User login
AVXS-101 may result in long-term motor improvements in SMA
CHARLOTTE, N.C. – AVXS-101, the Food and Drug Administration–approved therapy for spinal muscular atrophy (SMA), yields rapid, sustained improvements in CHOP INTEND scores, better survival, and motor function improvements at long-term follow-up, according to an analysis presented at the annual meeting of the Child Neurology Society. The results provide a clinical demonstration of continuous expression of the SMN protein, according to the investigators. In addition, AVXS-101 is associated with reduced health care utilization in treated infants, which could decrease costs, lessen the burden on patients and caregivers, and improve quality of life.
SMA1 is a progressive neurologic disease that causes loss of the lower motor neurons in the spinal cord and brainstem. Patients have increasing muscle weakness that leads to death or the need for permanent ventilation by age 2 years. The disease results from mutations in the SMN1 gene. AVXS-101 replaces the missing or nonfunctional SMN1 with a healthy copy of a human SMN gene.
AveXis, the company that developed the therapy, enrolled 12 patients with SMA1 in a phase 1/2a study between December 2014 and December 2015. All participants received one intravenous infusion of AVXS-101. Omar Dabbous, MD, vice president of global health economics, outcomes research, and real world evidence at AveXis in Bannockburn, Ill., and colleagues evaluated participants’ rates of event-free survival (i.e., absence of death or need for permanent ventilation), pulmonary or nutritional interventions, swallowing, hospitalization, and CHOP INTEND scores, as well as therapeutic safety at 2 years.
At study completion, all patients who had received a therapeutic dose had event-free survival. Seven participants did not need daily noninvasive ventilation. Eleven participants had stable or improved swallowing. All of the latter patients fed orally, and six fed exclusively by mouth. Eleven patients spoke.
Participants had a mean of 1.4 respiratory hospitalizations per year. Mean proportion of time participants spent hospitalized was 4.4%. Mean hospitalization rate per year was 2.1, and mean length of hospital stay was 6.7 days. In addition, participants’ CHOP INTEND scores increased from baseline by 9.8 points at 1 month and by 15.4 points at 3 months. Patients who received a therapeutic dose of AVXS-101 have maintained their motor milestones at long-term follow-up, which suggests that treatment effects persist over the long term. Adverse events included elevated serum aminotransferase levels, which were reduced by prednisolone.
Dr. Dabbous is an employee of AveXis, which developed AVXS-101.
SOURCE: Dabbous O et al. CNS 2019. Abstract 199.
CHARLOTTE, N.C. – AVXS-101, the Food and Drug Administration–approved therapy for spinal muscular atrophy (SMA), yields rapid, sustained improvements in CHOP INTEND scores, better survival, and motor function improvements at long-term follow-up, according to an analysis presented at the annual meeting of the Child Neurology Society. The results provide a clinical demonstration of continuous expression of the SMN protein, according to the investigators. In addition, AVXS-101 is associated with reduced health care utilization in treated infants, which could decrease costs, lessen the burden on patients and caregivers, and improve quality of life.
SMA1 is a progressive neurologic disease that causes loss of the lower motor neurons in the spinal cord and brainstem. Patients have increasing muscle weakness that leads to death or the need for permanent ventilation by age 2 years. The disease results from mutations in the SMN1 gene. AVXS-101 replaces the missing or nonfunctional SMN1 with a healthy copy of a human SMN gene.
AveXis, the company that developed the therapy, enrolled 12 patients with SMA1 in a phase 1/2a study between December 2014 and December 2015. All participants received one intravenous infusion of AVXS-101. Omar Dabbous, MD, vice president of global health economics, outcomes research, and real world evidence at AveXis in Bannockburn, Ill., and colleagues evaluated participants’ rates of event-free survival (i.e., absence of death or need for permanent ventilation), pulmonary or nutritional interventions, swallowing, hospitalization, and CHOP INTEND scores, as well as therapeutic safety at 2 years.
At study completion, all patients who had received a therapeutic dose had event-free survival. Seven participants did not need daily noninvasive ventilation. Eleven participants had stable or improved swallowing. All of the latter patients fed orally, and six fed exclusively by mouth. Eleven patients spoke.
Participants had a mean of 1.4 respiratory hospitalizations per year. Mean proportion of time participants spent hospitalized was 4.4%. Mean hospitalization rate per year was 2.1, and mean length of hospital stay was 6.7 days. In addition, participants’ CHOP INTEND scores increased from baseline by 9.8 points at 1 month and by 15.4 points at 3 months. Patients who received a therapeutic dose of AVXS-101 have maintained their motor milestones at long-term follow-up, which suggests that treatment effects persist over the long term. Adverse events included elevated serum aminotransferase levels, which were reduced by prednisolone.
Dr. Dabbous is an employee of AveXis, which developed AVXS-101.
SOURCE: Dabbous O et al. CNS 2019. Abstract 199.
CHARLOTTE, N.C. – AVXS-101, the Food and Drug Administration–approved therapy for spinal muscular atrophy (SMA), yields rapid, sustained improvements in CHOP INTEND scores, better survival, and motor function improvements at long-term follow-up, according to an analysis presented at the annual meeting of the Child Neurology Society. The results provide a clinical demonstration of continuous expression of the SMN protein, according to the investigators. In addition, AVXS-101 is associated with reduced health care utilization in treated infants, which could decrease costs, lessen the burden on patients and caregivers, and improve quality of life.
SMA1 is a progressive neurologic disease that causes loss of the lower motor neurons in the spinal cord and brainstem. Patients have increasing muscle weakness that leads to death or the need for permanent ventilation by age 2 years. The disease results from mutations in the SMN1 gene. AVXS-101 replaces the missing or nonfunctional SMN1 with a healthy copy of a human SMN gene.
AveXis, the company that developed the therapy, enrolled 12 patients with SMA1 in a phase 1/2a study between December 2014 and December 2015. All participants received one intravenous infusion of AVXS-101. Omar Dabbous, MD, vice president of global health economics, outcomes research, and real world evidence at AveXis in Bannockburn, Ill., and colleagues evaluated participants’ rates of event-free survival (i.e., absence of death or need for permanent ventilation), pulmonary or nutritional interventions, swallowing, hospitalization, and CHOP INTEND scores, as well as therapeutic safety at 2 years.
At study completion, all patients who had received a therapeutic dose had event-free survival. Seven participants did not need daily noninvasive ventilation. Eleven participants had stable or improved swallowing. All of the latter patients fed orally, and six fed exclusively by mouth. Eleven patients spoke.
Participants had a mean of 1.4 respiratory hospitalizations per year. Mean proportion of time participants spent hospitalized was 4.4%. Mean hospitalization rate per year was 2.1, and mean length of hospital stay was 6.7 days. In addition, participants’ CHOP INTEND scores increased from baseline by 9.8 points at 1 month and by 15.4 points at 3 months. Patients who received a therapeutic dose of AVXS-101 have maintained their motor milestones at long-term follow-up, which suggests that treatment effects persist over the long term. Adverse events included elevated serum aminotransferase levels, which were reduced by prednisolone.
Dr. Dabbous is an employee of AveXis, which developed AVXS-101.
SOURCE: Dabbous O et al. CNS 2019. Abstract 199.
