User login
IPD in children may be a signal of immunodeficiency
according to a systematic review published in JAMA Pediatrics.
Coen Butters, BMed, DCH, of the Royal Children’s Hospital in Melbourne, and coauthors wrote that, even with optimal vaccine coverage, there is still a group of children with increased susceptibility to invasive pneumococcal disease (IPD), and this could be a potential marker of primary immunodeficiency.
They conducted a systematic review of 17 studies of 6,002 children to examine the evidence on the incidence of primary immunodeficiency in children who presented with IPD but without any other risk factors or predisposing conditions.
Overall, the frequency of primary immunodeficiency in children presenting with IPD who did not have any other predisposing condition ranged from 1% to 26%.
One study of 162 children with IPD, which had an overall frequency of primary immunodeficiency of 10%, found that children older than 2 years were significantly more likely to have primary immunodeficiency than those aged under 2 years (26% vs. 3%; P less than .001).
Primary antibody deficiency was the most commonly diagnosed immunodeficiency in these children with IPD, accounting for 71% of cases. These deficiencies presented as hypogammaglobulinemia, specific pneumococcal antibody deficiency, X-linked agammaglobulinemia, and IgG2 deficiency.
The review also included four studies that looked at the frequency of mannose-binding lectin deficiency in 1,493 children with primary IPD. Two of these studies reported a prevalence of mannose-binding lectin deficiency ranging from 31% in children aged younger than 2 years to 41% in children younger than 1 year.
Five studies looked at the rate of primary immunodeficiency in children presenting with recurrent IPD. In addition to other predisposing conditions such as sickle cell disease, cancer, and anatomical breach in the blood-brain barrier, the three studies that screened for primary immunodeficiency found rates ranging from 10% to 67%. The most common conditions were complement deficiency, pneumococcal antibody deficiency, and a single case of TLR-signaling defect.
In a study of 162 children with primary IPD, screening for asplenia identified a single case of congenital asplenia. In another study of 2,498 cases of IPD, 22 patients had asplenia at presentation, half of whom died at presentation.
Dr. Butters and associates concluded that “this review’s findings suggests that existing data support the immune evaluation of children older than 2 years without a known predisposing condition who present with their first episode of Streptococcus pneumoniae meningitis, pneumonia, or recurrent IPD. Immune evaluation should include assessment for immunoglobulin deficiency, pneumococcal antibody deficiency, complement disorders, and asplenia.”
In an accompanying editorial, Stephen I. Pelton, MD, of the Maxwell Finland Laboratory for Infectious Diseases at Boston Medical Center, and coauthors wrote that in children with recurrent episodes of IPD caused by nonvaccine serotypes – particularly those aged over 5 years – evaluation for primary immunodeficiencies could uncover immune defects.
“Once identified, direct and indirect protection, penicillin prophylaxis, or a combination of these offers great potential for disease prevention and reduction of mortality and morbidity in children with [primary immunodeficiency],” they wrote.
No funding or conflicts of interest were declared for the study. Two of the editorialists declared research funding or honoraria from the pharmaceutical sector.
SOURCES: Butters C et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3203; Pelton SI et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3185.
according to a systematic review published in JAMA Pediatrics.
Coen Butters, BMed, DCH, of the Royal Children’s Hospital in Melbourne, and coauthors wrote that, even with optimal vaccine coverage, there is still a group of children with increased susceptibility to invasive pneumococcal disease (IPD), and this could be a potential marker of primary immunodeficiency.
They conducted a systematic review of 17 studies of 6,002 children to examine the evidence on the incidence of primary immunodeficiency in children who presented with IPD but without any other risk factors or predisposing conditions.
Overall, the frequency of primary immunodeficiency in children presenting with IPD who did not have any other predisposing condition ranged from 1% to 26%.
One study of 162 children with IPD, which had an overall frequency of primary immunodeficiency of 10%, found that children older than 2 years were significantly more likely to have primary immunodeficiency than those aged under 2 years (26% vs. 3%; P less than .001).
Primary antibody deficiency was the most commonly diagnosed immunodeficiency in these children with IPD, accounting for 71% of cases. These deficiencies presented as hypogammaglobulinemia, specific pneumococcal antibody deficiency, X-linked agammaglobulinemia, and IgG2 deficiency.
The review also included four studies that looked at the frequency of mannose-binding lectin deficiency in 1,493 children with primary IPD. Two of these studies reported a prevalence of mannose-binding lectin deficiency ranging from 31% in children aged younger than 2 years to 41% in children younger than 1 year.
Five studies looked at the rate of primary immunodeficiency in children presenting with recurrent IPD. In addition to other predisposing conditions such as sickle cell disease, cancer, and anatomical breach in the blood-brain barrier, the three studies that screened for primary immunodeficiency found rates ranging from 10% to 67%. The most common conditions were complement deficiency, pneumococcal antibody deficiency, and a single case of TLR-signaling defect.
In a study of 162 children with primary IPD, screening for asplenia identified a single case of congenital asplenia. In another study of 2,498 cases of IPD, 22 patients had asplenia at presentation, half of whom died at presentation.
Dr. Butters and associates concluded that “this review’s findings suggests that existing data support the immune evaluation of children older than 2 years without a known predisposing condition who present with their first episode of Streptococcus pneumoniae meningitis, pneumonia, or recurrent IPD. Immune evaluation should include assessment for immunoglobulin deficiency, pneumococcal antibody deficiency, complement disorders, and asplenia.”
In an accompanying editorial, Stephen I. Pelton, MD, of the Maxwell Finland Laboratory for Infectious Diseases at Boston Medical Center, and coauthors wrote that in children with recurrent episodes of IPD caused by nonvaccine serotypes – particularly those aged over 5 years – evaluation for primary immunodeficiencies could uncover immune defects.
“Once identified, direct and indirect protection, penicillin prophylaxis, or a combination of these offers great potential for disease prevention and reduction of mortality and morbidity in children with [primary immunodeficiency],” they wrote.
No funding or conflicts of interest were declared for the study. Two of the editorialists declared research funding or honoraria from the pharmaceutical sector.
SOURCES: Butters C et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3203; Pelton SI et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3185.
according to a systematic review published in JAMA Pediatrics.
Coen Butters, BMed, DCH, of the Royal Children’s Hospital in Melbourne, and coauthors wrote that, even with optimal vaccine coverage, there is still a group of children with increased susceptibility to invasive pneumococcal disease (IPD), and this could be a potential marker of primary immunodeficiency.
They conducted a systematic review of 17 studies of 6,002 children to examine the evidence on the incidence of primary immunodeficiency in children who presented with IPD but without any other risk factors or predisposing conditions.
Overall, the frequency of primary immunodeficiency in children presenting with IPD who did not have any other predisposing condition ranged from 1% to 26%.
One study of 162 children with IPD, which had an overall frequency of primary immunodeficiency of 10%, found that children older than 2 years were significantly more likely to have primary immunodeficiency than those aged under 2 years (26% vs. 3%; P less than .001).
Primary antibody deficiency was the most commonly diagnosed immunodeficiency in these children with IPD, accounting for 71% of cases. These deficiencies presented as hypogammaglobulinemia, specific pneumococcal antibody deficiency, X-linked agammaglobulinemia, and IgG2 deficiency.
The review also included four studies that looked at the frequency of mannose-binding lectin deficiency in 1,493 children with primary IPD. Two of these studies reported a prevalence of mannose-binding lectin deficiency ranging from 31% in children aged younger than 2 years to 41% in children younger than 1 year.
Five studies looked at the rate of primary immunodeficiency in children presenting with recurrent IPD. In addition to other predisposing conditions such as sickle cell disease, cancer, and anatomical breach in the blood-brain barrier, the three studies that screened for primary immunodeficiency found rates ranging from 10% to 67%. The most common conditions were complement deficiency, pneumococcal antibody deficiency, and a single case of TLR-signaling defect.
In a study of 162 children with primary IPD, screening for asplenia identified a single case of congenital asplenia. In another study of 2,498 cases of IPD, 22 patients had asplenia at presentation, half of whom died at presentation.
Dr. Butters and associates concluded that “this review’s findings suggests that existing data support the immune evaluation of children older than 2 years without a known predisposing condition who present with their first episode of Streptococcus pneumoniae meningitis, pneumonia, or recurrent IPD. Immune evaluation should include assessment for immunoglobulin deficiency, pneumococcal antibody deficiency, complement disorders, and asplenia.”
In an accompanying editorial, Stephen I. Pelton, MD, of the Maxwell Finland Laboratory for Infectious Diseases at Boston Medical Center, and coauthors wrote that in children with recurrent episodes of IPD caused by nonvaccine serotypes – particularly those aged over 5 years – evaluation for primary immunodeficiencies could uncover immune defects.
“Once identified, direct and indirect protection, penicillin prophylaxis, or a combination of these offers great potential for disease prevention and reduction of mortality and morbidity in children with [primary immunodeficiency],” they wrote.
No funding or conflicts of interest were declared for the study. Two of the editorialists declared research funding or honoraria from the pharmaceutical sector.
SOURCES: Butters C et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3203; Pelton SI et al. JAMA Pediatr. 2019 Sep 30. doi: 10.1001/jamapediatrics.2019.3185.
FROM JAMA PEDIATRICS
Targeted agents vs. chemoimmunotherapy as first-line treatment of CLL
SAN FRANCISCO – Should targeted agents replace chemoimmunotherapy (CIT) as first-line treatment for chronic lymphocytic leukemia (CLL)? A recent debate suggests there’s no consensus.

William G. Wierda, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston, and Jennifer R. Brown, MD, PhD, of Dana-Farber Cancer Institute in Boston, debated the topic at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Wierda argued that CLL patients should receive a BTK inhibitor or BCL2 inhibitor, with or without obinutuzumab, as first-line therapy because these targeted agents have been shown to provide better progression-free survival (PFS) than CIT, and the targeted therapies may prolong overall survival (OS) as well.
Dr. Brown countered that targeted agents don’t improve PFS for all CLL patients, improved PFS doesn’t always translate to improved OS, and targeted agents cost more than CIT.
No role for CIT as first-line treatment
“We have two approaches right now, with nonchemoimmunotherapy-based treatment,” Dr. Wierda said. “One approach, with small-molecule inhibitors, is to have a sustained and durable period of disease control, particularly with BTK inhibitors. The other strategy that has emerged is deep remissions with fixed-duration treatment with BCL2 small-molecule inhibitor-based therapy, which, I would argue, is better than being exposed to genotoxic chemoimmunotherapy.”
Dr. Wierda went on to explain that the BTK inhibitor ibrutinib has been shown to improve PFS, compared with CIT, in phase 3 trials.
In the iLLUMINATE trial, researchers compared ibrutinib plus obinutuzumab to chlorambucil plus obinutuzumab as first-line treatment in CLL. At a median follow-up of 31.3 months, the median PFS was not reached in the ibrutinib arm and was 19 months in the chlorambucil arm (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:43-56).
In the A041202 study, researchers compared ibrutinib alone (Ib) or in combination with rituximab (Ib-R) to bendamustine plus rituximab (BR) in untreated, older patients with CLL. The 2-year PFS estimates were 74% in the BR arm, 87% in the Ib arm, and 88% in the Ib-R arm (P less than .001 for BR vs. Ib or Ib-R; N Engl J Med. 2018; 379:2517-28).
In the E1912 trial, researchers compared Ib-R to fludarabine, cyclophosphamide, and rituximab (FCR) in younger, untreated CLL patients. The 3-year PFS was 89.4% with Ib-R and 72.9% with FCR (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43).
Dr. Wierda noted that the E1912 trial also showed superior OS with Ib-R. The 3-year OS rate was 98.8% with Ib-R and 91.5% with FCR (P less than .001). However, there was no significant difference in OS between the treatment arms in the A041202 trial or the iLLUMINATE trial.
“But I would argue that is, in part, because of short follow-up,” Dr. Wierda said. “The trials were all designed to look at progression-free survival, not overall survival. With longer follow-up, we may see differences in overall survival emerging.”
Dr. Wierda went on to say that fixed‐duration treatment with the BCL2 inhibitor venetoclax can improve PFS over CIT.
In the phase 3 CLL14 trial, researchers compared fixed-duration treatment with venetoclax plus obinutuzumab to chlorambucil plus obinutuzumab in previously untreated CLL patients with comorbidities. The estimated PFS at 2 years was 88.2% in the venetoclax group and 64.1% in the chlorambucil group (P less than .001; N Engl J Med. 2019; 380:2225-36).
“[There was] no difference in overall survival,” Dr. Wierda noted. “But, again, I would argue ... that follow-up is relatively limited. We may ultimately see a difference in overall survival.”
Based on these findings, Dr. Wierda made the following treatment recommendations:
- Any CLL patient with del(17p) or TP53 mutation, and older, unfit patients with unmutated IGHV should receive a BTK inhibitor, with or without obinutuzumab.
- All young, fit patients, and older, unfit patients with mutated IGHV should receive a BCL2 inhibitor plus obinutuzumab.
Dr. Wierda also noted that ibrutinib and venetoclax in combination have shown early promise for patients with previously untreated CLL (N Engl J Med. 2019; 380:2095-2103).
CIT still has a role as first-line treatment
Dr. Brown suggested that a PFS benefit may not be enough to recommend targeted agents over CIT. For one thing, the PFS benefit doesn’t apply to all patients, as the IGHV-mutated subgroup does equally well with CIT and targeted agents.

In the IGHV-mutated group from the E1912 trial, the 3-year PFS was 88% for patients who received Ib-R and those who received FCR (N Engl J Med. 2019 Aug 1;381:432-43). In the A041202 study, the 2-year PFS among IGHV-mutated patients was 87% in the BR arm, 86% in the Ib arm, and 88% in the Ib-R arm (N Engl J Med. 2018; 379:2517-28).
In the CLL14 trial, PFS rates were similar among IGHV-mutated patients who received chlorambucil plus obinutuzumab and IGHV-mutated or unmutated patients who received venetoclax and obinutuzumab (N Engl J Med. 2019; 380:2225-36).
Dr. Brown also noted that the overall improvement in PFS observed with ibrutinib and venetoclax doesn’t always translate to improved OS.
In the A041202 study, there was no significant difference in OS between the Ib, Ib-R, and BR arms (N Engl J Med. 2018; 379:2517-28). There was no significant difference in OS between the ibrutinib and chlorambucil arms in the iLLUMINATE trial (Lancet Oncol. 2019 Jan;20[1]:43-56). And there was no significant difference in OS between the venetoclax and chlorambucil arms in the CLL14 trial (N Engl J Med. 2019; 380:2225-36).
However, in the RESONATE-2 trial, ibrutinib provided an OS benefit over chlorambucil. The 2-year OS was 95% and 84%, respectively (P = .0145; Haematologica. Sept 2018;103:1502-10). Dr. Brown said the OS advantage in this study was due to the “very poor comparator of chlorambucil and very limited crossover.”
As Dr. Wierda mentioned, the OS rate was higher with Ib-R than with FCR in the E1912 trial. The 3-year OS rate was 98.8% and 91.5%, respectively (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43). Dr. Brown noted, however, that there were few deaths in this study, and many of them “were not clearly related to the disease or its treatment.”
Dr. Brown also pointed out that FCR has been shown to have curative potential in IGHV-mutated CLL in both the FCR300 trial (Blood. 2016 127:303-9) and the CLL8 trial (Blood. 2016 127:208-15).
Another factor to consider is the greater cost of targeted agents. One analysis suggested the per-patient lifetime cost of CLL treatment in the United States will increase from $147,000 to $604,000 as targeted therapies overtake CIT as first-line treatment (J Clin Oncol. 2017 Jan 10;35[2]:166-174).
“Given all of the above, chemoimmunotherapy is going to remain part of the treatment repertoire for CLL,” Dr. Brown said. “It’s our only known potential cure for the fit, mutated patients ... and can also result in prolonged treatment-free intervals for patients who are older. As we manage CLL as a chronic disease over a lifetime, we need to continue to have this in our armamentarium.”
Specifically, Dr. Brown said CIT is appropriate for patients who don’t have del(17p) or mutated TP53. FCR should be given to young, fit patients with IGHV-mutated CLL, and FCR or BR should be given to older patients and young, fit patients with IGHV-unmutated CLL.
Dr. Brown and Dr. Wierda reported financial ties to multiple pharmaceutical companies, including makers of CLL treatments.
SAN FRANCISCO – Should targeted agents replace chemoimmunotherapy (CIT) as first-line treatment for chronic lymphocytic leukemia (CLL)? A recent debate suggests there’s no consensus.
William G. Wierda, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston, and Jennifer R. Brown, MD, PhD, of Dana-Farber Cancer Institute in Boston, debated the topic at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Wierda argued that CLL patients should receive a BTK inhibitor or BCL2 inhibitor, with or without obinutuzumab, as first-line therapy because these targeted agents have been shown to provide better progression-free survival (PFS) than CIT, and the targeted therapies may prolong overall survival (OS) as well.
Dr. Brown countered that targeted agents don’t improve PFS for all CLL patients, improved PFS doesn’t always translate to improved OS, and targeted agents cost more than CIT.
No role for CIT as first-line treatment
“We have two approaches right now, with nonchemoimmunotherapy-based treatment,” Dr. Wierda said. “One approach, with small-molecule inhibitors, is to have a sustained and durable period of disease control, particularly with BTK inhibitors. The other strategy that has emerged is deep remissions with fixed-duration treatment with BCL2 small-molecule inhibitor-based therapy, which, I would argue, is better than being exposed to genotoxic chemoimmunotherapy.”
Dr. Wierda went on to explain that the BTK inhibitor ibrutinib has been shown to improve PFS, compared with CIT, in phase 3 trials.
In the iLLUMINATE trial, researchers compared ibrutinib plus obinutuzumab to chlorambucil plus obinutuzumab as first-line treatment in CLL. At a median follow-up of 31.3 months, the median PFS was not reached in the ibrutinib arm and was 19 months in the chlorambucil arm (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:43-56).
In the A041202 study, researchers compared ibrutinib alone (Ib) or in combination with rituximab (Ib-R) to bendamustine plus rituximab (BR) in untreated, older patients with CLL. The 2-year PFS estimates were 74% in the BR arm, 87% in the Ib arm, and 88% in the Ib-R arm (P less than .001 for BR vs. Ib or Ib-R; N Engl J Med. 2018; 379:2517-28).
In the E1912 trial, researchers compared Ib-R to fludarabine, cyclophosphamide, and rituximab (FCR) in younger, untreated CLL patients. The 3-year PFS was 89.4% with Ib-R and 72.9% with FCR (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43).
Dr. Wierda noted that the E1912 trial also showed superior OS with Ib-R. The 3-year OS rate was 98.8% with Ib-R and 91.5% with FCR (P less than .001). However, there was no significant difference in OS between the treatment arms in the A041202 trial or the iLLUMINATE trial.
“But I would argue that is, in part, because of short follow-up,” Dr. Wierda said. “The trials were all designed to look at progression-free survival, not overall survival. With longer follow-up, we may see differences in overall survival emerging.”
Dr. Wierda went on to say that fixed‐duration treatment with the BCL2 inhibitor venetoclax can improve PFS over CIT.
In the phase 3 CLL14 trial, researchers compared fixed-duration treatment with venetoclax plus obinutuzumab to chlorambucil plus obinutuzumab in previously untreated CLL patients with comorbidities. The estimated PFS at 2 years was 88.2% in the venetoclax group and 64.1% in the chlorambucil group (P less than .001; N Engl J Med. 2019; 380:2225-36).
“[There was] no difference in overall survival,” Dr. Wierda noted. “But, again, I would argue ... that follow-up is relatively limited. We may ultimately see a difference in overall survival.”
Based on these findings, Dr. Wierda made the following treatment recommendations:
- Any CLL patient with del(17p) or TP53 mutation, and older, unfit patients with unmutated IGHV should receive a BTK inhibitor, with or without obinutuzumab.
- All young, fit patients, and older, unfit patients with mutated IGHV should receive a BCL2 inhibitor plus obinutuzumab.
Dr. Wierda also noted that ibrutinib and venetoclax in combination have shown early promise for patients with previously untreated CLL (N Engl J Med. 2019; 380:2095-2103).
CIT still has a role as first-line treatment
Dr. Brown suggested that a PFS benefit may not be enough to recommend targeted agents over CIT. For one thing, the PFS benefit doesn’t apply to all patients, as the IGHV-mutated subgroup does equally well with CIT and targeted agents.
In the IGHV-mutated group from the E1912 trial, the 3-year PFS was 88% for patients who received Ib-R and those who received FCR (N Engl J Med. 2019 Aug 1;381:432-43). In the A041202 study, the 2-year PFS among IGHV-mutated patients was 87% in the BR arm, 86% in the Ib arm, and 88% in the Ib-R arm (N Engl J Med. 2018; 379:2517-28).
In the CLL14 trial, PFS rates were similar among IGHV-mutated patients who received chlorambucil plus obinutuzumab and IGHV-mutated or unmutated patients who received venetoclax and obinutuzumab (N Engl J Med. 2019; 380:2225-36).
Dr. Brown also noted that the overall improvement in PFS observed with ibrutinib and venetoclax doesn’t always translate to improved OS.
In the A041202 study, there was no significant difference in OS between the Ib, Ib-R, and BR arms (N Engl J Med. 2018; 379:2517-28). There was no significant difference in OS between the ibrutinib and chlorambucil arms in the iLLUMINATE trial (Lancet Oncol. 2019 Jan;20[1]:43-56). And there was no significant difference in OS between the venetoclax and chlorambucil arms in the CLL14 trial (N Engl J Med. 2019; 380:2225-36).
However, in the RESONATE-2 trial, ibrutinib provided an OS benefit over chlorambucil. The 2-year OS was 95% and 84%, respectively (P = .0145; Haematologica. Sept 2018;103:1502-10). Dr. Brown said the OS advantage in this study was due to the “very poor comparator of chlorambucil and very limited crossover.”
As Dr. Wierda mentioned, the OS rate was higher with Ib-R than with FCR in the E1912 trial. The 3-year OS rate was 98.8% and 91.5%, respectively (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43). Dr. Brown noted, however, that there were few deaths in this study, and many of them “were not clearly related to the disease or its treatment.”
Dr. Brown also pointed out that FCR has been shown to have curative potential in IGHV-mutated CLL in both the FCR300 trial (Blood. 2016 127:303-9) and the CLL8 trial (Blood. 2016 127:208-15).
Another factor to consider is the greater cost of targeted agents. One analysis suggested the per-patient lifetime cost of CLL treatment in the United States will increase from $147,000 to $604,000 as targeted therapies overtake CIT as first-line treatment (J Clin Oncol. 2017 Jan 10;35[2]:166-174).
“Given all of the above, chemoimmunotherapy is going to remain part of the treatment repertoire for CLL,” Dr. Brown said. “It’s our only known potential cure for the fit, mutated patients ... and can also result in prolonged treatment-free intervals for patients who are older. As we manage CLL as a chronic disease over a lifetime, we need to continue to have this in our armamentarium.”
Specifically, Dr. Brown said CIT is appropriate for patients who don’t have del(17p) or mutated TP53. FCR should be given to young, fit patients with IGHV-mutated CLL, and FCR or BR should be given to older patients and young, fit patients with IGHV-unmutated CLL.
Dr. Brown and Dr. Wierda reported financial ties to multiple pharmaceutical companies, including makers of CLL treatments.
SAN FRANCISCO – Should targeted agents replace chemoimmunotherapy (CIT) as first-line treatment for chronic lymphocytic leukemia (CLL)? A recent debate suggests there’s no consensus.
William G. Wierda, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston, and Jennifer R. Brown, MD, PhD, of Dana-Farber Cancer Institute in Boston, debated the topic at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Wierda argued that CLL patients should receive a BTK inhibitor or BCL2 inhibitor, with or without obinutuzumab, as first-line therapy because these targeted agents have been shown to provide better progression-free survival (PFS) than CIT, and the targeted therapies may prolong overall survival (OS) as well.
Dr. Brown countered that targeted agents don’t improve PFS for all CLL patients, improved PFS doesn’t always translate to improved OS, and targeted agents cost more than CIT.
No role for CIT as first-line treatment
“We have two approaches right now, with nonchemoimmunotherapy-based treatment,” Dr. Wierda said. “One approach, with small-molecule inhibitors, is to have a sustained and durable period of disease control, particularly with BTK inhibitors. The other strategy that has emerged is deep remissions with fixed-duration treatment with BCL2 small-molecule inhibitor-based therapy, which, I would argue, is better than being exposed to genotoxic chemoimmunotherapy.”
Dr. Wierda went on to explain that the BTK inhibitor ibrutinib has been shown to improve PFS, compared with CIT, in phase 3 trials.
In the iLLUMINATE trial, researchers compared ibrutinib plus obinutuzumab to chlorambucil plus obinutuzumab as first-line treatment in CLL. At a median follow-up of 31.3 months, the median PFS was not reached in the ibrutinib arm and was 19 months in the chlorambucil arm (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:43-56).
In the A041202 study, researchers compared ibrutinib alone (Ib) or in combination with rituximab (Ib-R) to bendamustine plus rituximab (BR) in untreated, older patients with CLL. The 2-year PFS estimates were 74% in the BR arm, 87% in the Ib arm, and 88% in the Ib-R arm (P less than .001 for BR vs. Ib or Ib-R; N Engl J Med. 2018; 379:2517-28).
In the E1912 trial, researchers compared Ib-R to fludarabine, cyclophosphamide, and rituximab (FCR) in younger, untreated CLL patients. The 3-year PFS was 89.4% with Ib-R and 72.9% with FCR (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43).
Dr. Wierda noted that the E1912 trial also showed superior OS with Ib-R. The 3-year OS rate was 98.8% with Ib-R and 91.5% with FCR (P less than .001). However, there was no significant difference in OS between the treatment arms in the A041202 trial or the iLLUMINATE trial.
“But I would argue that is, in part, because of short follow-up,” Dr. Wierda said. “The trials were all designed to look at progression-free survival, not overall survival. With longer follow-up, we may see differences in overall survival emerging.”
Dr. Wierda went on to say that fixed‐duration treatment with the BCL2 inhibitor venetoclax can improve PFS over CIT.
In the phase 3 CLL14 trial, researchers compared fixed-duration treatment with venetoclax plus obinutuzumab to chlorambucil plus obinutuzumab in previously untreated CLL patients with comorbidities. The estimated PFS at 2 years was 88.2% in the venetoclax group and 64.1% in the chlorambucil group (P less than .001; N Engl J Med. 2019; 380:2225-36).
“[There was] no difference in overall survival,” Dr. Wierda noted. “But, again, I would argue ... that follow-up is relatively limited. We may ultimately see a difference in overall survival.”
Based on these findings, Dr. Wierda made the following treatment recommendations:
- Any CLL patient with del(17p) or TP53 mutation, and older, unfit patients with unmutated IGHV should receive a BTK inhibitor, with or without obinutuzumab.
- All young, fit patients, and older, unfit patients with mutated IGHV should receive a BCL2 inhibitor plus obinutuzumab.
Dr. Wierda also noted that ibrutinib and venetoclax in combination have shown early promise for patients with previously untreated CLL (N Engl J Med. 2019; 380:2095-2103).
CIT still has a role as first-line treatment
Dr. Brown suggested that a PFS benefit may not be enough to recommend targeted agents over CIT. For one thing, the PFS benefit doesn’t apply to all patients, as the IGHV-mutated subgroup does equally well with CIT and targeted agents.
In the IGHV-mutated group from the E1912 trial, the 3-year PFS was 88% for patients who received Ib-R and those who received FCR (N Engl J Med. 2019 Aug 1;381:432-43). In the A041202 study, the 2-year PFS among IGHV-mutated patients was 87% in the BR arm, 86% in the Ib arm, and 88% in the Ib-R arm (N Engl J Med. 2018; 379:2517-28).
In the CLL14 trial, PFS rates were similar among IGHV-mutated patients who received chlorambucil plus obinutuzumab and IGHV-mutated or unmutated patients who received venetoclax and obinutuzumab (N Engl J Med. 2019; 380:2225-36).
Dr. Brown also noted that the overall improvement in PFS observed with ibrutinib and venetoclax doesn’t always translate to improved OS.
In the A041202 study, there was no significant difference in OS between the Ib, Ib-R, and BR arms (N Engl J Med. 2018; 379:2517-28). There was no significant difference in OS between the ibrutinib and chlorambucil arms in the iLLUMINATE trial (Lancet Oncol. 2019 Jan;20[1]:43-56). And there was no significant difference in OS between the venetoclax and chlorambucil arms in the CLL14 trial (N Engl J Med. 2019; 380:2225-36).
However, in the RESONATE-2 trial, ibrutinib provided an OS benefit over chlorambucil. The 2-year OS was 95% and 84%, respectively (P = .0145; Haematologica. Sept 2018;103:1502-10). Dr. Brown said the OS advantage in this study was due to the “very poor comparator of chlorambucil and very limited crossover.”
As Dr. Wierda mentioned, the OS rate was higher with Ib-R than with FCR in the E1912 trial. The 3-year OS rate was 98.8% and 91.5%, respectively (P less than .001; N Engl J Med. 2019 Aug 1;381:432-43). Dr. Brown noted, however, that there were few deaths in this study, and many of them “were not clearly related to the disease or its treatment.”
Dr. Brown also pointed out that FCR has been shown to have curative potential in IGHV-mutated CLL in both the FCR300 trial (Blood. 2016 127:303-9) and the CLL8 trial (Blood. 2016 127:208-15).
Another factor to consider is the greater cost of targeted agents. One analysis suggested the per-patient lifetime cost of CLL treatment in the United States will increase from $147,000 to $604,000 as targeted therapies overtake CIT as first-line treatment (J Clin Oncol. 2017 Jan 10;35[2]:166-174).
“Given all of the above, chemoimmunotherapy is going to remain part of the treatment repertoire for CLL,” Dr. Brown said. “It’s our only known potential cure for the fit, mutated patients ... and can also result in prolonged treatment-free intervals for patients who are older. As we manage CLL as a chronic disease over a lifetime, we need to continue to have this in our armamentarium.”
Specifically, Dr. Brown said CIT is appropriate for patients who don’t have del(17p) or mutated TP53. FCR should be given to young, fit patients with IGHV-mutated CLL, and FCR or BR should be given to older patients and young, fit patients with IGHV-unmutated CLL.
Dr. Brown and Dr. Wierda reported financial ties to multiple pharmaceutical companies, including makers of CLL treatments.
