change management protocols” depending on the cause of the concussion, study author Andrée-Anne Ledoux, PhD, a scientist at Children’s Hospital of Eastern Ontario Research Institute in Ottawa, Ontario, Canada, said in an interview. “That’s kind of good news. We’re applying the right management protocols with them.”
The data were published on December 4 in JAMA Network Open.
Secondary Analysis
The results stem from a planned secondary analysis of the prospective Predicting and Preventing Postconcussive Problems in Pediatrics study. Conducted from August 2013 to June 2015 at nine pediatric emergency departments in Canada, it included children of different ages (5 to < 18 years), genders, demographic characteristics, and comorbidities. All participants had a concussion.
The secondary analysis focused on study participants who were aged 5-12 years and had presented within 48 hours of injury. The primary outcome was symptom change, which was defined as current ratings minus preinjury ratings, across time (1, 2, 4, 8, and 12 weeks), measured using the Post-Concussion Symptom Inventory.
No significant differences in postinjury recovery curves were found between participants with sport-related concussions (SRC) and those with non-SRC. The latter injuries resulted from causes such as falls and objects dropped on heads. SRC and non-SRC showed a nonlinear association with time, with symptoms decreasing over time.
Perhaps surprisingly, the researchers also reported a higher rate of persisting symptoms after concussion (PSAC) following limited contact sports than following contact sports such as hockey, soccer, rugby, lacrosse, and football. Limited contact sports include activities such as bicycling, horseback riding, tobogganing, gymnastics, and cheerleading.
This finding suggests that the management of SRC may not require distinct strategies based on sports classification, the researchers wrote. “Instead, it may be more appropriate for clinicians to consider the specific dynamics of the activity, such as velocity and risk of falls from heights. This nuanced perspective can aid in assessing the likelihood of persisting symptoms.” The researchers urged more investigation of this question. “A larger sample with more information on injury height and velocity would be required to confirm whether an association exists.”
In addition, the researchers cited guidelines that include a recommendation for a gradual return to low to moderate physical and cognitive activity starting 24-48 hours after a concussion at a level that does not result in recurrence or exacerbation of symptoms.
“Children do need to return to their lives. They need to return to school,” said Ledoux. “They can have accommodations while they return to school, but just returning to school has huge benefits because you’re reintegrating the child into their typical lifestyle and socialization as well.”
A potential limitation of the study was its reliance on participants who had been seen in emergency departments and thus may have been experiencing more intense symptoms than those seen elsewhere.
The researchers also excluded cases of concussion resulting from assaults and motor vehicle crashes. This decision may explain why they didn’t reproduce the previous observation that patients with SRC tended to recover faster than those with concussions from other causes.
Injuries resulting from assaults and motor vehicle crashes can involve damage beyond concussions, Ledoux said. Including these cases would not allow for an apples-to-apples comparison of SRC and non-SRC.
‘Don’t Cocoon Kids’
The authors of an accompanying editorial wrote that the researchers had done “a beautiful job highlighting this important nuance.” Noncontact sports with seemingly little risk “actually carry substantial risks when one imagines the high-impact forces that can occur with a fall from height, albeit rare,” Scott Zuckerman, MD, MPH, assistant professor of neurological surgery at Vanderbilt University Medical Center, Nashville, Tennessee, and colleagues wrote.
The new analysis suggests a need to rethink a “somewhat archaic way of classifying sport risk, which may oversimplify how we categorize risk of brain and spine injuries.”
The commentary also noted how the researchers used the term PSAC to describe lingering symptoms instead of more widely used terms like “persistent postconcussive symptoms” or “postconcussive syndrome.”
“These traditional terms often connote a permanent syndrome or assumption that the concussion itself is solely responsible for 100% of symptoms, which can be harmful to a patient’s recovery,” the editorialists wrote. “Conversely, PSAC offers room for the clinician to discuss how other causes may be maintaining, magnifying, or mimicking concussion symptoms.”
Commenting on the findings, Richard Figler, MD, an orthopedic surgeon at the Cleveland Clinic, Cleveland, praised the researchers for addressing concussion in younger children, a field in which little research has been conducted. The research supports the current approaches to treatment. The approach has shifted toward easing children quickly and safely back into normal routines. “We don’t cocoon kids. We don’t send them to dark rooms,” Figler added.
He also commended the researchers’ decision to examine data about concussions linked to limited contact sports. In contact sports, participants may be more likely to anticipate and prepare for a hit. That’s not the case with injuries sustained in limited contact sports.
“Dodgeball is basically a sucker punch. That’s why these kids have so many concussions,” said Figler. “They typically don’t see the ball coming, or they can’t get out of the way, and they can’t tense themselves to take that blow.”
The Predicting and Preventing Postconcussive Problems in Pediatrics study was funded by the Canadian Institutes of Health Research and the Canadian Institutes of Health Research-Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team. Ledoux reported receiving grants from the Children’s Hospital of Eastern Ontario Foundation, Ontario Brain Institute, and University of Ottawa Brain and Mind Research Institute. She received nonfinancial support from Mobio Interactive outside the submitted work. Zuckerman reported receiving personal fees from the National Football League and Medtronic outside the submitted work. Figler had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Total Intravenous Anesthesia Enables Earlier Facial Nerve Monitoring Than Sevoflurane in Ear Surgery
TOPLINE:
Total intravenous anesthesia (TIVA) enables earlier intraoperative monitoring of facial nerve activity than sevoflurane anesthesia during ear surgery, with reduced patient-ventilator dyssynchrony and fewer requirements for postoperative antiemetics.
METHODOLOGY:
- Researchers evaluated the difference in the timeliness of intraoperative monitoring of facial nerve activity during ear surgery with TIVA vs sevoflurane anesthesia.
- They included 98 patients aged 18-74 years undergoing ear surgery between November 2021 and November 2022; patients were randomly assigned to receive either TIVA or sevoflurane during the procedure. Of these, 92 were included in the final analysis.
- Neuromuscular function was monitored quantitatively throughout anesthesia with train-of-four counts and train-of-four ratios.
- The time from the administration of rocuronium to the start of facial nerve monitoring was recorded.
- The primary outcome measure focused on the recovery index, defined as the time interval between a train-of-four ratio of 0.25 and 0.75; the key secondary outcome was the time to reach a train-of-four ratio of 0.25 from rocuronium administration.
TAKEAWAY:
- The time to reach a train-of-four ratio of 0.25 was achieved earlier with TIVA than with sevoflurane (34 minutes vs 51 minutes; P < .001).
- Patient-ventilator dyssynchrony occurred less frequently in the TIVA group than in the sevoflurane group (15% vs 39%; P = .01).
- Postoperative requests for antiemetics were less frequent in the TIVA group than in the sevoflurane group (2% vs 17%; P = .03).
IN PRACTICE:
“We suggest that TIVA may be a better choice than sevoflurane anesthesia to meet an earlier request” for intraoperative facial nerve monitoring by surgeons, the study authors wrote.
SOURCE:
The study was led by Yu Jeong Bang, MD, of the Department of Anesthesiology and Pain Medicine at Sungkyunkwan University School of Medicine, in Seoul, Republic of Korea. It was published online on November 27, 2024, in The Canadian Journal of Anesthesia.
LIMITATIONS:
A careful interpretation of results may be necessary when clinicians use balanced anesthesia, such as sevoflurane with adjuvants like opioids or nonopioids. The feasibility of intraoperative facial nerve monitoring was decided by the surgeon during surgery, and the lowest stimulation intensity threshold for electromyography amplitude was not detected, as it was not the focus of this study. Although patients requiring intraoperative facial nerve monitoring during ear surgery were enrolled, some did not undergo the procedure based on the surgeon’s judgment.
DISCLOSURES:
This study did not receive any funding. The authors disclosed no relevant conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Total intravenous anesthesia (TIVA) enables earlier intraoperative monitoring of facial nerve activity than sevoflurane anesthesia during ear surgery, with reduced patient-ventilator dyssynchrony and fewer requirements for postoperative antiemetics.
METHODOLOGY:
- Researchers evaluated the difference in the timeliness of intraoperative monitoring of facial nerve activity during ear surgery with TIVA vs sevoflurane anesthesia.
- They included 98 patients aged 18-74 years undergoing ear surgery between November 2021 and November 2022; patients were randomly assigned to receive either TIVA or sevoflurane during the procedure. Of these, 92 were included in the final analysis.
- Neuromuscular function was monitored quantitatively throughout anesthesia with train-of-four counts and train-of-four ratios.
- The time from the administration of rocuronium to the start of facial nerve monitoring was recorded.
- The primary outcome measure focused on the recovery index, defined as the time interval between a train-of-four ratio of 0.25 and 0.75; the key secondary outcome was the time to reach a train-of-four ratio of 0.25 from rocuronium administration.
TAKEAWAY:
- The time to reach a train-of-four ratio of 0.25 was achieved earlier with TIVA than with sevoflurane (34 minutes vs 51 minutes; P < .001).
- Patient-ventilator dyssynchrony occurred less frequently in the TIVA group than in the sevoflurane group (15% vs 39%; P = .01).
- Postoperative requests for antiemetics were less frequent in the TIVA group than in the sevoflurane group (2% vs 17%; P = .03).
IN PRACTICE:
“We suggest that TIVA may be a better choice than sevoflurane anesthesia to meet an earlier request” for intraoperative facial nerve monitoring by surgeons, the study authors wrote.
SOURCE:
The study was led by Yu Jeong Bang, MD, of the Department of Anesthesiology and Pain Medicine at Sungkyunkwan University School of Medicine, in Seoul, Republic of Korea. It was published online on November 27, 2024, in The Canadian Journal of Anesthesia.
LIMITATIONS:
A careful interpretation of results may be necessary when clinicians use balanced anesthesia, such as sevoflurane with adjuvants like opioids or nonopioids. The feasibility of intraoperative facial nerve monitoring was decided by the surgeon during surgery, and the lowest stimulation intensity threshold for electromyography amplitude was not detected, as it was not the focus of this study. Although patients requiring intraoperative facial nerve monitoring during ear surgery were enrolled, some did not undergo the procedure based on the surgeon’s judgment.
DISCLOSURES:
This study did not receive any funding. The authors disclosed no relevant conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Total intravenous anesthesia (TIVA) enables earlier intraoperative monitoring of facial nerve activity than sevoflurane anesthesia during ear surgery, with reduced patient-ventilator dyssynchrony and fewer requirements for postoperative antiemetics.
METHODOLOGY:
- Researchers evaluated the difference in the timeliness of intraoperative monitoring of facial nerve activity during ear surgery with TIVA vs sevoflurane anesthesia.
- They included 98 patients aged 18-74 years undergoing ear surgery between November 2021 and November 2022; patients were randomly assigned to receive either TIVA or sevoflurane during the procedure. Of these, 92 were included in the final analysis.
- Neuromuscular function was monitored quantitatively throughout anesthesia with train-of-four counts and train-of-four ratios.
- The time from the administration of rocuronium to the start of facial nerve monitoring was recorded.
- The primary outcome measure focused on the recovery index, defined as the time interval between a train-of-four ratio of 0.25 and 0.75; the key secondary outcome was the time to reach a train-of-four ratio of 0.25 from rocuronium administration.
TAKEAWAY:
- The time to reach a train-of-four ratio of 0.25 was achieved earlier with TIVA than with sevoflurane (34 minutes vs 51 minutes; P < .001).
- Patient-ventilator dyssynchrony occurred less frequently in the TIVA group than in the sevoflurane group (15% vs 39%; P = .01).
