User login
Cirrhosis, Child-Pugh score predict ERCP complications
Cirrhosis may increase the risk of complications from endoscopic retrograde cholangiopancreatography (ERCP), according to a retrospective study involving almost 700 patients.
The study also showed that Child-Pugh class was a better predictor of risk than Model for End-Stage Liver Disease (MELD) score, reported lead author Michelle Bernshteyn, MD, a third-year internal medicine resident at State University of New York, Syracuse , and colleagues.
“There remains a scarcity in the literature regarding complications and adverse effects after ERCP in cirrhotic patients, particularly those incorporating Child-Pugh class and MELD score or type of intervention as predictors,” Dr. Bernshteyn said during a virtual presentation at the American College of Gastroenterology annual meeting. “Furthermore, literature review demonstrates inconsistency among results.”
To gain clarity, Dr. Bernshteyn and colleagues reviewed electronic medical records from 692 patients who underwent ERCP, of whom 174 had cirrhosis and 518 did not. For all patients, the investigators analyzed demographics, comorbidities, indications for ERCP, type of sedation, type of intervention, and complications within a 30-day period. Complications included bleeding, pancreatitis, cholangitis, perforation, mortality caused by ERCP, and mortality from other causes. Patients with cirrhosis were further analyzed based on etiology of cirrhosis, Child-Pugh class, and MELD score.
The analysis revealed that complications were significantly more common in patients with cirrhosis than in those without cirrhosis (21.30% vs. 13.51%; P = .015). No specific complications were significantly more common in patients with cirrhosis than in those without cirrhosis.
In patients with cirrhosis, 41.18% of Child-Pugh class C patients had complications, compared with 15.15% of class B patients and 19.30% of class A patients (P = .010). In contrast, MELD scores were not significantly associated with adverse events.
Further analysis showed that, in patients without cirrhosis, diagnostic-only ERCP and underlying chronic obstructive pulmonary disease were associated with high rates of complications (P = .039 and P = .003, respectively). In patients with cirrhosis, underlying chronic obstructive pulmonary disease and hypertension predicted adverse events (P = .009 and P = .003, respectively).
“The results of our study reaffirm that liver cirrhosis has an impact on the occurrence of complications during ERCP,” Dr. Bernshteyn said. “Child-Pugh class seems to be more reliable as compared to MELD score in predicting complications of ERCP in cirrhosis patients,” she added. “However, we are also aware that Child-Pugh and MELD scores are complementary to each other while evaluating outcomes of any surgery in patients with cirrhosis.”
In 2017, Udayakumar Navaneethan, MD, a gastroenterologist at AdventHealth Orlando’s Center for Interventional Endoscopy, and an assistant professor at the University of Central Florida, Orlando, and colleagues published a national database study concerning the safety of ERCP in patients with liver cirrhosis.
“[The present] study is important as it highlights the fact that ERCP is associated with significant complications in cirrhotic patients compared to those without cirrhosis,” Dr. Navaneethan said when asked to comment. “Also, Child-Pugh score appeared to be more reliable than MELD score in predicting complications of ERCP in cirrhotic patients.”
He went on to explain relevance for practicing clinicians. “The clinical implications of the study are that a detailed risk-benefit discussion needs to be done with patients with liver cirrhosis, particularly with advanced liver disease Child-Pugh class C, irrespective of the etiology,” Dr. Navaneethan said. “ERCP should be performed when there is clear evidence that the benefits outweigh the risks.” The investigators and Dr. Navaneethan reported no conflicts of interest.
SOURCE: Bernshteyn M et al. ACG 2020, Abstract S0982.
Cirrhosis may increase the risk of complications from endoscopic retrograde cholangiopancreatography (ERCP), according to a retrospective study involving almost 700 patients.
The study also showed that Child-Pugh class was a better predictor of risk than Model for End-Stage Liver Disease (MELD) score, reported lead author Michelle Bernshteyn, MD, a third-year internal medicine resident at State University of New York, Syracuse , and colleagues.
“There remains a scarcity in the literature regarding complications and adverse effects after ERCP in cirrhotic patients, particularly those incorporating Child-Pugh class and MELD score or type of intervention as predictors,” Dr. Bernshteyn said during a virtual presentation at the American College of Gastroenterology annual meeting. “Furthermore, literature review demonstrates inconsistency among results.”
To gain clarity, Dr. Bernshteyn and colleagues reviewed electronic medical records from 692 patients who underwent ERCP, of whom 174 had cirrhosis and 518 did not. For all patients, the investigators analyzed demographics, comorbidities, indications for ERCP, type of sedation, type of intervention, and complications within a 30-day period. Complications included bleeding, pancreatitis, cholangitis, perforation, mortality caused by ERCP, and mortality from other causes. Patients with cirrhosis were further analyzed based on etiology of cirrhosis, Child-Pugh class, and MELD score.
The analysis revealed that complications were significantly more common in patients with cirrhosis than in those without cirrhosis (21.30% vs. 13.51%; P = .015). No specific complications were significantly more common in patients with cirrhosis than in those without cirrhosis.
In patients with cirrhosis, 41.18% of Child-Pugh class C patients had complications, compared with 15.15% of class B patients and 19.30% of class A patients (P = .010). In contrast, MELD scores were not significantly associated with adverse events.
Further analysis showed that, in patients without cirrhosis, diagnostic-only ERCP and underlying chronic obstructive pulmonary disease were associated with high rates of complications (P = .039 and P = .003, respectively). In patients with cirrhosis, underlying chronic obstructive pulmonary disease and hypertension predicted adverse events (P = .009 and P = .003, respectively).
“The results of our study reaffirm that liver cirrhosis has an impact on the occurrence of complications during ERCP,” Dr. Bernshteyn said. “Child-Pugh class seems to be more reliable as compared to MELD score in predicting complications of ERCP in cirrhosis patients,” she added. “However, we are also aware that Child-Pugh and MELD scores are complementary to each other while evaluating outcomes of any surgery in patients with cirrhosis.”
In 2017, Udayakumar Navaneethan, MD, a gastroenterologist at AdventHealth Orlando’s Center for Interventional Endoscopy, and an assistant professor at the University of Central Florida, Orlando, and colleagues published a national database study concerning the safety of ERCP in patients with liver cirrhosis.
“[The present] study is important as it highlights the fact that ERCP is associated with significant complications in cirrhotic patients compared to those without cirrhosis,” Dr. Navaneethan said when asked to comment. “Also, Child-Pugh score appeared to be more reliable than MELD score in predicting complications of ERCP in cirrhotic patients.”
He went on to explain relevance for practicing clinicians. “The clinical implications of the study are that a detailed risk-benefit discussion needs to be done with patients with liver cirrhosis, particularly with advanced liver disease Child-Pugh class C, irrespective of the etiology,” Dr. Navaneethan said. “ERCP should be performed when there is clear evidence that the benefits outweigh the risks.” The investigators and Dr. Navaneethan reported no conflicts of interest.
SOURCE: Bernshteyn M et al. ACG 2020, Abstract S0982.
Cirrhosis may increase the risk of complications from endoscopic retrograde cholangiopancreatography (ERCP), according to a retrospective study involving almost 700 patients.
The study also showed that Child-Pugh class was a better predictor of risk than Model for End-Stage Liver Disease (MELD) score, reported lead author Michelle Bernshteyn, MD, a third-year internal medicine resident at State University of New York, Syracuse , and colleagues.
“There remains a scarcity in the literature regarding complications and adverse effects after ERCP in cirrhotic patients, particularly those incorporating Child-Pugh class and MELD score or type of intervention as predictors,” Dr. Bernshteyn said during a virtual presentation at the American College of Gastroenterology annual meeting. “Furthermore, literature review demonstrates inconsistency among results.”
To gain clarity, Dr. Bernshteyn and colleagues reviewed electronic medical records from 692 patients who underwent ERCP, of whom 174 had cirrhosis and 518 did not. For all patients, the investigators analyzed demographics, comorbidities, indications for ERCP, type of sedation, type of intervention, and complications within a 30-day period. Complications included bleeding, pancreatitis, cholangitis, perforation, mortality caused by ERCP, and mortality from other causes. Patients with cirrhosis were further analyzed based on etiology of cirrhosis, Child-Pugh class, and MELD score.
The analysis revealed that complications were significantly more common in patients with cirrhosis than in those without cirrhosis (21.30% vs. 13.51%; P = .015). No specific complications were significantly more common in patients with cirrhosis than in those without cirrhosis.
In patients with cirrhosis, 41.18% of Child-Pugh class C patients had complications, compared with 15.15% of class B patients and 19.30% of class A patients (P = .010). In contrast, MELD scores were not significantly associated with adverse events.
Further analysis showed that, in patients without cirrhosis, diagnostic-only ERCP and underlying chronic obstructive pulmonary disease were associated with high rates of complications (P = .039 and P = .003, respectively). In patients with cirrhosis, underlying chronic obstructive pulmonary disease and hypertension predicted adverse events (P = .009 and P = .003, respectively).
“The results of our study reaffirm that liver cirrhosis has an impact on the occurrence of complications during ERCP,” Dr. Bernshteyn said. “Child-Pugh class seems to be more reliable as compared to MELD score in predicting complications of ERCP in cirrhosis patients,” she added. “However, we are also aware that Child-Pugh and MELD scores are complementary to each other while evaluating outcomes of any surgery in patients with cirrhosis.”
In 2017, Udayakumar Navaneethan, MD, a gastroenterologist at AdventHealth Orlando’s Center for Interventional Endoscopy, and an assistant professor at the University of Central Florida, Orlando, and colleagues published a national database study concerning the safety of ERCP in patients with liver cirrhosis.
“[The present] study is important as it highlights the fact that ERCP is associated with significant complications in cirrhotic patients compared to those without cirrhosis,” Dr. Navaneethan said when asked to comment. “Also, Child-Pugh score appeared to be more reliable than MELD score in predicting complications of ERCP in cirrhotic patients.”
He went on to explain relevance for practicing clinicians. “The clinical implications of the study are that a detailed risk-benefit discussion needs to be done with patients with liver cirrhosis, particularly with advanced liver disease Child-Pugh class C, irrespective of the etiology,” Dr. Navaneethan said. “ERCP should be performed when there is clear evidence that the benefits outweigh the risks.” The investigators and Dr. Navaneethan reported no conflicts of interest.
SOURCE: Bernshteyn M et al. ACG 2020, Abstract S0982.
FROM ACG 2020
Surgery may not be needed with locally advanced rectal cancer
A short course of radiation therapy followed by neoadjuvant chemotherapy resulted in a clinical complete response (CR) in almost half of 90 patients with locally advanced rectal cancer, allowing the majority of responders to skip surgical resection, a retrospective study indicates.
Specifically, at a median follow-up of 16.6 months for living patients, the initial clinical CR rate was 48% overall.
“While we do not have enough follow-up yet to know the late side-effect profile of this regimen, our preliminary results are promising,” Re-I Chin, MD, of Washington University School of Medicine, St. Louis, Missouri, told Medscape Medical News in an email.
The study was presented at the virtual 2020 meeting of the American Society of Radiation Oncology (ASTRO).
“Certainly, longer follow-up will be needed in this study, but none of the observed patients to date has experienced an unsalvageable failure,” said meeting discussant Amol Narang, MD, of Johns Hopkins University, Baltimore, Maryland.
He reminded conference attendees that, despite good evidence supporting equivalency in oncologic outcomes between short-course radiation and long-course chemoradiation, the former is “highly underutilized in the US” with a mere 1% usage rate between 2004 and 2014.
The current study’s short-course treatment approach was compared in this setting to long-course chemoradiation and adjuvant chemotherapy in the RAPIDO trial reported at the 2020 annual meeting of the American Society of Clinical Oncology (ASCO), Narang pointed out.
Short-course patients had a higher rate of pathological complete response (pCR) and a lower rate of treatment failure compared with patients who received long-course chemoradiation and adjuvant chemotherapy; both patient groups underwent total mesorectal excision — which is different from the current analysis. The RAPIDO investigators concluded that the approach featuring the short course “can be considered as a new standard of care.”
Narang said the data collectively “begs the question as to whether the superiority of long-course chemoradiation should really have to be demonstrated to justify its use.”
But Chin highlighted toxicity issues. “Historically, there have been concerns regarding toxicity with short-course radiation therapy since it requires larger doses of radiation given over a shorter period of time,” Chin explained. “But [the short course] is particularly convenient for patients since it saves them more than a month of daily trips to the radiation oncology department.”
Seven local failures
The single-center study involved patients with newly diagnosed, nonmetastatic rectal adenocarcinoma treated with short-course radiation therapy followed by chemotherapy in 2018 and 2019. Nearly all (96%) had locally advanced disease, with either a T3/T4 tumor or node-positive disease. Median tumor size was 4.6 cm.
“Many of the patients in the study had low lying tumors,” Chin reported, with a median distance from the anal verge of 7 cm.
Radiation therapy was delivered in 25 Gy given in five fractions over 5 consecutive days, with the option to boost the dose to 30 Gy or 35 Gy in five fractions if extra-mesorectal lymph nodes were involved. Conventional 3D or intensity-modulated radiation was used and all patients completed treatment.
The median interval between diagnosis of rectal cancer and initiation of radiation therapy was 1.4 months, while the median interval between completion of radiation to initiation of chemotherapy was 2.7 weeks.
The most common chemotherapy regimen was FOLFOX – consisting of leucovorin, fluorouracil (5-FU), and oxaliplatin – or modified FOLFOX. For patients who received six or more cycles of chemotherapy, the median time spent on treatment was 3.9 months.
For those who completed at least six cycles of chemotherapy, the overall clinical CR was 51%, and, for patients with locally advanced disease, the clinical CR rate was 49%. Five of the 43 patients who achieved an initial clinical CR still underwent surgical resection because of patient or physician preference. Among this small group of patients, 4 of the 5 achieved a pCR, and the remaining patient achieved a pathological partial response (pPR).
A total of 42 patients (47% of the group) achieved a partial response following the radiation plus chemotherapy paradigm, and one patient had progressive disease. All underwent surgical resection. One patient did not complete chemotherapy and did not get surgery and subsequently died.
This left 38 patients to be managed nonoperatively. In this nonoperative cohort, 79% of patients continued to have a clinical CR at the end of follow-up. Of the 7 patients with local failure, 6 were salvaged by surgery, one was salvaged by chemotherapy, and all 7 treatment failures had no evidence of disease at last follow-up.
Of the small group of 5 patients who achieved an initial clinical CR following short-course radiation therapy and neoadjuvant chemotherapy, there were no further events in this group, whereas, for patients who achieved only an initial partial response or who had progressive disease, 72% remained event-free.
Approximately half of those who achieved a clinical CR to the treatment regimen had no late gastrointestinal toxicities, and no grade 3 or 4 toxicities were observed. “Surgical resection of tumors — even without a permanent stoma — can result in significantly decreased bowel function, so our goal is to treat patients without surgery and maintain good bowel function,” Chin noted.
“For rectal cancer, both short-course radiation therapy and nonoperative management are emerging treatment paradigms that may be more cost-effective and convenient compared to long-course chemoradiation followed by surgery, [especially since] the COVID-19 pandemic...has spurred changes in clinical practices in radiation oncology,” she said.
Chin has disclosed no relevant financial relationships. Narang reports receiving research support from Boston Scientific.
This article first appeared on Medscape.com.
A short course of radiation therapy followed by neoadjuvant chemotherapy resulted in a clinical complete response (CR) in almost half of 90 patients with locally advanced rectal cancer, allowing the majority of responders to skip surgical resection, a retrospective study indicates.
Specifically, at a median follow-up of 16.6 months for living patients, the initial clinical CR rate was 48% overall.
“While we do not have enough follow-up yet to know the late side-effect profile of this regimen, our preliminary results are promising,” Re-I Chin, MD, of Washington University School of Medicine, St. Louis, Missouri, told Medscape Medical News in an email.
The study was presented at the virtual 2020 meeting of the American Society of Radiation Oncology (ASTRO).
“Certainly, longer follow-up will be needed in this study, but none of the observed patients to date has experienced an unsalvageable failure,” said meeting discussant Amol Narang, MD, of Johns Hopkins University, Baltimore, Maryland.
He reminded conference attendees that, despite good evidence supporting equivalency in oncologic outcomes between short-course radiation and long-course chemoradiation, the former is “highly underutilized in the US” with a mere 1% usage rate between 2004 and 2014.
The current study’s short-course treatment approach was compared in this setting to long-course chemoradiation and adjuvant chemotherapy in the RAPIDO trial reported at the 2020 annual meeting of the American Society of Clinical Oncology (ASCO), Narang pointed out.
Short-course patients had a higher rate of pathological complete response (pCR) and a lower rate of treatment failure compared with patients who received long-course chemoradiation and adjuvant chemotherapy; both patient groups underwent total mesorectal excision — which is different from the current analysis. The RAPIDO investigators concluded that the approach featuring the short course “can be considered as a new standard of care.”
Narang said the data collectively “begs the question as to whether the superiority of long-course chemoradiation should really have to be demonstrated to justify its use.”
But Chin highlighted toxicity issues. “Historically, there have been concerns regarding toxicity with short-course radiation therapy since it requires larger doses of radiation given over a shorter period of time,” Chin explained. “But [the short course] is particularly convenient for patients since it saves them more than a month of daily trips to the radiation oncology department.”
