User login
Jeff Evans has been editor of Rheumatology News/MDedge Rheumatology and the EULAR Congress News since 2013. He started at Frontline Medical Communications in 2001 and was a reporter for 8 years before serving as editor of Clinical Neurology News and World Neurology, and briefly as editor of GI & Hepatology News. He graduated cum laude from Cornell University (New York) with a BA in biological sciences, concentrating in neurobiology and behavior.
Axial spondyloarthritis survey raises importance of discussing treatment changes
More than half of patients with axial spondyloarthritis in a survey of ArthritisPower Registry participants said they discussed a change in treatment with their doctor at their most recent visit, and these discussions were about changing medication or increasing the dose in more than two-thirds of instances.
The cross-sectional survey, published in ACR Open Rheumatology, is believed to be the first “to look at treatment decision-making from the patient perspective, meaning this is our first quantitative analysis to examine how patients think about important disease management decisions and communicate with their doctor about their care,” W. Benjamin Nowell, PhD, director of patient-centered research at CreakyJoints and principal investigator of the ArthritisPower registry, said in a news release.
“This study makes it clear that there are unmet treatment needs in the axial spondyloarthritis [axSpA] patient community,” senior author Jessica A. Walsh, MD, rheumatologist and associate professor at the University of Utah, Salt Lake City, said in the release. “In the future, we need to identify the tools that this specific arthritis community needs to ensure that shared decision-making about disease management and treatment escalation is working effectively between the patient and the provider.”
Survey results
Of the survey’s 274 participants with physician-diagnosed axSpA, 57% said they discussed treatment change at their last physician visit, and nearly half of the time it was brought up by the patient. About 80% of patients in the survey said they researched treatment changes before the visit.
The most common discussion points were about changing medicines or increasing dose (69%), compared with reducing dose (28%) or switching treatments (39%). Another 12% of respondents entered free-text responses to an “other” option with things such as exercise, physical therapy, surgery, waiting on results, insurance, and pregnancy.
Close to half (47%) of the patients were taking biologic disease-modifying antirheumatic drugs (bDMARDs), followed by prescription NSAIDs (44%), steroids (16%), or conventional synthetic DMARDs (11%). Half of all patients said they also took prescription muscle relaxers, nerve pain medications or antidepressants, and opioids.
More than half (55%) of patients taking a bDMARD were at least somewhat satisfied with their treatment for axSpA, and about half were satisfied with their control of axSpA-related pain.
Of the 12% of patients in the survey who reported being very satisfied overall with their treatment, 77% were taking a bDMARD, and these bDMARD users said that they prioritized the prevention of long-term consequences and their physician’s advice in their decision-making process.
A large percentage – 43% – said they were somewhat or very dissatisfied with treatment, and nearly two-thirds of these patients had discussed treatment change at their last physician visit.
A large majority of patients who discussed a treatment change agreed to it (85%), most often because their disease was not controlled by their previous treatment or because they thought it could be better controlled by a change in treatment.
The survey respondents were about 50 years old on average, and most were women (87%) and White (85%). They experienced a delay in diagnosis averaging more than 10 years from first onset of axSpA symptoms to initial axSpA diagnosis by a physician.
The study was sponsored by Eli Lilly. The study was also indirectly partially supported by a grant from the Patient-Centered Outcomes Research Institute for ArthritisPower. Dr. Nowell reported receiving grants/contracts from AbbVie, Eli Lilly, and PCORI and is an employee of the Global Healthy Living Foundation. The GHLF receives grants, sponsorships, and contracts from pharmaceutical manufacturers and private foundations. Five authors are employees and shareholders of Eli Lilly. Two authors reported financial relationships with multiple pharmaceutical companies.
More than half of patients with axial spondyloarthritis in a survey of ArthritisPower Registry participants said they discussed a change in treatment with their doctor at their most recent visit, and these discussions were about changing medication or increasing the dose in more than two-thirds of instances.
The cross-sectional survey, published in ACR Open Rheumatology, is believed to be the first “to look at treatment decision-making from the patient perspective, meaning this is our first quantitative analysis to examine how patients think about important disease management decisions and communicate with their doctor about their care,” W. Benjamin Nowell, PhD, director of patient-centered research at CreakyJoints and principal investigator of the ArthritisPower registry, said in a news release.
“This study makes it clear that there are unmet treatment needs in the axial spondyloarthritis [axSpA] patient community,” senior author Jessica A. Walsh, MD, rheumatologist and associate professor at the University of Utah, Salt Lake City, said in the release. “In the future, we need to identify the tools that this specific arthritis community needs to ensure that shared decision-making about disease management and treatment escalation is working effectively between the patient and the provider.”
Survey results
Of the survey’s 274 participants with physician-diagnosed axSpA, 57% said they discussed treatment change at their last physician visit, and nearly half of the time it was brought up by the patient. About 80% of patients in the survey said they researched treatment changes before the visit.
The most common discussion points were about changing medicines or increasing dose (69%), compared with reducing dose (28%) or switching treatments (39%). Another 12% of respondents entered free-text responses to an “other” option with things such as exercise, physical therapy, surgery, waiting on results, insurance, and pregnancy.
Close to half (47%) of the patients were taking biologic disease-modifying antirheumatic drugs (bDMARDs), followed by prescription NSAIDs (44%), steroids (16%), or conventional synthetic DMARDs (11%). Half of all patients said they also took prescription muscle relaxers, nerve pain medications or antidepressants, and opioids.
