User login
Lucas Franki is an associate editor for MDedge News, and has been with the company since 2014. He has a BA in English from Penn State University and is an Eagle Scout.
EASL publishes new PBC guidelines
The European Association for the Study of the Liver has published a new guideline for the diagnosis, treatment, and management of primary biliary cholangitis.
PBC is likely in patients with persistent cholestatic symptoms or who have pruritis and fatigue. A diagnosis of PBC can be made if a patient has elevated alkaline phosphatase and antimitochondrial antibody, although elevated antimitochondrial antibody alone is not enough to diagnose PBC. Liver biopsy is not recommended, and liver imaging is not necessary to prove PBC but can be used to eliminate extrahepatic causes of cholestasis.
Pruritis, fatigue, and sicca complex are the most common symptoms of PBC and can significantly effect quality of life. Pruritis can be treated with cholestyramine or rifampicin. Clinicians should seek out and treat associated and alternate causes of fatigue and advise patients on strategies to avoid compounding fatigue problems. Sicca complex should be treated appropriately and, if patients develop refractory symptoms, referred to a specialist.
Complications of liver disease caused by PBC include osteoporosis, fat-soluble vitamin substitution, hyperlipidemia, varices, hepatocellular carcinoma, and need for liver transplant, though the need for liver transplant in PBC patient has decreased over time.
“Treatment guidelines facilitate a holistic life-long approach to the management of patients with PBC, and care pathways should be developed locally to capture the needs of patients. These can be subject to independent quality evaluation,” EASL concluded.
Find the full clinical guideline in the Journal of Hepatology (2017. doi: 10.1016/j.jhep.2017.03.022).
The European Association for the Study of the Liver has published a new guideline for the diagnosis, treatment, and management of primary biliary cholangitis.
PBC is likely in patients with persistent cholestatic symptoms or who have pruritis and fatigue. A diagnosis of PBC can be made if a patient has elevated alkaline phosphatase and antimitochondrial antibody, although elevated antimitochondrial antibody alone is not enough to diagnose PBC. Liver biopsy is not recommended, and liver imaging is not necessary to prove PBC but can be used to eliminate extrahepatic causes of cholestasis.
Pruritis, fatigue, and sicca complex are the most common symptoms of PBC and can significantly effect quality of life. Pruritis can be treated with cholestyramine or rifampicin. Clinicians should seek out and treat associated and alternate causes of fatigue and advise patients on strategies to avoid compounding fatigue problems. Sicca complex should be treated appropriately and, if patients develop refractory symptoms, referred to a specialist.
Complications of liver disease caused by PBC include osteoporosis, fat-soluble vitamin substitution, hyperlipidemia, varices, hepatocellular carcinoma, and need for liver transplant, though the need for liver transplant in PBC patient has decreased over time.
“Treatment guidelines facilitate a holistic life-long approach to the management of patients with PBC, and care pathways should be developed locally to capture the needs of patients. These can be subject to independent quality evaluation,” EASL concluded.
Find the full clinical guideline in the Journal of Hepatology (2017. doi: 10.1016/j.jhep.2017.03.022).
The European Association for the Study of the Liver has published a new guideline for the diagnosis, treatment, and management of primary biliary cholangitis.
PBC is likely in patients with persistent cholestatic symptoms or who have pruritis and fatigue. A diagnosis of PBC can be made if a patient has elevated alkaline phosphatase and antimitochondrial antibody, although elevated antimitochondrial antibody alone is not enough to diagnose PBC. Liver biopsy is not recommended, and liver imaging is not necessary to prove PBC but can be used to eliminate extrahepatic causes of cholestasis.
Pruritis, fatigue, and sicca complex are the most common symptoms of PBC and can significantly effect quality of life. Pruritis can be treated with cholestyramine or rifampicin. Clinicians should seek out and treat associated and alternate causes of fatigue and advise patients on strategies to avoid compounding fatigue problems. Sicca complex should be treated appropriately and, if patients develop refractory symptoms, referred to a specialist.