Seven local failures
The single-center study involved patients with newly diagnosed, nonmetastatic rectal adenocarcinoma treated with short-course radiation therapy followed by chemotherapy in 2018 and 2019. Nearly all (96%) had locally advanced disease, with either a T3/T4 tumor or node-positive disease. Median tumor size was 4.6 cm.
“Many of the patients in the study had low lying tumors,” Chin reported, with a median distance from the anal verge of 7 cm.
Radiation therapy was delivered in 25 Gy given in five fractions over 5 consecutive days, with the option to boost the dose to 30 Gy or 35 Gy in five fractions if extra-mesorectal lymph nodes were involved. Conventional 3D or intensity-modulated radiation was used and all patients completed treatment.
The median interval between diagnosis of rectal cancer and initiation of radiation therapy was 1.4 months, while the median interval between completion of radiation to initiation of chemotherapy was 2.7 weeks.
The most common chemotherapy regimen was FOLFOX – consisting of leucovorin, fluorouracil (5-FU), and oxaliplatin – or modified FOLFOX. For patients who received six or more cycles of chemotherapy, the median time spent on treatment was 3.9 months.
For those who completed at least six cycles of chemotherapy, the overall clinical CR was 51%, and, for patients with locally advanced disease, the clinical CR rate was 49%. Five of the 43 patients who achieved an initial clinical CR still underwent surgical resection because of patient or physician preference. Among this small group of patients, 4 of the 5 achieved a pCR, and the remaining patient achieved a pathological partial response (pPR).
A total of 42 patients (47% of the group) achieved a partial response following the radiation plus chemotherapy paradigm, and one patient had progressive disease. All underwent surgical resection. One patient did not complete chemotherapy and did not get surgery and subsequently died.
This left 38 patients to be managed nonoperatively. In this nonoperative cohort, 79% of patients continued to have a clinical CR at the end of follow-up. Of the 7 patients with local failure, 6 were salvaged by surgery, one was salvaged by chemotherapy, and all 7 treatment failures had no evidence of disease at last follow-up.
Of the small group of 5 patients who achieved an initial clinical CR following short-course radiation therapy and neoadjuvant chemotherapy, there were no further events in this group, whereas, for patients who achieved only an initial partial response or who had progressive disease, 72% remained event-free.
Approximately half of those who achieved a clinical CR to the treatment regimen had no late gastrointestinal toxicities, and no grade 3 or 4 toxicities were observed. “Surgical resection of tumors — even without a permanent stoma — can result in significantly decreased bowel function, so our goal is to treat patients without surgery and maintain good bowel function,” Chin noted.
“For rectal cancer, both short-course radiation therapy and nonoperative management are emerging treatment paradigms that may be more cost-effective and convenient compared to long-course chemoradiation followed by surgery, [especially since] the COVID-19 pandemic...has spurred changes in clinical practices in radiation oncology,” she said.
Chin has disclosed no relevant financial relationships. Narang reports receiving research support from Boston Scientific.
This article first appeared on Medscape.com.
A short course of radiation therapy followed by neoadjuvant chemotherapy resulted in a clinical complete response (CR) in almost half of 90 patients with locally advanced rectal cancer, allowing the majority of responders to skip surgical resection, a retrospective study indicates.
Specifically, at a median follow-up of 16.6 months for living patients, the initial clinical CR rate was 48% overall.
“While we do not have enough follow-up yet to know the late side-effect profile of this regimen, our preliminary results are promising,” Re-I Chin, MD, of Washington University School of Medicine, St. Louis, Missouri, told Medscape Medical News in an email.
The study was presented at the virtual 2020 meeting of the American Society of Radiation Oncology (ASTRO).
“Certainly, longer follow-up will be needed in this study, but none of the observed patients to date has experienced an unsalvageable failure,” said meeting discussant Amol Narang, MD, of Johns Hopkins University, Baltimore, Maryland.
He reminded conference attendees that, despite good evidence supporting equivalency in oncologic outcomes between short-course radiation and long-course chemoradiation, the former is “highly underutilized in the US” with a mere 1% usage rate between 2004 and 2014.
The current study’s short-course treatment approach was compared in this setting to long-course chemoradiation and adjuvant chemotherapy in the RAPIDO trial reported at the 2020 annual meeting of the American Society of Clinical Oncology (ASCO), Narang pointed out.
Short-course patients had a higher rate of pathological complete response (pCR) and a lower rate of treatment failure compared with patients who received long-course chemoradiation and adjuvant chemotherapy; both patient groups underwent total mesorectal excision — which is different from the current analysis. The RAPIDO investigators concluded that the approach featuring the short course “can be considered as a new standard of care.”
Narang said the data collectively “begs the question as to whether the superiority of long-course chemoradiation should really have to be demonstrated to justify its use.”
But Chin highlighted toxicity issues. “Historically, there have been concerns regarding toxicity with short-course radiation therapy since it requires larger doses of radiation given over a shorter period of time,” Chin explained. “But [the short course] is particularly convenient for patients since it saves them more than a month of daily trips to the radiation oncology department.”
Seven local failures
The single-center study involved patients with newly diagnosed, nonmetastatic rectal adenocarcinoma treated with short-course radiation therapy followed by chemotherapy in 2018 and 2019. Nearly all (96%) had locally advanced disease, with either a T3/T4 tumor or node-positive disease. Median tumor size was 4.6 cm.
“Many of the patients in the study had low lying tumors,” Chin reported, with a median distance from the anal verge of 7 cm.
Radiation therapy was delivered in 25 Gy given in five fractions over 5 consecutive days, with the option to boost the dose to 30 Gy or 35 Gy in five fractions if extra-mesorectal lymph nodes were involved. Conventional 3D or intensity-modulated radiation was used and all patients completed treatment.
The median interval between diagnosis of rectal cancer and initiation of radiation therapy was 1.4 months, while the median interval between completion of radiation to initiation of chemotherapy was 2.7 weeks.
The most common chemotherapy regimen was FOLFOX – consisting of leucovorin, fluorouracil (5-FU), and oxaliplatin – or modified FOLFOX. For patients who received six or more cycles of chemotherapy, the median time spent on treatment was 3.9 months.
For those who completed at least six cycles of chemotherapy, the overall clinical CR was 51%, and, for patients with locally advanced disease, the clinical CR rate was 49%. Five of the 43 patients who achieved an initial clinical CR still underwent surgical resection because of patient or physician preference. Among this small group of patients, 4 of the 5 achieved a pCR, and the remaining patient achieved a pathological partial response (pPR).
A total of 42 patients (47% of the group) achieved a partial response following the radiation plus chemotherapy paradigm, and one patient had progressive disease. All underwent surgical resection. One patient did not complete chemotherapy and did not get surgery and subsequently died.
This left 38 patients to be managed nonoperatively. In this nonoperative cohort, 79% of patients continued to have a clinical CR at the end of follow-up. Of the 7 patients with local failure, 6 were salvaged by surgery, one was salvaged by chemotherapy, and all 7 treatment failures had no evidence of disease at last follow-up.
Of the small group of 5 patients who achieved an initial clinical CR following short-course radiation therapy and neoadjuvant chemotherapy, there were no further events in this group, whereas, for patients who achieved only an initial partial response or who had progressive disease, 72% remained event-free.
Approximately half of those who achieved a clinical CR to the treatment regimen had no late gastrointestinal toxicities, and no grade 3 or 4 toxicities were observed. “Surgical resection of tumors — even without a permanent stoma — can result in significantly decreased bowel function, so our goal is to treat patients without surgery and maintain good bowel function,” Chin noted.
“For rectal cancer, both short-course radiation therapy and nonoperative management are emerging treatment paradigms that may be more cost-effective and convenient compared to long-course chemoradiation followed by surgery, [especially since] the COVID-19 pandemic...has spurred changes in clinical practices in radiation oncology,” she said.
Chin has disclosed no relevant financial relationships. Narang reports receiving research support from Boston Scientific.
This article first appeared on Medscape.com.
Burnout risk may be exacerbated by COVID crisis
New kinds of job stress multiply in unusual times
Clarissa Barnes, MD, a hospitalist at Avera McKennan Hospital in Sioux Falls, S.D., and until recently medical director of Avera’s LIGHT Program, a wellness-oriented service for doctors, nurse practitioners, and physician assistants, watched the COVID-19 crisis unfold up close in her community and her hospital. Sioux Falls traced its surge of COVID patients to an outbreak at a local meatpacking plant.

“In the beginning, we didn’t know much about the virus and its communicability, although we have since gotten a better handle on that,” she said. “We had questions: Should we give patients more fluids – or less? Steroids or not? In my experience as a hospitalist I never had patients die every day on my shift, but that was happening with COVID.” The crisis imposed serious stresses on frontline providers, and hospitalists were concerned about personal safety and exposure risk – not just for themselves but for their families.
“The first time I worked on the COVID unit, I moved into the guest room in our home, apart from my husband and our young children,” Dr. Barnes said. “Ultimately I caught the virus, although I have since recovered.” Her experience has highlighted how existing issues of job stress and burnout in hospital medicine have been exacerbated by COVID-19. Even physicians who consider themselves healthy may have little emotional reserve to draw upon in a crisis of this magnitude.
“We are social distancing at work, wearing masks, not eating together with our colleagues – with less camaraderie and social support than we used to have,” she said. “I feel exhausted and there’s no question that my colleagues and I have sacrificed a lot to deal with the pandemic.” Add to that the second front of the COVID-19 crisis, Dr. Barnes said, which is “fighting the medical information wars, trying to correct misinformation put out there by people. Physicians who have been on the front lines of the pandemic know how demoralizing it can be to have people negate your first-hand experience.”
The situation has gotten better in Sioux Falls, Dr. Barnes said, although cases have started rising in the state again. The stress, while not gone, is reduced. For some doctors, “COVID reminded us of why we do what we do. Some of the usual bureaucratic requirements were set aside and we could focus on what our patients needed and how to take care of them.”
Taking job stress seriously
Tiffani Panek, MA, SFHM, CLHM, administrator of the division of hospital medicine at Johns Hopkins Bayview Medical Center in Baltimore, said job stress is a major issue for hospitalist groups.

“We take it seriously here, and use a survey tool to measure morale in our group annually,” she said. “So far, knock on wood, Baltimore has not been one of the big hot spots, but we’ve definitely had waves of COVID patients.”
The Bayview hospitalist group has a diversified set of leaders, including a wellness director. “They’re always checking up on our people, keeping an eye on those who are most vulnerable. One of the stressors we hadn’t thought about before was for our people who live alone. With the isolation and lockdown, they haven’t been able to socialize, so we’ve made direct outreach, asking people how they were doing,” Ms. Panek said. “People know we’ve got their back – professionally and personally. They know, if there’s something we can do to help, we will do it.”
Bayview Medical Center has COVID-specific units and non-COVID units, and has tried to rotate hospitalist assignments because more than a couple days in a row spent wearing full personal protective equipment (PPE) is exhausting, Ms. Panek said. The group also allocated a respite room just outside the biocontainment unit, with a computer and opportunities for providers to just sit and take a breather – with appropriate social distancing. “It’s not fancy, but you just have to wear a mask, not full PPE.”
The Hopkins hospitalist group’s wellness director, Catherine Washburn, MD, also a working hospitalist, said providers are exhausted, and trying to transition to the new normal is a moving target.
“It’s hard for anyone to say what our lives will look like in 6 months,” she said. “People in our group have lost family members to COVID, or postponed major life events, like weddings. We acknowledge losses together as a group, and celebrate things worth celebrating, like babies or birthdays.”
Greatest COVID caseload
Joshua Case, MD, hospitalist medical director for 16 acute care hospitals of Northwell Health serving metropolitan New York City and Long Island, said his group’s hospitalists and other staff worked incredibly hard during the surge of COVID-19 patients in New York. “Northwell likely cared for more COVID patients than any other health care system in the U.S., if not the world.

“It’s vastly different now. We went from a peak of thousands of cases per day down to about 70-90 new cases a day across our system. We’re lucky our system recognized that COVID could be an issue early on, with all of the multifaceted stressors on patient care,” Dr. Case said. “We’ve done whatever we could to give people time off, especially as the census started to come down. We freed up as many supportive mental health services as we could, working with the health system’s employee assistance program.”
Northwell gave out numbers for the psychiatry department, with clinicians available 24/7 for a confidential call, along with outside volunteers and a network of trauma psychologists. “Our system also provided emergency child care for staff, including hospitalists, wherever we could, drawing upon community resources,” Dr. Case added.
“We recognize that we’re all in the same foxhole. That’s been a helpful attitude – recognizing that it’s okay to be upset in a crisis and to have trouble dealing with what’s going on,” he said. “We need to acknowledge that some of us are suffering and try to encourage people to face it head on. For a lot of physicians, especially those who were redeployed here from other departments, it was important just to have us ask if they were doing okay.”
Brian Schroeder, MHA, FACHE, FHM, assistant vice president for hospital and emergency medicine for Atrium Health, based in Charlotte, N.C., said one of the biggest sources of stress on his staff has been the constant pace of change – whether local hospital protocols, state policies, or guidelines from the Centers for Disease Control and Prevention. “The updating is difficult to keep up with. A lot of our physicians get worried and anxious that they’re not following the latest guidelines or correctly doing what they should be doing to care for COVID patients. One thing we’ve done to alleviate some of that fear and anxiety is through weekly huddles with our hospital teams, focusing on changes relevant to their work. We also have weekly ‘all-hands’ meetings for our 250 providers across 13 acute and four postacute facilities.”
Before COVID, it was difficult to get everyone together as one big group from hospitals up to 5 hours apart, but with the Microsoft Teams platform, they can all meet together.
“At the height of the pandemic, we’d convene weekly and share national statistics, organizational statistics, testing updates, changes to protocols,” Mr. Schroeder said. As the pace of change has slowed, these meetings were cut back to monthly. “Our physicians feel we are passing on information as soon as we get it. They know we’ll always tell them what we know.”
Sarah Richards, MD, assistant professor of internal medicine at the University of Nebraska, Omaha, who heads the Society of Hospital Medicine’s Well-Being Task Force, formed to address staff stress in the COVID environment, said there are things that health care systems can do to help mitigate job stress and burnout. But broader issues may need to be addressed at a national level. “SHM is trying to understand work-related stress – and to identify resources that could support doctors, so they can spend more of their time doing what they enjoy most, which is taking care of patients,” she said.
“We also recognize that people have had very different experiences, depending on geography, and at the individual level stressors are experienced very differently,” Dr. Richard noted. “One of the most common stressors we’ve heard from doctors is the challenge of caring for patients who are lonely and isolated in their hospital rooms, suffering and dying in new ways. In low-incidence areas, doctors are expressing guilt because they aren’t under as much stress as their colleagues. In high-incidence areas, doctors are already experiencing posttraumatic stress disorder.”
SHM’s Well-Being Task Force is working on a tool to help normalize these stressors and encourage open conversations about mental health issues. A guide called “HM COVID Check-in Guide for Self & Peers” is designed to help hospitalists break the culture of silence around well-being and burnout during COVID-19 and how people are handling and processing the pandemic experience. It is expected to be completed later this year, Dr. Richards said. Other SHM projects and resources for staff support are also in the works.
The impact on women doctors
In a recent Journal of Hospital Medicine article entitled “Collateral Damage: How COVID-19 is Adversely Impacting Women Physicians,” hospitalist Yemisi Jones, MD, medical director of continuing medical education at Cincinnati Children’s Hospital Medical Center, and colleagues argue that preexisting gender inequities in compensation, academic rank and leadership positions for physicians have made the COVID-19 crisis even more burdensome on female hospitalists.1

“Increased childcare and schooling obligations, coupled with disproportionate household responsibilities and an inability to work from home, will likely result in female hospitalists struggling to meet family needs while pandemic-related work responsibilities are ramping up,” they write. COVID may intensify workplace inequalities, with a lack of recognition of the undue strain that group policies place on women.
“Often women suffer in silence,” said coauthor Jennifer O’Toole, MD, MEd, director of education in the division of hospital medicine at Cincinnati Children’s Hospital Medical Center and program director of the internal medicine–pediatrics residency. “We are not always the best self-advocates, although many of us are working on that.”
When women in hospital medicine take leadership roles, these often tend to involve mutual support activities, taking care of colleagues, and promoting collaborative work environments, Dr. Jones added. The stereotypical example is the committee that organizes celebrations when group members get married or have babies.
These activities can take a lot of time, she said. “We need to pay attention to that kind of role in our groups, because it’s important to the cohesiveness of the group. But it often goes unrecognized and doesn’t translate into the currency of promotion and leadership in medicine. When women go for promotions in the future, how will what happened during the COVID crisis impact their opportunities?”
What is the answer to overcoming these systemic inequities? Start with making sure women are part of the leadership team, with responsibilities for group policies, schedules, and other important decisions. “Look at your group’s leadership – particularly the higher positions. If it’s not diverse, ask why. ‘What is it about the structure of our group?’ Make a more concerted effort in your recruitment and retention,” Dr. Jones said.
The JHM article also recommends closely monitoring the direct and indirect effects of COVID-19 on female hospitalists, inquiring specifically about the needs of women in the organization, and ensuring that diversity, inclusion, and equity efforts are not suspended during the pandemic. Gender-based disparities in pay also need a closer look, and not just one time but reviewed periodically and adjusted accordingly.
Mentoring for early career women is important, but more so is sponsorship – someone in a high-level leadership role in the group sponsoring women who are rising up the career ladder, Dr. O’Toole said. “Professional women tend to be overmentored and undersponsored.”