More than half (55%) of patients taking a bDMARD were at least somewhat satisfied with their treatment for axSpA, and about half were satisfied with their control of axSpA-related pain.
Of the 12% of patients in the survey who reported being very satisfied overall with their treatment, 77% were taking a bDMARD, and these bDMARD users said that they prioritized the prevention of long-term consequences and their physician’s advice in their decision-making process.
A large percentage – 43% – said they were somewhat or very dissatisfied with treatment, and nearly two-thirds of these patients had discussed treatment change at their last physician visit.
A large majority of patients who discussed a treatment change agreed to it (85%), most often because their disease was not controlled by their previous treatment or because they thought it could be better controlled by a change in treatment.
The survey respondents were about 50 years old on average, and most were women (87%) and White (85%). They experienced a delay in diagnosis averaging more than 10 years from first onset of axSpA symptoms to initial axSpA diagnosis by a physician.
The study was sponsored by Eli Lilly. The study was also indirectly partially supported by a grant from the Patient-Centered Outcomes Research Institute for ArthritisPower. Dr. Nowell reported receiving grants/contracts from AbbVie, Eli Lilly, and PCORI and is an employee of the Global Healthy Living Foundation. The GHLF receives grants, sponsorships, and contracts from pharmaceutical manufacturers and private foundations. Five authors are employees and shareholders of Eli Lilly. Two authors reported financial relationships with multiple pharmaceutical companies.
More than half of patients with axial spondyloarthritis in a survey of ArthritisPower Registry participants said they discussed a change in treatment with their doctor at their most recent visit, and these discussions were about changing medication or increasing the dose in more than two-thirds of instances.
The cross-sectional survey, published in ACR Open Rheumatology, is believed to be the first “to look at treatment decision-making from the patient perspective, meaning this is our first quantitative analysis to examine how patients think about important disease management decisions and communicate with their doctor about their care,” W. Benjamin Nowell, PhD, director of patient-centered research at CreakyJoints and principal investigator of the ArthritisPower registry, said in a news release.
“This study makes it clear that there are unmet treatment needs in the axial spondyloarthritis [axSpA] patient community,” senior author Jessica A. Walsh, MD, rheumatologist and associate professor at the University of Utah, Salt Lake City, said in the release. “In the future, we need to identify the tools that this specific arthritis community needs to ensure that shared decision-making about disease management and treatment escalation is working effectively between the patient and the provider.”
Survey results
Of the survey’s 274 participants with physician-diagnosed axSpA, 57% said they discussed treatment change at their last physician visit, and nearly half of the time it was brought up by the patient. About 80% of patients in the survey said they researched treatment changes before the visit.
The most common discussion points were about changing medicines or increasing dose (69%), compared with reducing dose (28%) or switching treatments (39%). Another 12% of respondents entered free-text responses to an “other” option with things such as exercise, physical therapy, surgery, waiting on results, insurance, and pregnancy.
Close to half (47%) of the patients were taking biologic disease-modifying antirheumatic drugs (bDMARDs), followed by prescription NSAIDs (44%), steroids (16%), or conventional synthetic DMARDs (11%). Half of all patients said they also took prescription muscle relaxers, nerve pain medications or antidepressants, and opioids.
More than half (55%) of patients taking a bDMARD were at least somewhat satisfied with their treatment for axSpA, and about half were satisfied with their control of axSpA-related pain.
Of the 12% of patients in the survey who reported being very satisfied overall with their treatment, 77% were taking a bDMARD, and these bDMARD users said that they prioritized the prevention of long-term consequences and their physician’s advice in their decision-making process.
A large percentage – 43% – said they were somewhat or very dissatisfied with treatment, and nearly two-thirds of these patients had discussed treatment change at their last physician visit.
A large majority of patients who discussed a treatment change agreed to it (85%), most often because their disease was not controlled by their previous treatment or because they thought it could be better controlled by a change in treatment.
The survey respondents were about 50 years old on average, and most were women (87%) and White (85%). They experienced a delay in diagnosis averaging more than 10 years from first onset of axSpA symptoms to initial axSpA diagnosis by a physician.
The study was sponsored by Eli Lilly. The study was also indirectly partially supported by a grant from the Patient-Centered Outcomes Research Institute for ArthritisPower. Dr. Nowell reported receiving grants/contracts from AbbVie, Eli Lilly, and PCORI and is an employee of the Global Healthy Living Foundation. The GHLF receives grants, sponsorships, and contracts from pharmaceutical manufacturers and private foundations. Five authors are employees and shareholders of Eli Lilly. Two authors reported financial relationships with multiple pharmaceutical companies.
FROM ACR OPEN RHEUMATOLOGY
Adalimumab biosimilar Cyltezo gets interchangeability designation
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
Temporary hold of mycophenolate helps immune response to SARS-CoV-2 vaccination
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
FROM ANNALS OF THE RHEUMATIC DISEASES
European agency recommends two new adalimumab biosimilars
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended marketing authorization this week for two new adalimumab biosimilars, Hukyndra and Libmyris.