REPORTING FROM CNS 2019
Serum test sheds light on Merkel cell carcinoma
LAS VEGAS – Merkel cell carcinoma, an extremely rare form of skin cancer, is often caused by a subclinical virus that routinely inhabits the skin. Now, a serum test of virus antibody levels is offering insight into the state of the disease, according to one dermatologist.
“If you have these antibodies, you have a better prognosis. You can follow those antibodies to test for recurrence or progression,” Isaac Brownell, MD, PhD, of the Dermatology Branch of the National Institutes of Health said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The cancer appears in the skin’s Merkel cells, which contribute to our sense of touch by helping us to discriminate textures. “When you put your hand in your pocket, and you can tell the difference between the front and back of a quarter,” he said, “you’re using the Merkel cells in your fingertips.”
Only about 2,500 cases of Merkel cell carcinoma appear in the United States each year, Dr. Brownell said. It appears more often in elderly white patients, is more common in men than women, and is more likely among immunosuppressed patients, whose risk is increased 15- to 20-fold. Cases are more common in sunnier regions – at least in men – and lesions frequently appear on the head, face, and neck.
Five-year survival is estimated at 51% if the cancer is localized, according to a 2016 study of 9,387 cases that Dr. Brownell highlighted. But survival declines dramatically if it has spread to lymph nodes or distant sites (Ann Surg Oncol. 2016 Oct;23[11]:3564-71).
In recent years, researchers have linked 80% of Merkel cell carcinoma cases to the Merkel cell polyomavirus, he said. The virus normally inhabits our skin with no ill effects, he said. “We all have this virus on our skin. It’s everywhere, and even children have antibodies,” he said. But mutations can lead to Merkel cell carcinoma.
Does it matter if cases are polyomavirus positive or polyomavirus negative? Not really, Dr. Brownell said, since the presence of the virus doesn’t appear to affect overall prognosis. However, he said, serum antibody testing can be helpful in polyomavirus-positive patients because it offers insight into prognosis and tumor burden. For example, “if the baseline titer falls and then starts to go up, they’re likely to have a recurrence, and you’ll want to look out for that,” he said.
Dr. Brownell offered another bit of advice: Be prepared to respond to patients who worry that they have a contagious virus and could be a danger to others. The proper answer, he said, is this: “You don’t have to worry about infecting people. Your tumor is not making the virus, you’re not infectious, and we have the virus on us already.”
For more information about the antibody test, visit merkelcell.org/sero.
Dr. Brownell reported having no relevant disclosures. SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Merkel cell carcinoma, an extremely rare form of skin cancer, is often caused by a subclinical virus that routinely inhabits the skin. Now, a serum test of virus antibody levels is offering insight into the state of the disease, according to one dermatologist.
“If you have these antibodies, you have a better prognosis. You can follow those antibodies to test for recurrence or progression,” Isaac Brownell, MD, PhD, of the Dermatology Branch of the National Institutes of Health said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The cancer appears in the skin’s Merkel cells, which contribute to our sense of touch by helping us to discriminate textures. “When you put your hand in your pocket, and you can tell the difference between the front and back of a quarter,” he said, “you’re using the Merkel cells in your fingertips.”
Only about 2,500 cases of Merkel cell carcinoma appear in the United States each year, Dr. Brownell said. It appears more often in elderly white patients, is more common in men than women, and is more likely among immunosuppressed patients, whose risk is increased 15- to 20-fold. Cases are more common in sunnier regions – at least in men – and lesions frequently appear on the head, face, and neck.
Five-year survival is estimated at 51% if the cancer is localized, according to a 2016 study of 9,387 cases that Dr. Brownell highlighted. But survival declines dramatically if it has spread to lymph nodes or distant sites (Ann Surg Oncol. 2016 Oct;23[11]:3564-71).
In recent years, researchers have linked 80% of Merkel cell carcinoma cases to the Merkel cell polyomavirus, he said. The virus normally inhabits our skin with no ill effects, he said. “We all have this virus on our skin. It’s everywhere, and even children have antibodies,” he said. But mutations can lead to Merkel cell carcinoma.
Does it matter if cases are polyomavirus positive or polyomavirus negative? Not really, Dr. Brownell said, since the presence of the virus doesn’t appear to affect overall prognosis. However, he said, serum antibody testing can be helpful in polyomavirus-positive patients because it offers insight into prognosis and tumor burden. For example, “if the baseline titer falls and then starts to go up, they’re likely to have a recurrence, and you’ll want to look out for that,” he said.
Dr. Brownell offered another bit of advice: Be prepared to respond to patients who worry that they have a contagious virus and could be a danger to others. The proper answer, he said, is this: “You don’t have to worry about infecting people. Your tumor is not making the virus, you’re not infectious, and we have the virus on us already.”
For more information about the antibody test, visit merkelcell.org/sero.
Dr. Brownell reported having no relevant disclosures. SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Merkel cell carcinoma, an extremely rare form of skin cancer, is often caused by a subclinical virus that routinely inhabits the skin. Now, a serum test of virus antibody levels is offering insight into the state of the disease, according to one dermatologist.
“If you have these antibodies, you have a better prognosis. You can follow those antibodies to test for recurrence or progression,” Isaac Brownell, MD, PhD, of the Dermatology Branch of the National Institutes of Health said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The cancer appears in the skin’s Merkel cells, which contribute to our sense of touch by helping us to discriminate textures. “When you put your hand in your pocket, and you can tell the difference between the front and back of a quarter,” he said, “you’re using the Merkel cells in your fingertips.”
Only about 2,500 cases of Merkel cell carcinoma appear in the United States each year, Dr. Brownell said. It appears more often in elderly white patients, is more common in men than women, and is more likely among immunosuppressed patients, whose risk is increased 15- to 20-fold. Cases are more common in sunnier regions – at least in men – and lesions frequently appear on the head, face, and neck.
Five-year survival is estimated at 51% if the cancer is localized, according to a 2016 study of 9,387 cases that Dr. Brownell highlighted. But survival declines dramatically if it has spread to lymph nodes or distant sites (Ann Surg Oncol. 2016 Oct;23[11]:3564-71).
In recent years, researchers have linked 80% of Merkel cell carcinoma cases to the Merkel cell polyomavirus, he said. The virus normally inhabits our skin with no ill effects, he said. “We all have this virus on our skin. It’s everywhere, and even children have antibodies,” he said. But mutations can lead to Merkel cell carcinoma.
Does it matter if cases are polyomavirus positive or polyomavirus negative? Not really, Dr. Brownell said, since the presence of the virus doesn’t appear to affect overall prognosis. However, he said, serum antibody testing can be helpful in polyomavirus-positive patients because it offers insight into prognosis and tumor burden. For example, “if the baseline titer falls and then starts to go up, they’re likely to have a recurrence, and you’ll want to look out for that,” he said.
Dr. Brownell offered another bit of advice: Be prepared to respond to patients who worry that they have a contagious virus and could be a danger to others. The proper answer, he said, is this: “You don’t have to worry about infecting people. Your tumor is not making the virus, you’re not infectious, and we have the virus on us already.”
For more information about the antibody test, visit merkelcell.org/sero.
