User login
Oral JAK Inhibitor's Clinical Place To Be Determined
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.

Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.

Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.

Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.
Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.
Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.
Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The new oral janus kinase inhibitor approved on Nov. 6 by the Food and Drug Administration is likely to be embraced by those rheumatoid arthritis patients who have found infection and infusions of biologics to be needling.
The Janus kinase inhibitor (JAK) tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis (RA), has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate.
Tofacitinib is a small-molecule inhibitor of the JAK pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
Dr. Eric L. Matteson said in an interview that he plans to offer this drug to patients with active disease. While some patients may embrace the idea of taking a pill, those who do not mind the needles because of their convenience and efficiency may opt to stay on their injected therapy. Of the nine biologic agents on the market currently for RA, four are infused and the others either are taken as subcutaneous injections or self-administered subcutaneously via a prefilled syringe. One of the four infused drugs is available as a prefilled self-administered syringe as well, with another biologic maker about to launch such a product.
Dr. Larry Greenbaum, a rheumatologist in Greenwood, Ind., recalled that before the introduction of biologics, "I thought patients would never accept parenteral medications, but almost all of them do accept these medications when they see how well they work. A pill will certainly be more welcome than an injection for most patients. But the Enbrel SureClick and the Humira Pen are very easy to use, so I don’t think patients are going to be breaking down the doors demanding this medication just so they don’t have to give themselves an injection!"
When asked where the new JAK inhibitor would fit into his own therapeutic lineup, Dr. Greenbaum noted that "the number of biologic medications is increasing all the time, and my conservative approach is usually to park the new medication at the bottom of the treatment algorithm until I have some compelling reason to use it sooner. No matter how good this medication is, it will have some very stiff competition from the available biologic drugs that work well and have long clinical track records."
In contrast, Dr. Karmela K. Chan, a rheumatologist in Pawtucket, R.I., said, "I definitely have patients who are completely opposed to any kind of injection, and, given a choice, they would rather take an oral drug. Several patients have asked me about switching from their injectable drug to an oral drug that they\'d already heard of.
"I don’t think I will switch most patients over. If something works, I tend not to want to mess with it. For new patients, I suspect my pitch will still be for the anti-TNF agent. I feel it is prudent to use agents that have been around longer. Another question will be how much more comfortable we are with how much information we have about potential adverse effects. However, I will most likely also present the option of the oral drug."
And then there is cost.
The JAK inhibitor will be expensive, just as the currently available biologic agents are. Pfizer has said that the recommended regimen of one 5-mg tablet twice a day will be priced at $2,000 a month, according to Dr. Matteson, chair of rheumatology and professor of medicine at the Mayo Clinic Medical School in Rochester, Minn. He noted that Pfizer is already offering a program to help patients cover their share of the copayment for the new drug.
Dr. Chan said that "without a doubt the cost will be an issue for patients. Cost of drug, side effect potential, efficaciousness, and convenience all factor into patients’ decisions.
"Also, yes, in our practice, I have been told multiple times that I could make more money if I put more people on infusions. We can buy and bill so we make more money that way, plus we make money off just the service of the infusion. But I think this issue of making money from infusions will perhaps not pass muster too much longer for the following reasons: Ethical physicians won’t infuse just to infuse (one would hope), and I think in the coming years fewer insurers will allow buy and bill. On top of that, I am not sure if Medicare reimbursement rates for the infusion service will change."
Dr. Matteson noted that the drug, to be marketed as Xeljanz, has shown efficacy compared with placebo in a number of studies considered by the Food and Drug Administration (FDA). The drug inhibits the protein kinase, which is important in cell-to-cell interaction and may be how the drug acts to decrease inflammation.
Only time will tell whether that decrease in inflammation will translate into reduced joint damage in RA patients or even into decreased risk for extra-articular manifestations of the disease, including cardiovascular disease, lung disease, and eye disease.
Treatment with anti–tumor necrosis factor (anti-TNF) drugs has been shown to lower the risk for cardiovascular disease associated with RA. But it took 5 years of post-marketing surveillance before rheumatologists began to recognize that benefit. Any similar effect with the JAK inhibitor may take just as long to become apparent, said Dr. Matteson.
In order to detect any effects, the FDA approved the drug with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a post-marketing study, according to the FDA’s statement announcing the approval.
Dr. Matteson noted that safety and efficacy trials of the drugs showed that two common side effects were headaches and diarrhea, severe enough to cause the patient to discontinue the drug.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The post-marketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.

Secukinumab Tames Hand, Foot Psoriasis
PRAGUE – The investigational anti-interleukin-17A biologic agent secukinumab holds particular promise for the treatment of psoriasis in two of its most challenging, tough-to-tame manifestations: hand and foot psoriasis and nail involvement, a study has shown.
A post hoc analysis of a previously reported phase II, double-blind, placebo-controlled, 12-week dose-finding study in 404 patients with moderate to severe plaque psoriasis focused in part on the 131 subjects with hand and/or foot involvement. The same regimen that achieved the best outcomes in the main study – 150 mg of secukinumab given subcutaneously at weeks 0, 1, 2, and 4 – showed the greatest beneficial effect for hand and foot psoriasis, Dr. Carle Paul reported at the annual congress of the European Academy of Dermatology and Venereology.
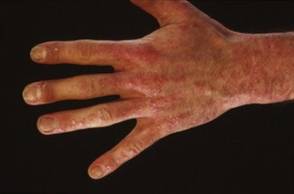
Fifty-four percent of patients randomized to this optimal secukinumab dosing regimen achieved a positive investigator global response of their moderate to severe hand and foot psoriasis at week 12 compared with 19% assigned to placebo. An investigator global response required at least a 2-point improvement on a 5-point scale along with a score of 0 or 1, meaning clear or only minimal disease, explained Dr. Paul, a dermatologist at the University of Toulouse (France).
The PASI 75 and PASI 90 rates in patients with hand and foot psoriasis – that is, the percentage of patients with at least a 75% or 90% improvement over baseline scores on the Psoriasis Area and Severity Index – were similar to the 55% and 32% rates observed in the overall study population, he added.
The phase II study also included 304 patients with nail psoriasis. At week 12, those patients treated with secukinumab at weeks 0, 1, 2, and 4 showed a mean 19% improvement on a composite fingernail involvement score, compared with a 14% worsening with placebo.
These findings are especially encouraging because hand, foot, and nail psoriasis affect an estimated 10%-55% of all psoriasis patients. Involvement at these sites causes considerable functional disability, including difficulty in walking and using the hands. In addition, hand, foot, and nail psoriasis are notoriously difficult to treat.
Secukinumab’s side effect pattern in the large phase II study was similar to that for placebo. Definitive safety and efficacy results will come from ongoing pivotal phase III clinical trials totaling more than 3,000 psoriasis patients worldwide. The results are anticipated next year.
In addition to being used for psoriasis, secukinumab is being developed as a novel treatment for rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Ongoing phase III studies are expected to report results in 2014. Also, phase II studies of secukinumab for multiple sclerosis are underway.