The biosimilars, both developed by STADA Arzneimittel AG, will be available as a 40-mg solution for injection in a pre-filled syringe and pre-filled pen and 80-mg solution for injection in a pre-filled syringe. Both biosimilars will have 15 indications:
- rheumatoid arthritis
- polyarticular juvenile idiopathic arthritis
- enthesitis-related arthritis
- ankylosing spondylitis
- axial spondyloarthritis without radiographic evidence of ankylosing spondylitis
- psoriatic arthritis
- chronic plaque psoriasis (adults and children)
- hidradenitis suppurativa
- Crohn’s disease (adults and children)
- ulcerative colitis (adults and children)
- uveitis (adults and children)
Data show that both Hukyndra and Libmyris are highly similar to the reference product Humira (adalimumab), a monoclonal antibody to tumor necrosis factor alpha, and have comparable quality, safety, and efficacy.
A version of this article first appeared on Medscape.com.
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended marketing authorization this week for two new adalimumab biosimilars, Hukyndra and Libmyris.
The biosimilars, both developed by STADA Arzneimittel AG, will be available as a 40-mg solution for injection in a pre-filled syringe and pre-filled pen and 80-mg solution for injection in a pre-filled syringe. Both biosimilars will have 15 indications:
- rheumatoid arthritis
- polyarticular juvenile idiopathic arthritis
- enthesitis-related arthritis
- ankylosing spondylitis
- axial spondyloarthritis without radiographic evidence of ankylosing spondylitis
- psoriatic arthritis
- chronic plaque psoriasis (adults and children)
- hidradenitis suppurativa
- Crohn’s disease (adults and children)
- ulcerative colitis (adults and children)
- uveitis (adults and children)
Data show that both Hukyndra and Libmyris are highly similar to the reference product Humira (adalimumab), a monoclonal antibody to tumor necrosis factor alpha, and have comparable quality, safety, and efficacy.
A version of this article first appeared on Medscape.com.
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended marketing authorization this week for two new adalimumab biosimilars, Hukyndra and Libmyris.
The biosimilars, both developed by STADA Arzneimittel AG, will be available as a 40-mg solution for injection in a pre-filled syringe and pre-filled pen and 80-mg solution for injection in a pre-filled syringe. Both biosimilars will have 15 indications:
- rheumatoid arthritis
- polyarticular juvenile idiopathic arthritis
- enthesitis-related arthritis
- ankylosing spondylitis
- axial spondyloarthritis without radiographic evidence of ankylosing spondylitis
- psoriatic arthritis
- chronic plaque psoriasis (adults and children)
- hidradenitis suppurativa
- Crohn’s disease (adults and children)
- ulcerative colitis (adults and children)
- uveitis (adults and children)
Data show that both Hukyndra and Libmyris are highly similar to the reference product Humira (adalimumab), a monoclonal antibody to tumor necrosis factor alpha, and have comparable quality, safety, and efficacy.
A version of this article first appeared on Medscape.com.
Low RA flare rate reported after Pfizer COVID vaccination
Patients with rheumatoid arthritis in remission had a rate of flare following vaccination with the Pfizer/BioNtech COVID-19 vaccine that appears to be on par with rates seen with other vaccines in patients with RA, according to results from a small Italian cohort study.
“Our data show a very low flare rate [7.8% (6 of 77)] after the BNT162b2 COVID-19 vaccine in patients with RA in remission and are consistent with previous findings about varicella-zoster virus (6.7%) and hepatitis B virus (2.2%) vaccinations,” Riccardo Bixio, MD, and colleagues from University of Verona (Italy) Hospital Trust wrote in ACR Open Rheumatology. “Because remission is not commonly obtained in the real world, we are aware that our findings may not be generalizable to all patients with RA receiving COVID-19 vaccination.”
Other studies of flare rate after COVID-19 vaccination in patients with a variety of rheumatic and musculoskeletal diseases have reported rates ranging from 5% to 17%, they said.
The 77 consecutive patients from the University of Verona center that conducted the study were all in clinical remission in the 3 months before vaccination based on a 28-joint Disease Activity Score based on C-reactive protein (DAS28-CRP) of less than 2.6, and all had discontinued antirheumatic therapies according to American College of Rheumatology COVID-19 recommendations. The researchers defined flares as agreement between patient and rheumatologist assessments and a DAS28-CRP increase of more than 1.2.
Five of the six people with a flare had it occur after the second dose at a mean of 2.6 days later, and all flares were resolved within 2 weeks using glucocorticoids with or without anti-inflammatory drugs. One flare was called severe. The overall disease activity of the cohort after 3 months was not significantly changed after vaccination.
In noting that five out of the six patients with flares had withdrawn or delayed antirheumatic therapies around the time of vaccination according to ACR recommendations, the authors wrote that “Even if there is no direct evidence that holding therapies could occur in a higher proportion of disease flares, we suggest that clinicians consider this possibility when counseling patients about COVID-19 vaccination.”
The authors had no outside funding for the study and had no potential conflicts of interest to disclose.
Patients with rheumatoid arthritis in remission had a rate of flare following vaccination with the Pfizer/BioNtech COVID-19 vaccine that appears to be on par with rates seen with other vaccines in patients with RA, according to results from a small Italian cohort study.
“Our data show a very low flare rate [7.8% (6 of 77)] after the BNT162b2 COVID-19 vaccine in patients with RA in remission and are consistent with previous findings about varicella-zoster virus (6.7%) and hepatitis B virus (2.2%) vaccinations,” Riccardo Bixio, MD, and colleagues from University of Verona (Italy) Hospital Trust wrote in ACR Open Rheumatology. “Because remission is not commonly obtained in the real world, we are aware that our findings may not be generalizable to all patients with RA receiving COVID-19 vaccination.”
