User login
Less is more when it comes to ketorolac for pain
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
PRACTICE CHANGER
Use a low dose (10 mg) of intravenous ketorolac for moderate to severe acute pain in adults because it is as effective as higher doses (15-30 mg).1
STRENGTH OF RECOMMENDATION
B: Based on a single, good-quality randomized controlled trial.
Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.

Alcohol use disorder: How best to screen and intervene
THE CASE
Ms. E, a 42-year-old woman, visited her new physician for a physical exam. When asked about alcohol intake, she reported that she drank 3 to 4 beers after work and sometimes 5 to 8 beers a day on the weekends. Occasionally, she exceeded those amounts, but she didn’t feel guilty about her drinking. She was often late to work and said her relationship with her boyfriend was strained. A review of systems was positive for fatigue, poor concentration, abdominal pain, and weight gain. Her body mass index was 41, pulse 100 beats/min, blood pressure 125/75 mm Hg, and she was afebrile. Her physical exam was otherwise within normal limits.
How would you proceed with this patient?
Alcohol use disorder (AUD) is a common and often untreated condition that is increasingly prevalent in the United States.1 The Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) characterizes AUD as a combination of signs and symptoms typifying alcohol abuse and dependence (discussed in a bit).2
Data from the 2015 National Survey on Drug Use and Health (NSDUH) showed 15.7 million Americans with AUD, affecting 6.2% of the population ages 18 years or older and 2.5% of adolescents ages 12 to 17 years.3
Alcohol use and AUD account for an estimated 3.8% of all global deaths and 4.6% of global disability-adjusted life years.4 AUD adversely affects several systems (TABLE 15), and patients with AUD are sicker and more likely to die younger than those without AUD.4 In the United States, prevalence of AUD has increased in recent years among women, older adults, racial minorities, and individuals with a low education level.6

Screening for AUD is reasonable and straightforward, although diagnosis and treatment of AUD in primary care settings may be challenging due to competing clinical priorities; lack of training, resources, and support; and skepticism about the efficacy of behavioral and pharmacologic treatments.7,8 However, family physicians are in an excellent position to diagnose and help address the complex biopsychosocial needs of patients with AUD, often in collaboration with colleagues and community organizations.
Signs and symptoms of AUD
In clinical practice, at least 2 of the following 11 behaviors or symptoms are required to diagnose AUD2:
- consuming larger amounts of alcohol over a longer period than intended
- persistent desire or unsuccessful efforts to cut down or control alcohol use
- making a significant effort to obtain, use, or recover from alcohol
In moderate-to-severe cases:
- cravings or urges to use alcohol
- recurrent failure to fulfill major work, school, or social obligations
- continued alcohol use despite recurrent social and interpersonal problems
- giving up social, occupational, and recreational activities due to alcohol
- using alcohol in physically dangerous situations
- continued alcohol use despite having physical or psychological problems
- tolerance to alcohol’s effects
- withdrawal symptoms.
Continue to: Patients meet criteria for mild AUD severity if...
Patients meet criteria for mild AUD severity if they exhibit 2 or 3 symptoms, moderate AUD with 4 or 5 symptoms, and severe AUD if there are 6 or more symptoms.2
Those who meet criteria for AUD and are able to stop using alcohol are deemed to be in early remission if the criteria have gone unfulfilled for at least 3 months and less than 12 months. Patients are considered to be in sustained remission if they have not met criteria for AUD at any time during a period of 12 months or longer.
How to detect AUD
Several clues in a patient’s history can suggest AUD (TABLE 29,10). Most imbibers are unaware of the dangers and may consider themselves merely “social drinkers.” Binge drinking may be an early indicator of vulnerability to AUD and should be assessed as part of a thorough clinical evaluation.11 The US Preventive Services Task Force (USPSTF) recommends (Grade B) that clinicians screen adults ages 18 years or older for alcohol misuse.12

Studies demonstrate that both genetic and environmental factors play important roles in the development of AUD.13 A family history of excessive alcohol use increases the risk of AUD. Comorbidity of AUD and other mental health conditions is extremely common. For example, high rates of association between major depressive disorder and AUD have been observed.14
Tools to use in screening and diagnosing AUD
Screening for AUD during an office visit can be done fairly quickly. While 96% of primary care physicians screen for alcohol misuse in some way, only 38% use 1 of the 3 tools recommended by the USPSTF15—the Alcohol Use Disorders Identification Test (AUDIT), the abbreviated AUDIT-C, or the National Institute on Alcohol Abuse and Alcoholism (NIAAA) single question screen—which detect the full spectrum of alcohol misuse in adults.12 Although the commonly used CAGE questionnaire is one of the most studied self-report tools, it has lower sensitivity at a lower level of alcohol intake.16
Continue to: The NIAAA single-question screen asks...
The NIAAA single-question screen asks how many times in the past year the patient had ≥4 drinks (women) or ≥5 drinks (men) in a day.15 The sensitivity and specificity of single-question screening are 82% to 87% and 61% to 79%, respectively, and the test has been validated in several different settings.12 The AUDIT screening tool, freely available from the World Health Organization, is a 10-item questionnaire that probes an individual’s alcohol intake, alcohol dependence, and adverse consequences of alcohol use. Administration of the AUDIT typically requires only 2 minutes. AUDIT-C17 is an abbreviated version of the AUDIT questionnaire that asks 3 consumption questions to screen for AUD.
It was found that AUDIT scores in the range of 8 to 15 indicate a medium-level alcohol problem, whereas a score of ≥16 indicates a high-level alcohol problem. The AUDIT-C is scored from 0 to 12, with ≥4 indicating a problem in men and ≥3
THE CASE
The physician had used the NIAAA single- question screen to determine that Ms. E drank more than 4 beers per day during social events and weekends, which occurred 2 to 3 times per month over the past year. She lives alone and said that she’d been seeing less and less of her boyfriend lately. Her score on the Patient Health Questionnaire (PHQ), which screens for depression, was 11, indicating moderate impairment. Her response on the CAGE questionnaire was negative for a problem with alcohol. However, her AUDIT score was 17, indicating a high-level alcohol problem. Based on these findings, her physician expressed concern that her alcohol use might be contributing to her symptoms and difficulties.
Although she did not have a history of increasing usage per day, a persistent desire to cut down, significant effort to obtain alcohol, or cravings, she was having work troubles and continued to drink even though it was straining relationships, promoting weight gain, and causing abdominal pain.
The physician asked her to schedule a return visit and ordered several blood studies. He also offered to connect her with a colleague with whom he collaborated who could speak with her about possible alcohol use disorders and depression.
Continue to: Selecting blood work in screening for AUD
Selecting blood work in screening for AUD
Lab tests used to measure hepatic injury due to alcohol include gamma-glutamyl-transferase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and macrocytic volume, although the indices of hepatic damage have low specificity. Elevated serum ethanol levels can reveal recent alcohol use, and vitamin deficiencies and other abnormalities can be used to differentiate other causes of hepatic inflammation and co-existing health issues (TABLE 310,18). A number of as-yet-unvalidated biomarkers are being studied to assist in screening, diagnosing, and treating AUD.18

What treatment approaches work for AUD?
Family physicians can efficiently and productively address AUD by using alcohol screening and brief intervention, which have been shown to reduce risky drinking. Reimbursement for this service is covered by such CPT codes as 99408, 99409, or H0049, or with other evaluation and management (E/M) codes by using modifier 25.
Treatment of AUD varies and should be customized to each patient’s needs, readiness, preferences, and resources. Individual and group counseling approaches can be effective, and medications are available for inpatient and outpatient settings. Psychotherapy options include brief interventions, 12-step programs (eg, Alcoholics Anonymous—https://www.aa.org/pages/en_US/find-aa-resources),motivational enhancement therapy, and cognitive behavioral therapy. Although it is beyond the scope of this article to describe these options in detail, resources are available for those who wish to learn more.19-21
Psychopharmacologic management includes US Food and Drug Administration (FDA)-approved medications such as disulfiram, naltrexone, and acamprosate, and off-label uses of other medications (TABLE 49). Not enough empiric evidence is available to judge the effectiveness of these medications in adolescents, and the FDA has not approved them for such use. Evidence from meta-analyses comparing naltrexone and acamprosate have shown naltrexone to be more efficacious in reducing heavy drinking and cravings, while acamprosate is effective in promoting abstinence.22,23 Naltrexone combined with behavioral intervention reduces the heavy drinking days and percentage of abstinence days.24

Current guideline recommendations from the American Psychiatric Association25 include:
- Naltrexone and acamprosate are recommended to treat patients with moderate-to-severe AUD in specific circumstances (eg, when nonpharmacologic approaches have failed to produce an effect or when patients prefer to use one of these medications).
- Topiramate and gabapentin are also suggested as medications for patients with moderate-to-severe AUD, but typically after first trying naltrexone and acamprosate.
- Disulfiram generally should not be used as first-line treatment. It produces physical reactions (eg, flushing) if alcohol is consumed within 12 to 24 hours of medication use.
Continue to: THE CASE
THE CASE
Ms. E was open to the idea of decreasing her alcohol use and agreed that she was depressed. Her lab tests at follow-up were normal other than an elevated AST/ALT of 90/80 U/L. S
She continued to get counseling for her AUD and for her comorbid depression in addition to taking a selective serotonin reuptake inhibitor. She is now in early remission for her alcohol use.
CORRESPONDENCE
Jaividhya Dasarathy, MD, Department of Family Medicine, Metro Health Medical Center, 2500 MetroHealth Drive, Cleveland, OH 44109; [email protected].
1. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72:757-766.
2. APA. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington DC; 2013.
3. HHS. Results from the 2015 National Survey on Drug Use and Health: summary of national findings. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.pdf. Accessed November 27, 2018.
4. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223-2233.
5. Chase V, Neild R, Sadler CW, et al. The medical complications of alcohol use: understanding mechanisms to improve management. Drug Alcohol Rev. 2005;24:253-265.
6. Grant BF, Chou SP, Saha TD, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001-2002 to 2012-2013: results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74:911-923.
7. Williams EC, Achtmeyer CE, Young JP, et al. Barriers to and facilitators of alcohol use disorder pharmacotherapy in primary care: a qualitative study in five VA clinics. J Gen Intern Med. 2018;33:258-267.
8. Zhang DX, Li ST, Lee QK, et al. Systematic review of guidelines on managing patients with harmful use of alcohol in primary healthcare settings. Alcohol Alcohol. 2017;52:595-609.
9. Wackernah RC, Minnick MJ, Clapp P. Alcohol use disorder: pathophysiology, effects, and pharmacologic options for treatment. Subst Abuse Rehabil. 2014;5:1-12.
10. Kattimani S, Bharadwaj B. Clinical management of alcohol withdrawal: a systematic review. Ind Psychiatry J. 2013;22:100-108.
11. Gowin JL, Sloan ME, Stangl BL, et al. Vulnerability for alcohol use disorder and rate of alcohol consumption. Am J Psychiatry. 2017;174:1094-1101.
12. Moyer VA; Preventive Services Task Force. Screening and behavioral counseling interventions in primary care to reduce alcohol misuse: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:210-218.
13. Tarter RE, Alterman AI, Edwards KL. Vulnerability to alcoholism in men: a behavior-genetic perspective. J Stud Alcohol. 1985;46:329-356.
14. Brière FN, Rohde P, Seeley JR, et al. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood [published online ahead of print, October 22, 2013]. Compr Psychiatry. 2014;55:526-533. doi: 10.1016/j.comppsych.2013.10.007.
15. Tan CH, Hungerford DW, Denny CH, et al. Screening for alcohol misuse: practices among U.S. primary care providers, DocStyles 2016. Am J Prev Med. 2018;54:173-180.
16. Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol. 2004;57:30-39.
17. Bush K, Kivlahan DR, McDonell MB, et al. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med. 1998;158:1789-1795.
18. Nanau RM, Neuman MG. Biomolecules and biomarkers used in diagnosis of alcohol drinking and in monitoring therapeutic interventions. Biomolecules. 2015;5:1339-1385.
19. Raddock M, Martukovich R, Berko E, et al. 7 tools to help patients adopt healthier behaviors. J Fam Pract. 2015;64:97-103.
20. AHRQ. Whitlock EP, Green CA, Polen MR, et al. Behavioral Counseling Interventions in Primary Care to Reduce Risky/Harmful Alcohol Use. 2004. https://www.ncbi.nlm.nih.gov/books/NBK42863/. Accessed November 17, 2018.
21. Miller WR, Baca C, Compton WM, et al. Addressing substance abuse in health care settings. Alcohol Clin Exp Res. 2006;30:292-302.
22. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108:275-293.
23. Rosner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22:11-23.
24. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003-2017.
25. Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association Practice Guideline for the Pharmacological Treatment of Patients With Alcohol Use Disorder. Am J Psychiatry. 2018;175:86-90.
THE CASE
Ms. E, a 42-year-old woman, visited her new physician for a physical exam. When asked about alcohol intake, she reported that she drank 3 to 4 beers after work and sometimes 5 to 8 beers a day on the weekends. Occasionally, she exceeded those amounts, but she didn’t feel guilty about her drinking. She was often late to work and said her relationship with her boyfriend was strained. A review of systems was positive for fatigue, poor concentration, abdominal pain, and weight gain. Her body mass index was 41, pulse 100 beats/min, blood pressure 125/75 mm Hg, and she was afebrile. Her physical exam was otherwise within normal limits.
How would you proceed with this patient?
Alcohol use disorder (AUD) is a common and often untreated condition that is increasingly prevalent in the United States.1 The Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) characterizes AUD as a combination of signs and symptoms typifying alcohol abuse and dependence (discussed in a bit).2
Data from the 2015 National Survey on Drug Use and Health (NSDUH) showed 15.7 million Americans with AUD, affecting 6.2% of the population ages 18 years or older and 2.5% of adolescents ages 12 to 17 years.3
Alcohol use and AUD account for an estimated 3.8% of all global deaths and 4.6% of global disability-adjusted life years.4 AUD adversely affects several systems (TABLE 15), and patients with AUD are sicker and more likely to die younger than those without AUD.4 In the United States, prevalence of AUD has increased in recent years among women, older adults, racial minorities, and individuals with a low education level.6
Screening for AUD is reasonable and straightforward, although diagnosis and treatment of AUD in primary care settings may be challenging due to competing clinical priorities; lack of training, resources, and support; and skepticism about the efficacy of behavioral and pharmacologic treatments.7,8 However, family physicians are in an excellent position to diagnose and help address the complex biopsychosocial needs of patients with AUD, often in collaboration with colleagues and community organizations.
Signs and symptoms of AUD
In clinical practice, at least 2 of the following 11 behaviors or symptoms are required to diagnose AUD2:
- consuming larger amounts of alcohol over a longer period than intended
- persistent desire or unsuccessful efforts to cut down or control alcohol use
- making a significant effort to obtain, use, or recover from alcohol
In moderate-to-severe cases:
- cravings or urges to use alcohol
- recurrent failure to fulfill major work, school, or social obligations
- continued alcohol use despite recurrent social and interpersonal problems
- giving up social, occupational, and recreational activities due to alcohol
- using alcohol in physically dangerous situations
- continued alcohol use despite having physical or psychological problems
- tolerance to alcohol’s effects
- withdrawal symptoms.
Continue to: Patients meet criteria for mild AUD severity if...
Patients meet criteria for mild AUD severity if they exhibit 2 or 3 symptoms, moderate AUD with 4 or 5 symptoms, and severe AUD if there are 6 or more symptoms.2
Those who meet criteria for AUD and are able to stop using alcohol are deemed to be in early remission if the criteria have gone unfulfilled for at least 3 months and less than 12 months. Patients are considered to be in sustained remission if they have not met criteria for AUD at any time during a period of 12 months or longer.
How to detect AUD
Several clues in a patient’s history can suggest AUD (TABLE 29,10). Most imbibers are unaware of the dangers and may consider themselves merely “social drinkers.” Binge drinking may be an early indicator of vulnerability to AUD and should be assessed as part of a thorough clinical evaluation.11 The US Preventive Services Task Force (USPSTF) recommends (Grade B) that clinicians screen adults ages 18 years or older for alcohol misuse.12
Studies demonstrate that both genetic and environmental factors play important roles in the development of AUD.13 A family history of excessive alcohol use increases the risk of AUD. Comorbidity of AUD and other mental health conditions is extremely common. For example, high rates of association between major depressive disorder and AUD have been observed.14
Tools to use in screening and diagnosing AUD
Screening for AUD during an office visit can be done fairly quickly. While 96% of primary care physicians screen for alcohol misuse in some way, only 38% use 1 of the 3 tools recommended by the USPSTF15—the Alcohol Use Disorders Identification Test (AUDIT), the abbreviated AUDIT-C, or the National Institute on Alcohol Abuse and Alcoholism (NIAAA) single question screen—which detect the full spectrum of alcohol misuse in adults.12 Although the commonly used CAGE questionnaire is one of the most studied self-report tools, it has lower sensitivity at a lower level of alcohol intake.16
Continue to: The NIAAA single-question screen asks...
The NIAAA single-question screen asks how many times in the past year the patient had ≥4 drinks (women) or ≥5 drinks (men) in a day.15 The sensitivity and specificity of single-question screening are 82% to 87% and 61% to 79%, respectively, and the test has been validated in several different settings.12 The AUDIT screening tool, freely available from the World Health Organization, is a 10-item questionnaire that probes an individual’s alcohol intake, alcohol dependence, and adverse consequences of alcohol use. Administration of the AUDIT typically requires only 2 minutes. AUDIT-C17 is an abbreviated version of the AUDIT questionnaire that asks 3 consumption questions to screen for AUD.
It was found that AUDIT scores in the range of 8 to 15 indicate a medium-level alcohol problem, whereas a score of ≥16 indicates a high-level alcohol problem. The AUDIT-C is scored from 0 to 12, with ≥4 indicating a problem in men and ≥3
THE CASE
The physician had used the NIAAA single- question screen to determine that Ms. E drank more than 4 beers per day during social events and weekends, which occurred 2 to 3 times per month over the past year. She lives alone and said that she’d been seeing less and less of her boyfriend lately. Her score on the Patient Health Questionnaire (PHQ), which screens for depression, was 11, indicating moderate impairment. Her response on the CAGE questionnaire was negative for a problem with alcohol. However, her AUDIT score was 17, indicating a high-level alcohol problem. Based on these findings, her physician expressed concern that her alcohol use might be contributing to her symptoms and difficulties.
Although she did not have a history of increasing usage per day, a persistent desire to cut down, significant effort to obtain alcohol, or cravings, she was having work troubles and continued to drink even though it was straining relationships, promoting weight gain, and causing abdominal pain.
The physician asked her to schedule a return visit and ordered several blood studies. He also offered to connect her with a colleague with whom he collaborated who could speak with her about possible alcohol use disorders and depression.
Continue to: Selecting blood work in screening for AUD
Selecting blood work in screening for AUD
Lab tests used to measure hepatic injury due to alcohol include gamma-glutamyl-transferase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and macrocytic volume, although the indices of hepatic damage have low specificity. Elevated serum ethanol levels can reveal recent alcohol use, and vitamin deficiencies and other abnormalities can be used to differentiate other causes of hepatic inflammation and co-existing health issues (TABLE 310,18). A number of as-yet-unvalidated biomarkers are being studied to assist in screening, diagnosing, and treating AUD.18
What treatment approaches work for AUD?
Family physicians can efficiently and productively address AUD by using alcohol screening and brief intervention, which have been shown to reduce risky drinking. Reimbursement for this service is covered by such CPT codes as 99408, 99409, or H0049, or with other evaluation and management (E/M) codes by using modifier 25.
Treatment of AUD varies and should be customized to each patient’s needs, readiness, preferences, and resources. Individual and group counseling approaches can be effective, and medications are available for inpatient and outpatient settings. Psychotherapy options include brief interventions, 12-step programs (eg, Alcoholics Anonymous—https://www.aa.org/pages/en_US/find-aa-resources),motivational enhancement therapy, and cognitive behavioral therapy. Although it is beyond the scope of this article to describe these options in detail, resources are available for those who wish to learn more.19-21
Psychopharmacologic management includes US Food and Drug Administration (FDA)-approved medications such as disulfiram, naltrexone, and acamprosate, and off-label uses of other medications (TABLE 49). Not enough empiric evidence is available to judge the effectiveness of these medications in adolescents, and the FDA has not approved them for such use. Evidence from meta-analyses comparing naltrexone and acamprosate have shown naltrexone to be more efficacious in reducing heavy drinking and cravings, while acamprosate is effective in promoting abstinence.22,23 Naltrexone combined with behavioral intervention reduces the heavy drinking days and percentage of abstinence days.24
Current guideline recommendations from the American Psychiatric Association25 include:
- Naltrexone and acamprosate are recommended to treat patients with moderate-to-severe AUD in specific circumstances (eg, when nonpharmacologic approaches have failed to produce an effect or when patients prefer to use one of these medications).
- Topiramate and gabapentin are also suggested as medications for patients with moderate-to-severe AUD, but typically after first trying naltrexone and acamprosate.
- Disulfiram generally should not be used as first-line treatment. It produces physical reactions (eg, flushing) if alcohol is consumed within 12 to 24 hours of medication use.
Continue to: THE CASE
THE CASE
Ms. E was open to the idea of decreasing her alcohol use and agreed that she was depressed. Her lab tests at follow-up were normal other than an elevated AST/ALT of 90/80 U/L. S
She continued to get counseling for her AUD and for her comorbid depression in addition to taking a selective serotonin reuptake inhibitor. She is now in early remission for her alcohol use.
CORRESPONDENCE
Jaividhya Dasarathy, MD, Department of Family Medicine, Metro Health Medical Center, 2500 MetroHealth Drive, Cleveland, OH 44109; [email protected].
THE CASE
Ms. E, a 42-year-old woman, visited her new physician for a physical exam. When asked about alcohol intake, she reported that she drank 3 to 4 beers after work and sometimes 5 to 8 beers a day on the weekends. Occasionally, she exceeded those amounts, but she didn’t feel guilty about her drinking. She was often late to work and said her relationship with her boyfriend was strained. A review of systems was positive for fatigue, poor concentration, abdominal pain, and weight gain. Her body mass index was 41, pulse 100 beats/min, blood pressure 125/75 mm Hg, and she was afebrile. Her physical exam was otherwise within normal limits.
How would you proceed with this patient?
Alcohol use disorder (AUD) is a common and often untreated condition that is increasingly prevalent in the United States.1 The Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) characterizes AUD as a combination of signs and symptoms typifying alcohol abuse and dependence (discussed in a bit).2
Data from the 2015 National Survey on Drug Use and Health (NSDUH) showed 15.7 million Americans with AUD, affecting 6.2% of the population ages 18 years or older and 2.5% of adolescents ages 12 to 17 years.3
Alcohol use and AUD account for an estimated 3.8% of all global deaths and 4.6% of global disability-adjusted life years.4 AUD adversely affects several systems (TABLE 15), and patients with AUD are sicker and more likely to die younger than those without AUD.4 In the United States, prevalence of AUD has increased in recent years among women, older adults, racial minorities, and individuals with a low education level.6
Screening for AUD is reasonable and straightforward, although diagnosis and treatment of AUD in primary care settings may be challenging due to competing clinical priorities; lack of training, resources, and support; and skepticism about the efficacy of behavioral and pharmacologic treatments.7,8 However, family physicians are in an excellent position to diagnose and help address the complex biopsychosocial needs of patients with AUD, often in collaboration with colleagues and community organizations.
Signs and symptoms of AUD
In clinical practice, at least 2 of the following 11 behaviors or symptoms are required to diagnose AUD2:
- consuming larger amounts of alcohol over a longer period than intended
- persistent desire or unsuccessful efforts to cut down or control alcohol use
- making a significant effort to obtain, use, or recover from alcohol
In moderate-to-severe cases:
- cravings or urges to use alcohol
- recurrent failure to fulfill major work, school, or social obligations
- continued alcohol use despite recurrent social and interpersonal problems
- giving up social, occupational, and recreational activities due to alcohol
- using alcohol in physically dangerous situations
- continued alcohol use despite having physical or psychological problems
- tolerance to alcohol’s effects
- withdrawal symptoms.
Continue to: Patients meet criteria for mild AUD severity if...
Patients meet criteria for mild AUD severity if they exhibit 2 or 3 symptoms, moderate AUD with 4 or 5 symptoms, and severe AUD if there are 6 or more symptoms.2
Those who meet criteria for AUD and are able to stop using alcohol are deemed to be in early remission if the criteria have gone unfulfilled for at least 3 months and less than 12 months. Patients are considered to be in sustained remission if they have not met criteria for AUD at any time during a period of 12 months or longer.
How to detect AUD
Several clues in a patient’s history can suggest AUD (TABLE 29,10). Most imbibers are unaware of the dangers and may consider themselves merely “social drinkers.” Binge drinking may be an early indicator of vulnerability to AUD and should be assessed as part of a thorough clinical evaluation.11 The US Preventive Services Task Force (USPSTF) recommends (Grade B) that clinicians screen adults ages 18 years or older for alcohol misuse.12
Studies demonstrate that both genetic and environmental factors play important roles in the development of AUD.13 A family history of excessive alcohol use increases the risk of AUD. Comorbidity of AUD and other mental health conditions is extremely common. For example, high rates of association between major depressive disorder and AUD have been observed.14
Tools to use in screening and diagnosing AUD
Screening for AUD during an office visit can be done fairly quickly. While 96% of primary care physicians screen for alcohol misuse in some way, only 38% use 1 of the 3 tools recommended by the USPSTF15—the Alcohol Use Disorders Identification Test (AUDIT), the abbreviated AUDIT-C, or the National Institute on Alcohol Abuse and Alcoholism (NIAAA) single question screen—which detect the full spectrum of alcohol misuse in adults.12 Although the commonly used CAGE questionnaire is one of the most studied self-report tools, it has lower sensitivity at a lower level of alcohol intake.16
Continue to: The NIAAA single-question screen asks...
The NIAAA single-question screen asks how many times in the past year the patient had ≥4 drinks (women) or ≥5 drinks (men) in a day.15 The sensitivity and specificity of single-question screening are 82% to 87% and 61% to 79%, respectively, and the test has been validated in several different settings.12 The AUDIT screening tool, freely available from the World Health Organization, is a 10-item questionnaire that probes an individual’s alcohol intake, alcohol dependence, and adverse consequences of alcohol use. Administration of the AUDIT typically requires only 2 minutes. AUDIT-C17 is an abbreviated version of the AUDIT questionnaire that asks 3 consumption questions to screen for AUD.
It was found that AUDIT scores in the range of 8 to 15 indicate a medium-level alcohol problem, whereas a score of ≥16 indicates a high-level alcohol problem. The AUDIT-C is scored from 0 to 12, with ≥4 indicating a problem in men and ≥3
THE CASE
The physician had used the NIAAA single- question screen to determine that Ms. E drank more than 4 beers per day during social events and weekends, which occurred 2 to 3 times per month over the past year. She lives alone and said that she’d been seeing less and less of her boyfriend lately. Her score on the Patient Health Questionnaire (PHQ), which screens for depression, was 11, indicating moderate impairment. Her response on the CAGE questionnaire was negative for a problem with alcohol. However, her AUDIT score was 17, indicating a high-level alcohol problem. Based on these findings, her physician expressed concern that her alcohol use might be contributing to her symptoms and difficulties.
Although she did not have a history of increasing usage per day, a persistent desire to cut down, significant effort to obtain alcohol, or cravings, she was having work troubles and continued to drink even though it was straining relationships, promoting weight gain, and causing abdominal pain.
The physician asked her to schedule a return visit and ordered several blood studies. He also offered to connect her with a colleague with whom he collaborated who could speak with her about possible alcohol use disorders and depression.
Continue to: Selecting blood work in screening for AUD
Selecting blood work in screening for AUD
Lab tests used to measure hepatic injury due to alcohol include gamma-glutamyl-transferase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and macrocytic volume, although the indices of hepatic damage have low specificity. Elevated serum ethanol levels can reveal recent alcohol use, and vitamin deficiencies and other abnormalities can be used to differentiate other causes of hepatic inflammation and co-existing health issues (TABLE 310,18). A number of as-yet-unvalidated biomarkers are being studied to assist in screening, diagnosing, and treating AUD.18
What treatment approaches work for AUD?
Family physicians can efficiently and productively address AUD by using alcohol screening and brief intervention, which have been shown to reduce risky drinking. Reimbursement for this service is covered by such CPT codes as 99408, 99409, or H0049, or with other evaluation and management (E/M) codes by using modifier 25.
Treatment of AUD varies and should be customized to each patient’s needs, readiness, preferences, and resources. Individual and group counseling approaches can be effective, and medications are available for inpatient and outpatient settings. Psychotherapy options include brief interventions, 12-step programs (eg, Alcoholics Anonymous—https://www.aa.org/pages/en_US/find-aa-resources),motivational enhancement therapy, and cognitive behavioral therapy. Although it is beyond the scope of this article to describe these options in detail, resources are available for those who wish to learn more.19-21
Psychopharmacologic management includes US Food and Drug Administration (FDA)-approved medications such as disulfiram, naltrexone, and acamprosate, and off-label uses of other medications (TABLE 49). Not enough empiric evidence is available to judge the effectiveness of these medications in adolescents, and the FDA has not approved them for such use. Evidence from meta-analyses comparing naltrexone and acamprosate have shown naltrexone to be more efficacious in reducing heavy drinking and cravings, while acamprosate is effective in promoting abstinence.22,23 Naltrexone combined with behavioral intervention reduces the heavy drinking days and percentage of abstinence days.24
Current guideline recommendations from the American Psychiatric Association25 include:
- Naltrexone and acamprosate are recommended to treat patients with moderate-to-severe AUD in specific circumstances (eg, when nonpharmacologic approaches have failed to produce an effect or when patients prefer to use one of these medications).
- Topiramate and gabapentin are also suggested as medications for patients with moderate-to-severe AUD, but typically after first trying naltrexone and acamprosate.
- Disulfiram generally should not be used as first-line treatment. It produces physical reactions (eg, flushing) if alcohol is consumed within 12 to 24 hours of medication use.
Continue to: THE CASE
THE CASE
Ms. E was open to the idea of decreasing her alcohol use and agreed that she was depressed. Her lab tests at follow-up were normal other than an elevated AST/ALT of 90/80 U/L. S
She continued to get counseling for her AUD and for her comorbid depression in addition to taking a selective serotonin reuptake inhibitor. She is now in early remission for her alcohol use.
CORRESPONDENCE
Jaividhya Dasarathy, MD, Department of Family Medicine, Metro Health Medical Center, 2500 MetroHealth Drive, Cleveland, OH 44109; [email protected].
1. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72:757-766.
2. APA. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington DC; 2013.
3. HHS. Results from the 2015 National Survey on Drug Use and Health: summary of national findings. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.pdf. Accessed November 27, 2018.
4. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223-2233.
5. Chase V, Neild R, Sadler CW, et al. The medical complications of alcohol use: understanding mechanisms to improve management. Drug Alcohol Rev. 2005;24:253-265.
6. Grant BF, Chou SP, Saha TD, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001-2002 to 2012-2013: results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74:911-923.
7. Williams EC, Achtmeyer CE, Young JP, et al. Barriers to and facilitators of alcohol use disorder pharmacotherapy in primary care: a qualitative study in five VA clinics. J Gen Intern Med. 2018;33:258-267.
8. Zhang DX, Li ST, Lee QK, et al. Systematic review of guidelines on managing patients with harmful use of alcohol in primary healthcare settings. Alcohol Alcohol. 2017;52:595-609.
9. Wackernah RC, Minnick MJ, Clapp P. Alcohol use disorder: pathophysiology, effects, and pharmacologic options for treatment. Subst Abuse Rehabil. 2014;5:1-12.
10. Kattimani S, Bharadwaj B. Clinical management of alcohol withdrawal: a systematic review. Ind Psychiatry J. 2013;22:100-108.
11. Gowin JL, Sloan ME, Stangl BL, et al. Vulnerability for alcohol use disorder and rate of alcohol consumption. Am J Psychiatry. 2017;174:1094-1101.
12. Moyer VA; Preventive Services Task Force. Screening and behavioral counseling interventions in primary care to reduce alcohol misuse: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:210-218.
13. Tarter RE, Alterman AI, Edwards KL. Vulnerability to alcoholism in men: a behavior-genetic perspective. J Stud Alcohol. 1985;46:329-356.
14. Brière FN, Rohde P, Seeley JR, et al. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood [published online ahead of print, October 22, 2013]. Compr Psychiatry. 2014;55:526-533. doi: 10.1016/j.comppsych.2013.10.007.
15. Tan CH, Hungerford DW, Denny CH, et al. Screening for alcohol misuse: practices among U.S. primary care providers, DocStyles 2016. Am J Prev Med. 2018;54:173-180.
16. Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol. 2004;57:30-39.
17. Bush K, Kivlahan DR, McDonell MB, et al. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med. 1998;158:1789-1795.
18. Nanau RM, Neuman MG. Biomolecules and biomarkers used in diagnosis of alcohol drinking and in monitoring therapeutic interventions. Biomolecules. 2015;5:1339-1385.
19. Raddock M, Martukovich R, Berko E, et al. 7 tools to help patients adopt healthier behaviors. J Fam Pract. 2015;64:97-103.
20. AHRQ. Whitlock EP, Green CA, Polen MR, et al. Behavioral Counseling Interventions in Primary Care to Reduce Risky/Harmful Alcohol Use. 2004. https://www.ncbi.nlm.nih.gov/books/NBK42863/. Accessed November 17, 2018.
21. Miller WR, Baca C, Compton WM, et al. Addressing substance abuse in health care settings. Alcohol Clin Exp Res. 2006;30:292-302.
22. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108:275-293.
23. Rosner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22:11-23.
24. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003-2017.
25. Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association Practice Guideline for the Pharmacological Treatment of Patients With Alcohol Use Disorder. Am J Psychiatry. 2018;175:86-90.
1. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM-5 alcohol use disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72:757-766.
2. APA. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington DC; 2013.
3. HHS. Results from the 2015 National Survey on Drug Use and Health: summary of national findings. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.pdf. Accessed November 27, 2018.
4. Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223-2233.
5. Chase V, Neild R, Sadler CW, et al. The medical complications of alcohol use: understanding mechanisms to improve management. Drug Alcohol Rev. 2005;24:253-265.
6. Grant BF, Chou SP, Saha TD, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001-2002 to 2012-2013: results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74:911-923.
7. Williams EC, Achtmeyer CE, Young JP, et al. Barriers to and facilitators of alcohol use disorder pharmacotherapy in primary care: a qualitative study in five VA clinics. J Gen Intern Med. 2018;33:258-267.
8. Zhang DX, Li ST, Lee QK, et al. Systematic review of guidelines on managing patients with harmful use of alcohol in primary healthcare settings. Alcohol Alcohol. 2017;52:595-609.
9. Wackernah RC, Minnick MJ, Clapp P. Alcohol use disorder: pathophysiology, effects, and pharmacologic options for treatment. Subst Abuse Rehabil. 2014;5:1-12.
10. Kattimani S, Bharadwaj B. Clinical management of alcohol withdrawal: a systematic review. Ind Psychiatry J. 2013;22:100-108.
11. Gowin JL, Sloan ME, Stangl BL, et al. Vulnerability for alcohol use disorder and rate of alcohol consumption. Am J Psychiatry. 2017;174:1094-1101.
12. Moyer VA; Preventive Services Task Force. Screening and behavioral counseling interventions in primary care to reduce alcohol misuse: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:210-218.
13. Tarter RE, Alterman AI, Edwards KL. Vulnerability to alcoholism in men: a behavior-genetic perspective. J Stud Alcohol. 1985;46:329-356.
14. Brière FN, Rohde P, Seeley JR, et al. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood [published online ahead of print, October 22, 2013]. Compr Psychiatry. 2014;55:526-533. doi: 10.1016/j.comppsych.2013.10.007.
15. Tan CH, Hungerford DW, Denny CH, et al. Screening for alcohol misuse: practices among U.S. primary care providers, DocStyles 2016. Am J Prev Med. 2018;54:173-180.
16. Aertgeerts B, Buntinx F, Kester A. The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol. 2004;57:30-39.
17. Bush K, Kivlahan DR, McDonell MB, et al. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med. 1998;158:1789-1795.
18. Nanau RM, Neuman MG. Biomolecules and biomarkers used in diagnosis of alcohol drinking and in monitoring therapeutic interventions. Biomolecules. 2015;5:1339-1385.
19. Raddock M, Martukovich R, Berko E, et al. 7 tools to help patients adopt healthier behaviors. J Fam Pract. 2015;64:97-103.
20. AHRQ. Whitlock EP, Green CA, Polen MR, et al. Behavioral Counseling Interventions in Primary Care to Reduce Risky/Harmful Alcohol Use. 2004. https://www.ncbi.nlm.nih.gov/books/NBK42863/. Accessed November 17, 2018.
21. Miller WR, Baca C, Compton WM, et al. Addressing substance abuse in health care settings. Alcohol Clin Exp Res. 2006;30:292-302.
22. Maisel NC, Blodgett JC, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108:275-293.
23. Rosner S, Leucht S, Lehert P, et al. Acamprosate supports abstinence, naltrexone prevents excessive drinking: evidence from a meta-analysis with unreported outcomes. J Psychopharmacol. 2008;22:11-23.
24. Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003-2017.
25. Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association Practice Guideline for the Pharmacological Treatment of Patients With Alcohol Use Disorder. Am J Psychiatry. 2018;175:86-90.