- Postoperative requests for antiemetics were less frequent in the TIVA group than in the sevoflurane group (2% vs 17%; P = .03).
IN PRACTICE:
“We suggest that TIVA may be a better choice than sevoflurane anesthesia to meet an earlier request” for intraoperative facial nerve monitoring by surgeons, the study authors wrote.
SOURCE:
The study was led by Yu Jeong Bang, MD, of the Department of Anesthesiology and Pain Medicine at Sungkyunkwan University School of Medicine, in Seoul, Republic of Korea. It was published online on November 27, 2024, in The Canadian Journal of Anesthesia.
LIMITATIONS:
A careful interpretation of results may be necessary when clinicians use balanced anesthesia, such as sevoflurane with adjuvants like opioids or nonopioids. The feasibility of intraoperative facial nerve monitoring was decided by the surgeon during surgery, and the lowest stimulation intensity threshold for electromyography amplitude was not detected, as it was not the focus of this study. Although patients requiring intraoperative facial nerve monitoring during ear surgery were enrolled, some did not undergo the procedure based on the surgeon’s judgment.
DISCLOSURES:
This study did not receive any funding. The authors disclosed no relevant conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Health Impacts of Micro- and Nanoplastics
In preparation for a future international treaty aimed at reducing plastic pollution, the French Parliamentary Office for the Evaluation of Scientific and Technological Choices presented the conclusions of a public hearing on the impact of plastics on various aspects of human health.
Increased Global Plastic Production
Philippe Bolo, a member of the French Democratic Party and the rapporteur for the public mission on the health impacts of plastics, spoke about the latest round of treaty negotiations, held from November 25 to December 1 in South Korea, attended by leading French and global experts about the impact of plastics on human health.
The hearing highlighted a sharp increase in plastic production. “It has doubled in the last 20 years and is expected to exceed 500 million tons in 2024,” Bolo said. This is about 60 kg per person. According to projections from the Organization for Economic Co-operation and Development, on its current trajectory, plastic production will reach 750 million tons by 2040 and surpass 1 billion tons before 2050, he said.
Minimal Plastic Waste Recycling
Around one third (32%) of plastics are used for packaging. “Therefore, most plastic production is still intended for single-use purposes,” he said. Plastic waste follows a similar growth trajectory, with volumes expected to rise from 360 million tons in 2020 to 617 million tons by 2040 unless action is taken. Very little of this waste is recycled, even in the most countries that are most advanced in terms of collection, sorting, and processing.
In France, for example, in 2018, only 0.6 million tons of the 3.6 million tons of plastic waste produced was truly recycled. This is less than one fifth (17%). Globally, less than 10% of plastic waste is recycled. In 2020, plastic waste that ended up in the environment represented 81 million tons, or 22% of the total. “Beyond waste, this leads to pollution by microplastics and nanoplastics, resulting from their fragmentation. All environments are affected: Seas, rivers, soils, air, and even living organisms,” Bolo said.
Methodological Challenges
However, measuring the impact of plastics on health faces methodological difficulties due to the wide variety of composition, size, and shape of plastics. Nevertheless, the French Standardization Association (Association Française de Normalisation) has conducted work to establish a characterization standard for microplastics in water, which serves as an international reference.
“It is also very difficult to know what we are ingesting,” Bolo said. “A study conducted in 2019 estimated that the average human absorbs 5 grams of plastics per week, the equivalent of a credit card.» Since then, other studies have revised this estimate downward, but no consensus has been reached.
A recent study across 109 countries, both industrialized and developing, found significant exposure, estimated at 500 mg/d, particularly in Southeast Asian countries, where it was due mainly to seafood consumption.
A study concluded that plastic water bottles contain 240,000 particles per liter, 90% of which are nanoplastics. These nanoparticles can pass through the intestinal barrier to enter the bloodstream and reach several organs including the heart, brain, and placenta, as well as the fetus.
Changes to the Microbiome
Microplastics also accumulate in organs. Thus, the amount of plastic in the lungs increases with age, suggesting that particles may persist in the body without being eliminated. The health consequences of this are still poorly understood, but exposure to plastics appears to cause changes in the composition of the intestinal microbiota. Pathobionts (commensal bacteria with harmful potential) have been found in both adults and children, which could contribute to dysbiosis of the gut microbiome. Furthermore, a decrease in butyrate, a short-chain fatty acid beneficial to health, has been observed in children’s intestines.
Inhaled nanoplastics may disrupt the mucociliary clearance mechanisms of the respiratory system. The toxicity of inhaled plastic particles was demonstrated as early as the 1970s among workers in the flocking industry. Some developed lung function impairments, shortness of breath, inflammation, fibrosis, and even lung cancer. Similar symptoms have been observed in workers in the textile and polyvinyl chloride industries.
A study published recently in The New England Journal of Medicine measured the amount of microplastics collected from carotid plaque of more than 300 patients who had undergone carotid endarterectomy for asymptomatic carotid artery disease. It found a 4.53 times higher risk for the primary endpoint, a composite of myocardial infarction, stroke, and all-cause mortality, among individuals with microplastics and nanoplastics in plaque compared with those without.
Health Affects High
The danger of plastics is also directly linked to the chemical substances they contain. A general scientific review looked at the health impacts of three chemicals used almost exclusively in plastics: Polybromodiphenyl ethers (PBDEs), used as flame retardants in textiles or electronics; bisphenol A (BPA), used in the lining of cans and bottles; and phthalates, particularly diethylhexyl phthalate (DEHP), used to make plastics more flexible.
The review highlighted strong epidemiological evidence linking fetal exposure to PBDEs during pregnancy to low birth weight and later exposure to delayed or impaired cognitive development in children and even a loss of IQ. Statistically significant evidence of disruption of thyroid function in adults was also found.
BPA is linked to genital malformations in female newborns exposed to BPA in utero, type 2 diabetes in adults, insulin resistance, and polycystic ovary syndrome in women. BPA exposure also increases the risk for obesity and hypertension in both children and adults, as well as the risk for cardiovascular disease in adults.
Finally, the review established links between exposure to DEHP and miscarriages, genital malformations in male newborns, delayed or impaired cognitive development in children, loss of IQ, delayed psychomotor development, early puberty in young girls, and endometriosis in young women. DEHP exposure also has multiple effects on cardiometabolic health, including insulin resistance, obesity, and elevated blood pressure.
The economic costs associated with the health impacts of these three substances have been estimated at $675 billion in the United States.
Bolo said that the solution to this plastic pollution is necessarily international. “We need an ambitious and legally binding treaty to reduce plastic production,” he said. “The damage is already done; we need to act to protect human health,” he concluded. The parliamentary office has made nine recommendations to the treaty negotiators.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
In preparation for a future international treaty aimed at reducing plastic pollution, the French Parliamentary Office for the Evaluation of Scientific and Technological Choices presented the conclusions of a public hearing on the impact of plastics on various aspects of human health.
Increased Global Plastic Production
Philippe Bolo, a member of the French Democratic Party and the rapporteur for the public mission on the health impacts of plastics, spoke about the latest round of treaty negotiations, held from November 25 to December 1 in South Korea, attended by leading French and global experts about the impact of plastics on human health.
The hearing highlighted a sharp increase in plastic production. “It has doubled in the last 20 years and is expected to exceed 500 million tons in 2024,” Bolo said. This is about 60 kg per person. According to projections from the Organization for Economic Co-operation and Development, on its current trajectory, plastic production will reach 750 million tons by 2040 and surpass 1 billion tons before 2050, he said.
Minimal Plastic Waste Recycling
Around one third (32%) of plastics are used for packaging. “Therefore, most plastic production is still intended for single-use purposes,” he said. Plastic waste follows a similar growth trajectory, with volumes expected to rise from 360 million tons in 2020 to 617 million tons by 2040 unless action is taken. Very little of this waste is recycled, even in the most countries that are most advanced in terms of collection, sorting, and processing.
In France, for example, in 2018, only 0.6 million tons of the 3.6 million tons of plastic waste produced was truly recycled. This is less than one fifth (17%). Globally, less than 10% of plastic waste is recycled. In 2020, plastic waste that ended up in the environment represented 81 million tons, or 22% of the total. “Beyond waste, this leads to pollution by microplastics and nanoplastics, resulting from their fragmentation. All environments are affected: Seas, rivers, soils, air, and even living organisms,” Bolo said.
Methodological Challenges
However, measuring the impact of plastics on health faces methodological difficulties due to the wide variety of composition, size, and shape of plastics. Nevertheless, the French Standardization Association (Association Française de Normalisation) has conducted work to establish a characterization standard for microplastics in water, which serves as an international reference.
“It is also very difficult to know what we are ingesting,” Bolo said. “A study conducted in 2019 estimated that the average human absorbs 5 grams of plastics per week, the equivalent of a credit card.» Since then, other studies have revised this estimate downward, but no consensus has been reached.
A recent study across 109 countries, both industrialized and developing, found significant exposure, estimated at 500 mg/d, particularly in Southeast Asian countries, where it was due mainly to seafood consumption.
A study concluded that plastic water bottles contain 240,000 particles per liter, 90% of which are nanoplastics. These nanoparticles can pass through the intestinal barrier to enter the bloodstream and reach several organs including the heart, brain, and placenta, as well as the fetus.
Changes to the Microbiome
Microplastics also accumulate in organs. Thus, the amount of plastic in the lungs increases with age, suggesting that particles may persist in the body without being eliminated. The health consequences of this are still poorly understood, but exposure to plastics appears to cause changes in the composition of the intestinal microbiota. Pathobionts (commensal bacteria with harmful potential) have been found in both adults and children, which could contribute to dysbiosis of the gut microbiome. Furthermore, a decrease in butyrate, a short-chain fatty acid beneficial to health, has been observed in children’s intestines.
Inhaled nanoplastics may disrupt the mucociliary clearance mechanisms of the respiratory system. The toxicity of inhaled plastic particles was demonstrated as early as the 1970s among workers in the flocking industry. Some developed lung function impairments, shortness of breath, inflammation, fibrosis, and even lung cancer. Similar symptoms have been observed in workers in the textile and polyvinyl chloride industries.
A study published recently in The New England Journal of Medicine measured the amount of microplastics collected from carotid plaque of more than 300 patients who had undergone carotid endarterectomy for asymptomatic carotid artery disease. It found a 4.53 times higher risk for the primary endpoint, a composite of myocardial infarction, stroke, and all-cause mortality, among individuals with microplastics and nanoplastics in plaque compared with those without.
Health Affects High
The danger of plastics is also directly linked to the chemical substances they contain. A general scientific review looked at the health impacts of three chemicals used almost exclusively in plastics: Polybromodiphenyl ethers (PBDEs), used as flame retardants in textiles or electronics; bisphenol A (BPA), used in the lining of cans and bottles; and phthalates, particularly diethylhexyl phthalate (DEHP), used to make plastics more flexible.
The review highlighted strong epidemiological evidence linking fetal exposure to PBDEs during pregnancy to low birth weight and later exposure to delayed or impaired cognitive development in children and even a loss of IQ. Statistically significant evidence of disruption of thyroid function in adults was also found.
BPA is linked to genital malformations in female newborns exposed to BPA in utero, type 2 diabetes in adults, insulin resistance, and polycystic ovary syndrome in women. BPA exposure also increases the risk for obesity and hypertension in both children and adults, as well as the risk for cardiovascular disease in adults.
Finally, the review established links between exposure to DEHP and miscarriages, genital malformations in male newborns, delayed or impaired cognitive development in children, loss of IQ, delayed psychomotor development, early puberty in young girls, and endometriosis in young women. DEHP exposure also has multiple effects on cardiometabolic health, including insulin resistance, obesity, and elevated blood pressure.