What are the answers?
Ultimately, listening is key to try to help people get through the pandemic, Dr. Washburn said. “People become burned out when they feel leadership doesn’t understand their needs or doesn’t hear their concerns. Our group leaders all do clinical work, so they are seen as one of us. They try very hard; they have listening ears. But listening is just the first step. Next step is to work creatively to get the identified needs met.”
A few years ago, Johns Hopkins developed training in enhanced communication in health care for all hospital providers, including nurses and doctors, encouraging them to get trained in how to actively listen and address their patients’ emotional and social experiences as well as disease, Dr. Washburn explained. Learning how to listen better to patients can enhance skills at listening to colleagues, and vice versa. “We recognize the importance of better communication – for reducing sentinel events in the hospital and also for preventing staff burnout.”
Dr. Barnes also does physician coaching, and says a lot of that work is helping people achieve clarity on their core values. “Healing patients is a core identify for physicians; we want to take care of people. But other things can get in the way of that, and hospitalist groups can work at minimizing those barriers. We also need to learn, as hospitalists, that we work in a group. You need to be creative in how you do your team building, especially now, when you can no longer get together for dinner. Whatever it is, how do we bring our team back together? The biggest source of support for many hospitalists, beyond their family, is the group.”
Dr. Case said there is a longer-term need to study the root causes of burnout in hospitalists and to identify the issues that cause job stress. “What is modifiable? How can we tackle it? I see that as big part of my job every day. Being a physician is hard enough as it is. Let’s work to resolve those issues that add needlessly to the stress.”
“I think the pandemic brought a magnifying glass to how important a concern staff stress is,” Ms. Panek said. Resilience is important.
“We were working in our group on creating a culture that values trust and transparency, and then the COVID crisis hit,” she said. “But you can still keep working on those things. We would not have been as good or as positive as we were in managing this crisis without that preexisting culture to draw upon. We always said it was important. Now we know that’s true.”
Reference
1. Jones Y et al. Collateral Damage: How COVID-19 Is Adversely Impacting Women Physicians. J Hosp Med. 2020 August;15(8):507-9.
New kinds of job stress multiply in unusual times
New kinds of job stress multiply in unusual times
Clarissa Barnes, MD, a hospitalist at Avera McKennan Hospital in Sioux Falls, S.D., and until recently medical director of Avera’s LIGHT Program, a wellness-oriented service for doctors, nurse practitioners, and physician assistants, watched the COVID-19 crisis unfold up close in her community and her hospital. Sioux Falls traced its surge of COVID patients to an outbreak at a local meatpacking plant.
“In the beginning, we didn’t know much about the virus and its communicability, although we have since gotten a better handle on that,” she said. “We had questions: Should we give patients more fluids – or less? Steroids or not? In my experience as a hospitalist I never had patients die every day on my shift, but that was happening with COVID.” The crisis imposed serious stresses on frontline providers, and hospitalists were concerned about personal safety and exposure risk – not just for themselves but for their families.
“The first time I worked on the COVID unit, I moved into the guest room in our home, apart from my husband and our young children,” Dr. Barnes said. “Ultimately I caught the virus, although I have since recovered.” Her experience has highlighted how existing issues of job stress and burnout in hospital medicine have been exacerbated by COVID-19. Even physicians who consider themselves healthy may have little emotional reserve to draw upon in a crisis of this magnitude.
“We are social distancing at work, wearing masks, not eating together with our colleagues – with less camaraderie and social support than we used to have,” she said. “I feel exhausted and there’s no question that my colleagues and I have sacrificed a lot to deal with the pandemic.” Add to that the second front of the COVID-19 crisis, Dr. Barnes said, which is “fighting the medical information wars, trying to correct misinformation put out there by people. Physicians who have been on the front lines of the pandemic know how demoralizing it can be to have people negate your first-hand experience.”
The situation has gotten better in Sioux Falls, Dr. Barnes said, although cases have started rising in the state again. The stress, while not gone, is reduced. For some doctors, “COVID reminded us of why we do what we do. Some of the usual bureaucratic requirements were set aside and we could focus on what our patients needed and how to take care of them.”
Taking job stress seriously
Tiffani Panek, MA, SFHM, CLHM, administrator of the division of hospital medicine at Johns Hopkins Bayview Medical Center in Baltimore, said job stress is a major issue for hospitalist groups.
“We take it seriously here, and use a survey tool to measure morale in our group annually,” she said. “So far, knock on wood, Baltimore has not been one of the big hot spots, but we’ve definitely had waves of COVID patients.”
The Bayview hospitalist group has a diversified set of leaders, including a wellness director. “They’re always checking up on our people, keeping an eye on those who are most vulnerable. One of the stressors we hadn’t thought about before was for our people who live alone. With the isolation and lockdown, they haven’t been able to socialize, so we’ve made direct outreach, asking people how they were doing,” Ms. Panek said. “People know we’ve got their back – professionally and personally. They know, if there’s something we can do to help, we will do it.”
Bayview Medical Center has COVID-specific units and non-COVID units, and has tried to rotate hospitalist assignments because more than a couple days in a row spent wearing full personal protective equipment (PPE) is exhausting, Ms. Panek said. The group also allocated a respite room just outside the biocontainment unit, with a computer and opportunities for providers to just sit and take a breather – with appropriate social distancing. “It’s not fancy, but you just have to wear a mask, not full PPE.”
The Hopkins hospitalist group’s wellness director, Catherine Washburn, MD, also a working hospitalist, said providers are exhausted, and trying to transition to the new normal is a moving target.
“It’s hard for anyone to say what our lives will look like in 6 months,” she said. “People in our group have lost family members to COVID, or postponed major life events, like weddings. We acknowledge losses together as a group, and celebrate things worth celebrating, like babies or birthdays.”
Greatest COVID caseload
Joshua Case, MD, hospitalist medical director for 16 acute care hospitals of Northwell Health serving metropolitan New York City and Long Island, said his group’s hospitalists and other staff worked incredibly hard during the surge of COVID-19 patients in New York. “Northwell likely cared for more COVID patients than any other health care system in the U.S., if not the world.
“It’s vastly different now. We went from a peak of thousands of cases per day down to about 70-90 new cases a day across our system. We’re lucky our system recognized that COVID could be an issue early on, with all of the multifaceted stressors on patient care,” Dr. Case said. “We’ve done whatever we could to give people time off, especially as the census started to come down. We freed up as many supportive mental health services as we could, working with the health system’s employee assistance program.”
Northwell gave out numbers for the psychiatry department, with clinicians available 24/7 for a confidential call, along with outside volunteers and a network of trauma psychologists. “Our system also provided emergency child care for staff, including hospitalists, wherever we could, drawing upon community resources,” Dr. Case added.
“We recognize that we’re all in the same foxhole. That’s been a helpful attitude – recognizing that it’s okay to be upset in a crisis and to have trouble dealing with what’s going on,” he said. “We need to acknowledge that some of us are suffering and try to encourage people to face it head on. For a lot of physicians, especially those who were redeployed here from other departments, it was important just to have us ask if they were doing okay.”
Brian Schroeder, MHA, FACHE, FHM, assistant vice president for hospital and emergency medicine for Atrium Health, based in Charlotte, N.C., said one of the biggest sources of stress on his staff has been the constant pace of change – whether local hospital protocols, state policies, or guidelines from the Centers for Disease Control and Prevention. “The updating is difficult to keep up with. A lot of our physicians get worried and anxious that they’re not following the latest guidelines or correctly doing what they should be doing to care for COVID patients. One thing we’ve done to alleviate some of that fear and anxiety is through weekly huddles with our hospital teams, focusing on changes relevant to their work. We also have weekly ‘all-hands’ meetings for our 250 providers across 13 acute and four postacute facilities.”
Before COVID, it was difficult to get everyone together as one big group from hospitals up to 5 hours apart, but with the Microsoft Teams platform, they can all meet together.
“At the height of the pandemic, we’d convene weekly and share national statistics, organizational statistics, testing updates, changes to protocols,” Mr. Schroeder said. As the pace of change has slowed, these meetings were cut back to monthly. “Our physicians feel we are passing on information as soon as we get it. They know we’ll always tell them what we know.”
Sarah Richards, MD, assistant professor of internal medicine at the University of Nebraska, Omaha, who heads the Society of Hospital Medicine’s Well-Being Task Force, formed to address staff stress in the COVID environment, said there are things that health care systems can do to help mitigate job stress and burnout. But broader issues may need to be addressed at a national level. “SHM is trying to understand work-related stress – and to identify resources that could support doctors, so they can spend more of their time doing what they enjoy most, which is taking care of patients,” she said.
“We also recognize that people have had very different experiences, depending on geography, and at the individual level stressors are experienced very differently,” Dr. Richard noted. “One of the most common stressors we’ve heard from doctors is the challenge of caring for patients who are lonely and isolated in their hospital rooms, suffering and dying in new ways. In low-incidence areas, doctors are expressing guilt because they aren’t under as much stress as their colleagues. In high-incidence areas, doctors are already experiencing posttraumatic stress disorder.”
SHM’s Well-Being Task Force is working on a tool to help normalize these stressors and encourage open conversations about mental health issues. A guide called “HM COVID Check-in Guide for Self & Peers” is designed to help hospitalists break the culture of silence around well-being and burnout during COVID-19 and how people are handling and processing the pandemic experience. It is expected to be completed later this year, Dr. Richards said. Other SHM projects and resources for staff support are also in the works.
The impact on women doctors
In a recent Journal of Hospital Medicine article entitled “Collateral Damage: How COVID-19 is Adversely Impacting Women Physicians,” hospitalist Yemisi Jones, MD, medical director of continuing medical education at Cincinnati Children’s Hospital Medical Center, and colleagues argue that preexisting gender inequities in compensation, academic rank and leadership positions for physicians have made the COVID-19 crisis even more burdensome on female hospitalists.1
“Increased childcare and schooling obligations, coupled with disproportionate household responsibilities and an inability to work from home, will likely result in female hospitalists struggling to meet family needs while pandemic-related work responsibilities are ramping up,” they write. COVID may intensify workplace inequalities, with a lack of recognition of the undue strain that group policies place on women.
“Often women suffer in silence,” said coauthor Jennifer O’Toole, MD, MEd, director of education in the division of hospital medicine at Cincinnati Children’s Hospital Medical Center and program director of the internal medicine–pediatrics residency. “We are not always the best self-advocates, although many of us are working on that.”
When women in hospital medicine take leadership roles, these often tend to involve mutual support activities, taking care of colleagues, and promoting collaborative work environments, Dr. Jones added. The stereotypical example is the committee that organizes celebrations when group members get married or have babies.
These activities can take a lot of time, she said. “We need to pay attention to that kind of role in our groups, because it’s important to the cohesiveness of the group. But it often goes unrecognized and doesn’t translate into the currency of promotion and leadership in medicine. When women go for promotions in the future, how will what happened during the COVID crisis impact their opportunities?”
What is the answer to overcoming these systemic inequities? Start with making sure women are part of the leadership team, with responsibilities for group policies, schedules, and other important decisions. “Look at your group’s leadership – particularly the higher positions. If it’s not diverse, ask why. ‘What is it about the structure of our group?’ Make a more concerted effort in your recruitment and retention,” Dr. Jones said.
The JHM article also recommends closely monitoring the direct and indirect effects of COVID-19 on female hospitalists, inquiring specifically about the needs of women in the organization, and ensuring that diversity, inclusion, and equity efforts are not suspended during the pandemic. Gender-based disparities in pay also need a closer look, and not just one time but reviewed periodically and adjusted accordingly.
Mentoring for early career women is important, but more so is sponsorship – someone in a high-level leadership role in the group sponsoring women who are rising up the career ladder, Dr. O’Toole said. “Professional women tend to be overmentored and undersponsored.”
What are the answers?
Ultimately, listening is key to try to help people get through the pandemic, Dr. Washburn said. “People become burned out when they feel leadership doesn’t understand their needs or doesn’t hear their concerns. Our group leaders all do clinical work, so they are seen as one of us. They try very hard; they have listening ears. But listening is just the first step. Next step is to work creatively to get the identified needs met.”
A few years ago, Johns Hopkins developed training in enhanced communication in health care for all hospital providers, including nurses and doctors, encouraging them to get trained in how to actively listen and address their patients’ emotional and social experiences as well as disease, Dr. Washburn explained. Learning how to listen better to patients can enhance skills at listening to colleagues, and vice versa. “We recognize the importance of better communication – for reducing sentinel events in the hospital and also for preventing staff burnout.”
Dr. Barnes also does physician coaching, and says a lot of that work is helping people achieve clarity on their core values. “Healing patients is a core identify for physicians; we want to take care of people. But other things can get in the way of that, and hospitalist groups can work at minimizing those barriers. We also need to learn, as hospitalists, that we work in a group. You need to be creative in how you do your team building, especially now, when you can no longer get together for dinner. Whatever it is, how do we bring our team back together? The biggest source of support for many hospitalists, beyond their family, is the group.”
Dr. Case said there is a longer-term need to study the root causes of burnout in hospitalists and to identify the issues that cause job stress. “What is modifiable? How can we tackle it? I see that as big part of my job every day. Being a physician is hard enough as it is. Let’s work to resolve those issues that add needlessly to the stress.”
“I think the pandemic brought a magnifying glass to how important a concern staff stress is,” Ms. Panek said. Resilience is important.
“We were working in our group on creating a culture that values trust and transparency, and then the COVID crisis hit,” she said. “But you can still keep working on those things. We would not have been as good or as positive as we were in managing this crisis without that preexisting culture to draw upon. We always said it was important. Now we know that’s true.”
Reference
1. Jones Y et al. Collateral Damage: How COVID-19 Is Adversely Impacting Women Physicians. J Hosp Med. 2020 August;15(8):507-9.
Clarissa Barnes, MD, a hospitalist at Avera McKennan Hospital in Sioux Falls, S.D., and until recently medical director of Avera’s LIGHT Program, a wellness-oriented service for doctors, nurse practitioners, and physician assistants, watched the COVID-19 crisis unfold up close in her community and her hospital. Sioux Falls traced its surge of COVID patients to an outbreak at a local meatpacking plant.
“In the beginning, we didn’t know much about the virus and its communicability, although we have since gotten a better handle on that,” she said. “We had questions: Should we give patients more fluids – or less? Steroids or not? In my experience as a hospitalist I never had patients die every day on my shift, but that was happening with COVID.” The crisis imposed serious stresses on frontline providers, and hospitalists were concerned about personal safety and exposure risk – not just for themselves but for their families.
“The first time I worked on the COVID unit, I moved into the guest room in our home, apart from my husband and our young children,” Dr. Barnes said. “Ultimately I caught the virus, although I have since recovered.” Her experience has highlighted how existing issues of job stress and burnout in hospital medicine have been exacerbated by COVID-19. Even physicians who consider themselves healthy may have little emotional reserve to draw upon in a crisis of this magnitude.
“We are social distancing at work, wearing masks, not eating together with our colleagues – with less camaraderie and social support than we used to have,” she said. “I feel exhausted and there’s no question that my colleagues and I have sacrificed a lot to deal with the pandemic.” Add to that the second front of the COVID-19 crisis, Dr. Barnes said, which is “fighting the medical information wars, trying to correct misinformation put out there by people. Physicians who have been on the front lines of the pandemic know how demoralizing it can be to have people negate your first-hand experience.”
The situation has gotten better in Sioux Falls, Dr. Barnes said, although cases have started rising in the state again. The stress, while not gone, is reduced. For some doctors, “COVID reminded us of why we do what we do. Some of the usual bureaucratic requirements were set aside and we could focus on what our patients needed and how to take care of them.”
Taking job stress seriously
Tiffani Panek, MA, SFHM, CLHM, administrator of the division of hospital medicine at Johns Hopkins Bayview Medical Center in Baltimore, said job stress is a major issue for hospitalist groups.
“We take it seriously here, and use a survey tool to measure morale in our group annually,” she said. “So far, knock on wood, Baltimore has not been one of the big hot spots, but we’ve definitely had waves of COVID patients.”
The Bayview hospitalist group has a diversified set of leaders, including a wellness director. “They’re always checking up on our people, keeping an eye on those who are most vulnerable. One of the stressors we hadn’t thought about before was for our people who live alone. With the isolation and lockdown, they haven’t been able to socialize, so we’ve made direct outreach, asking people how they were doing,” Ms. Panek said. “People know we’ve got their back – professionally and personally. They know, if there’s something we can do to help, we will do it.”
Bayview Medical Center has COVID-specific units and non-COVID units, and has tried to rotate hospitalist assignments because more than a couple days in a row spent wearing full personal protective equipment (PPE) is exhausting, Ms. Panek said. The group also allocated a respite room just outside the biocontainment unit, with a computer and opportunities for providers to just sit and take a breather – with appropriate social distancing. “It’s not fancy, but you just have to wear a mask, not full PPE.”
The Hopkins hospitalist group’s wellness director, Catherine Washburn, MD, also a working hospitalist, said providers are exhausted, and trying to transition to the new normal is a moving target.
“It’s hard for anyone to say what our lives will look like in 6 months,” she said. “People in our group have lost family members to COVID, or postponed major life events, like weddings. We acknowledge losses together as a group, and celebrate things worth celebrating, like babies or birthdays.”