The role of home BP monitoring: Answers to 10 common questions
National Health and Nutrition Examination Survey (NHANES) data from 2011 to 2014 revealed that 29% of adults in the United States have hypertension.1 Prevalence increases with age, so that 7% of adults ages 18 to 39 years, 32% of adults ages 40 to 59, and 65% of adults ages ≥60 years have the disease.1 This national survey data also showed that 53% of those given the diagnosis had uncontrolled hypertension, and that control of hypertension did not change significantly from 2009 to 2014.1
Elevated blood pressure (BP) has been the leading risk factor for death related to cardiovascular disease globally for the last 3 decades.2 Yet in 2 nationally representative samples, only 1 in 6 patients with documented BP ≥140/90 mm Hg received treatment intensification with new medication during primary care visits.3 Uncertainty about the representativeness of any single clinic BP measurement is a prominent reason for health care providers not to intensify therapy.4
Confirming the Dx outside the office. The 2015 US Preventive Services Task Force (USPSTF) guidelines on screening for hypertension state that, for most patients, a diagnosis of hypertension should be confirmed with out-of-office BP monitoring before initiating treatment.5 The USPSTF states that ambulatory BP monitoring (ABPM) is accurate for hypertension diagnosis and monitoring, and that home BP monitoring (HBPM) is an acceptable alternative, based on good quality evidence.
Access to ABPM, however, is often limited. In a 2015 survey of primary care clinics, only 25% of the 123 clinics that completed the questionnaire reported having access to it.6 Conversely, HBPM is widely available and acceptable to most patients. A recent NHANES survey showed that 43.5% of patients who were aware of their hypertension diagnosis engaged in HBPM.7
So what, exactly, should the role of HBPM be in the management of patients with hypertension? The evidence-based answers to the 10 questions that follow provide useful insights.
[polldaddy:10224678]
1. Can HBPM be used to confirm a Dx of hypertension?
Yes (Strength of recommendation [SOR] C).
In reviewing the diagnostic accuracy of various methods to confirm the diagnosis of hypertension, the USPSTF identified ABPM as the most accurate, followed by HBPM, with clinic BP measurements bringing up the rear.5 In adults ≥18 years of age, the USPSTF recommends obtaining BP measurements outside of the clinical setting for diagnostic confirmation before starting treatment unless the patient’s BP is ≥180/110 mm Hg, there is evidence of end-organ damage, or the patient has a diagnosis of secondary hypertension.5 The USPSTF recommends HBPM as an acceptable alternative to ABPM based on 6 studies including a total of 1253 participants.8 The percentage of patients with elevated office BP confirmed by HBPM to have hypertension was 45% to 84% across these 6 studies.
Sixteen studies from another systematic review evaluated the diagnostic accuracy of HBPM while using ABPM as a reference.9 This review found that HBPM had high specificity and negative predictive value, but low sensitivity and positive predictive value. There was moderate diagnostic agreement between HBPM and ABPM, with kappa statistic values of 0.37 to 0.73 across all studies.9
Continue to: In yet another study...
In yet another study, home BP and ambulatory BP measurements were identical when the same dual-mode device was used to measure both ambulatory and home BP.10
2. What are the diagnostic and treatment targets for home BP monitoring?
Treat patients if home BP is ≥130/80 mm Hg and categorize patients as normotensive if home BP is <125/76 mm Hg (SOR C). Monitor patients who are in between.
A 2017 joint statement from the American College of Cardiology/American Heart Association (ACC/AHA) Task Force states that the target BP for HBPM should be <130/80 mm Hg.11 The Joint National Commission (JNC) 8 issued BP goals of <140/90 mm Hg for adults <60 years of age and those with diabetes and/or chronic kidney disease, and a goal of <150/90 mm Hg for adults ≥60 years of age with no diabetes or chronic kidney disease,12 but much debate has recently surrounded these guidelines. JNC 8 does not provide a separate BP goal for HBPM.
Although based solely on evidence (and not patient-oriented outcomes), a home BP threshold of ≥135/85 mm Hg for the diagnosis and treatment of hypertension has been supported by the European Society of Hypertension consensus guidelines,13 results of a longitudinal study,14 meta-analyses of published studies, and a meta-analysis using individual subject data.15
Support for a home BP measurement of <125/76 mm Hg as normal is limited to a single cross-sectional study of 48 patients with 2 elevated office BP readings where the threshold of 125/76 mm Hg on home BP was shown to exclude 80% of patients diagnosed with hypertension by ambulatory readings.16 If home BP measurements are >125/76 mm Hgbut <135/85 mm Hg, 24-hour ambulatory BP monitoring is recommended to assess hypertension control.17
3. Does home BP monitoring improve hypertension control?
Yes, in the short term, but not in the long term (SOR C).
A meta-analysis of 13 comparative studies looking at HBPM alone vs usual care showed a small, but statistically significant, benefit of achieving target BP at 6 months with a relative risk ratio (RRR) of 1.3 (95% confidence interval [CI], 1.00-1.68; I2=77%).18 However, the pooled effects from 3 studies that measured the benefit of achieving a predefined BP target at the 12-month follow-up mark were not significant in this review (RRR=1.18; 95% CI, 0.95-1.46, I2=86%).18 The pooled effect from 19 studies from the same review showed that there was a statistically significant weighted mean difference of -3.9 mm Hg in systolic BP and a weighted mean difference of -2.4 mm Hg in diastolic BP at 6 months; however, the changes were no longer significant at the 12-month follow-up mark.18
Continue to: More than half of the studies included...
More than half of the studies included in the meta-analysis were of low quality, and none of the studies recruited patients based on differences in clinic BP and home BP patterns, but rather on controlled or uncontrolled hypertension status. The studies included in this meta-analysis measured final BP outcomes by measuring ambulatory BP or clinic BP.
Another systematic review of 19 studies and 7100 participants looking at how HBPM compared with ABPM as a measurement standard for BP control and patient outcomes found insufficient data to determine the benefit of using HBPM as a measurement standard for BP control.19
HBPM + added support. There was high-quality evidence from the meta-analysis that HBPM plus additional support vs usual care led to a reduction in BP and a higher proportion of patients achieving target BP.18 However, the additional support interventions in the studies were heterogeneous.
4. Should HBPM be used to detect a change in BP associated with medication alterations?
Yes (SOR B).
A 2008 meta-analysis20 and several other studies21,22 showed that HBPM has greater accuracy than office BP for identifying drug-induced BP changes. The 2008 meta-analysis looked at changes in office and home BP measurements produced by various antihypertensive drugs. In 7 studies that compared office BP measurements with home and ambulatory BP measurements, the 24-hour ambulatory BP measurements and home BP measurements showed less dramatic BP reductions with medications than clinic BP measurements.20 This meta-analysis included 30 studies with 6794 participants and showed that home BP readings fell 20% less than office BP readings; the difference was statistically significant. These findings suggest that treatment-attributable changes in home BP and clinic BP measurements are linearly related, with the treatment effect on home BP measurements being around 80% of the effect on clinic BP measurements.
5. Do home BP measurements correlate with clinical outcomes?
Yes, and better than office BP measurements do; however, most studies comparing home BP measurements with usual care while looking at clinical outcomes are observational or quasi-experimental (SOR B).
For example, a 2015 systematic review looking at associations between BP measurement type (office, home, and ambulatory) and patient mortality found 5 observational studies that showed that adding home or ambulatory BP information improved cardiovascular risk prediction models. Moreover, all-cause mortality was associated with home BP and ambulatory BP levels only and not with office BP levels.19 The number of participants in these 5 studies varied between 210 and 2051 with study duration between 2.4 and 12.3 years. Of note, every study had a distinct population, affecting the generalizability of the results.
Continue to: One quasi-experimental study...
One quasi-experimental study with 450 participants showed that home BP measurements were at least as good as ambulatory BP measurements at predicting end organ damage related to hypertension when organ damage was measured by cardiac echocardiography, detection of microalbuminuria, and carotid echocardiography.23 Similarly, a systematic review of 14 studies and 2485 participants comparing home, ambulatory, and office BP readings showed that home BP measurements’ association with left ventricular mass index is as good as that of ambulatory BP measurements, and superior to clinic BP readings.24
6. Does HBPM help improve medication adherence?
The jury is still out on this one (SOR B).
A 2006 systematic review of randomized controlled trials (RCTs) incorporating HBPM and evaluating medication adherence outcomes found that in 6 of the 11 studies identified, there was some improvement in medication adherence with HBPM.25 However, only 1 of the 6 studies in this review involved HBPM as the sole intervention; the remaining 5 studies employed additional adherence-enhancing strategies.
Another systematic review looking at HBPM vs usual care included 8 studies (3 of moderate quality and 5 of low quality) that measured medication adherence (using varying measures of adherence) of which only 3 studies showed some improvement in medication adherence with HBPM.18
7. Does HBPM reduce therapeutic inertia?
Yes (SOR B).
A meta-analysis of 15 studies showed that therapeutic inertia was less common with HBPM than with office BP monitoring alone; the relative risk for unchanged medication was 0.82 (95% CI, 0.68 to 0.99) with HBPM.26 However, 10 of the 15 studies were of low quality with a Jadad score ≤3.
8. Does HBPM, along with titration of treatment, improve BP outcomes?
Yes (SOR B).
Two RCTs that looked at self-monitoring of BP and self-titration of hypertensive medications showed significant reductions in BP levels.27,28 In a cluster RCT of home BP telemonitoring, in which the pharmacist adjusted antihypertensives based on transmitted BP measurements, hypertension control was significantly better in the intervention group than in the usual care group (57.2% vs 30%).29
Continue to: What are the recommended techniques for HBPM?
9. What are the recommended techniques for HBP
Patients should use a device that is validated, fully automated, and has an upper arm cuff (not a wrist monitor), according to a joint statement from the AHA, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association.17 (SOR C). (See validated BP monitor list at http://www.dableducational.org/sphygmomanometers/devices_2_sbpm.html.)
Patients should measure their BP in their nondominant arm after 5 minutes of rest with the arm at heart level, back supported, and feet flat on the ground. Patient technique and the accuracy of the home BP monitor should be checked annually. It is also recommended that patients check their BP 2 to 3 times every morning and evening. An average of 12 morning and evening measurements should be used for monitoring and treatment changes. An AHA informational sheet that shows how to measure BP properly can be found on their Web si
Several studies examining the accuracy of measuring BP over clothing did not find significant differences in BP measurements performed on a bare arm vs over a sleeve.30-33
10. What are the predictors of differences between home and office BP measurements?
Gender is one of the biggest predictors (SOR B).
A 2016 meta-analysis reported a total of 60 different hypothesized predictors of differences between home and clinic BP measurements (eg, gender, age, body mass index, systolic BP, diastolic BP). Masked hypertension was defined as a normal clinic BP reading and an elevated home BP reading. White coat hypertension was defined as an elevated clinic BP measurement with an acceptable home BP measurement. The researchers extracted odds ratios (ORs) for each study describing the association between patient characteristics and white coat or masked hypertension.34 Studies of masked hypertension diagnosed from HBPM showed male gender as the most significant predictor of home-clinic BP differences (OR=1.47, 95% CI, 1.18-1.75). In contrast, female gender was the only significant predictor of white coat hypertension (OR=3.38; 95% CI, 1.64-6.96) when comparing home BP with clinic BP measurements
Literature limitations and barriers to greater implementation
Most studies looking at HBPM outcomes have measured outcomes using ABPM or office BP measurements. The authors of studies using office BP as the outcome measure usually performed multiple BP measurements at often multiple office or clinic visits to calculate the true BP—a procedure that primary care practices rarely follow.35 Additionally, there are significant methodologic differences in HBPM and ABPM; home BP is measured at rest, while ambulatory BP is measured while the patient is mobile and functioning. There are insufficient prospective studies looking at HBPM effects on clinical and patient-oriented outcomes.
The evidence clearly supports using HBPM in the diagnosis of hypertension and suggests its benefit in hypertension management. However, there are significant barriers to incorporating HBPM into practice—barriers that are largely unaddressed in the literature.
For HBPM to be successful, patients need affordable validated home BP monitors covered by insurance that can translate home BP readings into usable information. Additional administrative and/or nursing assistance is required for patient education and support. Uploaded data need to be summarized in a way that is actionable and linked to the electronic health record.
Continue to: As the volume of patient-generated home date increases...
As the volume of patient-generated home data increases, there is a risk of information overload. Thus, meaningful summarization of the data is required to enable the physician, patient, and/or pharmacist to take prompt and effective action.
CORRESPONDENCE
Sonal J. Patil, MD, MSPH, Curtis W. and Ann H. Long Department of Family and Community Medicine, University of Missouri, MA306 Medical Sciences Building, DC032.00 Columbia, MO 65212; [email protected].
ACKNOWLEDGEMENTS
The authors are grateful to Dr. David R. Mehr for his valuable expertise in editing and proofreading this manuscript.
1. Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011-2014. NCHS Data Brief. 2015;(220):1-8.
2. Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardio-metabolic risk factors between 1980 and 2010: comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2:634-647.
3. Mu L, Mukamal KJ. Treatment intensification for hypertension in US ambulatory medical care. J Am Heart Assoc. 2016;5:e004188.
4. Kerr EA, Zikmund-Fisher BJ, Klamerus ML, et al. The role of clinical uncertainty in treatment decisions for diabetic patients with uncontrolled blood pressure. Ann Intern Med. 2008;148:717-727.
5. U.S. Preventive Services Task Force. Final recommendation statement: High blood pressure in adults: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/high-blood-pressure-in-adults-screening. Accessed July 19, 2017.
6. Woolsey S, Brown B, Ralls B, et al. Diagnosing hypertension in primary care clinics according to current guidelines. J Am Board Fam Med. 2017;30:170-177.
7. Ostchega Y, Zhang G, Kit BK, et al. Factors associated with home blood pressure monitoring among US adults: National Health and Nutrition Examination Survey, 2011-2014. Am J Hypertens. 2017;30:1126-1132.
8. Piper MA, Evans CV, Burda BU, et al. Diagnostic and predictive accuracy of blood pressure screening methods with consideration of rescreening intervals: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2015;162:192-204.
9. Stergiou GS, Bliziotis IA. Home blood pressure monitoring in the diagnosis and treatment of hypertension: a systematic review. Am J Hypertens. 2011;24:123-134.
10. Stergiou GS, Tzamouranis D, Nasothimiou EG, et al. Are there really differences between home and daytime ambulatory blood pressure? Comparison using a novel dual-mode ambulatory and home monitor. J Hum Hypertens. 2009;24:207-212.
11. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017;71:e13-e115.
12. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520. Erratum in JAMA. 2014;311:1809.
13. O’Brien E, Asmar R, Beilin L, et al. European Society of Hypertension recommendations for conventional, ambulatory and home blood pressure measurement. J Hypertens. 2003;21:821-848.
14. Tsunoda S, Kawano Y, Horio T, et al. Relationship between home blood pressure and longitudinal changes in target organ damage in treated hypertensive patients. Hypertens Res. 2002;25:167-173.
15. Staessen JA, Thijs L. Development of diagnostic thresholds for automated self-measurement of blood pressure in adults. First International Consensus Conference on Blood Pressure Self-Measurement. Blood Press Monit. 2000;5:101-109.
16. Mansoor GA, White WB. Self-measured home blood pressure in predicting ambulatory hypertension. Am J Hypertens. 2004;17(11 Pt 1):1017-1022.
17. Pickering TG, Miller NH, Ogedegbe G, et al. Call to action on use and reimbursement for home blood pressure monitoring: a joint scientific statement from the American Heart Association, American Society of Hypertension, and Preventive Cardiovascular Nurses Association. J Cardiovasc Nurs. 2008;23:299-323.
18. Uhlig K, Patel K, Ip S, Kitsios GD, Balk EM. Self-measured blood pressure monitoring in the management of hypertension: a systematic review and meta-analysis. Ann Intern Med. 2013;159:185-194.
19. Breaux-Shropshire TL, Judd E, Vucovich LA, et al. Does home blood pressure monitoring improve patient outcomes? A systematic review comparing home and ambulatory blood pressure monitoring on blood pressure control and patient outcomes. Integr Blood Press Control. 2015;8:43-49.
20. Ishikawa J, Carroll DJ, Kuruvilla S, et al. Changes in home versus clinic blood pressure with antihypertensive treatments: a meta-analysis. Hypertension. 2008;52:856-864.
21. Imai Y, Ohkubo T, Hozawa A, et al. Usefulness of home blood pressure measurements in assessing the effect of treatment in a single-blind placebo-controlled open trial. J Hypertens. 2001;19:179-185.
22. Vaur L, Dubroca I, Dutrey-Dupagne C, et al. Superiority of home blood pressure measurements over office measurements for testing antihypertensive drugs. Blood Press Monit. 1998;3:107-114.
23. Gaborieau V, Delarche N, Gosse P. Ambulatory blood pressure monitoring versus self-measurement of blood pressure at home: correlation with target organ damage. J Hypertens. 2008;26:1919-1927.
24. Bliziotis IA, Destounis A, Stergiou GS. Home versus ambulatory and office blood pressure in predicting target organ damage in hypertension: a systematic review and meta-analysis. J Hypertens. 2012;30:1289-1299.
25. Ogedegbe G, Schoenthaler A. A systematic review of the effects of home blood pressure monitoring on medication adherence. J Clin Hypertens (Greenwich). 2006;8:174-180.
26. Agarwal R, Bills JE, Hecht TJ, et al. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension. 2011;57:29-38.
27. McManus RJ, Mant J, Haque MS, et al. Effect of self-monitoring and medication self-titration on systolic blood pressure in hypertensive patients at high risk of cardiovascular disease: the TASMIN-SR randomized clinical trial. JAMA. 2014;312:799-808.
28. McManus RJ, Mant J, Bray EP, et al. Telemonitoring and self-management in the control of hypertension (TASMINH2): a randomised controlled trial. Lancet. 2010;376:163-172.
29. Margolis KL, Asche SE, Bergdall AR, et al. Effect of home blood pressure telemonitoring and pharmacist management on blood pressure control: The HyperLink Cluster Randomized Trial. JAMA. 2013;310:46-56.
30. Ma G, Sabin N, Dawes M. A comparison of blood pressure measurement over a sleeved arm versus a bare arm. CMAJ. 2008;178:585-589.
31. Kahan E, Yaphe J, Knaani-Levinz H, et al. Comparison of blood pressure measurements on the bare arm, below a rolled-up sleeve, or over a sleeve. Fam Pract. 2003;20:730-732.
32. Liebl M, Holzgreve H, Schulz M, et al. The effect of clothes on sphygmomanometric and oscillometric blood pressure measurement. Blood Press. 2004;13:279-282.
33. Holleman DR Jr., Westman EC, McCrory DC, et al. The effect of sleeved arms on oscillometric blood pressure measurement. J Gen Intern Med. 1993;8:325-326.
34. Sheppard JP, Fletcher B, Gill P, et al. Predictors of the home-clinic blood pressure difference: a systematic review and meta-analysis. Am J Hypertens. 2016;29:614-625.
35. Stergiou GS, Skeva II, Baibas NM, et al. Diagnosis of hypertension using home or ambulatory blood pressure monitoring: comparison with the conventional strategy based on repeated clinic blood pressure measurements. J Hypertens. 2000;18:1745-1751.
National Health and Nutrition Examination Survey (NHANES) data from 2011 to 2014 revealed that 29% of adults in the United States have hypertension.1 Prevalence increases with age, so that 7% of adults ages 18 to 39 years, 32% of adults ages 40 to 59, and 65% of adults ages ≥60 years have the disease.1 This national survey data also showed that 53% of those given the diagnosis had uncontrolled hypertension, and that control of hypertension did not change significantly from 2009 to 2014.1
Elevated blood pressure (BP) has been the leading risk factor for death related to cardiovascular disease globally for the last 3 decades.2 Yet in 2 nationally representative samples, only 1 in 6 patients with documented BP ≥140/90 mm Hg received treatment intensification with new medication during primary care visits.3 Uncertainty about the representativeness of any single clinic BP measurement is a prominent reason for health care providers not to intensify therapy.4
Confirming the Dx outside the office. The 2015 US Preventive Services Task Force (USPSTF) guidelines on screening for hypertension state that, for most patients, a diagnosis of hypertension should be confirmed with out-of-office BP monitoring before initiating treatment.5 The USPSTF states that ambulatory BP monitoring (ABPM) is accurate for hypertension diagnosis and monitoring, and that home BP monitoring (HBPM) is an acceptable alternative, based on good quality evidence.
Access to ABPM, however, is often limited. In a 2015 survey of primary care clinics, only 25% of the 123 clinics that completed the questionnaire reported having access to it.6 Conversely, HBPM is widely available and acceptable to most patients. A recent NHANES survey showed that 43.5% of patients who were aware of their hypertension diagnosis engaged in HBPM.7
So what, exactly, should the role of HBPM be in the management of patients with hypertension? The evidence-based answers to the 10 questions that follow provide useful insights.
[polldaddy:10224678]
1. Can HBPM be used to confirm a Dx of hypertension?
Yes (Strength of recommendation [SOR] C).
In reviewing the diagnostic accuracy of various methods to confirm the diagnosis of hypertension, the USPSTF identified ABPM as the most accurate, followed by HBPM, with clinic BP measurements bringing up the rear.5 In adults ≥18 years of age, the USPSTF recommends obtaining BP measurements outside of the clinical setting for diagnostic confirmation before starting treatment unless the patient’s BP is ≥180/110 mm Hg, there is evidence of end-organ damage, or the patient has a diagnosis of secondary hypertension.5 The USPSTF recommends HBPM as an acceptable alternative to ABPM based on 6 studies including a total of 1253 participants.8 The percentage of patients with elevated office BP confirmed by HBPM to have hypertension was 45% to 84% across these 6 studies.
Sixteen studies from another systematic review evaluated the diagnostic accuracy of HBPM while using ABPM as a reference.9 This review found that HBPM had high specificity and negative predictive value, but low sensitivity and positive predictive value. There was moderate diagnostic agreement between HBPM and ABPM, with kappa statistic values of 0.37 to 0.73 across all studies.9
Continue to: In yet another study...
In yet another study, home BP and ambulatory BP measurements were identical when the same dual-mode device was used to measure both ambulatory and home BP.10
2. What are the diagnostic and treatment targets for home BP monitoring?
Treat patients if home BP is ≥130/80 mm Hg and categorize patients as normotensive if home BP is <125/76 mm Hg (SOR C). Monitor patients who are in between.
A 2017 joint statement from the American College of Cardiology/American Heart Association (ACC/AHA) Task Force states that the target BP for HBPM should be <130/80 mm Hg.11 The Joint National Commission (JNC) 8 issued BP goals of <140/90 mm Hg for adults <60 years of age and those with diabetes and/or chronic kidney disease, and a goal of <150/90 mm Hg for adults ≥60 years of age with no diabetes or chronic kidney disease,12 but much debate has recently surrounded these guidelines. JNC 8 does not provide a separate BP goal for HBPM.
Although based solely on evidence (and not patient-oriented outcomes), a home BP threshold of ≥135/85 mm Hg for the diagnosis and treatment of hypertension has been supported by the European Society of Hypertension consensus guidelines,13 results of a longitudinal study,14 meta-analyses of published studies, and a meta-analysis using individual subject data.15
Support for a home BP measurement of <125/76 mm Hg as normal is limited to a single cross-sectional study of 48 patients with 2 elevated office BP readings where the threshold of 125/76 mm Hg on home BP was shown to exclude 80% of patients diagnosed with hypertension by ambulatory readings.16 If home BP measurements are >125/76 mm Hgbut <135/85 mm Hg, 24-hour ambulatory BP monitoring is recommended to assess hypertension control.17
3. Does home BP monitoring improve hypertension control?
Yes, in the short term, but not in the long term (SOR C).
A meta-analysis of 13 comparative studies looking at HBPM alone vs usual care showed a small, but statistically significant, benefit of achieving target BP at 6 months with a relative risk ratio (RRR) of 1.3 (95% confidence interval [CI], 1.00-1.68; I2=77%).18 However, the pooled effects from 3 studies that measured the benefit of achieving a predefined BP target at the 12-month follow-up mark were not significant in this review (RRR=1.18; 95% CI, 0.95-1.46, I2=86%).18 The pooled effect from 19 studies from the same review showed that there was a statistically significant weighted mean difference of -3.9 mm Hg in systolic BP and a weighted mean difference of -2.4 mm Hg in diastolic BP at 6 months; however, the changes were no longer significant at the 12-month follow-up mark.18
Continue to: More than half of the studies included...
More than half of the studies included in the meta-analysis were of low quality, and none of the studies recruited patients based on differences in clinic BP and home BP patterns, but rather on controlled or uncontrolled hypertension status. The studies included in this meta-analysis measured final BP outcomes by measuring ambulatory BP or clinic BP.
Another systematic review of 19 studies and 7100 participants looking at how HBPM compared with ABPM as a measurement standard for BP control and patient outcomes found insufficient data to determine the benefit of using HBPM as a measurement standard for BP control.19
HBPM + added support. There was high-quality evidence from the meta-analysis that HBPM plus additional support vs usual care led to a reduction in BP and a higher proportion of patients achieving target BP.18 However, the additional support interventions in the studies were heterogeneous.
4. Should HBPM be used to detect a change in BP associated with medication alterations?
Yes (SOR B).
A 2008 meta-analysis20 and several other studies21,22 showed that HBPM has greater accuracy than office BP for identifying drug-induced BP changes. The 2008 meta-analysis looked at changes in office and home BP measurements produced by various antihypertensive drugs. In 7 studies that compared office BP measurements with home and ambulatory BP measurements, the 24-hour ambulatory BP measurements and home BP measurements showed less dramatic BP reductions with medications than clinic BP measurements.20 This meta-analysis included 30 studies with 6794 participants and showed that home BP readings fell 20% less than office BP readings; the difference was statistically significant. These findings suggest that treatment-attributable changes in home BP and clinic BP measurements are linearly related, with the treatment effect on home BP measurements being around 80% of the effect on clinic BP measurements.
5. Do home BP measurements correlate with clinical outcomes?
Yes, and better than office BP measurements do; however, most studies comparing home BP measurements with usual care while looking at clinical outcomes are observational or quasi-experimental (SOR B).
For example, a 2015 systematic review looking at associations between BP measurement type (office, home, and ambulatory) and patient mortality found 5 observational studies that showed that adding home or ambulatory BP information improved cardiovascular risk prediction models. Moreover, all-cause mortality was associated with home BP and ambulatory BP levels only and not with office BP levels.19 The number of participants in these 5 studies varied between 210 and 2051 with study duration between 2.4 and 12.3 years. Of note, every study had a distinct population, affecting the generalizability of the results.
Continue to: One quasi-experimental study...
One quasi-experimental study with 450 participants showed that home BP measurements were at least as good as ambulatory BP measurements at predicting end organ damage related to hypertension when organ damage was measured by cardiac echocardiography, detection of microalbuminuria, and carotid echocardiography.23 Similarly, a systematic review of 14 studies and 2485 participants comparing home, ambulatory, and office BP readings showed that home BP measurements’ association with left ventricular mass index is as good as that of ambulatory BP measurements, and superior to clinic BP readings.24
6. Does HBPM help improve medication adherence?
The jury is still out on this one (SOR B).
A 2006 systematic review of randomized controlled trials (RCTs) incorporating HBPM and evaluating medication adherence outcomes found that in 6 of the 11 studies identified, there was some improvement in medication adherence with HBPM.25 However, only 1 of the 6 studies in this review involved HBPM as the sole intervention; the remaining 5 studies employed additional adherence-enhancing strategies.
Another systematic review looking at HBPM vs usual care included 8 studies (3 of moderate quality and 5 of low quality) that measured medication adherence (using varying measures of adherence) of which only 3 studies showed some improvement in medication adherence with HBPM.18
7. Does HBPM reduce therapeutic inertia?
Yes (SOR B).
A meta-analysis of 15 studies showed that therapeutic inertia was less common with HBPM than with office BP monitoring alone; the relative risk for unchanged medication was 0.82 (95% CI, 0.68 to 0.99) with HBPM.26 However, 10 of the 15 studies were of low quality with a Jadad score ≤3.
8. Does HBPM, along with titration of treatment, improve BP outcomes?
Yes (SOR B).
Two RCTs that looked at self-monitoring of BP and self-titration of hypertensive medications showed significant reductions in BP levels.27,28 In a cluster RCT of home BP telemonitoring, in which the pharmacist adjusted antihypertensives based on transmitted BP measurements, hypertension control was significantly better in the intervention group than in the usual care group (57.2% vs 30%).29
Continue to: What are the recommended techniques for HBPM?
9. What are the recommended techniques for HBP
Patients should use a device that is validated, fully automated, and has an upper arm cuff (not a wrist monitor), according to a joint statement from the AHA, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association.17 (SOR C). (See validated BP monitor list at http://www.dableducational.org/sphygmomanometers/devices_2_sbpm.html.)
Patients should measure their BP in their nondominant arm after 5 minutes of rest with the arm at heart level, back supported, and feet flat on the ground. Patient technique and the accuracy of the home BP monitor should be checked annually. It is also recommended that patients check their BP 2 to 3 times every morning and evening. An average of 12 morning and evening measurements should be used for monitoring and treatment changes. An AHA informational sheet that shows how to measure BP properly can be found on their Web si
Several studies examining the accuracy of measuring BP over clothing did not find significant differences in BP measurements performed on a bare arm vs over a sleeve.30-33
10. What are the predictors of differences between home and office BP measurements?
Gender is one of the biggest predictors (SOR B).
A 2016 meta-analysis reported a total of 60 different hypothesized predictors of differences between home and clinic BP measurements (eg, gender, age, body mass index, systolic BP, diastolic BP). Masked hypertension was defined as a normal clinic BP reading and an elevated home BP reading. White coat hypertension was defined as an elevated clinic BP measurement with an acceptable home BP measurement. The researchers extracted odds ratios (ORs) for each study describing the association between patient characteristics and white coat or masked hypertension.34 Studies of masked hypertension diagnosed from HBPM showed male gender as the most significant predictor of home-clinic BP differences (OR=1.47, 95% CI, 1.18-1.75). In contrast, female gender was the only significant predictor of white coat hypertension (OR=3.38; 95% CI, 1.64-6.96) when comparing home BP with clinic BP measurements
Literature limitations and barriers to greater implementation
Most studies looking at HBPM outcomes have measured outcomes using ABPM or office BP measurements. The authors of studies using office BP as the outcome measure usually performed multiple BP measurements at often multiple office or clinic visits to calculate the true BP—a procedure that primary care practices rarely follow.35 Additionally, there are significant methodologic differences in HBPM and ABPM; home BP is measured at rest, while ambulatory BP is measured while the patient is mobile and functioning. There are insufficient prospective studies looking at HBPM effects on clinical and patient-oriented outcomes.
The evidence clearly supports using HBPM in the diagnosis of hypertension and suggests its benefit in hypertension management. However, there are significant barriers to incorporating HBPM into practice—barriers that are largely unaddressed in the literature.
For HBPM to be successful, patients need affordable validated home BP monitors covered by insurance that can translate home BP readings into usable information. Additional administrative and/or nursing assistance is required for patient education and support. Uploaded data need to be summarized in a way that is actionable and linked to the electronic health record.
Continue to: As the volume of patient-generated home date increases...
As the volume of patient-generated home data increases, there is a risk of information overload. Thus, meaningful summarization of the data is required to enable the physician, patient, and/or pharmacist to take prompt and effective action.
CORRESPONDENCE
Sonal J. Patil, MD, MSPH, Curtis W. and Ann H. Long Department of Family and Community Medicine, University of Missouri, MA306 Medical Sciences Building, DC032.00 Columbia, MO 65212; [email protected].
ACKNOWLEDGEMENTS
The authors are grateful to Dr. David R. Mehr for his valuable expertise in editing and proofreading this manuscript.
National Health and Nutrition Examination Survey (NHANES) data from 2011 to 2014 revealed that 29% of adults in the United States have hypertension.1 Prevalence increases with age, so that 7% of adults ages 18 to 39 years, 32% of adults ages 40 to 59, and 65% of adults ages ≥60 years have the disease.1 This national survey data also showed that 53% of those given the diagnosis had uncontrolled hypertension, and that control of hypertension did not change significantly from 2009 to 2014.1
Elevated blood pressure (BP) has been the leading risk factor for death related to cardiovascular disease globally for the last 3 decades.2 Yet in 2 nationally representative samples, only 1 in 6 patients with documented BP ≥140/90 mm Hg received treatment intensification with new medication during primary care visits.3 Uncertainty about the representativeness of any single clinic BP measurement is a prominent reason for health care providers not to intensify therapy.4
Confirming the Dx outside the office. The 2015 US Preventive Services Task Force (USPSTF) guidelines on screening for hypertension state that, for most patients, a diagnosis of hypertension should be confirmed with out-of-office BP monitoring before initiating treatment.5 The USPSTF states that ambulatory BP monitoring (ABPM) is accurate for hypertension diagnosis and monitoring, and that home BP monitoring (HBPM) is an acceptable alternative, based on good quality evidence.
Access to ABPM, however, is often limited. In a 2015 survey of primary care clinics, only 25% of the 123 clinics that completed the questionnaire reported having access to it.6 Conversely, HBPM is widely available and acceptable to most patients. A recent NHANES survey showed that 43.5% of patients who were aware of their hypertension diagnosis engaged in HBPM.7
So what, exactly, should the role of HBPM be in the management of patients with hypertension? The evidence-based answers to the 10 questions that follow provide useful insights.
[polldaddy:10224678]
1. Can HBPM be used to confirm a Dx of hypertension?
Yes (Strength of recommendation [SOR] C).
In reviewing the diagnostic accuracy of various methods to confirm the diagnosis of hypertension, the USPSTF identified ABPM as the most accurate, followed by HBPM, with clinic BP measurements bringing up the rear.5 In adults ≥18 years of age, the USPSTF recommends obtaining BP measurements outside of the clinical setting for diagnostic confirmation before starting treatment unless the patient’s BP is ≥180/110 mm Hg, there is evidence of end-organ damage, or the patient has a diagnosis of secondary hypertension.5 The USPSTF recommends HBPM as an acceptable alternative to ABPM based on 6 studies including a total of 1253 participants.8 The percentage of patients with elevated office BP confirmed by HBPM to have hypertension was 45% to 84% across these 6 studies.
Sixteen studies from another systematic review evaluated the diagnostic accuracy of HBPM while using ABPM as a reference.9 This review found that HBPM had high specificity and negative predictive value, but low sensitivity and positive predictive value. There was moderate diagnostic agreement between HBPM and ABPM, with kappa statistic values of 0.37 to 0.73 across all studies.9
Continue to: In yet another study...
In yet another study, home BP and ambulatory BP measurements were identical when the same dual-mode device was used to measure both ambulatory and home BP.10
2. What are the diagnostic and treatment targets for home BP monitoring?
Treat patients if home BP is ≥130/80 mm Hg and categorize patients as normotensive if home BP is <125/76 mm Hg (SOR C). Monitor patients who are in between.
A 2017 joint statement from the American College of Cardiology/American Heart Association (ACC/AHA) Task Force states that the target BP for HBPM should be <130/80 mm Hg.11 The Joint National Commission (JNC) 8 issued BP goals of <140/90 mm Hg for adults <60 years of age and those with diabetes and/or chronic kidney disease, and a goal of <150/90 mm Hg for adults ≥60 years of age with no diabetes or chronic kidney disease,12 but much debate has recently surrounded these guidelines. JNC 8 does not provide a separate BP goal for HBPM.
Although based solely on evidence (and not patient-oriented outcomes), a home BP threshold of ≥135/85 mm Hg for the diagnosis and treatment of hypertension has been supported by the European Society of Hypertension consensus guidelines,13 results of a longitudinal study,14 meta-analyses of published studies, and a meta-analysis using individual subject data.15
Support for a home BP measurement of <125/76 mm Hg as normal is limited to a single cross-sectional study of 48 patients with 2 elevated office BP readings where the threshold of 125/76 mm Hg on home BP was shown to exclude 80% of patients diagnosed with hypertension by ambulatory readings.16 If home BP measurements are >125/76 mm Hgbut <135/85 mm Hg, 24-hour ambulatory BP monitoring is recommended to assess hypertension control.17
3. Does home BP monitoring improve hypertension control?
Yes, in the short term, but not in the long term (SOR C).
A meta-analysis of 13 comparative studies looking at HBPM alone vs usual care showed a small, but statistically significant, benefit of achieving target BP at 6 months with a relative risk ratio (RRR) of 1.3 (95% confidence interval [CI], 1.00-1.68; I2=77%).18 However, the pooled effects from 3 studies that measured the benefit of achieving a predefined BP target at the 12-month follow-up mark were not significant in this review (RRR=1.18; 95% CI, 0.95-1.46, I2=86%).18 The pooled effect from 19 studies from the same review showed that there was a statistically significant weighted mean difference of -3.9 mm Hg in systolic BP and a weighted mean difference of -2.4 mm Hg in diastolic BP at 6 months; however, the changes were no longer significant at the 12-month follow-up mark.18
Continue to: More than half of the studies included...
More than half of the studies included in the meta-analysis were of low quality, and none of the studies recruited patients based on differences in clinic BP and home BP patterns, but rather on controlled or uncontrolled hypertension status. The studies included in this meta-analysis measured final BP outcomes by measuring ambulatory BP or clinic BP.
Another systematic review of 19 studies and 7100 participants looking at how HBPM compared with ABPM as a measurement standard for BP control and patient outcomes found insufficient data to determine the benefit of using HBPM as a measurement standard for BP control.19
HBPM + added support. There was high-quality evidence from the meta-analysis that HBPM plus additional support vs usual care led to a reduction in BP and a higher proportion of patients achieving target BP.18 However, the additional support interventions in the studies were heterogeneous.
4. Should HBPM be used to detect a change in BP associated with medication alterations?
Yes (SOR B).
A 2008 meta-analysis20 and several other studies21,22 showed that HBPM has greater accuracy than office BP for identifying drug-induced BP changes. The 2008 meta-analysis looked at changes in office and home BP measurements produced by various antihypertensive drugs. In 7 studies that compared office BP measurements with home and ambulatory BP measurements, the 24-hour ambulatory BP measurements and home BP measurements showed less dramatic BP reductions with medications than clinic BP measurements.20 This meta-analysis included 30 studies with 6794 participants and showed that home BP readings fell 20% less than office BP readings; the difference was statistically significant. These findings suggest that treatment-attributable changes in home BP and clinic BP measurements are linearly related, with the treatment effect on home BP measurements being around 80% of the effect on clinic BP measurements.
5. Do home BP measurements correlate with clinical outcomes?
Yes, and better than office BP measurements do; however, most studies comparing home BP measurements with usual care while looking at clinical outcomes are observational or quasi-experimental (SOR B).
For example, a 2015 systematic review looking at associations between BP measurement type (office, home, and ambulatory) and patient mortality found 5 observational studies that showed that adding home or ambulatory BP information improved cardiovascular risk prediction models. Moreover, all-cause mortality was associated with home BP and ambulatory BP levels only and not with office BP levels.19 The number of participants in these 5 studies varied between 210 and 2051 with study duration between 2.4 and 12.3 years. Of note, every study had a distinct population, affecting the generalizability of the results.
Continue to: One quasi-experimental study...
One quasi-experimental study with 450 participants showed that home BP measurements were at least as good as ambulatory BP measurements at predicting end organ damage related to hypertension when organ damage was measured by cardiac echocardiography, detection of microalbuminuria, and carotid echocardiography.23 Similarly, a systematic review of 14 studies and 2485 participants comparing home, ambulatory, and office BP readings showed that home BP measurements’ association with left ventricular mass index is as good as that of ambulatory BP measurements, and superior to clinic BP readings.24
6. Does HBPM help improve medication adherence?
The jury is still out on this one (SOR B).
A 2006 systematic review of randomized controlled trials (RCTs) incorporating HBPM and evaluating medication adherence outcomes found that in 6 of the 11 studies identified, there was some improvement in medication adherence with HBPM.25 However, only 1 of the 6 studies in this review involved HBPM as the sole intervention; the remaining 5 studies employed additional adherence-enhancing strategies.
Another systematic review looking at HBPM vs usual care included 8 studies (3 of moderate quality and 5 of low quality) that measured medication adherence (using varying measures of adherence) of which only 3 studies showed some improvement in medication adherence with HBPM.18
7. Does HBPM reduce therapeutic inertia?
Yes (SOR B).
A meta-analysis of 15 studies showed that therapeutic inertia was less common with HBPM than with office BP monitoring alone; the relative risk for unchanged medication was 0.82 (95% CI, 0.68 to 0.99) with HBPM.26 However, 10 of the 15 studies were of low quality with a Jadad score ≤3.
8. Does HBPM, along with titration of treatment, improve BP outcomes?
Yes (SOR B).
Two RCTs that looked at self-monitoring of BP and self-titration of hypertensive medications showed significant reductions in BP levels.27,28 In a cluster RCT of home BP telemonitoring, in which the pharmacist adjusted antihypertensives based on transmitted BP measurements, hypertension control was significantly better in the intervention group than in the usual care group (57.2% vs 30%).29
Continue to: What are the recommended techniques for HBPM?
9. What are the recommended techniques for HBP
Patients should use a device that is validated, fully automated, and has an upper arm cuff (not a wrist monitor), according to a joint statement from the AHA, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association.17 (SOR C). (See validated BP monitor list at http://www.dableducational.org/sphygmomanometers/devices_2_sbpm.html.)
Patients should measure their BP in their nondominant arm after 5 minutes of rest with the arm at heart level, back supported, and feet flat on the ground. Patient technique and the accuracy of the home BP monitor should be checked annually. It is also recommended that patients check their BP 2 to 3 times every morning and evening. An average of 12 morning and evening measurements should be used for monitoring and treatment changes. An AHA informational sheet that shows how to measure BP properly can be found on their Web si
Several studies examining the accuracy of measuring BP over clothing did not find significant differences in BP measurements performed on a bare arm vs over a sleeve.30-33
10. What are the predictors of differences between home and office BP measurements?
Gender is one of the biggest predictors (SOR B).
A 2016 meta-analysis reported a total of 60 different hypothesized predictors of differences between home and clinic BP measurements (eg, gender, age, body mass index, systolic BP, diastolic BP). Masked hypertension was defined as a normal clinic BP reading and an elevated home BP reading. White coat hypertension was defined as an elevated clinic BP measurement with an acceptable home BP measurement. The researchers extracted odds ratios (ORs) for each study describing the association between patient characteristics and white coat or masked hypertension.34 Studies of masked hypertension diagnosed from HBPM showed male gender as the most significant predictor of home-clinic BP differences (OR=1.47, 95% CI, 1.18-1.75). In contrast, female gender was the only significant predictor of white coat hypertension (OR=3.38; 95% CI, 1.64-6.96) when comparing home BP with clinic BP measurements
Literature limitations and barriers to greater implementation
Most studies looking at HBPM outcomes have measured outcomes using ABPM or office BP measurements. The authors of studies using office BP as the outcome measure usually performed multiple BP measurements at often multiple office or clinic visits to calculate the true BP—a procedure that primary care practices rarely follow.35 Additionally, there are significant methodologic differences in HBPM and ABPM; home BP is measured at rest, while ambulatory BP is measured while the patient is mobile and functioning. There are insufficient prospective studies looking at HBPM effects on clinical and patient-oriented outcomes.
The evidence clearly supports using HBPM in the diagnosis of hypertension and suggests its benefit in hypertension management. However, there are significant barriers to incorporating HBPM into practice—barriers that are largely unaddressed in the literature.
For HBPM to be successful, patients need affordable validated home BP monitors covered by insurance that can translate home BP readings into usable information. Additional administrative and/or nursing assistance is required for patient education and support. Uploaded data need to be summarized in a way that is actionable and linked to the electronic health record.
Continue to: As the volume of patient-generated home date increases...
As the volume of patient-generated home data increases, there is a risk of information overload. Thus, meaningful summarization of the data is required to enable the physician, patient, and/or pharmacist to take prompt and effective action.
CORRESPONDENCE
Sonal J. Patil, MD, MSPH, Curtis W. and Ann H. Long Department of Family and Community Medicine, University of Missouri, MA306 Medical Sciences Building, DC032.00 Columbia, MO 65212; [email protected].
ACKNOWLEDGEMENTS
The authors are grateful to Dr. David R. Mehr for his valuable expertise in editing and proofreading this manuscript.
1. Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011-2014. NCHS Data Brief. 2015;(220):1-8.
2. Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardio-metabolic risk factors between 1980 and 2010: comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2:634-647.
3. Mu L, Mukamal KJ. Treatment intensification for hypertension in US ambulatory medical care. J Am Heart Assoc. 2016;5:e004188.
4. Kerr EA, Zikmund-Fisher BJ, Klamerus ML, et al. The role of clinical uncertainty in treatment decisions for diabetic patients with uncontrolled blood pressure. Ann Intern Med. 2008;148:717-727.
5. U.S. Preventive Services Task Force. Final recommendation statement: High blood pressure in adults: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/high-blood-pressure-in-adults-screening. Accessed July 19, 2017.
6. Woolsey S, Brown B, Ralls B, et al. Diagnosing hypertension in primary care clinics according to current guidelines. J Am Board Fam Med. 2017;30:170-177.
7. Ostchega Y, Zhang G, Kit BK, et al. Factors associated with home blood pressure monitoring among US adults: National Health and Nutrition Examination Survey, 2011-2014. Am J Hypertens. 2017;30:1126-1132.
8. Piper MA, Evans CV, Burda BU, et al. Diagnostic and predictive accuracy of blood pressure screening methods with consideration of rescreening intervals: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2015;162:192-204.
9. Stergiou GS, Bliziotis IA. Home blood pressure monitoring in the diagnosis and treatment of hypertension: a systematic review. Am J Hypertens. 2011;24:123-134.
10. Stergiou GS, Tzamouranis D, Nasothimiou EG, et al. Are there really differences between home and daytime ambulatory blood pressure? Comparison using a novel dual-mode ambulatory and home monitor. J Hum Hypertens. 2009;24:207-212.
11. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017;71:e13-e115.
12. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520. Erratum in JAMA. 2014;311:1809.
13. O’Brien E, Asmar R, Beilin L, et al. European Society of Hypertension recommendations for conventional, ambulatory and home blood pressure measurement. J Hypertens. 2003;21:821-848.
14. Tsunoda S, Kawano Y, Horio T, et al. Relationship between home blood pressure and longitudinal changes in target organ damage in treated hypertensive patients. Hypertens Res. 2002;25:167-173.
15. Staessen JA, Thijs L. Development of diagnostic thresholds for automated self-measurement of blood pressure in adults. First International Consensus Conference on Blood Pressure Self-Measurement. Blood Press Monit. 2000;5:101-109.
16. Mansoor GA, White WB. Self-measured home blood pressure in predicting ambulatory hypertension. Am J Hypertens. 2004;17(11 Pt 1):1017-1022.
17. Pickering TG, Miller NH, Ogedegbe G, et al. Call to action on use and reimbursement for home blood pressure monitoring: a joint scientific statement from the American Heart Association, American Society of Hypertension, and Preventive Cardiovascular Nurses Association. J Cardiovasc Nurs. 2008;23:299-323.
18. Uhlig K, Patel K, Ip S, Kitsios GD, Balk EM. Self-measured blood pressure monitoring in the management of hypertension: a systematic review and meta-analysis. Ann Intern Med. 2013;159:185-194.
19. Breaux-Shropshire TL, Judd E, Vucovich LA, et al. Does home blood pressure monitoring improve patient outcomes? A systematic review comparing home and ambulatory blood pressure monitoring on blood pressure control and patient outcomes. Integr Blood Press Control. 2015;8:43-49.
20. Ishikawa J, Carroll DJ, Kuruvilla S, et al. Changes in home versus clinic blood pressure with antihypertensive treatments: a meta-analysis. Hypertension. 2008;52:856-864.
21. Imai Y, Ohkubo T, Hozawa A, et al. Usefulness of home blood pressure measurements in assessing the effect of treatment in a single-blind placebo-controlled open trial. J Hypertens. 2001;19:179-185.
22. Vaur L, Dubroca I, Dutrey-Dupagne C, et al. Superiority of home blood pressure measurements over office measurements for testing antihypertensive drugs. Blood Press Monit. 1998;3:107-114.
23. Gaborieau V, Delarche N, Gosse P. Ambulatory blood pressure monitoring versus self-measurement of blood pressure at home: correlation with target organ damage. J Hypertens. 2008;26:1919-1927.
24. Bliziotis IA, Destounis A, Stergiou GS. Home versus ambulatory and office blood pressure in predicting target organ damage in hypertension: a systematic review and meta-analysis. J Hypertens. 2012;30:1289-1299.
25. Ogedegbe G, Schoenthaler A. A systematic review of the effects of home blood pressure monitoring on medication adherence. J Clin Hypertens (Greenwich). 2006;8:174-180.
26. Agarwal R, Bills JE, Hecht TJ, et al. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension. 2011;57:29-38.
27. McManus RJ, Mant J, Haque MS, et al. Effect of self-monitoring and medication self-titration on systolic blood pressure in hypertensive patients at high risk of cardiovascular disease: the TASMIN-SR randomized clinical trial. JAMA. 2014;312:799-808.
28. McManus RJ, Mant J, Bray EP, et al. Telemonitoring and self-management in the control of hypertension (TASMINH2): a randomised controlled trial. Lancet. 2010;376:163-172.
29. Margolis KL, Asche SE, Bergdall AR, et al. Effect of home blood pressure telemonitoring and pharmacist management on blood pressure control: The HyperLink Cluster Randomized Trial. JAMA. 2013;310:46-56.
30. Ma G, Sabin N, Dawes M. A comparison of blood pressure measurement over a sleeved arm versus a bare arm. CMAJ. 2008;178:585-589.
31. Kahan E, Yaphe J, Knaani-Levinz H, et al. Comparison of blood pressure measurements on the bare arm, below a rolled-up sleeve, or over a sleeve. Fam Pract. 2003;20:730-732.
32. Liebl M, Holzgreve H, Schulz M, et al. The effect of clothes on sphygmomanometric and oscillometric blood pressure measurement. Blood Press. 2004;13:279-282.
33. Holleman DR Jr., Westman EC, McCrory DC, et al. The effect of sleeved arms on oscillometric blood pressure measurement. J Gen Intern Med. 1993;8:325-326.
34. Sheppard JP, Fletcher B, Gill P, et al. Predictors of the home-clinic blood pressure difference: a systematic review and meta-analysis. Am J Hypertens. 2016;29:614-625.
35. Stergiou GS, Skeva II, Baibas NM, et al. Diagnosis of hypertension using home or ambulatory blood pressure monitoring: comparison with the conventional strategy based on repeated clinic blood pressure measurements. J Hypertens. 2000;18:1745-1751.
1. Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011-2014. NCHS Data Brief. 2015;(220):1-8.
2. Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardio-metabolic risk factors between 1980 and 2010: comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2:634-647.
3. Mu L, Mukamal KJ. Treatment intensification for hypertension in US ambulatory medical care. J Am Heart Assoc. 2016;5:e004188.
4. Kerr EA, Zikmund-Fisher BJ, Klamerus ML, et al. The role of clinical uncertainty in treatment decisions for diabetic patients with uncontrolled blood pressure. Ann Intern Med. 2008;148:717-727.
5. U.S. Preventive Services Task Force. Final recommendation statement: High blood pressure in adults: Screening. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/high-blood-pressure-in-adults-screening. Accessed July 19, 2017.
6. Woolsey S, Brown B, Ralls B, et al. Diagnosing hypertension in primary care clinics according to current guidelines. J Am Board Fam Med. 2017;30:170-177.
7. Ostchega Y, Zhang G, Kit BK, et al. Factors associated with home blood pressure monitoring among US adults: National Health and Nutrition Examination Survey, 2011-2014. Am J Hypertens. 2017;30:1126-1132.
8. Piper MA, Evans CV, Burda BU, et al. Diagnostic and predictive accuracy of blood pressure screening methods with consideration of rescreening intervals: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2015;162:192-204.
9. Stergiou GS, Bliziotis IA. Home blood pressure monitoring in the diagnosis and treatment of hypertension: a systematic review. Am J Hypertens. 2011;24:123-134.
10. Stergiou GS, Tzamouranis D, Nasothimiou EG, et al. Are there really differences between home and daytime ambulatory blood pressure? Comparison using a novel dual-mode ambulatory and home monitor. J Hum Hypertens. 2009;24:207-212.
11. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017;71:e13-e115.
12. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520. Erratum in JAMA. 2014;311:1809.
13. O’Brien E, Asmar R, Beilin L, et al. European Society of Hypertension recommendations for conventional, ambulatory and home blood pressure measurement. J Hypertens. 2003;21:821-848.
14. Tsunoda S, Kawano Y, Horio T, et al. Relationship between home blood pressure and longitudinal changes in target organ damage in treated hypertensive patients. Hypertens Res. 2002;25:167-173.
15. Staessen JA, Thijs L. Development of diagnostic thresholds for automated self-measurement of blood pressure in adults. First International Consensus Conference on Blood Pressure Self-Measurement. Blood Press Monit. 2000;5:101-109.
16. Mansoor GA, White WB. Self-measured home blood pressure in predicting ambulatory hypertension. Am J Hypertens. 2004;17(11 Pt 1):1017-1022.
17. Pickering TG, Miller NH, Ogedegbe G, et al. Call to action on use and reimbursement for home blood pressure monitoring: a joint scientific statement from the American Heart Association, American Society of Hypertension, and Preventive Cardiovascular Nurses Association. J Cardiovasc Nurs. 2008;23:299-323.
18. Uhlig K, Patel K, Ip S, Kitsios GD, Balk EM. Self-measured blood pressure monitoring in the management of hypertension: a systematic review and meta-analysis. Ann Intern Med. 2013;159:185-194.
19. Breaux-Shropshire TL, Judd E, Vucovich LA, et al. Does home blood pressure monitoring improve patient outcomes? A systematic review comparing home and ambulatory blood pressure monitoring on blood pressure control and patient outcomes. Integr Blood Press Control. 2015;8:43-49.
20. Ishikawa J, Carroll DJ, Kuruvilla S, et al. Changes in home versus clinic blood pressure with antihypertensive treatments: a meta-analysis. Hypertension. 2008;52:856-864.
21. Imai Y, Ohkubo T, Hozawa A, et al. Usefulness of home blood pressure measurements in assessing the effect of treatment in a single-blind placebo-controlled open trial. J Hypertens. 2001;19:179-185.
22. Vaur L, Dubroca I, Dutrey-Dupagne C, et al. Superiority of home blood pressure measurements over office measurements for testing antihypertensive drugs. Blood Press Monit. 1998;3:107-114.
23. Gaborieau V, Delarche N, Gosse P. Ambulatory blood pressure monitoring versus self-measurement of blood pressure at home: correlation with target organ damage. J Hypertens. 2008;26:1919-1927.
24. Bliziotis IA, Destounis A, Stergiou GS. Home versus ambulatory and office blood pressure in predicting target organ damage in hypertension: a systematic review and meta-analysis. J Hypertens. 2012;30:1289-1299.
25. Ogedegbe G, Schoenthaler A. A systematic review of the effects of home blood pressure monitoring on medication adherence. J Clin Hypertens (Greenwich). 2006;8:174-180.
26. Agarwal R, Bills JE, Hecht TJ, et al. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension. 2011;57:29-38.
27. McManus RJ, Mant J, Haque MS, et al. Effect of self-monitoring and medication self-titration on systolic blood pressure in hypertensive patients at high risk of cardiovascular disease: the TASMIN-SR randomized clinical trial. JAMA. 2014;312:799-808.
28. McManus RJ, Mant J, Bray EP, et al. Telemonitoring and self-management in the control of hypertension (TASMINH2): a randomised controlled trial. Lancet. 2010;376:163-172.
29. Margolis KL, Asche SE, Bergdall AR, et al. Effect of home blood pressure telemonitoring and pharmacist management on blood pressure control: The HyperLink Cluster Randomized Trial. JAMA. 2013;310:46-56.
30. Ma G, Sabin N, Dawes M. A comparison of blood pressure measurement over a sleeved arm versus a bare arm. CMAJ. 2008;178:585-589.
31. Kahan E, Yaphe J, Knaani-Levinz H, et al. Comparison of blood pressure measurements on the bare arm, below a rolled-up sleeve, or over a sleeve. Fam Pract. 2003;20:730-732.
32. Liebl M, Holzgreve H, Schulz M, et al. The effect of clothes on sphygmomanometric and oscillometric blood pressure measurement. Blood Press. 2004;13:279-282.
33. Holleman DR Jr., Westman EC, McCrory DC, et al. The effect of sleeved arms on oscillometric blood pressure measurement. J Gen Intern Med. 1993;8:325-326.
34. Sheppard JP, Fletcher B, Gill P, et al. Predictors of the home-clinic blood pressure difference: a systematic review and meta-analysis. Am J Hypertens. 2016;29:614-625.
35. Stergiou GS, Skeva II, Baibas NM, et al. Diagnosis of hypertension using home or ambulatory blood pressure monitoring: comparison with the conventional strategy based on repeated clinic blood pressure measurements. J Hypertens. 2000;18:1745-1751.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Migraine: Expanding our Tx arsenal
Migraine is a highly disabling primary headache disorder that affects more than 44 million Americans annually.1 The disorder causes pain, photophobia, phonophobia, and nausea that can last for hours, even days. Migraine headaches are 2 times more common in women than in men; although migraine is most common in people 30 to 39 years of age, all ages are affected.2,3 Frequency of migraine headache is variable; chronic migraineurs experience more than 15 headache days a month.

