User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Emergency Department Visits for Suicide Attempts Rise Across the United States
TOPLINE:
Emergency department (ED) visits in the United States for suicide attempts and intentional self-harm show an increasing trend from 2011 to 2020, with visits being most common among adolescents and the largest increase in visits being seen in adults aged 65 years or older.
METHODOLOGY:
- This study used data from the National Hospital Ambulatory Medical Care Survey, an annual nationwide cross-sectional survey, to track trends in ED visits for suicide attempts and intentional self-harm in the United States from 2011 to 2020.
- Researchers identified visits for suicide attempts and intentional self-harm, along with diagnoses of any co-occurring mental health conditions, using discharge diagnosis codes or reason-for-visit codes.
- The focus was to identify the percentages of ED visits for suicide attempts and intentional self-harm, with analyses done per 100,000 persons and for changes possibly linked to the COVID-19 pandemic in 2019-2020.
TAKEAWAY:
- The number of ED visits owing to suicide attempts and intentional self-harm increased from 1.43 million in 2011-2012 to 5.37 million in 2019-2020 (average annual percent change, 19.5%; 95% confidence interval, 16.9-22.2).
- The rate of ED visits for suicide attempts and intentional self-harm was higher among adolescents and young adults, particularly women, and lower among children.
- Despite a surge in ED visits for self-harm, less than 16% included a mental health evaluation, with visits among patients with mood disorders decreasing by 5.5% annually and those among patients with drug-related disorders increasing by 6.8% annually.
- In 2019-2020, those aged 15-20 years had the highest rate of ED visits (1552 visits per 100,000 persons), with a significant increase seen across all age groups; the largest increase was among those aged 65 years or older.
IN PRACTICE:
“Given that suicide attempts are the single greatest risk factor for suicide, evidence-based management of individuals presenting to emergency departments with suicide attempts and intentional self-harm is a critical component of comprehensive suicide prevention strategies,” the authors wrote.
SOURCE:
The investigation, led by Tanner J. Bommersbach, MD, MPH, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, Minnesota, was published online in The American Journal of Psychiatry.
LIMITATIONS:
Visits for suicide attempts and intentional self-harm were identified based on discharge diagnostic and reason-for-visit codes, which may have led to an underestimation of visits for suicide attempts. ED visits for suicidal vs nonsuicidal self-injury could not be distinguished due to reliance on discharge diagnostic codes. Visits for suicidal ideation, which was not the focus of the study, may have been miscoded as suicide attempts and intentional self-harm.
DISCLOSURES:
No funding source was reported for the study. Some authors received funding grants from various institutions, and one author disclosed receiving honoraria for service as a review committee member and serving as a stakeholder/consultant and as an advisory committee member for some institutes and agencies.
A version of this article appeared on Medscape.com.
TOPLINE:
Emergency department (ED) visits in the United States for suicide attempts and intentional self-harm show an increasing trend from 2011 to 2020, with visits being most common among adolescents and the largest increase in visits being seen in adults aged 65 years or older.
METHODOLOGY:
- This study used data from the National Hospital Ambulatory Medical Care Survey, an annual nationwide cross-sectional survey, to track trends in ED visits for suicide attempts and intentional self-harm in the United States from 2011 to 2020.
- Researchers identified visits for suicide attempts and intentional self-harm, along with diagnoses of any co-occurring mental health conditions, using discharge diagnosis codes or reason-for-visit codes.
- The focus was to identify the percentages of ED visits for suicide attempts and intentional self-harm, with analyses done per 100,000 persons and for changes possibly linked to the COVID-19 pandemic in 2019-2020.
TAKEAWAY:
- The number of ED visits owing to suicide attempts and intentional self-harm increased from 1.43 million in 2011-2012 to 5.37 million in 2019-2020 (average annual percent change, 19.5%; 95% confidence interval, 16.9-22.2).
- The rate of ED visits for suicide attempts and intentional self-harm was higher among adolescents and young adults, particularly women, and lower among children.
- Despite a surge in ED visits for self-harm, less than 16% included a mental health evaluation, with visits among patients with mood disorders decreasing by 5.5% annually and those among patients with drug-related disorders increasing by 6.8% annually.
- In 2019-2020, those aged 15-20 years had the highest rate of ED visits (1552 visits per 100,000 persons), with a significant increase seen across all age groups; the largest increase was among those aged 65 years or older.
IN PRACTICE:
“Given that suicide attempts are the single greatest risk factor for suicide, evidence-based management of individuals presenting to emergency departments with suicide attempts and intentional self-harm is a critical component of comprehensive suicide prevention strategies,” the authors wrote.
SOURCE:
The investigation, led by Tanner J. Bommersbach, MD, MPH, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, Minnesota, was published online in The American Journal of Psychiatry.
LIMITATIONS:
Visits for suicide attempts and intentional self-harm were identified based on discharge diagnostic and reason-for-visit codes, which may have led to an underestimation of visits for suicide attempts. ED visits for suicidal vs nonsuicidal self-injury could not be distinguished due to reliance on discharge diagnostic codes. Visits for suicidal ideation, which was not the focus of the study, may have been miscoded as suicide attempts and intentional self-harm.
DISCLOSURES:
No funding source was reported for the study. Some authors received funding grants from various institutions, and one author disclosed receiving honoraria for service as a review committee member and serving as a stakeholder/consultant and as an advisory committee member for some institutes and agencies.
A version of this article appeared on Medscape.com.
TOPLINE:
Emergency department (ED) visits in the United States for suicide attempts and intentional self-harm show an increasing trend from 2011 to 2020, with visits being most common among adolescents and the largest increase in visits being seen in adults aged 65 years or older.
METHODOLOGY:
- This study used data from the National Hospital Ambulatory Medical Care Survey, an annual nationwide cross-sectional survey, to track trends in ED visits for suicide attempts and intentional self-harm in the United States from 2011 to 2020.
- Researchers identified visits for suicide attempts and intentional self-harm, along with diagnoses of any co-occurring mental health conditions, using discharge diagnosis codes or reason-for-visit codes.
- The focus was to identify the percentages of ED visits for suicide attempts and intentional self-harm, with analyses done per 100,000 persons and for changes possibly linked to the COVID-19 pandemic in 2019-2020.
TAKEAWAY:
- The number of ED visits owing to suicide attempts and intentional self-harm increased from 1.43 million in 2011-2012 to 5.37 million in 2019-2020 (average annual percent change, 19.5%; 95% confidence interval, 16.9-22.2).
- The rate of ED visits for suicide attempts and intentional self-harm was higher among adolescents and young adults, particularly women, and lower among children.
- Despite a surge in ED visits for self-harm, less than 16% included a mental health evaluation, with visits among patients with mood disorders decreasing by 5.5% annually and those among patients with drug-related disorders increasing by 6.8% annually.
- In 2019-2020, those aged 15-20 years had the highest rate of ED visits (1552 visits per 100,000 persons), with a significant increase seen across all age groups; the largest increase was among those aged 65 years or older.
IN PRACTICE:
“Given that suicide attempts are the single greatest risk factor for suicide, evidence-based management of individuals presenting to emergency departments with suicide attempts and intentional self-harm is a critical component of comprehensive suicide prevention strategies,” the authors wrote.
SOURCE:
The investigation, led by Tanner J. Bommersbach, MD, MPH, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, Minnesota, was published online in The American Journal of Psychiatry.
LIMITATIONS:
Visits for suicide attempts and intentional self-harm were identified based on discharge diagnostic and reason-for-visit codes, which may have led to an underestimation of visits for suicide attempts. ED visits for suicidal vs nonsuicidal self-injury could not be distinguished due to reliance on discharge diagnostic codes. Visits for suicidal ideation, which was not the focus of the study, may have been miscoded as suicide attempts and intentional self-harm.
DISCLOSURES:
No funding source was reported for the study. Some authors received funding grants from various institutions, and one author disclosed receiving honoraria for service as a review committee member and serving as a stakeholder/consultant and as an advisory committee member for some institutes and agencies.
A version of this article appeared on Medscape.com.
DEA Training Mandate: 8 Hours of My Life I’d Like Back
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty. 
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes! 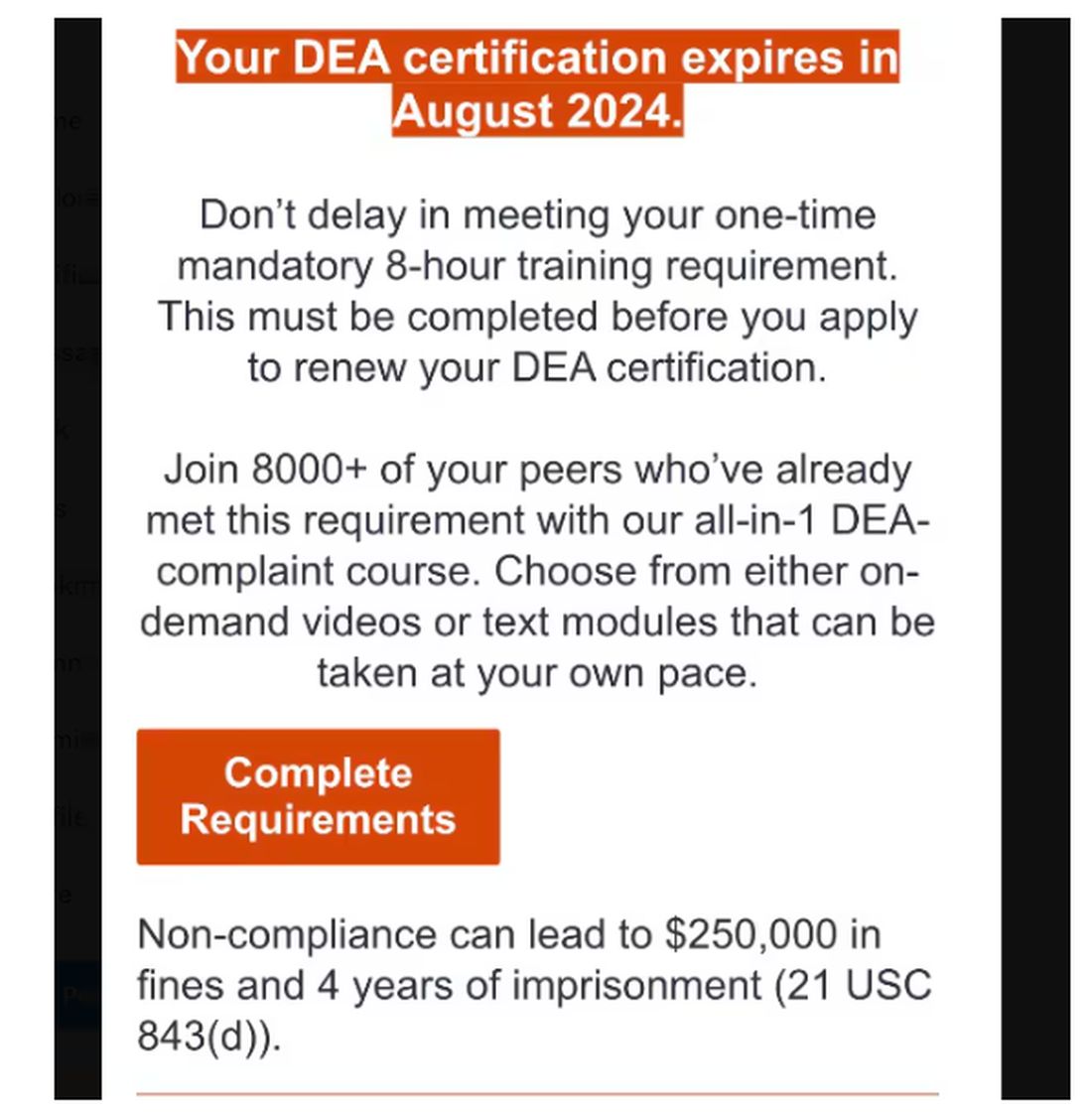
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
New Blood Test for Large Vessel Stroke Could Be a ‘Game Changer’
When combined with clinical scores, a “game-changing” blood test can expedite the diagnosis and treatment of large vessel occlusion (LVO) stroke, potentially saving many lives, new data suggested.
Using cutoff levels of two blood biomarkers, glial fibrillary acidic protein (GFAP; 213 pg/mL) and D-dimer (600 ng/mL), and the field assessment stroke triage for emergency destination (FAST-ED) (score, > 2), investigators were able to detect LVOs with 81% sensitivity and 93% specificity less than 6 hours from the onset of symptoms.
GFAP has previously been linked to brain bleeds and traumatic brain injury.
The test also ruled out all patients with brain bleeds, and investigators noted that it could also be used to detect intracerebral hemorrhage.
“We have developed a game-changing, accessible tool that could help ensure that more people suffering from stroke are in the right place at the right time to receive critical, life-restoring care,” senior author Joshua Bernstock, MD, PhD, MPH, a clinical fellow in the department of neurosurgery at Brigham and Women’s Hospital in Boston, said in a press release.
The findings were published online on May 17 in Stroke: Vascular and Interventional Neurology.
Early Identification Crucial
Acute LVO stroke is one of the most treatable stroke types because of the availability of endovascular thrombectomy (EVT). However, EVT requires specialized equipment and teams that represent a small subset of accredited stroke centers and an even smaller subset of emergency medical facilities, so early identification of LVO is crucial, the investigators noted.
Dr. Bernstock and his team developed the TIME trial to assess the sensitivity and specificity of the blood biomarkers and scale cutoff values for identifying LVO vs non-LVO stroke.
As part of the observational prospective cohort trial, investigators included consecutive patients admitted to the Brandon Regional Hospital Emergency Department in Brandon, Florida, between May 2021 and August 2022 if they were referred for a suspected stroke and the time from symptom onset was under 18 hours.
Patients were excluded if they received thrombolytic therapy before blood was collected or if it was anticipated that blood collection would be difficult.
Investigators gathered information on patients’ clinical data, hematology results, time since last known well, and imaging findings to construct a clinical diagnosis (LVO, non-LVO, ischemic stroke, hemorrhagic stroke, or transient ischemic attack [TIA]).
In addition to the National Institutes of Health Stroke Scale, patients were assessed with the FAST-ED, the Rapid Arterial oCclusion Evaluation (RACE), the Cincinnati Stroke Triage Assessment Tool, and the Emergency Medical Stroke Assessment.
Of 323 patients in the final study sample, 29 (9%) had LVO ischemic stroke, and 48 (15%) had non-LVO ischemic stroke. Another 13 (4%) had hemorrhagic stroke, 12 had TIA (3.7%), and the largest proportion of patients had stroke mimic (n = 220; 68%), which included encephalopathy, hyperglycemia, hypertensive emergency, migraine, posterior reversible encephalopathy syndrome, and undetermined.
The Case for Biomarkers
When investigators looked at those with LVO ischemic stroke, they found the concentration of plasma D-dimer was significantly higher than that in patients with non-LVO suspected stroke (LVO suspected stroke, 1213 ng/mL; interquartile range [IQR], 733-1609 vs non-LVO suspected stroke, 617 ng/mL; IQR, 377-1345; P < .001).
In addition, GFAP was significantly increased in the plasma of patients with hemorrhagic stroke vs all other patients with suspected stroke (hemorrhagic stroke, 1464 pg/mL; IQR, 292-2580 vs nonhemorrhagic suspected stroke, 48 pg/mL; IQR, 12-98; P < .005).
Combinations of the blood biomarkers with the scales FAST-ED or RACE showed the best performance for LVO detection, with a specificity of 94% (for either scale combination) and a sensitivity of 71% for both scales.
When investigators analyzed data for just those patients identified within 6 hours of symptom onset, the combination of biomarkers plus FAST-ED resulted in a specificity of 93% and a sensitivity of 81%.
