User login
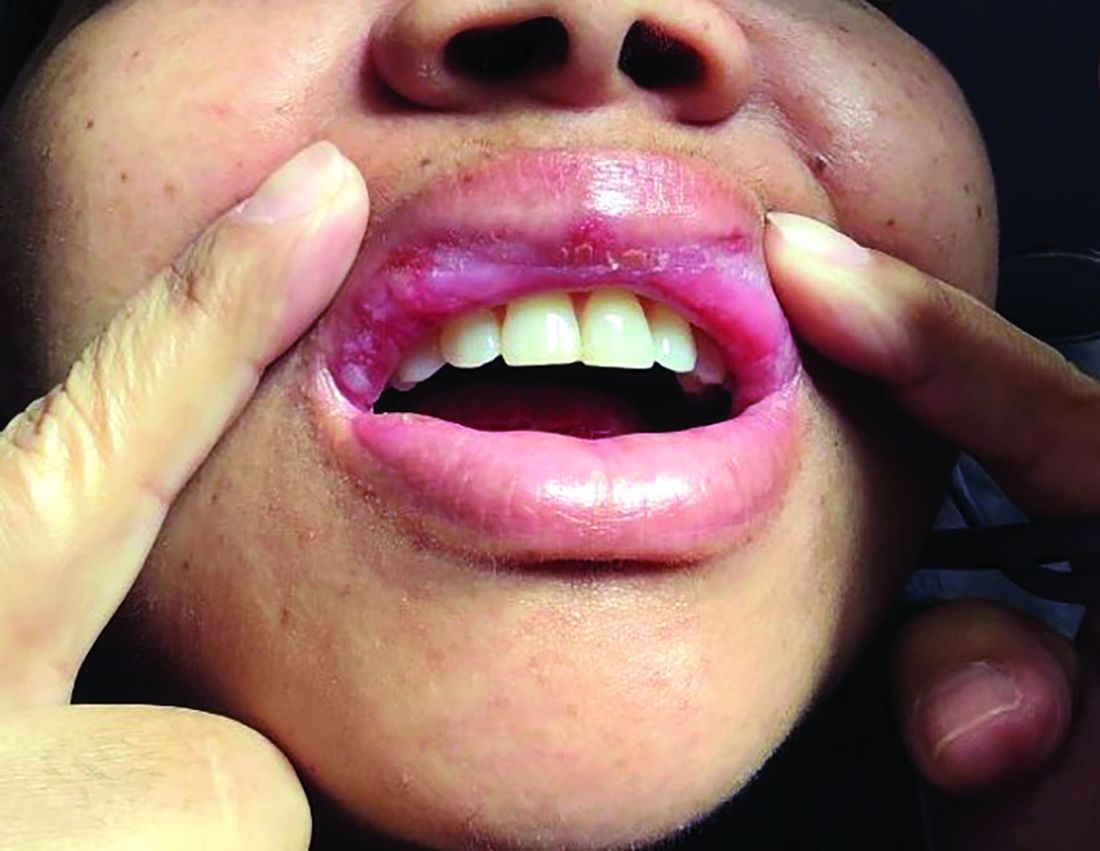
A 42-year-old woman presented with a few days of erosions on her buccal mucosa, tongue, and soft palate
in which lesions present in the same location upon repeated intake of the offending drug. The lesions typically present within 30 minutes to 8 hours of administration of the drug. These reactions can be considered allergic or pseudo-allergic, in which case, there is no notable adaptive immune response. CD8+ T cells appear to play a role in the epidermal injury via release of interferons and interactions with other inflammatory cells.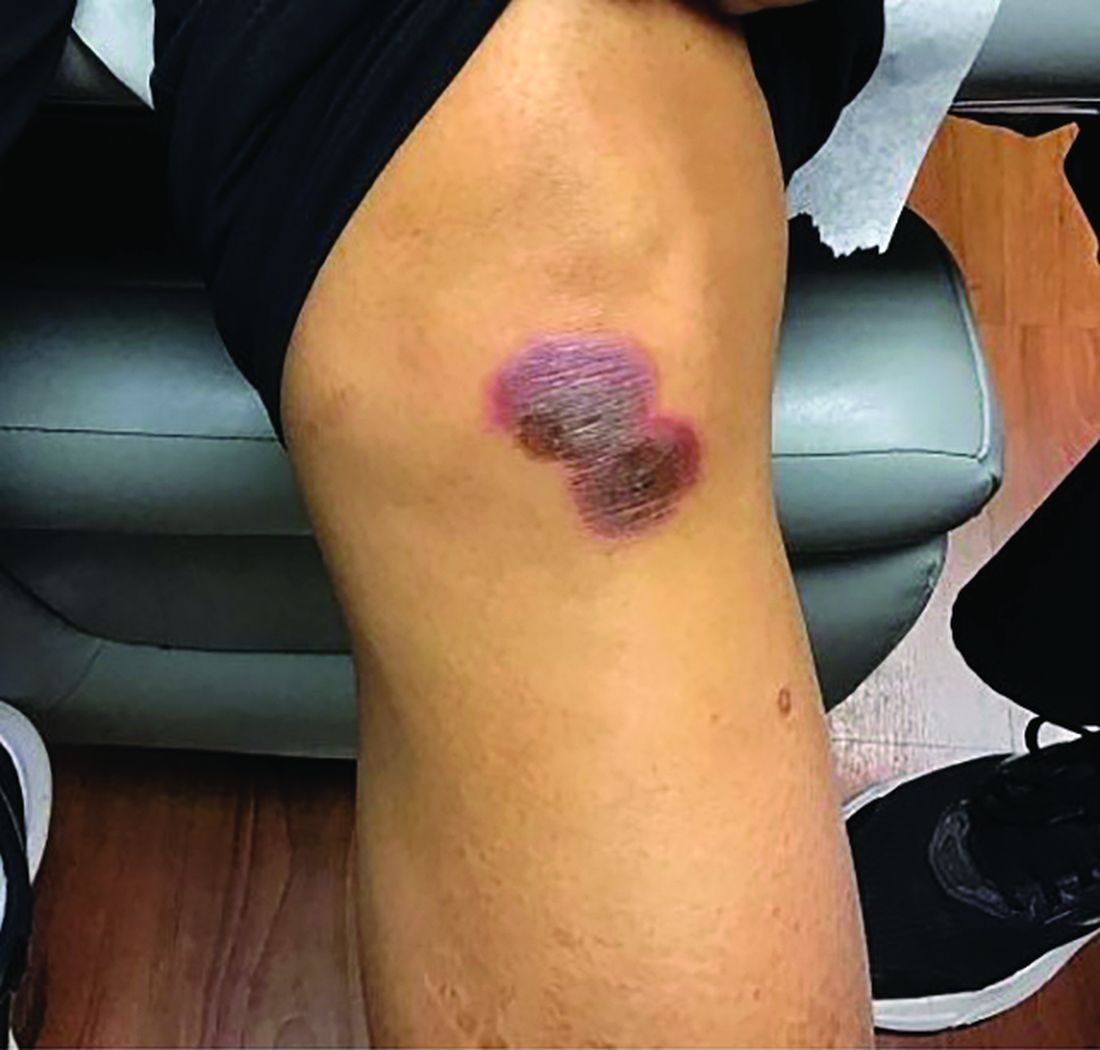
There are numerous drugs that can precipitate these findings. NSAIDs; antibiotics, such as tetracyclines, sulfonamides; and phenytoin are common offenders. In the case of our patient, naproxen was the offending medication.
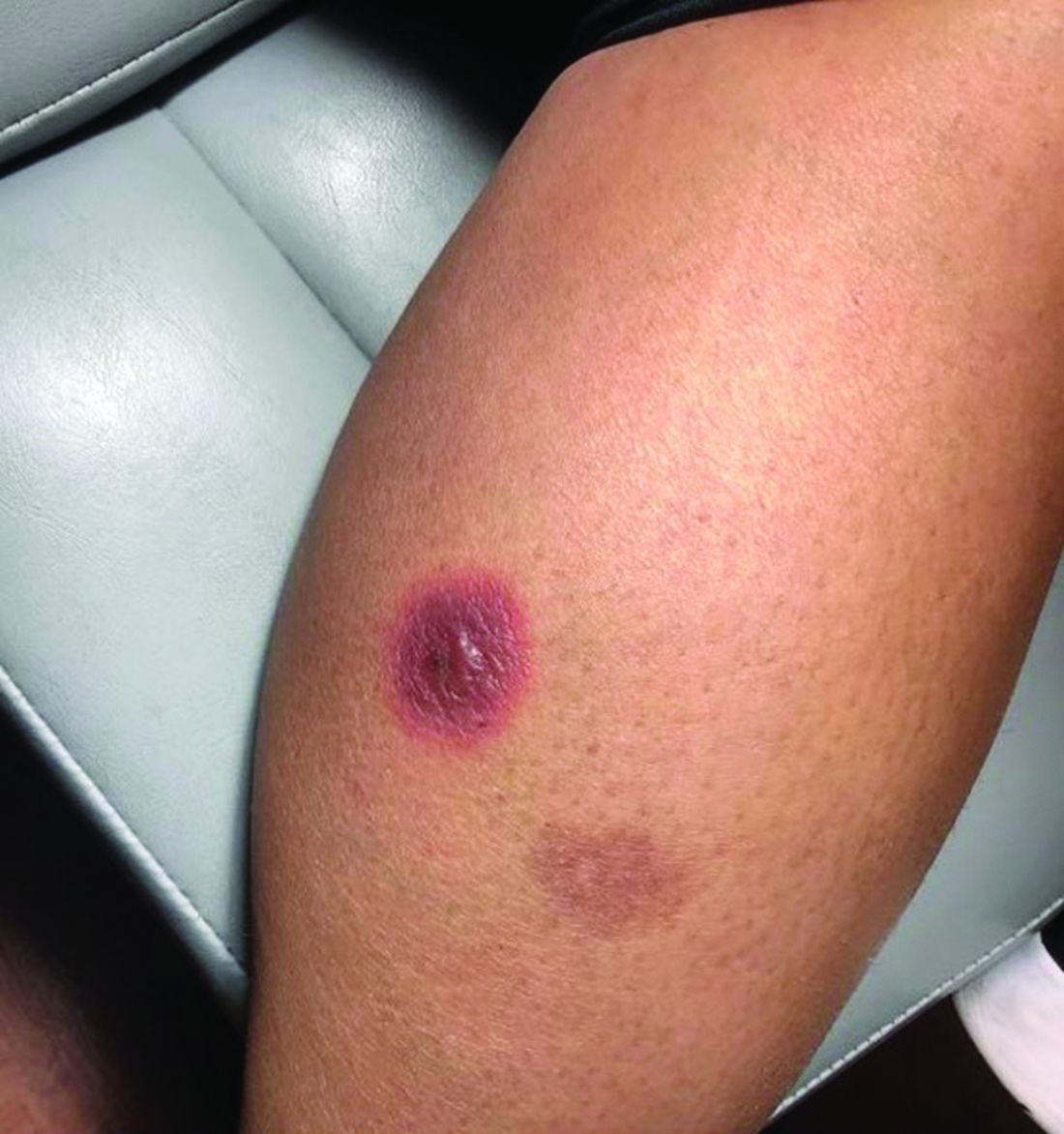
The classic presentation of FDE features annular, erythematous to violaceous macules on the skin or mucosa that can be asymptomatic or can produce burning, pain, or pruritus. The most common locations include the trunk and extremities, but the palms, soles, face, scalp, and mucosa can also be impacted. The oral mucosa seems to be the most common mucosal location. Intravenous administration of a drug is associated with more severe symptoms. Systemic symptoms are typically absent, and the eruption may initially be in one location, but may appear elsewhere upon repeated exposure to the offending medication.

The differential diagnosis includes arthropod bite reactions, urticaria, and erythema multiforme. Although FDEs are typically a clinical diagnosis, the histopathology will commonly show a vacuolar interface dermatitis. Furthermore, a variety of immune cells can be found, including neutrophilic, eosinophilic, and lymphocytic infiltrate. A combination of two or more histological patterns often favors the diagnosis of FDE.
Steroid creams can be prescribed to decrease the inflammatory reaction and improve symptoms; however, the definitive treatment of this condition is cessation of the offending agent. Postinflammatory hyperpigmentation is a common symptom after resolution of the condition, and it may take months to fade away. Further darkening can be prevented by practicing sun safety measures such as wearing sunblock, covering the affected areas, and avoiding prolonged sun exposure.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Igor Chaplik, DO, Aesthetix Dermatology, Fort Lauderdale. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Shaker G et al. Cureus. 2022 Aug 23;14(8):e28299.
Srivastava R et al. Indian J Dent. 2015 Apr-Jun;6(2):103-6.
Weyers W, Metze D. Dermatol Pract Concept. 2011 Jan 31;1(1):33-47.
in which lesions present in the same location upon repeated intake of the offending drug. The lesions typically present within 30 minutes to 8 hours of administration of the drug. These reactions can be considered allergic or pseudo-allergic, in which case, there is no notable adaptive immune response. CD8+ T cells appear to play a role in the epidermal injury via release of interferons and interactions with other inflammatory cells.
There are numerous drugs that can precipitate these findings. NSAIDs; antibiotics, such as tetracyclines, sulfonamides; and phenytoin are common offenders. In the case of our patient, naproxen was the offending medication.
The classic presentation of FDE features annular, erythematous to violaceous macules on the skin or mucosa that can be asymptomatic or can produce burning, pain, or pruritus. The most common locations include the trunk and extremities, but the palms, soles, face, scalp, and mucosa can also be impacted. The oral mucosa seems to be the most common mucosal location. Intravenous administration of a drug is associated with more severe symptoms. Systemic symptoms are typically absent, and the eruption may initially be in one location, but may appear elsewhere upon repeated exposure to the offending medication.
The differential diagnosis includes arthropod bite reactions, urticaria, and erythema multiforme. Although FDEs are typically a clinical diagnosis, the histopathology will commonly show a vacuolar interface dermatitis. Furthermore, a variety of immune cells can be found, including neutrophilic, eosinophilic, and lymphocytic infiltrate. A combination of two or more histological patterns often favors the diagnosis of FDE.
Steroid creams can be prescribed to decrease the inflammatory reaction and improve symptoms; however, the definitive treatment of this condition is cessation of the offending agent. Postinflammatory hyperpigmentation is a common symptom after resolution of the condition, and it may take months to fade away. Further darkening can be prevented by practicing sun safety measures such as wearing sunblock, covering the affected areas, and avoiding prolonged sun exposure.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Igor Chaplik, DO, Aesthetix Dermatology, Fort Lauderdale. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Shaker G et al. Cureus. 2022 Aug 23;14(8):e28299.
Srivastava R et al. Indian J Dent. 2015 Apr-Jun;6(2):103-6.
Weyers W, Metze D. Dermatol Pract Concept. 2011 Jan 31;1(1):33-47.
in which lesions present in the same location upon repeated intake of the offending drug. The lesions typically present within 30 minutes to 8 hours of administration of the drug. These reactions can be considered allergic or pseudo-allergic, in which case, there is no notable adaptive immune response. CD8+ T cells appear to play a role in the epidermal injury via release of interferons and interactions with other inflammatory cells.
There are numerous drugs that can precipitate these findings. NSAIDs; antibiotics, such as tetracyclines, sulfonamides; and phenytoin are common offenders. In the case of our patient, naproxen was the offending medication.
The classic presentation of FDE features annular, erythematous to violaceous macules on the skin or mucosa that can be asymptomatic or can produce burning, pain, or pruritus. The most common locations include the trunk and extremities, but the palms, soles, face, scalp, and mucosa can also be impacted. The oral mucosa seems to be the most common mucosal location. Intravenous administration of a drug is associated with more severe symptoms. Systemic symptoms are typically absent, and the eruption may initially be in one location, but may appear elsewhere upon repeated exposure to the offending medication.