Secukinumab is a fully human anti-interleukin-17A monoclonal antibody being developed by Novartis.
The phase II study and post hoc analysis were sponsored by Novartis. Dr. Paul is a recipient of research grants from and is a consultant to the company.
PRAGUE – The investigational anti-interleukin-17A biologic agent secukinumab holds particular promise for the treatment of psoriasis in two of its most challenging, tough-to-tame manifestations: hand and foot psoriasis and nail involvement, a study has shown.
A post hoc analysis of a previously reported phase II, double-blind, placebo-controlled, 12-week dose-finding study in 404 patients with moderate to severe plaque psoriasis focused in part on the 131 subjects with hand and/or foot involvement. The same regimen that achieved the best outcomes in the main study – 150 mg of secukinumab given subcutaneously at weeks 0, 1, 2, and 4 – showed the greatest beneficial effect for hand and foot psoriasis, Dr. Carle Paul reported at the annual congress of the European Academy of Dermatology and Venereology.
Fifty-four percent of patients randomized to this optimal secukinumab dosing regimen achieved a positive investigator global response of their moderate to severe hand and foot psoriasis at week 12 compared with 19% assigned to placebo. An investigator global response required at least a 2-point improvement on a 5-point scale along with a score of 0 or 1, meaning clear or only minimal disease, explained Dr. Paul, a dermatologist at the University of Toulouse (France).
The PASI 75 and PASI 90 rates in patients with hand and foot psoriasis – that is, the percentage of patients with at least a 75% or 90% improvement over baseline scores on the Psoriasis Area and Severity Index – were similar to the 55% and 32% rates observed in the overall study population, he added.
The phase II study also included 304 patients with nail psoriasis. At week 12, those patients treated with secukinumab at weeks 0, 1, 2, and 4 showed a mean 19% improvement on a composite fingernail involvement score, compared with a 14% worsening with placebo.
These findings are especially encouraging because hand, foot, and nail psoriasis affect an estimated 10%-55% of all psoriasis patients. Involvement at these sites causes considerable functional disability, including difficulty in walking and using the hands. In addition, hand, foot, and nail psoriasis are notoriously difficult to treat.
Secukinumab’s side effect pattern in the large phase II study was similar to that for placebo. Definitive safety and efficacy results will come from ongoing pivotal phase III clinical trials totaling more than 3,000 psoriasis patients worldwide. The results are anticipated next year.
In addition to being used for psoriasis, secukinumab is being developed as a novel treatment for rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Ongoing phase III studies are expected to report results in 2014. Also, phase II studies of secukinumab for multiple sclerosis are underway.
Secukinumab is a fully human anti-interleukin-17A monoclonal antibody being developed by Novartis.
The phase II study and post hoc analysis were sponsored by Novartis. Dr. Paul is a recipient of research grants from and is a consultant to the company.
PRAGUE – The investigational anti-interleukin-17A biologic agent secukinumab holds particular promise for the treatment of psoriasis in two of its most challenging, tough-to-tame manifestations: hand and foot psoriasis and nail involvement, a study has shown.
A post hoc analysis of a previously reported phase II, double-blind, placebo-controlled, 12-week dose-finding study in 404 patients with moderate to severe plaque psoriasis focused in part on the 131 subjects with hand and/or foot involvement. The same regimen that achieved the best outcomes in the main study – 150 mg of secukinumab given subcutaneously at weeks 0, 1, 2, and 4 – showed the greatest beneficial effect for hand and foot psoriasis, Dr. Carle Paul reported at the annual congress of the European Academy of Dermatology and Venereology.
Fifty-four percent of patients randomized to this optimal secukinumab dosing regimen achieved a positive investigator global response of their moderate to severe hand and foot psoriasis at week 12 compared with 19% assigned to placebo. An investigator global response required at least a 2-point improvement on a 5-point scale along with a score of 0 or 1, meaning clear or only minimal disease, explained Dr. Paul, a dermatologist at the University of Toulouse (France).
The PASI 75 and PASI 90 rates in patients with hand and foot psoriasis – that is, the percentage of patients with at least a 75% or 90% improvement over baseline scores on the Psoriasis Area and Severity Index – were similar to the 55% and 32% rates observed in the overall study population, he added.
The phase II study also included 304 patients with nail psoriasis. At week 12, those patients treated with secukinumab at weeks 0, 1, 2, and 4 showed a mean 19% improvement on a composite fingernail involvement score, compared with a 14% worsening with placebo.
These findings are especially encouraging because hand, foot, and nail psoriasis affect an estimated 10%-55% of all psoriasis patients. Involvement at these sites causes considerable functional disability, including difficulty in walking and using the hands. In addition, hand, foot, and nail psoriasis are notoriously difficult to treat.
Secukinumab’s side effect pattern in the large phase II study was similar to that for placebo. Definitive safety and efficacy results will come from ongoing pivotal phase III clinical trials totaling more than 3,000 psoriasis patients worldwide. The results are anticipated next year.
In addition to being used for psoriasis, secukinumab is being developed as a novel treatment for rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Ongoing phase III studies are expected to report results in 2014. Also, phase II studies of secukinumab for multiple sclerosis are underway.
Secukinumab is a fully human anti-interleukin-17A monoclonal antibody being developed by Novartis.
The phase II study and post hoc analysis were sponsored by Novartis. Dr. Paul is a recipient of research grants from and is a consultant to the company.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Fifty-four percent of patients with moderate to severe hand and foot psoriasis were rated clear or had only minimal disease at those sites at week 12 after being treated with secukinumab on weeks 0, 1, 2, and 4. Only 19% of placebo-treated subjects met that standard.
Data Source: This was a post hoc secondary analysis of 131 psoriasis patients with hand and foot involvement. They were part of a larger 404-patient, double-blind, randomized phase II study.
Disclosures: The phase II study and post hoc analysis were sponsored by Novartis. Dr. Paul is a recipient of research grants from and is a consultant to the company.

Game Changer? First JAK Inhibitor Approved for Rheumatoid Arthritis
The Janus kinase inhibitor tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis, has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate, the Food and Drug Administration announced on Nov. 6.
Tofacitinib is a small-molecule inhibitor of the Janus kinase (JAK) pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
It has been approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a postmarketing study, according to the FDA’s statement announcing the approval.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The postmarketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Pfizer is marketing the drug as Xeljanz. Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The Janus kinase inhibitor tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis, has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate, the Food and Drug Administration announced on Nov. 6.
Tofacitinib is a small-molecule inhibitor of the Janus kinase (JAK) pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
It has been approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a postmarketing study, according to the FDA’s statement announcing the approval.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The postmarketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Pfizer is marketing the drug as Xeljanz. Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
The Janus kinase inhibitor tofacitinib, a drug that has the promise to change the treatment experience for some patients with rheumatoid arthritis, has been approved to treat adults with moderately to severely active disease who have not responded adequately to or cannot tolerate methotrexate, the Food and Drug Administration announced on Nov. 6.