The economic costs associated with the health impacts of these three substances have been estimated at $675 billion in the United States.
Bolo said that the solution to this plastic pollution is necessarily international. “We need an ambitious and legally binding treaty to reduce plastic production,” he said. “The damage is already done; we need to act to protect human health,” he concluded. The parliamentary office has made nine recommendations to the treaty negotiators.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
In preparation for a future international treaty aimed at reducing plastic pollution, the French Parliamentary Office for the Evaluation of Scientific and Technological Choices presented the conclusions of a public hearing on the impact of plastics on various aspects of human health.
Increased Global Plastic Production
Philippe Bolo, a member of the French Democratic Party and the rapporteur for the public mission on the health impacts of plastics, spoke about the latest round of treaty negotiations, held from November 25 to December 1 in South Korea, attended by leading French and global experts about the impact of plastics on human health.
The hearing highlighted a sharp increase in plastic production. “It has doubled in the last 20 years and is expected to exceed 500 million tons in 2024,” Bolo said. This is about 60 kg per person. According to projections from the Organization for Economic Co-operation and Development, on its current trajectory, plastic production will reach 750 million tons by 2040 and surpass 1 billion tons before 2050, he said.
Minimal Plastic Waste Recycling
Around one third (32%) of plastics are used for packaging. “Therefore, most plastic production is still intended for single-use purposes,” he said. Plastic waste follows a similar growth trajectory, with volumes expected to rise from 360 million tons in 2020 to 617 million tons by 2040 unless action is taken. Very little of this waste is recycled, even in the most countries that are most advanced in terms of collection, sorting, and processing.
In France, for example, in 2018, only 0.6 million tons of the 3.6 million tons of plastic waste produced was truly recycled. This is less than one fifth (17%). Globally, less than 10% of plastic waste is recycled. In 2020, plastic waste that ended up in the environment represented 81 million tons, or 22% of the total. “Beyond waste, this leads to pollution by microplastics and nanoplastics, resulting from their fragmentation. All environments are affected: Seas, rivers, soils, air, and even living organisms,” Bolo said.
Methodological Challenges
However, measuring the impact of plastics on health faces methodological difficulties due to the wide variety of composition, size, and shape of plastics. Nevertheless, the French Standardization Association (Association Française de Normalisation) has conducted work to establish a characterization standard for microplastics in water, which serves as an international reference.
“It is also very difficult to know what we are ingesting,” Bolo said. “A study conducted in 2019 estimated that the average human absorbs 5 grams of plastics per week, the equivalent of a credit card.» Since then, other studies have revised this estimate downward, but no consensus has been reached.
A recent study across 109 countries, both industrialized and developing, found significant exposure, estimated at 500 mg/d, particularly in Southeast Asian countries, where it was due mainly to seafood consumption.
A study concluded that plastic water bottles contain 240,000 particles per liter, 90% of which are nanoplastics. These nanoparticles can pass through the intestinal barrier to enter the bloodstream and reach several organs including the heart, brain, and placenta, as well as the fetus.
Changes to the Microbiome
Microplastics also accumulate in organs. Thus, the amount of plastic in the lungs increases with age, suggesting that particles may persist in the body without being eliminated. The health consequences of this are still poorly understood, but exposure to plastics appears to cause changes in the composition of the intestinal microbiota. Pathobionts (commensal bacteria with harmful potential) have been found in both adults and children, which could contribute to dysbiosis of the gut microbiome. Furthermore, a decrease in butyrate, a short-chain fatty acid beneficial to health, has been observed in children’s intestines.
Inhaled nanoplastics may disrupt the mucociliary clearance mechanisms of the respiratory system. The toxicity of inhaled plastic particles was demonstrated as early as the 1970s among workers in the flocking industry. Some developed lung function impairments, shortness of breath, inflammation, fibrosis, and even lung cancer. Similar symptoms have been observed in workers in the textile and polyvinyl chloride industries.
A study published recently in The New England Journal of Medicine measured the amount of microplastics collected from carotid plaque of more than 300 patients who had undergone carotid endarterectomy for asymptomatic carotid artery disease. It found a 4.53 times higher risk for the primary endpoint, a composite of myocardial infarction, stroke, and all-cause mortality, among individuals with microplastics and nanoplastics in plaque compared with those without.
Health Affects High
The danger of plastics is also directly linked to the chemical substances they contain. A general scientific review looked at the health impacts of three chemicals used almost exclusively in plastics: Polybromodiphenyl ethers (PBDEs), used as flame retardants in textiles or electronics; bisphenol A (BPA), used in the lining of cans and bottles; and phthalates, particularly diethylhexyl phthalate (DEHP), used to make plastics more flexible.
The review highlighted strong epidemiological evidence linking fetal exposure to PBDEs during pregnancy to low birth weight and later exposure to delayed or impaired cognitive development in children and even a loss of IQ. Statistically significant evidence of disruption of thyroid function in adults was also found.
BPA is linked to genital malformations in female newborns exposed to BPA in utero, type 2 diabetes in adults, insulin resistance, and polycystic ovary syndrome in women. BPA exposure also increases the risk for obesity and hypertension in both children and adults, as well as the risk for cardiovascular disease in adults.
Finally, the review established links between exposure to DEHP and miscarriages, genital malformations in male newborns, delayed or impaired cognitive development in children, loss of IQ, delayed psychomotor development, early puberty in young girls, and endometriosis in young women. DEHP exposure also has multiple effects on cardiometabolic health, including insulin resistance, obesity, and elevated blood pressure.
The economic costs associated with the health impacts of these three substances have been estimated at $675 billion in the United States.
Bolo said that the solution to this plastic pollution is necessarily international. “We need an ambitious and legally binding treaty to reduce plastic production,” he said. “The damage is already done; we need to act to protect human health,” he concluded. The parliamentary office has made nine recommendations to the treaty negotiators.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
High-Volume Burn Resuscitation Increases Neurologic Risk
TOPLINE:
METHODOLOGY:
- Researchers conducted a single-center review of 5176 patients with burn injuries who were admitted to a verified American Burn Association center (2003-2017); 622 of them underwent head CT within 96 hours of admission, and 83 showed intracranial abnormalities.
- Of 42 patients (mean age, 49.7 years; 80.5% men) who were admitted within 24 hours of burn, 30 patients received < 200 mL/kg and 11 received > 200 mL/kg of total resuscitation fluids, with a median total body surface area (TBSA) of 20.0.
- The primary outcome assessed was the worsening of neurologic findings on imaging related to the volume of the resuscitation fluid administered; the secondary outcomes were the incidence of new or worsening intracranial abnormalities, including hemorrhage, edema, ischemia, or infarction.
TAKEAWAY:
- Neurologic findings worsened in 47.6% patients receiving < 200 mL/kg of fluid resuscitation and 85.7% of those receiving > 200 mL/kg (P =.064).
- Repeat imaging was performed in 21 (70.0%) patients receiving < 200 mL/kg and 7 (63.6%) patients receiving > 200 mL/kg of resuscitation who underwent follow-up imaging.
- The median TBSA was 16.5 in the < 200 mL/kg group and 53.2 in the > 200 mL/kg group (P <.001).
- Intracranial abnormalities were found in 31.3% patients with hemorrhage, 18.8% with worsening edema, and 43.8% with ischemia or infarction.
IN PRACTICE:
“Patients who received over 200 mL/kg of resuscitation had an increased progression of intracranial abnormalities when compared with patients receiving less volume resuscitation,” the authors wrote. “Neurologic changes prompting imaging in burn patients may be undetectable, and our study further highlights the need for routine evaluation with neurologic imaging when undergoing large-volume resuscitations.”
SOURCE:
The study was led by Connor L. Kenney, MD, Brooke Army Medical Center, San Antonio, and was published online on November 07, 2024, in the Journal of Surgical Research.
LIMITATIONS:
Study limitations included a small patient sample and unclear guidelines for obtaining head CT scans, making it difficult to distinguish between trauma-related brain changes and disease progression. Additionally, the study lacked data on hypotensive episodes and long-term neurologic outcomes.
DISCLOSURES:
This study did not receive any specific funding. The authors declared no conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers conducted a single-center review of 5176 patients with burn injuries who were admitted to a verified American Burn Association center (2003-2017); 622 of them underwent head CT within 96 hours of admission, and 83 showed intracranial abnormalities.
- Of 42 patients (mean age, 49.7 years; 80.5% men) who were admitted within 24 hours of burn, 30 patients received < 200 mL/kg and 11 received > 200 mL/kg of total resuscitation fluids, with a median total body surface area (TBSA) of 20.0.
- The primary outcome assessed was the worsening of neurologic findings on imaging related to the volume of the resuscitation fluid administered; the secondary outcomes were the incidence of new or worsening intracranial abnormalities, including hemorrhage, edema, ischemia, or infarction.
TAKEAWAY:
- Neurologic findings worsened in 47.6% patients receiving < 200 mL/kg of fluid resuscitation and 85.7% of those receiving > 200 mL/kg (P =.064).
- Repeat imaging was performed in 21 (70.0%) patients receiving < 200 mL/kg and 7 (63.6%) patients receiving > 200 mL/kg of resuscitation who underwent follow-up imaging.
- The median TBSA was 16.5 in the < 200 mL/kg group and 53.2 in the > 200 mL/kg group (P <.001).
- Intracranial abnormalities were found in 31.3% patients with hemorrhage, 18.8% with worsening edema, and 43.8% with ischemia or infarction.
IN PRACTICE:
“Patients who received over 200 mL/kg of resuscitation had an increased progression of intracranial abnormalities when compared with patients receiving less volume resuscitation,” the authors wrote. “Neurologic changes prompting imaging in burn patients may be undetectable, and our study further highlights the need for routine evaluation with neurologic imaging when undergoing large-volume resuscitations.”
SOURCE:
The study was led by Connor L. Kenney, MD, Brooke Army Medical Center, San Antonio, and was published online on November 07, 2024, in the Journal of Surgical Research.
LIMITATIONS:
Study limitations included a small patient sample and unclear guidelines for obtaining head CT scans, making it difficult to distinguish between trauma-related brain changes and disease progression. Additionally, the study lacked data on hypotensive episodes and long-term neurologic outcomes.
DISCLOSURES:
This study did not receive any specific funding. The authors declared no conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers conducted a single-center review of 5176 patients with burn injuries who were admitted to a verified American Burn Association center (2003-2017); 622 of them underwent head CT within 96 hours of admission, and 83 showed intracranial abnormalities.
- Of 42 patients (mean age, 49.7 years; 80.5% men) who were admitted within 24 hours of burn, 30 patients received < 200 mL/kg and 11 received > 200 mL/kg of total resuscitation fluids, with a median total body surface area (TBSA) of 20.0.
- The primary outcome assessed was the worsening of neurologic findings on imaging related to the volume of the resuscitation fluid administered; the secondary outcomes were the incidence of new or worsening intracranial abnormalities, including hemorrhage, edema, ischemia, or infarction.
TAKEAWAY:
- Neurologic findings worsened in 47.6% patients receiving < 200 mL/kg of fluid resuscitation and 85.7% of those receiving > 200 mL/kg (P =.064).
- Repeat imaging was performed in 21 (70.0%) patients receiving < 200 mL/kg and 7 (63.6%) patients receiving > 200 mL/kg of resuscitation who underwent follow-up imaging.
- The median TBSA was 16.5 in the < 200 mL/kg group and 53.2 in the > 200 mL/kg group (P <.001).