The differential diagnosis includes arthropod bite reactions, urticaria, and erythema multiforme. Although FDEs are typically a clinical diagnosis, the histopathology will commonly show a vacuolar interface dermatitis. Furthermore, a variety of immune cells can be found, including neutrophilic, eosinophilic, and lymphocytic infiltrate. A combination of two or more histological patterns often favors the diagnosis of FDE.
Steroid creams can be prescribed to decrease the inflammatory reaction and improve symptoms; however, the definitive treatment of this condition is cessation of the offending agent. Postinflammatory hyperpigmentation is a common symptom after resolution of the condition, and it may take months to fade away. Further darkening can be prevented by practicing sun safety measures such as wearing sunblock, covering the affected areas, and avoiding prolonged sun exposure.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Igor Chaplik, DO, Aesthetix Dermatology, Fort Lauderdale. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Shaker G et al. Cureus. 2022 Aug 23;14(8):e28299.
Srivastava R et al. Indian J Dent. 2015 Apr-Jun;6(2):103-6.
Weyers W, Metze D. Dermatol Pract Concept. 2011 Jan 31;1(1):33-47.
RVUs: A fair measure of your productivity?
This transcript has been edited for clarity.
The other day, I received a flowery, elaborate email from none other than a physician recruiter: “Beautiful parks, hiking, great schools, blah blah blah, worked RVU production bonus on top of base pay.”
That last part – RVUs. I’m lost. I hear mixed reviews from physicians who work in RVU-based systems. The entire thing seems overly complex and confusing, so let’s clear it up. I did my research, and I’m going to explain RVUs.
Types of RVUs
RVUs, or relative value units, are a standard set by Medicare, used to measure physician productivity and ultimately determine compensation. There are three types:
- Work RVUs (basically everything that happens during a patient encounter).
- Practice expense RVUs.
- Professional liability insurance RVUs.
Now, envision this equation. All three of those RVUs are each multiplied by a geographic practice cost index to come up with a total number, and then that is multiplied by the Medicare conversion factor, which right now is around $33 to $34, to come up with a total dollar amount.
Work RVUs make up the bulk of total RVUs and they get their value from CPT codes. That value is determined by CMS. The AMA’s Relative Value Scale Update Committee, or RUC, which is made up of 32 people from various medical and surgical subspecialties, regularly meets and makes recommendations on the value of various CPT codes.
Is specialty representation fair and balanced?
CMS historically has accepted a high percentage of RUC’s recommendations, so this is a very influential committee. This is also why RUC has led to some controversy, with some stating that there is a lack of primary care representation, and perhaps this is why CPT codes related to procedures tend to reimburse higher.
How does one weigh the value of an hour-long palliative conversation against the quick removal of a benign skin lesion? That’s a loaded question.
This is especially important if your salary, or at least part of it, is determined by total RVUs. You want to have a sense of the pros and cons of working in an RVU system and how this relates to your specialty, your practice, and your schedule.
An RVU-based system provides an objective measure on complex patient encounters, volume, and procedures, and it’s a somewhat unified measure. The cons are pretty clear because these models favor you seeing many patients and billing a lot, and often this favors employers over physicians.
Dr. Patel is a clinical instructor, department of pediatrics, at Columbia University, New York, and a pediatric hospitalist at Morgan Stanley Children’s Hospital of New York–Presbyterian. He reported a conflict of interest with Medumo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
The other day, I received a flowery, elaborate email from none other than a physician recruiter: “Beautiful parks, hiking, great schools, blah blah blah, worked RVU production bonus on top of base pay.”
That last part – RVUs. I’m lost. I hear mixed reviews from physicians who work in RVU-based systems. The entire thing seems overly complex and confusing, so let’s clear it up. I did my research, and I’m going to explain RVUs.
Types of RVUs
RVUs, or relative value units, are a standard set by Medicare, used to measure physician productivity and ultimately determine compensation. There are three types:
- Work RVUs (basically everything that happens during a patient encounter).
- Practice expense RVUs.
- Professional liability insurance RVUs.
Now, envision this equation. All three of those RVUs are each multiplied by a geographic practice cost index to come up with a total number, and then that is multiplied by the Medicare conversion factor, which right now is around $33 to $34, to come up with a total dollar amount.
Work RVUs make up the bulk of total RVUs and they get their value from CPT codes. That value is determined by CMS. The AMA’s Relative Value Scale Update Committee, or RUC, which is made up of 32 people from various medical and surgical subspecialties, regularly meets and makes recommendations on the value of various CPT codes.
Is specialty representation fair and balanced?
CMS historically has accepted a high percentage of RUC’s recommendations, so this is a very influential committee. This is also why RUC has led to some controversy, with some stating that there is a lack of primary care representation, and perhaps this is why CPT codes related to procedures tend to reimburse higher.
How does one weigh the value of an hour-long palliative conversation against the quick removal of a benign skin lesion? That’s a loaded question.
This is especially important if your salary, or at least part of it, is determined by total RVUs. You want to have a sense of the pros and cons of working in an RVU system and how this relates to your specialty, your practice, and your schedule.
An RVU-based system provides an objective measure on complex patient encounters, volume, and procedures, and it’s a somewhat unified measure. The cons are pretty clear because these models favor you seeing many patients and billing a lot, and often this favors employers over physicians.
Dr. Patel is a clinical instructor, department of pediatrics, at Columbia University, New York, and a pediatric hospitalist at Morgan Stanley Children’s Hospital of New York–Presbyterian. He reported a conflict of interest with Medumo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
The other day, I received a flowery, elaborate email from none other than a physician recruiter: “Beautiful parks, hiking, great schools, blah blah blah, worked RVU production bonus on top of base pay.”
That last part – RVUs. I’m lost. I hear mixed reviews from physicians who work in RVU-based systems. The entire thing seems overly complex and confusing, so let’s clear it up. I did my research, and I’m going to explain RVUs.
Types of RVUs
RVUs, or relative value units, are a standard set by Medicare, used to measure physician productivity and ultimately determine compensation. There are three types:
- Work RVUs (basically everything that happens during a patient encounter).
- Practice expense RVUs.
- Professional liability insurance RVUs.
Now, envision this equation. All three of those RVUs are each multiplied by a geographic practice cost index to come up with a total number, and then that is multiplied by the Medicare conversion factor, which right now is around $33 to $34, to come up with a total dollar amount.
Work RVUs make up the bulk of total RVUs and they get their value from CPT codes. That value is determined by CMS. The AMA’s Relative Value Scale Update Committee, or RUC, which is made up of 32 people from various medical and surgical subspecialties, regularly meets and makes recommendations on the value of various CPT codes.
Is specialty representation fair and balanced?
CMS historically has accepted a high percentage of RUC’s recommendations, so this is a very influential committee. This is also why RUC has led to some controversy, with some stating that there is a lack of primary care representation, and perhaps this is why CPT codes related to procedures tend to reimburse higher.
How does one weigh the value of an hour-long palliative conversation against the quick removal of a benign skin lesion? That’s a loaded question.
This is especially important if your salary, or at least part of it, is determined by total RVUs. You want to have a sense of the pros and cons of working in an RVU system and how this relates to your specialty, your practice, and your schedule.
An RVU-based system provides an objective measure on complex patient encounters, volume, and procedures, and it’s a somewhat unified measure. The cons are pretty clear because these models favor you seeing many patients and billing a lot, and often this favors employers over physicians.
Dr. Patel is a clinical instructor, department of pediatrics, at Columbia University, New York, and a pediatric hospitalist at Morgan Stanley Children’s Hospital of New York–Presbyterian. He reported a conflict of interest with Medumo.
A version of this article first appeared on Medscape.com.
‘Vaginal dryness’ can be fatal. No, really.
This transcript has been edited for clarity.
What do you mean, Dr. Rubin? How is vaginal dryness killing women? We minimize the term vaginal dryness. When women come to our offices and complain of a little vaginal dryness – or they don’t even come to our office to complain of it because the doctor can’t be bothered with a little vaginal dryness — what they don’t understand is that this “little vaginal dryness” is really something called genitourinary syndrome of menopause (GSM). They don’t know that because they’ve never heard of it, and you may have never heard of it either. In 2014, we changed the terms vaginal dryness and vulvovaginal atrophy or atrophic vaginitis to GSM to make it short and simple.
GSM – what does it mean? It’s not just a little vaginal dryness. It turns out that all of the genital and urinary symptoms from menopause just get worse over time. The bladder, the urethra, and the vagina have lots of hormone receptors, including estrogen and testosterone. When the body no longer makes those hormones, the system doesn’t work very well, and genital and urinary symptoms occur that just get worse over time without treatment. Unlike hot flashes, which tend to go away, GSM does not.
What are the symptoms of GSM? Some are sexual: a little vaginal dryness, pain with sex, and worsening orgasm. But there are also genital and urinary symptoms that get worse: itching, burning irritation, rawness, an awareness of their genitals that the patient has never had before. And as a urologist, we see frequency, urgency, and leakage.
The thing that kills women is recurrent urinary tract infections (UTIs). Did you know that UTIs account for 7 million visits and hospitalizations annually and 25% of all infections in older people? In fact, apparently one-third of the total Medicare expenditure is around UTIs. Not preventing UTIs is costing our health care system an enormous amount of money and resources.
Did you know we’ve had safe and effective treatment options for GSM since the 1970s? Vaginal hormones have existed since the 1970s, but we’re using them only for pain with sex and not for GSM. In fact, data show that by using vaginal hormones, we can prevent UTIs by more than 50%. We can save lives using safe, effective, local, low-dose vaginal hormone strategies. And they are safe and effective for all of our patients in pre- and post menopause.
There are five different treatment options: vaginal estrogen inserts, vaginal estrogen creams, vaginal dehydroepiandrosterone (DHEA), low-dose vaginal estrogen rings, and an oral pill option called ospemifene (Osphena). All are used to treat GSM and will only work if your patient actually uses them and continues to use them.
These treatments are safe. They are effective. They do not increase the level of systemic hormones in the bloodstream. I have many patients with breast cancer who use these products as well. The only patients you may want to talk to your oncology colleagues about is women on active aromatase inhibitors.
We have to understand that UTIs kill people and having GSM is debilitating, often requiring pain medication because it can hurt to sit or to wear pads and our patients’ quality of life is severely affected. So please consider learning how to treat GSM. It turns out you don’t have to do exams. You don’t have to do follow-up. You can give these therapies, and women can use them for life.
Now, if your patient has vaginal bleeding, of course they need to see their gynecologist. But this is something every primary care doctor can and should do. As a urologist, we prescribe a lot of tamsulosin (Flomax) for our male patients to help with urination. Vaginal estrogen or DHEA is basically like Flomax for women, but it prevents UTIs and actually works like sildenafil (Viagra) because it can help orgasm and reduce pain with sex.
You have access to affordable, safe, effective treatment options to treat GSM. So check them out and hopefully change the world.
Dr. Rubin is an assistant clinical professor in the department of urology at Georgetown University, Washington. She reported conflicts of interest with Sprout, Maternal Medical, Absorption Pharmaceuticals, GlaxoSmithKline, and Endo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
What do you mean, Dr. Rubin? How is vaginal dryness killing women? We minimize the term vaginal dryness. When women come to our offices and complain of a little vaginal dryness – or they don’t even come to our office to complain of it because the doctor can’t be bothered with a little vaginal dryness — what they don’t understand is that this “little vaginal dryness” is really something called genitourinary syndrome of menopause (GSM). They don’t know that because they’ve never heard of it, and you may have never heard of it either. In 2014, we changed the terms vaginal dryness and vulvovaginal atrophy or atrophic vaginitis to GSM to make it short and simple.
GSM – what does it mean? It’s not just a little vaginal dryness. It turns out that all of the genital and urinary symptoms from menopause just get worse over time. The bladder, the urethra, and the vagina have lots of hormone receptors, including estrogen and testosterone. When the body no longer makes those hormones, the system doesn’t work very well, and genital and urinary symptoms occur that just get worse over time without treatment. Unlike hot flashes, which tend to go away, GSM does not.
What are the symptoms of GSM? Some are sexual: a little vaginal dryness, pain with sex, and worsening orgasm. But there are also genital and urinary symptoms that get worse: itching, burning irritation, rawness, an awareness of their genitals that the patient has never had before. And as a urologist, we see frequency, urgency, and leakage.
The thing that kills women is recurrent urinary tract infections (UTIs). Did you know that UTIs account for 7 million visits and hospitalizations annually and 25% of all infections in older people? In fact, apparently one-third of the total Medicare expenditure is around UTIs. Not preventing UTIs is costing our health care system an enormous amount of money and resources.
Did you know we’ve had safe and effective treatment options for GSM since the 1970s? Vaginal hormones have existed since the 1970s, but we’re using them only for pain with sex and not for GSM. In fact, data show that by using vaginal hormones, we can prevent UTIs by more than 50%. We can save lives using safe, effective, local, low-dose vaginal hormone strategies. And they are safe and effective for all of our patients in pre- and post menopause.
There are five different treatment options: vaginal estrogen inserts, vaginal estrogen creams, vaginal dehydroepiandrosterone (DHEA), low-dose vaginal estrogen rings, and an oral pill option called ospemifene (Osphena). All are used to treat GSM and will only work if your patient actually uses them and continues to use them.
These treatments are safe. They are effective. They do not increase the level of systemic hormones in the bloodstream. I have many patients with breast cancer who use these products as well. The only patients you may want to talk to your oncology colleagues about is women on active aromatase inhibitors.
We have to understand that UTIs kill people and having GSM is debilitating, often requiring pain medication because it can hurt to sit or to wear pads and our patients’ quality of life is severely affected. So please consider learning how to treat GSM. It turns out you don’t have to do exams. You don’t have to do follow-up. You can give these therapies, and women can use them for life.
Now, if your patient has vaginal bleeding, of course they need to see their gynecologist. But this is something every primary care doctor can and should do. As a urologist, we prescribe a lot of tamsulosin (Flomax) for our male patients to help with urination. Vaginal estrogen or DHEA is basically like Flomax for women, but it prevents UTIs and actually works like sildenafil (Viagra) because it can help orgasm and reduce pain with sex.
You have access to affordable, safe, effective treatment options to treat GSM. So check them out and hopefully change the world.
Dr. Rubin is an assistant clinical professor in the department of urology at Georgetown University, Washington. She reported conflicts of interest with Sprout, Maternal Medical, Absorption Pharmaceuticals, GlaxoSmithKline, and Endo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
What do you mean, Dr. Rubin? How is vaginal dryness killing women? We minimize the term vaginal dryness. When women come to our offices and complain of a little vaginal dryness – or they don’t even come to our office to complain of it because the doctor can’t be bothered with a little vaginal dryness — what they don’t understand is that this “little vaginal dryness” is really something called genitourinary syndrome of menopause (GSM). They don’t know that because they’ve never heard of it, and you may have never heard of it either. In 2014, we changed the terms vaginal dryness and vulvovaginal atrophy or atrophic vaginitis to GSM to make it short and simple.
GSM – what does it mean? It’s not just a little vaginal dryness. It turns out that all of the genital and urinary symptoms from menopause just get worse over time. The bladder, the urethra, and the vagina have lots of hormone receptors, including estrogen and testosterone. When the body no longer makes those hormones, the system doesn’t work very well, and genital and urinary symptoms occur that just get worse over time without treatment. Unlike hot flashes, which tend to go away, GSM does not.
What are the symptoms of GSM? Some are sexual: a little vaginal dryness, pain with sex, and worsening orgasm. But there are also genital and urinary symptoms that get worse: itching, burning irritation, rawness, an awareness of their genitals that the patient has never had before. And as a urologist, we see frequency, urgency, and leakage.
The thing that kills women is recurrent urinary tract infections (UTIs). Did you know that UTIs account for 7 million visits and hospitalizations annually and 25% of all infections in older people? In fact, apparently one-third of the total Medicare expenditure is around UTIs. Not preventing UTIs is costing our health care system an enormous amount of money and resources.
Did you know we’ve had safe and effective treatment options for GSM since the 1970s? Vaginal hormones have existed since the 1970s, but we’re using them only for pain with sex and not for GSM. In fact, data show that by using vaginal hormones, we can prevent UTIs by more than 50%. We can save lives using safe, effective, local, low-dose vaginal hormone strategies. And they are safe and effective for all of our patients in pre- and post menopause.