Given that clinical stroke scales in patients with hemorrhagic stroke frequently suggest LVO and that these patients are not candidates for EVT, a tool capable of ruling out hemorrhage and identifying only nonhemorrhagic ischemic LVO is essential, the investigators noted.
“In stroke care, time is brain,” Dr. Bernstock said. “The sooner a patient is put on the right care pathway, the better they are going to do. Whether that means ruling out bleeds or ruling in something that needs an intervention, being able to do this in a prehospital setting with the technology that we built is going to be truly transformative.”
The study was funded by the Innovate UK grant and private funding. Dr. Bernstock has positions and equity in Pockit Diagnostics Ltd. and Treovir Inc. and is on the boards of Centile Bio and NeuroX1. Other disclosures are noted in the original article.
A version of this article appeared on Medscape.com.
When combined with clinical scores, a “game-changing” blood test can expedite the diagnosis and treatment of large vessel occlusion (LVO) stroke, potentially saving many lives, new data suggested.
Using cutoff levels of two blood biomarkers, glial fibrillary acidic protein (GFAP; 213 pg/mL) and D-dimer (600 ng/mL), and the field assessment stroke triage for emergency destination (FAST-ED) (score, > 2), investigators were able to detect LVOs with 81% sensitivity and 93% specificity less than 6 hours from the onset of symptoms.
GFAP has previously been linked to brain bleeds and traumatic brain injury.
The test also ruled out all patients with brain bleeds, and investigators noted that it could also be used to detect intracerebral hemorrhage.
“We have developed a game-changing, accessible tool that could help ensure that more people suffering from stroke are in the right place at the right time to receive critical, life-restoring care,” senior author Joshua Bernstock, MD, PhD, MPH, a clinical fellow in the department of neurosurgery at Brigham and Women’s Hospital in Boston, said in a press release.
The findings were published online on May 17 in Stroke: Vascular and Interventional Neurology.
Early Identification Crucial
Acute LVO stroke is one of the most treatable stroke types because of the availability of endovascular thrombectomy (EVT). However, EVT requires specialized equipment and teams that represent a small subset of accredited stroke centers and an even smaller subset of emergency medical facilities, so early identification of LVO is crucial, the investigators noted.
Dr. Bernstock and his team developed the TIME trial to assess the sensitivity and specificity of the blood biomarkers and scale cutoff values for identifying LVO vs non-LVO stroke.
As part of the observational prospective cohort trial, investigators included consecutive patients admitted to the Brandon Regional Hospital Emergency Department in Brandon, Florida, between May 2021 and August 2022 if they were referred for a suspected stroke and the time from symptom onset was under 18 hours.
Patients were excluded if they received thrombolytic therapy before blood was collected or if it was anticipated that blood collection would be difficult.
Investigators gathered information on patients’ clinical data, hematology results, time since last known well, and imaging findings to construct a clinical diagnosis (LVO, non-LVO, ischemic stroke, hemorrhagic stroke, or transient ischemic attack [TIA]).
In addition to the National Institutes of Health Stroke Scale, patients were assessed with the FAST-ED, the Rapid Arterial oCclusion Evaluation (RACE), the Cincinnati Stroke Triage Assessment Tool, and the Emergency Medical Stroke Assessment.
Of 323 patients in the final study sample, 29 (9%) had LVO ischemic stroke, and 48 (15%) had non-LVO ischemic stroke. Another 13 (4%) had hemorrhagic stroke, 12 had TIA (3.7%), and the largest proportion of patients had stroke mimic (n = 220; 68%), which included encephalopathy, hyperglycemia, hypertensive emergency, migraine, posterior reversible encephalopathy syndrome, and undetermined.
The Case for Biomarkers
When investigators looked at those with LVO ischemic stroke, they found the concentration of plasma D-dimer was significantly higher than that in patients with non-LVO suspected stroke (LVO suspected stroke, 1213 ng/mL; interquartile range [IQR], 733-1609 vs non-LVO suspected stroke, 617 ng/mL; IQR, 377-1345; P < .001).
In addition, GFAP was significantly increased in the plasma of patients with hemorrhagic stroke vs all other patients with suspected stroke (hemorrhagic stroke, 1464 pg/mL; IQR, 292-2580 vs nonhemorrhagic suspected stroke, 48 pg/mL; IQR, 12-98; P < .005).
Combinations of the blood biomarkers with the scales FAST-ED or RACE showed the best performance for LVO detection, with a specificity of 94% (for either scale combination) and a sensitivity of 71% for both scales.
When investigators analyzed data for just those patients identified within 6 hours of symptom onset, the combination of biomarkers plus FAST-ED resulted in a specificity of 93% and a sensitivity of 81%.
Given that clinical stroke scales in patients with hemorrhagic stroke frequently suggest LVO and that these patients are not candidates for EVT, a tool capable of ruling out hemorrhage and identifying only nonhemorrhagic ischemic LVO is essential, the investigators noted.
“In stroke care, time is brain,” Dr. Bernstock said. “The sooner a patient is put on the right care pathway, the better they are going to do. Whether that means ruling out bleeds or ruling in something that needs an intervention, being able to do this in a prehospital setting with the technology that we built is going to be truly transformative.”
The study was funded by the Innovate UK grant and private funding. Dr. Bernstock has positions and equity in Pockit Diagnostics Ltd. and Treovir Inc. and is on the boards of Centile Bio and NeuroX1. Other disclosures are noted in the original article.
A version of this article appeared on Medscape.com.
When combined with clinical scores, a “game-changing” blood test can expedite the diagnosis and treatment of large vessel occlusion (LVO) stroke, potentially saving many lives, new data suggested.
Using cutoff levels of two blood biomarkers, glial fibrillary acidic protein (GFAP; 213 pg/mL) and D-dimer (600 ng/mL), and the field assessment stroke triage for emergency destination (FAST-ED) (score, > 2), investigators were able to detect LVOs with 81% sensitivity and 93% specificity less than 6 hours from the onset of symptoms.
GFAP has previously been linked to brain bleeds and traumatic brain injury.
The test also ruled out all patients with brain bleeds, and investigators noted that it could also be used to detect intracerebral hemorrhage.
“We have developed a game-changing, accessible tool that could help ensure that more people suffering from stroke are in the right place at the right time to receive critical, life-restoring care,” senior author Joshua Bernstock, MD, PhD, MPH, a clinical fellow in the department of neurosurgery at Brigham and Women’s Hospital in Boston, said in a press release.
The findings were published online on May 17 in Stroke: Vascular and Interventional Neurology.
Early Identification Crucial
Acute LVO stroke is one of the most treatable stroke types because of the availability of endovascular thrombectomy (EVT). However, EVT requires specialized equipment and teams that represent a small subset of accredited stroke centers and an even smaller subset of emergency medical facilities, so early identification of LVO is crucial, the investigators noted.
Dr. Bernstock and his team developed the TIME trial to assess the sensitivity and specificity of the blood biomarkers and scale cutoff values for identifying LVO vs non-LVO stroke.
As part of the observational prospective cohort trial, investigators included consecutive patients admitted to the Brandon Regional Hospital Emergency Department in Brandon, Florida, between May 2021 and August 2022 if they were referred for a suspected stroke and the time from symptom onset was under 18 hours.
Patients were excluded if they received thrombolytic therapy before blood was collected or if it was anticipated that blood collection would be difficult.
Investigators gathered information on patients’ clinical data, hematology results, time since last known well, and imaging findings to construct a clinical diagnosis (LVO, non-LVO, ischemic stroke, hemorrhagic stroke, or transient ischemic attack [TIA]).
In addition to the National Institutes of Health Stroke Scale, patients were assessed with the FAST-ED, the Rapid Arterial oCclusion Evaluation (RACE), the Cincinnati Stroke Triage Assessment Tool, and the Emergency Medical Stroke Assessment.
Of 323 patients in the final study sample, 29 (9%) had LVO ischemic stroke, and 48 (15%) had non-LVO ischemic stroke. Another 13 (4%) had hemorrhagic stroke, 12 had TIA (3.7%), and the largest proportion of patients had stroke mimic (n = 220; 68%), which included encephalopathy, hyperglycemia, hypertensive emergency, migraine, posterior reversible encephalopathy syndrome, and undetermined.
The Case for Biomarkers
When investigators looked at those with LVO ischemic stroke, they found the concentration of plasma D-dimer was significantly higher than that in patients with non-LVO suspected stroke (LVO suspected stroke, 1213 ng/mL; interquartile range [IQR], 733-1609 vs non-LVO suspected stroke, 617 ng/mL; IQR, 377-1345; P < .001).
In addition, GFAP was significantly increased in the plasma of patients with hemorrhagic stroke vs all other patients with suspected stroke (hemorrhagic stroke, 1464 pg/mL; IQR, 292-2580 vs nonhemorrhagic suspected stroke, 48 pg/mL; IQR, 12-98; P < .005).
Combinations of the blood biomarkers with the scales FAST-ED or RACE showed the best performance for LVO detection, with a specificity of 94% (for either scale combination) and a sensitivity of 71% for both scales.
When investigators analyzed data for just those patients identified within 6 hours of symptom onset, the combination of biomarkers plus FAST-ED resulted in a specificity of 93% and a sensitivity of 81%.
Given that clinical stroke scales in patients with hemorrhagic stroke frequently suggest LVO and that these patients are not candidates for EVT, a tool capable of ruling out hemorrhage and identifying only nonhemorrhagic ischemic LVO is essential, the investigators noted.
“In stroke care, time is brain,” Dr. Bernstock said. “The sooner a patient is put on the right care pathway, the better they are going to do. Whether that means ruling out bleeds or ruling in something that needs an intervention, being able to do this in a prehospital setting with the technology that we built is going to be truly transformative.”
The study was funded by the Innovate UK grant and private funding. Dr. Bernstock has positions and equity in Pockit Diagnostics Ltd. and Treovir Inc. and is on the boards of Centile Bio and NeuroX1. Other disclosures are noted in the original article.
A version of this article appeared on Medscape.com.
FROM STROKE: VASCULAR AND INTERVENTIONAL NEUROLOGY
New Era? ‘Double Selective’ Antibiotic Spares the Microbiome
A new antibiotic uses a never-before-seen mechanism to deliver a direct hit on tough-to-treat infections while leaving beneficial microbes alone. The strategy could lead to a new class of antibiotics that attack dangerous bacteria in a powerful new way, overcoming current drug resistance while sparing the gut microbiome.
“The biggest takeaway is the double-selective component,” said co-lead author Kristen A. Muñoz, PhD, who performed the research as a doctoral student at University of Illinois at Urbana-Champaign (UIUC). “We were able to develop a drug that not only targets problematic pathogens, but because it is selective for these pathogens only, we can spare the good bacteria and preserve the integrity of the microbiome.”
The drug goes after Gram-negative bacteria — pathogens responsible for debilitating and even fatal infections like gastroenteritis, urinary tract infections, pneumonia, sepsis, and cholera. The arsenal of antibiotics against them is old, with no new classes specifically targeting these bacteria coming on the market since 1968.
Many of these bugs have become resistant to one or more antibiotics, with deadly consequences. And antibiotics against them can also wipe out beneficial gut bacteria, allowing serious secondary infections to flare up.
In a study published in Nature, the drug lolamicin knocked out or reduced 130 strains of antibiotic-resistant Gram-negative bacteria in cell cultures. It also successfully treated drug-resistant bloodstream infections and pneumonia in mice while sparing their gut microbiome.
With their microbiomes intact, the mice then fought off secondary infection with Clostridioides difficile (a leading cause of opportunistic and sometimes fatal infections in US health care facilities), while mice treated with other compounds that damaged their microbiome succumbed.
How It Works
Like a well-built medieval castle, Gram-negative bacteria are encased in two protective walls, or membranes. Dr. Muñoz and her team at UIUC set out to breach this defense by finding compounds that hinder the “Lol system,” which ferries lipoproteins between them.
From one compound they constructed lolamicin, which can stop Gram-negative pathogens — with little effect on Gram-negative beneficial bacteria and no effect on Gram-positive bacteria.
“Gram-positive bacteria do not have an outer membrane, so they do not possess the Lol system,” Dr. Muñoz said. “When we compared the sequences of the Lol system in certain Gram-negative pathogens to Gram-negative commensal [beneficial] gut bacteria, we saw that the Lol systems were pretty different.”
Tossing a monkey wrench into the Lol system may be the study’s biggest contribution to future antibiotic development, said Kim Lewis, PhD, professor of Biology and director of Antimicrobial Discovery Center at Northeastern University, Boston, who has discovered several antibiotics now in preclinical research. One, darobactin, targets Gram-negative bugs without affecting the gut microbiome. Another, teixobactin, takes down Gram-positive bacteria without causing drug resistance.
“Lolamicin hits a novel target. I would say that’s the most significant study finding,” said Dr. Lewis, who was not involved in the study. “That is rare. If you look at antibiotics introduced since 1968, they have been modifications of existing antibiotics or, rarely, new chemically but hitting the same proven targets. This one hits something properly new, and [that’s] what I found perhaps the most original and interesting.”
Kirk E. Hevener, PharmD, PhD, associate professor of Pharmaceutical Sciences at the University of Tennessee Health Science Center, Memphis, Tennessee, agreed. (Dr. Hevener also was not involved in the study.) “Lolamicin works by targeting a unique Gram-negative transport system. No currently approved antibacterials work in this way, meaning it potentially represents the first of a new class of antibacterials with narrow-spectrum Gram-negative activity and low gastrointestinal disturbance,” said Dr. Hevener, whose research looks at new antimicrobial drug targets.
The UIUC researchers noted that lolamicin has one drawback: Bacteria frequently developed resistance to it. But in future work, it could be tweaked, combined with other antibiotics, or used as a template for finding other Lol system attackers, they said.
“There is still a good amount of work cut out for us in terms of assessing the clinical translatability of lolamicin, but we are hopeful for the future of this drug,” Dr. Muñoz said.
Addressing a Dire Need
Bringing such a drug to market — from discovery to Food and Drug Administration approval — could take more than a decade, said Dr. Hevener. And new agents, especially for Gram-negative bugs, are sorely needed.
Not only do these bacteria shield themselves with a double membrane but they also “have more complex resistance mechanisms including special pumps that can remove antibacterial drugs from the cell before they can be effective,” Dr. Hevener said.
As a result, drug-resistant Gram-negative bacteria are making treatment of severe infections such as sepsis and pneumonia in health care settings difficult.
Bloodstream infections with drug-resistant Klebsiella pneumoniae have a 40% mortality rate, Dr. Lewis said. And microbiome damage caused by antibiotics is also widespread and deadly, wiping out communities of helpful, protective gut bacteria. That contributes to over half of the C. difficile infections that affect 500,000 people and kill 30,000 a year in the United States.
“Our arsenal of antibacterials that can be used to treat Gram-negative infections is dangerously low,” Dr. Hevener said. “Research will always be needed to develop new antibacterials with novel mechanisms of activity that can bypass bacterial resistance mechanisms.”
A version of this article appeared on Medscape.com.
A new antibiotic uses a never-before-seen mechanism to deliver a direct hit on tough-to-treat infections while leaving beneficial microbes alone. The strategy could lead to a new class of antibiotics that attack dangerous bacteria in a powerful new way, overcoming current drug resistance while sparing the gut microbiome.
“The biggest takeaway is the double-selective component,” said co-lead author Kristen A. Muñoz, PhD, who performed the research as a doctoral student at University of Illinois at Urbana-Champaign (UIUC). “We were able to develop a drug that not only targets problematic pathogens, but because it is selective for these pathogens only, we can spare the good bacteria and preserve the integrity of the microbiome.”