Dr. Brownell reported having no relevant disclosures. SDEF and this news organization are owned by the same parent company.
REPORTING FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
Cilofexor passes phase 2 for primary biliary cholangitis
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.

Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.
Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.
Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
REPORTING FROM THE LIVER MEETING 2019
Ataluren shows real-world benefit for nonsense mutation Duchenne muscular dystrophy
AUSTIN, TEX. – , according to new data.
“Participants in the STRIDE Registry [real-world patients] showed a reduction in functional decline over 48 weeks, compared with patients receiving placebo” in the trial, reported Abdallah Delage of PTC Therapeutics in Zug, Switzerland, and his associates.
Duchenne muscular dystrophy affects an estimated 1 in 3,600-6,000 male births globally, about 10%-15% of whom have nonsense mutation DMD. This mutation causes a truncated, nonfunctional dystrophin protein due to a premature stop codon, the authors explained. Ataluren “promotes ribosomal read-through of the premature stop codon to produce a full-length dystrophin protein,” they explained.
Ataluren is currently approved for ambulatory patients age 2 and older with nonsense mutation DMD in the European Union and several other European countries. Israel, Korea, Chile, and Ukraine have approved it for patients aged 5 and older.
The Strategic Targeting of Registries and International Database of Excellence (STRIDE) Registry contains real-world data from patients using ataluren as part of an ongoing multicenter observational postapproval safety study. The investigators are tracking patients for at least 5 years after enrollment in 14 countries where ataluren is approved or commercially available through early-access programs. Patients take 40 mg/kg daily: 10 mg/kg in the morning, 10 mg/kg midday, and 20 mg/kg in the evening.
The researchers compared outcomes in 216 patients in the STRIDE Registry with participants in a randomized controlled phase 3 study of ataluren involving 228 boys, aged 7-16, who received ataluren (n = 114) or placebo (n = 114) for 48 weeks. Patients were an average 9 years old in STRIDE and in both arms of the randomized controlled trial.
The STRIDE Registry participants, comprising 184 ambulatory and 26 nonambulatory patients at enrollment, had at least 48 weeks between their first and last assessment. All of the patients in the phase 3 study and 88.6% of the STRIDE Registry patients were receiving corticosteroids along with ataluren. The researchers compared the 184 ambulatory STRIDE participants with the participants of the randomized controlled trial for one primary and four secondary endpoints from baseline to 48 weeks.
For the primary endpoint, 6-minute walk distance, average distance was 35 meters shorter than baseline in STRIDE Registry participants (n = 66), 42.2 meters shorter in the patients receiving ataluren in the phase 3 study (n = 109), and 57.6 meters shorter in RCT patients receiving placebo in the phase 3 trial (n = 109).
A secondary endpoint, the time it took patients to walk or run 10 meters, increased 1.6 seconds from baseline to 48 weeks in STRIDE Registry participants (n = 61), 2.3 seconds in participants receiving ataluren in the phase 3 trial (n = 109), and 3.5 seconds in study participants receiving placebo (n = 110).
Another secondary endpoint, the change in time it took for patients to stand from supine position from baseline to 48 weeks, was 2.9 additional seconds for STRIDE participants (n = 55), 3.8 additional seconds in study participants receiving ataluren (n = 101), and 3.9 additional seconds in study participants receiving placebo (n = 96).
Two final secondary endpoints were the changes in time to climb four stairs and to descend four stairs from baseline to 48 weeks. STRIDE participants (n = 47) climbed four stairs 1.2 seconds more slowly at 48 weeks, compared with 2.7 seconds more slowly in the participants who received ataluren in the phase 3 trial (n = 105) and 4.5 seconds more slowly in those who received placebo. Descending four stairs took 0.5 more seconds at 48 weeks in STRIDE participants (n = 40), 2.2 more seconds in participants who received ataluren in the phase 3 trial (n = 106), and 4.0 more seconds in those who received placebo (n = 100).
At least one adverse event occurred in 20.7% of registry participants; seven of these were considered treatment related. Treatment-related side effects included abdominal pain, vomiting, headache, stomach ache, diarrhea, and increased serum lipids.
The study and STRIDE Registry is funded by PTC Therapeutics with TREAT-NMD and the Cooperative International Neuromuscular Research Group. Mr. Delage and five other authors are employees of PTC Therapeutics, and six authors had received speaker or consultancy fees or served on the advisory board of a variety of companies.
SOURCE: Delage A et al. AANEM 2019, Abstract 115.
AUSTIN, TEX. – , according to new data.
“Participants in the STRIDE Registry [real-world patients] showed a reduction in functional decline over 48 weeks, compared with patients receiving placebo” in the trial, reported Abdallah Delage of PTC Therapeutics in Zug, Switzerland, and his associates.
Duchenne muscular dystrophy affects an estimated 1 in 3,600-6,000 male births globally, about 10%-15% of whom have nonsense mutation DMD. This mutation causes a truncated, nonfunctional dystrophin protein due to a premature stop codon, the authors explained. Ataluren “promotes ribosomal read-through of the premature stop codon to produce a full-length dystrophin protein,” they explained.
Ataluren is currently approved for ambulatory patients age 2 and older with nonsense mutation DMD in the European Union and several other European countries. Israel, Korea, Chile, and Ukraine have approved it for patients aged 5 and older.
The Strategic Targeting of Registries and International Database of Excellence (STRIDE) Registry contains real-world data from patients using ataluren as part of an ongoing multicenter observational postapproval safety study. The investigators are tracking patients for at least 5 years after enrollment in 14 countries where ataluren is approved or commercially available through early-access programs. Patients take 40 mg/kg daily: 10 mg/kg in the morning, 10 mg/kg midday, and 20 mg/kg in the evening.
The researchers compared outcomes in 216 patients in the STRIDE Registry with participants in a randomized controlled phase 3 study of ataluren involving 228 boys, aged 7-16, who received ataluren (n = 114) or placebo (n = 114) for 48 weeks. Patients were an average 9 years old in STRIDE and in both arms of the randomized controlled trial.
The STRIDE Registry participants, comprising 184 ambulatory and 26 nonambulatory patients at enrollment, had at least 48 weeks between their first and last assessment. All of the patients in the phase 3 study and 88.6% of the STRIDE Registry patients were receiving corticosteroids along with ataluren. The researchers compared the 184 ambulatory STRIDE participants with the participants of the randomized controlled trial for one primary and four secondary endpoints from baseline to 48 weeks.
For the primary endpoint, 6-minute walk distance, average distance was 35 meters shorter than baseline in STRIDE Registry participants (n = 66), 42.2 meters shorter in the patients receiving ataluren in the phase 3 study (n = 109), and 57.6 meters shorter in RCT patients receiving placebo in the phase 3 trial (n = 109).
A secondary endpoint, the time it took patients to walk or run 10 meters, increased 1.6 seconds from baseline to 48 weeks in STRIDE Registry participants (n = 61), 2.3 seconds in participants receiving ataluren in the phase 3 trial (n = 109), and 3.5 seconds in study participants receiving placebo (n = 110).
Another secondary endpoint, the change in time it took for patients to stand from supine position from baseline to 48 weeks, was 2.9 additional seconds for STRIDE participants (n = 55), 3.8 additional seconds in study participants receiving ataluren (n = 101), and 3.9 additional seconds in study participants receiving placebo (n = 96).