- Intracranial abnormalities were found in 31.3% patients with hemorrhage, 18.8% with worsening edema, and 43.8% with ischemia or infarction.
IN PRACTICE:
“Patients who received over 200 mL/kg of resuscitation had an increased progression of intracranial abnormalities when compared with patients receiving less volume resuscitation,” the authors wrote. “Neurologic changes prompting imaging in burn patients may be undetectable, and our study further highlights the need for routine evaluation with neurologic imaging when undergoing large-volume resuscitations.”
SOURCE:
The study was led by Connor L. Kenney, MD, Brooke Army Medical Center, San Antonio, and was published online on November 07, 2024, in the Journal of Surgical Research.
LIMITATIONS:
Study limitations included a small patient sample and unclear guidelines for obtaining head CT scans, making it difficult to distinguish between trauma-related brain changes and disease progression. Additionally, the study lacked data on hypotensive episodes and long-term neurologic outcomes.
DISCLOSURES:
This study did not receive any specific funding. The authors declared no conflicts of interest.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Whipple Disease With Central Nervous System Involvement
Whipple Disease With Central Nervous System Involvement
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
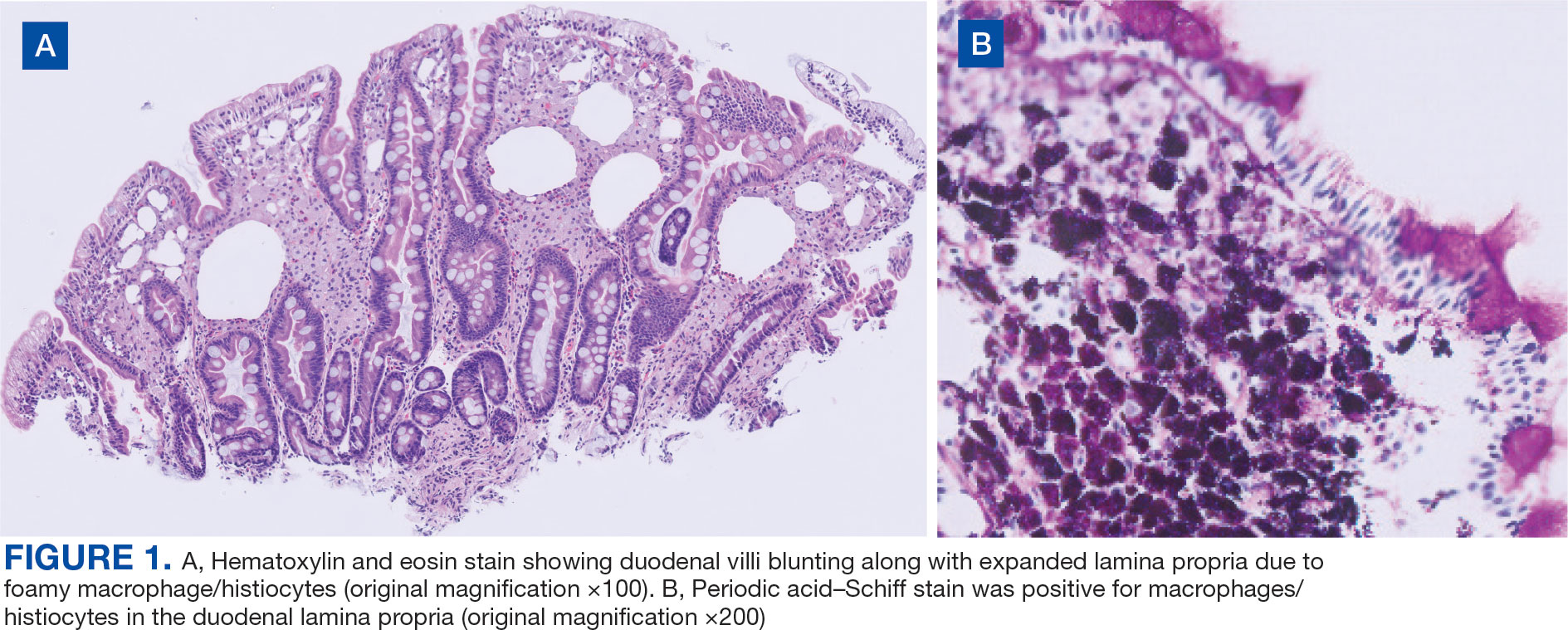
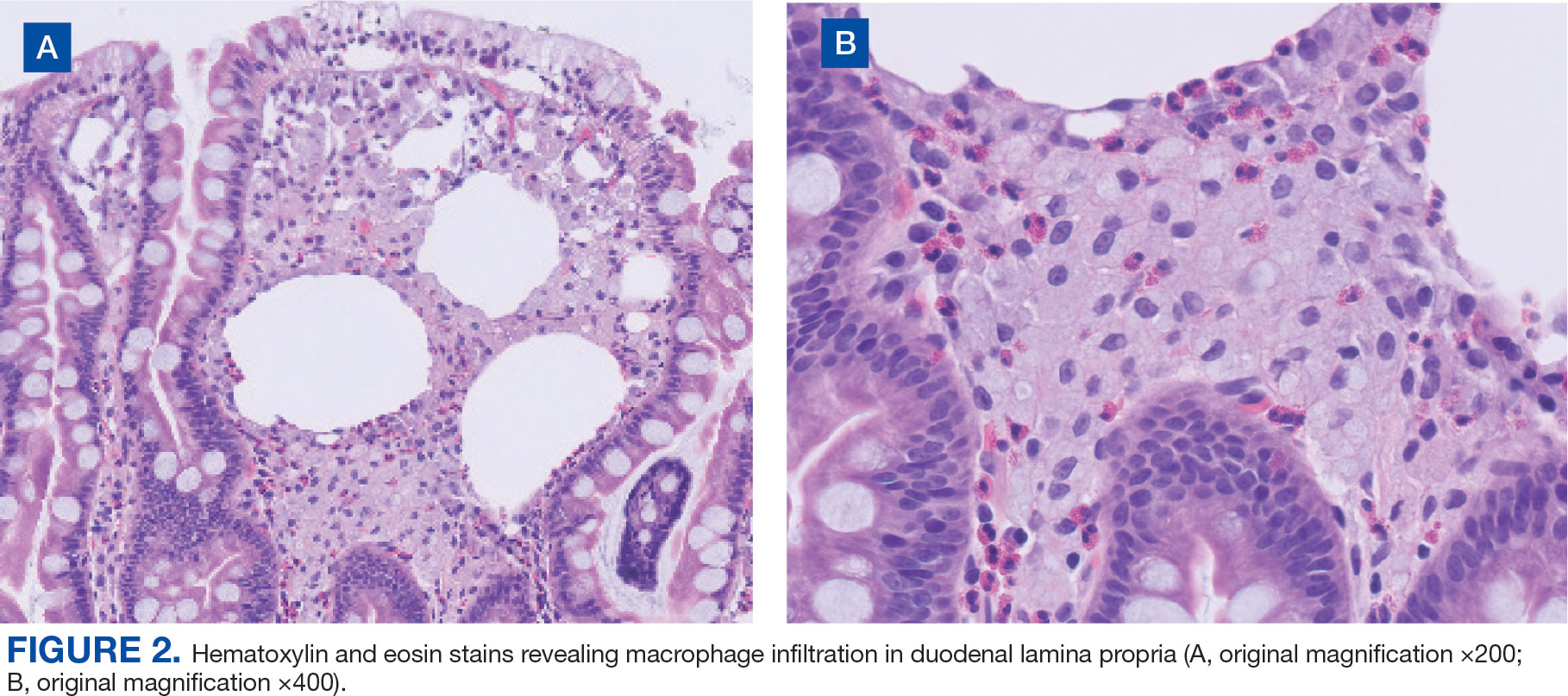
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
Whipple disease is a chronic, rare, infectious disease that manifests with systemic symptoms. This disease is caused by the gram-positive bacterium Tropheryma whipplei (T. whipplei). Common manifestations include gastrointestinal symptoms indicative of malabsorption, such as chronic diarrhea, unintentional weight loss (despite normal nutrient intake), and greasy, voluminous, foul-smelling stool. Other, less common manifestations include cardiovascular, endocrine, musculoskeletal, neurologic, and renal signs and symptoms. The prevalence of the disease is rare, affecting 3 in 1 million patients.1 This case highlights the importance of considering Whipple disease when treating patients with multiple symptoms and concurrent disease processes.
Case Presentation
A 53-year-old male with a medical history of hypertension, hyperlipidemia, hypothyroidism, and microcytic anemia presented with an 8-month history of persistent diarrhea associated with abdominal bloating, abdominal discomfort, and a 30-lb weight loss. He also reported fatigue, headaches, inability to concentrate, memory distortion, and visual disturbances involving flashes and floaters. The patient reported no fever, chills, nuchal rigidity, or prior neurologic symptoms. He reported intermittent bilateral hand and knee arthralgias. An autoimmune evaluation for arthralgia was negative, and a prior colonoscopy had been normal.
The patient’s hobbies included gardening, hiking, fishing, and deer hunting in Wyoming and Texas. He had spent time around cattle, dogs, and cats. He consumed alcohol twice weekly but reported no tobacco or illicit drug use or recent international travel. The patient’s family history was positive for rheumatoid arthritis, diabetes mellitus, and hypertension.
The patient’s vital signs were all within reference ranges, and lung auscultation revealed clear breathing sounds with no cardiac murmurs, gallops, or rubs. An abdominal examination revealed decreased bowel sounds, while the rest of the physical examination was otherwise normal.
Initial laboratory results showed that his sodium was 134 mEq/L (reference range, 136-145 mEq/L), hemoglobin was 9.3 g/dL (reference range for men, 14.0-18.0 g/dL), and hematocrit was 30.7% (reference range for men 42%-52%). His white blood cell (WBC) count and thyroid-stimulating hormone level were within normal limits. A cerebrospinal fluid (CSF) analysis revealed the following: WBCs 1.0/μL (0-5/μL), segmented neutrophils 10% (reference range, 7%), lymphocytes 80% (reference range, 40-80%), macrophages 10% (reference range, 2%), red blood cells 3 × 106 /μL (reference range, 4.3- 5.9 × 106 /µL), protein 23.5 mg/dL (reference range, 15-60 mg/dL), and glucose 44 mg/dL (reference range, 50-80 mg/dL).
Upper endoscopy with duodenal biopsy showed benign duodenal mucosa. Histopathologic evaluation revealed abundant foamy macrophages within lamina propria. Periodic acid–Schiff (PAS) stain was positive, diastase-resistant material was visualized within the macrophages (Figures 1 and 2). Polymerase chain reaction (PCR) testing of duodenal biopsy tissue was positive for T. whipplei. A lumbar puncture was performed, and PCR testing of CSF for T. whipplei was also positive. A stool PCR test was positive for Giardia. Transthoracic echocardiogram and brain magnetic resonance imaging were normal.
We treated the patient’s giardiasis with a single dose of oral tinidazole 2 g. To treat Whipple disease with central nervous system (CNS) involvement, we started the patient on ceftriaxone 2 g intravenous every 24 hours for 4 weeks, followed by oral trimethoprim and sulfamethoxazole (TMPSMX) 160/800 mg twice daily with an expected 1-year course.
Two months into TMP-SMX therapy, the patient developed an acute kidney injury with hyperkalemia (potassium, 5.5 mEq/L). We transitioned the therapy to doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily to complete 18 months of therapy. A lumbar puncture for CSF PCR and duodenal biopsy was planned for 6 months and 1 year after diagnosis.