There are five different treatment options: vaginal estrogen inserts, vaginal estrogen creams, vaginal dehydroepiandrosterone (DHEA), low-dose vaginal estrogen rings, and an oral pill option called ospemifene (Osphena). All are used to treat GSM and will only work if your patient actually uses them and continues to use them.
These treatments are safe. They are effective. They do not increase the level of systemic hormones in the bloodstream. I have many patients with breast cancer who use these products as well. The only patients you may want to talk to your oncology colleagues about is women on active aromatase inhibitors.
We have to understand that UTIs kill people and having GSM is debilitating, often requiring pain medication because it can hurt to sit or to wear pads and our patients’ quality of life is severely affected. So please consider learning how to treat GSM. It turns out you don’t have to do exams. You don’t have to do follow-up. You can give these therapies, and women can use them for life.
Now, if your patient has vaginal bleeding, of course they need to see their gynecologist. But this is something every primary care doctor can and should do. As a urologist, we prescribe a lot of tamsulosin (Flomax) for our male patients to help with urination. Vaginal estrogen or DHEA is basically like Flomax for women, but it prevents UTIs and actually works like sildenafil (Viagra) because it can help orgasm and reduce pain with sex.
You have access to affordable, safe, effective treatment options to treat GSM. So check them out and hopefully change the world.
Dr. Rubin is an assistant clinical professor in the department of urology at Georgetown University, Washington. She reported conflicts of interest with Sprout, Maternal Medical, Absorption Pharmaceuticals, GlaxoSmithKline, and Endo.
A version of this article first appeared on Medscape.com.
Treatment of the neck and lower face with botulinum toxin
.

The neck and the lower face are covered by thin layers of a vertical muscle, the anterior and posterior platysma muscle that is innervated by the cervical branch of the facial nerve. This muscle superficially blends with the muscles of the lower face, including the depressor anguli oris, depressor labii inferioris, mentalis, risorius, and orbicularis oris muscles. The inferior portion blends with the pectoralis and anterior deltoid muscles and lifts the skin of the neck.
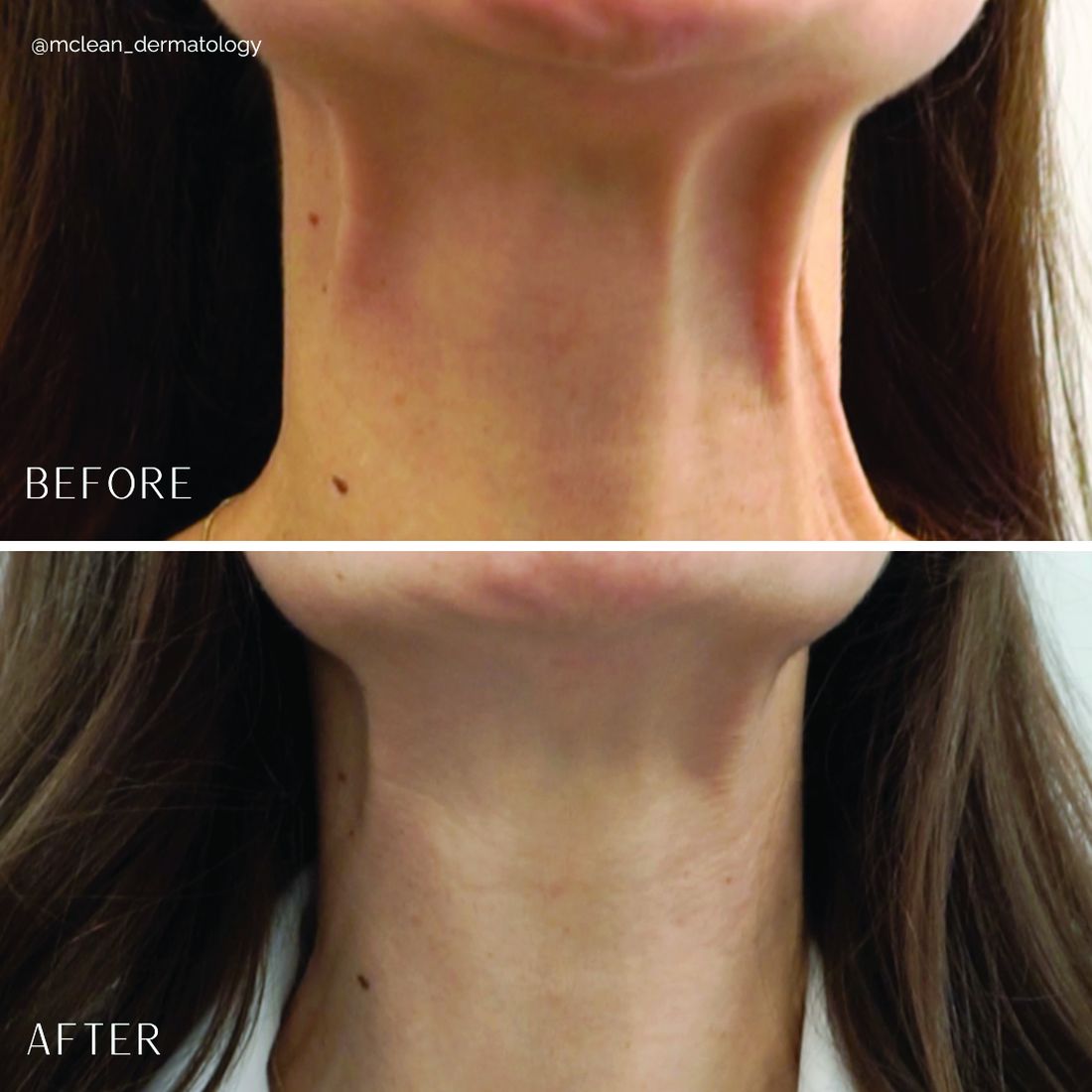
Treatment of the platysma muscle and bands with botulinum toxin is an effective treatment for aging and sagging of the lower face and neck. Although treatment techniques differ and there are currently no standardized guidelines, the treatment starts by having the patient contract the neck muscles (I have them sit upright, with their head completely straight and say “E” with force). After evaluating the tension of the muscle, the muscle should be grasped and pulled away from the neck. Botulinum toxin is injected perpendicular to the muscle, with a dose of approximately 2 units, 2 cm apart along the vertical muscle. Approximately 20-40 units are used for the anterior and lateral bands.
To balance the opposing forces of the depressors of the lower face and improve jowling and downturning of the mouth, 10-20 units are also injected subdermally 1 cm above and 1 cm below the mandibular border.
Understanding the anatomy of the face and neck is crucial to proper injection. Side effects from improper injection include dysphagia, dysphonia, asymmetric smile, and weakness of the neck muscles. It is also important to set realistic expectations and address other components of neck aging, including actinic damage, as well as submental and jowl fat. The manufacturer of onabotulinumtoxinA (Botox Cosmetic) recently announced positive results of a second phase 3 clinical trial evaluating onabotulinumtoxinA for the treatment of moderate to severe platysma prominence. Results of the multicenter, randomized, double blind, placebo-controlled study evaluated the safety and efficacy of one treatment versus placebo in 426 adults with moderate to severe platysmal prominence. The results showed statistically significant improvement of platysma prominence from baseline, based on investigator and patient assessments, with no new safety signals, according to the company. The company expects to submit phase 3 data to the Food and Drug Administration by the end of this year and if approved, it will be the first neurotoxin approved for the treatment of platysmal bands.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Brandt FS, Bellman B. Dermatol Surg. 1998 Nov;24(11):1232-4.
Matarasso A et al. Plast Reconstr Surg. 1999 Feb;103(2):645-52.
Rohrich RJ et al. Plast Reconstr Surg Glob Open. 2020 Jun 23;8(6):e2812.
.
The neck and the lower face are covered by thin layers of a vertical muscle, the anterior and posterior platysma muscle that is innervated by the cervical branch of the facial nerve. This muscle superficially blends with the muscles of the lower face, including the depressor anguli oris, depressor labii inferioris, mentalis, risorius, and orbicularis oris muscles. The inferior portion blends with the pectoralis and anterior deltoid muscles and lifts the skin of the neck.
Treatment of the platysma muscle and bands with botulinum toxin is an effective treatment for aging and sagging of the lower face and neck. Although treatment techniques differ and there are currently no standardized guidelines, the treatment starts by having the patient contract the neck muscles (I have them sit upright, with their head completely straight and say “E” with force). After evaluating the tension of the muscle, the muscle should be grasped and pulled away from the neck. Botulinum toxin is injected perpendicular to the muscle, with a dose of approximately 2 units, 2 cm apart along the vertical muscle. Approximately 20-40 units are used for the anterior and lateral bands.
To balance the opposing forces of the depressors of the lower face and improve jowling and downturning of the mouth, 10-20 units are also injected subdermally 1 cm above and 1 cm below the mandibular border.
Understanding the anatomy of the face and neck is crucial to proper injection. Side effects from improper injection include dysphagia, dysphonia, asymmetric smile, and weakness of the neck muscles. It is also important to set realistic expectations and address other components of neck aging, including actinic damage, as well as submental and jowl fat. The manufacturer of onabotulinumtoxinA (Botox Cosmetic) recently announced positive results of a second phase 3 clinical trial evaluating onabotulinumtoxinA for the treatment of moderate to severe platysma prominence. Results of the multicenter, randomized, double blind, placebo-controlled study evaluated the safety and efficacy of one treatment versus placebo in 426 adults with moderate to severe platysmal prominence. The results showed statistically significant improvement of platysma prominence from baseline, based on investigator and patient assessments, with no new safety signals, according to the company. The company expects to submit phase 3 data to the Food and Drug Administration by the end of this year and if approved, it will be the first neurotoxin approved for the treatment of platysmal bands.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Brandt FS, Bellman B. Dermatol Surg. 1998 Nov;24(11):1232-4.
Matarasso A et al. Plast Reconstr Surg. 1999 Feb;103(2):645-52.
Rohrich RJ et al. Plast Reconstr Surg Glob Open. 2020 Jun 23;8(6):e2812.
.
The neck and the lower face are covered by thin layers of a vertical muscle, the anterior and posterior platysma muscle that is innervated by the cervical branch of the facial nerve. This muscle superficially blends with the muscles of the lower face, including the depressor anguli oris, depressor labii inferioris, mentalis, risorius, and orbicularis oris muscles. The inferior portion blends with the pectoralis and anterior deltoid muscles and lifts the skin of the neck.
Treatment of the platysma muscle and bands with botulinum toxin is an effective treatment for aging and sagging of the lower face and neck. Although treatment techniques differ and there are currently no standardized guidelines, the treatment starts by having the patient contract the neck muscles (I have them sit upright, with their head completely straight and say “E” with force). After evaluating the tension of the muscle, the muscle should be grasped and pulled away from the neck. Botulinum toxin is injected perpendicular to the muscle, with a dose of approximately 2 units, 2 cm apart along the vertical muscle. Approximately 20-40 units are used for the anterior and lateral bands.
To balance the opposing forces of the depressors of the lower face and improve jowling and downturning of the mouth, 10-20 units are also injected subdermally 1 cm above and 1 cm below the mandibular border.
Understanding the anatomy of the face and neck is crucial to proper injection. Side effects from improper injection include dysphagia, dysphonia, asymmetric smile, and weakness of the neck muscles. It is also important to set realistic expectations and address other components of neck aging, including actinic damage, as well as submental and jowl fat. The manufacturer of onabotulinumtoxinA (Botox Cosmetic) recently announced positive results of a second phase 3 clinical trial evaluating onabotulinumtoxinA for the treatment of moderate to severe platysma prominence. Results of the multicenter, randomized, double blind, placebo-controlled study evaluated the safety and efficacy of one treatment versus placebo in 426 adults with moderate to severe platysmal prominence. The results showed statistically significant improvement of platysma prominence from baseline, based on investigator and patient assessments, with no new safety signals, according to the company. The company expects to submit phase 3 data to the Food and Drug Administration by the end of this year and if approved, it will be the first neurotoxin approved for the treatment of platysmal bands.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Brandt FS, Bellman B. Dermatol Surg. 1998 Nov;24(11):1232-4.
Matarasso A et al. Plast Reconstr Surg. 1999 Feb;103(2):645-52.
Rohrich RJ et al. Plast Reconstr Surg Glob Open. 2020 Jun 23;8(6):e2812.
Zuranolone: FAQs for clinicians and patients
The Food and Drug Administration approval of zuranolone for postpartum depression in August 2023 has raised many important questions (and opinions) about its future use in clinical practice.
At the UNC-Chapel Hill Center for Women’s Mood Disorders, we treat women and pregnant people throughout hormonal transitions, including pregnancy and the postpartum, and have been part of development, research, and now delivery of both brexanolone and zuranolone. While we are excited about new tools in the arsenal for alleviating maternal mental health, we also want to be clear that our work is far from complete and continued efforts to care for pregnant people and their families are imperative.

What is zuranolone?
Zuranolone (brand name Zurzuvae) is an oral medication developed by Sage Therapeutics and Biogen. It is a positive allosteric modulator of the GABAA receptor, the brain’s major inhibitory system. As a positive allosteric modulator, it increases the sensitivity of the GABAA receptor to GABA.
Zuranolone is very similar to brexanolone, a synthetic form of allopregnanolone, a neurosteroid byproduct of progesterone (see below). However, zuranolone is not an oral form of brexanolone – it was slightly modified to ensure good oral stability and bioavailability. It is metabolized by the hepatic enzyme CYP3A4 and has a half-life of 16-23 hours. Zurzuvae is currently produced in capsule form.
What does zuranolone treat?
Zuranolone is the first FDA-approved oral drug for postpartum depression (PPD). It follows brexanolone, an intravenous drug, which was the first FDA-approved medication for PPD. Though these are the first medications with specific approval for PDD, many other treatment options are currently available including therapy, SSRIs, serotonin norepinephrine reuptake inhibitors (SNRIs), and other treatments used in major depression.
How does zuranolone work?

Zuranolone is a neuroactive steroid, which means that it is a steroid that goes into and acts on the brain. Zuranolone binds to different GABA receptor subunits from those bound by other positive modulators, such as benzodiazepines (for example, lorazepam). As a synthetic form of allopregnanolone, a metabolite of progesterone which rises dramatically in pregnancy then drops during labor and delivery, zuranolone was originally thought to mitigate the response to this drop in patients that are vulnerable to it during the postpartum. An alternative proposed mechanism is that the increased GABAergic, inhibitory signaling with zuranolone may act directly to decrease depression irrespective of the exact mechanism by which the depression occurred.
How was it studied?
Zuranolone was studied in women with severe postpartum depression and had to meet criteria for major depressive disorder (MDD) no earlier than the third trimester of pregnancy (about 28 weeks’ gestation) and no later than 4 weeks post partum. Patients were excluded from these studies if they had a history of bipolar disorder, psychotic disorders, attempted suicide, or if they were at risk for suicide.
The two phase 3 clinical trials that led to FDA approval are ROBIN and SKYLARK. These studies measured the efficacy and safety of zuranolone at 30 mg and 50 mg, respectively, and met their end points of rapid improvement in depressive and anxiety symptoms in postpartum depression.
When will we be able to start using it?
It is anticipated that zuranolone will become commercially available in early 2024.
Who can prescribe it?

Those with medical licenses. Most people will likely receive treatment from their obstetric, family medicine, or psychiatric clinicians.
How much will it cost?
The manufacturers have not released this information as of August 2023.
What sort of doses and duration is recommended?
The current FDA recommended dose is 50 mg for 14 days, taken once per evening with a fatty meal. The dose can be reduced to 40 mg if there are central nervous system (CNS) depressant effects, and to 30 mg if the patient has severe hepatic or moderate-severe renal impairment. There are currently no studies on longer courses of treatment.
What happens if the patient relapses after a 14-day trial?