REPORTING FROM NCCN HEMATOLOGIC MALIGNANCIES
Novel cardiac troponin protocol rapidly rules out MI
PARIS – An accelerated rule-out pathway, reliant upon a single high-sensitivity cardiac troponin test upon presentation to the ED with suspected acute coronary syndrome, reduced length of stay and hospital admission rates without increasing cardiac events at 30 days or 1 year in a major Scottish study.
“We conclude that implementation of this early rule-out pathway is both effective and safe, and adoption of this pathway will have major benefits for patients and health care systems,” Nicholas L. Mills, MBChB, PhD, said in presenting the results of the HiSTORIC (High-Sensitivity Cardiac Troponin at Presentation to Rule Out Myocardial Infarction) trial at the annual congress of the European Society of Cardiology.
Indeed, in the Unites States, where more than 20 million people per year present to EDs with suspected ACS, the 3.3-hour reduction in length of stay achieved in the HiSTORIC trial by implementing the accelerated rule-out pathway would add up to a $3.6 billion annual savings in bed occupancy alone, according to Dr. Mills, who is chair of cardiology at the University of Edinburgh.
The HiSTORIC pathway incorporates separate thresholds for risk stratification and diagnosis. This strategy is based on an accumulation of persuasive evidence that the major advantage of high-sensitivity cardiac troponin testing is to rule out MI, rather than to rule it in, Dr. Mills explained.
HiSTORIC was a 2-year, prospective, stepped-wedge, cluster-randomized, controlled trial including 31,492 consecutive patients with suspected ACS who presented to seven participating hospitals in Scotland. Patients were randomized, at the hospital level, to one of two management pathways. The control group got a standard guideline-recommended strategy involving high-sensitivity cardiac troponin I testing upon presentation and again 6-12 hours later, with MI being ruled out if the troponin levels were not above the 99th percentile.
In contrast, the novel early rule-out strategy worked as follows: If the patient presented with at least 2 hours of symptoms and the initial troponin I level was below 5 ng/L, then MI was ruled out and the patient was triaged straightaway for outpatient management. If the level was above the 99th percentile, the patient was admitted for serial testing to be done 6-12 hours after symptom onset. And for an intermediate test result – that is, a troponin level between 5 ng/L and the 99th percentile – patients remained in the ED for retesting 3 hours from the time of presentation, and were subsequently admitted only if their troponin level was rising.
Using the accelerated rule-out strategy, two-thirds of patients were quickly discharged from the ED on the basis of a troponin level below 5 ng/mL, and another 7% were ruled out for MI and discharged from the ED after a 3-hour stay on the basis of their second test.
The primary efficacy outcome was length of stay from initial presentation to the ED to discharge. The duration was 10.1 hours with the guideline-recommended pathway and 6.8 hours with the accelerated rule-out pathway, for a statistically significant and clinically meaningful 3.3-hour difference. Moreover, the proportion of patients discharged directly from the ED without hospital admission increased from 53% to 74%, a 57% jump.
The primary safety outcome was the rate of MI or cardiac death post discharge. The rates at 30 days and 1 year were 0.4% and 2.6%, respectively, in the standard-pathway group, compared with 0.3% and 1.8% with the early rule-out pathway. Those between-group differences favoring the accelerated rule-out pathway weren’t statistically significant, but they provided reassurance that the novel pathway was safe.
Of note, this was the first-ever randomized trial to evaluate the safety and efficacy of an early rule-out pathway. Other rapid diagnostic pathways are largely based on observational experience and expert opinion, Dr. Mills said.
The assay utilized in the HiSTORIC trial was the Abbott Diagnostics Architect high sensitivity assay. The 5-ng/L threshold for early rule-out was chosen for the trial because an earlier study by Dr. Mills and coinvestigators showed that a level below that cutoff had a 99.6% negative predictive value for MI (Lancet. 2015 Dec 19;386[10012]:2481-8)
The early rule-out pathway was deliberately designed to be simple and pragmatic, according to the cardiologist. “One of the most remarkable observations in this trial was the adherence to the pathway. We prespecified three criteria to evaluate this and demonstrated adherence rates of 86%-92% for each of these criteria. This was despite the pathway being implemented in all consecutive patients at seven different hospitals and used by many hundreds of different clinicians.”
Discussant Hugo A. Katus, MD, called the HiSTORIC study “a really urgently needed and very well-conducted trial.”

“There were very consistently low MI and cardiac death rates at 30 days and 1 year. So this really works,” commented Dr. Katus, who is chief of internal medicine and director of the department of cardiovascular medicine at Heidelberg (Germany) University.
“Accelerated rule-out high-sensitivity cardiac troponin protocols are here to stay,” he declared.
However, Dr. Katus voiced a concern: “By early discharge as rule out, are other life-threatening conditions ignored?”
He raised this issue because of what he views as the substantial 1-year all-cause mortality and return-to-hospital rates of 5.8% and 39.2% in the standard-pathway group and 5.2% and 38.9% in the accelerated rule-out patients in HiSTORIC. An accelerated rule-out strategy should not prohibit a careful clinical work-up, he emphasized.
Dr. Mills discussed the results in a video interview.
The HiSTORIC trial was funded by the British Heart Foundation. Dr. Mills reported receiving research grants from Abbott Diagnostics and Siemens.
Simultaneous with Dr. Mills’ presentation of the HiSTORIC trial results at the ESC congress, an earlier study that formed the scientific basis for the investigators’ decision to employ distinct risk stratification and diagnostic thresholds for cardiac troponin testing was published online (Circulation. 2019 Sep 1. doi: 10.1161/CIRCULATIONAHA.119.042866). The actual HiSTORIC trial results will be published later.
Dr. Katus reported holding a patent for a cardiac troponin T test and serving as a consultant to AstraZeneca, Bayer, Boehringer Ingelheim, and Novo Nordisk.
PARIS – An accelerated rule-out pathway, reliant upon a single high-sensitivity cardiac troponin test upon presentation to the ED with suspected acute coronary syndrome, reduced length of stay and hospital admission rates without increasing cardiac events at 30 days or 1 year in a major Scottish study.
“We conclude that implementation of this early rule-out pathway is both effective and safe, and adoption of this pathway will have major benefits for patients and health care systems,” Nicholas L. Mills, MBChB, PhD, said in presenting the results of the HiSTORIC (High-Sensitivity Cardiac Troponin at Presentation to Rule Out Myocardial Infarction) trial at the annual congress of the European Society of Cardiology.
Indeed, in the Unites States, where more than 20 million people per year present to EDs with suspected ACS, the 3.3-hour reduction in length of stay achieved in the HiSTORIC trial by implementing the accelerated rule-out pathway would add up to a $3.6 billion annual savings in bed occupancy alone, according to Dr. Mills, who is chair of cardiology at the University of Edinburgh.
The HiSTORIC pathway incorporates separate thresholds for risk stratification and diagnosis. This strategy is based on an accumulation of persuasive evidence that the major advantage of high-sensitivity cardiac troponin testing is to rule out MI, rather than to rule it in, Dr. Mills explained.
HiSTORIC was a 2-year, prospective, stepped-wedge, cluster-randomized, controlled trial including 31,492 consecutive patients with suspected ACS who presented to seven participating hospitals in Scotland. Patients were randomized, at the hospital level, to one of two management pathways. The control group got a standard guideline-recommended strategy involving high-sensitivity cardiac troponin I testing upon presentation and again 6-12 hours later, with MI being ruled out if the troponin levels were not above the 99th percentile.
In contrast, the novel early rule-out strategy worked as follows: If the patient presented with at least 2 hours of symptoms and the initial troponin I level was below 5 ng/L, then MI was ruled out and the patient was triaged straightaway for outpatient management. If the level was above the 99th percentile, the patient was admitted for serial testing to be done 6-12 hours after symptom onset. And for an intermediate test result – that is, a troponin level between 5 ng/L and the 99th percentile – patients remained in the ED for retesting 3 hours from the time of presentation, and were subsequently admitted only if their troponin level was rising.
Using the accelerated rule-out strategy, two-thirds of patients were quickly discharged from the ED on the basis of a troponin level below 5 ng/mL, and another 7% were ruled out for MI and discharged from the ED after a 3-hour stay on the basis of their second test.
The primary efficacy outcome was length of stay from initial presentation to the ED to discharge. The duration was 10.1 hours with the guideline-recommended pathway and 6.8 hours with the accelerated rule-out pathway, for a statistically significant and clinically meaningful 3.3-hour difference. Moreover, the proportion of patients discharged directly from the ED without hospital admission increased from 53% to 74%, a 57% jump.
The primary safety outcome was the rate of MI or cardiac death post discharge. The rates at 30 days and 1 year were 0.4% and 2.6%, respectively, in the standard-pathway group, compared with 0.3% and 1.8% with the early rule-out pathway. Those between-group differences favoring the accelerated rule-out pathway weren’t statistically significant, but they provided reassurance that the novel pathway was safe.
Of note, this was the first-ever randomized trial to evaluate the safety and efficacy of an early rule-out pathway. Other rapid diagnostic pathways are largely based on observational experience and expert opinion, Dr. Mills said.
The assay utilized in the HiSTORIC trial was the Abbott Diagnostics Architect high sensitivity assay. The 5-ng/L threshold for early rule-out was chosen for the trial because an earlier study by Dr. Mills and coinvestigators showed that a level below that cutoff had a 99.6% negative predictive value for MI (Lancet. 2015 Dec 19;386[10012]:2481-8)
The early rule-out pathway was deliberately designed to be simple and pragmatic, according to the cardiologist. “One of the most remarkable observations in this trial was the adherence to the pathway. We prespecified three criteria to evaluate this and demonstrated adherence rates of 86%-92% for each of these criteria. This was despite the pathway being implemented in all consecutive patients at seven different hospitals and used by many hundreds of different clinicians.”
Discussant Hugo A. Katus, MD, called the HiSTORIC study “a really urgently needed and very well-conducted trial.”
“There were very consistently low MI and cardiac death rates at 30 days and 1 year. So this really works,” commented Dr. Katus, who is chief of internal medicine and director of the department of cardiovascular medicine at Heidelberg (Germany) University.
“Accelerated rule-out high-sensitivity cardiac troponin protocols are here to stay,” he declared.
However, Dr. Katus voiced a concern: “By early discharge as rule out, are other life-threatening conditions ignored?”
He raised this issue because of what he views as the substantial 1-year all-cause mortality and return-to-hospital rates of 5.8% and 39.2% in the standard-pathway group and 5.2% and 38.9% in the accelerated rule-out patients in HiSTORIC. An accelerated rule-out strategy should not prohibit a careful clinical work-up, he emphasized.
Dr. Mills discussed the results in a video interview.
The HiSTORIC trial was funded by the British Heart Foundation. Dr. Mills reported receiving research grants from Abbott Diagnostics and Siemens.
Simultaneous with Dr. Mills’ presentation of the HiSTORIC trial results at the ESC congress, an earlier study that formed the scientific basis for the investigators’ decision to employ distinct risk stratification and diagnostic thresholds for cardiac troponin testing was published online (Circulation. 2019 Sep 1. doi: 10.1161/CIRCULATIONAHA.119.042866). The actual HiSTORIC trial results will be published later.
Dr. Katus reported holding a patent for a cardiac troponin T test and serving as a consultant to AstraZeneca, Bayer, Boehringer Ingelheim, and Novo Nordisk.
PARIS – An accelerated rule-out pathway, reliant upon a single high-sensitivity cardiac troponin test upon presentation to the ED with suspected acute coronary syndrome, reduced length of stay and hospital admission rates without increasing cardiac events at 30 days or 1 year in a major Scottish study.
“We conclude that implementation of this early rule-out pathway is both effective and safe, and adoption of this pathway will have major benefits for patients and health care systems,” Nicholas L. Mills, MBChB, PhD, said in presenting the results of the HiSTORIC (High-Sensitivity Cardiac Troponin at Presentation to Rule Out Myocardial Infarction) trial at the annual congress of the European Society of Cardiology.
Indeed, in the Unites States, where more than 20 million people per year present to EDs with suspected ACS, the 3.3-hour reduction in length of stay achieved in the HiSTORIC trial by implementing the accelerated rule-out pathway would add up to a $3.6 billion annual savings in bed occupancy alone, according to Dr. Mills, who is chair of cardiology at the University of Edinburgh.
The HiSTORIC pathway incorporates separate thresholds for risk stratification and diagnosis. This strategy is based on an accumulation of persuasive evidence that the major advantage of high-sensitivity cardiac troponin testing is to rule out MI, rather than to rule it in, Dr. Mills explained.
HiSTORIC was a 2-year, prospective, stepped-wedge, cluster-randomized, controlled trial including 31,492 consecutive patients with suspected ACS who presented to seven participating hospitals in Scotland. Patients were randomized, at the hospital level, to one of two management pathways. The control group got a standard guideline-recommended strategy involving high-sensitivity cardiac troponin I testing upon presentation and again 6-12 hours later, with MI being ruled out if the troponin levels were not above the 99th percentile.
In contrast, the novel early rule-out strategy worked as follows: If the patient presented with at least 2 hours of symptoms and the initial troponin I level was below 5 ng/L, then MI was ruled out and the patient was triaged straightaway for outpatient management. If the level was above the 99th percentile, the patient was admitted for serial testing to be done 6-12 hours after symptom onset. And for an intermediate test result – that is, a troponin level between 5 ng/L and the 99th percentile – patients remained in the ED for retesting 3 hours from the time of presentation, and were subsequently admitted only if their troponin level was rising.
Using the accelerated rule-out strategy, two-thirds of patients were quickly discharged from the ED on the basis of a troponin level below 5 ng/mL, and another 7% were ruled out for MI and discharged from the ED after a 3-hour stay on the basis of their second test.
The primary efficacy outcome was length of stay from initial presentation to the ED to discharge. The duration was 10.1 hours with the guideline-recommended pathway and 6.8 hours with the accelerated rule-out pathway, for a statistically significant and clinically meaningful 3.3-hour difference. Moreover, the proportion of patients discharged directly from the ED without hospital admission increased from 53% to 74%, a 57% jump.
The primary safety outcome was the rate of MI or cardiac death post discharge. The rates at 30 days and 1 year were 0.4% and 2.6%, respectively, in the standard-pathway group, compared with 0.3% and 1.8% with the early rule-out pathway. Those between-group differences favoring the accelerated rule-out pathway weren’t statistically significant, but they provided reassurance that the novel pathway was safe.
Of note, this was the first-ever randomized trial to evaluate the safety and efficacy of an early rule-out pathway. Other rapid diagnostic pathways are largely based on observational experience and expert opinion, Dr. Mills said.
The assay utilized in the HiSTORIC trial was the Abbott Diagnostics Architect high sensitivity assay. The 5-ng/L threshold for early rule-out was chosen for the trial because an earlier study by Dr. Mills and coinvestigators showed that a level below that cutoff had a 99.6% negative predictive value for MI (Lancet. 2015 Dec 19;386[10012]:2481-8)
The early rule-out pathway was deliberately designed to be simple and pragmatic, according to the cardiologist. “One of the most remarkable observations in this trial was the adherence to the pathway. We prespecified three criteria to evaluate this and demonstrated adherence rates of 86%-92% for each of these criteria. This was despite the pathway being implemented in all consecutive patients at seven different hospitals and used by many hundreds of different clinicians.”
Discussant Hugo A. Katus, MD, called the HiSTORIC study “a really urgently needed and very well-conducted trial.”
“There were very consistently low MI and cardiac death rates at 30 days and 1 year. So this really works,” commented Dr. Katus, who is chief of internal medicine and director of the department of cardiovascular medicine at Heidelberg (Germany) University.
“Accelerated rule-out high-sensitivity cardiac troponin protocols are here to stay,” he declared.
However, Dr. Katus voiced a concern: “By early discharge as rule out, are other life-threatening conditions ignored?”
He raised this issue because of what he views as the substantial 1-year all-cause mortality and return-to-hospital rates of 5.8% and 39.2% in the standard-pathway group and 5.2% and 38.9% in the accelerated rule-out patients in HiSTORIC. An accelerated rule-out strategy should not prohibit a careful clinical work-up, he emphasized.
Dr. Mills discussed the results in a video interview.
The HiSTORIC trial was funded by the British Heart Foundation. Dr. Mills reported receiving research grants from Abbott Diagnostics and Siemens.
Simultaneous with Dr. Mills’ presentation of the HiSTORIC trial results at the ESC congress, an earlier study that formed the scientific basis for the investigators’ decision to employ distinct risk stratification and diagnostic thresholds for cardiac troponin testing was published online (Circulation. 2019 Sep 1. doi: 10.1161/CIRCULATIONAHA.119.042866). The actual HiSTORIC trial results will be published later.
Dr. Katus reported holding a patent for a cardiac troponin T test and serving as a consultant to AstraZeneca, Bayer, Boehringer Ingelheim, and Novo Nordisk.
REPORTING FROM THE ESC CONGRESS 2019

Clinical Pharmacists Improve Patient Outcomes and Expand Access to Care
The US is in the midst of a chronic disease crisis. According to the latest published data available, 60% of Americans have at least 1 chronic condition, and 42% have ≥ 2 chronic conditions.1 Estimates by the Health Resources and Services Administration (HRSA) indicate a current shortfall of 13 800 primary care physicians and a projected escalation of that shortage to be between 14 800 and 49 300 physicians by the year 2030.2
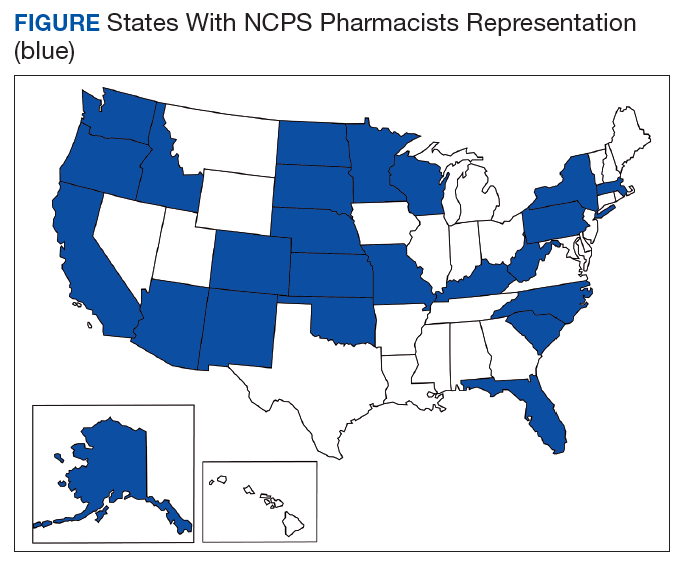
The US Public Health Service (USPHS) has used pharmacists since 1930 to provide direct patient care to underserved and vulnerable populations. Clinical pharmacists currently serve in direct patient care roles within the Indian Health Service (IHS), Federal Bureau of Prisons (BOP), Immigration and Customs Enforcement (ICE), and the United States Coast Guard (USCG) in many states (Figure). These pharmacists play a vital role in improving access to care and delivering quality care by managing acute and chronic diseases in collaborative practice settings and pharmacist-managed clinics.
It has previously been reported that in the face of physician shortages and growing demand for primary health care providers, pharmacists are well-equipped and motivated to meet this demand.3 A review of the previous 2 years of outcomes reported by clinical pharmacists certified through the USPHS National Clinical Pharmacy Specialist (NCPS) Committee are presented to demonstrate the impact of pharmacists in advancing the health of the populations they serve and to showcase a model for ameliorating the ongoing physician shortage.
Background
The USPHS NCPS Committee serves to promote uniform competency among clinical pharmacists by establishing national standards for protocols, collaborative practice agreements (CPAs), credentialing and privileging of pharmacists, and by collecting, reviewing, and publishing health care outcomes. The committee, whose constituents include pharmacist and physician subject matter experts from across USPHS agencies, reviews applications and protocols and certifies pharmacists (civilian and uniformed) to recognize an advanced scope of practice in managing various diseases and optimizing medication therapy. NCPScertified pharmacists manage a wide spectrum of diseases, including coagulopathy, asthma, diabetes mellitus (DM), hepatitis C, HIV, hypertension, pain, seizure disorders, and tobacco use disorders.
Clinical pharmacists practicing chronic disease management establish a clinical service in collaboration with 1 or more physicians, physician assistants, or nurse practitioners. In this collaborative practice, the health care practitioner(s) refer patients to be managed by a pharmacist for specific medical needs, such as anticoagulation management, or for holistic medication- focused care (eg, cardiovascular risk reduction, DM management, HIV, hepatitis, or mental health). The pharmacist may order and interpret laboratory tests, check vital signs, perform a limited physical examination, and gather other pertinent information from the patient and the medical record in order to provide the best possible care to the patient.
Medications may be started, stopped, or adjusted, education is provided, and therapeutic lifestyle interventions may be recommended. The pharmacist-run clinic provides the patient more frequent interaction with a health care professional (pharmacist) and focused disease management. As a result, pharmacists increase access to care and allow the medical team to handle a larger panel of patients as the practitioner delegates specified diseases to the pharmacist- managed clinic(s). The number of NCPS-certified pharmacists grew 46% from 2012 (n = 230) to 2017 (n = 336), reflecting an evolution of pharmacists’ practice to better meet the need of patients across the nation.
Methods
The NCPS Committee requires NCPS pharmacists to report data annually from all patients referred for pharmacist management for specific diseases in which they have been certified. The data reflect the patient’s clinical outcome goal status at the time of referral as well as the same status at the end of the reporting period or on release from the pharmacist-run clinic. These data describe the impact prescribing pharmacists have on patients reaching clinical outcome goals acting as the team member specializing in the medication selection and dosing aspect of care.
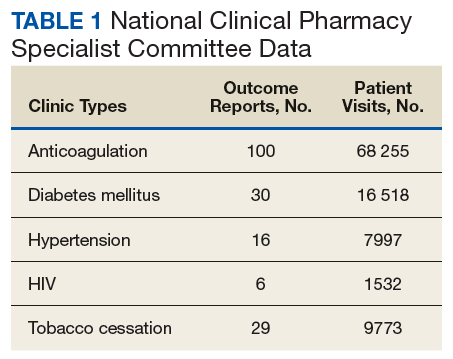
These records were reviewed for the fiscal year (FY) periods of October 1, 2015 to September 30, 2016 (FY 2016) and October 1, 2016 to September 30, 2017 (FY 2017). A systematic review of submitted reports resulted in 181 reports that included all requested data points for the disease as published here for FYs 2016 and 2017. These include 66 reports from FY 2016 and 115 reports from FY 2017; they cover 76 BOP and IHS facilities located across 24 states. Table 1 shows the number of outcome reports collected from 104 075 patient visits in pharmacist-run clinics in FYs 2016 and 2017.
Results
The following tables represent the standardized outcomes collected by NCPS-certified pharmacists providing direct patient care. Patients on anticoagulants (eg, warfarin) require special monitoring and education for drug interactions and adverse effects. NCPS-certified pharmacists were able to achieve a mean patient time in therapeutic range (TTR) of 67.6% (regardless of indication) over the 2 years (calculated per each facility by Rosendaal method of linear interpolation then combined in a weighted average per visit). The TTR produced by NCPS-certified pharmacists are consistent with Chest Guidelines and Expert Panel Report suggesting that TTR should be between 65% and 70%.4 Table 2 shows data from 100 reports with 68 255 patient visits for anticoagulation management.
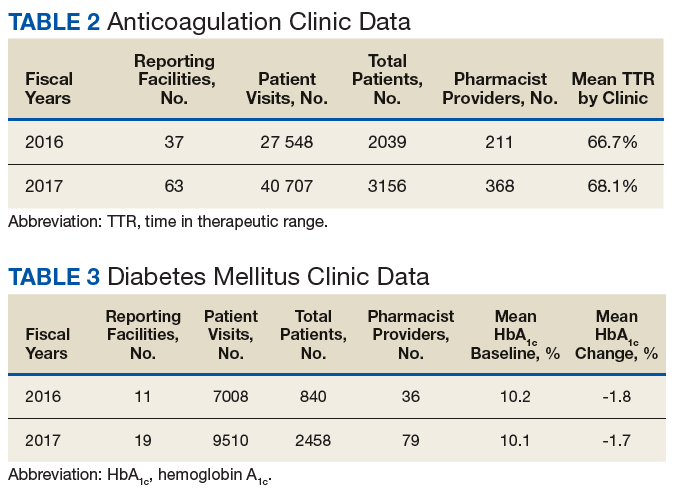
DM management can be complex and time-intensive. NCPS data indicate pharmacist intervention resulted in a mean decrease in hemoglobin A1c (HbA1c) of 1.8% from a baseline of 10.2% (decrease calculated per each facility then combined by weighted average per visit). Table 3 shows data from 30 reports with 16 518 patient visits for DM care.
In addition to diet and exercise, medication management plays a vital role in managing hypertension. Patients managed by an NCPS-certified pharmacist experienced a mean decrease in blood pressure from 144/83 to 133/77, putting them in goal for both systolic and diastolic ranges (decrease calculated per each facility then combined by weighted average per visit). Table 4 shows data from 16 reports and 7997 patient visits for treatment of hypertension.
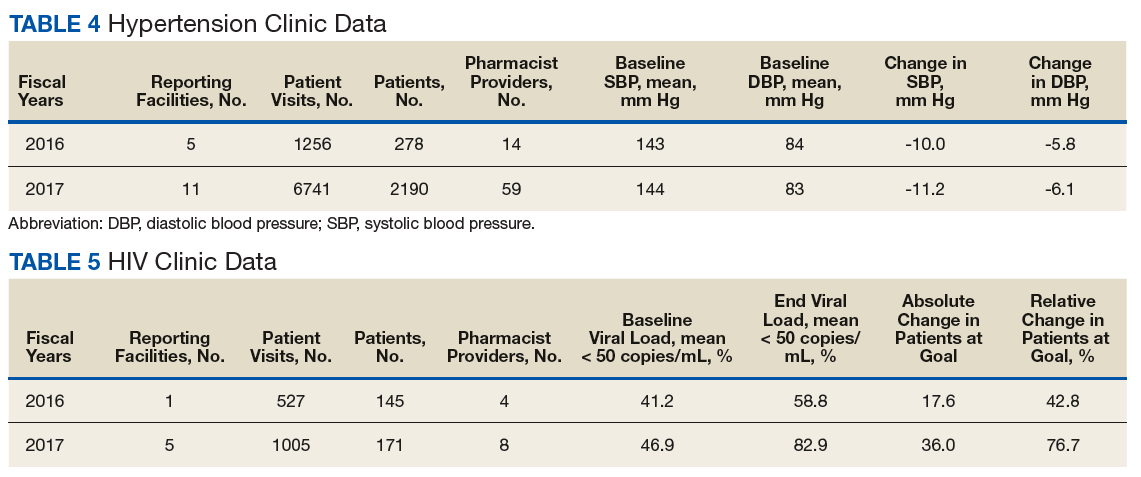
HIV viral suppression is vital in order to best manage patients with HIV and reduce the risk of transmission. Pharmacistled clinics have shown a 32.9% absolute improvement in patients at goal (viral load < 50 copies/mL), from a mean baseline of 46.0% to a mean final assessment of 71.6% of patients at goal (combined by weighted average visits). Table 5 shows data from 6 reports covering 1532 patient encounters for management of HIV.
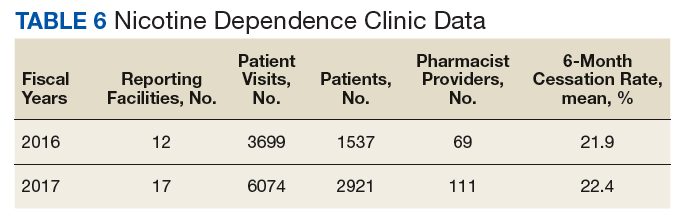
Nicotine dependence includes the use of cigarettes, cigars, pipe tobacco, chewing tobacco, and vaping products containing nicotine. NCPS-certified pharmacists have successfully helped patients improve their chance of quitting, with a 6-month quit rate of 22.2% (quit rate calculated per each facility then combined by weighted average by visits), which is higher than the national average of 9.4% as reported by the Centers for Disease and Control and Prevention. 5 Table 6 shows 29 reports covering 9773 patient visits for treatment of nicotine dependence.
Discussion
These data demonstrate the ability of advanced practice pharmacists in multiple locations within the federal sector to improve targeted clinical outcomes in patients with varying diseases. These results are strengthened by their varied origins as well as the improvements observed across the board. Limitations include the general lack of a comparable dataset, manual method of selfreporting by the individual facilities, and the relatively limited array of diseases reported. Although NCPS-certified pharmacists are currently providing care for patients with hepatitis C, asthma, seizure, pain and other diseases not reported here, there are insufficient data collected for FYs 2016 and 2017 to merit inclusion within this report.
Pharmacists are trusted, readily available medication experts. In a clinical role, NCPS-certified pharmacists have increased access to primary care services and demonstrated beneficial impact on important health outcomes as exhibited by the data reported above. Clinical pharmacy is a growing field, and NCPS has displayed continual growth in both the number of NCPS-certified pharmacists and the number of patient encounters performed by these providers. As more pharmacists in all settings collaborate with medical providers to offer high-quality clinical care, these providers will have more opportunity to delegate disease management. Continued reporting of clinical pharmacy outcomes is expected to increase confidence in pharmacists as primary care providers, increase utilization of pharmacy clinical services, and assist in easing the burden of primary care provider shortages across our nation.
Although these outcomes indicate demonstrable benefit in patient-centered outcomes, the need for ongoing assessment and continued improvement is not obviated. Future efforts may benefit from a comparison of alternative approaches to better facilitate the establishment of best practices. Alignment of clinical outcomes with the Centers for Medicare and Medicaid Services (CMS) Electronic Clinical Quality Measures, where applicable, also may prove beneficial by automating the reporting process and thereby decreasing the burden of reporting as well as providing an avenue for standard comparison across multiple populations. Clinical pharmacy interventions have positive outcomes based on the NCPS model, and the NCPS Committee invites other clinical settings to report outcomes data with which to compare.
Conclusion
The NCPS Committee has documented positive outcomes of clinical pharmacy intervention and anticipates growth of the pharmacy profession as additional states and health systems recognize the capacity of the pharmacist to provide high-quality, multidisciplinary patient care. Clinical pharmacists are prepared to address critical health care needs as the US continues to face a PCP shortage.2 The NCPS Committee challenges those participating in clinical pharmacy practice to report outcomes to amplify this body of evidence.