Discussion
Whipple disease is often overlooked when making a diagnosis due to the nonspecific nature of its associated signs and symptoms. Classic Whipple disease has 2 stages: an initial prodromal stage marked by intermittent arthralgias, followed by a second gastrointestinal stage that involves chronic diarrhea, abdominal pain, and weight loss.1-3 Infection can sometimes be misdiagnosed as seronegative rheumatoid arthritis and a definite diagnosis can be missed for extended periods, with 1 case taking up to 8 years to diagnose after the first joint manifestations.2,4,5 Blood culture-negative endocarditis has also been well documented.1-5
The most common CNS clinical manifestations of Whipple disease include cognitive changes (eg, dementia), ocular movement disturbances (eg, oculomasticatory myorhythmia, which is pathognomonic for Whipple disease), involuntary movements, and hypothalamic dysfunction.1,6 Other neurologic symptoms include seizures, ataxia, meningitis, and myelopathy. Cerebrospinal fluid studies vary, with some results being normal and others revealing elevated protein counts.1
Disease Course
A retrospective study by Compain and colleagues reports that Whipple disease follows 3 patterns of clinical CNS involvement: classic Whipple disease with neurologic involvement, Whipple disease with isolated neurologic involvement, and neurologic relapse of previously treated Whipple disease.6 Isolated neurologic involvement is roughly 4% to 8%.6-8 Previous studies showed that the average delay from the presentation of neurologic symptoms to diagnosis is about 30 months.9
Diagnosis can be made with histologic evaluation of duodenal tissue using hematoxylin-eosin and PAS stains, which reveal foamy macrophages in expanded duodenal lamina propria, along with a positive tissue PCR.1,5 The slow replication rate of T. whipplei limits the effectiveness of bacterial cultures. After adequate treatment, relapses are still possible and regularly involve the CNS.1,4
Treatment typically involves blood-brain barrier-crossing agents, such as 2 weeks of meropenem 1 g every 24 hours or 2 to 4 weeks of ceftriaxone 2 g every 24 hours, followed by 1 year of TMP-SMX 160/800 mg twice daily. Doxycycline 100 mg twice daily and hydroxychloroquine 200 mg orally 3 times daily have also been shown to be effective, as seen in our patient.
Mortality rates vary for patients with Whipple disease and CNS involvement. One study reported poor overall prognosis in patients with CNS involvement, with mortality rates as high as 27%.10 However, rates of early detection and appropriate treatment may be improving, with 1 case series reporting 11% mortality in 18 patients with Whipple disease.6
Diagnosis
Because Whipple disease mimics many other diseases, misdiagnosis as infectious and noninfectious etiologies is common. PAS stain and tissue PCR helped uncover Whipple disease in a patient erroneously diagnosed with refractory Crohn disease.11
Weight loss, diarrhea, arthralgias, and cognitive impairment can also be seen in celiac disease. However, dermatologic manifestations, metabolic bone disease, and vitamin deficiencies are characteristics of celiac disease and can help distinguish it from T. whipplei infection.12
Whipple disease can also be mistaken for tropical sprue. Both can manifest with chronic diarrhea and duodenal villous atrophy; however, tropical sprue is more prevalent in specific geographic areas, and clinical manifestations are primarily gastrointestinal. Weight loss, diarrhea, steatorrhea, and folate deficiency are unique findings in tropical sprue that help differentiate it from Whipple disease.13 Likewise, other infectious diseases can be misdiagnosed as Whipple disease. Duodenal villi blunting and positive PAS staining have been reported in a Mycobacterium avium complex intestinal infection in a patient with AIDS, leading to a misdiagnosis of Whipple disease.14
Some parasitic infections have gastrointestinal symptoms similar to those of Whipple disease and others, such as giardiasis, are known to occur concurrently with Whipple disease.15-17 Giardiasis can also account for weight loss, malabsorptive symptoms, and greasy diarrhea. One case report hypothesized that 1 disease may predispose individuals to the other, as they both affect villous architecture.17 Additional research is needed to determine where the case reports have left off and to explore the connection between the 2 conditions.
Conclusions
The diagnosis of Whipple disease is challenging and frequently missed due to the rare and protean nature of the disease. This case highlights the importance of clinical suspicion for Whipple disease, especially in patients presenting with chronic seronegative arthritis, gastrointestinal abnormalities, and cognitive changes. Furthermore, this case points to the importance of additional testing for Whipple disease, even when a concurrent infection, such as giardiasis, has been identified.
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
- Biagi F, Balduzzi D, Delvino P, Schiepatti A, Klersy C, Corazza GR. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-1348. doi:10.1007/s10096-015-2357-2
- Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356(1):55-66. doi:10.1056/NEJMra062477
- El-Abassi R, Soliman MY, Williams F, England JD. Whipple’s disease. J Neurol Sci. 2017;377:197-206. doi:10.1016/j.jns.2017.01.048
- Melas N, Amin R, Gyllemark P, Younes AH, Almer S. Whipple’s disease: the great masquerader-a high level of suspicion is the key to diagnosis. BMC Gastroenterol. 2021;21(1):128. doi:10.1186/s12876-021-01664-1
- Boumaza A, Azzouz EB, Arrindell J, Lepidi H, Mezouar S, Desnues B. Whipple’s disease and Tropheryma whipplei infections: from bench to bedside. Lancet Infect Dis. 2022;22(10):e280-e291. doi:10.1016/S1473-3099(22)00128-1
- Compain C, Sacre K, Puéchal X, et al. Central nervous system involvement in Whipple disease: clinical study of 18 patients and long-term follow-up. Medicine (Baltimore). 2013;92(6):324-330. doi:10.1097/MD.0000000000000010
- Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68(1):2-5. doi:10.1136/jnnp.68.1.2
- Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457. doi:10.1097/00005792-200211000-00005
- Durand DV, Lecomte C, Cathébras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne. Medicine (Baltimore). 1997;76(3):170-184. doi:10.1097/00005792-199705000-00003
- Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8(9):899-903.
- Klochan C, Anderson TA, Rose D, Dimitrov RK, Johnson RM. Nearly fatal case of Whipple’s disease in a patient mistakenly on anti-TNF therapy. ACG Case Rep J. 2013;1(1):25-28. doi:10.14309/crj.2013.11
- . Therrien A, Kelly CP, Silvester JA. Celiac disease: extraintestinal manifestations and associated conditions. J Clin Gastroenterol. 2020;54(1):8-21. doi:10.1097/MCG.0000000000001267
- Murray JA, Rubio-Tapia A. Diarrhoea due to small bowel diseases. Best Pract Res Clin Gastroenterol. 2012;26(5):581-600. doi:10.1016/j.bpg.2012.11.013
- Chirayath S, Bin Liaquat H, Bahirwani J, Labeeb A, Chaput K, Kaza C. Mycobacterium avium complex infection imitating Whipple disease in an immunocompromised patient with newly diagnosed acquired immunodeficiency syn - drome. ACG Case Rep J. 2021;8(5):e00588. doi:10.14309/crj.0000000000000588
- Fenollar F, Lepidi H, Gérolami R, Drancourt M, Raoult D. Whipple disease associated with giardiasis. J Infect Dis. 2003;188(6):828-834. doi:10.1086/378093
- Ruiz JAG, Simón PG, Aparicio Duque R, Mayor Jerez JL. Association between Whipple’s disease and Giardia lamblia infection. Rev Esp Enferm Dig. 2005;97(7)521-526. doi:10.4321/s1130-01082005000700007
- Gisbertz IA, Bergmans DC, van Marion-Kievit JA, Haak HR. Concurrent Whipple’s disease and Giardia lamblia infection in a patient presenting with weight loss. Eur J Intern Med. 2001;12(6):525-528. doi:10.1016/s0953-6205(01)00165-0
Whipple Disease With Central Nervous System Involvement
Whipple Disease With Central Nervous System Involvement

Smart Mattress to Reduce SUDEP?
LOS ANGELES — says one of the experts involved in its development.
When used along with a seizure detection device, Jong Woo Lee, MD, PhD, associate professor of neurology, Harvard Medical School, and Brigham and Women’s Hospital, both in Boston, Massachusetts, estimates the smart mattress could cut SUDEP by more than 50%.
In addition, early results from an observational study are backing this up, he said.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
Most SUDEP Cases Found Face Down
SUDEP is the leading cause of death in children with epilepsy and in otherwise healthy adult patients with epilepsy. When his fifth patient died of SUDEP, Lee decided it was time to try to tackle the high mortality rate associated with these unexpected deaths. “I desperately wanted to help, ” he said.
About 70% of SUDEP occurs during sleep, and victims are found face down, or in the prone position, 90% of the time, said Lee.
“Of course, the best way to prevent SUDEP is not to have a seizure, but once you have a seizure and once you’re face down, your risk for death goes up by somewhere between 30 and 100 times,” he explained.
Lee was convinced SUDEP could be prevented by simple interventions that stimulate the patient and turn them over. He noted the incidence of sudden infant death syndrome, “which has similar characteristics” to SUDEP, has been reduced by up to 75% through campaigns that simply advise placing babies on their backs.
“Most of SUDEP happens because your arousal system is knocked out and you just don’t take the breath that you’re supposed to. Just the act of turning people over and vibrating the bed will stimulate them,” he said.
However, it’s crucial that this be done quickly, said Lee. “When you look at patients who died on video and see the EEGs, everybody took their last breath within 3 minutes.”
Because the window of opportunity is so short, “we think that seizure detection devices alone are not going to really be effective because you just can’t get there or react within those 3 minutes.”
There are currently no products that detect the prone position or have the ability to reposition a patient quickly into the recovery sideways position.
Lee and his colleagues developed a smart system that can be embedded in a mattress that detects when someone is having a seizure, determines if that person is face down, and if so, safely stimulates and repositions them.
The mattress is made up of a series of programmable inflatable blocks or “cells” that have pressure, vibration, temperature, and humidity sensors embedded within. “Based on the pressure readings, we can figure out whether the patient is right side up, on their right side, on their left side, or face down,” said Lee.
If the person is face down, he or she can be repositioned within a matter of seconds. “Each of the cells can lift 1000 pounds,” he said. The mattress is “very comfortable,” said Lee, who has tried it out himself.
Eighteen normative control participants have been enrolled for development and training purposes. To date, 10 of these individuals, aged 18-53 years, weighing 100-182 lb, and with a height of 5 ft 2 in to 6 ft 1 in, underwent extensive formal testing on the prototype bed.
Researchers found the mattress responded quickly to different body positions and weights. “We were able to reposition everybody in around 20 seconds,” said Lee.
The overall accuracy of detecting the prone position was 96.8%. There were no cases of a supine or prone position being mistaken for each other.
Researchers are refining the algorithm to improve the accuracy for detecting the prone position and expect to have a completely functional prototype within a few years.
Big Step Forward
Commenting on the research, Daniel M. Goldenholz, MD, PhD, assistant professor, Division of Epilepsy, Harvard Beth Israel Deaconess Medical Center, Boston, said the study “is a big step forward in the race to provide an actionable tool to prevent SUDEP.”
The technology “appears to mostly be doing what it’s intended to do, with relatively minor technical errors being made,” he said.
However, it is not clear if this technology can truly save lives, said Goldenholz. “The data we have suggests that lying face down in bed after a seizure is correlated with SUDEP, but that does not mean that if we can simply flip people over, they for sure won’t die.”
Even if the new technology “works perfectly,” it’s still an open question, said Goldenholz. If it does save lives, “this will be a major breakthrough, and one that has been needed for a long time.”
However, even if it does not, he congratulates the team for trying to determine if reducing the prone position can help prevent SUDEP. He would like to see more “high-risk, high-reward” studies in the epilepsy field. “We are in so much need of new innovations.”