While there is no clear guidance, an open-label trial (The SHORELINE Study) demonstrated that a repeated 14-day administration can restore clinical response.
What are the side effects?

Common side effects include drowsiness, dizziness, lower energy, diarrhea, and symptoms similar to the common cold. Zuranolone can act like a CNS depressant and can lead to sedation and somnolence.
Are there any boxed warnings?
Because of the CNS depressant effects, zuranolone was given a boxed warning that patients should not drive or operate heavy machinery within 12 hours of taking the medication as it may lead to impairment. Similar to other antidepressants, there is also a warning that zuranolone may increase risk for suicidal thoughts in patients under 24 years old.
Can it be used with other medications?
Yes. In the original trials, women were allowed to remain on medications treating their depressive symptoms (such as SSRIs and SNRIs). According to the FDA, zuranolone can be used alone or with other antidepressants.
Are there any medicines to avoid?
We recommend caution with other medications which may increase sedation, such as benzodiazepines.
Can it be used with birth control?
Yes. In fact, because the outcomes on a fetus are not yet studied, it is recommended that patients be on concurrent birth control during treatment and for a week after cessation. This does not mean that zuranolone is known to cause issues with fetal development, but rather that we do not know at this time.
Can it be used in pregnancy?
As above, the outcomes on fetal development are not known at this time, nor are the effects of zuranolone on labor and delivery. More research will need to be done to understand if there is risk with taking zuranolone during pregnancy. It should be noted that allopregnanolone levels ordinarily reach quite high levels during pregnancy.
Long-term side effects?
Long-term side effects are unknown. The study duration of ROBIN and SKYLARK was 45 days.
Breastfeeding?
Use in lactation has not yet been studied. Continued research is needed.
Can it be used in mood changes related to other reproductive changes or diagnoses like premenstrual dysphoric disorder and perimenopause?
The mechanism by which zuranolone is thought to work – that is, during changes in reproductive hormones – is implicated in other reproductive transitions such as premenstrual dysphoric disorder and perimenopause when reproductive hormones are fluctuating, though at lower levels than in pregnancy. Research will be required to assess efficacy and safety; however, the mechanistic reasons is worth pursuing. Additionally, zuranolone has not been studied in postpartum psychosis.
Can zuranolone be used to treat other affective conditions besides postpartum depression? Bipolar disorder?
Whether it may be beneficial for patients with a depressive episode that is part of an underlying bipolar disorder or other psychiatric illness is not yet known.
Anxiety?
Along with depressive symptoms, women who received zuranolone in the clinical trials also had improvements in anxiety symptoms. These findings provide some hope that zuranolone may eventually be beneficial in patients with anxiety.
However, to date zuranolone has not been directly studied as a treatment for anxiety disorders (such as generalized anxiety disorder, panic disorder, etc.), so its efficacy for these illnesses is currently unknown.
Insomnia?
In a study of 153 postpartum women, randomized to placebo or zuranolone, scale questions for insomnia were improved in the group receiving zuranolone. This provides some hope that, if zuranolone is appropriate, concurrent polypharmacy with a sleep aid can be avoided. Additionally, future evaluation of use in insomnia outside of PPD may be warranted.
How is it different from brexanolone?
The two are slightly different molecules. Brexanolone is synthetically identical to allopregnanolone and zuranolone has been altered to be active and orally bioavailable.
Brexanolone is a 60-hour infusion that requires hospital admission at an approved health care site. Zuranolone is an oral at-home once-daily dosing treatment for 14 days. Zuranolone does not require enrollment in a risk evaluation and mitigation strategy for risk of excessive sedation and sudden loss of consciousness.
When would you consider zuranolone vs. brexanolone vs. other antidepressants?
Zuranolone and brexanolone are rapid-acting antidepressants with a response within 14 days or 60 hours, respectively. Antidepressants such as SSRIs/SNRIs are still available, well studied, and work, although take longer to reach clinical efficacy and are accompanied by potentially troubling side effects (for example, weight gain, sexual dysfunction).

Time to treatment effect should be considered when assessing severity of symptoms and functional impairment of the mother and the overall family unit. Brexanolone requires continuous monitoring which may be beneficial for women who are severely impaired and may benefit from frequent clinical monitoring. Brexanolone does not require a dose reduction with hepatic impairment, however, should be avoided in end-stage renal disease because of the potential accumulation of the solubilizing agent.
Where can I find more information?
Many states have maternal mental health consultation lines (examples include NCMATTERS here in North Carolina and MCPAP for Moms in Massachusetts) for clinicians (mental health, primary care, and obstetricians) that can be utilized for questions about prescribing. Postpartum Support International also has a clinician line for those without state services.
We plan to update this entry upon market release and access to new information.
Dr. Riddle and Dr. Nathan are assistant professors in the department of psychiatry at the University of North Carolina at Chapel Hill. Dr. Richardson is a perinatal psychiatry fellow, department of psychiatry, UNC-Chapel Hill. Dr. Rubinow is Distinguished Professor in the department of psychiatry, UNC-Chapel Hill. Dr. Meltzer-Brody is Assad Meymandi Distinguished Professor and Chair, department of psychiatry, UNC-Chapel Hill.
References
Deligiannidis KM et al. J Clin Psychiatry. 2023 Jan 30;84(1):22m14475. doi: 10.4088/JCP.22m14475.
Deligiannidis KM et al. . Obstetrics & Gynecology. 2023 May;141(5S):64S-65S. doi: 10.1097/01.AOG.0000930588.16136.3f.
Deligiannidis KM et al. Am J Psychiatry. 2023 Sep 1;180(9):668-75. doi: 10.1176/appi.ajp.20220785.
Deligiannidis KM et al. JAMA Psychiatry. 2021 Sep 1;78(9):951-59. doi: 10.1001/jamapsychiatry.2021.1559.
FDA Approves First Oral Treatment for Postpartum Depression. 2023 Aug 4. https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-treatment-postpartum-depression
ZURZUVAE – HIGHLIGHTS OF PRESCRIBING INFORMATION. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217369s000lbl.pdf
The Food and Drug Administration approval of zuranolone for postpartum depression in August 2023 has raised many important questions (and opinions) about its future use in clinical practice.
At the UNC-Chapel Hill Center for Women’s Mood Disorders, we treat women and pregnant people throughout hormonal transitions, including pregnancy and the postpartum, and have been part of development, research, and now delivery of both brexanolone and zuranolone. While we are excited about new tools in the arsenal for alleviating maternal mental health, we also want to be clear that our work is far from complete and continued efforts to care for pregnant people and their families are imperative.
What is zuranolone?
Zuranolone (brand name Zurzuvae) is an oral medication developed by Sage Therapeutics and Biogen. It is a positive allosteric modulator of the GABAA receptor, the brain’s major inhibitory system. As a positive allosteric modulator, it increases the sensitivity of the GABAA receptor to GABA.
Zuranolone is very similar to brexanolone, a synthetic form of allopregnanolone, a neurosteroid byproduct of progesterone (see below). However, zuranolone is not an oral form of brexanolone – it was slightly modified to ensure good oral stability and bioavailability. It is metabolized by the hepatic enzyme CYP3A4 and has a half-life of 16-23 hours. Zurzuvae is currently produced in capsule form.
What does zuranolone treat?
Zuranolone is the first FDA-approved oral drug for postpartum depression (PPD). It follows brexanolone, an intravenous drug, which was the first FDA-approved medication for PPD. Though these are the first medications with specific approval for PDD, many other treatment options are currently available including therapy, SSRIs, serotonin norepinephrine reuptake inhibitors (SNRIs), and other treatments used in major depression.
How does zuranolone work?
Zuranolone is a neuroactive steroid, which means that it is a steroid that goes into and acts on the brain. Zuranolone binds to different GABA receptor subunits from those bound by other positive modulators, such as benzodiazepines (for example, lorazepam). As a synthetic form of allopregnanolone, a metabolite of progesterone which rises dramatically in pregnancy then drops during labor and delivery, zuranolone was originally thought to mitigate the response to this drop in patients that are vulnerable to it during the postpartum. An alternative proposed mechanism is that the increased GABAergic, inhibitory signaling with zuranolone may act directly to decrease depression irrespective of the exact mechanism by which the depression occurred.
How was it studied?
Zuranolone was studied in women with severe postpartum depression and had to meet criteria for major depressive disorder (MDD) no earlier than the third trimester of pregnancy (about 28 weeks’ gestation) and no later than 4 weeks post partum. Patients were excluded from these studies if they had a history of bipolar disorder, psychotic disorders, attempted suicide, or if they were at risk for suicide.
The two phase 3 clinical trials that led to FDA approval are ROBIN and SKYLARK. These studies measured the efficacy and safety of zuranolone at 30 mg and 50 mg, respectively, and met their end points of rapid improvement in depressive and anxiety symptoms in postpartum depression.
When will we be able to start using it?
It is anticipated that zuranolone will become commercially available in early 2024.
Who can prescribe it?
Those with medical licenses. Most people will likely receive treatment from their obstetric, family medicine, or psychiatric clinicians.
How much will it cost?
The manufacturers have not released this information as of August 2023.
What sort of doses and duration is recommended?
The current FDA recommended dose is 50 mg for 14 days, taken once per evening with a fatty meal. The dose can be reduced to 40 mg if there are central nervous system (CNS) depressant effects, and to 30 mg if the patient has severe hepatic or moderate-severe renal impairment. There are currently no studies on longer courses of treatment.
What happens if the patient relapses after a 14-day trial?
While there is no clear guidance, an open-label trial (The SHORELINE Study) demonstrated that a repeated 14-day administration can restore clinical response.
What are the side effects?
Common side effects include drowsiness, dizziness, lower energy, diarrhea, and symptoms similar to the common cold. Zuranolone can act like a CNS depressant and can lead to sedation and somnolence.
Are there any boxed warnings?
Because of the CNS depressant effects, zuranolone was given a boxed warning that patients should not drive or operate heavy machinery within 12 hours of taking the medication as it may lead to impairment. Similar to other antidepressants, there is also a warning that zuranolone may increase risk for suicidal thoughts in patients under 24 years old.
Can it be used with other medications?
Yes. In the original trials, women were allowed to remain on medications treating their depressive symptoms (such as SSRIs and SNRIs). According to the FDA, zuranolone can be used alone or with other antidepressants.
Are there any medicines to avoid?
We recommend caution with other medications which may increase sedation, such as benzodiazepines.
Can it be used with birth control?
Yes. In fact, because the outcomes on a fetus are not yet studied, it is recommended that patients be on concurrent birth control during treatment and for a week after cessation. This does not mean that zuranolone is known to cause issues with fetal development, but rather that we do not know at this time.
Can it be used in pregnancy?
As above, the outcomes on fetal development are not known at this time, nor are the effects of zuranolone on labor and delivery. More research will need to be done to understand if there is risk with taking zuranolone during pregnancy. It should be noted that allopregnanolone levels ordinarily reach quite high levels during pregnancy.
Long-term side effects?
Long-term side effects are unknown. The study duration of ROBIN and SKYLARK was 45 days.
Breastfeeding?
Use in lactation has not yet been studied. Continued research is needed.
Can it be used in mood changes related to other reproductive changes or diagnoses like premenstrual dysphoric disorder and perimenopause?
The mechanism by which zuranolone is thought to work – that is, during changes in reproductive hormones – is implicated in other reproductive transitions such as premenstrual dysphoric disorder and perimenopause when reproductive hormones are fluctuating, though at lower levels than in pregnancy. Research will be required to assess efficacy and safety; however, the mechanistic reasons is worth pursuing. Additionally, zuranolone has not been studied in postpartum psychosis.
Can zuranolone be used to treat other affective conditions besides postpartum depression? Bipolar disorder?
Whether it may be beneficial for patients with a depressive episode that is part of an underlying bipolar disorder or other psychiatric illness is not yet known.
Anxiety?
Along with depressive symptoms, women who received zuranolone in the clinical trials also had improvements in anxiety symptoms. These findings provide some hope that zuranolone may eventually be beneficial in patients with anxiety.
However, to date zuranolone has not been directly studied as a treatment for anxiety disorders (such as generalized anxiety disorder, panic disorder, etc.), so its efficacy for these illnesses is currently unknown.
Insomnia?
In a study of 153 postpartum women, randomized to placebo or zuranolone, scale questions for insomnia were improved in the group receiving zuranolone. This provides some hope that, if zuranolone is appropriate, concurrent polypharmacy with a sleep aid can be avoided. Additionally, future evaluation of use in insomnia outside of PPD may be warranted.
How is it different from brexanolone?
The two are slightly different molecules. Brexanolone is synthetically identical to allopregnanolone and zuranolone has been altered to be active and orally bioavailable.
Brexanolone is a 60-hour infusion that requires hospital admission at an approved health care site. Zuranolone is an oral at-home once-daily dosing treatment for 14 days. Zuranolone does not require enrollment in a risk evaluation and mitigation strategy for risk of excessive sedation and sudden loss of consciousness.
When would you consider zuranolone vs. brexanolone vs. other antidepressants?
Zuranolone and brexanolone are rapid-acting antidepressants with a response within 14 days or 60 hours, respectively. Antidepressants such as SSRIs/SNRIs are still available, well studied, and work, although take longer to reach clinical efficacy and are accompanied by potentially troubling side effects (for example, weight gain, sexual dysfunction).
Time to treatment effect should be considered when assessing severity of symptoms and functional impairment of the mother and the overall family unit. Brexanolone requires continuous monitoring which may be beneficial for women who are severely impaired and may benefit from frequent clinical monitoring. Brexanolone does not require a dose reduction with hepatic impairment, however, should be avoided in end-stage renal disease because of the potential accumulation of the solubilizing agent.
Where can I find more information?
Many states have maternal mental health consultation lines (examples include NCMATTERS here in North Carolina and MCPAP for Moms in Massachusetts) for clinicians (mental health, primary care, and obstetricians) that can be utilized for questions about prescribing. Postpartum Support International also has a clinician line for those without state services.
We plan to update this entry upon market release and access to new information.
Dr. Riddle and Dr. Nathan are assistant professors in the department of psychiatry at the University of North Carolina at Chapel Hill. Dr. Richardson is a perinatal psychiatry fellow, department of psychiatry, UNC-Chapel Hill. Dr. Rubinow is Distinguished Professor in the department of psychiatry, UNC-Chapel Hill. Dr. Meltzer-Brody is Assad Meymandi Distinguished Professor and Chair, department of psychiatry, UNC-Chapel Hill.
References
Deligiannidis KM et al. J Clin Psychiatry. 2023 Jan 30;84(1):22m14475. doi: 10.4088/JCP.22m14475.
Deligiannidis KM et al. . Obstetrics & Gynecology. 2023 May;141(5S):64S-65S. doi: 10.1097/01.AOG.0000930588.16136.3f.
Deligiannidis KM et al. Am J Psychiatry. 2023 Sep 1;180(9):668-75. doi: 10.1176/appi.ajp.20220785.
Deligiannidis KM et al. JAMA Psychiatry. 2021 Sep 1;78(9):951-59. doi: 10.1001/jamapsychiatry.2021.1559.
FDA Approves First Oral Treatment for Postpartum Depression. 2023 Aug 4. https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-treatment-postpartum-depression
ZURZUVAE – HIGHLIGHTS OF PRESCRIBING INFORMATION. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217369s000lbl.pdf
The Food and Drug Administration approval of zuranolone for postpartum depression in August 2023 has raised many important questions (and opinions) about its future use in clinical practice.
At the UNC-Chapel Hill Center for Women’s Mood Disorders, we treat women and pregnant people throughout hormonal transitions, including pregnancy and the postpartum, and have been part of development, research, and now delivery of both brexanolone and zuranolone. While we are excited about new tools in the arsenal for alleviating maternal mental health, we also want to be clear that our work is far from complete and continued efforts to care for pregnant people and their families are imperative.
What is zuranolone?
Zuranolone (brand name Zurzuvae) is an oral medication developed by Sage Therapeutics and Biogen. It is a positive allosteric modulator of the GABAA receptor, the brain’s major inhibitory system. As a positive allosteric modulator, it increases the sensitivity of the GABAA receptor to GABA.