The drug goes after Gram-negative bacteria — pathogens responsible for debilitating and even fatal infections like gastroenteritis, urinary tract infections, pneumonia, sepsis, and cholera. The arsenal of antibiotics against them is old, with no new classes specifically targeting these bacteria coming on the market since 1968.
Many of these bugs have become resistant to one or more antibiotics, with deadly consequences. And antibiotics against them can also wipe out beneficial gut bacteria, allowing serious secondary infections to flare up.
In a study published in Nature, the drug lolamicin knocked out or reduced 130 strains of antibiotic-resistant Gram-negative bacteria in cell cultures. It also successfully treated drug-resistant bloodstream infections and pneumonia in mice while sparing their gut microbiome.
With their microbiomes intact, the mice then fought off secondary infection with Clostridioides difficile (a leading cause of opportunistic and sometimes fatal infections in US health care facilities), while mice treated with other compounds that damaged their microbiome succumbed.
How It Works
Like a well-built medieval castle, Gram-negative bacteria are encased in two protective walls, or membranes. Dr. Muñoz and her team at UIUC set out to breach this defense by finding compounds that hinder the “Lol system,” which ferries lipoproteins between them.
From one compound they constructed lolamicin, which can stop Gram-negative pathogens — with little effect on Gram-negative beneficial bacteria and no effect on Gram-positive bacteria.
“Gram-positive bacteria do not have an outer membrane, so they do not possess the Lol system,” Dr. Muñoz said. “When we compared the sequences of the Lol system in certain Gram-negative pathogens to Gram-negative commensal [beneficial] gut bacteria, we saw that the Lol systems were pretty different.”
Tossing a monkey wrench into the Lol system may be the study’s biggest contribution to future antibiotic development, said Kim Lewis, PhD, professor of Biology and director of Antimicrobial Discovery Center at Northeastern University, Boston, who has discovered several antibiotics now in preclinical research. One, darobactin, targets Gram-negative bugs without affecting the gut microbiome. Another, teixobactin, takes down Gram-positive bacteria without causing drug resistance.
“Lolamicin hits a novel target. I would say that’s the most significant study finding,” said Dr. Lewis, who was not involved in the study. “That is rare. If you look at antibiotics introduced since 1968, they have been modifications of existing antibiotics or, rarely, new chemically but hitting the same proven targets. This one hits something properly new, and [that’s] what I found perhaps the most original and interesting.”
Kirk E. Hevener, PharmD, PhD, associate professor of Pharmaceutical Sciences at the University of Tennessee Health Science Center, Memphis, Tennessee, agreed. (Dr. Hevener also was not involved in the study.) “Lolamicin works by targeting a unique Gram-negative transport system. No currently approved antibacterials work in this way, meaning it potentially represents the first of a new class of antibacterials with narrow-spectrum Gram-negative activity and low gastrointestinal disturbance,” said Dr. Hevener, whose research looks at new antimicrobial drug targets.
The UIUC researchers noted that lolamicin has one drawback: Bacteria frequently developed resistance to it. But in future work, it could be tweaked, combined with other antibiotics, or used as a template for finding other Lol system attackers, they said.
“There is still a good amount of work cut out for us in terms of assessing the clinical translatability of lolamicin, but we are hopeful for the future of this drug,” Dr. Muñoz said.
Addressing a Dire Need
Bringing such a drug to market — from discovery to Food and Drug Administration approval — could take more than a decade, said Dr. Hevener. And new agents, especially for Gram-negative bugs, are sorely needed.
Not only do these bacteria shield themselves with a double membrane but they also “have more complex resistance mechanisms including special pumps that can remove antibacterial drugs from the cell before they can be effective,” Dr. Hevener said.
As a result, drug-resistant Gram-negative bacteria are making treatment of severe infections such as sepsis and pneumonia in health care settings difficult.
Bloodstream infections with drug-resistant Klebsiella pneumoniae have a 40% mortality rate, Dr. Lewis said. And microbiome damage caused by antibiotics is also widespread and deadly, wiping out communities of helpful, protective gut bacteria. That contributes to over half of the C. difficile infections that affect 500,000 people and kill 30,000 a year in the United States.
“Our arsenal of antibacterials that can be used to treat Gram-negative infections is dangerously low,” Dr. Hevener said. “Research will always be needed to develop new antibacterials with novel mechanisms of activity that can bypass bacterial resistance mechanisms.”
A version of this article appeared on Medscape.com.
A new antibiotic uses a never-before-seen mechanism to deliver a direct hit on tough-to-treat infections while leaving beneficial microbes alone. The strategy could lead to a new class of antibiotics that attack dangerous bacteria in a powerful new way, overcoming current drug resistance while sparing the gut microbiome.
“The biggest takeaway is the double-selective component,” said co-lead author Kristen A. Muñoz, PhD, who performed the research as a doctoral student at University of Illinois at Urbana-Champaign (UIUC). “We were able to develop a drug that not only targets problematic pathogens, but because it is selective for these pathogens only, we can spare the good bacteria and preserve the integrity of the microbiome.”
The drug goes after Gram-negative bacteria — pathogens responsible for debilitating and even fatal infections like gastroenteritis, urinary tract infections, pneumonia, sepsis, and cholera. The arsenal of antibiotics against them is old, with no new classes specifically targeting these bacteria coming on the market since 1968.
Many of these bugs have become resistant to one or more antibiotics, with deadly consequences. And antibiotics against them can also wipe out beneficial gut bacteria, allowing serious secondary infections to flare up.
In a study published in Nature, the drug lolamicin knocked out or reduced 130 strains of antibiotic-resistant Gram-negative bacteria in cell cultures. It also successfully treated drug-resistant bloodstream infections and pneumonia in mice while sparing their gut microbiome.
With their microbiomes intact, the mice then fought off secondary infection with Clostridioides difficile (a leading cause of opportunistic and sometimes fatal infections in US health care facilities), while mice treated with other compounds that damaged their microbiome succumbed.
How It Works
Like a well-built medieval castle, Gram-negative bacteria are encased in two protective walls, or membranes. Dr. Muñoz and her team at UIUC set out to breach this defense by finding compounds that hinder the “Lol system,” which ferries lipoproteins between them.
From one compound they constructed lolamicin, which can stop Gram-negative pathogens — with little effect on Gram-negative beneficial bacteria and no effect on Gram-positive bacteria.
“Gram-positive bacteria do not have an outer membrane, so they do not possess the Lol system,” Dr. Muñoz said. “When we compared the sequences of the Lol system in certain Gram-negative pathogens to Gram-negative commensal [beneficial] gut bacteria, we saw that the Lol systems were pretty different.”
Tossing a monkey wrench into the Lol system may be the study’s biggest contribution to future antibiotic development, said Kim Lewis, PhD, professor of Biology and director of Antimicrobial Discovery Center at Northeastern University, Boston, who has discovered several antibiotics now in preclinical research. One, darobactin, targets Gram-negative bugs without affecting the gut microbiome. Another, teixobactin, takes down Gram-positive bacteria without causing drug resistance.
“Lolamicin hits a novel target. I would say that’s the most significant study finding,” said Dr. Lewis, who was not involved in the study. “That is rare. If you look at antibiotics introduced since 1968, they have been modifications of existing antibiotics or, rarely, new chemically but hitting the same proven targets. This one hits something properly new, and [that’s] what I found perhaps the most original and interesting.”
Kirk E. Hevener, PharmD, PhD, associate professor of Pharmaceutical Sciences at the University of Tennessee Health Science Center, Memphis, Tennessee, agreed. (Dr. Hevener also was not involved in the study.) “Lolamicin works by targeting a unique Gram-negative transport system. No currently approved antibacterials work in this way, meaning it potentially represents the first of a new class of antibacterials with narrow-spectrum Gram-negative activity and low gastrointestinal disturbance,” said Dr. Hevener, whose research looks at new antimicrobial drug targets.
The UIUC researchers noted that lolamicin has one drawback: Bacteria frequently developed resistance to it. But in future work, it could be tweaked, combined with other antibiotics, or used as a template for finding other Lol system attackers, they said.
“There is still a good amount of work cut out for us in terms of assessing the clinical translatability of lolamicin, but we are hopeful for the future of this drug,” Dr. Muñoz said.
Addressing a Dire Need
Bringing such a drug to market — from discovery to Food and Drug Administration approval — could take more than a decade, said Dr. Hevener. And new agents, especially for Gram-negative bugs, are sorely needed.
Not only do these bacteria shield themselves with a double membrane but they also “have more complex resistance mechanisms including special pumps that can remove antibacterial drugs from the cell before they can be effective,” Dr. Hevener said.
As a result, drug-resistant Gram-negative bacteria are making treatment of severe infections such as sepsis and pneumonia in health care settings difficult.
Bloodstream infections with drug-resistant Klebsiella pneumoniae have a 40% mortality rate, Dr. Lewis said. And microbiome damage caused by antibiotics is also widespread and deadly, wiping out communities of helpful, protective gut bacteria. That contributes to over half of the C. difficile infections that affect 500,000 people and kill 30,000 a year in the United States.
“Our arsenal of antibacterials that can be used to treat Gram-negative infections is dangerously low,” Dr. Hevener said. “Research will always be needed to develop new antibacterials with novel mechanisms of activity that can bypass bacterial resistance mechanisms.”
A version of this article appeared on Medscape.com.
Commonly Used Meds Tied to Lower Risk for Brain Aneurysm Rupture
(aSAH), a drug-wide association study suggested.
The blood pressure drug lisinopril; the cholesterol drug simvastatin; the diabetes drug metformin; and the drug tamsulosin, prescribed for an enlarged prostate, were all associated with decreased aSAH risk, investigators found.
Conversely, four other drugs were associated with an increased risk for this severely morbid, often deadly, condition.
“The motivation for this study was the fact that we can currently prevent bleeding from intracranial aneurysms only by invasive treatment of those aneurysms with inherent complication risks,” said study investigator Ynte Ruigrok, MD, PhD, associate professor of neurology and neurosurgery, University Medical Center Utrecht, Utrecht, the Netherlands. “Drugs to reduce or eliminate this risk are not yet available. This study is a first step in identifying such drugs.”
The findings were published online in Neurology.
Surprising Results
For the study, the researchers used the Secure Anonymized Information Linkage data bank in Wales to identify 4879 patients with aSAH between January 2000 and December 2019 and 43,911 patients without aSAH matched on age, sex, and year of database entry. Clustering resulted in 2023 unique drugs, of which 205 were commonly prescribed.
After adjusting for other factors such as high blood pressure, alcohol abuse, smoking, and a total number of health conditions, the results yielded two surprises, Dr. Ruigrok observed.
The first was a significant decrease in aSAH risk for current use of lisinopril, compared with nonuse (odds ratio [OR], 0.63; 95% confidence interval [CI], 0.44-0.90), and a nonsignificant decrease with current use of amlodipine (OR, 0.82; 95% CI, 0.65-1.04).
“Hypertension is a major risk factor for occurrence and bleeding from aneurysms. If there is indeed a specific blood pressure–lowering drug that not only has a blood pressure–lowering effect but also has additional protection against aSAH, then perhaps that drug should become the drug of choice in aneurysm patients in the future,” he said.
Notably, recent use of both drugs, defined as between 1 year and 3 months before the index date, was associated with an increased risk for aSAH. This trend was not found for other antihypertensives and was significant for amlodipine but not lisinopril.
The reasons are unclear, but “we trust the findings on lisinopril more,” Dr. Ruigrok said. “The findings on amlodipine may be due to confounding by indication, specifically caused by hypertension. Therefore, it is important to validate our findings in an independent research cohort, and we are in the process of doing so.”
The study’s second surprise was the antidiabetic drug metformin and cholesterol-lowering drug simvastatin were also associated with reduced aSAH risk, Dr. Ruigrok noted.
“We already knew from previous studies that diabetes and high cholesterol are protective factors for aSAH,” he said. “Our results suggest that perhaps not the conditions themselves are protective for aSAH but rather the drugs used to treat these conditions with are.”
The risk for a ruptured brain aneurysm among current users was 42% lower with metformin (OR, 0.58; 95% CI, 0.43-0.78), 22% lower with simvastatin (OR, 0.78; 95% CI, 0.64-0.96), and 45% lower with tamsulosin (OR, 0.55; 95% CI, 0.32-0.93).
An increased risk for aSAH was found only in current users of warfarin (OR, 1.35; 95% CI, 1.02-1.79), venlafaxine (OR, 1.67; 95% CI, 1.01-2.75), prochlorperazine (OR, 2.15; 95% CI, 1.45-3.18), and co-codamol (OR, 1.31; 95% CI, 1.10-1.56).
Other drugs within the classes of vitamin K antagonists, serotonin reuptake inhibitors, conventional antipsychotics, and compound analgesics did not show an association with aSAH.
The study was limited by the use of drug prescriptions, and patients may not take their drugs or use them incorrectly, noted the researchers, led by Jos P. Kanning, MSc, also with University Medical Center Utrecht.
The study was supported by the European Research Council. The authors reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
(aSAH), a drug-wide association study suggested.
The blood pressure drug lisinopril; the cholesterol drug simvastatin; the diabetes drug metformin; and the drug tamsulosin, prescribed for an enlarged prostate, were all associated with decreased aSAH risk, investigators found.
Conversely, four other drugs were associated with an increased risk for this severely morbid, often deadly, condition.
“The motivation for this study was the fact that we can currently prevent bleeding from intracranial aneurysms only by invasive treatment of those aneurysms with inherent complication risks,” said study investigator Ynte Ruigrok, MD, PhD, associate professor of neurology and neurosurgery, University Medical Center Utrecht, Utrecht, the Netherlands. “Drugs to reduce or eliminate this risk are not yet available. This study is a first step in identifying such drugs.”
The findings were published online in Neurology.
Surprising Results
For the study, the researchers used the Secure Anonymized Information Linkage data bank in Wales to identify 4879 patients with aSAH between January 2000 and December 2019 and 43,911 patients without aSAH matched on age, sex, and year of database entry. Clustering resulted in 2023 unique drugs, of which 205 were commonly prescribed.
After adjusting for other factors such as high blood pressure, alcohol abuse, smoking, and a total number of health conditions, the results yielded two surprises, Dr. Ruigrok observed.
The first was a significant decrease in aSAH risk for current use of lisinopril, compared with nonuse (odds ratio [OR], 0.63; 95% confidence interval [CI], 0.44-0.90), and a nonsignificant decrease with current use of amlodipine (OR, 0.82; 95% CI, 0.65-1.04).
“Hypertension is a major risk factor for occurrence and bleeding from aneurysms. If there is indeed a specific blood pressure–lowering drug that not only has a blood pressure–lowering effect but also has additional protection against aSAH, then perhaps that drug should become the drug of choice in aneurysm patients in the future,” he said.
Notably, recent use of both drugs, defined as between 1 year and 3 months before the index date, was associated with an increased risk for aSAH. This trend was not found for other antihypertensives and was significant for amlodipine but not lisinopril.
The reasons are unclear, but “we trust the findings on lisinopril more,” Dr. Ruigrok said. “The findings on amlodipine may be due to confounding by indication, specifically caused by hypertension. Therefore, it is important to validate our findings in an independent research cohort, and we are in the process of doing so.”
The study’s second surprise was the antidiabetic drug metformin and cholesterol-lowering drug simvastatin were also associated with reduced aSAH risk, Dr. Ruigrok noted.
“We already knew from previous studies that diabetes and high cholesterol are protective factors for aSAH,” he said. “Our results suggest that perhaps not the conditions themselves are protective for aSAH but rather the drugs used to treat these conditions with are.”