Recent estimates indicate that the cost of acute and chronic migraine headaches reaches approximately $78 million a year in the United States. 4 This high burden of disease has made effective migraine treatment options absolutely essential. Recent advances in our understanding of migraine pathophysiology have led to new therapeutic targets; there are now many novel treatment approaches on the horizon.
In this article, we review the diagnosis and management of migraine in detail. Our emphasis is on evidence-based approaches to acute and prophylactic treatment, including tried-and-true options and newly emerging therapies.
Neuronal dysfunction and a genetic predisposition
Although migraine was once thought to be caused by abnormalities of vasodilation, current research suggests that the disorder has its origins in primary neuronal dysfunction. There appears to be a genetic predisposition toward widespread neuronal hyperexcitability in migraineurs.5 In addition, hypothalamic neurons are thought to initiate migraine by responding to changes in brain homeostasis. Increased parasympathetic tone might activate meningeal pain receptors or lower the threshold for transmitting pain signals from the thalamus to the cortex.6
Prodromal symptoms and aura appear to originate from multiple areas across the brain, including the hypothalamus, cortex, limbic system, and brainstem. This widespread brain involvement might explain why some headache sufferers concurrently experience a variety of symptoms, including fatigue, depression, muscle pain, and an abnormal sensitivity to light, sound, and smell.6,7
Although the exact mechanisms behind each of these symptoms have yet to be defined precisely, waves of neuronal depolarization—known as cortical spreading depression—are suspected to cause migraine aura.8-10 Cortical spreading depression activates the trigeminal pain pathway and leads to the release of pro-inflammatory markers such as calcitonin gene-related protein (CGRP).6 A better understanding of these complex signaling pathways has helped provide potential therapeutic targets for new migraine drugs.
Diagnosis: Close patient inquiry is most helpful
The International Headache Society (IHS) criteria for primary headache disorders serve as the basis for the diagnosis of migraine and its subtypes, which include migraine without aura and migraine with aura. Due to variability of presentation, migraine with aura is further subdivided into migraine with typical aura (with and without headache), migraine with brainstem aura, hemiplegic migraine, and retinal migraine.11
Continue to: How is migraine defined?
How is migraine defined? Simply, migraine is classically defined as a unilateral, pulsating headache of moderate to severe intensity lasting 4 to 72 hours, associated with photophobia and phonophobia or nausea and vomiting, or both.11 Often visual in nature, aura is a set of neurologic symptoms that lasts for minutes and precedes the onset of the headache. The visual aura is often described as a scintillating scotoma that begins near the point of visual fixation and then spreads left or right. Other aura symptoms include tingling or numbness (second most common), speech disturbance (aphasia), motor changes and, in rare cases, a combination of these in succession. By definition, all of these symptoms fully resolve between attacks.11