Greatest COVID caseload
Joshua Case, MD, hospitalist medical director for 16 acute care hospitals of Northwell Health serving metropolitan New York City and Long Island, said his group’s hospitalists and other staff worked incredibly hard during the surge of COVID-19 patients in New York. “Northwell likely cared for more COVID patients than any other health care system in the U.S., if not the world.
“It’s vastly different now. We went from a peak of thousands of cases per day down to about 70-90 new cases a day across our system. We’re lucky our system recognized that COVID could be an issue early on, with all of the multifaceted stressors on patient care,” Dr. Case said. “We’ve done whatever we could to give people time off, especially as the census started to come down. We freed up as many supportive mental health services as we could, working with the health system’s employee assistance program.”
Northwell gave out numbers for the psychiatry department, with clinicians available 24/7 for a confidential call, along with outside volunteers and a network of trauma psychologists. “Our system also provided emergency child care for staff, including hospitalists, wherever we could, drawing upon community resources,” Dr. Case added.
“We recognize that we’re all in the same foxhole. That’s been a helpful attitude – recognizing that it’s okay to be upset in a crisis and to have trouble dealing with what’s going on,” he said. “We need to acknowledge that some of us are suffering and try to encourage people to face it head on. For a lot of physicians, especially those who were redeployed here from other departments, it was important just to have us ask if they were doing okay.”
Brian Schroeder, MHA, FACHE, FHM, assistant vice president for hospital and emergency medicine for Atrium Health, based in Charlotte, N.C., said one of the biggest sources of stress on his staff has been the constant pace of change – whether local hospital protocols, state policies, or guidelines from the Centers for Disease Control and Prevention. “The updating is difficult to keep up with. A lot of our physicians get worried and anxious that they’re not following the latest guidelines or correctly doing what they should be doing to care for COVID patients. One thing we’ve done to alleviate some of that fear and anxiety is through weekly huddles with our hospital teams, focusing on changes relevant to their work. We also have weekly ‘all-hands’ meetings for our 250 providers across 13 acute and four postacute facilities.”
Before COVID, it was difficult to get everyone together as one big group from hospitals up to 5 hours apart, but with the Microsoft Teams platform, they can all meet together.
“At the height of the pandemic, we’d convene weekly and share national statistics, organizational statistics, testing updates, changes to protocols,” Mr. Schroeder said. As the pace of change has slowed, these meetings were cut back to monthly. “Our physicians feel we are passing on information as soon as we get it. They know we’ll always tell them what we know.”
Sarah Richards, MD, assistant professor of internal medicine at the University of Nebraska, Omaha, who heads the Society of Hospital Medicine’s Well-Being Task Force, formed to address staff stress in the COVID environment, said there are things that health care systems can do to help mitigate job stress and burnout. But broader issues may need to be addressed at a national level. “SHM is trying to understand work-related stress – and to identify resources that could support doctors, so they can spend more of their time doing what they enjoy most, which is taking care of patients,” she said.
“We also recognize that people have had very different experiences, depending on geography, and at the individual level stressors are experienced very differently,” Dr. Richard noted. “One of the most common stressors we’ve heard from doctors is the challenge of caring for patients who are lonely and isolated in their hospital rooms, suffering and dying in new ways. In low-incidence areas, doctors are expressing guilt because they aren’t under as much stress as their colleagues. In high-incidence areas, doctors are already experiencing posttraumatic stress disorder.”
SHM’s Well-Being Task Force is working on a tool to help normalize these stressors and encourage open conversations about mental health issues. A guide called “HM COVID Check-in Guide for Self & Peers” is designed to help hospitalists break the culture of silence around well-being and burnout during COVID-19 and how people are handling and processing the pandemic experience. It is expected to be completed later this year, Dr. Richards said. Other SHM projects and resources for staff support are also in the works.
The impact on women doctors
In a recent Journal of Hospital Medicine article entitled “Collateral Damage: How COVID-19 is Adversely Impacting Women Physicians,” hospitalist Yemisi Jones, MD, medical director of continuing medical education at Cincinnati Children’s Hospital Medical Center, and colleagues argue that preexisting gender inequities in compensation, academic rank and leadership positions for physicians have made the COVID-19 crisis even more burdensome on female hospitalists.1
“Increased childcare and schooling obligations, coupled with disproportionate household responsibilities and an inability to work from home, will likely result in female hospitalists struggling to meet family needs while pandemic-related work responsibilities are ramping up,” they write. COVID may intensify workplace inequalities, with a lack of recognition of the undue strain that group policies place on women.
“Often women suffer in silence,” said coauthor Jennifer O’Toole, MD, MEd, director of education in the division of hospital medicine at Cincinnati Children’s Hospital Medical Center and program director of the internal medicine–pediatrics residency. “We are not always the best self-advocates, although many of us are working on that.”
When women in hospital medicine take leadership roles, these often tend to involve mutual support activities, taking care of colleagues, and promoting collaborative work environments, Dr. Jones added. The stereotypical example is the committee that organizes celebrations when group members get married or have babies.
These activities can take a lot of time, she said. “We need to pay attention to that kind of role in our groups, because it’s important to the cohesiveness of the group. But it often goes unrecognized and doesn’t translate into the currency of promotion and leadership in medicine. When women go for promotions in the future, how will what happened during the COVID crisis impact their opportunities?”
What is the answer to overcoming these systemic inequities? Start with making sure women are part of the leadership team, with responsibilities for group policies, schedules, and other important decisions. “Look at your group’s leadership – particularly the higher positions. If it’s not diverse, ask why. ‘What is it about the structure of our group?’ Make a more concerted effort in your recruitment and retention,” Dr. Jones said.
The JHM article also recommends closely monitoring the direct and indirect effects of COVID-19 on female hospitalists, inquiring specifically about the needs of women in the organization, and ensuring that diversity, inclusion, and equity efforts are not suspended during the pandemic. Gender-based disparities in pay also need a closer look, and not just one time but reviewed periodically and adjusted accordingly.
Mentoring for early career women is important, but more so is sponsorship – someone in a high-level leadership role in the group sponsoring women who are rising up the career ladder, Dr. O’Toole said. “Professional women tend to be overmentored and undersponsored.”
What are the answers?
Ultimately, listening is key to try to help people get through the pandemic, Dr. Washburn said. “People become burned out when they feel leadership doesn’t understand their needs or doesn’t hear their concerns. Our group leaders all do clinical work, so they are seen as one of us. They try very hard; they have listening ears. But listening is just the first step. Next step is to work creatively to get the identified needs met.”
A few years ago, Johns Hopkins developed training in enhanced communication in health care for all hospital providers, including nurses and doctors, encouraging them to get trained in how to actively listen and address their patients’ emotional and social experiences as well as disease, Dr. Washburn explained. Learning how to listen better to patients can enhance skills at listening to colleagues, and vice versa. “We recognize the importance of better communication – for reducing sentinel events in the hospital and also for preventing staff burnout.”
Dr. Barnes also does physician coaching, and says a lot of that work is helping people achieve clarity on their core values. “Healing patients is a core identify for physicians; we want to take care of people. But other things can get in the way of that, and hospitalist groups can work at minimizing those barriers. We also need to learn, as hospitalists, that we work in a group. You need to be creative in how you do your team building, especially now, when you can no longer get together for dinner. Whatever it is, how do we bring our team back together? The biggest source of support for many hospitalists, beyond their family, is the group.”
Dr. Case said there is a longer-term need to study the root causes of burnout in hospitalists and to identify the issues that cause job stress. “What is modifiable? How can we tackle it? I see that as big part of my job every day. Being a physician is hard enough as it is. Let’s work to resolve those issues that add needlessly to the stress.”
“I think the pandemic brought a magnifying glass to how important a concern staff stress is,” Ms. Panek said. Resilience is important.
“We were working in our group on creating a culture that values trust and transparency, and then the COVID crisis hit,” she said. “But you can still keep working on those things. We would not have been as good or as positive as we were in managing this crisis without that preexisting culture to draw upon. We always said it was important. Now we know that’s true.”
Reference
1. Jones Y et al. Collateral Damage: How COVID-19 Is Adversely Impacting Women Physicians. J Hosp Med. 2020 August;15(8):507-9.
‘Soak-and-smear’ AD protocol backed by evidence
The most effective initial step for clearing atopic dermatitis in infants and young children involves daily bathing, followed by immediate application of a moisturizer, topical steroid, or both, according to an expert speaking at the virtual annual Coastal Dermatology Symposium.
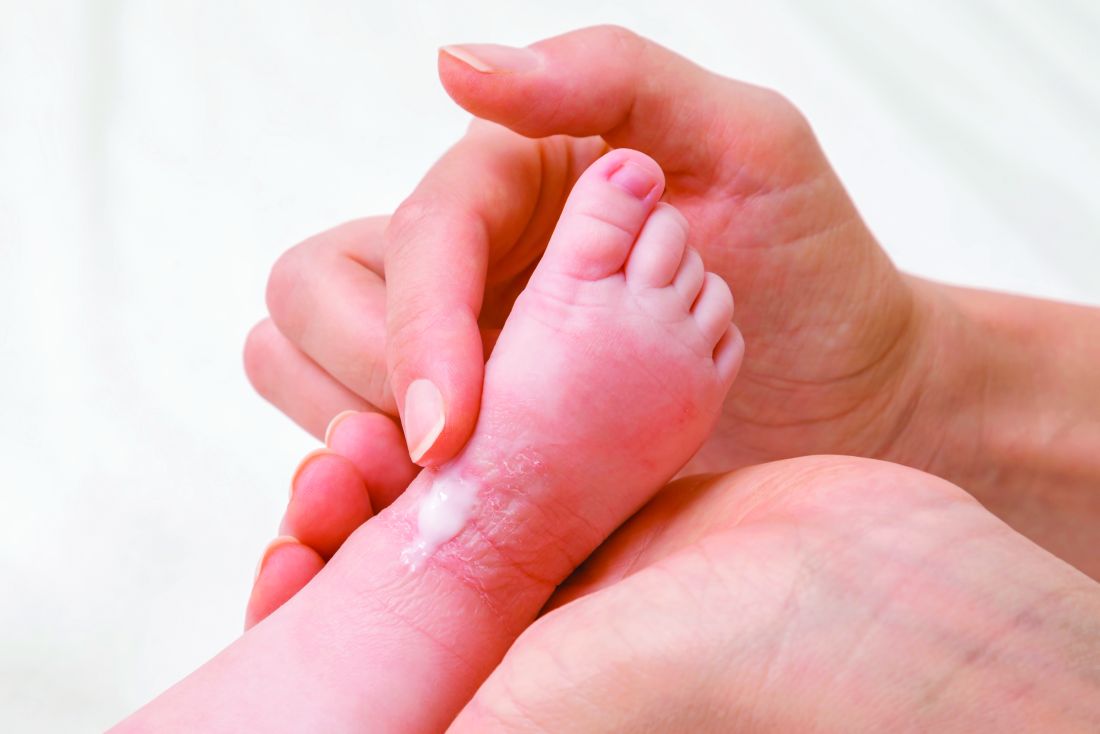
“If they are really severe, you can do it twice-daily, but there are several studies that show there is not a huge benefit of twice-daily over once-daily,” said Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland.
He called this technique “soak-and-smear.” The “smear” is performed immediately after the bath when the skin is still damp, he said. When clearing is the goal, and the child has moderate to severe atopic dermatitis (AD), 0.1% triamcinolone or a similar medium potency topical steroid can be applied, and after clearing, the steroid can be switched for a moisturizer, according to Dr. Simpson.
Rather than restricting application to areas of greatest skin involvement, “put it all over,” he advised.
The clearing regimen should be continued “for a couple of more days” after the lesions have resolved, with a return visit in about a week to confirm clearing and reinforce the next steps for keeping patients clear, he added.
The next steps depend on severity. According to Dr. Simpson, severity is defined less by the extent of skin involvement at the baseline examination than the speed at which symptoms return.
For those with only mild symptoms after an extended period of clearing, moisturizer might be sufficient to prevent a significant relapse. For children with a more rapid relapse, it will be necessary to reintroduce topical steroid either every day, every other day, or twice per week.
Whether with moisturizer or with topical steroids, the soak-and-smear technique has now been validated in a recently published crossover randomized trial.
In the trial, children aged 6 months to 11 years, with moderate to severe AD, were randomized to a twice-daily bath, called the “wet method,” versus a twice-weekly bath, called the “dry method.” Both groups received a cleanser and moisturizer along with a low-potency topical steroid as needed.
After 2 weeks, the 40 evaluable patients were crossed over to the opposite bathing technique. The wet, or soak-and-smear approach, was associated with a highly significant reduction in the primary endpoint of SCORing Atopic Dermatitis (SCORAD) index, compared with the dry method (95% confidence interval, 14.9-27.6; P less than .0001). In a secondary analysis, this translated into a 30% relative reduction in favor of the wet method.
In addition, there was improvement in a caregiver assessment of the Atopic Dermatitis Quickscore (ADQ). These data show that “twice-daily baths with topical steroids and moisturizer can help in more moderate to severe population,” said Dr. Simpson, who noted that he has participated in open-label studies with the same soak-and-smear technique that have produced similar results.
Once children are clear, Dr. Simpson recommends a maintenance strategy individualized for severity. In many cases, this will involve moisturizers applied after the bath, supplemented intermittently, such as once or twice per week, with topical steroids. However, if parents find themselves resorting to daily steroids to maintain control, “that’s when you incorporate the TCIs [topical calcineurin inhibitors].”
TCIs “can help you stay at twice-per-week topical steroids,” Dr. Simpson said at the meeting, jointly presented by the University of Louisville and Global Academy for Medical Education.
TCIs also help patients avoid steroid withdrawal, a particularly common phenomenon when topical steroids are applied repeatedly to the face. He recommended a proactive approach. By applying TCIs to areas where skin lesions frequently recur, such as the eyelids, flares can often be prevented.
Repeated applications of TCIs “is perfectly safe and effective, and there are many studies that show proactive treatment is very effective and can prevent you from having to use too much topical steroids” or move to a systemic steroid, Dr. Simpson said.
These steps have been highly effective for sustained control even in challenging cases of AD, but he emphasized the importance of explaining the rationale to parents and eliciting their adherence to these treatment steps. Writing out the instructions will reduce confusion and help parents keep their children clear, he added.
Lawrence F. Eichenfield, MD, professor of pediatrics and dermatology at the University of California, San Diego, agreed that this recently published crossover trial has been helpful in counseling parents about how to manage AD in their children.

“Many times, pediatricians tell parents to avoid bathing because they feel that bathing will dry out the skin,” he said. The crossover study, by showing better control of AD with frequent bathing, dispels that notion, although he is not convinced that bathing at this frequency is necessary.
“I have not advised anyone to do twice-daily bathing, with rare exceptions, on the basis on this study, but, basically, I think that whether people do daily bathing or every other day bathing, it is pretty reasonable that bathing might help as long as they are applying moisturizer immediately afterward,” he said.
Dr. Simpson reports financial relationships with AbbVie, Celgene Dermira, Genentech, GlaxoSmithKline, Incyte, Lilly, Medimmune, Pfizer, Regeneron/Sanofi, and Tioga.
This publication and Global Academy for Medical Education are owned by the same parent company.
The most effective initial step for clearing atopic dermatitis in infants and young children involves daily bathing, followed by immediate application of a moisturizer, topical steroid, or both, according to an expert speaking at the virtual annual Coastal Dermatology Symposium.
“If they are really severe, you can do it twice-daily, but there are several studies that show there is not a huge benefit of twice-daily over once-daily,” said Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland.
He called this technique “soak-and-smear.” The “smear” is performed immediately after the bath when the skin is still damp, he said. When clearing is the goal, and the child has moderate to severe atopic dermatitis (AD), 0.1% triamcinolone or a similar medium potency topical steroid can be applied, and after clearing, the steroid can be switched for a moisturizer, according to Dr. Simpson.
Rather than restricting application to areas of greatest skin involvement, “put it all over,” he advised.
The clearing regimen should be continued “for a couple of more days” after the lesions have resolved, with a return visit in about a week to confirm clearing and reinforce the next steps for keeping patients clear, he added.
The next steps depend on severity. According to Dr. Simpson, severity is defined less by the extent of skin involvement at the baseline examination than the speed at which symptoms return.
For those with only mild symptoms after an extended period of clearing, moisturizer might be sufficient to prevent a significant relapse. For children with a more rapid relapse, it will be necessary to reintroduce topical steroid either every day, every other day, or twice per week.
Whether with moisturizer or with topical steroids, the soak-and-smear technique has now been validated in a recently published crossover randomized trial.
In the trial, children aged 6 months to 11 years, with moderate to severe AD, were randomized to a twice-daily bath, called the “wet method,” versus a twice-weekly bath, called the “dry method.” Both groups received a cleanser and moisturizer along with a low-potency topical steroid as needed.
After 2 weeks, the 40 evaluable patients were crossed over to the opposite bathing technique. The wet, or soak-and-smear approach, was associated with a highly significant reduction in the primary endpoint of SCORing Atopic Dermatitis (SCORAD) index, compared with the dry method (95% confidence interval, 14.9-27.6; P less than .0001). In a secondary analysis, this translated into a 30% relative reduction in favor of the wet method.
In addition, there was improvement in a caregiver assessment of the Atopic Dermatitis Quickscore (ADQ). These data show that “twice-daily baths with topical steroids and moisturizer can help in more moderate to severe population,” said Dr. Simpson, who noted that he has participated in open-label studies with the same soak-and-smear technique that have produced similar results.