The risk for a ruptured brain aneurysm among current users was 42% lower with metformin (OR, 0.58; 95% CI, 0.43-0.78), 22% lower with simvastatin (OR, 0.78; 95% CI, 0.64-0.96), and 45% lower with tamsulosin (OR, 0.55; 95% CI, 0.32-0.93).
An increased risk for aSAH was found only in current users of warfarin (OR, 1.35; 95% CI, 1.02-1.79), venlafaxine (OR, 1.67; 95% CI, 1.01-2.75), prochlorperazine (OR, 2.15; 95% CI, 1.45-3.18), and co-codamol (OR, 1.31; 95% CI, 1.10-1.56).
Other drugs within the classes of vitamin K antagonists, serotonin reuptake inhibitors, conventional antipsychotics, and compound analgesics did not show an association with aSAH.
The study was limited by the use of drug prescriptions, and patients may not take their drugs or use them incorrectly, noted the researchers, led by Jos P. Kanning, MSc, also with University Medical Center Utrecht.
The study was supported by the European Research Council. The authors reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
(aSAH), a drug-wide association study suggested.
The blood pressure drug lisinopril; the cholesterol drug simvastatin; the diabetes drug metformin; and the drug tamsulosin, prescribed for an enlarged prostate, were all associated with decreased aSAH risk, investigators found.
Conversely, four other drugs were associated with an increased risk for this severely morbid, often deadly, condition.
“The motivation for this study was the fact that we can currently prevent bleeding from intracranial aneurysms only by invasive treatment of those aneurysms with inherent complication risks,” said study investigator Ynte Ruigrok, MD, PhD, associate professor of neurology and neurosurgery, University Medical Center Utrecht, Utrecht, the Netherlands. “Drugs to reduce or eliminate this risk are not yet available. This study is a first step in identifying such drugs.”
The findings were published online in Neurology.
Surprising Results
For the study, the researchers used the Secure Anonymized Information Linkage data bank in Wales to identify 4879 patients with aSAH between January 2000 and December 2019 and 43,911 patients without aSAH matched on age, sex, and year of database entry. Clustering resulted in 2023 unique drugs, of which 205 were commonly prescribed.
After adjusting for other factors such as high blood pressure, alcohol abuse, smoking, and a total number of health conditions, the results yielded two surprises, Dr. Ruigrok observed.
The first was a significant decrease in aSAH risk for current use of lisinopril, compared with nonuse (odds ratio [OR], 0.63; 95% confidence interval [CI], 0.44-0.90), and a nonsignificant decrease with current use of amlodipine (OR, 0.82; 95% CI, 0.65-1.04).
“Hypertension is a major risk factor for occurrence and bleeding from aneurysms. If there is indeed a specific blood pressure–lowering drug that not only has a blood pressure–lowering effect but also has additional protection against aSAH, then perhaps that drug should become the drug of choice in aneurysm patients in the future,” he said.
Notably, recent use of both drugs, defined as between 1 year and 3 months before the index date, was associated with an increased risk for aSAH. This trend was not found for other antihypertensives and was significant for amlodipine but not lisinopril.
The reasons are unclear, but “we trust the findings on lisinopril more,” Dr. Ruigrok said. “The findings on amlodipine may be due to confounding by indication, specifically caused by hypertension. Therefore, it is important to validate our findings in an independent research cohort, and we are in the process of doing so.”
The study’s second surprise was the antidiabetic drug metformin and cholesterol-lowering drug simvastatin were also associated with reduced aSAH risk, Dr. Ruigrok noted.
“We already knew from previous studies that diabetes and high cholesterol are protective factors for aSAH,” he said. “Our results suggest that perhaps not the conditions themselves are protective for aSAH but rather the drugs used to treat these conditions with are.”
The risk for a ruptured brain aneurysm among current users was 42% lower with metformin (OR, 0.58; 95% CI, 0.43-0.78), 22% lower with simvastatin (OR, 0.78; 95% CI, 0.64-0.96), and 45% lower with tamsulosin (OR, 0.55; 95% CI, 0.32-0.93).
An increased risk for aSAH was found only in current users of warfarin (OR, 1.35; 95% CI, 1.02-1.79), venlafaxine (OR, 1.67; 95% CI, 1.01-2.75), prochlorperazine (OR, 2.15; 95% CI, 1.45-3.18), and co-codamol (OR, 1.31; 95% CI, 1.10-1.56).
Other drugs within the classes of vitamin K antagonists, serotonin reuptake inhibitors, conventional antipsychotics, and compound analgesics did not show an association with aSAH.
The study was limited by the use of drug prescriptions, and patients may not take their drugs or use them incorrectly, noted the researchers, led by Jos P. Kanning, MSc, also with University Medical Center Utrecht.
The study was supported by the European Research Council. The authors reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM NEUROLOGY
In the Future, a Robot Intensivist May Save Your Life
This transcript has been edited for clarity.
They call it the “golden hour”: 60 minutes, give or take, when the chance to save the life of a trauma victim is at its greatest. If the patient can be resuscitated and stabilized in that time window, they stand a good chance of surviving. If not, well, they don’t.
But resuscitation is complicated. It requires blood products, fluids, vasopressors — all given in precise doses in response to rapidly changing hemodynamics. To do it right takes specialized training, advanced life support (ALS). If the patient is in a remote area or an area without ALS-certified emergency medical services, or is far from the nearest trauma center, that golden hour is lost. And the patient may be as well.
But we live in the future. We have robots in factories, self-driving cars, autonomous drones. Why not an autonomous trauma doctor? If you are in a life-threatening accident, would you want to be treated ... by a robot?
Enter “resuscitation based on functional hemodynamic monitoring,” or “ReFit,” introduced in this article appearing in the journal Intensive Care Medicine Experimental.
The idea behind ReFit is straightforward. Resuscitation after trauma should be based on hitting key hemodynamic targets using the tools we have available in the field: blood, fluids, pressors. The researchers wanted to develop a closed-loop system, something that could be used by minimally trained personnel. The input to the system? Hemodynamic data, provided through a single measurement device, an arterial catheter. The output: blood, fluids, and pressors, delivered intravenously.
The body (a prototype) of the system looks like this. You can see various pumps labeled with various fluids, electronic controllers, and so forth.
If that’s the body, then this is the brain – a ruggedized laptop interpreting a readout of that arterial catheter.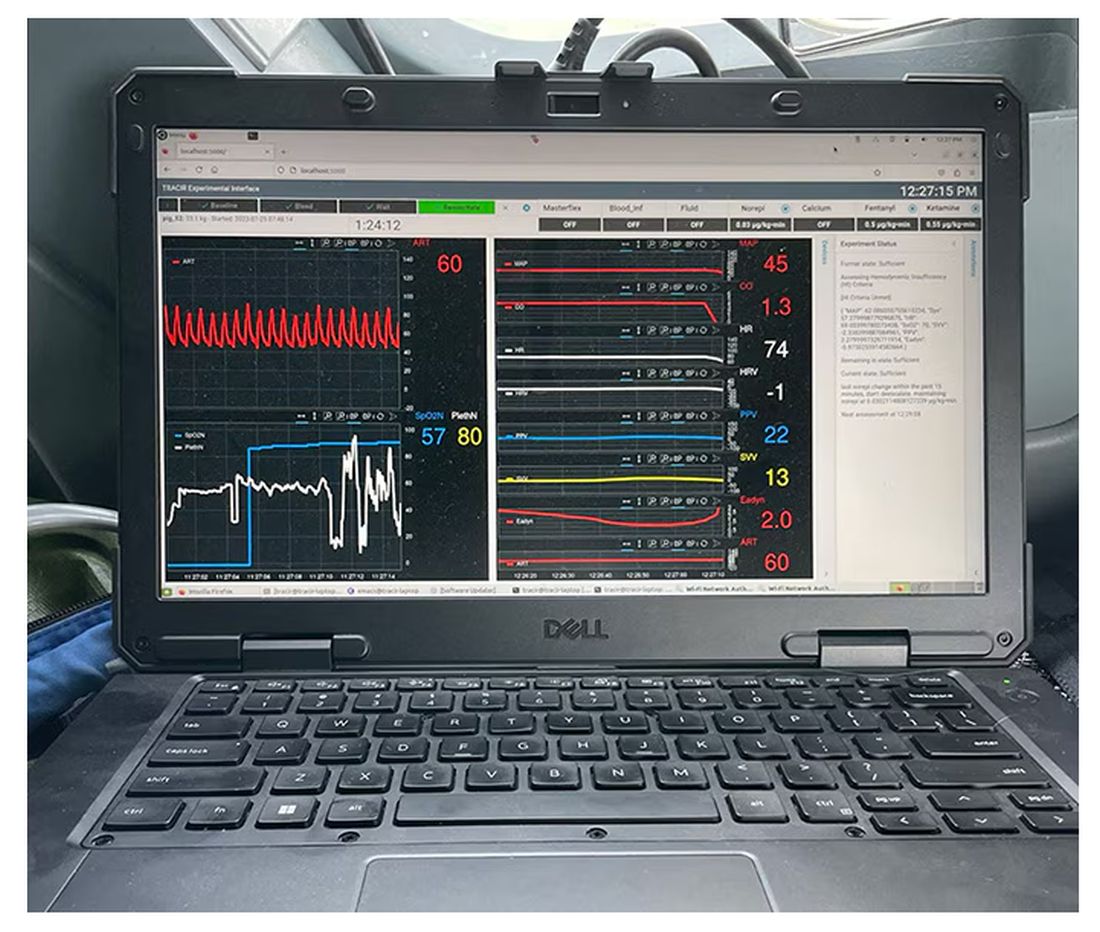
If that’s the brain, then the ReFit algorithm is the mind. The algorithm does its best to leverage all the data it can, so I want to walk through it in a bit of detail.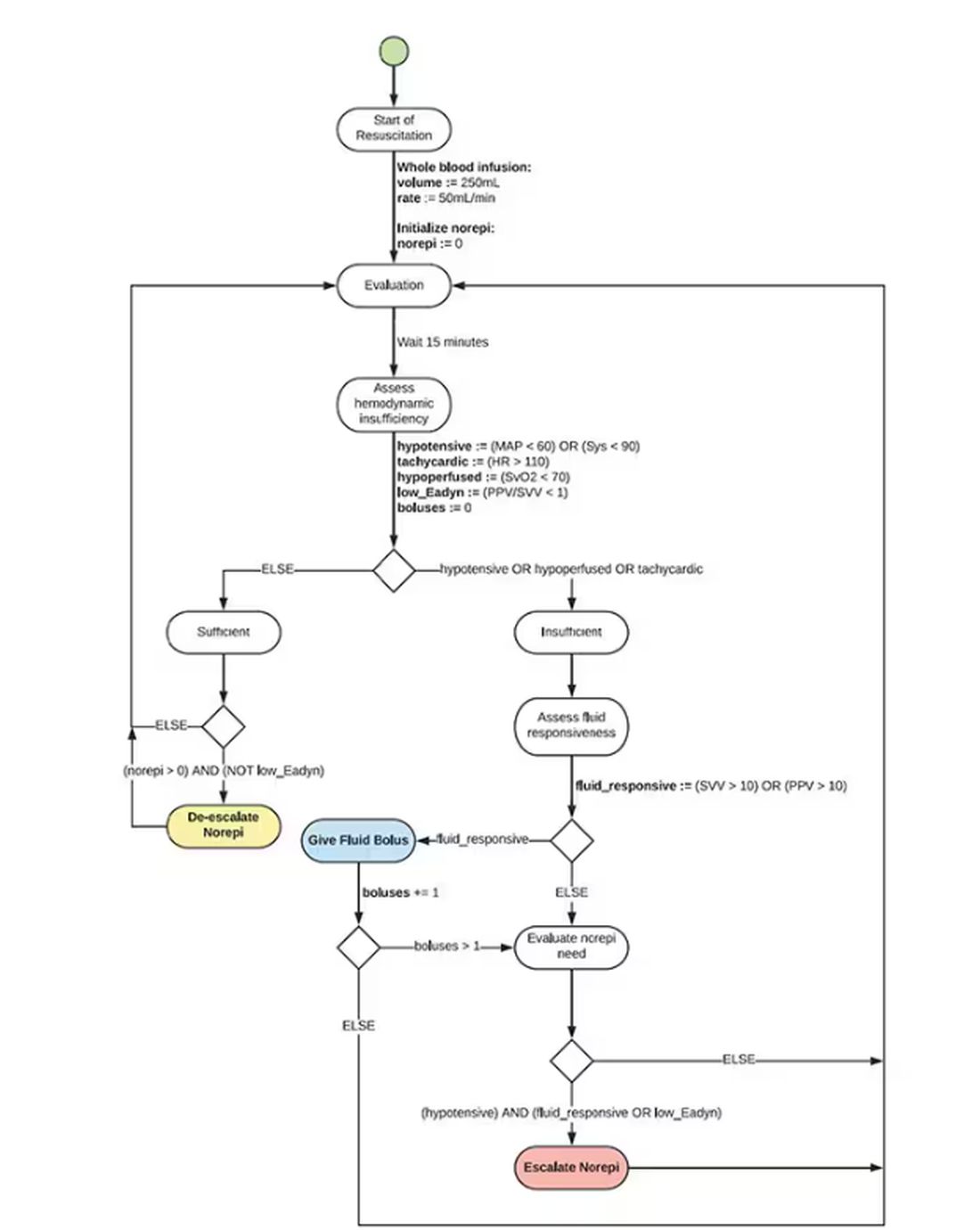
First, check to see whether the patient is stable, defined as a heart rate < 110 beats/min and a mean arterial pressure > 60 mm Hg. If not, you’re off to the races, starting with a bolus of whole blood.
Next, the algorithm gets really interesting. If the patient is still unstable, the computer assesses fluid responsiveness by giving a test dose of fluid and measuring the pulse pressure variation. Greater pulse pressure variation means more fluid responsiveness and the algorithm gives more fluid. Less pulse pressure variation leads the algorithm to uptitrate pressors — in this case, norepinephrine.
This cycle of evaluation and response keeps repeating. The computer titrates fluids and pressors up and down entirely on its own, in theory freeing the human team members to do other things, like getting the patient to a trauma center for definitive care.
So, how do you test whether something like this works? Clearly, you don’t want the trial run of a system like this to be used on a real human suffering from a real traumatic injury.
Once again, we have animals to thank for research advances — in this case, pigs. Fifteen pigs are described in the study. To simulate a severe, hemorrhagic trauma, they were anesthetized and the liver was lacerated. They were then observed passively until the mean arterial pressure had dropped to below 40 mm Hg.
This is a pretty severe injury. Three unfortunate animals served as controls, two of which died within the 3-hour time window of the study. Eight animals were plugged into the ReFit system.