Zuranolone is very similar to brexanolone, a synthetic form of allopregnanolone, a neurosteroid byproduct of progesterone (see below). However, zuranolone is not an oral form of brexanolone – it was slightly modified to ensure good oral stability and bioavailability. It is metabolized by the hepatic enzyme CYP3A4 and has a half-life of 16-23 hours. Zurzuvae is currently produced in capsule form.
What does zuranolone treat?
Zuranolone is the first FDA-approved oral drug for postpartum depression (PPD). It follows brexanolone, an intravenous drug, which was the first FDA-approved medication for PPD. Though these are the first medications with specific approval for PDD, many other treatment options are currently available including therapy, SSRIs, serotonin norepinephrine reuptake inhibitors (SNRIs), and other treatments used in major depression.
How does zuranolone work?
Zuranolone is a neuroactive steroid, which means that it is a steroid that goes into and acts on the brain. Zuranolone binds to different GABA receptor subunits from those bound by other positive modulators, such as benzodiazepines (for example, lorazepam). As a synthetic form of allopregnanolone, a metabolite of progesterone which rises dramatically in pregnancy then drops during labor and delivery, zuranolone was originally thought to mitigate the response to this drop in patients that are vulnerable to it during the postpartum. An alternative proposed mechanism is that the increased GABAergic, inhibitory signaling with zuranolone may act directly to decrease depression irrespective of the exact mechanism by which the depression occurred.
How was it studied?
Zuranolone was studied in women with severe postpartum depression and had to meet criteria for major depressive disorder (MDD) no earlier than the third trimester of pregnancy (about 28 weeks’ gestation) and no later than 4 weeks post partum. Patients were excluded from these studies if they had a history of bipolar disorder, psychotic disorders, attempted suicide, or if they were at risk for suicide.
The two phase 3 clinical trials that led to FDA approval are ROBIN and SKYLARK. These studies measured the efficacy and safety of zuranolone at 30 mg and 50 mg, respectively, and met their end points of rapid improvement in depressive and anxiety symptoms in postpartum depression.
When will we be able to start using it?
It is anticipated that zuranolone will become commercially available in early 2024.
Who can prescribe it?
Those with medical licenses. Most people will likely receive treatment from their obstetric, family medicine, or psychiatric clinicians.
How much will it cost?
The manufacturers have not released this information as of August 2023.
What sort of doses and duration is recommended?
The current FDA recommended dose is 50 mg for 14 days, taken once per evening with a fatty meal. The dose can be reduced to 40 mg if there are central nervous system (CNS) depressant effects, and to 30 mg if the patient has severe hepatic or moderate-severe renal impairment. There are currently no studies on longer courses of treatment.
What happens if the patient relapses after a 14-day trial?
While there is no clear guidance, an open-label trial (The SHORELINE Study) demonstrated that a repeated 14-day administration can restore clinical response.
What are the side effects?
Common side effects include drowsiness, dizziness, lower energy, diarrhea, and symptoms similar to the common cold. Zuranolone can act like a CNS depressant and can lead to sedation and somnolence.
Are there any boxed warnings?
Because of the CNS depressant effects, zuranolone was given a boxed warning that patients should not drive or operate heavy machinery within 12 hours of taking the medication as it may lead to impairment. Similar to other antidepressants, there is also a warning that zuranolone may increase risk for suicidal thoughts in patients under 24 years old.
Can it be used with other medications?
Yes. In the original trials, women were allowed to remain on medications treating their depressive symptoms (such as SSRIs and SNRIs). According to the FDA, zuranolone can be used alone or with other antidepressants.
Are there any medicines to avoid?
We recommend caution with other medications which may increase sedation, such as benzodiazepines.
Can it be used with birth control?
Yes. In fact, because the outcomes on a fetus are not yet studied, it is recommended that patients be on concurrent birth control during treatment and for a week after cessation. This does not mean that zuranolone is known to cause issues with fetal development, but rather that we do not know at this time.
Can it be used in pregnancy?
As above, the outcomes on fetal development are not known at this time, nor are the effects of zuranolone on labor and delivery. More research will need to be done to understand if there is risk with taking zuranolone during pregnancy. It should be noted that allopregnanolone levels ordinarily reach quite high levels during pregnancy.
Long-term side effects?
Long-term side effects are unknown. The study duration of ROBIN and SKYLARK was 45 days.
Breastfeeding?
Use in lactation has not yet been studied. Continued research is needed.
Can it be used in mood changes related to other reproductive changes or diagnoses like premenstrual dysphoric disorder and perimenopause?
The mechanism by which zuranolone is thought to work – that is, during changes in reproductive hormones – is implicated in other reproductive transitions such as premenstrual dysphoric disorder and perimenopause when reproductive hormones are fluctuating, though at lower levels than in pregnancy. Research will be required to assess efficacy and safety; however, the mechanistic reasons is worth pursuing. Additionally, zuranolone has not been studied in postpartum psychosis.
Can zuranolone be used to treat other affective conditions besides postpartum depression? Bipolar disorder?
Whether it may be beneficial for patients with a depressive episode that is part of an underlying bipolar disorder or other psychiatric illness is not yet known.
Anxiety?
Along with depressive symptoms, women who received zuranolone in the clinical trials also had improvements in anxiety symptoms. These findings provide some hope that zuranolone may eventually be beneficial in patients with anxiety.
However, to date zuranolone has not been directly studied as a treatment for anxiety disorders (such as generalized anxiety disorder, panic disorder, etc.), so its efficacy for these illnesses is currently unknown.
Insomnia?
In a study of 153 postpartum women, randomized to placebo or zuranolone, scale questions for insomnia were improved in the group receiving zuranolone. This provides some hope that, if zuranolone is appropriate, concurrent polypharmacy with a sleep aid can be avoided. Additionally, future evaluation of use in insomnia outside of PPD may be warranted.
How is it different from brexanolone?
The two are slightly different molecules. Brexanolone is synthetically identical to allopregnanolone and zuranolone has been altered to be active and orally bioavailable.
Brexanolone is a 60-hour infusion that requires hospital admission at an approved health care site. Zuranolone is an oral at-home once-daily dosing treatment for 14 days. Zuranolone does not require enrollment in a risk evaluation and mitigation strategy for risk of excessive sedation and sudden loss of consciousness.
When would you consider zuranolone vs. brexanolone vs. other antidepressants?
Zuranolone and brexanolone are rapid-acting antidepressants with a response within 14 days or 60 hours, respectively. Antidepressants such as SSRIs/SNRIs are still available, well studied, and work, although take longer to reach clinical efficacy and are accompanied by potentially troubling side effects (for example, weight gain, sexual dysfunction).
Time to treatment effect should be considered when assessing severity of symptoms and functional impairment of the mother and the overall family unit. Brexanolone requires continuous monitoring which may be beneficial for women who are severely impaired and may benefit from frequent clinical monitoring. Brexanolone does not require a dose reduction with hepatic impairment, however, should be avoided in end-stage renal disease because of the potential accumulation of the solubilizing agent.
Where can I find more information?
Many states have maternal mental health consultation lines (examples include NCMATTERS here in North Carolina and MCPAP for Moms in Massachusetts) for clinicians (mental health, primary care, and obstetricians) that can be utilized for questions about prescribing. Postpartum Support International also has a clinician line for those without state services.
We plan to update this entry upon market release and access to new information.
Dr. Riddle and Dr. Nathan are assistant professors in the department of psychiatry at the University of North Carolina at Chapel Hill. Dr. Richardson is a perinatal psychiatry fellow, department of psychiatry, UNC-Chapel Hill. Dr. Rubinow is Distinguished Professor in the department of psychiatry, UNC-Chapel Hill. Dr. Meltzer-Brody is Assad Meymandi Distinguished Professor and Chair, department of psychiatry, UNC-Chapel Hill.
References
Deligiannidis KM et al. J Clin Psychiatry. 2023 Jan 30;84(1):22m14475. doi: 10.4088/JCP.22m14475.
Deligiannidis KM et al. . Obstetrics & Gynecology. 2023 May;141(5S):64S-65S. doi: 10.1097/01.AOG.0000930588.16136.3f.
Deligiannidis KM et al. Am J Psychiatry. 2023 Sep 1;180(9):668-75. doi: 10.1176/appi.ajp.20220785.
Deligiannidis KM et al. JAMA Psychiatry. 2021 Sep 1;78(9):951-59. doi: 10.1001/jamapsychiatry.2021.1559.
FDA Approves First Oral Treatment for Postpartum Depression. 2023 Aug 4. https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-treatment-postpartum-depression
ZURZUVAE – HIGHLIGHTS OF PRESCRIBING INFORMATION. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217369s000lbl.pdf
Every click you make, the EHR is watching you
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.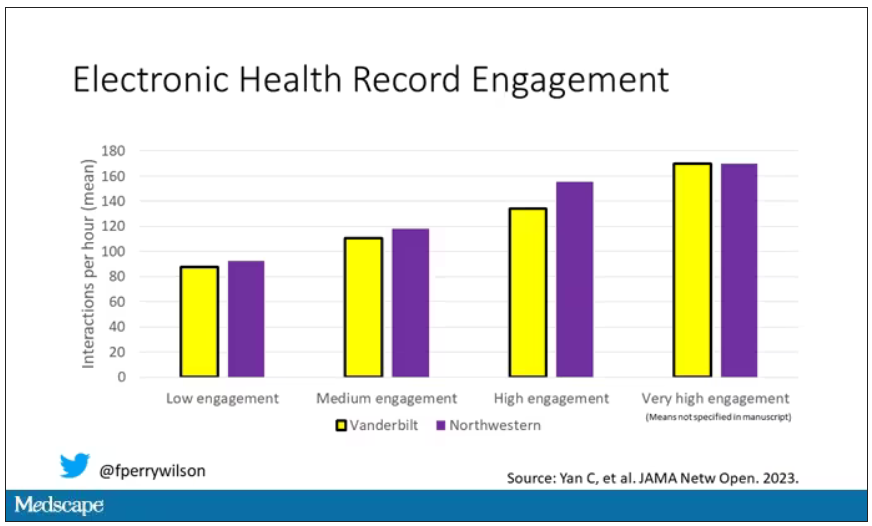
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.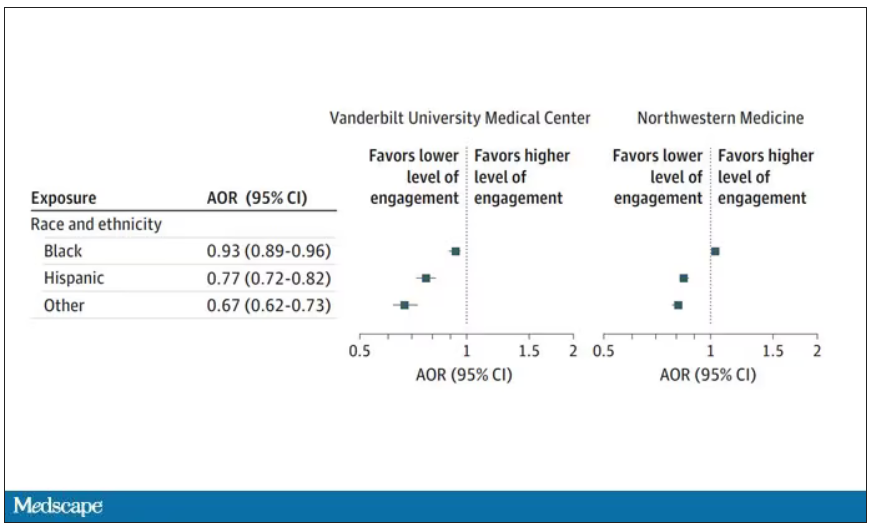
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.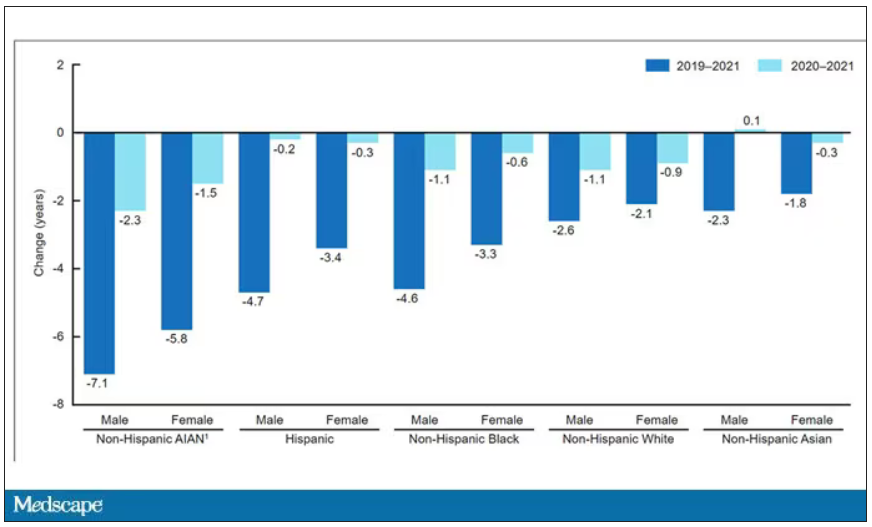
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
Take a closer look at sleep’s role in GERD
This transcript has been edited for clarity.
The ongoing longitudinal Nurses’ Health Study has served as an incredible database for evaluating disease states prospectively over decades, thanks to the robust input of its participants. Most recently, this allowed for an important analysis of the association between gastroesophageal reflux (GER) symptoms and sleep quality, the results of which were published in JAMA Network Open.
Approximately 49,000 women with a median age of 59 years (range, 48-69 years) provided data for this analysis. Starting in 2005, they were asked about their experience of GER symptoms. In 2017, they were also asked to respond to a questionnaire, a modified Pittsburgh Sleep Quality Index (PSQI). This is a tool we’ve used a lot in prospective studies looking at gastrointestinal diseases and sleep-related abnormalities. It’s unique in that it looks not only at sleep but also at next-day function and daytime sleepiness, which is important here for its implications related to reflux disease and sleep fragmentation.
For those with GER symptoms occurring once a week and more than once a week, the approximate relative risk increased by 30% and 53%, respectively. Clearly, the association of GER symptoms and relative sleep quality was really important.
It should be noted that the PSQI is a disease-independent, validated instrument. It’s not specific to GER disease or any diseases. It’s cross validated across 17 different languages. I think what’s most important about its use in the assessment here is the incorporation of next-day function and asking participants about daytime sleepiness, which we’ll discuss in more detail shortly.
The many causes of interrupted sleep
We’ve all experienced sleep fragmentation, whether in the form of having been on call during our medical training or common experiences like hearing a child cry in the night, a noisy truck pass by, or a dog barking. You may or may not remember that these happened the next day, but they’ve nonetheless interrupted your sleep efficiency.
When you transition laterally across the stages of sleep, that’s what establishes the circadian rhythm and ensures sleep hygiene. Typically, we require approximately 7 hours of restful sleep to do that. But if you fragment or interrupt this process, you more or less move your way erratically through the night, disrupting sleep hygiene and efficiency.
If you have a cognitive awakening during those disruptions, you may recall those events the next day. Or, you may not remember it at all, and such amnestic events are normal for some people with sleep disruptions.
You may also have a sensory arousal, whether it’s due to GER symptoms, auditory stimuli, bumping your toe, or whatever disruptive event. Any of these can cause you to lose that laterality of smooth transition through sleep.
Approximately 20% of the U.S. population have reported GER symptoms at least once a week. Incident data indicate that number may be increasing by as much as 5% a year. Much of that increase is tied to obesity. But nonetheless, it’s a problem on the rise.
It’s important to know this as we start to look at sleep. If GER is acting as a trigger to sleep disruption, you need to ask your patients with this condition about next-day function.
In particular, the next-day function questions to ask are, “How do you feel when you get up? Are you awake and refreshed? Do you have early fatigue? Do you drag yourself out of bed, have daytime somnolence, loss of concentration, or irritability?”