Tofacitinib is a small-molecule inhibitor of the Janus kinase (JAK) pathway of inflammatory cytokines that play a role in the pathogenesis of RA, and is the first drug in this class of oral drugs to be approved for RA.
It has been approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the serious risks associated with treatment, and a requirement that the manufacturer, Pfizer, conduct a postmarketing study, according to the FDA’s statement announcing the approval.
Approval was based on the results of seven studies, which found that patients with moderately to severely active RA had improvements in clinical response and physical functioning, when compared with those on placebo. Tofacitinib was associated with an increased risk of serious infections, including opportunistic infections; tuberculosis; cancers; and lymphoma, which are described in the boxed warning in the drug’s label, the FDA said.
Treatment was also associated with increases in cholesterol and liver enzymes, and decreased blood counts. The REMS consists of a Medication Guide that includes information for patients about the drug’s safety and a communication plan that will educate health care providers about the serious risks associated with the treatment.
The postmarketing study will compare two doses of tofacitinib with another approved treatment for RA.
At a meeting in May, the FDA’s Arthritis Advisory Committee voted 8 to 2 to recommend approval of tofacitinib for patients with RA, although panel members had lingering safety concerns.
Pfizer is marketing the drug as Xeljanz. Another JAK inhibitor, ruxolitinib (Jakafi), was approved to treat myelofibrosis in 2011.
Indirect Comparison Trial Favors Adalimumab for Psoriasis
PRAGUE – Recognizing the slim chances of a direct comparison study of two biologics for treatment of psoriasis, Dr. Kristian Reich and his coworkers have done the next best thing: they’ve conducted an indirect comparison study.
They used existing data from four randomized trials to compare two biologics adalimumab (Humira) and etanercept (Enbrel) and adjusted for between-trial differences in baseline patient demographics, treatment history, and numerous measures of psoriasis severity.

The analysis was supported by Abbott, the maker of adalimumab. The drug had a significantly better benefit-risk profile as measured by the number of days patients spent with at least a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) scores while also remaining free of study drug–related adverse events, Dr. Reich reported at the annual congress of the European Academy of Dermatology and Venereology.
The four phase III, double-blind, randomized, placebo-controlled, 12-week clinical trials included REVEAL (J. Am. Acad. Dermatol. 2008;56:106-15) and CHAMPION (Br. J. Dermatol. 2008;158:558-66), studies that pitted adalimumab against placebo in 1,302 subjects. The analysis also included the M10-114 and M10-315 trials, which included 390 patients randomized to briakinumab (Abbott), etanercept, or placebo.
Prior to propensity score weighting, participants in the adalimumab studies differed from those in the etanercept trials in terms of disease duration, percent body surface area involvement, extent of prior medication use, PASI scores, and other important variables. After matching, however, the two groups were closely similar across the board, explained Dr. Reich of the Hamburg (Germany) Dermatology Clinic.
Through 12 weeks of study participation, adalimumab-treated patients spent an average of 22.4 more days at PASI 75 with no study drug–related side effects than did those on placebo in the same two studies. In contrast, etanercept-treated patients spent 11.5 more days than did controls in this optimal state.
At week 4, the proportion of biologic-treated subjects who were PASI 75 responders free of moderate-to-severe study drug–related adverse events was 15.7% in the adalimumab group compared with 5.5% with etanercept. At week 8, these rates climbed to 46.3% in the adalimumab-treated patients and 16.2% in the etanercept group. By week 12, the figures were 58% for adalimumab compared with 39.4% with etanercept.
An obvious limitation of this indirect comparison is the potential for confounding due to unobserved baseline differences. But Dr. Reich said he and his coinvestigators adjusted and balanced for everything they could think of.
"To further adjust for potential differences between trials, outcomes were compared relative to the corresponding placebo arms after applying propensity weights and before comparing across trials. Thus, unobserved factors that equally impact outcomes on the drug and placebo arms would not bias this comparison of adalimumab and etanercept," the dermatologist said.
Dr. Reich has received research grants from and serves as a consultant to Abbott, which supported this analysis, as well as to numerous other pharmaceutical companies.
PRAGUE – Recognizing the slim chances of a direct comparison study of two biologics for treatment of psoriasis, Dr. Kristian Reich and his coworkers have done the next best thing: they’ve conducted an indirect comparison study.
They used existing data from four randomized trials to compare two biologics adalimumab (Humira) and etanercept (Enbrel) and adjusted for between-trial differences in baseline patient demographics, treatment history, and numerous measures of psoriasis severity.
The analysis was supported by Abbott, the maker of adalimumab. The drug had a significantly better benefit-risk profile as measured by the number of days patients spent with at least a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) scores while also remaining free of study drug–related adverse events, Dr. Reich reported at the annual congress of the European Academy of Dermatology and Venereology.
The four phase III, double-blind, randomized, placebo-controlled, 12-week clinical trials included REVEAL (J. Am. Acad. Dermatol. 2008;56:106-15) and CHAMPION (Br. J. Dermatol. 2008;158:558-66), studies that pitted adalimumab against placebo in 1,302 subjects. The analysis also included the M10-114 and M10-315 trials, which included 390 patients randomized to briakinumab (Abbott), etanercept, or placebo.
Prior to propensity score weighting, participants in the adalimumab studies differed from those in the etanercept trials in terms of disease duration, percent body surface area involvement, extent of prior medication use, PASI scores, and other important variables. After matching, however, the two groups were closely similar across the board, explained Dr. Reich of the Hamburg (Germany) Dermatology Clinic.
Through 12 weeks of study participation, adalimumab-treated patients spent an average of 22.4 more days at PASI 75 with no study drug–related side effects than did those on placebo in the same two studies. In contrast, etanercept-treated patients spent 11.5 more days than did controls in this optimal state.
At week 4, the proportion of biologic-treated subjects who were PASI 75 responders free of moderate-to-severe study drug–related adverse events was 15.7% in the adalimumab group compared with 5.5% with etanercept. At week 8, these rates climbed to 46.3% in the adalimumab-treated patients and 16.2% in the etanercept group. By week 12, the figures were 58% for adalimumab compared with 39.4% with etanercept.
An obvious limitation of this indirect comparison is the potential for confounding due to unobserved baseline differences. But Dr. Reich said he and his coinvestigators adjusted and balanced for everything they could think of.
"To further adjust for potential differences between trials, outcomes were compared relative to the corresponding placebo arms after applying propensity weights and before comparing across trials. Thus, unobserved factors that equally impact outcomes on the drug and placebo arms would not bias this comparison of adalimumab and etanercept," the dermatologist said.
Dr. Reich has received research grants from and serves as a consultant to Abbott, which supported this analysis, as well as to numerous other pharmaceutical companies.
PRAGUE – Recognizing the slim chances of a direct comparison study of two biologics for treatment of psoriasis, Dr. Kristian Reich and his coworkers have done the next best thing: they’ve conducted an indirect comparison study.
They used existing data from four randomized trials to compare two biologics adalimumab (Humira) and etanercept (Enbrel) and adjusted for between-trial differences in baseline patient demographics, treatment history, and numerous measures of psoriasis severity.