Acknowledgments
NCPS-certified pharmacists provided the outcomes detailed in this report. For document review and edits: Federal Bureau of Prison Publication Review Workgroup; RADM Ty Bingham, USPHS; CAPT Cindy Gunderson, USPHS; CAPT Kevin Brooks, USPHS.
1. Buttorff C, Ruder T, Bauman M. Multiple Chronic Conditions in the United States. Santa Monica, CA: Rand Corp; 2017.
2. Dall T, West T, Chakrabarti R, Reynolds R, Iacobucci W. The complexities of physician supply and demand: projections from 2016 to 2030, 2018 update. Association of American Medical Colleges. March 2018.
3. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. A report to the U.S. Surgeon General 2011. https://www .accp.com/docs/positions/misc/improving_patient_and _health_system_outcomes.pdf. Updated December 2011. Accessed September 11, 2019.
4. Lip G, Banerjee A, Boriani G, et al. Antithrombotic therapy for atrial fibrillation. CHEST guideline and Expert Panel Report. Chest. 2018;154(5):1121-1201.
5. Babb S, Marlarcher A, Schauer G, Asman K, Jamal A. Quitting smoking among adults—United States, 2000-2015. MMWR Morb Mortal Wkly Rep. 2017;65(52):1457-1464.
The US is in the midst of a chronic disease crisis. According to the latest published data available, 60% of Americans have at least 1 chronic condition, and 42% have ≥ 2 chronic conditions.1 Estimates by the Health Resources and Services Administration (HRSA) indicate a current shortfall of 13 800 primary care physicians and a projected escalation of that shortage to be between 14 800 and 49 300 physicians by the year 2030.2
The US Public Health Service (USPHS) has used pharmacists since 1930 to provide direct patient care to underserved and vulnerable populations. Clinical pharmacists currently serve in direct patient care roles within the Indian Health Service (IHS), Federal Bureau of Prisons (BOP), Immigration and Customs Enforcement (ICE), and the United States Coast Guard (USCG) in many states (Figure). These pharmacists play a vital role in improving access to care and delivering quality care by managing acute and chronic diseases in collaborative practice settings and pharmacist-managed clinics.
It has previously been reported that in the face of physician shortages and growing demand for primary health care providers, pharmacists are well-equipped and motivated to meet this demand.3 A review of the previous 2 years of outcomes reported by clinical pharmacists certified through the USPHS National Clinical Pharmacy Specialist (NCPS) Committee are presented to demonstrate the impact of pharmacists in advancing the health of the populations they serve and to showcase a model for ameliorating the ongoing physician shortage.
Background
The USPHS NCPS Committee serves to promote uniform competency among clinical pharmacists by establishing national standards for protocols, collaborative practice agreements (CPAs), credentialing and privileging of pharmacists, and by collecting, reviewing, and publishing health care outcomes. The committee, whose constituents include pharmacist and physician subject matter experts from across USPHS agencies, reviews applications and protocols and certifies pharmacists (civilian and uniformed) to recognize an advanced scope of practice in managing various diseases and optimizing medication therapy. NCPScertified pharmacists manage a wide spectrum of diseases, including coagulopathy, asthma, diabetes mellitus (DM), hepatitis C, HIV, hypertension, pain, seizure disorders, and tobacco use disorders.
Clinical pharmacists practicing chronic disease management establish a clinical service in collaboration with 1 or more physicians, physician assistants, or nurse practitioners. In this collaborative practice, the health care practitioner(s) refer patients to be managed by a pharmacist for specific medical needs, such as anticoagulation management, or for holistic medication- focused care (eg, cardiovascular risk reduction, DM management, HIV, hepatitis, or mental health). The pharmacist may order and interpret laboratory tests, check vital signs, perform a limited physical examination, and gather other pertinent information from the patient and the medical record in order to provide the best possible care to the patient.
Medications may be started, stopped, or adjusted, education is provided, and therapeutic lifestyle interventions may be recommended. The pharmacist-run clinic provides the patient more frequent interaction with a health care professional (pharmacist) and focused disease management. As a result, pharmacists increase access to care and allow the medical team to handle a larger panel of patients as the practitioner delegates specified diseases to the pharmacist- managed clinic(s). The number of NCPS-certified pharmacists grew 46% from 2012 (n = 230) to 2017 (n = 336), reflecting an evolution of pharmacists’ practice to better meet the need of patients across the nation.
Methods
The NCPS Committee requires NCPS pharmacists to report data annually from all patients referred for pharmacist management for specific diseases in which they have been certified. The data reflect the patient’s clinical outcome goal status at the time of referral as well as the same status at the end of the reporting period or on release from the pharmacist-run clinic. These data describe the impact prescribing pharmacists have on patients reaching clinical outcome goals acting as the team member specializing in the medication selection and dosing aspect of care.
These records were reviewed for the fiscal year (FY) periods of October 1, 2015 to September 30, 2016 (FY 2016) and October 1, 2016 to September 30, 2017 (FY 2017). A systematic review of submitted reports resulted in 181 reports that included all requested data points for the disease as published here for FYs 2016 and 2017. These include 66 reports from FY 2016 and 115 reports from FY 2017; they cover 76 BOP and IHS facilities located across 24 states. Table 1 shows the number of outcome reports collected from 104 075 patient visits in pharmacist-run clinics in FYs 2016 and 2017.
Results
The following tables represent the standardized outcomes collected by NCPS-certified pharmacists providing direct patient care. Patients on anticoagulants (eg, warfarin) require special monitoring and education for drug interactions and adverse effects. NCPS-certified pharmacists were able to achieve a mean patient time in therapeutic range (TTR) of 67.6% (regardless of indication) over the 2 years (calculated per each facility by Rosendaal method of linear interpolation then combined in a weighted average per visit). The TTR produced by NCPS-certified pharmacists are consistent with Chest Guidelines and Expert Panel Report suggesting that TTR should be between 65% and 70%.4 Table 2 shows data from 100 reports with 68 255 patient visits for anticoagulation management.
DM management can be complex and time-intensive. NCPS data indicate pharmacist intervention resulted in a mean decrease in hemoglobin A1c (HbA1c) of 1.8% from a baseline of 10.2% (decrease calculated per each facility then combined by weighted average per visit). Table 3 shows data from 30 reports with 16 518 patient visits for DM care.
In addition to diet and exercise, medication management plays a vital role in managing hypertension. Patients managed by an NCPS-certified pharmacist experienced a mean decrease in blood pressure from 144/83 to 133/77, putting them in goal for both systolic and diastolic ranges (decrease calculated per each facility then combined by weighted average per visit). Table 4 shows data from 16 reports and 7997 patient visits for treatment of hypertension.
HIV viral suppression is vital in order to best manage patients with HIV and reduce the risk of transmission. Pharmacistled clinics have shown a 32.9% absolute improvement in patients at goal (viral load < 50 copies/mL), from a mean baseline of 46.0% to a mean final assessment of 71.6% of patients at goal (combined by weighted average visits). Table 5 shows data from 6 reports covering 1532 patient encounters for management of HIV.
Nicotine dependence includes the use of cigarettes, cigars, pipe tobacco, chewing tobacco, and vaping products containing nicotine. NCPS-certified pharmacists have successfully helped patients improve their chance of quitting, with a 6-month quit rate of 22.2% (quit rate calculated per each facility then combined by weighted average by visits), which is higher than the national average of 9.4% as reported by the Centers for Disease and Control and Prevention. 5 Table 6 shows 29 reports covering 9773 patient visits for treatment of nicotine dependence.
Discussion
These data demonstrate the ability of advanced practice pharmacists in multiple locations within the federal sector to improve targeted clinical outcomes in patients with varying diseases. These results are strengthened by their varied origins as well as the improvements observed across the board. Limitations include the general lack of a comparable dataset, manual method of selfreporting by the individual facilities, and the relatively limited array of diseases reported. Although NCPS-certified pharmacists are currently providing care for patients with hepatitis C, asthma, seizure, pain and other diseases not reported here, there are insufficient data collected for FYs 2016 and 2017 to merit inclusion within this report.
Pharmacists are trusted, readily available medication experts. In a clinical role, NCPS-certified pharmacists have increased access to primary care services and demonstrated beneficial impact on important health outcomes as exhibited by the data reported above. Clinical pharmacy is a growing field, and NCPS has displayed continual growth in both the number of NCPS-certified pharmacists and the number of patient encounters performed by these providers. As more pharmacists in all settings collaborate with medical providers to offer high-quality clinical care, these providers will have more opportunity to delegate disease management. Continued reporting of clinical pharmacy outcomes is expected to increase confidence in pharmacists as primary care providers, increase utilization of pharmacy clinical services, and assist in easing the burden of primary care provider shortages across our nation.
Although these outcomes indicate demonstrable benefit in patient-centered outcomes, the need for ongoing assessment and continued improvement is not obviated. Future efforts may benefit from a comparison of alternative approaches to better facilitate the establishment of best practices. Alignment of clinical outcomes with the Centers for Medicare and Medicaid Services (CMS) Electronic Clinical Quality Measures, where applicable, also may prove beneficial by automating the reporting process and thereby decreasing the burden of reporting as well as providing an avenue for standard comparison across multiple populations. Clinical pharmacy interventions have positive outcomes based on the NCPS model, and the NCPS Committee invites other clinical settings to report outcomes data with which to compare.
Conclusion
The NCPS Committee has documented positive outcomes of clinical pharmacy intervention and anticipates growth of the pharmacy profession as additional states and health systems recognize the capacity of the pharmacist to provide high-quality, multidisciplinary patient care. Clinical pharmacists are prepared to address critical health care needs as the US continues to face a PCP shortage.2 The NCPS Committee challenges those participating in clinical pharmacy practice to report outcomes to amplify this body of evidence.
Acknowledgments
NCPS-certified pharmacists provided the outcomes detailed in this report. For document review and edits: Federal Bureau of Prison Publication Review Workgroup; RADM Ty Bingham, USPHS; CAPT Cindy Gunderson, USPHS; CAPT Kevin Brooks, USPHS.
The US is in the midst of a chronic disease crisis. According to the latest published data available, 60% of Americans have at least 1 chronic condition, and 42% have ≥ 2 chronic conditions.1 Estimates by the Health Resources and Services Administration (HRSA) indicate a current shortfall of 13 800 primary care physicians and a projected escalation of that shortage to be between 14 800 and 49 300 physicians by the year 2030.2
The US Public Health Service (USPHS) has used pharmacists since 1930 to provide direct patient care to underserved and vulnerable populations. Clinical pharmacists currently serve in direct patient care roles within the Indian Health Service (IHS), Federal Bureau of Prisons (BOP), Immigration and Customs Enforcement (ICE), and the United States Coast Guard (USCG) in many states (Figure). These pharmacists play a vital role in improving access to care and delivering quality care by managing acute and chronic diseases in collaborative practice settings and pharmacist-managed clinics.
It has previously been reported that in the face of physician shortages and growing demand for primary health care providers, pharmacists are well-equipped and motivated to meet this demand.3 A review of the previous 2 years of outcomes reported by clinical pharmacists certified through the USPHS National Clinical Pharmacy Specialist (NCPS) Committee are presented to demonstrate the impact of pharmacists in advancing the health of the populations they serve and to showcase a model for ameliorating the ongoing physician shortage.
Background
The USPHS NCPS Committee serves to promote uniform competency among clinical pharmacists by establishing national standards for protocols, collaborative practice agreements (CPAs), credentialing and privileging of pharmacists, and by collecting, reviewing, and publishing health care outcomes. The committee, whose constituents include pharmacist and physician subject matter experts from across USPHS agencies, reviews applications and protocols and certifies pharmacists (civilian and uniformed) to recognize an advanced scope of practice in managing various diseases and optimizing medication therapy. NCPScertified pharmacists manage a wide spectrum of diseases, including coagulopathy, asthma, diabetes mellitus (DM), hepatitis C, HIV, hypertension, pain, seizure disorders, and tobacco use disorders.
Clinical pharmacists practicing chronic disease management establish a clinical service in collaboration with 1 or more physicians, physician assistants, or nurse practitioners. In this collaborative practice, the health care practitioner(s) refer patients to be managed by a pharmacist for specific medical needs, such as anticoagulation management, or for holistic medication- focused care (eg, cardiovascular risk reduction, DM management, HIV, hepatitis, or mental health). The pharmacist may order and interpret laboratory tests, check vital signs, perform a limited physical examination, and gather other pertinent information from the patient and the medical record in order to provide the best possible care to the patient.
Medications may be started, stopped, or adjusted, education is provided, and therapeutic lifestyle interventions may be recommended. The pharmacist-run clinic provides the patient more frequent interaction with a health care professional (pharmacist) and focused disease management. As a result, pharmacists increase access to care and allow the medical team to handle a larger panel of patients as the practitioner delegates specified diseases to the pharmacist- managed clinic(s). The number of NCPS-certified pharmacists grew 46% from 2012 (n = 230) to 2017 (n = 336), reflecting an evolution of pharmacists’ practice to better meet the need of patients across the nation.
Methods
The NCPS Committee requires NCPS pharmacists to report data annually from all patients referred for pharmacist management for specific diseases in which they have been certified. The data reflect the patient’s clinical outcome goal status at the time of referral as well as the same status at the end of the reporting period or on release from the pharmacist-run clinic. These data describe the impact prescribing pharmacists have on patients reaching clinical outcome goals acting as the team member specializing in the medication selection and dosing aspect of care.
These records were reviewed for the fiscal year (FY) periods of October 1, 2015 to September 30, 2016 (FY 2016) and October 1, 2016 to September 30, 2017 (FY 2017). A systematic review of submitted reports resulted in 181 reports that included all requested data points for the disease as published here for FYs 2016 and 2017. These include 66 reports from FY 2016 and 115 reports from FY 2017; they cover 76 BOP and IHS facilities located across 24 states. Table 1 shows the number of outcome reports collected from 104 075 patient visits in pharmacist-run clinics in FYs 2016 and 2017.
Results
The following tables represent the standardized outcomes collected by NCPS-certified pharmacists providing direct patient care. Patients on anticoagulants (eg, warfarin) require special monitoring and education for drug interactions and adverse effects. NCPS-certified pharmacists were able to achieve a mean patient time in therapeutic range (TTR) of 67.6% (regardless of indication) over the 2 years (calculated per each facility by Rosendaal method of linear interpolation then combined in a weighted average per visit). The TTR produced by NCPS-certified pharmacists are consistent with Chest Guidelines and Expert Panel Report suggesting that TTR should be between 65% and 70%.4 Table 2 shows data from 100 reports with 68 255 patient visits for anticoagulation management.
DM management can be complex and time-intensive. NCPS data indicate pharmacist intervention resulted in a mean decrease in hemoglobin A1c (HbA1c) of 1.8% from a baseline of 10.2% (decrease calculated per each facility then combined by weighted average per visit). Table 3 shows data from 30 reports with 16 518 patient visits for DM care.
In addition to diet and exercise, medication management plays a vital role in managing hypertension. Patients managed by an NCPS-certified pharmacist experienced a mean decrease in blood pressure from 144/83 to 133/77, putting them in goal for both systolic and diastolic ranges (decrease calculated per each facility then combined by weighted average per visit). Table 4 shows data from 16 reports and 7997 patient visits for treatment of hypertension.
HIV viral suppression is vital in order to best manage patients with HIV and reduce the risk of transmission. Pharmacistled clinics have shown a 32.9% absolute improvement in patients at goal (viral load < 50 copies/mL), from a mean baseline of 46.0% to a mean final assessment of 71.6% of patients at goal (combined by weighted average visits). Table 5 shows data from 6 reports covering 1532 patient encounters for management of HIV.
Nicotine dependence includes the use of cigarettes, cigars, pipe tobacco, chewing tobacco, and vaping products containing nicotine. NCPS-certified pharmacists have successfully helped patients improve their chance of quitting, with a 6-month quit rate of 22.2% (quit rate calculated per each facility then combined by weighted average by visits), which is higher than the national average of 9.4% as reported by the Centers for Disease and Control and Prevention. 5 Table 6 shows 29 reports covering 9773 patient visits for treatment of nicotine dependence.
Discussion
These data demonstrate the ability of advanced practice pharmacists in multiple locations within the federal sector to improve targeted clinical outcomes in patients with varying diseases. These results are strengthened by their varied origins as well as the improvements observed across the board. Limitations include the general lack of a comparable dataset, manual method of selfreporting by the individual facilities, and the relatively limited array of diseases reported. Although NCPS-certified pharmacists are currently providing care for patients with hepatitis C, asthma, seizure, pain and other diseases not reported here, there are insufficient data collected for FYs 2016 and 2017 to merit inclusion within this report.
Pharmacists are trusted, readily available medication experts. In a clinical role, NCPS-certified pharmacists have increased access to primary care services and demonstrated beneficial impact on important health outcomes as exhibited by the data reported above. Clinical pharmacy is a growing field, and NCPS has displayed continual growth in both the number of NCPS-certified pharmacists and the number of patient encounters performed by these providers. As more pharmacists in all settings collaborate with medical providers to offer high-quality clinical care, these providers will have more opportunity to delegate disease management. Continued reporting of clinical pharmacy outcomes is expected to increase confidence in pharmacists as primary care providers, increase utilization of pharmacy clinical services, and assist in easing the burden of primary care provider shortages across our nation.
Although these outcomes indicate demonstrable benefit in patient-centered outcomes, the need for ongoing assessment and continued improvement is not obviated. Future efforts may benefit from a comparison of alternative approaches to better facilitate the establishment of best practices. Alignment of clinical outcomes with the Centers for Medicare and Medicaid Services (CMS) Electronic Clinical Quality Measures, where applicable, also may prove beneficial by automating the reporting process and thereby decreasing the burden of reporting as well as providing an avenue for standard comparison across multiple populations. Clinical pharmacy interventions have positive outcomes based on the NCPS model, and the NCPS Committee invites other clinical settings to report outcomes data with which to compare.
Conclusion
The NCPS Committee has documented positive outcomes of clinical pharmacy intervention and anticipates growth of the pharmacy profession as additional states and health systems recognize the capacity of the pharmacist to provide high-quality, multidisciplinary patient care. Clinical pharmacists are prepared to address critical health care needs as the US continues to face a PCP shortage.2 The NCPS Committee challenges those participating in clinical pharmacy practice to report outcomes to amplify this body of evidence.
Acknowledgments
NCPS-certified pharmacists provided the outcomes detailed in this report. For document review and edits: Federal Bureau of Prison Publication Review Workgroup; RADM Ty Bingham, USPHS; CAPT Cindy Gunderson, USPHS; CAPT Kevin Brooks, USPHS.
1. Buttorff C, Ruder T, Bauman M. Multiple Chronic Conditions in the United States. Santa Monica, CA: Rand Corp; 2017.
2. Dall T, West T, Chakrabarti R, Reynolds R, Iacobucci W. The complexities of physician supply and demand: projections from 2016 to 2030, 2018 update. Association of American Medical Colleges. March 2018.
3. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. A report to the U.S. Surgeon General 2011. https://www .accp.com/docs/positions/misc/improving_patient_and _health_system_outcomes.pdf. Updated December 2011. Accessed September 11, 2019.
4. Lip G, Banerjee A, Boriani G, et al. Antithrombotic therapy for atrial fibrillation. CHEST guideline and Expert Panel Report. Chest. 2018;154(5):1121-1201.
5. Babb S, Marlarcher A, Schauer G, Asman K, Jamal A. Quitting smoking among adults—United States, 2000-2015. MMWR Morb Mortal Wkly Rep. 2017;65(52):1457-1464.
1. Buttorff C, Ruder T, Bauman M. Multiple Chronic Conditions in the United States. Santa Monica, CA: Rand Corp; 2017.
2. Dall T, West T, Chakrabarti R, Reynolds R, Iacobucci W. The complexities of physician supply and demand: projections from 2016 to 2030, 2018 update. Association of American Medical Colleges. March 2018.
3. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. A report to the U.S. Surgeon General 2011. https://www .accp.com/docs/positions/misc/improving_patient_and _health_system_outcomes.pdf. Updated December 2011. Accessed September 11, 2019.
4. Lip G, Banerjee A, Boriani G, et al. Antithrombotic therapy for atrial fibrillation. CHEST guideline and Expert Panel Report. Chest. 2018;154(5):1121-1201.
5. Babb S, Marlarcher A, Schauer G, Asman K, Jamal A. Quitting smoking among adults—United States, 2000-2015. MMWR Morb Mortal Wkly Rep. 2017;65(52):1457-1464.
Osteoporosis remains a costly burden to older U.S. adults
ORLANDO – The burden of osteoporosis and fragility fractures in the United States remains high, particularly in older women and minorities, according to a speaker at the annual meeting of the American Society for Bone and Mineral Research.
For non-Hispanic Asian, non-Hispanic white, and Hispanic patients of various ethnic groups, as well as in women and older patients, osteoporosis and fragility fractures continue to be a problem, said Nicole C. Wright, PhD, MPH, of the department of epidemiology at the University of Alabama at Birmingham.
“It remains costly; it remains associated with more health care utilization,” Dr. Wright said. “We may be seeing some declines in some fragility fractures, but [we] are seeing increases in hip fractures.”
As part of the fourth edition of the U.S. Bone and Joint Initiative publication, “The Burden of Musculoskeletal Diseases in the United States,” Dr. Wright and colleagues examined the changes in osteoporosis burden between the third and fourth editions of the publication. They used data from the National Inpatient Sample (NIS) in 2013 and 2014 as well as the National Emergency Department Sample (NEDS) of national ED visits regardless of hospital admission status. In both databases, researchers analyzed data from adults aged 50 years or older where the primary discharge ICD-9 or ICD-10 code was a diagnosis of fracture.
Using National Health and Nutrition Examination Survey data, the researchers estimated an 11.0% osteoporosis prevalence for adults aged 50 years or older overall, a 16.5% prevalence in women, and a 5.1% prevalence in men as assessed by femoral neck and lumbar spine bone mineral density. Osteoporosis was most prevalent in Asian women (40.0%) and Asian men (7.5%), while there was a difference in prevalence in patients of Hispanic race depending on their origin; for example, Puerto Rican men had a higher prevalence of osteoporosis at 8.6%, compared with Hispanic men (2.3%) and non-Hispanic white men of other races (3.9%).
Of 19.5 million hospitalizations in the NIS database between 2013 and 2014, there were approximately 540,000 fragility fractures (2.8%), of which about 300,000 were hip fractures and about 100,000 discharges were for spine fractures, Dr. Wright said. In the NEDS database, the estimate of fragility fracture prevalence was 0.9% of 46.7 million ED visits between 2013 and 2014. Fracture prevalence was increased in women and in older age, with patients aged 80 years or older and those of non-Hispanic white race having the highest prevalence of hip fracture. However, she noted that NEDS data also showed higher prevalences of wrist and humerus fractures, which are not normally fractures that a patient visits the hospital as an inpatient for. “We need both data sets to ascertain fractures in the United States,” she said.
When examining fracture site trends over time, Dr. Wright and colleagues found hip fracture prevalence increased by 3.5% between 2010 and 2014, while there was a decrease of 11.9% in the prevalence of spine fractures over the same time period.
According to data from the Medical Expenditures Panel Survey, the direct cost of osteoporosis in aggregate was $73.6 billion between 2012 and 2014, which was 118% higher than between 1998 and 2000 when the costs were $28.1 billion. The costs were spread across ambulatory care, inpatient, and prescription costs equally, the researchers said.
Although the study was limited by examining fracture prevalence rather than incidence, the potential for missing some fractures based on methodology, and limited patient characteristics and follow-up information, the goal of the presentation was to highlight the new osteoporosis prevalence data and the continued burden of the disease.
“We hope that these new prevalence estimates continue to increase the awareness of osteoporosis and prevention,” she said.
Dr. Wright reported receiving grants from Amgen and serving as an expert witness for the law firm Norton Rose Fulbright and Pfizer.
SOURCE: Wright NC et al. ASBMR 2019, Abstract 1079.
ORLANDO – The burden of osteoporosis and fragility fractures in the United States remains high, particularly in older women and minorities, according to a speaker at the annual meeting of the American Society for Bone and Mineral Research.
For non-Hispanic Asian, non-Hispanic white, and Hispanic patients of various ethnic groups, as well as in women and older patients, osteoporosis and fragility fractures continue to be a problem, said Nicole C. Wright, PhD, MPH, of the department of epidemiology at the University of Alabama at Birmingham.
“It remains costly; it remains associated with more health care utilization,” Dr. Wright said. “We may be seeing some declines in some fragility fractures, but [we] are seeing increases in hip fractures.”
As part of the fourth edition of the U.S. Bone and Joint Initiative publication, “The Burden of Musculoskeletal Diseases in the United States,” Dr. Wright and colleagues examined the changes in osteoporosis burden between the third and fourth editions of the publication. They used data from the National Inpatient Sample (NIS) in 2013 and 2014 as well as the National Emergency Department Sample (NEDS) of national ED visits regardless of hospital admission status. In both databases, researchers analyzed data from adults aged 50 years or older where the primary discharge ICD-9 or ICD-10 code was a diagnosis of fracture.
Using National Health and Nutrition Examination Survey data, the researchers estimated an 11.0% osteoporosis prevalence for adults aged 50 years or older overall, a 16.5% prevalence in women, and a 5.1% prevalence in men as assessed by femoral neck and lumbar spine bone mineral density. Osteoporosis was most prevalent in Asian women (40.0%) and Asian men (7.5%), while there was a difference in prevalence in patients of Hispanic race depending on their origin; for example, Puerto Rican men had a higher prevalence of osteoporosis at 8.6%, compared with Hispanic men (2.3%) and non-Hispanic white men of other races (3.9%).
Of 19.5 million hospitalizations in the NIS database between 2013 and 2014, there were approximately 540,000 fragility fractures (2.8%), of which about 300,000 were hip fractures and about 100,000 discharges were for spine fractures, Dr. Wright said. In the NEDS database, the estimate of fragility fracture prevalence was 0.9% of 46.7 million ED visits between 2013 and 2014. Fracture prevalence was increased in women and in older age, with patients aged 80 years or older and those of non-Hispanic white race having the highest prevalence of hip fracture. However, she noted that NEDS data also showed higher prevalences of wrist and humerus fractures, which are not normally fractures that a patient visits the hospital as an inpatient for. “We need both data sets to ascertain fractures in the United States,” she said.
When examining fracture site trends over time, Dr. Wright and colleagues found hip fracture prevalence increased by 3.5% between 2010 and 2014, while there was a decrease of 11.9% in the prevalence of spine fractures over the same time period.
According to data from the Medical Expenditures Panel Survey, the direct cost of osteoporosis in aggregate was $73.6 billion between 2012 and 2014, which was 118% higher than between 1998 and 2000 when the costs were $28.1 billion. The costs were spread across ambulatory care, inpatient, and prescription costs equally, the researchers said.
Although the study was limited by examining fracture prevalence rather than incidence, the potential for missing some fractures based on methodology, and limited patient characteristics and follow-up information, the goal of the presentation was to highlight the new osteoporosis prevalence data and the continued burden of the disease.
“We hope that these new prevalence estimates continue to increase the awareness of osteoporosis and prevention,” she said.
Dr. Wright reported receiving grants from Amgen and serving as an expert witness for the law firm Norton Rose Fulbright and Pfizer.
SOURCE: Wright NC et al. ASBMR 2019, Abstract 1079.
ORLANDO – The burden of osteoporosis and fragility fractures in the United States remains high, particularly in older women and minorities, according to a speaker at the annual meeting of the American Society for Bone and Mineral Research.
For non-Hispanic Asian, non-Hispanic white, and Hispanic patients of various ethnic groups, as well as in women and older patients, osteoporosis and fragility fractures continue to be a problem, said Nicole C. Wright, PhD, MPH, of the department of epidemiology at the University of Alabama at Birmingham.
“It remains costly; it remains associated with more health care utilization,” Dr. Wright said. “We may be seeing some declines in some fragility fractures, but [we] are seeing increases in hip fractures.”
As part of the fourth edition of the U.S. Bone and Joint Initiative publication, “The Burden of Musculoskeletal Diseases in the United States,” Dr. Wright and colleagues examined the changes in osteoporosis burden between the third and fourth editions of the publication. They used data from the National Inpatient Sample (NIS) in 2013 and 2014 as well as the National Emergency Department Sample (NEDS) of national ED visits regardless of hospital admission status. In both databases, researchers analyzed data from adults aged 50 years or older where the primary discharge ICD-9 or ICD-10 code was a diagnosis of fracture.
Using National Health and Nutrition Examination Survey data, the researchers estimated an 11.0% osteoporosis prevalence for adults aged 50 years or older overall, a 16.5% prevalence in women, and a 5.1% prevalence in men as assessed by femoral neck and lumbar spine bone mineral density. Osteoporosis was most prevalent in Asian women (40.0%) and Asian men (7.5%), while there was a difference in prevalence in patients of Hispanic race depending on their origin; for example, Puerto Rican men had a higher prevalence of osteoporosis at 8.6%, compared with Hispanic men (2.3%) and non-Hispanic white men of other races (3.9%).
Of 19.5 million hospitalizations in the NIS database between 2013 and 2014, there were approximately 540,000 fragility fractures (2.8%), of which about 300,000 were hip fractures and about 100,000 discharges were for spine fractures, Dr. Wright said. In the NEDS database, the estimate of fragility fracture prevalence was 0.9% of 46.7 million ED visits between 2013 and 2014. Fracture prevalence was increased in women and in older age, with patients aged 80 years or older and those of non-Hispanic white race having the highest prevalence of hip fracture. However, she noted that NEDS data also showed higher prevalences of wrist and humerus fractures, which are not normally fractures that a patient visits the hospital as an inpatient for. “We need both data sets to ascertain fractures in the United States,” she said.
When examining fracture site trends over time, Dr. Wright and colleagues found hip fracture prevalence increased by 3.5% between 2010 and 2014, while there was a decrease of 11.9% in the prevalence of spine fractures over the same time period.
According to data from the Medical Expenditures Panel Survey, the direct cost of osteoporosis in aggregate was $73.6 billion between 2012 and 2014, which was 118% higher than between 1998 and 2000 when the costs were $28.1 billion. The costs were spread across ambulatory care, inpatient, and prescription costs equally, the researchers said.
Although the study was limited by examining fracture prevalence rather than incidence, the potential for missing some fractures based on methodology, and limited patient characteristics and follow-up information, the goal of the presentation was to highlight the new osteoporosis prevalence data and the continued burden of the disease.