Validated valuable questionnaires. To help with accurate and timely diagnosis, researchers have developed and validated simplified questionnaires that can be completed independently by patients presenting to primary care (TABLE 112,13):
- ID Migraine is a set of 3 questions that scores positive when a patient endorses at least 2 of the 3 symptoms. 12
- MS-Q is similar to the ID Migraine but includes 5 items. A score of ≥4 is a positive screen. 13
The sensitivity and specificity of MS-Q (0.93 and 0.81, respectively) are slightly higher than those of ID Migraine (0.81 and 0.75).13
Remember POUND. This mnemonic device can also be used during history-taking to aid in diagnostic accuracy. Migraine is highly likely (92%) in patients who endorse 4 of the following 5 symptoms and unlikely (17%) in those who endorse ≤2 symptoms14: Pulsatile quality of headache 4 to 72 hOurs in duration, Unilateral location, Nausea or vomiting, and Disabling intensity.

Differential Dx. Although the differential diagnosis of headache is broad (TABLE 214,15), the history alone can often guide clinicians towards the correct assessment. After taking the initial history (headache onset, location, duration, and associated symptoms), focus your attention on assessing the risk of intracranial pathology. This is best accomplished by assessing specific details of the history (TABLE 314) and findings on physical examination15:
- blood pressure measurement (seated, legs uncrossed, feet flat on the floor; having rested for 5 minutes; arm well supported)
- cranial nerve exam
- extremity strength testing
- eye exam (vision, extra-ocular muscles, visual fields, pupillary reactivity, and funduscopic exam)
- gait (tandem walk)
- reflexes.

Continue to: Further testing needed?
Further testing needed? Neuroimaging should be considered only in patients with an abnormal neurologic exam, atypical headache features, or certain risk factors, such as an immune deficiency. There is no role for electroencephalography or other diagnostic testing in migraine.16
Take a multipronged approach to treatment
As with other complex, chronic conditions, the treatment of migraine should take a multifaceted approach, including management of acute symptoms as well as prevention of future headaches. In 2015, the American Headache Society published a systematic review that specified particular treatment goals for migraine sufferers. 17 These goals include:
- headache reduction
- headache relief
- decreased disability from headache
- elimination of nausea and vomiting
- elimination of photophobia and phonophobia.
Our review, which follows, of therapeutic options focuses on the management of migraine in adults. Approaches in special populations (older adults, pregnant women, and children) are discussed afterward.
Pharmacotherapy for acute migraine
Acute migraine should be treated with an abortive medication at the onset of headache. The immediate goal is to relieve pain within 2 hours and prevent its recurrence within the subsequent 48 hours (TABLE 412,18-20).

In the general population, mild, infrequent migraines can be managed with acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs).21

Continue to: For moderate-to-severe migraine...
For moderate-to-severe migraine, triptans, which target serotonin receptors, are the drug of choice for most patients.21 Triptans are superior to placebo in achieving a pain-free state at 2 and 24 hours after administration; eletriptan has the most desirable outcome, with 68% of patients pain free at 2 hours and 54% pain free at 24 hours.22 Triptans are available as sublingual tablets and nasal sprays, as well as subcutaneous injections for patients with significant associated nausea and vomiting. Avoid prescribing triptans for patients with known vascular disease (eg, history of stroke, myocardial infarction, peripheral vascular disease, uncontrolled hypertension, or signs and symptoms of these conditions), as well as for patients with severe hepatic impairment.
Importantly, although triptans all have a similar mechanism of action, patients might respond differently to different drugs within the class. If a patient does not get adequate headache relief from an appropriate dosage of a given triptan during a particular migraine episode, a different triptan can be tried during the next migraine.22 Additionally, if a patient experiences an adverse effect from one triptan, this does not necessarily mean that a trial of another triptan at a later time is contraindicated.
For patients who have an incomplete response to migraine treatment or for those with frequent recurrence, the combination formulation of sumatriptan, 85 mg, and naproxen, 500 mg, showed the highest rate of resolution of headache within 2 hours compared with either drug alone.23 A similar result might be found by combining a triptan known to be effective for a patient and an NSAID other than naproxen. If migraine persists despite initial treatment of an attack, a different class of medication should be tried during the course of that attack to attain relief of symptoms of that migraine.21
When a patient is seen in an acute care setting (eg, emergency department, urgent care center) while suffering a migraine, additional treatment options are available. Intravenous (IV) anti-emetics are useful for relieving the pain of migraine and nausea, and can be used in combination with an IV NSAID (eg, ketorolac).21 The most effective anti-emetics are dopamine receptor type-2 blockers, including chlorpromazine, droperidol, metoclopramide, and prochlorperazine, which has the highest level of efficacy.24 Note that these medications do present the risk of a dystonic reaction; diphenhydramine is therefore often used in tandem to mitigate such a response.
Looking ahead. Although triptans are the current first-line therapy for acute migraine, their effectiveness is limited. Only 20% of patients report sustained relief of pain in the 2 to 24 hours after treatment, and the response can vary from episode to episode.25
Continue to: With better understading of the pathophysiology of migraine...
With better understanding of the pathophysiology of migraine, a host of novel anti-migraine drugs are on the horizon.
CGRP receptor antagonists. The neuropeptide CGRP, which mediates central and peripheral nervous system pain signaling, has been noted to be elevated during acute migraine attacks26; clinical trials are therefore underway to evaluate the safety and efficacy of CGRP receptor antagonists.18 These agents appear to be better tolerated than triptans, have fewer vascular and central nervous system adverse effects, and present less of a risk of medication overuse headache.18 Liver toxicity has been seen with some medications in this class and remains an important concern in their development.19
Phase 3 clinical trials for 1 drug in this class, ubrogepant, were completed in late 2017; full analysis of the data is not yet available. Primary outcomes being evaluated include relief of pain at 2 hours and relief from the most bothersome symptoms again at 2 hours.27
Selective serotonin-HT1f receptor agonists, such as lasmiditan, offer another potential approach. Although the exact mechanism of action of these agents is not entirely clear, clinical trials have supported their efficacy and safety.20 Importantly, ongoing trials are specifically targeting patients with known cardiovascular risk factors because they are most likely to benefit from the nonvasoconstrictive mechanism of action.28,29 Adverse effects reported primarily include dizziness, fatigue, and vertigo.
Strategies for managing recurrent episodic migraine
Because of the risk of medication overuse headache with acute treatment, daily preventive therapy for migraine is indicated for any patient with 30 :
- ≥6 headache days a month
- ≥4 headache days a month with some impairment
- ≥3 headache days a month with severe impairment.
Continue to: Treatment begins by having patients identify...
Treatment begins by having patients identify, and then avoid, migraine triggers (TABLE 5). This can be accomplished by having patients keep a headache diary, in which they can enter notations about personal and environmental situations that precede a headache.

For the individual patient, some triggers are modifiable; others are not. Helping a patient develop strategies for coping with triggers, rather than aiming for complete avoidance, might help her (him) manage those that are inescapable (eg stress, menstruation, etc).31 For many patients, however, this is not an adequate intervention and other approaches must be explored. When considering which therapy might be best for a given patient, evaluate her (his) comorbidities and assess that particular treatment for potential secondary benefits and the possibility of adverse effects. Pay attention to the choice of preventive therapy in women who are considering pregnancy because many available treatments are potentially teratogenic.
Oral medications. Oral agents from several classes of drugs can be used for migraine prophylaxis, including anti-epileptics,antidepressants, and antihypertensives (TABLE 620,29,30,32-41). Selected anti-epileptics (divalproex sodium, sodium valproate, topiramate) and beta-blockers (metoprolol, propranolol, and timolol) have the strongest evidence to support their use.32 Overall, regular use of prophylactic medications can reduce headache frequency by 50% for approximately 40% to 45% of patients who take them.29 However, adherence may be limited by adverse effects or perceived lack of efficacy, thus reducing their potential for benefit.42

OnabotulinumtoxinA. In patients with chronic migraine (≥15 headache days a month for at least 3 months) who have failed oral medications, the American Academy of Neurology (AAN) recommends the use of onabotulinumtoxinA.30 The treatment regimen comprises 31 injections at various sites on the head, neck, and shoulders every 3 months.33