The analysis was supported by Abbott, the maker of adalimumab. The drug had a significantly better benefit-risk profile as measured by the number of days patients spent with at least a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) scores while also remaining free of study drug–related adverse events, Dr. Reich reported at the annual congress of the European Academy of Dermatology and Venereology.
The four phase III, double-blind, randomized, placebo-controlled, 12-week clinical trials included REVEAL (J. Am. Acad. Dermatol. 2008;56:106-15) and CHAMPION (Br. J. Dermatol. 2008;158:558-66), studies that pitted adalimumab against placebo in 1,302 subjects. The analysis also included the M10-114 and M10-315 trials, which included 390 patients randomized to briakinumab (Abbott), etanercept, or placebo.
Prior to propensity score weighting, participants in the adalimumab studies differed from those in the etanercept trials in terms of disease duration, percent body surface area involvement, extent of prior medication use, PASI scores, and other important variables. After matching, however, the two groups were closely similar across the board, explained Dr. Reich of the Hamburg (Germany) Dermatology Clinic.
Through 12 weeks of study participation, adalimumab-treated patients spent an average of 22.4 more days at PASI 75 with no study drug–related side effects than did those on placebo in the same two studies. In contrast, etanercept-treated patients spent 11.5 more days than did controls in this optimal state.
At week 4, the proportion of biologic-treated subjects who were PASI 75 responders free of moderate-to-severe study drug–related adverse events was 15.7% in the adalimumab group compared with 5.5% with etanercept. At week 8, these rates climbed to 46.3% in the adalimumab-treated patients and 16.2% in the etanercept group. By week 12, the figures were 58% for adalimumab compared with 39.4% with etanercept.
An obvious limitation of this indirect comparison is the potential for confounding due to unobserved baseline differences. But Dr. Reich said he and his coinvestigators adjusted and balanced for everything they could think of.
"To further adjust for potential differences between trials, outcomes were compared relative to the corresponding placebo arms after applying propensity weights and before comparing across trials. Thus, unobserved factors that equally impact outcomes on the drug and placebo arms would not bias this comparison of adalimumab and etanercept," the dermatologist said.
Dr. Reich has received research grants from and serves as a consultant to Abbott, which supported this analysis, as well as to numerous other pharmaceutical companies.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Through 12 weeks of study participation, adalimumab-treated patients spent an average of 22.4 more days at PASI (Psoriasis Area and Severity Index) 75 with no study drug–related side effects than did those on placebo in the same two studies. In contrast, etanercept-treated patients spent 11.5 more days than did controls in this optimal state.
Data Source: This analysis involved an indirect comparison of data from participants in four phase III, randomized, double-blind, placebo-controlled trials of nearly 1,700 patients.
Disclosures: Abbott Laboratories funded the study. Dr. Reich has received research grants from and serves as a consultant to Abbott and other pharmaceutical companies developing dermatologic drugs.

Adalimumab for the Treatment of Pityriasis Rubra Pilaris: A Case Report
Psoriasis: Short-Course Clobetasol Enhances Etanercept Outcomes
PRAGUE – Adding short courses of clobetasol propionate foam to etanercept resulted in improved outcomes compared with etanercept alone in patients with moderate to severe plaque psoriasis in a large, multicenter phase III randomized trial.
Moreover, the boost in efficacy that resulted from add-on short-course therapy with the potent topical steroid came at no cost in terms of additional side effects, according to Dr. Mark G. Lebwohl, professor and chair of dermatology at Mount Sinai School of Medicine, New York.

He reported on 592 psoriasis patients who were randomized to etanercept at 50 mg twice weekly for 12 weeks alone or with the option of using clobetasol propionate foam twice daily for up to 2 weeks during weeks 11-12 as needed to clear lesions. Of patients with the opportunity to resort to short-course topical steroid therapy, 85% did so during week 11 and 82% exercised that option in week 12.
The primary end point was a 75% improvement in the Psoriasis Area and Severity Index score, or PASI 75, at week 12 compared with baseline as determined by blinded investigators. This occurred in 65% of patients in the dual-therapy group vs. 48% of those on etanercept alone.
The incremental improvement in PASI 90 seen in the etanercept plus clobetasol propionate group was similarly impressive: a 30% PASI 90 rate compared with 19% with etanercept alone, Dr. Lebwohl noted at the annual congress of the European Academy of Dermatology and Venereology.
Patient satisfaction scores were significantly higher in the dual-therapy arm as well. Of patients randomized to etanercept plus clobetasol propionate, 87% said they were satisfied or very satisfied with their therapy, vs. 78% on etanercept alone.
The rate of treatment-related adverse events leading to discontinuation of therapy was low in both study arms: 0.7% with etanercept plus short-course topical steroid therapy and 1.3% with etanercept alone. No serious treatment-related adverse events occurred in the dual-therapy group, and there was only one in patients assigned to etanercept alone.
This phase III study was sponsored by Amgen. Dr. Lebwohl reported that he serves as a consultant to Amgen as well as numerous other pharmaceutical companies involved in developing dermatologic drugs.
PRAGUE – Adding short courses of clobetasol propionate foam to etanercept resulted in improved outcomes compared with etanercept alone in patients with moderate to severe plaque psoriasis in a large, multicenter phase III randomized trial.
Moreover, the boost in efficacy that resulted from add-on short-course therapy with the potent topical steroid came at no cost in terms of additional side effects, according to Dr. Mark G. Lebwohl, professor and chair of dermatology at Mount Sinai School of Medicine, New York.
He reported on 592 psoriasis patients who were randomized to etanercept at 50 mg twice weekly for 12 weeks alone or with the option of using clobetasol propionate foam twice daily for up to 2 weeks during weeks 11-12 as needed to clear lesions. Of patients with the opportunity to resort to short-course topical steroid therapy, 85% did so during week 11 and 82% exercised that option in week 12.
The primary end point was a 75% improvement in the Psoriasis Area and Severity Index score, or PASI 75, at week 12 compared with baseline as determined by blinded investigators. This occurred in 65% of patients in the dual-therapy group vs. 48% of those on etanercept alone.
The incremental improvement in PASI 90 seen in the etanercept plus clobetasol propionate group was similarly impressive: a 30% PASI 90 rate compared with 19% with etanercept alone, Dr. Lebwohl noted at the annual congress of the European Academy of Dermatology and Venereology.
Patient satisfaction scores were significantly higher in the dual-therapy arm as well. Of patients randomized to etanercept plus clobetasol propionate, 87% said they were satisfied or very satisfied with their therapy, vs. 78% on etanercept alone.
The rate of treatment-related adverse events leading to discontinuation of therapy was low in both study arms: 0.7% with etanercept plus short-course topical steroid therapy and 1.3% with etanercept alone. No serious treatment-related adverse events occurred in the dual-therapy group, and there was only one in patients assigned to etanercept alone.