Two final secondary endpoints were the changes in time to climb four stairs and to descend four stairs from baseline to 48 weeks. STRIDE participants (n = 47) climbed four stairs 1.2 seconds more slowly at 48 weeks, compared with 2.7 seconds more slowly in the participants who received ataluren in the phase 3 trial (n = 105) and 4.5 seconds more slowly in those who received placebo. Descending four stairs took 0.5 more seconds at 48 weeks in STRIDE participants (n = 40), 2.2 more seconds in participants who received ataluren in the phase 3 trial (n = 106), and 4.0 more seconds in those who received placebo (n = 100).
At least one adverse event occurred in 20.7% of registry participants; seven of these were considered treatment related. Treatment-related side effects included abdominal pain, vomiting, headache, stomach ache, diarrhea, and increased serum lipids.
The study and STRIDE Registry is funded by PTC Therapeutics with TREAT-NMD and the Cooperative International Neuromuscular Research Group. Mr. Delage and five other authors are employees of PTC Therapeutics, and six authors had received speaker or consultancy fees or served on the advisory board of a variety of companies.
SOURCE: Delage A et al. AANEM 2019, Abstract 115.
AUSTIN, TEX. – , according to new data.
“Participants in the STRIDE Registry [real-world patients] showed a reduction in functional decline over 48 weeks, compared with patients receiving placebo” in the trial, reported Abdallah Delage of PTC Therapeutics in Zug, Switzerland, and his associates.
Duchenne muscular dystrophy affects an estimated 1 in 3,600-6,000 male births globally, about 10%-15% of whom have nonsense mutation DMD. This mutation causes a truncated, nonfunctional dystrophin protein due to a premature stop codon, the authors explained. Ataluren “promotes ribosomal read-through of the premature stop codon to produce a full-length dystrophin protein,” they explained.
Ataluren is currently approved for ambulatory patients age 2 and older with nonsense mutation DMD in the European Union and several other European countries. Israel, Korea, Chile, and Ukraine have approved it for patients aged 5 and older.
The Strategic Targeting of Registries and International Database of Excellence (STRIDE) Registry contains real-world data from patients using ataluren as part of an ongoing multicenter observational postapproval safety study. The investigators are tracking patients for at least 5 years after enrollment in 14 countries where ataluren is approved or commercially available through early-access programs. Patients take 40 mg/kg daily: 10 mg/kg in the morning, 10 mg/kg midday, and 20 mg/kg in the evening.
The researchers compared outcomes in 216 patients in the STRIDE Registry with participants in a randomized controlled phase 3 study of ataluren involving 228 boys, aged 7-16, who received ataluren (n = 114) or placebo (n = 114) for 48 weeks. Patients were an average 9 years old in STRIDE and in both arms of the randomized controlled trial.
The STRIDE Registry participants, comprising 184 ambulatory and 26 nonambulatory patients at enrollment, had at least 48 weeks between their first and last assessment. All of the patients in the phase 3 study and 88.6% of the STRIDE Registry patients were receiving corticosteroids along with ataluren. The researchers compared the 184 ambulatory STRIDE participants with the participants of the randomized controlled trial for one primary and four secondary endpoints from baseline to 48 weeks.
For the primary endpoint, 6-minute walk distance, average distance was 35 meters shorter than baseline in STRIDE Registry participants (n = 66), 42.2 meters shorter in the patients receiving ataluren in the phase 3 study (n = 109), and 57.6 meters shorter in RCT patients receiving placebo in the phase 3 trial (n = 109).
A secondary endpoint, the time it took patients to walk or run 10 meters, increased 1.6 seconds from baseline to 48 weeks in STRIDE Registry participants (n = 61), 2.3 seconds in participants receiving ataluren in the phase 3 trial (n = 109), and 3.5 seconds in study participants receiving placebo (n = 110).
Another secondary endpoint, the change in time it took for patients to stand from supine position from baseline to 48 weeks, was 2.9 additional seconds for STRIDE participants (n = 55), 3.8 additional seconds in study participants receiving ataluren (n = 101), and 3.9 additional seconds in study participants receiving placebo (n = 96).
Two final secondary endpoints were the changes in time to climb four stairs and to descend four stairs from baseline to 48 weeks. STRIDE participants (n = 47) climbed four stairs 1.2 seconds more slowly at 48 weeks, compared with 2.7 seconds more slowly in the participants who received ataluren in the phase 3 trial (n = 105) and 4.5 seconds more slowly in those who received placebo. Descending four stairs took 0.5 more seconds at 48 weeks in STRIDE participants (n = 40), 2.2 more seconds in participants who received ataluren in the phase 3 trial (n = 106), and 4.0 more seconds in those who received placebo (n = 100).
At least one adverse event occurred in 20.7% of registry participants; seven of these were considered treatment related. Treatment-related side effects included abdominal pain, vomiting, headache, stomach ache, diarrhea, and increased serum lipids.
The study and STRIDE Registry is funded by PTC Therapeutics with TREAT-NMD and the Cooperative International Neuromuscular Research Group. Mr. Delage and five other authors are employees of PTC Therapeutics, and six authors had received speaker or consultancy fees or served on the advisory board of a variety of companies.
SOURCE: Delage A et al. AANEM 2019, Abstract 115.
REPORTING FROM AANEM 2019
Better overall survival with nivolumab vs. chemo for advanced ESCC
BARCELONA – Nivolumab was associated with improved overall survival and a favorable safety profile, compared with chemotherapy, in patients with previously treated advanced esophageal squamous cell carcinoma (ESCC) in the open-label phase 3 ATTRACTION-3 study.
The overall survival (OS) benefit was observed regardless of tumor programmed death-ligand 1 (PD-L1) expression, Byoung Chul Cho, MD, reported at the European Society for Medical Oncology Congress.
The findings were reported online simultaneously in The Lancet Oncology.
Median OS at a minimum follow-up of 17.6 months was 10.9 vs. 8.4 months in 210 patients randomized to receive treatment with the PD-1 inhibitor nivolumab and 209 who received chemotherapy, respectively (hazard ratio, 0.77), said Dr. Cho of Yonsei Cancer Center, Yonsei University College of Medicine, Seoul, South Korea.
“Notably, there was a 13% and 10% improvement in overall survival rates at 12 months (47% vs. 34%) and 18 months (31% vs. 21%), respectively,” he said, also noting that the HRs for death favored nivolumab vs. chemotherapy across multiple prespecified subgroups, including those based on tumor PD-L1 expression (HRs, 0.69 and 0.84 for PD-L1 of 1% or greater and less than 1%, respectively).
No meaningful difference was seen in progression-free survival between the nivolumab and chemotherapy groups (12% vs. 7%; HR, 1.08), or in objective response rates (19% vs. 22%), he said.
“However, responses were substantially more durable with nivolumab, compared to chemotherapy; duration of response was 6.9 months with nivolumab vs. 3.9 months in the chemotherapy arm,” he said. “Notably, 21% of patients in the nivolumab arm were still in response, compared to only 6% in the chemotherapy arm.”