A 2010 large randomized controlled trial showed a decrease in the frequency of headache days for patients receiving onabotulinumtoxinA compared to placebo after a 24-week treatment period (7.8 fewer headache days a month, compared to 6.4 fewer in the placebo group).33 A recent systematic review also noted a reduction of 2 headache days a month compared with placebo; the authors cautioned, however, that data with which to evaluate onabotulinumtoxinA in comparison to other prophylactic agents are limited.43
Continue to: In both studies...
In both studies, the risk of adverse drug events due to onabotulinumtoxinA was high and led to a significant rate of discontinuation.33,43 Despite this, onabotulinumtoxinA remains the only Food and Drug Administration (FDA)–approved treatment for chronic migraine, making it reasonable to consider for appropriate patients.
Acupuncture. A 2016 Cochrane review found benefit for patients using acupuncture compared with sham acupuncture.34 When acupuncture was compared with prophylactic agents such as beta-blockers, calcium-channel blockers, and anti-epileptics, however, there was no significant difference between the procedure and pharmacotherapy. Patients willing and able to try acupuncture might see a reduction in the overall number of headaches. Acupuncture has few adverse effects; however, long-term data are lacking.34
Exercise is not supported by robust data for its role as a prophylactic treatment. It is generally considered safe in most populations, however, and can be pursued with little out-of-pocket cost.35
Cognitive behavioral therapy (CBT). The AAN recommends CBT, relaxation therapy, and biofeedback therapy. Accessibility of these services remains limited for many patients, and cost can be prohibitive.16
Supplements used to help prevent migraine include the root of Petasites hybridus (butterbur), magnesium, vitamin B2 (riboflavin), Tanacetum parthenium (feverfew), and coenzyme Q10.16 Although the strength of evidence for these therapies is limited by small trials, their overall risk of adverse effects is low, and they might be easier for patients to obtain than acupuncture or CBT.
Continue to: Butterbur, in particular...
Butterbur, in particular, has been found to be beneficial for migraine prevention in 2 small placebo-controlled trials. In a randomized controlled study of 245 patients P hybridus, (specifically, the German formulation, Petadolex), 75 mg BID, reduced the frequency of migraine attack by 48% at 4 months, compared to placebo (number needed to treat, 5.3).44 No difference was found at lower dosages. The most common reported adverse effect was burping.
Regrettably, unpurified butterbur extract contains pyrrolizidine alkaloids, potentially hepatotoxic and carcinogenic compounds. Because of variations in purification in production facilities in the United States, butterbur supplements might not have all of these compounds removed—and so should be used with caution.41
Magnesium. Studies evaluating the use of magnesium have demonstrated varied results; differences in methods and dosing have limited broad application of findings. As with most supplements considered for prophylactic treatment, magnesium dosing is poorly understood, and bioavailability varies in its different forms. Oral supplementation can be given as magnesium dicitrate, 600 mg/d.45
Recently, products containing various combinations of feverfew, coenzyme Q10, riboflavin, magnesium, and other supplements have shown benefit in early clinical trials.36,37
Neural stimulation. Over the past few years, a variety of transcutaneous nerve stimulator devices have gained FDA approval for use in migraine prophylaxis. The long-term safety and efficacy of these devices is not yet well understood, but they appear to provide headache relief in the short term and decrease the frequency of headache.38 Use of the noninvasive stimulators is limited today by high cost and poor coverage by US health care insurers.
Continue to: Newly available medical therapy
Newly available medical therapy. The FDA recently approved erenumab, a fully human monoclonal antibody for prevention of migraine in adults. This is the first drug in the CGRP antagonist class to be approved for this indication. Trials of this once-monthly, self-injectable drug show promising results for patients whose migraines have been refractory to other therapies.
A recent large trial evaluated 955 adults with migraine, randomizing them to receive erenumab, 70 mg; erenumab, 140 mg; or placebo over 28 weeks.39 The groups receiving erenumab had a nearly 2-fold higher odds of having their migraine reduced by 50%, compared with placebo (number needed to treat with the 140-mg dose, 4.27). Similar numbers of participants from all groups discontinued the study.39 Phase 3 trials that are not yet formally published have produced similarly beneficial results.40,46 The FDA has listed injection site reaction and constipation as the most reported adverse effects.40
Three other anti-CGRP antibodies are likely to be approved in the near future: fremanezumab, galcanezumab, and eptinezumab.
The approach to migraine in special populations
Management of acute and chronic migraine in children, pregnant women, and older adults requires special attention: Treatment approaches are different than they are for adults 19 to 65 years of age.
Pediatric patients. Migraine is the most common acute and recurrent headache syndrome in children. Headaches differ from those of adult migraine as a result of variations in brain maturation, plasticity, and cognitive development.47 Migraine attacks are often of shorter duration in children, lasting 1 to 2 hours, but can still be preceded by visual aura.48 Just as with adults, imaging, electroencephalography, lumbar puncture, and routine labs should be considered only if a child has an abnormal neurological exam or other concerning features (TABLE 214,15).
Continue to: The general approach to migraine treatment...
The general approach to migraine treatment in the pediatric population includes education of the child and family about symptom management. Acetaminophen, NSAIDs, and triptans are approved for abortive therapy in children and should be used for acute headache relief in the same way that they are used in adults. Oral rizatriptan, the most well studied triptan in the pediatric population, is approved for use in children as young as 6 years49; the pediatric dosage is 5 mg/d for patients weighing 20 to 39 kg and 10 mg/d for patients weighing more than 40 kg (same as the adult dosage).
Oral almotriptan and zolmitriptan are also approved for use in children 12 to 17 years of age. Usual dosages are: almotriptan, 12.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 25 mg/d); and zolmitriptan, 2.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 10 mg/d).50
For children who are unable to swallow pills or who are vomiting, a non-oral route of administration is preferable. Rizatriptan is available as an orally disintegrating tablet. Zolmitriptan is available in a nasal spray at a dose of 5 mg for children 12 years and older. Sumatriptan is not approved for use in patients younger than 18 years; however, recent studies have shown that it might have good efficacy and tolerability.50
Daily prophylactic treatment for recurrent migraine in the pediatric population is an evolving subject; published guidelines do not exist. It is reasonable to consider treatment using the same guidelines as those in place for adults.51 Topiramate, 1 to 2 mg/kg/d, is the only therapy approved by the FDA for episodic migraine preventive therapy in adolescents.50
Notably, a nonpharmacotherapeutic approach may be more effective for pediatric prevention. In 2017, a large double-blind, placebo-controlled trial investigated the use of amitriptyline, topiramate, and placebo for the treatment of recurrent migraine in children 8 to 17 years of age. An interim analysis of the 328 children enrolled found no significant differences in reduction of headache frequency with treatment compared with placebo over a 24-week period; the trial was stopped early due to futility.52
Continue to: The study did show...
The study did show, however, that reducing migraine triggers provided a high level of benefit to study participants. Stress is one of the most common migraine triggers in children; lack of sleep, exposure to a warm climate, and exposure to video games are also notable triggers.53 CBT may augment the efficacy of standard migraine medications in the pediatric population and may help prevent recurrence of episodes.54
Pregnancy. The treatment of migraine is different in pregnant women than it is in nonpregnant adults because of a concern over adverse effects on fetal development. For acute headache treatment, first-line therapies include trigger avoidance and acetaminophen, 1000 mg (maximum dosage, 4000 mg/d).55 If this is ineffective, a 10-mg dose of metoclopramide, as often as every 6 hours (not an FDA-approved indication), can be considered. During the second trimester, NSAIDs can be considered second-line therapy.
Triptans—specifically, sumatriptan and rizatriptan—can also be considered if first-line therapies fail.56 Triptan-exposed pregnant women with migraine have a rate of congenital malformations, spontaneous abortions, and prematurity that is similar to what is seen in pregnant women with migraine who have not been exposed to triptans. However, when triptan-exposed women are compared with healthy, non-migraine-suffering women, the rate of spontaneous abortion appears to be increased in the triptan-exposed population.57
Ergotamine is contraindicated during pregnancy because of its potential to induce uterine contractions and vasospasm, which can be detrimental to the fetus.56Nonpharmacotherapeutic interventions such as heat, ice, massage, rest, and avoidance of triggers are as successful in the pregnant population as in the nonpregnant population. For migraine prevention, coenzyme Q10, vitamins B2 and B6 (pyridoxine), and oral magnesium can be considered. Feverfew and butterbur should be avoided because of concerns about fetal malformation and preterm labor.58
Older adults. Choosing appropriate migraine therapy for older adults requires special consideration because of changes in drug metabolism and risks associated with drug adverse effects. Additionally, few studies of migraine drugs have included large populations of adults older than 65 years; medications should therefore be prescribed cautiously in this population, with particular attention to drug–drug interactions.
Continue to: Just as for younger adults...
Just as for younger adults, mild symptoms can be managed effectively with acetaminophen. NSAIDs may be used as well, but carry increased risks of gastric bleeding and elevation in blood pressure.59 The use of triptans is acceptable for the appropriate patient, but should be avoided in patients with known vascular disease.60 Antiemetics present an increased risk of extrapyramidal adverse effects in the elderly and should be used with caution at the lowest effective dosage.59 Novel mechanisms of action make some of the newer agents potentially safer for use in older adults when treating acute migraine.
For migraine prevention in older adults, particular attention should be paid to reducing triggers and minimizing polypharmacy.
More and more, successful treatment is within reach
With many clinical trials evaluating novel drugs underway, and additional studies contributing to our understanding of nonpharmacotherapeutic approaches to migraine treatment, improved headache control may become increasingly common over the next few years.
CORRESPONDENCE
Kathryn McGrath, MD, Department of Family and Community Medicine, Thomas Jefferson University, 1015 Walnut St, Philadelphia PA 19107; [email protected].
1. Stokes M, Becker WJ, Lipton RB, et al. Cost of health care among patients with chronic and episodic migraine in Canada and the USA: results from the International Burden of Migraine Study (IBMS). Headache. 2011;51:1058-1077.
2. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
3. Burch RC, Loder S, Loder E, et al. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache. 2015;55:21-34.
4. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81:479-484.
5. Ferrari MD, Klever RR, Terwindt GM, et al. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14:65-80.
6. Burstein R, Noseda R, Borsook D. Migraine: multiple processes, complex pathophysiology. J Neurosc. 2015;35:6619-6629.
7. Maniyar FH, Sprenger T, Monteith T, et al. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. 2013;137(Pt 1):232-241.
8. Cutrer FM, Sorensen AG, Weisskoff RM, et al. Perfusion‐weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25-31.
9. Hadjikhani N, Sanchez Del Rio MS, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687-4692.
10. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol. 2013;75:365-391.
11. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, (beta version). Cephalalgia. 2013;33:629-808.
12. Lipton RB, Dodick D, Sadovsky RE, et al; ID Migraine validation study. A self-administered screener for migraine in primary care: The ID Migraine™ validation study. Neurology. 2003;61:375-382.
13. Láinez MJ, Domínguez M, Rejas J, et al. Development and validation of the Migraine Screen Questionnaire (MS‐Q). Headache. 2005;45:1328-1338.
14. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006;296:1274-1283.
15. Becker WJ, Findlay T, Moga C, et al. Guideline for primary care management of headache in adults. Can Fam Physician. 2015;61:670-679.
16. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
17. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55:3-20.
18. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887-898.
19. Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533-552.
20. Färkkilä M, Diener HC, Géraud G, et al; COL MIG-202 study group. Efficacy and tolerability of lasmiditan, an oral 5-HT(1F) receptor agonist, for the acute treatment of migraine: a phase 2 randomised, placebo-controlled, parallel-group, dose-ranging study. Lancet Neurol. 2012;11:405-413.
21. Pringsheim T, Davenport WJ, Marmura MJ, et al. How to apply the AHS evidence assessment of the acute treatment of migraine in adults to your patient with migraine. Headache. 2016;56:1194-1200.
22. Thorlund K, Mills EJ, Wu P, et al. Comparative efficacy of triptans for the abortive treatment of migraine: a multiple treatment comparison meta-analysis. Cephalalgia. 2014;34:258-267.
23. Law S, Derry S, Moore RA. Sumatriptan plus naproxen for acute migraine attacks in adults. Cochrane Database Syst Rev. 2013;(10):CD008541.
24. Orr SL, Aubé M, Becker WJ, et al. Canadian Headache Society systematic review and recommendations on the treatment of migraine pain in emergency settings. Cephalalgia. 2015;35:271-284.
25. Ferrari MD, Goadsby PJ, Roon KI, et al. Triptans (serotonin, 5‐HT1B/1D agonists) in migraine: detailed results and methods of a meta‐analysis of 53 trials. Cephalalgia. 2002;22:633-658.
26. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48-56.
27. A phase 3, multicenter, randomized, double-blind, placebo-controlled single attack study to evaluate the efficacy, safety, and tolerability of oral ubrogepant in the acute treatment of migraine. https://clinicaltrials.gov/ct2/show/study/NCT02828020. Accessed November 16, 2018.
28. Rubio-Beltrán E, Labastida-Ramírez A, Villalón CM, et al. Is selective 5-HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. 2018;186:88-97.
29. Diener HC, Charles A, Goadsby PJ, et al. New therapeutic approaches for the prevention and treatment of migraine. Lancet Neurol. 2015;14:1010-1022.
30. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention. Headache. 2015;55 Suppl 2:103-122.
31. Martin PR. Behavioral management of migraine headache triggers: learning to cope with triggers. Curr Pain Headache Rep. 2010;14:221-227.
32. Loder E, Burch R, Rizzoli P. The 2012 AHS/AAN guidelines for prevention of episodic migraine: a summary and comparison with other recent clinical practice guidelines. Headache. 2012;52:930-945.
33. Dodick DW, Turkel CC, DeGryse RE, et al; PREEMPT Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double‐blind, randomized, placebo‐controlled phases of the PREEMPT clinical program. Headache. 2010;50:921-936.
34. Linde K, Allais G, Brinkhaus B, et al. Acupuncture for the prevention of episodic migraine. Cochrane Database Syst Rev. 2016(6):CD001218.
35. Varkey E, Cider Å, Carlsson J, et al. Exercise as migraine prophylaxis: a randomized study using relaxation and topiramate as controls. Cephalalgia. 2011;31:1428-1438.
36. Guilbot A, Bangratz M, Abdellah SA, et al. A combination of coenzyme Q10, feverfew and magnesium for migraine prophylaxis: a prospective observational study. BMC Complement Altern Med. 2017;17:433.
37. Dalla Volta G, Zavarize P, Ngonga G, et al. Combination of Tanacethum partenium, 5-hydrossitriptophan (5-Http) and magnesium in the prophylaxis of episodic migraine without aura (AURASTOP®) an observational study. Int J Neuro Brain Dis. 2017;4:1-4.
38. Puledda F, Goadsby PJ. An update on non‐pharmacological neuromodulation for the acute and preventive treatment of migraine. Headache. 2017;57:685-691.
39. Goadsby PJ, Reuter U, Hallström Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123-2132.
40. Reuter U. Efficacy and safety of erenumab in episodic migraine patients with 2-4 prior preventive treatment failures: Results from the Phase 3b LIBERTY study. Abstract 009, AAN 2018 Annual Meeting; April 24, 2018.
41. Diener HC, Freitag FG, Danesch U. Safety profile of a special butterbur extract from Petasites hybridus in migraine prevention with emphasis on the liver. Cephalalgia Reports. https://journals.sagepub.com/doi/10.1177/2515816318759304. 2018 May 2. Accessed December 15, 2018.
42. Kingston WS, Halker R. Determinants of suboptimal migraine diagnosis and treatment in the primary care setting. J Clin Outcomes Manag. 2017;24:319-324.
43. Herd CP, Tomlinson CL, Rick C, et al. Botulinum toxins for the prevention of migraine in adults. Cochrane Database of Syst Rev. 2018;6:CD011616.
44. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
45. Von Luckner A, Riederer F. Magnesium in migraine prophylaxis—is there an evidence‐based rationale? A systematic review. Headache. 2018;58:199-209.
46. Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16:425-434.
47. Sonal Sekhar M, Sasidharan S, Joseph S, et al. Migraine management: How do the adult and paediatric migraines differ? Saudi Pharm J. 2012;20:1-7.
48. Lewis DW. Pediatric migraine. In: Lewis DW. Clinician’s Manual on Treatment of Pediatric Migraine. London, UK: Springer Healthcare Ltd; 2010:15-26.
49. Ho TW, Pearlman E, Lewis D, et al. Efficacy and tolerability of rizatriptan in pediatric migraineurs: results from a randomized double-blind, placebo controlled trial using a novel adaptive enrichment design. Cephalagia. 2012;32:750-765.
50. Khrizman M, Pakalnis A. Management of pediatric migraine: current therapies. Pediatr Ann. 2018;47:e55-e60.
51. Lipton RB, Bigal ME, Diamond M, et al; AMPP Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
52. Powers SW, Coffey CS, Chamberlin LA, et al; CHAMP Investigators. Trial of amitriptyline, topiramate, and placebo for pediatric migraine. N Engl J Med. 2017;376:115-124.
53. Neut D, Fily A, Cuvellier JC, et al. The prevalence of triggers in paediatric migraine: a questionnaire study in 102 children and adolescents. J Headache Pain. 2012;13:61-65.
54. Ng QX, Venkatanarayanan N, Kumar L. A systematic review and meta‐analysis of the efficacy of cognitive behavioral therapy for the management of pediatric migraine. Headache. s2017;57:349-362.
55. Lipton RB, Baggish JS, Stewart WF, et al. Efficacy and safety of acetaminophen in the treatment of migraine: results of a randomized, double-blind, placebo-controlled, population-based study. Arch Intern Med. 2000;160:3486-3492.
56. Lucas S. Medication use in the treatment of migraine during pregnancy and lactation. Curr Pain Headache Rep. 2009;13:392-398.
57. Marchenko A, Etwel F, Olutunfesse O, et al. Pregnancy outcome following prenatal exposure to triptan medications: a meta-analysis. Headache. 2015:55:490-501.
58. Wells RE, Turner DP, Lee M, et al. Managing migraine during pregnancy and lactation. Curr Neurol Neurosci Rep. 2016;16:40.
59. Haan J, Hollander J, Ferrari MD. Migraine in the elderly: a review. Cephalalgia. 2007;27:97-106.
60. Gladstone JP, Eross EJ, Dodick DW. Migraine in special populations. Treatment strategies for children and adolescents, pregnant women, and the elderly. Postgrad Med. 2004;115:39-44,47-50.
Migraine is a highly disabling primary headache disorder that affects more than 44 million Americans annually.1 The disorder causes pain, photophobia, phonophobia, and nausea that can last for hours, even days. Migraine headaches are 2 times more common in women than in men; although migraine is most common in people 30 to 39 years of age, all ages are affected.2,3 Frequency of migraine headache is variable; chronic migraineurs experience more than 15 headache days a month.
Recent estimates indicate that the cost of acute and chronic migraine headaches reaches approximately $78 million a year in the United States. 4 This high burden of disease has made effective migraine treatment options absolutely essential. Recent advances in our understanding of migraine pathophysiology have led to new therapeutic targets; there are now many novel treatment approaches on the horizon.
In this article, we review the diagnosis and management of migraine in detail. Our emphasis is on evidence-based approaches to acute and prophylactic treatment, including tried-and-true options and newly emerging therapies.
Neuronal dysfunction and a genetic predisposition
Although migraine was once thought to be caused by abnormalities of vasodilation, current research suggests that the disorder has its origins in primary neuronal dysfunction. There appears to be a genetic predisposition toward widespread neuronal hyperexcitability in migraineurs.5 In addition, hypothalamic neurons are thought to initiate migraine by responding to changes in brain homeostasis. Increased parasympathetic tone might activate meningeal pain receptors or lower the threshold for transmitting pain signals from the thalamus to the cortex.6
Prodromal symptoms and aura appear to originate from multiple areas across the brain, including the hypothalamus, cortex, limbic system, and brainstem. This widespread brain involvement might explain why some headache sufferers concurrently experience a variety of symptoms, including fatigue, depression, muscle pain, and an abnormal sensitivity to light, sound, and smell.6,7
Although the exact mechanisms behind each of these symptoms have yet to be defined precisely, waves of neuronal depolarization—known as cortical spreading depression—are suspected to cause migraine aura.8-10 Cortical spreading depression activates the trigeminal pain pathway and leads to the release of pro-inflammatory markers such as calcitonin gene-related protein (CGRP).6 A better understanding of these complex signaling pathways has helped provide potential therapeutic targets for new migraine drugs.
Diagnosis: Close patient inquiry is most helpful
The International Headache Society (IHS) criteria for primary headache disorders serve as the basis for the diagnosis of migraine and its subtypes, which include migraine without aura and migraine with aura. Due to variability of presentation, migraine with aura is further subdivided into migraine with typical aura (with and without headache), migraine with brainstem aura, hemiplegic migraine, and retinal migraine.11
Continue to: How is migraine defined?
How is migraine defined? Simply, migraine is classically defined as a unilateral, pulsating headache of moderate to severe intensity lasting 4 to 72 hours, associated with photophobia and phonophobia or nausea and vomiting, or both.11 Often visual in nature, aura is a set of neurologic symptoms that lasts for minutes and precedes the onset of the headache. The visual aura is often described as a scintillating scotoma that begins near the point of visual fixation and then spreads left or right. Other aura symptoms include tingling or numbness (second most common), speech disturbance (aphasia), motor changes and, in rare cases, a combination of these in succession. By definition, all of these symptoms fully resolve between attacks.11
Validated valuable questionnaires. To help with accurate and timely diagnosis, researchers have developed and validated simplified questionnaires that can be completed independently by patients presenting to primary care (TABLE 112,13):
- ID Migraine is a set of 3 questions that scores positive when a patient endorses at least 2 of the 3 symptoms. 12
- MS-Q is similar to the ID Migraine but includes 5 items. A score of ≥4 is a positive screen. 13
The sensitivity and specificity of MS-Q (0.93 and 0.81, respectively) are slightly higher than those of ID Migraine (0.81 and 0.75).13
Remember POUND. This mnemonic device can also be used during history-taking to aid in diagnostic accuracy. Migraine is highly likely (92%) in patients who endorse 4 of the following 5 symptoms and unlikely (17%) in those who endorse ≤2 symptoms14: Pulsatile quality of headache 4 to 72 hOurs in duration, Unilateral location, Nausea or vomiting, and Disabling intensity.
Differential Dx. Although the differential diagnosis of headache is broad (TABLE 214,15), the history alone can often guide clinicians towards the correct assessment. After taking the initial history (headache onset, location, duration, and associated symptoms), focus your attention on assessing the risk of intracranial pathology. This is best accomplished by assessing specific details of the history (TABLE 314) and findings on physical examination15:
- blood pressure measurement (seated, legs uncrossed, feet flat on the floor; having rested for 5 minutes; arm well supported)
- cranial nerve exam
- extremity strength testing
- eye exam (vision, extra-ocular muscles, visual fields, pupillary reactivity, and funduscopic exam)
- gait (tandem walk)
- reflexes.
Continue to: Further testing needed?
Further testing needed? Neuroimaging should be considered only in patients with an abnormal neurologic exam, atypical headache features, or certain risk factors, such as an immune deficiency. There is no role for electroencephalography or other diagnostic testing in migraine.16
Take a multipronged approach to treatment
As with other complex, chronic conditions, the treatment of migraine should take a multifaceted approach, including management of acute symptoms as well as prevention of future headaches. In 2015, the American Headache Society published a systematic review that specified particular treatment goals for migraine sufferers. 17 These goals include:
- headache reduction
- headache relief
- decreased disability from headache
- elimination of nausea and vomiting
- elimination of photophobia and phonophobia.
Our review, which follows, of therapeutic options focuses on the management of migraine in adults. Approaches in special populations (older adults, pregnant women, and children) are discussed afterward.
Pharmacotherapy for acute migraine
Acute migraine should be treated with an abortive medication at the onset of headache. The immediate goal is to relieve pain within 2 hours and prevent its recurrence within the subsequent 48 hours (TABLE 412,18-20).
In the general population, mild, infrequent migraines can be managed with acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs).21
Continue to: For moderate-to-severe migraine...
For moderate-to-severe migraine, triptans, which target serotonin receptors, are the drug of choice for most patients.21 Triptans are superior to placebo in achieving a pain-free state at 2 and 24 hours after administration; eletriptan has the most desirable outcome, with 68% of patients pain free at 2 hours and 54% pain free at 24 hours.22 Triptans are available as sublingual tablets and nasal sprays, as well as subcutaneous injections for patients with significant associated nausea and vomiting. Avoid prescribing triptans for patients with known vascular disease (eg, history of stroke, myocardial infarction, peripheral vascular disease, uncontrolled hypertension, or signs and symptoms of these conditions), as well as for patients with severe hepatic impairment.
Importantly, although triptans all have a similar mechanism of action, patients might respond differently to different drugs within the class. If a patient does not get adequate headache relief from an appropriate dosage of a given triptan during a particular migraine episode, a different triptan can be tried during the next migraine.22 Additionally, if a patient experiences an adverse effect from one triptan, this does not necessarily mean that a trial of another triptan at a later time is contraindicated.
For patients who have an incomplete response to migraine treatment or for those with frequent recurrence, the combination formulation of sumatriptan, 85 mg, and naproxen, 500 mg, showed the highest rate of resolution of headache within 2 hours compared with either drug alone.23 A similar result might be found by combining a triptan known to be effective for a patient and an NSAID other than naproxen. If migraine persists despite initial treatment of an attack, a different class of medication should be tried during the course of that attack to attain relief of symptoms of that migraine.21
When a patient is seen in an acute care setting (eg, emergency department, urgent care center) while suffering a migraine, additional treatment options are available. Intravenous (IV) anti-emetics are useful for relieving the pain of migraine and nausea, and can be used in combination with an IV NSAID (eg, ketorolac).21 The most effective anti-emetics are dopamine receptor type-2 blockers, including chlorpromazine, droperidol, metoclopramide, and prochlorperazine, which has the highest level of efficacy.24 Note that these medications do present the risk of a dystonic reaction; diphenhydramine is therefore often used in tandem to mitigate such a response.
Looking ahead. Although triptans are the current first-line therapy for acute migraine, their effectiveness is limited. Only 20% of patients report sustained relief of pain in the 2 to 24 hours after treatment, and the response can vary from episode to episode.25
Continue to: With better understading of the pathophysiology of migraine...
With better understanding of the pathophysiology of migraine, a host of novel anti-migraine drugs are on the horizon.
CGRP receptor antagonists. The neuropeptide CGRP, which mediates central and peripheral nervous system pain signaling, has been noted to be elevated during acute migraine attacks26; clinical trials are therefore underway to evaluate the safety and efficacy of CGRP receptor antagonists.18 These agents appear to be better tolerated than triptans, have fewer vascular and central nervous system adverse effects, and present less of a risk of medication overuse headache.18 Liver toxicity has been seen with some medications in this class and remains an important concern in their development.19
Phase 3 clinical trials for 1 drug in this class, ubrogepant, were completed in late 2017; full analysis of the data is not yet available. Primary outcomes being evaluated include relief of pain at 2 hours and relief from the most bothersome symptoms again at 2 hours.27
Selective serotonin-HT1f receptor agonists, such as lasmiditan, offer another potential approach. Although the exact mechanism of action of these agents is not entirely clear, clinical trials have supported their efficacy and safety.20 Importantly, ongoing trials are specifically targeting patients with known cardiovascular risk factors because they are most likely to benefit from the nonvasoconstrictive mechanism of action.28,29 Adverse effects reported primarily include dizziness, fatigue, and vertigo.
Strategies for managing recurrent episodic migraine
Because of the risk of medication overuse headache with acute treatment, daily preventive therapy for migraine is indicated for any patient with 30 :
- ≥6 headache days a month
- ≥4 headache days a month with some impairment
- ≥3 headache days a month with severe impairment.
Continue to: Treatment begins by having patients identify...
Treatment begins by having patients identify, and then avoid, migraine triggers (TABLE 5). This can be accomplished by having patients keep a headache diary, in which they can enter notations about personal and environmental situations that precede a headache.
For the individual patient, some triggers are modifiable; others are not. Helping a patient develop strategies for coping with triggers, rather than aiming for complete avoidance, might help her (him) manage those that are inescapable (eg stress, menstruation, etc).31 For many patients, however, this is not an adequate intervention and other approaches must be explored. When considering which therapy might be best for a given patient, evaluate her (his) comorbidities and assess that particular treatment for potential secondary benefits and the possibility of adverse effects. Pay attention to the choice of preventive therapy in women who are considering pregnancy because many available treatments are potentially teratogenic.
Oral medications. Oral agents from several classes of drugs can be used for migraine prophylaxis, including anti-epileptics,antidepressants, and antihypertensives (TABLE 620,29,30,32-41). Selected anti-epileptics (divalproex sodium, sodium valproate, topiramate) and beta-blockers (metoprolol, propranolol, and timolol) have the strongest evidence to support their use.32 Overall, regular use of prophylactic medications can reduce headache frequency by 50% for approximately 40% to 45% of patients who take them.29 However, adherence may be limited by adverse effects or perceived lack of efficacy, thus reducing their potential for benefit.42
OnabotulinumtoxinA. In patients with chronic migraine (≥15 headache days a month for at least 3 months) who have failed oral medications, the American Academy of Neurology (AAN) recommends the use of onabotulinumtoxinA.30 The treatment regimen comprises 31 injections at various sites on the head, neck, and shoulders every 3 months.33
A 2010 large randomized controlled trial showed a decrease in the frequency of headache days for patients receiving onabotulinumtoxinA compared to placebo after a 24-week treatment period (7.8 fewer headache days a month, compared to 6.4 fewer in the placebo group).33 A recent systematic review also noted a reduction of 2 headache days a month compared with placebo; the authors cautioned, however, that data with which to evaluate onabotulinumtoxinA in comparison to other prophylactic agents are limited.43
Continue to: In both studies...
In both studies, the risk of adverse drug events due to onabotulinumtoxinA was high and led to a significant rate of discontinuation.33,43 Despite this, onabotulinumtoxinA remains the only Food and Drug Administration (FDA)–approved treatment for chronic migraine, making it reasonable to consider for appropriate patients.
Acupuncture. A 2016 Cochrane review found benefit for patients using acupuncture compared with sham acupuncture.34 When acupuncture was compared with prophylactic agents such as beta-blockers, calcium-channel blockers, and anti-epileptics, however, there was no significant difference between the procedure and pharmacotherapy. Patients willing and able to try acupuncture might see a reduction in the overall number of headaches. Acupuncture has few adverse effects; however, long-term data are lacking.34
Exercise is not supported by robust data for its role as a prophylactic treatment. It is generally considered safe in most populations, however, and can be pursued with little out-of-pocket cost.35
Cognitive behavioral therapy (CBT). The AAN recommends CBT, relaxation therapy, and biofeedback therapy. Accessibility of these services remains limited for many patients, and cost can be prohibitive.16
Supplements used to help prevent migraine include the root of Petasites hybridus (butterbur), magnesium, vitamin B2 (riboflavin), Tanacetum parthenium (feverfew), and coenzyme Q10.16 Although the strength of evidence for these therapies is limited by small trials, their overall risk of adverse effects is low, and they might be easier for patients to obtain than acupuncture or CBT.
Continue to: Butterbur, in particular...
Butterbur, in particular, has been found to be beneficial for migraine prevention in 2 small placebo-controlled trials. In a randomized controlled study of 245 patients P hybridus, (specifically, the German formulation, Petadolex), 75 mg BID, reduced the frequency of migraine attack by 48% at 4 months, compared to placebo (number needed to treat, 5.3).44 No difference was found at lower dosages. The most common reported adverse effect was burping.
Regrettably, unpurified butterbur extract contains pyrrolizidine alkaloids, potentially hepatotoxic and carcinogenic compounds. Because of variations in purification in production facilities in the United States, butterbur supplements might not have all of these compounds removed—and so should be used with caution.41
Magnesium. Studies evaluating the use of magnesium have demonstrated varied results; differences in methods and dosing have limited broad application of findings. As with most supplements considered for prophylactic treatment, magnesium dosing is poorly understood, and bioavailability varies in its different forms. Oral supplementation can be given as magnesium dicitrate, 600 mg/d.45
Recently, products containing various combinations of feverfew, coenzyme Q10, riboflavin, magnesium, and other supplements have shown benefit in early clinical trials.36,37
Neural stimulation. Over the past few years, a variety of transcutaneous nerve stimulator devices have gained FDA approval for use in migraine prophylaxis. The long-term safety and efficacy of these devices is not yet well understood, but they appear to provide headache relief in the short term and decrease the frequency of headache.38 Use of the noninvasive stimulators is limited today by high cost and poor coverage by US health care insurers.
Continue to: Newly available medical therapy
Newly available medical therapy. The FDA recently approved erenumab, a fully human monoclonal antibody for prevention of migraine in adults. This is the first drug in the CGRP antagonist class to be approved for this indication. Trials of this once-monthly, self-injectable drug show promising results for patients whose migraines have been refractory to other therapies.
A recent large trial evaluated 955 adults with migraine, randomizing them to receive erenumab, 70 mg; erenumab, 140 mg; or placebo over 28 weeks.39 The groups receiving erenumab had a nearly 2-fold higher odds of having their migraine reduced by 50%, compared with placebo (number needed to treat with the 140-mg dose, 4.27). Similar numbers of participants from all groups discontinued the study.39 Phase 3 trials that are not yet formally published have produced similarly beneficial results.40,46 The FDA has listed injection site reaction and constipation as the most reported adverse effects.40
Three other anti-CGRP antibodies are likely to be approved in the near future: fremanezumab, galcanezumab, and eptinezumab.
The approach to migraine in special populations
Management of acute and chronic migraine in children, pregnant women, and older adults requires special attention: Treatment approaches are different than they are for adults 19 to 65 years of age.
Pediatric patients. Migraine is the most common acute and recurrent headache syndrome in children. Headaches differ from those of adult migraine as a result of variations in brain maturation, plasticity, and cognitive development.47 Migraine attacks are often of shorter duration in children, lasting 1 to 2 hours, but can still be preceded by visual aura.48 Just as with adults, imaging, electroencephalography, lumbar puncture, and routine labs should be considered only if a child has an abnormal neurological exam or other concerning features (TABLE 214,15).
Continue to: The general approach to migraine treatment...
The general approach to migraine treatment in the pediatric population includes education of the child and family about symptom management. Acetaminophen, NSAIDs, and triptans are approved for abortive therapy in children and should be used for acute headache relief in the same way that they are used in adults. Oral rizatriptan, the most well studied triptan in the pediatric population, is approved for use in children as young as 6 years49; the pediatric dosage is 5 mg/d for patients weighing 20 to 39 kg and 10 mg/d for patients weighing more than 40 kg (same as the adult dosage).
Oral almotriptan and zolmitriptan are also approved for use in children 12 to 17 years of age. Usual dosages are: almotriptan, 12.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 25 mg/d); and zolmitriptan, 2.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 10 mg/d).50
For children who are unable to swallow pills or who are vomiting, a non-oral route of administration is preferable. Rizatriptan is available as an orally disintegrating tablet. Zolmitriptan is available in a nasal spray at a dose of 5 mg for children 12 years and older. Sumatriptan is not approved for use in patients younger than 18 years; however, recent studies have shown that it might have good efficacy and tolerability.50
Daily prophylactic treatment for recurrent migraine in the pediatric population is an evolving subject; published guidelines do not exist. It is reasonable to consider treatment using the same guidelines as those in place for adults.51 Topiramate, 1 to 2 mg/kg/d, is the only therapy approved by the FDA for episodic migraine preventive therapy in adolescents.50
Notably, a nonpharmacotherapeutic approach may be more effective for pediatric prevention. In 2017, a large double-blind, placebo-controlled trial investigated the use of amitriptyline, topiramate, and placebo for the treatment of recurrent migraine in children 8 to 17 years of age. An interim analysis of the 328 children enrolled found no significant differences in reduction of headache frequency with treatment compared with placebo over a 24-week period; the trial was stopped early due to futility.52
Continue to: The study did show...
The study did show, however, that reducing migraine triggers provided a high level of benefit to study participants. Stress is one of the most common migraine triggers in children; lack of sleep, exposure to a warm climate, and exposure to video games are also notable triggers.53 CBT may augment the efficacy of standard migraine medications in the pediatric population and may help prevent recurrence of episodes.54
Pregnancy. The treatment of migraine is different in pregnant women than it is in nonpregnant adults because of a concern over adverse effects on fetal development. For acute headache treatment, first-line therapies include trigger avoidance and acetaminophen, 1000 mg (maximum dosage, 4000 mg/d).55 If this is ineffective, a 10-mg dose of metoclopramide, as often as every 6 hours (not an FDA-approved indication), can be considered. During the second trimester, NSAIDs can be considered second-line therapy.
Triptans—specifically, sumatriptan and rizatriptan—can also be considered if first-line therapies fail.56 Triptan-exposed pregnant women with migraine have a rate of congenital malformations, spontaneous abortions, and prematurity that is similar to what is seen in pregnant women with migraine who have not been exposed to triptans. However, when triptan-exposed women are compared with healthy, non-migraine-suffering women, the rate of spontaneous abortion appears to be increased in the triptan-exposed population.57
Ergotamine is contraindicated during pregnancy because of its potential to induce uterine contractions and vasospasm, which can be detrimental to the fetus.56Nonpharmacotherapeutic interventions such as heat, ice, massage, rest, and avoidance of triggers are as successful in the pregnant population as in the nonpregnant population. For migraine prevention, coenzyme Q10, vitamins B2 and B6 (pyridoxine), and oral magnesium can be considered. Feverfew and butterbur should be avoided because of concerns about fetal malformation and preterm labor.58
Older adults. Choosing appropriate migraine therapy for older adults requires special consideration because of changes in drug metabolism and risks associated with drug adverse effects. Additionally, few studies of migraine drugs have included large populations of adults older than 65 years; medications should therefore be prescribed cautiously in this population, with particular attention to drug–drug interactions.
Continue to: Just as for younger adults...