For a window into what happens during this process, let’s take a look at the mean arterial pressure and heart rate readouts for one of the animals. You see that the blood pressure starts to fall precipitously after the liver laceration. The heart rate quickly picks up to compensate, raising the mean arterial pressure a bit, but this would be unsustainable with ongoing bleeding.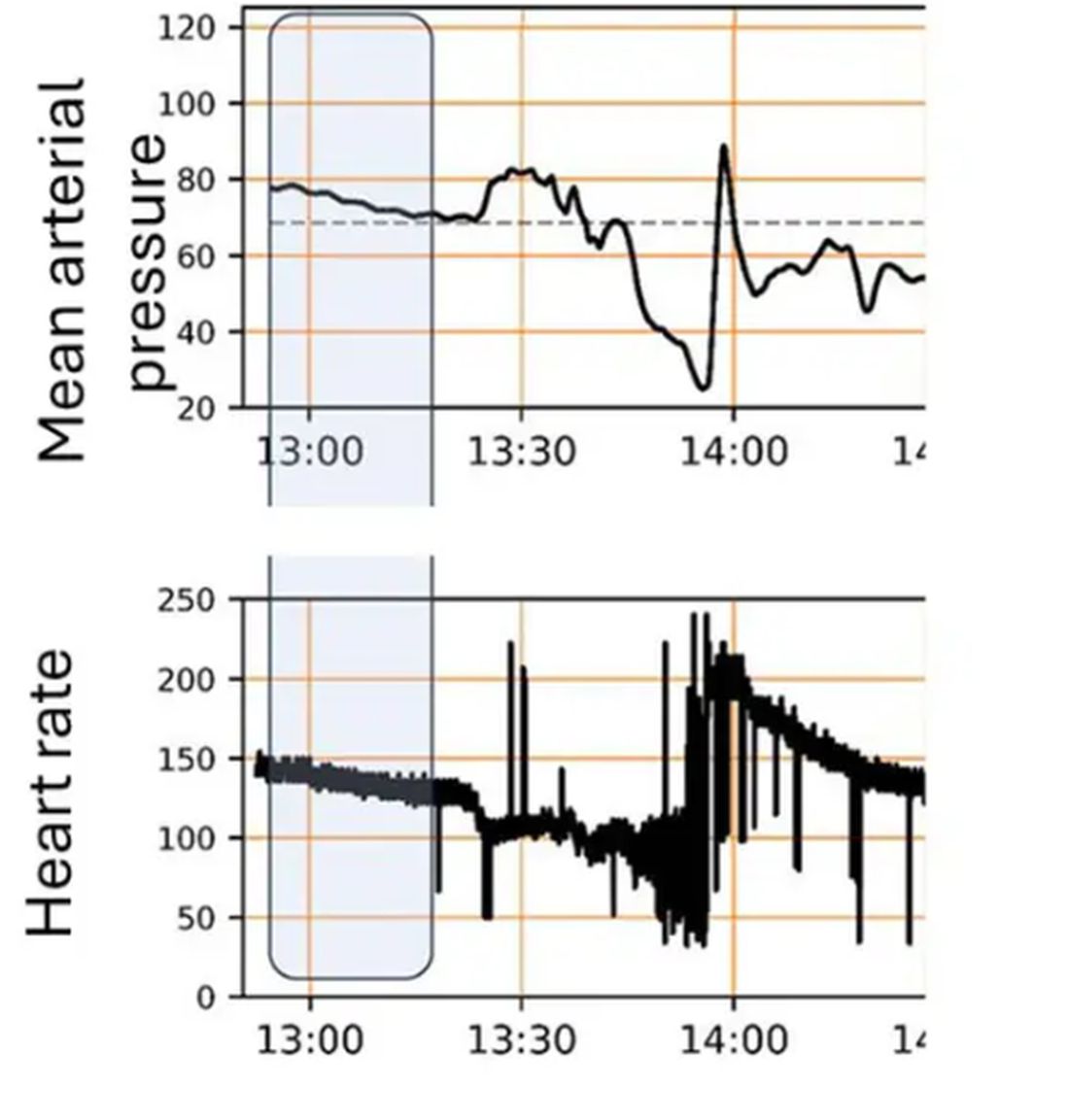
Here, the ReFit system takes over. Autonomously, the system administers two units of blood, followed by fluids, and then norepinephrine or further fluids per the protocol I described earlier. 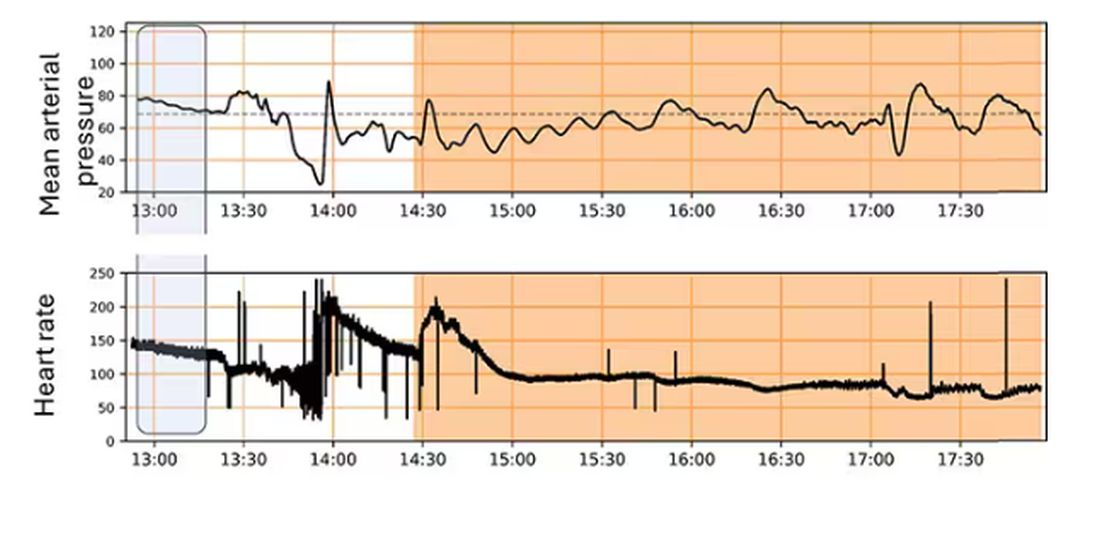
The practical upshot of all of this is stabilization, despite an as-yet untreated liver laceration.
Could an experienced ALS provider do this? Of course. But, as I mentioned before, you aren’t always near an experienced ALS provider.
This is all well and good in the lab, but in the real world, you actually need to transport a trauma patient. The researchers tried this also. To prove feasibility, four pigs were taken from the lab to the top of the University of Pittsburgh Medical Center, flown to Allegheny County Airport and back. Total time before liver laceration repair? Three hours. And all four survived.
It won’t surprise you to hear that this work was funded by the Department of Defense. You can see how a system like this, made a bit more rugged, a bit smaller, and a bit more self-contained could have real uses in the battlefield. But trauma is not unique to war, and something that can extend the time you have to safely transport a patient to definitive care — well, that’s worth its weight in golden hours.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
They call it the “golden hour”: 60 minutes, give or take, when the chance to save the life of a trauma victim is at its greatest. If the patient can be resuscitated and stabilized in that time window, they stand a good chance of surviving. If not, well, they don’t.
But resuscitation is complicated. It requires blood products, fluids, vasopressors — all given in precise doses in response to rapidly changing hemodynamics. To do it right takes specialized training, advanced life support (ALS). If the patient is in a remote area or an area without ALS-certified emergency medical services, or is far from the nearest trauma center, that golden hour is lost. And the patient may be as well.
But we live in the future. We have robots in factories, self-driving cars, autonomous drones. Why not an autonomous trauma doctor? If you are in a life-threatening accident, would you want to be treated ... by a robot?
Enter “resuscitation based on functional hemodynamic monitoring,” or “ReFit,” introduced in this article appearing in the journal Intensive Care Medicine Experimental.
The idea behind ReFit is straightforward. Resuscitation after trauma should be based on hitting key hemodynamic targets using the tools we have available in the field: blood, fluids, pressors. The researchers wanted to develop a closed-loop system, something that could be used by minimally trained personnel. The input to the system? Hemodynamic data, provided through a single measurement device, an arterial catheter. The output: blood, fluids, and pressors, delivered intravenously.
The body (a prototype) of the system looks like this. You can see various pumps labeled with various fluids, electronic controllers, and so forth.
If that’s the body, then this is the brain – a ruggedized laptop interpreting a readout of that arterial catheter.
If that’s the brain, then the ReFit algorithm is the mind. The algorithm does its best to leverage all the data it can, so I want to walk through it in a bit of detail.
First, check to see whether the patient is stable, defined as a heart rate < 110 beats/min and a mean arterial pressure > 60 mm Hg. If not, you’re off to the races, starting with a bolus of whole blood.
Next, the algorithm gets really interesting. If the patient is still unstable, the computer assesses fluid responsiveness by giving a test dose of fluid and measuring the pulse pressure variation. Greater pulse pressure variation means more fluid responsiveness and the algorithm gives more fluid. Less pulse pressure variation leads the algorithm to uptitrate pressors — in this case, norepinephrine.
This cycle of evaluation and response keeps repeating. The computer titrates fluids and pressors up and down entirely on its own, in theory freeing the human team members to do other things, like getting the patient to a trauma center for definitive care.
So, how do you test whether something like this works? Clearly, you don’t want the trial run of a system like this to be used on a real human suffering from a real traumatic injury.
Once again, we have animals to thank for research advances — in this case, pigs. Fifteen pigs are described in the study. To simulate a severe, hemorrhagic trauma, they were anesthetized and the liver was lacerated. They were then observed passively until the mean arterial pressure had dropped to below 40 mm Hg.
This is a pretty severe injury. Three unfortunate animals served as controls, two of which died within the 3-hour time window of the study. Eight animals were plugged into the ReFit system.
For a window into what happens during this process, let’s take a look at the mean arterial pressure and heart rate readouts for one of the animals. You see that the blood pressure starts to fall precipitously after the liver laceration. The heart rate quickly picks up to compensate, raising the mean arterial pressure a bit, but this would be unsustainable with ongoing bleeding.
Here, the ReFit system takes over. Autonomously, the system administers two units of blood, followed by fluids, and then norepinephrine or further fluids per the protocol I described earlier.
The practical upshot of all of this is stabilization, despite an as-yet untreated liver laceration.
Could an experienced ALS provider do this? Of course. But, as I mentioned before, you aren’t always near an experienced ALS provider.
This is all well and good in the lab, but in the real world, you actually need to transport a trauma patient. The researchers tried this also. To prove feasibility, four pigs were taken from the lab to the top of the University of Pittsburgh Medical Center, flown to Allegheny County Airport and back. Total time before liver laceration repair? Three hours. And all four survived.
It won’t surprise you to hear that this work was funded by the Department of Defense. You can see how a system like this, made a bit more rugged, a bit smaller, and a bit more self-contained could have real uses in the battlefield. But trauma is not unique to war, and something that can extend the time you have to safely transport a patient to definitive care — well, that’s worth its weight in golden hours.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
They call it the “golden hour”: 60 minutes, give or take, when the chance to save the life of a trauma victim is at its greatest. If the patient can be resuscitated and stabilized in that time window, they stand a good chance of surviving. If not, well, they don’t.
But resuscitation is complicated. It requires blood products, fluids, vasopressors — all given in precise doses in response to rapidly changing hemodynamics. To do it right takes specialized training, advanced life support (ALS). If the patient is in a remote area or an area without ALS-certified emergency medical services, or is far from the nearest trauma center, that golden hour is lost. And the patient may be as well.
But we live in the future. We have robots in factories, self-driving cars, autonomous drones. Why not an autonomous trauma doctor? If you are in a life-threatening accident, would you want to be treated ... by a robot?
Enter “resuscitation based on functional hemodynamic monitoring,” or “ReFit,” introduced in this article appearing in the journal Intensive Care Medicine Experimental.
The idea behind ReFit is straightforward. Resuscitation after trauma should be based on hitting key hemodynamic targets using the tools we have available in the field: blood, fluids, pressors. The researchers wanted to develop a closed-loop system, something that could be used by minimally trained personnel. The input to the system? Hemodynamic data, provided through a single measurement device, an arterial catheter. The output: blood, fluids, and pressors, delivered intravenously.
The body (a prototype) of the system looks like this. You can see various pumps labeled with various fluids, electronic controllers, and so forth.
If that’s the body, then this is the brain – a ruggedized laptop interpreting a readout of that arterial catheter.
If that’s the brain, then the ReFit algorithm is the mind. The algorithm does its best to leverage all the data it can, so I want to walk through it in a bit of detail.
First, check to see whether the patient is stable, defined as a heart rate < 110 beats/min and a mean arterial pressure > 60 mm Hg. If not, you’re off to the races, starting with a bolus of whole blood.
Next, the algorithm gets really interesting. If the patient is still unstable, the computer assesses fluid responsiveness by giving a test dose of fluid and measuring the pulse pressure variation. Greater pulse pressure variation means more fluid responsiveness and the algorithm gives more fluid. Less pulse pressure variation leads the algorithm to uptitrate pressors — in this case, norepinephrine.
This cycle of evaluation and response keeps repeating. The computer titrates fluids and pressors up and down entirely on its own, in theory freeing the human team members to do other things, like getting the patient to a trauma center for definitive care.
So, how do you test whether something like this works? Clearly, you don’t want the trial run of a system like this to be used on a real human suffering from a real traumatic injury.
Once again, we have animals to thank for research advances — in this case, pigs. Fifteen pigs are described in the study. To simulate a severe, hemorrhagic trauma, they were anesthetized and the liver was lacerated. They were then observed passively until the mean arterial pressure had dropped to below 40 mm Hg.
This is a pretty severe injury. Three unfortunate animals served as controls, two of which died within the 3-hour time window of the study. Eight animals were plugged into the ReFit system.
For a window into what happens during this process, let’s take a look at the mean arterial pressure and heart rate readouts for one of the animals. You see that the blood pressure starts to fall precipitously after the liver laceration. The heart rate quickly picks up to compensate, raising the mean arterial pressure a bit, but this would be unsustainable with ongoing bleeding.
Here, the ReFit system takes over. Autonomously, the system administers two units of blood, followed by fluids, and then norepinephrine or further fluids per the protocol I described earlier.
The practical upshot of all of this is stabilization, despite an as-yet untreated liver laceration.
Could an experienced ALS provider do this? Of course. But, as I mentioned before, you aren’t always near an experienced ALS provider.
This is all well and good in the lab, but in the real world, you actually need to transport a trauma patient. The researchers tried this also. To prove feasibility, four pigs were taken from the lab to the top of the University of Pittsburgh Medical Center, flown to Allegheny County Airport and back. Total time before liver laceration repair? Three hours. And all four survived.
It won’t surprise you to hear that this work was funded by the Department of Defense. You can see how a system like this, made a bit more rugged, a bit smaller, and a bit more self-contained could have real uses in the battlefield. But trauma is not unique to war, and something that can extend the time you have to safely transport a patient to definitive care — well, that’s worth its weight in golden hours.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Colchicine: A New Tool for Ischemic Stroke, CVD Event Recurrence?
BASEL, SWITZERLAND — However, the results did reveal a significant reduction in recurrent stroke and cardiovascular events in the per-protocol analysis and in the subgroup of patients with coronary artery disease.
“Although the primary endpoint was neutral, the CONVINCE results support the hypothesis that long-term anti-inflammatory therapy with colchicine may reduce recurrent stroke and cardiovascular events, specifically in stroke patients with atherosclerosis,” lead investigator Peter Kelly, MD, University College Dublin School of Medicine, Dublin, Ireland, concluded.
The results were presented at the European Stroke Organization Conference (ESOC) 2024.
Inflammation, Dr. Kelly said, plays an important role in the pathophysiology of atherosclerotic plaque, a major cause of cardiovascular events and ischemic strokes.
Colchicine, an established, widely available, low-cost drug that reduces inflammatory response, has been shown to reduce recurrent vascular events in patients with coronary artery disease.
The CONVINCE trial was conducted to see whether colchicine could show similar benefits in patients with non-severe, non-cardioembolic stroke or transient ischemic attack.
Conducted in 16 European countries and Canada, the CONVINCE trial included 3154 patients with a recent non-cardioembolic nondisabling ischemic stroke or high-risk transient ischemic attack. They were randomly assigned to receive colchicine (0.5 mg/d) or placebo.
Key exclusion criteria included evidence of atrial fibrillation or other source of cardioembolism, a defined cause of stroke other than atherosclerosis or small vessel disease, a glomerular filtration rate below 50 mL/min, and the use of drugs that interact with colchicine.