Those are key parameters we can use for looking back to the night before and gauging sleep efficiency. If you’re not asking those questions, you may miss out on identifying a patient having sleep fragmentation.
Sleep’s role in inflammatory disease processes
I now perform an interval assessment of this type not just in my patients with GER disease but across all my patients. I do so because sleep is physiologically important in so many ways.
In patients who have nonalcoholic fatty liver disease and a variety of other liver diseases, we’re finding an increased association with sleep fragmentation outside of sleep apnea.
The same is true with irritable bowel and other functional diseases.
When you have sleep fragmentation in inflammatory bowel disease, you turn on a variety of inflammatory proteins (e.g., C-reactive protein) and cytokines, such as interleukins and tumor necrosis factor alpha. These processes may actually tip somebody over to a pro-inflammatory state.
When it comes to what might be considered a relatively simpler condition like GER disease, Ronnie Fass and colleagues showed a number of years ago via Bernstein testing performed in patients with both fragmented and normal sleep that the sensory thresholds all get lowered in the former group. This is irrespective of whether you have a functional symptom or you’re awakened by bumping your toe, a headache, or having heartburn; your sensory thresholds are lower. As a result, the same stimulus provides a higher sense of awareness. By ramping up that awareness, you increase the interference with the next-day function.
We’ve shown that sleep fragmentation affects a variety of things, including immune function. This may be why many people get sick when they travel in between time zones.
There are also implications relating to things like obesity. When you have sleep dysfunction, you have effects on leptin and ghrelin, contrary to what you would normally want to have. This, in turn, causes adverse effects on stimulation or suppression of satiety or appetite. These are things that I counsel my patients about when I talk about reflux as well as those trying to lose weight.
Sleep disruption affects cortisol stimulation and has a significant correlation with type 2 diabetes, cardiovascular diseases, and even mortality statistics.
Advice for counseling patients
This latest analysis from the Nurses’ Health Study reminds us that a lot of people have reflux and a lot of people have sleep fragmentation. We need to do better in asking our patients if they have symptoms specific not only to reflux but also to potentially sleep-related complications.
The more we do that, the more we individualize patient treatment rather than treating them as a disease state. This, in turn, will allow us to practice personalized medicine. The more we can engage our patients with reflux disease by asking the right questions about next-day function, the better we can do in improving their outcomes.
It’s time for us all to open our eyes to the value of closing them. Let’s talk to our patients with reflux disease in a little bit of a different light, providing a new perspective on strategies we can use to mitigate and deal with those symptoms, thereby preventing the consequences of sleep fragmentation.
Hopefully, this overview gives you some guidance the next time you have a conversation with your patients. It will apply across many, many disease states, and in almost everything we do in gastroenterology.
David A. Johnson, MD, is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School, Norfolk, Va., and a past president of the American College of Gastroenterology. He reported advising with ISOTHRIVE and Johnson & Johnson.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
The ongoing longitudinal Nurses’ Health Study has served as an incredible database for evaluating disease states prospectively over decades, thanks to the robust input of its participants. Most recently, this allowed for an important analysis of the association between gastroesophageal reflux (GER) symptoms and sleep quality, the results of which were published in JAMA Network Open.
Approximately 49,000 women with a median age of 59 years (range, 48-69 years) provided data for this analysis. Starting in 2005, they were asked about their experience of GER symptoms. In 2017, they were also asked to respond to a questionnaire, a modified Pittsburgh Sleep Quality Index (PSQI). This is a tool we’ve used a lot in prospective studies looking at gastrointestinal diseases and sleep-related abnormalities. It’s unique in that it looks not only at sleep but also at next-day function and daytime sleepiness, which is important here for its implications related to reflux disease and sleep fragmentation.
For those with GER symptoms occurring once a week and more than once a week, the approximate relative risk increased by 30% and 53%, respectively. Clearly, the association of GER symptoms and relative sleep quality was really important.
It should be noted that the PSQI is a disease-independent, validated instrument. It’s not specific to GER disease or any diseases. It’s cross validated across 17 different languages. I think what’s most important about its use in the assessment here is the incorporation of next-day function and asking participants about daytime sleepiness, which we’ll discuss in more detail shortly.
The many causes of interrupted sleep
We’ve all experienced sleep fragmentation, whether in the form of having been on call during our medical training or common experiences like hearing a child cry in the night, a noisy truck pass by, or a dog barking. You may or may not remember that these happened the next day, but they’ve nonetheless interrupted your sleep efficiency.
When you transition laterally across the stages of sleep, that’s what establishes the circadian rhythm and ensures sleep hygiene. Typically, we require approximately 7 hours of restful sleep to do that. But if you fragment or interrupt this process, you more or less move your way erratically through the night, disrupting sleep hygiene and efficiency.
If you have a cognitive awakening during those disruptions, you may recall those events the next day. Or, you may not remember it at all, and such amnestic events are normal for some people with sleep disruptions.
You may also have a sensory arousal, whether it’s due to GER symptoms, auditory stimuli, bumping your toe, or whatever disruptive event. Any of these can cause you to lose that laterality of smooth transition through sleep.
Approximately 20% of the U.S. population have reported GER symptoms at least once a week. Incident data indicate that number may be increasing by as much as 5% a year. Much of that increase is tied to obesity. But nonetheless, it’s a problem on the rise.
It’s important to know this as we start to look at sleep. If GER is acting as a trigger to sleep disruption, you need to ask your patients with this condition about next-day function.
In particular, the next-day function questions to ask are, “How do you feel when you get up? Are you awake and refreshed? Do you have early fatigue? Do you drag yourself out of bed, have daytime somnolence, loss of concentration, or irritability?”
Those are key parameters we can use for looking back to the night before and gauging sleep efficiency. If you’re not asking those questions, you may miss out on identifying a patient having sleep fragmentation.
Sleep’s role in inflammatory disease processes
I now perform an interval assessment of this type not just in my patients with GER disease but across all my patients. I do so because sleep is physiologically important in so many ways.
In patients who have nonalcoholic fatty liver disease and a variety of other liver diseases, we’re finding an increased association with sleep fragmentation outside of sleep apnea.
The same is true with irritable bowel and other functional diseases.
When you have sleep fragmentation in inflammatory bowel disease, you turn on a variety of inflammatory proteins (e.g., C-reactive protein) and cytokines, such as interleukins and tumor necrosis factor alpha. These processes may actually tip somebody over to a pro-inflammatory state.
When it comes to what might be considered a relatively simpler condition like GER disease, Ronnie Fass and colleagues showed a number of years ago via Bernstein testing performed in patients with both fragmented and normal sleep that the sensory thresholds all get lowered in the former group. This is irrespective of whether you have a functional symptom or you’re awakened by bumping your toe, a headache, or having heartburn; your sensory thresholds are lower. As a result, the same stimulus provides a higher sense of awareness. By ramping up that awareness, you increase the interference with the next-day function.
We’ve shown that sleep fragmentation affects a variety of things, including immune function. This may be why many people get sick when they travel in between time zones.
There are also implications relating to things like obesity. When you have sleep dysfunction, you have effects on leptin and ghrelin, contrary to what you would normally want to have. This, in turn, causes adverse effects on stimulation or suppression of satiety or appetite. These are things that I counsel my patients about when I talk about reflux as well as those trying to lose weight.
Sleep disruption affects cortisol stimulation and has a significant correlation with type 2 diabetes, cardiovascular diseases, and even mortality statistics.
Advice for counseling patients
This latest analysis from the Nurses’ Health Study reminds us that a lot of people have reflux and a lot of people have sleep fragmentation. We need to do better in asking our patients if they have symptoms specific not only to reflux but also to potentially sleep-related complications.
The more we do that, the more we individualize patient treatment rather than treating them as a disease state. This, in turn, will allow us to practice personalized medicine. The more we can engage our patients with reflux disease by asking the right questions about next-day function, the better we can do in improving their outcomes.
It’s time for us all to open our eyes to the value of closing them. Let’s talk to our patients with reflux disease in a little bit of a different light, providing a new perspective on strategies we can use to mitigate and deal with those symptoms, thereby preventing the consequences of sleep fragmentation.
Hopefully, this overview gives you some guidance the next time you have a conversation with your patients. It will apply across many, many disease states, and in almost everything we do in gastroenterology.
David A. Johnson, MD, is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School, Norfolk, Va., and a past president of the American College of Gastroenterology. He reported advising with ISOTHRIVE and Johnson & Johnson.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
The ongoing longitudinal Nurses’ Health Study has served as an incredible database for evaluating disease states prospectively over decades, thanks to the robust input of its participants. Most recently, this allowed for an important analysis of the association between gastroesophageal reflux (GER) symptoms and sleep quality, the results of which were published in JAMA Network Open.
Approximately 49,000 women with a median age of 59 years (range, 48-69 years) provided data for this analysis. Starting in 2005, they were asked about their experience of GER symptoms. In 2017, they were also asked to respond to a questionnaire, a modified Pittsburgh Sleep Quality Index (PSQI). This is a tool we’ve used a lot in prospective studies looking at gastrointestinal diseases and sleep-related abnormalities. It’s unique in that it looks not only at sleep but also at next-day function and daytime sleepiness, which is important here for its implications related to reflux disease and sleep fragmentation.
For those with GER symptoms occurring once a week and more than once a week, the approximate relative risk increased by 30% and 53%, respectively. Clearly, the association of GER symptoms and relative sleep quality was really important.
It should be noted that the PSQI is a disease-independent, validated instrument. It’s not specific to GER disease or any diseases. It’s cross validated across 17 different languages. I think what’s most important about its use in the assessment here is the incorporation of next-day function and asking participants about daytime sleepiness, which we’ll discuss in more detail shortly.
The many causes of interrupted sleep
We’ve all experienced sleep fragmentation, whether in the form of having been on call during our medical training or common experiences like hearing a child cry in the night, a noisy truck pass by, or a dog barking. You may or may not remember that these happened the next day, but they’ve nonetheless interrupted your sleep efficiency.
When you transition laterally across the stages of sleep, that’s what establishes the circadian rhythm and ensures sleep hygiene. Typically, we require approximately 7 hours of restful sleep to do that. But if you fragment or interrupt this process, you more or less move your way erratically through the night, disrupting sleep hygiene and efficiency.
If you have a cognitive awakening during those disruptions, you may recall those events the next day. Or, you may not remember it at all, and such amnestic events are normal for some people with sleep disruptions.
You may also have a sensory arousal, whether it’s due to GER symptoms, auditory stimuli, bumping your toe, or whatever disruptive event. Any of these can cause you to lose that laterality of smooth transition through sleep.
Approximately 20% of the U.S. population have reported GER symptoms at least once a week. Incident data indicate that number may be increasing by as much as 5% a year. Much of that increase is tied to obesity. But nonetheless, it’s a problem on the rise.
It’s important to know this as we start to look at sleep. If GER is acting as a trigger to sleep disruption, you need to ask your patients with this condition about next-day function.
In particular, the next-day function questions to ask are, “How do you feel when you get up? Are you awake and refreshed? Do you have early fatigue? Do you drag yourself out of bed, have daytime somnolence, loss of concentration, or irritability?”
Those are key parameters we can use for looking back to the night before and gauging sleep efficiency. If you’re not asking those questions, you may miss out on identifying a patient having sleep fragmentation.
Sleep’s role in inflammatory disease processes
I now perform an interval assessment of this type not just in my patients with GER disease but across all my patients. I do so because sleep is physiologically important in so many ways.
In patients who have nonalcoholic fatty liver disease and a variety of other liver diseases, we’re finding an increased association with sleep fragmentation outside of sleep apnea.
The same is true with irritable bowel and other functional diseases.
When you have sleep fragmentation in inflammatory bowel disease, you turn on a variety of inflammatory proteins (e.g., C-reactive protein) and cytokines, such as interleukins and tumor necrosis factor alpha. These processes may actually tip somebody over to a pro-inflammatory state.
When it comes to what might be considered a relatively simpler condition like GER disease, Ronnie Fass and colleagues showed a number of years ago via Bernstein testing performed in patients with both fragmented and normal sleep that the sensory thresholds all get lowered in the former group. This is irrespective of whether you have a functional symptom or you’re awakened by bumping your toe, a headache, or having heartburn; your sensory thresholds are lower. As a result, the same stimulus provides a higher sense of awareness. By ramping up that awareness, you increase the interference with the next-day function.
We’ve shown that sleep fragmentation affects a variety of things, including immune function. This may be why many people get sick when they travel in between time zones.
There are also implications relating to things like obesity. When you have sleep dysfunction, you have effects on leptin and ghrelin, contrary to what you would normally want to have. This, in turn, causes adverse effects on stimulation or suppression of satiety or appetite. These are things that I counsel my patients about when I talk about reflux as well as those trying to lose weight.
Sleep disruption affects cortisol stimulation and has a significant correlation with type 2 diabetes, cardiovascular diseases, and even mortality statistics.
Advice for counseling patients
This latest analysis from the Nurses’ Health Study reminds us that a lot of people have reflux and a lot of people have sleep fragmentation. We need to do better in asking our patients if they have symptoms specific not only to reflux but also to potentially sleep-related complications.
The more we do that, the more we individualize patient treatment rather than treating them as a disease state. This, in turn, will allow us to practice personalized medicine. The more we can engage our patients with reflux disease by asking the right questions about next-day function, the better we can do in improving their outcomes.
It’s time for us all to open our eyes to the value of closing them. Let’s talk to our patients with reflux disease in a little bit of a different light, providing a new perspective on strategies we can use to mitigate and deal with those symptoms, thereby preventing the consequences of sleep fragmentation.
Hopefully, this overview gives you some guidance the next time you have a conversation with your patients. It will apply across many, many disease states, and in almost everything we do in gastroenterology.
David A. Johnson, MD, is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School, Norfolk, Va., and a past president of the American College of Gastroenterology. He reported advising with ISOTHRIVE and Johnson & Johnson.
A version of this article first appeared on Medscape.com.
Don’t fear POTS: Tips for diagnosis and treatment
This transcript has been edited for clarity.
Michelle L. O’Donoghue, MD, MPH: I’m here in Amsterdam at the European Society of Cardiology (ESC) Congress 2023. Joining me for a great discussion is my friend Dr. Pam Taub, who is a cardiologist and a professor of medicine at UC San Diego. She has a particular interest in postural orthostatic tachycardia syndrome (POTS), so that’s what we’ll be talking about today.
Thanks for joining me, Pam. When we think about POTS, for those who are not familiar with the term, what does it actually mean and how do you diagnose it?
No tilt table required
Pam R. Taub, MD: That’s typically associated with symptoms such as lightheadedness, dizziness, and cognitive difficulties such as brain fog. The diagnosis can be made by tilt-table testing, but it can also be made in the office with simple orthostats.
In my clinic, I have people lie down for 3-5 minutes. At the end of that period, you get a heart rate and blood pressure. Then you have them stand up for 3-5 minutes and then get heart rate and blood pressure, and you look at the differences. If the heart rate goes up by 30 points – so maybe they’re 80 beats/min when they’re lying down and when they stand up, it goes to 110 beats/min – that’s POTS, so very objective criteria. Typically, these people don’t have what we call orthostatic hypotension, where there is a significant decrease in the blood pressure. It’s more a heart rate issue.
Dr. O’Donoghue: How symptomatically do they usually present?
Dr. Taub: It’s a spectrum. Some people have mild symptoms. After they’re in the upright position for maybe 10 minutes, they get symptoms. There are some people who, when they go from a lying to standing position, they’re extremely symptomatic and can’t really do any activities. There are some people that are even wheelchair-bound because the symptoms are so debilitating. There’s a wide spectrum.
Dr. O’Donoghue: There has been more discussion, I feel like, about the rising prevalence of POTS as a diagnosis, and in particular since the COVID pandemic. What’s our understanding of the relationship between COVID and POTS and what the mechanism might be?
Dr. Taub: We’ve known that POTS can be triggered by a viral infection. Before COVID, we knew that in certain individuals that we think have an underlying genetic predisposition, usually some autoimmune substrate, when they get certain types of infections, whether it’s influenza or mononucleosis, they get POTS.