Just as for younger adults, mild symptoms can be managed effectively with acetaminophen. NSAIDs may be used as well, but carry increased risks of gastric bleeding and elevation in blood pressure.59 The use of triptans is acceptable for the appropriate patient, but should be avoided in patients with known vascular disease.60 Antiemetics present an increased risk of extrapyramidal adverse effects in the elderly and should be used with caution at the lowest effective dosage.59 Novel mechanisms of action make some of the newer agents potentially safer for use in older adults when treating acute migraine.
For migraine prevention in older adults, particular attention should be paid to reducing triggers and minimizing polypharmacy.
More and more, successful treatment is within reach
With many clinical trials evaluating novel drugs underway, and additional studies contributing to our understanding of nonpharmacotherapeutic approaches to migraine treatment, improved headache control may become increasingly common over the next few years.
CORRESPONDENCE
Kathryn McGrath, MD, Department of Family and Community Medicine, Thomas Jefferson University, 1015 Walnut St, Philadelphia PA 19107; [email protected].
Migraine is a highly disabling primary headache disorder that affects more than 44 million Americans annually.1 The disorder causes pain, photophobia, phonophobia, and nausea that can last for hours, even days. Migraine headaches are 2 times more common in women than in men; although migraine is most common in people 30 to 39 years of age, all ages are affected.2,3 Frequency of migraine headache is variable; chronic migraineurs experience more than 15 headache days a month.
Recent estimates indicate that the cost of acute and chronic migraine headaches reaches approximately $78 million a year in the United States. 4 This high burden of disease has made effective migraine treatment options absolutely essential. Recent advances in our understanding of migraine pathophysiology have led to new therapeutic targets; there are now many novel treatment approaches on the horizon.
In this article, we review the diagnosis and management of migraine in detail. Our emphasis is on evidence-based approaches to acute and prophylactic treatment, including tried-and-true options and newly emerging therapies.
Neuronal dysfunction and a genetic predisposition
Although migraine was once thought to be caused by abnormalities of vasodilation, current research suggests that the disorder has its origins in primary neuronal dysfunction. There appears to be a genetic predisposition toward widespread neuronal hyperexcitability in migraineurs.5 In addition, hypothalamic neurons are thought to initiate migraine by responding to changes in brain homeostasis. Increased parasympathetic tone might activate meningeal pain receptors or lower the threshold for transmitting pain signals from the thalamus to the cortex.6
Prodromal symptoms and aura appear to originate from multiple areas across the brain, including the hypothalamus, cortex, limbic system, and brainstem. This widespread brain involvement might explain why some headache sufferers concurrently experience a variety of symptoms, including fatigue, depression, muscle pain, and an abnormal sensitivity to light, sound, and smell.6,7
Although the exact mechanisms behind each of these symptoms have yet to be defined precisely, waves of neuronal depolarization—known as cortical spreading depression—are suspected to cause migraine aura.8-10 Cortical spreading depression activates the trigeminal pain pathway and leads to the release of pro-inflammatory markers such as calcitonin gene-related protein (CGRP).6 A better understanding of these complex signaling pathways has helped provide potential therapeutic targets for new migraine drugs.
Diagnosis: Close patient inquiry is most helpful
The International Headache Society (IHS) criteria for primary headache disorders serve as the basis for the diagnosis of migraine and its subtypes, which include migraine without aura and migraine with aura. Due to variability of presentation, migraine with aura is further subdivided into migraine with typical aura (with and without headache), migraine with brainstem aura, hemiplegic migraine, and retinal migraine.11
Continue to: How is migraine defined?
How is migraine defined? Simply, migraine is classically defined as a unilateral, pulsating headache of moderate to severe intensity lasting 4 to 72 hours, associated with photophobia and phonophobia or nausea and vomiting, or both.11 Often visual in nature, aura is a set of neurologic symptoms that lasts for minutes and precedes the onset of the headache. The visual aura is often described as a scintillating scotoma that begins near the point of visual fixation and then spreads left or right. Other aura symptoms include tingling or numbness (second most common), speech disturbance (aphasia), motor changes and, in rare cases, a combination of these in succession. By definition, all of these symptoms fully resolve between attacks.11
Validated valuable questionnaires. To help with accurate and timely diagnosis, researchers have developed and validated simplified questionnaires that can be completed independently by patients presenting to primary care (TABLE 112,13):
- ID Migraine is a set of 3 questions that scores positive when a patient endorses at least 2 of the 3 symptoms. 12
- MS-Q is similar to the ID Migraine but includes 5 items. A score of ≥4 is a positive screen. 13
The sensitivity and specificity of MS-Q (0.93 and 0.81, respectively) are slightly higher than those of ID Migraine (0.81 and 0.75).13
Remember POUND. This mnemonic device can also be used during history-taking to aid in diagnostic accuracy. Migraine is highly likely (92%) in patients who endorse 4 of the following 5 symptoms and unlikely (17%) in those who endorse ≤2 symptoms14: Pulsatile quality of headache 4 to 72 hOurs in duration, Unilateral location, Nausea or vomiting, and Disabling intensity.
Differential Dx. Although the differential diagnosis of headache is broad (TABLE 214,15), the history alone can often guide clinicians towards the correct assessment. After taking the initial history (headache onset, location, duration, and associated symptoms), focus your attention on assessing the risk of intracranial pathology. This is best accomplished by assessing specific details of the history (TABLE 314) and findings on physical examination15:
- blood pressure measurement (seated, legs uncrossed, feet flat on the floor; having rested for 5 minutes; arm well supported)
- cranial nerve exam
- extremity strength testing
- eye exam (vision, extra-ocular muscles, visual fields, pupillary reactivity, and funduscopic exam)
- gait (tandem walk)
- reflexes.
Continue to: Further testing needed?
Further testing needed? Neuroimaging should be considered only in patients with an abnormal neurologic exam, atypical headache features, or certain risk factors, such as an immune deficiency. There is no role for electroencephalography or other diagnostic testing in migraine.16
Take a multipronged approach to treatment
As with other complex, chronic conditions, the treatment of migraine should take a multifaceted approach, including management of acute symptoms as well as prevention of future headaches. In 2015, the American Headache Society published a systematic review that specified particular treatment goals for migraine sufferers. 17 These goals include:
- headache reduction
- headache relief
- decreased disability from headache
- elimination of nausea and vomiting
- elimination of photophobia and phonophobia.
Our review, which follows, of therapeutic options focuses on the management of migraine in adults. Approaches in special populations (older adults, pregnant women, and children) are discussed afterward.
Pharmacotherapy for acute migraine
Acute migraine should be treated with an abortive medication at the onset of headache. The immediate goal is to relieve pain within 2 hours and prevent its recurrence within the subsequent 48 hours (TABLE 412,18-20).
In the general population, mild, infrequent migraines can be managed with acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs).21
Continue to: For moderate-to-severe migraine...
For moderate-to-severe migraine, triptans, which target serotonin receptors, are the drug of choice for most patients.21 Triptans are superior to placebo in achieving a pain-free state at 2 and 24 hours after administration; eletriptan has the most desirable outcome, with 68% of patients pain free at 2 hours and 54% pain free at 24 hours.22 Triptans are available as sublingual tablets and nasal sprays, as well as subcutaneous injections for patients with significant associated nausea and vomiting. Avoid prescribing triptans for patients with known vascular disease (eg, history of stroke, myocardial infarction, peripheral vascular disease, uncontrolled hypertension, or signs and symptoms of these conditions), as well as for patients with severe hepatic impairment.
Importantly, although triptans all have a similar mechanism of action, patients might respond differently to different drugs within the class. If a patient does not get adequate headache relief from an appropriate dosage of a given triptan during a particular migraine episode, a different triptan can be tried during the next migraine.22 Additionally, if a patient experiences an adverse effect from one triptan, this does not necessarily mean that a trial of another triptan at a later time is contraindicated.
For patients who have an incomplete response to migraine treatment or for those with frequent recurrence, the combination formulation of sumatriptan, 85 mg, and naproxen, 500 mg, showed the highest rate of resolution of headache within 2 hours compared with either drug alone.23 A similar result might be found by combining a triptan known to be effective for a patient and an NSAID other than naproxen. If migraine persists despite initial treatment of an attack, a different class of medication should be tried during the course of that attack to attain relief of symptoms of that migraine.21
When a patient is seen in an acute care setting (eg, emergency department, urgent care center) while suffering a migraine, additional treatment options are available. Intravenous (IV) anti-emetics are useful for relieving the pain of migraine and nausea, and can be used in combination with an IV NSAID (eg, ketorolac).21 The most effective anti-emetics are dopamine receptor type-2 blockers, including chlorpromazine, droperidol, metoclopramide, and prochlorperazine, which has the highest level of efficacy.24 Note that these medications do present the risk of a dystonic reaction; diphenhydramine is therefore often used in tandem to mitigate such a response.
Looking ahead. Although triptans are the current first-line therapy for acute migraine, their effectiveness is limited. Only 20% of patients report sustained relief of pain in the 2 to 24 hours after treatment, and the response can vary from episode to episode.25
Continue to: With better understading of the pathophysiology of migraine...
With better understanding of the pathophysiology of migraine, a host of novel anti-migraine drugs are on the horizon.
CGRP receptor antagonists. The neuropeptide CGRP, which mediates central and peripheral nervous system pain signaling, has been noted to be elevated during acute migraine attacks26; clinical trials are therefore underway to evaluate the safety and efficacy of CGRP receptor antagonists.18 These agents appear to be better tolerated than triptans, have fewer vascular and central nervous system adverse effects, and present less of a risk of medication overuse headache.18 Liver toxicity has been seen with some medications in this class and remains an important concern in their development.19
Phase 3 clinical trials for 1 drug in this class, ubrogepant, were completed in late 2017; full analysis of the data is not yet available. Primary outcomes being evaluated include relief of pain at 2 hours and relief from the most bothersome symptoms again at 2 hours.27
Selective serotonin-HT1f receptor agonists, such as lasmiditan, offer another potential approach. Although the exact mechanism of action of these agents is not entirely clear, clinical trials have supported their efficacy and safety.20 Importantly, ongoing trials are specifically targeting patients with known cardiovascular risk factors because they are most likely to benefit from the nonvasoconstrictive mechanism of action.28,29 Adverse effects reported primarily include dizziness, fatigue, and vertigo.
Strategies for managing recurrent episodic migraine
Because of the risk of medication overuse headache with acute treatment, daily preventive therapy for migraine is indicated for any patient with 30 :
- ≥6 headache days a month
- ≥4 headache days a month with some impairment
- ≥3 headache days a month with severe impairment.
Continue to: Treatment begins by having patients identify...
Treatment begins by having patients identify, and then avoid, migraine triggers (TABLE 5). This can be accomplished by having patients keep a headache diary, in which they can enter notations about personal and environmental situations that precede a headache.
For the individual patient, some triggers are modifiable; others are not. Helping a patient develop strategies for coping with triggers, rather than aiming for complete avoidance, might help her (him) manage those that are inescapable (eg stress, menstruation, etc).31 For many patients, however, this is not an adequate intervention and other approaches must be explored. When considering which therapy might be best for a given patient, evaluate her (his) comorbidities and assess that particular treatment for potential secondary benefits and the possibility of adverse effects. Pay attention to the choice of preventive therapy in women who are considering pregnancy because many available treatments are potentially teratogenic.
Oral medications. Oral agents from several classes of drugs can be used for migraine prophylaxis, including anti-epileptics,antidepressants, and antihypertensives (TABLE 620,29,30,32-41). Selected anti-epileptics (divalproex sodium, sodium valproate, topiramate) and beta-blockers (metoprolol, propranolol, and timolol) have the strongest evidence to support their use.32 Overall, regular use of prophylactic medications can reduce headache frequency by 50% for approximately 40% to 45% of patients who take them.29 However, adherence may be limited by adverse effects or perceived lack of efficacy, thus reducing their potential for benefit.42
OnabotulinumtoxinA. In patients with chronic migraine (≥15 headache days a month for at least 3 months) who have failed oral medications, the American Academy of Neurology (AAN) recommends the use of onabotulinumtoxinA.30 The treatment regimen comprises 31 injections at various sites on the head, neck, and shoulders every 3 months.33
A 2010 large randomized controlled trial showed a decrease in the frequency of headache days for patients receiving onabotulinumtoxinA compared to placebo after a 24-week treatment period (7.8 fewer headache days a month, compared to 6.4 fewer in the placebo group).33 A recent systematic review also noted a reduction of 2 headache days a month compared with placebo; the authors cautioned, however, that data with which to evaluate onabotulinumtoxinA in comparison to other prophylactic agents are limited.43
Continue to: In both studies...
In both studies, the risk of adverse drug events due to onabotulinumtoxinA was high and led to a significant rate of discontinuation.33,43 Despite this, onabotulinumtoxinA remains the only Food and Drug Administration (FDA)–approved treatment for chronic migraine, making it reasonable to consider for appropriate patients.
Acupuncture. A 2016 Cochrane review found benefit for patients using acupuncture compared with sham acupuncture.34 When acupuncture was compared with prophylactic agents such as beta-blockers, calcium-channel blockers, and anti-epileptics, however, there was no significant difference between the procedure and pharmacotherapy. Patients willing and able to try acupuncture might see a reduction in the overall number of headaches. Acupuncture has few adverse effects; however, long-term data are lacking.34
Exercise is not supported by robust data for its role as a prophylactic treatment. It is generally considered safe in most populations, however, and can be pursued with little out-of-pocket cost.35
Cognitive behavioral therapy (CBT). The AAN recommends CBT, relaxation therapy, and biofeedback therapy. Accessibility of these services remains limited for many patients, and cost can be prohibitive.16
Supplements used to help prevent migraine include the root of Petasites hybridus (butterbur), magnesium, vitamin B2 (riboflavin), Tanacetum parthenium (feverfew), and coenzyme Q10.16 Although the strength of evidence for these therapies is limited by small trials, their overall risk of adverse effects is low, and they might be easier for patients to obtain than acupuncture or CBT.
Continue to: Butterbur, in particular...
Butterbur, in particular, has been found to be beneficial for migraine prevention in 2 small placebo-controlled trials. In a randomized controlled study of 245 patients P hybridus, (specifically, the German formulation, Petadolex), 75 mg BID, reduced the frequency of migraine attack by 48% at 4 months, compared to placebo (number needed to treat, 5.3).44 No difference was found at lower dosages. The most common reported adverse effect was burping.
Regrettably, unpurified butterbur extract contains pyrrolizidine alkaloids, potentially hepatotoxic and carcinogenic compounds. Because of variations in purification in production facilities in the United States, butterbur supplements might not have all of these compounds removed—and so should be used with caution.41
Magnesium. Studies evaluating the use of magnesium have demonstrated varied results; differences in methods and dosing have limited broad application of findings. As with most supplements considered for prophylactic treatment, magnesium dosing is poorly understood, and bioavailability varies in its different forms. Oral supplementation can be given as magnesium dicitrate, 600 mg/d.45
Recently, products containing various combinations of feverfew, coenzyme Q10, riboflavin, magnesium, and other supplements have shown benefit in early clinical trials.36,37
Neural stimulation. Over the past few years, a variety of transcutaneous nerve stimulator devices have gained FDA approval for use in migraine prophylaxis. The long-term safety and efficacy of these devices is not yet well understood, but they appear to provide headache relief in the short term and decrease the frequency of headache.38 Use of the noninvasive stimulators is limited today by high cost and poor coverage by US health care insurers.
Continue to: Newly available medical therapy
Newly available medical therapy. The FDA recently approved erenumab, a fully human monoclonal antibody for prevention of migraine in adults. This is the first drug in the CGRP antagonist class to be approved for this indication. Trials of this once-monthly, self-injectable drug show promising results for patients whose migraines have been refractory to other therapies.
A recent large trial evaluated 955 adults with migraine, randomizing them to receive erenumab, 70 mg; erenumab, 140 mg; or placebo over 28 weeks.39 The groups receiving erenumab had a nearly 2-fold higher odds of having their migraine reduced by 50%, compared with placebo (number needed to treat with the 140-mg dose, 4.27). Similar numbers of participants from all groups discontinued the study.39 Phase 3 trials that are not yet formally published have produced similarly beneficial results.40,46 The FDA has listed injection site reaction and constipation as the most reported adverse effects.40
Three other anti-CGRP antibodies are likely to be approved in the near future: fremanezumab, galcanezumab, and eptinezumab.
The approach to migraine in special populations
Management of acute and chronic migraine in children, pregnant women, and older adults requires special attention: Treatment approaches are different than they are for adults 19 to 65 years of age.
Pediatric patients. Migraine is the most common acute and recurrent headache syndrome in children. Headaches differ from those of adult migraine as a result of variations in brain maturation, plasticity, and cognitive development.47 Migraine attacks are often of shorter duration in children, lasting 1 to 2 hours, but can still be preceded by visual aura.48 Just as with adults, imaging, electroencephalography, lumbar puncture, and routine labs should be considered only if a child has an abnormal neurological exam or other concerning features (TABLE 214,15).
Continue to: The general approach to migraine treatment...
The general approach to migraine treatment in the pediatric population includes education of the child and family about symptom management. Acetaminophen, NSAIDs, and triptans are approved for abortive therapy in children and should be used for acute headache relief in the same way that they are used in adults. Oral rizatriptan, the most well studied triptan in the pediatric population, is approved for use in children as young as 6 years49; the pediatric dosage is 5 mg/d for patients weighing 20 to 39 kg and 10 mg/d for patients weighing more than 40 kg (same as the adult dosage).
Oral almotriptan and zolmitriptan are also approved for use in children 12 to 17 years of age. Usual dosages are: almotriptan, 12.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 25 mg/d); and zolmitriptan, 2.5 mg at onset, can repeat in 2 hours as needed (maximum dosage, 10 mg/d).50
For children who are unable to swallow pills or who are vomiting, a non-oral route of administration is preferable. Rizatriptan is available as an orally disintegrating tablet. Zolmitriptan is available in a nasal spray at a dose of 5 mg for children 12 years and older. Sumatriptan is not approved for use in patients younger than 18 years; however, recent studies have shown that it might have good efficacy and tolerability.50
Daily prophylactic treatment for recurrent migraine in the pediatric population is an evolving subject; published guidelines do not exist. It is reasonable to consider treatment using the same guidelines as those in place for adults.51 Topiramate, 1 to 2 mg/kg/d, is the only therapy approved by the FDA for episodic migraine preventive therapy in adolescents.50
Notably, a nonpharmacotherapeutic approach may be more effective for pediatric prevention. In 2017, a large double-blind, placebo-controlled trial investigated the use of amitriptyline, topiramate, and placebo for the treatment of recurrent migraine in children 8 to 17 years of age. An interim analysis of the 328 children enrolled found no significant differences in reduction of headache frequency with treatment compared with placebo over a 24-week period; the trial was stopped early due to futility.52
Continue to: The study did show...
The study did show, however, that reducing migraine triggers provided a high level of benefit to study participants. Stress is one of the most common migraine triggers in children; lack of sleep, exposure to a warm climate, and exposure to video games are also notable triggers.53 CBT may augment the efficacy of standard migraine medications in the pediatric population and may help prevent recurrence of episodes.54
Pregnancy. The treatment of migraine is different in pregnant women than it is in nonpregnant adults because of a concern over adverse effects on fetal development. For acute headache treatment, first-line therapies include trigger avoidance and acetaminophen, 1000 mg (maximum dosage, 4000 mg/d).55 If this is ineffective, a 10-mg dose of metoclopramide, as often as every 6 hours (not an FDA-approved indication), can be considered. During the second trimester, NSAIDs can be considered second-line therapy.
Triptans—specifically, sumatriptan and rizatriptan—can also be considered if first-line therapies fail.56 Triptan-exposed pregnant women with migraine have a rate of congenital malformations, spontaneous abortions, and prematurity that is similar to what is seen in pregnant women with migraine who have not been exposed to triptans. However, when triptan-exposed women are compared with healthy, non-migraine-suffering women, the rate of spontaneous abortion appears to be increased in the triptan-exposed population.57
Ergotamine is contraindicated during pregnancy because of its potential to induce uterine contractions and vasospasm, which can be detrimental to the fetus.56Nonpharmacotherapeutic interventions such as heat, ice, massage, rest, and avoidance of triggers are as successful in the pregnant population as in the nonpregnant population. For migraine prevention, coenzyme Q10, vitamins B2 and B6 (pyridoxine), and oral magnesium can be considered. Feverfew and butterbur should be avoided because of concerns about fetal malformation and preterm labor.58
Older adults. Choosing appropriate migraine therapy for older adults requires special consideration because of changes in drug metabolism and risks associated with drug adverse effects. Additionally, few studies of migraine drugs have included large populations of adults older than 65 years; medications should therefore be prescribed cautiously in this population, with particular attention to drug–drug interactions.
Continue to: Just as for younger adults...
Just as for younger adults, mild symptoms can be managed effectively with acetaminophen. NSAIDs may be used as well, but carry increased risks of gastric bleeding and elevation in blood pressure.59 The use of triptans is acceptable for the appropriate patient, but should be avoided in patients with known vascular disease.60 Antiemetics present an increased risk of extrapyramidal adverse effects in the elderly and should be used with caution at the lowest effective dosage.59 Novel mechanisms of action make some of the newer agents potentially safer for use in older adults when treating acute migraine.
For migraine prevention in older adults, particular attention should be paid to reducing triggers and minimizing polypharmacy.
More and more, successful treatment is within reach
With many clinical trials evaluating novel drugs underway, and additional studies contributing to our understanding of nonpharmacotherapeutic approaches to migraine treatment, improved headache control may become increasingly common over the next few years.
CORRESPONDENCE
Kathryn McGrath, MD, Department of Family and Community Medicine, Thomas Jefferson University, 1015 Walnut St, Philadelphia PA 19107; [email protected].
1. Stokes M, Becker WJ, Lipton RB, et al. Cost of health care among patients with chronic and episodic migraine in Canada and the USA: results from the International Burden of Migraine Study (IBMS). Headache. 2011;51:1058-1077.
2. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
3. Burch RC, Loder S, Loder E, et al. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache. 2015;55:21-34.
4. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81:479-484.
5. Ferrari MD, Klever RR, Terwindt GM, et al. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14:65-80.
6. Burstein R, Noseda R, Borsook D. Migraine: multiple processes, complex pathophysiology. J Neurosc. 2015;35:6619-6629.
7. Maniyar FH, Sprenger T, Monteith T, et al. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. 2013;137(Pt 1):232-241.
8. Cutrer FM, Sorensen AG, Weisskoff RM, et al. Perfusion‐weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25-31.
9. Hadjikhani N, Sanchez Del Rio MS, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687-4692.
10. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol. 2013;75:365-391.
11. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, (beta version). Cephalalgia. 2013;33:629-808.
12. Lipton RB, Dodick D, Sadovsky RE, et al; ID Migraine validation study. A self-administered screener for migraine in primary care: The ID Migraine™ validation study. Neurology. 2003;61:375-382.
13. Láinez MJ, Domínguez M, Rejas J, et al. Development and validation of the Migraine Screen Questionnaire (MS‐Q). Headache. 2005;45:1328-1338.
14. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006;296:1274-1283.
15. Becker WJ, Findlay T, Moga C, et al. Guideline for primary care management of headache in adults. Can Fam Physician. 2015;61:670-679.
16. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
17. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55:3-20.
18. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887-898.
19. Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533-552.
20. Färkkilä M, Diener HC, Géraud G, et al; COL MIG-202 study group. Efficacy and tolerability of lasmiditan, an oral 5-HT(1F) receptor agonist, for the acute treatment of migraine: a phase 2 randomised, placebo-controlled, parallel-group, dose-ranging study. Lancet Neurol. 2012;11:405-413.
21. Pringsheim T, Davenport WJ, Marmura MJ, et al. How to apply the AHS evidence assessment of the acute treatment of migraine in adults to your patient with migraine. Headache. 2016;56:1194-1200.
22. Thorlund K, Mills EJ, Wu P, et al. Comparative efficacy of triptans for the abortive treatment of migraine: a multiple treatment comparison meta-analysis. Cephalalgia. 2014;34:258-267.
23. Law S, Derry S, Moore RA. Sumatriptan plus naproxen for acute migraine attacks in adults. Cochrane Database Syst Rev. 2013;(10):CD008541.
24. Orr SL, Aubé M, Becker WJ, et al. Canadian Headache Society systematic review and recommendations on the treatment of migraine pain in emergency settings. Cephalalgia. 2015;35:271-284.
25. Ferrari MD, Goadsby PJ, Roon KI, et al. Triptans (serotonin, 5‐HT1B/1D agonists) in migraine: detailed results and methods of a meta‐analysis of 53 trials. Cephalalgia. 2002;22:633-658.
26. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48-56.
27. A phase 3, multicenter, randomized, double-blind, placebo-controlled single attack study to evaluate the efficacy, safety, and tolerability of oral ubrogepant in the acute treatment of migraine. https://clinicaltrials.gov/ct2/show/study/NCT02828020. Accessed November 16, 2018.
28. Rubio-Beltrán E, Labastida-Ramírez A, Villalón CM, et al. Is selective 5-HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. 2018;186:88-97.
29. Diener HC, Charles A, Goadsby PJ, et al. New therapeutic approaches for the prevention and treatment of migraine. Lancet Neurol. 2015;14:1010-1022.
30. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention. Headache. 2015;55 Suppl 2:103-122.
31. Martin PR. Behavioral management of migraine headache triggers: learning to cope with triggers. Curr Pain Headache Rep. 2010;14:221-227.
32. Loder E, Burch R, Rizzoli P. The 2012 AHS/AAN guidelines for prevention of episodic migraine: a summary and comparison with other recent clinical practice guidelines. Headache. 2012;52:930-945.
33. Dodick DW, Turkel CC, DeGryse RE, et al; PREEMPT Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double‐blind, randomized, placebo‐controlled phases of the PREEMPT clinical program. Headache. 2010;50:921-936.
34. Linde K, Allais G, Brinkhaus B, et al. Acupuncture for the prevention of episodic migraine. Cochrane Database Syst Rev. 2016(6):CD001218.
35. Varkey E, Cider Å, Carlsson J, et al. Exercise as migraine prophylaxis: a randomized study using relaxation and topiramate as controls. Cephalalgia. 2011;31:1428-1438.
36. Guilbot A, Bangratz M, Abdellah SA, et al. A combination of coenzyme Q10, feverfew and magnesium for migraine prophylaxis: a prospective observational study. BMC Complement Altern Med. 2017;17:433.
37. Dalla Volta G, Zavarize P, Ngonga G, et al. Combination of Tanacethum partenium, 5-hydrossitriptophan (5-Http) and magnesium in the prophylaxis of episodic migraine without aura (AURASTOP®) an observational study. Int J Neuro Brain Dis. 2017;4:1-4.
38. Puledda F, Goadsby PJ. An update on non‐pharmacological neuromodulation for the acute and preventive treatment of migraine. Headache. 2017;57:685-691.
39. Goadsby PJ, Reuter U, Hallström Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123-2132.
40. Reuter U. Efficacy and safety of erenumab in episodic migraine patients with 2-4 prior preventive treatment failures: Results from the Phase 3b LIBERTY study. Abstract 009, AAN 2018 Annual Meeting; April 24, 2018.
41. Diener HC, Freitag FG, Danesch U. Safety profile of a special butterbur extract from Petasites hybridus in migraine prevention with emphasis on the liver. Cephalalgia Reports. https://journals.sagepub.com/doi/10.1177/2515816318759304. 2018 May 2. Accessed December 15, 2018.
42. Kingston WS, Halker R. Determinants of suboptimal migraine diagnosis and treatment in the primary care setting. J Clin Outcomes Manag. 2017;24:319-324.
43. Herd CP, Tomlinson CL, Rick C, et al. Botulinum toxins for the prevention of migraine in adults. Cochrane Database of Syst Rev. 2018;6:CD011616.
44. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
45. Von Luckner A, Riederer F. Magnesium in migraine prophylaxis—is there an evidence‐based rationale? A systematic review. Headache. 2018;58:199-209.
46. Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16:425-434.
47. Sonal Sekhar M, Sasidharan S, Joseph S, et al. Migraine management: How do the adult and paediatric migraines differ? Saudi Pharm J. 2012;20:1-7.
48. Lewis DW. Pediatric migraine. In: Lewis DW. Clinician’s Manual on Treatment of Pediatric Migraine. London, UK: Springer Healthcare Ltd; 2010:15-26.
49. Ho TW, Pearlman E, Lewis D, et al. Efficacy and tolerability of rizatriptan in pediatric migraineurs: results from a randomized double-blind, placebo controlled trial using a novel adaptive enrichment design. Cephalagia. 2012;32:750-765.
50. Khrizman M, Pakalnis A. Management of pediatric migraine: current therapies. Pediatr Ann. 2018;47:e55-e60.
51. Lipton RB, Bigal ME, Diamond M, et al; AMPP Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
52. Powers SW, Coffey CS, Chamberlin LA, et al; CHAMP Investigators. Trial of amitriptyline, topiramate, and placebo for pediatric migraine. N Engl J Med. 2017;376:115-124.
53. Neut D, Fily A, Cuvellier JC, et al. The prevalence of triggers in paediatric migraine: a questionnaire study in 102 children and adolescents. J Headache Pain. 2012;13:61-65.
54. Ng QX, Venkatanarayanan N, Kumar L. A systematic review and meta‐analysis of the efficacy of cognitive behavioral therapy for the management of pediatric migraine. Headache. s2017;57:349-362.
55. Lipton RB, Baggish JS, Stewart WF, et al. Efficacy and safety of acetaminophen in the treatment of migraine: results of a randomized, double-blind, placebo-controlled, population-based study. Arch Intern Med. 2000;160:3486-3492.
56. Lucas S. Medication use in the treatment of migraine during pregnancy and lactation. Curr Pain Headache Rep. 2009;13:392-398.
57. Marchenko A, Etwel F, Olutunfesse O, et al. Pregnancy outcome following prenatal exposure to triptan medications: a meta-analysis. Headache. 2015:55:490-501.
58. Wells RE, Turner DP, Lee M, et al. Managing migraine during pregnancy and lactation. Curr Neurol Neurosci Rep. 2016;16:40.
59. Haan J, Hollander J, Ferrari MD. Migraine in the elderly: a review. Cephalalgia. 2007;27:97-106.
60. Gladstone JP, Eross EJ, Dodick DW. Migraine in special populations. Treatment strategies for children and adolescents, pregnant women, and the elderly. Postgrad Med. 2004;115:39-44,47-50.
1. Stokes M, Becker WJ, Lipton RB, et al. Cost of health care among patients with chronic and episodic migraine in Canada and the USA: results from the International Burden of Migraine Study (IBMS). Headache. 2011;51:1058-1077.
2. Smitherman TA, Burch R, Sheikh H, et al. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013;53:427-436.
3. Burch RC, Loder S, Loder E, et al. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache. 2015;55:21-34.
4. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81:479-484.
5. Ferrari MD, Klever RR, Terwindt GM, et al. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14:65-80.
6. Burstein R, Noseda R, Borsook D. Migraine: multiple processes, complex pathophysiology. J Neurosc. 2015;35:6619-6629.
7. Maniyar FH, Sprenger T, Monteith T, et al. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. 2013;137(Pt 1):232-241.
8. Cutrer FM, Sorensen AG, Weisskoff RM, et al. Perfusion‐weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25-31.
9. Hadjikhani N, Sanchez Del Rio MS, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687-4692.
10. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol. 2013;75:365-391.
11. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, (beta version). Cephalalgia. 2013;33:629-808.
12. Lipton RB, Dodick D, Sadovsky RE, et al; ID Migraine validation study. A self-administered screener for migraine in primary care: The ID Migraine™ validation study. Neurology. 2003;61:375-382.
13. Láinez MJ, Domínguez M, Rejas J, et al. Development and validation of the Migraine Screen Questionnaire (MS‐Q). Headache. 2005;45:1328-1338.
14. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006;296:1274-1283.
15. Becker WJ, Findlay T, Moga C, et al. Guideline for primary care management of headache in adults. Can Fam Physician. 2015;61:670-679.
16. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
17. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55:3-20.
18. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887-898.
19. Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533-552.
20. Färkkilä M, Diener HC, Géraud G, et al; COL MIG-202 study group. Efficacy and tolerability of lasmiditan, an oral 5-HT(1F) receptor agonist, for the acute treatment of migraine: a phase 2 randomised, placebo-controlled, parallel-group, dose-ranging study. Lancet Neurol. 2012;11:405-413.
21. Pringsheim T, Davenport WJ, Marmura MJ, et al. How to apply the AHS evidence assessment of the acute treatment of migraine in adults to your patient with migraine. Headache. 2016;56:1194-1200.
22. Thorlund K, Mills EJ, Wu P, et al. Comparative efficacy of triptans for the abortive treatment of migraine: a multiple treatment comparison meta-analysis. Cephalalgia. 2014;34:258-267.
23. Law S, Derry S, Moore RA. Sumatriptan plus naproxen for acute migraine attacks in adults. Cochrane Database Syst Rev. 2013;(10):CD008541.
24. Orr SL, Aubé M, Becker WJ, et al. Canadian Headache Society systematic review and recommendations on the treatment of migraine pain in emergency settings. Cephalalgia. 2015;35:271-284.
25. Ferrari MD, Goadsby PJ, Roon KI, et al. Triptans (serotonin, 5‐HT1B/1D agonists) in migraine: detailed results and methods of a meta‐analysis of 53 trials. Cephalalgia. 2002;22:633-658.
26. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48-56.
27. A phase 3, multicenter, randomized, double-blind, placebo-controlled single attack study to evaluate the efficacy, safety, and tolerability of oral ubrogepant in the acute treatment of migraine. https://clinicaltrials.gov/ct2/show/study/NCT02828020. Accessed November 16, 2018.
28. Rubio-Beltrán E, Labastida-Ramírez A, Villalón CM, et al. Is selective 5-HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. 2018;186:88-97.
29. Diener HC, Charles A, Goadsby PJ, et al. New therapeutic approaches for the prevention and treatment of migraine. Lancet Neurol. 2015;14:1010-1022.
30. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention. Headache. 2015;55 Suppl 2:103-122.
31. Martin PR. Behavioral management of migraine headache triggers: learning to cope with triggers. Curr Pain Headache Rep. 2010;14:221-227.
32. Loder E, Burch R, Rizzoli P. The 2012 AHS/AAN guidelines for prevention of episodic migraine: a summary and comparison with other recent clinical practice guidelines. Headache. 2012;52:930-945.
33. Dodick DW, Turkel CC, DeGryse RE, et al; PREEMPT Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double‐blind, randomized, placebo‐controlled phases of the PREEMPT clinical program. Headache. 2010;50:921-936.
34. Linde K, Allais G, Brinkhaus B, et al. Acupuncture for the prevention of episodic migraine. Cochrane Database Syst Rev. 2016(6):CD001218.
35. Varkey E, Cider Å, Carlsson J, et al. Exercise as migraine prophylaxis: a randomized study using relaxation and topiramate as controls. Cephalalgia. 2011;31:1428-1438.
36. Guilbot A, Bangratz M, Abdellah SA, et al. A combination of coenzyme Q10, feverfew and magnesium for migraine prophylaxis: a prospective observational study. BMC Complement Altern Med. 2017;17:433.
37. Dalla Volta G, Zavarize P, Ngonga G, et al. Combination of Tanacethum partenium, 5-hydrossitriptophan (5-Http) and magnesium in the prophylaxis of episodic migraine without aura (AURASTOP®) an observational study. Int J Neuro Brain Dis. 2017;4:1-4.
38. Puledda F, Goadsby PJ. An update on non‐pharmacological neuromodulation for the acute and preventive treatment of migraine. Headache. 2017;57:685-691.
39. Goadsby PJ, Reuter U, Hallström Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123-2132.
40. Reuter U. Efficacy and safety of erenumab in episodic migraine patients with 2-4 prior preventive treatment failures: Results from the Phase 3b LIBERTY study. Abstract 009, AAN 2018 Annual Meeting; April 24, 2018.
41. Diener HC, Freitag FG, Danesch U. Safety profile of a special butterbur extract from Petasites hybridus in migraine prevention with emphasis on the liver. Cephalalgia Reports. https://journals.sagepub.com/doi/10.1177/2515816318759304. 2018 May 2. Accessed December 15, 2018.
42. Kingston WS, Halker R. Determinants of suboptimal migraine diagnosis and treatment in the primary care setting. J Clin Outcomes Manag. 2017;24:319-324.
43. Herd CP, Tomlinson CL, Rick C, et al. Botulinum toxins for the prevention of migraine in adults. Cochrane Database of Syst Rev. 2018;6:CD011616.
44. Lipton RB, Göbel H, Einhäupl KM, et al. Petasites hybridus root (butterbur) is an effective preventive treatment for migraine. Neurology. 2004;63:2240-2244.
45. Von Luckner A, Riederer F. Magnesium in migraine prophylaxis—is there an evidence‐based rationale? A systematic review. Headache. 2018;58:199-209.
46. Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16:425-434.
47. Sonal Sekhar M, Sasidharan S, Joseph S, et al. Migraine management: How do the adult and paediatric migraines differ? Saudi Pharm J. 2012;20:1-7.
48. Lewis DW. Pediatric migraine. In: Lewis DW. Clinician’s Manual on Treatment of Pediatric Migraine. London, UK: Springer Healthcare Ltd; 2010:15-26.
49. Ho TW, Pearlman E, Lewis D, et al. Efficacy and tolerability of rizatriptan in pediatric migraineurs: results from a randomized double-blind, placebo controlled trial using a novel adaptive enrichment design. Cephalagia. 2012;32:750-765.
50. Khrizman M, Pakalnis A. Management of pediatric migraine: current therapies. Pediatr Ann. 2018;47:e55-e60.
51. Lipton RB, Bigal ME, Diamond M, et al; AMPP Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
52. Powers SW, Coffey CS, Chamberlin LA, et al; CHAMP Investigators. Trial of amitriptyline, topiramate, and placebo for pediatric migraine. N Engl J Med. 2017;376:115-124.
53. Neut D, Fily A, Cuvellier JC, et al. The prevalence of triggers in paediatric migraine: a questionnaire study in 102 children and adolescents. J Headache Pain. 2012;13:61-65.
54. Ng QX, Venkatanarayanan N, Kumar L. A systematic review and meta‐analysis of the efficacy of cognitive behavioral therapy for the management of pediatric migraine. Headache. s2017;57:349-362.
55. Lipton RB, Baggish JS, Stewart WF, et al. Efficacy and safety of acetaminophen in the treatment of migraine: results of a randomized, double-blind, placebo-controlled, population-based study. Arch Intern Med. 2000;160:3486-3492.
56. Lucas S. Medication use in the treatment of migraine during pregnancy and lactation. Curr Pain Headache Rep. 2009;13:392-398.
57. Marchenko A, Etwel F, Olutunfesse O, et al. Pregnancy outcome following prenatal exposure to triptan medications: a meta-analysis. Headache. 2015:55:490-501.
58. Wells RE, Turner DP, Lee M, et al. Managing migraine during pregnancy and lactation. Curr Neurol Neurosci Rep. 2016;16:40.
59. Haan J, Hollander J, Ferrari MD. Migraine in the elderly: a review. Cephalalgia. 2007;27:97-106.
60. Gladstone JP, Eross EJ, Dodick DW. Migraine in special populations. Treatment strategies for children and adolescents, pregnant women, and the elderly. Postgrad Med. 2004;115:39-44,47-50.
PRACTICE RECOMMENDATIONS
› Offer treatment with a triptan to adult patients with moderate-to-severe episodic migraine. A
› Consider prescribing topiramate, divalproex sodium, metoprolol, propranolol, or the herbal, Petasites hybridum, for the prevention of recurrent episodic migraine that has not responded to a reduction in headache triggers. A
› Add onabotulinumtoxinA injection to your therapeutic toolbox as an effective preventive treatment for chronic migraine (≥15 headache days a month for 3 months). B
› Recommend magnesium and feverfew as adjunctive preventive treatments for migraine. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Chemo-resistant reserve HSCs maintained in bone marrow
A small subpopulation of quiescent hematopoietic stem cells (HSCs) that restores the HSC pool and supports hematopoietic regeneration after chemotherapy has been identified, researchers reported.