Patients enrolled in the open label study had unresectable advanced or recurrent ESCC refractory or intolerant to one prior fluoropyrimidine/platinum-based therapy. They were randomized 1:1 to receive 240 mg of nivolumab every 2 weeks or investigators’ choice of paclitaxel or docetaxel.
Fewer treatment-related adverse events (TRAEs) were reported with nivolumab, Dr. Cho said.
Any grade TRAEs occurred in 66% vs. 95% of patients in the groups, respectively, and grade 3-4 TRAEs occurred in 18% vs. 63%. The majority of select TRAEs – defined as those with potential immunologic etiology, including endocrine, gastrointestinal, hepatic, pulmonary, renal, and skin effects – were grade 1 or 2, and the only difference between the nivolumab and chemotherapy groups with respect to those was in endocrine effects, which affected 11% vs. less than 1% of patients, respectively.
Grade 3/4 select TRAEs occurred in less than 2% of patients, Dr. Cho noted.
An exploratory analysis further showed significant overall improvement in health-related quality of life with nivolumab through week 42 on treatment, he added.
The findings are of note, because metastatic esophageal cancer has a 5-year relative survival rate of less than 8%, and ESCC accounts for about 90% of cases worldwide, he said, adding that current second-line chemotherapy options for ESCC offer poor long-term survival and are associated with toxicity.
Nivolumab, which showed promising antitumor activity and manageable toxicity for advanced ESCC in patients who were refractory to or intolerant of standard chemotherapies in the phase 2 ATTRACTION-1 study, is the first immune checkpoint inhibitor to demonstrate a statistically significant, clinically meaningful improvement in OS vs. chemotherapy in this setting, he said.
The findings of this final analysis of ATTRACTION-3, which shows a 23% reduction in the risk of death, a 2.5-month improvement in median OS, benefit across PD-L1 subgroups, and a favorable safety profile, suggest that nivolumab represents a new standard second-line treatment option for patients with advanced ESCC, he concluded.
ATTRACTION-3 was funded by Ono Pharmaceutical Co., in collaboration with Bristol-Myers Squibb. Dr. Cho reported relationships with Bristol-Myers Squibb, Ono Pharmaceutical, and others. He also reported stock ownership and/or patents with TheraCanVac and Champions Oncology.
SOURCE: Cho B et al. ESMO 2019, Abstract LBA11.
BARCELONA – Nivolumab was associated with improved overall survival and a favorable safety profile, compared with chemotherapy, in patients with previously treated advanced esophageal squamous cell carcinoma (ESCC) in the open-label phase 3 ATTRACTION-3 study.
The overall survival (OS) benefit was observed regardless of tumor programmed death-ligand 1 (PD-L1) expression, Byoung Chul Cho, MD, reported at the European Society for Medical Oncology Congress.
The findings were reported online simultaneously in The Lancet Oncology.
Median OS at a minimum follow-up of 17.6 months was 10.9 vs. 8.4 months in 210 patients randomized to receive treatment with the PD-1 inhibitor nivolumab and 209 who received chemotherapy, respectively (hazard ratio, 0.77), said Dr. Cho of Yonsei Cancer Center, Yonsei University College of Medicine, Seoul, South Korea.
“Notably, there was a 13% and 10% improvement in overall survival rates at 12 months (47% vs. 34%) and 18 months (31% vs. 21%), respectively,” he said, also noting that the HRs for death favored nivolumab vs. chemotherapy across multiple prespecified subgroups, including those based on tumor PD-L1 expression (HRs, 0.69 and 0.84 for PD-L1 of 1% or greater and less than 1%, respectively).
No meaningful difference was seen in progression-free survival between the nivolumab and chemotherapy groups (12% vs. 7%; HR, 1.08), or in objective response rates (19% vs. 22%), he said.
“However, responses were substantially more durable with nivolumab, compared to chemotherapy; duration of response was 6.9 months with nivolumab vs. 3.9 months in the chemotherapy arm,” he said. “Notably, 21% of patients in the nivolumab arm were still in response, compared to only 6% in the chemotherapy arm.”
Patients enrolled in the open label study had unresectable advanced or recurrent ESCC refractory or intolerant to one prior fluoropyrimidine/platinum-based therapy. They were randomized 1:1 to receive 240 mg of nivolumab every 2 weeks or investigators’ choice of paclitaxel or docetaxel.
Fewer treatment-related adverse events (TRAEs) were reported with nivolumab, Dr. Cho said.
Any grade TRAEs occurred in 66% vs. 95% of patients in the groups, respectively, and grade 3-4 TRAEs occurred in 18% vs. 63%. The majority of select TRAEs – defined as those with potential immunologic etiology, including endocrine, gastrointestinal, hepatic, pulmonary, renal, and skin effects – were grade 1 or 2, and the only difference between the nivolumab and chemotherapy groups with respect to those was in endocrine effects, which affected 11% vs. less than 1% of patients, respectively.
Grade 3/4 select TRAEs occurred in less than 2% of patients, Dr. Cho noted.
An exploratory analysis further showed significant overall improvement in health-related quality of life with nivolumab through week 42 on treatment, he added.
The findings are of note, because metastatic esophageal cancer has a 5-year relative survival rate of less than 8%, and ESCC accounts for about 90% of cases worldwide, he said, adding that current second-line chemotherapy options for ESCC offer poor long-term survival and are associated with toxicity.
Nivolumab, which showed promising antitumor activity and manageable toxicity for advanced ESCC in patients who were refractory to or intolerant of standard chemotherapies in the phase 2 ATTRACTION-1 study, is the first immune checkpoint inhibitor to demonstrate a statistically significant, clinically meaningful improvement in OS vs. chemotherapy in this setting, he said.
The findings of this final analysis of ATTRACTION-3, which shows a 23% reduction in the risk of death, a 2.5-month improvement in median OS, benefit across PD-L1 subgroups, and a favorable safety profile, suggest that nivolumab represents a new standard second-line treatment option for patients with advanced ESCC, he concluded.
ATTRACTION-3 was funded by Ono Pharmaceutical Co., in collaboration with Bristol-Myers Squibb. Dr. Cho reported relationships with Bristol-Myers Squibb, Ono Pharmaceutical, and others. He also reported stock ownership and/or patents with TheraCanVac and Champions Oncology.
SOURCE: Cho B et al. ESMO 2019, Abstract LBA11.
BARCELONA – Nivolumab was associated with improved overall survival and a favorable safety profile, compared with chemotherapy, in patients with previously treated advanced esophageal squamous cell carcinoma (ESCC) in the open-label phase 3 ATTRACTION-3 study.
The overall survival (OS) benefit was observed regardless of tumor programmed death-ligand 1 (PD-L1) expression, Byoung Chul Cho, MD, reported at the European Society for Medical Oncology Congress.
The findings were reported online simultaneously in The Lancet Oncology.
Median OS at a minimum follow-up of 17.6 months was 10.9 vs. 8.4 months in 210 patients randomized to receive treatment with the PD-1 inhibitor nivolumab and 209 who received chemotherapy, respectively (hazard ratio, 0.77), said Dr. Cho of Yonsei Cancer Center, Yonsei University College of Medicine, Seoul, South Korea.