Typically, when they get an infection, they start getting deconditioned. They don’t feel well, so they’re on bed rest. When they get long periods of bed rest, when they start to become active, they start to have overactivation of their sympathetic nervous system, and they have a large amount of cardiovascular deconditioning. It’s a cycle that is often triggered after an infection.
A huge increase of POTS has been seen after COVID-19 because we had so many people exposed to this virus. With COVID-19, there is a period where people don’t feel great and they are getting bed rest, so they’re getting deconditioned. We’ve seen so many patients referred for post-COVID POTS and also long COVID or the post-acute sequelae of COVID-19, where POTS is a part of that presentation.
Female sex and autoimmune conditions
Dr. O’Donoghue: We know that POTS seems to disproportionately affect women. Is that understood? Is it thought that that’s related to the perhaps the autoimmune component of that illness?
Dr. Taub: Yes. The theory is because women tend to have more autoimmune conditions, that’s why they’re more predisposed. There’s a large amount of genetic susceptibility. For instance, we know that there’s an association between POTS and conditions like Ehlers-Danlos syndrome and between POTS and mast cell activation. Some of those conditions are more prevalent in women as well.
Dr. O’Donoghue: I feel like many physicians don’t know how to manage POTS, and they’re actually a little fearful perhaps to take it on. Fortunately, there have been a growing number of POTS clinics with specialists that focus on that area. For the average practitioner who maybe can’t refer to a POTS clinic, how should they approach that?
Dr. Taub: The first thing is its diagnosis. When someone tells you that they have symptoms of orthostatic intolerance – so, activities that involve standing – you need to first have that on your differential diagnosis. You can make the diagnosis in the office with orthostats. You don’t need a tilt table. It’s sometimes helpful if you’re unsure about the diagnosis, but you can make the diagnosis.
Many times, you’re finding people that have very mild symptoms. You can treat that with some good lifestyle recommendations, such as increased hydration, increasing salt in their diet, and compression. And the exercise component is really important.
Many people with POTS are told to go exercise, go for a run, or go for a walk. That’s incorrect, because these people have symptoms when they’re in the upright position. The type of exercise they need to do initially is exercise in the lying or seated position – so exercises like rowing or a seated bike, and strength training. As they start to feel better, then they can do upright exercise.
You should never tell a person that has POTS to just initially start with upright exercise, because they’re going to feel so much worse and then they’re never going to want to exercise. It’s really important to give them the right exercise recommendations. I find that for many of these mild cases, if they do the right exercise and engage in the right lifestyle strategies, they get better.
Compression wear and drug therapy
Dr. O’Donoghue: When it comes to compression stockings, do you usually start with a particular length?
Dr. Taub: It’s interesting. There are many different compression stockings, medical grade. Through patients with POTS, I’ve gotten feedback on certain types of athletic wear that have built-in compression, and that’s a little bit easier for people to wear every day because they can do their errands and it doesn’t look like they’re wearing medical-grade compression stockings.
Basically, I’ve collected all the different recommendations that patients say help, and I give them a list. The medical-grade compression stockings sometimes are very challenging to put on, and sometimes people just need light compression or even just socks. Any kind of compression is going to help.
Dr. O’Donoghue: That’s a great tip, because I know there are many patients who refuse to wear the compression stockings. If there’s a fashionable alternative, that’s always good to reach for.
Dr. Taub: Another thing that patients have told me is that abdominal compression is also very helpful. There are many commercially available abdominal compression options, like shapewear. Many patients with POTS use that and that helps, too.
Dr. O’Donoghue: Good. For those patients with POTS that is refractory to the measures you’ve already discussed, what are the next steps after that?
Dr. Taub: Pharmacotherapy is very synergistic with lifestyle, and there are many different pharmacotherapy options. One of the first things that you want to think about is lowering that heart rate. The reason people feel horrible is because their heart rate is usually very high when they’re upright. If they’re upright for long periods of time and they’re having very high heart rates, they’re going to get really tired because it’s like they’re exercising for hours when they’re upright.
Heart rate lowering is the cornerstone of therapy. Traditionally, we’ve used beta-blockers for heart rate lowering. The problem is they also lower blood pressure. They can also cause fatigue, so not the ideal agent for patients with POTS.
One of the clinical trials that I led was with a drug called ivabradine, which selectively works on the SA node and decreases heart rate without affecting blood pressure. What’s really elegant about ivabradine is it has a more potent effect when the heart rate is higher. When the patient is standing, it’s going to have a more potent effect on heart rate lowering. It’s really well tolerated in patients with POTS. In our study, we showed an improvement in quality of life metrics. That’s one of the first-line drugs that I use for patients with POTS.
The other thing is some of them will also have a concomitant lowering of blood pressure. You can think about medications that increase blood pressure, like midodrine, fludrocortisone, and droxidopa. Sometimes that combination of a heart rate-lowering medication and a medication that increases blood pressure really works well.
Dr. O’Donoghue: That’s very helpful. I think that those kinds of practical tips are the ones that practitioners really want to reach for, because they need to have that algorithm in their mind to take on this condition. Thanks again for walking us through that.
I think it’s a very interesting space, and there’s more that we’re going to be learning over the next few years as we further flesh out these post-COVID cases and what we learn from that as well.
Dr. Taub: There are many clinical trials now starting in POTS, so it’s exciting.
Dr. O’Donoghue: Absolutely. Thank you again for joining me today. Signing off, this is Dr Michelle O’Donoghue.
Dr. O’Donoghue is a cardiologist at Brigham and Women’s Hospital and senior investigator with the TIMI Study Group. A strong believer in evidence-based medicine, she relishes discussions about the published literature. A native Canadian, Dr. O’Donoghue loves spending time outdoors with her family but admits with shame that she’s never strapped on hockey skates. She disclosed ties with Amgen, AstraZeneca Pharmaceuticals LP, CVS Minute Clinic, Eisai, GlaxoSmithKline, Janssen Pharmaceuticals, Merck, Novartis, and The Medicines Company. Dr. Taub is professor of Medicine, University of California San Diego Health, La Jolla. She disclosed ties with Amgen, Bayer, Boehringer Ingelheim, Medtronic, Merck, Novartis, Novo Nordisk, and Sanofi.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Michelle L. O’Donoghue, MD, MPH: I’m here in Amsterdam at the European Society of Cardiology (ESC) Congress 2023. Joining me for a great discussion is my friend Dr. Pam Taub, who is a cardiologist and a professor of medicine at UC San Diego. She has a particular interest in postural orthostatic tachycardia syndrome (POTS), so that’s what we’ll be talking about today.
Thanks for joining me, Pam. When we think about POTS, for those who are not familiar with the term, what does it actually mean and how do you diagnose it?
No tilt table required
Pam R. Taub, MD: That’s typically associated with symptoms such as lightheadedness, dizziness, and cognitive difficulties such as brain fog. The diagnosis can be made by tilt-table testing, but it can also be made in the office with simple orthostats.
In my clinic, I have people lie down for 3-5 minutes. At the end of that period, you get a heart rate and blood pressure. Then you have them stand up for 3-5 minutes and then get heart rate and blood pressure, and you look at the differences. If the heart rate goes up by 30 points – so maybe they’re 80 beats/min when they’re lying down and when they stand up, it goes to 110 beats/min – that’s POTS, so very objective criteria. Typically, these people don’t have what we call orthostatic hypotension, where there is a significant decrease in the blood pressure. It’s more a heart rate issue.
Dr. O’Donoghue: How symptomatically do they usually present?
Dr. Taub: It’s a spectrum. Some people have mild symptoms. After they’re in the upright position for maybe 10 minutes, they get symptoms. There are some people who, when they go from a lying to standing position, they’re extremely symptomatic and can’t really do any activities. There are some people that are even wheelchair-bound because the symptoms are so debilitating. There’s a wide spectrum.
Dr. O’Donoghue: There has been more discussion, I feel like, about the rising prevalence of POTS as a diagnosis, and in particular since the COVID pandemic. What’s our understanding of the relationship between COVID and POTS and what the mechanism might be?
Dr. Taub: We’ve known that POTS can be triggered by a viral infection. Before COVID, we knew that in certain individuals that we think have an underlying genetic predisposition, usually some autoimmune substrate, when they get certain types of infections, whether it’s influenza or mononucleosis, they get POTS.
Typically, when they get an infection, they start getting deconditioned. They don’t feel well, so they’re on bed rest. When they get long periods of bed rest, when they start to become active, they start to have overactivation of their sympathetic nervous system, and they have a large amount of cardiovascular deconditioning. It’s a cycle that is often triggered after an infection.
A huge increase of POTS has been seen after COVID-19 because we had so many people exposed to this virus. With COVID-19, there is a period where people don’t feel great and they are getting bed rest, so they’re getting deconditioned. We’ve seen so many patients referred for post-COVID POTS and also long COVID or the post-acute sequelae of COVID-19, where POTS is a part of that presentation.
Female sex and autoimmune conditions
Dr. O’Donoghue: We know that POTS seems to disproportionately affect women. Is that understood? Is it thought that that’s related to the perhaps the autoimmune component of that illness?
Dr. Taub: Yes. The theory is because women tend to have more autoimmune conditions, that’s why they’re more predisposed. There’s a large amount of genetic susceptibility. For instance, we know that there’s an association between POTS and conditions like Ehlers-Danlos syndrome and between POTS and mast cell activation. Some of those conditions are more prevalent in women as well.
Dr. O’Donoghue: I feel like many physicians don’t know how to manage POTS, and they’re actually a little fearful perhaps to take it on. Fortunately, there have been a growing number of POTS clinics with specialists that focus on that area. For the average practitioner who maybe can’t refer to a POTS clinic, how should they approach that?
Dr. Taub: The first thing is its diagnosis. When someone tells you that they have symptoms of orthostatic intolerance – so, activities that involve standing – you need to first have that on your differential diagnosis. You can make the diagnosis in the office with orthostats. You don’t need a tilt table. It’s sometimes helpful if you’re unsure about the diagnosis, but you can make the diagnosis.
Many times, you’re finding people that have very mild symptoms. You can treat that with some good lifestyle recommendations, such as increased hydration, increasing salt in their diet, and compression. And the exercise component is really important.
Many people with POTS are told to go exercise, go for a run, or go for a walk. That’s incorrect, because these people have symptoms when they’re in the upright position. The type of exercise they need to do initially is exercise in the lying or seated position – so exercises like rowing or a seated bike, and strength training. As they start to feel better, then they can do upright exercise.
You should never tell a person that has POTS to just initially start with upright exercise, because they’re going to feel so much worse and then they’re never going to want to exercise. It’s really important to give them the right exercise recommendations. I find that for many of these mild cases, if they do the right exercise and engage in the right lifestyle strategies, they get better.
Compression wear and drug therapy
Dr. O’Donoghue: When it comes to compression stockings, do you usually start with a particular length?
Dr. Taub: It’s interesting. There are many different compression stockings, medical grade. Through patients with POTS, I’ve gotten feedback on certain types of athletic wear that have built-in compression, and that’s a little bit easier for people to wear every day because they can do their errands and it doesn’t look like they’re wearing medical-grade compression stockings.
Basically, I’ve collected all the different recommendations that patients say help, and I give them a list. The medical-grade compression stockings sometimes are very challenging to put on, and sometimes people just need light compression or even just socks. Any kind of compression is going to help.
Dr. O’Donoghue: That’s a great tip, because I know there are many patients who refuse to wear the compression stockings. If there’s a fashionable alternative, that’s always good to reach for.
Dr. Taub: Another thing that patients have told me is that abdominal compression is also very helpful. There are many commercially available abdominal compression options, like shapewear. Many patients with POTS use that and that helps, too.
Dr. O’Donoghue: Good. For those patients with POTS that is refractory to the measures you’ve already discussed, what are the next steps after that?
Dr. Taub: Pharmacotherapy is very synergistic with lifestyle, and there are many different pharmacotherapy options. One of the first things that you want to think about is lowering that heart rate. The reason people feel horrible is because their heart rate is usually very high when they’re upright. If they’re upright for long periods of time and they’re having very high heart rates, they’re going to get really tired because it’s like they’re exercising for hours when they’re upright.
Heart rate lowering is the cornerstone of therapy. Traditionally, we’ve used beta-blockers for heart rate lowering. The problem is they also lower blood pressure. They can also cause fatigue, so not the ideal agent for patients with POTS.
One of the clinical trials that I led was with a drug called ivabradine, which selectively works on the SA node and decreases heart rate without affecting blood pressure. What’s really elegant about ivabradine is it has a more potent effect when the heart rate is higher. When the patient is standing, it’s going to have a more potent effect on heart rate lowering. It’s really well tolerated in patients with POTS. In our study, we showed an improvement in quality of life metrics. That’s one of the first-line drugs that I use for patients with POTS.
The other thing is some of them will also have a concomitant lowering of blood pressure. You can think about medications that increase blood pressure, like midodrine, fludrocortisone, and droxidopa. Sometimes that combination of a heart rate-lowering medication and a medication that increases blood pressure really works well.
Dr. O’Donoghue: That’s very helpful. I think that those kinds of practical tips are the ones that practitioners really want to reach for, because they need to have that algorithm in their mind to take on this condition. Thanks again for walking us through that.
I think it’s a very interesting space, and there’s more that we’re going to be learning over the next few years as we further flesh out these post-COVID cases and what we learn from that as well.
Dr. Taub: There are many clinical trials now starting in POTS, so it’s exciting.
Dr. O’Donoghue: Absolutely. Thank you again for joining me today. Signing off, this is Dr Michelle O’Donoghue.
Dr. O’Donoghue is a cardiologist at Brigham and Women’s Hospital and senior investigator with the TIMI Study Group. A strong believer in evidence-based medicine, she relishes discussions about the published literature. A native Canadian, Dr. O’Donoghue loves spending time outdoors with her family but admits with shame that she’s never strapped on hockey skates. She disclosed ties with Amgen, AstraZeneca Pharmaceuticals LP, CVS Minute Clinic, Eisai, GlaxoSmithKline, Janssen Pharmaceuticals, Merck, Novartis, and The Medicines Company. Dr. Taub is professor of Medicine, University of California San Diego Health, La Jolla. She disclosed ties with Amgen, Bayer, Boehringer Ingelheim, Medtronic, Merck, Novartis, Novo Nordisk, and Sanofi.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Michelle L. O’Donoghue, MD, MPH: I’m here in Amsterdam at the European Society of Cardiology (ESC) Congress 2023. Joining me for a great discussion is my friend Dr. Pam Taub, who is a cardiologist and a professor of medicine at UC San Diego. She has a particular interest in postural orthostatic tachycardia syndrome (POTS), so that’s what we’ll be talking about today.
Thanks for joining me, Pam. When we think about POTS, for those who are not familiar with the term, what does it actually mean and how do you diagnose it?
No tilt table required
Pam R. Taub, MD: That’s typically associated with symptoms such as lightheadedness, dizziness, and cognitive difficulties such as brain fog. The diagnosis can be made by tilt-table testing, but it can also be made in the office with simple orthostats.
In my clinic, I have people lie down for 3-5 minutes. At the end of that period, you get a heart rate and blood pressure. Then you have them stand up for 3-5 minutes and then get heart rate and blood pressure, and you look at the differences. If the heart rate goes up by 30 points – so maybe they’re 80 beats/min when they’re lying down and when they stand up, it goes to 110 beats/min – that’s POTS, so very objective criteria. Typically, these people don’t have what we call orthostatic hypotension, where there is a significant decrease in the blood pressure. It’s more a heart rate issue.
Dr. O’Donoghue: How symptomatically do they usually present?
Dr. Taub: It’s a spectrum. Some people have mild symptoms. After they’re in the upright position for maybe 10 minutes, they get symptoms. There are some people who, when they go from a lying to standing position, they’re extremely symptomatic and can’t really do any activities. There are some people that are even wheelchair-bound because the symptoms are so debilitating. There’s a wide spectrum.