He said he was “personally very inspired” by this work. “People are dying from this terrible disease, and this team is building what they hope might save lives.”
The study was funded by the National Institutes of Health. The mattress is being developed by Soterya. Lee reported no equity in Soterya. Goldenholz reported no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
LOS ANGELES — says one of the experts involved in its development.
When used along with a seizure detection device, Jong Woo Lee, MD, PhD, associate professor of neurology, Harvard Medical School, and Brigham and Women’s Hospital, both in Boston, Massachusetts, estimates the smart mattress could cut SUDEP by more than 50%.
In addition, early results from an observational study are backing this up, he said.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
Most SUDEP Cases Found Face Down
SUDEP is the leading cause of death in children with epilepsy and in otherwise healthy adult patients with epilepsy. When his fifth patient died of SUDEP, Lee decided it was time to try to tackle the high mortality rate associated with these unexpected deaths. “I desperately wanted to help, ” he said.
About 70% of SUDEP occurs during sleep, and victims are found face down, or in the prone position, 90% of the time, said Lee.
“Of course, the best way to prevent SUDEP is not to have a seizure, but once you have a seizure and once you’re face down, your risk for death goes up by somewhere between 30 and 100 times,” he explained.
Lee was convinced SUDEP could be prevented by simple interventions that stimulate the patient and turn them over. He noted the incidence of sudden infant death syndrome, “which has similar characteristics” to SUDEP, has been reduced by up to 75% through campaigns that simply advise placing babies on their backs.
“Most of SUDEP happens because your arousal system is knocked out and you just don’t take the breath that you’re supposed to. Just the act of turning people over and vibrating the bed will stimulate them,” he said.
However, it’s crucial that this be done quickly, said Lee. “When you look at patients who died on video and see the EEGs, everybody took their last breath within 3 minutes.”
Because the window of opportunity is so short, “we think that seizure detection devices alone are not going to really be effective because you just can’t get there or react within those 3 minutes.”
There are currently no products that detect the prone position or have the ability to reposition a patient quickly into the recovery sideways position.
Lee and his colleagues developed a smart system that can be embedded in a mattress that detects when someone is having a seizure, determines if that person is face down, and if so, safely stimulates and repositions them.
The mattress is made up of a series of programmable inflatable blocks or “cells” that have pressure, vibration, temperature, and humidity sensors embedded within. “Based on the pressure readings, we can figure out whether the patient is right side up, on their right side, on their left side, or face down,” said Lee.
If the person is face down, he or she can be repositioned within a matter of seconds. “Each of the cells can lift 1000 pounds,” he said. The mattress is “very comfortable,” said Lee, who has tried it out himself.
Eighteen normative control participants have been enrolled for development and training purposes. To date, 10 of these individuals, aged 18-53 years, weighing 100-182 lb, and with a height of 5 ft 2 in to 6 ft 1 in, underwent extensive formal testing on the prototype bed.
Researchers found the mattress responded quickly to different body positions and weights. “We were able to reposition everybody in around 20 seconds,” said Lee.
The overall accuracy of detecting the prone position was 96.8%. There were no cases of a supine or prone position being mistaken for each other.
Researchers are refining the algorithm to improve the accuracy for detecting the prone position and expect to have a completely functional prototype within a few years.
Big Step Forward
Commenting on the research, Daniel M. Goldenholz, MD, PhD, assistant professor, Division of Epilepsy, Harvard Beth Israel Deaconess Medical Center, Boston, said the study “is a big step forward in the race to provide an actionable tool to prevent SUDEP.”
The technology “appears to mostly be doing what it’s intended to do, with relatively minor technical errors being made,” he said.
However, it is not clear if this technology can truly save lives, said Goldenholz. “The data we have suggests that lying face down in bed after a seizure is correlated with SUDEP, but that does not mean that if we can simply flip people over, they for sure won’t die.”
Even if the new technology “works perfectly,” it’s still an open question, said Goldenholz. If it does save lives, “this will be a major breakthrough, and one that has been needed for a long time.”
However, even if it does not, he congratulates the team for trying to determine if reducing the prone position can help prevent SUDEP. He would like to see more “high-risk, high-reward” studies in the epilepsy field. “We are in so much need of new innovations.”
He said he was “personally very inspired” by this work. “People are dying from this terrible disease, and this team is building what they hope might save lives.”
The study was funded by the National Institutes of Health. The mattress is being developed by Soterya. Lee reported no equity in Soterya. Goldenholz reported no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
LOS ANGELES — says one of the experts involved in its development.
When used along with a seizure detection device, Jong Woo Lee, MD, PhD, associate professor of neurology, Harvard Medical School, and Brigham and Women’s Hospital, both in Boston, Massachusetts, estimates the smart mattress could cut SUDEP by more than 50%.
In addition, early results from an observational study are backing this up, he said.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
Most SUDEP Cases Found Face Down
SUDEP is the leading cause of death in children with epilepsy and in otherwise healthy adult patients with epilepsy. When his fifth patient died of SUDEP, Lee decided it was time to try to tackle the high mortality rate associated with these unexpected deaths. “I desperately wanted to help, ” he said.
About 70% of SUDEP occurs during sleep, and victims are found face down, or in the prone position, 90% of the time, said Lee.
“Of course, the best way to prevent SUDEP is not to have a seizure, but once you have a seizure and once you’re face down, your risk for death goes up by somewhere between 30 and 100 times,” he explained.
Lee was convinced SUDEP could be prevented by simple interventions that stimulate the patient and turn them over. He noted the incidence of sudden infant death syndrome, “which has similar characteristics” to SUDEP, has been reduced by up to 75% through campaigns that simply advise placing babies on their backs.
“Most of SUDEP happens because your arousal system is knocked out and you just don’t take the breath that you’re supposed to. Just the act of turning people over and vibrating the bed will stimulate them,” he said.
However, it’s crucial that this be done quickly, said Lee. “When you look at patients who died on video and see the EEGs, everybody took their last breath within 3 minutes.”
Because the window of opportunity is so short, “we think that seizure detection devices alone are not going to really be effective because you just can’t get there or react within those 3 minutes.”
There are currently no products that detect the prone position or have the ability to reposition a patient quickly into the recovery sideways position.
Lee and his colleagues developed a smart system that can be embedded in a mattress that detects when someone is having a seizure, determines if that person is face down, and if so, safely stimulates and repositions them.
The mattress is made up of a series of programmable inflatable blocks or “cells” that have pressure, vibration, temperature, and humidity sensors embedded within. “Based on the pressure readings, we can figure out whether the patient is right side up, on their right side, on their left side, or face down,” said Lee.
If the person is face down, he or she can be repositioned within a matter of seconds. “Each of the cells can lift 1000 pounds,” he said. The mattress is “very comfortable,” said Lee, who has tried it out himself.
Eighteen normative control participants have been enrolled for development and training purposes. To date, 10 of these individuals, aged 18-53 years, weighing 100-182 lb, and with a height of 5 ft 2 in to 6 ft 1 in, underwent extensive formal testing on the prototype bed.
Researchers found the mattress responded quickly to different body positions and weights. “We were able to reposition everybody in around 20 seconds,” said Lee.
The overall accuracy of detecting the prone position was 96.8%. There were no cases of a supine or prone position being mistaken for each other.
Researchers are refining the algorithm to improve the accuracy for detecting the prone position and expect to have a completely functional prototype within a few years.
Big Step Forward
Commenting on the research, Daniel M. Goldenholz, MD, PhD, assistant professor, Division of Epilepsy, Harvard Beth Israel Deaconess Medical Center, Boston, said the study “is a big step forward in the race to provide an actionable tool to prevent SUDEP.”
The technology “appears to mostly be doing what it’s intended to do, with relatively minor technical errors being made,” he said.
However, it is not clear if this technology can truly save lives, said Goldenholz. “The data we have suggests that lying face down in bed after a seizure is correlated with SUDEP, but that does not mean that if we can simply flip people over, they for sure won’t die.”
Even if the new technology “works perfectly,” it’s still an open question, said Goldenholz. If it does save lives, “this will be a major breakthrough, and one that has been needed for a long time.”
However, even if it does not, he congratulates the team for trying to determine if reducing the prone position can help prevent SUDEP. He would like to see more “high-risk, high-reward” studies in the epilepsy field. “We are in so much need of new innovations.”
He said he was “personally very inspired” by this work. “People are dying from this terrible disease, and this team is building what they hope might save lives.”
The study was funded by the National Institutes of Health. The mattress is being developed by Soterya. Lee reported no equity in Soterya. Goldenholz reported no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
FROM AES 2024
Sleep Apnea Linked to Heightened Mortality in Epilepsy
LOS ANGELES — according to a new analysis of over 2 million patient-years drawn from the Komodo Health Claims Database.
“A 10-year-old with uncontrolled epilepsy and central sleep apnea is about 200 times more likely to die than a general population 10-year-old. That’s comparable to a 10-year-old with {epilepsy and} congestive heart failure. Noncentral sleep apnea is comparable to being paralyzed. It’s a huge risk factor,” said poster presenter Dan Lloyd, advanced analytics lead at UCB, which sponsored the research.
The ordering of sleep apnea tests for patients with epilepsy is widely variable, according to Stefanie Dedeurwaerdere, PhD, who is the innovation and value creation lead at UCB. “Some doctors do that as a general practice, and some don’t. There’s no coherency in the way these studies are requested for epilepsy patients. We want to create some awareness around this topic,” she said, and added that treatment of sleep apnea may improve epileptic seizures.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
The study included mortality rates between January 2018 and December 2022, with a total of 2,355,410 patient-years and 968,993 patients, with an age distribution of 19.1% age 1 to less than 18 years, 23.7% age 18-35 years, and 57.2% age 36 years or older. Sleep apnea prevalences were 0.7% for central sleep apnea (CSA), 14.0% for obstructive sleep apnea (OSA), and 85.3% with no sleep apnea.
Among those aged 1-18 years, the standardized mortality ratio (SMR) for those with uncontrolled epilepsy was 27.7. For those with comorbid OSA, the SMR was 74.2, and for comorbid CSA, the SMR was 135.9. The association was less pronounced in older groups, dipping to 7.0, 11.3, and 19.5 in those aged 18-35 years, and 3.3, 3.1, and 2.8 among those aged 36 years or older.
Among the 1-18 age group, SMRs for other comorbidities included 132.3 for heart failure, 74.9 for hemiplegia/paraplegia, 55.3 for cerebrovascular disease, and 44.6 for chronic pulmonary disease.
Asked for comment, Gordon Buchanan, MD, PhD, welcomed the new work. “The results did not surprise me. I study sleep, epilepsy, and [sudden unexplained death in epilepsy (SUDEP)] in particular ... and every time I speak on these topics, someone asks me about risk of SUDEP in patients with sleep apnea. It’s great to finally have some data,” said Buchanan, a professor of neurology at the University of Iowa, Iowa City.
The authors found that patients undergoing continuous positive airway pressure (CPAP)/bi-level positive airway pressure therapy had a higher mortality risk than those not undergoing CPAP therapy but cautioned that uncontrolled confounders may be contributing to the effect.
Buchanan wondered if treatment with CPAP would be associated with a decreased mortality risk. “I think that would be interesting, but I know that, especially in children, it can be difficult to get them to remain compliant with CPAP. I think that would be interesting to know, if pushing harder to get the kids to comply with CPAP would reduce mortality,” he said.