Other studies of flare rate after COVID-19 vaccination in patients with a variety of rheumatic and musculoskeletal diseases have reported rates ranging from 5% to 17%, they said.
The 77 consecutive patients from the University of Verona center that conducted the study were all in clinical remission in the 3 months before vaccination based on a 28-joint Disease Activity Score based on C-reactive protein (DAS28-CRP) of less than 2.6, and all had discontinued antirheumatic therapies according to American College of Rheumatology COVID-19 recommendations. The researchers defined flares as agreement between patient and rheumatologist assessments and a DAS28-CRP increase of more than 1.2.
Five of the six people with a flare had it occur after the second dose at a mean of 2.6 days later, and all flares were resolved within 2 weeks using glucocorticoids with or without anti-inflammatory drugs. One flare was called severe. The overall disease activity of the cohort after 3 months was not significantly changed after vaccination.
In noting that five out of the six patients with flares had withdrawn or delayed antirheumatic therapies around the time of vaccination according to ACR recommendations, the authors wrote that “Even if there is no direct evidence that holding therapies could occur in a higher proportion of disease flares, we suggest that clinicians consider this possibility when counseling patients about COVID-19 vaccination.”
The authors had no outside funding for the study and had no potential conflicts of interest to disclose.
Patients with rheumatoid arthritis in remission had a rate of flare following vaccination with the Pfizer/BioNtech COVID-19 vaccine that appears to be on par with rates seen with other vaccines in patients with RA, according to results from a small Italian cohort study.
“Our data show a very low flare rate [7.8% (6 of 77)] after the BNT162b2 COVID-19 vaccine in patients with RA in remission and are consistent with previous findings about varicella-zoster virus (6.7%) and hepatitis B virus (2.2%) vaccinations,” Riccardo Bixio, MD, and colleagues from University of Verona (Italy) Hospital Trust wrote in ACR Open Rheumatology. “Because remission is not commonly obtained in the real world, we are aware that our findings may not be generalizable to all patients with RA receiving COVID-19 vaccination.”