Complications of liver disease caused by PBC include osteoporosis, fat-soluble vitamin substitution, hyperlipidemia, varices, hepatocellular carcinoma, and need for liver transplant, though the need for liver transplant in PBC patient has decreased over time.
“Treatment guidelines facilitate a holistic life-long approach to the management of patients with PBC, and care pathways should be developed locally to capture the needs of patients. These can be subject to independent quality evaluation,” EASL concluded.
Find the full clinical guideline in the Journal of Hepatology (2017. doi: 10.1016/j.jhep.2017.03.022).
FROM THE JOURNAL OF HEPATOLOGY
BMS recalls a lot of Eliquis 5 mg tablets
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
Mesh use for lap paraesophageal hernia repair held steady
Utilization of mesh in laparoscopic paraesophageal hernia repair (PEHR) remained steady from 2011 to 2014, despite a lack of evidence supporting its use, according to Francisco Schlottmann, MD, and his associates.
In an analysis of 9,590 laparoscopic PEHR performed from 2011 to 2014, 60.6% procedures were done without mesh and 39.4% were done with mesh. Over the 3-year study period, mesh utilization fell only 1.2% overall, with laparoscopic PEHR with mesh accounting for 39.4% of procedures in 2011 and 38.2% in 2014.
Patients who received mesh were slightly older and significantly more likely to be an inpatient admission. Postoperative urinary tract infection was less common in patients with mesh, occurring in 1% of patients, compared with 1.5% of patients without mesh. No significant difference in demographics was seen, and 30-day risk of comorbidity and mortality was the same. Mean length of stay was 2.7 days for PEHR with mesh and 2.5 days for PEHR without mesh.
“The use of mesh is associated with high expenses, and biomedical technology continues to offer newer and more expensive mesh products on the market. Given the progressive aging of the U.S. population, PEHR are expected to increase in the future. The indiscriminate and not supported by evidence use of mesh may determine unnecessary costs for the health care system,” the investigators noted.
Find the full study in the Journal of Gastrointestinal Surgery (2017 May 26. doi: 10.1007/s11605-017-3452-8).
Utilization of mesh in laparoscopic paraesophageal hernia repair (PEHR) remained steady from 2011 to 2014, despite a lack of evidence supporting its use, according to Francisco Schlottmann, MD, and his associates.
In an analysis of 9,590 laparoscopic PEHR performed from 2011 to 2014, 60.6% procedures were done without mesh and 39.4% were done with mesh. Over the 3-year study period, mesh utilization fell only 1.2% overall, with laparoscopic PEHR with mesh accounting for 39.4% of procedures in 2011 and 38.2% in 2014.
Patients who received mesh were slightly older and significantly more likely to be an inpatient admission. Postoperative urinary tract infection was less common in patients with mesh, occurring in 1% of patients, compared with 1.5% of patients without mesh. No significant difference in demographics was seen, and 30-day risk of comorbidity and mortality was the same. Mean length of stay was 2.7 days for PEHR with mesh and 2.5 days for PEHR without mesh.
“The use of mesh is associated with high expenses, and biomedical technology continues to offer newer and more expensive mesh products on the market. Given the progressive aging of the U.S. population, PEHR are expected to increase in the future. The indiscriminate and not supported by evidence use of mesh may determine unnecessary costs for the health care system,” the investigators noted.
Find the full study in the Journal of Gastrointestinal Surgery (2017 May 26. doi: 10.1007/s11605-017-3452-8).
Utilization of mesh in laparoscopic paraesophageal hernia repair (PEHR) remained steady from 2011 to 2014, despite a lack of evidence supporting its use, according to Francisco Schlottmann, MD, and his associates.
In an analysis of 9,590 laparoscopic PEHR performed from 2011 to 2014, 60.6% procedures were done without mesh and 39.4% were done with mesh. Over the 3-year study period, mesh utilization fell only 1.2% overall, with laparoscopic PEHR with mesh accounting for 39.4% of procedures in 2011 and 38.2% in 2014.