“We hope that these new prevalence estimates continue to increase the awareness of osteoporosis and prevention,” she said.
Dr. Wright reported receiving grants from Amgen and serving as an expert witness for the law firm Norton Rose Fulbright and Pfizer.
SOURCE: Wright NC et al. ASBMR 2019, Abstract 1079.
REPORTING FROM ASBMR 2019
Guttate Psoriasis Following Presumed Coxsackievirus A
There are 4 variants of psoriasis: plaque, guttate, pustular, and erythroderma (in order of prevalence).2 Guttate psoriasis is characterized by small, 2- to 10-mm, raindroplike lesions on the skin.1 It accounts for approximately 2% of total psoriasis cases and is commonly triggered by group A streptococcal pharyngitis or tonsillitis.3,4
Hand-foot-and-mouth disease (HFMD) is an illness most commonly caused by a coxsackievirus A infection but also can be caused by other enteroviruses.5,6 Coxsackievirus is a serotype of the Enterovirus species within the Picornaviridae family.7 Hand-foot-and-mouth disease is characterized by a brief fever and vesicular rashes on the palms, soles, or buttocks, as well as oropharyngeal ulcers.8 Typically, the rash is benign and short-lived.9 In rare cases, neurologic complications develop. There have been no reported cases of guttate psoriasis following a coxsackievirus A infection.
The involvement of coxsackievirus B in the etiopathogenesis of psoriasis has been previously reported.10 We report the case of guttate psoriasis following presumed coxsackievirus A HFMD.
Case Report
A 56-year-old woman presented with a vesicular rash on the hands, feet, and lips. The patient reported having a sore throat that started around the same time that the rash developed. The severity of the sore throat was rated as moderate. No fever was reported. One day prior, the patient’s primary care physician prescribed a tapered course of prednisone for the rash. The patient reported a medical history of herpes zoster virus, sunburn, and genital herpes. She was taking clonazepam and had a known allergy to penicillin.
Physical examination revealed erythematous vesicular and papular lesions on the extensor surfaces of the hands and feet. Vesicles also were noted on the vermilion border of the lip. Examination of the patient’s mouth showed blisters and shallow ulcerations in the oral cavity. A clinical diagnosis of coxsackievirus A HFMD was made, and the treatment plan included triamcinolone acetonide ointment 0.025% applied twice daily for 2 weeks and oral valacyclovir hydrochloride 1 g taken 3 times daily for 7 days. A topical emollient also was recommended for the lips when necessary. The lesions all resolved within a 2-week period with no sequela.
The patient returned 1 month later, citing newer red abdominal skin lesions. Fever was denied. She reported that both prescribed treatments had not been helping for the newer lesions. She noticed similar lesions on the groin and brought them to the attention of her gynecologist. Physical examination revealed salmon pink papules and plaques with silvery scaling involving the abdomen, bilateral upper extremities and ears, and scalp. The patient was then clinically diagnosed with guttate psoriasis. A shave biopsy of a representative lesion on the abdomen was performed. The treatment plan included betamethasone dipropionate cream 0.05% applied twice daily for 2 weeks, clobetasol propionate solution 0.05% applied twice daily for 14 days (for the scalp), and hydrocortisone valerate cream 0.2% applied twice daily for 14 days (for the groin).
The skin biopsy shown in the Figure was received in 10% buffered formalin, measuring 5×4×1 mm of skin. Sections showed an acanthotic epidermis with foci of spongiosis and hypergranulosis covered by mounds of parakeratosis infiltrated by neutrophils. Superficial perivascular and interstitial lymphocytic inflammation was present. Tortuous blood vessels within the papillary dermis also were present. Results showed psoriasiform dermatitis with mild spongiosis. Periodic acid–Schiff stain did not reveal any fungal organisms. These findings were consistent with a diagnosis of guttate psoriasis.
The patient then returned 1 month later mentioning continued flare-ups of the scalp as well as newer patches on the arms and hands that were less eruptive and faded more quickly. The plaques in the groin area had resolved. Physical examination showed fewer pink papules and plaques with silvery scaling on the abdomen, bilateral upper extremities and ears, and scalp. Topical medications were continued, and possible apremilast therapy for the psoriasis was discussed.
Comment
Enterovirus-derived HFMD likely is caused by coxsackie-virus A. Current evidence supports the theory that guttate psoriasis can be environmentally triggered in genetically susceptible individuals, often but not exclusively by a streptococcal infection. The causative agent elicits a T-cell–mediated reaction leading to increased type 1 helper T cells, IFN-γ, and IL-2 cytokine levels. HLA-Cw∗0602–positive patients are considered genetically susceptible and more likely to develop guttate psoriasis following an environmental trigger. Based on the coincidence in timing of both diagnoses, this reported case of guttate psoriasis may have been triggered by a coxsackievirus A infection.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23.
- Sarac G, Koca TT, Baglan T. A brief summary of clinical types of psoriasis. North Clin Istanb. 2016;1:79-82.
- Prinz JC. Psoriasis vulgaris—a sterile antibacterial skin reaction mediated by cross-reactive T cells? an immunological view of the pathophysiology of psoriasis. Clin Exp Dermatol. 2001;26:326-332.
- Telfer N, Chalmers RJ, Whale K, et al. The role of streptococcal infection in the initiation of guttate psoriasis. Arch Dermatol. 1992;128:39-42.
- Cabrerizo M, Tarragó D, Muñoz-Almagro C, et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China, 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Seitsonen J, Shakeel S, Susi P, et al. Structural analysis of coxsackievirus A7 reveals conformational changes associated with uncoating. J Virol. 2012;86:7207-7215.
- Wu Y, Yeo A, Phoon M, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Tesini BL. Hand-foot-and-mouth-disease (HFMD). May 2018. https://www.msdmanuals.com/professional/infectious-diseases/enteroviruses/hand-foot-and-mouth-disease-hfmd. Accessed September 25, 2019.
- Korzhova TP, Shyrobokov VP, Koliadenko VH, et al. Coxsackie B viral infection in the etiology and clinical pathogenesis of psoriasis [in Ukrainian]. Lik Sprava. 2001:54-58.
There are 4 variants of psoriasis: plaque, guttate, pustular, and erythroderma (in order of prevalence).2 Guttate psoriasis is characterized by small, 2- to 10-mm, raindroplike lesions on the skin.1 It accounts for approximately 2% of total psoriasis cases and is commonly triggered by group A streptococcal pharyngitis or tonsillitis.3,4
Hand-foot-and-mouth disease (HFMD) is an illness most commonly caused by a coxsackievirus A infection but also can be caused by other enteroviruses.5,6 Coxsackievirus is a serotype of the Enterovirus species within the Picornaviridae family.7 Hand-foot-and-mouth disease is characterized by a brief fever and vesicular rashes on the palms, soles, or buttocks, as well as oropharyngeal ulcers.8 Typically, the rash is benign and short-lived.9 In rare cases, neurologic complications develop. There have been no reported cases of guttate psoriasis following a coxsackievirus A infection.
The involvement of coxsackievirus B in the etiopathogenesis of psoriasis has been previously reported.10 We report the case of guttate psoriasis following presumed coxsackievirus A HFMD.
Case Report
A 56-year-old woman presented with a vesicular rash on the hands, feet, and lips. The patient reported having a sore throat that started around the same time that the rash developed. The severity of the sore throat was rated as moderate. No fever was reported. One day prior, the patient’s primary care physician prescribed a tapered course of prednisone for the rash. The patient reported a medical history of herpes zoster virus, sunburn, and genital herpes. She was taking clonazepam and had a known allergy to penicillin.
Physical examination revealed erythematous vesicular and papular lesions on the extensor surfaces of the hands and feet. Vesicles also were noted on the vermilion border of the lip. Examination of the patient’s mouth showed blisters and shallow ulcerations in the oral cavity. A clinical diagnosis of coxsackievirus A HFMD was made, and the treatment plan included triamcinolone acetonide ointment 0.025% applied twice daily for 2 weeks and oral valacyclovir hydrochloride 1 g taken 3 times daily for 7 days. A topical emollient also was recommended for the lips when necessary. The lesions all resolved within a 2-week period with no sequela.
The patient returned 1 month later, citing newer red abdominal skin lesions. Fever was denied. She reported that both prescribed treatments had not been helping for the newer lesions. She noticed similar lesions on the groin and brought them to the attention of her gynecologist. Physical examination revealed salmon pink papules and plaques with silvery scaling involving the abdomen, bilateral upper extremities and ears, and scalp. The patient was then clinically diagnosed with guttate psoriasis. A shave biopsy of a representative lesion on the abdomen was performed. The treatment plan included betamethasone dipropionate cream 0.05% applied twice daily for 2 weeks, clobetasol propionate solution 0.05% applied twice daily for 14 days (for the scalp), and hydrocortisone valerate cream 0.2% applied twice daily for 14 days (for the groin).
The skin biopsy shown in the Figure was received in 10% buffered formalin, measuring 5×4×1 mm of skin. Sections showed an acanthotic epidermis with foci of spongiosis and hypergranulosis covered by mounds of parakeratosis infiltrated by neutrophils. Superficial perivascular and interstitial lymphocytic inflammation was present. Tortuous blood vessels within the papillary dermis also were present. Results showed psoriasiform dermatitis with mild spongiosis. Periodic acid–Schiff stain did not reveal any fungal organisms. These findings were consistent with a diagnosis of guttate psoriasis.
The patient then returned 1 month later mentioning continued flare-ups of the scalp as well as newer patches on the arms and hands that were less eruptive and faded more quickly. The plaques in the groin area had resolved. Physical examination showed fewer pink papules and plaques with silvery scaling on the abdomen, bilateral upper extremities and ears, and scalp. Topical medications were continued, and possible apremilast therapy for the psoriasis was discussed.
Comment
Enterovirus-derived HFMD likely is caused by coxsackie-virus A. Current evidence supports the theory that guttate psoriasis can be environmentally triggered in genetically susceptible individuals, often but not exclusively by a streptococcal infection. The causative agent elicits a T-cell–mediated reaction leading to increased type 1 helper T cells, IFN-γ, and IL-2 cytokine levels. HLA-Cw∗0602–positive patients are considered genetically susceptible and more likely to develop guttate psoriasis following an environmental trigger. Based on the coincidence in timing of both diagnoses, this reported case of guttate psoriasis may have been triggered by a coxsackievirus A infection.
There are 4 variants of psoriasis: plaque, guttate, pustular, and erythroderma (in order of prevalence).2 Guttate psoriasis is characterized by small, 2- to 10-mm, raindroplike lesions on the skin.1 It accounts for approximately 2% of total psoriasis cases and is commonly triggered by group A streptococcal pharyngitis or tonsillitis.3,4
Hand-foot-and-mouth disease (HFMD) is an illness most commonly caused by a coxsackievirus A infection but also can be caused by other enteroviruses.5,6 Coxsackievirus is a serotype of the Enterovirus species within the Picornaviridae family.7 Hand-foot-and-mouth disease is characterized by a brief fever and vesicular rashes on the palms, soles, or buttocks, as well as oropharyngeal ulcers.8 Typically, the rash is benign and short-lived.9 In rare cases, neurologic complications develop. There have been no reported cases of guttate psoriasis following a coxsackievirus A infection.
The involvement of coxsackievirus B in the etiopathogenesis of psoriasis has been previously reported.10 We report the case of guttate psoriasis following presumed coxsackievirus A HFMD.
Case Report
A 56-year-old woman presented with a vesicular rash on the hands, feet, and lips. The patient reported having a sore throat that started around the same time that the rash developed. The severity of the sore throat was rated as moderate. No fever was reported. One day prior, the patient’s primary care physician prescribed a tapered course of prednisone for the rash. The patient reported a medical history of herpes zoster virus, sunburn, and genital herpes. She was taking clonazepam and had a known allergy to penicillin.
Physical examination revealed erythematous vesicular and papular lesions on the extensor surfaces of the hands and feet. Vesicles also were noted on the vermilion border of the lip. Examination of the patient’s mouth showed blisters and shallow ulcerations in the oral cavity. A clinical diagnosis of coxsackievirus A HFMD was made, and the treatment plan included triamcinolone acetonide ointment 0.025% applied twice daily for 2 weeks and oral valacyclovir hydrochloride 1 g taken 3 times daily for 7 days. A topical emollient also was recommended for the lips when necessary. The lesions all resolved within a 2-week period with no sequela.
The patient returned 1 month later, citing newer red abdominal skin lesions. Fever was denied. She reported that both prescribed treatments had not been helping for the newer lesions. She noticed similar lesions on the groin and brought them to the attention of her gynecologist. Physical examination revealed salmon pink papules and plaques with silvery scaling involving the abdomen, bilateral upper extremities and ears, and scalp. The patient was then clinically diagnosed with guttate psoriasis. A shave biopsy of a representative lesion on the abdomen was performed. The treatment plan included betamethasone dipropionate cream 0.05% applied twice daily for 2 weeks, clobetasol propionate solution 0.05% applied twice daily for 14 days (for the scalp), and hydrocortisone valerate cream 0.2% applied twice daily for 14 days (for the groin).
The skin biopsy shown in the Figure was received in 10% buffered formalin, measuring 5×4×1 mm of skin. Sections showed an acanthotic epidermis with foci of spongiosis and hypergranulosis covered by mounds of parakeratosis infiltrated by neutrophils. Superficial perivascular and interstitial lymphocytic inflammation was present. Tortuous blood vessels within the papillary dermis also were present. Results showed psoriasiform dermatitis with mild spongiosis. Periodic acid–Schiff stain did not reveal any fungal organisms. These findings were consistent with a diagnosis of guttate psoriasis.
The patient then returned 1 month later mentioning continued flare-ups of the scalp as well as newer patches on the arms and hands that were less eruptive and faded more quickly. The plaques in the groin area had resolved. Physical examination showed fewer pink papules and plaques with silvery scaling on the abdomen, bilateral upper extremities and ears, and scalp. Topical medications were continued, and possible apremilast therapy for the psoriasis was discussed.
Comment
Enterovirus-derived HFMD likely is caused by coxsackie-virus A. Current evidence supports the theory that guttate psoriasis can be environmentally triggered in genetically susceptible individuals, often but not exclusively by a streptococcal infection. The causative agent elicits a T-cell–mediated reaction leading to increased type 1 helper T cells, IFN-γ, and IL-2 cytokine levels. HLA-Cw∗0602–positive patients are considered genetically susceptible and more likely to develop guttate psoriasis following an environmental trigger. Based on the coincidence in timing of both diagnoses, this reported case of guttate psoriasis may have been triggered by a coxsackievirus A infection.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23.
- Sarac G, Koca TT, Baglan T. A brief summary of clinical types of psoriasis. North Clin Istanb. 2016;1:79-82.
- Prinz JC. Psoriasis vulgaris—a sterile antibacterial skin reaction mediated by cross-reactive T cells? an immunological view of the pathophysiology of psoriasis. Clin Exp Dermatol. 2001;26:326-332.
- Telfer N, Chalmers RJ, Whale K, et al. The role of streptococcal infection in the initiation of guttate psoriasis. Arch Dermatol. 1992;128:39-42.
- Cabrerizo M, Tarragó D, Muñoz-Almagro C, et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China, 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Seitsonen J, Shakeel S, Susi P, et al. Structural analysis of coxsackievirus A7 reveals conformational changes associated with uncoating. J Virol. 2012;86:7207-7215.
- Wu Y, Yeo A, Phoon M, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Tesini BL. Hand-foot-and-mouth-disease (HFMD). May 2018. https://www.msdmanuals.com/professional/infectious-diseases/enteroviruses/hand-foot-and-mouth-disease-hfmd. Accessed September 25, 2019.
- Korzhova TP, Shyrobokov VP, Koliadenko VH, et al. Coxsackie B viral infection in the etiology and clinical pathogenesis of psoriasis [in Ukrainian]. Lik Sprava. 2001:54-58.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23.
- Sarac G, Koca TT, Baglan T. A brief summary of clinical types of psoriasis. North Clin Istanb. 2016;1:79-82.
- Prinz JC. Psoriasis vulgaris—a sterile antibacterial skin reaction mediated by cross-reactive T cells? an immunological view of the pathophysiology of psoriasis. Clin Exp Dermatol. 2001;26:326-332.
- Telfer N, Chalmers RJ, Whale K, et al. The role of streptococcal infection in the initiation of guttate psoriasis. Arch Dermatol. 1992;128:39-42.
- Cabrerizo M, Tarragó D, Muñoz-Almagro C, et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China, 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Seitsonen J, Shakeel S, Susi P, et al. Structural analysis of coxsackievirus A7 reveals conformational changes associated with uncoating. J Virol. 2012;86:7207-7215.
- Wu Y, Yeo A, Phoon M, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Tesini BL. Hand-foot-and-mouth-disease (HFMD). May 2018. https://www.msdmanuals.com/professional/infectious-diseases/enteroviruses/hand-foot-and-mouth-disease-hfmd. Accessed September 25, 2019.
- Korzhova TP, Shyrobokov VP, Koliadenko VH, et al. Coxsackie B viral infection in the etiology and clinical pathogenesis of psoriasis [in Ukrainian]. Lik Sprava. 2001:54-58.
Practice Points
- Inquire about any illnesses preceding derma-tologic diseases.
- There may be additional microbial triggers for dermatologic diseases.

Photolichenoid Dermatitis: A Presenting Sign of Human Immunodeficiency Virus
Photolichenoid dermatitis is an uncommon eruptive dermatitis of variable clinical presentation. It has a histopathologic pattern of lichenoid inflammation and is best characterized as a photoallergic reaction.1 Photolichenoid dermatitis was first described in 1954 in association with the use of quinidine in the treatment of malaria.2 Subsequently, it has been associated with various medications, including trimethoprim-sulfamethoxazole, azithromycin, and nonsteroidal anti-inflammatory drugs.1,2 Photolichenoid dermatitis has been documented in patients with human immunodeficiency virus (HIV) with variable clinical presentations. Photolichenoid dermatitis in patients with HIV has been described both with and without an associated photosensitizing systemic agent, suggesting that HIV infection is an independent risk factor for the development of this eruption in patients with HIV.3-6
Case Report
A 62-year-old African man presented for evaluation of asymptomatic hypopigmented and depigmented patches in a photodistributed pattern. The eruption began the preceding summer when he noted a pink patch on the right side of the forehead. It progressed over 2 months to involve the face, ears, neck, and arms. His medical history was negative. The only medication he was taking was hydroxychloroquine, which was prescribed by another dermatologist when the patient first developed the eruption. The patient was unsure of the indication for the medication and admitted to poor compliance. A review of systems was negative. There was no personal or family history of autoimmune disease. A detailed sexual history and illicit drug history were not obtained. Physical examination revealed hypopigmented and depigmented patches, some with overlying erythema and collarettes of fine scale. The patches were photodistributed on the face, conchal bowls, neck, dorsal aspect of the hands, and extensor forearms (Figures 1 and 2). Macules of repigmentation were noted within some of the patches. There also were large hyperpigmented patches with peripheral hypopigmentation on the legs.
A punch biopsy taken from the left posterior neck revealed a patchy bandlike lymphocytic infiltrate in the superficial dermis with lymphocytes present at the dermoepidermal junction and scattered dyskeratotic keratinocytes extending into the mid spinous layer (Figure 3). Histopathologic findings were consistent with photolichenoid dermatitis.
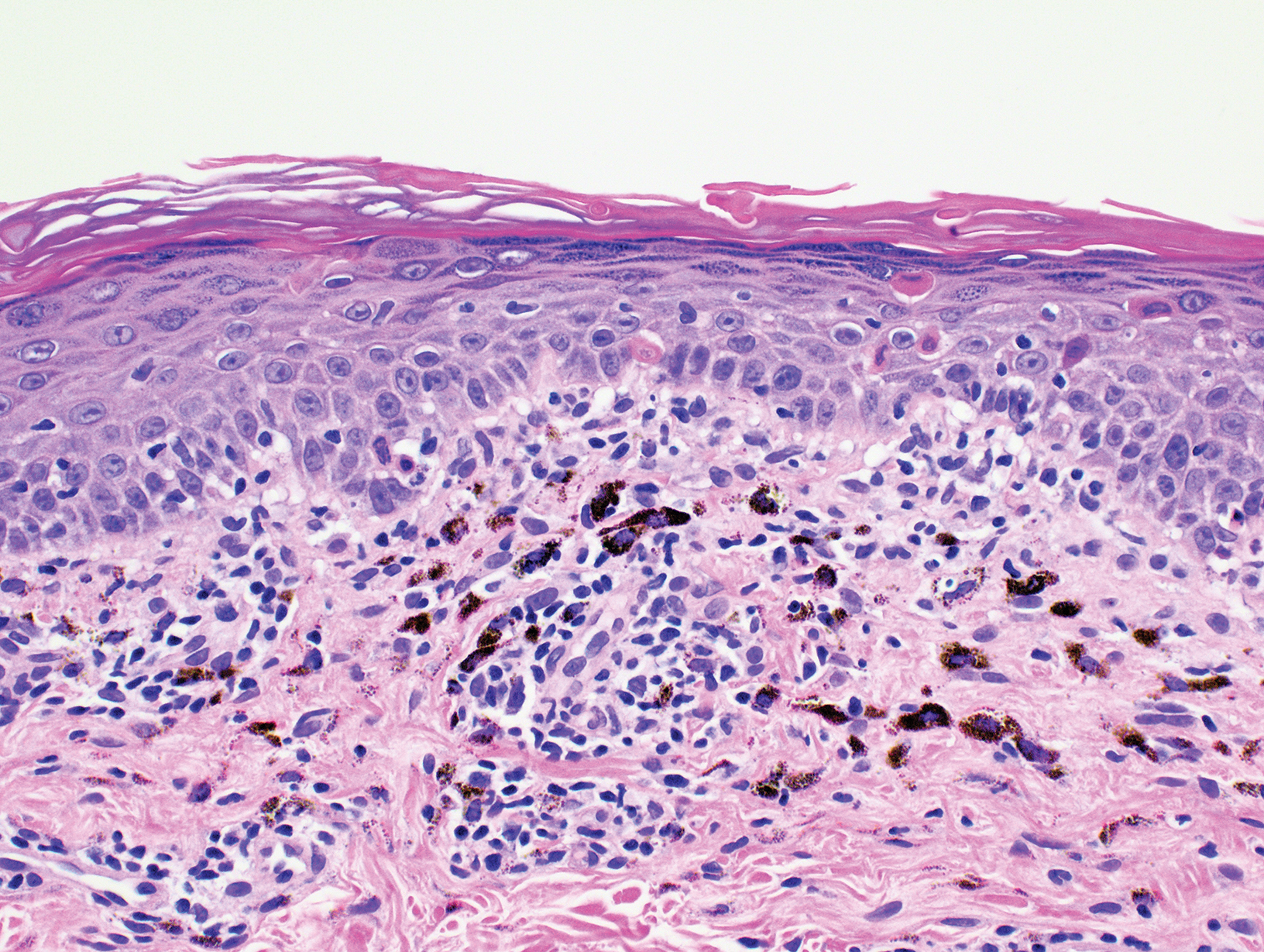
Laboratory workup revealed a normal complete blood cell count and complete metabolic panel. Other negative results included antinuclear antibody, anti-Ro antibody, anti-La antibody, QuantiFERON-TB Gold, syphilis IgG antibody, and hepatitis B surface antigen and antibody. Positive results included hepatitis B antibody, hepatitis C antibody, and HIV-2 antibody. The patient denied overt symptoms suggestive of an immunocompromised status, including fever, chills, weight loss, or diarrhea. Initial treatment included mid-potency topical steroids with continued progression of the eruption. Following histopathologic and laboratory results indicating photolichenoid eruption, treatment with hydroxychloroquine 200 mg twice daily was resumed. The patient was counseled on the importance of sun protection and was referred to an infectious disease clinic for treatment of HIV. He was ultimately lost to follow-up before further laboratory workup was obtained. Therefore, his CD4+ T-cell count and viral load were not obtained.
Comment
Prevalence of Photosensitive Eruptions
Photodermatitis is an uncommon clinical manifestation of HIV occurring in approximately 5% of patients who are HIV positive.3 Photosensitive eruptions previously described in association with HIV include porphyria cutanea tarda, pseudoporphyria, chronic actinic dermatitis, granuloma annulare, photodistributed dyspigmentation, and lichenoid photodermatitis.7 These HIV-associated photosensitive eruptions have been found to disproportionally affect patients of African and Native American descent.5,7,8 Therefore, a new photodistributed eruption in a patient of African or Native American descent should prompt evaluation of possible underlying HIV infection.
Presenting Sign of HIV Infection
We report a case of photolichenoid dermatitis presenting with loss of pigmentation as a presenting sign of HIV. The patient had no known history of HIV or prior opportunistic infections and was not taking any medications at the time of onset or presentation to clinic. Similar cases of photodistributed depigmentation with lichenoid inflammation on histopathology occurring in patients with HIV have been previously described.4-6,9 In these cases, most patients were of African descent with previously diagnosed advanced HIV and CD4 counts of less than 50 cells/mL3. The additional clinical findings of lichenoid papules and plaques were noted in several of these cases.5,6
Exposure to Photosensitizing Drugs
Photodermatitis in patients with HIV often is attributed to exposure to a photosensitizing drug. Many reported cases are retrospective and identify a temporal association between the onset of photodermatitis following the initiation of a photosensitizing drug. The most commonly implicated drugs have included nonsteroidal anti-inflammatory drugs, trimethoprim-sulfamethoxazole, and azithromycin. Other potential offenders may include saquinavir, dapsone, ketoconazole, and efavirenz.3,5 In cases in which temporal association with a new medication could not be identified, the photodermatitis often has been presumed to be due to polypharmacy and the potential synergistic effect of multiple photosensitizing drugs.3,5-8
Advanced HIV
There are several reported cases of photodermatitis occurring in patients who were not exposed to systemic photosensitizers. These patients had advanced HIV, meeting criteria for AIDS with a CD4 count of less than 200 cells/mL3. The majority of patients had an even lower CD4 count of less than 50 cells/mL3. Clinical presentations have included photodistributed lichenoid papules and plaques as well as depigmented patches.4,5,8,10
Evaluating HIV as a Risk Factor for Photodermatitis
Discerning the validity of the correlation between photodermatitis and HIV is difficult, as all previously reported cases are case reports and small retrospective case series.
Conclusion
This case represents an uncommon presentation of photolichenoid dermatitis as the presenting sign of HIV infection.10 Although most reported cases of photodermatitis in HIV are attributed to photosensitizing drugs, we propose that HIV may be an independent risk factor for the development of photodermatitis. We recommend consideration of HIV testing in patients who present with photodistributed depigmenting eruptions, even in the absence of a photosensitizing drug, particularly in patients of African and Native American descent.
- Collazo MH, Sanchez JL, Figueroa LD. Defining lichenoid photodermatitis. Int J Dermatol. 2009;48:239-242.
- Wechsler HL. Dermatitis medicamentosa; a lichen-planus-like eruption due to quinidine. AMA Arch Derm Syphilol. 1954;69:741-744.
- Bilu D, Mamelak AJ, Nguyen RH, et al. Clinical and epidemiologic characterization of photosensitivity in HIV-positive individuals. Photodermatol Photoimmunol Photomed. 2004;20:175-183.
- Philips RC, Motaparthi K, Krishnan B, et al. HIV photodermatitis presenting with widespread vitiligo-like depigmentation. Dermatol Online J. 2012;18:6.
- Berger TG, Dhar A. Lichenoid photoeruptions in human immunodeficiency virus infection. Arch Dermatol. 1994;130:609-613.
- Tran K, Hartman R, Tzu J, et al. Photolichenoid plaques with associated vitiliginous pigmentary changes. Dermatol Online J. 2011;17:13.
- Gregory N, DeLeo VA. Clinical manifestations of photosensitivity in patients with human immunodeficiency virus infection. Arch Dermatol. 1994;130:630-633.
- Vin-Christian K, Epstein JH, Maurer TA, et al. Photosensitivity in HIV-infected individuals. J Dermatol. 2000;27:361-369.
- Kigonya C, Lutwama F, Colebunders R. Extensive hypopigmentation after starting antiretroviral treatment in a human immunodeficiency virus (HIV)-seropositive African woman. Int J Dermatol. 2008;47:102-103.
- Pardo RJ, Kerdel FA. Hypertrophic lichen planus and light sensitivity in an HIV-positive patient. Int J Dermatol. 1988;27:642-644.
Photolichenoid dermatitis is an uncommon eruptive dermatitis of variable clinical presentation. It has a histopathologic pattern of lichenoid inflammation and is best characterized as a photoallergic reaction.1 Photolichenoid dermatitis was first described in 1954 in association with the use of quinidine in the treatment of malaria.2 Subsequently, it has been associated with various medications, including trimethoprim-sulfamethoxazole, azithromycin, and nonsteroidal anti-inflammatory drugs.1,2 Photolichenoid dermatitis has been documented in patients with human immunodeficiency virus (HIV) with variable clinical presentations. Photolichenoid dermatitis in patients with HIV has been described both with and without an associated photosensitizing systemic agent, suggesting that HIV infection is an independent risk factor for the development of this eruption in patients with HIV.3-6
Case Report
A 62-year-old African man presented for evaluation of asymptomatic hypopigmented and depigmented patches in a photodistributed pattern. The eruption began the preceding summer when he noted a pink patch on the right side of the forehead. It progressed over 2 months to involve the face, ears, neck, and arms. His medical history was negative. The only medication he was taking was hydroxychloroquine, which was prescribed by another dermatologist when the patient first developed the eruption. The patient was unsure of the indication for the medication and admitted to poor compliance. A review of systems was negative. There was no personal or family history of autoimmune disease. A detailed sexual history and illicit drug history were not obtained. Physical examination revealed hypopigmented and depigmented patches, some with overlying erythema and collarettes of fine scale. The patches were photodistributed on the face, conchal bowls, neck, dorsal aspect of the hands, and extensor forearms (Figures 1 and 2). Macules of repigmentation were noted within some of the patches. There also were large hyperpigmented patches with peripheral hypopigmentation on the legs.
A punch biopsy taken from the left posterior neck revealed a patchy bandlike lymphocytic infiltrate in the superficial dermis with lymphocytes present at the dermoepidermal junction and scattered dyskeratotic keratinocytes extending into the mid spinous layer (Figure 3). Histopathologic findings were consistent with photolichenoid dermatitis.