The primary endpoint was a composite of first recurrent ischemic stroke, myocardial infarction, cardiac arrest, or hospitalization for unstable angina. Study participants were followed-up over 36 months.
Results of the primary intention-to-treat analysis showed that the primary endpoint occurred in 153 patients randomized to low-dose colchicine (9.8%) versus 185 in the placebo group (11.8%). This translated into a hazard ratio (HR) of 0.84 (95% CI, 0.68-1.05; P = .12) — a nonsignificant result.
Reduced levels of C-reactive protein in the colchicine group showed the anti-inflammatory effect of treatment with colchicine, Dr. Kelly reported.
In a prespecified on-treatment analysis (excluding patients with major protocol violations), colchicine did show a significant benefit in the primary endpoint (HR, 0.80; 95% CI, 0.63-0.99).
A Novel Target for Stroke Treatment
In addition, significantly reduced rates of recurrent stroke or cardiovascular events were observed in the subgroup of patients with a history of coronary artery disease.
In an updated meta-analysis of existing colchicine studies including CONVINCE, there was a significant reduction in the risk for ischemic stroke (risk ratio, 0.73; 95% CI, 0.58-0.90).
“The signals of benefit of colchicine in secondary analyses are in line with findings from previous trials and indicate the potential of colchicine in prevention after stroke,” Dr. Kelly said.
He pointed out that the COVID pandemic reduced the planned follow-up time in the CONVINCE trial, which led to the study being underpowered for the primary analysis.
“Further trials are needed in all stroke subtypes, but with particular focus on patients with objective evidence of atherosclerosis,” he said.
Commenting on the findings, Mira Katan, MD, University Hospital of Basel, Switzerland, noted that inflammation represents a novel target for stroke treatment.
“We have never before looked at treating inflammation in stroke. Although the primary endpoint was not reached in the CONVINCE study, the on-treatment analysis and meta-analysis showed a risk reduction, and we know colchicine works in cardiology. I think this is a fantastic trial, giving us a new target for stroke therapy,” Dr. Katan said.
“I think we have a new tool, but of course we need further trials to confirm that,” she added.
The CONVINCE trial was supported by Health Research Board Ireland, Deutsche Forschungsgesellschaft, Fonds Wetenschappelijk Onderzoek (FWO), and the Irish Heart Foundation. Dr. Kelly received funding from the Irish Heart Foundation. Dr. Katan reported no relevant disclosures.
A version of this article appeared on Medscape.com.
BASEL, SWITZERLAND — However, the results did reveal a significant reduction in recurrent stroke and cardiovascular events in the per-protocol analysis and in the subgroup of patients with coronary artery disease.
“Although the primary endpoint was neutral, the CONVINCE results support the hypothesis that long-term anti-inflammatory therapy with colchicine may reduce recurrent stroke and cardiovascular events, specifically in stroke patients with atherosclerosis,” lead investigator Peter Kelly, MD, University College Dublin School of Medicine, Dublin, Ireland, concluded.
The results were presented at the European Stroke Organization Conference (ESOC) 2024.
Inflammation, Dr. Kelly said, plays an important role in the pathophysiology of atherosclerotic plaque, a major cause of cardiovascular events and ischemic strokes.
Colchicine, an established, widely available, low-cost drug that reduces inflammatory response, has been shown to reduce recurrent vascular events in patients with coronary artery disease.
The CONVINCE trial was conducted to see whether colchicine could show similar benefits in patients with non-severe, non-cardioembolic stroke or transient ischemic attack.
Conducted in 16 European countries and Canada, the CONVINCE trial included 3154 patients with a recent non-cardioembolic nondisabling ischemic stroke or high-risk transient ischemic attack. They were randomly assigned to receive colchicine (0.5 mg/d) or placebo.
Key exclusion criteria included evidence of atrial fibrillation or other source of cardioembolism, a defined cause of stroke other than atherosclerosis or small vessel disease, a glomerular filtration rate below 50 mL/min, and the use of drugs that interact with colchicine.
The primary endpoint was a composite of first recurrent ischemic stroke, myocardial infarction, cardiac arrest, or hospitalization for unstable angina. Study participants were followed-up over 36 months.
Results of the primary intention-to-treat analysis showed that the primary endpoint occurred in 153 patients randomized to low-dose colchicine (9.8%) versus 185 in the placebo group (11.8%). This translated into a hazard ratio (HR) of 0.84 (95% CI, 0.68-1.05; P = .12) — a nonsignificant result.
Reduced levels of C-reactive protein in the colchicine group showed the anti-inflammatory effect of treatment with colchicine, Dr. Kelly reported.
In a prespecified on-treatment analysis (excluding patients with major protocol violations), colchicine did show a significant benefit in the primary endpoint (HR, 0.80; 95% CI, 0.63-0.99).
A Novel Target for Stroke Treatment
In addition, significantly reduced rates of recurrent stroke or cardiovascular events were observed in the subgroup of patients with a history of coronary artery disease.
In an updated meta-analysis of existing colchicine studies including CONVINCE, there was a significant reduction in the risk for ischemic stroke (risk ratio, 0.73; 95% CI, 0.58-0.90).
“The signals of benefit of colchicine in secondary analyses are in line with findings from previous trials and indicate the potential of colchicine in prevention after stroke,” Dr. Kelly said.
He pointed out that the COVID pandemic reduced the planned follow-up time in the CONVINCE trial, which led to the study being underpowered for the primary analysis.
“Further trials are needed in all stroke subtypes, but with particular focus on patients with objective evidence of atherosclerosis,” he said.
Commenting on the findings, Mira Katan, MD, University Hospital of Basel, Switzerland, noted that inflammation represents a novel target for stroke treatment.
“We have never before looked at treating inflammation in stroke. Although the primary endpoint was not reached in the CONVINCE study, the on-treatment analysis and meta-analysis showed a risk reduction, and we know colchicine works in cardiology. I think this is a fantastic trial, giving us a new target for stroke therapy,” Dr. Katan said.
“I think we have a new tool, but of course we need further trials to confirm that,” she added.
The CONVINCE trial was supported by Health Research Board Ireland, Deutsche Forschungsgesellschaft, Fonds Wetenschappelijk Onderzoek (FWO), and the Irish Heart Foundation. Dr. Kelly received funding from the Irish Heart Foundation. Dr. Katan reported no relevant disclosures.
A version of this article appeared on Medscape.com.
BASEL, SWITZERLAND — However, the results did reveal a significant reduction in recurrent stroke and cardiovascular events in the per-protocol analysis and in the subgroup of patients with coronary artery disease.
“Although the primary endpoint was neutral, the CONVINCE results support the hypothesis that long-term anti-inflammatory therapy with colchicine may reduce recurrent stroke and cardiovascular events, specifically in stroke patients with atherosclerosis,” lead investigator Peter Kelly, MD, University College Dublin School of Medicine, Dublin, Ireland, concluded.
The results were presented at the European Stroke Organization Conference (ESOC) 2024.
Inflammation, Dr. Kelly said, plays an important role in the pathophysiology of atherosclerotic plaque, a major cause of cardiovascular events and ischemic strokes.
Colchicine, an established, widely available, low-cost drug that reduces inflammatory response, has been shown to reduce recurrent vascular events in patients with coronary artery disease.
The CONVINCE trial was conducted to see whether colchicine could show similar benefits in patients with non-severe, non-cardioembolic stroke or transient ischemic attack.
Conducted in 16 European countries and Canada, the CONVINCE trial included 3154 patients with a recent non-cardioembolic nondisabling ischemic stroke or high-risk transient ischemic attack. They were randomly assigned to receive colchicine (0.5 mg/d) or placebo.
Key exclusion criteria included evidence of atrial fibrillation or other source of cardioembolism, a defined cause of stroke other than atherosclerosis or small vessel disease, a glomerular filtration rate below 50 mL/min, and the use of drugs that interact with colchicine.
The primary endpoint was a composite of first recurrent ischemic stroke, myocardial infarction, cardiac arrest, or hospitalization for unstable angina. Study participants were followed-up over 36 months.
Results of the primary intention-to-treat analysis showed that the primary endpoint occurred in 153 patients randomized to low-dose colchicine (9.8%) versus 185 in the placebo group (11.8%). This translated into a hazard ratio (HR) of 0.84 (95% CI, 0.68-1.05; P = .12) — a nonsignificant result.
Reduced levels of C-reactive protein in the colchicine group showed the anti-inflammatory effect of treatment with colchicine, Dr. Kelly reported.
In a prespecified on-treatment analysis (excluding patients with major protocol violations), colchicine did show a significant benefit in the primary endpoint (HR, 0.80; 95% CI, 0.63-0.99).
A Novel Target for Stroke Treatment
In addition, significantly reduced rates of recurrent stroke or cardiovascular events were observed in the subgroup of patients with a history of coronary artery disease.
In an updated meta-analysis of existing colchicine studies including CONVINCE, there was a significant reduction in the risk for ischemic stroke (risk ratio, 0.73; 95% CI, 0.58-0.90).
“The signals of benefit of colchicine in secondary analyses are in line with findings from previous trials and indicate the potential of colchicine in prevention after stroke,” Dr. Kelly said.
He pointed out that the COVID pandemic reduced the planned follow-up time in the CONVINCE trial, which led to the study being underpowered for the primary analysis.
“Further trials are needed in all stroke subtypes, but with particular focus on patients with objective evidence of atherosclerosis,” he said.
Commenting on the findings, Mira Katan, MD, University Hospital of Basel, Switzerland, noted that inflammation represents a novel target for stroke treatment.
“We have never before looked at treating inflammation in stroke. Although the primary endpoint was not reached in the CONVINCE study, the on-treatment analysis and meta-analysis showed a risk reduction, and we know colchicine works in cardiology. I think this is a fantastic trial, giving us a new target for stroke therapy,” Dr. Katan said.
“I think we have a new tool, but of course we need further trials to confirm that,” she added.
The CONVINCE trial was supported by Health Research Board Ireland, Deutsche Forschungsgesellschaft, Fonds Wetenschappelijk Onderzoek (FWO), and the Irish Heart Foundation. Dr. Kelly received funding from the Irish Heart Foundation. Dr. Katan reported no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM ESOC 2024
New Administration Routes for Adrenaline in Anaphylaxis
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
IV Thrombolysis Offers No Benefit for Mild Stroke
BASEL, SWITZERLAND — , a new trial has concluded.
Results from the randomized controlled trial TEMPO-2 showed no benefit from treatment with tenecteplase following ischemic stroke. In addition, investigators found a small increased risk for symptomatic intracranial hemorrhage (ICH) and more deaths in the tenecteplase group compared with the control group.
The research suggests that although it makes sense to open up vessels in patients with minor stroke, they didn’t do better with thrombolysis.
“This is not the result we were hoping for, but I think the question of whether to treat these minor stroke patients who are not disabled has now been answered,” said lead investigator Shelagh B. Coutts, MD, University of Calgary, Alberta, Canada.
“After these results, I think we should scan these patients, admit them, give them dual antiplatelet therapy and IV fluids, and watch them like a hawk. If they deteriorate, we can intervene at that point.”
The findings were presented at the European Stroke Organization Conference (ESOC) 2024 annual meeting and published online simultaneously in The Lancet.
Very Little Data
Up to half of patients with ischemic stroke initially present with minimal symptoms, which are not disabling, investigators noted. Despite having low scores on the National Institutes of Health Stroke Scale (NIHSS) that typically range from 0 to 5, a third of these patients are dead or disabled at 90-day follow-up if thrombolysis is withheld.
Patients with minor deficits and evidence of an intracranial occlusion are a subpopulation at a high risk for early neurological deterioration, which most often occurs within the first 24 hours after presentation.
However, many physicians have concerns about giving thrombolysis to these patients because of the potential harm from bleeding in the absence of major deficits, and most trials of thrombolysis have excluded patients with minor stroke. That leaves very little high-quality data to guide practice for these patients.
Two previous studies have compared alteplase with antiplatelet agents in minor stroke, but no trial has specifically looked at the subset of patients with minor stroke who have intracranial occlusion. The TEMPO-2 trial was conducted to evaluate the use of tenecteplase in this patient population.
The multicenter, parallel group, open-label study was conducted at 48 hospitals in Australia, Austria, Brazil, Canada, Finland, Ireland, New Zealand, Singapore, Spain, and the United Kingdom.
The trial included patients with minor acute ischemic stroke (NIHSS score of 0-5) and intracranial occlusion or focal perfusion abnormality who were within 12 hours from stroke onset.
Patients received IV tenecteplase (0.25 mg/kg) or non-thrombolytic standard of care (control). Most patients in the control group were treated with dual antiplatelet therapy with aspirin and clopidogrel (57%) or aspirin monotherapy (23%).
The trial was stopped early for futility after 886 patients had been enrolled. The median NIHSS score was 2.
The primary outcome — a return to baseline functioning on the modified Rankin Scale score at 90 days — occurred in 75% of the control group and in 72% of the tenecteplase group (risk ratio [RR], 0.96; P = .29).
Although there were significantly more patients with early recanalization and an NIHSS score of 0 at day 5 or discharge after tenecteplase treatment, this did not translate into improved functional outcomes at 90 days.
More patients died in the tenecteplase group compared with the control group (5% vs 1%; adjusted hazard ratio, 3.8; P = .0085).
There were eight (2%) symptomatic ICHs in the tenecteplase group versus two (< 1%) in the control group (RR, 4.2; P = .059).
The ICH rate was not different in patients treated after 4.5 hours versus before 4.5 hours. The subgroup of patients treated at 4.5-12.0 hours showed weaker evidence of better outcomes with thrombolysis than those treated before 4.5 hours, suggesting that the 12-hour window for TEMPO-2 did not explain the absence of benefit seen with tenecteplase.
Patients in the control group did better than expected, which may have been the result of chance, patient selection, or greater use of dual antiplatelet therapy, researchers noted.
Despite higher recanalization rates in the tenecteplase group (48% vs 22%), there was no change in the rate of stroke progression between groups, with an 8% rate of progression seen overall in the study.
Noting that previous studies have shown that patients with minor stroke and intracranial occlusion are at a risk for both progression and disability, the authors suggested that good supportive care may have improved outcomes in both groups.
More Trials Needed
Commenting on the study at the ESOC meeting, Urs Fischer, MD, Basel University Hospital, Switzerland, said “What should we do for patients with mild stroke with vessel occlusion has been a huge unanswered question. The TEMPO-2 study did not show a benefit with thrombolysis, and there was a tendency toward an increased risk of ICH. This is an important finding.”
In an accompanying editorial, Simona Sacco, MD, University of L’Aquila, Italy, and Guillaume Turc, MD, Université Paris Cité, France, noted that different minor ischemic stroke populations pose different therapeutic challenges.
Observational data suggest a benefit of endovascular treatment for minor stroke with large vessel occlusion, and dedicated randomized controlled trials in this group are ongoing, they added.
Early dual antiplatelet treatment is now the recommended treatment of minor stroke and should therefore be the active comparator for non-cardioembolic strokes in future trials.
While TEMPO-2 did not prove that tenecteplase is better than the standard of care for the acute treatment of minor stroke, Dr. Sacco and Dr. Turc said the study confirms that tenecteplase is associated with a high rate of recanalization.
“Fast recanalization with intravenous thrombolysis, endovascular treatment, proper patient selection, and combination with dual antiplatelet treatment or early initiation of anticoagulants may translate into tangible clinical benefits for patients with minor ischemic stroke, which should be tested in future studies,” they wrote.