Once children are clear, Dr. Simpson recommends a maintenance strategy individualized for severity. In many cases, this will involve moisturizers applied after the bath, supplemented intermittently, such as once or twice per week, with topical steroids. However, if parents find themselves resorting to daily steroids to maintain control, “that’s when you incorporate the TCIs [topical calcineurin inhibitors].”
TCIs “can help you stay at twice-per-week topical steroids,” Dr. Simpson said at the meeting, jointly presented by the University of Louisville and Global Academy for Medical Education.
TCIs also help patients avoid steroid withdrawal, a particularly common phenomenon when topical steroids are applied repeatedly to the face. He recommended a proactive approach. By applying TCIs to areas where skin lesions frequently recur, such as the eyelids, flares can often be prevented.
Repeated applications of TCIs “is perfectly safe and effective, and there are many studies that show proactive treatment is very effective and can prevent you from having to use too much topical steroids” or move to a systemic steroid, Dr. Simpson said.
These steps have been highly effective for sustained control even in challenging cases of AD, but he emphasized the importance of explaining the rationale to parents and eliciting their adherence to these treatment steps. Writing out the instructions will reduce confusion and help parents keep their children clear, he added.
Lawrence F. Eichenfield, MD, professor of pediatrics and dermatology at the University of California, San Diego, agreed that this recently published crossover trial has been helpful in counseling parents about how to manage AD in their children.
“Many times, pediatricians tell parents to avoid bathing because they feel that bathing will dry out the skin,” he said. The crossover study, by showing better control of AD with frequent bathing, dispels that notion, although he is not convinced that bathing at this frequency is necessary.
“I have not advised anyone to do twice-daily bathing, with rare exceptions, on the basis on this study, but, basically, I think that whether people do daily bathing or every other day bathing, it is pretty reasonable that bathing might help as long as they are applying moisturizer immediately afterward,” he said.
Dr. Simpson reports financial relationships with AbbVie, Celgene Dermira, Genentech, GlaxoSmithKline, Incyte, Lilly, Medimmune, Pfizer, Regeneron/Sanofi, and Tioga.
This publication and Global Academy for Medical Education are owned by the same parent company.
The most effective initial step for clearing atopic dermatitis in infants and young children involves daily bathing, followed by immediate application of a moisturizer, topical steroid, or both, according to an expert speaking at the virtual annual Coastal Dermatology Symposium.
“If they are really severe, you can do it twice-daily, but there are several studies that show there is not a huge benefit of twice-daily over once-daily,” said Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland.
He called this technique “soak-and-smear.” The “smear” is performed immediately after the bath when the skin is still damp, he said. When clearing is the goal, and the child has moderate to severe atopic dermatitis (AD), 0.1% triamcinolone or a similar medium potency topical steroid can be applied, and after clearing, the steroid can be switched for a moisturizer, according to Dr. Simpson.
Rather than restricting application to areas of greatest skin involvement, “put it all over,” he advised.
The clearing regimen should be continued “for a couple of more days” after the lesions have resolved, with a return visit in about a week to confirm clearing and reinforce the next steps for keeping patients clear, he added.
The next steps depend on severity. According to Dr. Simpson, severity is defined less by the extent of skin involvement at the baseline examination than the speed at which symptoms return.
For those with only mild symptoms after an extended period of clearing, moisturizer might be sufficient to prevent a significant relapse. For children with a more rapid relapse, it will be necessary to reintroduce topical steroid either every day, every other day, or twice per week.
Whether with moisturizer or with topical steroids, the soak-and-smear technique has now been validated in a recently published crossover randomized trial.
In the trial, children aged 6 months to 11 years, with moderate to severe AD, were randomized to a twice-daily bath, called the “wet method,” versus a twice-weekly bath, called the “dry method.” Both groups received a cleanser and moisturizer along with a low-potency topical steroid as needed.
After 2 weeks, the 40 evaluable patients were crossed over to the opposite bathing technique. The wet, or soak-and-smear approach, was associated with a highly significant reduction in the primary endpoint of SCORing Atopic Dermatitis (SCORAD) index, compared with the dry method (95% confidence interval, 14.9-27.6; P less than .0001). In a secondary analysis, this translated into a 30% relative reduction in favor of the wet method.
In addition, there was improvement in a caregiver assessment of the Atopic Dermatitis Quickscore (ADQ). These data show that “twice-daily baths with topical steroids and moisturizer can help in more moderate to severe population,” said Dr. Simpson, who noted that he has participated in open-label studies with the same soak-and-smear technique that have produced similar results.
Once children are clear, Dr. Simpson recommends a maintenance strategy individualized for severity. In many cases, this will involve moisturizers applied after the bath, supplemented intermittently, such as once or twice per week, with topical steroids. However, if parents find themselves resorting to daily steroids to maintain control, “that’s when you incorporate the TCIs [topical calcineurin inhibitors].”
TCIs “can help you stay at twice-per-week topical steroids,” Dr. Simpson said at the meeting, jointly presented by the University of Louisville and Global Academy for Medical Education.
TCIs also help patients avoid steroid withdrawal, a particularly common phenomenon when topical steroids are applied repeatedly to the face. He recommended a proactive approach. By applying TCIs to areas where skin lesions frequently recur, such as the eyelids, flares can often be prevented.
Repeated applications of TCIs “is perfectly safe and effective, and there are many studies that show proactive treatment is very effective and can prevent you from having to use too much topical steroids” or move to a systemic steroid, Dr. Simpson said.
These steps have been highly effective for sustained control even in challenging cases of AD, but he emphasized the importance of explaining the rationale to parents and eliciting their adherence to these treatment steps. Writing out the instructions will reduce confusion and help parents keep their children clear, he added.
Lawrence F. Eichenfield, MD, professor of pediatrics and dermatology at the University of California, San Diego, agreed that this recently published crossover trial has been helpful in counseling parents about how to manage AD in their children.
“Many times, pediatricians tell parents to avoid bathing because they feel that bathing will dry out the skin,” he said. The crossover study, by showing better control of AD with frequent bathing, dispels that notion, although he is not convinced that bathing at this frequency is necessary.
“I have not advised anyone to do twice-daily bathing, with rare exceptions, on the basis on this study, but, basically, I think that whether people do daily bathing or every other day bathing, it is pretty reasonable that bathing might help as long as they are applying moisturizer immediately afterward,” he said.
Dr. Simpson reports financial relationships with AbbVie, Celgene Dermira, Genentech, GlaxoSmithKline, Incyte, Lilly, Medimmune, Pfizer, Regeneron/Sanofi, and Tioga.
This publication and Global Academy for Medical Education are owned by the same parent company.
FROM COASTAL DERM
Orbital Granuloma Formation Following Autoinjection of Paraffin Oil: Management Considerations
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.


After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.
After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.
After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
Practice Points
- The initial presentation of a foreign-body granulomatous process in a patient with surreptitious use of nonmedical filler can mimic infection; thus, careful history and diagnostic measures are paramount.
- Treatment of paraffin oil granuloma can be multifactorial and involves supportive care, systemic anti-inflammatory medications, time, and surgery.
- When a paraffin granuloma involves the orbital region, particular care is required to avoid long-term complications including cicatricial lagophthalmos, ectropion, or retractions, which can be mitigated with the help of oculoplastic surgery.

Tylosis in a Patient With Howel-Evans Syndrome: Management With Acitretin
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
Practice Points
- Keratoderma can be especially painful for patients and can have a great impact on their quality of life. For these patients, acitretin should be considered when topical therapies have failed.
- Howel-Evans syndrome is an autosomal-dominant condition that predominantly presents with plantar keratoderma and has a high risk for esophageal cancer.
Medicare fines half of hospitals for readmitting too many patients
Nearly half the nation’s hospitals, many of which are still wrestling with the financial fallout of the unexpected coronavirus, will get lower payments for all Medicare patients because of their history of readmitting patients, federal records show.
The penalties are the ninth annual round of the Hospital Readmissions Reduction Program created as part of the Affordable Care Act’s broader effort to improve quality and lower costs. The latest penalties are calculated using each hospital case history between July 2016 and June 2019, so the flood of coronavirus patients that have swamped hospitals this year were not included.
The Centers for Medicare & Medicaid Services announced in September it may suspend the penalty program in the future if the chaos surrounding the pandemic, including the spring’s moratorium on elective surgeries, makes it too difficult to assess hospital performance.
For this year, the penalties remain in effect. Retroactive to the federal fiscal year that began Oct. 1, Medicare will lower a year’s worth of payments to 2,545 hospitals, the data show. The average reduction is 0.69%, with 613 hospitals receiving a penalty of 1% or more.
Out of 5,267 hospitals in the country, Congress has exempted 2,176 from the threat of penalties, either because they are critical access hospitals – defined as the only inpatient facility in an area – or hospitals that specialize in psychiatric patients, children, veterans, rehabilitation or long-term care. Of the 3,080 hospitals CMS evaluated, 83% received a penalty.
The number and severity of penalties were comparable to those of recent years, although the number of hospitals receiving the maximum penalty of 3% dropped from 56 to 39. Because the penalties are applied to new admission payments, the total dollar amount each hospital will lose will not be known until after the fiscal year ends on July 30.
“It’s unfortunate that hospitals will face readmission penalties in fiscal year 2021,” said Akin Demehin, director of policy at the American Hospital Association. “Given the financial strain that hospitals are under, every dollar counts, and the impact of any penalty is significant.”
The penalties are based on readmissions of Medicare patients who initially came to the hospital with diagnoses of congestive heart failure, heart attack, pneumonia, chronic obstructive pulmonary disease, hip or knee replacement, or coronary artery bypass graft surgery. Medicare counts as a readmission any of those patients who ended up back in any hospital within 30 days of discharge, except for planned returns like a second phase of surgery.
A hospital will be penalized if its readmission rate is higher than expected given the national trends in any one of those categories.
The industry has disapproved of the program since its inception, complaining the measures aren’t precise and it unfairly punishes hospitals that treat low-income patients, who often don’t have the resources to ensure their recoveries are successful.
Michael Millenson, a health quality consultant who focuses on patient safety, said the penalties are a useful but imperfect mechanism to push hospitals to improve their care. The designers of the penalty system envisioned it as a way to neutralize the economic benefit hospitals get from readmitted patients under Medicare’s fee-for-service payment model, as they are otherwise paid for two stays instead of just one.
“Every industry complains the penalties are too harsh,” he said. “if you’re going to tell me we don’t need any economic incentives to do the right thing because we’re always doing the right thing – that’s not true.”
KHN (Kaiser Health News) is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
Nearly half the nation’s hospitals, many of which are still wrestling with the financial fallout of the unexpected coronavirus, will get lower payments for all Medicare patients because of their history of readmitting patients, federal records show.
The penalties are the ninth annual round of the Hospital Readmissions Reduction Program created as part of the Affordable Care Act’s broader effort to improve quality and lower costs. The latest penalties are calculated using each hospital case history between July 2016 and June 2019, so the flood of coronavirus patients that have swamped hospitals this year were not included.
The Centers for Medicare & Medicaid Services announced in September it may suspend the penalty program in the future if the chaos surrounding the pandemic, including the spring’s moratorium on elective surgeries, makes it too difficult to assess hospital performance.
For this year, the penalties remain in effect. Retroactive to the federal fiscal year that began Oct. 1, Medicare will lower a year’s worth of payments to 2,545 hospitals, the data show. The average reduction is 0.69%, with 613 hospitals receiving a penalty of 1% or more.
Out of 5,267 hospitals in the country, Congress has exempted 2,176 from the threat of penalties, either because they are critical access hospitals – defined as the only inpatient facility in an area – or hospitals that specialize in psychiatric patients, children, veterans, rehabilitation or long-term care. Of the 3,080 hospitals CMS evaluated, 83% received a penalty.
The number and severity of penalties were comparable to those of recent years, although the number of hospitals receiving the maximum penalty of 3% dropped from 56 to 39. Because the penalties are applied to new admission payments, the total dollar amount each hospital will lose will not be known until after the fiscal year ends on July 30.
“It’s unfortunate that hospitals will face readmission penalties in fiscal year 2021,” said Akin Demehin, director of policy at the American Hospital Association. “Given the financial strain that hospitals are under, every dollar counts, and the impact of any penalty is significant.”
The penalties are based on readmissions of Medicare patients who initially came to the hospital with diagnoses of congestive heart failure, heart attack, pneumonia, chronic obstructive pulmonary disease, hip or knee replacement, or coronary artery bypass graft surgery. Medicare counts as a readmission any of those patients who ended up back in any hospital within 30 days of discharge, except for planned returns like a second phase of surgery.
A hospital will be penalized if its readmission rate is higher than expected given the national trends in any one of those categories.
The industry has disapproved of the program since its inception, complaining the measures aren’t precise and it unfairly punishes hospitals that treat low-income patients, who often don’t have the resources to ensure their recoveries are successful.
Michael Millenson, a health quality consultant who focuses on patient safety, said the penalties are a useful but imperfect mechanism to push hospitals to improve their care. The designers of the penalty system envisioned it as a way to neutralize the economic benefit hospitals get from readmitted patients under Medicare’s fee-for-service payment model, as they are otherwise paid for two stays instead of just one.
“Every industry complains the penalties are too harsh,” he said. “if you’re going to tell me we don’t need any economic incentives to do the right thing because we’re always doing the right thing – that’s not true.”
KHN (Kaiser Health News) is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
Nearly half the nation’s hospitals, many of which are still wrestling with the financial fallout of the unexpected coronavirus, will get lower payments for all Medicare patients because of their history of readmitting patients, federal records show.
The penalties are the ninth annual round of the Hospital Readmissions Reduction Program created as part of the Affordable Care Act’s broader effort to improve quality and lower costs. The latest penalties are calculated using each hospital case history between July 2016 and June 2019, so the flood of coronavirus patients that have swamped hospitals this year were not included.
The Centers for Medicare & Medicaid Services announced in September it may suspend the penalty program in the future if the chaos surrounding the pandemic, including the spring’s moratorium on elective surgeries, makes it too difficult to assess hospital performance.
For this year, the penalties remain in effect. Retroactive to the federal fiscal year that began Oct. 1, Medicare will lower a year’s worth of payments to 2,545 hospitals, the data show. The average reduction is 0.69%, with 613 hospitals receiving a penalty of 1% or more.
Out of 5,267 hospitals in the country, Congress has exempted 2,176 from the threat of penalties, either because they are critical access hospitals – defined as the only inpatient facility in an area – or hospitals that specialize in psychiatric patients, children, veterans, rehabilitation or long-term care. Of the 3,080 hospitals CMS evaluated, 83% received a penalty.
The number and severity of penalties were comparable to those of recent years, although the number of hospitals receiving the maximum penalty of 3% dropped from 56 to 39. Because the penalties are applied to new admission payments, the total dollar amount each hospital will lose will not be known until after the fiscal year ends on July 30.
“It’s unfortunate that hospitals will face readmission penalties in fiscal year 2021,” said Akin Demehin, director of policy at the American Hospital Association. “Given the financial strain that hospitals are under, every dollar counts, and the impact of any penalty is significant.”
The penalties are based on readmissions of Medicare patients who initially came to the hospital with diagnoses of congestive heart failure, heart attack, pneumonia, chronic obstructive pulmonary disease, hip or knee replacement, or coronary artery bypass graft surgery. Medicare counts as a readmission any of those patients who ended up back in any hospital within 30 days of discharge, except for planned returns like a second phase of surgery.
A hospital will be penalized if its readmission rate is higher than expected given the national trends in any one of those categories.
The industry has disapproved of the program since its inception, complaining the measures aren’t precise and it unfairly punishes hospitals that treat low-income patients, who often don’t have the resources to ensure their recoveries are successful.
Michael Millenson, a health quality consultant who focuses on patient safety, said the penalties are a useful but imperfect mechanism to push hospitals to improve their care. The designers of the penalty system envisioned it as a way to neutralize the economic benefit hospitals get from readmitted patients under Medicare’s fee-for-service payment model, as they are otherwise paid for two stays instead of just one.
“Every industry complains the penalties are too harsh,” he said. “if you’re going to tell me we don’t need any economic incentives to do the right thing because we’re always doing the right thing – that’s not true.”
KHN (Kaiser Health News) is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
Blood test for Alzheimer’s disease comes to the clinic
, according to C2N Diagnostics, the company behind the test’s development. The availability of the noninvasive, easily administered test is being called a milestone in the early detection and diagnosis of Alzheimer’s disease.
The blood test “introduces a new option for patients, families, and the medical community that have eagerly awaited innovative tools to address Alzheimer’s troubling problems,” Joel B. Braunstein, MD, MBA, CEO of C2N Diagnostics, said in a press release.
“This is really an important advance,” said Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation (ADDF), which partially funded the development of the test, in a separate press release.
“You can now walk into your doctor’s office to get a blood test to help detect Alzheimer’s disease,” said Dr. Fillit. “This test answers a critical need for less costly and accessible diagnostic testing in memory and dementia care.”
A word of caution
However, Maria C. Carrillo, PhD, chief science officer, Alzheimer’s Association, highlighted the need for caution. The test is “very new,” experts have only “limited information” about it, and it is only available by prescription from a healthcare provider for patients with cognitive impairment, said Dr. Carrillo.
“The test is not [Food and Drug Administration] approved and it does not, on its own, diagnose Alzheimer’s disease,” added Dr. Carrillo. “Without FDA review, healthcare providers lack the agency’s guidance for how to use it when making decisions about a person’s health or treatment.”