Laboratory workup revealed a normal complete blood cell count and complete metabolic panel. Other negative results included antinuclear antibody, anti-Ro antibody, anti-La antibody, QuantiFERON-TB Gold, syphilis IgG antibody, and hepatitis B surface antigen and antibody. Positive results included hepatitis B antibody, hepatitis C antibody, and HIV-2 antibody. The patient denied overt symptoms suggestive of an immunocompromised status, including fever, chills, weight loss, or diarrhea. Initial treatment included mid-potency topical steroids with continued progression of the eruption. Following histopathologic and laboratory results indicating photolichenoid eruption, treatment with hydroxychloroquine 200 mg twice daily was resumed. The patient was counseled on the importance of sun protection and was referred to an infectious disease clinic for treatment of HIV. He was ultimately lost to follow-up before further laboratory workup was obtained. Therefore, his CD4+ T-cell count and viral load were not obtained.
Comment
Prevalence of Photosensitive Eruptions
Photodermatitis is an uncommon clinical manifestation of HIV occurring in approximately 5% of patients who are HIV positive.3 Photosensitive eruptions previously described in association with HIV include porphyria cutanea tarda, pseudoporphyria, chronic actinic dermatitis, granuloma annulare, photodistributed dyspigmentation, and lichenoid photodermatitis.7 These HIV-associated photosensitive eruptions have been found to disproportionally affect patients of African and Native American descent.5,7,8 Therefore, a new photodistributed eruption in a patient of African or Native American descent should prompt evaluation of possible underlying HIV infection.
Presenting Sign of HIV Infection
We report a case of photolichenoid dermatitis presenting with loss of pigmentation as a presenting sign of HIV. The patient had no known history of HIV or prior opportunistic infections and was not taking any medications at the time of onset or presentation to clinic. Similar cases of photodistributed depigmentation with lichenoid inflammation on histopathology occurring in patients with HIV have been previously described.4-6,9 In these cases, most patients were of African descent with previously diagnosed advanced HIV and CD4 counts of less than 50 cells/mL3. The additional clinical findings of lichenoid papules and plaques were noted in several of these cases.5,6
Exposure to Photosensitizing Drugs
Photodermatitis in patients with HIV often is attributed to exposure to a photosensitizing drug. Many reported cases are retrospective and identify a temporal association between the onset of photodermatitis following the initiation of a photosensitizing drug. The most commonly implicated drugs have included nonsteroidal anti-inflammatory drugs, trimethoprim-sulfamethoxazole, and azithromycin. Other potential offenders may include saquinavir, dapsone, ketoconazole, and efavirenz.3,5 In cases in which temporal association with a new medication could not be identified, the photodermatitis often has been presumed to be due to polypharmacy and the potential synergistic effect of multiple photosensitizing drugs.3,5-8
Advanced HIV
There are several reported cases of photodermatitis occurring in patients who were not exposed to systemic photosensitizers. These patients had advanced HIV, meeting criteria for AIDS with a CD4 count of less than 200 cells/mL3. The majority of patients had an even lower CD4 count of less than 50 cells/mL3. Clinical presentations have included photodistributed lichenoid papules and plaques as well as depigmented patches.4,5,8,10
Evaluating HIV as a Risk Factor for Photodermatitis
Discerning the validity of the correlation between photodermatitis and HIV is difficult, as all previously reported cases are case reports and small retrospective case series.
Conclusion
This case represents an uncommon presentation of photolichenoid dermatitis as the presenting sign of HIV infection.10 Although most reported cases of photodermatitis in HIV are attributed to photosensitizing drugs, we propose that HIV may be an independent risk factor for the development of photodermatitis. We recommend consideration of HIV testing in patients who present with photodistributed depigmenting eruptions, even in the absence of a photosensitizing drug, particularly in patients of African and Native American descent.
Photolichenoid dermatitis is an uncommon eruptive dermatitis of variable clinical presentation. It has a histopathologic pattern of lichenoid inflammation and is best characterized as a photoallergic reaction.1 Photolichenoid dermatitis was first described in 1954 in association with the use of quinidine in the treatment of malaria.2 Subsequently, it has been associated with various medications, including trimethoprim-sulfamethoxazole, azithromycin, and nonsteroidal anti-inflammatory drugs.1,2 Photolichenoid dermatitis has been documented in patients with human immunodeficiency virus (HIV) with variable clinical presentations. Photolichenoid dermatitis in patients with HIV has been described both with and without an associated photosensitizing systemic agent, suggesting that HIV infection is an independent risk factor for the development of this eruption in patients with HIV.3-6
Case Report
A 62-year-old African man presented for evaluation of asymptomatic hypopigmented and depigmented patches in a photodistributed pattern. The eruption began the preceding summer when he noted a pink patch on the right side of the forehead. It progressed over 2 months to involve the face, ears, neck, and arms. His medical history was negative. The only medication he was taking was hydroxychloroquine, which was prescribed by another dermatologist when the patient first developed the eruption. The patient was unsure of the indication for the medication and admitted to poor compliance. A review of systems was negative. There was no personal or family history of autoimmune disease. A detailed sexual history and illicit drug history were not obtained. Physical examination revealed hypopigmented and depigmented patches, some with overlying erythema and collarettes of fine scale. The patches were photodistributed on the face, conchal bowls, neck, dorsal aspect of the hands, and extensor forearms (Figures 1 and 2). Macules of repigmentation were noted within some of the patches. There also were large hyperpigmented patches with peripheral hypopigmentation on the legs.
A punch biopsy taken from the left posterior neck revealed a patchy bandlike lymphocytic infiltrate in the superficial dermis with lymphocytes present at the dermoepidermal junction and scattered dyskeratotic keratinocytes extending into the mid spinous layer (Figure 3). Histopathologic findings were consistent with photolichenoid dermatitis.
Laboratory workup revealed a normal complete blood cell count and complete metabolic panel. Other negative results included antinuclear antibody, anti-Ro antibody, anti-La antibody, QuantiFERON-TB Gold, syphilis IgG antibody, and hepatitis B surface antigen and antibody. Positive results included hepatitis B antibody, hepatitis C antibody, and HIV-2 antibody. The patient denied overt symptoms suggestive of an immunocompromised status, including fever, chills, weight loss, or diarrhea. Initial treatment included mid-potency topical steroids with continued progression of the eruption. Following histopathologic and laboratory results indicating photolichenoid eruption, treatment with hydroxychloroquine 200 mg twice daily was resumed. The patient was counseled on the importance of sun protection and was referred to an infectious disease clinic for treatment of HIV. He was ultimately lost to follow-up before further laboratory workup was obtained. Therefore, his CD4+ T-cell count and viral load were not obtained.
Comment
Prevalence of Photosensitive Eruptions
Photodermatitis is an uncommon clinical manifestation of HIV occurring in approximately 5% of patients who are HIV positive.3 Photosensitive eruptions previously described in association with HIV include porphyria cutanea tarda, pseudoporphyria, chronic actinic dermatitis, granuloma annulare, photodistributed dyspigmentation, and lichenoid photodermatitis.7 These HIV-associated photosensitive eruptions have been found to disproportionally affect patients of African and Native American descent.5,7,8 Therefore, a new photodistributed eruption in a patient of African or Native American descent should prompt evaluation of possible underlying HIV infection.
Presenting Sign of HIV Infection
We report a case of photolichenoid dermatitis presenting with loss of pigmentation as a presenting sign of HIV. The patient had no known history of HIV or prior opportunistic infections and was not taking any medications at the time of onset or presentation to clinic. Similar cases of photodistributed depigmentation with lichenoid inflammation on histopathology occurring in patients with HIV have been previously described.4-6,9 In these cases, most patients were of African descent with previously diagnosed advanced HIV and CD4 counts of less than 50 cells/mL3. The additional clinical findings of lichenoid papules and plaques were noted in several of these cases.5,6
Exposure to Photosensitizing Drugs
Photodermatitis in patients with HIV often is attributed to exposure to a photosensitizing drug. Many reported cases are retrospective and identify a temporal association between the onset of photodermatitis following the initiation of a photosensitizing drug. The most commonly implicated drugs have included nonsteroidal anti-inflammatory drugs, trimethoprim-sulfamethoxazole, and azithromycin. Other potential offenders may include saquinavir, dapsone, ketoconazole, and efavirenz.3,5 In cases in which temporal association with a new medication could not be identified, the photodermatitis often has been presumed to be due to polypharmacy and the potential synergistic effect of multiple photosensitizing drugs.3,5-8
Advanced HIV
There are several reported cases of photodermatitis occurring in patients who were not exposed to systemic photosensitizers. These patients had advanced HIV, meeting criteria for AIDS with a CD4 count of less than 200 cells/mL3. The majority of patients had an even lower CD4 count of less than 50 cells/mL3. Clinical presentations have included photodistributed lichenoid papules and plaques as well as depigmented patches.4,5,8,10
Evaluating HIV as a Risk Factor for Photodermatitis
Discerning the validity of the correlation between photodermatitis and HIV is difficult, as all previously reported cases are case reports and small retrospective case series.
Conclusion
This case represents an uncommon presentation of photolichenoid dermatitis as the presenting sign of HIV infection.10 Although most reported cases of photodermatitis in HIV are attributed to photosensitizing drugs, we propose that HIV may be an independent risk factor for the development of photodermatitis. We recommend consideration of HIV testing in patients who present with photodistributed depigmenting eruptions, even in the absence of a photosensitizing drug, particularly in patients of African and Native American descent.
- Collazo MH, Sanchez JL, Figueroa LD. Defining lichenoid photodermatitis. Int J Dermatol. 2009;48:239-242.
- Wechsler HL. Dermatitis medicamentosa; a lichen-planus-like eruption due to quinidine. AMA Arch Derm Syphilol. 1954;69:741-744.
- Bilu D, Mamelak AJ, Nguyen RH, et al. Clinical and epidemiologic characterization of photosensitivity in HIV-positive individuals. Photodermatol Photoimmunol Photomed. 2004;20:175-183.
- Philips RC, Motaparthi K, Krishnan B, et al. HIV photodermatitis presenting with widespread vitiligo-like depigmentation. Dermatol Online J. 2012;18:6.
- Berger TG, Dhar A. Lichenoid photoeruptions in human immunodeficiency virus infection. Arch Dermatol. 1994;130:609-613.
- Tran K, Hartman R, Tzu J, et al. Photolichenoid plaques with associated vitiliginous pigmentary changes. Dermatol Online J. 2011;17:13.
- Gregory N, DeLeo VA. Clinical manifestations of photosensitivity in patients with human immunodeficiency virus infection. Arch Dermatol. 1994;130:630-633.
- Vin-Christian K, Epstein JH, Maurer TA, et al. Photosensitivity in HIV-infected individuals. J Dermatol. 2000;27:361-369.
- Kigonya C, Lutwama F, Colebunders R. Extensive hypopigmentation after starting antiretroviral treatment in a human immunodeficiency virus (HIV)-seropositive African woman. Int J Dermatol. 2008;47:102-103.
- Pardo RJ, Kerdel FA. Hypertrophic lichen planus and light sensitivity in an HIV-positive patient. Int J Dermatol. 1988;27:642-644.
- Collazo MH, Sanchez JL, Figueroa LD. Defining lichenoid photodermatitis. Int J Dermatol. 2009;48:239-242.
- Wechsler HL. Dermatitis medicamentosa; a lichen-planus-like eruption due to quinidine. AMA Arch Derm Syphilol. 1954;69:741-744.
- Bilu D, Mamelak AJ, Nguyen RH, et al. Clinical and epidemiologic characterization of photosensitivity in HIV-positive individuals. Photodermatol Photoimmunol Photomed. 2004;20:175-183.
- Philips RC, Motaparthi K, Krishnan B, et al. HIV photodermatitis presenting with widespread vitiligo-like depigmentation. Dermatol Online J. 2012;18:6.
- Berger TG, Dhar A. Lichenoid photoeruptions in human immunodeficiency virus infection. Arch Dermatol. 1994;130:609-613.
- Tran K, Hartman R, Tzu J, et al. Photolichenoid plaques with associated vitiliginous pigmentary changes. Dermatol Online J. 2011;17:13.
- Gregory N, DeLeo VA. Clinical manifestations of photosensitivity in patients with human immunodeficiency virus infection. Arch Dermatol. 1994;130:630-633.
- Vin-Christian K, Epstein JH, Maurer TA, et al. Photosensitivity in HIV-infected individuals. J Dermatol. 2000;27:361-369.
- Kigonya C, Lutwama F, Colebunders R. Extensive hypopigmentation after starting antiretroviral treatment in a human immunodeficiency virus (HIV)-seropositive African woman. Int J Dermatol. 2008;47:102-103.
- Pardo RJ, Kerdel FA. Hypertrophic lichen planus and light sensitivity in an HIV-positive patient. Int J Dermatol. 1988;27:642-644.
Practice Points
- There are few reports in the literature of human immunodeficiency virus (HIV) presenting as a photolichenoid eruption.
- We report the case of a 62-year-old African man who presented with a new-onset photodistributed eruption and was subsequently diagnosed with HIV.
- This case supports testing for HIV in patients with a similar clinical presentation.
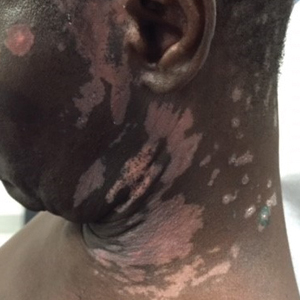
Cutaneous Mycobacterium haemophilum Infection Involving the Upper Extremities: Diagnosis and Management Guidelines
Infection with Mycobacterium haemophilum, a rare, slow-growing organism, most commonly presents as ulcerating cutaneous lesions and subcutaneous nodules in immunocompromised adults.1 The most common clinical presentation in adults includes cutaneous lesions, nodules, cysts, and papules, with signs and symptoms of erythema, pain, pruritus, and drainage.2 Disseminated disease states of septic arthritis, pulmonary infiltration, and osteomyelitis, though life-threatening, are less common manifestations reported in highly immunocompromised persons.3
Infection with M haemophilum presents a challenge to the dermatology community because it is infrequently suspected and misidentified, resulting in delayed diagnosis. Additionally, M haemophilum is an extremely fastidious organism that requires heme-supplemented culture media and a carefully regulated low temperature for many consecutive weeks to yield valid culture results.1 These features contribute to complications and delays in diagnosis of an already overlooked source of infection.
We discuss the clinical presentation, diagnosis, and treatment of 3 unusual cases of cutaneous M haemophilum infection involving the upper arms. The findings in these cases highlight the challenges inherent in diagnosis as well as the obstacles that arise in providing effective, long-term treatment of this infection.
Case Reports
Patient 1
A 69-year-old woman with a medical history of a single functioning kidney and moderate psoriasis managed with low-dosage methotrexate presented with an erythematous nonhealing wound on the left forearm that developed after she was scratched by a dog. The pustules, appearing as bright red, tender, warm abscesses, had been present for 3 months and were distributed on the left proximal and distal dorsal forearm (Figure 1A). The patient reported no recent travel, sick contacts, allergies, or new medications.
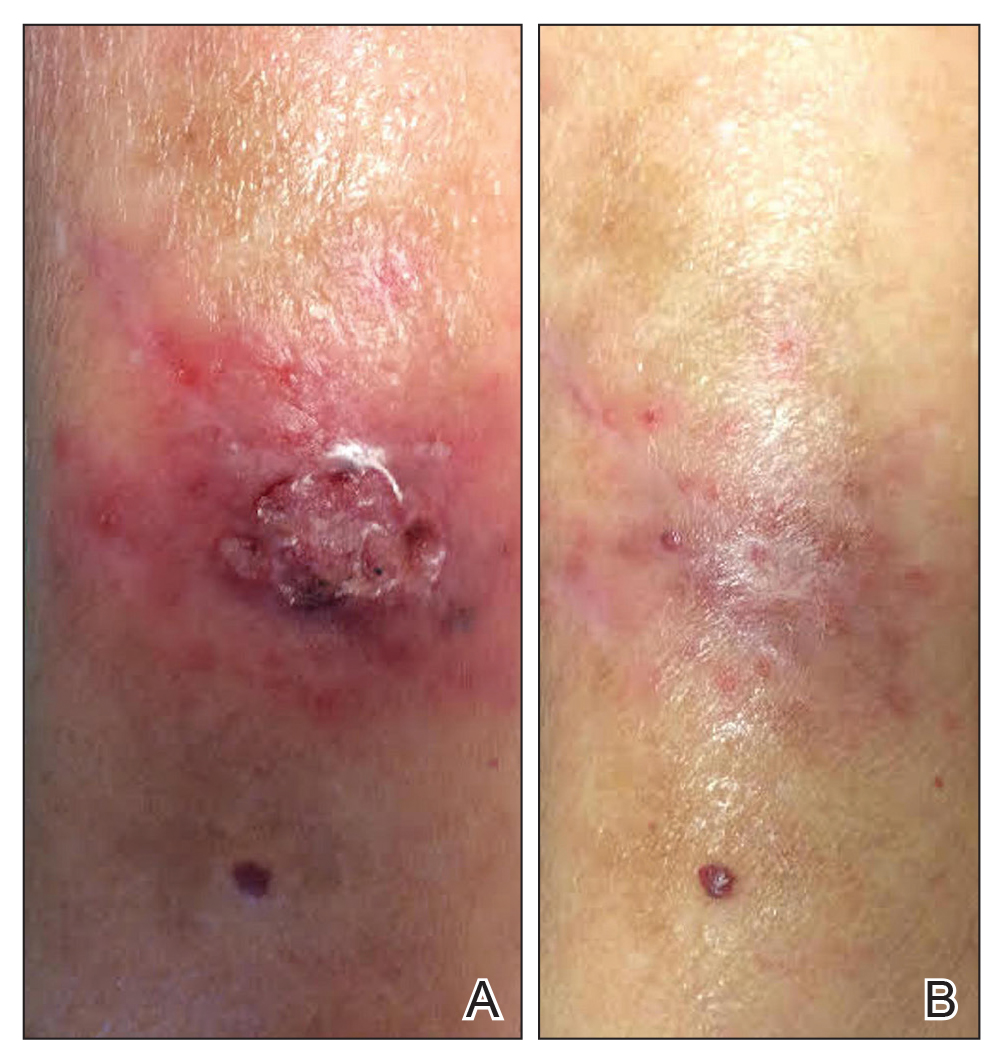
A shave biopsy was initially obtained. Swab specimens were sent for bacterial, fungal, and mycobacterial culture following discontinuation of methotrexate. Initial histopathologic analysis revealed aggregates of histiocytes and multinucleated giant cells within the dermis, surrounded by infiltrates of lymphocytes and neutrophils (Figure 2), consistent with a dermal noncaseating granulomatosis. Acid-fast bacilli (AFB), periodic acid–Schiff, Gram, and Grocott-Gomori methenamine-silver stains were negative for pathogenic microorganisms. There was no evidence of vasculitis.
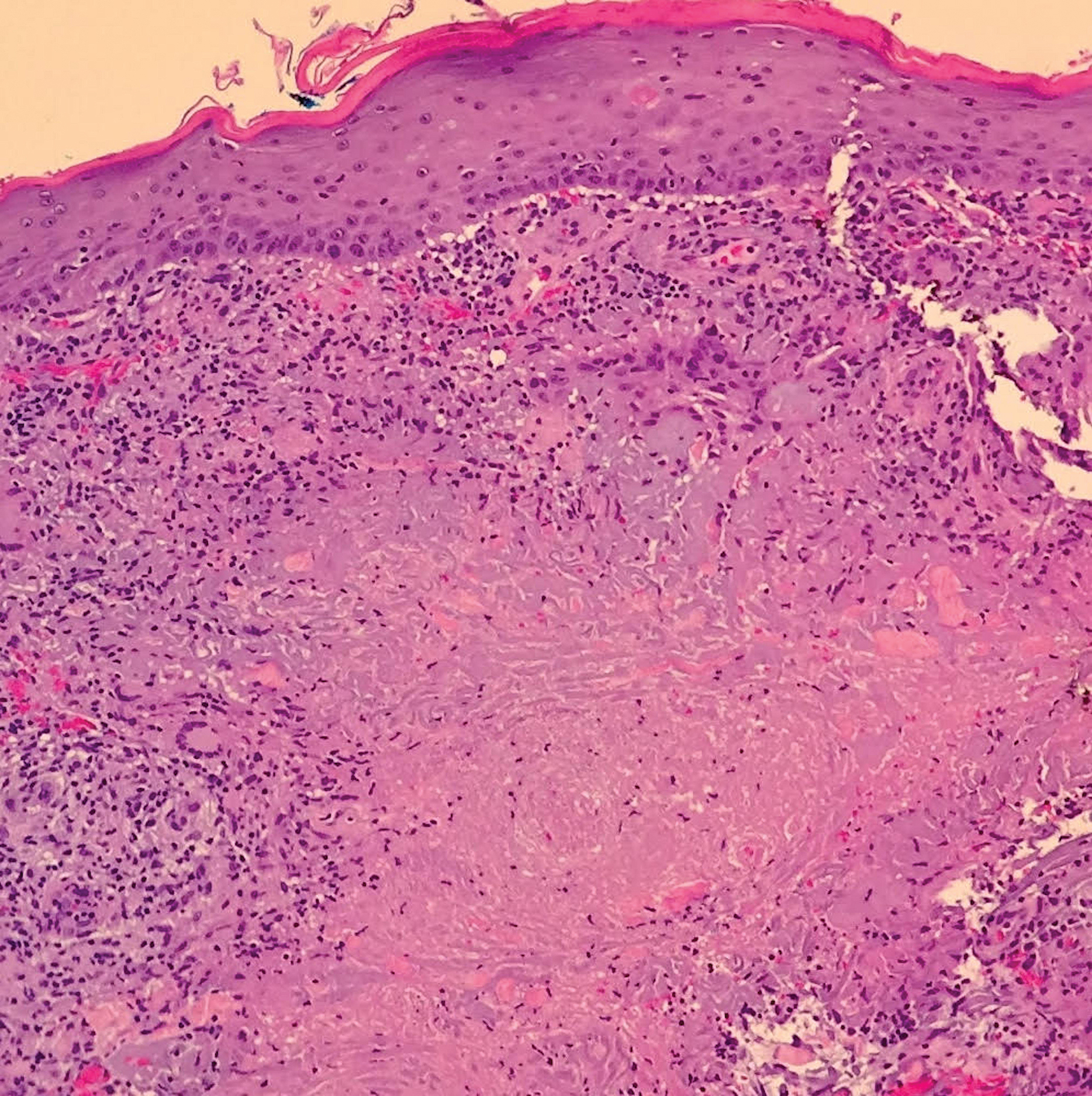
Despite negative special stains, an infectious cause was still suspected. Oral doxycycline monohydrate 100 mg twice daily, oral fluconazole 200 mg daily, and econazole cream 1% were prescribed because of concern for mycobacterial infection and initial growth of Candida parapsilosis in the swab culture.
A punch biopsy also was performed at this time for both repeat histopathologic analysis and tissue culture. Follow-up appointments were scheduled every 2 weeks. Staining by AFB of the repeat histopathologic specimen was negative.
The patient demonstrated symptomatic and aesthetic improvement (Figure 1B) during consecutive regular follow-up appointments while culture results were pending. No lesions appeared above the left elbow and she had no lymphadenopathy. Results of blood chemistry analyses and complete blood cell count throughout follow-up were normal.
The final tissue culture report obtained 7 weeks after initial presentation showed growth of M haemophilum despite a negative smear. The swab culture that initially was taken did not grow pathogenic organisms.
The patient was referred to an infectious disease specialist who confirmed that the atypical mycobacterial infection likely was the main source of the cutaneous lesions. She was instructed to continue econazole cream 1% and was given prescriptions for clarithromycin 500 mg twice daily, ciprofloxacin 500 mg twice daily, and rifampin 300 mg twice daily for a total duration of 12 to 18 months. The patient has remained on this triple-drug regimen and demonstrated improvement in the lesions. She has been off methotrexate while on antibiotic therapy.
Patient 2
A 79-year-old man with a medical history of chronic lymphocytic leukemia, basal cell carcinoma, and squamous cell carcinoma presented with a nonhealing, painful, red lesion on the left forearm of 1 week’s duration. Physical examination revealed a violaceous nontender plaque with erosions and desquamation that was initially diagnosed as a carbuncle. The patient reported a similar eruption on the right foot that was successfully treated with silver sulfadiazine by another physician.
Biopsy was performed by the shave method for histologic analysis and tissue culture. Doxycycline 100 mg twice daily was prescribed because of high suspicion of infection. Histologic findings revealed granulomatous inflammation with pseudoepitheliomatous hyperplasia, reported as squamous cell carcinoma. A second opinion confirmed suspicion of an infectious process; the patient remained on doxycycline. During follow-up, the lesion progressed to a 5-cm plaque studded with pustules and satellite papules. Multiple additional tissue cultures were performed over 2 months until “light growth” of M haemophilum was reported.
The patient showed minimal improvement on tetracycline antibiotics. His condition was complicated by a photosensitivity reaction to doxycycline on the left and right forearms, hands, and nose. Consequently, triamcinolone was prescribed, doxycycline was discontinued, and minocycline 100 mg twice daily and ciprofloxacin 500 mg twice daily were prescribed.
Nine months after initial presentation, the lesions were still present but remarkably improved. The antibiotic regimen was discontinued after 11 months.
Patient 3
A 77-year-old woman with a history of rheumatoid arthritis treated with methotrexate and abatacept as well as cutaneous T-cell lymphoma treated with narrowband UVB radiation presented to the emergency department with fever and an inflamed right forearm (Figure 3A). Initial bacterial cultures of the wound and blood were negative.
The patient was treated with vancomycin and discharged on cephalexin once she became afebrile. She was seen at our office the next week for further evaluation. We recommended that she discontinue all immunosuppressant medications. A 4-mm tissue biopsy for hematoxylin and eosin staining and a separate 4-mm punch biopsy for culture were performed while she was taking cephalexin. Histopathologic analysis revealed numerous neutrophilic abscesses; however, Gram, AFB, and fungal stains were negative.
Arm edema and pustules slowly resolved, but the eschar and verrucous plaques continued to slowly progress while the patient was off immunosuppression. She was kept off antibiotics until mycobacterial culture was positive at 4 weeks, at which time she was placed on doxycycline and clarithromycin. Final identification of M haemophilum was made at 6 weeks; consequently, doxycycline was discontinued and she was referred to infectious disease for multidrug therapy. She remained afebrile during the entire 6 weeks until cultures were final.
While immunosuppressants were discontinued and clarithromycin was administered, the plaque changed from an edematous pustular dermatitis to a verrucous crusted plaque. Neither epitrochlear nor axillary lymphadenopathy was noted during the treatment period. The infectious disease specialist prescribed azithromycin, ethambutol, and rifampin, which produced marked improvement (Figure 3B). The patient has remained off immunosuppressive therapy while on antibiotics.
Comment
Clinical Presentation and Diagnosis
Mycobacterium haemophilum is a rare infectious organism that affects primarily immunocompromised adults but also has been identified in immunocompetent adults and pediatric patients.2 Commonly affected immunosuppressed groups include solid organ transplant recipients, bone marrow transplant recipients, human immunodeficiency virus–positive patients, and patients with rheumatoid arthritis.
The infection typically presents as small violaceous papules and pustules that become painful and erythematous, with progression and draining ulceration in later stages.2 In our cases, all lesions tended to evolve into a verrucous plaque that slowly resolved with antibiotic therapy.
Due to the rarity of this infection, the initial differential diagnosis can include infection with other mycobacteria, Sporothrix, Staphylococcus aureus, and other fungal pathogens. Misdiagnosis is a common obstacle in the treatment of M haemophilum due to its rarity, often negative AFB stains, and slow growth on culture media; therefore, tissue culture is essential to successful diagnosis and management. The natural reservoir of M haemophilum is unknown, but infection has been associated with contaminated water sources.1 In one case (patient 1), symptoms developed after a dog scratch; the other 2 patients were unaware of injury to the skin.Laboratory diagnosis of M haemophilum is inherently difficult and protracted. The species is a highly fastidious and slow-growing Mycobacterium that requires cooler (30°C) incubation for many weeks on agar medium enriched with hemin or ferric ammonium citrate to obtain valid growth.1 To secure timely diagnosis, the organism’s slow agar growth warrants immediate tissue culture and biopsy when an immunocompromised patient presents with clinical features of atypical infection of an extremity. Mycobacterium haemophilum infection likely is underreported because of these difficulties in diagnosis.
Management
Although there are no standard guidelines for antibiotic treatment of M haemophilum, the current literature recommends triple-drug therapy with clarithromycin, ciprofloxacin, and rifamycin for at least 12 to 24 months.2
Upon clinical suspicion of an atypical Mycobacterium, we recommend a macrolide antibiotic over doxycycline, however, because this class of agents maintains broad coverage while being more specific for atypical mycobacteria. Although an atypical Mycobacterium was suspected early in the presentation in our cases, we discourage immediate use of triple-agent antibiotic therapy until laboratory evidence is procured to minimize antibiotic overuse in patients who do not have a final diagnosis. Single-agent therapy for prolonged treatment is discouraged for atypical mycobacterial infections because of the high risk of antibiotic resistance. Therapy should be tailored to the needs of the individual based on the extent of dissemination of disease and the severity of immunosuppression.1,2
Additionally, underlying disease that results in immunosuppression might necessitate treatment reevaluation (as occurred in our cases) requiring cessation of immunosuppressive drugs, extended careful monitoring, and pharmacotherapeutic readjustment through the course of treatment. The degree to which antibiotics contribute to eradication of M haemophilum is unknown; therefore, it is recommended that long-term antibiotic use and treatment aimed at recovering the immunocompromised state (eg, highly active antiretroviral therapy in a patient with AIDS) be implemented.2
Conclusion
Our 3 cases of M haemophilum infection involved the upper extremities of immunosuppressed patients older than 65 years. This propensity to affect the upper extremities could possibly be due to the lower temperature required for growth of M haemophilum. Initial histopathologic study showed granulomatous and neutrophilic infiltrates, yet histopathologic specimens from all 3 patients failed to display positive AFB staining, which delayed the initial antibiotic choice. In all cases, diagnosis was made by tissue culture after swab culture failed to grow the pathogen. Furthermore, the 3 cases took approximately 6 weeks to achieve final identification of the organism. Neither clinical lymphadenopathy nor systemic spread was noted in our patients; immunosuppression was discontinued when possible.