Other studies of flare rate after COVID-19 vaccination in patients with a variety of rheumatic and musculoskeletal diseases have reported rates ranging from 5% to 17%, they said.
The 77 consecutive patients from the University of Verona center that conducted the study were all in clinical remission in the 3 months before vaccination based on a 28-joint Disease Activity Score based on C-reactive protein (DAS28-CRP) of less than 2.6, and all had discontinued antirheumatic therapies according to American College of Rheumatology COVID-19 recommendations. The researchers defined flares as agreement between patient and rheumatologist assessments and a DAS28-CRP increase of more than 1.2.
Five of the six people with a flare had it occur after the second dose at a mean of 2.6 days later, and all flares were resolved within 2 weeks using glucocorticoids with or without anti-inflammatory drugs. One flare was called severe. The overall disease activity of the cohort after 3 months was not significantly changed after vaccination.
In noting that five out of the six patients with flares had withdrawn or delayed antirheumatic therapies around the time of vaccination according to ACR recommendations, the authors wrote that “Even if there is no direct evidence that holding therapies could occur in a higher proportion of disease flares, we suggest that clinicians consider this possibility when counseling patients about COVID-19 vaccination.”
The authors had no outside funding for the study and had no potential conflicts of interest to disclose.
FROM ACR OPEN RHEUMATOLOGY
NIH to study COVID vaccine booster in people with autoimmune disease
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
VEXAS: A novel rheumatologic, hematologic syndrome that’s making waves
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
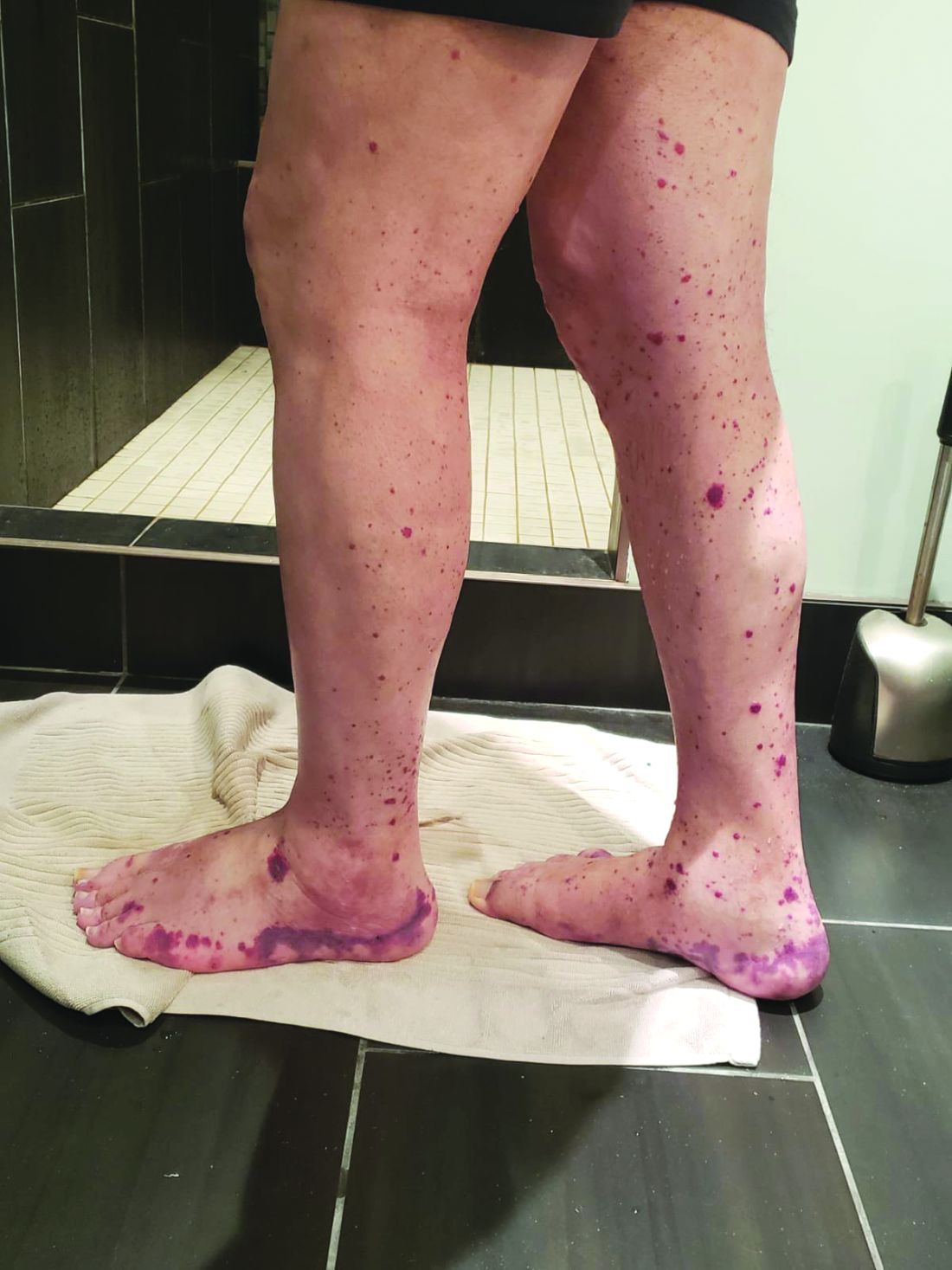
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
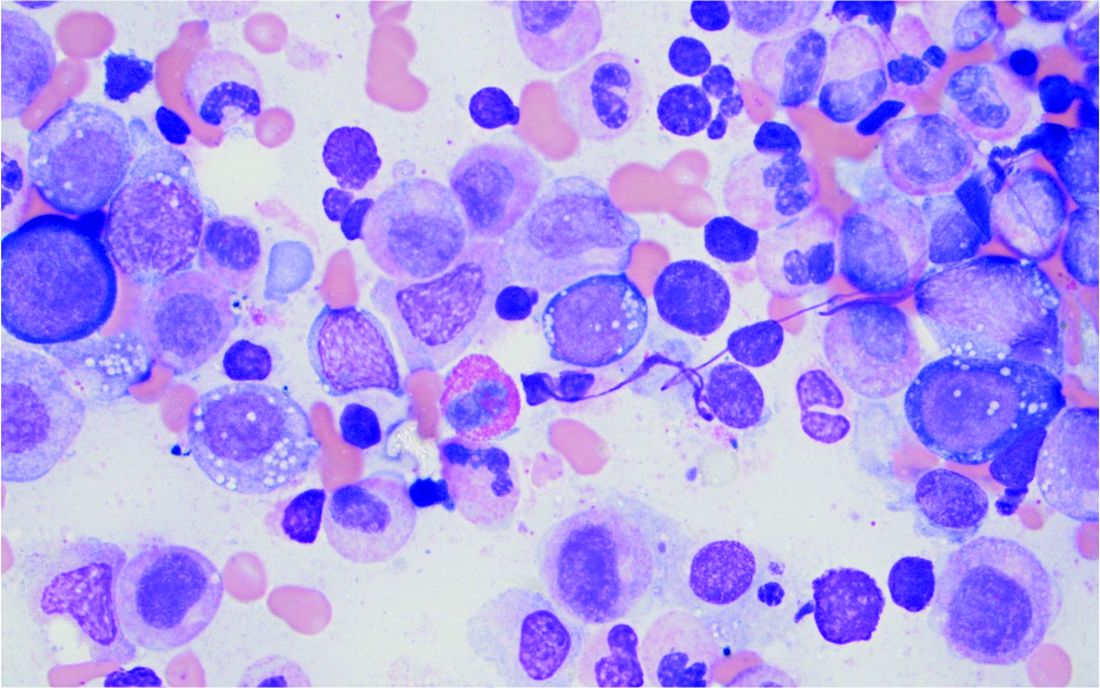
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
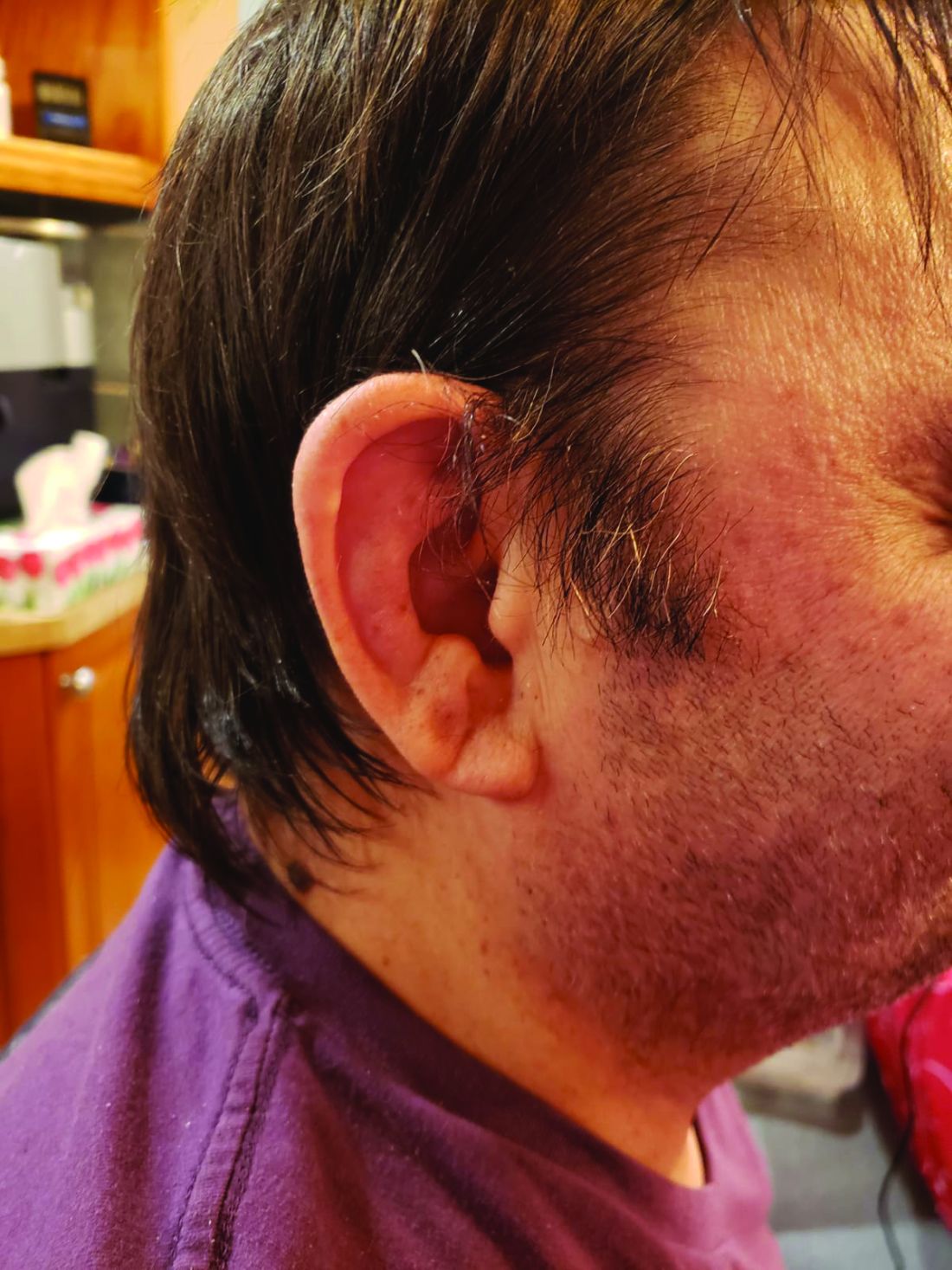
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
FDA warning letters target OTC cannabidiol product claims for pain relief
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.

In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
Dan Kastner wins Crafoord Prize in Polyarthritis