This phase III study was sponsored by Amgen. Dr. Lebwohl reported that he serves as a consultant to Amgen as well as numerous other pharmaceutical companies involved in developing dermatologic drugs.
PRAGUE – Adding short courses of clobetasol propionate foam to etanercept resulted in improved outcomes compared with etanercept alone in patients with moderate to severe plaque psoriasis in a large, multicenter phase III randomized trial.
Moreover, the boost in efficacy that resulted from add-on short-course therapy with the potent topical steroid came at no cost in terms of additional side effects, according to Dr. Mark G. Lebwohl, professor and chair of dermatology at Mount Sinai School of Medicine, New York.
He reported on 592 psoriasis patients who were randomized to etanercept at 50 mg twice weekly for 12 weeks alone or with the option of using clobetasol propionate foam twice daily for up to 2 weeks during weeks 11-12 as needed to clear lesions. Of patients with the opportunity to resort to short-course topical steroid therapy, 85% did so during week 11 and 82% exercised that option in week 12.
The primary end point was a 75% improvement in the Psoriasis Area and Severity Index score, or PASI 75, at week 12 compared with baseline as determined by blinded investigators. This occurred in 65% of patients in the dual-therapy group vs. 48% of those on etanercept alone.
The incremental improvement in PASI 90 seen in the etanercept plus clobetasol propionate group was similarly impressive: a 30% PASI 90 rate compared with 19% with etanercept alone, Dr. Lebwohl noted at the annual congress of the European Academy of Dermatology and Venereology.
Patient satisfaction scores were significantly higher in the dual-therapy arm as well. Of patients randomized to etanercept plus clobetasol propionate, 87% said they were satisfied or very satisfied with their therapy, vs. 78% on etanercept alone.
The rate of treatment-related adverse events leading to discontinuation of therapy was low in both study arms: 0.7% with etanercept plus short-course topical steroid therapy and 1.3% with etanercept alone. No serious treatment-related adverse events occurred in the dual-therapy group, and there was only one in patients assigned to etanercept alone.
This phase III study was sponsored by Amgen. Dr. Lebwohl reported that he serves as a consultant to Amgen as well as numerous other pharmaceutical companies involved in developing dermatologic drugs.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Of psoriasis patients on etanercept plus short-course clobetasol propionate foam, 65% achieved a PASI 75 score at week , compared with 48% of those randomized to etanercept alone.
Data Source: Data are from a phase 3, randomized, multicenter clinical trial involving 592 patients with moderate to severe plaque psoriasis.
Disclosures: This randomized trial was funded by Amgen. Dr. Lebwohl reported that he serves as a consultant to Amgen as well as numerous other pharmaceutical companies involved in developing dermatologic drugs.

Psoriasis Flares Rapidly Postpartum
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).

"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
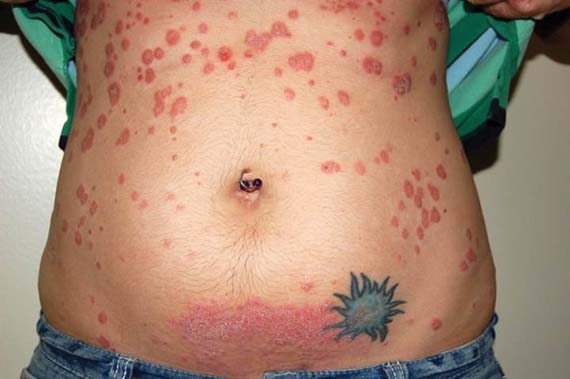
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
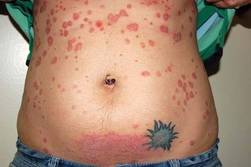
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).
"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).
"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
EXPERT ANALYSIS FROM THE SDEF WOMEN'S AND PEDIATRIC DERMATOLOGY SEMINAR
EULAR's Lupus Nephritis Guidelines Emphasize Early Biopsy
The European League Against Rheumatism has issued its first guidelines on management of lupus nephritis, and they advise renal biopsy at the first sign of kidney involvement, unlike the guidance issued by the American College of Rheumatology, which leaves timing of that testing up to the clinician’s judgment.
Other bright spots in EULAR’s guidelines, which it issued jointly with the European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) are their position that mycophenolic acid should be the first choice for immunosuppressive therapy, the precise recommendations for steroid dosage, stratification of treatment plans according to disease severity, and explicit advice on switching therapies after one drug has failed.
The European guidelines were published in the November issue of Annals of the Rheumatic Diseases (2012;71:1771-82) and are intended for rheumatologists, nephrologists, and internists managing adult and pediatric patients with lupus nephritis. The EULAR guidelines differ in several key ways from those issued by the American College of Rheumatology (ACR) in June (Arthritis Care Res. 2012;64:797-808).
The EULAR guidelines are unequivocal in their support for renal biopsy at any sign of renal involvement, including when unexplained renal insufficiency is accompanied by normal urinary findings. The ACR guidelines, by contrast, allow for more physician latitude in determining whether to biopsy.
"This can be an emotional issue because rheumatologists do not do biopsies and may try to avoid them," said the EULAR guidelines’ lead author, rheumatologist Dr. Dimitrios T. Boumpas, who is professor of medicine and director of internal medicine/rheumatology at the University of Crete in Heraklion, Greece, in an interview. "We felt that someone should make a position statement. Biopsy is simply best care. If you’re dealing with something severe like LN [lupus nephritis] and you avoid biopsy, that’s not good medicine."
Dr. Boumpas said the EULAR task force had also moved to put mycophenolic acid in the first position as an initial immunosuppressant treatment for most cases of class III-IV LN. Low-dose intravenous cyclophosphamide in combination with steroids is also recommended for this patient group.
There are no 5-year data for mycophenolic acid as there are for cyclophosphamide, Dr. Boumpas said. But the task force that developed the guidelines weighed data on safety and efficacy and found mycophenolic acid "as the clear first choice, while at the same time recognizing the limitations of the studies," he noted.
The ACR guidelines do not recommend use of azathioprine (AZA) as induction treatment. The EULAR recommendations acknowledge that AZA has been associated with a higher risk of renal flares, and call for its use in certain patients who have no adverse clinical or histological risk factors. Patients treated with AZA need close follow-up. "This is particularly important for countries without access to MPA [mycophenolic acid]," Dr. Boumpas said.
The EULAR guidelines also recommend switching to an alternative agent when patients fail to improve in 3-4 months or do not achieve partial response after 6-12 months, or a complete response after 2 years.
"This is based on evidence from both controlled trials and observational cohort studies, which highlight the fact that immunosuppressive agents, particularly cyclophosphamide, may take up to 2 years to achieve complete renal response," Dr. George K. Bertsias, also of the University of Crete in Heraklion and the first author of the guidelines, said in an interview. "On the other hand, lack of improvement at early time points (3-6 months) is associated with adverse prognosis and should evoke discussions for treatment intensification or switch."
This is a different timetable from that described in the ACR guidelines, which advocate switching after patients fail to respond after 6 months of treatment based on the treating physician’s clinical impression.