Mycobacterium haemophilum is an uncommon but potentially life-threatening infection that should be suspected in immunocompromised adults who present with atypical cellulitis of the extremities. The ultimate diagnosis often is delayed because the organism grows slowly (as long as 8 weeks) in tissue culture. For that reason, empiric antibiotic treatment, including a macrolide, should be considered in patients with disseminated or severe infection or critical immunosuppression and in those who do not demonstrate improvement in symptoms once immunosuppressants are withheld. A prolonged course of multiple-drug antibiotic therapy has proved to be effective for treating cutaneous infection with M haemophilum.
- Lindeboom JA, Bruijnesteijn van Coppenraet LE, van Soolingen D, et al. Clinical manifestations, diagnosis, and treatment of Mycobacterium haemophilum infections. Clin Microbiol Rev. 2011;24:701-717.
- Tangkosakul T, Hongmanee P, Malathum K. Cutaneous Mycobacterium haemophilum infections in immunocompromised patients in a tertiary hospital in Bangkok, Thailand: under-reported/under-recognized infection. JMM Case Rep. 2014;1:E002618.
- Sabeti S, Pourabdollah Tootkaboni M, Abdolahi M, et al. Mycobacterium haemophilum: a report of cutaneous infection in a patient with end-stage renal disease. Int J Mycobacteriol. 2016;5(suppl 1):S236.
Infection with Mycobacterium haemophilum, a rare, slow-growing organism, most commonly presents as ulcerating cutaneous lesions and subcutaneous nodules in immunocompromised adults.1 The most common clinical presentation in adults includes cutaneous lesions, nodules, cysts, and papules, with signs and symptoms of erythema, pain, pruritus, and drainage.2 Disseminated disease states of septic arthritis, pulmonary infiltration, and osteomyelitis, though life-threatening, are less common manifestations reported in highly immunocompromised persons.3
Infection with M haemophilum presents a challenge to the dermatology community because it is infrequently suspected and misidentified, resulting in delayed diagnosis. Additionally, M haemophilum is an extremely fastidious organism that requires heme-supplemented culture media and a carefully regulated low temperature for many consecutive weeks to yield valid culture results.1 These features contribute to complications and delays in diagnosis of an already overlooked source of infection.
We discuss the clinical presentation, diagnosis, and treatment of 3 unusual cases of cutaneous M haemophilum infection involving the upper arms. The findings in these cases highlight the challenges inherent in diagnosis as well as the obstacles that arise in providing effective, long-term treatment of this infection.
Case Reports
Patient 1
A 69-year-old woman with a medical history of a single functioning kidney and moderate psoriasis managed with low-dosage methotrexate presented with an erythematous nonhealing wound on the left forearm that developed after she was scratched by a dog. The pustules, appearing as bright red, tender, warm abscesses, had been present for 3 months and were distributed on the left proximal and distal dorsal forearm (Figure 1A). The patient reported no recent travel, sick contacts, allergies, or new medications.
A shave biopsy was initially obtained. Swab specimens were sent for bacterial, fungal, and mycobacterial culture following discontinuation of methotrexate. Initial histopathologic analysis revealed aggregates of histiocytes and multinucleated giant cells within the dermis, surrounded by infiltrates of lymphocytes and neutrophils (Figure 2), consistent with a dermal noncaseating granulomatosis. Acid-fast bacilli (AFB), periodic acid–Schiff, Gram, and Grocott-Gomori methenamine-silver stains were negative for pathogenic microorganisms. There was no evidence of vasculitis.
Despite negative special stains, an infectious cause was still suspected. Oral doxycycline monohydrate 100 mg twice daily, oral fluconazole 200 mg daily, and econazole cream 1% were prescribed because of concern for mycobacterial infection and initial growth of Candida parapsilosis in the swab culture.
A punch biopsy also was performed at this time for both repeat histopathologic analysis and tissue culture. Follow-up appointments were scheduled every 2 weeks. Staining by AFB of the repeat histopathologic specimen was negative.
The patient demonstrated symptomatic and aesthetic improvement (Figure 1B) during consecutive regular follow-up appointments while culture results were pending. No lesions appeared above the left elbow and she had no lymphadenopathy. Results of blood chemistry analyses and complete blood cell count throughout follow-up were normal.
The final tissue culture report obtained 7 weeks after initial presentation showed growth of M haemophilum despite a negative smear. The swab culture that initially was taken did not grow pathogenic organisms.
The patient was referred to an infectious disease specialist who confirmed that the atypical mycobacterial infection likely was the main source of the cutaneous lesions. She was instructed to continue econazole cream 1% and was given prescriptions for clarithromycin 500 mg twice daily, ciprofloxacin 500 mg twice daily, and rifampin 300 mg twice daily for a total duration of 12 to 18 months. The patient has remained on this triple-drug regimen and demonstrated improvement in the lesions. She has been off methotrexate while on antibiotic therapy.
Patient 2
A 79-year-old man with a medical history of chronic lymphocytic leukemia, basal cell carcinoma, and squamous cell carcinoma presented with a nonhealing, painful, red lesion on the left forearm of 1 week’s duration. Physical examination revealed a violaceous nontender plaque with erosions and desquamation that was initially diagnosed as a carbuncle. The patient reported a similar eruption on the right foot that was successfully treated with silver sulfadiazine by another physician.
Biopsy was performed by the shave method for histologic analysis and tissue culture. Doxycycline 100 mg twice daily was prescribed because of high suspicion of infection. Histologic findings revealed granulomatous inflammation with pseudoepitheliomatous hyperplasia, reported as squamous cell carcinoma. A second opinion confirmed suspicion of an infectious process; the patient remained on doxycycline. During follow-up, the lesion progressed to a 5-cm plaque studded with pustules and satellite papules. Multiple additional tissue cultures were performed over 2 months until “light growth” of M haemophilum was reported.
The patient showed minimal improvement on tetracycline antibiotics. His condition was complicated by a photosensitivity reaction to doxycycline on the left and right forearms, hands, and nose. Consequently, triamcinolone was prescribed, doxycycline was discontinued, and minocycline 100 mg twice daily and ciprofloxacin 500 mg twice daily were prescribed.
Nine months after initial presentation, the lesions were still present but remarkably improved. The antibiotic regimen was discontinued after 11 months.
Patient 3
A 77-year-old woman with a history of rheumatoid arthritis treated with methotrexate and abatacept as well as cutaneous T-cell lymphoma treated with narrowband UVB radiation presented to the emergency department with fever and an inflamed right forearm (Figure 3A). Initial bacterial cultures of the wound and blood were negative.
The patient was treated with vancomycin and discharged on cephalexin once she became afebrile. She was seen at our office the next week for further evaluation. We recommended that she discontinue all immunosuppressant medications. A 4-mm tissue biopsy for hematoxylin and eosin staining and a separate 4-mm punch biopsy for culture were performed while she was taking cephalexin. Histopathologic analysis revealed numerous neutrophilic abscesses; however, Gram, AFB, and fungal stains were negative.
Arm edema and pustules slowly resolved, but the eschar and verrucous plaques continued to slowly progress while the patient was off immunosuppression. She was kept off antibiotics until mycobacterial culture was positive at 4 weeks, at which time she was placed on doxycycline and clarithromycin. Final identification of M haemophilum was made at 6 weeks; consequently, doxycycline was discontinued and she was referred to infectious disease for multidrug therapy. She remained afebrile during the entire 6 weeks until cultures were final.
While immunosuppressants were discontinued and clarithromycin was administered, the plaque changed from an edematous pustular dermatitis to a verrucous crusted plaque. Neither epitrochlear nor axillary lymphadenopathy was noted during the treatment period. The infectious disease specialist prescribed azithromycin, ethambutol, and rifampin, which produced marked improvement (Figure 3B). The patient has remained off immunosuppressive therapy while on antibiotics.
Comment
Clinical Presentation and Diagnosis
Mycobacterium haemophilum is a rare infectious organism that affects primarily immunocompromised adults but also has been identified in immunocompetent adults and pediatric patients.2 Commonly affected immunosuppressed groups include solid organ transplant recipients, bone marrow transplant recipients, human immunodeficiency virus–positive patients, and patients with rheumatoid arthritis.
The infection typically presents as small violaceous papules and pustules that become painful and erythematous, with progression and draining ulceration in later stages.2 In our cases, all lesions tended to evolve into a verrucous plaque that slowly resolved with antibiotic therapy.
Due to the rarity of this infection, the initial differential diagnosis can include infection with other mycobacteria, Sporothrix, Staphylococcus aureus, and other fungal pathogens. Misdiagnosis is a common obstacle in the treatment of M haemophilum due to its rarity, often negative AFB stains, and slow growth on culture media; therefore, tissue culture is essential to successful diagnosis and management. The natural reservoir of M haemophilum is unknown, but infection has been associated with contaminated water sources.1 In one case (patient 1), symptoms developed after a dog scratch; the other 2 patients were unaware of injury to the skin.Laboratory diagnosis of M haemophilum is inherently difficult and protracted. The species is a highly fastidious and slow-growing Mycobacterium that requires cooler (30°C) incubation for many weeks on agar medium enriched with hemin or ferric ammonium citrate to obtain valid growth.1 To secure timely diagnosis, the organism’s slow agar growth warrants immediate tissue culture and biopsy when an immunocompromised patient presents with clinical features of atypical infection of an extremity. Mycobacterium haemophilum infection likely is underreported because of these difficulties in diagnosis.
Management
Although there are no standard guidelines for antibiotic treatment of M haemophilum, the current literature recommends triple-drug therapy with clarithromycin, ciprofloxacin, and rifamycin for at least 12 to 24 months.2
Upon clinical suspicion of an atypical Mycobacterium, we recommend a macrolide antibiotic over doxycycline, however, because this class of agents maintains broad coverage while being more specific for atypical mycobacteria. Although an atypical Mycobacterium was suspected early in the presentation in our cases, we discourage immediate use of triple-agent antibiotic therapy until laboratory evidence is procured to minimize antibiotic overuse in patients who do not have a final diagnosis. Single-agent therapy for prolonged treatment is discouraged for atypical mycobacterial infections because of the high risk of antibiotic resistance. Therapy should be tailored to the needs of the individual based on the extent of dissemination of disease and the severity of immunosuppression.1,2
Additionally, underlying disease that results in immunosuppression might necessitate treatment reevaluation (as occurred in our cases) requiring cessation of immunosuppressive drugs, extended careful monitoring, and pharmacotherapeutic readjustment through the course of treatment. The degree to which antibiotics contribute to eradication of M haemophilum is unknown; therefore, it is recommended that long-term antibiotic use and treatment aimed at recovering the immunocompromised state (eg, highly active antiretroviral therapy in a patient with AIDS) be implemented.2
Conclusion
Our 3 cases of M haemophilum infection involved the upper extremities of immunosuppressed patients older than 65 years. This propensity to affect the upper extremities could possibly be due to the lower temperature required for growth of M haemophilum. Initial histopathologic study showed granulomatous and neutrophilic infiltrates, yet histopathologic specimens from all 3 patients failed to display positive AFB staining, which delayed the initial antibiotic choice. In all cases, diagnosis was made by tissue culture after swab culture failed to grow the pathogen. Furthermore, the 3 cases took approximately 6 weeks to achieve final identification of the organism. Neither clinical lymphadenopathy nor systemic spread was noted in our patients; immunosuppression was discontinued when possible.
Mycobacterium haemophilum is an uncommon but potentially life-threatening infection that should be suspected in immunocompromised adults who present with atypical cellulitis of the extremities. The ultimate diagnosis often is delayed because the organism grows slowly (as long as 8 weeks) in tissue culture. For that reason, empiric antibiotic treatment, including a macrolide, should be considered in patients with disseminated or severe infection or critical immunosuppression and in those who do not demonstrate improvement in symptoms once immunosuppressants are withheld. A prolonged course of multiple-drug antibiotic therapy has proved to be effective for treating cutaneous infection with M haemophilum.
Infection with Mycobacterium haemophilum, a rare, slow-growing organism, most commonly presents as ulcerating cutaneous lesions and subcutaneous nodules in immunocompromised adults.1 The most common clinical presentation in adults includes cutaneous lesions, nodules, cysts, and papules, with signs and symptoms of erythema, pain, pruritus, and drainage.2 Disseminated disease states of septic arthritis, pulmonary infiltration, and osteomyelitis, though life-threatening, are less common manifestations reported in highly immunocompromised persons.3
Infection with M haemophilum presents a challenge to the dermatology community because it is infrequently suspected and misidentified, resulting in delayed diagnosis. Additionally, M haemophilum is an extremely fastidious organism that requires heme-supplemented culture media and a carefully regulated low temperature for many consecutive weeks to yield valid culture results.1 These features contribute to complications and delays in diagnosis of an already overlooked source of infection.
We discuss the clinical presentation, diagnosis, and treatment of 3 unusual cases of cutaneous M haemophilum infection involving the upper arms. The findings in these cases highlight the challenges inherent in diagnosis as well as the obstacles that arise in providing effective, long-term treatment of this infection.
Case Reports
Patient 1
A 69-year-old woman with a medical history of a single functioning kidney and moderate psoriasis managed with low-dosage methotrexate presented with an erythematous nonhealing wound on the left forearm that developed after she was scratched by a dog. The pustules, appearing as bright red, tender, warm abscesses, had been present for 3 months and were distributed on the left proximal and distal dorsal forearm (Figure 1A). The patient reported no recent travel, sick contacts, allergies, or new medications.
A shave biopsy was initially obtained. Swab specimens were sent for bacterial, fungal, and mycobacterial culture following discontinuation of methotrexate. Initial histopathologic analysis revealed aggregates of histiocytes and multinucleated giant cells within the dermis, surrounded by infiltrates of lymphocytes and neutrophils (Figure 2), consistent with a dermal noncaseating granulomatosis. Acid-fast bacilli (AFB), periodic acid–Schiff, Gram, and Grocott-Gomori methenamine-silver stains were negative for pathogenic microorganisms. There was no evidence of vasculitis.
Despite negative special stains, an infectious cause was still suspected. Oral doxycycline monohydrate 100 mg twice daily, oral fluconazole 200 mg daily, and econazole cream 1% were prescribed because of concern for mycobacterial infection and initial growth of Candida parapsilosis in the swab culture.
A punch biopsy also was performed at this time for both repeat histopathologic analysis and tissue culture. Follow-up appointments were scheduled every 2 weeks. Staining by AFB of the repeat histopathologic specimen was negative.
The patient demonstrated symptomatic and aesthetic improvement (Figure 1B) during consecutive regular follow-up appointments while culture results were pending. No lesions appeared above the left elbow and she had no lymphadenopathy. Results of blood chemistry analyses and complete blood cell count throughout follow-up were normal.
The final tissue culture report obtained 7 weeks after initial presentation showed growth of M haemophilum despite a negative smear. The swab culture that initially was taken did not grow pathogenic organisms.
The patient was referred to an infectious disease specialist who confirmed that the atypical mycobacterial infection likely was the main source of the cutaneous lesions. She was instructed to continue econazole cream 1% and was given prescriptions for clarithromycin 500 mg twice daily, ciprofloxacin 500 mg twice daily, and rifampin 300 mg twice daily for a total duration of 12 to 18 months. The patient has remained on this triple-drug regimen and demonstrated improvement in the lesions. She has been off methotrexate while on antibiotic therapy.
Patient 2
A 79-year-old man with a medical history of chronic lymphocytic leukemia, basal cell carcinoma, and squamous cell carcinoma presented with a nonhealing, painful, red lesion on the left forearm of 1 week’s duration. Physical examination revealed a violaceous nontender plaque with erosions and desquamation that was initially diagnosed as a carbuncle. The patient reported a similar eruption on the right foot that was successfully treated with silver sulfadiazine by another physician.
Biopsy was performed by the shave method for histologic analysis and tissue culture. Doxycycline 100 mg twice daily was prescribed because of high suspicion of infection. Histologic findings revealed granulomatous inflammation with pseudoepitheliomatous hyperplasia, reported as squamous cell carcinoma. A second opinion confirmed suspicion of an infectious process; the patient remained on doxycycline. During follow-up, the lesion progressed to a 5-cm plaque studded with pustules and satellite papules. Multiple additional tissue cultures were performed over 2 months until “light growth” of M haemophilum was reported.
The patient showed minimal improvement on tetracycline antibiotics. His condition was complicated by a photosensitivity reaction to doxycycline on the left and right forearms, hands, and nose. Consequently, triamcinolone was prescribed, doxycycline was discontinued, and minocycline 100 mg twice daily and ciprofloxacin 500 mg twice daily were prescribed.
Nine months after initial presentation, the lesions were still present but remarkably improved. The antibiotic regimen was discontinued after 11 months.
Patient 3
A 77-year-old woman with a history of rheumatoid arthritis treated with methotrexate and abatacept as well as cutaneous T-cell lymphoma treated with narrowband UVB radiation presented to the emergency department with fever and an inflamed right forearm (Figure 3A). Initial bacterial cultures of the wound and blood were negative.
The patient was treated with vancomycin and discharged on cephalexin once she became afebrile. She was seen at our office the next week for further evaluation. We recommended that she discontinue all immunosuppressant medications. A 4-mm tissue biopsy for hematoxylin and eosin staining and a separate 4-mm punch biopsy for culture were performed while she was taking cephalexin. Histopathologic analysis revealed numerous neutrophilic abscesses; however, Gram, AFB, and fungal stains were negative.
Arm edema and pustules slowly resolved, but the eschar and verrucous plaques continued to slowly progress while the patient was off immunosuppression. She was kept off antibiotics until mycobacterial culture was positive at 4 weeks, at which time she was placed on doxycycline and clarithromycin. Final identification of M haemophilum was made at 6 weeks; consequently, doxycycline was discontinued and she was referred to infectious disease for multidrug therapy. She remained afebrile during the entire 6 weeks until cultures were final.
While immunosuppressants were discontinued and clarithromycin was administered, the plaque changed from an edematous pustular dermatitis to a verrucous crusted plaque. Neither epitrochlear nor axillary lymphadenopathy was noted during the treatment period. The infectious disease specialist prescribed azithromycin, ethambutol, and rifampin, which produced marked improvement (Figure 3B). The patient has remained off immunosuppressive therapy while on antibiotics.
Comment
Clinical Presentation and Diagnosis
Mycobacterium haemophilum is a rare infectious organism that affects primarily immunocompromised adults but also has been identified in immunocompetent adults and pediatric patients.2 Commonly affected immunosuppressed groups include solid organ transplant recipients, bone marrow transplant recipients, human immunodeficiency virus–positive patients, and patients with rheumatoid arthritis.
The infection typically presents as small violaceous papules and pustules that become painful and erythematous, with progression and draining ulceration in later stages.2 In our cases, all lesions tended to evolve into a verrucous plaque that slowly resolved with antibiotic therapy.
Due to the rarity of this infection, the initial differential diagnosis can include infection with other mycobacteria, Sporothrix, Staphylococcus aureus, and other fungal pathogens. Misdiagnosis is a common obstacle in the treatment of M haemophilum due to its rarity, often negative AFB stains, and slow growth on culture media; therefore, tissue culture is essential to successful diagnosis and management. The natural reservoir of M haemophilum is unknown, but infection has been associated with contaminated water sources.1 In one case (patient 1), symptoms developed after a dog scratch; the other 2 patients were unaware of injury to the skin.Laboratory diagnosis of M haemophilum is inherently difficult and protracted. The species is a highly fastidious and slow-growing Mycobacterium that requires cooler (30°C) incubation for many weeks on agar medium enriched with hemin or ferric ammonium citrate to obtain valid growth.1 To secure timely diagnosis, the organism’s slow agar growth warrants immediate tissue culture and biopsy when an immunocompromised patient presents with clinical features of atypical infection of an extremity. Mycobacterium haemophilum infection likely is underreported because of these difficulties in diagnosis.
Management
Although there are no standard guidelines for antibiotic treatment of M haemophilum, the current literature recommends triple-drug therapy with clarithromycin, ciprofloxacin, and rifamycin for at least 12 to 24 months.2
Upon clinical suspicion of an atypical Mycobacterium, we recommend a macrolide antibiotic over doxycycline, however, because this class of agents maintains broad coverage while being more specific for atypical mycobacteria. Although an atypical Mycobacterium was suspected early in the presentation in our cases, we discourage immediate use of triple-agent antibiotic therapy until laboratory evidence is procured to minimize antibiotic overuse in patients who do not have a final diagnosis. Single-agent therapy for prolonged treatment is discouraged for atypical mycobacterial infections because of the high risk of antibiotic resistance. Therapy should be tailored to the needs of the individual based on the extent of dissemination of disease and the severity of immunosuppression.1,2
Additionally, underlying disease that results in immunosuppression might necessitate treatment reevaluation (as occurred in our cases) requiring cessation of immunosuppressive drugs, extended careful monitoring, and pharmacotherapeutic readjustment through the course of treatment. The degree to which antibiotics contribute to eradication of M haemophilum is unknown; therefore, it is recommended that long-term antibiotic use and treatment aimed at recovering the immunocompromised state (eg, highly active antiretroviral therapy in a patient with AIDS) be implemented.2
Conclusion
Our 3 cases of M haemophilum infection involved the upper extremities of immunosuppressed patients older than 65 years. This propensity to affect the upper extremities could possibly be due to the lower temperature required for growth of M haemophilum. Initial histopathologic study showed granulomatous and neutrophilic infiltrates, yet histopathologic specimens from all 3 patients failed to display positive AFB staining, which delayed the initial antibiotic choice. In all cases, diagnosis was made by tissue culture after swab culture failed to grow the pathogen. Furthermore, the 3 cases took approximately 6 weeks to achieve final identification of the organism. Neither clinical lymphadenopathy nor systemic spread was noted in our patients; immunosuppression was discontinued when possible.
Mycobacterium haemophilum is an uncommon but potentially life-threatening infection that should be suspected in immunocompromised adults who present with atypical cellulitis of the extremities. The ultimate diagnosis often is delayed because the organism grows slowly (as long as 8 weeks) in tissue culture. For that reason, empiric antibiotic treatment, including a macrolide, should be considered in patients with disseminated or severe infection or critical immunosuppression and in those who do not demonstrate improvement in symptoms once immunosuppressants are withheld. A prolonged course of multiple-drug antibiotic therapy has proved to be effective for treating cutaneous infection with M haemophilum.
- Lindeboom JA, Bruijnesteijn van Coppenraet LE, van Soolingen D, et al. Clinical manifestations, diagnosis, and treatment of Mycobacterium haemophilum infections. Clin Microbiol Rev. 2011;24:701-717.
- Tangkosakul T, Hongmanee P, Malathum K. Cutaneous Mycobacterium haemophilum infections in immunocompromised patients in a tertiary hospital in Bangkok, Thailand: under-reported/under-recognized infection. JMM Case Rep. 2014;1:E002618.
- Sabeti S, Pourabdollah Tootkaboni M, Abdolahi M, et al. Mycobacterium haemophilum: a report of cutaneous infection in a patient with end-stage renal disease. Int J Mycobacteriol. 2016;5(suppl 1):S236.
- Lindeboom JA, Bruijnesteijn van Coppenraet LE, van Soolingen D, et al. Clinical manifestations, diagnosis, and treatment of Mycobacterium haemophilum infections. Clin Microbiol Rev. 2011;24:701-717.
- Tangkosakul T, Hongmanee P, Malathum K. Cutaneous Mycobacterium haemophilum infections in immunocompromised patients in a tertiary hospital in Bangkok, Thailand: under-reported/under-recognized infection. JMM Case Rep. 2014;1:E002618.
- Sabeti S, Pourabdollah Tootkaboni M, Abdolahi M, et al. Mycobacterium haemophilum: a report of cutaneous infection in a patient with end-stage renal disease. Int J Mycobacteriol. 2016;5(suppl 1):S236.
Practice Points
- Mycobacterium haemophilum infections typically occur on the extremities of immunosuppressed patients.
- Acid-fast bacilli staining may be negative.
- Mycobacterial cultures may take up to 6 weeks for growth.
- Prolonged triple-antibiotic therapy and lowering of immunosuppression is ideal treatment.
Cutaneous Nocardiosis in an Immunocompromised Patient
Case Report
A 79-year-old man with chronic lymphocytic leukemia (CLL) who was being treated with ibrutinib presented to the emergency department with a dry cough, ataxia and falls, and vision loss. Physical examination was remarkable for diffuse crackles heard throughout the right lung and bilateral lower extremity weakness. Additionally, he had 4 pink mobile nodules on the left side of the forehead, right side of the chin, left submental area, and left postauricular scalp, which arose approximately 2 weeks prior to presentation. The left postauricular lesion had been tender at times and had developed a crust. The cutaneous lesions were all smaller than 2 cm.
The patient had a history of squamous cell carcinoma of the skin and was under the care of a dermatologist as an outpatient. His dermatologist had described him as an active gardener; he was noted to have healing abrasions on the forearms due to gardening raspberry bushes.
Computed tomography of the head revealed a 14-mm, ring-enhancing lesion in the left paramedian posterior frontal lobe with surrounding white matter vasogenic edema (Figure 1). Computed tomography of the chest revealed a peripheral mass on the right upper lobe measuring 6.3 cm at its greatest dimension (Figure 2).
Empiric antibiotic therapy with vancomycin and piperacillin-tazobactam was initiated. A dermatology consultation was placed by the hospitalist service; the consulting dermatologist noted that the patient had subepidermal nodules on the anterior thigh and abdomen, of which the patient had not been aware.
Clinically, the constellation of symptoms was thought to represent an infectious process or less likely metastatic malignancy. Biopsies of the nodule on the right side of the chin were performed and sent for culture and histologic examination. Sections from the anterior right chin showed compact orthokeratosis overlying a slightly spongiotic epidermis (Figure 3). Within the deep dermis, there was a dense mixed inflammatory infiltrate comprising predominantly neutrophils, with occasional eosinophils, lymphocytes, and histiocytes (Figure 4).
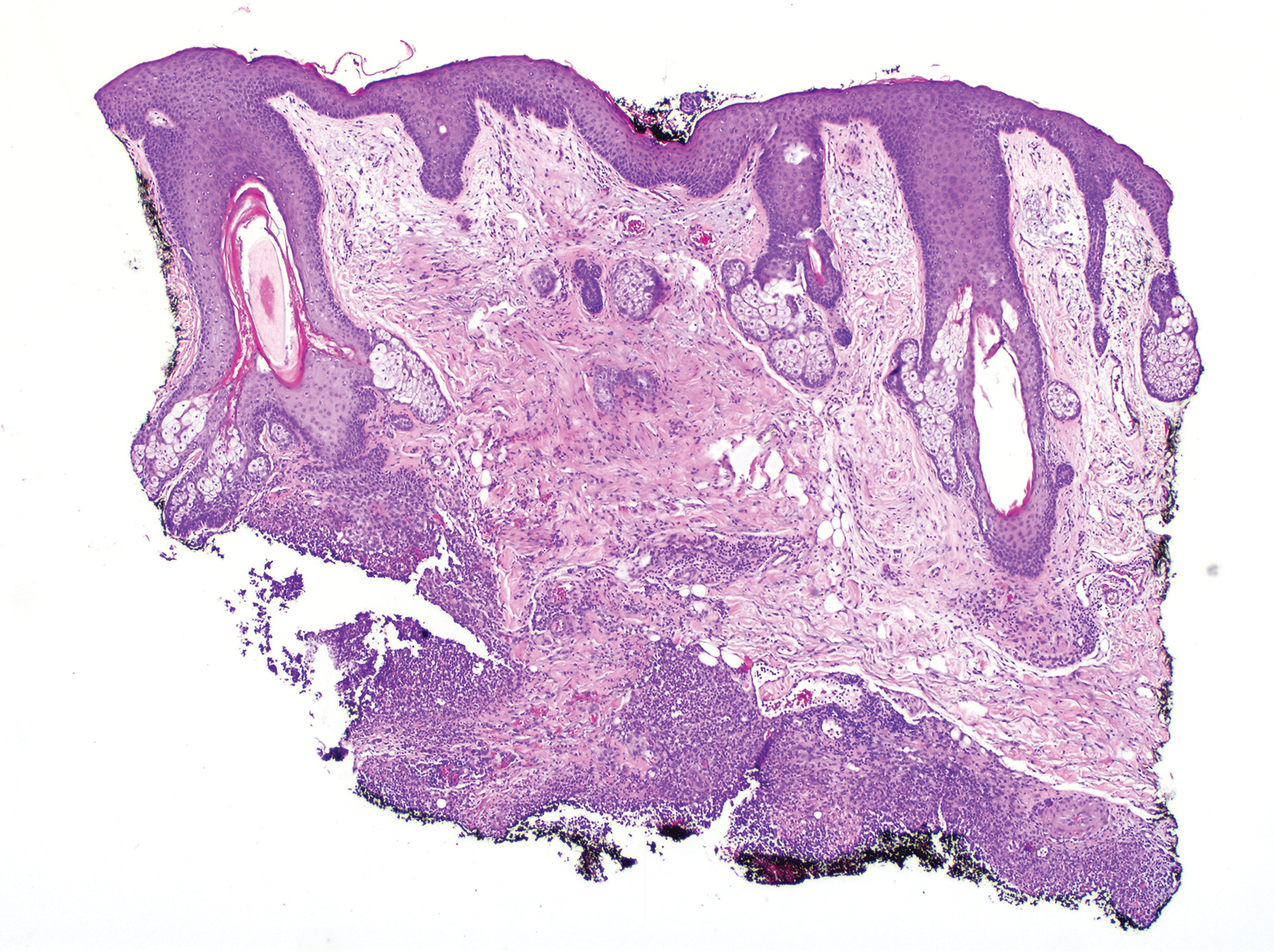
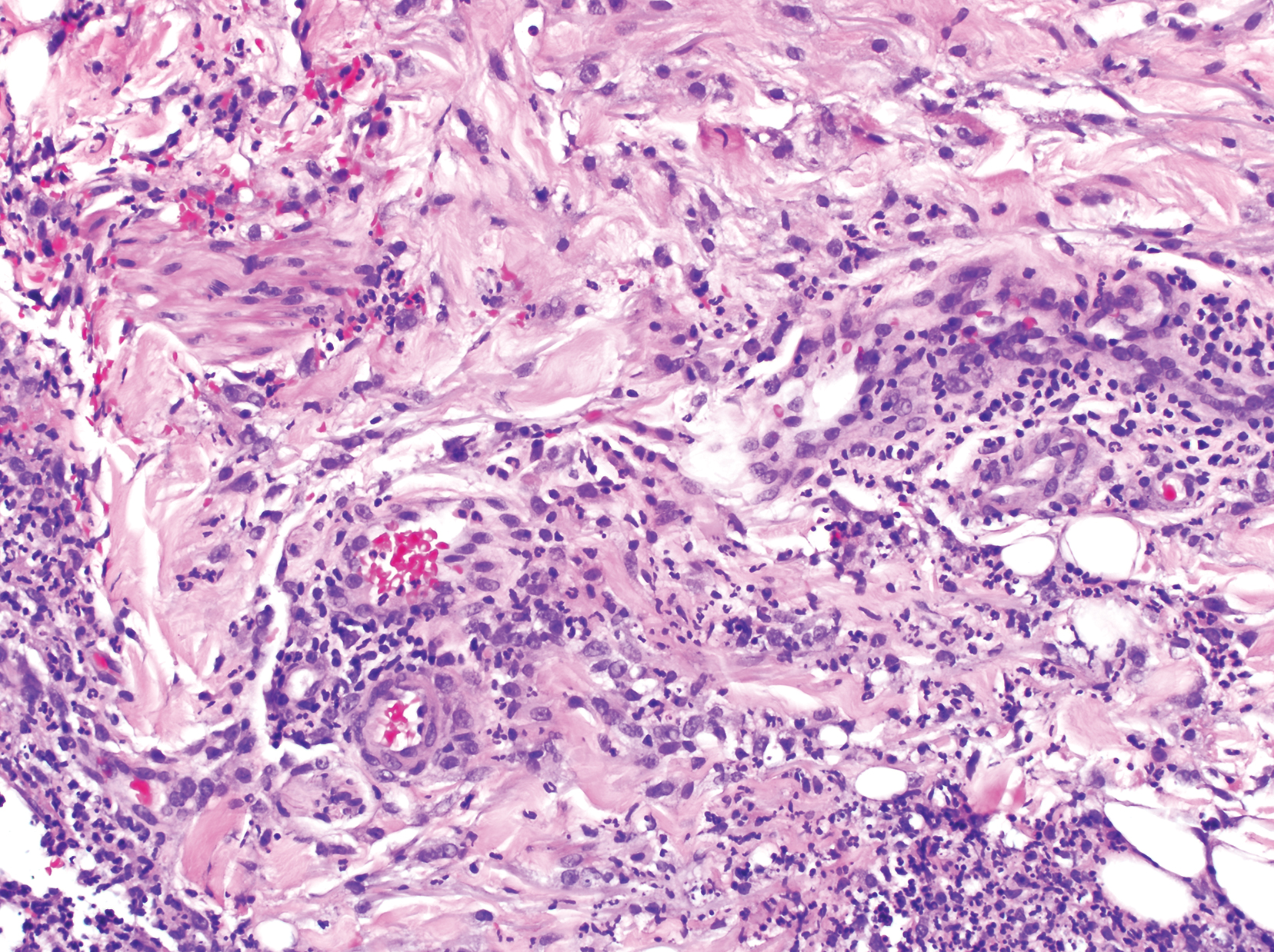
Gram stain revealed gram-variable, branching, bacterial organisms morphologically consistent with Nocardia. Grocott-Gomori methenamine-silver and periodic acid–Schiff stains also highlighted the bacterial organisms (Figure 5). An auramine-O stain was negative for acid-fast microorganisms. After 3 days on a blood agar plate, cultures of a specimen of the chin nodule grew branching filamentous bacterial organisms consistent with Nocardia.