Dr. Carrillo also noted that the test has only been studied in a limited number of individuals and that few data are available regarding underrepresented populations.
“As a result, it is not clear how accurate or generalizable the results are for all individuals and populations,” she noted.
Another factor to consider, said Dr. Carrillo, is that the test is not covered by insurance, including Medicare and Medicaid.
How it works
The test (PrecivityAD) is for use in patients with cognitive impairment. It requires a very small blood sample – as little as a teaspoon – from the patient’s forearm. The physician sends the sample to C2N Diagnostic’s specialized laboratory, where it is analyzed using mass spectrometry to measure concentrations of amyloid beta 42 and 40 and to detect the presence of apolipoprotein E isoforms.
The lab report, which is sent to the patient’s physician, details biomarker levels and provides an overall combined score, known as the Amyloid Probability Score, to assess the likelihood of low, intermediate, or high levels of amyloid plaque in the brain.
The company reports that, on the basis of data from 686 patients older than 60 years who had subjective cognitive impairment or dementia, the test correctly identified brain amyloid plaque status, as determined by quantitative amyloid positron-emission tomography (PET) scans, in 86% of the patients. In the analysis, the area under the curve for the receiver operating characteristic was 0.88.
The company notes that the test, the results of which require interpretation by a health care provider, is an important new tool to aid physicians in the evaluation process.
The new blood test is currently available in 45 states, the District of Columbia, and Puerto Rico.
C2N Diagnostics is moving ahead with development of a brain health panel to detect multiple blood-based markers for Alzheimer’s disease to aid in disease staging, treatment monitoring, and differential diagnosis.
The ADDF believes the path to approval of treatments of Alzheimer’s disease starts with a better diagnosis, Dr. Fillit said in his organization’s press release.
“Investing in biomarker research has been a core goal for the ADDF because reliable, accessible, and affordable biomarkers for Alzheimer’s diagnosis are critical to our ability to find drugs to prevent, slow, and even cure the disease. Our funding helped bring the first PET scan to market and now has helped bring the first blood test to market,” he said.
In addition to the ADDF, the National Institutes of Health, the GHR Foundation, and the BrightFocus Foundation contributed funding for the development of the amyloid blood test.
A version of this article originally appeared on Medscape.com.
, according to C2N Diagnostics, the company behind the test’s development. The availability of the noninvasive, easily administered test is being called a milestone in the early detection and diagnosis of Alzheimer’s disease.
The blood test “introduces a new option for patients, families, and the medical community that have eagerly awaited innovative tools to address Alzheimer’s troubling problems,” Joel B. Braunstein, MD, MBA, CEO of C2N Diagnostics, said in a press release.
“This is really an important advance,” said Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation (ADDF), which partially funded the development of the test, in a separate press release.
“You can now walk into your doctor’s office to get a blood test to help detect Alzheimer’s disease,” said Dr. Fillit. “This test answers a critical need for less costly and accessible diagnostic testing in memory and dementia care.”
A word of caution
However, Maria C. Carrillo, PhD, chief science officer, Alzheimer’s Association, highlighted the need for caution. The test is “very new,” experts have only “limited information” about it, and it is only available by prescription from a healthcare provider for patients with cognitive impairment, said Dr. Carrillo.
“The test is not [Food and Drug Administration] approved and it does not, on its own, diagnose Alzheimer’s disease,” added Dr. Carrillo. “Without FDA review, healthcare providers lack the agency’s guidance for how to use it when making decisions about a person’s health or treatment.”
Dr. Carrillo also noted that the test has only been studied in a limited number of individuals and that few data are available regarding underrepresented populations.
“As a result, it is not clear how accurate or generalizable the results are for all individuals and populations,” she noted.
Another factor to consider, said Dr. Carrillo, is that the test is not covered by insurance, including Medicare and Medicaid.
How it works
The test (PrecivityAD) is for use in patients with cognitive impairment. It requires a very small blood sample – as little as a teaspoon – from the patient’s forearm. The physician sends the sample to C2N Diagnostic’s specialized laboratory, where it is analyzed using mass spectrometry to measure concentrations of amyloid beta 42 and 40 and to detect the presence of apolipoprotein E isoforms.
The lab report, which is sent to the patient’s physician, details biomarker levels and provides an overall combined score, known as the Amyloid Probability Score, to assess the likelihood of low, intermediate, or high levels of amyloid plaque in the brain.
The company reports that, on the basis of data from 686 patients older than 60 years who had subjective cognitive impairment or dementia, the test correctly identified brain amyloid plaque status, as determined by quantitative amyloid positron-emission tomography (PET) scans, in 86% of the patients. In the analysis, the area under the curve for the receiver operating characteristic was 0.88.
The company notes that the test, the results of which require interpretation by a health care provider, is an important new tool to aid physicians in the evaluation process.
The new blood test is currently available in 45 states, the District of Columbia, and Puerto Rico.
C2N Diagnostics is moving ahead with development of a brain health panel to detect multiple blood-based markers for Alzheimer’s disease to aid in disease staging, treatment monitoring, and differential diagnosis.
The ADDF believes the path to approval of treatments of Alzheimer’s disease starts with a better diagnosis, Dr. Fillit said in his organization’s press release.
“Investing in biomarker research has been a core goal for the ADDF because reliable, accessible, and affordable biomarkers for Alzheimer’s diagnosis are critical to our ability to find drugs to prevent, slow, and even cure the disease. Our funding helped bring the first PET scan to market and now has helped bring the first blood test to market,” he said.
In addition to the ADDF, the National Institutes of Health, the GHR Foundation, and the BrightFocus Foundation contributed funding for the development of the amyloid blood test.
A version of this article originally appeared on Medscape.com.
, according to C2N Diagnostics, the company behind the test’s development. The availability of the noninvasive, easily administered test is being called a milestone in the early detection and diagnosis of Alzheimer’s disease.
The blood test “introduces a new option for patients, families, and the medical community that have eagerly awaited innovative tools to address Alzheimer’s troubling problems,” Joel B. Braunstein, MD, MBA, CEO of C2N Diagnostics, said in a press release.
“This is really an important advance,” said Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation (ADDF), which partially funded the development of the test, in a separate press release.
“You can now walk into your doctor’s office to get a blood test to help detect Alzheimer’s disease,” said Dr. Fillit. “This test answers a critical need for less costly and accessible diagnostic testing in memory and dementia care.”
A word of caution
However, Maria C. Carrillo, PhD, chief science officer, Alzheimer’s Association, highlighted the need for caution. The test is “very new,” experts have only “limited information” about it, and it is only available by prescription from a healthcare provider for patients with cognitive impairment, said Dr. Carrillo.
“The test is not [Food and Drug Administration] approved and it does not, on its own, diagnose Alzheimer’s disease,” added Dr. Carrillo. “Without FDA review, healthcare providers lack the agency’s guidance for how to use it when making decisions about a person’s health or treatment.”
Dr. Carrillo also noted that the test has only been studied in a limited number of individuals and that few data are available regarding underrepresented populations.
“As a result, it is not clear how accurate or generalizable the results are for all individuals and populations,” she noted.
Another factor to consider, said Dr. Carrillo, is that the test is not covered by insurance, including Medicare and Medicaid.
How it works
The test (PrecivityAD) is for use in patients with cognitive impairment. It requires a very small blood sample – as little as a teaspoon – from the patient’s forearm. The physician sends the sample to C2N Diagnostic’s specialized laboratory, where it is analyzed using mass spectrometry to measure concentrations of amyloid beta 42 and 40 and to detect the presence of apolipoprotein E isoforms.
The lab report, which is sent to the patient’s physician, details biomarker levels and provides an overall combined score, known as the Amyloid Probability Score, to assess the likelihood of low, intermediate, or high levels of amyloid plaque in the brain.
The company reports that, on the basis of data from 686 patients older than 60 years who had subjective cognitive impairment or dementia, the test correctly identified brain amyloid plaque status, as determined by quantitative amyloid positron-emission tomography (PET) scans, in 86% of the patients. In the analysis, the area under the curve for the receiver operating characteristic was 0.88.
The company notes that the test, the results of which require interpretation by a health care provider, is an important new tool to aid physicians in the evaluation process.
The new blood test is currently available in 45 states, the District of Columbia, and Puerto Rico.
C2N Diagnostics is moving ahead with development of a brain health panel to detect multiple blood-based markers for Alzheimer’s disease to aid in disease staging, treatment monitoring, and differential diagnosis.
The ADDF believes the path to approval of treatments of Alzheimer’s disease starts with a better diagnosis, Dr. Fillit said in his organization’s press release.
“Investing in biomarker research has been a core goal for the ADDF because reliable, accessible, and affordable biomarkers for Alzheimer’s diagnosis are critical to our ability to find drugs to prevent, slow, and even cure the disease. Our funding helped bring the first PET scan to market and now has helped bring the first blood test to market,” he said.
In addition to the ADDF, the National Institutes of Health, the GHR Foundation, and the BrightFocus Foundation contributed funding for the development of the amyloid blood test.
A version of this article originally appeared on Medscape.com.
How cannabis-based therapeutics could help fight COVID inflammation
Plagued by false starts, a few dashed hopes, but with perhaps a glimmer of light on the horizon, the race to find an effective treatment for COVID-19 continues. At last count, more than 300 treatments and 200 vaccines were in preclinical or clinical development (not to mention the numerous existing agents that are being evaluated for repurposing).
There is also a renewed interest in cannabinoid therapeutics — in particular, the nonpsychoactive agent cannabidiol (CBD) and the prospect of its modulating inflammatory and other disease-associated clinical indices, including SARS-CoV-2–induced viral load, hyperinflammation, the cytokine storm, and acute respiratory distress syndrome (ARDS).
Long hobbled by regulatory, political, and financial barriers, CBD’s potential ability to knock back COVID-19–related inflammation might just open doors that have been closed for years to CBD researchers.
Why CBD and why now?
CBD and the resulting therapeutics have been plagued by a complicated association with recreational cannabis use. It’s been just 2 years since CBD-based therapeutics moved into mainstream medicine — the US Food and Drug Administration (FDA) approved Epidiolex oral solution for the treatment of Lennox-Gastaut syndrome and Dravet syndrome, and in August, the FDA approved it for tuberous sclerosis complex.
CBD’s mechanism of action has not been fully elucidated, but on the basis of its role in immune responses — well described in research spanning more than two decades — it›s not surprising that cannabinoid researchers have thrown their hats into the COVID-19 drug development ring.
The anti-inflammatory potential of CBD is substantial and appears to be related to the fact that it shares 20 protein targets common to inflammation-related pathways, Jenny Wilkerson, PhD, research assistant professor at the University of Florida School of Pharmacy, Gainesville, Florida, explained to Medscape Medical News.
Among the various trials that are currently recruiting or are underway is one that is slated for completion this fall. CANDIDATE (Cannabidiol for COVID-19 Patients With Mild-to-Moderate COVID-19) is a randomized, controlled, double-blind study led by Brazilian researchers at the University of São Paulo. The study, which began recruitment this past August, enrolled 100 patients, 50 in the active treatment group (who received capsulated CBD 300 mg daily for 14 days plus pharmacologic therapy [antipyretics] and clinical measures) and 50 who received placebo.
The primary outcome is intended to help clarify the potential role of oral CBD for preventing COVID-19 disease progression, modifying disease-associated clinical indices, and modulating inflammatory parameters, such as the cytokine storm, according to lead investigator Jose Alexandre de Souza Crippa, MD, PhD, professor of neuropsychology at the Ribeirao Preto Medical School at the University of São Paulo in Brazil, in the description of the study on clinicaltrials.gov. Crippa declined to provide any additional information about the trial in an email to Medscape Medical News.
Calming or preventing the storm
While Crippa and colleagues wrap up their CBD trial in South America, several North American and Canadian researchers are seeking to clarify and address one of the most therapeutically challenging aspects of SARS-CoV-2 infection — the lung macrophage–orchestrated hyperinflammatory response.
Although hyperinflammation is not unique to SARS-CoV-2 infection, disease severity and COVID-19–related mortality have been linked to this rapid and prolonged surge of inflammatory cytokines (eg, interleukin 6 [IL-6], IL-10, tumor necrosis factors [TNF], and chemokines) and the cytokine storm.
“When you stimulate CB2 receptors (involved in fighting inflammation), you get a release of the same inflammatory cytokines that are involved in COVID,” Cecilia Costiniuk, MD, associate professor and researcher at the Research Institute of the McGill University Health Center, Montreal, Canada, told Medscape Medical News.
“So, if you can act on this receptor, you might be able to reduce the release of those damaging cytokines that are causing ARDS, lung damage, etc,” she explained. Targeting these inflammatory mediators has been a key strategy in research aimed at reducing COVID-19 severity and related mortality, which is where CBD comes into play.
“CBD is a very powerful immune regulator. It keeps the [immune] engine on, but it doesn’t push the gas pedal, and it doesn’t push the brake completely,” Babak Baban, PhD, professor and immunologist at the Dental College of Georgia at Augusta University, told Medscape Medical News.
To explore the effectiveness of CBD in reducing hyperactivated inflammatory reactions, Baban and colleagues examined the potential of CBD to ameliorate ARDS in a murine model. The group divided wild-type male mice into sham, control, and treatment groups.
The sham group received intranasal phosphate buffered saline; the treatment and control groups received a polyriboinosinic:polycytidylic acid (poly I:C) double-stranded RNA analogue (100 mcg daily for 3 days) to simulate the cytokine storm and clinical ARDS symptoms.
Following the second poly I:C dose, the treatment group received CBD 5 mg/kg intraperitoneally every other day for 6 days. The mice were sacrificed on day 8.
The study results, published in July in Cannabis and Cannabinoid Research, first confirmed that the poly I:C model simulated the cytokine storm in ARDS, reducing blood oxygen saturation by as much as 10% (from ±81.6% to ±72.2%).
Intraperitoneally administered CBD appeared to reverse these ARDS-like trends. “We observed a significant improvement in severe lymphopenia, a mild decline in the ratio of neutrophils to T cells, and significant reductions in levels of [inflammatory and immune factors] IL-6, IFN-gamma [interferon gamma], and in TNF-alpha after the second CBD dose,” Baban said.
There was also a marked downregulation in infiltrating neutrophils and macrophages in the lung, leading to partial restoration of lung morphology and structure. The investigators write that this suggests “a counter inflammatory role for CBD to limit ARDS progression.”
Additional findings from a follow-up study published in mid-October “provide strong data that CBD may partially assert its beneficial and protective impact through its regulation of the apelin peptide,” wrote Baban in an email to Medscape Medical News.
“Apelin may also be a reliable biomarker for early diagnosis of ARDS in general, and in COVID-19 in particular,” he wrote.
Questions remain concerning dose response and whether CBD alone or in combination with other phytocannabinoids is more effective for treating COVID-19. Timing is likewise unclear.
Baban explained that as a result of the biphasic nature of COVID-19, the “sweet spot” appears to be just before the innate immune response progresses into an inflammation-driven response and fibrotic lung damage occurs.
But Wilkerson isn’t as convinced. She said that as with a thermostat, the endocannabinoid system needs tweaking to get it in the right place, that is, to achieve immune homeostasis. The COVID cytokine storm is highly unpredictable, she added, saying, “Right now, the timing for controlling the COVID cytokine storm is really a moving target.”
Is safety a concern?
Safety questions are expected to arise, especially in relation to COVID-19. CBD is not risk free, and one size does not fit all. Human CBD studies report gastrointestinal and somnolent effects, as well as drug-drug interactions.
Findings from a recent systematic review of randomized, controlled CBD trials support overall tolerability, suggesting that serious adverse events are rare. Such events are believed to be related to drug-drug interactions rather than to CBD itself. On the flip side, it is nonintoxicating, and there does not appear to be potential for abuse.
“It’s generally well tolerated,” Wilkerson said. “There’ve now been several clinical trials in numerous patient population settings where basically the only time you really start to have issues is where you have patients on very select agents. But this is where a pharmacist would come into play.”
Costiniuk agreed: “Just because it’s cannabis, it doesn’t mean that there’s going to be strange or unusual effects; these people [ie, those with severe COVID-19] are in the hospital and monitored very closely.”
Delving into the weeds: What’s next?
Although non-COVID-19 cannabinoid researchers have encountered regulatory roadblocks, several research groups that have had the prescience to dive in at the right time are gaining momentum.
Baban’s team has connected with one of the nation’s few academic laboratories authorized to work with the SARS-CoV-2 virus and are awaiting protocol approval so that they can reproduce their research, this time using two CBD formulations (injectable and inhaled).
If findings are positive, they will move forward quickly to meet with the FDA, Baban said, adding that the team is also collaborating with two organizations to conduct human clinical trials in hopes of pushing up timing.
The initial article caught the eye of the World Health Organization, which included it in its global literature on the coronavirus resource section.
Israeli researchers have also been quite busy. InnoCan Pharma and Tel Aviv University are collaborating to explore the potential for CBD-loaded exosomes (minute extracellular particles that mediate intracellular communication, including via innate and adaptive immune responses). The group plans to use these loaded exosomes to target and facilitate recovery of COVID-19–damaged lung cells.
From a broader perspective, the prospects for harnessing cannabinoids for immune modulation will be more thoroughly explored in a special issue of Cannabis and Cannabinoid Research, which has extended its current call for papers, studies, abstracts, and conference proceedings until the end of December.
Like many of the therapeutic strategies under investigation for the treatment of COVID-19, studies in CBD may continue to raise more questions than answers.