The specific finding of heightened mortality associated with CSA is interesting, according to Buchanan. “We think of seizures propagating through the brain, maybe through direct synaptic connections or through spreading depolarization. So I think it would make sense that it would hit central regions that would then lead to sleep apnea.”
The relationship between OSA and epilepsy is likely complex. Epilepsy medications and special diets may influence body composition, which could in turn affect the risk for OSA, as could medications associated with psychiatric comorbidities, according to Buchanan.
The study is retrospective and based on claims data. It does not prove causation, and claims data do not fully capture mortality, which may lead to conservative SMR estimates. The researchers did not control for socioeconomic status, treatment status, and other comorbidities or conditions.
Lloyd and Dedeurwaerdere are employees of UCB, which sponsored the study. Buchanan had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
LOS ANGELES — according to a new analysis of over 2 million patient-years drawn from the Komodo Health Claims Database.
“A 10-year-old with uncontrolled epilepsy and central sleep apnea is about 200 times more likely to die than a general population 10-year-old. That’s comparable to a 10-year-old with {epilepsy and} congestive heart failure. Noncentral sleep apnea is comparable to being paralyzed. It’s a huge risk factor,” said poster presenter Dan Lloyd, advanced analytics lead at UCB, which sponsored the research.
The ordering of sleep apnea tests for patients with epilepsy is widely variable, according to Stefanie Dedeurwaerdere, PhD, who is the innovation and value creation lead at UCB. “Some doctors do that as a general practice, and some don’t. There’s no coherency in the way these studies are requested for epilepsy patients. We want to create some awareness around this topic,” she said, and added that treatment of sleep apnea may improve epileptic seizures.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
The study included mortality rates between January 2018 and December 2022, with a total of 2,355,410 patient-years and 968,993 patients, with an age distribution of 19.1% age 1 to less than 18 years, 23.7% age 18-35 years, and 57.2% age 36 years or older. Sleep apnea prevalences were 0.7% for central sleep apnea (CSA), 14.0% for obstructive sleep apnea (OSA), and 85.3% with no sleep apnea.
Among those aged 1-18 years, the standardized mortality ratio (SMR) for those with uncontrolled epilepsy was 27.7. For those with comorbid OSA, the SMR was 74.2, and for comorbid CSA, the SMR was 135.9. The association was less pronounced in older groups, dipping to 7.0, 11.3, and 19.5 in those aged 18-35 years, and 3.3, 3.1, and 2.8 among those aged 36 years or older.
Among the 1-18 age group, SMRs for other comorbidities included 132.3 for heart failure, 74.9 for hemiplegia/paraplegia, 55.3 for cerebrovascular disease, and 44.6 for chronic pulmonary disease.
Asked for comment, Gordon Buchanan, MD, PhD, welcomed the new work. “The results did not surprise me. I study sleep, epilepsy, and [sudden unexplained death in epilepsy (SUDEP)] in particular ... and every time I speak on these topics, someone asks me about risk of SUDEP in patients with sleep apnea. It’s great to finally have some data,” said Buchanan, a professor of neurology at the University of Iowa, Iowa City.
The authors found that patients undergoing continuous positive airway pressure (CPAP)/bi-level positive airway pressure therapy had a higher mortality risk than those not undergoing CPAP therapy but cautioned that uncontrolled confounders may be contributing to the effect.
Buchanan wondered if treatment with CPAP would be associated with a decreased mortality risk. “I think that would be interesting, but I know that, especially in children, it can be difficult to get them to remain compliant with CPAP. I think that would be interesting to know, if pushing harder to get the kids to comply with CPAP would reduce mortality,” he said.
The specific finding of heightened mortality associated with CSA is interesting, according to Buchanan. “We think of seizures propagating through the brain, maybe through direct synaptic connections or through spreading depolarization. So I think it would make sense that it would hit central regions that would then lead to sleep apnea.”
The relationship between OSA and epilepsy is likely complex. Epilepsy medications and special diets may influence body composition, which could in turn affect the risk for OSA, as could medications associated with psychiatric comorbidities, according to Buchanan.
The study is retrospective and based on claims data. It does not prove causation, and claims data do not fully capture mortality, which may lead to conservative SMR estimates. The researchers did not control for socioeconomic status, treatment status, and other comorbidities or conditions.
Lloyd and Dedeurwaerdere are employees of UCB, which sponsored the study. Buchanan had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
LOS ANGELES — according to a new analysis of over 2 million patient-years drawn from the Komodo Health Claims Database.
“A 10-year-old with uncontrolled epilepsy and central sleep apnea is about 200 times more likely to die than a general population 10-year-old. That’s comparable to a 10-year-old with {epilepsy and} congestive heart failure. Noncentral sleep apnea is comparable to being paralyzed. It’s a huge risk factor,” said poster presenter Dan Lloyd, advanced analytics lead at UCB, which sponsored the research.
The ordering of sleep apnea tests for patients with epilepsy is widely variable, according to Stefanie Dedeurwaerdere, PhD, who is the innovation and value creation lead at UCB. “Some doctors do that as a general practice, and some don’t. There’s no coherency in the way these studies are requested for epilepsy patients. We want to create some awareness around this topic,” she said, and added that treatment of sleep apnea may improve epileptic seizures.
The findings were presented at the American Epilepsy Society (AES) 78th Annual Meeting 2024.
The study included mortality rates between January 2018 and December 2022, with a total of 2,355,410 patient-years and 968,993 patients, with an age distribution of 19.1% age 1 to less than 18 years, 23.7% age 18-35 years, and 57.2% age 36 years or older. Sleep apnea prevalences were 0.7% for central sleep apnea (CSA), 14.0% for obstructive sleep apnea (OSA), and 85.3% with no sleep apnea.
Among those aged 1-18 years, the standardized mortality ratio (SMR) for those with uncontrolled epilepsy was 27.7. For those with comorbid OSA, the SMR was 74.2, and for comorbid CSA, the SMR was 135.9. The association was less pronounced in older groups, dipping to 7.0, 11.3, and 19.5 in those aged 18-35 years, and 3.3, 3.1, and 2.8 among those aged 36 years or older.
Among the 1-18 age group, SMRs for other comorbidities included 132.3 for heart failure, 74.9 for hemiplegia/paraplegia, 55.3 for cerebrovascular disease, and 44.6 for chronic pulmonary disease.
Asked for comment, Gordon Buchanan, MD, PhD, welcomed the new work. “The results did not surprise me. I study sleep, epilepsy, and [sudden unexplained death in epilepsy (SUDEP)] in particular ... and every time I speak on these topics, someone asks me about risk of SUDEP in patients with sleep apnea. It’s great to finally have some data,” said Buchanan, a professor of neurology at the University of Iowa, Iowa City.
The authors found that patients undergoing continuous positive airway pressure (CPAP)/bi-level positive airway pressure therapy had a higher mortality risk than those not undergoing CPAP therapy but cautioned that uncontrolled confounders may be contributing to the effect.
Buchanan wondered if treatment with CPAP would be associated with a decreased mortality risk. “I think that would be interesting, but I know that, especially in children, it can be difficult to get them to remain compliant with CPAP. I think that would be interesting to know, if pushing harder to get the kids to comply with CPAP would reduce mortality,” he said.
The specific finding of heightened mortality associated with CSA is interesting, according to Buchanan. “We think of seizures propagating through the brain, maybe through direct synaptic connections or through spreading depolarization. So I think it would make sense that it would hit central regions that would then lead to sleep apnea.”
The relationship between OSA and epilepsy is likely complex. Epilepsy medications and special diets may influence body composition, which could in turn affect the risk for OSA, as could medications associated with psychiatric comorbidities, according to Buchanan.
The study is retrospective and based on claims data. It does not prove causation, and claims data do not fully capture mortality, which may lead to conservative SMR estimates. The researchers did not control for socioeconomic status, treatment status, and other comorbidities or conditions.
Lloyd and Dedeurwaerdere are employees of UCB, which sponsored the study. Buchanan had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
FROM AES 2024
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
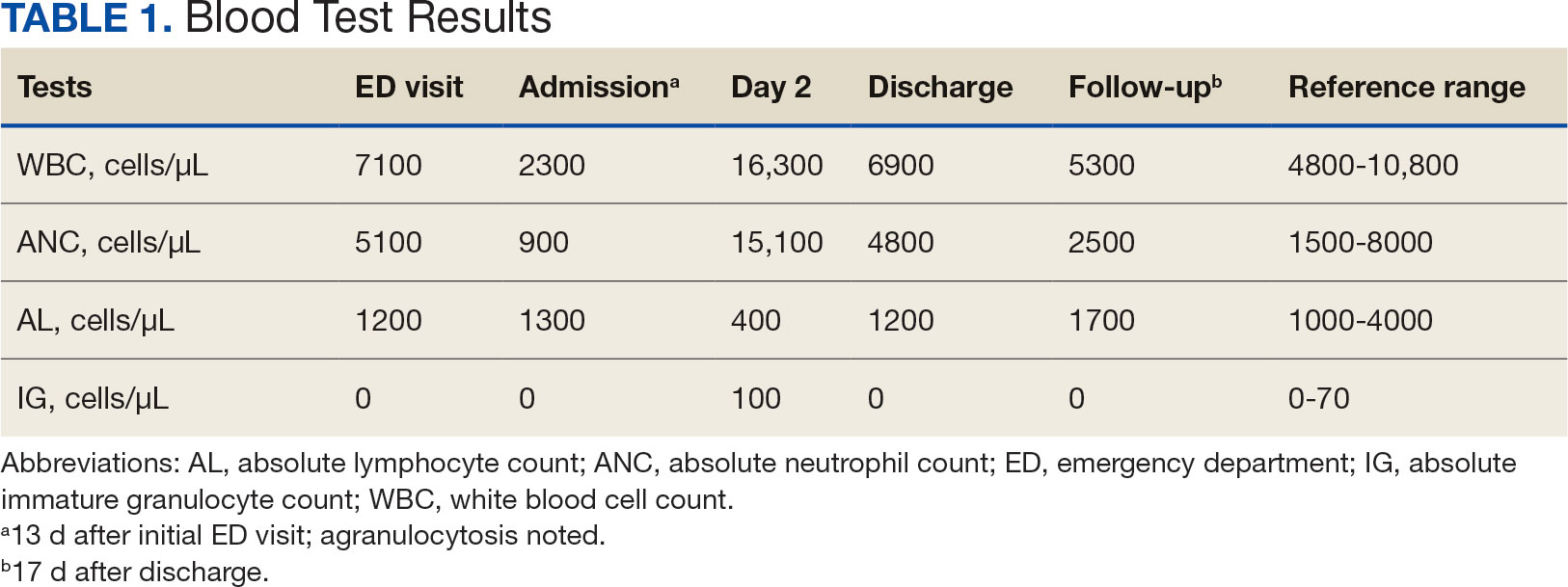
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.

The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.
The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
Acute agranulocytosis and aseptic meningitis are serious adverse effects (AEs) associated with sulfamethoxazole-trimethoprim. Acute agranulocytosis is a rare, potentially life-threatening blood dyscrasia characterized by a neutrophil count of < 500 cells per μL, with no relevant decrease in hemoglobin or platelet levels.1 Patients with agranulocytosis may be asymptomatic or experience severe sore throat, pharyngitis, or tonsillitis in combination with high fever, rigors, headaches, or malaise. These AEs are commonly classified as idiosyncratic and, in most cases, attributable to medications. If drug-induced agranulocytosis is suspected, the patient should discontinue the medication immediately.1
Meningitis is an inflammatory disease typically caused by viral or bacterial infections; however, it may also be attributed to medications or malignancy. Inflammation of the meninges with a negative bacterial cerebrospinal fluid culture is classified as aseptic meningitis. Distinguishing between aseptic and bacterial meningitis is crucial due to differences in illness severity, treatment options, and prognosis.2 Symptoms of meningitis may include fever, headache, nuchal rigidity, nausea, or vomiting.3 Several classes of medications can cause drug-induced aseptic meningitis (DIAM), but the most commonly reported antibiotic is sulfamethoxazole-trimethoprim.