Additionally, morphologically similar microorganisms were identified on a specimen of bronchoalveolar lavage (Figure 6). Blood cultures also returned positive for Nocardia. The specimen was sent to the South Dakota Public Health Laboratory (Pierre, South Dakota), which identified the organism as Nocardia asteroides. Given the findings in skin and the lungs, it was thought that the ring-enhancing lesion in the brain was most likely the result of Nocardia infection.
Antibiotic therapy was switched to trimethoprim-sulfamethoxazole. The patient’s mental status deteriorated; vital signs became unstable. He was transferred to the intensive care unit and was found to be hyponatremic, most likely a result of the brain lesion causing the syndrome of inappropriate antidiuretic hormone secretion. Mental status and clinical condition continued to deteriorate; the patient and his family decided to stop all aggressive care and move to a comfort-only approach. He was transferred to a hospice facility and died shortly thereafter.
Comment
Presentation and Diagnosis
Nocardiosis is an infrequently encountered opportunistic infection that typically targets skin, lungs, and the central nervous system (CNS). Nocardia species characteristically are gram-positive, thin rods that form beaded, right-angle, branching filaments.1 More than 50 Nocardia species have been clinically isolated.2
Definitive diagnosis requires culture. Nocardia grows well on nonselective media, such as blood or Löwenstein-Jensen agar; growth can be enhanced with 10% CO2. Growth can be slow, however, and takes from 48 hours to several weeks. Nocardia typically grows as buff or pigmented, waxy, cerebriform colonies at 3 to 5 days’ incubation.1
Cause of Infection
Nocardia species are commonly found in the environment—soil, plant matter, water, and decomposing organic material—as well as in the gastrointestinal tract and skin of animals. Infection has been reported in cattle, dogs, horses, swine, birds, cats, foxes, and a few other animals.2 A history of exposure, such as gardening or handling animals, should increase suspicion of Nocardia.3 Although infection is classically thought to affect immunocompromised patients, there are case reports of immunocompetent individuals developing disseminated infection.4-7 However, infected immunocompetent individuals typically have localized cutaneous infection, which often includes cellulitis, abscesses, or sporotrichoid patterns.2 Cutaneous infections typically are the result of direct inoculation of the skin through a penetrating injury.8
Disseminated nocardiosis can be caused by numerous species and generally is the result of primary pulmonary infection.9 In these cases, skin disease is present in approximately 10% of patients. Disseminated infection from cutaneous nocardiosis is uncommon; when it does occur, the most common site of dissemination is the CNS, resulting in abscess or cerebritis.10 Therefore, CNS involvement should always be ruled out on diagnosis in immunocompromised patients, even if neurologic symptoms are absent.9 Nearly 80% of patients with disseminated disease are, in fact, immunocompromised.8
Association With CLL
Chronic lymphocytic leukemia is associated with profound immunodeficiency caused by quantitative and qualitative aberrations in both innate and adaptive immunity. This perturbation of the immune system predisposes the patient to infection.11,12 Early in the course of CLL, a patient develops neutropenia, which predisposes to bacterial infection; later, the patient develops a sustained B- and T-cell immunodeficiency that predisposes to opportunistic infection.13 Treatment-naïve patients with CLL are commonly diagnosed with respiratory and urinary tract infections.12 Chronic lymphocytic leukemia patients treated with alemtuzumab or purine analogs have been reported to have the highest risk for major infection.14
Ibrutinib is a commonly used treatment of CLL because it induces apoptosis in B cells, which are abnormal in CLL. Ibrutinib functions by inhibiting the Bruton tyrosine kinase pathway, which is essential in B-cell production and maintenance.15 Studies have reported a high rate of infection in ibrutinib-treated CLL patients14,16; salvage ibrutinib therapy has been associated with higher infection risk than primary ibrutinib therapy.16,17 Long-term follow-up studies have shown a decreased rate of infection in ibrutinib-treated CLL after 2 years or longer of treatment, suggesting a reconstitution of normal B cells and humoral immunity with longer ibrutinib therapy.16,17
Many infections have been identified in association with ibrutinib therapy, including invasive aspergillosis, disseminated fusariosis, cerebral mucormycosis, disseminated cryptococcosis, and Pneumocystis jirovecii pneumonia.18-22 Disseminated nocardiosis has been reported in a few patients with CLL, though the treatment they received for CLL varied from case to case.23-25
Identification and Treatment
Clinical and microscopic identification of Nocardia organisms can be exceedingly difficult. Primary cutaneous nocardiosis clinically presents as tumors or nodules that often have a sporotrichoid pattern along the lymphatics. In disease that disseminates to skin, nocardiosis presents as vesiculopustules or abscesses. The biopsy specimen most often shows a dense dermal and subcutaneous infiltrate of neutrophils with abscess formation. Long-standing lesions might show chronic inflammation and nonspecific granulomas.
The appearance of Nocardia organisms is quite subtle on hematoxylin and eosin staining and can be easily missed. Special stains, such as Gram and Grocott-Gomori methenamine-silver stains as well as stains for acid-fast organisms, can be invaluable in diagnosing this disease. Biopsy in immunocompromised patients when nocardiosis is part of the differential diagnosis requires extra attention because the organisms can be gram variable and only partially acid fast, as was the case in our patient. Organisms typically will be positive with silver stains.
Trimethoprim-sulfamethoxazole typically is the first-line treatment of nocardiosis. Although prognosis is excellent when disease is confined to skin, disseminated infection has 25% mortality.8 Diagnosticians should maintain a high index of suspicion for the disease, especially in immunocompromised patients, because clinical and imaging findings can be nonspecific.
Conclusion
Our patient’s primary risk factor for nocardiosis was his immunocompromised state. In addition, he was an avid gardener, which increased his risk for exposure to the microorganism. Given the timing of disease progression, our case most likely represents primary cutaneous nocardiosis with dissemination to brain, lungs, and other organs, leading to death, and serves as a reminder to dermatologists and pathologists to establish a broad differential diagnosis when dealing with an infectious process in immunocompromised patients.
- Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Philadelphia, PA: Elsevier; 2016.
- McNeil MM, Brown JM. The medically important aerobic actinomycetes: epidemiology and microbiology. Clin Microbiol Rev. 1994;7:357-417.
- Grau Pérez M, Casabella Pernas A, de la Rosa Del Rey MDP, et al. Primary cutaneous nocardiosis: a pitfall in the diagnosis of skin infection. Infection. 2017;45:927-928.
- Oda R, Sekikawa Y, Hongo I. Primary cutaneous nocardiosis in an immunocompetent patient. Intern Med. 2017;56:469-470.
- Jiang Y, Huang A, Fang Q. Disseminated nocardiosis caused by Nocardia otitidiscaviarum in an immunocompetent host: a case report and literature review. Exp Ther Med. 2016;12:3339-3346.
- Cooper CJ, Said S, Popp M, et al. A complicated case of an immunocompetent patient with disseminated nocardiosis. Infect Dis Rep. 2014;6:5327.
- Kim MS, Choi H, Choi KC, et al. Primary cutaneous nocardiosis due to Nocardia vinacea: first case in an immunocompetent patient. Clin Exp Dermatol. 2011;36:812-814.
- Hall BJ, Hall JC, Cockerell CJ. Diagnostic Pathology. Nonneoplastic Dermatopathology. Salt Lake City, UT: Amirsys; 2012.
- Ambrosioni J, Lew D, Garbino J. Nocardiosis: updated clinical review and experience at a tertiary center. Infection. 2010;38:89-97.
- Bosamiya SS, Vaishnani JB, Momin AM. Sporotrichoid nocardiosis with cutaneous dissemination. Indian J Dermatol Venereol Leprol. 2011;77:535.
- Riches JC, Gribben JG. Understanding the immunodeficiency in chronic lymphocytic leukemia: potential clinical implications. Hematol Oncol Clin North Am. 2013;27:207-235.
- Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573-581.
- Tadmor T, Welslau M, Hus I. A review of the infection pathogenesis and prophylaxis recommendations in patients with chronic lymphocytic leukemia. Expert Rev Hematol. 2018;11:57-70.
- Williams AM, Baran AM, Meacham PJ, et al. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk Lymphoma. 2018;59:625-632.
- Dias AL, Jain D. Ibrutinib: a new frontier in the treatment of chronic lymphocytic leukemia by Bruton’s tyrosine kinase inhibition. Cardiovasc Hematol Agents Med Chem. 2013;11:265-271.
- Sun C, Tian X, Lee YS, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213-2219.
- Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497-2506.
- Arthurs B, Wunderle K, Hsu M, et al. Invasive aspergillosis related to ibrutinib therapy for chronic lymphocytic leukemia. Respir Med Case Rep. 2017;21:27-29.
- Chan TS, Au-Yeung R, Chim CS, et al. Disseminated fusarium infection after ibrutinib therapy in chronic lymphocytic leukaemia. Ann Hematol. 2017;96:871-872.
- Farid S, AbuSaleh O, Liesman R, et al. Isolated cerebral mucormycosis caused by Rhizomucor pusillus [published online October 4, 2017]. BMJ Case Rep. pii:bcr-2017-221473.
- Okamoto K, Proia LA, Demarais PL. Disseminated cryptococcal disease in a patient with chronic lymphocytic leukemia on ibrutinib. Case Rep Infect Dis. 2016;2016:4642831.
- Ahn IE, Jerussi T, Farooqui M, et al. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128:1940-1943.
- Roberts AL, Davidson RM, Freifeld AG, et al. Nocardia arthritidis as a cause of disseminated nocardiosis in a patient with chronic lymphocytic leukemia. IDCases. 2016;6:68-71.
- Rámila E, Martino R, Santamaría A, et al. Inappropriate secretion of antidiuretic hormone as the initial sign of central nervous system progression of nocardiosis in a patient with chronic lymphocytic leukemia. Haematologica. 1999;84:1155-1156.
- Phillips WB, Shields CL, Shields JA, et al. Nocardia choroidal abscess. Br J Ophthalmol. 1992;76:694-696.
Case Report
A 79-year-old man with chronic lymphocytic leukemia (CLL) who was being treated with ibrutinib presented to the emergency department with a dry cough, ataxia and falls, and vision loss. Physical examination was remarkable for diffuse crackles heard throughout the right lung and bilateral lower extremity weakness. Additionally, he had 4 pink mobile nodules on the left side of the forehead, right side of the chin, left submental area, and left postauricular scalp, which arose approximately 2 weeks prior to presentation. The left postauricular lesion had been tender at times and had developed a crust. The cutaneous lesions were all smaller than 2 cm.
The patient had a history of squamous cell carcinoma of the skin and was under the care of a dermatologist as an outpatient. His dermatologist had described him as an active gardener; he was noted to have healing abrasions on the forearms due to gardening raspberry bushes.
Computed tomography of the head revealed a 14-mm, ring-enhancing lesion in the left paramedian posterior frontal lobe with surrounding white matter vasogenic edema (Figure 1). Computed tomography of the chest revealed a peripheral mass on the right upper lobe measuring 6.3 cm at its greatest dimension (Figure 2).
Empiric antibiotic therapy with vancomycin and piperacillin-tazobactam was initiated. A dermatology consultation was placed by the hospitalist service; the consulting dermatologist noted that the patient had subepidermal nodules on the anterior thigh and abdomen, of which the patient had not been aware.
Clinically, the constellation of symptoms was thought to represent an infectious process or less likely metastatic malignancy. Biopsies of the nodule on the right side of the chin were performed and sent for culture and histologic examination. Sections from the anterior right chin showed compact orthokeratosis overlying a slightly spongiotic epidermis (Figure 3). Within the deep dermis, there was a dense mixed inflammatory infiltrate comprising predominantly neutrophils, with occasional eosinophils, lymphocytes, and histiocytes (Figure 4).
Gram stain revealed gram-variable, branching, bacterial organisms morphologically consistent with Nocardia. Grocott-Gomori methenamine-silver and periodic acid–Schiff stains also highlighted the bacterial organisms (Figure 5). An auramine-O stain was negative for acid-fast microorganisms. After 3 days on a blood agar plate, cultures of a specimen of the chin nodule grew branching filamentous bacterial organisms consistent with Nocardia.
Additionally, morphologically similar microorganisms were identified on a specimen of bronchoalveolar lavage (Figure 6). Blood cultures also returned positive for Nocardia. The specimen was sent to the South Dakota Public Health Laboratory (Pierre, South Dakota), which identified the organism as Nocardia asteroides. Given the findings in skin and the lungs, it was thought that the ring-enhancing lesion in the brain was most likely the result of Nocardia infection.
Antibiotic therapy was switched to trimethoprim-sulfamethoxazole. The patient’s mental status deteriorated; vital signs became unstable. He was transferred to the intensive care unit and was found to be hyponatremic, most likely a result of the brain lesion causing the syndrome of inappropriate antidiuretic hormone secretion. Mental status and clinical condition continued to deteriorate; the patient and his family decided to stop all aggressive care and move to a comfort-only approach. He was transferred to a hospice facility and died shortly thereafter.
Comment
Presentation and Diagnosis
Nocardiosis is an infrequently encountered opportunistic infection that typically targets skin, lungs, and the central nervous system (CNS). Nocardia species characteristically are gram-positive, thin rods that form beaded, right-angle, branching filaments.1 More than 50 Nocardia species have been clinically isolated.2
Definitive diagnosis requires culture. Nocardia grows well on nonselective media, such as blood or Löwenstein-Jensen agar; growth can be enhanced with 10% CO2. Growth can be slow, however, and takes from 48 hours to several weeks. Nocardia typically grows as buff or pigmented, waxy, cerebriform colonies at 3 to 5 days’ incubation.1
Cause of Infection
Nocardia species are commonly found in the environment—soil, plant matter, water, and decomposing organic material—as well as in the gastrointestinal tract and skin of animals. Infection has been reported in cattle, dogs, horses, swine, birds, cats, foxes, and a few other animals.2 A history of exposure, such as gardening or handling animals, should increase suspicion of Nocardia.3 Although infection is classically thought to affect immunocompromised patients, there are case reports of immunocompetent individuals developing disseminated infection.4-7 However, infected immunocompetent individuals typically have localized cutaneous infection, which often includes cellulitis, abscesses, or sporotrichoid patterns.2 Cutaneous infections typically are the result of direct inoculation of the skin through a penetrating injury.8
Disseminated nocardiosis can be caused by numerous species and generally is the result of primary pulmonary infection.9 In these cases, skin disease is present in approximately 10% of patients. Disseminated infection from cutaneous nocardiosis is uncommon; when it does occur, the most common site of dissemination is the CNS, resulting in abscess or cerebritis.10 Therefore, CNS involvement should always be ruled out on diagnosis in immunocompromised patients, even if neurologic symptoms are absent.9 Nearly 80% of patients with disseminated disease are, in fact, immunocompromised.8
Association With CLL
Chronic lymphocytic leukemia is associated with profound immunodeficiency caused by quantitative and qualitative aberrations in both innate and adaptive immunity. This perturbation of the immune system predisposes the patient to infection.11,12 Early in the course of CLL, a patient develops neutropenia, which predisposes to bacterial infection; later, the patient develops a sustained B- and T-cell immunodeficiency that predisposes to opportunistic infection.13 Treatment-naïve patients with CLL are commonly diagnosed with respiratory and urinary tract infections.12 Chronic lymphocytic leukemia patients treated with alemtuzumab or purine analogs have been reported to have the highest risk for major infection.14
Ibrutinib is a commonly used treatment of CLL because it induces apoptosis in B cells, which are abnormal in CLL. Ibrutinib functions by inhibiting the Bruton tyrosine kinase pathway, which is essential in B-cell production and maintenance.15 Studies have reported a high rate of infection in ibrutinib-treated CLL patients14,16; salvage ibrutinib therapy has been associated with higher infection risk than primary ibrutinib therapy.16,17 Long-term follow-up studies have shown a decreased rate of infection in ibrutinib-treated CLL after 2 years or longer of treatment, suggesting a reconstitution of normal B cells and humoral immunity with longer ibrutinib therapy.16,17
Many infections have been identified in association with ibrutinib therapy, including invasive aspergillosis, disseminated fusariosis, cerebral mucormycosis, disseminated cryptococcosis, and Pneumocystis jirovecii pneumonia.18-22 Disseminated nocardiosis has been reported in a few patients with CLL, though the treatment they received for CLL varied from case to case.23-25
Identification and Treatment
Clinical and microscopic identification of Nocardia organisms can be exceedingly difficult. Primary cutaneous nocardiosis clinically presents as tumors or nodules that often have a sporotrichoid pattern along the lymphatics. In disease that disseminates to skin, nocardiosis presents as vesiculopustules or abscesses. The biopsy specimen most often shows a dense dermal and subcutaneous infiltrate of neutrophils with abscess formation. Long-standing lesions might show chronic inflammation and nonspecific granulomas.
The appearance of Nocardia organisms is quite subtle on hematoxylin and eosin staining and can be easily missed. Special stains, such as Gram and Grocott-Gomori methenamine-silver stains as well as stains for acid-fast organisms, can be invaluable in diagnosing this disease. Biopsy in immunocompromised patients when nocardiosis is part of the differential diagnosis requires extra attention because the organisms can be gram variable and only partially acid fast, as was the case in our patient. Organisms typically will be positive with silver stains.
Trimethoprim-sulfamethoxazole typically is the first-line treatment of nocardiosis. Although prognosis is excellent when disease is confined to skin, disseminated infection has 25% mortality.8 Diagnosticians should maintain a high index of suspicion for the disease, especially in immunocompromised patients, because clinical and imaging findings can be nonspecific.
Conclusion
Our patient’s primary risk factor for nocardiosis was his immunocompromised state. In addition, he was an avid gardener, which increased his risk for exposure to the microorganism. Given the timing of disease progression, our case most likely represents primary cutaneous nocardiosis with dissemination to brain, lungs, and other organs, leading to death, and serves as a reminder to dermatologists and pathologists to establish a broad differential diagnosis when dealing with an infectious process in immunocompromised patients.
Case Report
A 79-year-old man with chronic lymphocytic leukemia (CLL) who was being treated with ibrutinib presented to the emergency department with a dry cough, ataxia and falls, and vision loss. Physical examination was remarkable for diffuse crackles heard throughout the right lung and bilateral lower extremity weakness. Additionally, he had 4 pink mobile nodules on the left side of the forehead, right side of the chin, left submental area, and left postauricular scalp, which arose approximately 2 weeks prior to presentation. The left postauricular lesion had been tender at times and had developed a crust. The cutaneous lesions were all smaller than 2 cm.
The patient had a history of squamous cell carcinoma of the skin and was under the care of a dermatologist as an outpatient. His dermatologist had described him as an active gardener; he was noted to have healing abrasions on the forearms due to gardening raspberry bushes.
Computed tomography of the head revealed a 14-mm, ring-enhancing lesion in the left paramedian posterior frontal lobe with surrounding white matter vasogenic edema (Figure 1). Computed tomography of the chest revealed a peripheral mass on the right upper lobe measuring 6.3 cm at its greatest dimension (Figure 2).
Empiric antibiotic therapy with vancomycin and piperacillin-tazobactam was initiated. A dermatology consultation was placed by the hospitalist service; the consulting dermatologist noted that the patient had subepidermal nodules on the anterior thigh and abdomen, of which the patient had not been aware.
Clinically, the constellation of symptoms was thought to represent an infectious process or less likely metastatic malignancy. Biopsies of the nodule on the right side of the chin were performed and sent for culture and histologic examination. Sections from the anterior right chin showed compact orthokeratosis overlying a slightly spongiotic epidermis (Figure 3). Within the deep dermis, there was a dense mixed inflammatory infiltrate comprising predominantly neutrophils, with occasional eosinophils, lymphocytes, and histiocytes (Figure 4).
Gram stain revealed gram-variable, branching, bacterial organisms morphologically consistent with Nocardia. Grocott-Gomori methenamine-silver and periodic acid–Schiff stains also highlighted the bacterial organisms (Figure 5). An auramine-O stain was negative for acid-fast microorganisms. After 3 days on a blood agar plate, cultures of a specimen of the chin nodule grew branching filamentous bacterial organisms consistent with Nocardia.
Additionally, morphologically similar microorganisms were identified on a specimen of bronchoalveolar lavage (Figure 6). Blood cultures also returned positive for Nocardia. The specimen was sent to the South Dakota Public Health Laboratory (Pierre, South Dakota), which identified the organism as Nocardia asteroides. Given the findings in skin and the lungs, it was thought that the ring-enhancing lesion in the brain was most likely the result of Nocardia infection.
Antibiotic therapy was switched to trimethoprim-sulfamethoxazole. The patient’s mental status deteriorated; vital signs became unstable. He was transferred to the intensive care unit and was found to be hyponatremic, most likely a result of the brain lesion causing the syndrome of inappropriate antidiuretic hormone secretion. Mental status and clinical condition continued to deteriorate; the patient and his family decided to stop all aggressive care and move to a comfort-only approach. He was transferred to a hospice facility and died shortly thereafter.
Comment
Presentation and Diagnosis
Nocardiosis is an infrequently encountered opportunistic infection that typically targets skin, lungs, and the central nervous system (CNS). Nocardia species characteristically are gram-positive, thin rods that form beaded, right-angle, branching filaments.1 More than 50 Nocardia species have been clinically isolated.2
Definitive diagnosis requires culture. Nocardia grows well on nonselective media, such as blood or Löwenstein-Jensen agar; growth can be enhanced with 10% CO2. Growth can be slow, however, and takes from 48 hours to several weeks. Nocardia typically grows as buff or pigmented, waxy, cerebriform colonies at 3 to 5 days’ incubation.1
Cause of Infection
Nocardia species are commonly found in the environment—soil, plant matter, water, and decomposing organic material—as well as in the gastrointestinal tract and skin of animals. Infection has been reported in cattle, dogs, horses, swine, birds, cats, foxes, and a few other animals.2 A history of exposure, such as gardening or handling animals, should increase suspicion of Nocardia.3 Although infection is classically thought to affect immunocompromised patients, there are case reports of immunocompetent individuals developing disseminated infection.4-7 However, infected immunocompetent individuals typically have localized cutaneous infection, which often includes cellulitis, abscesses, or sporotrichoid patterns.2 Cutaneous infections typically are the result of direct inoculation of the skin through a penetrating injury.8
Disseminated nocardiosis can be caused by numerous species and generally is the result of primary pulmonary infection.9 In these cases, skin disease is present in approximately 10% of patients. Disseminated infection from cutaneous nocardiosis is uncommon; when it does occur, the most common site of dissemination is the CNS, resulting in abscess or cerebritis.10 Therefore, CNS involvement should always be ruled out on diagnosis in immunocompromised patients, even if neurologic symptoms are absent.9 Nearly 80% of patients with disseminated disease are, in fact, immunocompromised.8
Association With CLL
Chronic lymphocytic leukemia is associated with profound immunodeficiency caused by quantitative and qualitative aberrations in both innate and adaptive immunity. This perturbation of the immune system predisposes the patient to infection.11,12 Early in the course of CLL, a patient develops neutropenia, which predisposes to bacterial infection; later, the patient develops a sustained B- and T-cell immunodeficiency that predisposes to opportunistic infection.13 Treatment-naïve patients with CLL are commonly diagnosed with respiratory and urinary tract infections.12 Chronic lymphocytic leukemia patients treated with alemtuzumab or purine analogs have been reported to have the highest risk for major infection.14
Ibrutinib is a commonly used treatment of CLL because it induces apoptosis in B cells, which are abnormal in CLL. Ibrutinib functions by inhibiting the Bruton tyrosine kinase pathway, which is essential in B-cell production and maintenance.15 Studies have reported a high rate of infection in ibrutinib-treated CLL patients14,16; salvage ibrutinib therapy has been associated with higher infection risk than primary ibrutinib therapy.16,17 Long-term follow-up studies have shown a decreased rate of infection in ibrutinib-treated CLL after 2 years or longer of treatment, suggesting a reconstitution of normal B cells and humoral immunity with longer ibrutinib therapy.16,17
Many infections have been identified in association with ibrutinib therapy, including invasive aspergillosis, disseminated fusariosis, cerebral mucormycosis, disseminated cryptococcosis, and Pneumocystis jirovecii pneumonia.18-22 Disseminated nocardiosis has been reported in a few patients with CLL, though the treatment they received for CLL varied from case to case.23-25
Identification and Treatment
Clinical and microscopic identification of Nocardia organisms can be exceedingly difficult. Primary cutaneous nocardiosis clinically presents as tumors or nodules that often have a sporotrichoid pattern along the lymphatics. In disease that disseminates to skin, nocardiosis presents as vesiculopustules or abscesses. The biopsy specimen most often shows a dense dermal and subcutaneous infiltrate of neutrophils with abscess formation. Long-standing lesions might show chronic inflammation and nonspecific granulomas.
The appearance of Nocardia organisms is quite subtle on hematoxylin and eosin staining and can be easily missed. Special stains, such as Gram and Grocott-Gomori methenamine-silver stains as well as stains for acid-fast organisms, can be invaluable in diagnosing this disease. Biopsy in immunocompromised patients when nocardiosis is part of the differential diagnosis requires extra attention because the organisms can be gram variable and only partially acid fast, as was the case in our patient. Organisms typically will be positive with silver stains.
Trimethoprim-sulfamethoxazole typically is the first-line treatment of nocardiosis. Although prognosis is excellent when disease is confined to skin, disseminated infection has 25% mortality.8 Diagnosticians should maintain a high index of suspicion for the disease, especially in immunocompromised patients, because clinical and imaging findings can be nonspecific.
Conclusion
Our patient’s primary risk factor for nocardiosis was his immunocompromised state. In addition, he was an avid gardener, which increased his risk for exposure to the microorganism. Given the timing of disease progression, our case most likely represents primary cutaneous nocardiosis with dissemination to brain, lungs, and other organs, leading to death, and serves as a reminder to dermatologists and pathologists to establish a broad differential diagnosis when dealing with an infectious process in immunocompromised patients.
- Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Philadelphia, PA: Elsevier; 2016.
- McNeil MM, Brown JM. The medically important aerobic actinomycetes: epidemiology and microbiology. Clin Microbiol Rev. 1994;7:357-417.
- Grau Pérez M, Casabella Pernas A, de la Rosa Del Rey MDP, et al. Primary cutaneous nocardiosis: a pitfall in the diagnosis of skin infection. Infection. 2017;45:927-928.
- Oda R, Sekikawa Y, Hongo I. Primary cutaneous nocardiosis in an immunocompetent patient. Intern Med. 2017;56:469-470.
- Jiang Y, Huang A, Fang Q. Disseminated nocardiosis caused by Nocardia otitidiscaviarum in an immunocompetent host: a case report and literature review. Exp Ther Med. 2016;12:3339-3346.
- Cooper CJ, Said S, Popp M, et al. A complicated case of an immunocompetent patient with disseminated nocardiosis. Infect Dis Rep. 2014;6:5327.