“Notably, there was a 13% and 10% improvement in overall survival rates at 12 months (47% vs. 34%) and 18 months (31% vs. 21%), respectively,” he said, also noting that the HRs for death favored nivolumab vs. chemotherapy across multiple prespecified subgroups, including those based on tumor PD-L1 expression (HRs, 0.69 and 0.84 for PD-L1 of 1% or greater and less than 1%, respectively).
No meaningful difference was seen in progression-free survival between the nivolumab and chemotherapy groups (12% vs. 7%; HR, 1.08), or in objective response rates (19% vs. 22%), he said.
“However, responses were substantially more durable with nivolumab, compared to chemotherapy; duration of response was 6.9 months with nivolumab vs. 3.9 months in the chemotherapy arm,” he said. “Notably, 21% of patients in the nivolumab arm were still in response, compared to only 6% in the chemotherapy arm.”
Patients enrolled in the open label study had unresectable advanced or recurrent ESCC refractory or intolerant to one prior fluoropyrimidine/platinum-based therapy. They were randomized 1:1 to receive 240 mg of nivolumab every 2 weeks or investigators’ choice of paclitaxel or docetaxel.
Fewer treatment-related adverse events (TRAEs) were reported with nivolumab, Dr. Cho said.
Any grade TRAEs occurred in 66% vs. 95% of patients in the groups, respectively, and grade 3-4 TRAEs occurred in 18% vs. 63%. The majority of select TRAEs – defined as those with potential immunologic etiology, including endocrine, gastrointestinal, hepatic, pulmonary, renal, and skin effects – were grade 1 or 2, and the only difference between the nivolumab and chemotherapy groups with respect to those was in endocrine effects, which affected 11% vs. less than 1% of patients, respectively.
Grade 3/4 select TRAEs occurred in less than 2% of patients, Dr. Cho noted.
An exploratory analysis further showed significant overall improvement in health-related quality of life with nivolumab through week 42 on treatment, he added.
The findings are of note, because metastatic esophageal cancer has a 5-year relative survival rate of less than 8%, and ESCC accounts for about 90% of cases worldwide, he said, adding that current second-line chemotherapy options for ESCC offer poor long-term survival and are associated with toxicity.
Nivolumab, which showed promising antitumor activity and manageable toxicity for advanced ESCC in patients who were refractory to or intolerant of standard chemotherapies in the phase 2 ATTRACTION-1 study, is the first immune checkpoint inhibitor to demonstrate a statistically significant, clinically meaningful improvement in OS vs. chemotherapy in this setting, he said.
The findings of this final analysis of ATTRACTION-3, which shows a 23% reduction in the risk of death, a 2.5-month improvement in median OS, benefit across PD-L1 subgroups, and a favorable safety profile, suggest that nivolumab represents a new standard second-line treatment option for patients with advanced ESCC, he concluded.
ATTRACTION-3 was funded by Ono Pharmaceutical Co., in collaboration with Bristol-Myers Squibb. Dr. Cho reported relationships with Bristol-Myers Squibb, Ono Pharmaceutical, and others. He also reported stock ownership and/or patents with TheraCanVac and Champions Oncology.
SOURCE: Cho B et al. ESMO 2019, Abstract LBA11.
REPORTING FROM ESMO 2019
Key clinical point: Nivolumab was associated with improved OS vs. chemotherapy, in previously treated advanced ESCC.
Major finding: Median OS was 10.9 vs. 8.4 months with nivolumab vs. chemotherapy, respectively (hazard ratio, 0.77).
Study details: A randomized, open-label, phase 3 study of 419 patients.
Disclosures: ATTRACTION-3 was funded by Ono Pharmaceutical Co., in collaboration with Bristol-Myers Squibb. Dr. Cho reported relationships with Bristol-Myers Squibb, Ono Pharmaceutical, and others. He reported stock ownership and/or patents with TheraCanVac and Champions Oncology.
Source: Cho B et al. ESMO 2019, Abstract LBA11.
Families face challenges of gene therapy
WASHINGTON– Gene therapy for the treatment of rare diseases continues to develop and new products are entering the pipeline; however, more work is needed to make the gene therapy experience easier on patients and their families, according to members of a panel at the NORD Rare Diseases & Orphan Product Breakthrough Summit, held by the National Organization for Rare Disorders.
Companies developing gene therapy cite their main challenges as identifying patients, developing clinical trials, coordinating treatment and supporting families, managing reimbursement, and manufacturing the treatment, said Mark Rothera, president and CEO of Orchard Therapeutics, developer of ex vivo autologous hematopoietic stem cell gene therapy.
For families of patients with rare diseases who are undergoing gene therapy, challenges include struggles such as language barriers, lack of wifi, and separation from other family members for extended periods, according to Amy Price, mother of a gene therapy recipient, as well as principal consultant to Rarallel and an advocate for metachromatic leukodystrophy.
Ms. Price cited a survey she conducted of families with children who underwent gene therapy. She collected data from 16 families about their initial visit as part of a gene therapy trial; the trials included 14 families in Milan; 1 in Bethesda, Md.; and 1 in Paris. The average age of the patients at the start of the trial was 3 years, with a range of 8 months to 11 years. The trials were conducted between 1990 and 2018.
Families participating in the trials spent an average of 5.5 months in the city where the trial was conducted, and an average of 48 days in an isolation ward with their child at the start of the study.
The five biggest challenges were financial well-being (cited by 60% of survey respondents), social isolation/being away from support system (60%), fear of the unknown/long-term treatment diagnosis (73%), family separation (67%), and caring for other children simultaneous during the trial period (60%).
In addition, patients averaged 12 follow-up visits, and the most common secondary challenges cited in the survey included time spent at the hospital, emotional and physical stress on the patient, fear of test results and outcomes, exhaustion, time away from work and school, and travel logistics.
Other stressors include language barriers and not being in children’s hospital, Ms. Price said.
Ms. Price proposed patient-focused solutions such as addressing cultural challenges, connecting families to local resources, and providing clinical follow-up locally to reduce the burden of travel to the trial site.
WASHINGTON– Gene therapy for the treatment of rare diseases continues to develop and new products are entering the pipeline; however, more work is needed to make the gene therapy experience easier on patients and their families, according to members of a panel at the NORD Rare Diseases & Orphan Product Breakthrough Summit, held by the National Organization for Rare Disorders.
Companies developing gene therapy cite their main challenges as identifying patients, developing clinical trials, coordinating treatment and supporting families, managing reimbursement, and manufacturing the treatment, said Mark Rothera, president and CEO of Orchard Therapeutics, developer of ex vivo autologous hematopoietic stem cell gene therapy.
For families of patients with rare diseases who are undergoing gene therapy, challenges include struggles such as language barriers, lack of wifi, and separation from other family members for extended periods, according to Amy Price, mother of a gene therapy recipient, as well as principal consultant to Rarallel and an advocate for metachromatic leukodystrophy.
Ms. Price cited a survey she conducted of families with children who underwent gene therapy. She collected data from 16 families about their initial visit as part of a gene therapy trial; the trials included 14 families in Milan; 1 in Bethesda, Md.; and 1 in Paris. The average age of the patients at the start of the trial was 3 years, with a range of 8 months to 11 years. The trials were conducted between 1990 and 2018.