DIAM is more prevalent in immunocompromised patients, such as those with a history of HIV/AIDS, organ transplant, collagen vascular disease, or malignancy, who may be prescribed sulfamethoxazoletrimethoprim for prophylaxis or treatment of infection.2 The case described in this article serves as a distinctive example of acute agranulocytosis complicated with aseptic meningitis caused by sulfamethoxazole-trimethoprim in an immunocompetent patient.
Case Presentation
A healthy male veteran aged 39 years presented to the Fargo Veterans Affairs Medical Center emergency department (ED) for worsening left testicular pain and increased urinary urgency and frequency for about 48 hours. The patient had no known medication allergies, was current on vaccinations, and his only relevant prescription was valacyclovir for herpes labialis. The evaluation included urinalysis, blood tests, and scrotal ultrasound. The urinalysis, blood tests, and vitals were unremarkable for any signs of systemic infection. The scrotal ultrasound was significant for left focal area of abnormal echogenicity with absent blood flow in the superior left testicle and a significant increase in blood flow around the left epididymis. Mild swelling in the left epididymis was present, with no significant testicular or scrotal swelling or skin changes observed. Urology was consulted and prescribed oral sulfamethoxazole-trimethoprim 800-160 mg every 12 hours for 30 days for the treatment of left epididymo-orchitis.
The patient returned to the ED 2 weeks later with fever, chills, headache, generalized body aches, urinary retention, loose stools, and nonspecific chest pressure. A serum blood test revealed significant neutropenia and leukopenia. The patient was admitted for observation, and sulfamethoxazole-trimethoprim was discontinued. The patient received sodium chloride intravenous (IV) fluid, oral potassium chloride supplementation, IV ondansetron, and analgesics, including oral acetaminophen, oral ibuprofen, and IV hydromorphone as needed. Repeated laboratory tests were completed with no specific findings; serum laboratory work, urinalysis, chest and abdominal X-rays, and echocardiogram were all unremarkable. The patient’s neutrophil count dropped from 5100 cells/µL at the initial ED presentation to 900 cells/µL (reference range, 1500-8000 cells/µL) (Table 1). Agranulocytosis quickly resolved after antibiotic discontinuation.
Upon further investigation, the patient took the prescribed sulfamethoxazole-trimethoprim for 10 days before stopping due to the resolution of testicular pain and epididymal swelling. The patient reported initial AEs of loose stools and generalized myalgias when first taking the medication. The patient restarted the antibiotic to complete the course of therapy after not taking it for 2 days. Within 20 minutes of restarting the medication, he experienced myalgias with pruritus, prompting him to return to the ED. Agranulocytosis and aseptic meningitis developed within 12 days after he was prescribed sulfamethoxazole-trimethoprim, though the exact timeframe is unknown.
The patient’s symptoms, except for a persistent headache, resolved during hospitalization. Infectious disease was consulted, and a lumbar puncture was performed due to prior fever and ongoing headaches to rule out a treatable cause of meningitis. The lumbar puncture showed clear spinal fluid, an elevated white blood cell count with neutrophil predominance, and normal protein and glucose levels. Cultures showed no aerobic, anaerobic, or fungal organisms. Herpes virus simplex and Lyme disease testing was not completed during hospitalization. Respiratory panel, legionella, and hepatitis A, B, and C tests were negative (Table 2). The negative laboratory test results strengthened the suspicion of aseptic meningitis caused by sulfamethoxazole-trimethoprim. The neurology consult recommended no additional treatments or tests.
The patient spontaneously recovered 2 days later, with a normalized complete blood count and resolution of headache. Repeat scrotal ultrasounds showed resolution of the left testicle findings. The patient was discharged and seen for a follow-up 14 days later. The final diagnosis requiring hospitalization was aseptic meningitis secondary to a sulfamethoxazole-trimethoprim.
Discussion
Sulfamethoxazole-trimethoprim is a commonly prescribed antibiotic for urinary tract infections, pneumocystis pneumonia, pneumocystis pneumonia prophylaxis, and methicillin-resistant Staphylococcus aureus skin and soft tissue infections. Empiric antibiotics for epididymo-orchitis caused by enteric organisms include levofloxacin or ofloxacin; however sulfamethoxazole-trimethoprim may be considered as alternative.5,6 Agranulocytosis induced by sulfamethoxazole-trimethoprim may occur due to the inhibition on folic acid metabolism, which makes the highly proliferating cells of the hematopoietic system more susceptible to neutropenia. Agranulocytosis typically occurs within 7 days of treatment initiation and generally resolves within a month after discontinuation of the offending agent.7 In this case, agranulocytosis resolved overnight, resulting in leukocytosis. One explanation for the rapid increase in white blood cell count may be the concurrent diagnosis of aseptic meningitis. Alternatively, the patient’s health and immunocompetence may have helped generate an adequate immune response. Medication-induced agranulocytosis is often a diagnosis of exclusion because it is typically difficult to diagnose.7 In more severe or complicated cases of agranulocytosis, filgrastim may be indicated.1
Sulfamethoxazole-trimethoprim is a lipophilic small-molecule medication that can cross the blood-brain barrier and penetrate the tissues of the bone, prostate, and central nervous system.8 Specifically, the medication can pass into the cerebrospinal fluid regardless of meningeal inflammation.9 The exact mechanism for aseptic meningitis caused by sulfamethoxazole-trimethoprim is unknown; however, it may accumulate in the choroid plexus, causing destructive inflammation of small blood vessels and resulting in aseptic meningitis.10 The onset of aseptic meningitis can vary from 10 minutes to 10 days after initiation of the medication. Pre-exposure to the medication typically results in earlier onset of symptoms, though patients do not need to have previously taken the medication to develop aseptic meningitis. Patients generally become afebrile with resolution of headache and mental status changes within 48 to 72 hours after stopping the medication or after about 5 to 7 half-lives of the medication are eliminated.11 Some patients may continue to experience worsening symptoms after discontinuation because the medication is already absorbed into the serum and tissues.
DIAM is an uncommon drug-induced hypersensitivity AE of the central nervous system. Diagnosing aseptic meningitis caused by sulfamethoxazole-trimethoprim can be challenging, as antibiotics are given to treat suspected infections, and the symptoms of aseptic meningitis can be hard to differentiate from those of infectious meningitis.11 Close monitoring between the initiation of the medication and the onset of clinical symptoms is necessary to assist in distinguishing between aseptic and infectious meningitis.3 If the causative agent is not discontinued, symptoms can quickly worsen, progressing from fever and headache to confusion, coma, and respiratory depression. A DIAM diagnosis can only be made with resolution of aseptic meningitis after stopping the contributory agent. If appropriate, this can be proven by rechallenging the medication in a controlled setting. The usual treatment for aseptic meningitis is supportive care, including hydration, antiemetics, electrolyte supplementation, and adequate analgesia.3
Differential diagnoses in this case included viral infection, meningitis, and allergic reaction to sulfamethoxazole-trimethoprim. The patient reported history of experiencing symptoms after restarting his antibiotic, raising strong suspicion for DIAM. Initiation of this antibiotic was the only recent medication change noted. Laboratory testing for infectious agents yielded negative results, including tests for aerobic and anaerobic bacteria as well as viral and fungal infections. A lumbar puncture and cerebrospinal fluid culture was clear, with no organisms shown on gram stain. Bacterial or viral meningitis was presumed less likely due to the duration of symptoms, correlation of symptoms coinciding with restarting the antibiotic, and negative cerebrospinal fluid culture results.
It was concluded that agranulocytosis and aseptic meningitis were likely induced by sulfamethoxazole-trimethoprim as supported by the improvement upon discontinuing the medication and subsequent worsening upon restarting it. Concurrent agranulocytosis and aseptic meningitis is rare, and there is typically no correlation between the 2 reactions. Since agranulocytosis may be asymptomatic, this case highlights the need to monitor blood cell counts in patients using sulfamethoxazole-trimethoprim. The possibility of DIAM should be considered in patients presenting with flu-like symptoms, and a lumbar puncture may be collected to rule out aseptic meningitis if the patient’s AEs are severe following the initiation of an antibiotic, particularly in immunosuppressed patients taking sulfamethoxazole-trimethoprim. This case is unusual because the patient was healthy and immunocompetent.
This case may not be generalizable and may be difficult to compare to other cases. Every case has patient-specific factors affecting subjective information, including the patient’s baseline, severity of symptoms, and treatment options. This report was based on a single patient case and corresponding results may be found in similar patient cases.
Conclusions
This case emphasizes the rare but serious AEs of acute agranulocytosis complicated with aseptic meningitis after prescribed sulfamethoxazole-trimethoprim. This is a unique case of an immunocompetent patient developing both agranulocytosis and aseptic meningitis after restarting the antibiotic to complete therapy. This case highlights the importance of monitoring blood cell counts and monitoring for signs and symptoms of aseptic meningitis, even during short courses of therapy. Further research is needed to recognize characteristics that may increase the risk for these AEs and to develop strategies for their prevention.
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
- Garbe E. Non-chemotherapy drug-induced agranulocytosis. Expert Opin Drug Saf. 2007;6(3):323-335. doi:10.1517/14740338.6.3.323
- Jha P, Stromich J, Cohen M, Wainaina JN. A rare complication of trimethoprim-sulfamethoxazole: drug induced aseptic meningitis. Case Rep Infect Dis. 2016;2016:3879406. doi:10.1155/2016/3879406
- Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4(2):285-297. doi:10.1517/14740338.4.2.285
- Jarrin I, Sellier P, Lopes A, et al. Etiologies and management of aseptic meningitis in patients admitted to an internal medicine department. Medicine (Baltimore). 2016;95(2):e2372. doi:10.1097/MD.0000000000002372
- Street EJ, Justice ED, Kopa Z, et al. The 2016 European guideline on the management of epididymo-orchitis. Int J STD AIDS. 2017;28(8):744-749. doi:10.1177/0956462417699356
- Kbirou A, Alafifi M, Sayah M, Dakir M, Debbagh A, Aboutaieb R. Acute orchiepididymitis: epidemiological and clinical aspects: an analysis of 152 cases. Ann Med Surg (Lond). 2022;75:103335. doi:10.1016/j.amsu.2022.103335
- Rattay B, Benndorf RA. Drug-induced idiosyncratic agranulocytosis - infrequent but dangerous. Front Pharmacol. 2021;12:727717. doi:10.3389/fphar.2021.727717
- Elmedani S, Albayati A, Udongwo N, Odak M, Khawaja S. Trimethoprim-sulfamethoxazole-induced aseptic meningitis: a new approach. Cureus. 2021;13(6):e15869. doi:10.7759/cureus.15869
- Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858-883. doi:10.1128/CMR.00007-10
- Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Intern Med. 1999;159(11):1185- 1194. doi:10.1001/archinte.159.11.1185
- Bruner KE, Coop CA, White KM. Trimethoprim-sulfamethoxazole-induced aseptic meningitis-not just another sulfa allergy. Ann Allergy Asthma Immunol. 2014;113(5):520-526. doi:10.1016/j.anai.2014.08.006
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim
Agranulocytosis and Aseptic Meningitis Induced by Sulfamethoxazole-Trimethoprim