- Kim MS, Choi H, Choi KC, et al. Primary cutaneous nocardiosis due to Nocardia vinacea: first case in an immunocompetent patient. Clin Exp Dermatol. 2011;36:812-814.
- Hall BJ, Hall JC, Cockerell CJ. Diagnostic Pathology. Nonneoplastic Dermatopathology. Salt Lake City, UT: Amirsys; 2012.
- Ambrosioni J, Lew D, Garbino J. Nocardiosis: updated clinical review and experience at a tertiary center. Infection. 2010;38:89-97.
- Bosamiya SS, Vaishnani JB, Momin AM. Sporotrichoid nocardiosis with cutaneous dissemination. Indian J Dermatol Venereol Leprol. 2011;77:535.
- Riches JC, Gribben JG. Understanding the immunodeficiency in chronic lymphocytic leukemia: potential clinical implications. Hematol Oncol Clin North Am. 2013;27:207-235.
- Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573-581.
- Tadmor T, Welslau M, Hus I. A review of the infection pathogenesis and prophylaxis recommendations in patients with chronic lymphocytic leukemia. Expert Rev Hematol. 2018;11:57-70.
- Williams AM, Baran AM, Meacham PJ, et al. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk Lymphoma. 2018;59:625-632.
- Dias AL, Jain D. Ibrutinib: a new frontier in the treatment of chronic lymphocytic leukemia by Bruton’s tyrosine kinase inhibition. Cardiovasc Hematol Agents Med Chem. 2013;11:265-271.
- Sun C, Tian X, Lee YS, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213-2219.
- Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497-2506.
- Arthurs B, Wunderle K, Hsu M, et al. Invasive aspergillosis related to ibrutinib therapy for chronic lymphocytic leukemia. Respir Med Case Rep. 2017;21:27-29.
- Chan TS, Au-Yeung R, Chim CS, et al. Disseminated fusarium infection after ibrutinib therapy in chronic lymphocytic leukaemia. Ann Hematol. 2017;96:871-872.
- Farid S, AbuSaleh O, Liesman R, et al. Isolated cerebral mucormycosis caused by Rhizomucor pusillus [published online October 4, 2017]. BMJ Case Rep. pii:bcr-2017-221473.
- Okamoto K, Proia LA, Demarais PL. Disseminated cryptococcal disease in a patient with chronic lymphocytic leukemia on ibrutinib. Case Rep Infect Dis. 2016;2016:4642831.
- Ahn IE, Jerussi T, Farooqui M, et al. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128:1940-1943.
- Roberts AL, Davidson RM, Freifeld AG, et al. Nocardia arthritidis as a cause of disseminated nocardiosis in a patient with chronic lymphocytic leukemia. IDCases. 2016;6:68-71.
- Rámila E, Martino R, Santamaría A, et al. Inappropriate secretion of antidiuretic hormone as the initial sign of central nervous system progression of nocardiosis in a patient with chronic lymphocytic leukemia. Haematologica. 1999;84:1155-1156.
- Phillips WB, Shields CL, Shields JA, et al. Nocardia choroidal abscess. Br J Ophthalmol. 1992;76:694-696.
- Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Philadelphia, PA: Elsevier; 2016.
- McNeil MM, Brown JM. The medically important aerobic actinomycetes: epidemiology and microbiology. Clin Microbiol Rev. 1994;7:357-417.
- Grau Pérez M, Casabella Pernas A, de la Rosa Del Rey MDP, et al. Primary cutaneous nocardiosis: a pitfall in the diagnosis of skin infection. Infection. 2017;45:927-928.
- Oda R, Sekikawa Y, Hongo I. Primary cutaneous nocardiosis in an immunocompetent patient. Intern Med. 2017;56:469-470.
- Jiang Y, Huang A, Fang Q. Disseminated nocardiosis caused by Nocardia otitidiscaviarum in an immunocompetent host: a case report and literature review. Exp Ther Med. 2016;12:3339-3346.
- Cooper CJ, Said S, Popp M, et al. A complicated case of an immunocompetent patient with disseminated nocardiosis. Infect Dis Rep. 2014;6:5327.
- Kim MS, Choi H, Choi KC, et al. Primary cutaneous nocardiosis due to Nocardia vinacea: first case in an immunocompetent patient. Clin Exp Dermatol. 2011;36:812-814.
- Hall BJ, Hall JC, Cockerell CJ. Diagnostic Pathology. Nonneoplastic Dermatopathology. Salt Lake City, UT: Amirsys; 2012.
- Ambrosioni J, Lew D, Garbino J. Nocardiosis: updated clinical review and experience at a tertiary center. Infection. 2010;38:89-97.
- Bosamiya SS, Vaishnani JB, Momin AM. Sporotrichoid nocardiosis with cutaneous dissemination. Indian J Dermatol Venereol Leprol. 2011;77:535.
- Riches JC, Gribben JG. Understanding the immunodeficiency in chronic lymphocytic leukemia: potential clinical implications. Hematol Oncol Clin North Am. 2013;27:207-235.
- Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573-581.
- Tadmor T, Welslau M, Hus I. A review of the infection pathogenesis and prophylaxis recommendations in patients with chronic lymphocytic leukemia. Expert Rev Hematol. 2018;11:57-70.
- Williams AM, Baran AM, Meacham PJ, et al. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk Lymphoma. 2018;59:625-632.
- Dias AL, Jain D. Ibrutinib: a new frontier in the treatment of chronic lymphocytic leukemia by Bruton’s tyrosine kinase inhibition. Cardiovasc Hematol Agents Med Chem. 2013;11:265-271.
- Sun C, Tian X, Lee YS, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213-2219.
- Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497-2506.
- Arthurs B, Wunderle K, Hsu M, et al. Invasive aspergillosis related to ibrutinib therapy for chronic lymphocytic leukemia. Respir Med Case Rep. 2017;21:27-29.
- Chan TS, Au-Yeung R, Chim CS, et al. Disseminated fusarium infection after ibrutinib therapy in chronic lymphocytic leukaemia. Ann Hematol. 2017;96:871-872.
- Farid S, AbuSaleh O, Liesman R, et al. Isolated cerebral mucormycosis caused by Rhizomucor pusillus [published online October 4, 2017]. BMJ Case Rep. pii:bcr-2017-221473.
- Okamoto K, Proia LA, Demarais PL. Disseminated cryptococcal disease in a patient with chronic lymphocytic leukemia on ibrutinib. Case Rep Infect Dis. 2016;2016:4642831.
- Ahn IE, Jerussi T, Farooqui M, et al. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128:1940-1943.
- Roberts AL, Davidson RM, Freifeld AG, et al. Nocardia arthritidis as a cause of disseminated nocardiosis in a patient with chronic lymphocytic leukemia. IDCases. 2016;6:68-71.
- Rámila E, Martino R, Santamaría A, et al. Inappropriate secretion of antidiuretic hormone as the initial sign of central nervous system progression of nocardiosis in a patient with chronic lymphocytic leukemia. Haematologica. 1999;84:1155-1156.
- Phillips WB, Shields CL, Shields JA, et al. Nocardia choroidal abscess. Br J Ophthalmol. 1992;76:694-696.
Practice Points
- Clinicians should consider a broad differential when dealing with infectious diseases in immunocompromised patients.
- Primary cutaneous nocardiosis classically presents as tumors or nodules with a sporotrichoid pattern along the lymphatics. Vesiculopustules and abscesses are seen in disseminated disease, which usually involves the skin, lungs, and/or central nervous system.
- Nocardia species are characteristically gram-positive, thin rods that form beaded, right-angle branching filaments.
- When nocardiosis is in the differential, special care should be taken, as organisms can be gram variable or only partially acid fast. Gram, Grocott-Gomori methenamine-silver, and acid-fast staining may be essential to making the diagnosis.
The Role of Vitamins and Supplements on Skin Appearance
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
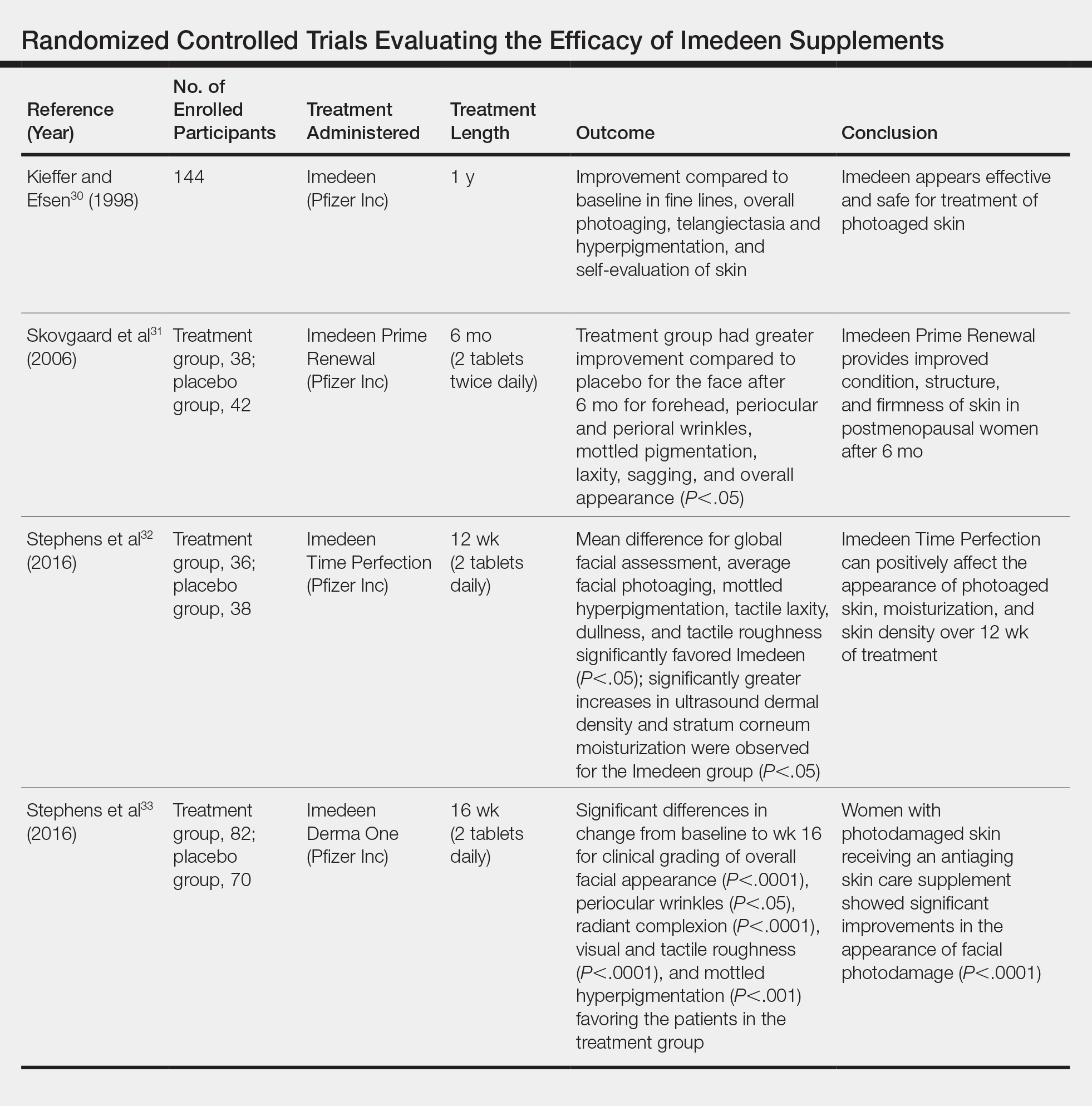
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
As the largest and most exposed organ in the body, the skin experiences trauma from both extrinsic and intrinsic aging factors, resulting in loss of elasticity, increased laxity, wrinkling, and rough-textured appearance.1 Chronologically aged skin appears dry, thin, and finely wrinkled; photoaged skin appears leathery with coarse wrinkles and uneven pigmentation.2 In recent years, numerous systemic nutrients have been proposed to improve skin appearance. This article reviews the efficacy of these vitamins and supplements.
Carotenoids
Carotenoids are a group of lipophilic molecules derived from vitamin A.3,4 Ingestion of carotenoids may play a role in photoprotection against UV radiation (UVR) by acting as acceptors of reactive oxygen species.4-6 Stahl et al7 investigated lycopene’s usefulness in protection against UVR-induced erythema. Over 10 weeks, 9 volunteers received 40 g of tomato paste containing 16 mg daily of lycopene while 10 controls received placebo. A solar simulator was used to induce erythema of the skin at weeks 0, 4, and 10. At week 10, erythema formation was 40% lower in the lycopene group compared to controls (P=.02).7
In another study assessing the photoprotective effects of a novel nutritional and phytonutrient blend of carotenoids, 36 women with Fitzpatrick skin types I and II were treated for 8 weeks.8 Presupplementation, UVR-induced erythema, and skin carotenoid concentrations were determined along with facial skin attributes and characteristics. Results showed protection against UVR-induced skin damage, with reductions in erythema at 3 minimal erythema doses (MEDs)(P=.01). Additionally, significant improvements were noted in facial skin elasticity, radiance, and overall appearance (all P<.05).8
In 2013, Meinke et al9 conducted an 8-week, double-blind, placebo-controlled study on 24 volunteers whose diets were supplemented with moderate amounts of carotenoids, including lutein, beta-carotene, and lycopene. Utilizing novel techniques to measure the skin’s ability to scavenge free radicals, they discovered that dietary carotenoids provided notable protection against stress-induced radical formation and increased baseline radical scavenging activity of the skin by 34%. The authors concluded that dietary supplementation could avoid premature skin aging.9
Vitamins C and E
Vitamin C is an essential vitamin that must be obtained through dietary sources.10 It functions as a free radical scavenger and is a necessary cofactor for the synthesis and stabilization of collagen.
A study evaluated the effect of UVR-induced oxidative stress and the association with vitamin C supplementation among 20 white patients with Fitzpatrick skin types II or III.11 The volunteers were treated with UVR on two 1-cm sites on the buttock. Six punch biopsies of these sites and 2 control biopsies from nonexposed skin were taken. Volunteers took vitamin C supplements (500 mg) for 8 weeks, and the exposure and biopsy were repeated. Researchers concluded that supplementation with vitamin C had no effect on the MED, with identical concentrations at baseline and after 8 weeks of supplementation. Additionally, there was no evidence that vitamin C affects UVR-induced oxidative stress.11
In 2007, Cosgrove et al12 conducted a study to assess the associations between nutrient intake and skin aging in more than 4000 women aged 40 to 74 years. Higher dietary vitamin C intakes were associated with a significantly lower likelihood of senile xerosis and wrinkled appearance (P<.009).12
Vitamin E is a lipid-soluble, membrane-bound vitamin, and its most active form is α-tocopherol.11,13 Vitamin E functions as an antioxidant and protects cellular membranes from lipid peroxidation by free radicals.13-15 Once oxidized, vitamin E can be regenerated to its reduced form by vitamin C.11 Their synergistic effects on skin protection have been studied extensively. A double-blind, placebo-controlled study of 10 patients compared 2 g of vitamin C combined with 1000 IU of vitamin E vs placebo.16 The patients’ skin reaction before and after 8 days of treatment were assessed by determination of MED and the cutaneous blood flow of skin irradiated with UV light. Results showed that the median MED of those taking vitamins increased from 80 to 96.5 mJ/cm2 (P<.01) and decreased for the placebo group. Investigators concluded that the combination of vitamins C and E reduces the sunburn reaction and leads to a reduction in the sequelae of UV-induced skin damage.16 A prospective, randomized, placebo-controlled study by Fuchs and Kern17 replicated these findings, also concluding that combinations of vitamins C and E provide improved photoprotective effects than either vitamin alone.
Vitamin D
Vitamin D is a fat-soluble vitamin obtained through dietary intake and exposure to UV light.3,18,19 Precursors of vitamin D require interaction with UV light for conversion into active forms. The highest concentrations of 7-dehydrocholesterol are found in keratinocytes in the basal cell and spinous cell layers of the skin where they are protected from UV light by melanin. As such, individuals with higher melanin content in their skin require more exposure to UV light to produce the same levels of vitamin D as those with less melanin,20 leading to a high rate of vitamin D deficiency in dark-skinned individuals. Because of their prodifferentiating and antiproliferative effects, vitamin D analogs have been very effective in the treatment of psoriasis.20,21 Vitamin D deficiency also has been implicated in the pathogenesis of vitiligo. A systematic review and meta-analysis conducted in 2016 found that a significant relationship existed between low 25-hydroxyvitamin D levels and vitiligo (P<.01), but no causal relationship could be established.22
A 2017 double-blind, placebo-controlled study performed by Scott et al23 aimed to elucidate the relationship between vitamin D concentrations and sunburn. Twenty adults received either placebo or high-dose vitamin D3 (200,000 IU) 1 hour after experimental sunburn induced by an erythemogenic dose of UVR. Investigators measured participants’ concentrations of the proinflammatory mediators tumor necrosis factor α and nitric oxide synthase via skin biopsy 48 hours later. Patients in the experimental group were found to have significantly reduced expression of both tumor necrosis factor α (P=.04) and nitric oxide synthase (P=.02). Additionally, participants with significantly higher vitamin D3 levels following supplementation (P=.007) demonstrated increased skin expression of the anti-inflammatory marker arginase-1 (P=.005) as well as a persistent reduction in skin redness (P=.02). Investigators concluded that vitamin D plays a large role in skin homeostasis and implicated vitamin D’s upregulation of arginase-1 as a potent mechanism of its anti-inflammatory effects.23
Collagen
As humans age, the density of collagen in the dermis decreases, leading to sagging and wrinkling of skin.24 Oral supplementation of collagen has been examined for its dermatologic benefits, primarily increasing the thickness and density of collagen in the dermal layer. In 2014, Proksch et al25 performed a double-blind, placebo-controlled trial in which 69 women were randomized to receive 2.5 or 5 g of collagen peptides or placebo for 8 weeks. Both treatment groups demonstrated improvements in skin elasticity as well as improved skin moisture and decreased skin evaporation; however, changes in the latter 2 qualities failed to reach statistical significance.25
The results of this study were replicated by Asserin et al.26 One hundred six female patients were randomly assigned to receive 10 g of collagen peptides or placebo daily for 8 weeks. The collagen group demonstrated significantly improved skin hydration (P=.003) and increased density of collagen in the dermis (P=.007) relative to placebo.26
In another randomized, double-blind, placebo-controlled study, 71 women consumed a 20-mL beverage containing either 3000 mg of collagen peptides or placebo for 12 weeks.27 Participants in the treatment group demonstrated significant decreases in periorbital wrinkles (P<.05) and enhanced facial skin moisture (P<.001) and elasticity (P<.001) after 12 weeks. Researchers concluded that oral supplementation with collagen peptides holds promise as a natural supplement to provide cutaneous antiaging properties.27
Ceramides
Ceramides are lipids composed of a sphingoid base conjugated to a fatty acid and serve as the main component of the stratum corneum of the skin. Ceramides are crucial for the maintenance of skin barrier integrity and for preventing transepidermal water loss.28 In a 3-month study of 51 women with dry skin, Guillou et al29 showed that a ceramide wheat extract capsule significantly increased corneometry measurements of skin hydration on the arms (P<.001) and the legs (P=.012) compared to placebo.
Mixed Supplements
The discovery that nutritional contents can affect skin appearance has energized the development of combination supplements containing multiple vitamins and micronutrients. Imedeen is a biomarine complex and antioxidant supplement with several different formulations, including Prime Renewal, Time Perfection, and Derma One (Pfizer Inc). The ingredients include a combination of a biomarine complex (blend of fish proteins and polysaccharides), lycopene, grape seed extract, vitamin C, vitamin E, and zinc. Several trials have been conducted to assess the efficacy of the supplements on improving the appearance of photodamaged and aged skin (Table).
A placebo-controlled, randomized study of 144 participants conducted by Kieffer and Efsen30 assessed the efficacy of Imedeen supplements over 12 months. The trial included a 3-month placebo-controlled study and 9-month uncontrolled continuation. Imedeen’s efficacy was measured using clinical evaluation, transepidermal water loss, self-evaluation, and photograph evaluation. After 1 year of treatment, improvement occurred in photograph evaluation of fine lines, overall photoaging, telangiectasia and hyperpigmentation, and self-evaluation of skin condition.30 Additional double-blind, placebo-controlled, randomized studies assessing the efficacy of Imedeen have shown increased dermal and epidermal thickness, improvement of stratum corneum moisturization, and improved overall facial complexion.31-33
Several combined supplements containing collagen peptide as the main ingredient have been created for use in skin care. Collagen is found in the extracellular matrix of the dermis and is responsible for the resiliency and strength of skin.34,35 Damage to the dermis can occur with prolonged UV light exposure and is seen histologically as disorganized collagen fibrils and grossly as wrinkles and photoaged skin.35,36
A study assessed the effect of BioCell Collagen (BioCell Technology, LLC), a supplement containing type II collagen, on skin aging.37 Twenty-six women underwent baseline visual assessments of their skin before taking 2 tablets of the supplement daily. Twelve weeks of supplementation led to significant reduction in global lines and wrinkles (13.2%; P=.028) as well as skin dryness and scaling (76%; P=.002). Assessment of collagen content at 6 weeks revealed a significant increase from baseline (6.3%; P=.002), though the difference after 12 weeks was not significant (3.5%; P=.134). The authors concluded that although preliminary data suggested that BioCell Collagen may reduce visible signs of aging, a controlled study was necessary to verify this finding.37
A single-blind, case-controlled study assessed a similar supplement, Celergen, that contained marine collagen peptides.38 Forty-one adults took 2 capsules each day for 60 days. Assessment of their skin physiology was conducted at the enrollment visit, 2 months later, and after the treatment period ended. Skin elasticity, transepidermal water loss, epidermal and dermal thickness, and density were measured. Investigators found that Celergen administration significantly enhanced skin elasticity and sebum production (P<.0001) but did not influence cutaneous moisture. The dermal thickness and homogenous distribution of collagen fibers were enhanced in 11 patients while properties of the epidermis remained unchanged. The study determined that supplementation remarkably improved skin elasticity, sebum production, and dermal ultrasonic markers.38
A double-blind, randomized, placebo-controlled study assessed a collagen- and antioxidant-containing supplement, Gold Collagen Forte, on skin properties.39 The treatment and placebo groups each consisted of 60 patients who consumed 1 bottle (50 mL) of the product each day for 90 days. Patients completed a self-assessment of their skin regarding photoaging, focusing on the crow’s-feet area and nasolabial folds, while skin elasticity was assessed with the SkinLab USB elasticity module. Results showed a significant increase in skin elasticity (+7.5%; P≤.001). Self-assessment results showed improvements in both the treatment and placebo groups, and investigators concluded that Gold Collagen Forte may have photoprotective effects and help improve skin health.39
Safety
Although trials have demonstrated vitamin supplementation to be safe and effective for skin enhancement, it is important to consider potential vitamin toxicities. High doses of vitamin C supplementation have been shown to cause damage via lipid peroxidation.40 In a study assessing if high levels of beta-carotene and vitamin E were associated with a lower risk for lung cancer, data showed that these supplements may actually have harmful effects.40,41 Additionally, consumption of high-dose dietary supplements has been associated with an increased risk for severe medical events, including disability and death among adolescents and young adults.42
Conclusion
Numerous trials have indicated that the use of systemic vitamins can have beneficial effects on the protection and appearance of skin. Photodamage from UV light–induced erythema can be decreased by carotenoids and vitamins C and E. Similarly, supplements that combine multiple nutrients with collagen have been shown to improve the appearance of aging skin by decreasing the prominence of wrinkles. Given the growing number of products and advertisements that exist in the supplement marketplace, it is crucial for clinicians to ground their recommendations to patients in the scientific data of robust studies.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
- Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. 2018;27:729-738.
- Rittié L, Fisher GJ. Natural and sun-induced aging of human skin. Cold Spring Harb Perspect Med. 2015;5:a015370.
- Draelos ZD. Nutrition and enhancing youthful-appearing skin. Clin Dermatol. 2010;28:400-408.
- Anunciato TP, da Rocha Filho PA. Carotenoids and polyphenols in nutricosmetics, nutraceuticals, and cosmeceuticals. J Cosmet Dermatol. 2012;11:51-54.
- Stahl W, Heinrich U, Jungmann H, et al. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795-798.
- Anstey AV. Systemic photoprotection with alpha-tocopherol (vitamin E) and beta-carotene. Clin Exp Dermatol. 2002;27:170-176.
- Stahl W, Heinrich U, Wiseman S, et al. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J Nutr. 2001;131:1449-1451.
- Wood SM, Mastaloudis AF, Hester SN, et al. Protective effects of a novel nutritional and phytonutrient blend on ultraviolet radiation-induced skin damage and inflammatory response through aging defense mechanisms. J Cosmet Dermatol. 2017;16:491-499.
- Meinke MC, Friedrich A, Tscherch K, et al. Influence of dietary carotenoids on radical scavenging capacity of the skin and skin lipids. Eur J Pharm Biopharm. 2013;84:365-373.
- Manela-Azulay M, Bagatin E. Cosmeceuticals vitamins. Clin Dermatol. 2009;27:469-474.
- McArdle F, Rhodes LE, Parslew R, et al. UVR-induced oxidative stress in human skin in vivo: effects of oral vitamin C supplementation. Free Radic Biol Med. 2002;33:1355-1362.
- Cosgrove MC, Franco OH, Granger SP, et al. Dietary nutrient intakes and skin-aging appearance among middle-aged American women. Am J Clin Nutr. 2007;86:1225-1231.
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667.
- Schagen SK, Zampeli VA, Makrantonaki E, et al. Discovering the link between nutrition and skin aging. Dermatoendocrinol. 2012;4:298-307.
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725-731.
- Eberlein-Konig B, Placzek M, Przybilla B. Protective effect against sunburn of combined systemic ascorbic acid (vitamin C) and d-alpha-tocopherol (vitamin E). J Am Acad Dermatol. 1998;38:45-48.
- Fuchs J, Kern H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: a clinical study using solar simulated radiation. Free Radic Biol Med. 1998;25:1006-1012.
- Shahriari M, Kerr PE, Slade K, et al. Vitamin D and the skin. Clin Dermatol. 2010;28:663-668.
- Soleymani T, Hung T, Soung J. The role of vitamin D in psoriasis: a review. Int J Dermatol. 2015;54:383-392.
- Lehmann B, Querings K, Reichrath J. Vitamin D and skin: new aspects for dermatology. Exp Dermatol. 2004;13(suppl 4):11-15.
- Kannan S, Lim HW. Photoprotection and vitamin D: a review. Photodermatol Photoimmunol Photomed. 2014;30:137-145.
- Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181-190.
- Scott JF, Das LM, Ahsanuddin S, et al. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J Invest Dermatol. 2017;137:2078-2086.
- Varani J, Dame MK, Rittie L, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861-1868.
- Proksch E, Segger D, Degwert J, et al. Oral supplementation of specific collagen peptides has beneficial effects on human skin physiology: a double-blind, placebo-controlled study. Skin Pharmacol Physiol. 2014;27:47-55.
- Asserin J, Lati E, Shioya T, et al. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J Cosmet Dermatol. 2015;14:291-301.
- Koizumi S, Inoue N, Shimizu M, et al. Effects of dietary supplementation with fish scales-derived collagen peptides on skin parameters and condition: a randomized, placebo-controlled, double-blind study. Int J Peptide Res Ther. 2018;24:397-402.
- Vollmer DL, West VA, Lephart ED. Enhancing skin health: by oral administration of natural compounds and minerals with implications to the dermal microbiome. Int J Mol Sci. 2018;19. doi:10.3390/ijms19103059.
- Guillou S, Ghabri S, Jannot C, et al. The moisturizing effect of a wheat extract food supplement on women’s skin: a randomized, double-blind placebo-controlled trial. Int J Cosmet Sci. 2011;33:138-143.
- Kieffer ME, Efsen J. Imedeen in the treatment of photoaged skin: an efficacy and safety trial over 12 months. J Eur Acad Dermatol Venereol. 1998;11:129-136.
- Skovgaard GR, Jensen AS, Sigler ML. Effect of a novel dietary supplement on skin aging in post-menopausal women. Eur J Clin Nutr. 2006;60:1201-1206.
- Stephens TJ, Sigler ML, Herndon JH Jr, et al. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen(®) Time Perfection(®) for improving the appearance of photodamaged skin. Clin Cosmet Investig Dermatol. 2016;9:63-70.
- Stephens TJ, Sigler ML, Hino PD, et al. A randomized, double-blind, placebo-controlled clinical trial evaluating an oral anti-aging skin care supplement for treating photodamaged skin. J Clin Aesthet Dermatol. 2016;9:25-32.
- El-Domyati M, Attia S, Saleh F, et al. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002;11:398-405.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Kang MC, Yumnam S, Kim SY. Oral intake of collagen peptide attenuates ultraviolet B irradiation-induced skin dehydration in vivo by regulating hyaluronic acid synthesis. Int J Mol Sci. 2018;19. doi:10.3390/ijms19113551.
- Schwartz SR, Park J. Ingestion of BioCell Collagen(®), a novel hydrolyzed chicken sternal cartilage extract; enhanced blood microcirculation and reduced facial aging signs. Clin Interv Aging. 2012;7:267-273.
- De Luca C, Mikhal’chik EV, Suprun MV, et al. Skin antiageing and systemic redox effects of supplementation with marine collagen peptides and plant-derived antioxidants: a single-blind case-control clinical study. Oxid Med Cell Longev. 2016;2016:4389410.
- Genovese L, Corbo A, Sibilla S. An insight into the changes in skin texture and properties following dietary intervention with a nutricosmeceutical containing a blend of collagen bioactive peptides and antioxidants. Skin Pharmacol Physiol. 2017;30:146-158.
- Hamishehkar H, Ranjdoost F, Asgharian P, et al. Vitamins, are they safe? Adv Pharm Bull. 2016;6:467-477.
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029-1035.
- Or F, Yongjoo K, Simms J, et al. Taking stock of dietary supplements’ harmful effects on children, adolescents, and young adults [published online June 3, 2019]. J Adolesc Health. S1054-139X(19)30163-6. doi:10.1016/j.jadohealth.2019.03.005.
Practice Points
- Multiple vitamins and supplements have demonstrated evidence in improving skin appearance.
- Carotenoids, along with vitamins C and E, have been shown to protect skin from UV-induced photodamage, while supplements containing collagen decrease the appearance of wrinkles.