For patients not responding to mycophenolic acid or cyclophosphamide, treatment may be switched from mycophenolic acid to cyclophosphamide or from cyclophosphamide to mycophenolic acid, according to the guidelines.
If switching fails, rituximab, a biological agent, may be given either as an add-on treatment or as monotherapy. Although randomized controlled trials have failed to demonstrate the superiority of rituximab over standard treatment in lupus nephritis, "there is culminating evidence from several uncontrolled studies and several groups worldwide that rituximab works in about half of patients with nephritis refractory to conventional immunosuppressive therapy," Dr. Boumpas said. "Since rituximab does not have adverse effects on the gonads – a significant issue in the care of young women with lupus – the committee decided to recommend it as an additional treatment resource."
The EULAR guidelines, in contrast to the ACR guidelines, contain specific dosing advice on steroids, advocating pulse steroids (500-1,000 mg of methylprednisolone daily for three doses) in combination with initial immunosuppressive therapy, followed by daily oral glucocorticoids (0.5-1.0 mg/kg per day), afterward tapering to the minimal amount necessary to control disease.
The EULAR guidelines also contain specific recommendations for patients planning pregnancy. In addition, they cover diagnosis and management of pediatric lupus nephritis, which largely follow adult recommendations. The pediatric recommendations are based on evidence in adults, and on nonrandomized evidence in children.
Work on the recommendations was funded by EULAR and the ERA-EDTA. The authors declared no conflicts of interest.
European Renal Association-European Dialysis and Transplant Association, ERA-EDTA, mycophenolic acid, immunosuppressive therapy, Annals of the Rheumatic Diseases, Dr. Dimitrios T. Boumpas,
The European League Against Rheumatism has issued its first guidelines on management of lupus nephritis, and they advise renal biopsy at the first sign of kidney involvement, unlike the guidance issued by the American College of Rheumatology, which leaves timing of that testing up to the clinician’s judgment.
Other bright spots in EULAR’s guidelines, which it issued jointly with the European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) are their position that mycophenolic acid should be the first choice for immunosuppressive therapy, the precise recommendations for steroid dosage, stratification of treatment plans according to disease severity, and explicit advice on switching therapies after one drug has failed.
The European guidelines were published in the November issue of Annals of the Rheumatic Diseases (2012;71:1771-82) and are intended for rheumatologists, nephrologists, and internists managing adult and pediatric patients with lupus nephritis. The EULAR guidelines differ in several key ways from those issued by the American College of Rheumatology (ACR) in June (Arthritis Care Res. 2012;64:797-808).
The EULAR guidelines are unequivocal in their support for renal biopsy at any sign of renal involvement, including when unexplained renal insufficiency is accompanied by normal urinary findings. The ACR guidelines, by contrast, allow for more physician latitude in determining whether to biopsy.
"This can be an emotional issue because rheumatologists do not do biopsies and may try to avoid them," said the EULAR guidelines’ lead author, rheumatologist Dr. Dimitrios T. Boumpas, who is professor of medicine and director of internal medicine/rheumatology at the University of Crete in Heraklion, Greece, in an interview. "We felt that someone should make a position statement. Biopsy is simply best care. If you’re dealing with something severe like LN [lupus nephritis] and you avoid biopsy, that’s not good medicine."
Dr. Boumpas said the EULAR task force had also moved to put mycophenolic acid in the first position as an initial immunosuppressant treatment for most cases of class III-IV LN. Low-dose intravenous cyclophosphamide in combination with steroids is also recommended for this patient group.
There are no 5-year data for mycophenolic acid as there are for cyclophosphamide, Dr. Boumpas said. But the task force that developed the guidelines weighed data on safety and efficacy and found mycophenolic acid "as the clear first choice, while at the same time recognizing the limitations of the studies," he noted.
The ACR guidelines do not recommend use of azathioprine (AZA) as induction treatment. The EULAR recommendations acknowledge that AZA has been associated with a higher risk of renal flares, and call for its use in certain patients who have no adverse clinical or histological risk factors. Patients treated with AZA need close follow-up. "This is particularly important for countries without access to MPA [mycophenolic acid]," Dr. Boumpas said.
The EULAR guidelines also recommend switching to an alternative agent when patients fail to improve in 3-4 months or do not achieve partial response after 6-12 months, or a complete response after 2 years.
"This is based on evidence from both controlled trials and observational cohort studies, which highlight the fact that immunosuppressive agents, particularly cyclophosphamide, may take up to 2 years to achieve complete renal response," Dr. George K. Bertsias, also of the University of Crete in Heraklion and the first author of the guidelines, said in an interview. "On the other hand, lack of improvement at early time points (3-6 months) is associated with adverse prognosis and should evoke discussions for treatment intensification or switch."
This is a different timetable from that described in the ACR guidelines, which advocate switching after patients fail to respond after 6 months of treatment based on the treating physician’s clinical impression.
For patients not responding to mycophenolic acid or cyclophosphamide, treatment may be switched from mycophenolic acid to cyclophosphamide or from cyclophosphamide to mycophenolic acid, according to the guidelines.
If switching fails, rituximab, a biological agent, may be given either as an add-on treatment or as monotherapy. Although randomized controlled trials have failed to demonstrate the superiority of rituximab over standard treatment in lupus nephritis, "there is culminating evidence from several uncontrolled studies and several groups worldwide that rituximab works in about half of patients with nephritis refractory to conventional immunosuppressive therapy," Dr. Boumpas said. "Since rituximab does not have adverse effects on the gonads – a significant issue in the care of young women with lupus – the committee decided to recommend it as an additional treatment resource."
The EULAR guidelines, in contrast to the ACR guidelines, contain specific dosing advice on steroids, advocating pulse steroids (500-1,000 mg of methylprednisolone daily for three doses) in combination with initial immunosuppressive therapy, followed by daily oral glucocorticoids (0.5-1.0 mg/kg per day), afterward tapering to the minimal amount necessary to control disease.
The EULAR guidelines also contain specific recommendations for patients planning pregnancy. In addition, they cover diagnosis and management of pediatric lupus nephritis, which largely follow adult recommendations. The pediatric recommendations are based on evidence in adults, and on nonrandomized evidence in children.
Work on the recommendations was funded by EULAR and the ERA-EDTA. The authors declared no conflicts of interest.
The European League Against Rheumatism has issued its first guidelines on management of lupus nephritis, and they advise renal biopsy at the first sign of kidney involvement, unlike the guidance issued by the American College of Rheumatology, which leaves timing of that testing up to the clinician’s judgment.
Other bright spots in EULAR’s guidelines, which it issued jointly with the European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) are their position that mycophenolic acid should be the first choice for immunosuppressive therapy, the precise recommendations for steroid dosage, stratification of treatment plans according to disease severity, and explicit advice on switching therapies after one drug has failed.
The European guidelines were published in the November issue of Annals of the Rheumatic Diseases (2012;71:1771-82) and are intended for rheumatologists, nephrologists, and internists managing adult and pediatric patients with lupus nephritis. The EULAR guidelines differ in several key ways from those issued by the American College of Rheumatology (ACR) in June (Arthritis Care Res. 2012;64:797-808).