Dr. O’Donoghue: There has been more discussion, I feel like, about the rising prevalence of POTS as a diagnosis, and in particular since the COVID pandemic. What’s our understanding of the relationship between COVID and POTS and what the mechanism might be?
Dr. Taub: We’ve known that POTS can be triggered by a viral infection. Before COVID, we knew that in certain individuals that we think have an underlying genetic predisposition, usually some autoimmune substrate, when they get certain types of infections, whether it’s influenza or mononucleosis, they get POTS.
Typically, when they get an infection, they start getting deconditioned. They don’t feel well, so they’re on bed rest. When they get long periods of bed rest, when they start to become active, they start to have overactivation of their sympathetic nervous system, and they have a large amount of cardiovascular deconditioning. It’s a cycle that is often triggered after an infection.
A huge increase of POTS has been seen after COVID-19 because we had so many people exposed to this virus. With COVID-19, there is a period where people don’t feel great and they are getting bed rest, so they’re getting deconditioned. We’ve seen so many patients referred for post-COVID POTS and also long COVID or the post-acute sequelae of COVID-19, where POTS is a part of that presentation.
Female sex and autoimmune conditions
Dr. O’Donoghue: We know that POTS seems to disproportionately affect women. Is that understood? Is it thought that that’s related to the perhaps the autoimmune component of that illness?
Dr. Taub: Yes. The theory is because women tend to have more autoimmune conditions, that’s why they’re more predisposed. There’s a large amount of genetic susceptibility. For instance, we know that there’s an association between POTS and conditions like Ehlers-Danlos syndrome and between POTS and mast cell activation. Some of those conditions are more prevalent in women as well.
Dr. O’Donoghue: I feel like many physicians don’t know how to manage POTS, and they’re actually a little fearful perhaps to take it on. Fortunately, there have been a growing number of POTS clinics with specialists that focus on that area. For the average practitioner who maybe can’t refer to a POTS clinic, how should they approach that?
Dr. Taub: The first thing is its diagnosis. When someone tells you that they have symptoms of orthostatic intolerance – so, activities that involve standing – you need to first have that on your differential diagnosis. You can make the diagnosis in the office with orthostats. You don’t need a tilt table. It’s sometimes helpful if you’re unsure about the diagnosis, but you can make the diagnosis.
Many times, you’re finding people that have very mild symptoms. You can treat that with some good lifestyle recommendations, such as increased hydration, increasing salt in their diet, and compression. And the exercise component is really important.
Many people with POTS are told to go exercise, go for a run, or go for a walk. That’s incorrect, because these people have symptoms when they’re in the upright position. The type of exercise they need to do initially is exercise in the lying or seated position – so exercises like rowing or a seated bike, and strength training. As they start to feel better, then they can do upright exercise.
You should never tell a person that has POTS to just initially start with upright exercise, because they’re going to feel so much worse and then they’re never going to want to exercise. It’s really important to give them the right exercise recommendations. I find that for many of these mild cases, if they do the right exercise and engage in the right lifestyle strategies, they get better.
Compression wear and drug therapy
Dr. O’Donoghue: When it comes to compression stockings, do you usually start with a particular length?
Dr. Taub: It’s interesting. There are many different compression stockings, medical grade. Through patients with POTS, I’ve gotten feedback on certain types of athletic wear that have built-in compression, and that’s a little bit easier for people to wear every day because they can do their errands and it doesn’t look like they’re wearing medical-grade compression stockings.
Basically, I’ve collected all the different recommendations that patients say help, and I give them a list. The medical-grade compression stockings sometimes are very challenging to put on, and sometimes people just need light compression or even just socks. Any kind of compression is going to help.
Dr. O’Donoghue: That’s a great tip, because I know there are many patients who refuse to wear the compression stockings. If there’s a fashionable alternative, that’s always good to reach for.
Dr. Taub: Another thing that patients have told me is that abdominal compression is also very helpful. There are many commercially available abdominal compression options, like shapewear. Many patients with POTS use that and that helps, too.
Dr. O’Donoghue: Good. For those patients with POTS that is refractory to the measures you’ve already discussed, what are the next steps after that?
Dr. Taub: Pharmacotherapy is very synergistic with lifestyle, and there are many different pharmacotherapy options. One of the first things that you want to think about is lowering that heart rate. The reason people feel horrible is because their heart rate is usually very high when they’re upright. If they’re upright for long periods of time and they’re having very high heart rates, they’re going to get really tired because it’s like they’re exercising for hours when they’re upright.
Heart rate lowering is the cornerstone of therapy. Traditionally, we’ve used beta-blockers for heart rate lowering. The problem is they also lower blood pressure. They can also cause fatigue, so not the ideal agent for patients with POTS.
One of the clinical trials that I led was with a drug called ivabradine, which selectively works on the SA node and decreases heart rate without affecting blood pressure. What’s really elegant about ivabradine is it has a more potent effect when the heart rate is higher. When the patient is standing, it’s going to have a more potent effect on heart rate lowering. It’s really well tolerated in patients with POTS. In our study, we showed an improvement in quality of life metrics. That’s one of the first-line drugs that I use for patients with POTS.
The other thing is some of them will also have a concomitant lowering of blood pressure. You can think about medications that increase blood pressure, like midodrine, fludrocortisone, and droxidopa. Sometimes that combination of a heart rate-lowering medication and a medication that increases blood pressure really works well.
Dr. O’Donoghue: That’s very helpful. I think that those kinds of practical tips are the ones that practitioners really want to reach for, because they need to have that algorithm in their mind to take on this condition. Thanks again for walking us through that.
I think it’s a very interesting space, and there’s more that we’re going to be learning over the next few years as we further flesh out these post-COVID cases and what we learn from that as well.
Dr. Taub: There are many clinical trials now starting in POTS, so it’s exciting.
Dr. O’Donoghue: Absolutely. Thank you again for joining me today. Signing off, this is Dr Michelle O’Donoghue.
Dr. O’Donoghue is a cardiologist at Brigham and Women’s Hospital and senior investigator with the TIMI Study Group. A strong believer in evidence-based medicine, she relishes discussions about the published literature. A native Canadian, Dr. O’Donoghue loves spending time outdoors with her family but admits with shame that she’s never strapped on hockey skates. She disclosed ties with Amgen, AstraZeneca Pharmaceuticals LP, CVS Minute Clinic, Eisai, GlaxoSmithKline, Janssen Pharmaceuticals, Merck, Novartis, and The Medicines Company. Dr. Taub is professor of Medicine, University of California San Diego Health, La Jolla. She disclosed ties with Amgen, Bayer, Boehringer Ingelheim, Medtronic, Merck, Novartis, Novo Nordisk, and Sanofi.
A version of this article appeared on Medscape.com.
Take two pills and make a donation
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.

My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.
My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.
My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Sleep irregularity
In discussions between health care providers and patients, the words “regularity” and “irregularity” come up primarily in reference to either constipation or menstrual cycles. However, the participants in a recent panel convened by the National Sleep Foundation think we should also be discussing irregularity when we are discussing sleep with our patients.
The sleep experts on the panel began by considering 40,000 papers that directly or tangentially dealt with the topic of irregular sleep patterns. The reviewers uncovered numerous references to an association between sleep irregularity and a wide variety of adverse health outcomes, including obesity and metabolic disorders, hypertension and other cardiovascular disorders, and elevations in several inflammatory markers. Not surprisingly, the investigators also found an abundance of references supporting an association between irregular sleep and a suite of mental health problems, including depression, mood disorders, lower self esteem, poor academic performance, and deficits in attention. For example, several of the studies the panel reviewed found that in college students, GPA was lower when their sleep pattern was irregular. There were some papers that found no significant association between irregular sleep and other adverse health outcomes, but none of the studies demonstrated an association with better or improved health outcomes.

There is currently no universally accepted definition of an irregular sleep pattern. The experts pointed to some papers that used a standard deviation of 1 hour from the patient’s usual bed time determined by averaging over an interval measured in weeks. You and I shouldn’t be surprised that irregular sleep is unhealthy, but the breadth of the panel’s findings is impressive.
Although it has been long in coming, sleep is finally beginning to get some attention by the media. The focus is usually on the optimal number of hours we need each night. This panel’s findings suggest that total sleep time is only part of the story, and may even be less important than the regularity of our sleep patterns.
For those of us in pediatrics, the place where irregularity raises its ugly head is with teenagers and weekends. Although the numbers are far from clear, the question remains of how effective is catch-up sleep after a week of too-early mornings and too-late bedtimes for the chronically under-slept adolescent.
In some studies in which patients had the demonstrable effects of sleep deprivation (e.g., metabolic and cardiovascular) there was some improvement when weekend sleep was extended by 1 or 2 hours, but none beyond 2 hours.
The panel’s findings, while certainly significant, merely add weight and nuance to the existing evidence of importance of sleep and the damage done by sleep deprivation. As one of the panel members has said, “Sleep is the third pillar of health, equally important as diet and exercise, if not more.” However, this message is not getting out, or at least it is not being heeded. Like obesity, our efforts as advisers to our patients isn’t working. Unfortunately, this is because our advice is often whispered and given halfheartedly.
There was some evidence of improvement as a result of the pandemic, when those fortunate enough to be able to work from home were taking advantage of the flexibility in their schedules and getting more sleep. But health care providers certainly can’t take responsibility for what was an accident of nature.
Those of you who have been reading Letters from Maine for the last 3 decades may tire of my beating the tired horse of sleep deprivation. But I will not be deterred. I see very little evidence among health care professionals in taking the importance of sleep seriously. Sure, they may include it buried in the list of potential contributors to their patient’s complaint, but I see very little effort to move it higher on their list of priorities and almost no movement toward making substantive recommendations and then reinforcing them with follow-up.
Like obesity, sleep deprivation is a societal problem. We can lay some of the blame on Thomas Edison, but Until that time you will continue to read columns like this one when I encounter significant studies on the importance of sleep.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
In discussions between health care providers and patients, the words “regularity” and “irregularity” come up primarily in reference to either constipation or menstrual cycles. However, the participants in a recent panel convened by the National Sleep Foundation think we should also be discussing irregularity when we are discussing sleep with our patients.
The sleep experts on the panel began by considering 40,000 papers that directly or tangentially dealt with the topic of irregular sleep patterns. The reviewers uncovered numerous references to an association between sleep irregularity and a wide variety of adverse health outcomes, including obesity and metabolic disorders, hypertension and other cardiovascular disorders, and elevations in several inflammatory markers. Not surprisingly, the investigators also found an abundance of references supporting an association between irregular sleep and a suite of mental health problems, including depression, mood disorders, lower self esteem, poor academic performance, and deficits in attention. For example, several of the studies the panel reviewed found that in college students, GPA was lower when their sleep pattern was irregular. There were some papers that found no significant association between irregular sleep and other adverse health outcomes, but none of the studies demonstrated an association with better or improved health outcomes.
There is currently no universally accepted definition of an irregular sleep pattern. The experts pointed to some papers that used a standard deviation of 1 hour from the patient’s usual bed time determined by averaging over an interval measured in weeks. You and I shouldn’t be surprised that irregular sleep is unhealthy, but the breadth of the panel’s findings is impressive.
Although it has been long in coming, sleep is finally beginning to get some attention by the media. The focus is usually on the optimal number of hours we need each night. This panel’s findings suggest that total sleep time is only part of the story, and may even be less important than the regularity of our sleep patterns.
For those of us in pediatrics, the place where irregularity raises its ugly head is with teenagers and weekends. Although the numbers are far from clear, the question remains of how effective is catch-up sleep after a week of too-early mornings and too-late bedtimes for the chronically under-slept adolescent.
In some studies in which patients had the demonstrable effects of sleep deprivation (e.g., metabolic and cardiovascular) there was some improvement when weekend sleep was extended by 1 or 2 hours, but none beyond 2 hours.
The panel’s findings, while certainly significant, merely add weight and nuance to the existing evidence of importance of sleep and the damage done by sleep deprivation. As one of the panel members has said, “Sleep is the third pillar of health, equally important as diet and exercise, if not more.” However, this message is not getting out, or at least it is not being heeded. Like obesity, our efforts as advisers to our patients isn’t working. Unfortunately, this is because our advice is often whispered and given halfheartedly.
There was some evidence of improvement as a result of the pandemic, when those fortunate enough to be able to work from home were taking advantage of the flexibility in their schedules and getting more sleep. But health care providers certainly can’t take responsibility for what was an accident of nature.
Those of you who have been reading Letters from Maine for the last 3 decades may tire of my beating the tired horse of sleep deprivation. But I will not be deterred. I see very little evidence among health care professionals in taking the importance of sleep seriously. Sure, they may include it buried in the list of potential contributors to their patient’s complaint, but I see very little effort to move it higher on their list of priorities and almost no movement toward making substantive recommendations and then reinforcing them with follow-up.
Like obesity, sleep deprivation is a societal problem. We can lay some of the blame on Thomas Edison, but Until that time you will continue to read columns like this one when I encounter significant studies on the importance of sleep.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
In discussions between health care providers and patients, the words “regularity” and “irregularity” come up primarily in reference to either constipation or menstrual cycles. However, the participants in a recent panel convened by the National Sleep Foundation think we should also be discussing irregularity when we are discussing sleep with our patients.
The sleep experts on the panel began by considering 40,000 papers that directly or tangentially dealt with the topic of irregular sleep patterns. The reviewers uncovered numerous references to an association between sleep irregularity and a wide variety of adverse health outcomes, including obesity and metabolic disorders, hypertension and other cardiovascular disorders, and elevations in several inflammatory markers. Not surprisingly, the investigators also found an abundance of references supporting an association between irregular sleep and a suite of mental health problems, including depression, mood disorders, lower self esteem, poor academic performance, and deficits in attention. For example, several of the studies the panel reviewed found that in college students, GPA was lower when their sleep pattern was irregular. There were some papers that found no significant association between irregular sleep and other adverse health outcomes, but none of the studies demonstrated an association with better or improved health outcomes.
There is currently no universally accepted definition of an irregular sleep pattern. The experts pointed to some papers that used a standard deviation of 1 hour from the patient’s usual bed time determined by averaging over an interval measured in weeks. You and I shouldn’t be surprised that irregular sleep is unhealthy, but the breadth of the panel’s findings is impressive.
Although it has been long in coming, sleep is finally beginning to get some attention by the media. The focus is usually on the optimal number of hours we need each night. This panel’s findings suggest that total sleep time is only part of the story, and may even be less important than the regularity of our sleep patterns.
For those of us in pediatrics, the place where irregularity raises its ugly head is with teenagers and weekends. Although the numbers are far from clear, the question remains of how effective is catch-up sleep after a week of too-early mornings and too-late bedtimes for the chronically under-slept adolescent.
In some studies in which patients had the demonstrable effects of sleep deprivation (e.g., metabolic and cardiovascular) there was some improvement when weekend sleep was extended by 1 or 2 hours, but none beyond 2 hours.
The panel’s findings, while certainly significant, merely add weight and nuance to the existing evidence of importance of sleep and the damage done by sleep deprivation. As one of the panel members has said, “Sleep is the third pillar of health, equally important as diet and exercise, if not more.” However, this message is not getting out, or at least it is not being heeded. Like obesity, our efforts as advisers to our patients isn’t working. Unfortunately, this is because our advice is often whispered and given halfheartedly.
There was some evidence of improvement as a result of the pandemic, when those fortunate enough to be able to work from home were taking advantage of the flexibility in their schedules and getting more sleep. But health care providers certainly can’t take responsibility for what was an accident of nature.
Those of you who have been reading Letters from Maine for the last 3 decades may tire of my beating the tired horse of sleep deprivation. But I will not be deterred. I see very little evidence among health care professionals in taking the importance of sleep seriously. Sure, they may include it buried in the list of potential contributors to their patient’s complaint, but I see very little effort to move it higher on their list of priorities and almost no movement toward making substantive recommendations and then reinforcing them with follow-up.
Like obesity, sleep deprivation is a societal problem. We can lay some of the blame on Thomas Edison, but Until that time you will continue to read columns like this one when I encounter significant studies on the importance of sleep.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].