Patients who received mesh were slightly older and significantly more likely to be an inpatient admission. Postoperative urinary tract infection was less common in patients with mesh, occurring in 1% of patients, compared with 1.5% of patients without mesh. No significant difference in demographics was seen, and 30-day risk of comorbidity and mortality was the same. Mean length of stay was 2.7 days for PEHR with mesh and 2.5 days for PEHR without mesh.
“The use of mesh is associated with high expenses, and biomedical technology continues to offer newer and more expensive mesh products on the market. Given the progressive aging of the U.S. population, PEHR are expected to increase in the future. The indiscriminate and not supported by evidence use of mesh may determine unnecessary costs for the health care system,” the investigators noted.
Find the full study in the Journal of Gastrointestinal Surgery (2017 May 26. doi: 10.1007/s11605-017-3452-8).
FROM THE JOURNAL OF GASTROINTESTINAL SURGERY
Sjögren’s syndrome most common extrahepatic PBC manifestation
Extrahepatic manifestations of primary biliary cholangitis (PBC) occur in 73% of patients, with Sjögren’s syndrome, thyroid dysfunction, and systemic sclerosis being the most common, according to a literature review from Sara Chalifoux, MD, and her associates.
Sjögren’s syndrome occurs in 3.5%-73% of PBC patients, usually presenting with dry eyes and oral complications. Sjögren’s treatment in PBC patients involves symptom management associated with exocrine gland infiltration.
Thyroid diseases are present in 5.6%-23.6% of PBC patients. Hashimoto’s thyroiditis is the most common hypothyroidism in PBC patients, presenting with symptoms such as constipation, bradycardia, oligomenorrhea, and inability to concentrate. Grave’s disease is the most common hyperthyroidism, presenting with symptoms such as palpitations, tremulousness, heat intolerance, and weight loss.
Systemic sclerosis occurs in 1.4%-12.3% of PBC patients. Multiple studies found that limited cutaneous systemic sclerosis was more common in PBC patients than was the diffuse form of the disease.
Other diseases that may have a connection to PBC but lack solid, compelling evidence to make a firm association include rheumatoid arthritis, systemic lupus erythematosus, and celiac disease. While many PBC patients have irritable bowel disorder, there is no significant association between the two conditions.
“The patient care team should include practitioners in rheumatology, endocrinology, pulmonology, and cardiology when indicated. Patients should follow up regularly with their primary care physicians. As some of these extrahepatic manifestations can lead to diseases with a poor prognosis, vigilant screening and close follow-up will lead to prompt identification and treatment,” the investigators noted.
The investigators reported no financial conflicts of interest.
Find the full study in Gut and Liver (doi: 10.5009/gnl16365).
Extrahepatic manifestations of primary biliary cholangitis (PBC) occur in 73% of patients, with Sjögren’s syndrome, thyroid dysfunction, and systemic sclerosis being the most common, according to a literature review from Sara Chalifoux, MD, and her associates.
Sjögren’s syndrome occurs in 3.5%-73% of PBC patients, usually presenting with dry eyes and oral complications. Sjögren’s treatment in PBC patients involves symptom management associated with exocrine gland infiltration.
Thyroid diseases are present in 5.6%-23.6% of PBC patients. Hashimoto’s thyroiditis is the most common hypothyroidism in PBC patients, presenting with symptoms such as constipation, bradycardia, oligomenorrhea, and inability to concentrate. Grave’s disease is the most common hyperthyroidism, presenting with symptoms such as palpitations, tremulousness, heat intolerance, and weight loss.
Systemic sclerosis occurs in 1.4%-12.3% of PBC patients. Multiple studies found that limited cutaneous systemic sclerosis was more common in PBC patients than was the diffuse form of the disease.
Other diseases that may have a connection to PBC but lack solid, compelling evidence to make a firm association include rheumatoid arthritis, systemic lupus erythematosus, and celiac disease. While many PBC patients have irritable bowel disorder, there is no significant association between the two conditions.
“The patient care team should include practitioners in rheumatology, endocrinology, pulmonology, and cardiology when indicated. Patients should follow up regularly with their primary care physicians. As some of these extrahepatic manifestations can lead to diseases with a poor prognosis, vigilant screening and close follow-up will lead to prompt identification and treatment,” the investigators noted.