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
“for establishing the concept of autoinflammatory diseases.” The prize, named after the donor Holger Crafoord because of his bout with severe rheumatoid arthritis toward the end of his life, is for 6 million Swedish kronor (approximately USD $700,000).
Dr. Kastner, scientific director at the U.S. National Human Genome Research Institute’s division of intramural research, received the award for identifying the mechanisms responsible for familial Mediterranean fever, tumor necrosis factor receptor–associated periodic syndrome, and other diagnoses within the group of autoinflammatory diseases.
“Dan Kastner is often called the father of autoinflammatory diseases, a title that he thoroughly deserves. His discoveries have taught us a great deal about the immune system and its functions, contributing to effective treatments that reduce the symptoms of diseases from which patients previously suffered enormously, sometimes leading to premature death,” Olle Kämpe, chair of the prize committee, said in a press announcement.
While the Crafoord Prize normally is awarded on a 3-year rotating basis for achievements in mathematics and astronomy, geosciences, and biosciences, the prize in polyarthritis is “only awarded when there has been scientific progress that motivates a prize,” according to the press release.
High-need, high-cost lupus patients described for first time
according to a retrospective analysis of hospitalization data from a tertiary care center.

“The identification of the HNHC [high-need, high-cost] cohort and the risk factors for hospitalizations for this cohort will help pave the way to develop programs that improve the quality of care for high-risk lupus patients and [at the same time] lower the cost of care for all lupus patients,” first author Allen Anandarajah, MBBS, and colleagues at the University of Rochester (N.Y.) wrote in Arthritis Care & Research.
Hospitalizations and readmissions are known to be common in patients with SLE, the authors said, and they “account for a large proportion of the direct costs associated with the care of this disease.”
“While HNHC cohorts have been described with other chronic diseases, this report is the first to describe the existence of such a cohort in the SLE population,” the researchers said.