This trial was funded by grants from the Canadian Institutes of Health Research, Heart and Stroke Foundation of Canada, and the British Heart Foundation. Boehringer Ingelheim provided tenecteplase for the study. Dr. Coutts reported no conflicts of interest. Dr. Sacco reported receiving grants for research from Novartis and Uriach; consulting fees from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; payment for lectures from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; and support for attending conferences from Lilly, Novartis, Teva, Lundbeck, and Pfizer. She is president elect of the European Stroke Organization and editor-in-chief of Cephalalgia. Dr. Turc reported payment for lectures from Guerbet France, is a member of the scientific advisory board of AI-Stroke, and is the Secretary General of the European Stroke Organisation.
A version of this article appeared on Medscape.com.
BASEL, SWITZERLAND — , a new trial has concluded.
Results from the randomized controlled trial TEMPO-2 showed no benefit from treatment with tenecteplase following ischemic stroke. In addition, investigators found a small increased risk for symptomatic intracranial hemorrhage (ICH) and more deaths in the tenecteplase group compared with the control group.
The research suggests that although it makes sense to open up vessels in patients with minor stroke, they didn’t do better with thrombolysis.
“This is not the result we were hoping for, but I think the question of whether to treat these minor stroke patients who are not disabled has now been answered,” said lead investigator Shelagh B. Coutts, MD, University of Calgary, Alberta, Canada.
“After these results, I think we should scan these patients, admit them, give them dual antiplatelet therapy and IV fluids, and watch them like a hawk. If they deteriorate, we can intervene at that point.”
The findings were presented at the European Stroke Organization Conference (ESOC) 2024 annual meeting and published online simultaneously in The Lancet.
Very Little Data
Up to half of patients with ischemic stroke initially present with minimal symptoms, which are not disabling, investigators noted. Despite having low scores on the National Institutes of Health Stroke Scale (NIHSS) that typically range from 0 to 5, a third of these patients are dead or disabled at 90-day follow-up if thrombolysis is withheld.
Patients with minor deficits and evidence of an intracranial occlusion are a subpopulation at a high risk for early neurological deterioration, which most often occurs within the first 24 hours after presentation.
However, many physicians have concerns about giving thrombolysis to these patients because of the potential harm from bleeding in the absence of major deficits, and most trials of thrombolysis have excluded patients with minor stroke. That leaves very little high-quality data to guide practice for these patients.
Two previous studies have compared alteplase with antiplatelet agents in minor stroke, but no trial has specifically looked at the subset of patients with minor stroke who have intracranial occlusion. The TEMPO-2 trial was conducted to evaluate the use of tenecteplase in this patient population.
The multicenter, parallel group, open-label study was conducted at 48 hospitals in Australia, Austria, Brazil, Canada, Finland, Ireland, New Zealand, Singapore, Spain, and the United Kingdom.
The trial included patients with minor acute ischemic stroke (NIHSS score of 0-5) and intracranial occlusion or focal perfusion abnormality who were within 12 hours from stroke onset.
Patients received IV tenecteplase (0.25 mg/kg) or non-thrombolytic standard of care (control). Most patients in the control group were treated with dual antiplatelet therapy with aspirin and clopidogrel (57%) or aspirin monotherapy (23%).
The trial was stopped early for futility after 886 patients had been enrolled. The median NIHSS score was 2.
The primary outcome — a return to baseline functioning on the modified Rankin Scale score at 90 days — occurred in 75% of the control group and in 72% of the tenecteplase group (risk ratio [RR], 0.96; P = .29).
Although there were significantly more patients with early recanalization and an NIHSS score of 0 at day 5 or discharge after tenecteplase treatment, this did not translate into improved functional outcomes at 90 days.
More patients died in the tenecteplase group compared with the control group (5% vs 1%; adjusted hazard ratio, 3.8; P = .0085).
There were eight (2%) symptomatic ICHs in the tenecteplase group versus two (< 1%) in the control group (RR, 4.2; P = .059).
The ICH rate was not different in patients treated after 4.5 hours versus before 4.5 hours. The subgroup of patients treated at 4.5-12.0 hours showed weaker evidence of better outcomes with thrombolysis than those treated before 4.5 hours, suggesting that the 12-hour window for TEMPO-2 did not explain the absence of benefit seen with tenecteplase.
Patients in the control group did better than expected, which may have been the result of chance, patient selection, or greater use of dual antiplatelet therapy, researchers noted.
Despite higher recanalization rates in the tenecteplase group (48% vs 22%), there was no change in the rate of stroke progression between groups, with an 8% rate of progression seen overall in the study.
Noting that previous studies have shown that patients with minor stroke and intracranial occlusion are at a risk for both progression and disability, the authors suggested that good supportive care may have improved outcomes in both groups.
More Trials Needed
Commenting on the study at the ESOC meeting, Urs Fischer, MD, Basel University Hospital, Switzerland, said “What should we do for patients with mild stroke with vessel occlusion has been a huge unanswered question. The TEMPO-2 study did not show a benefit with thrombolysis, and there was a tendency toward an increased risk of ICH. This is an important finding.”
In an accompanying editorial, Simona Sacco, MD, University of L’Aquila, Italy, and Guillaume Turc, MD, Université Paris Cité, France, noted that different minor ischemic stroke populations pose different therapeutic challenges.
Observational data suggest a benefit of endovascular treatment for minor stroke with large vessel occlusion, and dedicated randomized controlled trials in this group are ongoing, they added.
Early dual antiplatelet treatment is now the recommended treatment of minor stroke and should therefore be the active comparator for non-cardioembolic strokes in future trials.
While TEMPO-2 did not prove that tenecteplase is better than the standard of care for the acute treatment of minor stroke, Dr. Sacco and Dr. Turc said the study confirms that tenecteplase is associated with a high rate of recanalization.
“Fast recanalization with intravenous thrombolysis, endovascular treatment, proper patient selection, and combination with dual antiplatelet treatment or early initiation of anticoagulants may translate into tangible clinical benefits for patients with minor ischemic stroke, which should be tested in future studies,” they wrote.
This trial was funded by grants from the Canadian Institutes of Health Research, Heart and Stroke Foundation of Canada, and the British Heart Foundation. Boehringer Ingelheim provided tenecteplase for the study. Dr. Coutts reported no conflicts of interest. Dr. Sacco reported receiving grants for research from Novartis and Uriach; consulting fees from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; payment for lectures from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; and support for attending conferences from Lilly, Novartis, Teva, Lundbeck, and Pfizer. She is president elect of the European Stroke Organization and editor-in-chief of Cephalalgia. Dr. Turc reported payment for lectures from Guerbet France, is a member of the scientific advisory board of AI-Stroke, and is the Secretary General of the European Stroke Organisation.
A version of this article appeared on Medscape.com.
BASEL, SWITZERLAND — , a new trial has concluded.
Results from the randomized controlled trial TEMPO-2 showed no benefit from treatment with tenecteplase following ischemic stroke. In addition, investigators found a small increased risk for symptomatic intracranial hemorrhage (ICH) and more deaths in the tenecteplase group compared with the control group.
The research suggests that although it makes sense to open up vessels in patients with minor stroke, they didn’t do better with thrombolysis.
“This is not the result we were hoping for, but I think the question of whether to treat these minor stroke patients who are not disabled has now been answered,” said lead investigator Shelagh B. Coutts, MD, University of Calgary, Alberta, Canada.
“After these results, I think we should scan these patients, admit them, give them dual antiplatelet therapy and IV fluids, and watch them like a hawk. If they deteriorate, we can intervene at that point.”
The findings were presented at the European Stroke Organization Conference (ESOC) 2024 annual meeting and published online simultaneously in The Lancet.
Very Little Data
Up to half of patients with ischemic stroke initially present with minimal symptoms, which are not disabling, investigators noted. Despite having low scores on the National Institutes of Health Stroke Scale (NIHSS) that typically range from 0 to 5, a third of these patients are dead or disabled at 90-day follow-up if thrombolysis is withheld.
Patients with minor deficits and evidence of an intracranial occlusion are a subpopulation at a high risk for early neurological deterioration, which most often occurs within the first 24 hours after presentation.
However, many physicians have concerns about giving thrombolysis to these patients because of the potential harm from bleeding in the absence of major deficits, and most trials of thrombolysis have excluded patients with minor stroke. That leaves very little high-quality data to guide practice for these patients.
Two previous studies have compared alteplase with antiplatelet agents in minor stroke, but no trial has specifically looked at the subset of patients with minor stroke who have intracranial occlusion. The TEMPO-2 trial was conducted to evaluate the use of tenecteplase in this patient population.
The multicenter, parallel group, open-label study was conducted at 48 hospitals in Australia, Austria, Brazil, Canada, Finland, Ireland, New Zealand, Singapore, Spain, and the United Kingdom.
The trial included patients with minor acute ischemic stroke (NIHSS score of 0-5) and intracranial occlusion or focal perfusion abnormality who were within 12 hours from stroke onset.
Patients received IV tenecteplase (0.25 mg/kg) or non-thrombolytic standard of care (control). Most patients in the control group were treated with dual antiplatelet therapy with aspirin and clopidogrel (57%) or aspirin monotherapy (23%).
The trial was stopped early for futility after 886 patients had been enrolled. The median NIHSS score was 2.
The primary outcome — a return to baseline functioning on the modified Rankin Scale score at 90 days — occurred in 75% of the control group and in 72% of the tenecteplase group (risk ratio [RR], 0.96; P = .29).
Although there were significantly more patients with early recanalization and an NIHSS score of 0 at day 5 or discharge after tenecteplase treatment, this did not translate into improved functional outcomes at 90 days.
More patients died in the tenecteplase group compared with the control group (5% vs 1%; adjusted hazard ratio, 3.8; P = .0085).
There were eight (2%) symptomatic ICHs in the tenecteplase group versus two (< 1%) in the control group (RR, 4.2; P = .059).
The ICH rate was not different in patients treated after 4.5 hours versus before 4.5 hours. The subgroup of patients treated at 4.5-12.0 hours showed weaker evidence of better outcomes with thrombolysis than those treated before 4.5 hours, suggesting that the 12-hour window for TEMPO-2 did not explain the absence of benefit seen with tenecteplase.
Patients in the control group did better than expected, which may have been the result of chance, patient selection, or greater use of dual antiplatelet therapy, researchers noted.
Despite higher recanalization rates in the tenecteplase group (48% vs 22%), there was no change in the rate of stroke progression between groups, with an 8% rate of progression seen overall in the study.
Noting that previous studies have shown that patients with minor stroke and intracranial occlusion are at a risk for both progression and disability, the authors suggested that good supportive care may have improved outcomes in both groups.
More Trials Needed
Commenting on the study at the ESOC meeting, Urs Fischer, MD, Basel University Hospital, Switzerland, said “What should we do for patients with mild stroke with vessel occlusion has been a huge unanswered question. The TEMPO-2 study did not show a benefit with thrombolysis, and there was a tendency toward an increased risk of ICH. This is an important finding.”
In an accompanying editorial, Simona Sacco, MD, University of L’Aquila, Italy, and Guillaume Turc, MD, Université Paris Cité, France, noted that different minor ischemic stroke populations pose different therapeutic challenges.
Observational data suggest a benefit of endovascular treatment for minor stroke with large vessel occlusion, and dedicated randomized controlled trials in this group are ongoing, they added.
Early dual antiplatelet treatment is now the recommended treatment of minor stroke and should therefore be the active comparator for non-cardioembolic strokes in future trials.
While TEMPO-2 did not prove that tenecteplase is better than the standard of care for the acute treatment of minor stroke, Dr. Sacco and Dr. Turc said the study confirms that tenecteplase is associated with a high rate of recanalization.
“Fast recanalization with intravenous thrombolysis, endovascular treatment, proper patient selection, and combination with dual antiplatelet treatment or early initiation of anticoagulants may translate into tangible clinical benefits for patients with minor ischemic stroke, which should be tested in future studies,” they wrote.
This trial was funded by grants from the Canadian Institutes of Health Research, Heart and Stroke Foundation of Canada, and the British Heart Foundation. Boehringer Ingelheim provided tenecteplase for the study. Dr. Coutts reported no conflicts of interest. Dr. Sacco reported receiving grants for research from Novartis and Uriach; consulting fees from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; payment for lectures from Novartis, Allergan-AbbVie, Teva, Lilly, Lundbeck, Pfizer, Novo Nordisk, Abbott, and AstraZeneca; and support for attending conferences from Lilly, Novartis, Teva, Lundbeck, and Pfizer. She is president elect of the European Stroke Organization and editor-in-chief of Cephalalgia. Dr. Turc reported payment for lectures from Guerbet France, is a member of the scientific advisory board of AI-Stroke, and is the Secretary General of the European Stroke Organisation.
A version of this article appeared on Medscape.com.
FROM ESOC 2024
Follow-Up Outcomes Data Often Missing for FDA Drug Approvals Based on Surrogate Markers
Over the past few decades, the US Food and Drug Administration (FDA) has increasingly relied on surrogate measures such as blood tests instead of clinical outcomes for medication approvals. But critics say the agency lacks consistent standards to ensure the surrogate aligns with clinical outcomes that matter to patients — things like improvements in symptoms and gains in function.
Sometimes those decisions backfire. Consider: In July 2021, the FDA approved aducanumab for the treatment of Alzheimer’s disease, bucking the advice of an advisory panel for the agency that questioned the effectiveness of the medication. Regulators relied on data from the drugmaker, Biogen, showing the monoclonal antibody could reduce levels of amyloid beta plaques in blood — a surrogate marker officials hoped would translate to clinical benefit.
The FDA’s decision triggered significant controversy, and Biogen in January announced it is pulling it from the market this year, citing disappointing sales.
Although the case of aducanumab might seem extreme, given the stakes — Alzheimer’s remains a disease without an effective treatment — it’s far from unusual.
“When we prescribe a drug, there is an underlying assumption that the FDA has done its due diligence to confirm the drug is safe and of benefit,” said Reshma Ramachandran, MD, MPP, MHS, a researcher at Yale School of Medicine, New Haven, Connecticut, and a coauthor of a recent review of surrogate outcomes. “In fact, we found either no evidence or low-quality evidence.” Such markers are associated with clinical outcomes. “We just don’t know if they work meaningfully to treat the patient’s condition. The results were pretty shocking for us,” she said.
The FDA in 2018 released an Adult Surrogate Endpoint Table listing markers that can be used as substitutes for clinical outcomes to more quickly test, review, and approve new therapies. The analysis found the majority of these endpoints lacked subsequent confirmations, defined as published meta-analyses of clinical studies to validate the association between the marker and a clinical outcome important to patients.
In a paper published in JAMA, Dr. Ramachandran and her colleagues looked at 37 surrogate endpoints for nearly 3 dozen nononcologic diseases in the table.
Approval with surrogate markers implies responsibility for postapproval or validation studies — not just lab measures or imaging findings but mortality, morbidity, or improved quality of life, said Joshua D. Wallach, PhD, MS, assistant professor in the department of epidemiology at the Emory Rollins School of Public Health in Atlanta and lead author of the JAMA review.
Dr. Wallach said surrogate markers are easier to measure and do not require large and long trials. But the FDA has not provided clear rules for what makes a surrogate marker valid in clinical trials.
“They’ve said that at a minimum, it requires meta-analytical evidence from studies that have looked at the correlation or the association between the surrogate and the clinical outcome,” Dr. Wallach said. “Our understanding was that if that’s a minimum expectation, we should be able to find those studies in the literature. And the reality is that we were unable to find evidence from those types of studies supporting the association between the surrogate and the clinical outcome.”
Physicians generally do not receive training about the FDA approval process and the difference between biomarkers, surrogate markers, and clinical endpoints, Dr. Ramachandran said. “Our study shows that things are much more uncertain than we thought when it comes to the prescribing of new drugs,” she said.