The investigators reported no financial conflicts of interest.
Find the full study in Gut and Liver (doi: 10.5009/gnl16365).
Extrahepatic manifestations of primary biliary cholangitis (PBC) occur in 73% of patients, with Sjögren’s syndrome, thyroid dysfunction, and systemic sclerosis being the most common, according to a literature review from Sara Chalifoux, MD, and her associates.
Sjögren’s syndrome occurs in 3.5%-73% of PBC patients, usually presenting with dry eyes and oral complications. Sjögren’s treatment in PBC patients involves symptom management associated with exocrine gland infiltration.
Thyroid diseases are present in 5.6%-23.6% of PBC patients. Hashimoto’s thyroiditis is the most common hypothyroidism in PBC patients, presenting with symptoms such as constipation, bradycardia, oligomenorrhea, and inability to concentrate. Grave’s disease is the most common hyperthyroidism, presenting with symptoms such as palpitations, tremulousness, heat intolerance, and weight loss.
Systemic sclerosis occurs in 1.4%-12.3% of PBC patients. Multiple studies found that limited cutaneous systemic sclerosis was more common in PBC patients than was the diffuse form of the disease.
Other diseases that may have a connection to PBC but lack solid, compelling evidence to make a firm association include rheumatoid arthritis, systemic lupus erythematosus, and celiac disease. While many PBC patients have irritable bowel disorder, there is no significant association between the two conditions.
“The patient care team should include practitioners in rheumatology, endocrinology, pulmonology, and cardiology when indicated. Patients should follow up regularly with their primary care physicians. As some of these extrahepatic manifestations can lead to diseases with a poor prognosis, vigilant screening and close follow-up will lead to prompt identification and treatment,” the investigators noted.
The investigators reported no financial conflicts of interest.
Find the full study in Gut and Liver (doi: 10.5009/gnl16365).
FROM GUT AND LIVER
ABP 501 equivalent in efficacy to adalimumab for moderate to severe RA
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Adequate ACR 20 response occurred in 74.6% of patients receiving ABP 501 and in 72.4% of patients receiving adalimumab.
Data source: A randomized, double-blind equivalence study across 100 medical centers in 12 countries of 526 patients with moderate to severe RA.
Disclosures: The study was funded by Amgen. Dr. Cohen and five of his associated reported conflicts of interest. Two investigators are employees of Amgen.
FDA approves Rebinyn for hemophilia B treatment
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
FDA approves generic Strattera for pediatric, adult ADHD patients
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
Alzheimer’s mortality in U.S. grew from 1999 to 2014
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.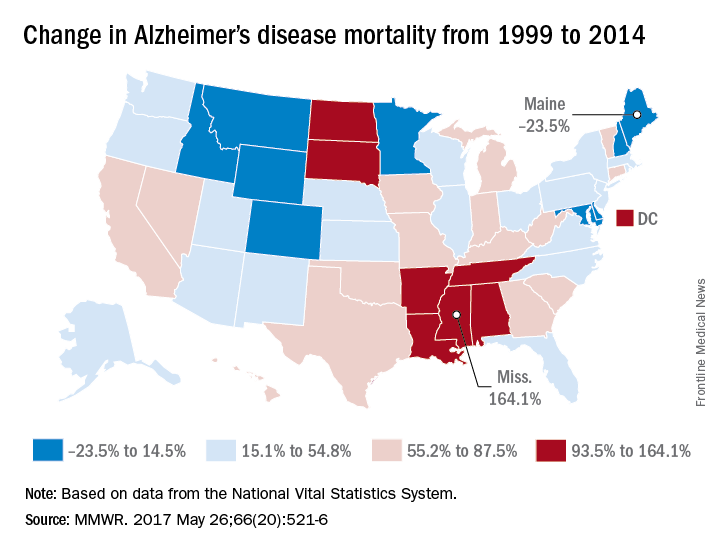
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
FROM MMWR
FDA approves sarilumab for DMARD-intolerant RA patients
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.
FDA approves first specific treatment for giant cell arteritis
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.