Families participating in the trials spent an average of 5.5 months in the city where the trial was conducted, and an average of 48 days in an isolation ward with their child at the start of the study.
The five biggest challenges were financial well-being (cited by 60% of survey respondents), social isolation/being away from support system (60%), fear of the unknown/long-term treatment diagnosis (73%), family separation (67%), and caring for other children simultaneous during the trial period (60%).
In addition, patients averaged 12 follow-up visits, and the most common secondary challenges cited in the survey included time spent at the hospital, emotional and physical stress on the patient, fear of test results and outcomes, exhaustion, time away from work and school, and travel logistics.
Other stressors include language barriers and not being in children’s hospital, Ms. Price said.
Ms. Price proposed patient-focused solutions such as addressing cultural challenges, connecting families to local resources, and providing clinical follow-up locally to reduce the burden of travel to the trial site.
WASHINGTON– Gene therapy for the treatment of rare diseases continues to develop and new products are entering the pipeline; however, more work is needed to make the gene therapy experience easier on patients and their families, according to members of a panel at the NORD Rare Diseases & Orphan Product Breakthrough Summit, held by the National Organization for Rare Disorders.
Companies developing gene therapy cite their main challenges as identifying patients, developing clinical trials, coordinating treatment and supporting families, managing reimbursement, and manufacturing the treatment, said Mark Rothera, president and CEO of Orchard Therapeutics, developer of ex vivo autologous hematopoietic stem cell gene therapy.
For families of patients with rare diseases who are undergoing gene therapy, challenges include struggles such as language barriers, lack of wifi, and separation from other family members for extended periods, according to Amy Price, mother of a gene therapy recipient, as well as principal consultant to Rarallel and an advocate for metachromatic leukodystrophy.
Ms. Price cited a survey she conducted of families with children who underwent gene therapy. She collected data from 16 families about their initial visit as part of a gene therapy trial; the trials included 14 families in Milan; 1 in Bethesda, Md.; and 1 in Paris. The average age of the patients at the start of the trial was 3 years, with a range of 8 months to 11 years. The trials were conducted between 1990 and 2018.
Families participating in the trials spent an average of 5.5 months in the city where the trial was conducted, and an average of 48 days in an isolation ward with their child at the start of the study.
The five biggest challenges were financial well-being (cited by 60% of survey respondents), social isolation/being away from support system (60%), fear of the unknown/long-term treatment diagnosis (73%), family separation (67%), and caring for other children simultaneous during the trial period (60%).
In addition, patients averaged 12 follow-up visits, and the most common secondary challenges cited in the survey included time spent at the hospital, emotional and physical stress on the patient, fear of test results and outcomes, exhaustion, time away from work and school, and travel logistics.
Other stressors include language barriers and not being in children’s hospital, Ms. Price said.
Ms. Price proposed patient-focused solutions such as addressing cultural challenges, connecting families to local resources, and providing clinical follow-up locally to reduce the burden of travel to the trial site.
EXPERT ANALYSIS FROM NORD 2019
Adiposis Dolorosa Pain Management
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
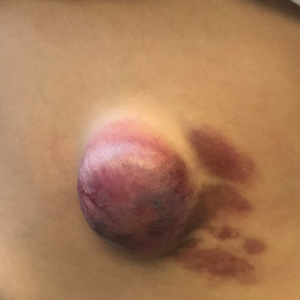
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
Hidradenitis Suppurativa for the Dermatologic Hospitalist
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Practice Points
- Hidradenitis suppurativa (HS) is a common dermatologic condition that frequently presents in emergency and inpatient settings.
- Anemia, leukocytosis, neutrophilia, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level are common markers of chronic inflammation in HS patients and might not signify infection.
- Acute management of HS should focus on anti-inflammatory and antibiotic regimens, with increasing severity dictating the need for more aggressive therapy.
- Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting emergency department and inpatient care utilization.

Experts address barriers to genetic screening
WASHINGTON – Early diagnosis and intervention for genetic diseases using the latest carrier screening can allow families to be prepared and informed prior to pregnancy, said Aishwarya Arjunan, MS, MPH, a clinical product specialist for carrier screening at Myriad Women’s Health, part of a diagnostic testing company based in Salt Lake City, Utah.
“Rare diseases are responsible for 35% of deaths in the first year of life,” she said in a panel discussion at the Rare Diseases and Orphan Products Breakthrough Summit sponsored by the National Organization for Rare Disorders.
Most patients with rare diseases go through a “diagnostic odyssey” lasting an average of 8 years before they receive an accurate diagnosis, she said. During this time, data suggest that they have likely been misdiagnosed three times and have seen more than 10 specialists, she added.
Barriers to genetic screening include limited access to genetics professionals, lack of patient and provider education about screening, issues of insurance coverage and reimbursement, coding challenges, and misperceptions about the perceived impact of screening, noted Jodie Vento, manager of the Center for Rare Disease Therapy at the Children’s Hospital of Pittsburgh.
The genetic carrier screening options, often referred to as panethnic expanded carrier screening, represents a change from previous screening protocols based on ethnicity, said Ms. Arjunan. However, guidelines for screening based on ethnicity “misses a significant percentage of pregnancies affected by serious conditions and widens the health disparity gap,” she said.
By contrast, expanded carrier screening allows for standardization of care that gives couples and families information to make decisions and preparations.
Current genetic testing strategies include single gene testing, in which a single gene of interest is tested; multigene panel testing, in which a subset of clinically important genes are tested; whole-exome sequencing, in which the DNA responsible for coding proteins is tested; and whole-genome sequencing, in which the entire human genome is tested for genetic disorders.
Improving access to genetic testing involves a combination of provider education, changes in payer policies, action by advocacy groups, and adjustment of societal guidelines, said Ms. Arjunan. However, the advantages of expanded carrier screening are many and include guiding patients to expert care early and setting up plans for long-term care and follow-up, she noted. In addition, early identification through screening can help patients reduce or eliminate the diagnostic odyssey and connect with advocacy and community groups for support, she concluded.
The presenters had no financial conflicts to disclose.
WASHINGTON – Early diagnosis and intervention for genetic diseases using the latest carrier screening can allow families to be prepared and informed prior to pregnancy, said Aishwarya Arjunan, MS, MPH, a clinical product specialist for carrier screening at Myriad Women’s Health, part of a diagnostic testing company based in Salt Lake City, Utah.
“Rare diseases are responsible for 35% of deaths in the first year of life,” she said in a panel discussion at the Rare Diseases and Orphan Products Breakthrough Summit sponsored by the National Organization for Rare Disorders.
Most patients with rare diseases go through a “diagnostic odyssey” lasting an average of 8 years before they receive an accurate diagnosis, she said. During this time, data suggest that they have likely been misdiagnosed three times and have seen more than 10 specialists, she added.
Barriers to genetic screening include limited access to genetics professionals, lack of patient and provider education about screening, issues of insurance coverage and reimbursement, coding challenges, and misperceptions about the perceived impact of screening, noted Jodie Vento, manager of the Center for Rare Disease Therapy at the Children’s Hospital of Pittsburgh.
The genetic carrier screening options, often referred to as panethnic expanded carrier screening, represents a change from previous screening protocols based on ethnicity, said Ms. Arjunan. However, guidelines for screening based on ethnicity “misses a significant percentage of pregnancies affected by serious conditions and widens the health disparity gap,” she said.