These “reserve” HSCs were resistant to chemotherapy, whereas “primed” HSCs were sensitive to DNA damage from chemotherapy, the researchers said.
Both reserve and active stem cells were maintained in the bone marrow by specific niches, researchers demonstrated in experiments described in detail in Cell Reports.
Of note, primitive reserve HSCs were maintained in a bone marrow region that enriches N-cadherin expressing (N-cad+) bone and marrow stromal progenitor cells. According to investigators, those N-cad+ cells protected primitive reserve HSCs from chemotherapy-related stress, which survived to support hematopoietic regeneration after myeloablation.
These findings advance understanding of HSC biology, and could open new avenues for treating blood diseases and autoimmune disorders.
“In effect, we showed that hematopoietic stem cells have functionally distinct subpopulations: one that acts under normal conditions, and the other that acts under times of stress,” study senior author Linheng Li, PhD, a researcher at Stowers Institute for Medical Research in Kansas City, Mo., said in a statement.
Hematopoietic stem cells are heterogeneous, with some maintained in an active state, and others in a quiescent state that is related to their self-renewal capacity and linked to low metabolic activity, sometimes called HSC dormancy or hibernation, Dr. Li and his coinvestigators explained in their report.
However, despite their quiescence, the majority of those HSCs will not survive chemotherapeutic stress, they added.
A small subpopulation of cells termed reserve HSCs can survive stress related to 5-fluorouracil chemotherapy in mice and following transplantation, Dr. Li and his colleagues demonstrated in experiments described in the paper. By contrast, a much larger population of quiescent cells, primed HSCs, were chemotherapy sensitive.
“Most likely, primed HSCs reflect a transitional state between [reserve HSCs] and proliferating [short-term HSCs],” Dr. Li and his colleagues said in their report.
In one key experiment, Dr. Li and his coinvestigators used a cell surface marker to isolate reserve HSCs and primed HSCs, which were transplanted into mice. Following engraftment, the mice were treated with 5-fluorouracil. They found that reserve HSCs, reflected by their derived blood cells, were unaffected by treatment, whereas derivatives of primed HSCs declined.
In another experiment, the researchers labeled HSCs with fluorescent tags to identify their location in the bone marrow. They found reserve cells concentrated in a specific bone marrow niche adjacent to N-cad+ cells.
The N-cad+ cells were bone and marrow stromal progenitor cells (BMSPCs), giving rise to osteoblasts, adipocytes, and chondrocytes, according to investigators.
Ablating the N-cad+ niche cells impaired reserve HSC maintenance in both homeostasis and regeneration, investigators added.
The work was supported by the Stowers Institute for Medical Research, the National Cancer Institute, and other grant support. The researchers reported that they had no competing interests related to the study.
SOURCE: Zhao M et al. Cell Rep. 2019 Jan 15;26(3):652-69.e6.
A small subpopulation of quiescent hematopoietic stem cells (HSCs) that restores the HSC pool and supports hematopoietic regeneration after chemotherapy has been identified, researchers reported.
These “reserve” HSCs were resistant to chemotherapy, whereas “primed” HSCs were sensitive to DNA damage from chemotherapy, the researchers said.
Both reserve and active stem cells were maintained in the bone marrow by specific niches, researchers demonstrated in experiments described in detail in Cell Reports.
Of note, primitive reserve HSCs were maintained in a bone marrow region that enriches N-cadherin expressing (N-cad+) bone and marrow stromal progenitor cells. According to investigators, those N-cad+ cells protected primitive reserve HSCs from chemotherapy-related stress, which survived to support hematopoietic regeneration after myeloablation.
These findings advance understanding of HSC biology, and could open new avenues for treating blood diseases and autoimmune disorders.
“In effect, we showed that hematopoietic stem cells have functionally distinct subpopulations: one that acts under normal conditions, and the other that acts under times of stress,” study senior author Linheng Li, PhD, a researcher at Stowers Institute for Medical Research in Kansas City, Mo., said in a statement.
Hematopoietic stem cells are heterogeneous, with some maintained in an active state, and others in a quiescent state that is related to their self-renewal capacity and linked to low metabolic activity, sometimes called HSC dormancy or hibernation, Dr. Li and his coinvestigators explained in their report.
However, despite their quiescence, the majority of those HSCs will not survive chemotherapeutic stress, they added.
A small subpopulation of cells termed reserve HSCs can survive stress related to 5-fluorouracil chemotherapy in mice and following transplantation, Dr. Li and his colleagues demonstrated in experiments described in the paper. By contrast, a much larger population of quiescent cells, primed HSCs, were chemotherapy sensitive.
“Most likely, primed HSCs reflect a transitional state between [reserve HSCs] and proliferating [short-term HSCs],” Dr. Li and his colleagues said in their report.
In one key experiment, Dr. Li and his coinvestigators used a cell surface marker to isolate reserve HSCs and primed HSCs, which were transplanted into mice. Following engraftment, the mice were treated with 5-fluorouracil. They found that reserve HSCs, reflected by their derived blood cells, were unaffected by treatment, whereas derivatives of primed HSCs declined.
In another experiment, the researchers labeled HSCs with fluorescent tags to identify their location in the bone marrow. They found reserve cells concentrated in a specific bone marrow niche adjacent to N-cad+ cells.
The N-cad+ cells were bone and marrow stromal progenitor cells (BMSPCs), giving rise to osteoblasts, adipocytes, and chondrocytes, according to investigators.
Ablating the N-cad+ niche cells impaired reserve HSC maintenance in both homeostasis and regeneration, investigators added.
The work was supported by the Stowers Institute for Medical Research, the National Cancer Institute, and other grant support. The researchers reported that they had no competing interests related to the study.
SOURCE: Zhao M et al. Cell Rep. 2019 Jan 15;26(3):652-69.e6.
A small subpopulation of quiescent hematopoietic stem cells (HSCs) that restores the HSC pool and supports hematopoietic regeneration after chemotherapy has been identified, researchers reported.
These “reserve” HSCs were resistant to chemotherapy, whereas “primed” HSCs were sensitive to DNA damage from chemotherapy, the researchers said.
Both reserve and active stem cells were maintained in the bone marrow by specific niches, researchers demonstrated in experiments described in detail in Cell Reports.
Of note, primitive reserve HSCs were maintained in a bone marrow region that enriches N-cadherin expressing (N-cad+) bone and marrow stromal progenitor cells. According to investigators, those N-cad+ cells protected primitive reserve HSCs from chemotherapy-related stress, which survived to support hematopoietic regeneration after myeloablation.
These findings advance understanding of HSC biology, and could open new avenues for treating blood diseases and autoimmune disorders.
“In effect, we showed that hematopoietic stem cells have functionally distinct subpopulations: one that acts under normal conditions, and the other that acts under times of stress,” study senior author Linheng Li, PhD, a researcher at Stowers Institute for Medical Research in Kansas City, Mo., said in a statement.
Hematopoietic stem cells are heterogeneous, with some maintained in an active state, and others in a quiescent state that is related to their self-renewal capacity and linked to low metabolic activity, sometimes called HSC dormancy or hibernation, Dr. Li and his coinvestigators explained in their report.
However, despite their quiescence, the majority of those HSCs will not survive chemotherapeutic stress, they added.
A small subpopulation of cells termed reserve HSCs can survive stress related to 5-fluorouracil chemotherapy in mice and following transplantation, Dr. Li and his colleagues demonstrated in experiments described in the paper. By contrast, a much larger population of quiescent cells, primed HSCs, were chemotherapy sensitive.
“Most likely, primed HSCs reflect a transitional state between [reserve HSCs] and proliferating [short-term HSCs],” Dr. Li and his colleagues said in their report.
In one key experiment, Dr. Li and his coinvestigators used a cell surface marker to isolate reserve HSCs and primed HSCs, which were transplanted into mice. Following engraftment, the mice were treated with 5-fluorouracil. They found that reserve HSCs, reflected by their derived blood cells, were unaffected by treatment, whereas derivatives of primed HSCs declined.
In another experiment, the researchers labeled HSCs with fluorescent tags to identify their location in the bone marrow. They found reserve cells concentrated in a specific bone marrow niche adjacent to N-cad+ cells.
The N-cad+ cells were bone and marrow stromal progenitor cells (BMSPCs), giving rise to osteoblasts, adipocytes, and chondrocytes, according to investigators.
Ablating the N-cad+ niche cells impaired reserve HSC maintenance in both homeostasis and regeneration, investigators added.
The work was supported by the Stowers Institute for Medical Research, the National Cancer Institute, and other grant support. The researchers reported that they had no competing interests related to the study.
SOURCE: Zhao M et al. Cell Rep. 2019 Jan 15;26(3):652-69.e6.
FROM CELL REPORTS
Key clinical point:
Major finding: Reserve HSCs were resistant to chemotherapy, whereas by contrast, primed HSCs were chemosensitive.
Study details: A series of preclinical experiments designed to characterize HSCs and document the effects of chemotherapy-related stress.
Disclosures: The work was supported by the Stowers Institute for Medical Research, the National Cancer Institute, and other grant support. The researchers reported that they had no competing interests related to the study.
Source: Zhao M et al. Cell Rep. 2019 Jan 15;26(3):652-69.e6.
PCSK9 inhibition isn’t the answer for high Lp(a)
CHICAGO – according to the results of the ANITSCHKOW study, Erik S. Stroes, MD, PhD, reported at the American Heart Association scientific sessions.