To see if a small group of SLE patients would constitute the majority of hospitalizations and consequently the costs of such care, Dr. Anandarajah and associates analyzed data from 202 SLE patients and their 467 hospitalizations at the University of Rochester–affiliated Strong Memorial Hospital during July 1, 2013, to June 30, 2016. The patients had a mean age of 46 years and included 183 females. A total of 46.5% were White, 43.1% were African American, 6.9% were Hispanic, and 3.5% were of Asian descent. These patients had median lengths of stay of 7 days per SLE patient and 4 days per admission, with median costs of $19,271 per patient and $14,375 per admission.
The researchers identified 44 patients (22%) who accounted for 275 admissions (59%) during the 3-year period. This group’s median of 4 admissions per patient was significantly higher than the median of 1 recorded in all the other hospitalized SLE patients, as was its number of readmissions within 30 days (105 total and median of 1 vs. 11 total and median of 0). The high-risk SLE patients spent a significantly greater amount of time in the hospital than did other patients (median of 30 days vs. 5 days), and their median cost was more than six times as great ($95,262 vs. $14,360). High-risk patients’ median cost per admission also was significantly greater ($19,376 vs. $12,833).
Infections were the most common cause of hospitalization among both high-risk patients and others (28% vs. 23%, respectively) and the rate of involvement of different organ systems as a cause for hospitalization were similar between the groups, except that patients at lower risk significantly more often had gynecologic/obstetric concerns (10% vs. 2%) or nervous system involvement (16% vs. 5%), and high-risk patients were significantly more likely to have gastrointestinal complaints (20% vs. 8%).
Clinically, high-risk patients had significantly higher median scores on the Systemic Lupus International Collaborating Clinics Damage Index and the Comorbidity Index, as well as a significantly higher median level of double-stranded DNA. However, they had no differences in complement factor levels or body mass index.
The high-risk patients also were younger (mean of 42 vs. 46 years) and were diagnosed at a younger mean age (26 vs. 31 years). More high-risk patients were African American (55% vs. 40%) and were more likely to live in areas identified with poverty (50% vs. 29%).
A multivariate analysis that controlled for relevant confounders showed that high-risk patients had a 10 percentage point lower medication possession ratio, which is an indicator of whether a patient had adequate medication supply in a given time frame. High-risk patients overall had a higher average number of medications to treat lupus.
“Our findings underscore the importance of identifying HNHC SLE patients when designing and implementing interventions to lower hospitalizations and improve the quality of care for lupus patients. Furthermore, it is imperative that we develop programs to address the modifiable social and behavioral factors in addition to providing high-quality clinical care targeted for this group,” the researchers wrote.
Some of the limitations in the generalizability of the results include the use of data from a large tertiary medical center serving a large catchment area, with a consequently sicker group of patients, and the potential to miss readmissions to other nearby hospitals. However, “as one of the few centers [in the region] that provides in-patient rheumatology care ... it is less likely that patients would have sought care elsewhere,” they noted.
The study involved no outside source of funding, and the authors had no relevant conflicts of interest.
SOURCE: Anandarajah A et al. Arthritis Care Res. 2020 Nov 17. doi: 10.1002/acr.24510.
according to a retrospective analysis of hospitalization data from a tertiary care center.
“The identification of the HNHC [high-need, high-cost] cohort and the risk factors for hospitalizations for this cohort will help pave the way to develop programs that improve the quality of care for high-risk lupus patients and [at the same time] lower the cost of care for all lupus patients,” first author Allen Anandarajah, MBBS, and colleagues at the University of Rochester (N.Y.) wrote in Arthritis Care & Research.
Hospitalizations and readmissions are known to be common in patients with SLE, the authors said, and they “account for a large proportion of the direct costs associated with the care of this disease.”
“While HNHC cohorts have been described with other chronic diseases, this report is the first to describe the existence of such a cohort in the SLE population,” the researchers said.
To see if a small group of SLE patients would constitute the majority of hospitalizations and consequently the costs of such care, Dr. Anandarajah and associates analyzed data from 202 SLE patients and their 467 hospitalizations at the University of Rochester–affiliated Strong Memorial Hospital during July 1, 2013, to June 30, 2016. The patients had a mean age of 46 years and included 183 females. A total of 46.5% were White, 43.1% were African American, 6.9% were Hispanic, and 3.5% were of Asian descent. These patients had median lengths of stay of 7 days per SLE patient and 4 days per admission, with median costs of $19,271 per patient and $14,375 per admission.
The researchers identified 44 patients (22%) who accounted for 275 admissions (59%) during the 3-year period. This group’s median of 4 admissions per patient was significantly higher than the median of 1 recorded in all the other hospitalized SLE patients, as was its number of readmissions within 30 days (105 total and median of 1 vs. 11 total and median of 0). The high-risk SLE patients spent a significantly greater amount of time in the hospital than did other patients (median of 30 days vs. 5 days), and their median cost was more than six times as great ($95,262 vs. $14,360). High-risk patients’ median cost per admission also was significantly greater ($19,376 vs. $12,833).
Infections were the most common cause of hospitalization among both high-risk patients and others (28% vs. 23%, respectively) and the rate of involvement of different organ systems as a cause for hospitalization were similar between the groups, except that patients at lower risk significantly more often had gynecologic/obstetric concerns (10% vs. 2%) or nervous system involvement (16% vs. 5%), and high-risk patients were significantly more likely to have gastrointestinal complaints (20% vs. 8%).