By contrast, expanded carrier screening allows for standardization of care that gives couples and families information to make decisions and preparations.
Current genetic testing strategies include single gene testing, in which a single gene of interest is tested; multigene panel testing, in which a subset of clinically important genes are tested; whole-exome sequencing, in which the DNA responsible for coding proteins is tested; and whole-genome sequencing, in which the entire human genome is tested for genetic disorders.
Improving access to genetic testing involves a combination of provider education, changes in payer policies, action by advocacy groups, and adjustment of societal guidelines, said Ms. Arjunan. However, the advantages of expanded carrier screening are many and include guiding patients to expert care early and setting up plans for long-term care and follow-up, she noted. In addition, early identification through screening can help patients reduce or eliminate the diagnostic odyssey and connect with advocacy and community groups for support, she concluded.
The presenters had no financial conflicts to disclose.
WASHINGTON – Early diagnosis and intervention for genetic diseases using the latest carrier screening can allow families to be prepared and informed prior to pregnancy, said Aishwarya Arjunan, MS, MPH, a clinical product specialist for carrier screening at Myriad Women’s Health, part of a diagnostic testing company based in Salt Lake City, Utah.
“Rare diseases are responsible for 35% of deaths in the first year of life,” she said in a panel discussion at the Rare Diseases and Orphan Products Breakthrough Summit sponsored by the National Organization for Rare Disorders.
Most patients with rare diseases go through a “diagnostic odyssey” lasting an average of 8 years before they receive an accurate diagnosis, she said. During this time, data suggest that they have likely been misdiagnosed three times and have seen more than 10 specialists, she added.
Barriers to genetic screening include limited access to genetics professionals, lack of patient and provider education about screening, issues of insurance coverage and reimbursement, coding challenges, and misperceptions about the perceived impact of screening, noted Jodie Vento, manager of the Center for Rare Disease Therapy at the Children’s Hospital of Pittsburgh.
The genetic carrier screening options, often referred to as panethnic expanded carrier screening, represents a change from previous screening protocols based on ethnicity, said Ms. Arjunan. However, guidelines for screening based on ethnicity “misses a significant percentage of pregnancies affected by serious conditions and widens the health disparity gap,” she said.
By contrast, expanded carrier screening allows for standardization of care that gives couples and families information to make decisions and preparations.
Current genetic testing strategies include single gene testing, in which a single gene of interest is tested; multigene panel testing, in which a subset of clinically important genes are tested; whole-exome sequencing, in which the DNA responsible for coding proteins is tested; and whole-genome sequencing, in which the entire human genome is tested for genetic disorders.
Improving access to genetic testing involves a combination of provider education, changes in payer policies, action by advocacy groups, and adjustment of societal guidelines, said Ms. Arjunan. However, the advantages of expanded carrier screening are many and include guiding patients to expert care early and setting up plans for long-term care and follow-up, she noted. In addition, early identification through screening can help patients reduce or eliminate the diagnostic odyssey and connect with advocacy and community groups for support, she concluded.
The presenters had no financial conflicts to disclose.
EXPERT ANALYSIS FROM NORD 2019
Neonatal Consultations: Vascular Lumps, Bumps, and Tumors in the Neonate
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).

Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
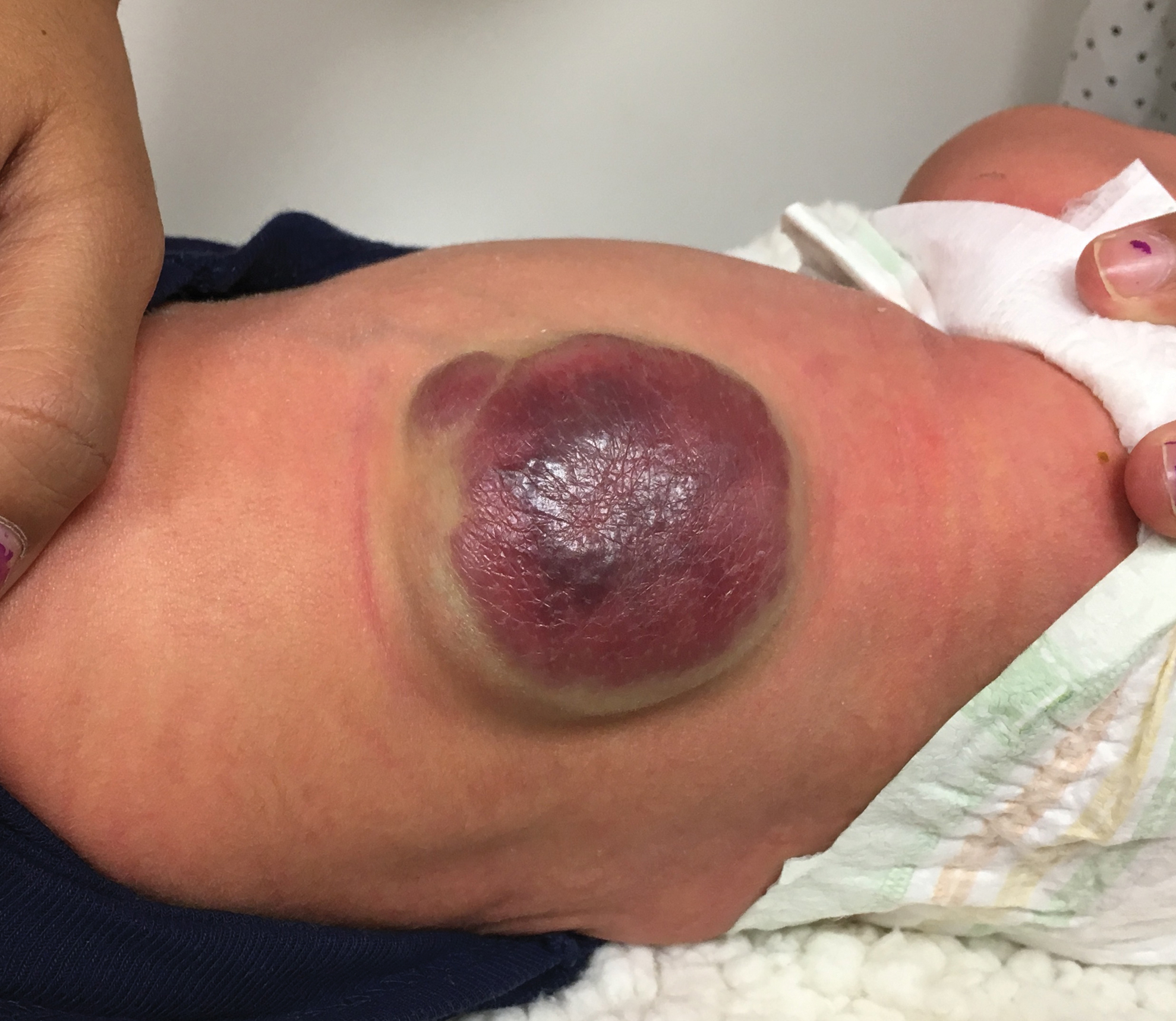
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).
Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).
Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.