“The reality is that for now we don’t have any drugs to significantly lower elevated Lp(a),” he said. “We can identify patients with elevated Lp(a), but we don’t have a clue how to treat them.”
Elevated Lp(a) is a highly prevalent lipid abnormality. It induces arterial wall inflammation, a known predictor of future cardiovascular events. The monoclonal antibodies that inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9) dramatically reduce LDL cholesterol and also reduce arterial wall inflammation. In the published studies, PCSK9 inhibitors also reduced Lp(a) by an average of 27%; however, most participants in those studies had isolated high LDL with a normal or slightly elevated Lp(a).
ANITSCHKOW was the first double-blind, randomized, placebo-controlled study to look at the effects of a PCSK9 inhibitor – in this case, evolocumab (Repatha) – in patients with severe elevations in both LDL and Lp(a). The results proved disappointing yet informative, according to Dr. Stroes, professor of internal medicine and a vascular medicine specialist at the University of Amsterdam.
The 16-week, 14-site trial included 128 Dutch, American, and Canadian patients with a mean baseline LDL of 146 mg/dL and a median Lp(a) of 202 nmol/L who were randomized to monthly subcutanous injections of evolocumab at 420 mg or placebo. All participants had evidence of significant arterial wall inflammation at baseline as measured by PET-CT. Of the subjects, 54% were on statin therapy.
Evolucumab achieved a placebo-subtracted 61% reduction in LDL to 60 mg/dL but a mere 14% reduction in Lp(a) to 188 nmol/L, still far in excess of the 50 nmol/L cutoff defining elevated Lp(a).
The primary endpoint was change in arterial wall inflammation from baseline to week 16 as measured using PET-CT. Based upon the results of other studies showing a 3.3% drop in arterial wall inflammation for every 10% reduction in LDL, Dr. Stroes and his coinvestigators expected to see a 20% decrease in arterial wall inflammation in the evolocumab group. Instead, they found a mere 8.4% reduction, which wasn’t significantly different than in placebo-treated controls. And there was no difference in arterial wall inflammation between the group on concomitant statin therapy and those who weren’t.
The implication is that the residual Lp(a) elevation despite PCSK9 inhibitor therapy might explain the discrepancy, compared with previous studies in which LDL lowering did reduce arterial wall inflammation, according to Dr. Stroes.
“Persistent arterial wall inflammation on PET-CT after evolocumab, potentially related to persistent Lp(a) elevation, implies the need for additional therapies to decrease the proinflammatory state in Lp(a) elevation,” he observed.
Lp(a) in the spotlight
An elevated Lp(a) of 50 nmol/L or more is present in 20% of the general population, according to a Danish study. More than 70% of a person’s Lp(a) level is genetically driven. And a genetically driven elevated Lp(a) has been shown to be associated with a twofold to fourfold increased risk of cardiovascular events.
Moreover, other investigators have shown that a severely elevated Lp(a) (greater than 180 nmol/L) poses a cardiovascular risk comparable with that of heterozygous familial hypercholesterolemia and is present in 1 in 100 individuals.
“We spend a lot of time on familial hypercholesterolemia, and we should. But mind you, this severe Lp(a) elevation is more frequent than heterozygous FH,” Dr. Stroes said.
Session cochair Robert H. Eckel, MD, asked the audience for a show of hands by those who regularly measure Lp(a) in their patients. Very few hands were raised.
“I measure Lp(a) frequently, and I think it’s a very important risk factor,” declared Dr. Eckel, professor of medicine and director of the lipid clinic at University of Colorado Hospital, Aurora.
The ANITSCHKOW study was sponsored by Amgen. Dr. Stroes reported receiving institutional research grants from and serving as a paid speaker for Amgen, Merck, Novartis, and Regeneron.
CHICAGO – according to the results of the ANITSCHKOW study, Erik S. Stroes, MD, PhD, reported at the American Heart Association scientific sessions.
“The reality is that for now we don’t have any drugs to significantly lower elevated Lp(a),” he said. “We can identify patients with elevated Lp(a), but we don’t have a clue how to treat them.”
Elevated Lp(a) is a highly prevalent lipid abnormality. It induces arterial wall inflammation, a known predictor of future cardiovascular events. The monoclonal antibodies that inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9) dramatically reduce LDL cholesterol and also reduce arterial wall inflammation. In the published studies, PCSK9 inhibitors also reduced Lp(a) by an average of 27%; however, most participants in those studies had isolated high LDL with a normal or slightly elevated Lp(a).
ANITSCHKOW was the first double-blind, randomized, placebo-controlled study to look at the effects of a PCSK9 inhibitor – in this case, evolocumab (Repatha) – in patients with severe elevations in both LDL and Lp(a). The results proved disappointing yet informative, according to Dr. Stroes, professor of internal medicine and a vascular medicine specialist at the University of Amsterdam.
The 16-week, 14-site trial included 128 Dutch, American, and Canadian patients with a mean baseline LDL of 146 mg/dL and a median Lp(a) of 202 nmol/L who were randomized to monthly subcutanous injections of evolocumab at 420 mg or placebo. All participants had evidence of significant arterial wall inflammation at baseline as measured by PET-CT. Of the subjects, 54% were on statin therapy.
Evolucumab achieved a placebo-subtracted 61% reduction in LDL to 60 mg/dL but a mere 14% reduction in Lp(a) to 188 nmol/L, still far in excess of the 50 nmol/L cutoff defining elevated Lp(a).
The primary endpoint was change in arterial wall inflammation from baseline to week 16 as measured using PET-CT. Based upon the results of other studies showing a 3.3% drop in arterial wall inflammation for every 10% reduction in LDL, Dr. Stroes and his coinvestigators expected to see a 20% decrease in arterial wall inflammation in the evolocumab group. Instead, they found a mere 8.4% reduction, which wasn’t significantly different than in placebo-treated controls. And there was no difference in arterial wall inflammation between the group on concomitant statin therapy and those who weren’t.
The implication is that the residual Lp(a) elevation despite PCSK9 inhibitor therapy might explain the discrepancy, compared with previous studies in which LDL lowering did reduce arterial wall inflammation, according to Dr. Stroes.
“Persistent arterial wall inflammation on PET-CT after evolocumab, potentially related to persistent Lp(a) elevation, implies the need for additional therapies to decrease the proinflammatory state in Lp(a) elevation,” he observed.
Lp(a) in the spotlight
An elevated Lp(a) of 50 nmol/L or more is present in 20% of the general population, according to a Danish study. More than 70% of a person’s Lp(a) level is genetically driven. And a genetically driven elevated Lp(a) has been shown to be associated with a twofold to fourfold increased risk of cardiovascular events.
Moreover, other investigators have shown that a severely elevated Lp(a) (greater than 180 nmol/L) poses a cardiovascular risk comparable with that of heterozygous familial hypercholesterolemia and is present in 1 in 100 individuals.
“We spend a lot of time on familial hypercholesterolemia, and we should. But mind you, this severe Lp(a) elevation is more frequent than heterozygous FH,” Dr. Stroes said.
Session cochair Robert H. Eckel, MD, asked the audience for a show of hands by those who regularly measure Lp(a) in their patients. Very few hands were raised.
“I measure Lp(a) frequently, and I think it’s a very important risk factor,” declared Dr. Eckel, professor of medicine and director of the lipid clinic at University of Colorado Hospital, Aurora.
The ANITSCHKOW study was sponsored by Amgen. Dr. Stroes reported receiving institutional research grants from and serving as a paid speaker for Amgen, Merck, Novartis, and Regeneron.
CHICAGO – according to the results of the ANITSCHKOW study, Erik S. Stroes, MD, PhD, reported at the American Heart Association scientific sessions.
“The reality is that for now we don’t have any drugs to significantly lower elevated Lp(a),” he said. “We can identify patients with elevated Lp(a), but we don’t have a clue how to treat them.”
Elevated Lp(a) is a highly prevalent lipid abnormality. It induces arterial wall inflammation, a known predictor of future cardiovascular events. The monoclonal antibodies that inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9) dramatically reduce LDL cholesterol and also reduce arterial wall inflammation. In the published studies, PCSK9 inhibitors also reduced Lp(a) by an average of 27%; however, most participants in those studies had isolated high LDL with a normal or slightly elevated Lp(a).
ANITSCHKOW was the first double-blind, randomized, placebo-controlled study to look at the effects of a PCSK9 inhibitor – in this case, evolocumab (Repatha) – in patients with severe elevations in both LDL and Lp(a). The results proved disappointing yet informative, according to Dr. Stroes, professor of internal medicine and a vascular medicine specialist at the University of Amsterdam.
The 16-week, 14-site trial included 128 Dutch, American, and Canadian patients with a mean baseline LDL of 146 mg/dL and a median Lp(a) of 202 nmol/L who were randomized to monthly subcutanous injections of evolocumab at 420 mg or placebo. All participants had evidence of significant arterial wall inflammation at baseline as measured by PET-CT. Of the subjects, 54% were on statin therapy.
Evolucumab achieved a placebo-subtracted 61% reduction in LDL to 60 mg/dL but a mere 14% reduction in Lp(a) to 188 nmol/L, still far in excess of the 50 nmol/L cutoff defining elevated Lp(a).
The primary endpoint was change in arterial wall inflammation from baseline to week 16 as measured using PET-CT. Based upon the results of other studies showing a 3.3% drop in arterial wall inflammation for every 10% reduction in LDL, Dr. Stroes and his coinvestigators expected to see a 20% decrease in arterial wall inflammation in the evolocumab group. Instead, they found a mere 8.4% reduction, which wasn’t significantly different than in placebo-treated controls. And there was no difference in arterial wall inflammation between the group on concomitant statin therapy and those who weren’t.
The implication is that the residual Lp(a) elevation despite PCSK9 inhibitor therapy might explain the discrepancy, compared with previous studies in which LDL lowering did reduce arterial wall inflammation, according to Dr. Stroes.
“Persistent arterial wall inflammation on PET-CT after evolocumab, potentially related to persistent Lp(a) elevation, implies the need for additional therapies to decrease the proinflammatory state in Lp(a) elevation,” he observed.
Lp(a) in the spotlight
An elevated Lp(a) of 50 nmol/L or more is present in 20% of the general population, according to a Danish study. More than 70% of a person’s Lp(a) level is genetically driven. And a genetically driven elevated Lp(a) has been shown to be associated with a twofold to fourfold increased risk of cardiovascular events.
Moreover, other investigators have shown that a severely elevated Lp(a) (greater than 180 nmol/L) poses a cardiovascular risk comparable with that of heterozygous familial hypercholesterolemia and is present in 1 in 100 individuals.
“We spend a lot of time on familial hypercholesterolemia, and we should. But mind you, this severe Lp(a) elevation is more frequent than heterozygous FH,” Dr. Stroes said.
Session cochair Robert H. Eckel, MD, asked the audience for a show of hands by those who regularly measure Lp(a) in their patients. Very few hands were raised.
“I measure Lp(a) frequently, and I think it’s a very important risk factor,” declared Dr. Eckel, professor of medicine and director of the lipid clinic at University of Colorado Hospital, Aurora.
The ANITSCHKOW study was sponsored by Amgen. Dr. Stroes reported receiving institutional research grants from and serving as a paid speaker for Amgen, Merck, Novartis, and Regeneron.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point: Evolocumab has no effect on arterial wall inflammation in patients with severely elevated Lp(a).
Major finding: Median Lp(a) declined modestly from 202 nmol/L to 188 nmol/L in response to evolocumab.
Study details: This multicenter, 16-week, double-blind, placebo-controlled study included 128 patients with both elevated LDL and Lp(a).
Disclosures: The ANITSCHKOW study was sponsored by Amgen. The presenter reported receiving institutional research grants from and serving as a paid speaker for Amgen, Merck, Novartis, and Regeneron.
Flu activity increases after 2 weeks of declines
according to the Centers for Disease Control and Prevention.
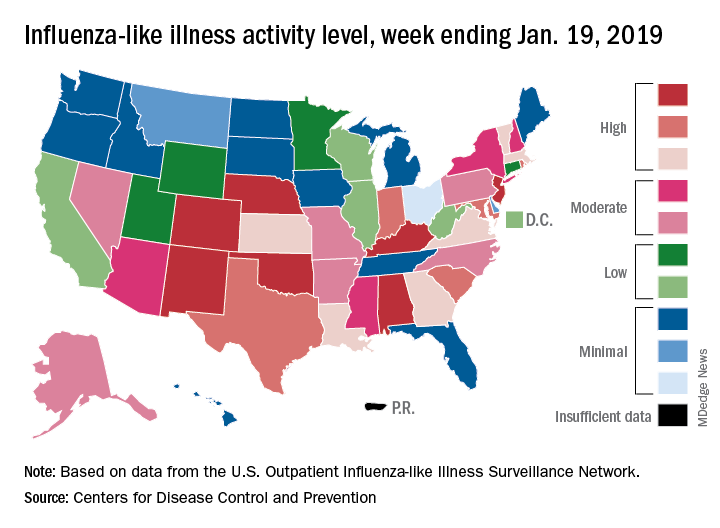
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.3% for the most recent measurement period, the CDC’s influenza division reported Jan 25. The previous 2-week decline had seen ILI visits dip down to 3.1% for the week ending Jan. 12 after hitting a season high of 4%.
To go along with the national increase in visits, more states reported high levels of flu activity. For the week ending Jan. 19, seven states were at level 10 on the CDC’s 1-10 scale, compared with four the previous week, and there were 18 states in the high range (levels 8-10), compared with 9 the week before, the CDC said.
Three flu-related pediatric deaths were reported in the week ending Jan. 19, although all three occurred during earlier weeks. The total number of pediatric deaths for the 2018-2019 season is now up to 22. Deaths among all ages, which are reported a week later, totaled 118 for the week ending Jan. 12, with 63% of reporting complete. There were 144 deaths during the week ending Jan. 5, with reporting 86% complete. During the second full week of 2018, in the middle of the very severe 2017-2018 season, there were 1,537 flu-related deaths, CDC data show.
Women with RA have reduced chance of live birth after assisted reproduction treatment
Women with rheumatoid arthritis who undergo assisted reproduction treatment have a decreased likelihood of live births versus women without rheumatoid arthritis, according to authors of a recent Denmark-wide cohort study.
The problem might be caused by an impaired chance of embryo implantation, authors of the study reported in the Annals of the Rheumatic Diseases.
Corticosteroids prescribed before embryo transfer might have improved the likelihood of live birth in these women with rheumatoid arthritis, according to the investigators, led by Professor Bente Mertz Nørgård of the center for clinical epidemiology at the Odense (Denmark) University Hospital.
However, findings with regard to that potential effect of corticosteroids were “not unambiguous,” said Prof. Nørgård and her coauthors said in their report.
The cohort study, based on Danish registry data from 1994-2017, included 1,149 embryo transfers in women and rheumatoid arthritis with 198,941 embryo transfers in women without rheumatoid arthritis.
Live births per embryo transfer were less likely in women with rheumatoid arthritis versus those they were in women with no rheumatoid arthritis, with an adjusted odds ratio of 0.78 (95% confidence interval, 0.65-0.92), according to investigators.
Chances of biochemical and clinical pregnancies were also lower in women with rheumatoid arthritis, with odds ratios of 0.81 (95% CI, 0.68-0.95) and 0.82 (95% CI, 0.59-1.15), respectively, the investigators found in an analysis of secondary outcomes in the study.
Corticosteroids prescribed before embryo transfer increased odds of live birth, with an adjusted odds ratio of 1.32 (95% CI, 0.85-2.05), though the underlying reason why corticosteroids were prescribed could not be established in this data set, investigators cautioned.
“The impact of corticosteroid prior to embryo transfer, found in our study, could be due to a suppression of ‘abnormalities in the immune system’ in women with RA, but we have to underline that this is speculative,” Prof. Nørgård and her colleagues said in a discussion of their results.
Future investigations are needed to clarify the role of corticosteroids in women with rheumatoid arthritis undergoing assisted reproduction treatment, they added.
Support for the study came from the Research Foundation of the Region of Southern Denmark and the Free Research Foundation at Odense University Hospital. Dr. Nørgård and her coauthors said they had no competing interests related to the research.
SOURCE: Nørgård BM et al. Ann Rheum Dis. 2019 Jan 12. doi: 10.1136/annrheumdis-2018-214619.
Women with rheumatoid arthritis who undergo assisted reproduction treatment have a decreased likelihood of live births versus women without rheumatoid arthritis, according to authors of a recent Denmark-wide cohort study.
The problem might be caused by an impaired chance of embryo implantation, authors of the study reported in the Annals of the Rheumatic Diseases.
Corticosteroids prescribed before embryo transfer might have improved the likelihood of live birth in these women with rheumatoid arthritis, according to the investigators, led by Professor Bente Mertz Nørgård of the center for clinical epidemiology at the Odense (Denmark) University Hospital.
However, findings with regard to that potential effect of corticosteroids were “not unambiguous,” said Prof. Nørgård and her coauthors said in their report.
The cohort study, based on Danish registry data from 1994-2017, included 1,149 embryo transfers in women and rheumatoid arthritis with 198,941 embryo transfers in women without rheumatoid arthritis.
Live births per embryo transfer were less likely in women with rheumatoid arthritis versus those they were in women with no rheumatoid arthritis, with an adjusted odds ratio of 0.78 (95% confidence interval, 0.65-0.92), according to investigators.
Chances of biochemical and clinical pregnancies were also lower in women with rheumatoid arthritis, with odds ratios of 0.81 (95% CI, 0.68-0.95) and 0.82 (95% CI, 0.59-1.15), respectively, the investigators found in an analysis of secondary outcomes in the study.
Corticosteroids prescribed before embryo transfer increased odds of live birth, with an adjusted odds ratio of 1.32 (95% CI, 0.85-2.05), though the underlying reason why corticosteroids were prescribed could not be established in this data set, investigators cautioned.
“The impact of corticosteroid prior to embryo transfer, found in our study, could be due to a suppression of ‘abnormalities in the immune system’ in women with RA, but we have to underline that this is speculative,” Prof. Nørgård and her colleagues said in a discussion of their results.
Future investigations are needed to clarify the role of corticosteroids in women with rheumatoid arthritis undergoing assisted reproduction treatment, they added.
Support for the study came from the Research Foundation of the Region of Southern Denmark and the Free Research Foundation at Odense University Hospital. Dr. Nørgård and her coauthors said they had no competing interests related to the research.
SOURCE: Nørgård BM et al. Ann Rheum Dis. 2019 Jan 12. doi: 10.1136/annrheumdis-2018-214619.
Women with rheumatoid arthritis who undergo assisted reproduction treatment have a decreased likelihood of live births versus women without rheumatoid arthritis, according to authors of a recent Denmark-wide cohort study.
The problem might be caused by an impaired chance of embryo implantation, authors of the study reported in the Annals of the Rheumatic Diseases.
Corticosteroids prescribed before embryo transfer might have improved the likelihood of live birth in these women with rheumatoid arthritis, according to the investigators, led by Professor Bente Mertz Nørgård of the center for clinical epidemiology at the Odense (Denmark) University Hospital.
However, findings with regard to that potential effect of corticosteroids were “not unambiguous,” said Prof. Nørgård and her coauthors said in their report.
The cohort study, based on Danish registry data from 1994-2017, included 1,149 embryo transfers in women and rheumatoid arthritis with 198,941 embryo transfers in women without rheumatoid arthritis.
Live births per embryo transfer were less likely in women with rheumatoid arthritis versus those they were in women with no rheumatoid arthritis, with an adjusted odds ratio of 0.78 (95% confidence interval, 0.65-0.92), according to investigators.
Chances of biochemical and clinical pregnancies were also lower in women with rheumatoid arthritis, with odds ratios of 0.81 (95% CI, 0.68-0.95) and 0.82 (95% CI, 0.59-1.15), respectively, the investigators found in an analysis of secondary outcomes in the study.
Corticosteroids prescribed before embryo transfer increased odds of live birth, with an adjusted odds ratio of 1.32 (95% CI, 0.85-2.05), though the underlying reason why corticosteroids were prescribed could not be established in this data set, investigators cautioned.
“The impact of corticosteroid prior to embryo transfer, found in our study, could be due to a suppression of ‘abnormalities in the immune system’ in women with RA, but we have to underline that this is speculative,” Prof. Nørgård and her colleagues said in a discussion of their results.
Future investigations are needed to clarify the role of corticosteroids in women with rheumatoid arthritis undergoing assisted reproduction treatment, they added.
Support for the study came from the Research Foundation of the Region of Southern Denmark and the Free Research Foundation at Odense University Hospital. Dr. Nørgård and her coauthors said they had no competing interests related to the research.
SOURCE: Nørgård BM et al. Ann Rheum Dis. 2019 Jan 12. doi: 10.1136/annrheumdis-2018-214619.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Women with rheumatoid arthritis (RA) who undergo assisted reproduction treatment have decreased chances of live births, compared with women without RA.
Major finding: The odds ratio for live births per embryo transfer in women with RA, as compared to women without RA, was 0.78 (95% confidence interval, 0.65-0.92).
Study details: A nationwide cohort study including 1,149 embryo transfers in women with rheumatoid arthritis and 198,941 without rheumatoid arthritis
Disclosures: Study authors had no disclosures. Support for the study came from the Research Foundation of the Region of Southern Denmark and the Free Research Foundation at Odense (Denmark) University Hospital.
Source: Nørgård BM et al. Ann Rheum Dis. 2019 Jan 12. doi: 10.1136/annrheumdis-2018-214619.
Impaired clot lysis associated with mild bleeding symptoms
Patients with self-reported mild bleeding symptoms may have impaired clot lysis, according to investigators. This finding is remarkable because it contrasts with known bleeding disorders, such as hemophilia, which are associated with enhanced clot lysis, reported lead author Minka J.A. Vries, MD, of the Cardiovascular Research Institute Maastricht (CARIM) at Maastricht (the Netherlands) University and her colleagues.
The observational study, which included 335 patients undergoing elective surgery at Maastricht University Medical Center, was conducted to better understand lysis capacity, which is challenging to assess in a clinical setting. Although the Euglobulin Lysis Time (ELT) is often used in the clinic, it cannot determine the influence of hemostatic proteins or formation of a fibrin clot under physiological conditions.
“In the more recently developed lysis assays,” the investigators wrote in Thrombosis Research, “the turbidity lysis assay and the tissue plasminogen activator–rotational thromboelastometry (tPA-ROTEM) [assay], all plasma proteins are present and fibrin is formed under more physiological conditions for the measurement of fibrinolysis.” These two tests were used in the present study.
Of the 335 adult patients, 240 had self-reported mild bleeding symptoms, and 95 did not. Patients with bleeding disorders, thrombocytopenia, or anemia were excluded, as were pregnant women and those taking blood thinners or NSAIDs. Along with assessing time parameters of fibrinolysis, clot-associated proteins were measured for possible imbalances.
“We hypothesized that clot lysis capacity is enhanced in patients with mild bleeding symptoms,” the investigators wrote, based on other bleeding disorders. Surprisingly, the results told a different story.
After adjusting for sex, BMI, and age, patients with bleeding symptoms had lower tPA-ROTEM lysis speed (beta −0.35; P = .007) and longer tPA-ROTEM lysis time (beta 0.29; P = .022) than did patients without bleeding symptoms. The investigators found that tPA-ROTEM measurements depended on factor II, factor XII, alpha2-antiplasmin, plasminogen, thrombin activatable fibrinolysis inhibitor (TAFI), and plasminogen activator inhibitor–1 (PAI-1) level. In contrast, turbidity lysis assay measurements were not significantly different between groups. This latter assay was influenced by alpha2-antiplasmin, TAFI, and PAI-1.
“We did not find evidence for systemic hyperfibrinolytic capacity in patients reporting mild bleeding symptoms in comparison to patients not reporting bleeding symptoms,” the investigators concluded. “tPA-ROTEM even suggested a slower clot lysis in these patients. Though this may appear counterintuitive, our results are in line with two papers assessing systemic clot lysis in mild bleeders.”
While this phenomenon gains supporting evidence, it remains poorly understood.
“We have no good explanation for these findings,” the investigators noted.
This study was funded by the Sint Annadal Foundation Maastricht, Maastricht University Medical Centre, CTMM INCOAG Maastricht, Cardiovascular Research Institute Maastricht, and the British Heart Foundation. No conflicts of interest were reported.
SOURCE: Vries MJA et al. Thromb Res. 2018 Dec 4. doi: 10.1016/j.thromres.2018.12.004.
Patients with self-reported mild bleeding symptoms may have impaired clot lysis, according to investigators. This finding is remarkable because it contrasts with known bleeding disorders, such as hemophilia, which are associated with enhanced clot lysis, reported lead author Minka J.A. Vries, MD, of the Cardiovascular Research Institute Maastricht (CARIM) at Maastricht (the Netherlands) University and her colleagues.
The observational study, which included 335 patients undergoing elective surgery at Maastricht University Medical Center, was conducted to better understand lysis capacity, which is challenging to assess in a clinical setting. Although the Euglobulin Lysis Time (ELT) is often used in the clinic, it cannot determine the influence of hemostatic proteins or formation of a fibrin clot under physiological conditions.
“In the more recently developed lysis assays,” the investigators wrote in Thrombosis Research, “the turbidity lysis assay and the tissue plasminogen activator–rotational thromboelastometry (tPA-ROTEM) [assay], all plasma proteins are present and fibrin is formed under more physiological conditions for the measurement of fibrinolysis.” These two tests were used in the present study.
Of the 335 adult patients, 240 had self-reported mild bleeding symptoms, and 95 did not. Patients with bleeding disorders, thrombocytopenia, or anemia were excluded, as were pregnant women and those taking blood thinners or NSAIDs. Along with assessing time parameters of fibrinolysis, clot-associated proteins were measured for possible imbalances.
“We hypothesized that clot lysis capacity is enhanced in patients with mild bleeding symptoms,” the investigators wrote, based on other bleeding disorders. Surprisingly, the results told a different story.
After adjusting for sex, BMI, and age, patients with bleeding symptoms had lower tPA-ROTEM lysis speed (beta −0.35; P = .007) and longer tPA-ROTEM lysis time (beta 0.29; P = .022) than did patients without bleeding symptoms. The investigators found that tPA-ROTEM measurements depended on factor II, factor XII, alpha2-antiplasmin, plasminogen, thrombin activatable fibrinolysis inhibitor (TAFI), and plasminogen activator inhibitor–1 (PAI-1) level. In contrast, turbidity lysis assay measurements were not significantly different between groups. This latter assay was influenced by alpha2-antiplasmin, TAFI, and PAI-1.
“We did not find evidence for systemic hyperfibrinolytic capacity in patients reporting mild bleeding symptoms in comparison to patients not reporting bleeding symptoms,” the investigators concluded. “tPA-ROTEM even suggested a slower clot lysis in these patients. Though this may appear counterintuitive, our results are in line with two papers assessing systemic clot lysis in mild bleeders.”
While this phenomenon gains supporting evidence, it remains poorly understood.
“We have no good explanation for these findings,” the investigators noted.
This study was funded by the Sint Annadal Foundation Maastricht, Maastricht University Medical Centre, CTMM INCOAG Maastricht, Cardiovascular Research Institute Maastricht, and the British Heart Foundation. No conflicts of interest were reported.
SOURCE: Vries MJA et al. Thromb Res. 2018 Dec 4. doi: 10.1016/j.thromres.2018.12.004.
Patients with self-reported mild bleeding symptoms may have impaired clot lysis, according to investigators. This finding is remarkable because it contrasts with known bleeding disorders, such as hemophilia, which are associated with enhanced clot lysis, reported lead author Minka J.A. Vries, MD, of the Cardiovascular Research Institute Maastricht (CARIM) at Maastricht (the Netherlands) University and her colleagues.
The observational study, which included 335 patients undergoing elective surgery at Maastricht University Medical Center, was conducted to better understand lysis capacity, which is challenging to assess in a clinical setting. Although the Euglobulin Lysis Time (ELT) is often used in the clinic, it cannot determine the influence of hemostatic proteins or formation of a fibrin clot under physiological conditions.
“In the more recently developed lysis assays,” the investigators wrote in Thrombosis Research, “the turbidity lysis assay and the tissue plasminogen activator–rotational thromboelastometry (tPA-ROTEM) [assay], all plasma proteins are present and fibrin is formed under more physiological conditions for the measurement of fibrinolysis.” These two tests were used in the present study.
Of the 335 adult patients, 240 had self-reported mild bleeding symptoms, and 95 did not. Patients with bleeding disorders, thrombocytopenia, or anemia were excluded, as were pregnant women and those taking blood thinners or NSAIDs. Along with assessing time parameters of fibrinolysis, clot-associated proteins were measured for possible imbalances.
“We hypothesized that clot lysis capacity is enhanced in patients with mild bleeding symptoms,” the investigators wrote, based on other bleeding disorders. Surprisingly, the results told a different story.
After adjusting for sex, BMI, and age, patients with bleeding symptoms had lower tPA-ROTEM lysis speed (beta −0.35; P = .007) and longer tPA-ROTEM lysis time (beta 0.29; P = .022) than did patients without bleeding symptoms. The investigators found that tPA-ROTEM measurements depended on factor II, factor XII, alpha2-antiplasmin, plasminogen, thrombin activatable fibrinolysis inhibitor (TAFI), and plasminogen activator inhibitor–1 (PAI-1) level. In contrast, turbidity lysis assay measurements were not significantly different between groups. This latter assay was influenced by alpha2-antiplasmin, TAFI, and PAI-1.
“We did not find evidence for systemic hyperfibrinolytic capacity in patients reporting mild bleeding symptoms in comparison to patients not reporting bleeding symptoms,” the investigators concluded. “tPA-ROTEM even suggested a slower clot lysis in these patients. Though this may appear counterintuitive, our results are in line with two papers assessing systemic clot lysis in mild bleeders.”
While this phenomenon gains supporting evidence, it remains poorly understood.
“We have no good explanation for these findings,” the investigators noted.
This study was funded by the Sint Annadal Foundation Maastricht, Maastricht University Medical Centre, CTMM INCOAG Maastricht, Cardiovascular Research Institute Maastricht, and the British Heart Foundation. No conflicts of interest were reported.
SOURCE: Vries MJA et al. Thromb Res. 2018 Dec 4. doi: 10.1016/j.thromres.2018.12.004.
FROM THROMBOSIS RESEARCH
Key clinical point: Patients with self-reported mild bleeding symptoms may have impaired clot lysis, in contrast with known bleeding disorders.
Major finding: Patients with mild bleeding had longer whole blood tissue plasminogen activator-rotational thromboelastometry lysis times (P = .022) than did patients without symptoms.
Study details: An observational study of 335 adult patients undergoing elective surgery.
Disclosures: This study was funded by the Sint Annadal Foundation, Maastricht University Medical Center, CTMM INCOAG Maastricht, Cardiovascular Research Institute Maastricht, and the British Heart Foundation. No conflicts of interest were reported.
Source: Vries MJA et al. Thromb Res. 2018 Dec 4. doi: 10.1016/j.thromres.2018.12.004.
Join the Conversation on SVSConnect
Transitioning toward retirement or looking for postgrad training opportunities? Or maybe you need some advice on good “power meals” to have during your busy days? These are only a few of the many threads attracting comments on SVSConnect, your online community. Members can discuss, collaborate, network and even share documents with one another. Log in with your SVS credentials and add your thoughts. If you’re not signed up, do so today.
Transitioning toward retirement or looking for postgrad training opportunities? Or maybe you need some advice on good “power meals” to have during your busy days? These are only a few of the many threads attracting comments on SVSConnect, your online community. Members can discuss, collaborate, network and even share documents with one another. Log in with your SVS credentials and add your thoughts. If you’re not signed up, do so today.
Transitioning toward retirement or looking for postgrad training opportunities? Or maybe you need some advice on good “power meals” to have during your busy days? These are only a few of the many threads attracting comments on SVSConnect, your online community. Members can discuss, collaborate, network and even share documents with one another. Log in with your SVS credentials and add your thoughts. If you’re not signed up, do so today.