Still, Wilkerson is optimistic. “Taken together, these studies along with countless others suggest that the complex pharmacophore of Cannabis sativa may hold therapeutic utility to treat lung inflammation, such as what is seen in a COVID-19 cytokine storm,» she told Medscape Medical News. “I’m very excited to see what comes out of the research.”
Baban, Wilkerson, and Costiniuk have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Plagued by false starts, a few dashed hopes, but with perhaps a glimmer of light on the horizon, the race to find an effective treatment for COVID-19 continues. At last count, more than 300 treatments and 200 vaccines were in preclinical or clinical development (not to mention the numerous existing agents that are being evaluated for repurposing).
There is also a renewed interest in cannabinoid therapeutics — in particular, the nonpsychoactive agent cannabidiol (CBD) and the prospect of its modulating inflammatory and other disease-associated clinical indices, including SARS-CoV-2–induced viral load, hyperinflammation, the cytokine storm, and acute respiratory distress syndrome (ARDS).
Long hobbled by regulatory, political, and financial barriers, CBD’s potential ability to knock back COVID-19–related inflammation might just open doors that have been closed for years to CBD researchers.
Why CBD and why now?
CBD and the resulting therapeutics have been plagued by a complicated association with recreational cannabis use. It’s been just 2 years since CBD-based therapeutics moved into mainstream medicine — the US Food and Drug Administration (FDA) approved Epidiolex oral solution for the treatment of Lennox-Gastaut syndrome and Dravet syndrome, and in August, the FDA approved it for tuberous sclerosis complex.
CBD’s mechanism of action has not been fully elucidated, but on the basis of its role in immune responses — well described in research spanning more than two decades — it›s not surprising that cannabinoid researchers have thrown their hats into the COVID-19 drug development ring.
The anti-inflammatory potential of CBD is substantial and appears to be related to the fact that it shares 20 protein targets common to inflammation-related pathways, Jenny Wilkerson, PhD, research assistant professor at the University of Florida School of Pharmacy, Gainesville, Florida, explained to Medscape Medical News.
Among the various trials that are currently recruiting or are underway is one that is slated for completion this fall. CANDIDATE (Cannabidiol for COVID-19 Patients With Mild-to-Moderate COVID-19) is a randomized, controlled, double-blind study led by Brazilian researchers at the University of São Paulo. The study, which began recruitment this past August, enrolled 100 patients, 50 in the active treatment group (who received capsulated CBD 300 mg daily for 14 days plus pharmacologic therapy [antipyretics] and clinical measures) and 50 who received placebo.
The primary outcome is intended to help clarify the potential role of oral CBD for preventing COVID-19 disease progression, modifying disease-associated clinical indices, and modulating inflammatory parameters, such as the cytokine storm, according to lead investigator Jose Alexandre de Souza Crippa, MD, PhD, professor of neuropsychology at the Ribeirao Preto Medical School at the University of São Paulo in Brazil, in the description of the study on clinicaltrials.gov. Crippa declined to provide any additional information about the trial in an email to Medscape Medical News.
Calming or preventing the storm
While Crippa and colleagues wrap up their CBD trial in South America, several North American and Canadian researchers are seeking to clarify and address one of the most therapeutically challenging aspects of SARS-CoV-2 infection — the lung macrophage–orchestrated hyperinflammatory response.
Although hyperinflammation is not unique to SARS-CoV-2 infection, disease severity and COVID-19–related mortality have been linked to this rapid and prolonged surge of inflammatory cytokines (eg, interleukin 6 [IL-6], IL-10, tumor necrosis factors [TNF], and chemokines) and the cytokine storm.
“When you stimulate CB2 receptors (involved in fighting inflammation), you get a release of the same inflammatory cytokines that are involved in COVID,” Cecilia Costiniuk, MD, associate professor and researcher at the Research Institute of the McGill University Health Center, Montreal, Canada, told Medscape Medical News.
“So, if you can act on this receptor, you might be able to reduce the release of those damaging cytokines that are causing ARDS, lung damage, etc,” she explained. Targeting these inflammatory mediators has been a key strategy in research aimed at reducing COVID-19 severity and related mortality, which is where CBD comes into play.
“CBD is a very powerful immune regulator. It keeps the [immune] engine on, but it doesn’t push the gas pedal, and it doesn’t push the brake completely,” Babak Baban, PhD, professor and immunologist at the Dental College of Georgia at Augusta University, told Medscape Medical News.
To explore the effectiveness of CBD in reducing hyperactivated inflammatory reactions, Baban and colleagues examined the potential of CBD to ameliorate ARDS in a murine model. The group divided wild-type male mice into sham, control, and treatment groups.
The sham group received intranasal phosphate buffered saline; the treatment and control groups received a polyriboinosinic:polycytidylic acid (poly I:C) double-stranded RNA analogue (100 mcg daily for 3 days) to simulate the cytokine storm and clinical ARDS symptoms.
Following the second poly I:C dose, the treatment group received CBD 5 mg/kg intraperitoneally every other day for 6 days. The mice were sacrificed on day 8.
The study results, published in July in Cannabis and Cannabinoid Research, first confirmed that the poly I:C model simulated the cytokine storm in ARDS, reducing blood oxygen saturation by as much as 10% (from ±81.6% to ±72.2%).
Intraperitoneally administered CBD appeared to reverse these ARDS-like trends. “We observed a significant improvement in severe lymphopenia, a mild decline in the ratio of neutrophils to T cells, and significant reductions in levels of [inflammatory and immune factors] IL-6, IFN-gamma [interferon gamma], and in TNF-alpha after the second CBD dose,” Baban said.
There was also a marked downregulation in infiltrating neutrophils and macrophages in the lung, leading to partial restoration of lung morphology and structure. The investigators write that this suggests “a counter inflammatory role for CBD to limit ARDS progression.”
Additional findings from a follow-up study published in mid-October “provide strong data that CBD may partially assert its beneficial and protective impact through its regulation of the apelin peptide,” wrote Baban in an email to Medscape Medical News.
“Apelin may also be a reliable biomarker for early diagnosis of ARDS in general, and in COVID-19 in particular,” he wrote.
Questions remain concerning dose response and whether CBD alone or in combination with other phytocannabinoids is more effective for treating COVID-19. Timing is likewise unclear.
Baban explained that as a result of the biphasic nature of COVID-19, the “sweet spot” appears to be just before the innate immune response progresses into an inflammation-driven response and fibrotic lung damage occurs.
But Wilkerson isn’t as convinced. She said that as with a thermostat, the endocannabinoid system needs tweaking to get it in the right place, that is, to achieve immune homeostasis. The COVID cytokine storm is highly unpredictable, she added, saying, “Right now, the timing for controlling the COVID cytokine storm is really a moving target.”
Is safety a concern?
Safety questions are expected to arise, especially in relation to COVID-19. CBD is not risk free, and one size does not fit all. Human CBD studies report gastrointestinal and somnolent effects, as well as drug-drug interactions.
Findings from a recent systematic review of randomized, controlled CBD trials support overall tolerability, suggesting that serious adverse events are rare. Such events are believed to be related to drug-drug interactions rather than to CBD itself. On the flip side, it is nonintoxicating, and there does not appear to be potential for abuse.
“It’s generally well tolerated,” Wilkerson said. “There’ve now been several clinical trials in numerous patient population settings where basically the only time you really start to have issues is where you have patients on very select agents. But this is where a pharmacist would come into play.”
Costiniuk agreed: “Just because it’s cannabis, it doesn’t mean that there’s going to be strange or unusual effects; these people [ie, those with severe COVID-19] are in the hospital and monitored very closely.”
Delving into the weeds: What’s next?
Although non-COVID-19 cannabinoid researchers have encountered regulatory roadblocks, several research groups that have had the prescience to dive in at the right time are gaining momentum.
Baban’s team has connected with one of the nation’s few academic laboratories authorized to work with the SARS-CoV-2 virus and are awaiting protocol approval so that they can reproduce their research, this time using two CBD formulations (injectable and inhaled).
If findings are positive, they will move forward quickly to meet with the FDA, Baban said, adding that the team is also collaborating with two organizations to conduct human clinical trials in hopes of pushing up timing.
The initial article caught the eye of the World Health Organization, which included it in its global literature on the coronavirus resource section.
Israeli researchers have also been quite busy. InnoCan Pharma and Tel Aviv University are collaborating to explore the potential for CBD-loaded exosomes (minute extracellular particles that mediate intracellular communication, including via innate and adaptive immune responses). The group plans to use these loaded exosomes to target and facilitate recovery of COVID-19–damaged lung cells.
From a broader perspective, the prospects for harnessing cannabinoids for immune modulation will be more thoroughly explored in a special issue of Cannabis and Cannabinoid Research, which has extended its current call for papers, studies, abstracts, and conference proceedings until the end of December.
Like many of the therapeutic strategies under investigation for the treatment of COVID-19, studies in CBD may continue to raise more questions than answers.
Still, Wilkerson is optimistic. “Taken together, these studies along with countless others suggest that the complex pharmacophore of Cannabis sativa may hold therapeutic utility to treat lung inflammation, such as what is seen in a COVID-19 cytokine storm,» she told Medscape Medical News. “I’m very excited to see what comes out of the research.”
Baban, Wilkerson, and Costiniuk have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Plagued by false starts, a few dashed hopes, but with perhaps a glimmer of light on the horizon, the race to find an effective treatment for COVID-19 continues. At last count, more than 300 treatments and 200 vaccines were in preclinical or clinical development (not to mention the numerous existing agents that are being evaluated for repurposing).
There is also a renewed interest in cannabinoid therapeutics — in particular, the nonpsychoactive agent cannabidiol (CBD) and the prospect of its modulating inflammatory and other disease-associated clinical indices, including SARS-CoV-2–induced viral load, hyperinflammation, the cytokine storm, and acute respiratory distress syndrome (ARDS).
Long hobbled by regulatory, political, and financial barriers, CBD’s potential ability to knock back COVID-19–related inflammation might just open doors that have been closed for years to CBD researchers.
Why CBD and why now?
CBD and the resulting therapeutics have been plagued by a complicated association with recreational cannabis use. It’s been just 2 years since CBD-based therapeutics moved into mainstream medicine — the US Food and Drug Administration (FDA) approved Epidiolex oral solution for the treatment of Lennox-Gastaut syndrome and Dravet syndrome, and in August, the FDA approved it for tuberous sclerosis complex.
CBD’s mechanism of action has not been fully elucidated, but on the basis of its role in immune responses — well described in research spanning more than two decades — it›s not surprising that cannabinoid researchers have thrown their hats into the COVID-19 drug development ring.
The anti-inflammatory potential of CBD is substantial and appears to be related to the fact that it shares 20 protein targets common to inflammation-related pathways, Jenny Wilkerson, PhD, research assistant professor at the University of Florida School of Pharmacy, Gainesville, Florida, explained to Medscape Medical News.
Among the various trials that are currently recruiting or are underway is one that is slated for completion this fall. CANDIDATE (Cannabidiol for COVID-19 Patients With Mild-to-Moderate COVID-19) is a randomized, controlled, double-blind study led by Brazilian researchers at the University of São Paulo. The study, which began recruitment this past August, enrolled 100 patients, 50 in the active treatment group (who received capsulated CBD 300 mg daily for 14 days plus pharmacologic therapy [antipyretics] and clinical measures) and 50 who received placebo.
The primary outcome is intended to help clarify the potential role of oral CBD for preventing COVID-19 disease progression, modifying disease-associated clinical indices, and modulating inflammatory parameters, such as the cytokine storm, according to lead investigator Jose Alexandre de Souza Crippa, MD, PhD, professor of neuropsychology at the Ribeirao Preto Medical School at the University of São Paulo in Brazil, in the description of the study on clinicaltrials.gov. Crippa declined to provide any additional information about the trial in an email to Medscape Medical News.
Calming or preventing the storm
While Crippa and colleagues wrap up their CBD trial in South America, several North American and Canadian researchers are seeking to clarify and address one of the most therapeutically challenging aspects of SARS-CoV-2 infection — the lung macrophage–orchestrated hyperinflammatory response.
Although hyperinflammation is not unique to SARS-CoV-2 infection, disease severity and COVID-19–related mortality have been linked to this rapid and prolonged surge of inflammatory cytokines (eg, interleukin 6 [IL-6], IL-10, tumor necrosis factors [TNF], and chemokines) and the cytokine storm.
“When you stimulate CB2 receptors (involved in fighting inflammation), you get a release of the same inflammatory cytokines that are involved in COVID,” Cecilia Costiniuk, MD, associate professor and researcher at the Research Institute of the McGill University Health Center, Montreal, Canada, told Medscape Medical News.
“So, if you can act on this receptor, you might be able to reduce the release of those damaging cytokines that are causing ARDS, lung damage, etc,” she explained. Targeting these inflammatory mediators has been a key strategy in research aimed at reducing COVID-19 severity and related mortality, which is where CBD comes into play.
“CBD is a very powerful immune regulator. It keeps the [immune] engine on, but it doesn’t push the gas pedal, and it doesn’t push the brake completely,” Babak Baban, PhD, professor and immunologist at the Dental College of Georgia at Augusta University, told Medscape Medical News.
To explore the effectiveness of CBD in reducing hyperactivated inflammatory reactions, Baban and colleagues examined the potential of CBD to ameliorate ARDS in a murine model. The group divided wild-type male mice into sham, control, and treatment groups.
The sham group received intranasal phosphate buffered saline; the treatment and control groups received a polyriboinosinic:polycytidylic acid (poly I:C) double-stranded RNA analogue (100 mcg daily for 3 days) to simulate the cytokine storm and clinical ARDS symptoms.
Following the second poly I:C dose, the treatment group received CBD 5 mg/kg intraperitoneally every other day for 6 days. The mice were sacrificed on day 8.
The study results, published in July in Cannabis and Cannabinoid Research, first confirmed that the poly I:C model simulated the cytokine storm in ARDS, reducing blood oxygen saturation by as much as 10% (from ±81.6% to ±72.2%).
Intraperitoneally administered CBD appeared to reverse these ARDS-like trends. “We observed a significant improvement in severe lymphopenia, a mild decline in the ratio of neutrophils to T cells, and significant reductions in levels of [inflammatory and immune factors] IL-6, IFN-gamma [interferon gamma], and in TNF-alpha after the second CBD dose,” Baban said.
There was also a marked downregulation in infiltrating neutrophils and macrophages in the lung, leading to partial restoration of lung morphology and structure. The investigators write that this suggests “a counter inflammatory role for CBD to limit ARDS progression.”
Additional findings from a follow-up study published in mid-October “provide strong data that CBD may partially assert its beneficial and protective impact through its regulation of the apelin peptide,” wrote Baban in an email to Medscape Medical News.
“Apelin may also be a reliable biomarker for early diagnosis of ARDS in general, and in COVID-19 in particular,” he wrote.
Questions remain concerning dose response and whether CBD alone or in combination with other phytocannabinoids is more effective for treating COVID-19. Timing is likewise unclear.
Baban explained that as a result of the biphasic nature of COVID-19, the “sweet spot” appears to be just before the innate immune response progresses into an inflammation-driven response and fibrotic lung damage occurs.
But Wilkerson isn’t as convinced. She said that as with a thermostat, the endocannabinoid system needs tweaking to get it in the right place, that is, to achieve immune homeostasis. The COVID cytokine storm is highly unpredictable, she added, saying, “Right now, the timing for controlling the COVID cytokine storm is really a moving target.”
Is safety a concern?
Safety questions are expected to arise, especially in relation to COVID-19. CBD is not risk free, and one size does not fit all. Human CBD studies report gastrointestinal and somnolent effects, as well as drug-drug interactions.
Findings from a recent systematic review of randomized, controlled CBD trials support overall tolerability, suggesting that serious adverse events are rare. Such events are believed to be related to drug-drug interactions rather than to CBD itself. On the flip side, it is nonintoxicating, and there does not appear to be potential for abuse.
“It’s generally well tolerated,” Wilkerson said. “There’ve now been several clinical trials in numerous patient population settings where basically the only time you really start to have issues is where you have patients on very select agents. But this is where a pharmacist would come into play.”
Costiniuk agreed: “Just because it’s cannabis, it doesn’t mean that there’s going to be strange or unusual effects; these people [ie, those with severe COVID-19] are in the hospital and monitored very closely.”
Delving into the weeds: What’s next?
Although non-COVID-19 cannabinoid researchers have encountered regulatory roadblocks, several research groups that have had the prescience to dive in at the right time are gaining momentum.
Baban’s team has connected with one of the nation’s few academic laboratories authorized to work with the SARS-CoV-2 virus and are awaiting protocol approval so that they can reproduce their research, this time using two CBD formulations (injectable and inhaled).
If findings are positive, they will move forward quickly to meet with the FDA, Baban said, adding that the team is also collaborating with two organizations to conduct human clinical trials in hopes of pushing up timing.
The initial article caught the eye of the World Health Organization, which included it in its global literature on the coronavirus resource section.
Israeli researchers have also been quite busy. InnoCan Pharma and Tel Aviv University are collaborating to explore the potential for CBD-loaded exosomes (minute extracellular particles that mediate intracellular communication, including via innate and adaptive immune responses). The group plans to use these loaded exosomes to target and facilitate recovery of COVID-19–damaged lung cells.
From a broader perspective, the prospects for harnessing cannabinoids for immune modulation will be more thoroughly explored in a special issue of Cannabis and Cannabinoid Research, which has extended its current call for papers, studies, abstracts, and conference proceedings until the end of December.