The EULAR guidelines are unequivocal in their support for renal biopsy at any sign of renal involvement, including when unexplained renal insufficiency is accompanied by normal urinary findings. The ACR guidelines, by contrast, allow for more physician latitude in determining whether to biopsy.
"This can be an emotional issue because rheumatologists do not do biopsies and may try to avoid them," said the EULAR guidelines’ lead author, rheumatologist Dr. Dimitrios T. Boumpas, who is professor of medicine and director of internal medicine/rheumatology at the University of Crete in Heraklion, Greece, in an interview. "We felt that someone should make a position statement. Biopsy is simply best care. If you’re dealing with something severe like LN [lupus nephritis] and you avoid biopsy, that’s not good medicine."
Dr. Boumpas said the EULAR task force had also moved to put mycophenolic acid in the first position as an initial immunosuppressant treatment for most cases of class III-IV LN. Low-dose intravenous cyclophosphamide in combination with steroids is also recommended for this patient group.
There are no 5-year data for mycophenolic acid as there are for cyclophosphamide, Dr. Boumpas said. But the task force that developed the guidelines weighed data on safety and efficacy and found mycophenolic acid "as the clear first choice, while at the same time recognizing the limitations of the studies," he noted.
The ACR guidelines do not recommend use of azathioprine (AZA) as induction treatment. The EULAR recommendations acknowledge that AZA has been associated with a higher risk of renal flares, and call for its use in certain patients who have no adverse clinical or histological risk factors. Patients treated with AZA need close follow-up. "This is particularly important for countries without access to MPA [mycophenolic acid]," Dr. Boumpas said.
The EULAR guidelines also recommend switching to an alternative agent when patients fail to improve in 3-4 months or do not achieve partial response after 6-12 months, or a complete response after 2 years.
"This is based on evidence from both controlled trials and observational cohort studies, which highlight the fact that immunosuppressive agents, particularly cyclophosphamide, may take up to 2 years to achieve complete renal response," Dr. George K. Bertsias, also of the University of Crete in Heraklion and the first author of the guidelines, said in an interview. "On the other hand, lack of improvement at early time points (3-6 months) is associated with adverse prognosis and should evoke discussions for treatment intensification or switch."
This is a different timetable from that described in the ACR guidelines, which advocate switching after patients fail to respond after 6 months of treatment based on the treating physician’s clinical impression.
For patients not responding to mycophenolic acid or cyclophosphamide, treatment may be switched from mycophenolic acid to cyclophosphamide or from cyclophosphamide to mycophenolic acid, according to the guidelines.
If switching fails, rituximab, a biological agent, may be given either as an add-on treatment or as monotherapy. Although randomized controlled trials have failed to demonstrate the superiority of rituximab over standard treatment in lupus nephritis, "there is culminating evidence from several uncontrolled studies and several groups worldwide that rituximab works in about half of patients with nephritis refractory to conventional immunosuppressive therapy," Dr. Boumpas said. "Since rituximab does not have adverse effects on the gonads – a significant issue in the care of young women with lupus – the committee decided to recommend it as an additional treatment resource."
The EULAR guidelines, in contrast to the ACR guidelines, contain specific dosing advice on steroids, advocating pulse steroids (500-1,000 mg of methylprednisolone daily for three doses) in combination with initial immunosuppressive therapy, followed by daily oral glucocorticoids (0.5-1.0 mg/kg per day), afterward tapering to the minimal amount necessary to control disease.
The EULAR guidelines also contain specific recommendations for patients planning pregnancy. In addition, they cover diagnosis and management of pediatric lupus nephritis, which largely follow adult recommendations. The pediatric recommendations are based on evidence in adults, and on nonrandomized evidence in children.
Work on the recommendations was funded by EULAR and the ERA-EDTA. The authors declared no conflicts of interest.
European Renal Association-European Dialysis and Transplant Association, ERA-EDTA, mycophenolic acid, immunosuppressive therapy, Annals of the Rheumatic Diseases, Dr. Dimitrios T. Boumpas,
European Renal Association-European Dialysis and Transplant Association, ERA-EDTA, mycophenolic acid, immunosuppressive therapy, Annals of the Rheumatic Diseases, Dr. Dimitrios T. Boumpas,
FROM ANNALS OF THE RHEUMATIC DISEASES
Psoriasis Plus Diabetes Equals Heightened Vascular Risk
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Patients with diabetes and psoriasis had 5-year risks of new-onset microvascular and macrovascular diabetic complications that were 14% and 13% greater, respectively, than in matched psoriasis-free patients with diabetes.
Data Source: This was a retrospective study of 6,164 psoriasis patients with diabetes and an equal number of matched psoriasis-free patients with diabetes.
Disclosures: Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
Experience Proved Rituximab Best for Vasculitis
NEWPORT BEACH, CALIF. – Rituximab is a good remission induction agent in patients with small vessel vasculitis, such as Wegener’s granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome, according to Dr. Leonard Calabrese, chair of clinical immunology and professor of medicine at the Cleveland Clinic.
It appears to be a better choice in some patients than cyclophosphamide, the traditional option, although cyclophosphamide remains the standard of care for remission induction in patients with antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis, he said at Perspectives in Rheumatic Diseases 2012.
The Food and Drug Administration approved rituximab for small vessel vasculitis in 2011 based on a trial that pitted cyclophosphamide induction and azathioprine maintenance against a 6-month regimen of rituximab, with patients in both arms of the study receiving prednisone. They all had severe but not fulminant Wegener’s granulomatosis or microscopic polyangiitis; over half had renal involvement.
Of the 99 rituximab patients, 63 (64%) reached the primary end point of steroid-free remission at 6 months, compared with 52 (53%) of the 98 cyclophosphamide patients. (N. Engl. J. Med. 2010;363:221-32).
At 18 months follow-up, 36% of rituximab patients remained in remission without treatment.
"All the trends in the trial favored rituximab. Patients who came in with [new] disease had equal outcomes. If they had relapsed on [cyclophosphamide], rituximab was significantly better," Dr. Calabrese said.
Given the results, and the fact that rituximab might have fewer long-term side effects, it "is favored as the initial therapy for patients who would otherwise be offered cyclophosphamide. Clearly, rituximab is the drug of choice for [cyclophosphamide] relapsers," he said.
Also based on the results, Dr. Calabrese said he is comfortable stopping therapy and observing patients "with new-onset moderate disease that melts away" with rituximab.
But for patients with a relapsing course or who respond only partially to rituximab, "I don’t favor observation. I think repeated rituximab or an antimetabolite is in order. Most the time, I’ve been doing repeated rituximab," he said.
The optimal rituximab patient remains unclear. "There’s more work to be done on the [study] results. We are looking for predictors," he said.
Meanwhile, "cyclophosphamide and apheresis are still my go-to therapies for patients who are ventilator dependent or have a pulmonary or renal presentation," Dr. Calabrese said.