Surrogate Markers on the Rise
Dr. Wallach’s group looked for published meta-analyses compiling randomized controlled trials reporting surrogate endpoints for more than 3 dozen chronic nononcologic conditions, including type 2 diabetes, Alzheimer’s, kidney disease, HIV, gout, and lupus. They found no meta-analyses at all for 59% of the surrogate markers, while for those that were studied, few reported high-strength evidence of an association with clinical outcomes.
The findings echo previous research. In a 2020 study in JAMA Network Open, researchers tallied primary endpoints for all FDA approvals of new drugs and therapies during three 3-year periods: 1995-1997, 2005-2007, and 2015-2017. The proportion of products whose approvals were based on the use of clinical endpoints decreased from 43.8% in 1995-1997 to 28.4% in 2005-2007 to 23.3% in 2015-2017. The share based on surrogate endpoints rose from 43.3% to roughly 60% over the same interval.
A 2017 study in the Journal of Health Economics found the use of “imperfect” surrogate endpoints helped support the approval of an average of 16 new drugs per year between 2010 and 2014 compared with six per year from 1998 to 2008.
Similar concerns about weak associations between surrogate markers and drugs used to treat cancer have been documented before, including in a 2020 study published in eClinicalMedicine. The researchers found the surrogate endpoints in the FDA table either were not tested or were tested but proven to be weak surrogates.
“And yet the FDA considered these as good enough not only for accelerated approval but also for regular approval,” said Bishal Gyawali, MD, PhD, associate professor in the department of oncology at Queen’s University, Kingston, Ontario, Canada, who led the group.
The use of surrogate endpoints is also increasing in Europe, said Huseyin Naci, MHS, PhD, associate professor of health policy at the London School of Economics and Political Science in England. He cited a cohort study of 298 randomized clinical trials (RCTs) in JAMA Oncology suggesting “contemporary oncology RCTs now largely measure putative surrogate endpoints.” Dr. Wallach called the FDA’s surrogate table “a great first step toward transparency. But a key column is missing from that table, telling us what is the basis for which the FDA allows drug companies to use the recognized surrogate markers. What is the evidence they are considering?”
If the agency allows companies the flexibility to validate surrogate endpoints, postmarketing studies designed to confirm the clinical utility of those endpoints should follow.
“We obviously want physicians to be guided by evidence when they’re selecting treatments, and they need to be able to interpret the clinical benefits of the drug that they’re prescribing,” he said. “This is really about having the research consumer, patients, and physicians, as well as industry, understand why certain markers are considered and not considered.”
Dr. Wallach reported receiving grants from the FDA (through the Yale University — Mayo Clinic Center of Excellence in Regulatory Science and Innovation), National Institute on Alcohol Abuse and Alcoholism (1K01AA028258), and Johnson & Johnson (through the Yale University Open Data Access Project); and consulting fees from Hagens Berman Sobol Shapiro LLP and Dugan Law Firm APLC outside the submitted work. Dr. Ramachandran reported receiving grants from the Stavros Niarchos Foundation and FDA; receiving consulting fees from ReAct Action on Antibiotic Resistance strategy policy program outside the submitted work; and serving in an unpaid capacity as chair of the FDA task force for the nonprofit organization Doctors for America and in an unpaid capacity as board president for Universities Allied for Essential Medicines North America.
A version of this article appeared on Medscape.com.
Over the past few decades, the US Food and Drug Administration (FDA) has increasingly relied on surrogate measures such as blood tests instead of clinical outcomes for medication approvals. But critics say the agency lacks consistent standards to ensure the surrogate aligns with clinical outcomes that matter to patients — things like improvements in symptoms and gains in function.
Sometimes those decisions backfire. Consider: In July 2021, the FDA approved aducanumab for the treatment of Alzheimer’s disease, bucking the advice of an advisory panel for the agency that questioned the effectiveness of the medication. Regulators relied on data from the drugmaker, Biogen, showing the monoclonal antibody could reduce levels of amyloid beta plaques in blood — a surrogate marker officials hoped would translate to clinical benefit.
The FDA’s decision triggered significant controversy, and Biogen in January announced it is pulling it from the market this year, citing disappointing sales.
Although the case of aducanumab might seem extreme, given the stakes — Alzheimer’s remains a disease without an effective treatment — it’s far from unusual.
“When we prescribe a drug, there is an underlying assumption that the FDA has done its due diligence to confirm the drug is safe and of benefit,” said Reshma Ramachandran, MD, MPP, MHS, a researcher at Yale School of Medicine, New Haven, Connecticut, and a coauthor of a recent review of surrogate outcomes. “In fact, we found either no evidence or low-quality evidence.” Such markers are associated with clinical outcomes. “We just don’t know if they work meaningfully to treat the patient’s condition. The results were pretty shocking for us,” she said.
The FDA in 2018 released an Adult Surrogate Endpoint Table listing markers that can be used as substitutes for clinical outcomes to more quickly test, review, and approve new therapies. The analysis found the majority of these endpoints lacked subsequent confirmations, defined as published meta-analyses of clinical studies to validate the association between the marker and a clinical outcome important to patients.
In a paper published in JAMA, Dr. Ramachandran and her colleagues looked at 37 surrogate endpoints for nearly 3 dozen nononcologic diseases in the table.
Approval with surrogate markers implies responsibility for postapproval or validation studies — not just lab measures or imaging findings but mortality, morbidity, or improved quality of life, said Joshua D. Wallach, PhD, MS, assistant professor in the department of epidemiology at the Emory Rollins School of Public Health in Atlanta and lead author of the JAMA review.
Dr. Wallach said surrogate markers are easier to measure and do not require large and long trials. But the FDA has not provided clear rules for what makes a surrogate marker valid in clinical trials.
“They’ve said that at a minimum, it requires meta-analytical evidence from studies that have looked at the correlation or the association between the surrogate and the clinical outcome,” Dr. Wallach said. “Our understanding was that if that’s a minimum expectation, we should be able to find those studies in the literature. And the reality is that we were unable to find evidence from those types of studies supporting the association between the surrogate and the clinical outcome.”
Physicians generally do not receive training about the FDA approval process and the difference between biomarkers, surrogate markers, and clinical endpoints, Dr. Ramachandran said. “Our study shows that things are much more uncertain than we thought when it comes to the prescribing of new drugs,” she said.
Surrogate Markers on the Rise
Dr. Wallach’s group looked for published meta-analyses compiling randomized controlled trials reporting surrogate endpoints for more than 3 dozen chronic nononcologic conditions, including type 2 diabetes, Alzheimer’s, kidney disease, HIV, gout, and lupus. They found no meta-analyses at all for 59% of the surrogate markers, while for those that were studied, few reported high-strength evidence of an association with clinical outcomes.
The findings echo previous research. In a 2020 study in JAMA Network Open, researchers tallied primary endpoints for all FDA approvals of new drugs and therapies during three 3-year periods: 1995-1997, 2005-2007, and 2015-2017. The proportion of products whose approvals were based on the use of clinical endpoints decreased from 43.8% in 1995-1997 to 28.4% in 2005-2007 to 23.3% in 2015-2017. The share based on surrogate endpoints rose from 43.3% to roughly 60% over the same interval.
A 2017 study in the Journal of Health Economics found the use of “imperfect” surrogate endpoints helped support the approval of an average of 16 new drugs per year between 2010 and 2014 compared with six per year from 1998 to 2008.
Similar concerns about weak associations between surrogate markers and drugs used to treat cancer have been documented before, including in a 2020 study published in eClinicalMedicine. The researchers found the surrogate endpoints in the FDA table either were not tested or were tested but proven to be weak surrogates.
“And yet the FDA considered these as good enough not only for accelerated approval but also for regular approval,” said Bishal Gyawali, MD, PhD, associate professor in the department of oncology at Queen’s University, Kingston, Ontario, Canada, who led the group.
The use of surrogate endpoints is also increasing in Europe, said Huseyin Naci, MHS, PhD, associate professor of health policy at the London School of Economics and Political Science in England. He cited a cohort study of 298 randomized clinical trials (RCTs) in JAMA Oncology suggesting “contemporary oncology RCTs now largely measure putative surrogate endpoints.” Dr. Wallach called the FDA’s surrogate table “a great first step toward transparency. But a key column is missing from that table, telling us what is the basis for which the FDA allows drug companies to use the recognized surrogate markers. What is the evidence they are considering?”
If the agency allows companies the flexibility to validate surrogate endpoints, postmarketing studies designed to confirm the clinical utility of those endpoints should follow.
“We obviously want physicians to be guided by evidence when they’re selecting treatments, and they need to be able to interpret the clinical benefits of the drug that they’re prescribing,” he said. “This is really about having the research consumer, patients, and physicians, as well as industry, understand why certain markers are considered and not considered.”
Dr. Wallach reported receiving grants from the FDA (through the Yale University — Mayo Clinic Center of Excellence in Regulatory Science and Innovation), National Institute on Alcohol Abuse and Alcoholism (1K01AA028258), and Johnson & Johnson (through the Yale University Open Data Access Project); and consulting fees from Hagens Berman Sobol Shapiro LLP and Dugan Law Firm APLC outside the submitted work. Dr. Ramachandran reported receiving grants from the Stavros Niarchos Foundation and FDA; receiving consulting fees from ReAct Action on Antibiotic Resistance strategy policy program outside the submitted work; and serving in an unpaid capacity as chair of the FDA task force for the nonprofit organization Doctors for America and in an unpaid capacity as board president for Universities Allied for Essential Medicines North America.
A version of this article appeared on Medscape.com.
Over the past few decades, the US Food and Drug Administration (FDA) has increasingly relied on surrogate measures such as blood tests instead of clinical outcomes for medication approvals. But critics say the agency lacks consistent standards to ensure the surrogate aligns with clinical outcomes that matter to patients — things like improvements in symptoms and gains in function.
Sometimes those decisions backfire. Consider: In July 2021, the FDA approved aducanumab for the treatment of Alzheimer’s disease, bucking the advice of an advisory panel for the agency that questioned the effectiveness of the medication. Regulators relied on data from the drugmaker, Biogen, showing the monoclonal antibody could reduce levels of amyloid beta plaques in blood — a surrogate marker officials hoped would translate to clinical benefit.
The FDA’s decision triggered significant controversy, and Biogen in January announced it is pulling it from the market this year, citing disappointing sales.
Although the case of aducanumab might seem extreme, given the stakes — Alzheimer’s remains a disease without an effective treatment — it’s far from unusual.
“When we prescribe a drug, there is an underlying assumption that the FDA has done its due diligence to confirm the drug is safe and of benefit,” said Reshma Ramachandran, MD, MPP, MHS, a researcher at Yale School of Medicine, New Haven, Connecticut, and a coauthor of a recent review of surrogate outcomes. “In fact, we found either no evidence or low-quality evidence.” Such markers are associated with clinical outcomes. “We just don’t know if they work meaningfully to treat the patient’s condition. The results were pretty shocking for us,” she said.
The FDA in 2018 released an Adult Surrogate Endpoint Table listing markers that can be used as substitutes for clinical outcomes to more quickly test, review, and approve new therapies. The analysis found the majority of these endpoints lacked subsequent confirmations, defined as published meta-analyses of clinical studies to validate the association between the marker and a clinical outcome important to patients.
In a paper published in JAMA, Dr. Ramachandran and her colleagues looked at 37 surrogate endpoints for nearly 3 dozen nononcologic diseases in the table.
Approval with surrogate markers implies responsibility for postapproval or validation studies — not just lab measures or imaging findings but mortality, morbidity, or improved quality of life, said Joshua D. Wallach, PhD, MS, assistant professor in the department of epidemiology at the Emory Rollins School of Public Health in Atlanta and lead author of the JAMA review.
Dr. Wallach said surrogate markers are easier to measure and do not require large and long trials. But the FDA has not provided clear rules for what makes a surrogate marker valid in clinical trials.
“They’ve said that at a minimum, it requires meta-analytical evidence from studies that have looked at the correlation or the association between the surrogate and the clinical outcome,” Dr. Wallach said. “Our understanding was that if that’s a minimum expectation, we should be able to find those studies in the literature. And the reality is that we were unable to find evidence from those types of studies supporting the association between the surrogate and the clinical outcome.”
Physicians generally do not receive training about the FDA approval process and the difference between biomarkers, surrogate markers, and clinical endpoints, Dr. Ramachandran said. “Our study shows that things are much more uncertain than we thought when it comes to the prescribing of new drugs,” she said.
Surrogate Markers on the Rise
Dr. Wallach’s group looked for published meta-analyses compiling randomized controlled trials reporting surrogate endpoints for more than 3 dozen chronic nononcologic conditions, including type 2 diabetes, Alzheimer’s, kidney disease, HIV, gout, and lupus. They found no meta-analyses at all for 59% of the surrogate markers, while for those that were studied, few reported high-strength evidence of an association with clinical outcomes.
The findings echo previous research. In a 2020 study in JAMA Network Open, researchers tallied primary endpoints for all FDA approvals of new drugs and therapies during three 3-year periods: 1995-1997, 2005-2007, and 2015-2017. The proportion of products whose approvals were based on the use of clinical endpoints decreased from 43.8% in 1995-1997 to 28.4% in 2005-2007 to 23.3% in 2015-2017. The share based on surrogate endpoints rose from 43.3% to roughly 60% over the same interval.
A 2017 study in the Journal of Health Economics found the use of “imperfect” surrogate endpoints helped support the approval of an average of 16 new drugs per year between 2010 and 2014 compared with six per year from 1998 to 2008.
Similar concerns about weak associations between surrogate markers and drugs used to treat cancer have been documented before, including in a 2020 study published in eClinicalMedicine. The researchers found the surrogate endpoints in the FDA table either were not tested or were tested but proven to be weak surrogates.
“And yet the FDA considered these as good enough not only for accelerated approval but also for regular approval,” said Bishal Gyawali, MD, PhD, associate professor in the department of oncology at Queen’s University, Kingston, Ontario, Canada, who led the group.
The use of surrogate endpoints is also increasing in Europe, said Huseyin Naci, MHS, PhD, associate professor of health policy at the London School of Economics and Political Science in England. He cited a cohort study of 298 randomized clinical trials (RCTs) in JAMA Oncology suggesting “contemporary oncology RCTs now largely measure putative surrogate endpoints.” Dr. Wallach called the FDA’s surrogate table “a great first step toward transparency. But a key column is missing from that table, telling us what is the basis for which the FDA allows drug companies to use the recognized surrogate markers. What is the evidence they are considering?”
If the agency allows companies the flexibility to validate surrogate endpoints, postmarketing studies designed to confirm the clinical utility of those endpoints should follow.
“We obviously want physicians to be guided by evidence when they’re selecting treatments, and they need to be able to interpret the clinical benefits of the drug that they’re prescribing,” he said. “This is really about having the research consumer, patients, and physicians, as well as industry, understand why certain markers are considered and not considered.”
Dr. Wallach reported receiving grants from the FDA (through the Yale University — Mayo Clinic Center of Excellence in Regulatory Science and Innovation), National Institute on Alcohol Abuse and Alcoholism (1K01AA028258), and Johnson & Johnson (through the Yale University Open Data Access Project); and consulting fees from Hagens Berman Sobol Shapiro LLP and Dugan Law Firm APLC outside the submitted work. Dr. Ramachandran reported receiving grants from the Stavros Niarchos Foundation and FDA; receiving consulting fees from ReAct Action on Antibiotic Resistance strategy policy program outside the submitted work; and serving in an unpaid capacity as chair of the FDA task force for the nonprofit organization Doctors for America and in an unpaid capacity as board president for Universities Allied for Essential Medicines North America.
A version of this article appeared on Medscape.com.
FROM JAMA