Like many of the therapeutic strategies under investigation for the treatment of COVID-19, studies in CBD may continue to raise more questions than answers.
Still, Wilkerson is optimistic. “Taken together, these studies along with countless others suggest that the complex pharmacophore of Cannabis sativa may hold therapeutic utility to treat lung inflammation, such as what is seen in a COVID-19 cytokine storm,» she told Medscape Medical News. “I’m very excited to see what comes out of the research.”
Baban, Wilkerson, and Costiniuk have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Sublingual apomorphine alleviates off episodes in Parkinson’s disease
, long-term follow-up of a phase 3 study has shown. Besides the usual adverse effects with apomorphine, the sublingual film was associated with more oral adverse effects than seen with the injectable drug. However, it may have some advantages over subcutaneous apomorphine injections in terms of administration during off episodes.
The study was presented at the Movement Disorder Society 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
For example, the new formulation is more convenient than carrying an injection. It comes in a small, tear-open packet that contains a medication strip patients place under their tongues.
“When a patient is in the off state, depending on how off they are, they could have a little difficulty opening the strip [packet], but anyone can open the strip for them,” said lead author Rajesh Pahwa, MD, professor of neurology and chief of the Parkinson and Movement Disorder Division at the University of Kansas Medical Center in Kansas City. “On the other hand with the subcutaneous, they have to give the injection themselves and a stranger or someone is not going to help them with that.”
Open-label safety and efficacy study
The aims of this open-label, 48-week follow-up were to add new patients to assess safety and tolerability over the long term and to see if continued benefit from a previous 12-week double-blind study was still present at 1 year for patients in the earlier study.
This multicenter study (NCT02542696) included “rollover” patients (n = 78 for safety; n = 70 for efficacy) from the previous phase 2/3 double-blind trial, as well as new patients with no prior exposure to apomorphine sublingual film (n = 347 for safety; n = 275 for efficacy).
New patients experienced one or more off episodes per day with a daily off time of 2 hours or more per day while on stable doses of levodopa/carbidopa. All had clinically meaningful responses to levodopa/carbidopa and were judged by the investigator to be Stage 1-3 by modified Hoehn and Yahr scale rating during ON periods.
Rollover patients completed the prior study and had no major changes in their anti-Parkinson’s medications since then. Mouth cankers or sores were exclusion criteria for either group. New subjects could not have received subcutaneous apomorphine within 7 days of a screening visit.
The demographics and baseline characteristics of the new and rollover groups were similar (approximately 64 years; 65%-71% male; 96% White; 8.3-9.6 years since diagnosis; 3.9 to 4.1 off episodes/day, and total mean daily levodopa dose of 1120 to 1478 mg).
Assessing only the group of new patients, the investigators reported that 80% had a Hoehn and Yahr score of 2 or 2.5 when in the ON state and a Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III predose score of 41.8.
At the beginning of this study, patients in an off period received titrated doses of 10-35 mg of sublingual apomorphine in 5 mg increments during sequential office visits until they achieved a tolerable full ON within 45 minutes of a dose. They then entered a 48-week safety and efficacy phase, during which they self-administered the drug at home up to five times daily for off episodes with a minimum of 2 hours between doses. The investigators could adjust the doses for safety or lack of efficacy.
Two-thirds of new patients and three-quarters of rollovers received doses in the 10-20 mg range. The highest dose in the study of 35 mg was used by only 8%-9% of patients, but the highest approved and marketed dose is 30 mg.
Long-term benefits
Onset of efficacy was achieved by 15 minutes after dose for both new and rollover patients, and maximal efficacy occurred by 30 minutes. Results were very similar at 24, 36, and 48 weeks. The investigators did not perform statistical analyses.
Across study weeks 1, 12, 24, 36, and 48, between 77% and 92% of new patients and between 65% and 77% of rollover patients self-reported full ON within 30 minutes. “The long-term benefits are maintained over a year as far as the speed of onset and the duration,” Dr. Pahwa said.
Treatment-emergent adverse events occurred in about half of the new and the rollover patient groups in the titration phase and in 71%-81% of patients during the long-term safety phase. Nearly all were mild to moderate in severity.
A large number of participants withdrew from this long-term safety phase because of adverse events – 90 (33%) of new enrollees and 16 (23%) of rollover patients. Only 4% dropped out for lack of efficacy, all in the new enrollee group. Because the sublingual formulation is delivered under the tongue, patients in that group had more oral side effects, Dr. Pahwa said. Otherwise, “the side effects were very similar to the subcutaneous delivery.”
Treatment-emergent adverse events specific to sublingual apomorphine included oral mucosal erythema, lip or tongue swelling, and mouth ulceration (6% to 7% of patients each). Occurring less often were glossodynia, oral candidiasis, stomatitis, and tongue ulceration (2% each).
These were in addition to adverse events typically occurring with subcutaneous apomorphine, which are nausea, falls, dizziness, somnolence, dyskinesia, syncope, and yawning.
There are no head-to-head comparisons of sublingual versus subcutaneous delivery of apomorphine. But based on experience, Dr. Pahwa said, “With the subcutaneous, you have a slightly faster onset of action compared to the sublingual. However, sublingual has a slightly longer duration of benefit.”
He predicted that patients may prefer using an injection for a faster benefit or a sublingual for a slightly longer benefit.
More therapeutic options are welcome
Commenting on the study, Ray Dorsey, MD, professor of neurology at the University of Rochester (N.Y.), said that, for people with more advanced Parkinson’s disease “there’s usually a caregiver who’s injecting someone with an off period, as opposed to sublingual, which seems like a much easier way of administering a drug, especially for people with motor fluctuations.”
He noted that adverse events that led to premature discontinuation from the study “are concerning about the overall tolerability of the drug, which also will be determined in clinical practice, and will likely influence its overall utility.”
However, more therapeutic options are welcome because “the number of people with advanced Parkinson’s disease is going to grow and grow substantially,” he said. “So having therapies that help people with more advanced Parkinson’s disease ... many of whom don’t reach the clinic ... are going to be increasingly important.”
The study was supported by Sunovion. Dr. Pahwa and Dr. Dorsey reported conflicts of interest with numerous sources in industry.
A version of this article originally appeared on Medscape.com.
, long-term follow-up of a phase 3 study has shown. Besides the usual adverse effects with apomorphine, the sublingual film was associated with more oral adverse effects than seen with the injectable drug. However, it may have some advantages over subcutaneous apomorphine injections in terms of administration during off episodes.
The study was presented at the Movement Disorder Society 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
For example, the new formulation is more convenient than carrying an injection. It comes in a small, tear-open packet that contains a medication strip patients place under their tongues.
“When a patient is in the off state, depending on how off they are, they could have a little difficulty opening the strip [packet], but anyone can open the strip for them,” said lead author Rajesh Pahwa, MD, professor of neurology and chief of the Parkinson and Movement Disorder Division at the University of Kansas Medical Center in Kansas City. “On the other hand with the subcutaneous, they have to give the injection themselves and a stranger or someone is not going to help them with that.”
Open-label safety and efficacy study
The aims of this open-label, 48-week follow-up were to add new patients to assess safety and tolerability over the long term and to see if continued benefit from a previous 12-week double-blind study was still present at 1 year for patients in the earlier study.
This multicenter study (NCT02542696) included “rollover” patients (n = 78 for safety; n = 70 for efficacy) from the previous phase 2/3 double-blind trial, as well as new patients with no prior exposure to apomorphine sublingual film (n = 347 for safety; n = 275 for efficacy).
New patients experienced one or more off episodes per day with a daily off time of 2 hours or more per day while on stable doses of levodopa/carbidopa. All had clinically meaningful responses to levodopa/carbidopa and were judged by the investigator to be Stage 1-3 by modified Hoehn and Yahr scale rating during ON periods.
Rollover patients completed the prior study and had no major changes in their anti-Parkinson’s medications since then. Mouth cankers or sores were exclusion criteria for either group. New subjects could not have received subcutaneous apomorphine within 7 days of a screening visit.
The demographics and baseline characteristics of the new and rollover groups were similar (approximately 64 years; 65%-71% male; 96% White; 8.3-9.6 years since diagnosis; 3.9 to 4.1 off episodes/day, and total mean daily levodopa dose of 1120 to 1478 mg).
Assessing only the group of new patients, the investigators reported that 80% had a Hoehn and Yahr score of 2 or 2.5 when in the ON state and a Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III predose score of 41.8.
At the beginning of this study, patients in an off period received titrated doses of 10-35 mg of sublingual apomorphine in 5 mg increments during sequential office visits until they achieved a tolerable full ON within 45 minutes of a dose. They then entered a 48-week safety and efficacy phase, during which they self-administered the drug at home up to five times daily for off episodes with a minimum of 2 hours between doses. The investigators could adjust the doses for safety or lack of efficacy.
Two-thirds of new patients and three-quarters of rollovers received doses in the 10-20 mg range. The highest dose in the study of 35 mg was used by only 8%-9% of patients, but the highest approved and marketed dose is 30 mg.
Long-term benefits
Onset of efficacy was achieved by 15 minutes after dose for both new and rollover patients, and maximal efficacy occurred by 30 minutes. Results were very similar at 24, 36, and 48 weeks. The investigators did not perform statistical analyses.
Across study weeks 1, 12, 24, 36, and 48, between 77% and 92% of new patients and between 65% and 77% of rollover patients self-reported full ON within 30 minutes. “The long-term benefits are maintained over a year as far as the speed of onset and the duration,” Dr. Pahwa said.
Treatment-emergent adverse events occurred in about half of the new and the rollover patient groups in the titration phase and in 71%-81% of patients during the long-term safety phase. Nearly all were mild to moderate in severity.
A large number of participants withdrew from this long-term safety phase because of adverse events – 90 (33%) of new enrollees and 16 (23%) of rollover patients. Only 4% dropped out for lack of efficacy, all in the new enrollee group. Because the sublingual formulation is delivered under the tongue, patients in that group had more oral side effects, Dr. Pahwa said. Otherwise, “the side effects were very similar to the subcutaneous delivery.”
Treatment-emergent adverse events specific to sublingual apomorphine included oral mucosal erythema, lip or tongue swelling, and mouth ulceration (6% to 7% of patients each). Occurring less often were glossodynia, oral candidiasis, stomatitis, and tongue ulceration (2% each).
These were in addition to adverse events typically occurring with subcutaneous apomorphine, which are nausea, falls, dizziness, somnolence, dyskinesia, syncope, and yawning.
There are no head-to-head comparisons of sublingual versus subcutaneous delivery of apomorphine. But based on experience, Dr. Pahwa said, “With the subcutaneous, you have a slightly faster onset of action compared to the sublingual. However, sublingual has a slightly longer duration of benefit.”
He predicted that patients may prefer using an injection for a faster benefit or a sublingual for a slightly longer benefit.
More therapeutic options are welcome
Commenting on the study, Ray Dorsey, MD, professor of neurology at the University of Rochester (N.Y.), said that, for people with more advanced Parkinson’s disease “there’s usually a caregiver who’s injecting someone with an off period, as opposed to sublingual, which seems like a much easier way of administering a drug, especially for people with motor fluctuations.”
He noted that adverse events that led to premature discontinuation from the study “are concerning about the overall tolerability of the drug, which also will be determined in clinical practice, and will likely influence its overall utility.”
However, more therapeutic options are welcome because “the number of people with advanced Parkinson’s disease is going to grow and grow substantially,” he said. “So having therapies that help people with more advanced Parkinson’s disease ... many of whom don’t reach the clinic ... are going to be increasingly important.”
The study was supported by Sunovion. Dr. Pahwa and Dr. Dorsey reported conflicts of interest with numerous sources in industry.
A version of this article originally appeared on Medscape.com.
, long-term follow-up of a phase 3 study has shown. Besides the usual adverse effects with apomorphine, the sublingual film was associated with more oral adverse effects than seen with the injectable drug. However, it may have some advantages over subcutaneous apomorphine injections in terms of administration during off episodes.
The study was presented at the Movement Disorder Society 23rd International Congress of Parkinson’s Disease and Movement Disorders (Virtual) 2020.
For example, the new formulation is more convenient than carrying an injection. It comes in a small, tear-open packet that contains a medication strip patients place under their tongues.
“When a patient is in the off state, depending on how off they are, they could have a little difficulty opening the strip [packet], but anyone can open the strip for them,” said lead author Rajesh Pahwa, MD, professor of neurology and chief of the Parkinson and Movement Disorder Division at the University of Kansas Medical Center in Kansas City. “On the other hand with the subcutaneous, they have to give the injection themselves and a stranger or someone is not going to help them with that.”
Open-label safety and efficacy study
The aims of this open-label, 48-week follow-up were to add new patients to assess safety and tolerability over the long term and to see if continued benefit from a previous 12-week double-blind study was still present at 1 year for patients in the earlier study.
This multicenter study (NCT02542696) included “rollover” patients (n = 78 for safety; n = 70 for efficacy) from the previous phase 2/3 double-blind trial, as well as new patients with no prior exposure to apomorphine sublingual film (n = 347 for safety; n = 275 for efficacy).
New patients experienced one or more off episodes per day with a daily off time of 2 hours or more per day while on stable doses of levodopa/carbidopa. All had clinically meaningful responses to levodopa/carbidopa and were judged by the investigator to be Stage 1-3 by modified Hoehn and Yahr scale rating during ON periods.
Rollover patients completed the prior study and had no major changes in their anti-Parkinson’s medications since then. Mouth cankers or sores were exclusion criteria for either group. New subjects could not have received subcutaneous apomorphine within 7 days of a screening visit.
The demographics and baseline characteristics of the new and rollover groups were similar (approximately 64 years; 65%-71% male; 96% White; 8.3-9.6 years since diagnosis; 3.9 to 4.1 off episodes/day, and total mean daily levodopa dose of 1120 to 1478 mg).
Assessing only the group of new patients, the investigators reported that 80% had a Hoehn and Yahr score of 2 or 2.5 when in the ON state and a Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III predose score of 41.8.
At the beginning of this study, patients in an off period received titrated doses of 10-35 mg of sublingual apomorphine in 5 mg increments during sequential office visits until they achieved a tolerable full ON within 45 minutes of a dose. They then entered a 48-week safety and efficacy phase, during which they self-administered the drug at home up to five times daily for off episodes with a minimum of 2 hours between doses. The investigators could adjust the doses for safety or lack of efficacy.
Two-thirds of new patients and three-quarters of rollovers received doses in the 10-20 mg range. The highest dose in the study of 35 mg was used by only 8%-9% of patients, but the highest approved and marketed dose is 30 mg.
Long-term benefits
Onset of efficacy was achieved by 15 minutes after dose for both new and rollover patients, and maximal efficacy occurred by 30 minutes. Results were very similar at 24, 36, and 48 weeks. The investigators did not perform statistical analyses.
Across study weeks 1, 12, 24, 36, and 48, between 77% and 92% of new patients and between 65% and 77% of rollover patients self-reported full ON within 30 minutes. “The long-term benefits are maintained over a year as far as the speed of onset and the duration,” Dr. Pahwa said.
Treatment-emergent adverse events occurred in about half of the new and the rollover patient groups in the titration phase and in 71%-81% of patients during the long-term safety phase. Nearly all were mild to moderate in severity.
A large number of participants withdrew from this long-term safety phase because of adverse events – 90 (33%) of new enrollees and 16 (23%) of rollover patients. Only 4% dropped out for lack of efficacy, all in the new enrollee group. Because the sublingual formulation is delivered under the tongue, patients in that group had more oral side effects, Dr. Pahwa said. Otherwise, “the side effects were very similar to the subcutaneous delivery.”
Treatment-emergent adverse events specific to sublingual apomorphine included oral mucosal erythema, lip or tongue swelling, and mouth ulceration (6% to 7% of patients each). Occurring less often were glossodynia, oral candidiasis, stomatitis, and tongue ulceration (2% each).
These were in addition to adverse events typically occurring with subcutaneous apomorphine, which are nausea, falls, dizziness, somnolence, dyskinesia, syncope, and yawning.
There are no head-to-head comparisons of sublingual versus subcutaneous delivery of apomorphine. But based on experience, Dr. Pahwa said, “With the subcutaneous, you have a slightly faster onset of action compared to the sublingual. However, sublingual has a slightly longer duration of benefit.”
He predicted that patients may prefer using an injection for a faster benefit or a sublingual for a slightly longer benefit.
More therapeutic options are welcome
Commenting on the study, Ray Dorsey, MD, professor of neurology at the University of Rochester (N.Y.), said that, for people with more advanced Parkinson’s disease “there’s usually a caregiver who’s injecting someone with an off period, as opposed to sublingual, which seems like a much easier way of administering a drug, especially for people with motor fluctuations.”
He noted that adverse events that led to premature discontinuation from the study “are concerning about the overall tolerability of the drug, which also will be determined in clinical practice, and will likely influence its overall utility.”
However, more therapeutic options are welcome because “the number of people with advanced Parkinson’s disease is going to grow and grow substantially,” he said. “So having therapies that help people with more advanced Parkinson’s disease ... many of whom don’t reach the clinic ... are going to be increasingly important.”
The study was supported by Sunovion. Dr. Pahwa and Dr. Dorsey reported conflicts of interest with numerous sources in industry.
A version of this article originally appeared on Medscape.com.
FROM MOVEMENT DISORDERS SOCIETY 2020