Clinically, high-risk patients had significantly higher median scores on the Systemic Lupus International Collaborating Clinics Damage Index and the Comorbidity Index, as well as a significantly higher median level of double-stranded DNA. However, they had no differences in complement factor levels or body mass index.
The high-risk patients also were younger (mean of 42 vs. 46 years) and were diagnosed at a younger mean age (26 vs. 31 years). More high-risk patients were African American (55% vs. 40%) and were more likely to live in areas identified with poverty (50% vs. 29%).
A multivariate analysis that controlled for relevant confounders showed that high-risk patients had a 10 percentage point lower medication possession ratio, which is an indicator of whether a patient had adequate medication supply in a given time frame. High-risk patients overall had a higher average number of medications to treat lupus.
“Our findings underscore the importance of identifying HNHC SLE patients when designing and implementing interventions to lower hospitalizations and improve the quality of care for lupus patients. Furthermore, it is imperative that we develop programs to address the modifiable social and behavioral factors in addition to providing high-quality clinical care targeted for this group,” the researchers wrote.
Some of the limitations in the generalizability of the results include the use of data from a large tertiary medical center serving a large catchment area, with a consequently sicker group of patients, and the potential to miss readmissions to other nearby hospitals. However, “as one of the few centers [in the region] that provides in-patient rheumatology care ... it is less likely that patients would have sought care elsewhere,” they noted.
The study involved no outside source of funding, and the authors had no relevant conflicts of interest.
SOURCE: Anandarajah A et al. Arthritis Care Res. 2020 Nov 17. doi: 10.1002/acr.24510.
according to a retrospective analysis of hospitalization data from a tertiary care center.
“The identification of the HNHC [high-need, high-cost] cohort and the risk factors for hospitalizations for this cohort will help pave the way to develop programs that improve the quality of care for high-risk lupus patients and [at the same time] lower the cost of care for all lupus patients,” first author Allen Anandarajah, MBBS, and colleagues at the University of Rochester (N.Y.) wrote in Arthritis Care & Research.
Hospitalizations and readmissions are known to be common in patients with SLE, the authors said, and they “account for a large proportion of the direct costs associated with the care of this disease.”
“While HNHC cohorts have been described with other chronic diseases, this report is the first to describe the existence of such a cohort in the SLE population,” the researchers said.
To see if a small group of SLE patients would constitute the majority of hospitalizations and consequently the costs of such care, Dr. Anandarajah and associates analyzed data from 202 SLE patients and their 467 hospitalizations at the University of Rochester–affiliated Strong Memorial Hospital during July 1, 2013, to June 30, 2016. The patients had a mean age of 46 years and included 183 females. A total of 46.5% were White, 43.1% were African American, 6.9% were Hispanic, and 3.5% were of Asian descent. These patients had median lengths of stay of 7 days per SLE patient and 4 days per admission, with median costs of $19,271 per patient and $14,375 per admission.
The researchers identified 44 patients (22%) who accounted for 275 admissions (59%) during the 3-year period. This group’s median of 4 admissions per patient was significantly higher than the median of 1 recorded in all the other hospitalized SLE patients, as was its number of readmissions within 30 days (105 total and median of 1 vs. 11 total and median of 0). The high-risk SLE patients spent a significantly greater amount of time in the hospital than did other patients (median of 30 days vs. 5 days), and their median cost was more than six times as great ($95,262 vs. $14,360). High-risk patients’ median cost per admission also was significantly greater ($19,376 vs. $12,833).
Infections were the most common cause of hospitalization among both high-risk patients and others (28% vs. 23%, respectively) and the rate of involvement of different organ systems as a cause for hospitalization were similar between the groups, except that patients at lower risk significantly more often had gynecologic/obstetric concerns (10% vs. 2%) or nervous system involvement (16% vs. 5%), and high-risk patients were significantly more likely to have gastrointestinal complaints (20% vs. 8%).
Clinically, high-risk patients had significantly higher median scores on the Systemic Lupus International Collaborating Clinics Damage Index and the Comorbidity Index, as well as a significantly higher median level of double-stranded DNA. However, they had no differences in complement factor levels or body mass index.
The high-risk patients also were younger (mean of 42 vs. 46 years) and were diagnosed at a younger mean age (26 vs. 31 years). More high-risk patients were African American (55% vs. 40%) and were more likely to live in areas identified with poverty (50% vs. 29%).
A multivariate analysis that controlled for relevant confounders showed that high-risk patients had a 10 percentage point lower medication possession ratio, which is an indicator of whether a patient had adequate medication supply in a given time frame. High-risk patients overall had a higher average number of medications to treat lupus.
“Our findings underscore the importance of identifying HNHC SLE patients when designing and implementing interventions to lower hospitalizations and improve the quality of care for lupus patients. Furthermore, it is imperative that we develop programs to address the modifiable social and behavioral factors in addition to providing high-quality clinical care targeted for this group,” the researchers wrote.
Some of the limitations in the generalizability of the results include the use of data from a large tertiary medical center serving a large catchment area, with a consequently sicker group of patients, and the potential to miss readmissions to other nearby hospitals. However, “as one of the few centers [in the region] that provides in-patient rheumatology care ... it is less likely that patients would have sought care elsewhere,” they noted.
The study involved no outside source of funding, and the authors had no relevant conflicts of interest.
SOURCE: Anandarajah A et al. Arthritis Care Res. 2020 Nov 17. doi: 10.1002/acr.24510.
FROM ARTHRITIS CARE & RESEARCH