Azathioprine, methotrexate, leflunomide, and mycophenolate are among the options for step-down therapy when patients are treated traditionally with cyclophosphamide for remission, but findings from a recent study showed "there’s no doubt that azathioprine is more effective than mycophenolate for remission maintenance. Azathioprine, in those who can tolerate it, is quite good," he said (JAMA 2010;304:2381-8).
When patients don’t have life-threatening target organ involvement – no red blood–cell casts in the urine, no hypoxic lung involvement, and normal creatinines and liver functions – methotrexate alone might be the best option.
Such patients "tend to be young and have upper-airway-limited disease. These people are very good methotrexate candidates, 20-25 mg/day with high dose glucocorticoids," he said.
For mild, limited disease, "methotrexate works great," he said.
Dr. Calabrese is a consultant for Aventis, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer. He is a speaker for Amgen. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
NEWPORT BEACH, CALIF. – Rituximab is a good remission induction agent in patients with small vessel vasculitis, such as Wegener’s granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome, according to Dr. Leonard Calabrese, chair of clinical immunology and professor of medicine at the Cleveland Clinic.
It appears to be a better choice in some patients than cyclophosphamide, the traditional option, although cyclophosphamide remains the standard of care for remission induction in patients with antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis, he said at Perspectives in Rheumatic Diseases 2012.
The Food and Drug Administration approved rituximab for small vessel vasculitis in 2011 based on a trial that pitted cyclophosphamide induction and azathioprine maintenance against a 6-month regimen of rituximab, with patients in both arms of the study receiving prednisone. They all had severe but not fulminant Wegener’s granulomatosis or microscopic polyangiitis; over half had renal involvement.
Of the 99 rituximab patients, 63 (64%) reached the primary end point of steroid-free remission at 6 months, compared with 52 (53%) of the 98 cyclophosphamide patients. (N. Engl. J. Med. 2010;363:221-32).
At 18 months follow-up, 36% of rituximab patients remained in remission without treatment.
"All the trends in the trial favored rituximab. Patients who came in with [new] disease had equal outcomes. If they had relapsed on [cyclophosphamide], rituximab was significantly better," Dr. Calabrese said.
Given the results, and the fact that rituximab might have fewer long-term side effects, it "is favored as the initial therapy for patients who would otherwise be offered cyclophosphamide. Clearly, rituximab is the drug of choice for [cyclophosphamide] relapsers," he said.
Also based on the results, Dr. Calabrese said he is comfortable stopping therapy and observing patients "with new-onset moderate disease that melts away" with rituximab.
But for patients with a relapsing course or who respond only partially to rituximab, "I don’t favor observation. I think repeated rituximab or an antimetabolite is in order. Most the time, I’ve been doing repeated rituximab," he said.
The optimal rituximab patient remains unclear. "There’s more work to be done on the [study] results. We are looking for predictors," he said.
Meanwhile, "cyclophosphamide and apheresis are still my go-to therapies for patients who are ventilator dependent or have a pulmonary or renal presentation," Dr. Calabrese said.
Azathioprine, methotrexate, leflunomide, and mycophenolate are among the options for step-down therapy when patients are treated traditionally with cyclophosphamide for remission, but findings from a recent study showed "there’s no doubt that azathioprine is more effective than mycophenolate for remission maintenance. Azathioprine, in those who can tolerate it, is quite good," he said (JAMA 2010;304:2381-8).
When patients don’t have life-threatening target organ involvement – no red blood–cell casts in the urine, no hypoxic lung involvement, and normal creatinines and liver functions – methotrexate alone might be the best option.
Such patients "tend to be young and have upper-airway-limited disease. These people are very good methotrexate candidates, 20-25 mg/day with high dose glucocorticoids," he said.
For mild, limited disease, "methotrexate works great," he said.
Dr. Calabrese is a consultant for Aventis, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer. He is a speaker for Amgen. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
NEWPORT BEACH, CALIF. – Rituximab is a good remission induction agent in patients with small vessel vasculitis, such as Wegener’s granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome, according to Dr. Leonard Calabrese, chair of clinical immunology and professor of medicine at the Cleveland Clinic.
It appears to be a better choice in some patients than cyclophosphamide, the traditional option, although cyclophosphamide remains the standard of care for remission induction in patients with antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis, he said at Perspectives in Rheumatic Diseases 2012.
The Food and Drug Administration approved rituximab for small vessel vasculitis in 2011 based on a trial that pitted cyclophosphamide induction and azathioprine maintenance against a 6-month regimen of rituximab, with patients in both arms of the study receiving prednisone. They all had severe but not fulminant Wegener’s granulomatosis or microscopic polyangiitis; over half had renal involvement.
Of the 99 rituximab patients, 63 (64%) reached the primary end point of steroid-free remission at 6 months, compared with 52 (53%) of the 98 cyclophosphamide patients. (N. Engl. J. Med. 2010;363:221-32).
At 18 months follow-up, 36% of rituximab patients remained in remission without treatment.
"All the trends in the trial favored rituximab. Patients who came in with [new] disease had equal outcomes. If they had relapsed on [cyclophosphamide], rituximab was significantly better," Dr. Calabrese said.
Given the results, and the fact that rituximab might have fewer long-term side effects, it "is favored as the initial therapy for patients who would otherwise be offered cyclophosphamide. Clearly, rituximab is the drug of choice for [cyclophosphamide] relapsers," he said.
Also based on the results, Dr. Calabrese said he is comfortable stopping therapy and observing patients "with new-onset moderate disease that melts away" with rituximab.
But for patients with a relapsing course or who respond only partially to rituximab, "I don’t favor observation. I think repeated rituximab or an antimetabolite is in order. Most the time, I’ve been doing repeated rituximab," he said.
The optimal rituximab patient remains unclear. "There’s more work to be done on the [study] results. We are looking for predictors," he said.
Meanwhile, "cyclophosphamide and apheresis are still my go-to therapies for patients who are ventilator dependent or have a pulmonary or renal presentation," Dr. Calabrese said.
Azathioprine, methotrexate, leflunomide, and mycophenolate are among the options for step-down therapy when patients are treated traditionally with cyclophosphamide for remission, but findings from a recent study showed "there’s no doubt that azathioprine is more effective than mycophenolate for remission maintenance. Azathioprine, in those who can tolerate it, is quite good," he said (JAMA 2010;304:2381-8).
When patients don’t have life-threatening target organ involvement – no red blood–cell casts in the urine, no hypoxic lung involvement, and normal creatinines and liver functions – methotrexate alone might be the best option.
Such patients "tend to be young and have upper-airway-limited disease. These people are very good methotrexate candidates, 20-25 mg/day with high dose glucocorticoids," he said.
For mild, limited disease, "methotrexate works great," he said.
Dr. Calabrese is a consultant for Aventis, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer. He is a speaker for Amgen. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
EXPERT ANALYSIS FROM PERSPECTIVES IN RHEUMATIC DISEASES 2012